Neoplasia
Cancer is the second leading cause of death in the United States; only cardiovascular diseases exact a higher toll. Even more agonizing than the associated mortality is the emotional and physical suffering inflicted by neoplasms. Patients and the public often ask, “When will there be a cure for cancer?” The answer to this simple question is difficult, because cancer is not one disease but rather many disorders that share a profound growth dysregulation. Some cancers, such as Hodgkin lymphoma, are highly curable, whereas others, such as cancer of the pancreas, are virtually always fatal. The only hope for controlling cancer lies in learning more about its pathogenesis, and great strides have been made in understanding the molecular basis of cancer. This chapter deals with the basic biology of neoplasia—the nature of benign and malignant neoplasms and the molecular basis of neoplastic transformation. The host response to tumors and the clinical features of neoplasia also are discussed.
Before we discuss the features of cancer cells and the mechanisms of carcinogenesis, it is useful to summarize the fundamental and shared characteristics of cancers:
• Cancer is a genetic disorder caused by DNA mutations. Most pathogenic mutations are either induced by exposure to mutagens or occur spontaneously as part of aging. In addition, cancers frequently show epigenetic changes, such as focal increases in DNA methylation and alterations in histone modifications, which may themselves stem from acquired mutations in genes that regulate such modifications. These genetic and epigenetic changes alter the expression or function of key genes that regulate fundamental cellular processes, such as growth, survival, and senescence.
• Genetic alterations in cancer cells are heritable, being passed to daughter cells upon cell division. As a result, cells harboring these alterations are subject to Darwinian selection (survival of the fittest, arguably the most important scientific concept in biology). Cells bearing mutations that provide a growth or survival advantage outcompete their neighbors and thus come to dominate the population. At the time of tumor initiation, these selective advantages are conferred on a single cell, and as a result all tumors are clonal (i.e., the progeny of one cell). However, even beyond the point of initiation, Darwinian selection continues to shape the evolution of cancers by favoring the emergence of genetically distinct subclones with more aggressive characteristics, an important concept referred to as progression and discussed in more detail later in this chapter.
• Mutations and epigenetic alterations impart to cancer cells a set of properties that are referred to collectively as cancer hallmarks. These properties produce the cellular phenotypes that dictate the natural history of cancers as well as their response to various therapies. The molecular underpinnings of each hallmark of cancer are discussed in later sections.
Basic research has elucidated many of the cellular and molecular abnormalities that give rise to cancer and govern its pernicious behavior. These insights are in turn leading to a revolution in the diagnosis and treatment of cancer, an emerging triumph of biomedical science.
Nomenclature
Neoplasia literally means “new growth.” Neoplastic cells are said to be transformed because they continue to replicate, apparently oblivious to the regulatory influences that control normal cells. Neoplasms therefore enjoy a degree of autonomy and tend to increase in size regardless of their local environment. Their autonomy is by no means complete, however. All neoplasms depend on the host for their nutrition and blood supply. Neoplasms derived from hormone responsive tissues often also require endocrine support, and such dependencies sometimes can be exploited therapeutically.
In common medical usage, a neoplasm often is referred to as a tumor, and the study of tumors is called oncology (from oncos, “tumor,” and logos, “study of”). Among tumors, the division of neoplasms into benign and malignant categories is based on a judgment of a tumor's potential clinical behavior.
• A tumor is said to be benign when its microscopic and gross characteristics are considered to be relatively innocent, implying that it will remain localized and is amenable to local surgical removal. Affected patients generally survive. Of note, however, benign tumors can produce more than localized lumps, and sometimes they produce significant morbidity or are even lethal.
• Malignant, as applied to a neoplasm, implies that the lesion can invade and destroy adjacent structures and spread to distant sites (metastasize) to cause death. Malignant tumors are collectively referred to as cancers, derived from the Latin word for “crab”—that is, they adhere to any part that they seize in an obstinate manner, similar to a crab's behavior. Not all cancers pursue so deadly a course. The most aggressive are also some of the most curable, but the designation malignant constitutes a red flag.
All tumors, benign and malignant, have two basic components: (1) the parenchyma, made up of transformed or neoplastic cells, and (2) the supporting, host-derived, non-neoplastic stroma, made up of connective tissue, blood vessels, and host-derived inflammatory cells. The parenchyma of the neoplasm largely determines its biologic behavior, and it is this component from which the tumor derives its name. The stroma is crucial to the growth of the neoplasm, since it carries the blood supply and provides support for the growth of parenchymal cells. Although the biologic behavior of tumors largely reflects the behavior of the parenchymal cells, there has been a growing realization that stromal cells and neoplastic cells carry on a two-way conversation that influences the growth of the tumor.
Benign Tumors
In general, benign tumors are designated by attaching the suffix -oma to the cell type from which the tumor arises. For example, a benign tumor arising in fibrous tissue is a fibroma; a benign cartilaginous tumor is a chondroma. More varied and complex nomenclature is applied to benign epithelial tumors. The term adenoma is generally applied not only to benign epithelial neoplasms that produce glandlike structures, but also to benign epithelial neoplasms that are derived from glands but lack a glandular growth pattern. Thus, a benign epithelial neoplasm arising from renal tubule cells and growing in a glandlike pattern is termed an adenoma, as is a mass of benign epithelial cells that produces no glandular patterns but has its origin in the adrenal cortex. Papillomas are benign epithelial neoplasms, growing on any surface, that produce microscopic or macroscopic fingerlike fronds. A polyp is a mass that projects above a mucosal surface, as in the gut, to form a macroscopically visible structure (Fig. 6.1). Although this term commonly is used for benign tumors, some malignant tumors also may grow as polyps, whereas other polyps (such as nasal polyps) are not neoplastic but inflammatory in origin. Cystadenomas are hollow cystic masses that typically arise in the ovary.

Malignant Tumors
The nomenclature of malignant tumors essentially follows that of benign tumors, with certain additions and exceptions.
• Malignant neoplasms arising in “solid” mesenchymal tissues or its derivatives are called sarcomas, whereas those arising from the mesenchymal cells of the blood are called leukemias or lymphomas. Sarcomas are designated based on their cell-type composition, which presumably reflects their cell of origin. Thus, a malignant neoplasm comprised of fat-like cells is a liposarcoma, and a malignant neoplasm composed of chondrocyte-like cells is a chondrosarcoma.
• While the epithelia of the body are derived from all three germ cell layers, malignant neoplasms of epithelial cells are called carcinomas regardless of the tissue of origin. Thus, malignant neoplasms arising in the renal tubular epithelium (mesoderm), the skin (ectoderm), and lining epithelium of the gut (endoderm) are all considered carcinomas. Furthermore, mesoderm may give rise to carcinomas (epithelial), sarcomas (mesenchymal), and hematolymphoid tumors (leukemias and lymphomas).
• Carcinomas are subdivided further. Carcinomas that grow in a glandular pattern are called adenocarcinomas, and those that produce squamous cells are called squamous cell carcinomas. Sometimes the tissue or organ of origin can be identified, as in the designation of renal cell adenocarcinoma, but it is not uncommon for tumors to show little or no differentiation. Such tumors are referred to as poorly differentiated or undifferentiated carcinoma.
The transformed cells in a neoplasm, whether benign or malignant, usually resemble each other, consistent with their origin from a single transformed progenitor cell. In some unusual instances, however, the tumor cells undergo divergent differentiation, creating so-called “mixed tumors”. Mixed tumors are still of monoclonal origin, but the progenitor cell in such tumors has the capacity to differentiate down more than one lineage. The best example is mixed tumor of salivary gland. These tumors have obvious epithelial components dispersed throughout a fibromyxoid stroma, sometimes harboring islands of cartilage or bone (Fig. 6.2). All of these diverse elements are thought to derive from a single transformed epithelial progenitor cell, and the preferred designation for these neoplasms is pleomorphic adenoma. Fibroadenoma of the female breast is another common mixed tumor. This benign tumor contains a mixture of proliferating ductal elements (adenoma) embedded in loose fibrous tissue (fibroma). Unlike pleomorphic adenoma, only the fibrous component is neoplastic, but the term fibroadenoma remains in common usage.
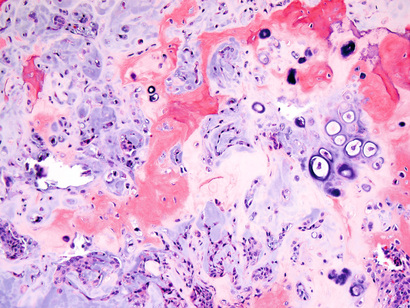
Teratoma is a special type of mixed tumor that contains recognizable mature or immature cells or tissues derived from more than one germ cell layer, and sometimes all three. Teratomas originate from totipotential germ cells such as those that normally reside in the ovary and testis and that are sometimes abnormally present in midline embryonic rests. Germ cells have the capacity to differentiate into any of the cell types found in the adult body; not surprisingly, therefore, they may give rise to neoplasms that contain elements resembling bone, epithelium, muscle, fat, nerve, and other tissues, all thrown together in a helter-skelter fashion.
The specific names of the more common neoplasms are presented in Table 6.1. Some glaring inconsistencies may be noted. For example, the terms lymphoma, mesothelioma, melanoma, and seminoma are used for malignant neoplasms. Unfortunately for students, these exceptions are firmly entrenched in medical terminology.
Table 6.1
Nomenclature of Tumors
| Tissue of Origin | Benign | Malignant |
| One Parenchymal Cell Type | ||
| Connective tissue and derivatives | Fibroma | Fibrosarcoma |
| Lipoma | Liposarcoma | |
| Chondroma | Chondrosarcoma | |
| Osteoma | Osteogenic sarcoma | |
| Endothelium and related cell types | ||
| Blood vessels | Hemangioma | Angiosarcoma |
| Lymph vessels | Lymphangioma | Lymphangiosarcoma |
| Mesothelium | Mesothelioma | |
| Brain coverings | Meningioma | Invasive meningioma |
| Blood cells and related cell types | ||
| Hematopoietic cells | Leukemias | |
| Lymphoid tissue | Lymphomas | |
| Muscle | ||
| Smooth | Leiomyoma | Leiomyosarcoma |
| Striated | Rhabdomyoma | Rhabdomyosarcoma |
| Skin | ||
| Stratified squamous | Squamous cell papilloma | Squamous cell or epidermoid carcinoma |
| Basal cells of skin or adnexa | Basal cell carcinoma | |
| Tumors of melanocytes | Nevus | Malignant melanoma |
| Epithelial lining of glands or ducts | Adenoma | Adenocarcinoma |
| Papilloma | Papillary carcinomas | |
| Cystadenoma | Cystadenocarcinoma | |
| Lung | Bronchial adenoma | Bronchogenic carcinoma |
| Kidney | Renal tubular adenoma | Renal cell carcinoma |
| Liver | Liver cell adenoma | Hepatocellular carcinoma |
| Bladder | Urothelial papilloma | Urothelial carcinoma |
| Placenta | Hydatidiform mole | Choriocarcinoma |
| Testicle | Seminoma Embryonal carcinoma | |
| More Than One Neoplastic Cell Type—Mixed Tumors, Usually Derived From One Germ Cell Layer | ||
| Salivary glands | Pleomorphic adenoma (mixed tumor of salivary gland) | Malignant mixed tumor of salivary gland |
| Renal anlage | Wilms tumor | |
| More Than One Neoplastic Cell Type Derived From More Than One Germ Cell Layer—Teratogenous | ||
| Totipotential cells in gonads or in embryonic rests | Mature teratoma, dermoid cyst | Immature teratoma, teratocarcinoma |

There also are other instances of confusing terminology:
• Hamartoma is a mass of disorganized tissue indigenous to the particular site, such as the lung or the liver. While traditionally considered developmental malformations, many hamartomas have clonal chromosomal aberrations that are acquired through somatic mutations and on this basis are now considered to be neoplastic.
• Choristoma is a congenital anomaly consisting of a heterotopic nest of cells. For example, a small nodule of well-developed and normally organized pancreatic tissue may be found in the submucosa of the stomach, duodenum, or small intestine. The designation -oma, connoting a neoplasm, imparts to these lesions an undeserved gravity, as they are usually of trivial significance.
Although the terminology of neoplasms is regrettably complex, an understanding of the nomenclature is important because it is the language by which a tumor's nature and significance is communicated among physicians in different disciplines involved in cancer care.
Characteristics of Benign and Malignant Neoplasms
There are three fundamental features by which most benign and malignant tumors can be distinguished: differentiation and anaplasia, local invasion, and metastasis. In general, rapid growth also signifies malignancy, but many malignant tumors grow slowly and as a result growth rate is not a reliable discriminator between good and bad actors. Nothing is more important to the patient with a tumor than being told: “It is benign.” Although some neoplasms defy easy characterization, in most instances the determination of benign versus malignant is made with remarkable accuracy using long-established clinical and anatomic criteria.
Differentiation and Anaplasia
Differentiation refers to the extent to which neoplasms resemble their parenchymal cells of origin, both morphologically and functionally; lack of differentiation is called anaplasia. In general, benign neoplasms are composed of well-differentiated cells that closely resemble their normal counterparts. A lipoma is made up of mature fat cells laden with cytoplasmic lipid vacuoles, and a chondroma is made up of mature cartilage cells that synthesize their usual cartilaginous matrix—evidence of morphologic and functional differentiation. In well-differentiated benign tumors, mitoses are usually rare and are of normal configuration.
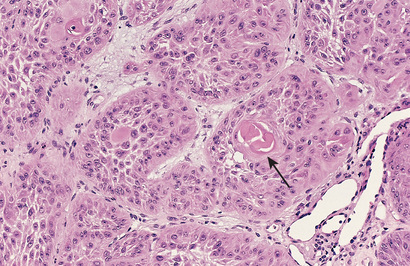
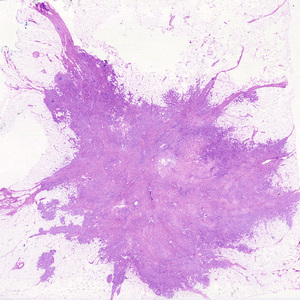
By contrast, while malignant neoplasms exhibit a wide range of parenchymal cell differentiation, most exhibit morphologic alterations that betray their malignant nature. In well-differentiated cancers, these features may be quite subtle (Fig. 6.3). For example, well-differentiated adenocarcinoma of the thyroid gland may contain normal-appearing follicles, its malignant potential being only revealed by invasion into adjacent tissues or metastasis. The stroma carrying the blood supply is crucial to the growth of tumors but does not aid in the separation of benign from malignant ones. The amount of stromal connective tissue does, however, determine the consistency of a neoplasm. Certain cancers induce a dense, abundant fibrous stroma (desmoplasia), making them hard, so-called “scirrhous tumors”.
Tumors composed of undifferentiated cells are said to be anaplastic, a feature that is a reliable indicator of malignancy. The term anaplasia literally means “backward formation”—implying dedifferentiation, or loss of the structural and functional differentiation of normal cells. It is now known, however, that at least some cancers arise from stem cells in tissues; in these tumors, failure of differentiation of transformed stem cells, rather than dedifferentiation of specialized cells, accounts for their anaplastic appearance. Recent studies also indicate that in some cases, dedifferentiation of apparently mature cells occurs during carcinogenesis. Anaplastic cells often display the following morphologic features:
• Pleomorphism (i.e., variation in size and shape) (Fig. 6.4)
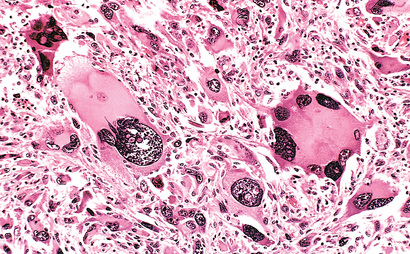
• Nuclear abnormalities, consisting of extreme hyperchromatism (dark-staining), variation in nuclear size and shape, or unusually prominent single or multiple nucleoli. Enlargement of nuclei may result in an increased nuclear-to-cytoplasmic ratio that approaches 1 : 1 instead of the normal 1 : 4 or 1 : 6. Nucleoli may attain astounding sizes, sometimes approaching the diameter of normal lymphocytes.
• Tumor giant cells may be formed. These are considerably larger than neighboring cells and may possess either one enormous nucleus or several nuclei.
• Atypical mitoses, which may be numerous. Anarchic multiple spindles may produce tripolar or quadripolar mitotic figures (Fig. 6.5).
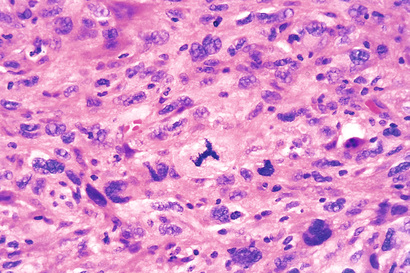
• Loss of polarity, such that anaplastic cells lack recognizable patterns of orientation to one another. Such cells may grow in sheets, with total loss of communal structures, such as glands or stratified squamous architecture.
Well-differentiated tumor cells are likely to retain the functional capabilities of their normal counterparts, whereas anaplastic tumor cells are much less likely to have specialized functional activities. For example, benign neoplasms and even well-differentiated cancers of endocrine glands frequently elaborate the hormones characteristic of their cell of origin. Similarly, well-differentiated squamous cell carcinomas produce keratin (see Fig. 6.3), just as well-differentiated hepatocellular carcinomas secrete bile. In other instances, unanticipated functions emerge. Some cancers may express fetal proteins not produced by comparable cells in the adult. Cancers of nonendocrine origin may produce so-called “ectopic hormones.” For example, certain lung carcinomas may produce adrenocorticotropic hormone (ACTH), parathyroid hormone–like hormone, insulin, glucagon, and others. More is said about these so-called “paraneoplastic” phenomena later.
Also of relevance in the discussion of differentiation and anaplasia is dysplasia, referring to disorderly proliferation. Dysplastic epithelium is recognized by a loss in the uniformity of individual cells and in their architectural orientation. Dysplastic cells exhibit considerable pleomorphism and often possess abnormally large, hyperchromatic nuclei. Mitotic figures are more abundant than usual and frequently appear in abnormal locations within the epithelium. In dysplastic stratified squamous epithelium, mitoses are not confined to the basal layers, where they normally occur, but may be seen throughout the epithelium. In addition, there is considerable architectural anarchy. For example, the usual progressive maturation of tall cells in the basal layer to flattened squames on the surface may be lost and replaced by a disordered hodgepodge of dark basal-appearing cells. When dysplastic changes are severe and involve the entire thickness of the epithelium, the lesion is referred to as carcinoma in situ, a preinvasive stage of cancer (Fig. 6.6).
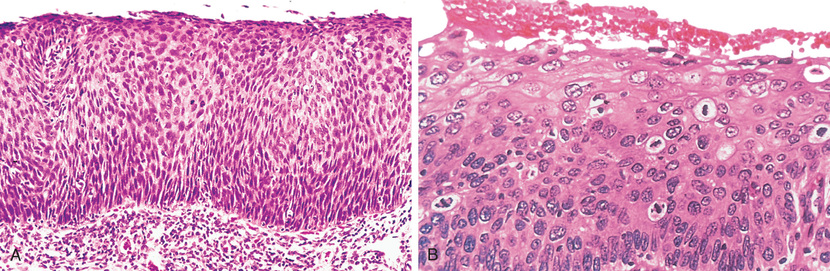
It is important to appreciate that dysplasia is not synonymous with cancer. Mild to moderate dysplasias that do not involve the entire thickness of the epithelium sometimes regress completely, particularly if inciting causes are removed. However, dysplasia is often noted adjacent to frankly malignant neoplasms (e.g., in cigarette smokers with lung cancer), and in general the presence of dysplasia marks a tissue as being at increased risk for developing an invasive cancer.
Local Invasion
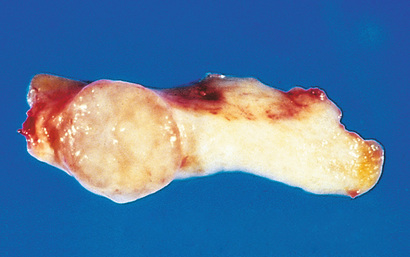
The growth of cancers is accompanied by progressive infiltration, invasion, and destruction of surrounding tissues, whereas most benign tumors grow as cohesive expansile masses that remain localized to their sites of origin. Because benign tumors grow and expand slowly, they usually develop a rim of compressed fibrous tissue (Figs. 6.7 and 6.8). This capsule consists largely of extracellular matrix that is deposited by stromal cells such as fibroblasts, which are activated by hypoxic damage to parenchymal cells resulting from compression by the expanding tumor. Encapsulation creates a tissue plane that makes the tumor discrete, moveable (non-fixed), and readily excisable by surgical enucleation. However, it is important to recognize that not all benign neoplasms are encapsulated. For example, the leiomyoma of the uterus is discretely demarcated from the surrounding smooth muscle by a zone of compressed and attenuated normal myometrium, but lacks a capsule. A few benign tumors are neither encapsulated nor discretely defined; lack of demarcation is particularly likely in benign vascular neoplasms such as hemangiomas, which understandably may be difficult to excise. These exceptions are pointed out only to emphasize that although encapsulation is the rule in benign tumors, the lack of a capsule does not mean that a tumor is malignant. Sadly, because of their uncivilized nature, tumor cells sometimes do not follow the rules set by humans. We will see such deviations many times in this chapter.
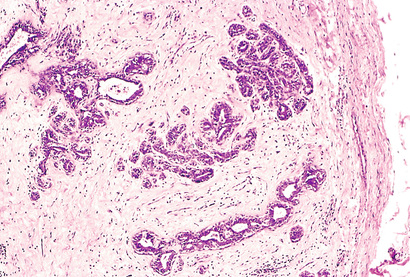
Next to the development of metastases, invasiveness is the feature that most reliably distinguishes cancers from benign tumors (Figs. 6.9 and 6.10). Cancers lack well-defined capsules. There are instances in which a slowly growing malignant tumor deceptively appears to be encased by the stroma of the surrounding host tissue, but microscopic examination reveals tiny crablike feet penetrating the margin and infiltrating adjacent structures. This infiltrative mode of growth makes it necessary to remove a wide margin of surrounding normal tissue when surgical excision of a malignant tumor is attempted. Surgical pathologists carefully examine the margins of resected tumors to ensure that they are devoid of cancer cells (clean margins).
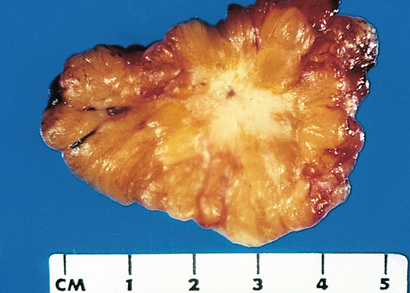
Metastasis
Metastasis is defined by the spread of a tumor to sites that are physically discontinuous with the primary tumor and unequivocally marks a tumor as malignant, as by definition benign neoplasms do not metastasize. The invasiveness of cancers permits them to penetrate into blood vessels, lymphatics, and body cavities, providing opportunities for spread (Fig. 6.11). Overall, approximately 30% of patients with newly diagnosed solid tumors (excluding skin cancers other than melanomas) present with clinically evident metastases. An additional 20% have occult (hidden) metastases at the time of diagnosis.

In general, the more anaplastic and the larger the primary neoplasm, the more likely is metastatic spread, but as with most rules there are exceptions. Extremely small cancers have been known to metastasize; conversely, some large and ominous-looking lesions may not. While all malignant tumors can metastasize, some do so very infrequently. For example, basal cell carcinomas of the skin and most primary tumors of the central nervous system are highly locally invasive but rarely metastasize. It is evident then that the properties of local invasion and metastasis are sometimes separable.
A special circumstance involves so-called “blood cancers”, the leukemias and lymphomas. These tumors are derived from blood-forming cells that normally have the capacity to enter the bloodstream and travel to distant sites; as a result, with only rare exceptions, leukemias and lymphomas are taken to be disseminated diseases at diagnosis and are always considered to be malignant.
Malignant neoplasms disseminate by one of three pathways: (1) seeding within body cavities, (2) lymphatic spread, or (3) hematogenous spread. Spread by seeding occurs when neoplasms invade a natural body cavity. This mode of dissemination is particularly characteristic of cancers of the ovary, which often cover the peritoneal surfaces widely. The implants literally may glaze all peritoneal surfaces and yet not invade the underlying tissues. Here is an instance where the ability to reimplant and grow at sites distant from the primary tumor seems to be separable from the capacity to invade. Neoplasms of the central nervous system, such as a medulloblastoma or ependymoma, may penetrate the cerebral ventricles and be carried by the cerebrospinal fluid to reimplant on the meningeal surfaces, either within the brain or in the spinal cord.
Lymphatic spread is more typical of carcinomas, whereas hematogenous spread is favored by sarcomas. There are numerous interconnections, however, between the lymphatic and vascular systems, so all forms of cancer may disseminate through either or both systems. The pattern of lymph node involvement depends principally on the site of the primary neoplasm and the natural pathways of local lymphatic drainage. Lung carcinomas arising in the respiratory passages metastasize first to the regional bronchial lymph nodes and then to the tracheobronchial and hilar nodes. Carcinoma of the breast usually arises in the upper outer quadrant and first spreads to the axillary nodes. However, medial breast lesions may drain through the chest wall to the nodes along the internal mammary artery. Thereafter, in both instances, the supraclavicular and infraclavicular nodes may be seeded. In some cases, the cancer cells seem to travel in lymphatic channels within the immediately proximate nodes to be trapped in subsequent lymph nodes, producing so-called “skip metastases.” The cells may traverse all of the lymph nodes ultimately to reach the vascular compartment by way of the thoracic duct.
A “sentinel lymph node” is the first regional lymph node that receives lymph flow from a primary tumor. It can be identified by injection of blue dyes or radiolabeled tracers near the primary tumor. Biopsy of sentinel lymph nodes allows determination of the extent of spread of tumor and can be used to plan treatment.
Of note, although enlargement of nodes near a primary neoplasm should arouse concern for metastatic spread, it does not always imply cancerous involvement. The necrotic products of the neoplasm and tumor antigens often evoke immunologic responses in the nodes, such as hyperplasia of the follicles (lymphadenitis) and proliferation of macrophages in the subcapsular sinuses (sinus histiocytosis). Thus, histopathologic verification of tumor within an enlarged lymph node is required.
While hematogenous spread is the favored pathway for sarcomas, carcinomas use it as well. As might be expected, arteries are penetrated less readily than are veins. With venous invasion, the bloodborne cells follow the venous flow draining the site of the neoplasm, with tumor cells often stopping in the first capillary bed they encounter. Since all portal area drainage flows to the liver, and all caval blood flows to the lungs, the liver and lungs are the most frequently involved secondary sites in hematogenous dissemination. Cancers arising near the vertebral column often embolize through the paravertebral plexus; this pathway probably is involved in the frequent vertebral metastases of carcinomas of the thyroid and prostate glands.
Certain carcinomas have a propensity to grow within veins. Renal cell carcinoma often invades the renal vein to grow in a snakelike fashion up the inferior vena cava, sometimes reaching the right side of the heart. Hepatocellular carcinomas often penetrate and grow within the radicles of portal and hepatic veins, eventually reaching the main venous channels. Remarkably, such intravenous growth may not be accompanied by widespread dissemination.
Many observations suggest that the anatomic localization of a neoplasm and its venous drainage cannot wholly explain the systemic distributions of metastases. For example, prostatic carcinoma preferentially spreads to bone, bronchogenic carcinoma tends to involve the adrenal glands and the brain, and neuroblastoma spreads to the liver and bones. Conversely, skeletal muscles, although rich in capillaries, are rarely sites of tumor metastases. The molecular basis of such tissue-specific homing of tumor cells is discussed later.
Thus, numerous features of tumors (Fig. 6.12) usually permit the differentiation of benign and malignant neoplasms.
 Summary
Summary
Characteristics of Benign and Malignant Tumors
• Benign and malignant tumors can be distinguished from one another based on the degree of differentiation, rate of growth, local invasiveness, and distant spread.
• Benign tumors resemble the tissue of origin and are well differentiated; malignant tumors are poorly or completely undifferentiated (anaplastic).
• Benign tumors tend to be slow growing, whereas malignant tumors generally grow faster.
• Benign tumors are well circumscribed and have a capsule; malignant tumors are poorly circumscribed and invade the surrounding normal tissues.
• Benign tumors remain localized to the site of origin, whereas malignant tumors are locally invasive and metastasize to distant sites.

Epidemiology
Study of cancer occurrence in populations has contributed substantially to knowledge about its origins. The now well-established concept that cigarette smoking is causally associated with lung cancer arose primarily from epidemiologic studies. A comparison of the incidence rates for colon cancer and dietary patterns in the Western world and in Africa led to the recognition that dietary fat and fiber content may figure importantly in the causation of this cancer. Major insights into the causes of cancer can be obtained by epidemiologic studies that relate particular environmental, racial (possibly hereditary), and cultural influences to the occurrence of specific neoplasms. Certain diseases associated with an increased risk for developing cancer also provide clues to the pathogenesis of cancer. The following discussion first summarizes the overall incidence of cancer to provide insight into the magnitude of the cancer problem and then reviews factors relating to the patient and the environment that influence the predisposition to cancer.
Cancer Incidence
For the year 2012, the World Health Organization (WHO) estimated that there were about 14.1 million new cancer cases worldwide, leading to 8.2 million deaths (approximately 22,500 deaths per day). Moreover, due to increasing population size, by the year 2035 the WHO projects that the numbers of cancer cases and deaths worldwide will increase to 24 million and 14.6 million, respectively (based on current mortality rates). Additional perspective on the likelihood of developing a specific form of cancer can be gained from national incidence and mortality data. In the United States, it is estimated that the year 2016 will be marked by approximately 1.69 million new cases of cancer and 595,000 cancer deaths. Incidence data for the most common forms of cancer, with the major killers identified, are presented in Fig. 6.13.
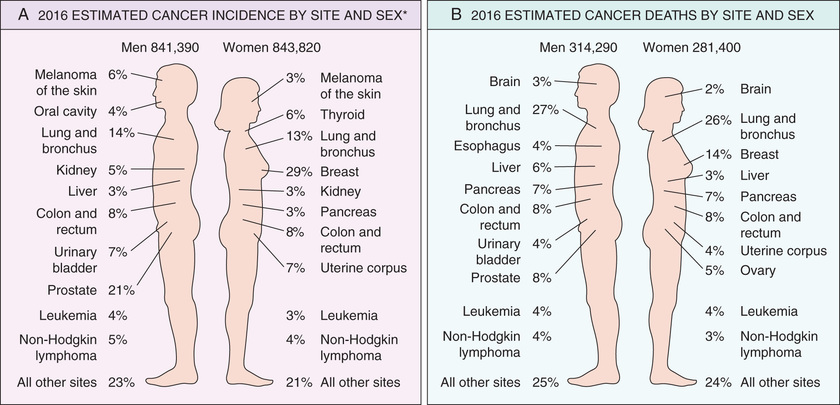
Over several decades, the death rates for many forms of cancer have changed. Since 1995, the incidence of cancer in men and women in the United States has been roughly stable, but the cancer death rate has decreased by roughly 20% in men and 10% in women. Among men, 80% of the decrease is accounted for by lower death rates for cancers of the lung, prostate, and colon; among women, nearly 60% of the decrease is due to reductions in death rates from breast and colorectal cancers. Decreased use of tobacco products is responsible for the reduction in lung cancer deaths, while improved detection and treatment are responsible for the decrease in death rates for colorectal, female breast, and prostate cancers.
The last half-century has also seen a sharp decline in death rates from cervical cancer and gastric cancer in the United States. The decrease in cervical cancer is directly attributable to widespread use of the Papanicolaou (PAP) smear test for early detection of this tumor and its precursor lesions. The deployment of the human papillomavirus (HPV) vaccine may nearly eliminate this cancer in coming years. The cause of the decline in death rates for cancers of the stomach is obscure; it may be related to decreasing exposure to unknown dietary carcinogens.
Environmental Factors
Environmental exposures appear to be the dominant risk factors for many common cancers, suggesting that a high fraction of cancers are potentially preventable. This notion is supported by the geographic variation in death rates from specific forms of cancer, which is thought to stem mainly from differences in environmental exposures. For instance, death rates from breast cancer are about four to five times higher in the United States and Europe than in Japan. Conversely, the death rate for stomach carcinoma in men and women is about seven times higher in Japan than in the United States. Liver cell carcinoma is relatively infrequent in the United States but is the most lethal cancer among many African populations. Nearly all the evidence indicates that these geographic differences have environmental rather than genetic origins. For example, Nisei (second-generation Japanese living in the United States) have mortality rates for certain forms of cancer that are intermediate between those in natives of Japan and in Americans who have lived in the United States for many generations. The two rates come closer with each passing generation.
There is no paucity of environmental factors that contribute to cancer. They lurk in the ambient environment, in the workplace, in food, and in personal practices. They can be as universal as sunlight or be largely restricted to urban settings (e.g., asbestos) or particular occupations (Table 6.2). The most important environmental exposures linked to cancer include the following:
• Diet. Certain features of diet have been implicated as predisposing influences. More broadly, obesity, currently epidemic in the United States, is associated with a modestly increased risk for developing many different cancers.
• Smoking. Smoking, particularly of cigarettes, has been implicated in cancer of the mouth, pharynx, larynx, esophagus, pancreas, bladder, and, most significantly, the lung, as 90% of lung cancer deaths are related to smoking.
• Alcohol consumption. Alcohol abuse is an independent risk factor for cancers of the oropharynx, larynx, esophagus, and (due to alcoholic cirrhosis) liver. Moreover, alcohol and tobacco smoking synergistically increase the risk for developing cancers of the upper airways and upper digestive tract.
• Reproductive history. There is strong evidence that lifelong cumulative exposure to estrogen stimulation, particularly if unopposed by progesterone, increases the risk for developing cancers of the endometrium and breast, both of which are estrogen-responsive tissues.
• Infectious agents. It is estimated that infectious agents cause approximately 15% of cancers worldwide.
Table 6.2
Occupational Cancers
| Agents or Groups of Agents | Human Cancers for Which Reasonable Evidence Is Available | Typical Use or Occurrence |
| Arsenic and arsenic compounds | Lung carcinoma, skin carcinoma | By-product of metal smelting; component of alloys, electrical and semiconductor devices, medications and herbicides, fungicides, and animal dips |
| Asbestos | Lung, esophageal, gastric, and colon carcinoma; mesothelioma | Formerly used for many applications because of fire, heat, and friction resistance; still found in existing construction as well as fire-resistant textiles, friction materials (i.e., brake linings), underlayment and roofing papers, and floor tiles |
| Benzene | Acute myeloid leukemia | Principal component of light oil; despite known risk, many applications exist in printing and lithography, paint, rubber, dry cleaning, adhesives and coatings, and detergents; formerly widely used as solvent and fumigant |
| Beryllium and beryllium compounds | Lung carcinoma | Missile fuel and space vehicles; hardener for lightweight metal alloys, particularly in aerospace applications and nuclear reactors |
| Cadmium and cadmium compounds | Prostate carcinoma | Uses include yellow pigments and phosphors; found in solders; used in batteries and as alloy and in metal platings and coatings |
| Chromium compounds | Lung carcinoma | Component of metal alloys, paints, pigments, and preservatives |
| Nickel compounds | Lung and oropharyngeal carcinoma | Nickel plating; component of ferrous alloys, ceramics, and batteries; by-product of stainless-steel arc welding |
| Radon and its decay products | Lung carcinoma | From decay of minerals containing uranium; potentially serious hazard in quarries and underground mines |
| Vinyl chloride | Hepatic angiosarcoma | Refrigerant; monomer for vinyl polymers; adhesive for plastics; formerly inert aerosol propellant in pressurized containers |
Thus, there is no escape: it seems that everything people do to earn a livelihood, to subsist, or to enjoy life turns out to be illegal, immoral, or fattening, or—most disturbing—possibly carcinogenic!
Age and Cancer
In general, the frequency of cancer increases with age. Most cancer deaths occur between 55 and 75 years of age; the rate declines, along with the population base, after 75 years of age. The rising incidence with age may be explained by the accumulation of somatic mutations that drive the emergence of malignant neoplasms (discussed later). The decline in immune competence that accompanies aging also may be a factor.
Although cancer preferentially affects older adults, it also is responsible for slightly more than 10% of all deaths among children younger than 15 years of age (Chapter 7). The major lethal cancers in children are leukemias, tumors of the central nervous system, lymphomas, and soft-tissue and bone sarcomas. As discussed later, study of several childhood tumors, such as retinoblastoma, has provided fundamental insights into the pathogenesis of malignant transformation.
Acquired Predisposing Conditions
Acquired conditions that predispose to cancer include disorders associated with chronic inflammation, immunodeficiency states, and precursor lesions. Many chronic inflammatory conditions create a fertile “soil” for the development of malignant tumors (Table 6.3). Tumors arising in the context of chronic inflammation are mostly carcinomas, but also include mesothelioma and several kinds of lymphoma. By contrast, immunodeficiency states mainly predispose to virus-induced cancers, including specific types of lymphoma and carcinoma and some sarcoma-like proliferations.
Table 6.3
Chronic Inflammatory States and Cancer
| Pathologic Condition | Associated Neoplasm(s) | Etiologic Agent |
| Asbestosis, silicosis | Mesothelioma, lung carcinoma | Asbestos fibers, silica particles |
| Inflammatory bowel disease | Colorectal carcinoma | |
| Lichen sclerosis | Vulvar squamous cell carcinoma | |
| Pancreatitis | Pancreatic carcinoma | Alcoholism, germ line mutations (e.g., in the trypsinogen gene) |
| Chronic cholecystitis | Gallbladder cancer | Bile acids, bacteria, gallbladder stones |
| Reflux esophagitis, Barrett esophagus | Esophageal carcinoma | Gastric acid |
| Sjögren syndrome, Hashimoto thyroiditis | MALT lymphoma | |
| Opisthorchis, cholangitis | Cholangiocarcinoma, colon carcinoma | Liver flukes (Opisthorchis viverrini) |
| Gastritis/ulcers | Gastric adenocarcinoma, MALT lymphoma | Helicobacter pylori |
| Hepatitis | Hepatocellular carcinoma | Hepatitis B and/or C virus |
| Osteomyelitis | Carcinoma in draining sinuses | Bacterial infection |
| Chronic cervicitis | Cervical carcinoma | Human papillomavirus |
| Chronic cystitis | Bladder carcinoma | Schistosomiasis |
Precursor lesions are localized disturbances of epithelial differentiation that are associated with an elevated risk for developing carcinoma. They may arise secondary to chronic inflammation or hormonal disturbances (in endocrine-sensitive tissues), or may occur spontaneously. Molecular analyses have shown that precursor lesions often possess some of the genetic lesions found in their associated cancers (discussed later). However, progression to cancer is not inevitable, and it is important to recognize precursor lesions because their removal or reversal lowers cancer risk.
Many different precursor lesions have been described; among the most common are the following:
• Squamous metaplasia and dysplasia of bronchial mucosa, seen in in habitual smokers—a risk factor for lung carcinoma (Chapter 13)
• Endometrial hyperplasia and dysplasia, seen in women with unopposed estrogenic stimulation—a risk factor for endometrial carcinoma (Chapter 19)
• Leukoplakia of the oral cavity, vulva, and penis, which may progress to squamous cell carcinoma (Chapters 15, 18, and 19)
• Villous adenoma of the colon, associated with a high risk for progression to colorectal carcinoma (Chapter 15)
In this context it also may be asked, “What is the risk for malignant change in a benign neoplasm?”—or, stated differently, “Are benign tumors precancers?” In general the answer is no, but inevitably there are exceptions, and perhaps it is better to say that each type of benign tumor is associated with a particular level of risk, ranging from high to virtually nonexistent. As cited earlier, adenomas of the colon as they enlarge can undergo malignant transformation in up to 50% of cases; by contrast, malignant change is extremely rare in leiomyomas of the uterus.
Interactions Between Environmental and Genetic Factors
Cancer behaves like an inherited trait in some families, usually due to germ line mutations that affect the function of a gene that suppresses cancer (a so-called “tumor suppressor gene,” discussed later). What then can be said about the influence of heredity on sporadic malignant neoplasms, which constitute roughly 95% of the cancers in the United States?
While the evidence suggests that sporadic cancers can largely be attributed to environmental factors or acquired predisposing conditions, lack of family history does not preclude an inherited component. It may in fact be difficult to tease out hereditary and genetic contributions because these factors often interact. Such interactions may be particularly complex when tumor development is affected by small contributions from multiple genes. Furthermore, genetic factors may alter the risk for developing environmentally induced cancers. Instances where this holds true often involve inherited variation in enzymes such as components of the cytochrome P-450 system that metabolize procarcinogens to active carcinogens. Conversely, environmental factors can influence the risk for developing cancer, even in individuals who inherit well-defined “cancer genes.” For instance, breast cancer risk in females who inherit mutated copies of the BRCA1 or BRCA2 tumor suppressor genes (discussed later) is almost three-fold higher for women born after 1940 than for women born before that year, perhaps because of changes in reproductive behavior or increases in obesity in more recent times.
Summary
Epidemiology of Cancer
• The incidence of cancer varies with age, geographic factors, and genetic background. The geographic variation in cancer incidence results mostly from different environmental exposures. Cancer can occur at any age, but is most common in older adults.
• Environmental factors implicated in carcinogenesis include infectious agents, smoking, alcohol, diet, obesity, reproductive history, and exposure to carcinogens.
• Cancer risk rises in certain tissues in the setting of increased cellular proliferation caused by chronic inflammation or hormonal stimulation.
• Epithelial cell linings may develop morphologic changes that signify an increased risk for developing cancer; such lesions are referred to as precursor lesions.
• The risk for developing cancer is modified by interactions between environmental exposures and genetic variants.
Cancer Genes
It could be argued that the proliferation of literature on the molecular basis of cancer has outpaced the growth of even the most malignant of tumors. Researchers and students alike can easily get lost in the growing forest of information. But it has become eminently clear that cancer is a disease caused by mutations that alter the function of of a finite subset of the 20,000 or so human genes. For simplicity, we will refer to these genes as cancer genes. Cancer genes can be defined as genes that are recurrently affected by genetic aberrations in cancers, presumably because they contribute directly to the malignant behavior of cancer cells. Causative mutations that give rise to cancer genes may be acquired by the action of environmental agents, such as chemicals, radiation, or viruses, may occur spontaneously, or may be inherited in the germ line. If such mutations drive carcinogenesis, a key prediction is that each cell in an individual tumor should share mutations that were present in the founding cell at the time of transformation. This expectation has been realized in all tumors that have been systematically analyzed by genomic sequencing, providing strong support for the hypothesis that cancer is at its root a genetic disease.
Cancer genes number in the hundreds and new ones are still being discovered. Not only are these genes numerous, but many have unpronounceable acronyms for names that are difficult to remember, even for the aficionado. One way to try to simplify this complexity is to consider that cancer genes fall into one of four major functional classes:
• Oncogenes are genes that induce a transformed phenotype when expressed in cells by promoting increased cell growth. A major discovery in cancer was the realization that oncogenes are mutated or overexpressed versions of normal cellular genes, which are called proto-oncogenes. Most oncogenes encode transcription factors, factors that participate in pro-growth signaling pathways, or factors that enhance cell survival. They are considered dominant genes because a mutation involving a single allele is sufficient to produce a pro-oncogenic effect.
• Tumor suppressor genes are genes that normally prevent uncontrolled growth and, when mutated or lost from a cell, allow the transformed phenotype to develop. Often both normal alleles of tumor suppressor genes must be damaged for transformation to occur. Tumor suppressor genes can be placed into two general groups, “governors” that act as important brakes on cellular proliferation, and “guardians” that are responsible for sensing genomic damage. Some guardian genes initiate and choreograph a complex “damage control response” that leads to the cessation of proliferation or, if the damage is too great to be repaired, or induce apoptosis.
• Genes that regulate apoptosis primarily act by enhancing cell survival, rather than stimulating proliferation per se. Understandably, genes of this class that protect against apoptosis are often overexpressed in cancer cells, whereas those that promote apoptosis tend to be underexpressed or functionally inactivated by mutations.
• To this list may now be added genes that regulate interactions between tumor cells and host cells, as these genes are also recurrently mutated or functionally altered in certain cancers. Particularly important are genes that enhance or inhibit recognition of tumors cells by the host immune system.
In most instances, the mutations that give rise to cancer genes are acquired during life and are confined to the cancer cells. However, causative mutations sometimes are inherited in the germ line and are therefore present in every cell in the body, placing the affected individual at high risk for developing cancer. Understandably, in families in which these germ line mutations are passed from generation to generation, cancer behaves like an inherited trait (Table 6.4). We will touch on important familial cancer syndromes and associated genes and cancers later in this chapter.
Table 6.4
Inherited Predisposition to Cancer
| Inherited Predisposition | Gene(s) |
| Autosomal Dominant Cancer Syndromes | |
| Retinoblastoma | RB |
| Li-Fraumeni syndrome (various tumors) | TP53 |
| Melanoma | CDKN2A |
| Familial adenomatous polyposis/colon cancer | APC |
| Neurofibromatosis 1 and 2 | NF1, NF2 |
| Breast and ovarian tumors | BRCA1, BRCA2 |
| Multiple endocrine neoplasia 1 and 2 | MEN1, RET |
| Hereditary nonpolyposis colon cancer | MSH2, MLH1, MSH6 |
| Nevoid basal cell carcinoma syndrome | PTCH1 |
| Autosomal Recessive Syndromes of Defective DNA Repair | |
| Xeroderma pigmentosum | Diverse genes involved in nucleotide excision repair |
| Ataxia-telangiectasia | ATM |
| Bloom syndrome | BLM |
| Fanconi anemia | Diverse genes involved in repair of DNA cross-links |

Presented next is a discussion of the varied genetic lesions that underlie altered cancer gene expression and function.
Genetic Lesions in Cancer
The genetic changes found in cancers vary from point mutations involving single nucleotides to abnormalities large enough to produce gross changes in chromosome structure. In certain neoplasms, genetic abnormalities are nonrandom and highly characteristic. Specific chromosomal abnormalities have been identified in most leukemias and lymphomas and in an increasing number of nonhematopoietic tumors, while other tumors are characterized by particular point mutations. It is believed that all recurrent genetic changes alter the activity of one or more cancer genes in a fashion that gives the affected cells a selective advantage, presumably by contributing to one or more of the hallmarks of cancer.
Driver and Passenger Mutations
In the following sections, we briefly review the types of mutations that are commonly found in cancers. Before doing so, however, we must first touch on the important concept of driver mutations and passenger mutations. Driver mutations are mutations that alter the function of cancer genes and thereby directly contribute to the development or progression of a given cancer. They are usually acquired, but as mentioned earlier, occasionally inherited. By contrast, passenger mutations are acquired mutations that are neutral in terms of fitness and do not affect cellular behavior; they just come along for the proverbial ride. Because they occur at random, passenger mutations are sprinkled throughout the genome, whereas driver mutations tend to be tightly clustered within cancer genes. It is now appreciated that particularly in cancers caused by carcinogen exposure, such as melanoma and smoking-related lung cancer, passenger mutations greatly outnumber driver mutations.
Despite their apparently innocuous nature, passenger mutations have nevertheless proven to be important in several ways:
• In carcinogen-associated cancers, mutational analysis has provided definitive evidence that most genomic damage is directly caused by the carcinogen in question. For example, before sequencing of melanoma genomes, the causative role of sun exposure in this cancer was debated. This is no longer so, as most melanomas have thousands of mutations of a type that is specifically linked to damage caused by ultraviolet light.
• A second, more nefarious effect of passenger mutations is that they create genetic variants that, while initially neutral, may provide tumor cells with a selective advantage in the setting of therapy. The evidence for this comes from DNA sequence analyses of tumors at the time of recurrence after drug therapy; in many instances, mutations that lead directly to drug resistance are found in most tumor cells. Generally, the same resistance mutations can also be found before therapy, but only in a very small fraction of cells. In such instances, it appears that the selective pressure of therapy “converts” a neutral passenger mutation into a driver mutation, to the benefit of the tumor and the detriment of the patient.
Point Mutations
Point mutations can either activate or inactivate the protein products of the affected genes depending on their precise position and consequence. Point mutations that convert proto-oncogenes into oncogenes generally produce a gain-of-function by altering amino acid residues in a domain that normally holds the protein's activity in check. A cardinal example is point mutations that convert the RAS gene into a cancer gene, one of the most comment events in human cancers. By contrast, point mutations (as well as larger aberrations, such as insertions and deletions) in tumor suppressor genes reduce or disable the function of the encoded protein. The tumor suppressor gene that is most commonly affected by point mutations in cancer is TP53, a prototypical “guardian” type tumor suppressor gene (discussed later).
Gene Rearrangements
Gene rearrangements may be produced by chromosomal translocations or inversions. Specific chromosomal translocations and inversions are highly associated with certain malignancies, particularly neoplasms derived from hematopoietic cells and other kinds of mesenchymal cells. These rearrangements can activate proto-oncogenes in two ways:
• Some gene rearrangements result in overexpression of proto-oncogenes by removing them from their normal regulatory elements and placing them under control of an inappropriate, highly active promoter or enhancer. Two different kinds of B cell lymphoma provide illustrative examples of this mechanism. In more than 90% of cases of Burkitt lymphoma, the cells have a translocation, usually between chromosomes 8 and 14, that leads to overexpression of the MYC gene on chromosome 8 by juxtaposition with immunoglobulin heavy chain gene regulatory elements on chromosome 14 (Fig. 6.14). In follicular lymphoma, a reciprocal translocation between chromosomes 14 and 18 leads to overexpression of the anti-apoptotic gene, BCL2, on chromosome 18, also driven by immunoglobulin gene regulatory elements.
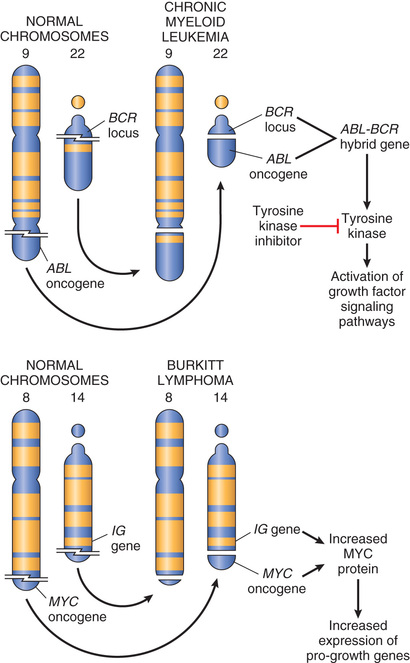
• Other oncogenic gene rearrangements create fusion genes encoding novel chimeric proteins. Most notable is the Philadelphia (Ph) chromosome in chronic myeloid leukemia, consisting of a balanced reciprocal translocation between chromosomes 9 and 22 (see Fig. 6.14). As a consequence, the derivative chromosome 22 (the Philadelphia chromosome) appears smaller than normal. This cytogenetic change is seen in more than 90% of cases of chronic myeloid leukemia and results in the fusion of portions of the BCR gene on chromosome 22 and the ABL gene on chromosome 9. The few Philadelphia chromosome–negative cases harbor a cryptic (cytogenetically silent) BCR-ABL fusion gene, the presence of which is the sine qua non of chronic myeloid leukemia. As discussed later, the BCR-ABL fusion gene encodes a novel tyrosine kinase with potent transforming activity.
Lymphoid tumors are most commonly associated with recurrent gene rearrangements. This relationship exists because normal lymphocytes express special enzymes that purposefully introduce DNA breaks during the processes of immunoglobulin or T cell receptor gene recombination. Repair of these DNA breaks is error-prone, and the resulting mistakes sometimes result in gene rearrangements that activate proto-oncogenes. Two other types of mesenchymal tumors, myeloid neoplasms (acute myeloid leukemias and myeloproliferative disorders) and sarcomas, also frequently possess gene rearrangements. Unlike lymphoid neoplasms, the cause of the DNA breaks that lead to gene rearrangements in myeloid neoplasms and sarcomas is unknown. In general, the rearrangements that are seen in myeloid neoplasms and sarcomas create fusion genes that encode either hyperactive tyrosine kinases (akin to BCR-ABL) or novel oncogenic transcription factors. A well-characterized example of the latter is the (11;22)(q24;q12) translocation in Ewing sarcoma. This rearrangement creates a fusion gene encoding a chimeric oncoprotein composed of portions of two different transcription factors called EWS and FLI1.
Identification of pathogenic gene rearrangements in carcinomas has lagged because karyotypically evident translocations and inversions (which point to the location of important oncogenes) are rare in carcinomas. However, advances in DNA sequencing have revealed recurrent cryptic pathogenic gene rearrangements in carcinomas as well. As with hematologic malignancies and sarcomas, gene rearrangements in solid tumors can contribute to carcinogenesis either by increasing expression of an oncogene or by generation of a novel fusion gene. Examples will be discussed along with specific cancers in other chapters. As with a fusion gene such as BCR-ABL, some of the fusion genes in solid tumors also provide drug targets (e.g., EML-ALK in lung cancer; Chapter 13).
Deletions
Deletions are another prevalent abnormality in tumor cells. Deletion of specific regions of chromosomes may result in the loss of particular tumor suppressor genes. Tumor suppressors generally require inactivation of both alleles in order for them to contribute to carcinogenesis. A common mechanism for this is an inactivating point mutation in one allele, followed by deletion of the other, nonmutated allele. As discussed later, deletions involving 13q14, the site of the RB gene, are associated with retinoblastoma, and deletion of 17p is associated with loss of TP53, arguably the most important tumor suppressor gene.
Gene Amplifications
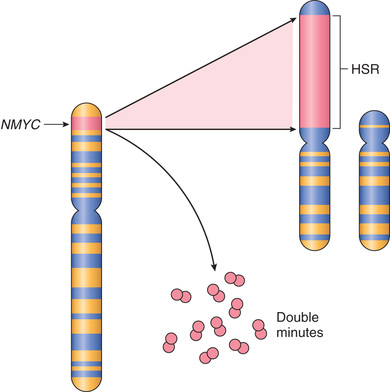
Proto-oncogenes may be converted to oncogenes by gene amplification, with consequent overexpression and hyperactivity of otherwise normal proteins. Such amplification may produce several hundred copies of the gene, a change in copy number that can be readily detected by molecular hybridization with appropriate DNA probes. In some cases, the amplified genes produce chromosomal changes that can be identified microscopically. Two mutually exclusive patterns are seen: multiple small, extrachromosomal structures called double minutes; and homogeneously staining regions. The latter derive from the insertion of the amplified genes into new chromosomal locations, which may be distant from the normal location of the involved genes; because regions containing amplified genes lack a normal banding pattern, they appear homogeneous in a G-banded karyotype. Two clinically important examples of amplification involve the NMYC gene in neuroblastoma and the HER2 gene in breast cancers. NMYC is amplified in 25% to 30% of neuroblastomas, and the amplification is associated with poor prognosis (Fig. 6.15). HER2 (also known as ERBB2) amplification occurs in about 20% of breast cancers, and antibody therapy directed against the receptor encoded by the HER2 gene has proved effective in this subset of tumors.
Aneuploidy
Aneuploidy is defined as a number of chromosomes that is not a multiple of the haploid state; for humans, that is a chromosome number that is not a multiple of 23. Aneuploidy is remarkably common in cancers, particularly carcinomas, and was proposed as a cause of carcinogenesis over 100 years ago. Aneuploidy frequently results from errors of the mitotic checkpoint, the major cell cycle control mechanism that acts to prevent mistakes in chromosome segregation. The mitotic checkpoint prevents aneuploidy by inhibiting the irreversible transition to anaphase until all of the replicated chromosomes have made productive attachments to spindle microtubules. Complete absence of the mitotic checkpoint leads to rapid cell death as a consequence of abnormal chromosome segregation.
Mechanistic data establishing aneuploidy as a cause of carcinogenesis, rather than a consequence, have been difficult to generate. However, statistical approaches made possible by detailed analysis of cancer cells suggest (as might be expected) that aneuploidy tends to increase the copy number of key oncogenes and decrease the copy number of potent tumor suppressors. For example, chromosome 8, which almost never is lost and often is present in increased copies in tumor cells, is where the MYC oncogene is located. By contrast, portions of chromosome 17, where the TP53 gene is located, are often lost and are infrequently gained. Thus, tumor development and progression may be molded by changes in chromosome numbers that enhance the dosage of oncogenes while restricting the activity of tumor suppressor genes.
MicroRNAs and Cancer
As discussed in Chapter 1, microRNAs (miRNAs) are noncoding, single-stranded RNAs, approximately 22 nucleotides in length, that function as negative regulators of genes. They inhibit gene expression posttranscriptionally by repressing translation or, in some cases, by messenger RNA (mRNA) cleavage. In view of their important functions in control of cell growth, differentiation, and survival, it is not surprising that accumulating evidence indicates that miRNAs also can contribute to carcinogenesis. Specifically, if the target of a miRNA is a tumor suppressor gene, then overactivity of the miRNA can reduce the tumor suppressor protein. Such miRNAs are sometimes referred to as oncomIRs. Conversely, if an miRNA inhibits the translation of an oncogene, a reduction in the quantity or function of that miRNA will lead to overproduction of the oncogene product. Such relationships have already been established by miRNA profiling of several human tumors. For example, downregulation or deletion of certain miRNAs in some leukemias and lymphomas results in increased expression of BCL2, an anti-apoptotic gene. Thus, by negatively regulating BCL2, such miRNAs behave as tumor suppressor genes. Dysregulation of other miRNAs that control the expression of the RAS and MYC oncogenes also has been detected in lung tumors and in certain B-cell leukemias, respectively.
Epigenetic Modifications and Cancer
You will recall from Chapter 1 that epigenetics refers to reversible, heritable changes in gene expression that occur without mutation. Such changes involve posttranslational modifications of histones and DNA methylation, both of which affect gene expression. In normal, differentiated cells, the major portion of the genome is not expressed. These regions of the genome are silenced by DNA methylation and histone modifications. On the other hand, cancer cells are characterized by a global DNA hypomethylation and selective promoter-localized hypermethylation. Indeed, it has become evident during the past several years that tumor suppressor genes are sometimes silenced by hypermethylation of promoter sequences, rather than by mutation. In addition, genome-wide hypomethylation has been shown to cause chromosomal instability and can induce tumors in mice. Thus, epigenetic changes may influence carcinogenesis in many ways. As an added wrinkle, deep sequencing of cancer genomes has identified mutations in genes that regulate epigenetic modifications in many cancers. Thus, certain genetic changes in cancers may be selected because they lead to alterations of the “epigenome” that favor cancer growth and survival.
The epigenetic state of particular cell types—a feature described as the epigenetic context—also dictates their response to signals that control growth and differentiation. As mentioned earlier, epigenetic modifications regulate gene expression, allowing cells with the same genetic makeup (e.g., a neuron and a keratinocyte) to have completely different appearances and functions. In some instances, the epigenetic state of a cell dramatically affects its response to otherwise identical signals. For example, the NOTCH1 gene has an oncogenic role in T-cell leukemia, yet acts as a tumor suppressor in squamous cell carcinomas. As would be expected, this dichotomy exists because activated NOTCH1 turns on pro-growth genes in T-cell progenitors and tumor suppressor genes in keratinocytes.
Summary
Genetic Lesions in Cancer
• Mutations in cancer cells fall into two major classes, driver (pathogenic) mutations and passenger (neutral) mutations.
• Passenger mutations may become driver mutations if selective pressure on the tumor changes, for example, in the setting of treatment with an effective therapeutic drug.
• Tumor cells may acquire driver mutations through several means, including point mutations and nonrandom chromosomal abnormalities that contribute to malignancy; these include gene rearrangements, deletions, and amplifications.
• Gene rearrangements (usually caused by translocations, but sometimes by inversions of other more complex events) contribute to carcinogenesis by overexpression of oncogenes or generation of novel fusion proteins with altered signaling capacity.
• Deletions frequently affect tumor suppressor genes, whereas gene amplification increases the expression of oncogenes.
• Overexpression of miRNAs can contribute to carcinogenesis by reducing the expression of tumor suppressors, while deletion or loss of expression of miRNAs can lead to overexpression of proto-oncogenes.
• Tumor suppressor genes and DNA repair genes also may be silenced by epigenetic changes, which involve reversible, heritable changes in gene expression that occur not by mutation but by methylation of the promoter.
Carcinogenesis: A Multistep Process
Fortunately, in most if not all instances, no single mutation is sufficient to transform a normal cell into a cancer cell. Carcinogenesis is thus a multistep process resulting from the accumulation of multiple genetic alterations that collectively give rise to the transformed phenotype and all of its associated hallmarks, discussed later. As mentioned earlier, the presence of driver mutations in some non-neoplastic precursor lesions suggest the need for additional mutations for transition to a full blown cancer and thus support this model.
Beyond tumor initiation from a single founding cell, it is important to recognize that cancers continue to undergo Darwinian selection and therefore continue to evolve (Fig. 6.16). It is well established that during their course cancers generally become more aggressive and acquire greater malignant potential, a phenomenon referred to as tumor progression. At the molecular level, tumor progression most likely results from mutations that accumulate independently in different cells. Some of these mutations may be lethal, but others may affect the function of cancer genes, thereby making the affected cells more adept at growth, survival, invasion, metastasis, or immune evasion. Due to this selective advantage, subclones that acquire these mutations may come to dominate one area of a tumor, either at the primary site or at sites of metastasis. As a result of continuing mutation and Darwinian selection, even though malignant tumors are monoclonal in origin they are typically genetically heterogeneous by the time of their clinical presentation. In advanced tumors exhibiting genetic instability, the extent of genetic heterogeneity may be enormous.
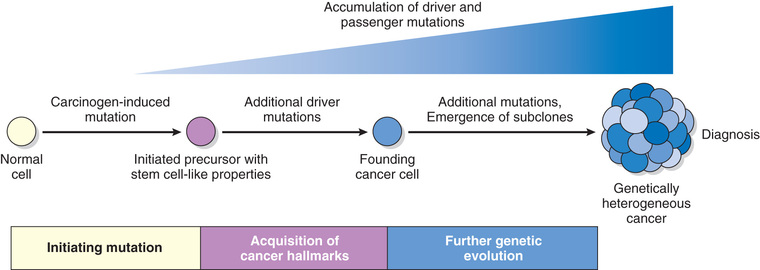
Genetic evolution shaped by darwinian selection can explain the two most pernicious properties of cancers: the tendency over time for cancers to become both more aggressive and less responsive to therapy. Thus, genetic heterogeneity has implications not only for cancer progression but also for its response to therapy. Experience has shown that when tumors recur after chemotherapy, the recurrent tumor is almost always resistant to the original drug regimen if it is given again. Experimental data suggest that this acquired resistance stems from the outgrowth of subclones that have, by chance, mutations (or epigenetic alterations) that impart drug resistance.
Hallmarks of Cancer
This overview serves as background for a more detailed consideration of the molecular pathogenesis of cancer. As mentioned earlier, bona fide cancer genes number in the hundreds, at a minimum. While it is traditional to describe the function of cancer genes one gene at a time, the blizzard of mutated genes emerging from the sequencing of cancer genomes has blanketed the landscape and revealed the limitations of trying to grasp the fundamental properties of cancer, gene by gene. A much more tractable and conceptually satisfying way to think about the biology of cancer is to consider the common phenotypic and biological properties of cancer cells. It appears that all cancers display eight fundamental changes in cell physiology, which are considered the hallmarks of cancer. These changes are illustrated in Fig. 6.17 and consist of the following:
• Self-sufficiency in growth signals
• Insensitivity to growth-inhibitory signals
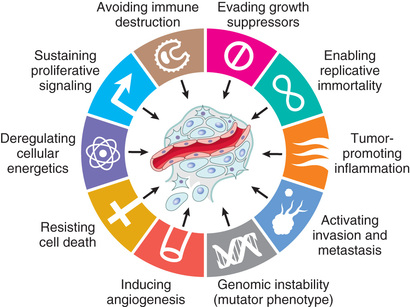
The acquisition of the genetic and epigenetic alterations that confer these hallmarks may be accelerated by cancer-promoting inflammation and by genomic instability. These are considered enabling characteristics because they promote cellular transformation and subsequent tumor progression.
Mutations in genes that regulate some or all of these cellular traits are seen in every cancer; accordingly, these traits form the basis of the following discussion of the molecular origins of cancer. Of note, by convention, gene symbols are italicized but their protein products are not (e.g., RB gene and RB protein, TP53 and p53, MYC and MYC).
Self-Sufficiency in Growth Signals
The self-sufficiency in growth that characterizes cancer cells generally stems from gain-of-function mutations that convert proto-oncogenes to oncogenes. Oncogenes encode proteins called oncoproteins that promote cell growth, even in the absence of normal growth-promoting signals. To appreciate how oncogenes drive inappropriate cell growth, it is helpful to review briefly the sequence of events that characterize normal cell proliferation (introduced in Chapter 1). Under physiologic conditions, cell proliferation can be readily resolved into the following steps:
1. Binding of a growth factor to its specific receptor on the cell membrane
2. Transient and limited activation of the growth factor receptor, which in turn activates several signal-transducing proteins on the inner leaflet of the plasma membrane
3. Transmission of the transduced signal across the cytosol to the nucleus by second messengers or a cascade of signal transduction molecules
4. Induction and activation of nuclear regulatory factors that initiate and regulate DNA transcription and the biosynthesis of other cellular components that are needed for cell division, such as organelles, membrane components, and ribosomes
5. Entry and progression of the cell into the cell cycle, resulting ultimately in cell division
The mechanisms that endow cancer cells with the ability to proliferate can be grouped according to their role in the growth factor–induced signal transduction cascade and cell cycle regulation. Indeed, each one of the listed steps is susceptible to corruption in cancer cells.
Growth Factors
Cancers may secrete their own growth factors or induce stromal cells to produce growth factors in the tumor microenvironment. Most soluble growth factors are made by one cell type and act on a neighboring cell to stimulate proliferation (paracrine action). Normally, cells that produce the growth factor do not express the cognate receptor, preventing the formation of positive feedback loops within the same cell. This “rule” may be broken by cancer cells in several different ways.
• Some cancer cells acquire growth self-sufficiency by acquiring the ability to synthesize the same growth factors to which they are responsive. For example, many glioblastomas secrete platelet-derived growth factor (PDGF) and express the PDGF receptor, and many sarcomas make both transforming growth factor-α (TGF-α) and its receptor. Similar autocrine loops are fairly common in many types of cancer.
• Another mechanism by which cancer cells acquire growth self-sufficiency is by interaction with stroma. In some cases, tumor cells send signals to activate normal cells in the supporting stroma, which in turn produce growth factors that promote tumor growth.
Growth Factor Receptors
The next group in the sequence of signal transduction is growth factor receptors. Some growth factor receptors have an intrinsic tyrosine kinase activity that is activated by growth factor binding, while others signal by stimulating the activity of downstream proteins. Many of the myriad growth factor receptors function as oncoproteins when they are mutated or if they overexpressed. The best-documented examples of overexpression involve the epidermal growth factor (EGF) receptor family. ERBB1, the EGF receptor, is overexpressed in 80% of squamous cell carcinomas of the lung, 50% or more of glioblastomas, and 80% to 100% of epithelial tumors of the head and neck. As mentioned earlier, the gene encoding a related receptor, HER2 (ERBB2), is amplified in approximately 20% of breast cancers and in a smaller fraction of adenocarcinomas of the lung, ovary, stomach, and salivary glands. These tumors are exquisitely sensitive to the mitogenic effects of small amounts of growth factors. The significance of HER2 in the pathogenesis of breast cancers is illustrated dramatically by the clinical benefit derived from blocking the extracellular domain of this receptor with anti-HER2 antibodies, an elegant example of “bench to bedside” medicine. In other instances, tyrosine kinase activity is stimulated by point mutations or small indels that lead to subtle but functionally important changes in protein structure, or gene rearrangements that create fusion genes encoding chimeric receptors. In each of these cases, the mutated receptors are constitutively active, delivering mitogenic signals to cells even in the absence of growth factors. These types of mutations are most common in leukemias, lymphomas, and certain forms of sarcoma.
Downstream Signal-Transducing Proteins
Cancer cells often acquire growth autonomy as a result of mutations in genes that encode components of signaling pathways downstream of growth factor receptors. The signaling proteins that couple growth factor receptors to their nuclear targets are activated by ligand binding to growth factor receptors. The signals are trasnmitted to the nucleus through various signal transduction molecules. Two important oncoproteins in the category of signaling molecules are RAS and ABL. Each of these is discussed briefly next.
RAS
RAS is the most commonly mutated oncogene in human tumors. Approximately 30% of all human tumors contain mutated RAS genes, and the frequency is even higher in some specific cancers (e.g., pancreatic adenocarcinoma). RAS is a member of a family of small G proteins that bind guanosine nucleotides (guanosine triphosphate [GTP] and guanosine diphosphate [GDP]). Signaling by RAS involves the following sequential steps:
• Normally, RAS flips back and forth between an excited signal-transmitting state and a quiescent state. RAS is inactive when bound to GDP; stimulation of cells by growth factors such as EGF and PDGF leads to exchange of GDP for GTP and subsequent conformational changes that generate active RAS (Fig. 6.18). This excited signal-emitting state is short-lived, however, because the intrinsic guanosine triphosphatase (GTPase) activity of RAS hydrolyzes GTP to GDP, releasing a phosphate group and returning the protein to its quiescent GDP-bound state. The GTPase activity of activated RAS is magnified dramatically by a family of GTPase-activating proteins (GAPs), which act as molecular brakes that prevent uncontrolled RAS activation by favoring hydrolysis of GTP to GDP.
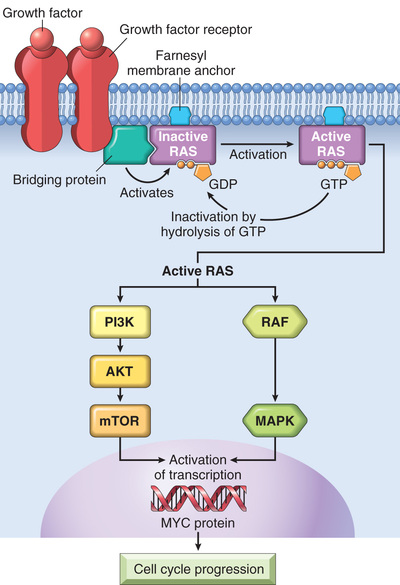
• Activated RAS stimulates downstream regulators of proliferation by several interconnected pathways that converge on the nucleus and alter the expression of genes that regulate growth, such as MYC. While details of the signaling cascades (some of which are illustrated in Fig. 6.18) downstream of RAS are not discussed here, an important point is that mutational activation of these signaling intermediates mimics the growth promoting effects of activated RAS. For example, BRAF, which lies in the so-called “RAF/ERK/MAP kinase pathway” is mutated in more than 60% of melanomas and is associated with unregulated cell proliferation. Mutations of phosphatidly inositol-3 kinase (PI3 kinase) in the PI3K/AKT pathway also occur with high frequency in some tumor types, with similary consequences.
RAS most commonly is activated by point mutations in amino acid residues that are either within the GTP-binding pocket or in the enzymatic region that carries out GTP hydrolysis. Both kinds of mutations interfere with breakdown of GTP, which is essential to inactivate RAS. RAS is thus trapped in its activated, GTP-bound form, and the cell is forced into a continuously proliferating state. It follows from this scenario that the consequences of activating mutations in RAS should be mimicked by loss-of-function mutations in GAPs, which would lead to a failure to simulate GTP hydrolysis and thereby restrain RAS. Indeed, the GAP neurofibromin-1 (NF1) is mutated in the cancer-prone familial disorder neurofibromatosis type 1 (Chapter 22) and is a bona fide tumor suppressor. Similarly, another important tumor suppressor called PTEN is a negative inhibitor of PI3 kinase and is frequently mutated in carcinomas, certain leukemias, and other cancers as well.
ABL
Several non–receptor tyrosine kinases function as signal transduction molecules. In this group, ABL is the best defined with respect to carcinogenesis.
The ABL proto-oncoprotein has tyrosine kinase activity that is dampened by internal negative regulatory domains. As discussed earlier (see Fig. 6.14), in chronic myeloid leukemia and certain acute leukemias, a part of the ABL gene is translocated from its normal abode on chromosome 9 to chromosome 22, where it fuses with part of the breakpoint cluster region (BCR) gene. This fusion gene encodes as BCR-ABL hybrid protein that contains the ABL tyrosine kinase domain and a BCR domain that self-associates, an event that unleashes a constitutive tyrosine kinase activity. Of interest, the BCR-ABL protein activates all of the signals that are downstream of RAS, making it a potent stimulator of cell growth.
The crucial role of BCR-ABL in cancer has been confirmed by the dramatic clinical response of patients with chronic myeloid leukemia to BCR-ABL kinase inhibitors. The prototype of this kind of drug, imatinib mesylate (Gleevec), galvanized interest in design of drugs that target specific molecular lesions found in various cancers (so-called “targeted therapy”). BCR-ABL also is an example of the concept of oncogene addiction, wherein a tumor is profoundly dependent on a single signaling molecule. BCR-ABL fusion gene formation is an early, perhaps initiating, event that drives leukemogenesis. Development of leukemia probably requires other collaborating mutations, but the transformed cell continues to depend on BCR-ABL for signals that mediate growth and survival. BCR-ABL signaling can be seen as the central lodgepole around which the transformed state is “built”. If the lodgepole is removed by inhibition of the BCR-ABL kinase, the structure collapses. In view of this level of dependency, it is not surprising that acquired resistance of tumors to BCR-ABL inhibitors often is due to the outgrowth of a subclone with a mutation in BCR-ABL that prevents binding of the drug to the BCR-ABL protein.
Nuclear Transcription Factors
The ultimate consequence of signaling through oncoproteins such as RAS or ABL is inappropriate and continuous stimulation of nuclear transcription factors that drive the expression of growth-promoting genes. Growth autonomy may thus be a consequence of mutations affecting genes that regulate DNA transcription. A host of oncoproteins, including products of the MYC, MYB, JUN, FOS, and REL oncogenes, function as transcription factors that regulate the expression of growth-promoting genes, such as cyclins. Of these, MYC is involved most commonly in human tumors.
Dysregulation of MYC promotes tumorigenesis by simultaneously promoting the progression of cells through the cell cycle and enhancing alterations in metabolism that support cell growth. MYC primarily functions by activating the transcription of other genes. Genes activated by MYC include several growth-promoting genes, including cyclin-dependent kinases (CDKs), whose products drive cells into the cell cycle (discussed next), and genes that control pathways that produce the building blocks (e.g., amino acids, lipids, nucleotides) that are needed for cell growth and division. As mentioned earlier (see Fig. 6.14), dysregulation of MYC results from a (8;14) translocation in Burkitt lymphoma, a highly aggressive B-cell tumor. MYC also is amplified in breast, colon, lung, and many other cancers, while the related NMYC and LMYC genes are amplified in neuroblastomas and small cell cancers of lung, respectively.
Cyclins and Cyclin-Dependent Kinases
As mentioned in Chapter 1, growth factors transduce signals that stimulate the orderly progression of cells through the various phases of the cell cycle, the process by which cells replicate their DNA in preparation for cell division. You will recall that progression of cells through the cell cycle is orchestrated by cyclin-dependent kinases (CDKs), which are activated by binding to cyclins, so called because of the cyclic nature of their production and degradation. The CDK-cyclin complexes phosphorylate crucial target proteins that drive cells forward through the cell cycle. While cyclins arouse the CDKs, CDK inhibitors (CDKIs), of which there are many, silence the CDKs and exert negative control over the cell cycle. Expression of these inhibitors is downregulated by mitogenic signaling pathways, thus promoting the progression of the cell cycle.
There are two main cell cycle checkpoints, one at the G1/S transition and the other at the G2/M transition, each of which is tightly regulated by a balance of growth-promoting and growth-suppressing factors, as well as by sensors of DNA damage (Chapter 1). If activated, these DNA-damage sensors transmit signals that arrest cell cycle progression and, if cell damage cannot be repaired, initiate apoptosis. Once cells pass through the G1/S checkpoint, they are committed to undergo cell division. Understandably, then, defects in the G1/S checkpoint are particularly important in cancer, since these lead directly to increased cell division. Indeed, all cancers appear to have genetic lesions that disable the G1/S checkpoint, causing cells to continually reenter the S phase. For unclear reasons, particular lesions vary widely in frequency across tumor types, but they fall into two major categories.
• Gain-of-function mutations involving CDK4 or D cyclins. Mishaps increasing the expression of cyclin D or CDK4 are common events in neoplastic transformation. The cyclin D genes are overexpressed in many cancers, including those affecting the breast, esophagus, liver, and a subset of lymphomas and plasma cell tumors. Amplification of the CDK4 gene occurs in melanomas, sarcomas, and glioblastomas. Mutations affecting cyclins B and E and other CDKs also occur, but they are much less frequent than those affecting cyclin D and CDK4.
• Loss-of-function mutations involving CDKIs. CDKIs frequently are disabled by mutation or gene silencing in many human malignancies. For example, germline mutations of CDKN2A, a gene that encodes the CDK inhibitor p16, are present in 25% of melanoma-prone kindreds, and acquired deletion or inactivation of CDKN2A is seen in 75% of pancreatic carcinomas, 40% to 70% of glioblastomas, 50% of esophageal cancers and certain leukemias, and 20% of non–small cell lung carcinomas, soft-tissue sarcomas, and bladder cancers.
A final consideration of importance in a discussion of growth-promoting signals is that the increased production of oncoproteins does not by itself lead to sustained proliferation of cancer cells. There are two built-in mechanisms, cell senescence and apoptosis, that oppose oncogene-mediated cell growth. As discussed later, genes that regulate these two braking mechanisms must be disabled to allow the action of oncogenes to proceed unopposed.
Summary
Self-Sufficiency in Growth Signals
• Proto-oncogenes: normal cellular genes whose products promote cell proliferation
• Oncogenes: mutant or overexpressed versions of proto-oncogenes that function autonomously without a requirement for normal growth-promoting signals
• Oncoproteins promote uncontrolled cell proliferation by several mechanisms:
• Stimulus-independent expression of growth factor and its receptor, setting up an autocrine loop of cell proliferation (e.g., PDGF–PDGF receptor in brain tumors)
• Mutations in genes encoding growth factor receptors or tyrosine kinases leading to constitutive signaling
• Amplification of EGF receptor family genes such as HER2 in breast cancer
• Fusion of portions of the ABL tyrosine kinase gene and the BCR protein gene, creating a BCR-ABL fusion gene encoding a constitutively active tyrosine kinase, in certain leukemias
• Mutations in genes encoding signaling molecules
• RAS commonly is mutated in human cancers and normally flips between resting GDP-bound state and active GTP-bound state; mutations block hydrolysis of GTP to GDP, leading to unchecked signaling
• Overproduction or unregulated activity of transcription factors
• Translocation of MYC in some lymphomas leads to overexpression and unregulated expression of its target genes controlling cell cycling and survival
• Mutations that activate cyclin genes or inactivate negative regulators of cyclins and cyclin-dependent kinases
• Complexes of cyclins with CDKs drive the cell cycle by phosphorylating various substrates and normally are controlled by CDK inhibitors. Mutations in genes encoding cyclins, CDKs, and CDK inhibitors result in uncontrolled cell cycle progression and are found in a wide variety of cancers including melanomas and brain, lung, and pancreatic cancers.
Insensitivity to Growth Inhibitory Signals: Tumor Suppressor Genes
Isaac Newton theorized that every action has an equal and opposite reaction. Although Newton was not a cancer biologist, his formulation holds true for cell growth. Whereas oncogenes encode proteins that promote cell growth, the products of tumor suppressor genes apply brakes to cell proliferation. Disruption of such genes renders cells refractory to growth inhibition and mimics the growth-promoting effects of oncogenes. The following discussion describes tumor suppressor genes, their products, and possible mechanisms by which loss of their function contributes to unregulated cell growth.
In principle, anti-growth signals can prevent cell proliferation by several complementary mechanisms. The signal may cause dividing cells to enter G0 (quiescence), where they remain until external cues prod their reentry into the proliferative pool. Alternatively, the cells may enter a postmitotic, differentiated pool and lose replicative potential. Nonreplicative senescence, alluded to earlier, is another mechanism of escape from sustained cell growth. And, as a last-ditch effort, the cells may be programmed for death by apoptosis. As we will see, tumor suppressor genes have all these “tricks” in their toolbox designed to halt wayward cells from becoming malignant.
RB: Governor of the Cell Cycle
RB, a key negative regulator of the cell cycle, is directly or indirectly inactivated in most human cancers. The retinoblastoma gene (RB) was the first tumor suppressor gene to be discovered and is now considered the prototype of this family of cancer genes. As with many advances in medicine, the discovery of tumor suppressor genes was accomplished by the study of a rare disease—in this case, retinoblastoma, an uncommon childhood tumor. Approximately 60% of retinoblastomas are sporadic, while the remaining ones are familial, the predisposition to develop the tumor being transmitted as an autosomal dominant trait. To account for the sporadic and familial occurrence of an identical tumor, Knudson, in 1974, proposed his now famous two-hit hypothesis, which in molecular terms can be stated as follows:
• Two mutations (hits) are required to produce retinoblastoma. These involve the RB gene, which has been mapped to chromosomal locus 13q14. Both of the normal alleles of the RB locus must be inactivated (hence the two hits) for the development of retinoblastoma (Fig. 6.19).
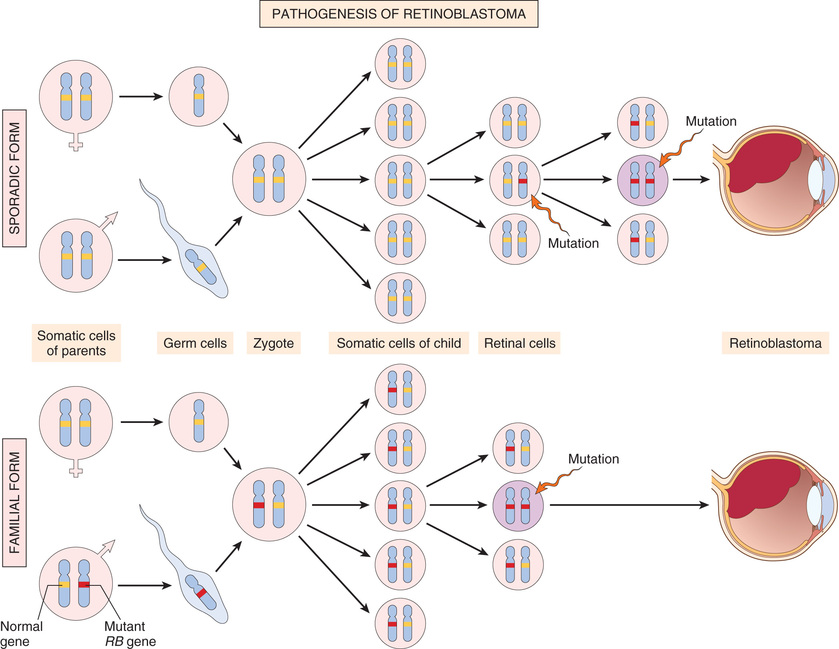
• In familial cases, children inherit one defective copy of the RB gene in the germ line; the other copy is normal. Retinoblastoma develops when the normal RB gene is lost in retinoblasts as a result of somatic mutation. Because in retinoblastoma families a single germ line mutation is sufficient to transmit disease risk, the trait has an autosomal dominant inheritance pattern.
• In sporadic cases, both normal RB alleles are lost by somatic mutation in one of the retinoblasts. The end result is the same: a retinal cell that has lost both of the normal copies of the RB gene becomes cancerous.
From the above, it is evident that although the risk for developing retinoblastoma in retinoblastoma families is inherited as a dominant trait, at the level of the cell, one intact RB gene is all that is needed for normal function.
Although the loss of normal RB genes initially was discovered in retinoblastomas, it is now evident that biallelic loss of this gene is a fairly common feature of several tumors, including breast cancer, small cell cancer of the lung, and bladder cancer. Patients with familial retinoblastoma also are at greatly increased risk for developing osteosarcomas and some soft-tissue sarcomas.
The function of the RB protein is to regulate the G1/S checkpoint, the portal through which cells must pass before DNA replication commences. Although each phase of the cell cycle circuitry is monitored carefully, the transition from G1 to S is an extremely important checkpoint in the cell cycle “clock.” In the G1 phase, diverse signals are integrated to determine whether the cell should progress through the cell cycle, or exit the cell cycle and differentiate. The RB gene product, RB, is a DNA-binding protein that serves as a point of integration for these diverse signals, which ultimately act by altering the phosphorylation state of RB. Specifically, signals that promote cell cycle progression lead to the phosphorylation and inactivation of RB, while those that block cell cycle progression act by maintaining RB in an active hypophosphorylated state.
To appreciate this crucial role of RB in the cell cycle, it is helpful to review the mechanisms that enforce the G1/S transition.
• The initiation of DNA replication (S phase) requires the activity of cyclin E/CDK2 complexes, and expression of cyclin E is dependent on the E2F family of transcription factors. Early in G1, RB is in its hypophosphorylated active form, and it binds to and inhibits the E2F family of transcription factors, preventing transcription of cyclin E. Hypophosphorylated RB blocks E2F-mediated transcription in at least two ways (Fig. 6.20). First, it sequesters E2F, preventing it from interacting with other transcriptional activators. Second, RB recruits chromatin remodeling proteins, such as histone deacetylases and histone methyltransferases, which bind to the promoters of E2F-responsive genes such as cyclin E. These enzymes modify chromatin at the promoters to make DNA insensitive to transcription factors.
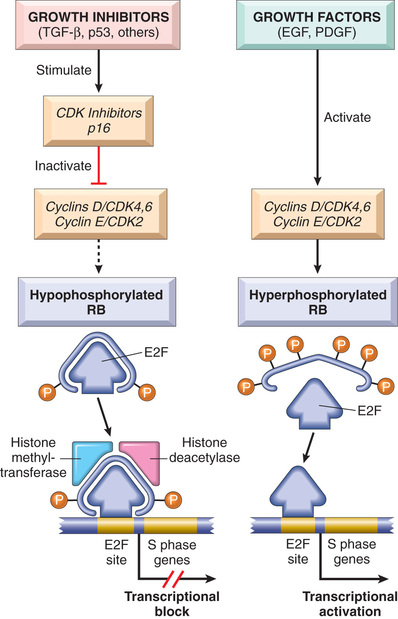
• This situation is changed on mitogenic signaling. Growth factor signaling leads to cyclin D expression and activation of cyclin D–CDK4/6 complexes. The level of cyclin D-CDK4/6 activity is tempered by antagonists such as p16, which is itself subject to regulation by growth inhibitors such as TGFβ that serve to set a threshold for mitogenic responses. If the stimulus is sufficently strong, cyclin D-CDK4/6 complexes phosphorylate RB, inactivating the protein and releasing E2F to induce target genes such as cyclin E. Cyclin E/CDK complexes then stimulate DNA replication and progression through the cell cycle. When the cells enter S phase, they are committed to divide without additional growth factor stimulation. During the ensuing M phase, the phosphate groups are removed from RB by cellular phosphatases, regenerating the hypophosphorylated form of RB.
• E2F is not the sole target of RB. The versatile RB protein binds to a variety of other transcription factors that regulate cell differentiation. For example, RB stimulates myocyte-, adipocyte-, melanocyte-, and macrophage-specific transcription factors. Thus, the RB pathway couples control of cell cycle progression at G1 with differentiation, which may explain how differentiation is associated with exit from the cell cycle.
In view of the centrality of RB to the control of the cell cycle, an interesting question is why RB is not mutated in every cancer. In fact, mutations in other genes that control RB phosphorylation can mimic the effect of RB loss and are commonly found in many cancers that have normal RB genes. For example, mutational activation of CDK4 and overexpression of cyclin D favor cell proliferation by facilitating RB phosphorylation and inactivation. Indeed, cyclin D is overexpressed in many tumors because of amplification or translocation of the cyclin D1 gene. Mutational inactivation of genes encoding CDKIs also can drive the cell cycle by removing important brakes on cyclin/CDK activity. As mentioned earlier, the CDKN2A gene, which encodes the CDK inhibitor p16, is an extremely common target of deletion or mutational inactivation in human tumors.
It is now accepted that loss of normal cell cycle control is central to malignant transformation and that at least one of the four key regulators of the cell cycle (p16, cyclin D, CDK4, RB) is mutated in most human cancers. Notably, in cancers caused by certain oncogenic viruses (discussed later), this is achieved through direct targeting of RB by viral proteins. For example, the human papillomavirus (HPV) E7 protein binds to the hypophosphorylated form of RB, preventing it from inhibiting the E2F transcription factors. Thus, RB is functionally deleted, leading to uncontrolled growth.
Summary
RB: Governor of the Cell Cycle
• Like other tumor suppressor genes, both copies of RB must be dysfunctional for tumor development to occur.
• In cases of familial retinoblastoma, one defective copy of the RB gene is present in the germ line, so that only one additional somatic mutation is needed to completely eliminate RB function.
• RB exerts anti-proliferative effects by controlling the G1-to-S transition of the cell cycle. In its active form, RB is hypophosphorylated and binds to E2F transcription factors. This interaction prevents transcription of genes like cyclin E that are needed for DNA replication, and so the cells are arrested in G1.
• Growth factor signaling leads to cyclin D expression, activation of cyclin D–CDK4/6 complexes, inactivation of RB by phosphorylation, and thus release of E2F.
• Loss of cell cycle control is fundamental to malignant transformation. Almost all cancers have a disabled G1 checkpoint due to mutation of either RB or genes that affect RB function, such as cyclin D, CDK4, and CDKIs.
• Many oncogenic DNA viruses, like HPV, encode proteins (e.g., E7) that bind RB and render it nonfunctional.
TP53: Guardian of the Genome
The p53-encoding tumor suppressor gene, TP53, is the most commonly mutated gene in human cancer. The p53 protein is a transcription factor that thwarts neoplastic transformation by three interlocking mechanisms: activation of temporary cell cycle arrest (termed quiescence), induction of permanent cell cycle arrest (termed senescence), or triggering of programmed cell death (termed apoptosis). If RB is a “sensor” of external signals, p53 can be viewed as a central monitor of internal stress, directing the stressed cells toward one of these pathways.
A variety of stresses trigger the p53 response pathways, including anoxia, inappropriate pro-growth stimuli (e.g., unbridled MYC or RAS activity), and DNA damage. By managing the DNA damage response, p53 plays a central role in maintaining the integrity of the genome, as discussed next.
In nonstressed, healthy cells, p53 has a short half-life (20 minutes) because of its association with MDM2, a protein that targets p53 for destruction. When the cell is stressed, for example, by an assault on its DNA, “sensors” that include protein kinases such as ATM (ataxia telangiectasia mutated) are activated. These activated sensors catalyze posttranslational modifications in p53 that release it from MDM2, increasing its half-life and enhancing its ability to drive the transcription of target genes. Hundreds of genes whose transcription is triggered by p53 have been found. These genes suppress neoplastic transformation by three mechanisms:
• p53-mediated cell cycle arrest may be considered the primordial response to DNA damage (Fig. 6.21). It occurs late in the G1 phase and is caused mainly by p53-dependent transcription of the CDKI gene CDKN1A (p21). The p21 protein inhibits cyclin–CDK complexes and prevents phosphorylation of RB, thereby arresting cells in the G1 phase. Such a pause in cell cycling is welcome, because it gives the cells “breathing time” to repair DNA damage. The p53 protein also induces expression of DNA damage repair genes. If DNA damage is repaired successfully, p53 upregulates transcription of MDM2, leading to its own destruction and relief of the cell cycle block. If the damage cannot be repaired, the cell may enter p53-induced senescence or undergo p53-directed apoptosis.
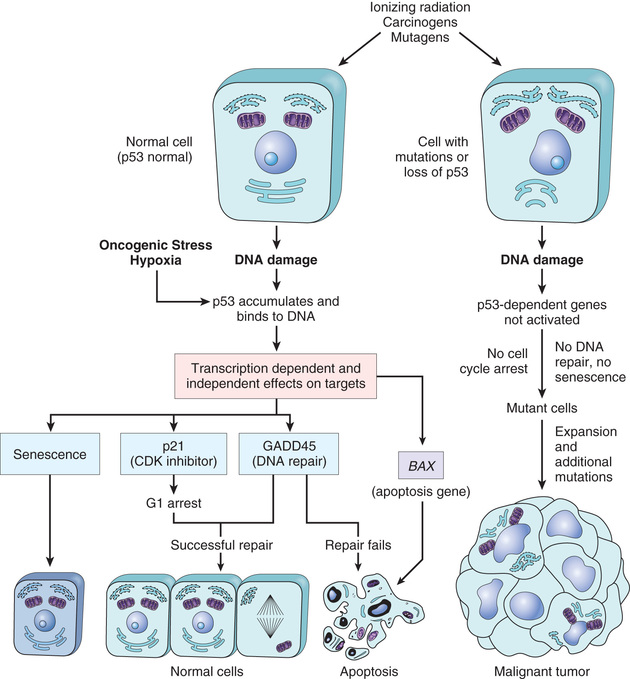
• p53-induced senescence is a form of permanent cell cycle arrest characterized by specific changes in morphology and gene expression that differentiate it from quiescence or reversible cell cycle arrest. Senescence requires activation of p53 and/or Rb and expression of their mediators, such as the CDKIs. The mechanisms of senescence are unclear but seem to involve global chromatin changes, which drastically and permanently alter gene expression.
• p53-induced apoptosis of cells with irreversible DNA damage is the ultimate protective mechanism against neoplastic transformation. It is mediated by upregulation of several pro-apoptotic genes, including BAX and PUMA (discussed later).
To summarize, p53 is activated by stresses such as DNA damage and assists in DNA repair by causing G1 arrest and inducing the expression of DNA repair genes. A cell with damaged DNA that cannot be repaired is directed by p53 to either enter senescence or undergo apoptosis (see Fig. 6.21). In view of these activities, p53 has been rightfully called the “guardian of the genome.” With loss of normal p53 function, DNA damage goes unrepaired, mutations become fixed in dividing cells, and the cell turns onto a one-way street leading to malignant transformation.
Confirming the importance of TP53 in controlling carcinogenesis, more than 70% of human cancers have a defect in this gene, and the remaining malignant neoplasms often have defects in genes upstream or downstream of TP53. Biallelic abnormalities of the TP53 gene are found in virtually every type of cancer, including carcinomas of the lung, colon, and breast—the three leading causes of cancer deaths. In most cases, mutations affecting both TP53 alleles are acquired in somatic cells. In other tumors, such as certain sarcomas, the TP53 gene is intact but p53 function is lost because of amplification and overexpression of the MDM2 gene, which encodes a potent inhibitor of p53. Less commonly, patients inherit a mutant TP53 allele; the resulting disorder is called the Li-Fraumeni syndrome. As in the case with familial retinoblastoma, inheritance of one mutant TP53 allele predisposes affected individuals to develop malignant tumors because only one additional hit is needed to inactivate the second, normal allele. Patients with the Li-Fraumeni syndrome have a 25-fold greater chance of developing a malignant tumor by 50 years of age compared with the general population. In contrast to tumors developing in patients who inherit a mutant RB allele, the spectrum of tumors that develop in patients with the Li-Fraumeni syndrome is much more varied; the most common types are sarcomas, breast cancer, leukemia, brain tumors, and carcinomas of the adrenal cortex. Compared with individuals diagnosed with sporadic tumors, patients with Li-Fraumeni syndrome develop tumors at a younger age and may develop multiple primary tumors.
As with RB, normal p53 also can be rendered nonfunctional by certain oncogenic DNA viruses. Proteins encoded by oncogenic HPVs, certain polyoma viruses, and hepatitis B virus bind to p53 and nullify its protective function. Thus, transforming DNA viruses subvert two of the best-understood tumor suppressors, RB and p53.
Summary
TP53: Guardian of the Genome
• TP53 encodes p53, the central monitor of stress in the cell, which can be activated by anoxia, inappropriate oncogene signaling, or DNA damage. Activated p53 controls the expression and activity of genes involved in cell cycle arrest, DNA repair, cellular senescence, and apoptosis.
• DNA damage leads to activation of p53 by phosphorylation. Activated p53 drives transcription of CDKN1A (p21), which prevents RB phosphorylation, thereby causing a G1-S block in the cell cycle. This pause allows the cells to repair DNA damage.
• If DNA damage cannot be repaired, p53 induces cellular senescence or apoptosis.
• Of human tumors, 70% demonstrate biallelic mutations in TP53. Patients with the rare Li-Fraumeni syndrome inherit one defective copy of TP53 in the germ line, such that only one additional mutation is required to lose normal p53 function. Li-Fraumeni syndrome patients are prone to develop a wide variety of tumors.
• As with RB, p53 can be incapacitated when bound by proteins encoded by oncogenic DNA viruses such as HPV.
Transforming Growth Factor-β Pathway
Although much is known about the circuitry that applies brakes to the cell cycle, the molecules that transmit anti-proliferative signals to cells are less well characterized. Best-known is TGF-β, a member of a family of dimeric growth factors that includes bone morphogenetic proteins and activins. In most normal epithelial, endothelial, and hematopoietic cells, TGF-β is a potent inhibitor of proliferation. It regulates cellular processes by binding to a complex composed of TGF-β receptors I and II. Dimerization of the receptor upon ligand binding leads to a cascade of events that result in the transcriptional activation of CDKIs with growth-suppressing activity, as well as repression of growth-promoting genes such as MYC and CDK4.
In many forms of cancer, the growth-inhibiting effects of the TGF-β pathways are impaired by mutations affecting TGF-β signaling. These mutations may alter the type II TGF-β receptor or SMAD molecules that serve to transduce anti-proliferative signals from the receptor to the nucleus. Mutations affecting the type II receptor are seen in cancers of the colon, stomach, and endometrium. Mutational inactivation of SMAD4, 1 of the 10 proteins known to be involved in TGF-β signaling, is common in pancreatic cancers. In other cancers, by contrast, loss of TGF-β–mediated growth control occurs at a level downstream of the core TGF-β signaling pathway; for example, there may be loss of p21 expression and/or overexpression of MYC. These tumor cells can then use other elements of the TGF-β–induced program, including immune system suppression or promotion of angiogenesis, to facilitate tumor progression. Thus, TGF-β can function to prevent or promote tumor growth, depending on the state of other genes in the cell. Indeed, in many late-stage tumors, TGF-β signaling activates epithelial-to-mesenchymal transition (EMT), a process that promotes migration, invasion, and metastasis, as discussed later.
Contact Inhibition, NF2, and APC
When cancer cells are grown in the laboratory, their proliferation fails to be inhibited when they come in contact with each other. This is in sharp contrast to nontransformed cells, which stop proliferating once they form confluent monolayers. The mechanisms that govern contact inhibition are only now being discovered. Cell–cell contacts in many tissues are mediated by homodimeric interactions between transmembrane proteins called cadherins. E-cadherin (E for epithelial) mediates cell–cell contact in epithelial layers. Two mechanisms have been proposed to explain how E-cadherin maintains contact inhibition:
• One mechanism is mediated by the tumor suppressor gene NF2. Its product, neurofibromin-2, more commonly called merlin, acts downstream of E-cadherin in a signling pathway that helps fo maintain contact inhibition. Homozygous loss of NF2 is known to cause certain neural tumors, and germ line mutations in NF2 are associated with a tumor-prone hereditary condition called neurofibromatosis type 2.
• A second mechanism by which E-cadherin may regulate contact inhibition involves its ability to bind β-catenin, another signaling protein. β-catenin is a key component of the WNT signaling pathway (described below), which has broad but as of yet incompletely understood roles in regulating the morphology and organization of epithelial cells lining structures such as the gut.
A further clue to the important of E-cadherin and β-catenin in epithelial cancers is illustrated by the rare hereditary disease adenomatous polyposis coli (APC). This disorder is characterized by the development of numerous adenomatous polyps in the colon that have a very high incidence of transformation into colonic cancers. The polyps consistently show loss of a tumor suppressor gene called APC (named for the disease), which exerts anti-proliferative effects in an unusual manner. APC encodes a cytoplasmic protein whose dominant function is to promote the degradation of β-catenin, which has several functions. In addition to binding E-cadherin, β-catenin also is a key component of the WNT signaling pathway (illustrated in Fig. 6.22). WNTs are soluble factors that bind WNT receptors, which in turn transmit signals that prevent the APC-mediated degradation of β-catenin, allowing it to translocate to the nucleus, where it acts as a transcriptional activator. In quiescent cells that have not been exposed to WNT, cytoplasmic β-catenin is degraded by a destruction complex, of which APC is an integral part. With loss of APC (e.g., in colon cancers), β-catenin degradation is prevented, and the WNT signaling response is inappropriately activated even in the absence of WNT factors. In colonic epithelium this leads to increased transcription of growth-promoting genes, such as cyclin D1 and MYC, as well as transcriptional regulators, such as TWIST and SLUG, which repress E-cadherin expression and thus reduce contact inhibition.
APC behaves as a typical tumor suppressor gene. Individuals born with one mutated allele typically are found to have hundreds to thousands of adenomatous polyps in the colon by their teens or twenties; these polyps show loss of the other APC allele. Almost invariably, one or more polyps undergo malignant transformation, as discussed later. APC mutations are seen in 70% to 80% of sporadic colon cancers. Colonic cancers with normal APC genes sometimes show activating mutations of β-catenin that render them refractory to the degrading action of APC.
Summary
TGF-β, Contact Inhibition, and APC-β-Catenin Pathways
• TGF-β inhibits proliferation of many cell types by activation of growth-inhibiting genes such as CDKIs and suppression of growth-promoting genes such as MYC and those encoding cyclins.
• TGF-β function is compromised in many tumors by mutations in its receptors (colon, stomach, endometrium) or by mutational inactivation of SMAD genes that transduce TGF-β signaling (pancreas).
• E-cadherin maintains contact inhibition, which is lost in malignant cells.
• The APC gene exerts anti-proliferative actions by regulating the destruction of the cytoplasmic protein β-catenin. With a loss of APC, β-catenin is not destroyed, and it translocates to the nucleus, where it acts as a growth-promoting transcription factor.
• In familial adenomatous polyposis syndrome, inheritance of a germ line mutation in the APC gene and sporadic loss of the sole normal allele causes the development of hundreds of colonic polyps at a young age. Inevitably, one or more of these polyps evolves into a colonic cancer. Somatic loss of both alleles of the APC gene is seen in approximately 70% of sporadic colon cancers.
Altered Cellular Metabolism
Even in the presence of ample oxygen, cancer cells demonstrate a distinctive form of cellular metabolism characterized by high levels of glucose uptake and increased conversion of glucose to lactose (fermentation) via the glycolytic pathway. This phenomenon, called the Warburg effect and also known as aerobic glycolysis, has been recognized for many years (indeed, Otto Warburg received the Nobel Prize in 1931 for its discovery). Clinically, the “glucose-hunger” of tumors is used to visualize tumors via positron emission tomography (PET) scanning, in which patients are injected with 18F-fluorodeoxyglucose, a glucose derivative that is preferentially taken up into tumor cells (as well as normal, actively dividing tissues such as the bone marrow). Most tumors are PET-positive, and rapidly growing ones are markedly so.
Warburg's discovery was largely neglected for many years, but over the past decade, metabolism has become one of the most active areas of cancer research. Metabolic pathways (like signaling pathways) in normal and cancer cells are still being elucidated, and the details are complex, but at the heart of the Warburg effect lies a simple question: why is it advantageous for a cancer cell to rely on seemingly inefficient glycolysis (which generates two molecules of ATP per molecule of glucose) instead of oxidative phosphorylation (which generates up to 36 molecules of ATP per molecule of glucose)? While pondering this question, it is important to recognize that rapidly proliferating normal cells, such as in embryonic tissues and lymphocytes during immune responses, also rely on aerobic fermentation. Thus, “Warburg metabolism” is not cancer specific, but instead is a general property of growing cells that becomes “fixed” in cancer cells.
The answer to this riddle is simple: Aerobic glycolysis provides rapidly dividing tumor cells with metabolic intermediates that are needed for the synthesis of cellular components, whereas mitochondrial oxidative phosphorylation does not. The reason growing cells rely on aerobic glycolysis becomes readily apparent when one considers that a growing cell has a strict biosynthetic requirement; it must duplicate all of its cellular components—DNA, RNA, proteins, lipid, and organelles—before it can divide and produce two daughter cells. While oxidative phosphorylation yields abundant ATP, it fails to produce any carbon moieties that can be used to build the cellular components needed for growth (proteins, lipids, and nucleic acids). Even cells that are not actively growing must shunt some metabolic intermediates away from oxidative phosphorylation in order to synthesize macromolecules that are needed for cellular maintenance.
By contrast, in actively growing cells only a small fraction of the cellular glucose is shunted through the oxidative phosphorylation pathway, such that on average each molecule of glucose metabolized produces approximately four molecules of ATP. Presumably, this balance (heavily biased toward aerobic fermentation, with a bit of oxidative phosphorylation) hits a metabolic “sweet spot” that is optimal for growth. It follows that growing cells do rely on mitochondrial metabolism. However, a major function of mitochondria in growing cells is not to generate ATP, but rather to carry out reactions that generate metabolic intermediates that can be shunted off and used as precursors in the synthesis of cellular building blocks. For example, lipid biosynthesis requires acetyl-CoA, and acetyl-CoA is largely synthesized in growing cells from intermediates such as citrate that are generated in mitochondria.
So how is this profound reprogramming of metabolism, the Warburg effect, triggered in growing normal and malignant cells? As might be guessed, metabolic reprogramming is produced by signaling cascades downstream of growth factor receptors, the very same pathways that are deregulated by mutations in oncogenes and tumors suppressor genes in cancers. Thus, whereas in rapidly dividing normal cells aerobic glycolysis ceases when the tissue is no longer growing, in cancer cells this reprogramming persists due to the action of oncogenes and the loss of tumor suppressor gene function. Some of the important points of cross-talk between pro–growth signaling factors and cellular metabolism are shown in Fig. 6.23 and include the following:
• Growth factor receptor signaling. In addition to transmitting growth signals to the nucleus, signals from growth factor receptors also influence metabolism by upregulating glucose uptake and inhibiting the activity of pyruvate kinase, which catalyzes the last step in the glycolytic pathway, the conversion of phosphoenolpyruvate to pyruvate. This creates a damming effect that leads to the buildup of upstream glycolytic intermediates, which are siphoned off for synthesis of DNA, RNA, and protein.
• RAS signaling. Signals downstream of RAS upregulate the activity of glucose transporters and multiple glycolytic enzymes, thus increasing glycolysis; promote shunting of mitochondrial intermediates to pathways leading to lipid biosynthesis; and stimulate factors that are required for protein synthesis.
• MYC. As mentioned earlier, pro-growth pathways upregulate expression of the transcription factor MYC, which drives changes in gene expression that support anabolic metabolism and cell growth. Among the MYC-regulated genes are those for several glycolytic enzymes and glutaminase, which is required for mitochondrial utilization of glutamine, a key source of carbon moieties needed for biosynthesis of cellular building blocks.
The flipside of the coin is that tumor suppressors often inhibit metabolic pathways that support growth. We have already discussed the “braking” effect of the tumor suppressors NF1 and PTEN on signals downstream of growth factor receptors and RAS, allowing them to oppose the Warburg effect. Indeed, it may be that many (and perhaps all) tumor suppressors that induce growth arrest suppress the Warburg effect. For example, p53, arguably the most important tumor suppressor, upregulates target genes that collectively inhibit glucose uptake, glycolysis, lipogenesis, and the generation of NADPH (a key cofactor needed for the biosynthesis of macromolecules). Thus, it is increasingly clear that the functions of many oncoproteins and tumor suppressors are inextricably intertwined with cellular metabolism.
Beyond the Warburg effect, there are two other links between metabolism and cancer that are of sufficient importance to merit brief mention, autophagy and an unusual set of oncogenic mutations that lead to the creation of oncometabolites, small molecules that appear to directly contribute to the transformed state.
Autophagy
Autophagy is a state of severe nutrient deficiency in which cells not only arrest their growth, but also cannibalize their own organelles, proteins, and membranes as carbon sources for energy production (Chapter 2). If this adaptation fails, the cells die. Tumor cells often seem to be able to grow under marginal environmental conditions without triggering autophagy, suggesting that the pathways that induce autophagy are deranged. In keeping with this, several genes that promote autophagy are tumor suppressors. Whether autophagy is always bad from the vantage point of the tumor, however, remains a matter of active investigation and debate. For example, under conditions of severe nutrient deprivation, tumor cells may use autophagy to become “dormant,” a state of metabolic hibernation that allows cells to survive hard times for long periods. Such cells are believed to be resistant to therapies that kill actively dividing cells, and could therefore be responsible for therapeutic failures. Thus, autophagy may be a tumor's friend or foe depending on how the signaling pathways that regulate it are “wired” in a given tumor.
Oncometabolism
Another surprising group of genetic alterations discovered through tumor genome sequencing studies are mutations in enzymes that participate in the Krebs cycle. Of these, mutations in isocitrate dehydrogenase (IDH) have garnered the most interest, as they have revealed a new mechanism of oncogenesis termed oncometabolism (Fig. 6.24).
The proposed steps in the oncogenic pathway involving IDH are as follows:
• IDH acquires a mutation that leads to a specific amino acid substitution involving residues in the active site of the enzyme. As a result, the mutated protein loses it ability to function as an isocitrate dehydrogenase and instead acquires a new enzymatic activity that catalyzes the production of 2-hydroxglutarate (2-HG).
• 2-HG in turn acts as an inhibitor of several other enzymes that are members of the TET family, including TET2.
• TET2 is one of several factors that regulate DNA methylation, which you will recall is an epigenetic modification that controls normal gene expression and often goes awry in cancer. According to the model, loss of TET2 activity leads to abnormal patterns of DNA methylation.
• Abnormal DNA methylation in turn leads to misexpression of currently unknown cancer genes, which drive cellular transformation and oncogenesis.
Thus, according to this scenario, mutated IDH acts as an oncoprotein by producing 2-HG, which is considered a prototypical oncometabolite. Oncogenic IDH mutations have now been described in a diverse collection of cancers, including a sizable fraction of cholangiocarcinomas, gliomas, acute myeloid leukemias, and sarcomas. Of clinical significance, because the mutated IDH proteins have an altered structure, it has been possible to develop drugs that inhibit mutated IDH and not the normal IDH enzyme. These drugs are now being tested in cancer patients and have produced encouraging therapeutic responses. This developing story is a remarkable example of how detailed understanding of oncogenic mechanisms can yield entirely new kinds of anti-cancer drugs.
Summary
Altered Cellular Metabolism
• Warburg metabolism is a form of pro-growth metabolism favoring glycolysis over oxidative phosphorylation. It is induced in normal cells by exposure to growth factors and becomes fixed in cancer cells due to the action of certain driver mutations.
• Many oncoproteins (RAS, MYC, mutated growth factor receptors) induce or contribute to Warburg metabolism, and many tumor suppressors (PTEN, NF1, p53) oppose it.
• Stress may induce cells to consume their components in a process called autophagy. Cancer cells may accumulate mutations to avoid autophagy, or may corrupt the process to provide nutrients for continued growth and survival.
• Some oncoproteins such as mutated IDH act by causing the formation of high levels of “oncometabolites” that alter the epigenome, thereby leading to changes in gene expression that are oncogenic.