Liver and Gallbladder
The Liver and Bile Ducts
The normal adult liver weighs 1400 to 1600 gm. It has a dual blood supply, with the portal vein providing 60% to 70% of hepatic blood flow and the hepatic artery supplying the remaining 30% to 40%. The portal vein and the hepatic artery enter the inferior aspect of the liver through the hilum, or porta hepatis. Within the liver, the branches of the portal veins, hepatic arteries, and bile ducts travel in parallel within portal tracts, ramifying variably through 10 to 12 orders of branches.
The most common terminology used to describe the hepatic microarchitecture is based on the lobular model (Fig. 16.1). This model divides the liver into lobules 1- to 2-mm in diameter that are centered on a terminal tributary of the hepatic vein and demarcated by portal tracts at their periphery. These lobules are often drawn as hexagonal structures, though in humans the shapes are far more variable; nonetheless, it is a useful simplification. A second model divides the liver into triangular acini (see Fig. 16.1) based on the position of hepatocytes relative to their blood supply. The hepatocytes in the vicinity of the terminal hepatic vein are called centrilobular; those near the portal tract are periportal. Division of the lobular parenchyma into zones is an important concept because each zone differs with respect to its metabolic activities and susceptibility to certain forms of hepatic injury.
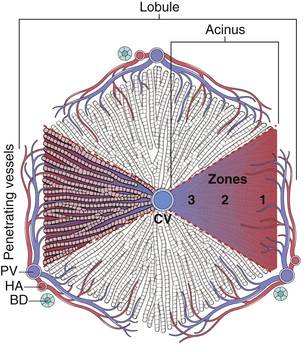
Within the lobule, hepatocytes are organized into anastomosing sheets or “plates” extending from portal tracts to the terminal hepatic veins. Between the trabecular plates of hepatocytes are vascular sinusoids. Blood traverses the sinusoids and exits into the terminal hepatic veins through numerous orifices in the vein wall. Hepatocytes are thus bathed by well-mixed portal venous blood on one side and hepatic arterial blood on the other. The sinusoids are lined by a fenestrated endothelium that overlies a perisinusoidal space (the space of Disse) into which abundant hepatocyte microvilli protrude. Attached to the luminal face of the sinusoids are scattered Kupffer cells, specialized long-lived tissue macrophages that arise early in embryogenesis. Another specialized cell type, the hepatic stellate cell, is found in the space of Disse and has a role in the storage of vitamin A. Between abutting hepatocytes are bile canaliculi, channels 1 to 2 µm in diameter that are formed by grooves in the plasma membranes of adjacent hepatocytes and are separated from the vascular space by tight junctions. These channels drain successively into the intralobular canals of Hering, periportal bile ductules, and finally into the terminal bile ducts within the portal tracts.
General Features of Liver Disease
The major primary diseases of the liver are viral hepatitis, alcoholic liver disease, nonalcoholic fatty liver disease (NAFLD), and hepatocellular carcinoma (HCC). The liver also is frequently damaged secondarily in a variety of common disorders, such as cardiac disease, disseminated cancer, and extrahepatic infections. The functional reserve of the liver masks the clinical impact of mild liver damage, but severe diffuse liver disease often has life-threatening consequences.
With the rare exception of fulminant hepatic failure, liver disease is an insidious process in which the signs and symptoms of hepatic decompensation appear weeks, months, or even years after the onset of injury. The hepatic injury may be imperceptible to the patient and be manifest only by laboratory test abnormalities (Table 16.1), and liver injury and healing also may be subclinical. Hence, individuals with hepatic abnormalities who are referred to hepatologists most frequently have chronic liver disease.
Table 16.1
Laboratory Evaluation of Liver Disease
| Test Category | Blood Measurement* |
| Hepatocyte integrity | Cytosolic hepatocellular enzymes† Serum aspartate aminotransferase (AST) Serum alanine aminotransferase (ALT) Serum lactate dehydrogenase (LDH) |
| Biliary excretory function | Substances normally secreted in bile† Serum bilirubin Total: unconjugated plus conjugated Direct: conjugated only Urine bilirubin Serum bile acids Plasma membrane enzymes (from damage to bile canaliculus)† Serum alkaline phosphatase Serum γ-glutamyl transpeptidase (GGT) |
| Hepatocyte function | Proteins secreted into the blood Serum albumin‡ Prothrombin time (PT)† Partial thromboplastin time (PTT)† Hepatocyte metabolism Serum ammonia† Aminopyrine breath test (hepatic demethylation)‡ |
Mechanisms of Injury and Repair
Injured hepatocytes may show several potentially reversible changes, such as accumulation of fat and bilirubin (cholestasis); when injury is not reversible, hepatocytes die by necrosis or apoptosis. Necrosis (Fig. 16.2) is commonly seen following hepatic injury caused by hypoxia and ischemia. Apoptotic cell death (Fig. 16.3) predominates in viral, autoimmune, and drug- and toxin-induced hepatitides.
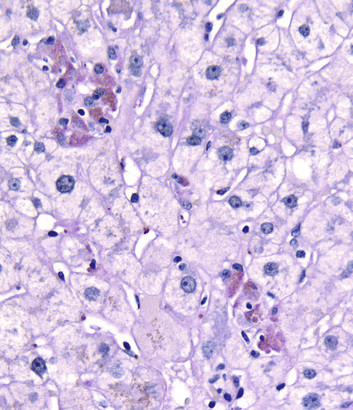
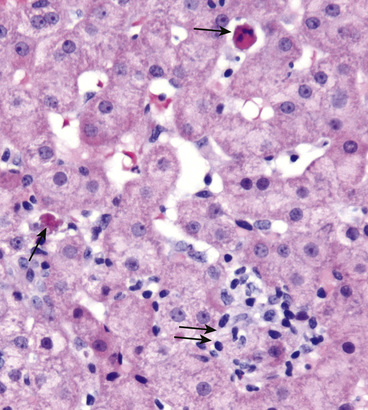
Widespread death of hepatocytes may produce confluent necrosis. This may be seen in acute toxic or ischemic injuries or in severe chronic viral or autoimmune hepatitis. Confluent necrosis begins as a zone of hepatocyte dropout around the central vein. With increasing severity necrosis “bridges” central veins and portal tracts or adjacent portal tracts.
Regeneration of lost hepatocytes takes place primarily by mitotic replication of hepatocytes adjacent to those that have died. In more severe forms of acute liver injury hepatic stem cells located in a niche near the canal of Hering may also begin to divide, but the contribution of stem cells to the replenishment of hepatocytes in the setting of acute liver damage remains uncertain. In longstanding chronic liver diseases, however, there is clear evidence that stem cell proliferation and differentiation make significant contributions to parenchymal restoration, probably following the replicative senescence of preexisting hepatocytes. The differentiating progeny of these tissue stem cells produce duct-like structures, called ductular reactions, a morphologic marker of stem cell–mediated liver regeneration.
Scar formation may follow very severe acute injury, but occurs more often as a reaction to chronic injury. The principal cell type involved in scar deposition is the perisinusoidal hepatic stellate cell. Following liver injury, stellate cells may become activated and convert into highly fibrogenic myofibroblasts, which produce the fibrous scar. Stellate cell activation involves complex interactions between Kupffer cells, hepatocytes, and inflammatory cells. When there is severe injury that causes death of large number of hepatocytes and the drop out of liver cells, there may be collapse of the underlying reticulin, precluding orderly regeneration of hepatocytes. In such cases, there is activation of stellate cells, and the areas of liver cell loss are replaced by fibrous septae. Eventually, these fibrous septa encircle surviving, regenerating hepatocytes in late-stage chronic liver disease, many forms of which are described as cirrhosis.
Inflammation and immunologic reactions are involved in many forms of liver disease. Systemic inflammation alters the metabolic and biosynthetic activities of the liver, leading to increased secretion of acute-phase reactants such as C-reactive protein, serum amyloid A protein (a precursor of some forms of amyloid) and hepcidin, a key regulator of iron metabolism (Chapter 12). As we will discuss, adaptive immune cells play a critical role in viral hepatitis, with CD4+ and CD8+ T cells being particularly important in the eradication of virus-infected hepatocytes and, in chronic disease, liver injury.
Liver Failure
The most severe clinical consequence of liver disease is liver failure. It primarily occurs in three clinical scenarios: acute, chronic, and acute-on-chronic liver failure.
Acute Liver Failure
Acute liver failure is defined as a liver disease that produces hepatic encephalopathy within 6 months of the initial diagnosis. The condition is known as fulminant liver failure when the encephalopathy develops within 2 weeks of the onset of jaundice, and as subfulminant liver failure when the encephalopathy develops within 3 months. In the United States, accidental or deliberate ingestion of acetaminophen accounts for almost 50% of cases of acute liver failure, while autoimmune hepatitis, other drugs and toxins, and acute hepatitis A and B infections account for the remainder of cases. In Asia, acute hepatitis B and E predominate as causes of acute liver failure.
 Morphology
Morphology
The clinical syndrome of acute liver failure is reflected anatomically and histologically as massive hepatic necrosis. The liver is small and shrunken due to loss of parenchyma (Fig. 16.4A). Microscopically, there are large zones of destruction surrounding occasional islands of regenerating hepatocytes (Fig. 16.4B). Scar is mostly absent given the acute nature of the process.

Clinical Features
Acute liver failure manifests with nausea, vomiting, jaundice, and fatigue, which are followed by the onset of life-threatening encephalopathy, coagulation defects, and portal hypertension associated with ascites. Typically, transaminase levels in the serum are elevated into the thousands. The liver is initially enlarged by swelling and edema related to inflammation, but then as parenchyma is destroyed the liver shrinks dramatically. Eventually, as hepatocytes are lost, serum transaminase values level off and then decline rapidly as their source disappears. Worsening jaundice, coagulopathy, and encephalopathy develop; with unabated progression, the end result is multiorgan failure and, without transplantation, death. Manifestations of acute liver failure include the following:
• Jaundice and icterus (yellow discoloration of the skin and sclera, respectively) due to retention of bilirubin, and cholestasis due to systemic retention of not only bilirubin but also other solutes eliminated in bile.
• Hepatic encephalopathy encompasses a spectrum of disturbances in consciousness ranging from subtle behavioral abnormalities, to confusion and stupor, to coma and death. Encephalopathy may develop over days, weeks, or a few months after acute injury. Fluctuating neurologic signs, including rigidity, hyperreflexia, and asterixis, may develop. Asterixis refers to a nonrhythmic rapid extension-flexion movement of the head and extremities, best seen as “flapping” of the hands when the arms are held in extension with dorsiflexed wrists. Elevated ammonia levels in blood and the central nervous system correlate with impaired neuronal function and brain edema.
• Coagulopathy. The liver is the source of a number of coagulation factors that decline in the face of liver failure, leading to easy bruising and bleeding. Paradoxically, disseminated intravascular coagulation (Chapter 12) also may occur due to failure of the damaged liver to remove activated coagulation factors.
• Portal hypertension arises when there is diminished flow through the portal venous system, which may occur because of obstruction at the prehepatic, intrahepatic, or posthepatic level. While it can occur in acute live failure, portal hypertension is more commonly seen with chronic liver failure and is discussed later. In acute liver failure, the obstruction is usually intrahepatic and the major clinical consequences are ascites and hepatic encephalopathy. In chronic liver disease, portal hypertension develops over months to years, and its effects are more complex and widespread (see later).
• Hepatorenal syndrome is a form of renal failure occurring in individuals with liver failure in whom there are no intrinsic morphologic or functional causes for kidney dysfunction. Sodium retention, impaired free-water excretion, and decreased renal perfusion and glomerular filtration rate are the main renal functional abnormalities. There is decreased renal perfusion pressure due to systemic vasodilation, activation of the renal sympathetic nervous system and vasoconstriction of the afferent renal arterioles, and increased activation of the renin-angiotensin axis, causing vasoconstriction that further decreases glomerular filtration. The syndrome's onset begins with a decrease in urine output and rising blood urea nitrogen and creatinine levels.
Chronic Liver Failure and Cirrhosis
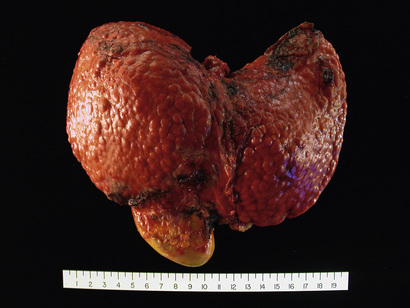
Cirrhosis is the morphologic change most often associated with chronic liver disease; it refers to the diffuse transformation of the liver into regenerative parenchymal nodules surrounded by fibrous bands (Fig. 16.5). The leading causes of chronic liver failure worldwide include chronic hepatitis B, chronic hepatitis C, non-alcoholic fatty liver disease (NAFLD), and alcoholic liver disease. While cirrhosis is a common feature of a number of chronic liver diseases, it is not a specific entity, and it is important to recognize that not all chronic liver disease terminates in cirrhosis, and that not all cirrhosis leads to end-stage liver disease. For example, chronic biliary tract diseases often do not give rise to cirrhosis even at end stage, whereas patients with treated autoimmune hepatitis or cured hepatitis C may have adequate liver function in the face of cirrhosis. Even in diseases that are likely to give rise to cirrhosis, such as untreated viral hepatitis, alcoholic liver disease, NAFLD, and metabolic diseases, the morphology and pathophysiology of cirrhosis in each may be different.
Morphology
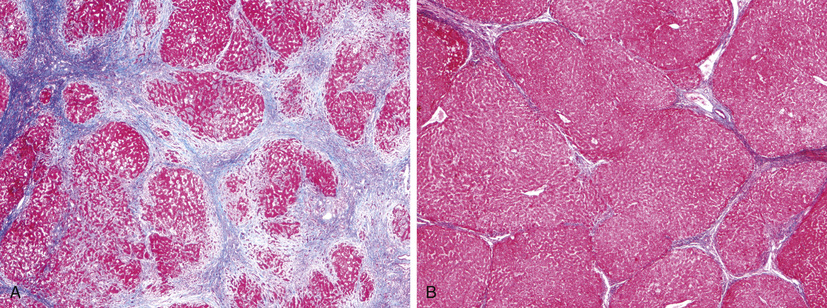
Liver failure in chronic liver disease is most often associated with cirrhosis, which is marked by the diffuse transformation of the entire liver into regenerative parenchymal nodules surrounded by fibrous bands. The nodular nature of the process is readily evident both grossly (Fig. 16.5) and microscopically (Fig. 16.6, A). The size of the nodules, the pattern of scarring (linking of portal tracts to each other vs. linking of portal tracts to central veins), the degree of parenchymal loss, and the frequency of vascular thrombosis (particularly of the portal vein) all vary between diseases and even, in some cases, between individuals with the same disease.
As mentioned earlier, stem cell activation and differentiation gives rise to duct-like structures, the so called ductular reactions. In chronic liver disease, ductular reactions increase with disease progression and are usually most prominent in cirrhosis. Ductular reactions may incite some of the scarring in chronic liver disease and thus may have a negative effect on progressive liver disease.
Regression of fibrosis and even of fully established cirrhosis may follow disease remission or cure. Scars become thinner, more densely compacted, and eventually start to fragment (see Fig. 16.6, B). As fibrous septa break apart, adjacent nodules of regenerating parenchyma coalesce into larger islands. All cirrhotic livers show elements of both progression and regression, with the balance being dictated by the severity and persistence of the underlying disease.
Clinical Features
About 40% of individuals with cirrhosis are asymptomatic until the most advanced stages of the disease. Even at late stages, they present with nonspecific clinical manifestations, such as anorexia, weight loss, weakness, and, eventually signs and symptoms of liver failure discussed earlier. Jaundice, encephalopathy, and coagulopathy may result from chronic liver disease, much the same as in acute liver failure. However, there are some significant additional features:
• Chronic severe jaundice can lead to pruritus (itching), the intensity of which may be so profound that patients scratch their skin raw and risk repeated bouts of potentially life-threatening infection. In some patients, severe pruritus is the primary indication for liver transplantation. Pruritus also is frequently seen in other disorders associated with cholestasis, suggesting that it is somehow related to the build up of bile salts in the body, but its precise pathogenesis is unknown.
• Portal hypertension is more frequent and manifests in more complex ways in chronic liver failure than in acute liver failure (Fig. 16.7). Portosystemic shunts develop when blood flow is reversed from the portal to systemic circulation. These shunts are principally produced by dilation of collateral vessels. Most notably, venous bypasses develop wherever the systemic and the portal circulations share common capillary beds, the most clinically important of which are esophagogastric varices, which appear in about 40% of individuals with advanced-stage liver disease. These often cause massive, frequently fatal hematemesis, particularly when there is compounding coagulopathy. Portal hypertension often occurs and may lead to congestive splenomegaly, which can lower the platelet count due to sequestration of these elements in the expanded red pulp.
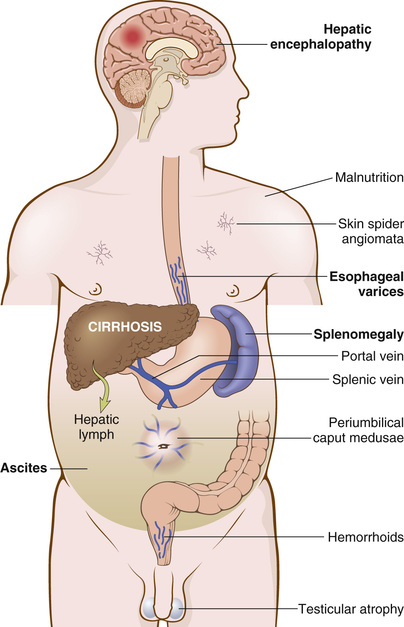
• Hyperestrogenemia due to impaired estrogen metabolism in male patients with chronic liver failure can give rise to palmar erythema (a reflection of local vasodilatation) and spider angiomas of the skin. Such male hyperestrogenemia also leads to hypogonadism and gynecomastia. Hypogonadism also may occur in women from disruption of hypothalamic-pituitary axis functioning.
• Most chronic liver diseases predispose to development of hepatocellular carcinoma (HCC, discussed later).
The course and severity of chronic liver disease with cirrhosis vary widely from patient to patient. Even in instances in which cirrhosis regresses following disease remission or cure, portal hypertension may persist due to the presence of irreversible shunts. The causes of death are liver failure (as in acute liver disease) and HCC. Clinical and laboratory findings are the main criteria used to gauge prognosis and disease progression. It some centers, portal venous wedge pressures are measured to assess the degree of vascular obstruction. Liver biopsy findings correlate with the presence and severity of portal hypertension. For example, specimens with thin fibrous septa and large islands of regenerated parenchyma are unlikely to be associated with portal hypertension, whereas broad bands of fibrosis and loss of parenchyma portend the development of portal hypertension and end-stage liver disease.
Acute-on-Chronic Liver Failure
Some individuals after years of stable, well-compensated, chronic disease suddenly develop signs of acute liver failure. In such patients, there is often established cirrhosis with extensive vascular shunting, or large volumes of functioning liver parenchyma with a borderline vascular supply, both of which leave the liver vulnerable to superimposed, potentially lethal insults. The short-term mortality of patients with this form of liver failure is around 50%.
Hepatic insults that cause sudden decompensation of patients with chronic liver disease include: hepatitis D superinfection in those with chronic hepatitis B; emergence of resistance to medical therapy in those with viral hepatitis; development of ascending bacterial cholangitis in patients with primary sclerosing cholangitis; or replacement of liver parenchyma by primary or metastatic carcinoma. In other instances the cause may be a systemic disorder, such as sepsis, acute cardiac failure or a superimposed toxic injury that tips a well-compensated cirrhotic patient into liver failure.
 Summary
Summary
Liver Failure
• Liver failure may follow acute injury or chronic injury, or it may occur as an acute insult superimposed on otherwise well-compensated chronic liver disease.
• The mnemonic for causes of acute liver failure are as follows:
• A: acetaminophen, hepatitis A, autoimmune hepatitis
• D: drugs/toxins, hepatitis D
• E: hepatitis E, esoteric causes (Wilson disease, Budd-Chiari syndrome)
• F: fatty change of the microvesicular type (fatty liver of pregnancy, valproate, tetracycline, Reye syndrome)
• Potentially fatal sequelae of liver failure include coagulopathy, encephalopathy, portal hypertension and ascites, hepatorenal syndrome, and portopulmonary hypertension.
Infectious Disorders
Viral Hepatitis
The terminology for acute and chronic viral hepatitis can be confusing, because the same word, hepatitis, can be used to describe several different entities; careful attention to context can clarify its meaning in any situation. Firstly, hepatitis is the name applied to viruses (hepatitis A, B, C, D, and E virus) that are hepatotropic, that is, have a specific affinity for the liver. Secondly, hepatitis is applied to patterns of acute and chronic hepatic injuries that are produced not only by hepatotropic viruses, but also by damage produced by other viruses such as EBV, CMV , and yellow fever as well as autoimmune reactions, drugs, and toxins. In this section, we will focus on the main features of hepatotropic viruses, which are summarized in Table 16.2, and we will then discuss the clinicopathologic characteristics of acute and chronic viral hepatitis.
Table 16.2
The Hepatitis Viruses
| Virus | Hepatitis A (HAV) | Hepatitis B (HBV) | Hepatitis C (HCV) | Hepatitis D (HDV) | Hepatitis E (HEV) |
| Viral genome | ssRNA | partially dsDNA | ssRNA | Circular defective ssRNA | ssRNA |
| Viral family | Hepatovirus; related to picornavirus | Hepadnavirus | Flaviviridae | Subviral particle in Deltaviridae family | Calicivirus |
| Route of transmission | Fecal-oral (contaminated food or water) | Parenteral, sexual contact, perinatal | Parenteral; intranasal cocaine use is a risk factor | Parenteral | Fecal-oral |
| Incubation period | 2–6 weeks | 2–26 weeks (mean 8 weeks) | 4–26 weeks (mean 9 weeks) | Same as HBV | 4–5 weeks |
| Frequency of chronic liver disease | Never | 5%–10% | >80% | 10% (coinfection); 90%–100% for superinfection | In immunocompromised hosts only |
| Diagnosis | Detection of serum IgM antibodies | Detection of HBsAg or antibody to HBcAg; PCR for HBV DNA | ELISA for antibody detection; PCR for HCV RNA | Detection of IgM and IgG antibodies, HDV RNA in serum, or HDAg in liver biopsy | Detection of serum IgM and IgG antibodies; PCR for HEV RNA |

Hepatitis A Virus (HAV)
HAV usually is a benign self-limited infection that does not cause chronic hepatitis and rarely (in about 0.1% of cases) produces fulminant hepatitis. HAV has an incubation period of 3-6 weeks. It is typically cleared by the host immune response, so it does not establish a carrier state. The infection occurs throughout the world and is endemic in countries with poor hygiene and sanitation.
Acute HAV tends to cause a febrile illness associated with jaundice and nonspecific symptoms such as fatigue and loss of appetite. Overall, HAV accounts for about 25% of clinically evident acute hepatitis worldwide.
HAV is a small, nonenveloped, positive-strand RNA picornavirus that occupies its own genus, Hepatovirus. Ultrastructurally, HAV is an icosahedral capsid 27 nm in diameter. The receptor for HAV is HAVcr-1, a membrane glycoprotein that also may serve as a receptor for Ebola virus. HAV is spread by ingestion of contaminated water and food and is shed in the stool for 2 to 3 weeks before and 1 week after the onset of jaundice. Thus, close personal contact with an infected individual or fecal-oral contamination accounts for most cases and explains outbreaks in institutional settings such as schools and nurseries, as well as water-borne epidemics in places where people live in overcrowded, unsanitary conditions. HAV can also be detected in serum and saliva of infected individuals.
In developed countries, sporadic infections may be contracted by the consumption of raw or steamed shellfish that have concentrated the virus from seawater contaminated with human sewage. Infected workers in the food industry are another source of outbreaks. HAV itself does not seem to be cytopathic. The cellular immune response, particularly that involving cytotoxic CD8+ T cells, plays a key role in HAV-mediated hepatocellular injury.
Because HAV viremia is transient, blood-borne transmission is very rare; therefore, donated blood is not specifically screened for this virus. IgM antibody against HAV appears in blood at the onset of symptoms and is a reliable marker of acute infection (Fig. 16.8). Fecal shedding of the virus ends as the IgM titer rises. The IgM response usually declines in a few months followed by the appearance of IgG anti-HAV that persists for years, often conferring lifelong immunity. However, there are no routinely available tests for IgG anti-HAV; the presence of IgG anti-HAV is inferred from the difference between total and IgM anti-HAV. The HAV vaccine, available since 1992, is effective in preventing infection.

Hepatitis B Virus (HBV)
The outcome of HBV infection varies widely, from (1) acute hepatitis with recovery and clearance of the virus; (2) nonprogressive chronic hepatitis; (3) progressive chronic disease ending in cirrhosis; (4) fulminant hepatitis with massive liver necrosis; and (5) an asymptomatic “healthy” carrier state. HBV-induced chronic liver disease is also an important precursor for the development of HCC. The approximate frequencies of various clinical outcomes of HBV infection are depicted in Fig. 16.9.
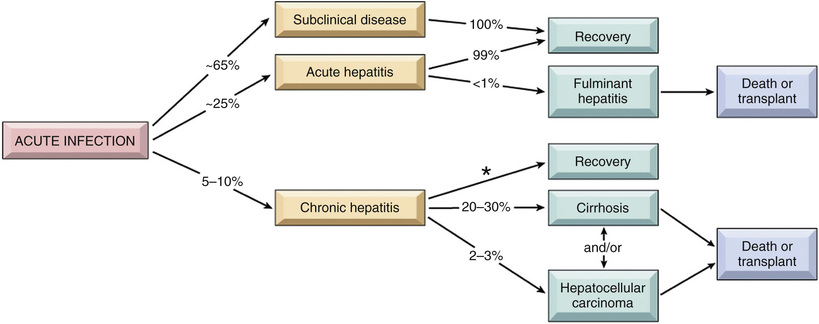
Liver disease due to HBV infection is an enormous global health problem. One-third of the world's population (2 billion individuals) has been infected with HBV, and 400 million individuals have chronic infections. Seventy-five percent of chronic carriers live in Asia and the Western Pacific rim. The global prevalence of chronic hepatitis B infection varies from greater than 8% in parts of Africa to less than 2% in Western Europe, North America, and Australia.
The mode of transmission of HBV also varies with the geographic locale. In high-prevalence regions of the world, perinatal transmission during childbirth accounts for 90% of cases. In areas with intermediate prevalence, horizontal transmission, especially in early childhood, dominates. Spread among children usually occurs through minor breaks in the skin or mucous membranes following physical contact with infected individuals. In low-prevalence areas, unprotected sex and intravenous drug abuse (sharing of needles and syringes) are the chief modes of spread. Transfusion-related spread has been reduced greatly by screening of donated blood for HBsAg and by cessation of the practice of paying blood donors. Vaccination induces a protective antibody response in 95% of infants, children, and adolescents. Universal vaccination has had notable success in Taiwan and Gambia, but has yet to be adopted worldwide.
HBV has a prolonged incubation period (2–26 weeks). Unlike HAV, HBV remains in the blood during active episodes of acute and chronic hepatitis. Approximately 70% of adults with newly acquired HBV have mild or no symptoms and do not develop jaundice. The remaining 30% have nonspecific constitutional symptoms such as anorexia, fever, jaundice, and right upper-quadrant pain. In most cases, the infection is self-limited and resolves without treatment, but chronic disease develops in 10% of infected individuals. Fulminant hepatitis is rare, occurring in approximately 0.1% to 0.5% of acutely infected individuals.
HBV is a member of Hepadnaviridae, a family of DNA viruses that cause hepatitis in multiple animal species. The HBV genome is a partially double-stranded, 3200-nucleotide, circular DNA with four open reading frames, which encode the following proteins:
• Nucleocapsid “core” protein (HBcAg, hepatitis B core antigen) and a longer polypeptide with a precore and core region, designated HBeAg (hepatitis B e antigen). The precore region directs the secretion of the HBeAg polypeptide into blood, whereas HBcAg remains in hepatocytes, where it participates in the assembly of virions.
• Envelope glycoproteins (HBsAg, hepatitis B surface antigen). Infected hepatocytes synthesize and secrete massive quantities of noninfective envelope glycoproteins (mainly small HBsAg).
• A polymerase (Pol) with both DNA polymerase activity and reverse transcriptase activity, which enables genomic replication to occur through a unique DNA → RNA → DNA cycle via an intermediate RNA template. This unusual polymerase is the target of drugs used to treat hepatitis B infection (described later).
• HBx protein, which is required for virus replication and which may act as a transcriptional transactivator for viral genes and a wide variety of host genes. It has been implicated in the pathogenesis of HBV-associated liver cancer.
The course of the disease can be followed clinically by monitoring certain serum markers (Fig. 16.10).
• HBsAg appears before the onset of symptoms, peaks during symptomatic disease, and then usually declines to undetectable levels in 12 weeks (though it may occasionally persist as long as 24 weeks).
• Anti-HBs antibody appears after the acute disease is over and usually is not detected until a few weeks to several months after HBsAg disappears. Anti-HBs may persist for life and confers protection, which is the rationale for current vaccines containing HBsAg.
• HBeAg and HBV DNA appear in serum soon after HBsAg and signify ongoing viral replication. Persistence of HBeAg is an indicator of progression to chronic hepatitis. The appearance of anti-HBe antibodies implies that an acute infection has peaked and is on the wane.
• IgM anti-HBc becomes detectable in serum shortly before the onset of symptoms, concurrent with the onset of elevated serum aminotransferase levels (indicative of hepatocyte destruction). Over a period of months, the IgM anti-HBc antibody is replaced by IgG anti-HBc. As in the case of anti-HAV, there is no direct assay for IgG anti-HBc; its presence is inferred from decline of IgM anti-HBc in the face of rising total anti-HBc.
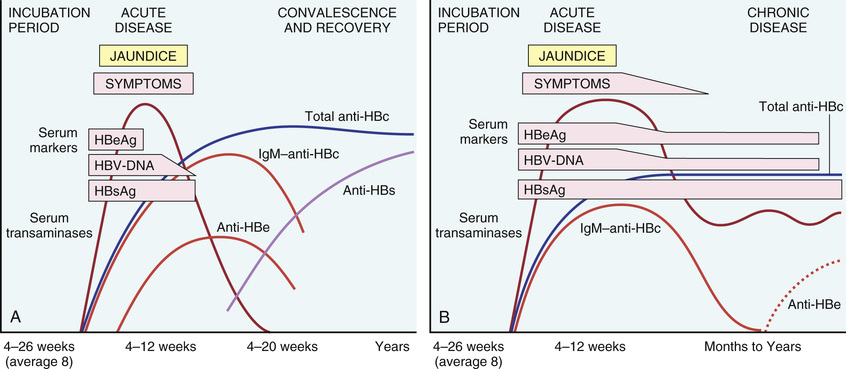
Occasionally, mutated strains of HBV emerge that do not produce HBeAg but are replication competent and express HBcAg. In such patients, the HBeAg may be low or undetectable despite the presence of serum HBV DNA. A second ominous development is the appearance of HBV mutants in vaccinated individuals that replicate in the presence of normally protective anti-HBs antibodies.
The host immune response is the main determinant of the outcome of the infection. Innate immune mechanisms protect the host during initial phases of the infection, and a strong response by virus-specific CD4+ and CD8+ interferon γ–producing cells is associated with the resolution of acute infection. HBV generally is not directly hepatotoxic, and most hepatocyte injury is caused by CD8+ cytotoxic T cells attacking infected cells.
Patient age at the time of infection is the best predictor of chronicity. In general, the younger the age at the time of HBV infection, the higher the chance of chronic infection. Treatment of chronic hepatitis B with viral polymerase inhibitors and interferon can slow disease progression, reduce liver damage, and prevent liver cirrhosis or liver cancer but does not eliminate the infection. As a result, treatment sometimes fails due to emergence of viruses bearing mutations that lead to drug resistance.
Hepatitis C Virus (HCV)
HCV is a major cause of liver disease, with approximately 170 million individuals affected worldwide. Approximately 4.1 million Americans (1.6% of the population) have chronic HCV infection. Notably, there has been a decrease in the annual incidence of infection from a mid-1980s peak of over 230,000 new infections per year to 30,000 new infections per year currently, due primarily to a reduction in transfusion-associated cases as a result of effective screening procedures. Until recently, the number of patients with chronic infection appeared likely to continue to increase, but new therapies (discussed later) are changing the outlook for the better.
According to data from the Centers for Disease Control and Prevention (CDC), the most common risk factors for HCV infection are as follows:
Currently, transmission of HCV by blood transfusion is close to zero in the United States; the risk for acquiring HCV by needle stick is about six times higher than that for HIV (1.8 vs. 0.3%). For children, the major route of infection is vertical perinatal transmission from the mother. Some patients have multiple risk factors, but one-third of individuals have no identifiable risk factors, an enduring mystery.
HCV, discovered in 1989, is a member of the Flaviviridae family. Just as in the case of HIV, an understanding of viral replication and assembly has facilitated the development of highly effective anti-HCV drugs (described below). HCV is a small, enveloped, single-stranded RNA virus with a 9600-base genome encoding a single polyprotein that is processed by several proteases into 10 functional proteins. Included among these viral proteins is a viral protease that is needed for complete processing of the polyprotein; NS5A, a protein that is essential for assembly of HCV into mature virions; and a viral RNA polymerase that is necessary for replication of the viral genome (Fig. 16.11). Because of the low fidelity of the HCV RNA polymerase, the viral genome is inherently unstable, giving rise to new genetic variants at a high pace. This has led to the appearance of six major HCV genotypes worldwide, each with one or more “subspecies.” Infections in most individuals are due to a virus of a single genotype, but new genetic variants are generated in the host as long as viral replication persists. As a result, each patient usually comes to be infected with a population of divergent but closely related HCV variants known as quasispecies.
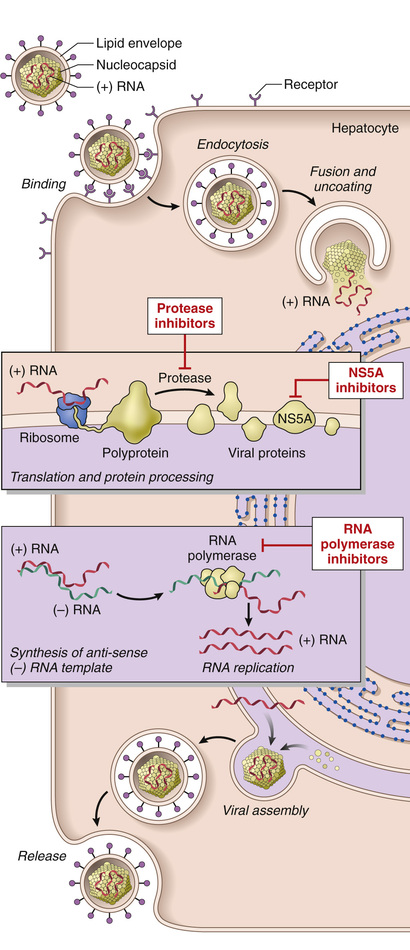
The incubation period for HCV hepatitis ranges from 4 to 26 weeks, with a mean of 9 weeks. In about 85% of individuals, the acute infection is asymptomatic and goes unrecognized. HCV RNA is detectable in blood for 1 to 3 weeks, coincident with elevations in serum transaminases (Fig. 16.12). In symptomatic acute HCV infection, anti-HCV antibodies are detected in only 50% to 70% of patients; in the remaining patients, the anti-HCV antibodies emerge after 3 to 6 weeks. The clinical course of acute HCV hepatitis is milder than that of HBV. It is not known why only a small minority of individuals is capable of clearing HCV infection.
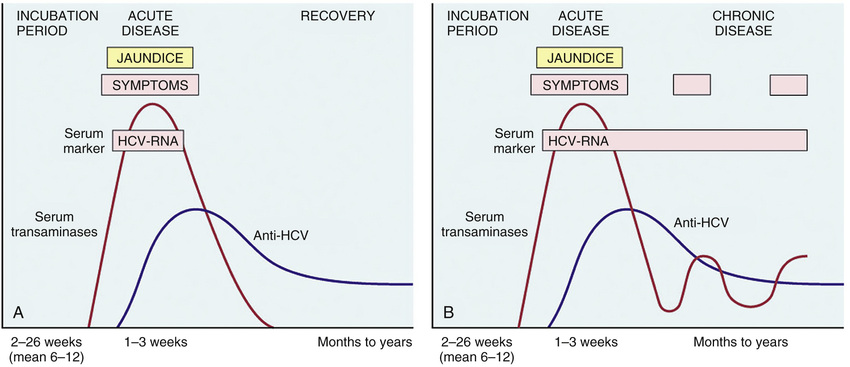
Persistent infection and chronic hepatitis are the hallmarks of HCV infection, despite the generally asymptomatic nature of the acute illness. In contrast to HBV, chronic disease occurs in the majority of HCV-infected individuals (80%–90%), and cirrhosis eventually occurs in as many as one-third. The mechanisms leading to chronicity are not well understood, but it is clear that the virus uses multiple strategies to evade host anti-viral immunity. In addition to rapid generation of genetic variants, which may allow the virus to elude neutralizing antibodies, HCV encodes proteins that inhibit Toll-like receptor and interferon signaling in hepatocytes, activites that would otherwise allow hepatocytes to resist viral infection.
In chronic HCV infection, circulating HCV RNA persists in 90% of patients despite the presence of neutralizing antibodies (see Fig. 16.12). Hence, testing for HCV RNA must be done to confirm the diagnosis of chronic HCV infection. A characteristic clinical feature of chronic HCV infection is episodic elevations in serum aminotransferases separated by periods of normal or near-normal enzyme levels. However, even HCV-infected patients with normal transaminases are at high risk for developing permanent liver damage, and anyone with detectable serum HCV RNA needs treatment and long-term medical follow-up.
Fortunately, recent years have seen dramatic improvements in treatment of HCV infection that stem from development of drugs that specifically target the viral protease, RNA polymerase, and NS5A protein, all of which are required for production of virus (Fig. 16.11). Combination therapy with these drugs (a strategy akin to triple drug therapy for HIV) has proven to be remarkably effective. The goal of current treatment is to eradicate HCV RNA, which is defined by the absence of detectable HCV RNA in the blood 6 months after treatment is stopped and is associated with a high probability of cure. Currently, over 95% of HCV infections are curable, and this can be expected to improve further as new anti-viral drugs become available. The major downside of these advances is their very high cost; a curative course of drug therapy costs over $100,000, and it is estimated that treatment of HCV infections in the United States alone may generate expenses of over $50 billion over the next 5 years.
Hepatitis D Virus (HDV)
Also called the delta agent, HDV is a unique RNA virus that is dependent for its life cycle on HBV. Infection with HDV arises in the following settings:
• Coinfection by HDV and HBV. The HBV must become established first to provide the HBsAg, which is necessary for production of complete HDV virions. Coinfection with HBV and HDV is associated with higher rates of severe acute hepatitis and fulminant liver failure, particularly in intravenous drug abusers, and higher rates of progression to chronic infection, which is often complicated by emergence of liver cancer.
• Superinfection of a chronic HBV carrier by HDV. The superinfection presents 30 to 50 days later as severe acute hepatitis in a previously unrecognized HBV carrier or as an exacerbation of preexisting chronic hepatitis B. Chronic HDV infection occurs in 80% to 90% of such patients. The superinfection may have two phases: an acute phase with active HDV replication and suppression of HBV with high ALT levels, followed by a chronic phase in which HDV replication decreases, HBV replication increases, ALT levels fluctuate, and the disease progresses to cirrhosis and hepatocellular cancer.
HDV infection occurs worldwide and affects an estimated 15 million individuals (about 5% of the 300 million individuals infected by HBV). Its prevalence varies, being highest in the Amazon basin, Africa, the Middle East, and Southern Italy, and lowest in Southeast Asia and China. In most western countries, it is largely restricted to intravenous drug abusers and those who have had multiple blood transfusions.
HDV RNA is detectable in the blood and liver at the time of onset of acute symptomatic disease. IgM anti-HDV is a reliable indicator of recent HDV exposure, but is frequently short-lived. Acute coinfection by HDV and HBV is associated with the presence of IgM against HDAg and HBcAg (denoting new infection with hepatitis B). With chronic delta hepatitis arising from HDV superinfection, HBsAg is present in serum, and anti-HDV antibodies (IgG and IgM) persist for months or longer. Because of its dependency on HBV, HDV infection is prevented by vaccination against HBV.
Hepatitis E Virus (HEV)
HEV is an enterically transmitted, water-borne infection that usually produces a self-limiting disease. The virus typically infects young to middle-aged adults. HEV is a zoonotic disease with animal reservoirs that include monkeys, cats, pigs, and dogs. Epidemics have been reported in Asia and the Indian subcontinent, sub-Saharan Africa, and Mexico, and sporadic cases are seen in Western nations, particularly where pig farming is common and in travelers returning from regions of high incidence. Of greater importance, HEV infection accounts for 30% to 60% of cases of sporadic acute hepatitis in India, exceeding the frequency of HAV. A characteristic feature of HEV infection is the high mortality rate among pregnant women, approaching 20%. In most cases, HEV is not associated with chronic liver disease or persistent viremia. The average incubation period following exposure is 4 to 5 weeks.
Discovered in 1983, HEV is an unenveloped, positive-stranded RNA virus in the Hepevirus genus. Virions are shed in stool during the acute illness. Before the onset of clinical illness, HEV RNA and HEV virions can be detected by PCR in stool and serum. The onset of rising serum aminotransferases, clinical illness, and elevated IgM anti-HEV titers are virtually simultaneous. Symptoms resolve in 2 to 4 weeks, during which time the IgM titers fall and IgG anti-HEV titers rise.
Clinicopathologic Syndromes of Viral Hepatitis
As already discussed, infection with hepatitis viruses produces a wide range of outcomes. Acute infection by each of the hepatotropic viruses may be symptomatic or asymptomatic. HAV and HEV do not cause chronic hepatitis, and only a small number of HBV-infected adults develop chronic hepatitis. In contrast, HCV is notorious for producing chronic infections. Fulminant hepatitis is unusual and is seen primarily with HAV, HBV, or HDV infections. Although HBV and HCV are responsible for most cases of chronic hepatitis, there are many other causes of similar clinicopathologic presentations, including autoimmune hepatitis and drug- and toxin-induced hepatitis (discussed later). Therefore, serologic and molecular studies are essential for the diagnosis of viral hepatitis and for distinguishing between the various types.
Acute Asymptomatic Infection With Recovery.
Patients in this group are identified incidentally on the basis of elevated serum transaminases or the presence of anti-viral antibodies. HAV and HBV infections, particularly in childhood, are frequently subclinical.
Acute Symptomatic Infection With Recovery.
Whichever virus is involved, acute disease follows a similar course, consisting of: (1) an incubation period of variable length (see Table 16.2); (2) a symptomatic preicteric phase; (3) a symptomatic icteric phase; and (4) convalescence. Peak infectivity occurs during the last asymptomatic days of the incubation period and the early days of acute symptoms.
Fulminant Hepatic Failure.
Viral hepatitis is responsible for about 12% of cases of fulminant hepatic failure; of these, two-thirds are caused by HBV infection and the rest by HAV. Survival for more than 1 week may permit recovery to occur via replication of residual hepatocytes. Activation of the stem/progenitor cells in the canals of Hering gives rise to very prominent ductular reactions but is usually insufficient to accomplish full restitution. Fulminant hepatic failure that follows acute viral hepatitis is treated supportively. Liver transplantation is the only option for patients whose disease does not resolve, as death from secondary infections and failure of other organs is otherwise inevitable.
Chronic Hepatitis.
Chronic hepatitis is defined as persistent or relapsing hepatic disease for a period of more than 6 months. The clinical features are extremely variable and are not predictive of outcome. In some patients, the only signs of chronic disease are elevations of serum transaminases. Laboratory studies also may reveal prolongation of the prothrombin time and, in some instances, hyperglobulinemia, hyperbilirubinemia, and mild elevations in alkaline phosphatase levels. In symptomatic individuals, the most common finding is fatigue; less commonly, there is malaise, loss of appetite, and bouts of mild jaundice. In precirrhotic chronic hepatitis, physical findings are few, the most common being spider angiomas, palmar erythema, mild hepatomegaly, hepatic tenderness, and mild splenomegaly. Occasionally, in cases of HBV and HCV, immune complex disease develops that results in vasculitis (subcutaneous or visceral, Chapter 10) and glomerulonephritis (Chapter 14). Cryoglobulinemia is found in about 35% of individuals with chronic hepatitis C.
The Carrier State.
A carrier is an individual who is chronically infected with a hepatropic virus and has no or subclinical evidence of liver disease. In both cases, particularly the latter, these individuals constitute reservoirs for infection. In the case of HBV, “healthy carriers” typically have serum studies that show an absence of HBeAg, the presence of anti-HBe, normal aminotransferases, and low or undetectable serum HBV DNA and liver biopsies showing a lack of significant inflammation or parenchymal injury. HBV infection acquired early in life in endemic areas (such as Southeast Asia, China, and sub-Saharan Africa) gives rise to a carrier state in more than 90% of cases, whereas in nonendemic regions the carrier state is rare. By contrast, it has been estimated that HCV infection in the United States produces a carrier state in 10% to 40% of cases.
HIV and Chronic Viral Hepatitis.
Because of their similar transmission modes and overlapping risk factors, coinfection of HIV and hepatitis viruses is a common clinical problem. In the United States, 10% of HIV-infected individuals are coinfected with HBV and 25% with HCV, and, when untreated, chronic HBV and HCV infection are important causes of morbidity and mortality in HIV-infected individuals, even in those who receive effective anti-HIV therapy. Similarly, in individuals who progress to acquired immunodeficiency syndrome (AIDS), liver disease is the second most common cause of death. However, in adequately treated immunocompetent HIV patients, the severity and progression of HBV and HCV infection and response to anti–hepatitis virus therapy resembles that seen in non-HIV–infected individuals.
Morphology
Clinical assessment of chronic hepatitis sometimes requires liver biopsy in addition to clinical and serologic data. Liver biopsy is helpful in confirming the clinical diagnosis, excluding common concomitant conditions (e.g., fatty liver disease, hemochromatosis), assessing histologic features associated with an increased risk for malignancy (discussed later), grading the extent of hepatocyte injury and inflammation, and staging the progression of scarring. Historically, histologic grading and staging of chronic hepatitis in liver biopsy specimens have been central to determinations of whether to attempt treatment of the underlying disease; however, with the new, highly effective targeted antiviral therapies for hepatitis C, fewer pretreatment biopsies are being performed.
The general morphologic features of acute and chronic viral hepatitis are depicted schematically in Fig. 16.13. The morphologic changes in acute and chronic viral hepatitis are shared among the hepatotropic viruses and can be mimicked by drug reactions or autoimmune hepatitis.

Acute viral hepatitis. Grossly, livers involved by mild acute hepatitis appear normal or slightly mottled. At the other end of the spectrum, massive hepatic necrosis may produce a greatly shrunken liver, as discussed earlier. Microscopically, there is considerable morphologic overlap in acute hepatitis caused by various hepatropic viruses. As is typical of many viral infections, mononuclear cells predominate in all phases of viral hepatitis. A subtle difference is that the mononuclear infiltrate in hepatitis A may be especially rich in plasma cells. Most parenchymal injury is scattered throughout the hepatic lobule as “spotty necrosis” or lobular hepatitis. Portal inflammation in acute hepatitis is minimal or absent. As discussed earlier, hepatocyte injury may result in necrosis or apoptosis. In the former, the cytoplasm appears empty, with only scattered wisps of cytoplasmic remnants, and eventual rupture of cell membranes leads to “dropout” of hepatocytes. In their place, collapsed sinusoidal collagen reticulin framework remains behind along with scavenger macrophages. With apoptosis, hepatocytes shrink, becoming intensely eosinophilic, and their nuclei become pyknotic and fragmented; effector T cells may be present in the immediate vicinity.
In severe acute hepatitis, confluent necrosis of hepatocytes is seen around central veins. In these areas, there may be cellular debris, collapsed reticulin fibers, congestion/hemorrhage, and variable inflammation. With increasing severity, there is central-portal bridging necrosis, followed by parenchymal collapse. In its most severe form, massive hepatic necrosis and fulminant liver failure ensue.
Chronic viral hepatitis. The defining histologic feature of chronic viral hepatitis is mononuclear portal infiltration. It may be mild to severe and variable from one portal tract to the other. There is often interface hepatitis as well, in addition to lobular hepatitis, distinguished by its location at the interface between hepatocellular parenchyma and portal tract stroma. The hallmark of progressive chronic liver damage is scarring. At first, only portal tracts exhibit fibrosis, but in some patients, with time, fibrous septa—bands of dense scar—will extend between portal tracts. In the most severe cases, continued scarring and nodule formation leads to the development of cirrhosis, as discussed earlier.
Certain histologic features point to specific viral etiologies in chronic hepatitis. In chronic hepatitis B, “ground-glass” hepatocytes (cells with endoplasmic reticulum swollen by HBsAg) are a diagnostic hallmark, and the presence of viral antigen in these cells can be confirmed by immunostaining (Fig. 16.14). Liver biopsies involved by chronic hepatitis C quite commonly show large lymphoid aggregates (Fig. 16.15). Often, hepatitis C, particularly genotype 3, is associated with fatty change in scattered hepatocytes. Bile duct injury is also prominent in some cases of hepatitis C and may mimic the histologic changes seen in primary biliary cholangitis (see later); clinical parameters distinguish these two diseases easily, however.


Summary
Viral Hepatitis
• In the alphabet of hepatotropic viruses, some easy mnemonic devices may be useful:
• The vowels (hepatitis A and E) never cause chronic hepatitis, only AcutE hepatitis.
• Only the consonants (hepatitis B, C, D) have the potential to cause chronic disease (C for consonant and for chronic).
• Hepatitis B can be transmitted by blood, birthing, and “bonking” (as they say in the United Kingdom).
• Hepatitis C is the single virus that is more often chronic than not (almost never detected acutely; 85% or more of patients develop chronic hepatitis, 20% of whom will develop cirrhosis).
• Hepatitis D, the delta agent, is a defective virus, requiring hepatitis B coinfection for its own capacity to infect and replicate.
• Hepatitis E is endemic in equatorial regions and frequently epidemic.
• The inflammatory cells in both acute and chronic viral hepatitis are mainly T cells; it is the pattern of injury that is different, not the nature of the infiltrate.
• Patients with long-standing HBV or HCV infections are at increased risk for development of HCC.
Bacterial, Parasitic, and Helminthic Infections
A multitude of organisms can infect the liver and biliary tree, including bacteria, fungi, helminths and other parasites, and protozoa. Infectious organisms can reach the liver through several pathways:
• Ascending infection, via the gut and biliary tract (ascending cholangitis)
• Vascular seeding, most often through the portal system via the gastrointestinal tract
• Direct invasion, from an adjacent source (e.g., bacterial cholecystitis)
Bacteria that may establish an infection in the liver via the blood include Staphylococcus aureus in toxic shock syndrome, Salmonella typhi in typhoid fever, and Treponema pallidum in secondary or tertiary syphilis. Ascending infections are most common in the setting of partial or complete biliary tract obstruction and are typically caused by gut flora, which may colonize the static bile in the ducts. Whatever the source of the bacteria, with pyogenic organisms intrahepatic abscesses may develop, producing fever, right upper-quadrant pain, and tender hepatomegaly. Although antibiotic therapy may sterilize small abscesses, surgical drainage is often necessary for larger lesions. More commonly, extrahepatic bacterial infections, particularly sepsis, induce mild hepatic inflammation and varying degrees of hepatocellular cholestasis indirectly, without establishing an infectious nidus in the liver.
Other non-viral infectious agents cause liver disease with important or unusual pathogenic features that merit specific comment. These include the following:
• Schistosomiasis, most commonly found in Asia, Africa, and South America, is one of the most common causes of noncirrhotic portal hypertension worldwide. Adult worms in the gut produce numerous eggs, some of which find their way into the portal circulation, where they lodge and induce a granulomatous reaction associated with marked fibrosis.
• Entamoeba histolytica, an important cause of dysentery (Chapter 15), sometimes ascends to the liver through portal circulation and produces secondary foci of infection that can progress to large necrotic areas called amebic liver abscesses. Amebic abscesses are more common in the right lobe of the liver. The abscess cavity contains necrotic liver cells, but unlike pyogenic abscesses, neutrophils are absent.
• Liver fluke infection, most common in Southeast Asia, is associated with a high rate of cholangiocarcinoma. Responsible organisms include Fasciola hepatica, Opisthorcis species, and Clonorchis sinensis.
• Echinococcal infections may cause the formation of intrahepatic hydatid cysts that produce symptoms due to pressure on surrounding structures or following rupture.
Autoimmune Hepatitis
Autoimmune hepatitis is a chronic, progressive hepatitis with all the features of autoimmune diseases in general: genetic predisposition, association with other autoimmune diseases, the presence of autoantibodies, and therapeutic response to immunosuppression. Risk for autoimmune hepatitis is associated with certain HLA alleles, such as the DRB1* allele in Caucasians, but as in other autoimmune disorders the mechanistic basis for this relationship is unclear. Triggers for the immune reaction may include viral infections or drug or toxin exposures.
Clinicopathologic Features
The annual incidence is highest among white northern Europeans at 1.9 in 100,000, but all ethnic groups are susceptible. There is a female predominance (78%). Autoimmune hepatitis is classified into two types, based on the patterns of circulating antibodies.
• Type 1, more common in middle-age and older individuals, is characterized by the presence of anti-nuclear (ANA), anti–smooth muscle actin (SMA), anti-mitochondrial (AMA), and anti–soluble liver antigen/liver-pancreas antigen (anti-SLA/LP) antibodies.
• Type 2, usually seen in children and teenagers, is chararcterized by the presence of anti–liver kidney microsome-1 antibodies and anti–liver cytosol-1 antibodies.
Morphology
Although autoimmune hepatitis shares patterns of injury with acute or chronic viral hepatitis, the time course of histologic progression differs. In viral hepatitis, fibrosis typically follows many years of slowly accumulating parenchymal injury, whereas in autoimmune hepatitis, there is an early phase of severe parenchymal destruction followed rapidly by scarring. For unclear reasons, this early wave of hepatocyte damage is often subclinical. The following features are typical of autoimmune hepatitis:
An acute clinical illness is a common presentation (40%); sometimes the disease is fulminant, progressing to hepatic encephalopathy within 8 weeks of onset. Mortality for patients with severe untreated autoimmune hepatitis is approximately 40% within 6 months of diagnosis, and cirrhosis develops in at least 40% of survivors. Hence, diagnosis and intervention are imperative. Immunosuppressive therapy is usually effective, leading to remission in 80% of patients and enabling long-term survival. End-stage disease is an indication for liver transplantation. The 10-year survival rate after liver transplant is 75%, but recurrence in the transplanted organ occurs in 20% of cases.
Summary
• There are two primary types of autoimmune hepatitis:
• Type 1 autoimmune hepatitis is most often seen in middle-age women and is characteristically associated with anti-nuclear and anti–smooth muscle antibodies.
• Type 2 autoimmune hepatitis is most often seen in children or teenagers and is associated with anti–liver kidney microsomal autoantibodies.
• Autoimmune hepatitis may either develop with a rapidly progressive acute disease or follow a more indolent path; if untreated, both are likely to lead to liver failure.
• Plasma cells are a prominent and characteristic component of the inflammatory infiltrate in biopsy specimens showing autoimmune hepatitis.
Drug- and Toxin-Induced Liver Injury
As the major drug metabolizing and detoxifying organ in the body, the liver is subject to injury from an enormous array of therapeutic and environmental chemicals. Injury may result from direct toxicity, may occur through hepatic conversion of a xenobiotic compound to an active toxin, or may be produced by immune mechanisms, such as by the drug or a metabolite acting as a hapten to convert a cellular protein into an immunogen. A diagnosis of drug- or toxin-induced liver injury may be made on the basis of a temporal association of liver damage with drug or toxin exposure, recovery (usually) upon removal of the inciting agent, and exclusion of other potential causes. Exposure to a toxin or therapeutic agent should always be included in the differential diagnosis of any form of liver disease.
Principles of drug and toxic injury are discussed in Chapter 8. Here it suffices to note that drug reactions may be predictable (intrinsic) or unpredictable (idiosyncratic). Predictable drug or toxin reactions affect all individuals in a dose-dependent fashion. Unpredictable reactions depend on idiosyncrasies of the host, particularly the propensity to mount an immune response to the antigenic stimulus or the rate at which the agent can be metabolized. Both classes of injury may be immediate or take weeks to months to develop (Table 16.3).
• A classic, predictable hepatotoxin is acetaminophen, now the most common cause of acute liver failure necessitating transplantation in the United States. The toxic agent is not acetaminophen itself but rather toxic metabolites produced by the cytochrome P-450 system. The damage begins in centrilobular hepatocytes but extends to encompass entire lobules in the most severe cases.
• Examples of drugs that can cause idiosyncratic reactions include chlorpromazine, an agent that causes cholestasis in patients who are slow to metabolize it, and halothane and its derivatives, which can cause a fatal immune-mediated hepatitis after repeated exposure.
Table 16.3
Patterns of Injury in Drug- and Toxin-Induced Hepatic Injury
| Pattern of Injury | Morphologic Findings | Examples of Associated Agents |
| Cholestatic | Bland hepatocellular cholestasis, without inflammation | Contraceptive and anabolic steroids, antibiotics, HAART |
| Cholestatic hepatitis | Cholestasis with lobular necrosis and inflammation; may show bile duct destruction | Antibiotics, phenothiazines, statins |
| Hepatocellular necrosis | Spotty hepatocyte necrosis | Methyldopa, phenytoin |
| Massive necrosis | Acetaminophen, halothane | |
| Chronic hepatitis | Isoniazid | |
| Fatty liver disease | Large and small droplet fat | Ethanol, corticosteroids, methotrexate, total parenteral nutrition |
| “Microvesicular steatosis” (diffuse small droplet fat) | Valproate, tetracycline, aspirin (Reye syndrome), HAART | |
| Steatohepatitis with Mallory-Denk bodies | Ethanol, amiodarone | |
| Fibrosis and cirrhosis | Periportal and pericellular fibrosis | Alcohol, methotrexate, enalapril, vitamin A and other retinoids |
| Granulomas | Noncaseating epithelioid granulomas | Sulfonamides, amiodarone, isoniazid |
| Fibrin ring granulomas | Allopurinol | |
| Vascular lesions | Sinusoidal obstruction syndrome (veno-occlusive disease): obliteration of central veins | High-dose chemotherapy, bush teas |
| Budd-Chiari syndrome | Oral contraceptives | |
| Peliosis hepatis: blood-filled cavities, not lined by endothelial cells | Anabolic steroids, tamoxifen |

Alcoholic and Nonalcoholic Fatty Liver Disease
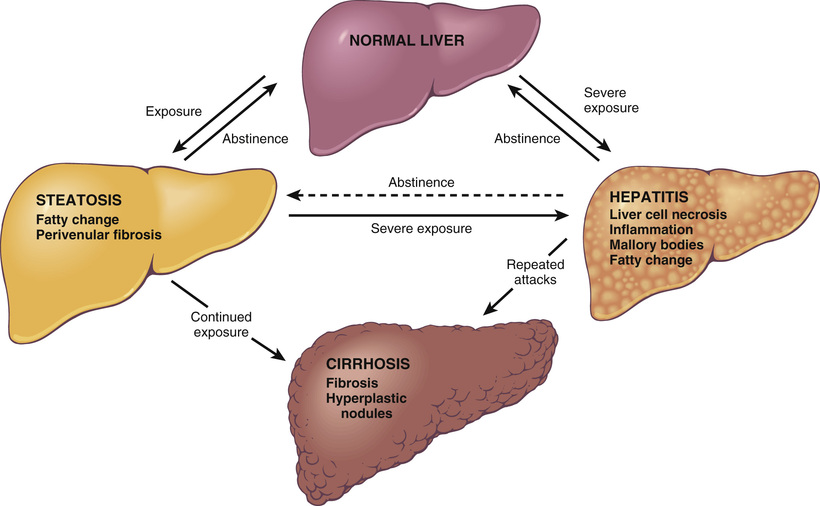
Alcohol is a well-known cause of fatty liver disease in adults and can manifest histologically as steatosis, steatohepatitis, and cirrhosis. In recent years, it has become evident that another entity, so-called “nonalcoholic fatty liver disease (NAFLD),” can mimic the entire spectrum of hepatic changes associated with alcohol abuse. Since the morphologic changes of alcoholic and NAFLD are indistinguishable, they are discussed together, followed by the pathogenesis and distinctive clinical features of each entity.
Morphology
Three types of liver alterations are observed in fatty liver disease: steatosis (fatty change), hepatitis (alcoholic or steatohepatitis), and fibrosis.
Hepatocellular steatosis. Hepatocellular fat accumulation typically begins in centrilobular hepatocytes. The lipid droplets range from small (microvesicular) to large (macrovesicular), the largest filling and expanding the cell and displacing the nucleus. As steatosis becomes more extensive, the lipid accumulation spreads outward from the central vein to hepatocytes in the midlobule and then the periportal regions (Fig. 16.16). Macroscopically, fatty livers with widespread steatosis are large (weighing 4–6 kg or more), soft, yellow, and greasy.

Steatohepatitis. These changes typically are more pronounced with alcohol use than in NAFLD, but can be seen in either:
• Hepatocyte ballooning. Single or scattered foci of cells undergo swelling and necrosis; as with steatosis, these features are most prominent in the centrilobular regions (Fig. 16.17A).

• Mallory-Denk bodies. These consist of tangled skeins of intermediate filaments (including ubiquitinylated keratins 8 and 18) and are visible as eosinophilic cytoplasmic inclusions in degenerating hepatocytes (see Fig. 16.17B).
• Neutrophil infiltration. Predominantly neutrophilic infiltration may permeate the lobule and accumulate around degenerating hepatocytes, particularly those containing Mallory-Denk bodies. Lymphocytes and macrophages also may be seen in portal tracts or parenchyma.
Steatofibrosis. Fatty liver disease of all kinds has a distinctive pattern of scarring. Like other changes, fibrosis appears first in the centrilobular region as central vein sclerosis. Perisinusoidal scarring appears next in the space of Disse of the centrilobular region and then spreads outward, encircling individual or small clusters of hepatocytes in a chicken wire fence pattern (see Fig. 16.16). Tendrils of fibrosis eventually link to portal tracts and then condense to create central portal fibrous septa. As these become more prominent, the liver takes on a nodular, cirrhotic appearance. Because in most cases the underlying cause persists, the continual subdivision of established nodules by new, perisinusoidal scarring leads to a classic micronodular or Laennec cirrhosis. Early in the course, the liver is yellow-tan, fatty, and enlarged, but with persistent damage over the course of years the liver is transformed into a brown, shrunken, nonfatty organ composed of cirrhotic nodules that are usually less than 0.3 cm in diameter—smaller than is typical for most chronic viral hepatitis. The end-stage cirrhotic liver may enter into a “burned-out” phase devoid of fatty change and other typical features. A majority of cases of cryptogenic cirrhosis, without clear etiology, are now recognized as “burned-out” NAFLD.
Alcoholic Liver Disease
Excessive ethanol consumption causes more than 60% of chronic liver disease in Western countries and accounts for 40% to 50% of deaths due to cirrhosis. Among the most important adverse effects of chronic alcohol consumption are the overlapping forms of alcohol-related fatty liver disease already discussed: (1) hepatic steatosis, (2) alcoholic hepatitis, and (3) fibrosis and cirrhosis, collectively referred to as alcoholic liver disease (Fig. 16.18).
Between 90% and 100% of heavy drinkers develop fatty liver (i.e., hepatic steatosis), and of those, 10% to 35% develop alcoholic hepatitis, whereas only 8% to 20% of chronic alcoholics develop cirrhosis. Steatosis, alcoholic hepatitis, and fibrosis may develop sequentially or independently, so they do not necessarily represent a sequential continuum of changes. Hepatocellular carcinoma arises in 10% to 20% of patients with alcoholic cirrhosis.
Pathogenesis
Short-term ingestion of as much as 80 g of ethanol per day (5–6 beers or 8–9 ounces of 80-proof liquor) generally produces mild reversible hepatic changes, such as fatty liver. Chronic intake of 40 to 80 g/day is considered a borderline risk factor for severe injury. For reasons that may relate to decreased gastric metabolism of ethanol and differences in body composition, women are more susceptible than men to hepatic injury. It seems that how often and what one drinks may affect the risk for liver disease development. For example, binge drinking causes more liver injury than that associated with steady, lower-level consumption. Since not everyone who drinks gets all the listed complications, individual, possibly genetic, risk factors must exist, but no reliable markers of susceptibility are known. In the absence of a clear understanding of the factors that influence liver damage, it is difficult to state what constitutes a safe level of alcohol consumption.
Hepatocellular steatosis is caused by alcohol through several mechanisms. First, metabolism of ethanol by alcohol dehydrogenase and acetaldehyde dehydrogenase generates large amounts of nicotinamide-adenine dinucleotide (NADH), which increases shunting of substrates away from catabolism and toward lipid biosynthesis. Second, ethanol impairs the assembly and secretion of lipoproteins. The net effect is to cause the accumulation of intracellular lipids.
The cause of alcoholic hepatitis is uncertain, but it may stem from one or more of the following toxic byproducts of ethanol and its metabolites:
• Acetaldehyde (a major metabolite of ethanol) induces lipid peroxidation and acetaldehyde-protein adduct formation, which may disrupt cytoskeleton and membrane function.
• Alcohol directly affects mitochondrial function and membrane fluidity.
• Reactive oxygen species generated during oxidation of ethanol by the microsomal ethanol oxidizing system react with and damage membranes and proteins. Reactive oxygen species also are produced by neutrophils, which infiltrate areas of hepatocyte necrosis.
Because generation of acetaldehyde and free radicals is maximal in the centrilobular region, this region is most susceptible to toxic injury. Pericellular and sinusoidal fibrosis develop first in this area of the lobule. Concurrent viral hepatitis, particularly hepatitis C, is a major accelerator of liver disease in alcoholics. The prevalence of hepatitis C among individuals with alcoholic liver disease is about 30% (and vice versa).
For unknown reasons, cirrhosis develops in only a small fraction of chronic alcoholics. With complete abstinence, at least partial regression of scarring occurs, and the micronodular liver transforms though parenchymal regeneration into a macronodular cirrhotic organ (see Figure 16.6); rarely, there is regression of cirrhosis altogether.
Clinical Features
Alcoholic steatosis may be innocuous or give rise to hepatomegaly with mild elevations of serum bilirubin and alkaline phosphatase. Severe hepatic compromise is unusual. Alcohol withdrawal and the provision of an adequate diet are sufficient treatment.
It is estimated that 15 to 20 years of excessive drinking are necessary to develop alcoholic cirrhosis, but alcoholic hepatitis can occur after just weeks or months of alcohol abuse. The onset is typically acute and often follows a bout of particularly heavy drinking. Symptoms and laboratory abnormalities range from minimal to severe. Most patients present with malaise, anorexia, weight loss, upper-abdominal discomfort, tender hepatomegaly, and fever. Typical findings include hyperbilirubinemia, elevated serum alkaline phosphatase levels, and neutrophilic leukocytosis. Serum alanine and aspartate aminotransferases are elevated but usually remain below 500 U/mL. The outlook is unpredictable; each bout of alcoholic hepatitis carries a 10% to 20% risk for death. With repeated bouts, cirrhosis appears in about one-third of patients within a few years.
The manifestations of alcoholic cirrhosis are similar to those of other forms of cirrhosis. In chronic alcoholics, ethanol may be the major source of calories in the diet, displacing other nutrients and leading to malnutrition and vitamin deficiencies (e.g., thiamine, vitamin B12). Compounding these effects is impaired digestive function, primarily related to chronic gastric and intestinal mucosal damage and pancreatitis.
The long-term outlook for alcoholic patients with liver disease is variable. The most important aspect of treatment is abstinence from alcohol. The 5-year survival rate approaches 90% in abstainers who are free of jaundice, ascites, and hematemesis, but drops to 50% to 60% in individuals who continue to imbibe. Among those with end-stage alcoholic liver disease, the immediate causes of death are as follows:
• Massive gastrointestinal hemorrhage
• Intercurrent infection (to which affected individuals are predisposed)
Summary
Alcoholic Liver Disease
• Alcoholic liver disease has three main manifestations, hepatic steatosis, alcoholic hepatitis, and cirrhosis, which may occur alone or in combination.
• Cirrhosis typically develops after more than 10 years of heavy drinking, but only occurs in a small proportion of chronic alcoholics; alcoholic cirrhosis has similar clinical signs and symptoms as cirrhosis caused by viral hepatitis.
• The multiple pathologic effects of alcohol include changes in lipid metabolism, decreased export of lipoproteins, and cell injury caused by reactive oxygen species and metabolites of alcohol.
Nonalcoholic Fatty Liver Disease
NAFLD is a common condition in which fatty liver disease develops in individuals who do not drink alcohol. The liver can show any of the three types of changes discussed earlier (steatosis, steatohepatitis, and cirrhosis), though on average inflammation is less prominent than in alcoholic liver disease. The term nonalcoholic steatohepatitis (NASH) is used to describe overt clinical features of liver injury, such as elevated transaminases, and the histologic features of hepatitis already discussed. NAFLD is consistently associated with insulin resistance and the metabolic syndrome (Chapter 8). Other commonly associated abnormalities are as follows:
• Type 2 diabetes (or family history of the condition)
• Obesity, primarily central obesity (body mass index >30 kg/m2 in whites and >25 kg/m2 in Asians)
• Dyslipidemia (hypertriglyceridemia, low high-density lipoprotein cholesterol, high low-density lipoprotein cholesterol)
Pathogenesis
The key initiating events in NAFLD appear to be the development of obesity and insulin resistance, the latter within both adipose tissue and the liver. These factors combine to increase the mobilization of free fatty acids from adipose tissue, which are taken up by hepatocytes, and to stimulate the synthesis of fatty acids within hepatocytes. It is estimated that over half of the lipid found in hepatocytes in NAFLD is derived from adipose tissue, with most of the remainder coming from de novo synthesis in liver cells. Precisely how the accumulation of lipid in hepatocytes predisposes to the development of NASH is not known and may involve several interrelated mechanisms. Excessive intrahepatic lipids and their metabolic intermediates enhance insulin resistance in the liver and sensitize hepatocytes to the toxic effects of inflammatory cytokines, which are produced in increased amounts in the setting of the metabolic syndrome. In addition, hepatocytes in patients with NASH show evidence of inflammasome activation, possibly due to direct or indirect effects of particular lipids, leading to local release of the pro-inflammatory cytokine IL-1. Other products of lipid metabolism appear to be directly toxic to hepatocytes; proposed mechanisms include increased production of reactive oxygen species, induction of ER stress, and disruption of mitochondrial function. Liver injury resulting from these various insults causes stellate cell activation, collagen deposition, and hepatic fibrosis, which along with ongoing hepatocyte damage lead to full-blown NASH.
Clinical Features
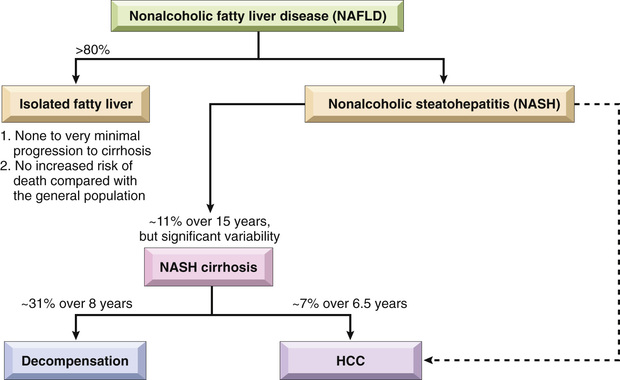
NAFLD is the most common cause of incidental elevation of serum transaminases. Most individuals with steatosis are asymptomatic; patients with active steatohepatitis or fibrosis may also be asymptomatic, but some may have fatigue, malaise, right upper-quadrant discomfort, or more severe symptoms of chronic liver disease. Liver biopsy is required to identify NASH and distinguish it from uncomplicated NAFLD. Fortunately, the frequency of progression from steatosis to active steatohepatitis and then from active steatohepatitis to cirrhosis is low (Fig. 16.19). Nevertheless, NAFLD is considered to be a significant contributor to the pathogenesis of “cryptogenic” cirrhosis. Because they share common risk factors, the incidence of coronary artery disease also is increased in patients with NAFLD.
Current therapy is directed toward obesity reduction and reversal of insulin resistance. Lifestyle modifications that lead to weight loss (diet and exercise) appear to be the most effective form of treatment.
Pediatric NAFLD is becoming an increasing problem as obesity and metabolic syndrome approach epidemic proportions. In children, the appearance of the histologic lesions is somewhat different, as inflammation and scarring tend to be more prominent in the portal tracts and periportal regions, and mononuclear infiltrates rather than neutrophilic infiltrates predominate.
Summary
Nonalcoholic Fatty Liver Disease
• Nonalcoholic fatty liver disease (NAFLD) is associated with the metabolic syndrome, obesity, type 2 diabetes, and dyslipidemia and/or hypertension.
• NAFLD may show all the changes associated with alcoholic liver disease: steatosis, nonalcoholic steatohepatitis (NASH), and cirrhosis, although the features of steatohepatitis (such as hepatocyte ballooning, Mallory-Denk bodies, and neutrophilic infiltration) often are less prominent than they are in alcohol-related injury.
• Pediatric NAFLD is increasingly being recognized as the obesity epidemic spreads to pediatric age groups, although its histologic pattern differs somewhat from that seen in adults.
Inherited Metabolic Liver Diseases
Although there are many inherited metabolic liver diseases, only some relatively common, pathogenically interesting entities are discussed here: hereditary hemochromatosis, Wilson disease, and alpha-1-anti-trypsin (α1AT) deficiency.
Hemochromatosis
Hemochromatosis is caused by excessive absoprtion of iron, which is primarily deposited in parenchymal organs such as the liver and pancreas, as well as in the heart, joints, and endocrine organs. It results most commonly from an inherited disorder, hereditary hemochromatosis. When iron accumulation occurs as a consequence of parenteral administration of iron, usually in the form of transfusions, it is called acquired hemochromatosis. Secondary iron overload also can complicate diseases that are associated with persistent ineffective erythropoiesis, particularly thalassemia and myelodysplastic syndromes (Chapter 12).
As discussed in Chapter 12, the total body iron pool ranges from 2 to 6 gm in normal adults; about 0.5 gm is stored in hepatocytes. In severe hemochromatosis, total iron may exceed 50 gm, one-third of which accumulates in the liver. Fully developed cases exhibit (1) micronodular cirrhosis; (2) diabetes mellitus (up to 80% of patients); and (3) abnormal skin pigmentation (up to 80% of patients).
Pathogenesis
Because there is no regulated iron excretion from the body, the total body content of iron is tightly regulated by intestinal absorption. As discussed in Chapter 12, hepcidin is a circulating peptide hormone that acts as a key negative regulator of intestinal iron uptake. Diverse mutations in several genes have been described in hereditary hemochromatosis, all of which lower hepcidin levels or diminish hepcidin function. Whatever the underlying defect, the net result is an increase in intestinal absorption of dietary iron, leading to an accumulation of 0.5 to 1 gm of iron per year.
The most frequently mutated gene in patients with hereditary hemochromatosis is HFE, which is located on chromosome 6 close to the HLA gene cluster. HFE encodes an HLA class I–like molecule that regulates the synthesis of hepcidin in hepatocytes. The most common HFE mutation is a cysteine-to-tyrosine substitution at amino acid 282 (C282Y). This mutation, which inactivates the HFE protein, is present in over 70% of patients diagnosed with hereditary hemochromatosis and is most common in European populations. Several other mutations can also give rise to hemochromatosis, including other mutations in HFE as well as mutations in transferrin receptor 2 and in hepcidin itself. The associated clinical condition is milder with some of these alternative mutations and more severe with others, sometimes manifesting in young adults or even during childhood.
Whatever the underlying cause, the onset of disease typically occurs after 20 gm of stored iron have accumulated. Excessive iron appears to be directly toxic to host tissues. Mechanisms of liver injury include the following:
• Lipid peroxidation via iron-catalyzed free radical reactions
• Stimulation of collagen formation by activation of hepatic stellate cells
• DNA damage by reactive oxygen species, leading to lethal cell injury or predisposition to HCC
The deleterious effects of iron on cells that are not fatally injured are reversible, and removal of excess iron with therapy promotes recovery of tissue function.
Morphology
The morphologic changes in severe hemochromatosis are characterized principally by (1) tissue deposition of hemosiderin in the following organs (in decreasing order of severity): liver, pancreas, myocardium, pituitary gland, adrenal gland, thyroid and parathyroid glands, joints, and skin; (2) cirrhosis; and (3) pancreatic fibrosis. In the liver, iron becomes evident first as golden-yellow hemosiderin granules in the cytoplasm of periportal hepatocytes, which can be histochemically stained with Prussian blue (Fig. 16.20). With increasing iron load, there is progressive deposition in the rest of the lobule, the bile duct epithelium, and Kupffer cells. At this stage, the liver typically is slightly enlarged and chocolate brown. Fibrous septa develop slowly, linking portal tracts to each other and leading ultimately to cirrhosis in an intensely pigmented (very dark brown to black) liver.

The pancreas also becomes pigmented, acquires diffuse interstitial fibrosis, and may show parenchymal atrophy. Hemosiderin is found in the acinar and the islet cells and sometimes in the interstitial fibrous stroma. The heart often is enlarged, with hemosiderin granules within the myocardial fibers. The pigmentation may induce a striking brown coloration of the myocardium. A delicate interstitial fibrosis may appear. Although skin pigmentation is partially attributable to hemosiderin deposition in dermal macrophages and fibroblasts, most of the coloration results from increased epidermal melanin production. The combination of these pigments renders the skin slate-gray. With hemosiderin deposition in the joint synovial linings, an acute synovitis may develop. There is also excessive deposition of calcium pyrophosphate, which damages the articular cartilage and sometimes produces disabling polyarthritis, referred to as pseudogout. With the onset of cirrhosis, the testes may become atrophic.
Clinical Features
Symptoms usually appear earlier in men than in women since menstrual bleeding limits the accumulation of iron until menopause. This results in a male-to-female ratio of clinically significant iron overload of approximately 5 : 1 to 7 : 1. In the most common form caused by HFE mutations, symptoms usually appear in the fifth and sixth decades of life in men and later in women. With population screening, it has become clear that homozygosity for the most common HFE mutation (C282Y) shows variable penetrance; thus disease development is not inevitable, presumably because other genetic and environmental factors influence the rate of iron accumulation.
The principal manifestations include hepatomegaly, abdominal pain, skin pigmentation (particularly in sun-exposed areas), deranged glucose homeostasis or frank diabetes mellitus due to destruction of pancreatic islets, cardiac dysfunction (arrhythmias, cardiomyopathy), and atypical arthritis. In some patients, the presenting complaint is hypogonadism (e.g., amenorrhea in the female, impotence and loss of libido in the male). As noted, clinically apparent disease is more common in males and rarely becomes evident before 40 years of age. Death may result from cirrhosis or cardiac disease. In those with untreated disease, the risk for HCC is increased 200-fold, presumably because of ongoing liver damage and the genotoxic effects of oxidants generated by iron in the liver.
Fortunately, hemochromatosis can be diagnosed long before irreversible tissue damage has occurred. Screening of family members of probands is important. Heterozygotes also accumulate excessive iron, but not to a level that causes significant tissue damage. Currently most patients with hemochromatosis are diagnosed in the subclinical, precirrhotic stage due to routine serum iron measurements (as part of another diagnostic workup). Regular phlebotomy results in steady removal of excess tissue iron, and with this simple treatment life expectancy is normal.
Wilson Disease
Wilson disease is an autosomal recessive disorder caused by mutation of the ATP7B gene, which results in impaired copper excretion into bile and a failure to incorporate copper into ceruloplasmin. This disorder is marked by the accumulation of toxic levels of copper in many tissues and organs, principally the liver, brain, and eye. Normally, 40% to 60% of ingested copper (2–5 mg/day) is absorbed in the duodenum and proximal small intestine, from where it is transported complexed with albumin and histidine to the liver. Here, free copper dissociates and is taken up by hepatocytes, where copper is incorporated into enzymes and α2-globulin (apoceruloplasmin) to form ceruloplasmin, which is secreted into the blood. Ceruloplasmin carries 90% to 95% of plasma copper. Circulating ceruloplasmin is eventually desialylated, endocytosed by the liver, and degraded within lysosomes, after which the released copper is excreted into bile. This degradation/excretion pathway is the primary route for copper elimination.
The ATP7B gene, located on chromosome 13, encodes a transmembrane copper-transporting ATPase that is expressed on the hepatocyte canalicular membrane. The overwhelming majority of patients with Wilson disease are compound heterozygotes with different loss-of-function mutations affecting each ATP7B allele. The overall frequency of mutated alleles is 1 : 100, and the prevalence of the disease is approximately 1 : 30,000 to 1 : 50,000. Loss of ATP7B protein function impairs the transport of copper into the bile and the incorporation of copper into ceruloplasmin, which is not secreted in its apoceruloplasmin form. These abnormalities lead to copper accumulation in the liver and a decrease in plasma ceruloplasmin. Accumulating copper causes liver injury through the production of reactive oxygen species by the Fenton reaction (Chapter 3). Eventually, non-ceruloplasmin–bound copper is released from injured hepatocytes into the circulation, causing red cell hemolysis and allowing copper to deposit in other tissues, such as the brain, corneas, kidneys, bones, joints, and parathyroid glands. Concomitantly, urinary excretion of copper increases markedly from its normal minuscule levels.
Morphology
The liver often bears the brunt of injury. The hepatic changes are variable, ranging from relatively minor to massive and mimic many other disease processes. There may be mild to moderate fatty change (steatosis) associated with focal hepatocyte necrosis. Acute, fulminant hepatitis can mimic acute viral hepatitis. Chronic hepatitis in Wilson disease exhibits moderate to severe inflammation and hepatocyte necrosis, areas of fatty change, and features of steatohepatitis (hepatocyte ballooning with prominent Mallory-Denk bodies). In advanced cases, cirrhosis may be seen. Copper deposition in hepatocytes can be demonstrated by special stains (rhodamine stain for copper, orcein stain for copper-associated protein).
Toxic injury to the brain primarily affects the basal ganglia. Nearly all patients with neurologic involvement develop eye lesions called Kayser-Fleischer rings, green to brown deposits of copper in Desçemet membrane in the limbus of the cornea.
Clinical Features
The age at onset and the clinical presentation of Wilson disease are extremely variable. Symptoms usually appear between 6 and 40 years of age. Acute or chronic liver disease are common presenting features. Neuropsychiatric manifestations are the initial features in most of the remaining cases and stem from deposition of copper in the basal ganglia.
The diagnosis of Wilson disease is based on low levels of serum ceruloplasmin, an increase in hepatic copper content (the most sensitive test), and increased urinary excretion of copper (the most specific test). Hepatic copper content in excess of 250 µg per gram dry weight of liver is taken to be diagnostic, but is only about 80% sensitive. In those with lower liver copper levels, the diagnosis depends on other abnormalities, such as elevated urinary copper, low serum ceruloplasmin, and the presence of Kayser-Fleischer rings. Unlike hereditary hemochromatosis, where the limited number of genetic variants makes genetic testing fairly simple, the large number of different causative mutations in ATP7B7 complicates the use of DNA sequencing as a diagnostic test. Serum copper levels also are of no diagnostic value, as they may be low, normal, or elevated, depending on the stage of the liver disease.
Early recognition and long-term copper chelation therapy (with D-penicillamine or Trientine) or zinc-based therapy (which inhibits copper uptake in the gut) has dramatically altered the usual progressive downhill course. Individuals with hepatitis or advanced cirrhosis require liver transplantation, which can be curative.
α1-Anti-Trypsin Deficiency
α1-Anti-trypsin deficiency is an autosomal recessive disorder marked by very low levels of circulating α1-anti-trypsin (α1AT) that is caused by mutations that lead to misfolding of α1AT. The major function of α1AT is to inhibit proteases, particularly neutrophil elastase, cathepsin G, and proteinase 3, which are released from neutrophils at sites of inflammation. α1AT deficiency leads to the development of pulmonary emphysema because the activity of destructive proteases is not inhibited (Chapter 13). It also causes liver disease as a consequence of hepatocellular accumulation of the misfolded α1AT protein.
α1AT is a small 394–amino acid plasma glycoprotein synthesized predominantly by hepatocytes. The gene, located on chromosome 14, is very polymorphic. At least 75 α1AT variants have been identified, denoted alphabetically by their relative migration on an isoelectric gel. The general notation is “Pi” for “protease inhibitor” and an alphabetic letter for the position on the gel; two letters denote the genotype of the two alleles. The most common genotype is PiMM, occurring in 90% of individuals (the “wild-type”).
The most common clinically significant mutation is PiZ; PiZZ homozygotes have circulating α1AT levels that are only 10% of normal. These individuals are at high risk for developing clinical disease. Variant alleles are codominant, and, consequently, PiMZ heterozygotes have intermediate plasma levels of α1AT. Among individuals of northern European descent, the PiZZ state affects 1 in 1800 live births. Because of the early presentation of the liver disease, α1AT deficiency is the most commonly diagnosed genetic hepatic disorder in infants and children.
Pathogenesis
The PiZ polypeptide is prone to misfolding and aggregation due to a single amino acid glutamine-to-lysine substitution at residue 342 (E342K). This in turn creates endoplasmic reticulum stress and triggers the unfolded protein response, which ultimately leads to apoptosis. It is worth emphasizing that the liver damage is caused by protein misfolding, whereas lung damage leading to emphysema stems from the loss of α1AT function and excessive protease activity. Although all individuals with the PiZZ genotype accumulate α1AT-Z in the endoplasmic reticulum of hepatocytes, only 10% to 15% develop overt clinical liver disease; thus, other genetic factors or environmental factors must also play a role in the development of liver disease.
Morphology
α1-Anti-trypsin deficiency is characterized by the presence of round-to-oval cytoplasmic globular inclusions in hepatocytes that are strongly periodic acid–Schiff (PAS) positive and diastase resistant (Fig. 16.21). Periportal hepatocytes are most affected in early and in mild forms of the disease, with central lobular hepatocytes being affected later or in more severe disease. Other pathologic features vary, ranging from hepatitis to fibrosis to full-blown cirrhosis.

Clinical Features
Neonatal hepatitis with cholestatic jaundice appears in 10% to 20% of newborns with α1AT deficiency. In adolescence, presenting symptoms may be related to hepatitis or cirrhosis. Attacks of hepatitis may subside with apparent complete recovery, or they may become chronic and lead progressively to cirrhosis. Alternatively, the disease may remain silent until cirrhosis appears in middle to later adult life. HCC develops in 2% to 3% of PiZZ adults, usually in the setting of cirrhosis. The definitive treatment, for severe hepatic disease is liver transplantation. In patients with pulmonary disease, avoidance of cigarette smoking is crucial, because smoking results in accumulation of neutrophils and release of elastase in the lung that is not inactivated because of lack of α1AT. The unopposed action of neutrophil derived proteases destroys elastic fibers in alveolar walls, leading to emphysema (Chapter 13).
Summary
Inherited Metabolic Liver Disease
• Hemochromatosis is most commonly caused by mutations in the HFE gene and less commonly by mutations in other genes, all of which result in decreased hepcidin levels or function and increased intestinal iron uptake. It is characterized by accumulation of iron in the liver, pancreas, and other tissues.
• Wilson disease is caused by loss-of-function mutations in the metal ion transporter ATP7B, which results in accumulation of copper in the liver, brain (particularly basal ganglia), and eyes (Kayser-Fleischer rings).
• Wilson disease effects on the liver are protean, showing changes ranging from acute massive hepatic necrosis, to fatty liver disease, to chronic hepatitis and cirrhosis.
• α1AT deficiency is a disease in which mutations in α1AT lead to its misfolding, causing liver toxicity and a functional deficit of α1AT in the plasma. This deficiency places affected individuals at high risk for emphysema, particularly smokers, due to the unopposed effects of proteases released from neutrophils.
Cholestatic Syndromes
Hepatic bile serves two major functions: (1) the emulsification of dietary fat in the lumen of the gut through the detergent action of bile salts, and (2) the elimination of bilirubin, excess cholesterol, xenobiotics, and other waste products that are insufficiently water-soluble to be excreted into urine. Processes that interfere with excretion of bile lead to jaundice and icterus due to retention of bilirubin, and to cholestasis (discussed later).
Jaundice may occur in settings of increase bilirubin production (e.g., extravascular red cell hemolysis), hepatocyte dysfunction (e.g., hepatitis), or obstruction of the flow of bile (e.g., an impacted gallstone), any of which can disturb the equilibrium between bilirubin production and clearance (summarized in Table 16.4). The metabolism of bilirubin by the liver occurs in four steps: uptake from the circulation; intracellular storage; conjugation with glucuronic acid; and biliary excretion. These are discussed next.
Table 16.4
Major Causes of Jaundice
| Predominantly Unconjugated Hyperbilirubinemia |
| Excess Production of Bilirubin |
| Reduced Hepatic Uptake |
| Impaired Bilirubin Conjugation |
| Predominantly Conjugated Hyperbilirubinemia |
| Decreased Hepatocellular Excretion |
| Impaired Intrahepatic or Extrahepatic Bile Flow |

Bilirubin and Bile Formation
Bilirubin is the end product of heme degradation (Fig. 16.22). Approximately 85% of daily production (0.2–0.3 gm) is derived from the breakdown of senescent red cells by macrophages in the spleen, liver, and bone marrow. The remainder is derived from the turnover of hepatic heme or hemoproteins (e.g., the P-450 cytochromes) and from destruction of red cell precursors in the bone marrow (Chapter 12). Whatever the source, intracellular heme oxygenase oxidizes heme to biliverdin (step 1 in Fig. 16.22), which is immediately reduced to bilirubin by biliverdin reductase. Bilirubin thus formed is released and binds to serum albumin (step 2), which is critical since bilirubin is virtually insoluble in aqueous solutions at physiologic pH and also highly toxic to tissues. Albumin carries bilirubin to the liver, where bilirubin is taken up into hepatocytes (step 3) and conjugated with one or two molecules of glucuronic acid by bilirubin uridine diphosphate (UDP)–glucuronyltransferase (UGT1A1, step 4) in the endoplasmic reticulum. Water-soluble, nontoxic bilirubin glucuronides are then excreted into the bile. Most bilirubin glucuronides are deconjugated in the gut lumen by bacterial β-glucuronidases and degraded to colorless urobilinogens (step 5). The urobilinogens and the residue of intact pigment are largely excreted in feces. Approximately 20% of the urobilinogens formed are reabsorbed in the ileum and colon, returned to the liver, and reexcreted into bile. A small amount of reabsorbed urobilinogen is excreted in the urine.

Two-thirds of the organic materials in bile are bile salts, which are formed by the conjugation of bile acids with taurine or glycine. Bile acids, the major catabolic products of cholesterol, are a family of water-soluble sterols with carboxylated side chains. The primary human bile acids are cholic acid and chenodeoxycholic acid. Bile acids are highly effective detergents. Their primary physiologic role is to solubilize water-insoluble lipids secreted by hepatocytes into bile, and also to solubilize dietary lipids in the gut lumen. Ninety-five percent of secreted bile acids, conjugated or unconjugated, are reabsorbed from the gut lumen and recirculate to the liver (enterohepatic circulation), thus helping to maintain a large endogenous pool of bile acids for digestive and excretory purposes.
Pathophysiology of Jaundice
Both unconjugated bilirubin and conjugated bilirubin (bilirubin glucuronides) may accumulate systemically. As discussed earlier, unconjugated bilirubin is virtually insoluble and tightly bound to albumin. As a result, it cannot be excreted in the urine, even when blood levels are high. Normally, a very small amount of unconjugated bilirubin is present as a free anion in plasma. If unconjugated bilirubin levels rise, this unbound fraction may diffuse into tissues, particularly the brain in infants, and produce toxic injury. The unbound plasma fraction increases in severe hemolytic disease or when protein-binding drugs displace bilirubin from albumin. Hence, hemolytic disease of the newborn (erythroblastosis fetalis) may lead to accumulation of unconjugated bilirubin in the brain, which can cause severe neurologic damage, referred to as kernicterus (Chapter 7). In contrast, conjugated bilirubin is water-soluble, nontoxic, and only loosely bound to albumin. Because of its solubility and weak association with albumin, excess conjugated bilirubin in plasma can be excreted in urine.
Serum bilirubin levels in the normal adult vary between 0.3 and 1.2 mg/dL. Jaundice becomes evident when the serum bilirubin levels rise above 2 to 2.5 mg/dL; levels as high as 30 to 40 mg/dL can occur with severe disease. Causes of conjugated and unconjugated hyperbilirubinemia differ, and so measurement of both forms is of value in evaluating a patient with jaundice.
Defects in Hepatocellular Bilirubin Metabolism
Neonatal Jaundice
Because the hepatic machinery for conjugating and excreting bilirubin does not fully mature until about 2 weeks of age, almost every newborn develops transient and mild unconjugated hyperbilirubinemia, termed neonatal jaundice or physiologic jaundice of the newborn. This may be exacerbated by breastfeeding, due to the action of bilirubin-deconjugating enzymes in breast milk. Nevertheless, sustained jaundice in the newborn is abnormal and is discussed later in the “Neonatal Cholestasis” section.
Hereditary Hyperbilirubinemias
Jaundice also may result from inborn errors of metabolism, including the following:
• Gilbert syndrome is a common (7% of the population) inherited condition that manifests as fluctuating unconjugated hyperbilirubinemia of variable severity. The primary cause is mildly decreased hepatic levels of glucuronosyltransferase attributed to a mutation in the encoding gene, UGT1A1; polymorphisms in the gene may play a role in the variable expression of this disorder. Gilbert syndrome is not associated with any morbidity. By contrast, severe glucuronosyltransferase deficiency causes a rare disorder called Crigler-Najjar Syndrome Type 1 that is fatal in infancy.
• Dubin-Johnson syndrome results from an autosomal recessive defect in the transport protein responsible for hepatocellular excretion of bilirubin glucuronides across the canalicular membrane. Affected individuals exhibit conjugated hyperbilirubinemia. Other than having a darkly pigmented liver (from polymerized epinephrine metabolites, not bilirubin) and hepatomegaly, patients are normal.
Cholestasis
Cholestasis is a condition caused by extrahepatic or intrahepatic obstruction of bile channels or by defects in hepatocyte bile secretion. Patients may have jaundice, pruritus, skin xanthomas (focal accumulation of cholesterol), or symptoms related to intestinal malabsorption, including nutritional deficiencies of the fat-soluble vitamins A, D, or K. A characteristic laboratory finding is elevated serum alkaline phosphatase and γ-glutamyl transpeptidase (GGT), enzymes that are present on the apical membranes of hepatocytes and cholangiocytes.
Morphology
The morphologic features of cholestasis depend on its severity, duration, and underlying cause. Common to both obstructive and nonobstructive cholestasis is the accumulation of bile pigment within the hepatic parenchyma (Fig. 16.23). Elongated green-brown plugs of bile are visible in dilated bile canaliculi. Rupture of canaliculi leads to extravasation of bile, which is quickly phagocytosed by Kupffer cells. Droplets of bile pigment also accumulate within hepatocytes, which can take on a fine, foamy appearance referred to as feathery degeneration. Occasional apoptotic hepatocytes also may be seen.

Bile Duct Obstruction and Ascending Cholangitis
The most common cause of bile duct obstruction in adults is extrahepatic cholelithiasis (gallstones, discussed later), followed by malignant obstructions, and postsurgical strictures. Obstructive conditions in children include biliary atresia, cystic fibrosis, choledochal cysts (a cystic anomaly of the extrahepatic biliary tree), and syndromes in which there are insufficient intrahepatic bile ducts (paucity of bile duct syndromes). The initial morphologic features of cholestasis have been discussed and are entirely reversible with correction of the obstruction. Prolonged obstruction can lead to biliary cirrhosis, discussed later.
Ascending cholangitis, secondary bacterial infection of the biliary tree, may complicate duct obstruction. Enteric organisms such as coliforms and enterococci are common culprits. Cholangitis usually presents with fever, chills, abdominal pain, and jaundice. The most severe form of cholangitis is suppurative cholangitis, in which purulent bile fills and distends bile ducts. Since sepsis rather than cholestasis tends to dominate this potentially grave process, prompt diagnostic evaluation and intervention are imperative.
Since extrahepatic biliary obstruction is frequently amenable to surgical treatment, correct and prompt diagnosis is imperative. In contrast, cholestasis due to diseases of the intrahepatic biliary tree or hepatocellular secretory failure (collectively termed intrahepatic cholestasis) is not benefited by surgery (short of transplantation), and the patient's condition may be worsened by an operative procedure. It is thus important to establish the underlying basis for jaundice and cholestasis.
Morphology
Acute biliary obstruction, either intrahepatic or extrahepatic, causes distention of upstream bile ducts, which often become dilated. In addition, ductular reactions appear at the portal-parenchymal interface along with stromal edema and neutrophils. The hallmark of superimposed infection (ascending cholangitis) is the influx of periductular neutrophils into the bile duct epithelium and lumen (Fig. 16.24).

Left uncorrected, the inflammation and ductular reactions resulting from chronic biliary obstruction initiate periportal fibrosis, eventually generating secondary or obstructive biliary cirrhosis (Fig. 16.25). Cholestatic features in the parenchyma may be prominent. These take the form of extensive feathery degeneration of periportal hepatocytes, a type of cytoplasmic swelling often associated with Mallory-Denk bodies and bile infarcts caused by the detergent effects of extravasated bile.

Neonatal Cholestasis
Prolonged conjugated hyperbilirubinemia in the neonate, termed neonatal cholestasis (as opposed to the already discussed neonatal jaundice) affects approximately 1 in 2500 live births. The major conditions causing it are (1) cholangiopathies, primarily biliary atresia (discussed later), and (2) a variety of disorders causing conjugated hyperbilirubinemia in the neonate, collectively referred to as neonatal hepatitis.
Neonatal hepatitis is not a specific entity, nor does it necessarily have an inflammatory basis. Rather it is an indication to conduct a diligent search for recognizable toxic, metabolic, and infectious liver diseases, as greater than 85% of cases have identifiable causes.
Differentiation of biliary atresia from nonobstructive neonatal cholestasis is very important, since definitive treatment of biliary atresia requires surgical intervention (Kasai procedure), whereas surgery may adversely affect a child with other disorders. Fortunately, discrimination can be made on the basis of clinical data in about 90% of cases. In 10% of cases, liver biopsy may be necessary to distinguish neonatal hepatitis from an identifiable cholangiopathy. Affected infants have jaundice, dark urine, light or acholic stools, and hepatomegaly. Variable degrees of hepatic synthetic dysfunction may be identified, such as hypoprothrombinemia.
Morphology
The morphologic features of neonatal hepatitis (Fig. 16.26) include striking giant-cell transformation of hepatocytes, associated with lobular disarray, focal liver cell apoptosis and prominent hepatocellular and canalicular cholestasis. In some cases, this parenchymal pattern of injury also is accompanied by ductular reaction and fibrosis of portal tracts.

Biliary Atresia
Biliary atresia is defined as a complete or partial obstruction of the extrahepatic biliary tree that occurs within the first 3 months of life. It underlies approximately one-third of cases of neonatal cholestasis and is the single most frequent cause of death from liver disease in early childhood. Approximately 50% to 60% of children referred for liver transplantation have biliary atresia.
Pathogenesis
Two major forms of biliary atresia are recognized; these are based on the presumed timing of luminal obliteration.
• The fetal form accounts for as many as 20% of cases and is commonly associated with other developmental anomalies involving the thoracic and abdominal organs, including malrotation of abdominal viscera, interrupted inferior vena cava, polysplenia, and congenital heart disease.
• Much more common is the perinatal form of biliary atresia, in which an apparently normally developed biliary tree is injured and obstructed following birth. The etiology of perinatal biliary atresia is unknown; viral infection and toxic exposures are considered prime suspects.
Morphology
The salient features of biliary atresia include inflammation and fibrosing stricture of the hepatic or common bile ducts; in some individuals, periductular inflammation also extends into the intrahepatic bile ducts, leading to progressive destruction of the intrahepatic biliary tree as well. When biliary atresia is unrecognized or uncorrected, cirrhosis develops within 3 to 6 months of birth.
There is considerable variability in the pattern of biliary atresia. When the disease is limited to the common duct or right and/or left hepatic bile ducts with patent intrahepatic branches, the disease is surgically correctable (Kasai procedure). Unfortunately, in 90% of patients the obstruction also involves bile ducts at or above the porta hepatis. These cases are not correctable, since there are no patent bile ducts amenable to surgical anastomosis.
Clinical Features
Infants with biliary atresia present with neonatal cholestasis, but exhibit normal birth weight and postnatal weight gain. There is a slight female predominance. Initially stools are normal, but they become acholic as the disease progresses. Ascending cholangitis and/or intrahepatic progression of the disease may impede attempts at surgical resection of the obstruction and bypass of the biliary tree. Transplantation of a donor liver and its accompanying bile ducts is the primary hope for saving these young patients. Without surgical intervention, death usually occurs within 2 years of birth.
Autoimmune Cholangiopathies
Autoimmune cholangiopathies comprise two distinct immunologically-mediated disorders that involve intrahepatic bile ducts: primary biliary cholangitis and primary sclerosing cholangitis. The salient features of these are listed in Table 16.5.
Table 16.5
Main Features of Primary Biliary Cholangitis and Primary Sclerosing Cholangitis
| Parameter | Primary Biliary Cholangitis | Primary Sclerosing Cholangitis |
| Age | Median age 50 years | Median age 30 years |
| Gender | 90% female | 70% male |
| Clinical course | Progressive | Unpredictable, but progressive |
| Associated conditions | Sjögren syndrome (70%) | Inflammatory bowel disease (70%) |
| Scleroderma (5%) | Pancreatitis (≤25%) | |
| Thyroid disease (20%) | Idiopathic fibrosing diseases (retroperitoneal fibrosis) | |
| Serology | 95% AMA-positive | 0%–5% AMA-positive (low titer) |
| 20% ANA-positive | 6% ANA-positive | |
| 40% ANCA-positive | 65% ANCA-positive | |
| Radiology | Normal | Strictures and beading of large bile ducts; pruning of smaller ducts |
| Duct lesion | Florid duct lesions and loss of small ducts only | Inflammatory destruction of extrahepatic and large intrahepatic ducts; fibrotic obliteration of medium and small intrahepatic ducts |

Primary Biliary Cholangitis
Primary biliary cholangitis (PBC) is an autoimmune disease whose primary feature is nonsuppurative, inflammatory destruction of small- and medium-sized intrahepatic bile ducts. Large intrahepatic ducts and the extrahepatic biliary tree are not involved. Previously, this disease was known as primary biliary cirrhosis, but most patients do not progress to this stage, and the name primary biliary cholangitis is now preferred.
PBC is primarily a disease of middle-age women, with a female-to-male ratio of 6:1. Its peak incidence is between 40 and 50 years of age. The disease is most prevalent in Northern European countries (England and Scotland) and the Northern United States (Minnesota), where the prevalence is as high as 400 per 1 million cases. Recent increases in incidence and prevalence along with geographic clustering suggest that both environmental and genetic factors are important in its pathogenesis. Family members of PBC patients have an increased risk for developing the disease.
Pathogenesis
PBC is thought to be an autoimmune disorder, but as with other autoimmune diseases the triggers that initiate PBC are unknown. Anti-mitochondrial antibodies are the most characteristic finding in PBC. T cells specific for certain mitochondrial enzymes are another feature of the disease, supporting the notion of an immune-mediated process. Other findings suggestive of altered immunity include aberrant expression of MHC class II molecules on bile duct epithelial cells, accumulation of autoreactive T cells around bile ducts, and the frequent presence of other autoantibodies against nuclear pore proteins centromeric proteins, and other cellular components.
Morphology
Interlobular bile ducts are actively destroyed by lymphoplasmacytic inflammation with or without granulomas (the florid duct lesion) (Fig. 16.27). Some biopsy specimens, however, do not have active lesions and only show the absence of bile ducts in portal tracts. The disease is quite patchy in distribution; it is common to see a single bile duct under immune attack in one level of a biopsy specimen, while other nearby ducts, are unaffected. Ductular reactions follow on this duct injury, and these in turn participate in the development of portal-portal septal fibrosis.


In the absence of treatment, the disease follows one of two paths to end-stage disease. In the first, most classic pathway, there is increasingly widespread duct loss, slowly leading to established cirrhosis and eventually to profound cholestasis. Alternatively, some patients eventually developed prominent portal hypertension rather than severe cholestasis. Fortunately, both of these outcomes are now rarely seen.
Clinical Features
Most patients are diagnosed while asymptomatic following a workup triggered by the identification of an elevated serum alkaline phosphatase level or severe itching. Hypercholesterolemia is common. Anti-mitochondrial antibodies are present in 90% to 95% of patients. They are highly characteristic of PBC, although other autoantibodies may be seen in a small number of cases. The disease is confirmed by liver biopsy, which is considered diagnostic if a florid duct lesion is present. When symptoms appear, their onset is insidious, with patients typically complaining of slowly increasing fatigue and pruritus.
In recent years, early treatment with oral ursodeoxycholic acid has dramatically improved outcomes by slowing disease progression. Its mechanism of action remains unclear, but is presumably related to the ability of ursodeoxycholate to enter the bile acid pool and alter the biochemical composition of bile.
With time, even with treatment, secondary features may emerge, including skin hyperpigmentation, xanthelasmas, steatorrhea, and vitamin D malabsorption–related osteomalacia and/or osteoporosis. Individuals with PBC may also have extrahepatic manifestations of autoimmunity, including the sicca complex of dry eyes and mouth (Sjögren syndrome), systemic sclerosis, thyroiditis, rheumatoid arthritis, Raynaud phenomenon, and celiac disease. Liver transplantation is the best treatment for individuals with advanced liver disease.
Primary Sclerosing Cholangitis
Primary sclerosing cholangitis (PSC) is characterized by inflammation and obliterative fibrosis of intrahepatic and extrahepatic bile ducts, leading to dilation of preserved segments. Irregular biliary strictures and dilations cause the characteristic “beading” of the intrahepatic and extrahepatic biliary tree seen by MRI. Inflammatory bowel disease (Chapter 15), most commonly ulcerative colitis, coexists in approximately 70% of individuals with PSC. Conversely, the prevalence of PSC in individuals with ulcerative colitis is about 4%. Like inflammatory bowel disease, PSC tends to occur in the third through fifth decades of life and has a 2 : 1 male predominance (see Table 16.5).
Pathogenesis
Several features of PSC suggest immunologically mediated injury to bile ducts. T cells in the periductal stroma, the presence of autoantibodies, an association with HLA-B8 and other MHC alleles, and clinical linkage to ulcerative colitis all support an underlying, immunologic process. First-degree relatives of patients with PSC are at increased risk for developing the disease, suggesting that genetic factors also contribute.
In one model, it is proposed that T cells activated in the damaged mucosa of patients with ulcerative colitis migrate to the liver, where they recognize a cross-reacting bile duct antigen and initiate an autoimmune assault on bile ducts. Autoantibody profiles in PSC are not as characteristic as in PBC, but atypical perinuclear anti-neutrophil cytoplasmic antibodies (pANCA) that recognize a nuclear envelope protein are found in up to 80% of patients. The pathogenic relationship of pANCA to PSC is unknown.
Morphology
Morphologic changes differ between large ducts (intrahepatic and extrahepatic) and smaller intrahepatic ducts. Large duct inflammation resembles that seen in ulcerative colitis, taking the form of neutrophils infiltrating into the epithelium superimposed on a chronic inflammatory background. Inflamed areas develop strictures as scarring narrows the lumen. The smaller ducts, however, often have little inflammation and show a striking circumferential, “onion skin” fibrosis around an atrophic duct lumen (Fig. 16.29), which eventually is obliterated, leaving a “tombstone” scar. Because the likelihood of sampling small-duct lesions on a random needle biopsy is small, diagnosis depends on radiologic imaging of the extrahepatic and large intrahepatic ducts. As the disease progresses, the liver becomes markedly cholestatic, culminating in cirrhosis. Biliary intraepithelial neoplasia often appear in the setting of chronic inflammation and cholangiocarcinoma develops in up to 7% of patients, usually with a fatal outcome.

Clinical Features
Patients may come to attention only because of persistent elevation of serum alkaline phosphatase, particularly in those with ulcerative colitis who are being routinely screened. Alternatively, progressive fatigue, pruritus, and jaundice may develop. Acute bouts of ascending cholangitis may also signal the presence or progression of PSC. Chronic pancreatitis and chronic cholecystitis due to involvement of the pancreatic ducts and gallbladder are also seen. In some patients, sclerosing cholangitis is associated with autoimmune pancreatitis. In such cases PSC may be one manifestation of IgG4 related chronic disease (Chapter 5).
PSC follows a protracted course of 5 to 17 years, and severely afflicted patients have symptoms typical of chronic cholestatic liver disease, including steatorrhea. Unlike with PBC, there is no satisfactory medical treatment. A variety of immunosuppressive agents have been tried, but none has been proven to alter the disease course. Endoscopic dilation with sphincterotomy or stenting is used to relieve obstruction. Liver transplantation is the only definitive treatment for individuals with end-stage liver disease.
Summary
Cholestatic Diseases
• Cholestasis occurs with impaired excretion of bile, leading to jaundice and accumulation of bile pigment in the hepatic parenchyma. Causes include mechanical or inflammatory obstruction or destruction of the bile ducts or metabolic defects in hepatocyte bile secretion.
• Large bile duct obstruction is most commonly associated with gallstones and malignancies involving the head of the pancreas. Chronic obstruction can lead to cirrhosis.
• Neonatal cholestasis is not a specific entity; it is variously associated with cholangiopathies such as biliary atresia and a variety of disorders causing conjugated hyperbilirubinemia in the neonate, collectively referred to as neonatal hepatitis.
• Primary biliary cholangitis is an autoimmune disease with progressive, inflammatory, often granulomatous, destruction of small to medium intrahepatic bile ducts. It most often occurs in middle-age women and is associated with anti-mitochondrial antibodies and often with other autoimmune diseases, such as Sjögren syndrome and Hashimoto thyroiditis.
• Primary sclerosing cholangitis is an autoimmune disease with progressive inflammatory and sclerosing destruction of intrahepatic and extrahepatic bile ducts of all sizes. Diagnosis is made by radiologic imaging of the biliary tree. It most often occurs in younger men and has a strong association with inflammatory bowel disease, particularly ulcerative colitis.
Circulatory Disorders
Hepatic circulatory disorders can be grouped according to whether the disorder leads to abnormalities in the inflow, flow-through, or outflow of blood (Fig. 16.30).
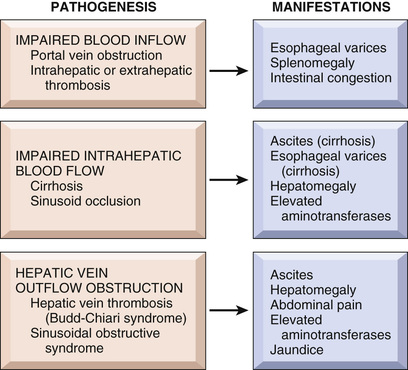
Impaired Blood Flow Into the Liver
Hepatic Artery Compromise
Liver infarcts are rare, thanks to the double blood supply to the liver. Nonetheless, thrombosis or obstruction of an intrahepatic branch of the hepatic artery by embolism (Fig. 16.31), neoplasia, or an inflammatory process such as polyarteritis nodosa (Chapter 10) may produce an infarct, which may either be pale or hemorrhagic if suffused with blood from the portal circulation. Blockage of the main hepatic artery may not produce ischemic necrosis of the organ, particularly if the liver is otherwise normal, as retrograde arterial flow through accessory vessels and the portal venous supply is usually sufficient to sustain the liver parenchyma.
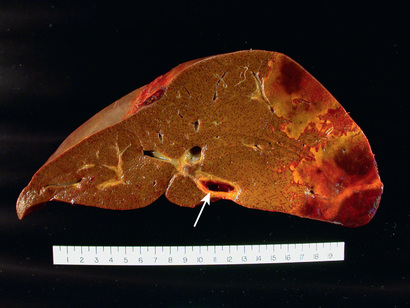
Portal Vein Obstruction and Thrombosis
Blockage of the extrahepatic portal vein may cause only vague symptoms or may be a catastrophic and potentially lethal event; most cases fall somewhere in between. Occlusive disease of the portal vein or its major radicles typically produces abdominal pain and other manifestations of portal hypertension, principally esophageal varices that are prone to rupture. Ascites is not common (because the block is presinusoidal), but, when present, is often massive and intractable.
Extrahepatic portal vein obstruction may be idiopathic (approximately one-third of cases) or may arise from a number of conditions. Some of the most common settings for development of extrahepatic portal vein obstruction include the following:
• Cirrhosis, which is associated with portal vein thrombosis in 25% of patients, some of whom have other risk factors as well
• Hypercoagulable states, including myeloproliferative neoplasms such as polycythemia vera (Chapter 12), inherited thrombophilias such as factor V Leiden (Chapter 4), and miscellaneous hypercoagulable conditions such as paroxysmal nocturnal hemoglobinuria and anti-phospholipid antibody syndrome
• Inflammatory processes involving the splenic vein or portal vein, such as pancreatitis and intraabdominal sepsis
Obstruction of intrahepatic portal vein radicles may be caused by acute thrombosis. The thrombosis does not cause ischemic infarction but instead results in a sharply demarcated area of red-blue discoloration called infarct of Zahn. There is no necrosis, only severe hepatocellular atrophy and marked congestion of distended sinusoids. The most common cause of small portal vein branch obstruction is schistosomiasis; the eggs of the parasites lodge in and obstruct the smallest portal vein branches. The other disorders producing this pattern of injury are now collectively referred to as obliterative portal venopathy, which often presents as noncirrhotic portal hypertension. Causes of obliterative portal venopathy are not well understood. It occurs in both untreated and treated HIV disease, and may in some instances be a complication of anti-retroviral therapy.
Impaired Blood Flow Through the Liver
The most common intrahepatic cause of blood flow obstruction is cirrhosis, as discussed earlier. In addition, physical occlusion of the sinusoids occurs in sickle cell disease, disseminated intravascular coagulation, eclampsia, and intrasinusoidal metastasis of solid tumors. If severe and unabated, all of these disorders may produce sufficient obstruction of blood flow to cause massive necrosis of hepatocytes and fulminant hepatic failure.
Hepatic Venous Outflow Obstruction
Hepatic Vein Thrombosis
Occlusive events can occur in any caliber of hepatic vein branches. If it occurs in the smallest intrahepatic branches, it produces sinusoidal obstruction syndrome (formally known as veno-occlusive disease). A rare, but well-known cause of this syndrome is consumption of pyrrolizidine alkaloid–containing Jamaican bush tea, but it now occurs primarily following allogeneic hematopoietic stem cell transplantation, usually within the first 3 weeks, or in cancer patients receiving chemotherapy, in whom it can have as high as 30% mortality.
The obstruction of two or more major hepatic veins produces liver enlargement, pain, and ascites, a condition known as Budd-Chiari syndrome. Obstruction of a single main hepatic vein by thrombosis is clinically silent. Hepatic damage is the consequence of increased intrahepatic blood pressure. Hepatic vein thrombosis is associated with the same hypercoagulable states as portal vein thrombosis as well as intraabdominal cancers, particularly HCC. As is often the case in those afflicted with various thrombotic disorders, Budd-Chiari syndrome often occurs in patients with several risk factors, such as pregnancy or oral contraceptive use combined with an underlying thrombophilic disorder.
Morphology
In Budd-Chiari syndrome, the liver is swollen and red-purple and has a tense capsule (Fig. 16.32). There may be areas of hemorrhagic collapse alternating with areas of preserved or regenerating parenchyma, depending on which small and large hepatic veins are obstructed. Microscopically, the affected hepatic parenchyma reveals severe centrilobular congestion and necrosis. Centrilobular fibrosis develops in instances in which the thrombosis is more slowly developing. The major veins may contain fresh occlusive thrombi or, in chronic cases, organized adherent thrombi.

The mortality of untreated acute hepatic vein thrombosis is high. The condition is rare, and treatments are largely empiric. They include anti-coagulation to prevent clot propagation; angioplasty to restore the patency of occluded veins; thrombolysis; and creation of portovenous shunts, using either invasive radiologic approaches or surgery, in order to decompress the liver. The chronic form is far less lethal, and more than two-thirds of patients are alive after 5 years.
Passive Congestion and Centrilobular Necrosis
These hepatic manifestations of systemic circulatory compromise—passive congestion and centrilobular necrosis—are considered together because they represent a morphologic continuum. Both changes are commonly seen at autopsy, as there is an element of preterminal circulatory failure in virtually every nontraumatic death.
Morphology
Right-sided cardiac decompensation leads to passive congestion of the liver. The liver is slightly enlarged, tense, and cyanotic, with rounded edges. Microscopically there is congestion of centrilobular sinusoids. With time, centrilobular hepatocytes become atrophic, resulting in markedly attenuated liver cell plates. Left-sided cardiac failure or shock may lead to hepatic hypoperfusion and hypoxia, causing ischemic coagulative necrosis of hepatocytes in the central region of the lobule (centrilobular necrosis).
The combination of hypoperfusion and retrograde congestion acts synergistically to cause centrilobular hemorrhagic necrosis. The liver takes on a variegated mottled appearance, reflecting hemorrhage and necrosis in the centrilobular regions (Fig. 16.33A). This finding is known as nutmeg liver due to its resemblance to the cut surface of a nutmeg. There typically is a sharp demarcation between viable periportal and necrotic or atrophic pericentral regions that are suffused with blood (see Fig. 16.33B). Uncommonly, with sustained chronic severe congestive heart failure, centrilobular fibrosis (cardiac sclerosis) or even cirrhosis develops.

Summary
Circulatory Disorders
• Circulatory disorders of the liver can be caused by impaired blood inflow, defects in intrahepatic blood flow, and obstruction of blood outflow.
• Portal vein obstruction by intrahepatic or extrahepatic thrombosis may cause portal hypertension, esophageal varices, and ascites.
• The most common cause of impaired intrahepatic blood flow is cirrhosis.
• Obstructions of blood outflow include hepatic vein thrombosis (Budd-Chiari syndrome) and sinusoidal obstruction syndrome, previously known as veno-occlusive disease.
Nodules and Tumors
Hepatic masses come to attention for a variety of reasons. They may generate epigastric fullness and discomfort or be detected by routine physical examination or radiographic studies for other indications. Hepatic masses include nodular hyperplasias and true neoplasms.
Focal Nodular Hyperplasia
Solitary or multiple hyperplastic hepatocellular nodules that may develop in the noncirrhotic liver are called focal nodular hyperplasias. These lesions arise from local alterations in hepatic parenchymal blood supply, such as arterio-venous malformations or inflammatory or posttraumatic obliteration of portal vein radicles and compensatory augmentation of arterial blood supply.
Morphology
Focal nodular hyperplasia appears as a well-demarcated, poorly encapsulated nodule ranging up to many centimeters in diameter (Fig. 16.34A). It presents as a mass lesion in an otherwise normal liver, most frequently in young to middle-age adults. Typically, there is a central gray-white, depressed stellate scar from which fibrous septa radiate to the periphery (see Fig. 16.34B).

Microscopically, the central scar contains large abnormal vessels and ductular reactions along the spokes of scar. A vascular lesion is probably the initiating insult, as it is believed that hypoperfused parenchyma collapses to produce the septa, whereas hyperperfused regions undergo hyperplasia. The hyperplastic regions are composed of normal hepatocytes separated by thickened sinusoidal plates.
Benign Neoplasms
Cavernous hemangiomas are the most common benign liver tumors (Chapter 10). The chief clinical significance of cavernous hemangiomas is that they must be distinguished radiographically or intraoperatively from metastatic tumors.
Hepatocellular Adenomas
Benign neoplasms developing from hepatocytes are called hepatocellular adenomas (Fig. 16.35). They may be detected incidentally as a hepatic mass on abdominal imaging or when they cause symptoms. The most common symptom is pain, which may be caused by pressure placed on the liver capsule by the expanding mass or hemorrhagic necrosis of the tumor as it outstrips its blood supply. Hepatocellular adenomas occasionally rupture, an event that may lead to life-threatening intraabdominal bleeding.
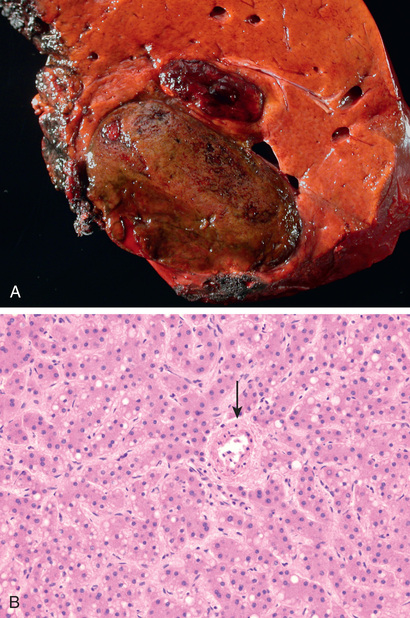
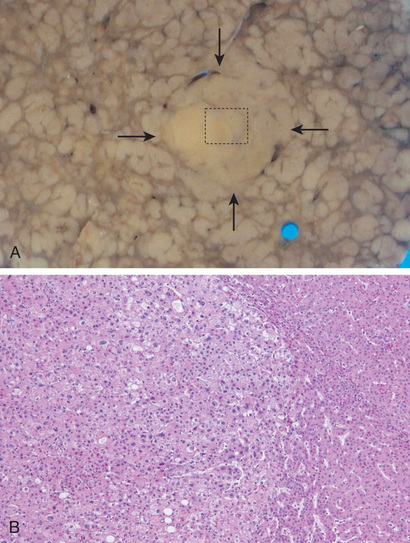
Hepatic adenomas can be subclassified molecularly into tumors at low, intermediate, and high risk for malignant transformation. Sex hormone exposure (e.g., oral contraceptive pills, anabolic steroids) markedly increases the frequency of all types of hepatic adenoma, and cessation of exposure to sex hormones often—but not always—leads to tumor regression, clearly linking sex hormones to the growth and survival of tumor cells in some cases.
Malignant Neoplasms
Malignant tumors occurring in the liver can be primary or metastatic. The latter are far more common. Our discussion here is focused on primary hepatic tumors. Most primary liver cancers arise from hepatocytes and are termed hepatocellular carcinoma (HCC). Much less common are cancers of bile duct origin, cholangiocarcinomas. Other types of primary liver cancers, such as hepatoblastoma (a childhood hepatocellular tumor) and angiosarcoma, are too rare to merit further discussion.
Hepatocellular Carcinoma (HCC)
Globally, HCC, also erroneously known as hepatoma, accounts for approximately 5.4% of all cancers, but its incidence varies widely in different parts of the world. More than 85% of cases occur in countries with high rates of chronic HBV infection. The incidence of HCC is highest in Asia (southeast China, Korea, Taiwan) and sub-Saharan Africa, areas in which HBV is transmitted vertically and, as already discussed, the carrier state starts in infancy. Moreover, many of these populations are exposed to aflatoxin, which when combined with HBV infection increases the risk for HCC dramatically. The peak incidence of HCC in these areas is between 20 and 40 years of age and, in almost 50% of cases, the tumor appears in the absence of cirrhosis.
In Western counties, the incidence of HCC is rapidly rising, largely owing to the increased prevalence of hepatitis C. The number of new HCC cases tripled in the United States in recent decades, but its incidence is still 8-fold to 30-fold lower than in some Asian countries. It is hoped that new, effective treatments for hepatitis C infection will stem the rising tide of HCC in the United States. In Western populations, HCC rarely manifests before 60 years of age, and in almost 90% of cases the malignancy emerges after cirrhosis becomes established. There is a pronounced male predominance throughout the world, about 3 : 1 in low-incidence areas and as high as 8 : 1 in high-incidence areas.
Pathogenesis
Chronic liver diseases are the most common setting for emergence of HCC. While usually identified in a background of cirrhosis, cirrhosis is not required for hepatocarcinogenesis. Rather, progression to cirrhosis and to hepatocellular cancer take place in parallel over many years to decades.
The most important underlying factors in hepatocarcinogenesis are viral infections (HBV, HCV) and toxic injuries (aflatoxin, alcohol). Thus, where HBV and HCV are endemic, there is a very high incidence of HCC. Coinfection further increases risk. Aflatoxin is a mycotoxin produced by Aspergillus species that contaminates staple food crops in Africa and Asia. Aflatoxin metabolites are present in the urine of individuals who consume these foods, as are aflatoxin-albumin adducts in serum. These biomarkers identify populations at risk and have helped to confirm the importance of aflatoxin in hepatocarcinogenesis. As discussed earlier, aflatoxin synergizes with HBV (and perhaps also with HCV) to increase risk further.
Other HCC risk factors all share the ability to cause chronic liver injury associated with varying degrees of inflammation. These factors include:
• Alcohol consumption, which synergistically increases risk with HBV, HCV, and possibly even cigarette smoking.
• Inherited disorders, particularly hereditary hemochromatosis and α1AT deficiency, and to a lesser degree Wilson disease
• Metabolic syndrome and its attendant obesity, diabetes mellitus, and NAFLD, all of which increase the risk for HCC.
As with all cancers, HCC is induced by acquired driver mutations in oncogenes and tumor suppressor genes. No single, universal sequence of molecular or genetic alterations leads to emergence of HCC. Gain of function mutations in beta-catenin and loss of function mutations in p53 are the two most common driver mutations. Beta-catenin mutations are identified in up to 40% of HCCs. These tumors are more likely to be unrelated to HBV and to demonstrate genomic instability. Inactivation of TP53 is present in up to 60% of HCCs. These tumors are strongly associated with exposure to aflatoxin, which appears in many cases to be directly responsible for the causative TP53 mutations.
HCC often appears to arise from premalignant precursors lesions. Hepatic adenoma has already been discussed, some of which carry beta-catenin–activating mutations. Chronic liver disease is associated with cellular dysplasias called large-cell change and small-cell change. These may be found at any stage of chronic liver disease, before or after development of cirrhosis, and serve to indicate which patients need more aggressive cancer surveillance. Dysplastic nodules are usually found in cirrhosis, either radiologically or in resected specimens (including explants). Low-grade dysplastic nodules may or may not undergo transformation to higher-grade lesions, but they indicate a higher risk for HCC. High-grade dysplastic nodules are probably the most important precursor of HCC in viral hepatitis and alcoholic liver disease. Overt HCC is often found in high-grade dysplastic nodules in biopsy or resection specimens.
Morphology
HCC may appear grossly as (1) a unifocal (usually large) mass (Fig. 16.37); (2) multifocal, widely distributed nodules of variable size; or (3) a diffusely infiltrative cancer, permeating widely and sometimes involving the entire liver. Sometimes HCCs arise within dysplastic nodules (Fig. 16.37), eventually overgrowing these precursor lesions. Intrahepatic metastases by either vascular invasion or direct extension become more likely once tumors reach 3 cm in size. These metastases are usually small, satellite tumor nodules around a larger primary mass. Vascular invasion is also the most likely route for extrahepatic metastasis, especially by the hepatic venous system, usually only in advanced cases. Occasionally, long, snakelike masses of tumor invade the portal vein (causing portal hypertension) or inferior vena cava; in the latter instance, the tumor may extend all the way up into the right ventricle. Lymph node metastases are less common.

HCCs range from well differentiated to highly anaplastic lesions. Well-differentiated HCCs are composed of cells that look like normal hepatocytes and grow as thick trabeculae (recapitulating liver cell plates) or in pseudoglandular patterns that recapitulate poorly formed, ectatic bile canaliculi (see Fig. 16.37).
Clinical Features
The clinical manifestations of HCC are varied and in Western populations are often masked by symptoms related to underlying cirrhosis or chronic hepatitis. In areas of high incidence, such as tropical Africa where aflatoxin exposure is common, patients usually have no clinical history of liver disease (although cirrhosis may be detected at autopsy). In both populations, most patients have ill-defined upper-abdominal pain, malaise, fatigue, weight loss, and sometimes awareness of an abdominal mass or abdominal fullness. Jaundice, fever, and gastrointestinal or esophageal variceal bleeding are inconstant findings.
Laboratory studies may provide clues but are rarely conclusive. Elevated serum levels of α-fetoprotein are found in 50% of individuals with advanced HCC, but this is neither a sensitive nor specific marker for premalignant or early well-differentiated cancers. Better tests for detection of small tumors are imaging studies, such as ultrasonography, computed tomography, and magnetic resonance imaging. Increasing arterialization during the development and progression of HCC can be identified by imaging and is so characteristic that its detection can be diagnostic, precluding the need for tissue biopsy.
The natural history of HCC involves the progressive enlargement of the primary mass until it disturbs hepatic function or metastasizes, most commonly to the lungs. Death usually occurs from (1) cachexia, (2) gastrointestinal or esophageal variceal bleeding, (3) liver failure with hepatic coma, or rarely (4) rupture of the tumor with fatal hemorrhage. The 5-year survival of large tumors is dismal, and the majority of patients die within 2 years of diagnosis.
With implementation of screening procedures and advances in imaging, the detection of HCCs less than 2 cm in diameter has increased in countries where such facilities are available. These small tumors can be removed surgically or ablated (e.g., through embolization, microwave radiation, or freezing) with good outcomes. If relatively small HCCs arise in the setting of advanced stage (cirrhotic) chronic liver disease, liver transplantation is a better option and may be curative. Radiofrequency ablation and chemoembolization are used for local control of unresectable tumors. The kinase inhibitor sorafenib can prolong the life of individuals with advanced-stage HCC.
Cholangiocarcinoma
Cholangiocarcinoma (CCA), the second most common primary malignant tumor of the liver after HCC, arises from intrahepatic and extrahepatic bile ducts. It accounts for 3% of gastrointestinal cancers in the United States, where there are approximately 2000 to 3000 new cases each year. However, in some regions of southeast Asia such as northeastern Thailand, Laos, and Cambodia where infestation with liver flukes is endemic, cholangiocarcinoma is much more common, occurring at rates 30 to 40 times higher than in areas of Asia without liver fluke infestation.
All risk factors for cholangiocarcinoma cause chronic inflammation and cholestasis, which presumably promote occurrence of somatic mutations or epigenetic alterations in cholangiocytes. The risk factors include infestation by liver flukes (particularly Opisthorchis and Clonorchis species), chronic inflammatory disease of the large bile ducts (such as primary sclerosing cholangitis), hepatolithiasis, and fibropolycystic liver disease. As with HCC, rates of cholangiocarcinoma also are elevated in patients with hepatitis B and C and NAFLD.
Morphology
Extrahepatic cholangiocarcinomas are generally small lesions at the time of diagnosis, as they cause obstruction of the biliary tract early in their course. Most tumors appear as firm, gray nodules within the bile duct wall; some may be diffusely infiltrative, while others are papillary or polypoid. Intrahepatic cholangiocarcinomas occur in noncirrhotic livers (Fig. 16.38A) and may track along the intrahepatic portal tract system or produce a single massive tumor.

Cholangiocarcinomas are typical mucin-producing adenocarcinomas. Most are well to moderately differentiated, growing as glandular/tubular structures lined by malignant epithelial cells (see Fig. 16.38B). They typically incite marked desmoplasia. Lymphovascular invasion and perineural invasion are both common and often lead to extensive intrahepatic and extrahepatic metastases.
Summary
Liver Tumors
• The liver is the most common site of metastatic cancers from primary tumors of the colon, lung, and breast.
• Hepatocellular adenomas are benign tumors of hepatocytes. Most can be subclassified on the basis of molecular changes with varying degrees of malignant potential. They are associated with use of oral contraceptives and androgens.
• The two main types of malignant tumors are hepatocellular carcinomas and cholangiocarcinomas; HCCs are much more common.
• HCC is a common tumor in regions of Asia and Africa, and its incidence is increasing in the United States.
• The main etiologic agents for HCC are hepatitis B and C, alcoholic cirrhosis, hemochromatosis, and exposure to aflatoxins. In the Western population, about 90% of HCCs develop in cirrhotic livers; in Asia, almost 50% of cases develop in noncirrhotic livers.
• The chronic inflammation and cellular regeneration associated with viral hepatitis are predisposing factors for the development of carcinomas.
• HCC may be unifocal or multifocal, tends to invade blood vessels, and recapitulates normal liver architecture to varying degrees.
• Cholangiocarcinoma is a tumor of intrahepatic or extrahepatic bile ducts that is relatively common in areas where liver flukes, such as Opisthorchis and Clonorchis species, are endemic.
Gallbladder
Gallstone Disease
Gallstones afflict 10% to 20% of adults residing in Western countries in the Northern Hemisphere, 20% to 40% in Latin American countries, and only 3% to 4% in Asian countries. In the United States, about 1 million new cases of gallstones are diagnosed annually, and two-thirds of individuals so affected undergo surgery, resulting in the removal of as much as 25 to 50 million tons of stones per year! There are two main types of gallstones: cholesterol stones, containing crystalline cholesterol monohydrate (80% of stones in Western countries), and pigment stones, made of bilirubin calcium salts.
Pathogenesis
Bile formation is the only significant pathway for elimination of excess cholesterol from the body, either as free cholesterol or as bile salts. Cholesterol is rendered water-soluble by aggregation with bile salts and lecithins. When cholesterol concentrations exceed the solubilizing capacity of bile (supersaturation), cholesterol can no longer remain dispersed and crystallizes out of solution. Cholesterol gallstone formation is enhanced by hypomobility of the gallbladder (stasis), which promotes nucleation, and by mucus hypersecretion, with consequent trapping of the crystals, thereby enhancing their aggregation into stones.
Pigment stones form when the bile contains a high concentration of unconjugated bilirubin in the biliary tree, as may occur in patients with chronic extravascular red cell hemolysis or with certain infections of the biliary tract, such as liver flukes. The precipitates are primarily insoluble calcium bilirubinate salts.
Prevalence and Risk Factors
The major risk factors for gallstones are listed in Table 16.6. In up to 80% of individuals with gallstones, the only identifiable risk factors are age and gender. Some elaboration on these risk factors follows:
• Age and gender. The prevalence of gallstones increases throughout life. In the United States, less than 5% to 6% of the population younger than 40 years of age has stones, in contrast with 25% to 30% of those older than 80 years of age. The prevalence in women of all ages is about twice as high as in men.
• Ethnic and geographic. Cholesterol gallstone prevalence approaches 50% to 75% in certain Native American populations (Pima, Hopi, and Navajo), whereas pigment stones are rare. The high prevalence in these populations seems to be related to biliary cholesterol hypersecretion.
• Heredity. In addition to ethnicity, a positive family history imparts increased risk, as do a variety of inborn errors of metabolism, such as those associated with impaired bile salt synthesis and secretion.
• Environment. Estrogens increase hepatic cholesterol uptake and synthesis, leading to excess biliary secretion of cholesterol. These effects explain the increased risk for gallstone disease with oral contraceptive use and with pregnancy. Obesity, rapid weight loss, and treatment with the hypocholesterolemic agent clofibrate also are strongly associated with increased biliary cholesterol secretion and risk for gallstone disease.
• Acquired disorders. Any condition in which gallbladder motility is reduced predisposes to gallstones, such as pregnancy, rapid weight loss, and spinal cord injury. In most cases, however, gallbladder hypomotility is present without obvious cause.
Morphology
Cholesterol stones arise exclusively in the gallbladder and consist of 50% to 100% cholesterol. Pure cholesterol stones are pale yellow; increasing proportions of calcium carbonate, phosphates, and bilirubin impart gray-white to black discoloration (Fig. 16.39). They are ovoid and firm; they can occur singly, but most often there are several, with faceted surfaces resulting from their apposition. Most cholesterol stones are radiolucent, although as many as 20% may contain sufficient calcium carbonate to be radiopaque.
Pigment stones may arise anywhere in the biliary tree and are classified into black and brown stones. In general, black pigment stones are found in sterile gallbladder bile, while brown stones are found in infected intrahepatic or extrahepatic ducts. The stones contain calcium salts of unconjugated bilirubin and lesser amounts of other calcium salts, mucin glycoproteins, and cholesterol. Black stones are usually small, numerous, and fragile to the touch (Fig. 16.40). Brown stones tend to be single or few in number and to have a soft, greasy, soaplike consistency owing to the presence of fatty acid salts released from biliary lecithins by bacterial phospholipases. Because of calcium carbonates and phosphates, 50% to 75% of black stones are radiopaque. Brown stones, which contain calcium soaps, are radiolucent.

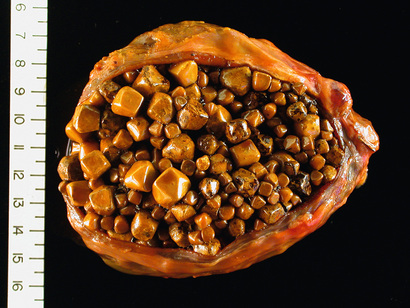

Clinical Features
Gallstones may be present for decades without causing any symptoms, and 70% to 80% of individuals with gallstones remain asymptomatic throughout life. In an unfortunate minority, however, the symptoms are striking. There is usually right upper-quadrant or epigastric pain, often excruciating, which may be constant or, less commonly, spasmodic. Such “biliary” pain is caused by gallbladder or biliary tree obstruction or by inflammation of the gallbladder itself. More severe complications include empyema, perforation, fistulas, inflammation of the biliary tree, obstructive cholestasis and pancreatitis. The larger the stone, the less likely it is to enter the cystic or common ducts to produce obstruction; thus, the very small stones, or “gravel,” are dangerous. Occasionally a large stone may erode directly into an adjacent loop of small bowel, generating intestinal obstruction (gallstone ileus).
Cholecystitis
Inflammation of the gallbladder may be acute, chronic, or acute superimposed on chronic, and almost always occurs in association with gallstones. In the United States, cholecystitis is one of the most common indications for abdominal surgery. Its epidemiologic distribution closely parallels that of gallstones.
Acute Calculous Cholecystitis
Acute inflammation of a gallbladder that contains stones is termed acute calculous cholecystitis and is precipitated in 90% of cases by obstruction of the gallbladder neck or cystic duct. It is the most common major complication of gallstones and the most frequent indication for emergency cholecystectomy. Manifestations of obstruction may appear with remarkable suddenness and constitute a surgical emergency. In some cases, however, symptoms may be mild and resolve without intervention.
Acute calculous cholecystitis initially results from chemical irritation and inflammation of the gallbladder wall due to obstruction of bile outflow. Gallbladder injury in the setting of bile obstruction stems from several sources, including: phospholipases derived from the mucosa hydrolyze biliary lecithin to lysolecithin, which is toxic to the mucosa; the normally protective glycoprotein mucous layer is disrupted, exposing the mucosal epithelium to the detergent action of bile salts.; prostaglandins released within the wall of the distended gallbladder enhance mucosal and mural inflammation; and distention and increased intraluminal pressure may compromise blood flow to the mucosa. All of these effects occur in the absence of bacterial infection, which may be superimposed later.
Acute Acalculous Cholecystitis
Between 5% and 12% of gallbladders removed for acute cholecystitis contain no gallstones. Most cases occur in seriously ill patients. Some of the most common predisposing insults are as follows:
Other contributing factors include dehydration, gallbladder stasis and sludging, vascular compromise, and bacterial contamination.
Chronic Cholecystitis
Chronic cholecystitis may be the sequel to repeated bouts of acute cholecystitis, but in most instances it develops without any antecedent history of acute attacks. Like acute cholecystitis, it is almost always associated with gallstones. However, gallstones do not seem to be an essential part of the initiation of inflammation or the development of pain, because chronic acalculous cholecystitis causes symptoms and morphologic alterations similar to those seen in the calculous form. Rather, supersaturation of bile appears to predispose to both chronic inflammation and, in most instances, stone formation. Microorganisms, usually E. coli and enterococci, can be cultured from the bile in only about one-third of cases. Unlike acute calculous cholecystitis, obstruction of gallbladder outflow by stones is not requisite in chronic cholecystitis. Most gallbladders removed at elective surgery for gallstones show features of chronic cholecystitis, making it likely that biliary symptoms emerge after long-term coexistence of gallstones and low-grade inflammation.
Morphology
In acute cholecystitis, the gallbladder usually is enlarged and tense, and has a bright red or blotchy, violaceous color, the latter imparted by subserosal hemorrhages. The serosa frequently is covered by a fibrinous or, in severe cases, a fibrinopurulent exudate. In 90% of cases, stones are present, often obstructing the neck of the gallbladder or the cystic duct. The gallbladder lumen is filled with cloudy or turbid bile that may contain fibrin, blood, and pus. When the contained exudate is mostly pus, the condition is referred to as empyema of the gallbladder. In mild cases, the gallbladder wall is thickened, edematous, and hyperemic. In more severe cases, the gallbladder wall is green-black and necrotic—a condition termed gangrenous cholecystitis. On histologic examination, the inflammatory reactions are not distinctive and consist of some combination of the usual patterns of acute inflammation (i.e., edema, leukocytic infiltration, vascular congestion, abscess formation, gangrenous necrosis).
The morphologic changes in chronic cholecystitis are extremely variable and sometimes subtle. The mere presence of stones within the gallbladder, even in the absence of acute inflammation, often is taken as sufficient justification for a diagnosis. The gallbladder may be contracted, of normal size, or enlarged. Mucosal ulcerations are infrequent; the submucosa and subserosa often are thickened from fibrosis. In the absence of superimposed acute cholecystitis, collections of lymphocytes in the wall are the only sign of inflammation (Fig. 16.41A). Outpouchings of mucosal epithelium through the wall of the gallbladder (Rokitansky-Aschoff sinuses) may be quite prominent (see Fig. 16.41B).

Clinical Features
Acute calculous cholecystitis presents with biliary pain that lasts for more than 6 hours. The pain is severe, usually steady, upper abdominal in location, and often radiates to the right shoulder. Fever, nausea, leukocytosis, and prostration are classic; the presence of conjugated hyperbilirubinemia suggests obstruction of the common bile duct. The right subcostal region is markedly tender and rigid as a result of spasm of the abdominal muscles; occasionally a tender, distended gallbladder can be palpated. Mild attacks usually subside spontaneously over 1 to 10 days; however, recurrence is common. Approximately 25% of symptomatic patients are sufficiently ill to require surgical intervention.
The diagnosis of acute cholecystitis usually is based on the detection of gallstones by ultrasonography, typically accompanied by evidence of a thickened gallbladder wall. Attention to this disorder is important because of the potential for the following serious complications:
• Bacterial superinfection leading to cholangitis or sepsis
• Gallbladder perforation and local abscess formation
• Gallbladder rupture leading to diffuse peritonitis
• Biliary enteric (cholecystenteric) fistula, with drainage of bile into adjacent organs, entry of air and bacteria into the biliary tree, and potentially gallstone-induced intestinal obstruction (ileus)
• Aggravation of preexisting medical illness, with cardiac, pulmonary, renal, or liver decompensation
Symptoms arising from acute acalculous cholecystitis usually are obscured by another serious medical or surgical condition, which sets the stage for the development of cholecystitis. The diagnosis therefore rests on a high index of suspicion. Chronic cholecystitis lacks the striking manifestations of the acute forms and is usually characterized by recurrent attacks of steady epigastric or right upper-quadrant pain. Nausea, vomiting, and intolerance for fatty foods are frequent accompaniments. Chronic cholecystitis is a pathologic diagnosis based on the examination of the resected gallbladder. Beyond signs and symptoms mentioned above, its principal importance may lie in the association of gallstones and chronic inflammation with carcinoma of the gallbladder (discussed next).
Carcinoma of the Gallbladder
Carcinoma of the gallbladder is the most common malignancy of the extrahepatic biliary tract. It is slightly more common in women and occurs most frequently in the seventh decade of life. The incidence in the United States is 1 in 50,000. Only rarely is it discovered at a resectable stage, and the mean 5-year survival rate has remained unchanged at about 5% to 12% over the past many years. The most important risk factor associated with gallbladder carcinoma is gallstones (cholelithiasis), which are present in 95% of cases. Presumably, gallbladders containing stones or infectious agents develop cancer as a result of chronic inflammation, a known enabler of malignancy in several organs (Chapter 6). Carcinogenic derivatives of bile acids also are suspected to play a role. Primary sclerosing cholangitis is also a risk factor.
Morphology
Carcinomas of the gallbladder show two patterns of growth: infiltrating and exophytic. The infiltrating pattern is more common and usually appears as a poorly defined area of diffuse wall thickening and induration. The exophytic pattern grows into the lumen as an irregular, cauliflower mass, but at the same time invades the underlying wall (Fig. 16.42). Most carcinomas of the gallbladder are adenocarcinomas. About 5% are squamous cell carcinomas or have adenosquamous differentiation.

Clinical Features
Preoperative diagnosis of carcinoma of the gallbladder is the exception rather than the rule, occurring in fewer than 20% of patients. Presenting symptoms are insidious and typically indistinguishable from those associated with cholelithiasis: abdominal pain, jaundice, anorexia, nausea and vomiting. There is no satisfactory treatment of gall bladder cancer due to the advanced stage at the time of diagnosis. Only 10% are diagnosed at a stage that is early enough to attempt curative surgery.
Summary
Gallbladder Diseases
• Gallbladder diseases include cholelithiasis and acute and chronic cholecystitis, and gall bladder cancer.
• Gallstone formation is a common condition in Western countries. The great majority of the gallstones are cholesterol stones. Pigmented stones containing bilirubin and calcium are most common in Asian countries, due to the higher incidence of chronic hemolytic disorders and liver fluke infestations in these locales.
• Risk factors for the development of cholesterol stones are advancing age, female gender, estrogen use, obesity, and heredity.
• Cholecystitis almost always occurs in association with cholelithiasis, although in about 10% of cases it occurs in the absence of gallstones.
• Acute calculous cholecystitis is the most common reason for emergency cholecystectomy.
• Gall bladder cancer is almost always associated with gall stones. Because of the advanced stage at diagnosis, it has a very poor prognosis.