Opioids
Basic Mechanisms
Introduction
The use of opium as a drug dates back to thousands of years BC, and use of this extract of the exudate of Papaver somniferum has been traced through many ancient civilizations, including Persia, Egypt, and Mesopotamia. Archeology hints that Neanderthals used the opium poppy more than 30,000 years ago. Homer in The Odyssey calls it “…a drug that had the power of robbing grief and anger of their sting and banishing all painful memories….” Morphine, the main active agent in opium, has become the “gold standard” analgesic to which all other opioids are compared. Molecular cloning of the three main (or “classic”) opiate receptors—mu, delta, and kappa—and ongoing development of different agonists and antagonists for the receptors have allowed many studies on the physiological roles of the opioid receptors. However, the development of novel potent analgesics acting on opioid receptors, which potentially lack the typical mu receptor–mediated side effects, has not yet been achieved. In addition, studies over the past decade have revealed that morphine and other mu opioids do not always produce the same degree of analgesia and tolerance in all conditions. Thus, distinct mu agonists can differentially activate and regulate mu receptor activity; furthermore, opioid analgesia can be altered by the presence of inflammation and also by nerve damage. This chapter examines current knowledge on the molecular aspects of opioid receptors, the molecular mechanisms of action of opioids at their receptors, and their activity in the spinal cord and brain relevant to pain relief. We also report on data on the mechanisms by which opioid controls can be altered in different pain states and will attempt to relate this basic research to opioid therapy in patients.
Opioid Receptors: Molecular Aspects
Molecular Components of the Opioid System: Receptors, Peptides, and Their Genes
From the very early days of opioid research it seemed obvious that opiate alkaloids act on the nervous system. The existence of specific receptors was demonstrated by the presence of high-affinity and saturable binding sites in brain membrane preparations (Pert and Snyder 1973, Simon et al 1973, Terenius 1973). Naloxone, a synthetic morphine derivative, was found to block morphine activity and was considered the prototypical opioid antagonist. From there, any biological activity that was reversed by naloxone was considered opioid in nature. Synthetic chemistry provided novel alkaloid compounds with opioid activity that revealed several classes of opioid receptors (Martin et al 1976). Ultimately, three major opioid receptor subtypes emerged from pharmacological studies, referred to as mu, delta, and kappa receptors. Their molecular characterization, however, waited almost another 20 years because of the paucity and strongly hydrophobic properties of these membrane receptor proteins.
The first opioid receptor to be characterized at the molecular level was a mouse delta receptor. Molecular cloning of this receptor was achieved by expression cloning (Evans et al 1992, Kieffer et al 1992). Isolation of this cDNA represented a milestone in opioid research (Barnard 1993, Brownstein 1993) and opened the way to functional exploration of the opioid system by mutagenesis approaches in vitro and in vivo. A first step was molecular identification of an opioid receptor gene family, including mu, delta, and kappa, as well as the closely related ORL1 receptors (Taylor and Dickenson 1998). Their genes have now been cloned in many species, including humans, rodents, amphibians, and zebra fish. In humans and in mice the four genes are highly homologous in their intron/exon organization, possibly deriving from a common ancestor (for review, see Kieffer 1995, 1997; see also Table 30-1).
Table 30-1
The Opioid Receptor Gene Family

Four homologous genes have been identified in the mammalian genome. Because of strong sequence homology and similar genomic organization, the ORL receptor gene is part of the opioid receptor gene family. This receptor, however, does not show high-affinity binding for opioid ligands. All the genomic information is available at http://www.ncbi.nlm.nih.gov.
Compound B, (1-[(3R,4R)-1-cyclooctylmethyl-3-hydroxymethyl-4-piperidyl]-3-ethyl-1,3-dihydro-2H-benzimidazol-2-one); CTAP, (D-Phe-Cys-Tyr-D-Trp-Arg-Thr-Pen-Thr-NH2); DAMGO, ([D-Ala2,N-Me-Phe4,Gly-ol5]enkephalin); DPDPE, ([D-Pen2,D-Pen5]-enkephalin); OFQ, orphanin FQ).
In the early 1970s, demonstration of opioid binding sites launched the search for endogenous ligands. Met- and leu-enkephalins, two closely related pentapeptides, were first purified from brain and sequenced (Hughes et al 1975). Many more peptides have since been isolated from nervous tissue, the pituitary gland, and the adrenals (Akil et al 1984). These peptides share a common N-terminal sequence—YGGFL/M—considered the opioid pharmacophore and partially overlapping the morphine structure (Barnard 1993). Three genes encoding those peptides were cloned in the early 1980s (Nakanishi et al 1979, Comb et al 1982, Kakidani et al 1982). The genes encode large precursor proteins, proopiomelanocortin, preproenkephalin, and preprodynorphin. Proopiomelanocortin produces the largest opioid peptide, β-endorphin, as well as peptides with non-opioid activities. The preproenkephalin and preprodynorphin precursors are processed to generate several copies of enkephalin and dynorphin peptides, respectively. All members of this large family of opioid peptides act as agonists at mu, delta, and kappa receptors with nanomolar affinity and limited receptor selectivity (Akil et al 1998). The discovery of endogenous opioid peptides further broadened the panel of available opioid ligands, and synthesis of a vast plethora of enkephalin derivatives completed the large repertoire of alkaloid-type opioids (Corbett et al 1993) to study mu, delta, and kappa receptor function.
Opioid Receptors: Structure–Activity
Opioid receptors belong to the superfamily of G protein–coupled receptors (GPCRs). This receptor family comprises several hundred members in the human genome (Lagerstrom and Schioth 2008). GPCRs contain seven hydrophobic transmembrane domains interconnected by short loops and display an extracellular N-terminal domain and an intracellular C-terminal tail (Fig. 30-1A). A classic serpentine representation the delta receptor is shown in Figure 30-1B.
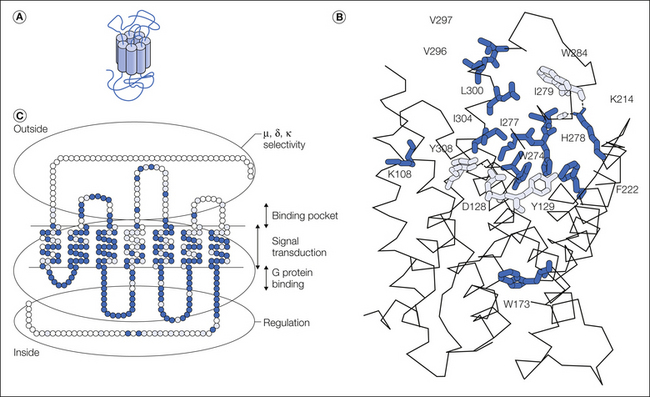
Figure 30-1 Opioid receptor structure and signaling.
A, Functional domains of opioid receptors. A serpentine representation of the delta opioid receptor is shown. Amino acid residues that differ across mu, delta and kappa receptor are shown as open circles, while conserved or identical residues are indicted as grey and black circles, respectively. B, Opioid receptor signaling. The agonist (A)-bound receptor may activate G protein (Gi/o)-dependent (E1) or –independent (E2) effectors. The active receptor/effector complexes modify neuronal excitability and cell transcription in an agonist-specific and cell-dependent manner. C, Ligand-specific signaling complexes at the mu opioid receptor from both in vitro and in vivo studies. Treatment with morphine or DAMGO elicits differential signaling and trafficking of the mu opioid receptor. Activation of the mu opioid receptor by the low-internalizing agonist morphine or the high-internalizing agonist DAMGO leads to the formation of distinct signaling complexes, resulting for example in distinct desensitization mechanisms (PKC- or GRK2-dependent pathway). (adapted from Pradhan et al 2012)
Mu, delta, and kappa receptors are highly homologous, with transmembrane domains and intracellular loops best conserved (86%–100%). The extracellular loops, however, as well as N- or C-terminal tails, differ largely. Sequence comparisons, combined with mutagenesis experiments, have led to the identification of receptor domains with specific functions (Fig. 30-1C). Structure–activity relationships in opioid receptors have been evaluated in great detail in several reviews (see Befort and Kieffer 1997, Law et al 1999a, Chaturvedi et al 2000, Décaillot and Kieffer 2003).
Receptor Structure
The first structure of a GPCR, the bovine rhodopsin, was solved by x-ray cristallography (Palczewski et al 2000) and was long used as a template for structure-function predictions at GPCRs. Another 7 years were required until first GPCRs bound to hormones or neurotransmitters were crystallized and their structure solved at atomic level (Audet and Bouvier 2012). In 2012, 20 years after opioid receptor cloning, a structure was reported for each member of the opioid receptor gene family. The receptors were crystallized under an inactive form, were bound to an antagonist, and structures of the mu receptor-beta-funaltrexamine (Manglik et al 2012), the delta receptor-naltrindole (Granier et al 2012), the kappa receptor-JDTic (Wu et al 2012), and the ORL-1 receptor-C-24 (Thompson et al 2012) complexes are now available (PBD accession numbers 4DKL, 4EJ4, ADJH and 4EA3, respectively). The four receptors share a conserved wide-open binding pocket contrasting with buried pockets of other GPCRs crystallized so far. Mu receptors form intimate oligomeric pairs within the crystal (Manglik et al 2012), supporting the view that opioid receptors may function as homo- or heterodimers, or even larger oligomers (Jordan and Devi 1999). This breakthrough in opioid research reveals ligand-binding modes and enables the development of structure-based approaches to design better drugs. A next challenge will be to elucidate receptor activation mechanisms and understand receptor signaling at structural level.
The Binding Site
Depending on the ligand type, the binding site of GPCRs is either located in extracellular domains (e.g., thrombin receptor) or buried within the seven-helix bundle (small biogenic amine receptors). Alternatively, the binding site can overlap both the external and transmembrane regions, as is the case for peptidic GPCRs, including opioid receptors.
The extracellular loops (e1, e2, and e3) in opioid receptors establish first contact with the ligand approaching the binding site and are important for mu/delta/kappa selectivity. The study of chimeric mu receptors incorporating delta, kappa, or even angiotensin II receptor domains led to the proposal that e1 and e3 are important determinants in the mu receptor for the high-affinity binding of mu-selective compounds. E3 in the delta receptor was dissected by both loss-of-function and binding rescue experiments and was shown to be the most critical site in this receptor for high-affinity binding of delta-selective ligands. In kappa receptors, acidic amino acid residues of e2 seem to represent a unique feature that would favor the binding of basic dynorphin peptides, and e3 also contributes to the recognition of small non-peptidic kappa-selective compounds. Altogether, the extracellular domains of opioid receptors are considered to be both anchoring points for large opioid ligands and gates filtering opioid entry into the binding pocket.
In contrast to extracellular domains, transmembrane (Tm) domains are highly conserved and form an opioid binding pocket that is similar across mu, delta, and kappa receptors. Three-dimensional computer models have highlighted a binding pocket penetrating the upper half of the helical bundle that consists of two subsites: a large hydrophobic domain is formed by aromatic residues spanning Tm3–Tm7, whereas a hydrophilic area lies over Tm3 and Tm7. Amino acid residues were tested by site-directed mutagenesis, and data from studies on the delta receptor, for example, confirmed the implication that the hydrophobic pocket is formed by Y129 (Tm3); W173 (Tm4); F222 (Tm5); W274, I277, I278, H279, and W284 (Tm6); and L300, I304, and Y308 (Tm7). The hydroxyl groups in Y129 (Tm3) and Y308 (Tm7), as well as the carboxyl group of D128 (Tm3), delimit the hydrophilic part of the site, and D128 was proposed to be the counter ion for the universal protonated amine present in every opioid ligand (for discussion, see Décaillot and Kieffer 2004). The binding crevice of all three receptors was investigated by cysteine accessibility scanning of Tm6. The outward half of the helix was found to be water accessible in each case, consistent with the notion of a binding pocket penetrating the helical bundle halfway.
Receptor Activation
Several site-directed mutagenesis experiments provided the first hints on activation determinants in opioid receptors. Most interesting are mutations inducing constitutive activation of the receptor (i.e., ligand-independent activity). In the delta receptor, D128 (Tm3) replaced by Q, A, K, or H enhances spontaneous activity of the receptor (Befort et al 1999, Cavalli et al 1999). Furthermore, the Y308F mutant (Tm7) is also a constitutively activated mutant (CAM) receptor. Three-dimensional modeling indicates a possible hydrogen bond between D128 and Y308, which suggests that a Tm3–Tm7 interhelical interaction could contribute to maintain the receptor in an inactive conformation (Befort et al 1999). Interestingly, the mutation of a conserved S in Tm4 unexpectedly transformed the classic antagonists—in particular, naloxone—into agonists in mu, delta, and kappa receptors, thus suggesting a role of Tm4 in the activation process (Claude et al 1996, Law et al 1999). These site-directed mutagenesis studies, however, remain limited to agonist binding domains of the receptor.
Recently, a random mutagenesis approach was used in an attempt to visualize the entire activation process without any preconceived model-guided assumption (Décaillot et al 2003). A collection of about 3000 delta receptor mutants was generated randomly and screened for constitutive activity with a reporter gene–signaling assay. Thirty CAM receptors were isolated and point mutations were found distributed throughout the receptor protein, thus indicating that many receptor domains could contribute to receptor activation. Mutations within the helical bundle, as well as e3, were analyzed on a receptor three-dimensional model. Strikingly, activating mutations clustered in space and revealed an activation path throughout the receptor protein. From this study, a mechanism was proposed in which the opioid ligand would bind to e3 and destabilize Tm6–Tm7 interactions at the extracellular face of the receptor. Entering the binding pocket, the amphiphilic agonist would disrupt the strong hydrophobic and hydrophilic interactions that maintain Tm3–Tm6–Tm7 tightly packed in the inactive receptor. Tm3 would move toward Tm4, whereas Tm6 and Tm7 would separate from each other. This helical movement would propagate to the cytoplasmic face of the receptor and break an ionic lock between Tm6 and Tm7. The resulting structural modifications of i3 and C-terminal domains proximal to the membrane would favor G-protein activation. The latter step is consistent with the previous observation that peptides competing with i3, but not i2, impair delta receptor coupling (Georgoussi et al 1997). Many of the residues found mutated in the random mutagenesis study are conserved in mu, delta, and kappa receptors, as well as in other GPCRs, thus suggesting that this mechanism may apply broadly.
Receptor Signaling
Opioid receptors are coupled to Go/Gi inhibitory proteins, and modulation of many G-protein effectors has been demonstrated in both transfected cells and native tissue (for review, see Law et al 2000, Williams et al 2001). Opioids inhibit voltage-dependent calcium channels or activate inwardly rectifying potassium channels, thereby decreasing neuronal excitability. Opioids also inhibit the cyclic adenosine monophosphate pathway and activate mitogen-activated protein kinase cascades, both activities affecting cytoplasmic events and transcriptional activity of the cell. Finally, crosstalk between the mu opioid receptor and the insulin receptor was recently demonstrated (Li et al 2003), thus broadening the panel of opioid receptor–associated signaling cascades. Consistent with their highly homologous intracellular loops, all three opioid receptors show similar coupling properties, although some differences in the ratios of mu-, delta-, or kappa-activated Go/Gis have been observed in heterologous expression systems (Law et al 1999b). Opioid receptors also stimulate G protein-independent signaling pathways, notably via β–arrestins (Shukla et al 2011), and activate phosphorylation cascades that ultimately modify gene transcription and durably affect cell physiology. Overall opioid receptor activation leads to inhibit neuronal activity (Fig. 30-1B) and a main goal in the field of opioid receptor signaling, as for GPCR signaling in general, is the identification of signaling pathways that indeed operate in vivo and control specific behavioral responses and drug effects. Whether this is the case in neuronal networks remains to be clarified.
Research over the last decade has indicated that G protein-coupled receptors exist in multiple conformations and that agonists can stabilize different active states. The distinct receptor conformations induced by ligands result in distinct receptor-effector complexes, which produce varying levels of activation or inhibition of subsequent signaling cascades. As a consequence, the agonist-receptor-effector complex–rather than the receptor itself–is the key determinant for subsequent cellular and in vivo signaling (Fig. 30-1B). This concept, referred to ligand-directed signaling or biased agonism, has important biological and therapeutic implications (Galandrin et al 2007). Many in vitro studies have shown biased agonism at mu, delta, and kappa receptors (see Fig. 30-1C for mu). Importantly, in vivo consequences of this phenomenon have recently been demonstrated (reviewed in Pradhan et al 2012). For the mu and delta opioid receptors, agonist-specific signaling and trafficking events are observed in response to both acute and repeated drug administration in vivo. The ability of mu agonists to internalize the receptor influences drug analgesic efficacy and tolerance, as well as addictive behaviors (Kim et al 2008, Berger et al 2011). Similarly, internalizing properties of delta agonists strongly influence the development of tolerance, which evolves into either generalized tolerance or pain-specific tolerance (Pradhan et al 2010, Pradhan et al 2011). For the kappa opioid receptor, ligand-specific signaling responses have been proposed to differentially mediate analgesic and dysphoric effects, which potentially are dissociable at the level of downstream effectors (Bruchas and Chavkin 2010).
Overall, biased agonism is one of the several mechanisms by which opioid ligands can produce diverse physiological effects and may underlie the highly complex opioid pharmacological heterogeneity. Future studies will indicate whether the concept of ligand-dependent responses for a given receptor translates into tailor-made pharmacotherapies where advantageous drug effects are selectively targeted over adverse effects. Most important to cell physiology is rapid termination of receptor signaling, and several regulatory processes are known to follow agonist-induced receptor activation. Such processes include phosphorylation of intracellular receptor determinants by a number of protein kinases, binding of arrestin to the phosphorylated domains, uncoupling of receptors from G proteins, rapid receptor endocytosis, and receptor recycling or down-regulation. All these phenomena lead to desensitization of receptor signaling. Receptor truncation or point mutagenesis experiments in opioid receptors have demonstrated the important role of the C-terminal tail in all the regulatory events (for delta receptor, see Décaillot and Kieffer 2004). The correlation between these distinct events has been further investigated, particularly in mu receptor mutants (for review, see Law et al 1999), and data indicate that phosphorylation is not obligatory for internalization or that down-regulation does not necessarily correlate with desensitization. Therefore, many distinct molecular mechanisms concomitantly contribute to the modulation of opioid receptor activities, all of which involve interaction of intracellular receptor domains with specific cytoplasmic proteins. These clearly differ from cell to cell and, for a large part, remain to be discovered (Brady and Limbird 2002). Obviously, the large differences in the C-terminal structures of opioid receptors should lead to distinct mu, delta, and kappa receptor physiology despite their similar binding and transduction abilities (Fig. 30-1C). A good example is the observation that after agonist-induced internalization, mu receptors efficiently recycle to the cell surface, whereas delta receptors are committed to lysosomal degradation, two distinct endocytic fates that can be modified by C-terminal swapping (Tanowitz and von Zastrow 2003).
Opioid Receptor Diversity: The Molecular Basis for Pharmacological Subtypes
Opioid receptor pharmacology is complex, and the existence of multiple mu (Pasternak 1993), delta (Traynor and Elliott 1993, Zaki et al 1996), and kappa (Traynor 1989) receptor types has been proposed from a wide plethora of in vitro and in vivo experiments. Gene cloning has provided three receptor genes only, and the molecular basis for pharmacological diversity remains a matter of debate (Zaki et al 1996, Befort and Kieffer 1997). Homology cloning, as well as bioinformatic search in mammalian genomes, shows no evidence of additional closely related opioid receptor genes. Alternative splicing could potentially generate receptor protein variants from the three known opioid receptor genes. Reverse transcriptase polymerase chain reaction experiments have allowed the detection of alternative transcripts in several cell lines or tissues for all three receptors, with their abundance definitely being lower than that of the three known transcripts. Delta and kappa receptor mRNA variants encode truncated—and thus probably non-functional—receptors (for review, see Gavériaux-Ruff and Kieffer 1999). Alternative mu receptor mRNA molecules encode receptors with variable C termini, and some alterations in the binding or signaling properties of the putative encoded receptors have been reported in transfected systems (Bolan et al 2004).
To date, it has been extremely difficult to establish the biological relevance of these alternative transcripts in vivo and to correlate their existence with the multiple opioid receptor subtypes described earlier by the pharmacology. On the other hand, increasing evidence supports the notion that the three known mu, delta, and kappa receptor proteins may adopt multiple active conformations that would contribute to their pharmacological heterogeneity. In an early report, extensive site-directed mutagenesis of the delta receptor opioid binding site showed unique sets of interactions for each of the 12 opioid compounds under study (Befort et al 1996). These data suggested the existence of multiple binding sites, which opened the way to the possible existence of multiple ligand–receptor conformational states. This was later substantiated functionally with the discovery of ligand-dependent receptor responses. Particularly striking was the finding that in contrast to the peptidic mu agonist DAMGO, morphine was unable to induce mu receptor internalization whereas both compounds efficiently signaled within the cell (Arden et al 1995, Keith et al 1996). Further studies in transfected cells confirmed that receptor activation and subsequent regulation are strongly agonist dependent and that agonist efficacy does not necessarily correlate with their ability to induce receptor phosphorylation, internalization, or desensitization (von Zastrow et al 2003). The best possible interpretation of these data is that multiple active conformations of the receptor do exist and are being stabilized by individual ligands, a notion that has been proposed and investigated for other GPCRs (for a recent example, see Beinborn et al 2004). An important implication is that the ligand–receptor complex, rather than the receptor itself, determines the ultimate physiological cellular response. Therefore, a single receptor protein (e.g., the mu receptor) is able to trigger distinct pharmacological responses depending on the bound ligand.
At least as important as the interacting ligand is the cellular environment of the receptor. Many proteins directly interact with the receptor, thereby modulating receptor conformation and activity. Beyond the known Go/Gi alpha proteins calmodulin, kinases, and arrestins, other regulatory proteins, multidomain scaffolding units, or chaperone molecules potentially influence opioid receptor pharmacology, as was demonstrated for several other GPCRs (Brady and Limbird 2002). Proteomic approaches have recently led to the identification of novel interacting partners for opioid receptor C-terminal tails, including signaling (phospholipase A2), cytoskeletal (filamin and periplakin), and sorting (GASP) proteins (for review, see Contet et al 2004). This research field is expanding rapidly and the importance of these interactions on opioid binding and signaling is being investigated.
Another potential receptor modulator is the receptor itself. The possibility that GPCRs may exist as dimeric or oligomeric complexes has gained much support in recent years. The co-expression of delta and kappa receptors in transfected cells produced delta–kappa heterodimers, as revealed by co-immunoprecipitation experiments, and most importantly, resulted in substantial modifications of ligand binding (Jordan and Devi 1999). In this experimental setting, the decreased binding of selective mu and delta ligands with a concomitant increase in the binding of non-selective opioids was consistent with a kappa-2 pharmacology. Similarly, mu and delta receptor co-expression led to an alteration in opioid binding and signaling that was attributed to receptor dimerization (for review, see Devi 2001, Levac et al 2002). Taken together, the data suggest that the physical association of opioid receptors—either as homodimers or as heterodimers—creates novel receptor entities with unique pharmacological activities that can potentially enlarge opioid receptor heterogeneity. Whether receptor dimerization or interaction with other protein partners truly modulates opioid pharmacology in vivo remains an important question.
Finally, the entire cellular context adds another level of complexity to biological responses to agonists. First, G-alpha subunits, considered the classic signaling effectors, differ across cell types. Second, the number of possible G protein–associated signaling pathways has expanded dramatically beyond the classic second-messenger generating systems (Marinissen and Gutkind 2001). Altogether, the variable combinations of G-alpha proteins and the nature of associated intracellular signaling networks necessarily generate cell-specific responses that represent another source of pharmacological heterogeneity, particularly in the analysis of complex responses such as nociceptive behavior.
In conclusion, molecular approaches have provided a novel prospect of the opioid receptor that should be viewed as a dynamic multicomponent unit rather than as a single protein entity. The specific biological response is elicited and determined by the ligand-bound receptor within its neuronal microenvironment. The possibility of multiple active conformations of the receptor, which are influenced by the chemical nature of the agonist and modulated by interacting membrane or intracellular protein partners, forms the basis for enormous opioid receptor diversity. Hence, there is no doubt that several mu, delta, and kappa pharmacological profiles or “subtypes” can arise from activation of the three known mu, delta, and kappa receptor proteins, depending on the experimental setting.
In fact, as in vivo molecular pharmacology develops, it is likely that the complexity of opioid responses will extend well beyond the previously reported pharmacological subtypes. Particularly intriguing was the recent observation that morphine efficiently internalizes mu receptors in dendrites but not in the cell bodies of nucleus accumbens neurons (Haberstock-Debic et al 2003), a finding that could not be anticipated from previous studies of classic expression systems or from experiments using brain homogenates and whole animals. Cellular compartmentalization—and therefore the precise localization of the receptor—is yet an additional source of pharmacological heterogeneity.
The finding of agonist-driven responses at the mu receptor has important therapeutic implications. Gene knockout experiments have definitively demonstrated that mu receptors mediate all morphine activities (Matthes et al 1996, Kieffer and Gavériaux-Ruff 2002), thereby ruling out the possibility that the beneficial (analgesia) and non-desirable (tolerance, dependence, respiratory depression) activities of the prototypical opiate could be mediated by two separate molecular entities—and hence distinct therapeutic targets. Mu receptor signaling, however, is dissociable from mu receptor phosphorylation, internalization, or down-regulation (see above). Because morphine signaling triggers low levels of receptor regulation, there has been much speculation on the possibility that sustained morphine signaling may in fact induce the cellular and neuronal adaptations responsible for tolerance and dependence (Kieffer and Evans 2002). There are therefore at least two “types” of active mu receptors that could be defined as either highly regulated or poorly regulated receptors and be represented by DAMGO/mu and morphine/mu receptor complexes. Although most presently available mu agonists seem to induce profound tolerance in vivo (Evans et al 2000), the intriguing possibility exists that targeting the highly regulated receptor conformation may lead to a potent non-addictive analgesic drug. Because we are only starting to understand the molecular basis for mu opioid receptor heterogeneity, rational design of such compounds at present represents a far distant goal.
Opioid Physiology and Pain
Deleting Opioid Receptor or Peptide Genes in Vivo: Consequences on Pain Pharmacology
Homologous recombination techniques have led to the production of mice with targeted gene deletions, and mouse lines lacking each genetic component of the opioid system have been created, including the knockout of mu, delta, or kappa receptor genes, as well as endorphin/proopiomelanocortin, preproenkephalin, or preprodynorphin genes (for review, see Kieffer and Gavériaux-Ruff 2002). All the mutant mice are viable and fertile, as are triple-mutant mice lacking all the opioid receptor genes, thus indicating that the opioid system is not essential for survival. The spontaneous behavior of these knockout lines has been analyzed by many investigators, as well as the responses of receptor-deficient mice to prototypal opiate or non-opioid drugs (Gavériaux-Ruff and Kieffer 2002, Kieffer and Gavériaux-Ruff 2002, Pintar and Kieffer 2004). Here we will summarize a number of findings that relate to pain research.
One important issue was the clarification of in vivo molecular targets for clinically useful opiates or for opioid compounds that have been classically used in pharmacology to study mu, delta, and kappa receptor function. A significant finding was the abolition of all morphine effects in mice lacking mu receptors, which indicated that a single gene product is essential for the wide spectrum of both the beneficial and adverse activities of morphine. Furthermore, the strong decrease in U50,488H hypolocomotion, analgesia, and aversion in kappa receptor–deficient mice and the maintained U50,488H analgesia in mice lacking either mu or delta receptors confirmed the selective activity of this prototypal kappa agonist in vivo. This also demonstrated that kappa receptor–mediated analgesia is independent from mu- and delta receptor–mediated analgesia, at least in models of acute thermal pain. Finally, examination of the analgesic activities of DPDPE and deltorphin, considered the reference selective delta agonists, has been very informative. Results from studies performed in separate laboratories investigating delta agonist activities in either mu or delta receptor knockout mice have been extremely diverse. Data showed either maintained or decreased antinociceptive properties of the two compounds in both mutant animals (Kieffer and Gavériaux-Ruff 2002). One recent study reported a direct comparison of the two mouse lines in response to DPDPE and deltorphin treatment (Scherrer et al 2004). In the tail immersion test there was no analgesia in the mu null mutant and full analgesia in the delta null mutant, the latter being reversed by the mu antagonist CTOP. In the hot plate test, the two compounds produced full analgesia in the delta knockout mice and lower but significant analgesia in the mu mutants. In this test also, deltorphin elicited weak but measurable analgesia in the mu/kappa double-null mutant, in which only delta receptors were expressed. Together, these data suggest that mu rather than delta receptors are recruited by delta agonists in spinal analgesia and that both mu and delta receptors contribute to supraspinal analgesia. Although the conclusions apply only to behavioral models of acute thermal pain, these data have revealed the need to develop more selective non-peptidic delta agonists to further validate delta receptors as possible therapeutic targets in pain control. Conclusions on opioid pharmacology from knockout mice data are shown in Figure 30-2.
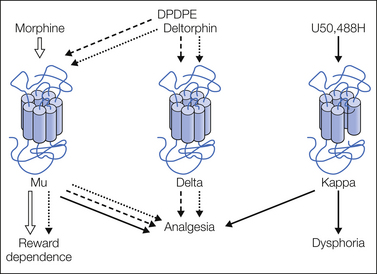
Figure 30-2 Opioid receptor gene knockout and opioid pharmacology.
Pharmacological activities of prototypal opioid receptor agonists have been tested on mice lacking the mu-, delta-, or the kappa-opioid receptor gene. Main conclusions from the data are summarized in this scheme. Mu receptors are essential for all the biological activities of morphine. Kappa receptors are responsible for the analgesic and aversive properties of the reference U50,488H compound. Analgesic activities of the currently used delta agonists (DPDPE, deltorphin) require both mu and delta receptors. (Reproduced from Kieffer BL, Gavériaux-Ruff C 2002 Exploring the opioid system by gene knockout. Progress in Neurobiology 66:285–306. Copyright 2002 Elsevier Ltd.)
Another important issue is the respective contribution of each component of the opioid system to the regulation of physiological pain. Nociceptive responses to a number of acute noxious stimuli were examined in all the knockout lines and are summarized in Table 30-2. Taken together, the data show mostly enhanced pain sensitivity, concordant with the notion of an antinociceptive opioid tone. Some paradoxical responses have also been described—for example, decreased writhing or inflammatory hyperalgesia in the mu receptor knockout and reduced formalin irritation in the preproenkephalin null mutants (see Table 30-2)—which remain unexplained.
Table 30-2
Phenotypes of Mice Lacking Genes of the Opioid System in Behavioral Models of Pain
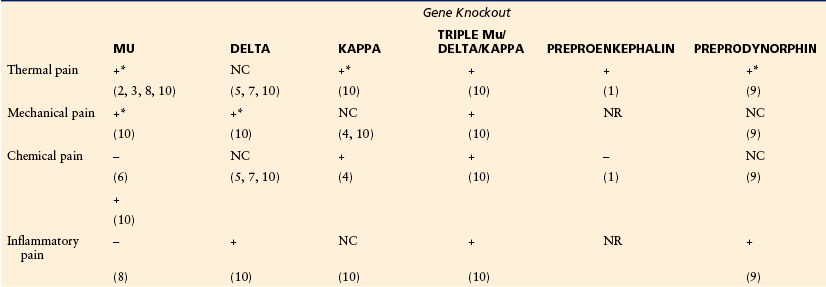
This table summarizes modifications of nociceptive responses in knockout animals. +, increased pain; –, decreased pain; NC, no change; NR, not reported. Asterisks indicate that in some reports (not indicated in the table), no change was found (for details, see Kieffer and Gavériauz-Ruff 2002). Thermal pain was assessed by using a tail flick, hot plate, or plantar assay. Mechanical pain was measured with the paw pressure test or von Frey filaments. Chemical pain was examined by either acetic acid writhing or the early phase of the formalin test. Inflammatory pain was measured in the late phase of the formalin test or following treatment with Freund’s complete adjuvant. Most mutants show increased pain sensitivity in accordance with the notion of an antinociceptive opioid tone. Some paradoxical responses are reported (decreased pain). Preprodynorphin null mutants have been examined in a model of neuropathic pain (not displayed in the table), and a biphasic phenotype was observed. Pain responses in mice lacking bendorphin have not yet been reported.
(1) Konig et al 1996, (2) Sora et al 1997, (3) Matthes et al 1998, (4) Simonin et al 1998, (5) Zhu et al 1999, (6) Sora et al 1999, (7) Filliol et al 2000, (8) Qiu et al 2000, (9) Wang et al 2001, (10) Martin et al 2003.
Also of importance are reports from many investigators using mu, delta, or kappa knockout mice suggesting that the three receptors regulate distinct pain modalities. Recently, a direct comparative study of the three mutant lines clarified this issue. Responses of single and combinatorial opioid receptor knockout mice were compared in several models of acute pain, and the distinct patterns of tonic antinociceptive activities were confirmed (Martin et al 2003):
Gender differences were reported in all the phenotypes, and together, the data were consistent with previous pharmacology. Importantly, deletion of all three opioid receptors (triple knockout) strongly enhanced pain sensitivity in all the tests and for both genders, thus suggesting that although the specific contribution of each opioid receptor is subtle, the opioid system as a whole exerts a significant inhibitory tone on physiological pain.
The development of chronic pain in mutant mice has been addressed only recently. Most striking is the fact that delta opioid receptor knockout mice showed no, or only subtle, modification of pain thresholds in models of acute pain but displayed increased pain responses in models of neuropathic (Nadal et al 2006) and inflammatory pain (Gavériaux-Ruff et al 2008). These data clearly establish the existence of an endogenous delta opioid receptor activity that reduces chronic pain. Also, knockout mice were insensitive to nortriptyline, a tricyclic antidepressant drug that fully reverses mechanical allodynia in an animal model of neuropathic pain (Benbouzid et al 2008). The latter data suggest that delta opioid receptors reduce chronic pain downstream of aminergic systems. Finally, a conditional knockout approach was applied to the delta opioid receptor gene. Using Nav1.8-Cre mice, the receptor was mainly deleted in primary nociceptive afferent neurons of dorsal root ganglia and left intact at all other sites of nociceptive pathways. Conditional mutant mice showed significantly enhanced inflammatory pain and strongly reduced antihyperalgesic effects of SNC80. The latter study demonstrates that activity of a highly restricted receptor population of the peripheral nervous system is sufficient to reduce chronic pain and mediate delta opioid analgesia (Gavériaux-Ruff et al 2011).
Finally, mice lacking preprodynorphin have been studied in both inflammatory and neuropathic pain models (Wang et al 2001), and the data indicate a biphasic role of prodynorphin in the control of nociceptive responses that may result from the dual opioid (kappa) and non-opioid (N-methyl-D-aspartate [NMDA]) activities of the peptide.
In conclusion, analysis of the opioid system in knockout mice provides genetic evidence that mu, delta, and kappa receptors, activated by endogenous peptides, all contribute to modulate pain. These mutant mice show altered responses in many other types of behaviors, including addictive (for review, see Kieffer and Gavériaux-Ruff 2002) and emotional behavior (Filliol et al 2000). Extending data from pharmacology, genetic manipulations unambiguously reveal unique contributions of each opioid receptor and peptide in opioid biology. In the future, the development of conditional gene knockout and the sophistication of mouse models of chronic pain should reveal the specific opioid receptor populations that control abnormal pain states, as well as the sites of opioid peptide release.
Opioid Receptor and Peptide Actions: Pathways Relevant to Pain
Coupling of opioid receptors to K+ and Ca2+ ion channels is believed to be a main mechanism whereby either exogenous or endogenous opioids produce analgesia. Ultimately, activation of all three opioid receptors, as well as the ORL receptor, and subsequent modification of ion channel activities will inhibit neuronal and cellular activity. The location of the receptors on synaptic circuits will have a bearing on what this leads to in terms of function in integrated systems. Thus, the opening of potassium channels—the most well-documented effect of opioid receptor activation—will inhibit release of transmitters if the receptors are on presynaptic terminals and will inhibit neuronal firing if the opioid receptor is found post-synaptically on neuronal cell bodies.
The nature of the neuron is another issue. Opioid receptor activation can cause positive effects whereby inhibition of one neuron allows other neurons downstream to become active. Thus, in integrated systems, opioids do not simply produce inhibition, and this is likely to contribute to low-dose excitations of neurons, supraspinal analgesic mechanisms, emesis, and the rewarding effects of opioids. This disinhibition occurs through opioid inhibition of inhibitory neurons, such as γ-aminobutyric acid (GABA)-containing cells. In this way, reduction of inhibition leads to facilitation in a circuit.
Activation of the opioid system by receptor agonists can provide considerable antinociception as demonstrated by various preclinical studies (Yaksh and Noueihed 1985; Dickenson 1994, 1995; Mao et al 1995; Ossipov et al 1995a, 1995b) and its widespread clinical manipulation (McQuay et al 1992). Endogenous release of the endorphins and enkephalins from intrinsic spinal neurons is stimulated only by high-intensity stimuli (Lucas and Yaksh 1990), and under normal conditions, even though the opioid antagonist naloxone does not produce hyperalgesia (Yaksh 1989), data from knockout studies reveal opioid-mediated tonic control.
The discovery that the endogenous opioid peptides, in particular, the enkephalins, are inactivated by two metallopeptidases—neutral endopeptidase and aminopeptidase N—led to the production of what has been termed “dual inhibitors.” These are single molecules with the ability to block both enzymes and thereby produce protection of a large proportion of the available endogenous opioids of the enkephalin family. These compounds have been used as a way of developing “physiological” analgesics that may lack some of the side effects of morphine by protecting the endogenous opioids that are being released during synaptic activity rather than simply activating a receptor. This approach has led to a number of compounds being produced, and most recently, a series of dual aminophosphinic inhibitors of the two enkephalin-catabolizing enzymes have been designed. These types of compounds are effective in tests such as the hot plate, rat tail flick, writhing, and formalin tests. Under these conditions, extracellular levels of the endogenous enkephalins’ brain areas related to pain are increased. Overall, the compounds produce effects that are about 40% of the maximal analgesia and can be antagonized by prior injection of naloxone. Since the increase in endogenous enkephalin levels parallels the antinociceptive responses, it is clear that the reason for the subtle tone is the rapid metabolism of these peptides (Le Guen et al 2003).
Other than studies on endogenous opioid systems, studies on activation of the opioid receptor system through exogenous agonists have provided data that suggest a substantial amount of plasticity in persistent pain states. Although such changes are beneficial in the presence of inflammation, neuropathic pain caused by peripheral nerve damage more often than not displays reduced sensitivity to opioids. This is evident both preclinically (Mao et al 1995, Ossipov et al 1995a), where the route of application is critical (Suzuki et al 1999), and clinically (Portenoy et al 1990, Jadad et al 1992) and is surrounded by much controversy. Mechanisms by which opioid sensitivity may be reduced after nerve injury include a reduction in spinal opioid receptors (Porreca et al 1998), non-opioid receptor–expressing Aβ fiber–mediated allodynia, increased cholecystokinin (CCK) antagonism of opioid actions (Nichols et al 1995), and NMDA-mediated dorsal horn neuronal hyperexcitability, which probably requires a greater opioid inhibitory counteraction (Dickenson 1997). These issues will be discussed later in this section.
Spinal Analgesia
Autoradiographic and immunohistochemical techniques have demonstrated that within the spinal cord, opioid receptors are located mostly in the superficial dorsal horn (laminae I and II), with a smaller population in deeper layers (Besse et al 1990b, Rahman et al 1998). The contribution of mu, delta, and kappa receptors to the total opiate binding throughout the spinal cord is estimated at 70, 24, and 6%, respectively (Besse et al 1990a, 1990b), at a predominantly (>70%) presynaptic location on the central terminals of only small-diameter nociceptive primary afferents. This is likely to include both C and Aδ fibers but exclude large-diameter A fibers. Opioid receptors are synthesized in small-diameter dorsal root ganglion (DRG) cell bodies and transported centrally and peripherally. This implies that the main mechanism of spinal opioid analgesia, whether it be mediated endogenously or exogenously, is via activation of presynaptic opioid receptors, which act to selectively decrease release of transmitter from nociceptive afferents and thus nociceptive transmission, with innocuous evoked activity being left intact. Indeed, spinally applied morphine can reduce release of substance P and calcitonin gene–related peptide after noxious stimulation (Go and Yaksh 1987), and excitatory but not inhibitory lamina II synaptic transmission is inhibited by a presynaptic opioid mechanism (Kohno et al 1999). Opioid receptors are also found in the periphery since after synthesis they are moved both to the central and peripheral endings of small fibers and their expression is increased in nociceptive primary afferents during inflammation. Endogenously, opiates are released from immune cells, and exogenous agonists developed for peripheral application have been shown to be antinociceptive in inflammatory states (Machelska et al 1999), where their restriction to outside the blood–brain barrier may allow reduced side effects (Janson and Stein 2003).
The remaining 30% of opioid receptors are located post-synaptically on interneurons and the dendrites of projection cells (Besse et al 1990a) visualized functionally as agonist-stimulated receptor internalization (Trafton et al 2000). Any opioid-mediated cell hyperpolarization leading to inhibition of firing of neurons with wide–dynamic range input will not exert nociceptive-specific effects, and from electrophysiological studies a small inhibition of Aβ fiber–evoked responses can be observed (Dickenson 1994, Dickenson and Suzuki 1999). Since the inhibitory effect is much less pronounced than that observed on the C fiber–evoked response, this confirms that the predominant site of action of spinal opioids is via presynaptic opioid receptors on the central terminals of nociceptive afferents (Ossipov et al 1995a, 1995b). This anatomical location of opioid receptors on fine (Aδ and C fiber) afferent terminals means that tactile information, transmitted by Aβ fibers, is mostly unimpaired by opioids since only the post-synaptic receptors will control input from these large fibers that converge onto lamina V wide–dynamic range neurons (Fig. 30-3). Thus, dynamic allodynias thought to be mediated by these fibers may be less well controlled than noxious (Aδ and C) and static allodynias (likely to be Aδ mediated; see Dickenson and Sullivan 1986, Dickenson and Suzuki 1999, Field et al 1999).
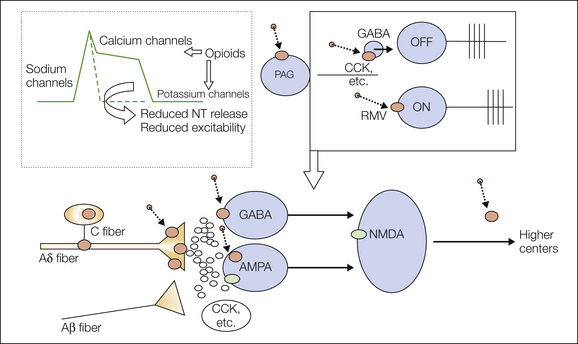
Figure 30-3 Opioid analgesia.
The diagram shows the sites and mechanisms of opioid analgesia at the spinal and supraspinal levels. The diagram at the top left depicts the cellular effects of opioid receptor activation in which the opening of potassium channels or the closing of calcium channels will attenuate the excitability of terminals or neurons, depending on the pre- or post-synaptic locations of the receptors. Below, the production of opioid receptors (larger brown circles) in the dorsal root ganglion cells of fine fibers and in post-synaptic spinal neurons allows opioids (smaller brown circles) to reduce activation of spinal neurons by peripheral input. Above, the effects of opioids on ON and OFF cells in the rostromedial medulla (RVM) are shown; opioids activate OFF cells (via γ-aminobutyric acid [GABA] neurons) and inhibit ON cells, thereby changing spinal descending controls. AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; CCK, cholecystokinin; NMDA, N-methyl-D-aspartate; NT, neurotransmitter; PAG, periaqueductal gray.
The importance of the spinal actions of opioids is evidenced by the rapidity with which the original animal behavioral studies on spinal delivery led to effective human applications. A large number of electrophysiological and behavioral studies have shown that mu, delta, and kappa receptor agonists and nociceptin acting at the ORL1 receptor cause antinociception and inhibition of spinal nociceptive neurons after intrathecal application (Yaksh and Noueihed 1985, Dickenson 1994, Taylor and Dickenson 1998). In normal animals, opioids are selective for noxious-evoked activity, and there are clear rank orders of potency of drugs within a receptor class. The mechanisms by which these effects are produced have been discussed in the preceding sections. In broad terms, the most potent opioids are the mu ligands, presumably reflecting the fact that mu opioid sites are the largest pool in the spinal cord. Delta opioids, nociceptin, and some kappa opioids then follow in order of potency. One important factor is the inverse relationship between lipophilicity and potency for a range of synthetic opioids acting at the mu receptor (McQuay et al 1989, Dickenson et al 1990). Here, the opioid with the highest potency was morphine, which is the least lipophilic. Drugs with high systemic potency as a result of their lipophilicity (e.g., fentanyl) were relatively ineffective after spinal application, probably because of non-specific binding of the lipophilic opioids in the lipid-rich fiber tracts capping the cord or as a consequence of vascular redistribution reducing the dose of opioid reaching opioid receptors in the superficial spinal cord. This is probably one of the principal reasons for the effectiveness of peptides given spinally since they are unlikely to redistribute far from the opioid receptors. Overall, there is a remarkable correlation between animal and human data on effective doses of different opioids given spinally (Yaksh 1997).
High levels of opioid receptors and endogenous opioids are present in the spinal cord. Orphanin FQ–like immunoreactivity and ORL1 receptors are located in the dorsal horn of the spinal cord and in the midbrain and brain stem sites, in some cases alongside the enkephalins, dynorphins, and endomorphins. Nociceptin has been identified in the spinal cord in areas adjacent but mostly separate from the location of the other endogenous opioids (Taylor and Dickenson 1998). The spinal level of all the opioid peptides is not altered by rhizotomy, thus indicating that they are all derived from intrinsic spinal neurons or descending pathways from the brain (Riedl et al 1996). Whereas the enkephalins and endorphins are inhibitory, as are mu and delta synthetic opioids (with the exception of low-dose mu facilitation), dynorphin—the endogenous kappa opioid receptor agonist—has a number of effects that differ from the typical opioid actions. It produces facilitation of some neurons and inhibition of other nociceptive neurons when applied spinally (Knox and Dickenson 1987), and spinal application of a kappa receptor antagonist both increases and decreases individual neuronal activity in normal animals but is considerably more effective in animals with inflammation (Stanfa and Dickenson 1994). What the consequences are for pain transmission is not yet understood.
Recent studies on the mechanisms by which the brain stem controls spinal activity in relation to nerve injury have shown that enhanced spinal levels of dynorphin can contribute to enhanced pain (Lai et al 2001, Wang et al 2001). Whether the effects of dynorphin are mediated by kappa opioid receptors is doubtful in this context, but this is an interesting example of an opioid peptide being pro-nociceptive. The functions of nociceptin in the processing of noxious events are also controversial (Henderson and McKnight 1997, Darland et al 1998, Taylor and Dickenson 1998, Heinricher 2003), although the discrepancies may be resolved on the basis of differential spinal and supraspinal actions of the peptide. In general, when administered into supraspinal sites, nociceptin produces hyperalgesia, whereas spinal administration has clearly been shown to be inhibitory and to produce analgesia (Darland et al 1998, Taylor and Dickenson 1998). These spinal antinociceptive effects of the peptide are observed in behavioral and in vivo and in vitro electrophysiological studies. Thus, nociceptin suppresses spinal reflexes and dorsal horn neuronal activity. Intrathecal orphanin FQ has also been shown to be antinociceptive in the tail flick test at doses that do not induce motor deficits or sedation (Darland et al 1998, Taylor and Dickenson 1998). The recent description of antagonists of the ORL1 receptor should help in clarifying the function of this receptor (Koizumi et al 2004).
Supraspinal Analgesia
Output from the spinal cord is attenuated by opioids. Opioid mechanisms at a number of other supraspinal sites such as the thalamic levels, the amygdala, and the sensory cortex are likely to play key roles in the overall analgesic state. However, from both animal and patient data, it is clear that spinal routes can almost entirely abolish pain responses and therefore prevent supraspinal activation by noxious input. As pain-related activity arrives at higher centers, input from the spinal cord to the parabrachial area, the central gray, and the amygdala may contribute mostly to the emotional aspects of pain, whereas input to the thalamus generates the sensory aspects (Rahman et al 2003). Higher centers play key roles in cognition, memory, attention, punishment, and other processes, and thus research on the ways in which centers involved in the higher processing of pain interact with opioids and sensory transmission may well yield further targets for therapy. One circuit may well be this type of loop whereby areas important in anxiety and attention that will probably be altered in chronic pain patients can facilitate spinal events (Hunt and Mantyh 2001, Rahman et al 2003). The ability not only to image pain but also to examine drug effects in humans will be a powerful tool for understanding the ways in which analgesics alter central nervous system (CNS) function (Rogers et al 2004).
These different projection pathways tend to emanate from different spinal nociceptive neurons such that the affective pathway arises predominantly from lamina I whereas the sensory–discriminative pathway arises more from lamina V. Importantly, in animals both neuronal populations are suppressed by opioids, thus confirming that any dissociation of the emotional and sensory aspects of pain produced by opioids must arise from brain mechanisms.
Important supraspinal sites of opioid action are the midbrain and brain stem structures (i.e., the periaqueductal gray [PAG] and the rostroventral medulla ([RVM]). In fact, the first studies on opioid sites of action pointed to the brain and in particular to sites accessible from the ventricles. Exogenous injection of morphine into either of these sites causes antinociception via increased activity in the inhibitory descending controls terminating in the dorsal horn of the spinal cord (for reviews, see Yaksh 1997, Fields 2000, Heinricher 2003, Heinricher and Neubert 2004). Electrical stimulation or application of glutamate to these sites, which would activate neurons, also causes antinociception, so morphine would appear to act through disinhibition to increase outflow from both sites (see Fig. 30-3). An influential hypothesis to explain this circuitry concerns the electrophysiological identification of two major populations of RVM output neurons: ON cells, whose activity coincides with spinal nociceptive reflexes, and OFF cells, whose activity is related to suppression of these reflexes. Infusion of morphine into the RVM is associated with a pronounced reduction in the activity of ON cells and increased activity of OFF cells, thereby supporting the pro- and antinociceptive labels assigned to ON and OFF cells, respectively:
There are also important interactions between serotonin (5-HT) and nitric oxide (NO) in the PAG. NO appears to be required for the 5-HT–mediated inhibition of PAG output and reversal of antinociception (Hamalainen and Lovick 1997). The circuitry between the PAG and the RVM is complex; when combined with the ascending pathways, a feedback loop in the modulation of nociceptive information becomes apparent.
Fibers descending from the RVM to the dorsal horn of the spinal cord are mostly serotonergic, enkephalinergic, glycinergic, and GABAergic. The nucleus raphe magnus contained within the RVM and the noradrenergic nuclei (locus coeruleus, subcoeruleus, A5 and A7 cell groups) are major PAG relays for noradrenergic and serotonergic descending pathways, respectively, to the dorsal horn (Kwiat and Basbaum 1992). Rather than the RVM being a homogeneous population of serotonergic neurons, GABA- (and glycine)-releasing neurons are now thought to constitute a significant proportion of spinally projecting RVM fibers (Antal et al 1996). The pharmacology of noradrenergic and serotonergic modulation in the dorsal horn is complex, but opioids can also interact with noradrenergic mechanisms, and many studies have shown that the effector mechanism and the location of the major noradrenaline target receptor—the α2 adrenoceptor—are very similar to those of opioid receptors.
In this context, tramadol, a dual serotonin and noradrenaline uptake inhibitor yet a weak opioid, has been succeeded by the new drug tapentadol, which produces analgesia through a dual mechanism of action within a single molecule. Tapentadol has quite low affinity for the mu opioid receptor, where it acts as an agonist, but the same molecule also acts as a noradrenaline reuptake inhibitor and thus increases levels of the transmitter, which in turn leads to analgesia through activation of the inhibitory α2 adrenoceptors. This novel dual central mechanism of action been termed MOR-NRI (Tzschentke et al 2007). There are a number of preclinical studies on its mechanisms, as well as phase II/III clinical studies, and the drug is a powerful analgesic, presumably because of these dual actions.
Opioids and Neuropathic Pain
The role of opioids in neuropathic pain is another good example of how opinions change. Neuropathic pain arising from peripheral nerve injury is a clinical disorder that appears to involve various peripheral and central components of the sensory systems (see references in Fields and Rowbotham 1994). The original clinical study on this issue suggested a lack of efficacy with fixed doses of opioids (Arner and Meyerson 1988). Since the dose–response curves for most opioids in animals are clear and steep, it is perhaps not surprising that many animal studies tend to concur with the present clinical view that opioids can be effective after nerve injury. Thus, opioid dose escalation in patients was shown to produce good analgesia (Portenoy et al 1990). Further studies showed that in general, morphine could be effective in a group of patients with neuropathy (Rowbotham et al 1991), although the analgesia attained was less than that in a group with nociceptive pain (Jadad et al 1992). Resolution of this problem has important clinical implications. This is based on findings that systemic morphine has an inhibitory effect (but reduced in many studies) on the behavioral or neuronal responses in rats subjected to nerve injury in comparison to control animals. Taking the literature as a whole, the anti-allodynic and antinociceptive abilities of morphine in behavioral studies involving neuropathy are somewhat variable. This again is not unexpected and appears to be dependent on the model of neuropathy; on what was measured in the behavioral assessment, as well as the nature of the particular stimulus; and on the dose and route of morphine administration (Attal et al 1991, Bian et al 1995, Ossipov et al 1995b, Dickenson and Suzuki 1999). This in turn may well apply to the clinical situation, where some patients clearly do gain benefit with opioids, but it is not known whether particular neuropathic syndromes or symptoms have differential sensitivity to opioids. This may well depend on the doses achieved.
Among the various factors that relate to opioid efficacy after nerve injury is the modality used to assess nociceptive behavior; this can vary as a result of both the nature of the stimulus and its intensity, as illustrated by mechanical allodynia and thermal hyperalgesia. These stimuli are likely to be processed via different neuronal circuits and pathways. In patients and in animals, mechanical allodynia, both static and dynamic, is evident following nerve damage, although dynamic allodynia is harder than static allodynia to assess in animals. Static allodynia is evoked by increasing pressure on the skin (such as with von Frey forces) and has been shown in neuropathic pain patients to not depend on Aβ fibers since it survives compression ischemia, which interrupts conduction in large myelinated fibers. Static allodynia is, however, dependent on capsaicin-sensitive Aδ fibers. Dynamic mechanical allodynia, as induced by light stroking of the skin, appears to be signaled by large-diameter myelinated Aβ sensory neurons (Field et al 1999). Morphine blocks static allodynia after systemic administration in nerve ligation models, whereas spinal administration is ineffective. In contrast, in the model of diabetes-induced neuropathy, both spinal and systemic morphine was effective for static allodynia, whereas the dynamic component was left intact (Field et al 1999). The latter study, which used identical doses of subcutaneous morphine, is in accordance with the reductions in evoked neuronal responses to static von Frey stimuli that have been demonstrated in spinal neuronal activities (Dickenson and Suzuki 1999). Not only do large A fibers not possess opioid receptors, but the responses of dorsal horn neurons to Aδ- and C-fiber (but not Aβ-fiber) stimulation are also blocked by morphine (Doi and Jurna 1982, Dickenson and Suzuki 1999).
It has also been demonstrated that stroking of the hindpaw ipsilateral to chronic constriction induces Fos expression at the spinal cord level in the superficial and deep dorsal horn. Such expression was not seen in control animals and was insensitive to morphine, in contrast to that evoked by noxious heat (Le Guen and Besson 2001). The lack of effect of morphine on stroking-evoked Fos expression in the dorsal horn supports the hypothesis that tactile allodynia is related to the activation of large primary afferent fibers with low opioid sensitivity. Thus, the positive effect of morphine on static but not dynamic allodynia is probably due to opioid receptors on Aδ and C fibers and not Aβ sensory neurons. Although this may mean a lack of effect on dynamic allodynia, this mechanism preserves low-threshold tactile sensitivity when opioids are used for other pain states.
Considering the animal data, it appears that there is a degree of inconsistency between the inhibitory effects of systemically administered morphine observed behaviorally and those seen on neuronal measures. Thus, intrathecal morphine administration was more effective than the systemic route of administration in producing inhibition of the evoked neuronal responses (electrical/mechanical/thermal stimuli) of spinal nerve–ligated rats (Suzuki et al 1999). The spinal neurons of spinal nerve–ligated rats thus exhibited reduced sensitivity to systemic morphine in comparison to normal and sham-operated rats. The systemic route of administration produced marked side effects (e.g., respiratory depression) in the anesthetized animals that limited dose escalation. Interestingly, intrathecal morphine administration produced greater attenuation of neuronal activity in spinal nerve–ligated rats than in controls, which does not agree with a number of previous behavioral studies (Lee et al 1995, Mao et al 1995). Hence there appears to be a degree of discrepancy between the results of some previous behavioral studies and current electrophysiological findings. Behavioral studies assess withdrawal thresholds as measures of allodynia (Lee et al 1995, Wegert et al 1997). This clearly contrasts with electrophysiological approaches that are based on the ability of morphine to produce inhibition of the suprathreshold evoked neuronal responses to electrical, mechanical, or thermal stimuli. It is feasible that morphine may not exert an effect on the withdrawal thresholds to mechanical/thermal stimuli while still having inhibitory effects on the suprathreshold firing of spinal neurons. This is likely if increases in the stimulus intensity recruit first large and then fine fibers (given the different opioid sensitivity of these fiber types). Furthermore, reductions in the suprathreshold responses of neurons after morphine administration will probably reduce the sensory response to the stimulus but may still allow a level of activity in the spinal circuitry on motor neurons that exceeds the levels required to elicit a withdrawal reflex. Be that as it may, there are clear demonstrations of morphine attenuating low-threshold input after nerve injury in some studies (Suzuki et al 1999, Erichsen and Blackburn-Munro 2002, Zhao et al 2004).
Opioids produce their actions through spinal cord and supraspinal opioid mechanisms (see above). Neuronal functions at these levels may all be disturbed by neuropathy, including the presynaptic opioid receptor sites on terminals of fine afferent fibers. There are high levels of spinal cord opioid receptors around C-fiber terminal zones in lamina I and the substantia gelatinosa and lower levels in deeper laminae (Besse et al 1990a, 1990b). These receptors are synthesized in the cell bodies of small afferent fibers in the DRG and transported to the peripheral and central terminals, with the latter being the site for modulation of neurotransmitter release. Down-regulation of mu-opioid receptors occurs in rat and monkey DRG neurons and in the dorsal horn after complete or partial sciatic nerve injury. Thus, peripheral nerve section causes a substantial reduction in spinal opioid receptors, mostly a result of disturbed axonal transport (Lombard and Besson 1989, Besse et al 1992). These studies on nerve transection in animals may well be relevant to postamputation pain in humans. Partial nerve damage (frequently associated with neuropathic pain) appears to lead to a much smaller and spatially restricted reduction in opioid receptors (Porreca et al 1998). In the case of less severe peripheral nerve insults, where axonal transport remains partially intact, there will also be only a limited loss of opioid receptors, which is likely to be correlated with reduced opioid effects rather than opioid resistance.
An additional issue is that calcium channel activity, vital for transmitter release, is also up-regulated after nerve injury (Vanegas and Schaible 2000, Dickenson et al 2001, Matthews and Dickenson 2002). Thus, nerve injury leads not only to reduced presynaptic opioid receptors but also to changes in calcium channels that result in increased transmitter release, which will directly oppose the inhibitory actions of opioids at presynaptic sites. As peripheral neurons become more active, action potentials arrive in their central terminals and calcium channels are activated. Results with drugs that block neuronal voltage-sensitive calcium channels would also suggest that there is an increase in central neuronal excitability after both inflammation and nerve damage that involves the N-, P-, and T-type calcium channels (Vanegas and Schaible 2000). In models of neuropathy there is up-regulation of the α2δ1 subunit with correlation to both allodynia and the action of gabapentin, which acts on this protein (Luo et al 2002). However, it is the N-type channels that exhibit the most marked expression and functional changes after nerve injury (Matthews and Dickenson 2001, Cizkova et al 2002). Thus, if a combination of a modest reduction in opioid receptor number and increased activity of calcium channels coincided, the presynaptic actions of opioids would be markedly attenuated. In support of this idea, morphine responsivity can be restored after nerve injury by reducing afferent input (Ossipov et al 1995b).
There are also functional changes that can influence the population of post-synaptic opioid receptors through alterations in the activity of spinal neurons. It is clear that in animals with reduced presynaptic opioid receptors, the post-synaptic actions of opioids require higher doses of systemic morphine than normal (Lombard and Besson 1989). This is likely to arise from the fact that the post-synaptic receptors account for only about 30% of the total spinal receptor population.
Another complication is activity of the neurons. A major drive in nerve injury and other pain states is increased excitation of spinal neurons because of activation of NMDA receptors (Dickenson 1994) as a result of peripheral nerve injury and suprathreshold stimuli. Both wind-up and spinal long-term potentiation, which rely on activation of the NMDA receptor, involve short- and long-term increased spinal cord neuronal excitability. The balance therefore shifts toward excitation, and consequently a greater degree of activation of post-synaptic opioid receptors would be required to counter the excitation. This may simply require more opioid drug. This concept is further supported by a number of different studies showing an increase in the efficacy of morphine in pain models in the presence of NMDA receptor antagonists (Dickenson et al 2001) and by putative block of calcium channel function (Matthews and Dickenson 2002) and the application of growth factors (Cahill et al 2003). Thus, after nerve injury, pathology can result in not only reduced presynaptic opioid control of transmitter release but also a coincident need for a greater contribution of post-synaptic opioid receptors to control the hyperexcitability of spinal neurons. In this context it is interesting to note that morphine, in combination with gabapentin (with actions on the α2δ1 subunit of calcium channels) produces greater than additive actions in both preclinical and clinical investigations (Matthews and Dickenson 2002, Gilron et al 2005).
Overall, the results of the preclinical investigations discussed provide a basis for the difficulties encountered clinically surrounding the efficacy of opioids in the treatment of neuropathic pain (for which their effectiveness remains controversial). Opinion is now leading to a consensus that they do indeed have effectiveness but that dose increases may be needed. Indeed, the route of administration has clearly been shown in electrophysiological, immunohistochemical, and behavioral studies (Suzuki et al 1999, Catheline et al 2001, Zhao et al 2004) to have an impact on the relative effectiveness of opioids (see also Eisenach and Lindner 2004). This may translate into better understanding of their use in the clinical management of pain from nerve injury.
Opioids and Inflammation
In states of inflammation, opioids can be more effective than in normal animals, and this is likely to relate to the effectiveness of opioids in patients after trauma and after surgery when tissue is damaged. The antinociceptive effects of systemic opioids in many models of inflammatory nociception have been reported. These models range from a few hours of localized inflammation to models of generalized arthritis with much longer time courses (Kayser and Guilbaud 1983, Neil et al 1986, Millan et al 1987, Stein et al 1988, Joris et al 1990, Kayser et al 1991). With a variety of behavioral tests the antinociceptive potency of opioids is found to be greater than that with acute noxious stimuli in normal animals. Behavioral (Hylden et al 1991) and electrophysiological (Stanfa et al 1992) approaches reveal that these increases in potency are rapid. Thus, only a few hours after the induction of peripheral inflammation, spinal opioid receptor agonists have enhanced potency against noxious stimuli. The enhancement in the spinal potency of mu, delta, and kappa receptor agonists varies with the receptor: morphine exhibits a far greater increase in spinal potency than do delta or kappa opioids, which show only relatively modest increases in potency. This altered potency of spinal opioids in inflammation could arise from a change in either the number or the affinity of spinal opioid receptors. There is little evidence of any marked change in spinal opioid receptors, even after weeks of inflammation (Stanfa and Dickenson 1993). Nociceptin is also more effective when given spinally by intrathecal administration after peripheral inflammation (Taylor and Dickenson 1998), and here there is an up-regulation of the receptor in the superficial spinal cord induced by inflammation (Jia et al 1998). Interestingly, RNA for the nociceptin precursor can be detected in DRGs within 30 minutes of inflammation (Andoh et al 1997), whereas it is practically absent in normal animals.
One factor that could make an important contribution to the enhanced potency of systemic opioids in inflammation is opioid actions at sites in the inflamed peripheral tissue. For instance, naloxone administered directly into an inflamed paw is able to antagonize the actions of systemically administered opioids (Stein et al 1988, Kayser et al 1991, Janson and Stein 2003). Although it has been long held that opioids act exclusively within the CNS (with regard to analgesia), there are opioid receptors outside the CNS, and many studies show that most agonists at the various opioid receptors can produce analgesic effects in the periphery. In normal conditions these actions are minimal, but under inflammatory conditions (in both animals and humans), opioids are able to access opioid receptors at sites of tissue damage. Since opioid receptors are synthesized in the cell bodies of fine fibers, it is not surprising that they can be transported from DRGs toward the peripheral sensory nerve endings. Studies show that the opioid receptors increase in expression and coupling after inflammation, and meanwhile, immune cells that contain endogenous opioid peptides build up within the inflamed tissue. A number of conditions—ranging from stress to the application of cytokines—can trigger the release of opioid peptides to interact with opioid receptors on peripheral neurons and produce local analgesia. This may lead to novel approaches for the development of peripherally acting analgesics, and some clinical data show this peripheral analgesia to be associated with reduced central side effects.
The issues that remain relate both to the level of efficacy of peripheral opioids (given the major effects of central opioid analgesia after inflammation) and to the need to avoid peripheral side effects of opioids such as constipation and nausea. Indeed, in this context, another approach has been to use centrally acting opioids with peripheral antagonists to circumvent the side effects produced by opioids (Bates et al 2004). The relative utility of these approaches will be fully gauged only by controlled clinical studies (Janson and Stein 2003).
Opioids in Cancer Pain
The advent of models of cancer pain in animals has now provided the opportunity to assess the sensitivity of opioids to cancer pain in preclinical settings (Luger et al 2002). A number of translational studies based on behavioral, neurochemical, and in vivo neuronal recordings have started to reveal some of the pathophysiological mechanisms underlying cancer pain, with the most numerous studies examining cancer-induced bone pain (CIBP). Opioids, despite their side effects, are the mainstay of analgesic control of severe pain from malignancy in the bone, and thus a number of studies have examined their actions in CIBP models. Studies on the analgesic effects of morphine further support the concept that CIBP is a unique pain state in that the efficacy of acute systemic administration is less than that observed in inflammatory models (Luger et al 2002, Wacnick et al 2003). Further studies using acute opioid administration confirm that high doses are needed to reduce pain behavior (Vermeirsch et al 2004), congruent with clinical observations of high doses of opioids being necessary to combat incident pain in cancer.
In an attempt to monitor the efficacy of morphine over time against CIBP via a chronic dosing schedule (Urch et al 2005), it was reported that morphine effectively reduced pain behavior, but comparisons showed that the analgesic effects were wearing off between doses. When a systemic dose of morphine was injected acutely on the last day of treatment to the chronic morphine group, the opioid reduced pain behavior, but it was less efficacious in this group than in chronically treated animals at this time point. Thus, the moderate effects of acute administration support the use of a chronic opioid treatment regimen for CIBP. This may explain why higher acute systemic doses were needed to attenuate pain in other studies, where investigations were carried out at later postoperative days when the pain had reached a more severe quality.
Parallel electrophysiological characterization has revealed that the hyperexcitability of superficial dorsal horn neurons was attenuated, though not reversed, in animals with chronic treatments (Urch et al 2005), so even under chronic morphine treatment there is still greater access of low-threshold stimuli to brain regions involved in pain processing, which may relate to the problems of controlling incident pain with opioids in the clinic.
Anti-Opioid Systems
A neurotransmitter system that produces opposite actions following tissue and nerve injury is CCK. CCK is a peptide with a variety of roles in CNS function that include anxiety and food intake. There is good evidence that in normal animals, physiological levels of CCK can interfere with the actions of morphine at the level of the spinal cord (Watkins et al 1984, Magnuson et al 1990, Stanfa and Dickenson 1993, Stanfa et al 1994, Wiesenfeld-Hallin et al 2002). The CCK receptors are mainly of the CCKB type (now known as CCK2) in the rat CNS and the CCK1 type in the periphery (confusingly apparently reversed in humans). This anti-opioid action is enhanced after nerve injury (Nichols et al 1995). Peripheral nerve injury has been shown to induce an up-regulation of CCK mRNA in ipsilateral DRG neurons. The mechanism by which CCK diminishes the antinociceptive effect of mu opioids is unknown, but CCK is capable of mobilizing calcium from intracellular stores via post-receptor mechanisms, which would lead to physiological antagonism of the suppression of calcium entry into nerve terminals following opioid receptor activation (Wang et al 1992).
A further mechanism proposed is that CCK, acting on the CCK2 receptor, decreases the availability of enkephalins. There is other evidence that enkephalins can synergize with morphine in the production of analgesia via their delta receptors and that CCK reduces enkephalin levels with a negative effect on morphine analgesia (Ossipov et al 1994, Vanderah et al 1994). This further emphasizes the interaction between opioid systems and CCK2 receptors. In addition, the weak CCK receptor antagonist proglumide was shown to enhance opioid analgesia (Watkins et al 1984) and has been demonstrated to be effective in humans, whereas L-365,260, a selective antagonist of the CCK2 receptor, failed to augment the analgesic effect of morphine in human subjects with chronic neuropathic pain (McCleane 2003).
In vivo and in vitro evidence suggests that another peptide, neuropeptide FF (NPFF), reduces both opioid analgesia and the inhibition of calcium currents induced by a number of opioids. There are actions at both spinal and supraspinal sites. These results suggest that activation of both NPFF1 and NPFF2 receptors leads to behavioral and cellular anti-opioid effects that are similar to those caused by CCK (Roumy et al 2003). The lack of good antagonists of NPFF receptors means that this peptide is less well studied than CCK.
Orphanin FQ/nociceptin, the endogenous ligand for the ORL1 receptor, has been reported to have no effect or to produce hyperalgesia, analgesia, or anti-hyperalgesia. These issues have been discussed previously, but the hyperalgesia seen with the activation of supraspinal structures appears to be mediated through the physiological opposition of opioid actions that is likely to result from neuronal disinhibition since the peptide always has direct inhibitory actions. This “anti-opioid” effect appears to occur with the endogenous opioid systems, perhaps explaining the hyperalgesia seen in some studies (Taylor and Dickenson 1998, Heinricher 2003).
Spinal dynorphin has been hypothesized to contribute to some of the states of hyperalgesia that can result from tissue and nerve injury or, indeed, from sustained morphine exposure (Ossipov et al 2004). There is evidence that these actions of dynorphin are not mediated through kappa opioid systems but rather result from actions that lead to spinal release of excitatory amino acids and prostaglandins (Koetzner et al 2004). The association of pain behavior with the expression of spinal dynorphin is based on the ability of the peptide to mimic many of the characteristics of chronic, neuropathic pain when administered spinally. One of the mechanisms that leads to the release of dynorphin at the spinal level is activation of pro-nociceptive descending pathways from the brain stem (Lai et al 2001, Wang et al 2001).
A series of recent studies has emphasized the importance of descending facilitatory controls on spinal sensory processing. These systems emanate from the same areas as those where opioids induce supraspinal analgesia, namely, the RVM. Thus, a descending facilitatory pathway has been described that involves ascending information from lamina I of the spinal cord and then a relay from the parabrachial region that activates pro-nociceptive pathways from the RVM, which finally activates a spinal 5-HT3 receptor–mediated spinal facilitation (Rahman et al 2003). This RVM area is a major site of action of morphine at supraspinal sites, and here again the balance shifts toward excitation in the context of persistent pain. A wealth of data show that direct activation of RVM “ON” cells by CCK is sufficient to produce thermal hyperalgesia, thus suggesting that the pro-nociceptive and anti-opioid effects of this peptide in the brain stem are mediated by actions on different RVM cell classes. Data also show that activation of descending nociceptive facilitatory pathways is important in the maintenance of neuropathic pain, and at least some of these pathways appear to be dependent on sustained afferent input from injured nerves to these brain stem sites that increases after nerve injury. That the abnormal tonic activity of descending facilitation mechanisms contributes to the painful sequelae of peripheral nerve injury is interesting in its own right. However, the fact that these RVM sites are opioid sensitive and are altered after nerve injury suggests that supraspinal (as well as spinal) changes may also contribute to the changed effectiveness of opioids after nerve damage (Kovelowski et al 2000).
Opioid-Induced Hyperalgesia
Despite opiate analgesics being the first line for pain relief in a variety of painful conditions ranging from acute pain and postoperative pain to chronic pain, sustained opioid consumption can unexpectedly produce paradoxical pain that is characterized by nociceptive sensitization (lowered pain thresholds) and analgesic tolerance (opiate dose escalation), referred to as opioid-induced hyperalgesia (OIH). OIH is reported in patients (Arner et al 1988, Hay et al 2009) and the laboratory (Mao and Meyer 2001).
Consideration of the anti-opioid systems described above lends credence to the concept that opioids may induce neuroadaptive alterations in pain transmission as well as in the modulatory circuitry, which then leads to OIH. Thus, enhanced neurokinin 1 receptor–mediated transmission (King et al 2005) and spinal dynorphin expression (Vanderah et al 2001, Gardell et al 2002) are among the mechanisms proposed to be essential for the development of OIH, which of course occurs in the absence of peripheral pathology. Enhanced descending facilitation to the spinal cord from higher CNS centers may lead to spinal cord hyperexcitability (Suzuki et al 2006), and this too has been seen in both the development and maintenance of OIH (Vanderah et al 2001). Neuronal recordings in OIH show reduced thresholds and enhanced responses of spinal cord neurons (Suzuki et al 2006).
Morphine Metabolites
Morphine is an example of a drug that has active metabolites. Thus, glucuronidation of morphine leads to two major metabolites: morphine-3-glucuronide (M3G) and morphine-6-glucuronide (M6G) (Sullivan et al 1989). These metabolites have quite different actions in that M6G is more potent than morphine itself and, although the degree of this enhanced action is variable from study to study, on average it is about 10-fold more effective in tests of analgesia (Sullivan et al 1989). Why this is the case is not obvious at present since the affinity of M6G for the mu receptor is not very different from that of morphine. Even so, M6G remains more effective than morphine even when applied directly to the CNS such as to the spinal cord. However, the M3G metabolite has no affinity for the mu receptor and therefore has no opioid actions. Nevertheless, it has been suggested that M3G is a factor that contributes to reduced opioid sensitivity based on two behavioral studies (Smith et al 1990, Gong et al 1992). In the latter study, when the metabolite was given by the intraventricular route, it caused marked behavioral agitation that interfered with the behavioral tests.
In contrast to these studies, there is electrophysiological evidence (where non-specific effects are less likely to interfere with the results) that even with dose ratios of 100:1 of the metabolite to morphine, M3G has absolutely no effect on the spinal antinociceptive effects of morphine (Hewett et al 1993). Since the spinal site of action of morphine is a major contributor to the systemic analgesia produced by the opioid and to renal insufficiency (where the metabolite will accumulate), opiate effects tend to be enhanced. It is therefore highly unlikely that M3G is an important factor in cases of pain poorly responsive to opioids.
Other Opioids
Biochemical studies describe ketobemidone and methadone as having high non-competitive affinity for the NMDA receptor in the rat spinal cord and cortex, with potency comparable to that of dextromethorphan and ketamine (Ebert et al 1995). Racemic methadone, as well as its d- and l-isomers, exerts NMDA receptor antagonism at both the spinal and cortical levels (Gorman et al 1997). In vivo studies show that methadone produces consistent analgesia in various experimental and neuropathic pain models (Shimoyama et al 1997, Carpenter et al 2000, Bulka et al 2002), but often the effects seen are reversible by naloxone. There is no agreement on the clinical implication of the pharmacological properties of methadone or indeed other d-isomers of opioids from these studies.
Side Effects of Opioids
The large numbers of opioid receptors in regions such as the solitary tract and adjacent areas are probably related to the respiratory effects of opiates: cough suppression and nausea and vomiting.
Opiates acting in the brain stem reduce the sensitivity of the respiratory centers to PCO2, and this is the most common cause of death from overdose with the street use of opiates.
Opiates activate the chemoreceptor trigger zone in the medulla to cause nausea and vomiting; cough suppression also occurs because of the inhibitory effects of opiates on brain stem nuclei in the cough reflex pathway. Dextromethorphan is the non-opiate isomer of levorphanol and is an effective cough suppressant.
Actions in the monoamine nuclei (such as the well-demonstrated effects of opioids on noradrenergic transmission in the locus coeruleus and enhancement of dopamine release in the ventral tegmental area) are likely to be associated with reward processes and therefore relate to dependence. Psychological dependence does not seem to occur to any great extent in the presence of pain.
The relative extent of the unwanted effects caused by selective agonists at the different opioid receptors is of great importance in determining whether non-mu opioids will have better spectra of action than morphine. However, there are good indications that the delta and kappa receptor agonists cause less respiratory depression than mu and that prolonged protection of the enkephalins by peptidase inhibitors has no dependence liability (Le Guen et al 2003).
Peripheral Side Effects
A number of side effects of opiates are due to their actions on opiate receptors outside the CNS. Opiates constrict the pupils by acting on the oculomotor nucleus and cause constipation by inducing maintained contraction of the smooth muscle of the gut, which reduces motility. This diminished propulsion, coupled with opiates reducing secretion in the gut, underlies its antidiarrheal effect. Opiates contract sphincters throughout the gastrointestinal tract. Although these effects are predominantly peripheral actions, there are central contributions as well. Morphine can also release histamine from mast cells, which can produce irritation and bronchospasm in extreme cases. Opiates have minimal cardiovascular effects at therapeutic doses.
Conclusion
The recent advances in our understanding of opioid function and dysfunction and a clearer feel for the factors that can influence the efficacy of opioids form a basis for improving the clinical outcomes of opioid use. One important issue is the variability in opioid actions based on gender studies in animals and humans (Fillingim and Gear 2004). Opioids and their receptors are part of the integrated functional pharmacological repertoire of neurons in the nervous systems; consequently, alteration in the status of opioid receptors and activation of other transmitter systems will interact to modulate the function of the CNS. Modern genomic and post-genomic techniques in integrated systems are adding to our understanding of opioid functions. This knowledge can be harnessed by means of combination therapy, and by recognizing the potential for plasticity in opioid actions, clinical use of these drugs can be improved.
The references for this chapter can be found at www.expertconsult.com.
References
Akil H., Owens C., Gutstein H., et al. Endogenous opioids: overview and current issues. Drug and Alcohol Dependence. 1998;51:127–140.
Akil H., Watson S.J., Young E., et al. Endogenous opioids: biology and function. Annual Review of Neuroscience. 1984;7:223–255.
Andoh T., Itoh M., Kuraishi Y. Nociceptin gene expression in rat dorsal root ganglia induced by peripheral inflammation. Neuroreport. 1997;8:2793–2796.
Antal M., Petko M., Polgar E., et al. Direct evidence of an extensive GABAergic innervation of the spinal dorsal horn by fibres descending from the rostral ventromedial medulla. Neuroscience. 1996;73:509–518.
Arden J.R., Segredo V., Wang Z., et al. Phosphorylation and agonist-specific intracellular trafficking of an epitope-tagged mu-opioid receptor expressed in HEK 293 cells. Journal of Neurochemistry. 1995;65:1636–1645.
Arner S., Meyerson B.A. Lack of analgesic effect of opioids on neuropathic and idiopathic forms of pain. Pain. 1988;33:11–23.
Arner S., Rawal N., Gustafsson L.L. Clinical experience of long-term treatment with epidural and intrathecal opioids—a nationwide survey. Acta Anaesthesiologica Scandinavica. 1988;32:253–259.
Attal N., Chen Y.L., Kayser V., et al. Behavioural evidence that systemic morphine may modulate a phasic pain-related behaviour in a rat model of peripheral mononeuropathy. Pain. 1991;47:65–70.
Barnard E.A. Pipe dreams realized. Current Biology. 1993;3:211–214.
Bates J.J., Foss J.F., Murphy D.B. Are peripheral opioid antagonists the solution to opioid side effects? Anesthesia and Analgesia. 2004;98:116–122.
Befort K., Kieffer B.L. Structure–activity relationships in the delta opioid receptor. Pain Reviews. 1997;4:100–121.
Befort K., Tabbara L., Kling D., et al. Role of aromatic transmembrane residues of the delta-opioid receptor in ligand recognition. Journal of Biological Chemistry. 1996;271:10161–10168.
Befort K., Zilliox C., Filliol D., et al. Constitutive activation of the delta opioid receptor by mutations in transmembrane domains III and VII. Journal of Biological Chemistry. 1999;274:18574–18581.
Beinborn M., Ren Y., Blaker M., et al. Ligand function at constitutively active receptor mutants is affected by two distinct yet interacting mechanisms. Molecular Pharmacology. 2004;65:753–760.
Berger A.C., Whistler J.L. Morphine-induced mu opioid receptor trafficking enhances reward yet prevents compulsive drug use. EMBO Mol Med. 2011;3:385–397.
Besse D., Lombard M.C., Perrot S., et al. Regulation of opioid binding sites in the superficial dorsal horn of the rat spinal cord following loose ligation of the sciatic nerve: comparison with sciatic nerve section and lumbar dorsal rhizotomy. Neuroscience. 1992;50:921–933.
Besse D., Lombard M.C., Zajac J.M., et al. Pre- and postsynaptic distribution of mu, delta and kappa opioid receptors in the superficial layers of the cervical dorsal horn of the rat spinal cord. Brain Research. 1990;521:15–22.
Besse D., Lombard M.C., Zajac J.M., et al. Pre- and postsynaptic location of mu, delta and kappa opioid receptors in the superficial layers of the dorsal horn of the rat spinal cord. Progress in Clinical and Biological Research. 1990;328:183–186.
Bian D., Nichols M.L., Ossipov M.H., et al. Characterization of the antiallodynic efficacy of morphine in a model of neuropathic pain in rats. Neuroreport. 1995;6:1981–1984.
Bolan E.A., Pan Y.X., Pasternak G.W. Functional analysis of MOR-1 splice variants of the mouse mu opioid receptor gene Oprm. Synapse. 2004;51:11–18.
Brady A.E., Limbird L.E. G protein–coupled receptor interacting proteins: emerging roles in localization and signal transduction. Cell Signalling. 2002;14:297–309.
Brownstein M.J. A brief history of opiates, opioid peptides, and opioid receptors. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:5391–5393.
Bruchas M.R., Land B.B., Chavkin C. The dynorphin/kappa opioid system as a modulator of stress-induced and pro-addictive behaviors. Brain Res. 2010;1314:44–55.
Bulka A., Plesan A., Xu X.-J., et al. Reduced tolerance to the anti-hyperalgesic effect of methadone in comparison to morphine in a rat model of mononeuropathy. Pain. 2002;95:103–109.
Cahill C.M., Dray A., Coderre T.J. Intrathecal nerve growth factor restores opioid effectiveness in an animal model of neuropathic pain. Neuropharmacology. 2003;45:543–552.
Carpenter K.J., Chapman V., Dickenson A.H. Neuronal inhibitory effects of methadone are predominantly opioid receptor mediated in the rat spinal cord in vivo. European Journal of Pain. 2000;4:19–26.
Catheline G., Le Guen S., Besson J.M. Intravenous morphine does not modify dorsal horn touch-evoked allodynia in the mononeuropathic rat: a Fos study. Pain. 2001;92:389–398.
Cavalli A., Babey A.M., Loh H.H. Altered adenylyl cyclase responsiveness subsequent to point mutations of Asp 128 in the third transmembrane domain of the delta-opioid receptor. Neuroscience. 1999;93:1025–1031.
Chaturvedi K., Christoffers K.H., Singh K., et al. Structure and regulation of opioid receptors. Biopolymers. 2000;55:334–346.
Cizkova D., Marsala J., Lukacova N., et al. Localization of N-type Ca2+ channels in the rat spinal cord following chronic constrictive nerve injury. Experimental Brain Research. 2002;147:456–463.
Claude P.A., Wotta D.R., Zhang X.H., et al. Mutation of a conserved serine in TM4 of opioid receptors confers full agonistic properties to classical antagonists. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:5715–5719.
Comb M., Seeburg P.H., Adelman J., et al. Primary structure of the human Met- and Leu-enkephalin precursor and its mRNA. Nature. 1982;295:663–666.
Contet C., Kieffer B.L., Befort K. Mu opioid receptor: a gateway to drug addiction. Current Opinion in Neurobiology. 2004;14:370–378.
Corbett A.D., Paterson S.J., Kosterlitz H.W. Selectivity of ligands for opioid receptors. In: Herz A., ed. Handbook of experimental pharmacology. Opioids I. Berlin: Springer-Verlag; 1993:645–673.
Darland T., Heinricher M.M., Grandy D.K. Orphanin FQ/nociceptin: a role in pain and analgesia, but so much more. Trends in Neuroscience. 1998;21:215–221.
Décaillot F., Kieffer B.L. In vitro and in vivo mutagenesis: insights into delta receptor structure and function. In: Chang K.-J., Porreca F., Woods J.H., eds. The delta receptor. New York: Marcel Dekker; 2004:41–60.
Décaillot F.M., Befort K., Filliol D., et al. Opioid receptor random mutagenesis reveals a mechanism for G protein–coupled receptor activation. Nature Structural Biology. 2003;10:629–636.
Devi L.A. Heterodimerization of G-protein–coupled receptors: pharmacology, signaling and trafficking. Trends in Pharmacological Sciences. 2001;22:532–537.
Dickenson A., Where and how do opioids act?. Gebhart G., Hammond D., Jensen T., eds. Proceedings of the 7th World Congress on Pain. Progress in pain research and management, vol 2. Seattle: IASP Press; 1994:525–552.
Dickenson A.H. Spinal cord pharmacology of pain. British Journal of Anaesthesia. 1995;75:193–200.
Dickenson A.H. Mechanisms of hypersensitivity: excitatory amino acid mechanisms and their control. In: Dickenson A.H., Besson J.-M., eds. The pharmacology of pain. Berlin: Springer-Verlag; 1997:167–210.
Dickenson A.H., Matthews E., Suzuki R., Central nervous system mechanisms of pain in peripheral neuropathy. Hansson P.T., Fields H.L., Hill R.G., et al, eds. Neuropathic pain pathophysiology and treatment. Progress in pain research and management, vol 21. Seattle: IASP Press; 2001:85–106.
Dickenson A.H., Sullivan A.F. Electrophysiological studies on the effects of intrathecal morphine on nociceptive neurones in the rat dorsal horn. Pain. 1986;24:211–222.
Dickenson A.H., Sullivan A.F., McQuay H.J. Intrathecal etorphine, fentanyl and buprenorphine on spinal nociceptive neurones in the rat. Pain. 1990;42:227–234.
Dickenson A.H., Suzuki R. Function and dysfunction of opioid receptors in the spinal cord. In: Kalso E., McQuay H.J., Weisenfeld-Hallin Z., eds. Opioid sensitivity of chronic non-cancer pain. Seattle: IASP Press; 1999:17–44.
Doi T., Jurna I. Analgesic effect of intrathecal morphine demonstrated in ascending nociceptive activity in the rat spinal cord and in effectiveness of caerulein and cholecystokinin octapeptide. Brain Research. 1982;234:399–407.
Dondio G., Ronzoni S., Farina C., et al. Selective delta opioid receptor agonists for inflammatory and neuropathic pain. Farmaco. 2001;56:117–119.
Ebert B., Andersen S., Krogsgaard-Larsen P. Ketobemidone, methadone and pethidine are non-competitive N-methyl-D-aspartate (NMDA) antagonists in the rat cortex and spinal cord. Neuroscience Letters. 1995;187:165–168.
Eisenach J., Lindner M.D. Did experimenter bias conceal the efficacy of spinal opioids in previous studies with the spinal nerve ligation model of neuropathic pain? Anesthesiology. 2004;100:765–767.
Erichsen H.K., Blackburn-Munro G. Pharmacological characterisation of the spared nerve injury model of neuropathic pain. Pain. 2002;98:151–161.
Evans C.J., Keith D.E., Jr., Morrison H., et al. Cloning of a delta opioid receptor by functional expression. Science. 1992;258:1952–1955.
Evans C.J., Monteillet-Agius G., Saliminejad N., et al. Opiate drugs: “guilt by association,”. Molecular Psychiatry. 2000;5:122–123.
Field M.J., McCleary S., Hughes J., et al. Gabapentin and pregabalin, but not morphine and amitriptyline, block both static and dynamic components of mechanical allodynia induced by streptozocin in the rat. Pain. 1999;80:391–398.
Fields H., Rowbotham M., Multiple mechanisms of neuropathic pain: a clinical perspective. Gebhart G., Hammond D., Jensen T., eds. Proceedings of the 7th World Congress on Pain. Progress in pain research and management, vol 2. Seattle: IASP Press; 1994:437–454.
Fields H.L. Pain modulation: expectation, opioid analgesia and virtual pain. Progress in Brain Research. 2000;122:245–253.
Filliol D., Ghozland S., Chluba J., et al. Mice deficient for delta- and mu-opioid receptors exhibit opposing alterations of emotional responses. Nature Genetics. 2000;25:195–200.
Fillingim R.B., Gear R.W. Sex differences in opioid analgesia: clinical and experimental findings. European Journal of Pain. 2004;8:413–425.
Galandrin S., Oligny-Longpre G., Bouvier M. The evasive nature of drug efficacy: implications for drug discovery. Trends Pharmacol Sci. 2007;28:423–430.
Gardell L.R., Wang R., Burgess S.E., et al. Sustained morphine exposure induces a spinal dynorphin-dependent enhancement of excitatory transmitter release from primary afferent fibers. Journal of Neuroscience. 2002;22:6747–6755.
Gavériaux-Ruff C., Kieffer B.L. Opioid receptors: gene structure and function. In: Stein C., ed. Opioids in pain control: basic and clinical aspects. Cambridge: Cambridge University Press; 1999:1–19.
Gavériaux-Ruff C., Kieffer B.L. Opioid receptor genes inactivated in mice: the highlights. Neuropeptides. 2002;36:62–71.
Georgoussi Z., Merkouris M., Mullaney I., et al. Selective interactions of mu-opioid receptors with pertussis toxin–sensitive G proteins: involvement of the third intracellular loop and the c-terminal tail in coupling. Biochimica et Biophysica Acta. 1997;1359:263–274.
Gilron I., Bailey J.M., Tu D., et al. Morphine, gabapentin, or their combination for neuropathic pain. New England Journal of Medicine. 2005;352:1324–1334.
Go V.L., Yaksh T.L. Release of substance P from the cat spinal cord. Journal of Physiology. 1987;391:141–167.
Gong Q.L., Hedner J., Bjorkman R., et al. Morphine-3-glucuronide may functionally antagonize morphine-6-glucuronide induced antinociception and ventilatory depression in the rat. Pain. 1992;48:249–255.
Gorman A.L., Elliot K.J., Inturrisi C.E. The d- and l-isomers of methadone bind to the non-competitive site on the N-methyl-D-aspartate (NMDA) receptor in rat forebrain and spinal cord. Neuroscience Letters. 1997;223:5–8.
Granier S., Manglik A., Kruse A.C., et al. Structure of the delta-opioid receptor bound to naltrindole. Nature. 2012;485:400–404.
Haberstock-Debic H., Wein M., Barrot M., et al. Morphine acutely regulates opioid receptor trafficking selectively in dendrites of nucleus accumbens neurons. Journal of Neuroscience. 2003;23:4324–4332.
Hamalainen M.M., Lovick T.A. Involvement of nitric oxide and serotonin in modulation of antinociception and pressor responses evoked by stimulation in the dorsolateral region of the periaqueductal gray matter in the rat. Neuroscience. 1997;80:821–827.
Hay J.L., White J.M., Bochner F., et al. Hyperalgesia in opioid-managed chronic pain and opioid-dependent patients. Journal of Pain. 2009;10:316–322.
Heinricher M.M. Orphanin FQ/nociceptin: from neural circuitry to behavior. Life Sciences. 2003;73:813–822.
Heinricher M.M., Neubert M.J. Neural basis for the hyperalgesic action of cholecystokinin in the rostral ventromedial medulla. Journal of Neurophysiology. 2004;92:1982–1989.
Henderson G., McKnight A.T. The orphan opioid receptor and its endogenous ligand—nociceptin/orphanin FQ. Trends in Pharmacological Sciences. 1997;18:293–300.
Hewett K., Dickenson A.H., McQuay H.J. Lack of effect of morphine-3-glucuronide on the spinal antinociceptive actions of morphine in the rat: an electrophysiological study. Pain. 1993;53:59–63.
Hughes J., Smith T.W., Kosterlitz H.W., et al. Identification of two related pentapeptides from the brain with potent opiate agonist activity. Nature. 1975;258:577–580.
Hunt S., Mantyh P. The molecular dynamics of pain control. Nature Reviews. Neuroscience. 2001;2:83–91.
Hylden J.L., Thomas D.A., Iadarola M.J., et al. Spinal opioid analgesic effects are enhanced in a model of unilateral inflammation/hyperalgesia: possible involvement of noradrenergic mechanisms. European Journal of Pharmacology. 1991;194:135–143.
Jadad A.R., Carroll D., Glynn C.J., et al. Morphine responsiveness of chronic pain: double-blind randomised crossover study with patient-controlled analgesia. Lancet. 1992;339:1367–1371.
Janson W., Stein C. Peripheral opioid analgesia. Current Pharmaceutical Biotechnology. 2003;4:270–274.
Jia Y., Linden D.R., Serie J.R., et al. Nociceptin/orphanin FQ binding increases in superficial laminae of the rat spinal cord during persistent peripheral inflammation. Neuroscience Letters. 1998;250:21–24.
Jordan B.A., Devi L.A. G-protein–coupled receptor heterodimerization modulates receptor function. Nature. 1999;399:697–700.
Joris J., Costello A., Dubner R., et al. Opiates suppress carrageenan-induced edema and hyperthermia at doses that inhibit hyperalgesia. Pain. 1990;43:95–103.
Kakidani H., Furutani Y., Takahashi H., et al. Cloning and sequence analysis of cDNA for porcine beta-neo-endorphin/dynorphin precursor. Nature. 1982;298:245–249.
Kayser V., Chen Y.L., Guilbaud G. Behavioural evidence for a peripheral component in the enhanced antinociceptive effect of a low dose of systemic morphine in carrageenan-induced hyperalgesic rats. Brain Research. 1991;560:237–244.
Kayser V., Guilbaud G. The analgesic effects of morphine, but not those of the enkephalinase inhibitor thiorphan, are enhanced in arthritic rats. Brain Research. 1983;267:131–138.
Keith D.E., Murray S.R., Zaki P.A., et al. Morphine activates opioid receptors without causing their rapid internalization. Journal of Biological Chemistry. 1996;271:19021–19024.
Kieffer B.L. Recent advances in molecular recognition and signal transduction of active peptides: receptors for opioid peptides. Molecular Neurobiology. 1995;15:615–635.
Kieffer B.L. Molecular aspects of opioid receptors. In: Dickenson A., Besson J.M., eds. Handbook of experimental pharmacology. The pharmacology of pain. Berlin: Springer-Verlag; 1997:281–303.
Kieffer B.L., Befort K., Gavériaux-Ruff C., et al. The delta-opioid receptor: isolation of a cDNA by expression cloning and pharmacological characterization. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:12048–12052.
Kieffer B.L., Evans C.J. Opioid tolerance—in search of the holy grail. Cell. 2002;108:587–590.
Kieffer B.L., Gavériaux-Ruff C. Exploring the opioid system by gene knockout. Progress in Neurobiology. 2002;66:285–306.
Kim J.A., Bartlett S., He L., et al. Morphine-induced receptor endocytosis in a novel knockin mouse reduces tolerance and dependence. Curr Biol. 2008;18:129–135.
King T., Gardell L.R., Wang R., et al. Role of NK-1 neurotransmission in opioid-induced hyperalgesia. Pain. 2005;116:276–288.
Knox R.J., Dickenson A.H. Effects of selective and non-selective kappa-opioid receptor agonists on cutaneous C-fibre–evoked responses of rat dorsal horn neurones. Brain Research. 1987;415:21–29.
Koetzner L., Hua X.Y., Lai J., et al. Nonopioid actions of intrathecal dynorphin evoke spinal excitatory amino acid and prostaglandin E2 release mediated by cyclooxygenase-1 and -2. Journal of Neuroscience. 2004;24:1451–1458.
Kohno T., Kumamoto E., Higashi H., et al. Actions of opioids on excitatory and inhibitory transmission in substantia gelatinosa of adult rat spinal cord. Journal of Physiology. 1999;518:803–813.
Koizumi M., Sakoori K., Midorikawa N., et al. The NOP (ORL1) receptor antagonist compound B stimulates mesolimbic dopamine release and is rewarding in mice by a non-NOP-receptor–mediated mechanism. British Journal of Pharmacology. 2004;143:53–62.
Konig M., Zimmer A.M., Steiner H., et al. Pain responses, anxiety and aggression in mice deficient in pre-proenkephalin. Nature. 1996;383:535–538.
Kovelowski C.J., Ossipov M.H., Sun H., et al. Supraspinal cholecystokinin may drive tonic descending facilitation mechanisms to maintain neuropathic pain in the rat. Pain. 2000;87:265–273.
Kwiat G., Basbaum A. The origin of brainstem noradrenergic and serotonergic projections to the spinal cord dorsal horn of the rat. Somatosensory & Motor Research. 1992;9:157–173.
Lagerstrom M.C., Schioth H.B. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat Rev Drug Discov. 2008;7:339–357.
Lai J., Ossipov M.H., Vanderah T.W., et al. Neuropathic pain: the paradox of dynorphin. Molecular Interventions. 2001;1:160–167.
Law P.Y., Loh H.H. Regulation of opioid receptor activities. Journal of Pharmacology and Experimental Therapeutics. 1999;289:607–624.
Law P.Y., Wong Y.H., Loh H.H. Mutational analysis of the structure and function of opioid receptors. Biopolymers. 1999;51:440–455.
Law P.Y., Wong Y.H., Loh H.H. Molecular mechanisms and regulation of opioid receptor signaling. Annual Review of Pharmacology and Toxicology. 2000;40:389–430.
Lee Y.W., Chaplan S.R., Yaksh T.L. Systemic and supraspinal, but not spinal, opiates suppress allodynia in a rat neuropathic pain model. Neuroscience Letters. 1995;199:111–114.
Le Guen S., Besson J.M. Intravenous morphine does not modify dorsal horn touch-evoked allodynia in the mononeuropathic rat: a Fos study. Pain. 2001;92:389–398.
Le Guen S., Mas Nieto M., Canestrelli C., et al. Pain management by a new series of dual inhibitors of enkephalin degrading enzymes: long lasting antinociceptive properties and potentiation by CCK2 antagonist or methadone. Pain. 2003;104:139–148.
Levac B.A., O’Dowd B.F., George S.R. Oligomerization of opioid receptors: generation of novel signaling units. Current Opinion in Pharmacology. 2002;2:76–81.
Li Y., Eitan S., Wu J., et al. Morphine induces desensitization of insulin receptor signaling. Molecular and Cellular Biology. 2003;23:6255–6266.
Lombard M.C., Besson J.M. Attempts to gauge the relative importance of pre- and postsynaptic effects of morphine on the transmission of noxious messages in the dorsal horn of the rat spinal cord. Pain. 1989;37:335–345.
Lucas D., Yaksh T.L. Release in vivo of Met-enkephalin and encrypted forms of Met-enkephalin from brain and spinal cord of the anesthetized cat. Peptides. 1990;11:1119–1125.
Luger N.M., Sabino M.A., Schwei M.J., et al. Efficacy of systemic morphine suggests a fundamental difference in the mechanisms that generate bone cancer vs inflammatory pain. Pain. 2002;99:397–406.
Luo Z.D., Calcutt N.A., Higuera E.S., et al. Injury type–specific calcium channel alpha 2 delta-1 subunit up-regulation in rat neuropathic pain models correlates with antiallodynic effects of gabapentin. Journal of Pharmacology and Experimental Therapeutics. 2002;303:1199–1205.
Machelska H., Binder W., Stein C., Opioid receptors in the periphery. Kalso E., McQuay H., Wiesenfeld-Hallin Z., eds. Opioid sensitivity of chronic noncancer pain. Progress in pain research and management, vol 14. Seattle: IASP Press; 1999:45–58.
Magnuson D.S., Sullivan A.F., Simonnet G., et al. Differential interactions of cholecystokinin and FLFQPQRF-NH2 with mu and delta opioid antinociception in the rat spinal cord. Neuropeptides. 1990;16:213–218.
Manglik A., Kruse A.C., Kobilka T.S., et al. Crystal structure of the micro-opioid receptor bound to a morphinan antagonist. Nature. 2012;485:321–326.
Mao J., Mayer D.J. Spinal cord neuroplasticity following repeated opioid exposure and its relation to pathological pain. Annals of the New York Academy of Sciences. 2001;933:175–184.
Mao J., Price D.D., Mayer D.J. Experimental mononeuropathy reduces the antinociceptive effects of morphine: implications for common intracellular mechanisms involved in morphine tolerance and neuropathic pain. Pain. 1995;61:353–364.
Marinissen M.J., Gutkind J.S. G-protein–coupled receptors and signaling networks: emerging paradigms. Trends in Pharmacological Sciences. 2001;22:368–376.
Martin M., Matifas A., Maldonado R., et al. Acute antinociceptive responses in single and combinatorial opioid receptor knockout mice: distinct mu, delta and kappa tones. European Journal of Neuroscience. 2003;17:701–708.
Martin W.R., Eades C.G., Thompson J.A., et al. The effects of morphine- and nalorphine-like drugs in the nondependent and morphine-dependent chronic spinal dog. Journal of Pharmacology and Experimental Therapeutics. 1976;197:517–532.
Matthes H.W., Maldonado R., Simonin F., et al. Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu-opioid-receptor gene. Nature. 1996;383:819–823.
Matthes H.W.D., Smadja C., Valverde O., et al. Activity of the delta-opioid receptor is partially reduced while activity of the kappa-receptor is maintained in mutant mice lacking the mu-receptor. Journal of Neuroscience. 1998;18:7285–7295.
Matthews E., Dickenson A. Effects of spinally delivered N- and P-type voltage-dependent calcium channel antagonists on dorsal horn neuronal responses in a rat model of neuropathy. Pain. 2001;92:235–246.
Matthews E., Dickenson A.H. A combination of gabapentin and morphine mediates enhanced inhibitory effects on dorsal horn neuronal responses in a rat model of neuropathy. Anesthesiology. 2002;96:633–640.
McCleane G.J. A randomised, double blind, placebo controlled crossover study of the cholecystokinin 2 antagonist L-365,260 as an adjunct to strong opioids in chronic human neuropathic pain. Neuroscience Letters. 2003;338:151–154.
McQuay H.J., Jadad A.R., Carroll D., et al. Opioid sensitivity of chronic pain: a patient-controlled analgesia method. Anaesthesia. 1992;47:757–767.
McQuay H.J., Sullivan A.F., Smallman K., et al. Intrathecal opioids, potency and lipophilicity. Pain. 1989;36:111–115.
Millan M.J., Czlonkowski A., Pilcher C.W., et al. A model of chronic pain in the rat: functional correlates of alterations in the activity of opioid systems. Journal of Neuroscience. 1987;7:77–87.
Nakanishi S., Inoue A., Kita T., et al. Nucleotide sequence of cloned cDNA for bovine corticotropin-beta-lipotropin precursor. Nature. 1979;278:423–427.
Neil A., Kayser V., Gacel G., et al. Opioid receptor types and antinociceptive activity in chronic inflammation: both kappa- and mu-opiate agonistic effects are enhanced in arthritic rats. European Journal of Pharmacology. 1986;130:203–208.
Neubert M.J., Kincaid W., Heinricher M.M. Nociceptive facilitating neurons in the rostral ventromedial medulla. Pain. 2004;110:158–165.
Nichols M.L., Bian D., Ossipov M.H., et al. Regulation of antiallodynic efficacy by CCK in a model of neuropathic pain in rats. Journal of Pharmacology and Experimental Therapeutics. 1995;275:1339–1345.
Ossipov M.H., Kovelowski C.J., Vanderah T., et al. Naltrindole, an opioid delta antagonist, blocks the enhancement of morphine-antinociception induced by a CCKB antagonist in the rat. Neuroscience Letters. 1994;181:9–12.
Ossipov M.H., Lai J., King T., et al. Antinociceptive and nociceptive actions of opioids. Journal of Neurobiology. 2004;61:126–148.
Ossipov M.H., Lopez Y., Nichols M.L., et al. The loss of antinociceptive efficacy of spinal morphine in rats with nerve ligation injury is prevented by reducing spinal afferent drive. Neuroscience Letters. 1995;199:87–90.
Ossipov M.H., Lopez Y., Nichols M.L., et al. Inhibition by spinal morphine of the tail-flick response is attenuated in rats with nerve ligation injury. Neuroscience Letters. 1995;199:83–86.
Palczewski K., Kumasaka T., Hori T., et al. Crystal structure of rhodopsin: a G protein–coupled receptor. Science. 2000;289:739–745.
Pasternak G.W. Pharmacological mechanisms of opioid analgesics. Clinical Neuropharmacology. 1993;16:1–18.
Pert C.B., Snyder S.H. Opiate receptor: demonstration in nervous tissue. Science. 1973;179:1011–1014.
Pintar J., Kieffer B.L. Genetic analysis of the endogenous opioid system. In: Offermans S., Hein L., eds. Handbook of experimental pharmacology. Transgenic models in pharmacology. Berlin: Springer-Verlag, 2004.
Porreca F., Tang Q.B., Bian D., et al. Spinal opioid mu receptor expression in lumbar spinal cord of rats following nerve injury. Brain Research. 1998;795:197–203.
Portenoy R.K., Foley K.M., Inturrisi C.E. The nature of opioid responsiveness and its implications for neuropathic pain: new hypotheses derived from studies of opioid infusions. Pain. 1990;43:273–286.
Pradhan A.A., et al. Ligand-Directed Signaling Within the Opioid Receptor Family. Br J Pharmacol. 2012.
Pradhan A.A., Befort K., Nozaki C., Gaveriaux-Ruff C., Kieffer B.L. The delta opioid receptor: an evolving target for the treatment of brain disorders. Trends Pharmacol Sci. 2010.
Pradhan A.A., Walwyn W., Nozaki C., et al. Ligand-directed trafficking of the delta-opioid receptor in vivo: two paths toward analgesic tolerance. J Neurosci. 2010;30:16459–16468.
Qiu C., Sora I., Ren K., et al. Enhanced delta-opioid receptor–mediated antinociception in mu-opioid receptor–deficient mice. European Journal of Pharmacology. 2000;387:163–169.
Rahman W., Dashwood M.R., Fitzgerald M., et al. Postnatal development of multiple opioid receptors in the spinal cord and development of spinal morphine analgesia. Brain Research. Developmental Brain Research. 1998;108:239–254.
Rahman W., Suzuki R., Dickenson A.H. Pains, brains and spinal gains: facilitatory mechanisms underlying altered pain states. Journal of Palliative Medicine and Palliative Care. 2003;2:82–89.
Riedl M., Shuster S., Vulchanova L., et al. Orphanin FQ/nociceptin-immunoreactive nerve fibers parallel those containing endogenous opioids in rat spinal cord. Neuroreport. 1996;7:1369–1372.
Rogers R., Wise R.G., Painter D.J., et al. An investigation to dissociate the analgesic and anesthetic properties of ketamine using functional magnetic resonance imaging. Anesthesiology. 2004;100:292–301.
Roumy M., Garnier M., Zajac J.M. Neuropeptide FF receptors 1 and 2 exert an anti-opioid activity in acutely dissociated rat dorsal raphe and periventricular hypothalamic neurones. Neuroscience Letters. 2003;348:159–162.
Rowbotham M.C., Reisner-Keller L.A., Fields H.L. Both intravenous lidocaine and morphine reduce the pain of postherpetic neuralgia. Neurology. 1991;41:1024–1028.
Scherrer G., Befort K., Contet C., et al. The delta agonists DPDPE and deltorphin II recruit predominantly mu receptors to produce thermal analgesia: a parallel study of mu, delta and combinatorial opioid receptor knockout mice. European Journal of Neuroscience. 2004;19:2239–2248.
Shacham S., Topf M., Avisar N., et al. Modeling the 3D structure of GPCRs from sequence. Medicinal Research Reviews. 2001;21:472–483.
Shimoyama N., Shimoyama M., Elliot K., et al. d-Methadone is antinociceptive in the rat formalin test. Journal of Pharmacology and Experimental Therapeutics. 1997;282:648–652.
Shukla A.K., Xiao K., Lefkowitz R.J. l Emerging paradigms of beta-arrestin-dependent seven transmembrane receptor signaling. Trends Biochem Sci. 2011;36:457–469.
Simon E.J., Hiller J.M., Edelman I. Stereospecific binding of the potent narcotic analgesic (3H) etorphine to rat-brain homogenate. Proceedings of the National Academy of Sciences of the United States of America. 1973;70:1947–1949.
Simonin F., Valverde O., Smadja C., et al. Disruption of the kappa-opioid receptor gene in mice enhances sensitivity to chemical visceral pain, impairs pharmacological actions of the selective kappa-agonist U-50,488H and attenuates morphine withdrawal. EMBO Journal. 1998;16:886–897.
Smith M.T., Watt J.A., Cramond T. Morphine-3-glucuronide—a potent antagonist of morphine analgesia. Life Sciences. 1990;47:579–585.
Sora I., Li X.F., Funada M., et al. Visceral chemical nociception in mice lacking mu-opioid receptors: effects of morphine, SNC80 and U-50,488. European Journal of Pharmacology. 1999;366:R3–R5.
Sora I., Takahashi N., Funada M., et al. Opiate receptor knockout mice define mu receptor roles in endogenous nociceptive responses and morphine-induced analgesia. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:1544–1549.
Stanfa L.C., Dickenson A.H. Cholecystokinin as a factor in the enhanced potency of spinal morphine following carrageenin inflammation. British Journal of Pharmacology. 1993;108:967–973.
Stanfa L.C., Dickenson A.H. Electrophysiological studies on the spinal roles of endogenous opioids in carrageenan inflammation. Pain. 1994;56:185–191.
Stanfa L., Dickenson A., Xu X.J., et al. Cholecystokinin and morphine analgesia: variations on a theme. Trends in Pharmacological Sciences. 1994;15:65–66.
Stanfa L.C., Sullivan A.F., Dickenson A.H. Alterations in neuronal excitability and the potency of spinal mu, delta and kappa opioids after carrageenan-induced inflammation. Pain. 1992;50:345–354.
Stein C., Millan M.J., Yassouridis A., et al. Antinociceptive effects of mu- and kappa-agonists in inflammation are enhanced by a peripheral opioid receptor-specific mechanism. European Journal of Pharmacology. 1988;155:255–264.
Sullivan A.F., McQuay H.J., Bailey D., et al. The spinal antinociceptive actions of morphine metabolites morphine-6-glucuronide and normorphine in the rat. Brain Research. 1989;482:219–224.
Suzuki R., Chapman V., Dickenson A. The effectiveness of spinal and systemic morphine on rat dorsal horn neuronal responses in the spinal nerve ligation model of neuropathic pain. Pain. 1999;80:215–228.
Suzuki R., Porreca F., Dickenson A.H. Evidence for spinal dorsal horn hyperexcitability in rats following sustained morphine exposure. Neuroscience Letters. 2006;407:156–161.
Tanowitz M., von Zastrow M. A novel endocytic recycling signal that distinguishes the membrane trafficking of naturally occurring opioid receptors. Journal of Biological Chemistry. 2003;278:45978–45986.
Taylor F., Dickenson A. Nociceptin/orphanin FQ. A new opioid, a new analgesic? Neuroreport. 1998;9:R65–R70.
Terenius L. Stereospecific interaction between narcotic analgesics and a synaptic plasma membrane fraction of rat cerebral cortex. Acta Pharmacologica et Toxicologica. 1973;32:317–320.
Thompson A.A., Liu S., Chun E., et al. Structure of the nociception/orphanin FQ receptor in a complex with a peptide mimetic. Nature. 2012;485:395–399.
Trafton J.A., Abbadie C., Marek K., et al. Postsynaptic signaling via the μ-opioid receptor: responses of dorsal horn neurons to exogenous opioids and noxious stimulation. Journal of Neuroscience. 2000;20:8578–8584.
Traynor J. Subtypes of the kappa-opioid receptor: fact or fiction? Trends in Pharmacological Sciences. 1989;10:52–53.
Traynor J.R., Elliott J. Delta-opioid receptor subtypes and cross-talk with mu-receptors. Trends in Pharmacological Sciences. 1993;14:84–86.
Tzschentke T.M., Christoph T., Kögel B., et al. (-)-(1R,2R)-3-(3 dimethylamino 1-ethyl-2-methyl-propyl)-phenol hydrochloride (tapentadol HCl): a novel mu-opioid receptor agonist/norepinephrine reuptake inhibitor with broad spectrum analgesic properties. Journal of Pharmacology and Experimental Therapeutics. 2007;323:265–276.
Urch C.E., Donovan-Rodriguez T., Gordon-Williams R., et al. Efficacy of chronic morphine in a rat model of cancer-induced bone pain: behavior and in dorsal horn pathophysiology. Journal of Pain. 2005;6:837–845.
Vanderah T.W., Lai J., Yamamura H.I., et al. Antisense oligodeoxynucleotide to the CCKB receptor produces naltrindole- and [Leu5]enkephalin antiserum-sensitive enhancement of morphine antinociception. Neuroreport. 1994;5:2601–2605.
Vanderah T.W., Suenaga N.M., Ossipov M.H., et al. Tonic descending facilitation from the rostral ventromedial medulla mediates opioid-induced abnormal pain and antinociceptive tolerance. Journal of Neuroscience. 2001;21:279–286.
Vanegas H., Schaible H. Effects of antagonists to high-threshold calcium channels upon spinal mechanisms of pain, hyperalgesia and allodynia. Pain. 2000;85:9–18.
Vermeirsch H., Nuydens R.M., Salmon P.L., et al. Bone cancer pain model in mice: evaluation of pain behavior, bone destruction and morphine sensitivity. Pharmacology, Biochemistry, and Behavior. 2004;79:243–251.
von Zastrow M., Svingos A., Haberstock-Debic H., et al. Regulated endocytosis of opioid receptors: cellular mechanisms and proposed roles in physiological adaptation to opiate drugs. Current Opinion in Neurobiology. 2003;13:348–353.
Wang J., Ren M., Han J. Mobilization of calcium from intracellular stores as one of the mechanisms underlying the antiopioid effect of cholecystokinin octapeptide. Peptides. 1992;13:947–951.
Wang Z., Gardell L.R., Ossipov M.H., et al. Pronociceptive actions of dynorphin maintain chronic neuropathic pain. Journal of Neuroscience. 2001;21:1779–1786.
Watkins L.R., Kinscheck I.B., Mayer D.J. Potentiation of opiate analgesia and apparent reversal of morphine tolerance by proglumide. Science. 1984;224:395–396.
Wegert S., Ossipov M.H., Nichols M.L., et al. Differential activities of intrathecal MK-801 or morphine to alter responses to thermal and mechanical stimuli in normal or nerve-injured rats. Pain. 1997;71:57–64.
Wiesenfeld-Hallin Z., Xu X.J., Hokfelt T. The role of spinal cholecystokinin in chronic pain states. Pharmacology and Toxicology. 2002;91:398–403.
Williams J.T., Christie M.J., Manzoni O. Cellular and synaptic adaptations mediating opioid dependence. Physiological Reviews. 2001;81:299–343.
Wu H., Wacher D., Mileni M., et al. Structure of the human kappa-opioid receptor in complex with JDTic. Nature. 2012;485:327–332.
Yaksh T.L. Behavioral and autonomic correlates of the tactile evoked allodynia produced by spinal glycine inhibition: effects of modulatory receptor systems and excitatory amino acid antagonists. Pain. 1989;37:111–123.
Yaksh T. Pharmacology and mechanisms of opioid analgesic activity. In: Yaksh T., Lynch C., Zapol W., et al, eds. Anesthesia: biologic foundations. Philadelphia: Lippincott-Raven; 1997:921–934.
Yaksh T.L., Noueihed R. The physiology and pharmacology of spinal opiates. Annual Review of Pharmacology and Toxicology. 1985;25:433–462.
Zaki P.A., Bilsky E.J., Vanderah T.W., et al. Opioid receptor types and subtypes: the delta receptor as a model. Annual Review of Pharmacology and Toxicology. 1996;36:379–401.
Zhao C., Tall J.M., Meyer R.A., et al. Antiallodynic effects of systemic and intrathecal morphine in the spared nerve injury model of neuropathic pain in rats. Anesthesiology. 2004;100:905–911.
Zhu Y., King M.A., Schuller A.G., et al. Retention of supraspinal delta-like analgesia and loss of morphine tolerance in delta opioid receptor knockout mice. Neuron. 1999;24:243–252.
Akil H., Watson S.J., Young E., et al. Endogenous opioids: biology and function. Annual Review of Neuroscience. 1984;7:223–255.
Bates J.J., Foss J.F., Murphy D.B. Are peripheral opioid antagonists the solution to opioid side effects? Anesthesia and Analgesia. 2004;98:116–122.
Befort K., Kieffer B.L. Structure–activity relationships in the delta opioid receptor. Pain Reviews. 1997;4:100–121.
Besse D., Lombard M.C., Zajac J.M., et al. Pre- and postsynaptic distribution of mu, delta and kappa opioid receptors in the superficial layers of the cervical dorsal horn of the rat spinal cord. Brain Research. 1990;521:15–22.
Décaillot F.M., Befort K., Filliol D., et al. Opioid receptor random mutagenesis reveals a mechanism for G protein–coupled receptor activation. Nature Structural Biology. 2003;10:629–636.
Dickenson A., Where and how do opioids act?. Gebhart G., Hammond D., Jensen T., eds. Proceedings of the 7th World Congress on Pain. Progress in pain research and management, vol 2. Seattle: IASP Press; 1994:525–552.
Dickenson A.H. Spinal cord pharmacology of pain. British Journal of Anaesthesia. 1995;75:193–200.
Evans C.J., Keith D.E., Jr., Morrison H., et al. Cloning of a delta opioid receptor by functional expression. Science. 1992;258:1952–1955.
Gilron I., Bailey J.M., Tu D., et al. Morphine, gabapentin, or their combination for neuropathic pain. New England Journal of Medicine. 2005;352:1324–1334.
Heinricher M.M., Neubert M.J. Neural basis for the hyperalgesic action of cholecystokinin in the rostral ventromedial medulla. Journal of Neurophysiology. 2004;92:1982–1989.
Hylden J.L., Thomas D.A., Iadarola M.J., et al. Spinal opioid analgesic effects are enhanced in a model of unilateral inflammation/hyperalgesia: possible involvement of noradrenergic mechanisms. European Journal of Pharmacology. 1991;194:135–143.
Kieffer B.L. Molecular aspects of opioid receptors. In: Dickenson A., Besson J.M., eds. Handbook of experimental pharmacology. The pharmacology of pain. Berlin: Springer-Verlag; 1997:281–303.
Mao J., Price D.D., Mayer D.J. Experimental mononeuropathy reduces the antinociceptive effects of morphine: implications for common intracellular mechanisms involved in morphine tolerance and neuropathic pain. Pain. 1995;61:353–364.
Neil A., Kayser V., Gacel G., et al. Opioid receptor types and antinociceptive activity in chronic inflammation: both kappa- and mu-opiate agonistic effects are enhanced in arthritic rats. European Journal of Pharmacology. 1986;130:203–208.
Ossipov M.H., Lopez Y., Nichols M.L., et al. The loss of antinociceptive efficacy of spinal morphine in rats with nerve ligation injury is prevented by reducing spinal afferent drive. Neuroscience Letters. 1995;199:87–90.
Pert C.B., Snyder S.H. Opiate receptor: demonstration in nervous tissue. Science. 1973;179:1011–1014.
Urch C.E., Donovan-Rodriguez T., Gordon-Williams R., et al. Efficacy of chronic morphine in a rat model of cancer-induced bone pain: behavior and in dorsal horn pathophysiology. Journal of Pain. 2005;6:837–845.
Vanderah T.W., Suenaga N.M., Ossipov M.H., et al. Tonic descending facilitation from the rostral ventromedial medulla mediates opioid-induced abnormal pain and antinociceptive tolerance. Journal of Neuroscience. 2001;21:279–286.
Wacnik P.W., Kehl L.J., Trempe T.M., et al. Tumor implantation in mouse humerus evokes movement-related hyperalgesia exceeding that evoked by intramuscular carrageenan. Pain Jan. 2003;101(1-2):175–186.
Williams J.T., Christie M.J., Manzoni O. Cellular and synaptic adaptations mediating opioid dependence. Physiological Reviews. 2001;81:299–343.