ENDOCRINE EMERGENCIES
THOMAS BUCKLEY and MARGARET MURPHY
INTRODUCTION
The endocrine system is comprised of glands capable of synthesising and releasing chemical messengers known as hormones. One of the key roles of the endocrine system is to maintain an optimal internal environment throughout the life span, and, in the context of acute or critical illness, to initiate adaptive responses when emergency demands occur. Endocrine emergencies constitute only a fraction of emergency department (ED) workload, and, as such, healthcare professionals working in pre-hospital and emergency care may have limited experience in early detection and management of such emergencies. The most frequent endocrine emergencies are related to diabetes. With early detection and early interventions, diabetic emergencies may be successfully managed. In this chapter diabetic ketoacidosis is discussed, as well as other, less common endocrine emergencies, with emphasis on early assessment and initial management strategies.
Anatomy and physiology
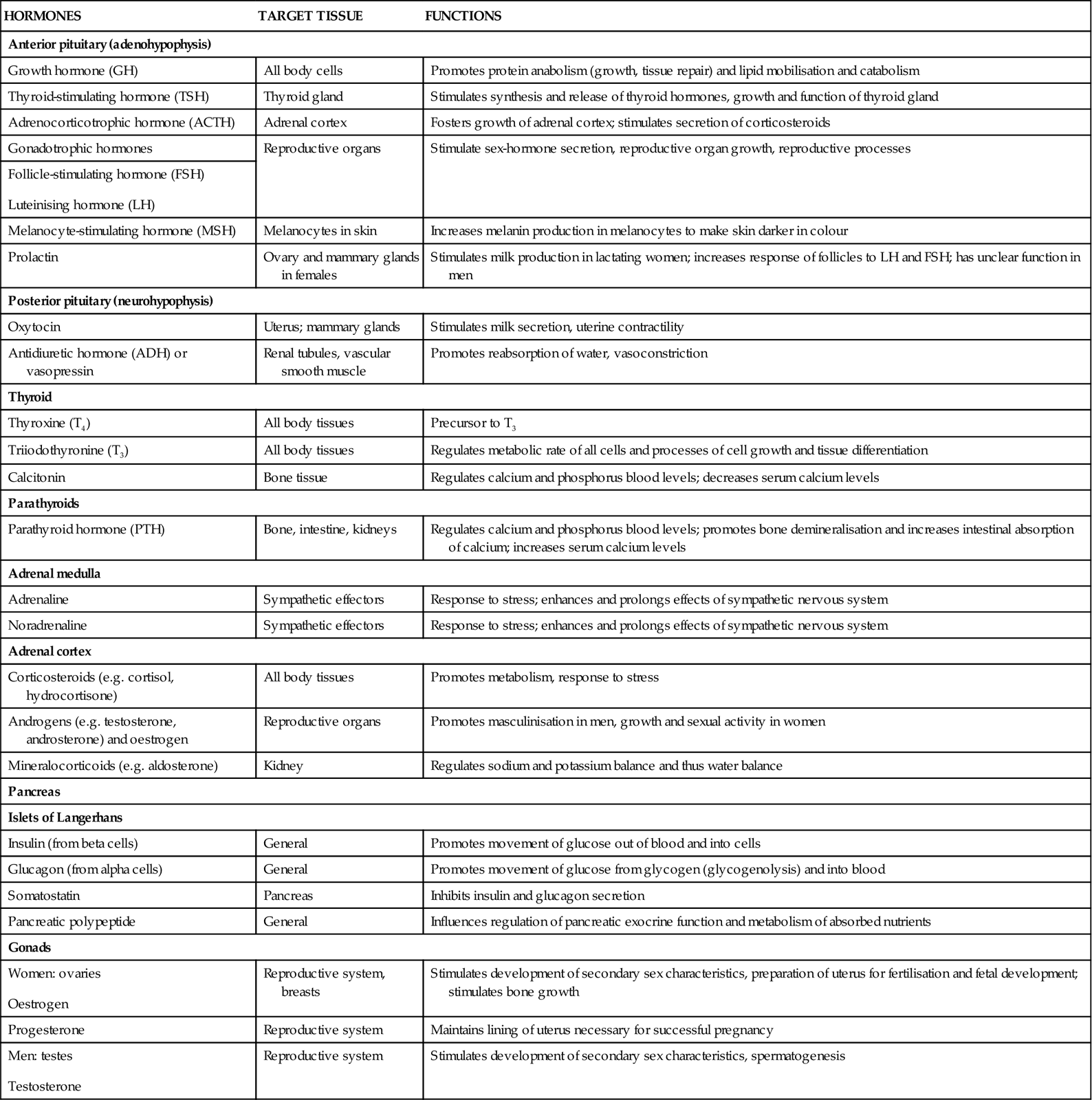
The endocrine system consists of the hypothalamus, pituitary, pineal gland, thyroid, parathyroid, adrenals, pancreas, testes (in males) and ovaries (in females) (Fig 26.1). Hormone molecules are transported via blood to their target tissues, where each hormone exerts its characteristic regulatory function at a cellular and molecular level.1 An overview of the major endocrine glands, hormones produced and functions is presented in Table 26.1.
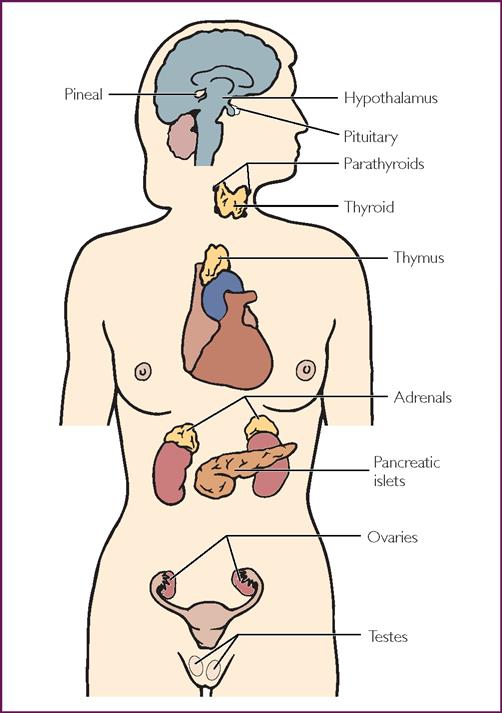
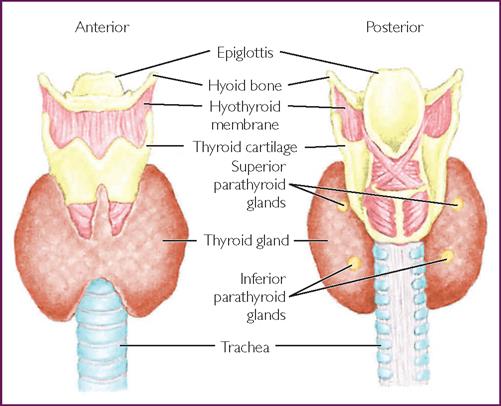
The parathyroid glands actually lie on the posterior surface of the thyroid gland.
TABLE 26.1
Primary endocrine glands and hormones2
| HORMONES | TARGET TISSUE | FUNCTIONS |
| Anterior pituitary (adenohypophysis) | ||
| Growth hormone (GH) | All body cells | Promotes protein anabolism (growth, tissue repair) and lipid mobilisation and catabolism |
| Thyroid-stimulating hormone (TSH) | Thyroid gland | Stimulates synthesis and release of thyroid hormones, growth and function of thyroid gland |
| Adrenocorticotrophic hormone (ACTH) | Adrenal cortex | Fosters growth of adrenal cortex; stimulates secretion of corticosteroids |
| Gonadotrophic hormones | Reproductive organs | Stimulate sex-hormone secretion, reproductive organ growth, reproductive processes |
|
Follicle-stimulating hormone (FSH) Luteinising hormone (LH) | ||
| Melanocyte-stimulating hormone (MSH) | Melanocytes in skin | Increases melanin production in melanocytes to make skin darker in colour |
| Prolactin | Ovary and mammary glands in females | Stimulates milk production in lactating women; increases response of follicles to LH and FSH; has unclear function in men |
| Posterior pituitary (neurohypophysis) | ||
| Oxytocin | Uterus; mammary glands | Stimulates milk secretion, uterine contractility |
| Antidiuretic hormone (ADH) or vasopressin | Renal tubules, vascular smooth muscle | Promotes reabsorption of water, vasoconstriction |
| Thyroid | ||
| Thyroxine (T4) | All body tissues | Precursor to T3 |
| Triiodothyronine (T3) | All body tissues | Regulates metabolic rate of all cells and processes of cell growth and tissue differentiation |
| Calcitonin | Bone tissue | Regulates calcium and phosphorus blood levels; decreases serum calcium levels |
| Parathyroids | ||
| Parathyroid hormone (PTH) | Bone, intestine, kidneys | Regulates calcium and phosphorus blood levels; promotes bone demineralisation and increases intestinal absorption of calcium; increases serum calcium levels |
| Adrenal medulla | ||
| Adrenaline | Sympathetic effectors | Response to stress; enhances and prolongs effects of sympathetic nervous system |
| Noradrenaline | Sympathetic effectors | Response to stress; enhances and prolongs effects of sympathetic nervous system |
| Adrenal cortex | ||
| Corticosteroids (e.g. cortisol, hydrocortisone) | All body tissues | Promotes metabolism, response to stress |
| Androgens (e.g. testosterone, androsterone) and oestrogen | Reproductive organs | Promotes masculinisation in men, growth and sexual activity in women |
| Mineralocorticoids (e.g. aldosterone) | Kidney | Regulates sodium and potassium balance and thus water balance |
| Pancreas | ||
| Islets of Langerhans | ||
| Insulin (from beta cells) | General | Promotes movement of glucose out of blood and into cells |
| Glucagon (from alpha cells) | General | Promotes movement of glucose from glycogen (glycogenolysis) and into blood |
| Somatostatin | Pancreas | Inhibits insulin and glucagon secretion |
| Pancreatic polypeptide | General | Influences regulation of pancreatic exocrine function and metabolism of absorbed nutrients |
| Gonads | ||
|
Women: ovaries Oestrogen |
Reproductive system, breasts | Stimulates development of secondary sex characteristics, preparation of uterus for fertilisation and fetal development; stimulates bone growth |
| Progesterone | Reproductive system | Maintains lining of uterus necessary for successful pregnancy |
|
Men: testes Testosterone |
Reproductive system | Stimulates development of secondary sex characteristics, spermatogenesis |
Most hormones are proteins and are synthesised and produced in endocrine glands throughout the body. Hormone release occurs in response to altered cellular environment and are regulated by one or more of the following mechanisms:
The most important regulatory mechanism is the negative feedback system where the endocrine system is controlled through negative feedback loops. An example of negative feedback is where thyroid-stimulating hormone (TSH) released from the anterior pituitary stimulates the synthesis and secretion of thyroid hormones. TSH secretion is regulated by thyrotropin-releasing hormone primarily in the hypothalamus and is inhibited by the thyroid-secreted hormones thyroxine (T4) and, to a lesser extent, triiodothyronine (T3).
Negative feedback systems maintain a delicate balance to ensure that hormone levels remain within physiological levels. Pathological conditions occur when there is a lack of negative-feedback inhibition resulting in excessive hormonal levels. Alerted renal and liver function, and external factors, such as pain, fear and stress, all influence hormone release. Neural stimulation, for example sympathetic activation during stress, results in increased levels of hormones such as adrenaline and glucagon resulting in increasing heart rate, blood pressure and serum blood sugar levels. When the stress reduces, adrenaline and glucagon levels decrease and symptoms subside. Intrinsic rhythms (e.g. circadian rhythms) vary from hours to weeks and provide another method of hormone control.3,4
Once secreted into the circulatory system, hormones may be either water soluble or lipid soluble or circulate in a free or active form. Water-soluble hormones usually have a short half-life (e.g. insulin with a half-life of 3–5 minutes), whereas lipid-soluble hormones (e.g. cortisol or thyroxine) have a considerably longer half-life. Some hormones, such as cortisol, also have small amounts of unbound, circulating, free cortisol in addition to the bound form.1,3
Hypothalamus
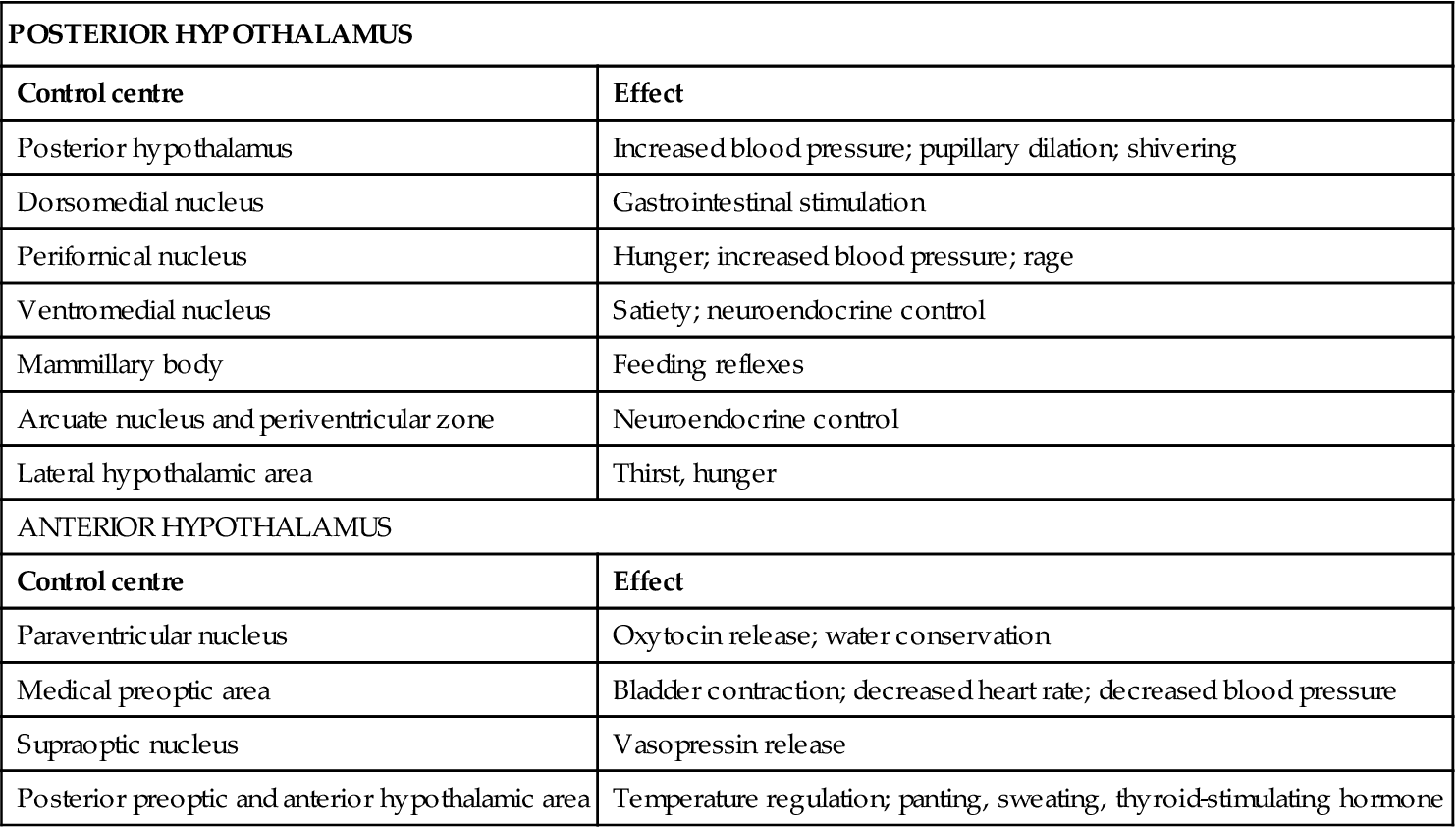
The hypothalamus creates part of the walls and floor of the third ventricle of the brain and has been described as an automatic nervous centre (Fig 26.2). Although the hypothalamus is small in size, centres in the anterior and posterior hypothalamus are responsible for performing numerous vital functions, most of which relate either directly or indirectly to the regulation of visceral activities (Table 26.2). The hypothalamus is responsible for limbic (emotional) regulation, as well as instinctual functions.1,3
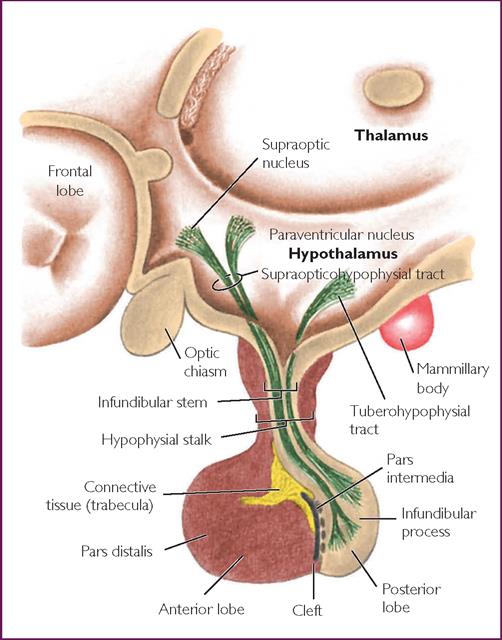
TABLE 26.2
Control centres of the hypothalamus
| POSTERIOR HYPOTHALAMUS | |
| Control centre | Effect |
| Posterior hypothalamus | Increased blood pressure; pupillary dilation; shivering |
| Dorsomedial nucleus | Gastrointestinal stimulation |
| Perifornical nucleus | Hunger; increased blood pressure; rage |
| Ventromedial nucleus | Satiety; neuroendocrine control |
| Mammillary body | Feeding reflexes |
| Arcuate nucleus and periventricular zone | Neuroendocrine control |
| Lateral hypothalamic area | Thirst, hunger |
| ANTERIOR HYPOTHALAMUS | |
| Control centre | Effect |
| Paraventricular nucleus | Oxytocin release; water conservation |
| Medical preoptic area | Bladder contraction; decreased heart rate; decreased blood pressure |
| Supraoptic nucleus | Vasopressin release |
| Posterior preoptic and anterior hypothalamic area | Temperature regulation; panting, sweating, thyroid-stimulating hormone |
The hypothalamus lies close to the pituitary and is linked to the brain by nerves and blood vessels. It produces neurosecretory chemicals that regulate anterior pituitary action through the stimulation or suppression of various hormones. These hormones are responsible for the regulation of other endocrine glands via the negative feedback loop. As the hypothalamus is one of the most vital structures of the body, dysfunction can have a serious effect on the autonomic, somatic or psychic functions of the body.1,3
Pituitary
The pituitary is located on the inferior aspect of the brain in the region of the diencephalon, and lies within the sella turcica of the middle cranial fossa (Fig 26.2). Pituitary secretions are controlled by the hypothalamus, as well as negative feedback from target glands. It is rounded and pea-shaped, measuring approximately 1 cm in diameter and is attached to the hypothalamus by the infundibular stalk.1 The pituitary gland is structurally and functionally divided into the anterior and posterior regions. The anterior region contains secretory cells, and secretes trophic hormones—the term trophic meaning ‘food’. The anterior pituitary hormones do not target food; rather they result in hypertrophy of their targets when levels are high, and result in atrophy of target organs when levels are low. Hormones that are secreted by the anterior pituitary include growth hormone, adrenocorticotrophic hormone (ACTH), thyroid-stimulating hormone (TSH), prolactin, follicle-stimulating hormone and luteinising hormone.1
The posterior pituitary consists of neural cells that serve as a supporting structure for nerve fibres and nerve endings, and secretes two hormones into the circulation: antidiuretic hormone (ADH) and oxytocin. Both ADH and oxytocin are produced by the hypothalamus and stored in the pituitary’s posterior lobe until required.1,6
Thyroid
The thyroid is the largest of the endocrine glands. It is butterfly shaped and positioned just below the larynx, partially surrounding the trachea (Fig 26.3). It consists of two lobes that lie on either side of the trachea which are connected anteriorly by a broad isthmus.1,3 The initiating hormone is thyrotropin-releasing hormone (TRH) which is synthesised and stored in the hypothalamus and released into the hypothalamic–pituitary portal system, circulates to the pituitary and stimulates the release of TSH. TSH stimulates the thyroid gland to produce thyroid hormone, a process that requires iodide. Ninety per cent of thyroid hormone is in the form of thyroxine (T4), and the remainder triiodothyronine (T3). These hormones are essential for proper growth and development, neurological function and the determination of basal metabolic rate (BMR).1 Generally, thyroid hormones exert a number of permissive effects on many organs but abnormally high or low levels can exert pronounced effects.
Adrenals
The adrenal glands (also called the suprarenal glands) are paired organs located in the retroperitoneal area above the upper pole of each kidney and embedded against the muscles of the back in a protective layer of fat. The adrenal glands are generally pyramidal in appearance, measuring approximately 50 mm long, 30 mm wide and 10 mm deep.4 Like the pituitary, the adrenal glands have dual origin. Each gland consists of an outer cortical layer and an inner medullary layer. They are functionally two different endocrine tissues, located in the same organ, and each secretes different hormones that are regulated by different control systems.
The bulk of the gland is made up by the adrenal cortex, which is divided into three zones. These are the outer zona glomerulosa, intermediate zona fasciculata and inner zona reticularis. The adrenal cortex secretes more than 50 steroid hormones, classified as glucocorticoids, mineralocorticoids and androgens.1,3 Cortisol, the most abundant glucocorticoid, is necessary for the maintenance of life and protection from stress. Cortisol has a half-life of approximately 90 minutes and increases blood glucose levels through the promotion of hepatic gluconeogenesis by facilitating conversion of amino acids to glucose and inhibiting protein synthesis.7 Pathophysiologically, high levels of aldosterone have been associated with hypertension, atherosclerosis and heart failure.8 Aldosterone is a potent mineralocorticoid that maintains extracellular fluid volume, regulated primarily by the renin–angiotensin–aldesterone system to alter serum sodium levels.
The adrenal medulla is composed of tightly packed clusters of cells, innervated by sympathetic neurons that are arranged around blood vessels. Impulses are initiated from the hypothalamus via the spinal cord when the sympathetic division of the autonomic nervous system (ANS) is stimulated. The cells of the medulla secrete the catecholamines adrenaline and noradrenaline in a ratio of approximately 4 : 1. Approximately 30% of adrenaline is secreted from the adrenal medulla; the remainder comes from nerve terminals. Adrenaline is up to 10 times more potent than noradrenaline, although the latter has a longer duration of action.1,3 Once released, catecholamines remain in the plasma for a very short duration, just several seconds, but result in increased cardiac output and heart rate, dilated coronary blood vessels, increased mental alertness, increased respiratory rate and elevated metabolic rate. Activation of the adrenal medulla together with sympathetic division of the ANS prepares the body for greater physical performance, the ‘fight or flight’ response.
Excessive stimulation of the adrenal medulla can result in depletion of the body’s energy reserves and a high level of corticosteroid secretion from the adrenal cortex. This can significantly impair the immune system. The major functions of catecholamines are summarised in Table 26.3.
TABLE 26.3
| CLASS AND FUNCTION | ALPHA-ADRENERGIC | BETA-ADRENERGIC | DOPAMINERGIC |
| Agonist | Noradrenaline | Adrenaline | Dopamine |
| Antagonist | Phentolamine | Propranolol | Haloperidol |
| Actions | |||
|
Cardiac Smooth muscle Metabolic |
Contracts |
Inotropic and chronotropic Relaxes Lipolysis Glycogenolysis Gluconeogenesis |
Inotropic Mixed |
| Molecular | Decreases cAMP | Increases cAMP | Increases cAMP |
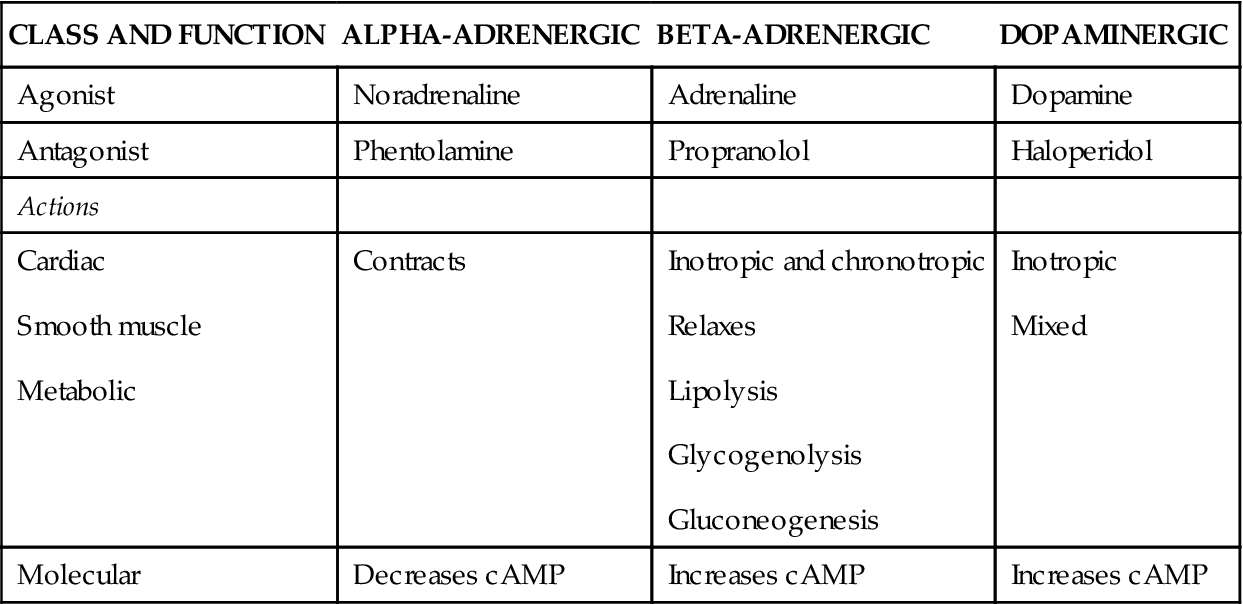
cAMP: cyclic adenosine monophosphate; this is a second messenger and regulates the effects of adrenaline and glucagon
Pancreas
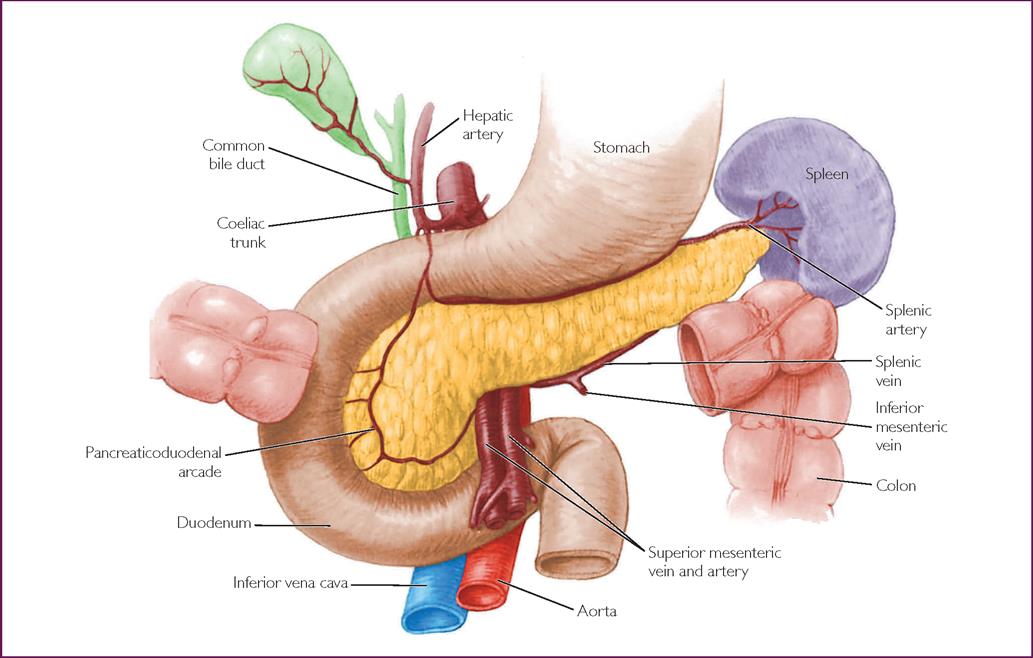
The pancreas is situated behind the stomach and anterior to the first and second lumbar vertebrae, in the retroperitoneal space. The body of the pancreas extends horizontally across the abdominal wall with the head in the curve of the abdomen and the tail touching the spleen. It is a soft, lobular gland (Fig 26.4). The pancreas has both endocrine and exocrine functions. Acini are exocrine cells that release amylase, lipase and other enzymes that aid digestion. The endocrine portion of the pancreas consists of scattered clusters of cells called the pancreatic islets, or islets of Langerhans. The islets account for less than 2% of the gland and consist of three types of hormone-secreting cells that produce hormones responsible for serum glucose regulation. Alpha cells produce and secrete glucagon, beta cells produce and secrete insulin and delta cells secrete somatostatin, which inhibits glucagon and insulin release.
Insulin, synthesised from the precursor proinsulin, is stimulated by increased serum glucose levels, the amino acids arginine and lysine, serum free-fatty acids and parasympathetic stimulation. Conversely, insulin secretion reduces in response to low serum glucose, high levels of insulin through the beta-cell negative feedback system and sympathetic stimulation. The primary role of insulin is to facilitate glucose uptake into cells; it is an anabolic hormone, promoting synthesis of proteins, lipids and nucleic acids resulting in increased metabolism (Table 26.4).
TABLE 26.4
Action of insulin10
| LIVER | ADIPOSE TISSUE | MUSCLE | |
| Anticatabolic effects |
↓ glycogenolysis ↓ gluconeogenesis ↓ ketogenesis |
↓ lipolysis |
↓ protein catabolism ↓ amino acid output ↓ amino acid oxidation |
| Anabolic effects |
↑ glycogen synthesis ↑ fatty acid synthesis |
↑ glycerol synthesis ↑ fatty acid synthesis |
↑ amino acid uptake ↑ protein synthesis ↑ glycogen synthesis |

Glucagon acts primarily in the liver, stimulating glycogenolysis and gluconeogenesis in response to hypoglycaemia, resulting in increased blood glucose.
Diabetes
Diabetes is a chronic disease caused by relative or absolute insulin insufficiency. Diabetes is the fastest growing chronic condition in Australia. In Australia, 280 people develop diabetes every day. Over 100,000 Australians have developed diabetes in the past year. Almost 1.1 million Australians currently have diagnosed diabetes.11 This includes:
• 120,000 people with type 1 diabetes
• 956,000 people with type 2 diabetes
• 23,600 women with gestational diabetes.11
Type 1 diabetes is characterised by destruction of insulin-producing beta cells caused by an autoimmune abnormality. Onset is usually rapid, although latent autoimmune diabetes (LADA) is a more slowly progressive autoimmune diabetes in adults. The exact cause of type 1 diabetes remains unknown and incidence has a strong family link. Type 2 diabetes is the most common form of diabetes in Australia, affecting 85–90% of all people with diabetes.11,12 Traditionally, type 2 diabetes has been associated with later adulthood, although incidence is increasing in children. Type 2 diabetes may range from predominant insulin resistance to a predominant insulin secretory defect, with or without insulin resistance.13 Gestational diabetes refers to glucose intolerance with onset during pregnancy. Between 3% and 8% of pregnant women develop gestational diabetes around the 24th to 28th week of pregnanacy. Women who develop gestational diabetes are at higher risk of developing type 2 diabetes.11,12
Other specific and less-common types of type 2 diabetes include:3,13
• genetic defects of beta cell function (e.g. MODY1–MODY6 sulfonylurea receptor [KCNJ11] genes)
• genetic defects in insulin action (e.g. type A insulin resistance and leprechaunism)
• diseases of the exocrine pancreas (e.g. pancreatitis, cystic fibrosis, haemochromatosis); endocrinopathies (e.g. Cushing syndrome)
• drug-induced or chemical-induced diabetes (e.g. glucocorticoids)
• infections (e.g. congenital rubella, cytomegalovirus)
• uncommon but specific forms of immune-mediated diabetes (e.g. ‘stiff-man’ syndrome and anti-insulin-receptor antibodies)
• other genetic syndromes sometimes associated with diabetes (e.g. Down syndrome, Wolfram syndrome, Turner’s syndrome and myotonic dystrophy).
Diabetic ketoacidosis
Diabetic ketoacidosis (DKA) is an acute, life-threatening condition characterised by hyperglycaemia, the presense of ketones in urine or blood and acidosis (arterial pH < 7.3 or HCO3− <16). It has long been assumed that DKA is pathognomic of type 1 diabetes with most presentations occurring in individuals with type 1 disease. However, it is now recognised that it can also occur in type 2 diabetes, especially in African-American and ethnic minority populations.3,11–13 Among patients who develop DKA, mortality rates are between 5% and 15%. Seventy per cent of diabetes-related deaths in children are attributed to DKA. It occurs in 20–30% of all new-onset presentations of diabetes; therefore, acute presentation often results in a diagnosis of diabetes.11,13
Hyperglycaemia is a result of severe insulin deficiency, either absolute or relative, which impairs peripheral glucose uptake and promotes fat breakdown. Relative glucagon excess promotes hepatic gluconeogenesis. Overall, metabolism in DKA shifts from the normal fed state characterised by carbohydrate metabolism to a fasting state characterised by fat metabolism (Fig 26.5). Secondary consequences of the primary metabolic derangements include ketosis, caused by a switch to fat metabolism, leading to free fatty acid oxidation in the liver. As a result the ketone bodies acetoacetic acid and 3-hydroxybutyric acid are formed. The dissociation of the ketone bodies (weak acids) results in acidosis due to depletion of extracellular and cellular buffers. As a consequence of hyperglycaemic state, renal threshold is surpassed and glucose is secreted in the urine, known as osmotic diuresis. This hyperglycaemia-induced osmotic diuresis depletes sodium, potassium, phosphates and water as well as ketones and glucose, resulting in absolute dehydration and electrolyte depletion.

Clinical features
Diabetic ketoacidosis tends to occur in leaner, young, type 1 diabetes patients and although symptoms may be present for several days, the presentation of DKA is usually rapid and predictable. DKA may be precipitated by factors that result in increased circulating levels of stress hormones, such as adrenaline, growth hormone and cortisol, and the resulting increase in insulin resistance reduces the effectiveness of any residual insulin production or injected insulin. Examples of precipitating causes may include infection, myocardial infarction, cerebrovascular causes, influenza, surgical emergencies, cocaine use or stress.14,15 These conditions often present with their own complex set of signs and symptoms, and therefore the possibility of DKA needs to always be considered in any diabetic patient presenting with the potential triggers listed above. Additionally, errors of insulin administration or patient manipulation with insulin treatment may be the precipitant of an acute DKA episode.
After prolonged insulin deficiency, hyperglycaemia leads to thirst, polyuria, dehydration, electrolyte depletion and metabolic acidosis. Patients with DKA often complain of non-specific symptoms such as malaise and fatigue. Nausea, vomiting and abdominal pain are common and although it is not fully understood why these symptoms occur, it is thought to be the result of delayed gastric emptying, ileus, subacute pancreatitis, oesophagitis with ulceration or bowel ischaemia.15,16
Physical signs include evidence of dehydration such as tachycardia; hypotension may be the result of volume depletion, sepsis or both. Additionally, skin examination may reveal diminished skin turgor, and dry mucous membranes due to dehydration. The most frequent cardiac rhythm is sinus tachycardia; however, dysrhythmias can occur secondary to electrolyte disturbances. The patient may also be tachypnoeic with Kussmaul respirations and a fruity odour to their breath as a result of exhaled acetone.13,15,16 The clinical features of DKA are summarised in Table 26.5.
TABLE 26.5
Clinical manifestations of diabetic ketoacidosis
| SYSTEM | CLINICAL MANIFESTATIONS |
| Neurological | Lethargy, malaise and fatigue, possible confusion and coma in extreme cases |
| Respiratory | Kussmaul respirations, fruity acetone breath |
| Cardiovascular | Tachycardia, hypotension, dysrhythmias |
| Integumentary | Flushed skin, dry mucous membranes, poor skin turgor, fever if infection |
| Renal | Thirst, polyuria, ketonuria, glucosuria |
| Gastrointestinal | Nausea, vomiting, abdominal cramps or pain |
Assessment and diagnosis
The first priority is to establish the severity of the presenting problem and hence the need for immediate intervention. The initial clinical examination follows the ABCD mnemonic to assess the potential or actual threat to Airway, Breathing, Circulation and Disability. Interventions may need to be initiated if these parameters are compromised. A focused secondary assessment then follows, usually directed by the presenting signs and symptoms.
In determining the severity of the patient’s illness and the need for intervention, it is important to determine the history of the presenting illness. Most patients presenting with DKA have a known history of diabetes, making differential diagnosis fairly uncomplicated. However, as patients may also have signs and symptoms related to the precipitant factor, or trigger, of the DKA, laboratory tests will be needed to confirm DKA. The signs and symptoms may develop rapidly, but there is usually a history of being unwell for days, predominantly with gastrointestinal symptoms, such as vomiting, excessive thirst or urination and abdominal pain. In more advanced stages the patient may be confused or obtunded. The patient’s breathing pattern may be described as rapid, referred to as Kussmaul’s respirations with a distinctive ‘sweet-smelling’ odour. Dehydration, occasionally associated with hypotension, tachycardia and delayed capillary refill, may also be present. However, there may also be excessive urinary output in a hypoperfused patient with significant weight loss (up to 5 kg/week).
Bedside testing of capillary blood glucose and beta-hydroxybutyrate levels using point-of-care testing, in patients with the above presentation, will confirm the diagnosis of diabetic ketoacidosis (see Ch 17, p. 332, for information on blood glucose testing techniques). A diligent search for the precipitant is also essential. These causes may include infection, cessation of insulin, trauma or acute myocardial infarction (may be painless), to name a few. Other bedside testing that may assist this process include urinalysis, electrocardiogram and vital signs monitoring.
Laboratory investigations
Laboratory tests should be obtained as early as possible. Diagnosis of DKA is confirmed in the presence of four clinical features:
1. Hyperglycaemia with serum glucose higher than 13.8 mmol/L
3. Metabolic acidosis with pH < 7.3
4. Dehydration.15
Other possible laboratory findings are summarised in Table 26.6.15,16,17
TABLE 26.6
Laboratory findings in diabetic ketoacidosis (DKA)
| LABORATORY ASSESSMENT | POSSIBLE FINDING | CLINICAL RELEVANCE |
| Sodium | For each 5.5 mmol/L increase in glucose from normal, serum sodium is lowered by 1.6 mmol/L | Increased sodium results from extravascular water movement to intervascular space due to the osmotic effect of hyperglycaemia |
| Potassium | Initial serum potassium levels may be normal or slightly elevated | Potassium levels can rapidly decrease after commencement of insulin as potassium moves into the cells |
| Beta-hydroxybutyrate | Levels greater than 0.5 mmol/L are considered abnormal, and levels of 3 mmol/L correlate with need for DKA treatment |
Beta-hydroxybutyrate, the more common ketone body, is not detected by Ketostix. Serum or capillary beta-hydroxybutyrate can be used to follow response to treatment. |
| Osmolality | Blood levels may be increased due to intravascular dehydration | DKA patients who are comatosed typically have values > 330 mOsm/kg. If the osmolality is less than this in a patient who is comatose, other causes of obtundation should be investigated |
| Amylase | Hyperamylasaemia | Hyperamylasaemia may be seen even in absence of pancreatitis. It is not understood why this occurs, but it occurs in up to 75% of patients with DKA |
| White cell count | Elevated white cell count > 15 × 109/L | Elevated white cell count may suggest underlying infection |
| Urea and creatinine | Elevated | Serum levels may be elevated due to dehydration |
Patient management
While DKA generally requires specialist treatment, early assessment and initial treatments should commence as soon as possible in the pre-hospital setting. Once initial treatments have been commenced, most patients with DKA can be nursed in high-dependency units or even general medical wards. Only patients with severe DKA or critical illness that precipitated the event usually require admission to the intensive care unit.
While most institutions have DKA management protocols to guide overall management, the generally agreed principles of DKA management are to correct fluid depletion, decrease the blood glucose level, correct the electrolyte imbalance and treat the precipitating causes.13,15,16 To ensure efficacy and safety of treatments, patients with DKA will require close monitoring of clinical and metabolic status to monitor response to treatments and recognise changes to the patient’s condition. For example, onset of headaches or decreased level of consciousness may indicate the development of cerebral oedema and the need for more-focused assessment and interventions in such patients.
Fluid depletion
Intravascular fluid depletion in adults with DKA can be significant. In the absence of heart failure, the fluid of choice in the resuscitation phase is sodium chloride 0.9%. If the patient is hypotensive (systolic blood pressure below 90 mmHg) 500 mL sodium chloride 0.9% is given intravenously over 10–15 minutes while requesting senior medical review.13 This rehydration therapy can be commenced by pre-hospital responders when treating adult patients. When the systolic blood pressure is above 90 mmHg fluid replacement for most people of average weight is 1000 mL within the first hour.16 Fluids should then be reduced to 1000 mL over 2 hours for 2 consecutive hours and after that, titrated to maintain adequate blood pressure, pulse, urinary output and mental status. Correcting intravascular dehydration will reduce plasma osmolarity and blood glucose levels. Care needs to be taken in cases of younger patients with DKA as rapid fluid infusion may result in cerebral oedema, which has a high mortality rate. For this reason many pre-hospital guidelines do not advocate this therapy. As a guide, the aim should be to reduce serum osmolarity not more than 3 mOsm/L/h or decrease sodium concentration by less than 1 mmol/hr to avoid potential cerebral oedema due to large fluid shifts. Additionally, if serum sodium rises above 155 mmol/L, switching to 0.45% sodium chloride may need to be considered, although the optimal time to use 0.45% sodium chloride remains uncertain.16
Many DKA protocols will recommend commencing glucose (5% dextrose) at 80–120 mL/h to prevent hypoglycaemia when blood glucose levels approach 15 mmol/L.14,18 The glucose rate is adjusted to achieve a target blood glucose level (BGL) of 10–15 mmol/L. Occasionally 10% glucose may be needed. Normal saline will be continued if hydration status requires.
Blood glucose level
Intravenous insulin will lower blood glucose concentration through increased glucose utilisation in peripheral tissues and a decrease in hepatic glucose production. Insulin will decrease ketone release, thereby correcting metabolic acidosis. Initial commencement of insulin infusion at a rate of approximately 5 units/hour (or 0.1 units/kg/h) is generally recommended to encourage a steady fall in blood glucose levels. However, infusions may need to be increased if initial rates do not reduce blood glucose levels after 1–2 hours. It is imperative that insulin therapy is continued until ketonaemia is resolved.
Blood glucose levels should be monitored at least hourly, with the aim to prevent blood sugar levels falling below 10 mmol/L, and 5% dextrose should be added to the fluid replacement regimen once blood sugar levels approach 12–14 mmol/L.14,18 It is recommended that a mechanical pump or syringe driver is used to deliver the infusion. All intravenous tubing must be flushed with the prepared insulin solution prior to patient administration to ensure tubing is coated with insulin solution as insulin adheres to the tubing. If intravenous infusion is not possible, then intramuscular-route infusion may be considered and can be successful in delivering 8–10 units of insulin hourly.13
The transition from intravenous insulin to subcutaneous injections can be initiated once glucose level is < 11 mmol/L, serum bicarbonate level > 18 mmol/L, venous pH > 7.30 and the patient has been eating and drinking for at least 24 hours.14,16 Transition to subcutaneous insulin can be challenging, and involvement of the diabetes team is recommended. Normal diet should be resumed prior to ceasing the infusion. The subcutaneous insulin or anti hyperglycaemic therapy is given with the next meal. An overlap time between the intravenous and subcutaneous insulin will be allowed depending on the type of insulin. Cease infusion 2 hours after giving regular insulin (i.e Actrapid, Humulin R, Mixtard 30/70, Mixtard 50/50, Humulin 30/70) or 1 hour after oral anti-hyperglycaemic therapy or rapid-acting insulin (i.e. Novarapid, Humalog, Apidra, Nova Mix 30, Humalog Mix 25, Humalog Mix 50).
Electrolyte imbalance
Total body depletion of potassium can occur in DKA, despite initial normal or slightly elevated serum levels (Table 26.6). To prevent acute hypokalaemia after fluid and insulin therapy, potassium replacement should be considered once the patient’s serum levels are known and adequate renal output established. The aim is to maintain serum potassium levels at between 4 and 5 mmol/L. Most DKA protocols will advocate commencing potassium replacement at a rate of 20 mmol/h if potassium levels are < 3.5 mmol/L, adjusted in the succeeding hours to a replacement rate of 10 mmol/h to maintain a serum potassium level of 3.5–5.5 mmol/L.13
Cease potassium infusion when the serum potassium is greater than 5.5 mmol/L.
While there are many slight variations on how to replace potassium, initial and 2-hourly monitoring of serum potassium and, if possible, continuous electrocardiogram are recommended during the resuscitation and treatment phases of DKA management. An example guide to potassium replacement is presented in Box 26.1.
Bicarbonate replacement in patients with initial metabolic acidosis remains controversial, with no clear benefit in clinical trials. However, in patients with severe acidosis (i.e. pH < 7), infusion of sodium bicarbonate may be considered after consultation with an endocrinologist when pH is < 7.0 and the HCO3− is <5.16,18 It is important to note that infusion of sodium bicarbonate is likely to lower serum potassium levels significantly. Sodium bicarbonate may be administered at a rate of 1–2 mmol/kg or 70–100 mL (8.4%) over 20–30 minutes.13
Although serum phosphate depletion is frequently observed in patients with DKA, replacement is not routinely recommended with no clear benefit and a potential increased risk of hypocalcaemia and hypomagnesaemia.18 Serum phosphate level is often normal at presentation and may decrease with insulin therapy. Phosphate replacement may be considered necessary in patients with cardiac dysfunction, anaemia or respiratory depression if their serum phosphate level is less than 1.0 mg/dL. Replacement in such circumstances may consists of 20–30 mEq/L potassium phosphate in intra-venous fluids.18 In less urgent circumstances, if phosphate replacement is considered necessary, oral supplementation may be safer.16
Pre-hospital responders may be confronted by patients in a poor conscious state presenting with hyperventilation to produce compensatory respiratory alkalosis. Some intensive care paramedic guidelines may allow for drug-facilitated intubation to assist with airway protection and ventilation maintenance. See Chapter 17, p. 318, for intubation techniques. In such instances the preservation of the compensatory alkalosis is imperative with end-tidal carbon dioxide values determined before any therapy is provided.
Investigations and consider precipitating causes
Identification of the precipitating cause is important both to prevent further occurrence and if appropriate to treat confounding illness. This will involve the following investigations:
• Baseline arterial blood gases (hypoxia, lactic acidosis) (see Ch 17, p. 329, for techniques)
• Electrolytes (Ca, Mg, PO4, urea, creatinine)
• Liver function tests, amylase, lipase
• Septic screen (blood cultures, CRP, chest X-ray, MSU)
• Pregnancy test in females of child-bearing age
• Serum cortisol, if adrenal insufficiency is considered
• Alcohol and drug screen if drug use is suspected because acidosis will not resolve.
Assessment by a diabetes educator as early as possible after presentation is also highly recommended, as it is estimated that ongoing diabetes education and follow-up care may prevent unnecessary hospital admissions in about 50% of presentations. Co-existing illness such as pelvic or rectal abscess, pneumonia and silent myocardial infarction should be excluded prior to discharge.15 Special concern exists in the pregnant woman presenting with DKA. Fetal mortality rate may be as high as 30% and up to 60% with ketoacidosis coma. Fetal death mainly occurs in women with overt diabetes, but may occur in gestational diabetes.15
Hyperglycaemic, hyperosmolar syndrome
Hyperglycaemic, hyperosmolar syndrome (HHS) is characterised by hyperglycaemia and high plasma osmolality without significant ketoacidosis (Fig 26.6). In the HHS state, blood sugar levels rise slowly and patients become progressively unwell. In most cases, sufficient insulin exists to prevent ketone formation and therefore metabolic acidosis is normally not present, except in extreme cases. In contrast to DKA, patients with HHS present with an increase in serum bicarbonate levels, usually exceeding 18 mmol/L, and serum sodium levels frequently 140 mmol/L or higher. Serum osmolality levels will also be significantly elevated, in the region of 350 mOsm/kg, and serum blood glucose levels will usually be < 50–60 mmol/L.

The HHS state occurs in type 2 diabetes and usually has gradual onset occurring over several weeks, not days. Patients presenting with HHS usually have an underlying medical condition exacerbating often-undiagnosed type 2 diabetes. Traditionally, patients presenting with HHS are usually middle-aged or elderly, but awareness of increased reports of type 2 diabetes in younger adults and children make presentations in these groups possible.
Differentiation between HHS and DKA may be difficult initially (Table 26.7). However, the treatment approach initially is similar to DKA, with a few modifications:
• If serum sodium is greater than 155 mmol/L, it is recommended to use 0.45% sodium chloride as initial fluid replacement.13,14
• As with DKA, insulin replacement is usually required initially but blood sugar levels tend to fall rapidly with hydration; after recovery, not all patients will require insulin treatment.
For many presenting with HHS, this may be their first diagnosis with type 2 diabetes and therefore the diabetes team should be alerted early and involved in the patient’s ongoing and future care.
TABLE 26.7
Comparison of hyperosmolar, hyperglycaemic syndrome (HHS) and diabetic ketoacidosis (DKA)19
| CLINICAL PICTURE | HHS | DKA |
| General |
More dehydrated, not acidotic Frequently comatose No hyperventilation |
More acidotic and less dehydrated Rarely comatose Hyperventilation |
| Age frequency | Usually elderly | Younger patients |
| Type of diabetes mellitus | Type 2 or non-insulin-dependent | Type 1 or insulin-dependent |
| Previous history of diabetes mellitus | In only 50% | Almost always |
| Prodromes | Several days duration | Less than 1 day |
| Neurological symptoms and signs | Very common | Rare |
| Underlying renal or cardiovascular disease | About 85% | About 15% |
| Laboratory findings | ||
|
Blood glucose Plasma ketones Serum sodium Serum potassium Serum bicarbonate Anion gap Blood pH Serum osmolality Serum urea Free fatty acids |
More than 800 mg/dL Less than large in undiluted specimen Normal, elevated, low Normal or elevated More than 16 mEq 0–12 mEq Normal More than 350 mOsm/L Higher than DKA Less than 1000 mEq/L |
Usually less than 800 mg/dL Positive in several dilutions Usually low Elevated, normal or low Less than 10 mEq More than 12 mEq Less than 7.35 Less than 330 mOsm/L Not as high as in HHNS More than 1500 mEq/L |
| Complications | ||
| Thrombosis | Frequent | Very rare |
| Mortality | 20–50% | 1–10% |
| Diabetes treatment after recovery | Diet alone or oral agents (sometimes) | Always insulin |
Hypoglycaemia
Hypoglycaemia most commonly occurs in type 1 diabetes, although it may also occur in type 2 diabetics.13,16 Most episodes of hypoglycaemia are related to insulin treatment, although sulfonylurea drugs may also cause hypoglycaemic episodes.20 Sulfonylurea medications act by binding to a high-affinity receptor on the surface of the pancreatic islet beta cells, potentiating normal glucose-stimulated insulin release in the presence of a pancreas with functioning beta cells.13 An inevitable consequence of tight glycaemic control, most hypoglycaemic episodes are managed by the patients themselves, a family member or ambulance services.
Hypoglycaemia should be considered in any unresponsive patient until proven otherwise, and insulin overdose considered in patients who present with hypoglycaemia that requires continuing doses of intravenous glucose to maintain blood glucose above 5 mmol/L. A lack of dietary intake, increased physical stress, liver disease, changes in insulin or oral medication regimens, pregnancy, pancreatitis, pituitary insufficiency, Addison’s disease, alcohol ingestion and drugs such as non-steroidal anti-inflammatory drugs, phenytoin, thyroid hormones and propanolol can all contribute to hypoglycaemia.13,15 It is also worth noting that beta-blockers can mask the adrenergic warning symptoms, making symptoms sudden and unexpected in patients on such therapy.13
Definitions of hypoglycaemia
The normal blood glucose level (BGL) is 4.0–8.0 mmol/L. When the blood glucose level is < 2.5 mmol/L this is defined as severe hypoglycaemia.16 A blood glucose level of < 3.5 mmol/L is considered moderate hypoglycaemia.13
Additionally, hypoglycaemic symptoms may occur when a very high blood glucose level falls too rapidly (e.g. a blood glucose level of 16.7 mmol/L falling quickly to 10 mmol/L). This is especially true for patients with chronically elevated blood sugar levels.21 Symptoms tend to be grouped as either:
• autonomic (i.e. sweating, warm sensation, anxiety, nausea, palpations and possibly hunger) or
• neurological (i.e. tiredness, visual disturbance, drowsiness, altered behaviour, confusion and, if untreated, seizures or coma).
Autonomic-related symptoms frequently occur with blood sugar levels around 3.5 mmol/L, whereas neurological symptoms tend to be present with blood sugar levels closer to 2.5 mmol/L. A combination of sweating and reduced activity, along with depleted energy reserves, predisposes hypoglycaemic patients to hypothermia. Close attention to rewarming and protecting the patient from further heat loss is required during initial management.
Treatment
Mild to moderate hypoglycaemia
If the patient is conscious and cooperative, a readily available and fast-acting glucose-containing food or drink (60–130 mL fruit juice, 75–150 mL lemonade, jelly beans, honey, 15 g of glucose in adults) may be considered, followed by a lower-glycaemic-load carbohydrate meal (sandwich, dried fruit).13
Severe hypoglycaemia
In episodes of severe hypoglycaemia, the person is likely to be unconscious or confused, requiring assistance. Treatment recommendations for hypoglycaemia in adults are glucose 50% given intravenously (into the antecubital vein if possible), 0.5–2 mg intramuscular (IM) glucagon or subcutaneous (SC) glucagon 1 mg (treatment of choice for non-healthcare professionals), depending on availability and clinical setting.13 The IM route has a quicker release rate and rectifies the blood sugar levels more quickly, while the SC route is slower and more sustained. Response would be expected within 5–6 minutes of injection (either glucose or glucagon). However, prolonged hypoglycaemia associated with a seizure may take several hours for recovery of full consciousness and cognition. In the pre-hospital setting, use of glucose 50% is not advocated and glucose 10% by intravenous infusion (see below) or glucagon are preferred treatment choices.
Following successful reversal of hypoglycaemia, blood glucose should be monitored every 1–2 hours initially, and then revert to the patient’s usual testing regimen. Consultation with the diabetic specialist team is advisable to determine the cause of the hypoglycaemia; consider medication dose changes and commence education to prevent further episodes. Most patients are usually discharged home if the cause of the hypoglycaemic event can be identified unless they are on oral antihyperglycaemic medications that have a longer half-life.
Alcoholic ketocidosis
Alcoholic acidosis usually occurs in patients with chronic alcoholism in the setting of prolonged fasting, protracted vomiting and large alcohol ingestion.22 The patient usually presents with severe hypoglycaemia but will have concurrent accumulation of ketoacids and lactic acid. Insulin deficiency, depleted glycogen stores and volume depletion provide an appropriate milieu for the development of alcoholic ketoacidosis. Hypoglycaemia occurs, as gluconeogenesis is inhibited by ethanol and glycogenolysis is exhausted by a significant fasting state. If insulin levels are decreased, metabolism of glucose is altered leading to the utilisation of fat and muscle tissue for energy. This results in ketosis which, together with profound dehydration, continues the cycle of ketosis and acidosis.22,23
Management
The focus of treatment in the ED is rehydration with Normal saline and correction of hypoglycaemia with 5% dextrose. This treatment modality is usually sufficient to reverse the acidosis. Adjunctive therapy includes parental thiamine administration. Thiamine is often given before glucose administration to prevent precipitating Wernicke’s encephalopathy, a neurological syndrome associated with ataxia, ophthalmoplegia and mental status changes.17 Vital signs and neurological assessment are imperative to the nursing care of these patients, together with accurate monitoring of fluid, hydration and electrolyte status. Monitor also for signs of alcohol withdrawal.
Adrenal insufficiency
Adrenal insufficiency is a condition that occurs when glucocorticoid production is inadequate to meet metabolic requirements (Fig 26.7).19 It may be caused by structural or functional lesions of the adrenal cortex (primary adrenal insufficiency) or anterior pituitary gland/hypothalamus (secondary adrenal insufficiency).
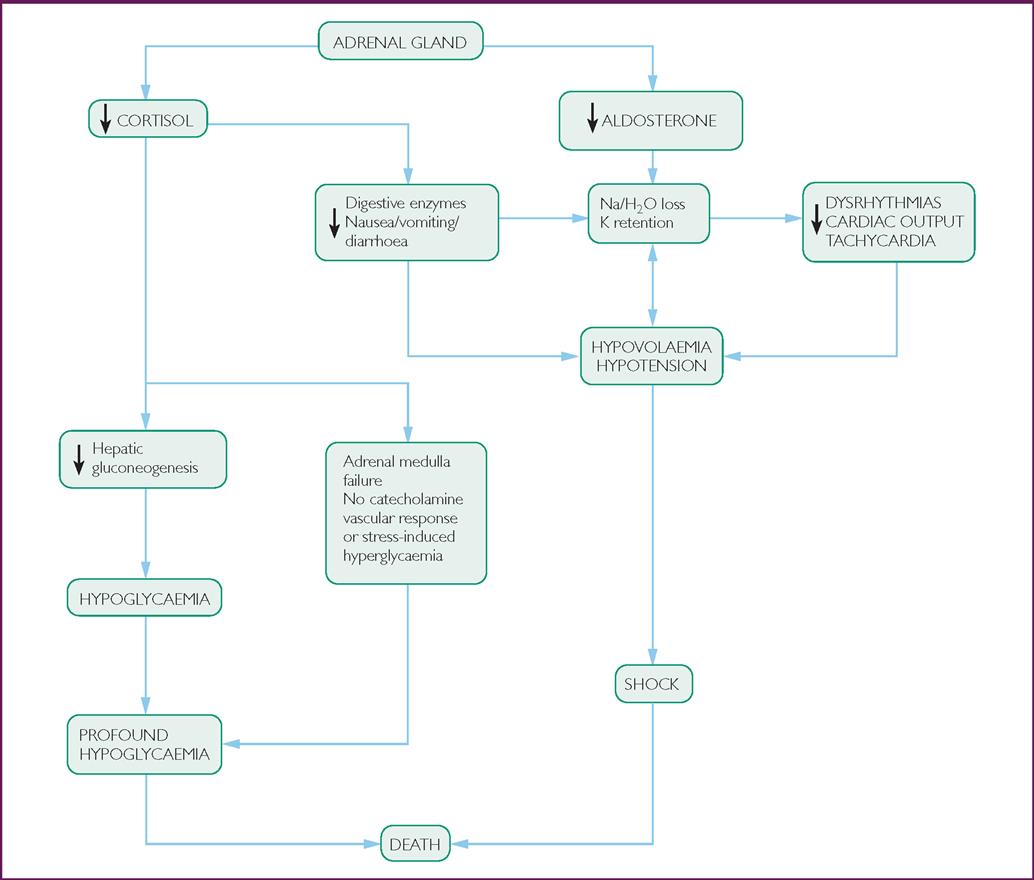
The presenting signs and symptoms can vary from non-specific clinical features, such as tiredness, nausea, anorexia, lethargy, mild hypotension, fever and abdominal pain, through to potentially life-threatening cardiovascular collapse. Historical indicators of the potential for adrenal insufficiency to occur include history of long-term glucocorticoid treatment and/or known adrenal failure.16
Precipitating factors and aetiology
The aetiology of adrenal insufficiency is also diverse. It may result from abrupt withdrawal of glucocorticoid therapy or the onset of an acute illness or stressor, such as infection or trauma in steroid-dependent patients. Causes that are specific to the adrenal gland include autoimmune disorders, adrenal haemorrhage or infiltrates (carcinoma, sarcoidosis), bilateral adrenalectomy or drugs (ketoconazole). Pituitary and hypothalamic causes of adrenal insufficiency may be due to tumours, apoplexy or granulomatous disease.14
Clinical management
As with all emergency presentations, the clinical assessment follows the ABCD mnemonic and interventions are initiated if a threat to any of these parameters is established. Fluid resuscitation with intravenous Normal saline 0.9% is usually required, titrated to the clinical response. Hydrocortisone infusion is commenced immediately at 4 mg/h and maintained for 24 hours.13,14
Following the primary survey, the healthcare should continue with a focused assessment to identify any precipitating event. Broad-spectrum antibiotic therapy may be indicated if infection is expected. Having stabilised the patient, blood is sampled for random cortisol and adrenocorticotrophic hormone (ACTH) levels. This test is time-sensitive and needs to reach the laboratory within 30 minutes. The blood sample must be placed on ice for transport. Diagnosis is confirmed by performing a short ACTH stimulation (Synacthen) test. Hydrocortisone therapy will be slowly reduced over the following 24–48 hours. The maintenance dose of glucocorticoid is usually hydrocortisone 10 mg twice per day.14
Discharge advice
A patient with adrenal insufficiency should be educated on the following key features of self-care on discharge. Patient education on glucocorticoid therapy is very important following an acute crisis. This education will include instructions to increase glucocorticoid dose at times of intercurrent illness. Wearing a ‘medical alert’ bracelet or necklace is advocated. Instructions on the use of an emergency injection pack (hydrocortisone), particularly when away from medical care, may also be included as part of the discharge advice. Above all, the early recognition of signs and symptoms suggestive of an adrenal crisis (nausea, vomiting, dehydration, feeling faint) are emphasised.
Acute pituitary apoplexy
This rare disorder is caused by infarction or haemorrhage of the pituitary gland associated with trauma, hypertension, anticoagulation, cardiac surgery and a large number of other conditions. Acute pituitary apoplexy in uncommon, affecting both males and females equally with an incidence of 0.6–9.1%.14
Clinical features
The clinical signs and symptoms are similar to those seen in raised intracranial pressure and include headache, vomiting, photophobia and altered level of consciousness. Neuro-ophthalamic signs, such as visual field defects and loss of vision, may also be present.16
Treatment
As with all emergency presentations, the clinical assessment follows the ABCD mnemonic and the initiation of interventions to support these systems. Hydrocortisone therapy is started immediately by administering hydrocortisone 4 mg/h intravenously.13,14 Blood levels are taken prior to commencing steroid therapy. They include the following:
Diagnosis is confirmed by urgent magnetic resonance imaging or computed tomography. A critical referral is then made for a neurosurgical consult, with operative management associated with improved outcomes especially in visual acuity.14
Thyroid storm
Thyroid emergencies are very rare but can be life-threatening.24 Abnormality, either hyperactivity or hypoactivity, can result in multisystem symptoms. Thyroid storm (hyperactivity or thyrotoxicosis) commonly occurs as a complication of Graves’ disease, primarily in women aged between 30 and 40 years. The hypothalamic–pituitary–thyroid counterregulatory system is responsible for normal thyroid function.
Precipitating factors and aetiology
Thyroid crisis occurs when any part of the circuit malfunctions. It is unusual for untreated hyperthyroidism to present as thyroid storm, as there are usually precipitating events. Hyperthyroidism can be divided into three categories: true (i.e. excessive thyroid hormones), drug-induced or thyroid injury. True hyperthyroidism is characterised by an overactive thyroid gland and excessive production of thyroid hormones. In Graves’ disease, thyroid-stimulating immunoglobulins increase thyroid activity. Tumours and thyroid nodules also increase thyroid activity. A type of thyroid toxicosis occurs with an increased amount of circulating hormones without concurrent overactivity from the thyroid, as in thyroiditis or ingestion of thyroid hormones. Drugs such as iodine and iodine-containing agents such as amiodarone and lithium may potentially induce hyperthyroidism. Other thyroid-specific precipitation factors in thyroid storm may include thyroid injury such as palpation, infarction or an adenoma.3,14,16
Clinical features
Thyroid storm, an endocrine emergency first described in 1926, remains a diagnostic and therapeutic challenge.24 Thyroid storm occurs with rapid elevation in thyroid hormone levels and is a clinical diagnosis. General precipitating factors include: infection, non-thyroidal trauma or surgery, psychosis, parturition, myocardial infarction or other medical problems.14 A concise medical history is imperative to the treatment of these patients, as it is unusual for untreated hyperthyroidism to present as thyroid storm. Assessment should include history of recent illnesses and medications. Red flags to observe for include recent weight loss despite increased appetite and increased caloric intake; abdominal pain is also a common complaint. The patient may be restless with a reduced attention span and prone to changing discussion topics frequently. Tremors and manic behaviours are also common. In the late stages, an alteration in mental status occurs, which can progress to coma. In extreme cases hyperthermia can occur with temperatures reaching 40.5–41°C, as well as tachycardia with heart rates of 200–300 beats/minute, which increase the risk of cardiac failure and arrest.24 Rales secondary to cardiac failure may be heard. The skin can progress from warm and diaphoretic to hot and dry as dehydration worsens. Increased gastric motility can cause nausea, vomiting and diarrhoea. Hepatic tenderness, jaundice and thinning of the hair may also occur. Goitre, an enlarged thyroid gland, develops as the condition progresses. Eyes become protuberant, periorbital oedema develops and the patient has a staring gaze with heavy eyelids. The clinical manifestations of thyroid storm are summarised in Table 26.8.
TABLE 26.8
Clinical manifestations of thyroid storm
| SYSTEM | CLINICAL MANIFESTATIONS |
| Neurological | Nervousness, restlessness, tremors, confusion |
| Pulmonary | Tachycardia, dysrhythmias, hypertension |
| Respiratory | Shortness of breath, dyspnoea, rales, congestive heart failure |
| Gastrointestinal | Hyperactive bowel, abdominal pain, decreased appetite, weight loss, diarrhoea, jaundice |
| Ocular | Exophthalmos, lid lag, staring gaze |
| Integumentary | Hyperthermia, flushed, diaphoresis, poor turgor |
Treatment
Treatment should be initiated promptly targeting all steps of thyroid hormone function, release and action.25 Thyroid hormone levels can help differentiate the causative factor; however, the hormone levels are frequently not readily available, making differentiation between thyroid storm and thyrotoxicosis difficult. If thyroid storm is suspected, therapy needs to be aggressive and rapid so that hormone levels are reduced and haemodynamic integrity is preserved. The treatment principles are to decrease the production and release of thyroid hormones, inhibit the peripheral effects of thyroid hormones using beta-blockers and identify and deal with the underlying precipitating factors.
Propanolol can be given intravenously to reduce the heart rate. Younger patients and those in an acute crisis often require larger than normal doses. The propanolol acts to block the conversion of the T4 to T3. In cases where beta-blockers are contraindicated (e.g. in diabetes, pregnancy and asthma), digitalis may be used.14,16
Propylthiouracil, which inhibits thyroid hormone synthesis, and methimazole may be used to further block hormone synthesis. This can be given orally or through a gastric tube. Onset of action occurs within an hour; however, full potential is not reached for 3–6 weeks.3,14 Iodine may be given 1 hour after antithyroid medications to slow the release of stored thyroid hormones from the thyroid gland. Iodine can be given orally or by intravenous infusion. Patients with Graves’ disease and thyroid crisis metabolise and use cortisol faster than normal. Glucocorticosteroids, such as dexamethasone or hydrocortisone, have been found to increase survival rates, as they prevent adrenal compromise and inhibit T4 and T3 conversion.25,26
Approaches to supportive therapy may include fluid resuscitation if required and reduction in core body temperature.
Myxoedema coma
Myxoedema coma is a rare but potentially serious complication of untreated hypothyroidism.
Precipitating factors and aetiology
The incidence of myxoedema coma is greater in men than in women. Precipitating causes include hypothermia, infection, myocardial infarction or congestive heart failure, cerebral vascular accidents, drug-induced respiratory depression (e.g. sedatives, anaesthetics or tranquillisers), trauma or gastroinstestinal blood loss.14,16 The disease usually progresses insidiously over months to years, with coma developing when the patient is subjected to stress. It is known to also affect elderly women who have long-standing hypothyroidism and those with undiagnosed hypothyroidism. The latter tends to occur in the winter months. Survival rates are increased when prompt hormone replacement with intense supportive care is received. Mortality rates, however, approach 50%.27
Clinical features
A hypoactive thyroid results in reduced metabolic rate and activity. Pronounced fatigue, decreased activity tolerance, episodes of shortness of breath and weight gain may be displayed. Tongue swelling or macroglossia are common complaints. Patients may display elements of confusion and be slow to answer questions (Table 26.9). This alteration in mental status can result in coma. Psychiatric symptoms such as hallucinations, paranoia, depression, combativeness and decreased concern for personal appearance may also present, and are often referred to as ‘myxoedema madness’.
TABLE 26.9
Clinical manifestations of myxoedema coma
| SYSTEM | CLINICAL MANIFESTATIONS |
| Neurological | Confusion, lethargy, coma |
| Pulmonary | Decreased stroke volume, decreased cardiac output, bradycardia, peripheral vasoconstriction, inverted T waves, prolonged QT interval |
| Respiratory | Macroglossia, obesity-related sleep apnoea, pneumonia, hypoventilation, hypercarbia |
| Gastrointestinal | Hypoglycaemia, constipation |
| Renal | Decreased renal blood flow, decreased sodium reabsorption, hyponatraemia |
Angio-oedema can result in airway occlusion in an unconscious or semiconscious patient. Weak respiratory effort with decreased respiratory drive results in alveolar hypoventilation and can predispose patients to infections. Alveolar hypoventilation also results in hypercarbia, which can cause alteration in mental status. Obesity-related sleep apnoea could further compromise the respiratory system (Table 26.9).
Cardiac changes include bradycardia, decreased stroke volume and decreased cardiac output. Widespread ST and T-wave changes may be evident, with prolonged QT intervals. Body temperature will be low and the skin pale and cool due to peripheral vasoconstriction. In myxoedema coma, the glomerular filtration rate and renal blood flow decreases. Generalised non-pitting oedema as a result of increased insulin sensitivity and decreased oral intake and constipation may also occur. Biochemical abnormalities may include hyponatraemia, increased creatine phosphokinase and lactate dehydrogenase, hypoglycaemia and normocytic or macrocytic anaemia.16 Thyroid-stimulating hormones may be modestly low in primary hypothyroidism or low in secondary hypothyroidism, but free thyroxine levels are usually low.16
Treatment
Even with prompt treatment, mortality can be as high as 30% in myxoedema coma. Care is primarily focused on hormone replacement after initial ABC assessment and stabilisation. Hormone replacement may take the form of intravenous T4 (levothyroxine). Large doses are used to saturate empty sites and replenish the circulating levels. T4 avoids adverse cardiac effects that might occur with a sudden increase in T3. Some doctors may recommend a combination of both T3 and T4. As myxoedema coma is a rare condition, a lack of clinical trials to suggest best methods is lacking, and management is likely to be guided by an endocrinology specialist. Electrocardiogram monitoring during careful titration of thyroid replacement is recommended.14
Cushing’s syndrome
Cushing’s syndrome is a rare endocrine disorder involving the hypothalamic–pituitary–adrenal glands, which results in excessive cortisol levels.28,29 Cortisol, a steroid hormone produced by the adrenal gland and normally produced in response to stress, can trigger physiological changes resulting in a wide range of health problems including hypertension, hyperglycaemia, muscle wastage and osteoporosis. Causes of Cushing’s syndrome include:
• iatrogenic factors—prolonged administration of a glucocorticoid such as prednisone to treat conditions such as asthma or rheumatoid arthritis
• ectopic factors—production of cortisol by adrenal adenoma or carcinoma (e.g. lung cancer can produce ACTH which when added to the normal pituitary production of ACTH leads to excessive cortisol secretion)
• problems in the hypothalamic–pituitary–adrenal axis resulting in excessive cortisol secretion from the adrenal glands due to overstimulation of the adrenal glands by ACTH.29
Features that best discriminate Cushing’s syndrome from other common conditions
• Skin—Easy bruising, facial plethora, purple striae
• Musculoskeletal system—Proximal muscle weakness and/or myopathy; early osteoporosis with or without vertebral fractures or osteonecrosis of femoral or humeral head
Other features
• Skin and hair—Thin skin, poor wound healing, hirsutism or scalp thinning
• Body habitus—Weight gain and central obesity; dorsocervical fat pad (‘buffalo hump’), supraclavicular fat pads, facial fullness (‘moon face’)
• Reproductive system—Menstrual irregularity, infertility
• Psychiatric effects—Depression, psychosis, irritability, insomnia, fatigue
• Cardiovascular effects—Congestive cardiac failure, hypertension, thrombosis (including deep vein thrombosis and myocardial infarction)
• Immune system—Immunosuppression causing recurrent and atypical infection, including tuberculosis.29
Assessment and investigations
The diagnosis of Cushing’s syndrome is generally based on laboratory investigations. Serum ACTH levels determine the aetiology of the excessive cortisol secretion.
• An elevated ACTH indicates ACTH-dependent disease, with further pituitary investigations required to identify the source.
• Normal ACTH levels identify the need for abdominal computed tomography scanning to investigate the possibility of an adrenal tumour.
Other laboratory tests indicated include:
• serum glucose to establish if hyperglycaemia is present
• full blood count and white blood cell count may reveal elevated levels of both, but a decrease in lymphocytes
• electrolytes, particularly serum potassium which may be decreased
• 24-hour urine collection may also be commenced to measure cortisol levels.28,29
Treatment
Treatment modalities are centred on removing the precipitating cause. Endogenous Cushing’s syndrome requires surgical removal of the tumour causing the oversecretion of cortisol. Patients will require glucocortisol replacement therapy for a period of time post surgery due to adrenal insufficiency. Pharmacological blockade of cortisol production may be the treatment of choice in some patient populations, using metyrapone or mitotane. If the cause is iatrogenic due to long-term glucocorticoid therapy, the prescribed dose of steroids is gradually reduced.
Metyrapone
• Action—reversibly inhibits the biosynthesis of cortisol, corticosterone and aldosterone in the adrenal cortex.
• to establish the diagnosis of adrenocortical hyperfunction in Cushing’s syndrome
• following pituitary surgery.
• Contraindications—adrenocortical insufficiency.
• Adverse reactions—nausea, vomiting, dizziness, light-headedness, abdominal pain.
• Dosage—500–1000 mg orally 3 times a day.13
Mitotane
Mitotane is not registered for use in Australia but is available via the Special Access Scheme. Dosage is 500 mg orally initially, increasing up to 4–6 g daily.13
Mrs Riley, a 40-year-old woman, presents to the emergency department (ED) by ambulance following a 000 call by her husband. She was found by her husband to be drowsy and disorientated following a collapse.
The following handover was given by the paramedic:
• 40-year-old female found by her husband with a decreased level of consciousness.
• Patient has been recently treated for a tooth abscess by her dentist for which she was taking flucloxacillin 500 mg 6-hourly. Yesterday she complained of abdominal pain and persistent vomiting and has been unable to tolerate even clear fluids for the past 48 hours.
Her observations by the paramedics on arrival were:
The treatment initiated was oxygen therapy and the establishment of an intravenous line. 1 L NaCl 0.9% was commenced. Haemodynamic monitoring was initiated by the paramedics. En route the patient’s condition deteriorated and on arrival in ED her Glasgow Coma Scale score was 7 and she was tolerating an oral adjunct (Guedel’s airway).
She has no known allergies. She was previously healthy with no known medical conditions except hypertension.
In ED the patient was allocated a triage category 1 and directed to the resuscitation area. The first priority was to establish the severity of Mrs Riley’s symptoms and hence the need for medical intervention. This determination was made within seconds of the primary assessment. It was established that there was an actual threat to her airway, breathing, circulation and disability; interventions were required immediately. This included the establishment of a definitive airway via endotracheal intubation, and ventilatory support to achieve adequate oxygenation and ventilation. Mrs Riley was sedated and paralysed using rapid sequence induction (thiopentone and suxamethonium) to facilitate the intubation process. Circulatory support was established by infusing 3 L Normal saline 0.9% via a large-bore cannula to correct the hypotension. A blood glucose level was recorded as ‘HI’ using point-of-care testing. Following the establishment of hyperglycaemia in this acutely ill patient, the next bedside test was serum ketone monitoring. The result was a serum ketone level of 4.1 mmol/L.
Ketosis and hyperglycaemia are distinct metabolic problems. When these abnormal states occur together, diabetic ketoacidosis (DKA) may result. In determining the severity of Mrs Riley’s illness (DKA), it was necessary to consider a combination of clinical assessment findings, historical factors, diagnostic and laboratory testing and the outcome of interventions. A secondary assessment outlying these findings is given in Table 26.CS1.
TABLE 26.CS1
| CLINICAL FINDING | FINDING/INTERVENTION | |
| E |
Exposure Expose patient to identify all injuries: recent tooth extraction |
Potential site of infection Commenced on antibiotic therapy Septic screening, including chest X-ray |
| F |
Fluids The circulatory system was assessed and the following vital signs established: • blood pressure 110/70 mmHg • heart rate 112 beats/minute • respiratory rate—ventilated • GCS 3 (sedated and paralysed) • pupils (equal and reacting to light) • SpO2 99% • Temperature 37.9°C |
Circulatory support was established Fluid resuscitation: 3 L NaCl 0.9% were infused in the first 2 hours followed by 1 L NaCl 0.9% over the next hour Indwelling catheter was inserted, which initially drained 650 mL dilute urine. Hourly urinary measures were commenced to ensure a urinary output of 0.5 mL/kg/h ECG recorded—Sinus tachycardia with no acute changes Haemodynamic monitoring was commenced via arterial line and continuous cardiac monitoring |
| Continued | ||
| G | Glucose |
Formal blood sugar 28.4 mmol/L Serum ketones 4.1 mmol/L Commenced on insulin infusion therapy(50 units Actrapid in 50 mL NaCl infusing at 5 units/h) |
| H | History |
Hypertension Not a known diabetic (gestational diabetes only) Referred by GP and seen in ED 2 days prior with: • an impacted infected left tooth • persistent vomiting with 3 kg loss in 1 week • Blood pressure 115/50 mmHg |
| H | Head-to-toe assessment | |
| Head/neck |
Left upper molar extraction CT head/facial bones: NAD | |
| Respiratory |
Clear Ventilated: SIMV ABG: severe acidosis pH: 6.7 PaCO2 28.4 mmHg HCO3– 4.9 mmol/L BE −27.1 mmol/L Lactate: 0.9 mmol/L | |
| Cardiovascular |
Cool peripheries Dual heart sounds CK < 20 µmol/L (normal limits are: male 60–120 µmol/L, female 40–90 µmol/L) Troponin negative | |
| Abdomen |
Soft and non-tender Bowel sounds present Beta-hCG negative LFTs normal Amylase and lipase normal | |
| Renal |
Very good urinary output Potassium 4.2 mmol/K (potassium replacement commenced at 10 mmol/h) Sodium 139 mmol/L; corrected sodium 153 mmol/L Anion gap 23 mmol/L | |
| I | Inspect the back | Nil abnormality found |
ABG: arterial blood gas; BE: base excess; CK: creatine kinase; CT: computed tomography; GCS: Glasgow Coma Scale; hCG: human chorionic gonadotrophin; HCO3–: bicarbonate level; LFT: liver function test; PaCO2: arterial pressure of carbon dioxide; SIMV: synchronised intermittent mandatory ventilation; SpO2: peripheral oxygen saturation
With respect to the circulation and metabolic derangements that Mrs Riley suffered, the management goals were to:
• replace salt and fluid losses
• restrain lipolysis and inhibit glucose production with insulin
The greatest dangers to Mrs Riley are acidosis and electrolyte disturbances, especially hypokalaemia, not hyperglycaemia. The first priority was rapid fluid resuscitation to improve tissue perfusion and the tissue’s response to insulin. Fluids also decrease the blood sugar level by 30%. Correcting Mrs Riley’s hypotension would also decrease the secretion of counterregulatory hormones and improve acidosis. As there was clear clinical evidence of ketoacidosis, the next priority was the commencement of an insulin infusion. The initial infusion rate was 5 units/h. The blood glucose level progressively decreased from 28.4 mmol/L by 4.5–5.5 mmol/L over the next few hours. When Mrs Riley’s blood glucose level dropped to 14 mmol/L, a 5% dextrose infusion was commenced at 80 mL/h. This allowed the blood glucose level to be maintained until the acidosis was reversed by the insulin therapy. The next most-critical management step was the recognition of the need to replace potassium. Potassium replacement was commenced at 10 mmol/h, and close monitoring was initiated in anticipation of a further fall in serum potassium due to the commencement of insulin therapy and the correction of the acidosis. Identification of the precipitating factor was the next vital step. This was identified as the infected and impacted left tooth. She was commenced on ceftriaxone, metronidazole and gentamycin. Mrs Riley was then transferred to the intensive care unit 4 hours after presenting to the ED.
During her ICU stay, she was further resuscitated and stabilised. Her acidosis and hyperglycaemia were corrected slowly. She was successfully extubated 3 days later. The maxillofacial and endocrine teams were consulted on her care. Mrs Riley was discharged home on insulin 9 days later.
Questions
1. What are the main characteristics of DKA?
2. What are the key physical signs seen in patients presenting with DKA?
3. What is the first priority in managing the patient in the above case?
4. What are the overarching key principles of managing this patient presenting with DKA?
5. What are the most common errors when managing a patient with DKA?
 Answers to Case Study Questions can be found on evolve
Answers to Case Study Questions can be found on evolve