Genetics of Pain
Jeffrey S. Mogil, Mitchell B. Max† and Inna Belfer
Individual Differences in Pain
The robust interindividual variability observed in sensitivity to pain, the propensity for chronic pain conditions to develop, and the response to analgesic manipulations is a scientific and clinical challenge. Laboratory studies have documented impressive individual differences in thresholds, tolerance, and pain scale ratings of noxious experimental stimuli (Lanier 1943, Kim et al 2004, Nielsen et al 2005). Impressive correlations between pain ratings and simultaneously obtained measures of cortical activation with functional magnetic resonance imaging or positron emission tomography (Zubieta et al 2001, Coghill et al 2003) suggest that these individual differences truly reflect variable perception and not variable scale utilization. Epidemiological studies of chronic pain syndromes known to develop after specific traumatic or infectious insults consistently reveal that chronic pain will eventually develop in only a small fraction of patients (Veldman et al 1993, Andersen et al 1995, Cluff and Rowbotham 1998). Thus, these insults are not themselves sufficient to produce chronic pain; some factor intrinsic to the recipient of the injury is also to blame. The explanation is very likely to be a classic example of genetic–environmental interaction: both the injury and some innate or acquired propensity are necessary. A potentially useful fact is that individual differences in laboratory pain sensitivity are predictive of clinical pain severity and response to treatment (Edwards 2005).
Impressive interindividual variability has been documented in response to experimental and clinical administration of analgesics as well, including opioids (Lasagna and Beecher 1954, Wolff et al 1965, Levine et al 1981, Aubrun et al 2003), placebo (Wolff et al 1965, Levine et al 1981, Amanzio et al 2001), and non-steroidal anti-inflammatory drugs (Wolff et al 1965, Day et al 1988, Walker et al 1997).
An understanding of the basis of such individual differences would have a number of benefits for therapeutic development efforts, prediction of risk, and individual tailoring of existing therapies. Obviously, environmental factors are important in the final explanation, both in terms of their unique additive effects and in their interaction with pain-relevant genetic factors. This review, however, aims to describe only techniques and recent progress in pain and the genetics of analgesia in laboratory animals and humans.
Pain Genetics in Laboratory Animals
In theory, many genetic approaches are as tractable in humans as in laboratory animals. For example, identification of trait-relevant genes by linkage mapping or association study can easily be performed in humans. Why then bother studying pain genetics in laboratory rats and mice? One reason is the statistical power and experimental simplicity afforded by controlled crosses, which obviously can be attempted only in laboratory animals. Starting with inbred progenitors (see below), a single set of grandparental breeders can easily beget hundreds of the genetically segregating F2 hybrid or backcross mice needed for mapping complex traits by linkage. Simultaneous evaluation of the many existing inbred strains themselves can now also be used for gene identification by haplotype mapping. By contrast, even the largest human pedigrees are modest in size and genetically complicated (with more than two possible alleles at each locus) in comparison. Single-gene association studies in humans remain poorly replicable (Lohmueller et al 2003), and the powerful method of full-genome, single nucleotide polymorphism (SNP)–based linkage disequilibrium mapping (i.e., genome-wide association studies [GWASs]) has not quite yet been applied to pain, at least not in the peer-reviewed literature.
If we are to use laboratory animals to study pain genetics, we must ask to what extent are data derived from such animals relevant to our species. There is no particular reason to believe that specific DNA sequence variants common in Mus musculus, say, would be preserved in human beings, the lineages of the two species having diverged more than 100 million years ago. However, there is ample reason to expect that genes relevant to pain in mice would also be relevant to pain in humans. On completion of the human and mouse genome sequences, Mural and colleagues (2002) performed a detailed analysis of the genes on mouse chromosome 16. Of the 731 predicted genes, only 14 (1.9%) had no homologues in the human genome. The evolutionarily ancient role of nociception suggests that mammals ought to be quite similar in genetic and physiological processing of this particular biological trait, even though humans probably have a more sophisticated cognitive and emotional dimension to their pain.
Although any number of rat strain differences of relevance to pain have been demonstrated (see Xu and Wiesenfeld-Hallin 2004, Fecho et al 2005, Fecho and Valtschanoff 2006, LaCroix-Fralish et al 2005, Paulson et al 2005, Avsaroglu et al 2007, Terner et al 2006, Herradon et al 2007, Rode et al 2007), the mouse has clearly become the default laboratory subject for genetic research. One reason is the much larger number of commercially available inbred mouse strains (Beck et al 2000) than inbred rat strains. Another is the relatively more dense genetic map in the mouse, which led to completed sequencing of the mouse genome (Waterston et al 2002) several years before that of the rat (Gibbs et al 2004). Probably the most important reason for ascendance of the mouse in pain genetics research was the unique ability in this species to create transgenic knockout mice (although rat knockouts can now be constructed; Jacob et al 2010), which caused intense interest in the normative responses of the inbred mouse strains in which such mutations are created.
Approaches to Pain Genetics in the Mouse
There are two related problems to which pain genetics is addressed. One type of pain genetics asks the question, which genes are relevant to pain? This is merely a restatement of the fundamental question of reductionist pain biology, which aims to define the molecular “players”: the proteins playing a role in mediation of pain perception and modulation. There are considerable advantages to identifying pain-relevant proteins by identifying the genes coding for them in that genetic approaches offer simplicity (there are only ~30,000 genes but many more proteins) and unparalleled specificity (typically, DNA or mRNA sequences of >15 nucleotides unambiguously define one and only one gene). The second type of pain genetics asks the question, of the pain-relevant genes, which are responsible for individual differences in sensitivity to pain and analgesia and for differential susceptibility and/or expression of painful pathologies? This second question represents classic, mendelian genetics as applied to pain traits. Along the way to answering these questions, one can adopt either a “bottom-up” (genotype → phenotype) or a “top-down” (phenotype → genotype) strategy (Fig. 10-1). With a bottom-up strategy, one focuses on a particular gene (usually identified via the known role of its protein product in pain) and studies the relationship between expression of that gene and some systems-level pain phenomenon. That is, one can measure or alter expression of the mRNA transcripts of individual genes to provide evidence of involvement of these genes—and thus their proteins—in some aspect of pain physiology. With a top-down strategy, one examines populations showing contrasting systems-level phenotypes (e.g., different strains, nerve-damaged versus intact animals) and tries to find the genes responsible for the differences. The responsible genes will produce either proteins with a change or deletion of an amino acid or acids in the populations being compared or proteins that are alternatively spliced to reflect different exons. In other cases, the gene variants might not affect protein structure at all but rather the basal expression levels of these proteins. The various methods in current use are discussed briefly; their respective advantages and disadvantages in the study of pain genetics have been addressed previously (Mogil and McCarson 2000).
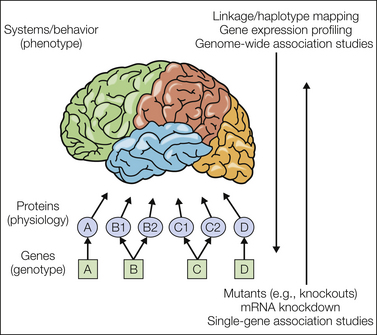
Figure 10-1 “Bottom-up” and “top-down” strategies in genetics.
Systems- or behavioral-level neural phenomena (traits or phenotypes, represented in the figure by the brain) are produced by the coordinated activity of proteins interacting with each other and with small molecules and ions (i.e., physiology). Proteins are produced by genes in either a one-to-one manner (as in genes A and D) or via alternative splicing in which one gene can produce multiple proteins (as in genes B and C). Not pictured are protein–protein interactions and protein–gene interactions in which proteins can affect the expression of their own gene or others. Environmental factors also produce protein-level changes, which can then affect gene expression, including via epigenetic regulation. Until recently, biology consisted of the study of only two of these three levels. Today, we are able to associate genotype with phenotype specifically without directly measuring or altering proteins. Two broad strategies can be used. In “bottom-up” approaches, genes are individually targeted and their expression is either reduced or ablated completely to study the resultant effect on the organism’s phenotype. In “top-down” approaches, contrasting phenotypes are compared, and genes whose expression and/or sequence also contrast are sought. In other words, one can use top-down strategies to find the genes relevant to the phenotype and use bottom-up strategies to study how the genotype affects the phenotype.
Transgenic Knockout Mice and Other Mutants
By far the most common bottom-up approach to pain genetics is the creation and testing of transgenic knockout mice. These are genetically engineered null mutants in which a single gene is effectively ablated via homologous recombination of embryonic stem cells with a transgenic targeting vector (see Capecchi 1989). Such knockout mice are compared with “wild-type” and heterozygous littermates, and if the genetic “lesion” results in an aberrant behavioral and/or biochemical pain phenotype (e.g., altered sensitivity on an algesiometric assay), this demonstration provides evidence, subject to certain caveats (see Lariviere et al 2001), that the gene is required for normal expression of the trait. A large number of knockout mice have been tested for nociceptive and analgesic sensitivity; we maintain a web database of findings from such experiments called the Pain Genes Database (LaCroix-Fralish et al 2007). As of this writing, the pain-related phenotypes of 322 mutants have been documented (see http://paingeneticslab.ca/ 4105/06_02_pain_genetics_database.asp). A comprehensive review of transgenic studies of pain is beyond the scope of this chapter, however, and findings using this technique will appear elsewhere in this volume. To give the reader a sense, Table 10-1 lists simply the number of genes currently demonstrated to affect pain, hypersensitivity after inflammatory and neuropathic injury, and morphine analgesia via the transgenic technique. It should also be noted that spontaneously occurring mutations (of coat color genes and genes causing obvious phenotypic abnormalities) have been and continue to be studied for their relevance to pain (see Mogil et al 1996b; also see Nissenbaum et al 2010 for review). Great sums of money have been spent on the deliberate induction, via chemical mutagenesis, of random point mutations combined with systematic screening for phenotypic alterations. Three public mutagenesis projects (Hrabe de Angelis et al 2000, Nolan et al 2000, Sayah et al 2000) included a nociceptive assay (hot plate test), but data describing only one pain-relevant mutant were ever published (Cook et al 2007). A genome-wide screen of Drosophila mutations recently identified a thermal avoidance gene in flies that appears to play a similar role in humans (Neely et al 2010) (see below).
Table 10-1
Number of Genes Whose Null Mutation Affects Pain, Morphine Analgesia, or Hypersensitivity States
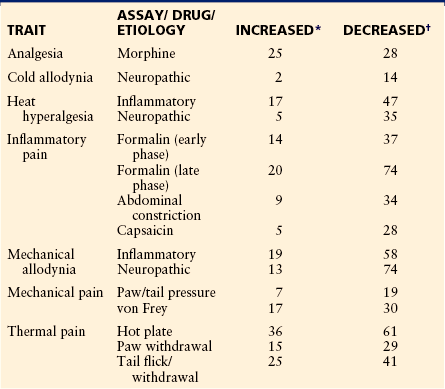
*Null mutant (knockout) mouse significantly more sensitive than wild-type mouse.
†Null mutant (knockout) mouse significantly less sensitive than wild-type mouse.
Data from Pain Genes Database. Available at http://paingeneticslab.ca/4105/06_02_pain_genetics_database.asp. Search performed October 17, 2010.
mRNA Knockdown
In the original version of this bottom-up approach, called antisense knockdown, an oligonucleotide is synthesized to be complementary (i.e., in an antisense orientation) to an mRNA transcript of the gene of interest and injected into a target tissue. By a mechanism involving steric hindrance in ribosomes and/or enzymatic degradation, the antisense oligonucleotide prevents (to some degree, hence “knockdown” instead of “knockout”) successful translation of the native mRNA into protein (Wahlestedt 1994). Although the antisense approach has several major advantages over knockout mice (see Mogil and McCarson 2000), it is technically difficult and has been used fairly sparsely in pain research. When successful, however, it has provided impressive demonstrations of the role of particular genes in pain (e.g., Young et al 1998, Porreca et al 1999, Joshi et al 2000).
More recently, mRNA knockdown in vitro and in vivo has been achieved with the use of small interfering RNA (siRNA; see Rohl and Kurreck 2006, Kim and Rossi 2007), which provides much greater translational block efficiency. The role of any number of genes in pain has been demonstrated via in vivo knockdown using siRNA, known as RNA interference (Dorn et al 2004; Tan et al 2005, 2010; Christoph et al 2006; Lin et al 2006; Altier et al 2007; Dong et al 2007; Gabra et al 2007; Kasama et al 2007; Kobayashi et al 2007; Deval et al 2008; Ohara et al 2008; Tumati et al 2008; Vit et al 2008; Xu et al 2008; Garraway et al 2009; LaCroix-Fralish et al 2009; Ndong et al 2009; Chen et al 2010; Chu et al 2010; Gao et al 2010; Kiguchi et al 2010; Kramer et al 2010; Lan et al 2010; Patte-Mensah et al 2010; Takahashi et al 2010; Wei et al 2010; Wu et al 2010).
Selective Breeding
Selective breeding is the oldest of the top-down approaches and has been performed in agriculture and animal husbandry for millennia. Also known as artificial selection, the technique as applied scientifically involves two-way breeding of a genetically heterogeneous stock of animals based on their response in regard to a trait of interest (see Crabbe et al 1990). Only extreme responders are used to beget the next generation of offspring, and the resultant “high” and “low” lines will start to diverge phenotypically for all heritable traits. The great advantage of the technique is that all trait-relevant genes will eventually become fixed in alternate allelic states in the two lines such that very robust phenotypic differences are demonstrated. Of greatest relevance to pain genetics are three selective breeding projects: the HA/LA rat lines selected for high and low autotomy after nerve transection (Devor and Raber 1990), the HA/LA mouse lines selected for high and low analgesia after forced swim stress (Panocka et al 1986), and the HAR/LAR mouse lines selected for high and low analgesic response to levorphanol (and in later generations, morphine) (Belknap et al 1983). Much has been learned from these lines (see Mogil et al 1996b); however, it remains difficult to identify the fixed genes, and this approach is rarely used today.
Inbred Strains and Linkage Mapping
The use of inbred strains for analysis of classic pain genetics is the dominant modern top-down strategy. Inbred strains are produced by repeated brother × sister mating over at least 20 generations, a breeding scheme that eliminates all genetic heterozygosity and renders individual members of the strain isogenic (i.e., clones) to all others, barring the very rare occurrence of new mutations (Beck et al 2000). Many existing inbred mouse strains are the descendents of European and East Asian “fancy mice” bred at the turn of the 20th century by Abbie Lathrop and William Castle, and excellent genealogical records are available in many cases (Beck et al 2000). A comparison of randomly selected inbred strains allows one to survey alleles of trait-relevant genes existing in the European and East Asian “fancy mouse” population of the early 1900s fixed into a homozygous state in every strain but into a different state in phenotypically contrasting strains. That is, each laboratory inbred strain is a different fixed “mosaic” of M. musculus domesticus, M. musculus castaneus, and M. musculus musculus alleles (Wade et al 2002). Inbred strains derived from stocks of Japanese wild mice (M. musculus molossinus) may have even greater genetic variability (Koide et al 2000). Because they are isogenic, inbred strains are tremendously useful for establishing the heritability of traits since within-strain variability is by definition of environmental origin and between-strain variability is very probably genetic. An inbred “strain survey” can identify the most robust pair-wise strain differences, and these extreme-responding strains are the ideal progenitors of linkage-mapping populations.
Linkage mapping, which when applied to complex traits is known as quantitative trait locus (QTL) mapping, searches for genomic regions that are co-inherited with trait variability in genetically segregating populations (e.g., F2 intercross, backcross, recombinant inbred strains) (see Lander and Schork 1994). As a practical matter, one correlates the phenotypic responses of, say, F2 hybrid mice with their inherited genotype at non-coding, polymorphic DNA markers (microsatellites or SNPs), and where such genetic “linkage” is established then infers the existence of a trait-relevant gene or genes in the genomic vicinity of the markers. By contrast to all other strategies in current use, QTL mapping allows one to identify genes containing the very polymorphisms that are the cause of the original strain difference since what is being examined is not gene expression but the DNA sequence itself. The disadvantage of QTL mapping is that identifying the broad genomic region linked to the trait (i.e., the QTL) is far easier than “positional cloning,” or identifying the gene and polymorphism responsible (Nadeau and Frankel 2000). Positional cloning has historically been attempted by creating advanced mapping populations such as congenic strains (Darvasi 1998), followed by cloning and sequencing.
A very useful advance is haplotype mapping, also known as in silico QTL mapping (Grupe et al 2001; also see Wang et al 2005). Because DNA variants are not inherited independently but rather in “blocks” known as haplotypes (see later) and since sequencing in multiple inbred mouse strains has already occurred, one can now simply search for statistical associations between strain means and haplotypic patterns. With a sufficiently high number of strains, significant linkages can be evinced, and the mapping resolution is extremely good (generally within the genes themselves or very close to them). Despite many false positives, this technique has been used profitably in mouse pain genetics in recent years.
Gene Expression Profiling with Microarrays
The most recently developed top-down strategy is microarray or “gene chip” expression profiling. The technique, developed by Pat Brown’s laboratory at Stanford (Schena et al 1995) and made widely available by commercial entities such as Affymetrix, Inc., allows quantification of mRNA expression in a massively parallel fashion (see Watson and Akil 1999). Other related techniques, such as differential display polymerase chain reaction (Reeves et al 1995) and massively parallel signature sequencing (Brenner et al 2000), allow the same sort of analysis. To run a profiling experiment, one isolates the total RNA from at least two contrasting tissue sources (e.g., dorsal root ganglia from a rat in neuropathic pain versus dorsal root ganglia from a control rat), and after a series of reverse transcription, amplification, and labeling steps, hybridizes the samples to a nylon or glass array. In the common Affymetrix version, the glass “chip” contains synthesized oligonucleotides corresponding to many thousands of known and unknown gene transcripts in catalogued locations. The fluorescent probes are excited by a laser and detected with a scanning confocal microscope; the intensity and color of fluorescent labeling at each location correspond to the absolute and relative (between groups) abundance of that particular gene’s mRNA in the tissue.
To date, approximately 30 microarray studies directly focused on chronic pain and using standard assays have been published (Bonilla et al 2002; Costigan et al 2002; Ko et al 2002; Kubo et al 2002; Sun et al 2002; Wang et al 2002; Xiao et al 2002; Valder et al 2003; Yang et al 2004a, 2007; Anseloni et al 2005; Barr et al 2005; Nesic et al 2005; Ren et al 2005; LaCroix-Fralish et al 2006; Rodriguez Parkitna et al 2006; Gao et al 2007; Geranton et al 2007; Griffin et al 2007; Levin et al 2008; Nishida et al 2008; Yukhananov and Kissin 2008; Kim et al 2009; Maratou et al 2009; Persson et al 2009; Vega-Avelaira et al 2009; Nissenbaum et al 2010; Okumura et al 2010; Zhang et al 2010). The results of all these investigations (also see Kim et al 2001, Sun et al 2002, Hammer et al 2010) illustrate the power of and problems with the approach. Depending on the criteria used to establish “significance,” these studies have identified up to hundreds of genes up- or down-regulated by the injury. On the plus side, many of these genes were not previously known to play any role whatsoever in chronic pain, and thus the heuristic potential is immense. Most investigators were surprised, for example, by the large number of “hits” among genes relevant to neuroinflammation (Costigan et al 2002, Wang et al 2002). Various forms of cluster analysis applied to microarray data may identify groups of genes that are co-regulated (Griffin et al 2003), an impossible task with other approaches. However, it is difficult to evaluate the relative importance (and cause-and-effect relationships) of these hundreds of genes and also very difficult to determine which of them are specifically related to pain versus the nerve injury itself. It is also difficult (without confirmation by other techniques) to separate true from false positives. We recently performed a meta-analysis of the first 20 published chronic pain–relevant microarray studies (LaCroix-Fralish et al 2011).
Findings from Animal Pain Genetics
As mentioned, the major aim of this chapter is to review the genetics of individual differences since the molecular genetics of pain is addressed elsewhere in this volume. Therefore, in this final section we discuss findings of two types: (1) general principles of pain genetics derived largely from selective breeding and inbred strain comparisons and (2) results of pain-relevant QTL and haplotype mapping studies. Interested readers are directed to other reviews as well (Mogil 1999, 2004; Mogil et al 1996b; LaCroix-Fralish and Mogul 2009).
How Heritable Is Pain in Rats and Mice?
Heritability (h2; see later for further discussion) is the proportion of trait variance attributable to genetic inheritance. Heritability can be estimated in laboratory animals either by using panels of inbred strains or by assessing the response to artificial selection (see Crabbe et al 1990). A large number of studies noting pain-relevant strain differences have compared the thermal nociception–sensitive and opioid analgesia–resistant C57BL/6 mouse strain with the nociception-resistant and opioid analgesia–sensitive DBA/2 strain (see Belknap and O’Toole 1991, Mogil et al 1996b). These strains do not represent the extreme responders among commonly available mouse strains, however, and in any case pair-wise comparisons do not provide much power to estimate heritability. In the only systematic inbred strain surveys to date, the heritability of response to 22 common algesiometric assays has been found to range from h2 = 0.30–0.76, with a median value of 0.46 (Mogil et al 1999a, Lariviere et al 2002). This range of heritability is highly similar to that obtained in large human twin studies of experimental pain published in recent years (Norbury et al 2007, Nielsen et al 2008). Using the same set of strains, the heritability of the efficacy of eight analgesics was found to be slightly lower, h2 = 0.12–0.45, with a median value of 0.29 (Wilson et al 2003a, 2003b). The three existing selective breeding studies of relevance to pain (see earlier) have yielded heritability estimates of h2 = 0.30 for autotomy (Raber and Devor 2002), h2 = 0.32 for levorphanol analgesia (Belknap et al 1983), and h2 > 0.32 for swim stress–induced analgesia (Marek et al 1993).
Genetic Correlations
One interesting use of both selected lines and inbred strains involves the investigation of genetic correlations (see Crabbe et al 1990). Most genes are known or thought to be pleiotropic, that is, affecting more than one trait. If selection or chance has fixed the alleles of a gene into a homozygous state, that fixation will probably affect multiple traits. Genetic correlation is demonstrated if lines selected for their performance on one trait are observed to differ on another or if the same distribution of phenotypic responses on two traits is observed among a panel of inbred strains.
As might be expected, the sensitivity of inbred strains and selected lines on certain nociceptive assays is genetically correlated with sensitivity on other nociceptive assays, but not all other nociceptive assays. Inbred mouse strains sensitive to tail flick test nociception are by and large the same mouse strains sensitive to paw withdrawal (Hargreaves’) test nociception. By contrast, a largely different set of strains are sensitive to formalin test nociception (Mogil et al 1999b). In our analysis of 22 nociceptive assays in a common set of 12 strains, we provided evidence using multivariate analysis of five “clusters” of assays: (1) thermal nociception, (2) ongoing nociception from chemical stimuli, (3) mechanical hypersensitivity, (4) thermal hypersensitivity, and (5) afferent-dependent (featuring initial ongoing nociception) thermal hypersensitivity (Lariviere et al 2002). Since variability measured on tests within a cluster should be mediated by common genes but variability on tests in different clusters by different genes, we believe that these clusters represent fundamental “types” of pain in the mouse. This is a nice example of how a genetic approach can address a non-genetic question. Species specificity may apply, however, since similar studies using rat strains have yielded somewhat different results (Shir et al 2001, Xu et al 2001).
Another remarkable genetic correlation pertains to analgesia of different modalities. Such correlation was hinted at by findings in selected lines, for example, when mice selected for high and low stress-induced analgesia were found to be high and low responders, selectively, to levorphanol, morphine, and the κ-opioid agonist U50,488 (Marek et al 1993, Mogil et al 1995). We recently evaluated the analgesic potency of five neurochemically distinct analgesics in 12 inbred strains: morphine; U50,488; clonidine; epibatidine; and the cannabinoid agonist WIN55,212-2 (Wilson et al 2003b). Although these drugs all activate descending pain-inhibitory mechanisms in the central nervous system, they bind to five distinct molecular sites. Thus, we were surprised to discover a remarkably high degree of genetic correlation among them (r = 0.33–0.68). This finding has important implications. First, it appears that a “master” analgesia variability gene or set of genes must exist, and it is highly unlikely that this gene is related to the binding site of each drug. If this is true, current attempts to explain variable analgesic drug action (i.e., pharmacogenetics) solely by searching for polymorphisms in the binding site gene will be incomplete. In a follow-up study, a similar genetic correlation was observed between acetylsalicylic acid and indomethacin analgesia, but acetaminophen analgesia appeared to be genetically distinct (Wilson et al 2003a).
A related genetic correlation is the repeatedly noted correlation between baseline nociceptive sensitivity and subsequent sensitivity to inhibition of the stimulus by analgesics (Mogil et al 1996a, Elmer et al 1998, Wilson et al 2003b). That is, mouse strains initially sensitive to nociception are also resistant to analgesia, and vice versa. This principle works within a number of nociceptive modalities and suggests that the “master” analgesia gene or genes referred to above may in fact be ones with a primary action on nociception, which may subsequently affect analgesia as well. Elmer and colleagues (1998) hypothesized that this can be explained in terms of genetic differences in effective stimulus intensity affecting fractional receptor occupancy of the analgesic.
Qualitative Strain Differences
The vast majority of documented strain differences of relevance to pain are quantitative in nature, differences in degree rather than in kind. In a few intriguing cases, however, qualitative strain differences have been uncovered, thus suggesting that different genotypes might be processing pain via different mechanisms. For example, we and others have noted that mediation of stress-induced analgesia by either opioid (naloxone-reversible) or non-opioid mechanisms is strain dependent (Urca et al 1985, Mogil and Belknap 1997), and the involvement of N-methyl-D-aspartate (NMDA) receptors in κ-opioid analgesia appears to be strain dependent as well (unpublished data). Rady and Fujimoto (1999) demonstrated that heroin produces analgesia via μ-, κ-, or δ-opioid receptors, depending on the mouse strain in question. There even exists evidence of genetic effects on the anatomy of the pain-relevant neurocircuitry. For example, Sprague-Dawley rats from Harlan Sprague-Dawley, Inc., feature bilateral innervation of the dorsal horn by noradrenergic locus coeruleus neurons, but a different substrain of Sprague-Dawley rats from the now defunct Sasco, Inc., feature unilateral innervation of the ventral horn (Clark and Proudfit 1992). A recent study has demonstrated strain differences in rat and mouse sciatic nerve anatomy, including even the number of lumbar vertebrae (Rigaud et al 2008). Finally, we have recently observed differences among mouse strains in apparent dorsal root ganglion size that are so large that the ganglia are visible to the naked eye (unpublished data).
Quantitative Trait Locus and Haplotype Mapping Studies
A number of pain-related traits have been subjected to QTL and/or haplotype mapping, three nociceptive modalities (thermal nociception, formalin test, autotomy following denervation) (Mogil et al 1997a, 2005, 2006; Seltzer et al 2001; Devor et al 2005, 2007; Wilson et al 2008; Nissenbaum et al 2008, 2010; Smith et al 2008; LaCroix-Fralish et al 2009; Fortin et al 2010; Li et al 2010), multiple analgesic modalities (morphine; stress-induced analgesia; U50,488; nitrous oxide; WIN55,212-2; clonidine) (Quock et al 1996, Bergeson et al 2001, Mogil et al 2003, Smith et al 2008), and morphine hyperalgesia (Liang et al 2006a, 2006b). The results are summarized in Table 10-2. In a number of cases, results from mouse QTL/haplotype mapping have been replicated successfully in humans as well (either in the same study or later by other groups) (Mogil et al 1997a, 2005; Chu et al 2009; Nissenbaum et al 2010), thus attesting to the usefulness of mouse genetics.
Table 10-2
Quantitative Trait Loci for Pain-Related Traits
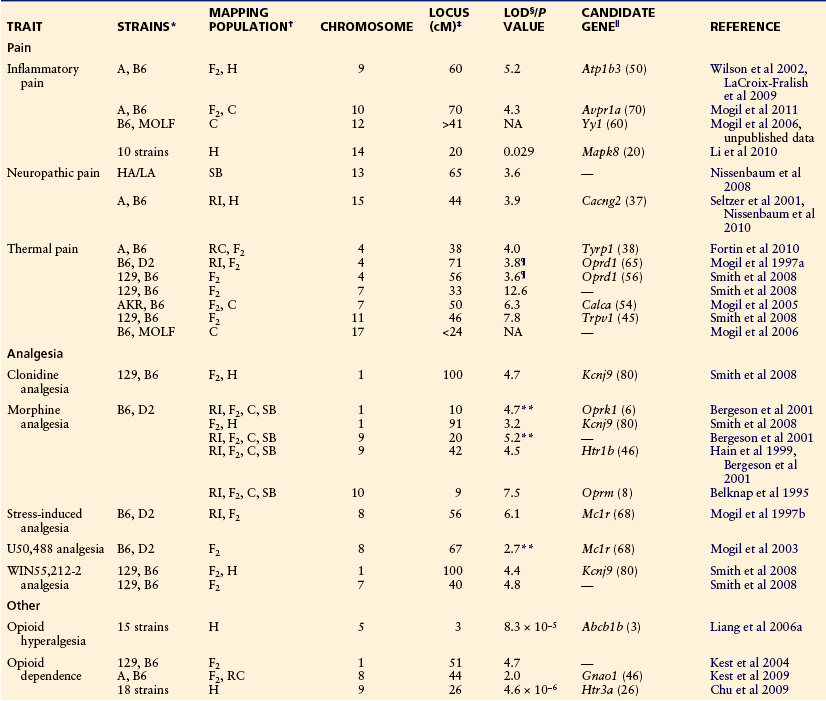
*129, 129P3/J; A, A/J; AKR, AKR/J; B6, C57BL/6J; D2, DBA/2J; HA/LA, selectively bred “high autotomy”/“low autotomy” rat strains; MOLF, MOLF/EiJ.
†Mapping populations used (in chronological order in each study): C, congenic strains in which a chromosomal segment (or segments) from one strain are bred onto the genetic background of another; F2, F2 hybrid (intercross); H, haplotype analysis; RC, recombinant congenic strain panel consisting of congenic strains from an F2 intercross; RI, recombinant inbred strain panel consisting of re-inbred strains from an F2 intercross; SB, short-term marker-assisted selective breeding.
‡Location in centimorgans (cM; approximately 1 million base pairs) from the proximal end of the chromosome of the peak statistical evidence for genetic linkage. The 95% confidence intervals in this type of study are generally very large, however, and average more than 10 cM. For linkages identified with only congenic strains, peaks are not relevant, only ranges.
§Logarithm of the odds (LOD) score for linkage of morphine analgesia to the defined genomic region. The conservative threshold for “significant” linkage is 4.3, but all listed LOD scores are significant by permutation analysis. NA, not applicable in the case of linkages obtained with congenics; NS, not significant but strong trends toward significance.
‖The gene and the protein that it codes that may underlie the quantitative trait loci, as supported by evidence presented in the cited reference. The gene’s chromosomal location in centimorgans is presented in parentheses.
Pain Genetics Studies in Humans
Based on the impressive initial results of genetic studies of pain in rodents, the reader might reverse our opening question and ask why one needs to do human studies at all. By inbreeding mice until they are homozygous at every locus, one can magnify the effects of polymorphisms on the phenotype. The mouse researcher can also rigorously control the environment and the pain-provoking stimulus. If almost all human and mouse genes are homologous, why is it not sufficient to examine every mouse gene for effects on pain?
The behavioral and genetic methods used in these rodent studies have revealed hundreds or possibly even thousands of novel pain candidates (see LaCroix-Fralish et al 2011). However, this abundance of riches presents new problems that human studies might help solve. First, how should one prioritize these targets for physiological study or for the development of new drugs that mimic or antagonize them? Second, are there subtle differences in physiology between mice and humans that would lead to an erroneous choice of analgesic drug targets? Putative analgesics such as neurokinin 1 and glycine-site NMDA receptor antagonists have worked well in rats but have failed in clinical pain conditions, costing drug companies tens of millions of dollars. Finally, humans but not rodents can describe important phenomena such as pain quality, ongoing or stimulus-independent pain (see Mogil 2009, Mogil et al 2010; but see also Sufka 1994, King et al 2009, Langford et al 2010), and relationships between pain and mood.
The following discussion suggests several ways that clinical researchers can solve some of these problems. About half of the molecules studied intensively by behavioral genetics laboratories are encoded by genes that appear to have common genetic polymorphisms that affect the amount or structure of mRNA/protein. If either consequence occurs, we will term this a “functional” polymorphism, which might therefore alter a clinical phenotype (Cravchik and Goldman 2000, Venter et al 2001). One might be able to prioritize this half of the genes on a rodent-derived pain candidate gene list by studying the effects of the genetic variants on a clinical pain outcome. Such a study, somewhat analogous to transgenic and linkage mapping studies in animals, is much less expensive than developing a new drug and then carrying out toxicology studies and a clinical trial. The roughly half of human genes with only rare functionally significant variants will not be informative unless one screens large numbers of patients to find the variant subset.
Some Definitions
Disease Genes versus Pain Genes
If one searches PubMed or Medline for the words “pain,” “human,” and “polymorphism,” most of the resulting reports will describe genes that cause a visible injury that gives rise to pain—a herniated lumbar disc, occluded coronary artery, or a tumor. Pain researchers are not particularly interested in these “disease genes,” but rather in genes that affect the processing of pain, given a uniform injury. Even though this distinction sounds clear when one contrasts a collagen gene variant predisposing to intervertebral disc herniation (Ala-Kokko 2002) with an NMDA receptor polymorphism affecting spinal dorsal horn pain transmission, it may not be obvious whether genes coding for hypo- or hypersecretion of inflammatory mediators at an injury site are “disease genes” or “pain genes.”
Mendelian Disorders versus Complex Genetic Disorders
Human diseases and disorders can be roughly classified into three categories in terms of their association with genetic factors: single-gene disease, multifactorial disease, and non-genetic factorial disease. The onset of a single-gene disease is induced by a mutation in a certain gene. The prevalence in the general population is low, and heritability follows a simple mendelian model: one (dominant) or two copies (recessive) of the altered gene, without the need for any other mutations, alter the phenotype. A handful of mendelian disorders of pain processing have been described. The best-known examples are the various hereditary sensory and autonomic neuropathies (HSANs) types I–V (Nagasako et al 2003), in which peripheral nerve degeneration accounts for the deficit in pain sensation. In one subclass, HSAN IV, various mutations that impair the function of the nerve growth factor receptor TrkA (NTRK1) have been identified as the cause (Indo et al 1996). Congenital indifference to pain (HSAN V), a rare autosomal recessive disorder characterized by insensitivity to all modalities of pain except neuropathic pain, is associated with missense mutations in SCN9A (Nav1.7 channel). In fact, this same gene is responsible for three human pain disorders: nonsense (loss-of-function) mutations cause a complete absence of pain (Cox et al 2006, Goldberg et al 2007), whereas activating (gain-of-function) mutations cause severe episodic pain in paroxysmal extreme pain disorder (Fertleman et al 2006) and primary erythromelalgia (Yang et al 2004b). All these mutations are rare with very low allelic frequencies, but a recent study showed that a common polymorphism in SCN9A (rs6746030 A allele) is associated with altered pain perception in the general population in several chronic pain models (Reimann et al 2010).
Another class of rare mendelian pain disorders consists of the familial hemiplegic migraines (FHMs). Various mutations of the gene coding for the α1a subunit of the voltage-gated calcium channel, CACNA1A, have been identified as the cause of FHM type 1 (Ducros et al 2002). The genes responsible for FHM types 2 and 3 are now known to be SCN1A, which encodes the Nav1.1 sodium channel (Dichgans et al 2005), and ATP1A2, which encodes the α2 subunit of Na+,K+-ATPase (DeFusco et al 2003), respectively. A very recent study has identified the KCNK18 gene, which encodes the TWIK-related spinal cord potassium channel (TRESK), in familial migraine with aura. These findings have all led to the hypothesis that migraines are related to dysfunction of ion transport. However, the relationship between these genes and common migraine is unclear. Association studies involving these and related genes and common migraine have failed (Peroutka et al 1997, Lea et al 2001, Nyholt et al 2008), although de novo mutations in ATP1A2 and CACNA1A have been shown to be frequent in early-onset sporadic hemiplegic migraine (Riant et al 2010). One might refrain from calling any of these “pain genes,” however, because the encoded channel abnormalities probably initiate the migraine attack rather than affecting processing of the nociceptive input during the attack.
Non-parametric linkage strategies using affected sibling pairs have proved effective in examining the genetic contribution to suspected mendelian disorders (McPeek 1999). It is possible that other mendelian pain disorders are waiting to be discovered by a pain researcher who brings a pain questionnaire and a thermal probe into a genetics clinic. However, we speculate that most such strong single-gene effects have been reported already. The various aspects of pain processing are more likely to be “complex genetic traits,” phenotypes determined by small contributions from many genes (independently or through gene–gene interactions) and genetic–environmental interactions. Contributions of such genes, which often alter the risk for common diseases by 25–300% (Lohmueller et al 2003), are small enough that their influence is rarely obvious in a family pedigree but common enough to account for a large proportion of the total disease burden caused by Alzheimer’s disease, Crohn’s disease, diabetes, and other disorders. The remainder of this chapter discusses approaches that one might take to show that pain disorders are complex genetic traits and to identify the genetic polymorphisms responsible.
Twin Studies of Heritability
Monozygotic (MZ) twins have identical genotypes at each of their genes, whereas dizygotic (DZ) twins, like fraternal siblings, have about half their genes in common. In the mid-1920s, geneticists began to compare the variance of traits in MZ versus DZ twin studies to estimate heritability (h2), or the proportion of the variance in a trait that is determined by heredity versus environment (Spector 2000). For traits measured on a continuous scale,
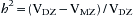
where VDZ and VMZ are, respectively, variances of the trait in DZ and MZ twins (Cavalli-Sforza and Bodmer 1971). An analogous method is used for dichotomous traits. A trait completely determined by genes would have identical magnitudes in each member of MZ twin pairs, and hence VMZ = 0, VDZ = 1, and h2 = 1.0. For a trait without genetic influence, VDZ and VMZ represent environmental variance entirely. If we assume that MZ and DZ twins are treated with similar uniformity (a questionable assumption), these variances are equal and h2 = 0.
Table 10-3 shows heritability values for physical characteristics, some well-studied medical diseases, and several pain traits. Apart from the well-replicated migraine data, most of these pain traits have been examined in only one study each.
Table 10-3
Heritability Estimates from Twin Studies Comparing Monozygotic and Dizygotic Twins
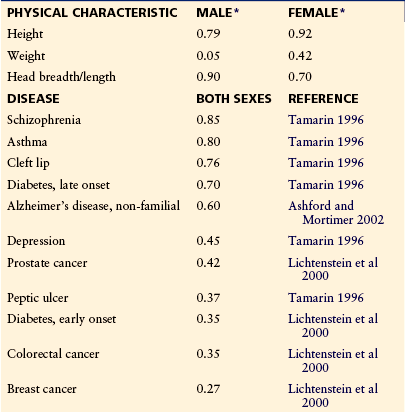
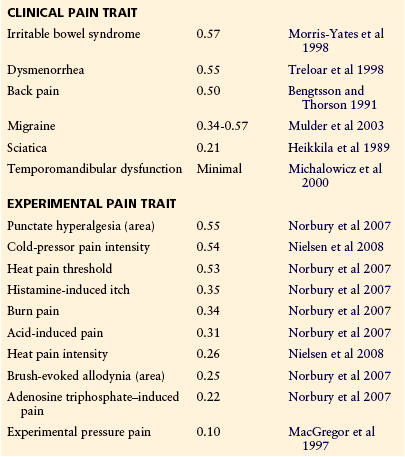
*From Osborne RH, De George FV 1959 Genetic basis of morphological variation. Harvard University Press, Cambridge, Mass.
Advantages of Twin Studies of Heritability
Disadvantages of Twin Studies of Pain Heritability
The heritability measures of 50% or more of the phenotypic variance in back pain, migraine, dysmenorrhea, and irritable bowel syndrome are comparable to many other medical diseases considered deserving of major genotyping investment. Unlike the rodent studies described previously, in which experimental injuries and sensory stimuli were identical and pain expression was the dependent variable, the clinical pain heritability for back pain, migraine, irritable bowel syndrome, and dysmenorrhea may just reflect a heritable feature of the disease-related structural changes in the spine, cerebral vessels, or abdominal viscera in the presence of uniform pain-processing mechanisms. Assuming the multifactorial nature of most human pain disorders influenced by both diverse environmental factors (e.g., trauma, lifestyle, and stress) and a complex array of multiple genetic polymorphisms, twin and family linkage studies would require several thousands of subjects to have sufficient power for accurate measurement of the heritability of each disorder (Risch 2000).
Association Studies: Candidate Genes and Whole-Genome Searches
“Association study” is a term that refers to a standard case–control approach in unrelated individuals to test whether a genetic variant affects the risk for a disease phenotype as opposed to the more common genotype. Lately, many clinical pain researchers have used association study designs with very modest numbers of subjects to identify and characterize polymorphisms in specific genes associated with pain processing, for example, in the contribution of the catechol O-methyltransferase (COMT) (Diatchenko et al 2006b, Nackley and Diatchenko 2010), β2-adrenergic receptor (Diatchenko et al 2006a), and serotonin transporter (Herken et al 2001) genes to the risk for temporomandibular pain disorder. Pharmacogenetic studies have revealed a predictive value of common polymorphisms in CYP2D6, which normally converts tramadol to the potent opioid analgesic (+)O-desmethyltramadol, for response to tramadol (Stamer et al 2003) and to codeine, whose efficacy is largely determined by metabolism to morphine by cytochrome P450 2D6 (Poulsen et al 1998). Other reports include genetic studies involving migraine (Sandor et al 2002), back pain (Solovieva 2004), post-herpetic neuralgia (Sato et al 2002), fibromyalgia (Offenbaecher et al 1999), complex regional pain syndrome (van de Beek et al 2003), dysmenorrhea (Wu et al 2000), and prostatic pain (Shoskes et al 2002). Many of these studies are small, involve only a single polymorphism, and await replication. Other have used multiple independent cohorts for replication and functional follow-up studies on associated markers using animal models and cell lines. These studies have not only demonstrated statistical association between the gene marker and pain phenotype but also suggested mechanisms through which the genes affect pain. For example, guanosine triphosphate cyclohydrolase (GCH1) is the rate-limiting enzyme for tetrahydrobiopterin (BH4) synthesis; BH4 is an essential co-factor for catecholamine, serotonin, and nitric oxide production. In humans, common polymorphisms in GCH1 were found to be associated with less sciatica pain and reduced experimental pain sensitivity (Tegeder et al 2006). Forskolin-stimulated immortalized leukocytes from homozygote carriers up-regulated GCH1 less than did controls.
Markers of choice for genetic association studies are SNPs since they are most abundant throughout the human genome, may have functional effect and high frequency in the population, and account for most of common inherited variability (Pettersson et al 2009).
Like other types of case–control studies and unlike the traditional family-based pedigree and sibling linkage studies (Risch and Merikangas 1996), genetic association studies are sensitive enough to detect small changes in risk. However, unlike traditional linkage studies, in which several markers are sufficient to scan an entire chromosome, each locus tested in an association study informs the investigator only about disease susceptibility mutations on a tiny stretch of DNA surrounding the locus in question.
This can be explained by the ancestral history of common genotypes. Modern humans arose from a few founder individuals in Africa several hundred thousand years ago, thus limiting genotypes to a few patterns that have recombined in each generation at an approximate rate of one crossover per chromosome. Recombination tends to occur at high-probability “hot spots” (Wall and Pritchard 2003), with short stretches of DNA throughout the genome left unchanged from the founders’ nucleotide sequences (Fig. 10-2). West Africans have about 30,000 of these preserved “haplotype blocks” averaging 10,000 base pairs in length. European and Asian populations went through a later “population bottleneck” of a few founders who had emigrated from Africa approximately 20,000 years ago. Because fewer generations were available for recombination, such populations now have about 10,000 larger preserved haplotype blocks averaging about 30,000 base pairs each (Gabriel et al 2002). Isolated populations who went through later bottlenecks, such as residents of Iceland, Finland, Sardinia, or the American Amish, have larger preserved haplotype blocks. Over each of these DNA segments, approximately 90% of individual chromosomes have one of the two to five most common haplotypes for that segment (see Fig. 10-2).
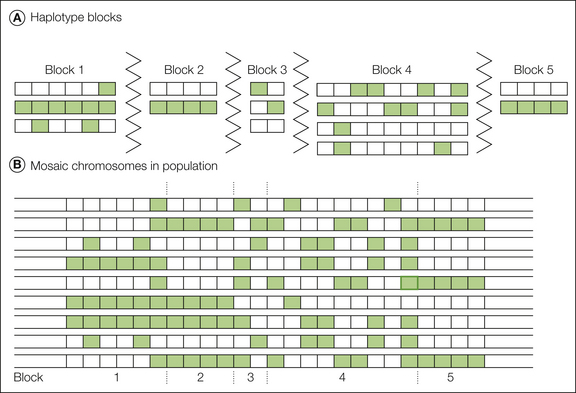
Figure 10-2 Haplotype blocks.
Green and white represent two alternative alleles, or single nucleotide polymorphisms (SNPs). The figure omits the more than 99% of nucleotides that are identical in almost all individuals. A, When common variants are considered, the haplotype block paradigm portrays the genome as a series of short segments separated by recombination hot spots (zigzag lines in the figure). Within each block there is little or no evidence of recombination, only a small number of distinct haplotypes is present in the population, and not all combinations of the various alleles are found. B, Most chromosomes in the population are a mosaic arrangement of one of the variants from each block. (Adapted from Cardon LR, Abecasis GR 2003 Using haplotype blocks to map human complex trait loci. Trends in Genetics 19:135–140. Copyright 2003, Elsevier Ltd.)
This recent discovery of preserved haplotype blocks is a boon to association studies. If one genotypes any six common polymorphisms within a block (tagging SNPs), one can accurately predict all common variants in that haplotype block, including untested common variants that may be the true disease risk alleles (Gabriel et al 2002). Many candidate genes are made up of one to five blocks, largely dependent on gene length. Use of 300,000 to 2.5 million SNP assays to identify every haplotype block in the individual will allow one to search for unsuspected haplotype blocks that may be associated with a disease phenotype. Toward this end, the 1000 Genomes Project Consortium (2002) has sequenced 179 individuals to identify the location, allele frequency, and local haplotype structure of approximately 15 million SNPs (along with other types of variants).
Steps in Planning Genetic Association Studies
Understanding the Determinants of Sample Size
Performing candidate gene studies is like playing the lottery. Though encouraged by results in mouse pain genetics, we do not know for certain whether there are major prizes to be won in the human pain gene lottery, that is, whether there are common polymorphisms with a large enough effect on pain phenotypes to be detected. Such polymorphisms have been discovered and replicated as risk factors for some, but not all major medical diseases (Lohmueller et al 2003). Assuming that such prizes await the pain geneticist, study of any particular candidate gene has a very small chance of success, and until it is replicated, an association is more likely to be a chance than a real finding. True associations with a phenotype probably occur for a small minority of the 30,000 human genes, most of which have never been studied. Pain genetics may surpass the lottery for astute players who understand how the two games differ in their investment-to-reward curves. In the lottery, the chance of payoff has a linear relationship to the number of tickets purchased. There is an economy of scale in “buying” hundreds or thousands of gene assays (Risch and Merikangas 1996, Camp 1997). Figure 10-3 shows that as one linearly increases the sample size, one can exponentially increase the number of independent tests of candidate loci! With only an eight-fold increase in sample size over the test of a single genetic locus, one can test every gene or haplotype block in the genome and correct for as many as 1,000,000 simultaneous tests with a Bonferroni adjustment. These statistical considerations are also pertinent to studies that use a clinical phenotype to scan the complete set of messenger RNA or proteins for a few molecules whose change in amount or function is associated with pain.
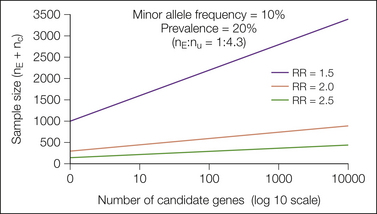
Figure 10-3 Linear increases in sample size allow exponential increases in the numbers of independent genetic loci that one can test simultaneously, which provides a major incentive to collect large cohorts of patients in genetic association studies. N is plotted against the number of independent polymorphisms tested in a scan of up to 10,000 loci. The injury or disease is assumed to produce an incidence of chronic pain equal to 20% in patients exposed to at least one copy of the minor allele (i.e., a dominant model; see Belfer et al 2004 for calculations for co-dominant and recessive models). The three curves in the panel correspond to a relative risk (RR) of 1.5, 2.0, and 2.5, and the population frequency of the minor alleles is 10%. nE, number exposed to the uncommon allele; nU, number unexposed. (Figure provided courtesy Dr. Tianxia Wu; also in Max MB 2004 Assessing pain candidate gene studies. Pain 109:1–3.)
Figures 10-3 and 10-4 show that the major driver of sample size in a genetic study is the size of the relative risk (RR) that one wishes to detect. If the polymorphism increases the risk for the disorder by 2.5 or more, which is the case for the common apolipoprotein E ε4 polymorphism in Alzheimer’s disease or NOD2 in Crohn’s disease, several hundred well-studied patients may be sufficient to examine a large number of candidate genes or even the whole genome. If RR is less than 1.5, as has been the case with many common polymorphisms that have replicated associations with common diseases (Lohmueller et al 2003), sample sizes in the thousands may be needed. Part of this uncertainty is completely up to nature—functionally important variants in pain genes are either common or not—but as will be discussed later, RR values conferred by candidate genes may be increased by optimal definition of the phenotype and adjustment for co-variates.
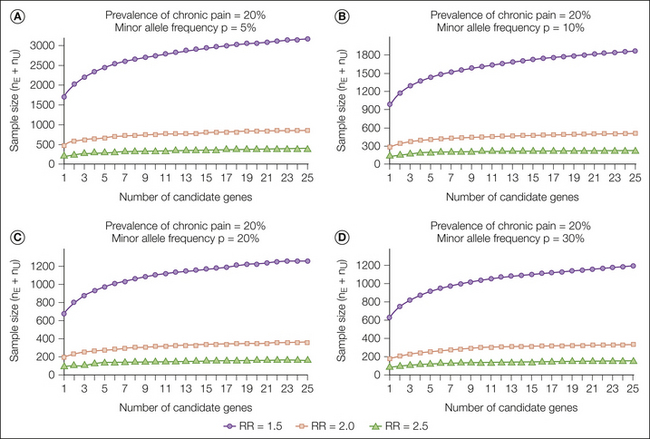
Figure 10-4 Number of subjects required to detect an association between candidate polymorphisms and increased risk for chronic pain versus the number of independent polymorphisms tested.
The injury or disease is assumed to produce an incidence of chronic pain of 20% in patients unexposed to the (candidate) less common allele. The three curves in each panel correspond to a relative risk (RR) of 1.5, 2.0, and 2.5 conferred by exposure to at least one copy of the uncommon allele. The four panels shown represent the population frequency of the minor allele as 5% (A), 10% (B), 20% (C), and 30% (D). nE, number exposed to the uncommon allele; nU, number unexposed. (Reproduced from Belfer I, Wu T, Kingman A, et al 2004 Candidate gene studies of human pain mechanisms: a method for optimizing choice of polymorphisms and sample size. Anesthesiology 100:1562–1572. Copyright 2004, Lippincott Williams & Wilkins. This paper also includes sample size tables for when the prevalence of chronic pain is 10% or 30%.)
Choosing a Pain Phenotype for Genetic Studies
At this moment, no one knows which pain phenotypes will be optimal for genetic studies. Considerations in choosing a pain phenotype are discussed in the following sections.
Availability of Large Numbers of Patients
As discussed previously, 1 association study with 1000 patients is worth far more than 10 studies of 100 patients. Therefore, there are major advantages in choosing common pain conditions. If one is collecting patient populations with mixed ethnicity, care must be taken to avoid “population stratification bias.” This type of bias may occur if (1) the polymorphism to be tested has different frequencies in the major ethnic groups that make up the study population and (2) the prevalence of the phenotype of interest differs among these ethnic groups (Cardon and Palmer 2003). This opens up the possibility of either a false positive or false negative for the association. Current recommendations include low- and high-tech remedies. The low-tech remedy is to analyze allele frequencies and outcomes in the ethnic groups and, if they are not different, pool the results. The high-tech remedy is called “genomic control” (Chen et al 2003). One defines ethnicity more subtly than conventional classifications by typing patients for 100–200 markers throughout the genome (ancestry-informative markers [AIMs]) and then correct for biases in either direction (Enoch et al 2006).
Specificity of Pain Type
Geneticists agree that an overly broad choice of phenotype is the most common fatal flaw of clinical studies. Moreover, the mouse studies cited previously have already shown that responses to three to five distinct classes of pain stimuli have different patterns of inheritance across inbred mouse strains, and this conclusion was further supported by the human twin data of Nielsen and colleagues (2008). This suggests that different types of pain have different underlying genetic mechanisms and that pooling different clinical pain syndromes may sacrifice power. On the other hand, finding common “intermediate phenotypes” (also known as endophenotypes) (Carlson et al 2004) that overlap between pain disorders may help overcome this difficulty by clustering patients and improving power to detect the association signal. Quantitative sensory testing, evaluation of autonomic function, and psychosocial profiling of patients with different pain syndromes may identify useful intermediate phenotypes for genetic association studies.
Pain in Multisomatoform Disorders
Many patients evaluated by the medical system because of multiple unexplained pain complaints lack any evidence of a structural lesion. Some investigators have suggested that “multisomatoform” pain disorders such as fibromyalgia, irritable bowel syndrome, chronic tension-type headache, interstitial cystitis, and temporomandibular dysfunction share neural abnormalities that amplify sensory stimuli and increase the risk for mood disorders (Kroenke and Harris 2001). The finding that polymorphisms of the monoamine-degrading enzyme COMT may alter the intensity of experimental pain (Zubieta et al 2003) and the risk for temporomandibular pain disorder (Diatchenko et al 2005) may encourage additional genetic approaches to these puzzling disorders. Recent studies have revealed multiple genetic (related to catecholamine and opioid systems) and psychological factors (e.g., somatization and catastrophizing) contributing to and predictive of multisomatoform pain (Slade et al 2007).
Availability for Real-Time versus Retrospective Assessment
Convenience, economy, and the need for large numbers of patients make it appealing to gather DNA and clinical data from an existing database of patients known to have suffered a painful illness or injury at various times in the past. Prospective collection of patients before or shortly after the injury is more difficult but avoids the substantial errors in memory for pain and makes possible collection of data about mood, sleep, analgesic use, and other co-variates and co-morbid conditions. A prospective longitudinal design, with several time points, would be ideal since it provides data on both the causality and stability of phenotypes.
Knowledge of Prognostic Factors
The signal-to-noise ratio of any experimental effect improves as one explains other sources of variance and removes them from the general error term. Therefore, one can optimize the resolving power of genetic studies either by choosing a condition with well-studied predictors such as postoperative dental pain, post-herpetic neuralgia, or sciatica that persists after discectomy or by characterizing these prognostic factors in the course of the genetic study. This statistical sharpening of the phenotype tends to increase the RR conferred by a functional polymorphism, thereby decreasing the required sample size.
Prioritizing Candidate Genes
At the time of this writing, the study of hundreds or thousands of candidate pain genes strains the researcher’s resources for genotyping and the statistical power to do so in many independent tests. Even with genotyping costs dropping and pre-designed assays becoming available for most SNPs, one may still wish to use a systematic process of selecting high- and lower-priority candidate genes. One should avoid the temptation to carry out whatever assays of candidate genes are available in nearby laboratories. Unless one has specified a priority list of candidate genes beforehand, others may discount positive associations with a candidate gene of interest because so many other tests were done simultaneously. Belfer and co-workers (2004) described a systematic method for prioritization with which they compiled a list of the 200 molecules that had been mentioned in scientific papers and reviews on pain mechanisms. They searched the literature to score each molecule from 0–3 points for three characteristics that might enhance success in an association study of neuropathic pain patients: relevance to neuropathic pain processing, frequency of a variant in the gene, and strength of evidence that the polymorphism affects protein expression or any clinical phenotype. This exercise showed that many of the commonly discussed pain-processing molecules have polymorphisms that affect function and are common enough to examine in cohorts of several hundred pain patients. For the most common polymorphisms, one can readily accrue enough patients to examine the effects of homozygosity for the uncommon variant or interactions between polymorphisms of two candidate genes. For example, for an uncommon variant with a population allelic frequency, p, of 30% (i.e., 30% of chromosomes bear the uncommon variant), p2, or 9% of the subjects, will be homozygous for the uncommon variant. The proportion of subjects with at least one copy of the variant will be 1 − (1 − p)2 = 51%, and (51%)2 = 26% will have at least one copy of the uncommon variant of two such candidate genes.
For this gene prioritization exercise, the published literature was sufficient to collect a short list of well-known pain candidates. The animal genetics described earlier are generating many more. Linkage mapping studies in mouse models of pain may offer several arguments for examining the gene in humans. For example, a recent study by Nissenbaum and colleagues (2010) in which two fine-mapping strategies were used in a mouse model mapped a pain-related quantitative trait locus to a 4.2-megabase interval on chromosome 15. This interval included 155 genes. Bio-informatics and whole-genome microarray expression analysis were used to narrow the list of candidates and ultimately to pinpoint the CACNG2 gene, which encodes for stargazin, a protein involved in the trafficking of glutamatergic α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors. A common polymorphism in CACNG2 was then found to be associated with human neuropathic pain (Nissenbaum et al 2010).
Microarray studies in animal pain models are generating hundreds of new pain candidates that have not previously been mentioned in the scientific literature. Some of these candidates, including GCH1 described previously and KCNS1—a recently discovered pain gene encoding a potassium channel subunit involved in neuronal excitability—were confirmed in human pain cohorts and replicated in several pain conditions (Tegeder et al 2006, Costigan et al 2010). One can apply a similar prioritization algorithm to these new candidate genes with only slightly greater difficulty than in the example of Belfer and colleagues (2004). Polymorphism frequency can readily be found in genomic databases, but there is not yet any systematic catalog of the functional impact of most of the millions of polymorphisms in the genome. Bio-informatics tools that can predict the effect of changes in amino acids on protein structure and identify gene-regulatory sequences may help the investigator guess the functional impact of such changes (Belfer et al 2004).
Recent studies using a comparative genomic approach have shown success in identifying evolutionarily conserved pain candidate genes. For example, whole-genome, neuron-specific RNAi knockdown in Drosophila uncovered hundreds of novel candidate genes for thermal nociception. One of the screen hits was the calcium channel subunit straightjacket/α2δ3. Mice mutant for the stj ortholog CACNA2D3 (α2δ3) also exhibit impaired behavioral heat pain sensitivity. In humans, α2δ3 SNP variants are associated with reduced sensitivity to acute noxious heat and lower levels of chronic back pain (Neely et al 2010). These data reinforce the extraordinary conservation of the neurobiological mechanisms of nociception, from its manifestation as avoidance of damage in primitive creatures such as flies to the complex sensation of pain in humans.
“Pain Chip” and Genome-wide Association Studies
By moving from single-candidate to multiple-target genetic study one can use customized pre-designed SNP arrays with informative (tagging) or functional markers across genes previously linked to pain processing. For example, the “Pain Research Panel” of 350 candidate genes and 200 AIMs for population structure analysis was genotyped in a large cohort of chronic pain patients, and the analysis identified multiple genes associated with pain phenotypes and psychological risk factors (Smith et al 2009). Since pain is a complex trait and multiple genes with limited effect size may contribute to its variability, genotyping of many targets simultaneously is more efficient than single-gene genotyping in time and money and provides data for association, pathway, and population admixture analyses. Disadvantages of this approach include a larger sample size required and gene selection bias (previous evidence on selected genes). Genome-wide screening may overcome the selection bias limitation and tremendously facilitate pain research in several ways, including identifying entirely novel pain mediators; focusing ongoing programs of drug development in preclinical models on specific pathways or mediators of human pain; extracting up to 50% of the variance from studies of non-genetic pain risk factors; providing a better understanding of the risks, mechanisms, and treatment of pain in individual patients (thus eventually leading to “personalized pain medicine”); and finally, improving the classification of pain disorders (Max et al 2008). To date, only one pain-related GWAS study has been published. A large study of 2731 migraine patients and 10,747 population-matched controls identified the rs1835740 allele on chromosome 8q22.1 to be highly associated with migraine (P = 5.38 × 10−12) . The association was replicated in 3202 cases and 40,062 controls, and the allele was correlated with expression (in a lymphoblastoid cell line) of a nearby gene called MTDH (astrocyte elevated gene 1) (Antilla et al 2010).
Next-Generation Sequencing
Completion of the human genome sequence, deposition of millions of SNPs into public databases, rapid improvements in SNP genotyping technology, and the International HapMap Project provided a basis for GWAS, and mapping causal genes with modest effects has become possible. However, because it is reasonable to suppose that both common and rare alleles contribute to common disease and quantitative traits, detection of rare alleles by recently developed high-throughput next-generation sequencing should be attempted for pain phenotypes in advance of or in conjunction with GWAS (McClellan and King 2010). The underlying assumption in the use of sequencing to complement genetic studies is that individuals at the extremes of a phenotype will be harboring alleles that have high effect size and are easily determined to be functional (stop codon, missense, or splicing mutations) to pinpoint the pathways and positively identify novel “pain genes.” Although whole-genome sequencing may in the future be used as an approach to identify candidate genes for pain sensitivity by finding rare functional variants in extreme cases (with the highest and lowest degree of the pain phenotype), the high per-sample costs and the intensive use of sequencing resources currently make this approach untenable. An alternative approach is to specifically sequence only the exons and promoter regions of every annotated gene in these extreme samples. This has the advantages of lower cost per subject, lower sequencing capacity requirements, and in a bio-informatics sense, easier identification of possibly functional variants that can be tested. Once candidate genes have been identified by sequencing, follow-up studies may reanalyze GWAS data on these genes to look for common alleles that might be functional and have a modulating effect on the phenotype.
Conclusions
Genetic methods have become an established tool in the dissection of pain mechanisms in animal models and have led to the discovery of novel pain-processing molecules. The finding of substantial inter-strain diversity of pain responses in rodents suggests that common polymorphisms that alter pain processing were not deleterious to survival in the wild populations from which the mouse strains were originally derived. This balance in pain gene polymorphisms is likely to exist in other mammals, including humans.
Animal and human experiments might best be used complementarily. The principles established in animal pain genetics will probably continue to guide many aspects of clinical approaches to pain genetics. Whenever clinical pain studies confirm an animal finding or discover a novel association, animal models can then be used to dissect the mechanism by which particular variants affect pain processing. The well-established discipline of pharmacogenetics has already proved valuable in tailoring treatments to individual pain patients and is readily adaptable to new classes of drugs and pain mechanisms.
Acknowledgment
We thank Dr. Colin Hodgkinson (NIH/NIAAA) for helpful discussions and comments.
The references for this chapter can be found at www.expertconsult.com.
References
1000 Genomes Project Consortium T. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073.
Ala-Kokko L. Genetic risk factors for lumbar disc disease. Annals of Medicine. 2002;34:42–47.
Altier C., Dale C.S., Kisilevsky A.E., et al. Differential role of N-type calcium channel splice isoforms in pain. Journal of Neuroscience. 2007;27:6363–6373.
Amanzio M., Pollo A., Maggi G., et al. Response variability to analgesics: a role for non-specific activation of endogenous opioids. Pain. 2001;90:205–215.
Andersen G., Vestergaard K., Ingeman-Nielsen M., et al. Incidence of central post-stroke pain. Pain. 1995;61:187–193.
Anseloni V.C.Z., He F., Novikova S.I., et al. Alterations in stress-associated behaviors and neurochemical markers in adult rats after neonatal short-lasting local inflammatory insult. Neuroscience. 2005;131:635–645.
Antilla V., Stefansson H., Kallela M., For the International Headache Genetics Consortium. Genome-wide association study of migraine implicates a common susceptibility variant on 8q22.1. Nature Genetics. 2010;42:869–874.
Ashford J.W., Mortimer J.A. Non-familial Alzheimer’s disease is mainly due to genetic factors. Journal of Alzheimer’s Disease. 2002;4:169–177.
Aubrun F., Langeron O., Quesnel C., et al. Relationships between measurement of pain using visual analog score and morphine requirements during postoperative intravenous morphine titration. Anesthesiology. 2003;98:1415–1421.
Avsaroglu H., van der Sar A.S., van Lith H.A., et al. Differences in response to anaesthetics and analgesics between inbred rat strains. Laboratory Animals. 2007;41:337–344.
Barr G.A., Gao P., Wang S., et al. Microarray analysis of gene expression following the formalin test in the infant rat. Pain. 2005;117:6–18.
Beck J.A., Lloyd S., Hafezparast M., et al. Genealogies of mouse inbred strains. Nature Genetics. 2000;24:23–25.
Belfer I., Wu T., Kingman A., et al. Candidate gene studies of human pain mechanisms: a method for optimizing choice of polymorphisms and sample size. Anesthesiology. 2004;100:1562–1572.
Belknap J.K., Haltli N.R., Goebel D.M., et al. Selective breeding for high and low levels of opiate-induced analgesia in mice. Behavior Genetics. 1983;13:383–396.
Belknap J.K., Mogil J.S., Helms M.L., et al. Localization to chromosome 10 of a locus influencing morphine analgesia in crosses derived from C57BL/6 and DBA/2 mouse strains. Life Sciences. 1995;57:PL117–PL124.
Belknap J.K., O’Toole L.A. Studies of genetic differences in response to opioid drugs. In: Harris R.A., Crabbe J.C., eds. The genetic basis of alcohol and drug actions. New York: Springer; 1991:225–252.
Bengtsson B., Thorson J. Back pain: a study of twins. Acta Geneticae et Medicae Gemmellologiae. 1991;40:83–90.
Bergeson S.E., Helms M.L., O’Toole L.A., et al. Quantitative trait loci influencing morphine antinociception in four mapping populations. Mammalian Genome. 2001;12:546–553.
Bonilla I.E., Tanabe K., Strittmatter S.M. Small proline-rich repeat protein 1A is expressed by axotomized neurons and promotes axonal outgrowth. Journal of Neuroscience. 2002;22:1303–1315.
Brenner S., Johnson M., Bridgham J., et al. Gene expression analysis by massively parallel signature sequencing (MPSS) on microbead arrays. Nature Biotechnology. 2000;18:630–634.
Camp N.J. Genomewide transmission/disequilibrium testing—consideration of the genotypic relative risks at disease loci. American Journal of Human Genetics. 1997;61:1424–1430.
Capecchi M.R. Altering the genome by homologous recombination. Science. 1989;244:1288–1292.
Cardon L.R., Abecasis G.R. Using haplotype blocks to map human complex trait loci. Trends in Genetics. 2003;19:135–140.
Cardon L.R., Palmer L.J. Population stratification and spurious allelic association. Lancet. 2003;361:598–604.
Carlson C.S., Eberle M.A., Kruglyak L., et al. Mapping complex disease loci in whole-genome association studies. Nature. 2004;429:446–452.
Cavalli-Sforza L.L., Bodmer W.F. The genetics of human populations. San Francisco: Freeman; 1971.
Chen H.S., Zhu X., Zhao H., et al. Qualitative semi-parametric test for genetic associations in case-control designs under structured populations. Annals of Human Genetics. 2003;67:250–264.
Chen Y., Yang C., Wang Z.J. Ca2+/calmodulin-dependent protein kinase IIα is required for the initiation and maintenance of opioid-induced hyperalgesia. Journal of Neuroscience. 2010;30:38–46.
Christoph T., Grunweller A., Mika J., et al. Silencing of vanilloid receptor TRPV1 by RNAi reduces neuropathic and visceral pain in vivo. Biochemical and Biophysical Research Communications. 2006;350:238–243.
Chu L.F., Liang D.-Y., Li X., et al. From mouse to man: the 5-HT3 receptor modulates physical dependence on opioid narcotics. Pharmacogenetics and Genomics. 2009;19:193–205.
Chu Y.X., Zhang Y., Zhang Y.Q., et al. Involvement of microglial P2X7 receptors and downstream signaling pathways in long-term potentiation of spinal nociceptive responses. Brain, Behavior, and Immunity. 2010;24:1176–1189.
Clark F.M., Proudfit H.K. Anatomical evidence for genetic differences in the innervation of the rat spinal cord by noradrenergic locus coeruleus neurons. Brain Research. 1992;591:44–53.
Cluff R.S., Rowbotham M.C. Pain caused by herpes zoster infection. Neurologic Clinics. 1998;16:813–832.
Coghill R.C., McHaffie J.G., Yen Y- F. Neural correlates of interindividual differences in the subjective experience of pain. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:8538–8542.
Cook M.N., Dunning J.P., Wiley R.G., et al. Neurobehavioral mutants identified in an ENU-mutagenesis project. Mammalian Genome. 2007;18:559–572.
Costigan M., Befort K., Karchewski L., et al. Replicate high-density rat genome oligonucleotide microarrays reveal hundreds of regulated genes in the dorsal root ganglion after peripheral nerve injury. BMC Neuroscience. 2002;3:16.
Costigan M., Belfer I., Griffin R.S., et al. Multiple chronic pain states are associated with a common amino acid–changing allele in KCNS1. Brain. 2010;133:2519–2527.
Cox J.J., Reimann F., Nicholas A.K., et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature. 2006;444:894–898.
Crabbe J.C., Phillips T.J., Kosobud A., et al. Estimation of genetic correlation: interpretation of experiments using selectively bred and inbred animals. Alcoholism. Clinical and Experimental Research. 1990;14:141–151.
Cravchik A., Goldman D. Neurochemical individuality: genetic diversity among human dopamine and serotonin receptors and transporters. Archives of General Psychiatry. 2000;57:1105–1114.
Darvasi A. Experimental strategies for the genetic dissection of complex traits in animal models. Nature Genetics. 1998;18:19–24.
Day R.O., Graham G.G., Williams K.M., et al. Variability in response to NSAIDs: fact or fiction? Drugs. 1988;36:643–651.
DeFusco M., Marconi R., Silvestri L., et al. Haploinsufficiency of ATP1A2 encoding the Na+/K+ pump α2 subunit associated with familial hemiplegic migraine type 2. Nature Genetics. 2003;33:192–196.
Deval E., Noel J., Lay N., et al. ASIC3, a sensor of acidic and primary inflammatory pain. EMBO Journal. 2008;27:2047–3055.
Devor M., Gilad A., Arbilly M., et al. pain1: a neuropathic pain QTL on mouse chromosome 15 in a C3HxC58 backcross. Pain. 2005;116:289–293.
Devor M., Gilad A., Arbilly M., et al. Sex-specific variability and a ‘cage effect’ independently mask a neuropathic pain quantitative trait locus detected in a whole genome scan. European Journal of Neuroscience. 2007;26:681–688.
Devor M., Raber P. Heritability of symptoms in an experimental model of neuropathic pain. Pain. 1990;42:51–67.
Diatchenko L., Anderson A.D., Slade G.D., et al. Three major haplotypes of the β2 adrenergic receptor define psychological profile, blood pressure, and the risk for development of a common musculoskeletal pain disorder. American Journal of Medical Genetics. 2006;141B:449–462.
Diatchenko L., Nackley A.G., Slade G.D., et al. Catechol-O-methyltransferase gene polymorphisms are associated with multiple pain-evoking stimuli. Pain. 2006;125:216–224.
Diatchenko L., Slade G.D., Nackley A.G., et al. Genetic basis for individual variations in pain perception and the development of a chronic pain condition. Human Molecular Genetics. 2005;14:135–143.
Dichgans M., Freilinger T., Eckstein G., et al. Mutation in the neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine. Lancet. 2005;366:371–377.
Dong X.W., Goregoaker S., Engler H., et al. Small interfering RNA–mediated selective knockdown of Na(V)1.8 tetrodotoxin-resistant sodium channel reverses mechanical allodynia in neuropathic rats. Neuroscience. 2007;146:812–821.
Dorn G., Patel S., Wotherspoon G., et al. siRNA relieves chronic neuropathic pain. Nucleic Acids Research. 2004;32:e49.
Ducros A., Tournier-Lasserve E., Bousser M.G. The genetics of migraine. Lancet Neurology. 2002;1:285–293.
Edwards R.R. Individual differences in endogenous pain modulation as a risk factor for chronic pain. Neurology. 2005;65:437–443.
Elmer G.I., Pieper J.O., Negus S.S., et al. Genetic variance in nociception and its relationship to the potency of morphine-induced analgesia in thermal and chemical tests. Pain. 1998;75:129–140.
Enoch M.A., Shen P.H., Xu K., et al. Using ancestry-informative markers to define populations and detect population stratification. Journal of Psychopharmacology. 2006;20:19–26.
Fecho K., Nackley A.G., Wu Y., et al. Basal and carrageenan-induced pain behavior in Sprague-Dawley, Lewis and Fischer rats. Physiology & Behavior. 2005;85:177–186.
Fecho K., Valtschanoff J.G. Acute inflammatory and neuropathic pain in Lewis and Fischer rats. Journal of Neuroendocrinology. 2006;18:504–513.
Fertleman C.R., Baker M.D., Parker K.A., et al. SCN9A mutations in paroxysmal extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes. Neuron. 2006;52:767–774.
Fortin A., Diez E., Ritchie J., et al. Positional cloning of a quantitative trait locus contributing to pain sensitivity: possible mediation by Tyrp1. Genes, Brain, and Behavior. 2010;9:856–867.
Gabra B.H., Kessler F.K., Ritter J.K., et al. Decrease in N-methyl-D-aspartic acid receptor-NR2B subunit levels by intrathecal short-hairpin RNA blocks group I metabotropic glutamate receptor–mediated hyperalgesia. Journal of Pharmacology and Experimental Therapeutics. 2007;322:186–194.
Gabriel S.B., Schaffner S.F., Nguyen H., et al. The structure of haplotype blocks in the human genome. Science. 2002;296:2225–2229.
Gao Y.Z., Guo S.Y., Yin Q.Z., et al. An individual variation study of electroacupuncture analgesia in rats using microarray. American Journal of Chinese Medicine. 2007;35:767–778.
Gao Y.J., Zhang L., Ji R.R. Spinal injection of TNF-alpha–activated astrocytes produces persistent pain symptom mechanical allodynia by releasing monocyte chemoattractant protein-1. Glia. 2010;58:1871–1880.
Garraway S.M., Xu Q., Inturrisi C.E. siRNA-mediated knockdown of the NR1 subunit gene of the NMDA receptor attenuates formalin-induced pain behaviors in adult rats. Journal of Pain. 2009;10:380–390.
Geranton S.M., Morenilla-Palao C., Hunt S.P. A role for transcriptional repressor methyl-CpG–binding protein 2 and plasticity-related gene serum- and glucocorticoid-inducible kinase 1 in the induction of inflammatory pain states. Journal of Neuroscience. 2007;27:6163–6173.
Gibbs R.A., Weinstock G.M., Metzker M.L., et al. Genome sequence of the brown Norway rat yields insights into mammalian evolution. Nature. 2004;428:493–521.
Goldberg Y.P., MacFarlane J., MacDonald M.L., et al. Loss-of-function mutations in the Nav1.7 gene underlie congenital indifference to pain in multiple human populations. Clinical Genetics. 2007;71:311–319.
Griffin R.S., Costigan M., Brenner G.J., et al. Complement induction in spinal cord microglia results in anaphylatoxin C5a–mediated pain hypersensitivity. Journal of Neuroscience. 2007;27:8699–8708.
Griffin R.S., Mills C.D., Costigan M., et al. Exploiting microarrays to reveal differential gene expression in the nervous system. Genome Biology. 2003;4:105.
Grupe A., Germer S., Usuka J., et al. In silico mapping of complex disease-related traits in mice. Science. 2001;292:1915–1918.
Hain H.S., Belknap J.K., Mogil J.S. Pharmacogenetic evidence for the involvement of 5-hydroxytryptamine (serotonin)-1B receptors in the mediation of morphine antinociceptive sensitivity. Journal of Pharmacology and Experimental Therapeutics. 1999;291:444–449.
Hammer P., Banck M.S., Amberg R., et al. mRNA-seq with agnostic splice site discovery for nervous system transcriptomics tested in chronic pain. Genome Research. 2010;20:847–860.
Heikkila J.K., Koskenvuo M., Heliovaara M., et al. Genetic and environmental factors in sciatica: evidence from a nationwide panel of 9365 adult twin pairs. Annals of Medicine. 1989;21:393–398.
Herken H., Erdal E., Mutlu N., et al. Possible association of temporomandibular joint pain and dysfunction with a polymorphism in the serotonin transporter gene. American Journal of Orthodontics and Dentofacial Orthopedics. 2001;120:308–313.
Herradon G., Ezquerra L., Nguyen T., et al. Changes in BDNF gene expression correlate with rat strain differences in neuropathic pain. Neuroscience Letters. 2007;420:273–276.
Hrabe de Angelis M., Flaswinkel H., Fuchs H., et al. Genome-wide, large-scale production of mutant mice by ENU mutagenesis. Nature. 2000;25:444–447.
Indo Y., Tsurata Y., Karim M.A., et al. Mutations in the TRKA/NGF receptor gene in patients with congenital insensitivity to pain with anhidrosis. Nature Genetics. 1996;13:485–488.
Jacob H.J., Lazar J., Dwinell M.R., et al. Gene targeting in the rat: advances and opportunities. Trends in Genetics. 2010;26:510–518.
Joshi S.K., Su X., Porreca F., et al. κ-Opioid receptor agonists modulate visceral nociception at a novel, peripheral site of action. Journal of Neuroscience. 2000;20:5874–5879.
Kasama S., Kawakubo M., Suzuki T., et al. RNA interference–mediated knock-down of transient receptor potential vanilloid 1 prevents forepaw inflammatory hyperalgesia in rat. European Journal of Neuroscience. 2007;25:2956–2963.
Kendler K.S. Twin studies of psychiatric illness: an update. Archives of General Psychiatry. 2001;58:1005–1014.
Kest B., Palmese C.A., Juni A., et al. Mapping of a quantitative trait locus for morphine withdrawal sensitivity. Mammalian Genome. 2004;15:610–617.
Kest B., Smith S.B., Schorscher-Petcu A., et al. Gnao1 (Gαo protein) is a likely genetic contributor to variation in physical dependence on opioids in mice. Neuroscience. 2009;162:1255–1264.
Kiguchi N., Maeda T., Kobayashi Y., et al. Macrophage inflammatory protein-1alpha mediates the development of neuropathic pain following peripheral nerve injury through interleukin-1beta up-regulation. Pain. 2010;149:305–315.
Kim D.H., Rossi J.J. Strategies for silencing human disease using RNA interference. Nature Reviews. Genetics. 2007;8:173–184.
Kim D.S., Lee S.J., Park S.Y., et al. Differentially expressed genes in rat dorsal root ganglia following peripheral nerve injury. Neuroreport. 2001;12:3401–3405.
Kim D.S., Figueroa K.W., Li K.W., et al. Profiling of dynamically changed gene expression in dorsal root ganglia post peripheral nerve injury and a critical role of injury-induced glial fibrillary acetic protein in maintenance of pain behaviors. Pain. 2009;143:114–122.
Kim H., Neubert J.K., San Miguel A., et al. Genetic influence on variability in human acute experimental pain sensitivity associated with gender, ethnicity and psychological temperament. Pain. 2004;109:488–496.
King T., Vera-Portocarrero L., Gutierrez T., et al. Unmasking the tonic-aversive state in neuropathic pain. Nature Neuroscience. 2009;12:1361–1363.
Ko J., Na D.S., Lee Y.H., et al. cDNA microarray analysis of the differential gene expression in the neuropathic pain and electroacupuncture treatment models. Journal of Biochemistry and Molecular Biology. 2002;35:420–427.
Kobayashi H., Kitamura T., Sekiguchi M., et al. Involvement of EphB1 receptor/EphrinB2 ligand in neuropathic pain. Spine. 2007;32:1592–1598.
Koide T., Moriwaki K., Ikeda K., et al. Multi-phenotype behavioral characterization of inbred strains derived from wild stocks of Mus musculus. Mammalian Genome. 2000;11:664–670.
Kramer P.R., Puri J., Bellinger L.L. Knockdown of FcγRIII in an arthritic temporomandibular joint reduced the nociceptive response. Arthritis and Rheumatism. 2010;62:3109–3118.
Kroenke K., Harris L. Symptoms research: a fertile field. Annals of Internal Medicine. 2001;134:801–802.
Kroenke K., Spitzer R.L., deGruy F.V., 3rd., et al. A symptom checklist to screen for somatoform disorders in primary care. Psychosomatics. 1998;39:263–272.
Kubo T., Yamashita T., Yamaguchi A., et al. Analysis of genes induced in peripheral nerve after axotomy using cDNA microarrays. Journal of Neurochemistry. 2002;82:1129–1136.
LaCroix-Fralish M.L., Austin J.S., Liu G., et al. Patterns of pain: meta-analysis of microarray studies of pain. Pain. 2011;152:1888–1898.
LaCroix-Fralish M.L., Ledoux J.B., Mogil J.S. The Pain Genes Database: an interactive web browser of pain-related transgenic knockout studies. Pain. 2007;131:3.e1–3.e4.
LaCroix-Fralish M.L., Mo G., Smith S.B., et al. The β3 subunit of the Na+, K+-ATPase affects pain sensitivity. Pain. 2009;144:294–302.
LaCroix-Fralish M.L., Mogil J.S. Progress in genetic studies of pain and analgesia. Annual Review of Pharmacology and Toxicology. 2009;49:97–121.
LaCroix-Fralish M.L., Mogil J.S., Weinstein J.N., et al. The magnitude of mechanical allodynia in a rodent model of lumbar radiculopathy is dependent on strain and sex. Spine. 2005;30:1821–1827.
LaCroix-Fralish M.L., Tawfik V.L., Tanga F.Y., et al. Differential spinal cord gene expression in rodent models of radicular and neuropathic pain. Anesthesiology. 2006;104:1283–1292.
Lan L.S., Ping Y.J., Na W.L., et al. Down-regulation of Toll-like receptor 4 gene expression by short interfering RNA attenuates bone cancer pain in a rat model. Molecular Pain. 2010;6:2.
Lander E.S., Schork N.J. Genetic dissection of complex traits. Science. 1994;265:2037–2048.
Langford D.L., Bailey A.L., Chanda M.L., et al. Coding of facial expressions of pain in the laboratory mouse. Nature Methods. 2010;7:447–449.
Lanier L.H. Variability in the pain threshold. Science. 1943;97:49–50.
Lariviere W.R., Chesler E.J., Mogil J.S. Transgenic studies of pain and analgesia: mutation or background phenotype? Journal of Pharmacology and Experimental Therapeutics. 2001;297:467–473.
Lariviere W.R., Wilson S.G., Laughlin T.M., et al. Heritability of nociception. III. Genetic relationships among commonly used assays of nociception and hypersensitivity. Pain. 2002;97:75–86.
Lasagna L., Beecher H.K. The optimal dose of morphine. Journal of the American Medical Association. 1954;156:230–234.
Lea R.A., Curtain R.P., Hutchins C., et al. Investigation of the CACNA1A gene as a candidate for typical migraine susceptibility. American Journal of Medical Genetics. 2001;105:707–712.
Levin M.E., Jin J.G., Ji R.R., et al. Complement activation in the peripheral nervous system following the spinal nerve ligation model of neuropathic pain. Pain. 2008;137:182–201.
Levine J.D., Gordon N.C., Smith R., et al. Analgesic responses to morphine and placebo in individuals with postoperative pain. Pain. 1981;10:379–389.
Li X., Sahbaie P., Zheng M., et al. Expression genetics identifies spinal mechanisms supporting formalin late phase behaviors. Molecular Pain. 2010;6:11.
Liang D.Y., Liao G., Lighthall G.K., et al. Genetic variants of the P-glycoprotein gene Abcb1b modulate opioid-induced hyperalgesia, tolerance and dependence. Pharmacogenetics and Genomics. 2006;16:825–835.
Liang D.Y., Liao G., Wang J., et al. A genetic analysis of opioid-induced hyperalgesia in mice. Anesthesiology. 2006;104:1054–1062.
Lichtenstein P., Holm N.V., Verkasalo P.K., et al. Environmental and heritable factors in the causation of cancer—analyses of cohorts of twins from Sweden, Denmark, and Finland. New England Journal of Medicine. 2000;343:78–85.
Lin C.R., Amaya F., Barrett L., et al. Prostaglandin E2 receptor EP4 contributes to inflammatory pain hypersensitivity. Journal of Pharmacology and Experimental Therapeutics. 2006;319:1096–1103.
Lohmueller K.E., Pearce C.L., Pike M., et al. Meta-analysis of genetic association studies supports a contribution of common variants to susceptibility to common disease. Nature Genetics. 2003;33:177–182.
MacGregor A.J., Griffiths G.O., Baker J., et al. Determinants of pressure pain threshold in adult twins: evidence that shared environmental influences predominate. Pain. 1997;73:253–257.
MacGregor A.J., Snieder H., Schork N.J., et al. Twins. Novel uses to study complex traits and genetic diseases. Trends in Genetics. 2000;16:131–134.
Maratou K., Wallace V.C.J., Hasnie F.S., et al. Comparison of dorsal root ganglion gene expression in rat models of traumatic and HIV-associated neuropathic pain. European Journal of Pain. 2009;13:387–398.
Marek P., Mogil J.S., Belknap J.K., et al. Levorphanol and swim stress–induced analgesia in selectively bred mice: evidence for genetic commonalities. Brain Research. 1993;608:353–357.
Max M.B. Assessing pain candidate gene studies. Pain. 2004;109:1–3.
Max M.B., Stewart W.F., The molecular epidemiology of pain: a new discipline for drug discovery. Nature Reviews. Drug Discovery. 2008;7:647–658.
McClellan J., King M C. Genetic heterogeneity in human disease. Cell. 2010;141:210–217.
McPeek M.S. Optimal allele-sharing statistics for genetic mapping using affected relatives. Genetic Epidemiology. 1999;16:225–249.
Michalowicz B.S., Pihlstrom B.L., Hodges J.S., et al. No heritability of temporomandibular joint signs and symptoms. Journal of Dental Research. 2000;79:1573–1578.
Mogil J.S. The genetic mediation of individual differences in sensitivity to pain and its inhibition. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:7744–7751.
Mogil J.S. The genetics of pain, vol 28. In: Mogil J.S., ed. Progress in pain research and management. Seattle: IASP Press, 2004.
Mogil J.S. Animal models of pain: progress and challenges. Nature Reviews. Neuroscience. 2009;10:283–294.
Mogil J.S., Belknap J.K. Sex and genotype determine the selective activation of neurochemically-distinct mechanisms of swim stress–induced analgesia. Pharmacology, Biochemistry, and Behavior. 1997;56:61–66.
Mogil J.S., Davis K.D., Derbyshire S.W. The necessity of animal models in pain research. Pain. 2010;151:12–17.
Mogil J.S., Kest B., Sadowski B., et al. Differential genetic mediation of sensitivity to morphine in genetic models of opiate antinociception: influence of nociceptive assay. Journal of Pharmacology and Experimental Therapeutics. 1996;276:532–544.
Mogil J.S., Marek P., Flodman P., et al. One or two genetic loci mediate high opiate analgesia in selectively bred mice. Pain. 1995;60:125–135.
Mogil J.S., McCarson K.E. Finding pain genes: bottom-up and top-down approaches. Journal of Pain. 2000;1(suppl 1):66–80.
Mogil J.S., Meirmeister F., Seifert F., et al. Variable sensitivity to noxious heat is mediated by differential expression of the CGRP gene. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:12938–12943.
Mogil J.S., Richards S.P., O’Toole L.A., et al. Genetic sensitivity to hot-plate nociception in DBA/2J and C57BL/6J inbred mouse strains: possible sex-specific mediation by δ2-opioid receptors. Pain. 1997;70:267–277.
Mogil J.S., Richards S.P., O’Toole L.A., et al. Identification of a sex-specific quantitative trait locus mediating nonopioid stress-induced analgesia in female mice. Journal of Neuroscience. 1997;17:7995–8002.
Mogil J.S., Ritchie J., Sotocinal S.G., et al. Screening for pain phenotypes: analysis of three congenic mouse strains on a battery of nine nociceptive assays. Pain. 2006;126:24–34.
Mogil J.S., Sternberg W.F., Marek P., et al. The genetics of pain and pain inhibition. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:3048–3055.
Mogil J.S., Sorge R.E., LaCroix-Fralish M.L., et al. Pain sensitivity and vasopressin analgesia are mediated by a gene-sex-environment interaction. Nature Neuroscience. 2011;14:1569–1573.
Mogil J.S., Wilson S.G., Bon K., et al. Heritability of nociception. I. Responses of eleven inbred mouse strains on twelve measures of nociception. Pain. 1999;80:67–82.
Mogil J.S., Wilson S.G., Bon K., et al. Heritability of nociception. II. “Types” of nociception revealed by genetic correlation analysis. Pain. 1999;80:83–93.
Mogil J.S., Wilson S.G., Chesler E.J., et al. The melanocortin-1 receptor gene mediates female-specific mechanisms of analgesia in mice and humans. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:4867–4872.
Morris-Yates A., Talley N.J., Boyce P.M., et al. Evidence of a genetic contribution to functional bowel disorder. American Journal of Gastroenterology. 1998;93:1311–1317.
Mulder E.J., Van Baal C., Gaist D., et al. Genetic and environmental influences on migraine: a twin study across six countries. Twin Research. 2003;6:422–431.
Mural R.J., Adams M.D., Myers E.W., et al. A comparison of whole-genome shotgun-derived mouse chromosome 16 and the human genome. Science. 2002;296:1661–1671.
Nackley A.G., Diatchenko L. Assessing potential functionality of catechol-O-methyltransferase (COMT) polymorphisms associated with pain sensitivity and temporomandibular joint disorders. Methods in Molecular Biology. 2010;617:375–393.
Nadeau J.H., Frankel W.N. The roads from phenotypic variation to gene discovery: mutagenesis versus QTLs. Nature Genetics. 2000;25:381–384.
Nagasako E.M., Oaklander A.L., Dworkin R.H. Congenital insensitivity to pain: an update. Pain. 2003;101:213–219.
Ndong C., Pradhan A., Puma C., et al. Role of rat sensory neuron-specific receptor (rSNSR1) in inflammatory pain: contribution of TRPV1 to SNSR signaling in the pain pathway. Pain. 2009;143:130–137.
Neely G.G., Hess A., Costigan M., et al. A genome-wide Drosophila screen for heat nociception identifies α2δ3 as an evolutionary conserved pain gene. Cell. 2010;143:628–638.
Nesic O., Lee J., Johnson K.M., et al. Transcriptional profiling of spinal cord injury–induced central neuropathic pain. Journal of Neurochemistry. 2005;95:998–1014.
Nielsen C.S., Price D.D., Vassend O., et al. Characterizing individual differences in heat-pain sensitivity. Pain. 2005;119:65–74.
Nielsen C.S., Stubhaug A., Price D.D., et al. Individual differences in pain sensitivity: genetic and environmental contributions. Pain. 2008;136:21–29.
Nishida K., Kuchiiwa S., Oiso S., et al. Up-regulation of matrix metalloproteinase-3 in the dorsal root ganglion of rats with paclitaxel-induced neuropathy. Cancer Science. 2008;99:1618–1625.
Nissenbaum J., Devor M., Seltzer Z., et al. Susceptibility to chronic pain following nerve injury is genetically affected by CACNG2. Genome Research. 2010;20:1180–1190.
Nissenbaum J., Shpigler H., Pisanté A., et al. pain 2: a neuropathic pain QTL identified on rat chromosome 2. Pain. 2008;135:92–97.
Nolan P.M., Peters J., Strivens M., et al. A systematic, genome-wide, phenotype-driven mutagenesis programme for gene function studies in the mouse. Nature. 2000;25:440–443.
Norbury T.A., MacGregor A.J., Urwin J., et al. Heritability of responses to painful stimuli in women: a classical twin study. Brain. 2007;130:3041–3049.
Nyholt D.R., LaForge K.S., Kallela M., et al. A high-density association screen of 155 ion transport genes for involvement with common migraine. Human Molecular Genetics. 2008;17:3318–3331.
Offenbaecher M., Bondy B., de Jonge S., et al. Possible association of fibromyalgia with a polymorphism in the serotonin transporter gene regulatory region. Arthritis and Rheumatism. 1999;42:2482–2488.
Ohara P.T., Vit J.P., Bhargava A., et al. Evidence for a role of connexin 43 in trigeminal pain using RNA interference in vivo. Journal of Neurophysiology. 2008;100:3064–3073.
Okumura M., Iwata K., Yasuda K., et al. Alternation of gene expression in trigeminal ganglion neurons following complete Freund’s adjuvant or capsaicin injection into the rat face. Journal of Molecular Neuroscience. 2010;42:200–209.
Osborne R.H., De George F.V. Genetic basis of morphological variation. Cambridge, Mass: Harvard University Press; 1959.
Panocka I., Marek P., Sadowski B. Inheritance of stress-induced analgesia in mice. Selective breeding study. Brain Research. 1986;397:152–155.
Patte-Mensah C., Meyer L., Schaeffer V., et al. Selective regulation of 3 alpha-hydroxysteroid oxido-reductase expression in dorsal root ganglion neurons: a possible mechanism to cope with peripheral nerve injury–induced chronic pain. Pain. 2010;150:522–534.
Paulson P.E., Gorman A.L., Yezierski R.P., et al. Differences in forebrain activation in two strains of rat at rest and after spinal cord injury. Experimental Neurology. 2005;196:413–421.
Peroutka S.J., Wilhoit T.L., Boatwright M., et al. Polymorphisms within the CACNL1A4 gene are not associated with migraine without aura or migraine with aura. Headache. 1997;37:326–327.
Persson A.K., Gebauer M., Jordan S., et al. Correlational analysis for identifying genes whose regulation contributes to chronic neuropathic pain. Molecular Pain. 2009;5:7.
Pettersson F.H., Anderson C.A., Clarke G.M., et al. Marker selection for genetic case-control association studies. Nature Protocols. 2009;4:743–752.
Porreca F., Lai J., Bian D., et al. A comparison of the potential role of the tetrodotoxin-insensitive sodium channels, PN3/SNS and NaN/SNS2, in rat models of chronic pain. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:7640–7644.
Poulsen L., Riishede L., Brosen K., et al. Codeine in post-operative pain. Study of the influence of sparteine phenotype and serum concentrations of morphine and morphine-6-glucuronide. European Journal of Clinical Pharmacology. 1998;54:451–454.
Quock R.M., Mueller J.L., Vaughn L.K., et al. Nitrous oxide antinociception in BXD recombinant inbred mouse strains and identification of quantitative trait loci. Brain Research. 1996;725:23–29.
Raber P., Devor M. Social variables affect phenotype in the neuroma model of neuropathic pain. Pain. 2002;97:139–150.
Rady J.J., Elmer G.I., Fujimoto J.M. Opioid receptor selectivity of heroin given intracerebroventricularly differs in six strains of inbred mice. Journal of Pharmacology and Experimental Therapeutics. 1999;288:438–445.
Reeves S.A., Rubio M.P., Louis D.N. General method for PCR amplification and direct sequencing of mRNA differential display products. BioTechniques. 1995;18:18–20.
Reimann F., Cox J.J., Belfer I., et al. Pain perception is altered by a nucleotide polymorphism in SCN9A. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:5148–5153.
Ren K., Novikova S.I., He F., et al. Neonatal local noxious insult affects gene expression in the spinal dorsal horn of adult rats. Molecular Pain. 2005;1:27.
Riant F., Ducros A., Ploton C., et al. De novo mutations in ATP1A2 and CACNA1A are frequent in early-onset sporadic hemiplegic migraine. Neurology. 2010;75:967–972.
Rigaud M., Gemes G., Barabas M.E., et al. Species and strain differences in rodent sciatic nerve anatomy: implications for studies of neuropathic pain. Pain. 2008;136:188–201.
Risch N., Merikangas K. The future of genetic studies of complex human diseases. Science. 1996;273:1516–1517.
Risch N.J. Searching for genetic determinants in the new millennium. Nature. 2000;405:847–856.
Rocha E.M., Prkachin K.M., Beaumont S.L., et al. Pain reactivity and somatization in kindergarten-age children. Journal of Pediatric Psychology. 2003;28:47–57.
Rode F., Thomsen M., Brolos T., et al. The importance of genetic background on pain behaviours and pharmacological sensitivity in the rat spared nerve injury model of peripheral neuropathic pain. European Journal of Pharmacology. 2007;564:103–111.
Rodriguez Parkitna J., Korostynski M., Kaminska-Chowaniec D., et al. Comparison of gene expression profiles in neuropathic and inflammatory pain. Journal of Physiology and Pharmacology. 2006;57:401–414.
Rohl T., Kurreck J. RNA interference in pain research. Journal of Neurochemistry. 2006;99:371–380.
Sandor P.S., Ambrosini A., Agosti R.M. Genetics of migraine: possible links to neurophysiological abnormalities. Headache. 2002;42:365–377.
Sato M., Ohashi J., Tsuchiya N., et al. Association of HLA-A*3303-B*4403-DRB1*1302 haplotype, but not of TNFA promoter and NKp30 polymorphism, with postherpetic neuralgia (PHN) in the Japanese population. Genes and Immunity. 2002;3:477–481.
Sayah D.M., Khan A.H., Gasperoni T.L., et al. A genetic screen for novel behavioral mutations in mice. Molecular Psychiatry. 2000;5:369–377.
Schena M., Shalon D., Davis R.W., et al. Quantitative monitoring of gene expression patterns with a complimentary DNA microarray. Science. 1995;270:467–470.
Seltzer Z., Wu T., Max M.B., et al. Mapping a gene for neuropathic pain–related behavior following peripheral neurectomy in the mouse. Pain. 2001;93:101–106.
Shir Y., Zeltser R., Vatine J.J., et al. Correlation of intact sensibility and neuropathic pain–related behaviors in eight inbred and outbred rat strains and selection lines. Pain. 2001;90:75–82.
Shoskes D.A., Albakri Q., Thomas K., et al. Cytokine polymorphisms in men with chronic prostatitis/chronic pelvic pain syndrome: association with diagnosis and treatment response. Journal of Urology. 2002;168:331–335.
Slade G.D., Diatchenko L., Bhalang K., et al. Influence of psychological factors on risk of temporomandibular disorders. Journal of Dental Research. 2007;86:1120–1125.
Smith S.B., Fillingim R.B., Slade G.D., et al. Candidate genes associate with psychological risk factors for chronic pain in OPPERA, a multi-site population-based cohort study. In Society of Neuroscience Abstracts. Chicago: Society for Neuroscience; 2009.
Smith S.B., Marker C.L., Perry C., et al. Quantitative trait locus and computational mapping identifies Kcnj9 (GIRK3) as a candidate gene affecting analgesia from multiple drug classes. Pharmacogenetics and Genomics. 2008;18:231–241.
Solovieva S., Leino-Arjas P., Saarela J., et al. Possible association of interleukin 1 gene locus polymorphisms with low back pain. Pain. 2004;109:8–19.
Spector T.D. The history of twin and sibling-pair studies. In: Spector T.D., Snieder H., MacGregor A.J., eds. Advances in twin and sib-pair analysis. Cambridge, United Kingdom: Greenwich Medical Media; 2000:1–10.
Stamer U.M., Lehnen K., Hothker F., et al. Impact of CYP2D6 genotype on postoperative tramadol analgesia. Pain. 2003;105:231–238.
Sufka K.J. Conditioned place preference paradigm: a novel approach for analgesic drug assessment against chronic pain. Pain. 1994;58:355–366.
Sun H., Xu J., Della Penna K.B., et al. Dorsal horn–enriched genes identified by DNA microarray, in situ hybridization and immunohistochemistry. BMC Neuroscience. 2002;3:11.
Takahashi T., Aoki Y., Okubo K., et al. Upregulation of Ca(v)3.2 T-type calcium channels targeted by endogenous hydrogen sulfide contributes to maintenance of neuropathic pain. Pain. 2010;150:183–191.
Tamarin R.H. Principles of genetics, ed 5. Dubuque, Iowa: William C. Brown; 1996.
Tan P.H., Chia Y.Y., Chow L.H., et al. Gene knockdown of the N-methyl-D-aspartate receptor NR1 subunit with subcutaneous small interfering RNA reduces inflammation-induced nociception in rats. Anesthesiology. 2010;112:1482–1493.
Tan P.H., Yang L.C., Shih H.C., et al. Gene knockdown with intrathecal siRNA of NMDA receptor NR2B subunit reduces formalin-induced nociception in the rat. Gene Therapy. 2005;12:59–66.
Tegeder I., Costigan M., Griffin R.S., et al. GTP cyclohydrolase and tetrahydrobiopterin regulate pain sensitivity and persistence. Nature Medicine. 2006;12:1269–1277.
Terner J.M., Barrett A.C., Lomas L.M., et al. Influence of low doses of naltrexone on morphine antinociception and morphine tolerance in male and female rats of four strains. Pain. 2006;122:90–101.
Treloar S.A., Martin N.G., Heath A.C. Longitudinal genetic analysis of menstrual flow, pain, and limitation in a sample of Australian twins. Behavior Genetics. 1998;28:107–116.
Tumati S., Milnes T.L., Yamamura H.I., et al. Intrathecal Raf-1–selective siRNA attenuates sustained morphine-mediated thermal hyperalgesia. European Journal of Pharmacology. 2008;601:207–208.
Urca G., Segev S., Sarne Y. Footshock-induced analgesia: its opioid nature depends on the strain of rat. Brain Research. 1985;329:109–116.
Valder C.R., Liu J.J., Song Y.H., et al. Coupling gene chip analyses and rat genetic variances in identifying potential target genes that may contribute to neuropathic allodynia development. Journal of Neurochemistry. 2003;87:560–573.
van de Beek W.J., Roep B.O., van der Slik A.R., et al. Susceptibility loci for complex regional pain syndrome. Pain. 2003;103:93–97.
Vega-Avelaira D., Geranton S.M., Fitzgerald M. Differential regulation of immune responses and macrophage/neuron interactions in the dorsal root ganglion in young and adult rats following nerve injury. Molecular Pain. 2009;5:70.
Veldman P.H.J.M., Reynen H.M., Arntz I.E., et al. Signs and symptoms of reflex sympathetic dystrophy: prospective study of 829 patients. Lancet. 1993;342:1012–1016.
Venter J.C., Adams M.D., Myers E.W., et al. The sequence of the human genome. Science. 2001;291:1304–1351.
Vit J.P., O’Hara P.T., Bhargava A., et al. Silencing the Kir4.1 potassium channel subunit in satellite glial cells of the rat trigeminal ganglion results in pain-like behavior in the absence of nerve injury. Journal of Neuroscience. 2008;28:4161–4171.
Wade C.M., Kulbokas E.J., III., Kirby A.W., et al. The mosaic structure of variation in the laboratory mouse genome. Nature. 2002;420:574–578.
Wahlestedt C. Antisense oligonucleotide strategies in neuropharmacology. Trends in Pharmacological Sciences. 1994;15:42–46.
Walker J.S., Sheather-Reid R.B., Carmody J.J., et al. Nonsteroidal antiinflammatory drugs in rheumatoid arthritis and osteoarthritis. Arthritis and Rheumatism. 1997;40:1944–1954.
Wall J.D., Pritchard J.K. Haplotype blocks and linkage disequilibrium in the human genome,. Nature Reviews, Genetics. 2003;4:587–597.
Wang H., Sun H., Della Penna K.B., et al. Chronic neuropathic pain is accompanied by global changes in gene expression and shares pathobiology with neurodegenerative diseases. Neuroscience. 2002;114:529–546.
Wang J., Liao G., Usuka J., et al. Computational genetics: from mouse to human. Trends in Genetics. 2005;21:526–532.
Waterston R.H., Linblad-Toh K., Birney E., for the Mouse Genome Sequencing Consortium, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562.
Watson S.J., Akil H. Gene chips and arrays revealed: a primer on their power and their uses. Biological Psychiatry. 1999;45:533–543.
Wei F., Dubner R., Zou S., et al. Molecular depletion of descending serotonin unmasks its novel facilitatory role in the development of persistent pain. Journal of Neuroscience. 2010;30:8624–8636.
Wilson S.G., Bryant C.D., Lariviere W.R., et al. The heritability of antinociception II: pharmacogenetic mediation of three over-the-counter analgesics in mice. Journal of Pharmacology and Experimental Therapeutics. 2003;305:755–764.
Wilson S.G., Chesler E.J., Hain H.S., et al. Identification of quantitative trait loci for chemical/inflammatory nociception in mice. Pain. 2002;96:385–391.
Wilson S.G., Smith S.B., Chesler E.J., et al. The heritability of antinociception: common pharmacogenetic mediation of five neurochemically distinct analgesics. Journal of Pharmacology and Experimental Therapeutics. 2003;304:547–559.
Wolff B.B., Kantor T.G., Jarvik M.E., et al. Response of experimental pain to analgesic drugs. I. Morphine, aspirin, and placebo. Clinical Pharmacology and Therapeutics. 1965;7:224–238.
Wu D., Wang X., Chen D., et al. Metabolic gene polymorphisms and risk of dysmenorrhea. Epidemiology. 2000;11:648–653.
Wu F.X., Bian J.J., Miao X.R., et al. Intrathecal siRNA against Toll-like receptor 4 reduces nociception in a rat model of neuropathic pain. International Journal of Medical Sciences. 2010;7:251–259.
Xiao H.S., Huang Q.H., Zhang F.X., et al. Identification of gene expression profile of dorsal root ganglion in the rat peripheral axotomy model of neuropathic pain. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:8360–8365.
Xu Q., Garraway S.M., Weyerbacher A.R., et al. Activation of the neuronal extracellular signal–regulated kinase 2 in the spinal cord dorsal horn is required for complete Freund’s adjuvant–induced pain hypersensitivity. Journal of Neuroscience. 2008;28:14087–14096.
Xu X.J., Plesan A., Yu W., et al. Possible impact of genetic differences on the development of neuropathic pain–like behaviors after unilateral sciatic nerve ischemic injury in rats. Pain. 2001;89:135–145.
Xu X.J., Wiesenfeld-Hallin Z. Individual differences in pain: rat models. In: Mogil J.S., ed. Progress in pain research and management. Seattle: IASP Press; 2004:107–121.
Yang H.Y., Mitchell K., Keller J.M., et al. Peripheral inflammation increases Scya2 expression in sensory ganglia and cytokine and endothelial related gene expression in inflamed tissue. Journal of Neurochemistry. 2007;103:1628–1643.
Yang L., Zhang F.X., Huang F., et al. Peripheral nerve injury induces trans-synaptic modification of channels, receptors and signal pathways in rat dorsal spinal cord. European Journal of Neuroscience. 2004;19:871–883.
Yang Y., Wang Y., Li S., et al. Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. Journal of Medical Genetics. 2004;41:171–174.
Young M.R., Blackburn-Munro G., Dickinson T., et al. Antisense ablation of type I metabotropic glutamate receptor mGluR1 inhibits spinal nociceptive transmission. Journal of Neuroscience. 1998;18:10180–10188.
Yukhananov R., Kissin I. Persistent changes in spinal cord gene expression after recovery from inflammatory hyperalgesia: a preliminary study on pain memory. BMC Neuroscience. 2008;9:32.
Zhang F.X., Liu X.J., Gong L.Q., et al. Inhibition of inflammatory pain by activating B-type natriuretic peptide signal pathway in nociceptive sensory neurons. Journal of Neuroscience. 2010;30:10927–10938.
Zubieta J.K., Heitzeg M.M., Smith Y.R., et al. COMT val158met genotype affects μ-opioid neurotransmitter responses to a pain stressor. Science. 2003;299:1240–1243.
Zubieta J.K., Smith Y.R., Bueller J.A., et al. Regional mu opioid receptor regulation of sensory and affective dimensions of pain. Science. 2001;293:311–315.
†Deceased.