Cyclooxygenase Inhibitors
Basic Aspects
Mode of Action
Inhibition of prostaglandin production through blockade of cyclooxygenases (COXs) was identified in the early 1970s as the major mechanism of action of aspirin/acetylsalicylic acid and the pharmacologically related non-steroidal anti-inflammatory drugs (NSAIDs) (Vane 1971). A variety of stimuli induce the formation of arachidonic acid from phospholipids of the cell membrane, mainly through cytosolic phospholipase A2. Arachidonic acid then serves as the substrate for prostaglandin G/H synthases, colloquially also called COXs, which convert arachidonic acid into prostaglandin G2 (PGG2) and PGH2 in a two-step process consisting of an initial COX reaction and a second hydroperoxidase reaction (Fig. 32-1). In the late 1980s and early 1990s, the first cDNA of COX enzymes from seminal vesicles was sequenced (Merlie et al 1988, DeWitt et al 1989). Molecular biologists subsequently discovered a second COX isoform whose expression was regulated by cytokines and glucocorticoids (Kujubu et al 1991, O’Banion et al 1991). Both isoforms were found to be encoded by separate genes and were called prostaglandin G/H synthase-1 and -2, or COX-1 and COX-2 (Fig. 32-2). They differ in their regional expression and temporal inducibility, which forms the basis of their different functions in health and disease (Vane et al 1994). COX-1 is constitutively expressed in most tissues and provides the tonic supply of prostanoids required for homeostasis in many organs, organ systems, and cells, including the upper gastrointestinal (GI) tract, platelets, and kidney. COX-2, on the other hand, is under the transcriptional control of pro- and anti-inflammatory cytokines. In many cells such as macrophages it becomes expressed only in response to inflammatory stimuli (Raz et al 1989, 1990), whereas immunosuppressants such as glucocorticoids reduce its expression (Masferrer et al 1992, Yamagata et al 1993). Such concepts fostered the idea of selective inhibition of COX-2 being sufficient for analgesic/anti-inflammatory effects but sparing unwanted effects in the GI tract and possibly in other organ systems. This dichotomous picture of constitutive COX-1 and inducible COX-2 is, however, not consistently followed in all organs. Constitutive or inflammation-independent expression of COX-2 has, for example, been observed in endothelial cells and in the kidney. Inhibition of constitutive COX-2 expression at these sites may contribute to the undesired actions of NSAIDs and coxibs, such as arterial hypertension (Forman et al 2005) and increased cardiovascular risk (Bombardier et al 2000, Bresalier et al 2005).
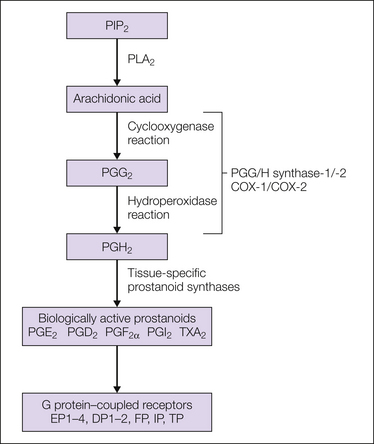
Figure 32-1 Classic prostanoid biosynthesis pathway.
Prostanoid biosynthesis starts with the release of arachidonic acid from cell membranes, mainly through cytosolic phospholipase A2 (PLA2)-dependent hydrolysis. Arachidonic acid is subsequently metabolized by the either one of the two cyclooxygenases (COX-1, COX-2). Prostaglandin H2 (PGH2) is biologically largely inactive but serves as a substrate for tissue-specific prostaglandin synthases, which produce the different biologically active prostanoids. These prostanoids exert their biological action through binding to specific G protein–coupled receptors. PIP2, phosphatidylinositol 4,5,-bisphosphate; TXA2, thromboxane A2.
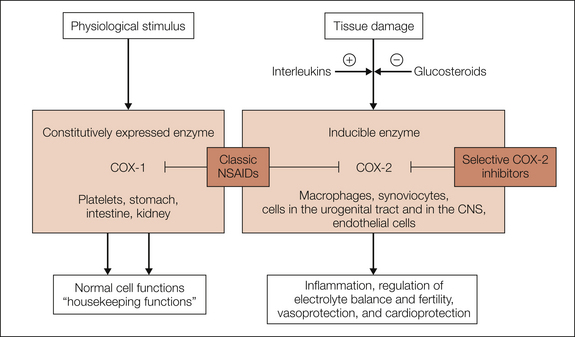
Figure 32-2 Simplified description of the physiological and pathophysiological roles of cyclooxygenase-1 (COX-1) and COX-2.
COX-1 is expressed constitutively in most tissues and fulfills housekeeping functions by producing prostaglandins. COX-2 is an inducible isoenzyme that is expressed in inflammatory cells (e.g., macrophages and synoviocytes) after exposure to pro-inflammatory cytokines and is down-regulated by glucocorticoids. In the kidney (macula densa) and other areas of the urogenital tract and in the central nervous system (CNS), COX-2 is already significantly expressed even in the absence of inflammation. Induction of expression of COX-2 in peripheral and central nervous tissues appears to be most prominent in connection with inflammatory painful reactions. Both enzymes are blocked by classic acidic antipyretic analgesics (non-steroidal anti-inflammatory drugs [NSAIDs]).
Cloning and subsequent heterologous expression of both enzymes allowed high-throughput screening of large compound libraries in the quest for selective inhibitors. Subsequent testing of established COX inhibitors on recombinant enzymes, as well as in isoform-specific ex vivo assays, revealed that most existing drugs were non-specific inhibitors of both enzymes (Patrignani et al 1994), but some experimental compounds proved to be selective (Masferrer et al 1994, Riendeau et al 1997).
Heterologous expression of both enzymes also paved the path for crystallographic analysis of both COXs. These studies provided important insight into the structural basis of their enzymatic activity (Picot et al 1994) and their interaction with inhibitors (Loll et al 1995, Kurumbail et al 1996). It was found that COX-1 and COX-2 insert as homodimers into lipid membranes (Smith and Dewitt 1996) and form a hydrophobic channel that allows arachidonic acid to reach the catalytic center of the enzymes (Picot et al 1994). Most known COX inhibitors inhibit the catalytic site or the channel in a competitive manner. Only aspirin/acetylsalicylic acid blocks this access irreversibly through covalent acetylation of a serine residue (S530) close to the catalytic center (Roth et al 1975). This irreversible action forms the basis for its unique long-lasting inhibitory action on platelet aggregation. The availability of structural information also permitted structure-based rational drug design approaches. It was discovered that differences in the structure of COX-1 and COX-2 were sufficiently large to enable selective inhibition of COX-2 (Kurumbail et al 1996). A side pocket present in the channel formed by the COX-2 enzyme but absent from COX-1 turned out to be particularly relevant for the design of subtype-selective blockers. A third COX isoform, called COX-3 (Chandrasekharan et al 2002), is not encoded by a separate gene but results from alternative splicing of the COX-1 gene and retention of intron 1. COX-3 mRNA has, however, been detected only in dogs and is apparently not present in humans (Dinchuk et al 2003).
Although the role of COX inhibition and diminished prostanoid formation is generally accepted as the predominant mechanism of action of NSAIDs and coxibs, their role is much less clear in acetaminophen/paracetamol, dipyrone/metamizole, and related compounds. These drugs show only limited inhibition of COXs in vitro and largely lack anti-inflammatory action. They may, however, still inhibit COXs in vivo (e.g., through active metabolites). Recent evidence from ex vivo studies indeed suggests that acetaminophen/paracetamol possesses inhibitory actions on COX-2 in humans comparable even to those of coxibs (Hinz et al 2008). The combination of antipyretic and analgesic actions of acetaminophen/paracetamol is clearly consistent with an inhibitory effect of acetaminophen/paracetamol on prostaglandin formation. Its non-acidic (chemically neutral) nature may facilitate permeation of acetaminophen/paracetamol across the blood–brain barrier into the central nervous system (CNS), where an inhibitory action on COX-2–dependent production of prostaglandins would exert analgesic and antipyretic effects. The apparent lack of a clear anti-inflammatory action of acetaminophen/paracetamol may, on the other hand, be ascribed to the absence of their enrichment in inflamed tissue (see also below) and to the presence of higher arachidonic acid or peroxide concentrations in inflamed tissue, which would render competitive inhibition less effective.
Cyclooxygenase Products as Mediators of Hyperalgesia
Prostaglandins, in particular PGE2 and PGI2 (also called prostacyclin), increase the sensitivity of peripheral nociceptor endings to noxious stimuli (Fig. 32-3A). They facilitate activation of the receptors and ion channels involved in the nociceptive transduction process, such as the transient potential channel TRPV1 (Caterina et al 1997) and certain voltage-gated Na+ channels, in particular, Nav1.8 (Akopian et al 1996). These actions occur through G protein–coupled receptors specific for the different subtypes of prostaglandins and through the subsequent activation of different protein kinases.
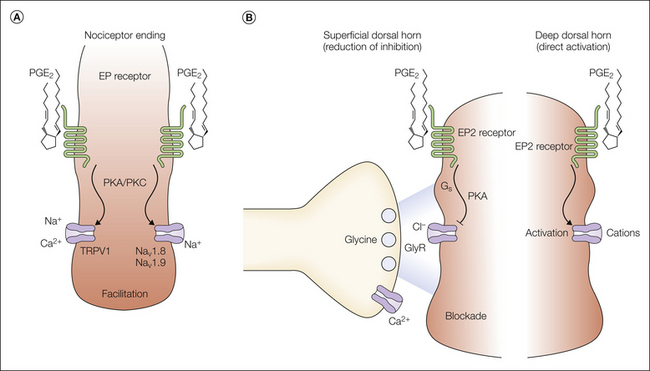
Figure 32-3 Molecular mechanisms of prostaglandin E2 (PGE2)-mediated nociceptive sensitization.
A, Schematic representation of a polymodal (nociceptive) C fiber. PGE2 has been shown to facilitate the activation of membrane responses to capsaicin and noxious heat, thus suggesting that the capsaicin receptor TRPV1 (transient receptor potential vanilloid 1) is one possible target of PGE2. The tetrodotoxin-resistant Na+ channels Nav1.8 and Nav1.9 may represent another target involved in peripheral sensitization processes. B, The spinal cord dorsal horn represents a second major site of the pain-sensitizing action of PGE2. Two possible molecular mechanisms have thus far been proposed at this site. PGE2 reduces the action of the major inhibitory neurotransmitter glycine in the superficial layers of the dorsal horn and—at higher concentrations—leads to direct depolarization of deep dorsal horn neurons. EP2, PGE2 receptor; GlyR, glycine receptor; PKA, protein kinase A; PKC, protein kinase C.
Application of PGE2 to isolated dorsal root ganglion (DRG) cells (Lopshire and Nicol 1997) or to HEK 293 cells transfected with TRPV1 (Rathee et al 2002) dramatically increases current responses to capsaicin and heat, two prototypical activators of TRPV1. This phenomenon is likely to underlie the thermal hyperalgesia in inflammatory disease states (Caterina et al 2000, Davis et al 2000). It is tempting to speculate that a leftward shift in TRPV1 activation by several degrees could render TRPV1 susceptible to activation by physiological temperatures. Such a process could contribute to the generation of spontaneous pain.
Tetrodotoxin-resistant Na+ channels, especially Nav1.8, represent another potential substrate for peripheral nociceptive sensitization by prostaglandins. These channels become more readily activated in the presence of a number of inflammatory mediators, including PGE2 (England et al 1996, Gold et al 1996). In particular, Nav1.8 is selectively expressed in nociceptive small- and medium-sized DRG neurons (Akopian et al 1996). Modulation of these Na+ channels involves activation of adenylyl cyclase and increases in cyclic adenosine monophosphate, which possibly leads to protein kinase A–dependent phosphorylation of the channels. Via this mechanism, the prostaglandins produced during inflammatory responses may significantly increase the excitability of nociceptive nerve fibers and also contribute to the recruitment of nociceptors. A significant proportion of nociceptors in healthy (uninflamed and uninjured) tissue are mechanically insensitive and are not activated even by strong stimuli (Schaible and Schmidt 1988a, Kress et al 1992). Following tissue trauma and the release of prostaglandins, these silent nociceptors become excitable to pressure, changes in temperature, and tissue acidosis (Neugebauer et al 1995), which contributes to the generation and maintenance of hyperalgesia (Schaible and Schmidt 1988b).
The sensitizing actions of prostaglandins are not restricted to peripheral nociceptor endings but also occur at spinal sites (Fig. 32-3B). Intrathecal injection of PGE2 causes sensitization to thermal and mechanical stimuli. Expression of COX-2 (Beiche et al 1996, Samad et al 2001) and inducible prostaglandin E synthase (mPGES1) (Claveau et al 2003, Guay et al 2004) increases in the CNS in response to peripheral inflammation. In rodents, hyperalgesia can be partially prevented or reversed by intrathecally injected NSAIDs (Malmberg and Yaksh 1992a, 1992b) or coxibs (Reinold et al 2005), an action that is accompanied by a decrease in spinal PGE2 concentrations (Malmberg and Yaksh 1995). Different cellular mechanisms of action have been proposed for the pro-nociceptive effects of PGE2 in the spinal cord. Baba and colleagues (2001) have shown that PGE2 can directly depolarize deep dorsal horn neurons, and Ahmadi and associates (2002) demonstrated that PGE2 reduces inhibitory glycinergic neurotransmission through a post-synaptic mechanism. This latter action is triggered by activation of PGE2 receptors of the EP2 subtype (Reinold et al 2005). Activation of these receptors leads to the protein kinase A–dependent phosphorylation and inhibition of a certain isoform of glycine receptors that contain the α3 subunit (GlyRα3) (Harvey et al 2004). This subunit is expressed in the superficial layers of the spinal dorsal horn, where nociceptive afferents terminate and where glycinergic neurotransmission is inhibited by PGE2. Evidence of the in vivo relevance of this centrally mediated form of inflammatory pain sensitization comes from several studies using mice carrying targeted mutations in genes critically involved in these pathways. Mice lacking neuronal protein kinase A showed less thermal hyperalgesia after injection of PGE2 into the spinal canal (Malmberg et al 1997). Specific disruption of COX-2 in neural cells (through the use of nestin-driven cre expression) protects mice from mechanical hyperalgesia after peripheral inflammation (Vardeh et al 2007). Mice deficient in either GlyRα3 or the EP2 receptor not only lack PGE2-mediated inhibition of glycinergic neurotransmission in the spinal cord dorsal horn but also largely lack the pro-nociceptive effects of spinal PGE2 (Harvey et al 2004, Reinold et al 2005). Subsequent work has studied the phenotypes of GlyRα3-deficient mice in more detail. These studies showed that disruption of the GlyRα3 gene did not affect nociceptive responses in the formalin test and after peripheral nerve lesions (Hosl et al 2006), which is consistent with the specific involvement of prostaglandins in inflammatory pain. Similar unchanged nociceptive responses were also reported for carrageenan-induced cutaneous inflammation and kaolin/carrageenan-induced arthritis or iodoacetate osteoarthritis (Harvey et al 2009).
The contribution of a spinal site of action of COX inhibitors in humans has recently been addressed in two studies that tested the action of intrathecally injected ketorolac against experimental pain in human volunteers (Eisenach et al 2010b) and postoperative and chronic pain in patients (Eisenach et al 2010a). Intrathecal ketorolac was well tolerated, but its analgesic effects were much less than expected based on preclinical data from animal experiments. It showed some efficacy against sunburn-induced hyperalgesia in volunteers but did not induce significant analgesia against postoperative pain. Only in a subset of chronic pain patients whose spinal prostaglandin levels were increased most significantly did intrathecal ketorolac reduce spinal PGE2 levels and show analgesic efficacy.
Diminished Prostaglandin Production As a Source of Other Desired Effects of Cyclooxygenase Inhibitors
Diminished formation of prostanoids can also explain most of the anti-inflammatory and antipyretic actions of antipyretic analgesics. Edema formation and plasma extravasation are promoted in particular by PGI2 (Murata et al 1997). Antipyresis originates from reduced activation of EP3 receptors in the hypothalamus (Ushikubi et al 1998). There is good evidence that the pro-pyretic PGE2 does not originate from neural sources as the pro-nociceptive PGE2 does but instead originates from endothelial cells that reside in the walls of cerebral blood vessels and produce PGE2 through COX-2 (Vardeh et al 2007). Inhibition of platelet aggregation may also be considered a desired effect of antipyretic analgesics. It occurs through reduced formation of COX-1–dependent thromboxane in platelets. Inhibition of platelet aggregation to a degree relevant for the prevention of arterial thrombosis is primarily seen with low doses of aspirin/acetylsalicylic acid. Inhibition by other reversible COX inhibitors is probably too transient to exert clinically relevant therapeutic effects (Patrono and Baigent 2009). With respect to cardiovascular risks, increases in blood pressure and the reduced anti-aggregatory and vasodilating effects of PGI2 probably counteract the transient beneficial effects from transient thromboxane inhibition by classic NSAIDs (Grosser et al 2006). The transient nature of inhibition of platelet aggregation does, however, not exclude increased bleeding risk in predisposed patients receiving NSAIDs or dipyrone/metamizole.
Undesired Effects of Cyclooxygenase Inhibitors
Classic side effects of COX inhibitors include ulcers of the upper GI tract, asthma, increases in blood pressure, kidney damage, and bleeding. Inhibition of constitutive prostaglandin formation in the GI tract and in the kidneys is a major cause of the undesired effects of the classic NSAIDs, in particular, GI tract toxicity and renal impairment. PGE2 protects the gastric mucosa by reducing acid and promoting mucous secretion via activation of EP1 and EP3 receptors (Takeuchi et al 1999, Suzuki et al 2001, Kato et al 2005). The GI tract toxicity of NSAIDs is probably aggravated by accumulation of these acidic compounds in mucosal cells through a process called ion trapping (see also below), and part of the mucosal damage evoked by orally administered NSAIDs may also be derived from COX-independent toxicity, which is supported by the observation that gastric ulcers still develop in COX-1–deficient mice after oral administration of indomethacin (Langenbach et al 1995). Asthma attacks induced by aspirin/acetylsalicylic acid and by NSAIDs have for a long time been attributed to the so-called leukotriene shift—a shift of arachidonic acid metabolism from COX-mediated production of prostaglandins to increased lipoxygenase-dependent leukotriene production. More recent evidence suggests that under conditions of airway inflammation, certain prostaglandins, in particular, PGD2 and PGE2, exert a protective function by down-modulating the immune responses (Hammad et al 2007) or by dilating the airways (Tilley et al 2003). Blockade of their formation would then cause acute bronchoconstriction. In the kidney, prostaglandins regulate perfusion, filtration, and salt reuptake, actions that contribute to the significant increases in arterial blood pressure that are already seen after short-term use of classic NSAIDs. Long-term use or abuse of NSAIDs has been the leading cause of terminal renal failure for several decades.
In addition to these classic side effects, the potential cardiovascular toxicity of coxibs and NSAIDs has gained significant attention after the withdrawal of rofecoxib from the market because of its association with increased cardiovascular risk (Bombardier et al 2000, Bresalier et al 2005). Initially, it was thought that the increased cardiovascular toxicity would be a problem specific to coxibs, most likely caused by a coxib-induced imbalance of protective (PGI2) and potentially harmful (thromboxane) mediators. There is now widespread consensus that the increase in cardiovascular risk results from inhibition of PGI2 formation and occurs independent of the presence or absence of additional blockade of thromboxane formation (Grosser et al 2006).
Interactions of Antipyretic Analgesics with Non-prostanoid Mediators
Although diminished prostanoid formation provides a plausible explanation for many of the desired and undesired actions of NSAIDs and coxibs and possibly at least for part of the analgesic and antipyretic effects of classic non-acidic antipyretic analgesics, these drugs also interfere with the production of other mediators such as endocannabinoids, which potentially also contribute to their in vivo actions (Fig. 32-4). Several reports suggest that COXs serve other enzymatic functions in addition to the formation of prostanoids. COX-2, but not COX-1, metabolizes the two endocannabinoids arachidonoyl ethanol amide (AEA or anandamide) (Yu et al 1997) and 2-arachidonoyl glycerol (2-AG) to generate prostaglandin ethanolamides (prostamides) and prostaglandin glyceryl ester (Kozak et al 2002). Blockade of COX-2 might thus not only reduce tissue concentrations of prostanoids but also increase levels of endocannabinoids. Increases in spinal endocannabinoid tone might indeed contribute to the spinal analgesic action of indomethacin and flurbiprofen (Guhring et al 2002, Ates et al 2003). Furthermore, there is evidence of biological activity of prostamides (Woodward et al 2008) and possibly also prostaglandin glyceryl esters (Nirodi et al 2004, Sang et al 2007). Inhibition of COX-2 could thus interfere with nociception not only through diminished formation of prostanoids but also through reduced degradation of endocannabinoids or reduced formation of prostamides or prostaglandin glyceryl esters.
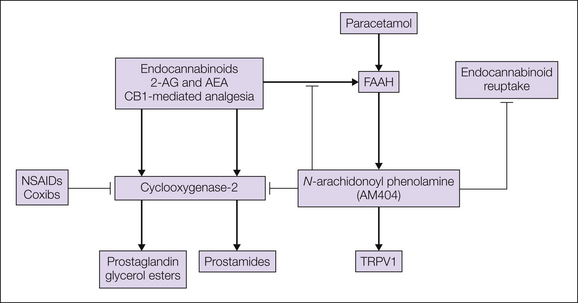
Figure 32-4 Possible interactions of cyclooxygenase (COX) inhibitors and acetaminophen/paracetamol with the endocannabinoid system.
Because COX-2 not only produces prostanoids but also degrades endocannabinoids, inhibition of it may not only reduce prostanoid concentrations but also increase endocannabinoid concentrations. A specific interaction of the endocannabinoid system apparently exists for acetaminophen/paracetamol, which is metabolized by the fatty acid amide hydrolase (FAAH) to N-arachidonoyl phenolamide (AM404). AM404 possibly facilitates endocannabinoid actions by inhibiting their degradation by FAAH and COX-2 and their reuptake by yet to be identified cannabinoid transporters. AEA, arachidonoyl ethanol amide; 2-AG, 2-arachidonoyl glycerol; CB1, endocannabinoid receptor 1; NSAIDs, non-steroidal anti-inflammatory drugs; TRPV1, transient receptor potential vanilloid 1.
A possible contribution of the endocannabinoid system to the analgesic activity of antipyretic analgesics has been studied particularly extensively for acetaminophen/paracetamol. At least in the rodent brain and spinal cord, acetaminophen/paracetamol is converted to N-arachidonoyl phenolamine (AM404) through metabolism by fatty acid amide hydrolase (FAAH) and conjugation with arachidonic acid (Hogestatt et al 2005). AM404 interferes with a variety of enzymes and ion channels. It is a known inhibitor of anandamide reuptake (Beltramo et al 1997) and degradation by FAAH (Glaser et al 2003). It binds CB1 receptors, activates TRPV1 channels (Yue et al 2004), and inhibits COX-1 and COX-2 (Hogestatt et al 2005). Interestingly, recent data indicate that the analgesic actions of acetaminophen/paracetamol are in fact lost in mice lacking CB1 receptors and are antagonized by pharmacological blockade of CB1 receptors (Mallet et al 2008). Whether this CB1 receptor–dependent analgesia by acetaminophen/paracetamol is due to reduced endocannabinoid metabolism by COX-2, inhibition of cannabinoid transport or degradation, or other still unidentified processes is not known at present. Other intriguing aspects of acetaminophen/paracetamol pharmacology include the contribution of TRPV1 activation to its analgesic antipyretic actions (Gavva et al 2007, Mallet et al 2010). An alternative explanation for its efficacy was recently published, namely, that nociceptive input to the spinal cord is blocked by metabolites of paracetamol/acetaminophen acting on transient receptor potential ankyrin 1 (TRPA1) (Andersson et al 2011).
Aspirin/acetylsalicylic acid not only blocks the formation of prostanoids from arachidonic acids but also redirects the metabolic activity of COX-2 to the formation of so-called aspirin-triggered lipoxins (ATLs), which exert anti-inflammatory effects (Chiang et al 2005). One of these ATLs reduces inflammatory hyperalgesia both after systemic and after intrathecal injection. Possible cellular mechanisms of the spinal anti-hyperalgesia include a reduction in the activation of spinal astrocytes (Svensson et al 2007).
Biodistribution of Antipyretic Analgesics
The desired and undesired actions of antipyretic analgesics are not only determined by their molecular actions on COXs and possibly other enzymes but also strongly influenced by their pharmacokinetic properties, in particular, by their biodistribution (Brune et al 2010). Most non-selective COX inhibitors (classic NSAIDs) are weak acids with pKa values between 3.5 and 5.5. This physicochemical property predisposes NSAIDs to become enriched (“trapped”) inside cells neighboring an acidic environment, such as cells in the stomach wall, the urinary tract, and inflamed tissues (Fig. 32-5). Since this pattern of biodistribution also reflects the major sites of action of NSAIDs, it is tempting to speculate that the acidic nature of NSAIDs contributes to the distinct actions and side effects of NSAIDs and non-acidic antipyretic COX inhibitors and coxibs. The higher lipophilicity of the non-acidic compounds, such as acetaminophen/paracetamol, dipyrone/metamizole, and most coxibs, may facilitate their penetration into the CNS.
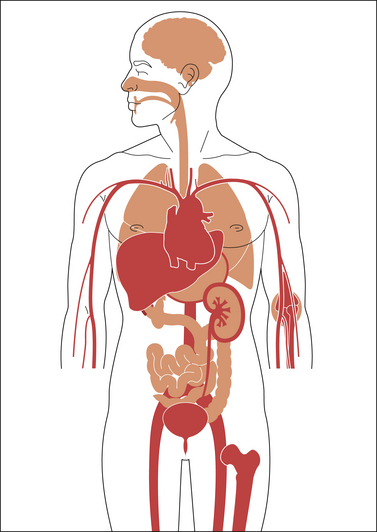
Figure 32-5 Schematic representation of the distribution of acidic antipyretic analgesics in the human body (transposition of data from animal experiments to human conditions).
Dark areas indicate high concentrations of the acidic antipyretic analgesics: in the stomach and upper gastrointestinal tract wall, blood, liver, bone marrow and spleen (not shown), inflamed tissue (e.g., joints), and the kidney (cortex > medulla). Some acidic antipyretic analgesics are excreted in part unchanged in urine and achieve high concentration in this body fluid; others encounter the enterohepatic circulation and are found in high concentrations as conjugates in bile.
Properties of NSAIDs and Coxibs in Clinical Use
This section deals with clinically used COX inhibitors (NSAIDs and coxibs). Table 32-1 summarizes the most relevant desired and undesired effects of these drugs. The pharmacokinetic properties and therapeutic doses of individual compounds are summarized in Table 32-2 (NSAIDs) and Table 32-3 (coxibs). Table 32-2 also contains data on aspirin/acetylsalicylic acid, which differs in many respects from the other NSAIDs (see below). Most of the data in Tables 32-2 and 32-3 are derived from Brune et al 2010.
Table 32-1
Most Relevant Desired and Undesired Actions of Classic Non-selective NSAIDs
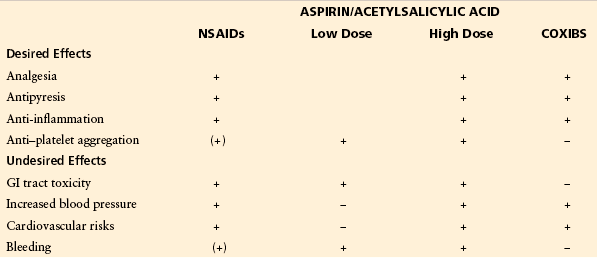
GI, gastrointestinal; NSAIDs, non-steroidal anti-inflammatory drugs.
Table 32-2
Acidic Antipyretic Analgesics (Anti-inflammatory Antipyretic Analgesics, NSAIDs): Chemical Classes, Structures, Physicochemical and Pharmacological Data, Therapeutic Dosage
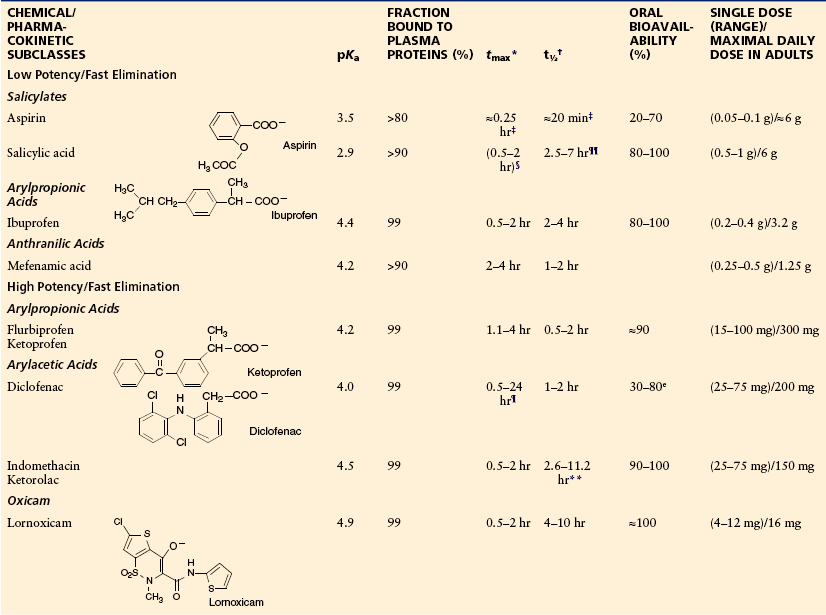
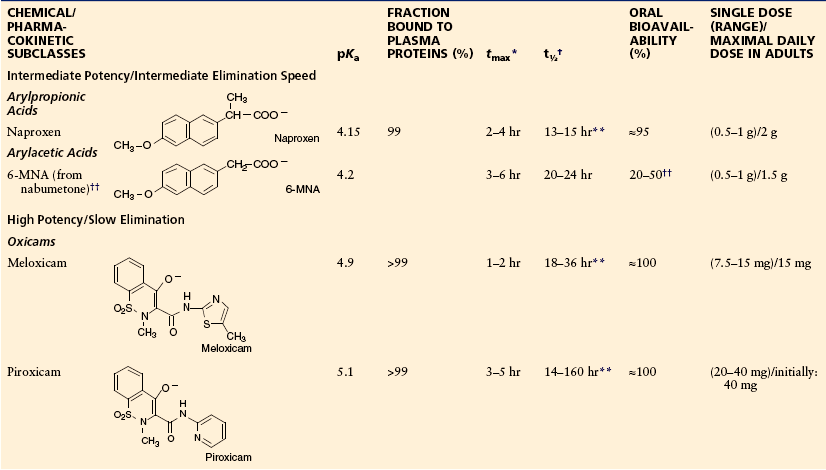
*Time to reach maximum plasma concentration after oral administration.
†Terminal half-life of elimination.
‡Of aspirin, the prodrug of salicylic acid.
§Depending on the galenic formulation.
¶Monolithic acid–resistant tablet or similar form.
**EHC, enterohepatic circulation.
††6-MNA, active metabolite of nabumetone.
The authors have taken all efforts to ensure correct information on drug doses; they do however not take any legal responsibility for incorrect data. Single doses and maximum daily doses may differ between countries.
Table 32-3
Coxibs: Chemical Classes, Structures, Physicochemical and Pharmacological Data, Therapeutic Dosage
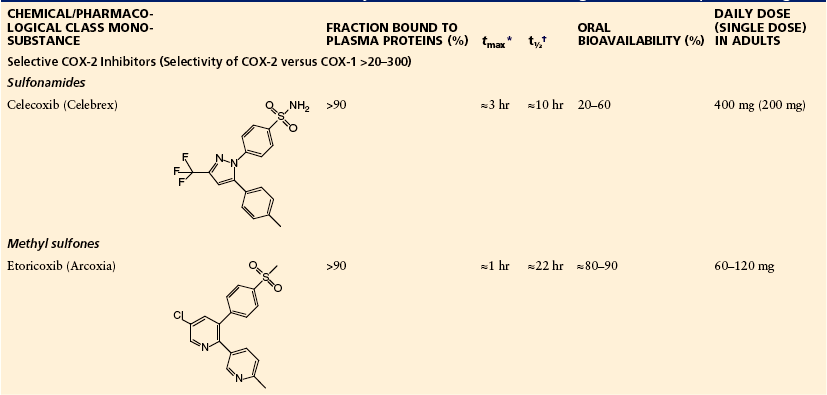
*Time to reach maximum plasma concentration after oral administration.
†Terminal half-life of elimination, dependent on liver function with phenazone.
The authors have taken all efforts to ensure correct information on drug doses; they do however not take any legal responsibility for incorrect data. Single doses and maximum daily doses may differ between countries.
NSAIDs
Classic (non-selective) COX inhibitors differ in their potency, that is, the dose necessary to achieve a certain effect, which ranges from a few milligrams (lornoxicam) to about 1 g (e.g., salicylic acid). They also differ in their pharmacokinetic characteristics: speed of absorption (time to peak, tmax, which may also depend on the galenic formulation used), maximal plasma concentrations (cmax), elimination half-life (t½), and oral bioavailability (AUCrel). Based on these characteristics we propose here that NSAIDs be classified as (1) NSAIDs with a short elimination half-life and low potency, (2) NSAIDs with a short elimination half-life and high potency, and (3) NSAIDs with a long elimination half-life.
NSAIDs with a Short Elimination Half-Life (Low Potency)
The prototype of this class is ibuprofen. Depending on the galenic formulation, fast or slow absorption (and onset of action) may be achieved (Laska et al 1986). Fast absorption is seen, for example, when it is given as a lysin salt (Geisslinger et al 1989). The bioavailability of ibuprofen is close to 100% and elimination is always fast, even in patients suffering from mild or severe impairment of liver or kidney function. Therefore, ibuprofen is used in single doses of between 200 mg and 1 g. A maximum dose of 3.2 g/day (United States) or 2.4 g (Europe) is recommended for rheumatoid arthritis. Ibuprofen (at low doses) appears to be particularly useful for the treatment of acute occasional inflammatory pain. It may also be used, though with less benefit, for chronic rheumatic diseases (high doses). At high doses the otherwise harmless compound increases in toxicity (Kaufman et al 1993).
Ibuprofen is also used as a pure S-enantiomer because only this enantiomer is a (direct) COX inhibitor. On the other hand, the R-enantiomer, which accounts for 50% of the usual racemic mixture, is converted to the S-enantiomer in humans (Rudy et al 1991). It is unknown whether use of the pure S-enantiomer offers any benefits (Mayer and Testa 1997).
NSAIDs with a Short Elimination Half-Life (High Potency)
Drugs in this group are prevalent in the treatment of musculoskeletal pain. The most widely used compound worldwide is diclofenac, which appears to be less active on COX-1 than on COX-2 (Tegeder et al 1999). This preferential inhibition of COX-2 may contribute to its relatively low toxicity in the GI tract (Henry et al 1996).
One limitation of diclofenac results from the usual galenic formulation, which consists of a monolithic acid–resistant encapsulation. This may cause retarded absorption of the active ingredient because of retention of such monolithic formulations in the stomach for hours or even days. Moreover, diclofenac is subject to considerable first-pass metabolism, which results in limited (about 50%) oral bioavailability. Consequently, lack of therapeutic effect may require adaptation of the dosage or change of the drug. New galenic formulations (microencapsulation, salts, etc.) have remedied some of these deficits. The slightly higher incidence of liver toxicity with diclofenac may result from the high degree of first-pass metabolism, which leads to a host of reactive metabolites (Boelsterli 2003).
This group contains other important drugs such as flurbiprofen, indomethacin, lornoxicam, and ketorolac (very potent), as well as ketoprofen and fenoprofen (less active). All show high oral bioavailability and good effectiveness, but also a relatively high risk for unwanted drug effects (Henry et al 1996). Drugs in this group are widely used for arthritis and osteoarthritis. Ketorolac is used after surgery in some countries. The long-lasting inhibition of COX-1 may cause bleeding in postoperative patients.
NSAIDs with a Long Elimination Half-Life
Some forms of pain, migraine, menstrual cramps, osteoarthritis, and rheumatoid arthritis appear to be adequate indications for drugs with a long elimination half-life. These drugs exert lasting pain reduction, but also more undesired drug effects. Naproxen and several oxicams (meloxicam, piroxicam, and tenoxicam) fall into this group. These compounds owe their slow elimination to slow metabolism together with a high degree of enteropathic circulation. The long half-life (days) does not make these drugs less suitable for the treatment of acute pain of short duration, but their main indication is inflammatory pain likely to persist for days, such as pain in patients with rheumatoid arthritis or bone metastases. The high potency and long persistence in the body may be the reason for the somewhat higher incidence of serious adverse drug effects in the GI tract and kidney (Henry et al 1996).
Aspirin/Acetylsalicylic Acid
Aspirin/acetylsalicylic acid deserves special discussion. Aspirin/acetylsalicylic acid is about 100 times more potent as a COX inhibitor than its metabolite salicylic acid is. As outlined above, the parent compound acetylates a serin residue in the active center of COX-1 and COX-2 and inactivates both COXs permanently. Most cells, however, compensate for this inactivation by de novo synthesis of the enzyme, with the exception of thrombocytes, in which a single dose of aspirin/acetylsalicylic acid blocks COX-1 activity irreversibly and hence the body’s thromboxane synthesis for several days (the turnover time of thrombocytes). When low doses are given, aspirin/acetylsalicylic acid acetylates COX-1 and blocks thromboxane synthesis in all platelets passing through the capillary bed of the GI tract but does not interfere with prostacyclin synthesis in endothelial cells outside the gut. This selectivity is due to the rapid cleavage of aspirin/acetylsalicylic acid, which leaves little if any intact aspirin/acetylsalicylic acid in the post-hepatic systemic circulation. The endothelial cells thus continue to release prostacyclin and maintain their antithrombotic activity. The dominant indication for aspirin/acetylsalicylic acid is for the prevention of thromboembolic events. Even at these low doses, acetylsalicylic acid may still cause bleeding from existing ulcers as a result of its long-lasting platelet inhibitory effect, as well as topical irritation of the gastric mucosa. Because of the high risk for bleeding, use of aspirin/acetylsalicylic acid for analgesia has largely been abandoned and only a few very specific indications in pediatrics (Kawasaki’s syndrome and acute rheumatic fever) have remained in addition to antithrombotic therapy. Furthermore, aspirin/acetylsalicylic acid should not be used by pregnant women (premature bleeding, closure of the ductus arteriosus) or by children before puberty (Reye’s syndrome with severe liver and brain damage).
Preferential and Selective Cyclooxygenase-2 Inhibitors
Following the discovery that the prostaglandins responsible for inflammation and the accompanying pain and hyperalgesia are predominantly produced by COX-2, selective COX-2 inhibitors were developed (Flower 2003). At present, two classes are on the market. They are both non-acidic and show relatively homogeneous distribution throughout the body. The characteristics of COX-2–selective compounds are detailed in Table 32-3.
Depending on the speed of absorption, bioavailability, and terminal elimination half-life, these drugs are useful for acute and prolonged pain states. Both compounds have been shown to be associated with reduced GI toxicity in comparison to the classic non-selective (and acidic) compounds (FitzGerald and Patrono 2001, Flower 2003). On the other hand, some patients experience less analgesic and anti-inflammatory activity than with non-selective acidic compounds. All these compounds interfere with prostanoid production in the kidney and blood vessels and thereby cause increases in blood pressure and water retention. They may also interfere with blood pressure control in hypertensive patients (White and Campbell 2010). Celecoxib inhibits CYP2D6, which causes interactions with cardiovascular (e.g., metoprolol) and psychotropic drugs (Werner et al 2003). The methyl sulfone etoricoxib is metabolized by drug-oxidizing enzymes and consequently has only few drug interactions (e.g., with methotrexate).
Etoricoxib is absorbed quickly and eliminated slowly, which may make it particularly suitable for fast but long-lasting pain relief; however, long persistence in the body may also have disadvantages (see Brune et al 2009). Finally, it should be borne in mind that blockade of COX-2 may be problematic in patients at risk for cardiovascular thromboembolic reactions. It could be shown that endothelium-derived PGI2 is partly produced by COX-2 (for review see FitzGerald and Patrono 2001). Recent evidence indicates that all inhibitors of COX-2—selective and non-selective—may increase the risk for myocardial infarction and stroke (Bombardier et al 2000, Bresalier et al 2005, Solomon et al 2005, Warner and Mitchell 2008). As a consequence, COX inhibitors should be given at the lowest dose and for the shortest period necessary.
It has been claimed that nabumetone, etodolac, and meloxicam are particularly well tolerated in the GI tract because they inhibit predominantly COX-2. These results are not fully accepted. The active metabolite of nabumetone shows no selectivity for COX-2, and the selectivity of etodolac and meloxicam is not superior to that of diclofenac when tested ex vivo in humans (Patrignani et al 1994, Riendeau et al 1997).
Non-Acidic Antipyretic Analgesics
Acetaminophen/paracetamol is representative of the members of this group remaining on the market. Its typical desired and undesired effects are summarized in Table 32-4, and its pharmacokinetic and pharmacodynamic data are presented in Table 32-5. In vitro, acetaminophen/paracetamol is a very weak, possibly indirect inhibitor of COXs, but evidence that it is indeed working through COX inhibition comes from ex vivo experiments showing that acetaminophen/paracetamol is a preferential COX-2 inhibitor in vivo. In doses of up to 4 g/day it reduces PGE2 production in humans to below 80% (Hinz et al 2008). The weak COX inhibition by acetaminophen/paracetamol appears to result from its low efficacy, which limits its use to mild pain. Increasing the dose above 4 g/day will cause serious liver damage (Bridger et al 1998). Moreover, it is now well documented that use of acetaminophen/paracetamol is associated with increased blood pressure (Sudano et al 2010) and an increase in cardiovascular events (Chan et al 2006). In addition, acetaminophen/paracetamol given during pregnancy has been linked with cryptorchidism (reduced fertility of the male offspring; Kristensen et al 2011) and with asthma (Etminan et al 2009, Scialli et al 2010). Finally, acetaminophen/paracetamol increases the risk for GI tract ulcers when given together with aspirin/acetylsalicylic acid (Rahme et al 2008, Shaheen et al 2010).
Table 32-4
Most Relevant Desired and Undesired Actions of Acetaminophen/Paracetamol and Dipyrone/Metamizole
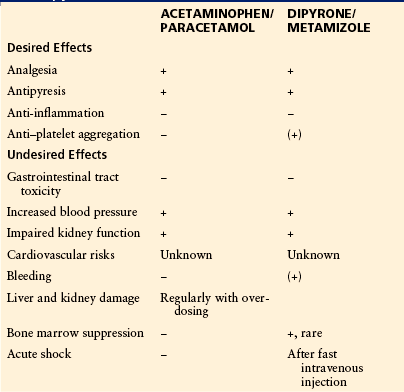
Table 32-5
Non-acidic Antipyretic Analgesics: Chemical Classes, Structures, Physicochemical and Pharmacological Data, Therapeutic Dosage
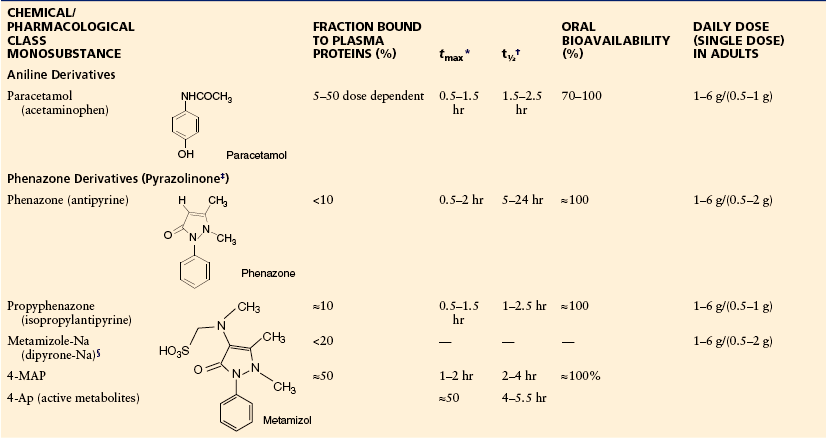
4-AP, 4-aminophenazone; 4-MAP, 4-methylaminophenazone.
*Time to reach maximum plasma concentration after oral administration.
†Terminal half-life of elimination, dependent on liver function with phenazone.
‡Terms such as pyrazole and, incorrectly, pyrazolone are also in use.
§Noraminopyrinemethanosulfonate-Na.
The authors have taken all efforts to ensure correct information on drug doses; they do however not take any legal responsibility for incorrect data. Single doses and maximum daily doses may differ between countries.
After overdosage, acetaminophen/paracetamol is metabolized to highly toxic nucleophilic benzoquinones, which bind covalently to DNA and structural proteins in parenchymal cells (e.g., in the liver and kidney), where these reactive intermediates are produced (Fig. 32-6) (for review, see Seeff et al 1986). In the absence of early proper treatment, the consequence is potentially fatal liver damage. If detected early (within the first 12 hours after intake), intoxication can be treated with N-acetylcysteine, which regenerates the detoxifying mechanisms. Acetaminophen/paracetamol should not be taken by patients with seriously impaired liver function. The remaining indications for acetaminophen/paracetamol are fever and mild forms of pain, such as in the context of viral respiratory infections. Acetaminophen/paracetamol is still used in children, but despite its somewhat lower toxicity in juvenile patients, lethal cases of (involuntary) overdosage have occurred.
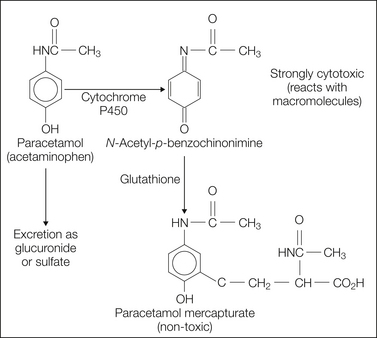
Figure 32-6 Schematic diagram of the metabolism of acetaminophen/paracetamol.
At therapeutic doses most of the acetaminophen/paracetamol is metabolized in the liver in a phase II reaction and excreted as glucuronide or sulfate. At higher doses the responsible enzymes become saturated and acetaminophen/paracetamol is metabolized in the liver via a P450-dependent mechanism, which leads to the formation of N-acetyl-p-benzoquinone imine, a highly cell-toxic metabolite. This metabolite can initially be detoxified via a glutathione-dependent step to acetaminophen mercapturate. At doses beyond 100 mg/kg, the glutathione becomes exhausted and N-acetyl-p-benzoquinone imine now reacts with macromolecules in hepatocytes, which leads to cell death and acute liver failure.
Phenazone and Its Derivatives
Following the discovery of phenazone some 130 years ago, drug companies tried to improve two aspects of this compound to produce a more potent drug and to develop a water-soluble derivative that can be given intravenously. The best-known results of these attempts are dipyrone/metamizole and propyphenazone (see Tables 32-4 and 32-5). The two compounds differ from phenazone in their potency, elimination half-life (Levy et al 1995), and water solubility (dipyrone is a water-soluble prodrug of methyl aminophenazone).
Phenazone, propyphenazone, and dipyrone/metamizole are used in many countries as the prevailing antipyretic analgesics (Latin America, many countries in Asia, Eastern and Central Europe). Dipyrone/metamizole has been accused of causing agranulocytosis. The incidence, however, is generally very low (one case per million treatment periods) (Levy 1986, Kaufman et al 1991).
All antipyretic analgesics have been claimed to cause Stevens–Johnson syndrome and Lyell’s syndrome, as well as shock reactions. New data indicate that the incidence of these events is on the same order of magnitude as, for example, penicillins (Roujeau et al 1995, Mockenhaupt et al 1996, Kaufman 2003). All non-acidic phenazone derivatives lack anti-inflammatory activity; they are devoid of GI tract and (acute) renal toxicity. Dipyrone/metamizole is safe at overdosage, which is in contrast to acetaminophen/paracetamol (Wolhoff et al 1983). If one compares aspirin/acetylsalicylic acid, acetaminophen/paracetamol, and propyphenazone for the same indication (e.g., occasional headache), it is obvious that aspirin/acetylsalicylic acid is more dangerous than either propyphenazone or acetaminophen/paracetamol.
The references for this chapter can be found at www.expertconsult.com.
References
Ahmadi S., Lippross S., Neuhuber W.L., et al. PGE2 selectively blocks inhibitory glycinergic neurotransmission onto rat superficial dorsal horn neurons. Nature Neuroscience. 2002;5:34–40.
Akopian A.N., Sivilotti L., Wood J.N. A tetrodotoxin-resistant voltage-gated sodium channel expressed by sensory neurons. Nature. 1996;379:257–262.
Andersson D.A., Gentry C., Alenmyr L., et al. TRPA1 mediates spinal antinociception induced by acetaminophen and the cannabinoid Δ(9)-tetrahydrocannabiorcol. Nature Communications. 2011;2:551.
Ates M., Hamza M., Seidel K., et al. Intrathecally applied flurbiprofen produces an endocannabinoid-dependent antinociception in the rat formalin test. European Journal of Neuroscience. 2003;17:597–604.
Baba H., Kohno T., Moore K.A., et al. Direct activation of rat spinal dorsal horn neurons by prostaglandin E2. Journal of Neuroscience. 2001;21:1750–1756.
Beiche F., Scheuerer S., Brune K., et al. Up-regulation of cyclooxygenase-2 mRNA in the rat spinal cord following peripheral inflammation. FEBS Letters. 1996;390:165–169.
Beltramo M., Stella N., Calignano A., et al. Functional role of high-affinity anandamide transport, as revealed by selective inhibition. Science. 1997;277:1094–1097.
Boelsterli U.A. Diclofenac-induced liver injury: a paradigm of idiosyncratic drug toxicity. Toxicology and Applied Pharmacology. 2003;192:307–322.
Bombardier C., Laine L., Reicin A., et al. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. VIGOR Study Group. New England Journal of Medicine. 2000;343:1520–1528. 1522 p following 1528
Bresalier R.S., Sandler R.S., Quan H., et al. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. New England Journal of Medicine. 2005;352:1092–1102.
Bridger S., Henderson K., Glucksman E., et al. Deaths from low dose paracetamol poisoning. British Medical Journal. 1998;316:1724–1725.
Brune K., Hinz B., Otterness I. Aspirin and acetaminophen: should they be available over the counter? Current Rheumatology Reports. 2009;11:36–40.
Brune K., Renner B., Hinz B. Using pharmacokinetic principles to optimize pain therapy. Nature Reviews. Rheumatology. 2010;6:589–598.
Caterina M.J., Leffler A., Malmberg A.B., et al. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science. 2000;288:306–313.
Caterina M.J., Schumacher M.A., Tominaga M., et al. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389:816–824.
Chan A.T., Manson J.E., Albert C.M., et al. Nonsteroidal antiinflammatory drugs, acetaminophen, and the risk of cardiovascular events. Circulation. 2006;113:1578–1587.
Chandrasekharan N.V., Dai H., Roos K.L., et al. COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: cloning, structure, and expression. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:13926–13931.
Chiang N., Arita M., Serhan C.N. Anti-inflammatory circuitry: lipoxin, aspirin-triggered lipoxins and their receptor ALX. Prostaglandins, Leukotrienes, and Essential Fatty Acids. 2005;73:163–177.
Claveau D., Sirinyan M., Guay J., et al. Microsomal prostaglandin E synthase-1 is a major terminal synthase that is selectively up-regulated during cyclooxygenase-2–dependent prostaglandin E2 production in the rat adjuvant-induced arthritis model. Journal of Immunology. 2003;170:4738–4744.
Davis J.B., Gray J., Gunthorpe M.J., et al. Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature. 2000;405:183–187.
DeWitt D.L., el-Harith E.A., Smith W.L. Molecular cloning of prostaglandin G/H synthase. Advances in Prostaglandin, Thromboxane, and Leukotriene Research. 1989;19:454–457.
Dinchuk J.E., Liu R.Q., Trzaskos J.M. COX-3: in the wrong frame in mind. Immunology Letters. 2003;86:121.
Eisenach J.C., Curry R., Rauck R., et al. Role of spinal cyclooxygenase in human postoperative and chronic pain. Anesthesiology. 2010;112:1225–1233.
Eisenach J.C., Curry R., Tong C., et al. Effects of intrathecal ketorolac on human experimental pain. Anesthesiology. 2010;112:1216–1224.
England S., Bevan S., Docherty R.J. PGE2 modulates the tetrodotoxin-resistant sodium current in neonatal rat dorsal root ganglion neurones via the cyclic AMP–protein kinase A cascade. Journal of Physiology. 1996;495:429–440.
Etminan M., Sadatsafavi M., Jafari S., et al. Acetaminophen use and the risk of asthma in children and adults: a systematic review and metaanalysis. Chest. 2009;136:1316–1323.
FitzGerald G.A., Patrono C. The coxibs, selective inhibitors of cyclooxygenase-2. New England Journal of Medicine. 2001;345:433–442.
Flower R.J. The development of COX2 inhibitors. Nature Reviews. Drug Discovery. 2003;2:179–191.
Forman J.P., Stampfer M.J., Curhan G.C. Non-narcotic analgesic dose and risk of incident hypertension in US women. Hypertension. 2005;46:500–507.
Gavva N.R., Bannon A.W., Hovland D.N., Jr., et al. Repeated administration of vanilloid receptor TRPV1 antagonists attenuates hyperthermia elicited by TRPV1 blockade. Journal of Pharmacology and Experimental Therapeutics. 2007;323:128–137.
Geisslinger G., Dietzel K., Bezler H., et al. Therapeutically relevant differences in the pharmacokinetical and pharmaceutical behavior of ibuprofen lysinate as compared to ibuprofen acid. International Journal of Clinical Pharmacology, Therapy, and Toxicology. 1989;27:324–328.
Glaser S.T., Abumrad N.A., Fatade F., et al. Evidence against the presence of an anandamide transporter. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:4269–4274.
Gold M.S., Reichling D.B., Shuster M.J., et al. Hyperalgesic agents increase a tetrodotoxin-resistant Na+ current in nociceptors. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:1108–1112.
Grosser T., Fries S., FitzGerald G.A. Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities. Journal of Clinical Investigation. 2006;116:4–15.
Guay J., Bateman K., Gordon R., et al. Carrageenan-induced paw edema in rat elicits a predominant prostaglandin E2 (PGE2) response in the central nervous system associated with the induction of microsomal PGE2 synthase-1. Journal of Biological Chemistry. 2004;279:24866–24872.
Guhring H., Hamza M., Sergejeva M., et al. A role for endocannabinoids in indomethacin-induced spinal antinociception. European Journal of Pharmacology. 2002;454:153–163.
Hammad H., Kool M., Soullie T., et al. Activation of the D prostanoid 1 receptor suppresses asthma by modulation of lung dendritic cell function and induction of regulatory T cells. Journal of Experimental Medicine. 2007;204:357–367.
Harvey R.J., Depner U.B., Wassle H., et al. GlyRα3: an essential target for spinal PGE2-mediated inflammatory pain sensitization. Science. 2004;304:884–887.
Harvey V.L., Caley A., Muller U.C., et al. A selective role for α3 subunit glycine receptors in inflammatory pain. Frontiers in Molecular Neuroscience. 2009;2:14.
Henry D., Lim L.L., Garcia Rodriguez L.A., et al. Variability in risk of gastrointestinal complications with individual non-steroidal anti-inflammatory drugs: results of a collaborative meta-analysis. British Medical Journal. 1996;312:1563–1566.
Hinz B., Cheremina O., Brune K. Acetaminophen (paracetamol) is a selective cyclooxygenase-2 inhibitor in man. FASEB Journal. 2008;22:383–390.
Hogestatt E.D., Jonsson B.A., Ermund A., et al. Conversion of acetaminophen to the bioactive N-acylphenolamine AM404 via fatty acid amide hydrolase–dependent arachidonic acid conjugation in the nervous system. Journal of Biological Chemistry. 2005;280:31405–31412.
Hosl K., Reinold H., Harvey R.J., et al. Spinal prostaglandin E receptors of the EP2 subtype and the glycine receptor alpha3 subunit, which mediate central inflammatory hyperalgesia, do not contribute to pain after peripheral nerve injury or formalin injection. Pain. 2006;126:46–53.
Kato S., Aihara E., Yoshii K., et al. Dual action of prostaglandin E2 on gastric acid secretion through different EP-receptor subtypes in the rat. American Journal of Physiology. Gastrointestinal and Liver Physiology. 2005;289:G64–G69.
Kaufman W.D. Risk of anaphylaxis in a hospital population in relation to the use of various drugs: an international study. Pharmacoepidemiology and Drug Safety. 2003;12:195–202.
Kaufman D.W., Kelly J.P., Levy M., et al. The drug etiology of agranulocytosis: an aplastic anemia. New York: Oxford University Press; 1991.
Kaufman D.W., Kelly J.P., Sheehan J.E., et al. Nonsteroidal anti-inflammatory drug use in relation to major upper gastrointestinal bleeding. Clinical Pharmacology and Therapeutics. 1993;53:485–494.
Kozak K.R., Crews B.C., Morrow J.D., et al. Metabolism of the endocannabinoids, 2-arachidonylglycerol and anandamide, into prostaglandin, thromboxane, and prostacyclin glycerol esters and ethanolamides. Journal of Biological Chemistry. 2002;277:44877–44885.
Kress M., Koltzenburg M., Reeh P.W., et al. Responsiveness and functional attributes of electrically localized terminals of cutaneous C-fibers in vivo and in vitro. Journal of Neurophysiology. 1992;68:581–595.
Kristensen D.M., Hass U., Lesné L., et al. Intrauterine exposure to mild analgesics is a risk factor for development of male reproductive disorders in human and rat. Human Reproduction. 2011;26:235–244.
Kujubu D.A., Fletcher B.S., Varnum B.C., et al. TIS10, a phorbol ester tumor promoter–inducible mRNA from Swiss 3T3 cells, encodes a novel prostaglandin synthase/cyclooxygenase homologue. Journal of Biological Chemistry. 1991;266:12866–12872.
Kurumbail R.G., Stevens A.M., Gierse J.K., et al. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature. 1996;384:644–648.
Langenbach R., Morham S.G., Tiano H.F., et al. Prostaglandin synthase 1 gene disruption in mice reduces arachidonic acid–induced inflammation and indomethacin-induced gastric ulceration. Cell. 1995;83:483–492.
Laska E.M., Sunshine A., Marrero I., et al. The correlation between blood levels of ibuprofen and clinical analgesic response. Clinical Pharmacology and Therapeutics. 1986;40:1–7.
Levy M. Risks of agranulocytosis and aplastic anemia. A first report of their relation to drug use with special reference to analgesics. The International Agranulocytosis and Aplastic Anemia Study. JAMA: Journal of the American Medical Association. 1986;256:1749–1757.
Levy M., Zylber-Katz E., Rosenkranz B. Clinical pharmacokinetics of dipyrone and its metabolites. Clinical Pharmacokinetics. 1995;28:216–234.
Loll P.J., Picot D., Garavito R.M. The structural basis of aspirin activity inferred from the crystal structure of inactivated prostaglandin H2 synthase. Nature Structural Biology. 1995;2:637–643.
Lopshire J.C., Nicol G.D. Activation and recovery of the PGE2-mediated sensitization of the capsaicin response in rat sensory neurons. Journal of Neurophysiology. 1997;78:3154–3164.
Mallet C., Barrière D.A., Ermund A., et al. TRPV1 in brain is involved in acetaminophen-induced antinociception. PLoS One. 5(9), 2010. pii: e12748
Mallet C., Daulhac L., Bonnefont J., et al. Endocannabinoid and serotonergic systems are needed for acetaminophen-induced analgesia. Pain. 2008;139:190–200.
Malmberg A.B., Brandon E.P., Idzerda R.L., et al. Diminished inflammation and nociceptive pain with preservation of neuropathic pain in mice with a targeted mutation of the type I regulatory subunit of cAMP-dependent protein kinase. Journal of Neuroscience. 1997;17:7462–7470.
Malmberg A.B., Yaksh T.L. Antinociceptive actions of spinal nonsteroidal anti-inflammatory agents on the formalin test in the rat. Journal of Pharmacology and Experimental Therapeutics. 1992;263:136–146.
Malmberg A.B., Yaksh T.L. Hyperalgesia mediated by spinal glutamate or substance P receptor blocked by spinal cyclooxygenase inhibition. Science. 1992;257:1276–1279.
Malmberg A.B., Yaksh T.L. Cyclooxygenase inhibition and the spinal release of prostaglandin E2 and amino acids evoked by paw formalin injection: a microdialysis study in unanesthetized rats. Journal of Neuroscience. 1995;15:2768–2776.
Masferrer J.L., Seibert K., Zweifel B., et al. Endogenous glucocorticoids regulate an inducible cyclooxygenase enzyme. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:3917–3921.
Masferrer J.L., Zweifel B.S., Manning P.T., et al. Selective inhibition of inducible cyclooxygenase 2 in vivo is antiinflammatory and nonulcerogenic. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:3228–3232.
Mayer J.M., Testa B. Pharmacodynamics, pharmacokinetics and toxicity of ibuprofen enantiomers. Drugs of the Future. 1997;22:1347–1366.
Merlie J.P., Fagan D., Mudd J., et al. Isolation and characterization of the complementary DNA for sheep seminal vesicle prostaglandin endoperoxide synthase (cyclooxygenase). Journal of Biological Chemistry. 1988;263:3550–3553.
Mockenhaupt M., Schlingmann J., Schroeder W., et al. Evaluation of non-steroidal anti-inflammatory drugs (NSAIDs) and muscle relaxants as risk factors for Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN). Pharmacoepidemiology and Drug Safety. 1996;5:116.
Murata T., Ushikubi F., Matsuoka T., et al. Altered pain perception and inflammatory response in mice lacking prostacyclin receptor. Nature. 1997;388:678–682.
Neugebauer V., Weiretter F., Schaible H.G. Involvement of substance P and neurokinin-1 receptors in the hyperexcitability of dorsal horn neurons during development of acute arthritis in rat’s knee joint. Journal of Neurophysiology. 1995;73:1574–1583.
Nirodi C.S., Crews B.C., Kozak K.R., et al. The glyceryl ester of prostaglandin E2 mobilizes calcium and activates signal transduction in RAW264.7 cells. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:1840–1845.
O’Banion M.K., Sadowski H.B., Winn V., et al. A serum- and glucocorticoid-regulated 4-kilobase mRNA encodes a cyclooxygenase-related protein. Journal of Biological Chemistry. 1991;266:23261–23267.
Patrignani P., Panara M.R., Greco A., et al. Biochemical and pharmacological characterization of the cyclooxygenase activity of human blood prostaglandin endoperoxide synthases. Journal of Pharmacology and Experimental Therapeutics. 1994;271:1705–1712.
Patrono C., Baigent C. Low-dose aspirin, coxibs, and other NSAIDs: a clinical mosaic emerges. Molecular Interventions. 2009;9:31–39.
Picot D., Loll P.J., Garavito R.M. The x-ray crystal structure of the membrane protein prostaglandin H2 synthase-1. Nature. 1994;367:243–249.
Rahme E., Barkun A., Nedjar H., et al. Hospitalizations for upper and lower GI events associated with traditional NSAIDs and acetaminophen among the elderly in Quebec, Canada. American Journal of Gastroenterology. 2008;103:872–882.
Rathee P.K., Distler C., Obreja O., et al. PKA/AKAP/VR-1 module: a common link of Gs-mediated signaling to thermal hyperalgesia. Journal of Neuroscience. 2002;22:4740–4745.
Raz A., Wyche A., Fu J., et al. Regulation of prostanoids synthesis in human fibroblasts and human blood monocytes by interleukin-1, endotoxin, and glucocorticoids. Advances in Prostaglandin, Thromboxane, and Leukotriene Research. 1990;20:22–27.
Raz A., Wyche A., Needleman P. Temporal and pharmacological division of fibroblast cyclooxygenase expression into transcriptional and translational phases. Proceedings of the National Academy of Sciences of the United States of America. 1989;86:1657–1661.
Reinold H., Ahmadi S., Depner U.B., et al. Spinal inflammatory hyperalgesia is mediated by prostaglandin E receptors of the EP2 subtype. Journal of Clinical Investigation. 2005;115:673–679.
Riendeau D., Charleson S., Cromlish W., et al. Comparison of the cyclooxygenase-1 inhibitory properties of nonsteroidal anti-inflammatory drugs (NSAIDs) and selective COX-2 inhibitors, using sensitive microsomal and platelet assays. Canadian Journal of Physiology and Pharmacology. 1997;75:1088–1095.
Roth G.J., Stanford N., Majerus P.W. Acetylation of prostaglandin synthase by aspirin. Proceedings of the National Academy of Sciences of the United States of America. 1975;72:3073–3076.
Roujeau J.C., Kelly J.P., Naldi L., et al. Medication use and the risk of Stevens-Johnson syndrome or toxic epidermal necrolysis. New England Journal of Medicine. 1995;333:1600–1607.
Rudy A.C., Knight P.M., Brater D.C., et al. Stereoselective metabolism of ibuprofen in humans: administration of R-, S- and racemic ibuprofen. Journal of Pharmacology and Experimental Therapeutics. 1991;259:1133–1139.
Samad T.A., Moore K.A., Sapirstein A., et al. Interleukin-1beta–mediated induction of COX-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature. 2001;410:471–475.
Sang N., Zhang J., Chen C. COX-2 oxidative metabolite of endocannabinoid 2-AG enhances excitatory glutamatergic synaptic transmission and induces neurotoxicity. Journal of Neurochemistry. 2007;102:1966–1977.
Schaible H.G., Schmidt R.F. Excitation and sensitization of fine articular afferents from cat’s knee joint by prostaglandin E2. Journal of Physiology. 1988;403:91–104.
Schaible H.G., Schmidt R.F. Time course of mechanosensitivity changes in articular afferents during a developing experimental arthritis. Journal of Neurophysiology. 1988;60:2180–2195.
Scialli A.R., Ang R., Breitmeyer J., et al. Childhood asthma and use during pregnancy of acetaminophen. A critical review. Reproductive Toxicology. 2010;30:508–519.
Seeff L.B., Cuccherini B.A., Zimmerman H.J., et al. Acetaminophen hepatotoxicity in alcoholics. A therapeutic misadventure. Annals of Internal Medicine. 1986;104:399–404.
Shaheen S.O., Newson R.B., Smith G.D., et al. Prenatal paracetamol exposure and asthma: further evidence against confounding. International Journal of Epidemiology. 2010;39:790–794.
Smith W.L., Dewitt D.L. Prostaglandin endoperoxide H synthases-1 and -2. Advances in Immunology. 1996;62:167–215.
Solomon S.D., McMurray J.J., Pfeffer M.A., et al. Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. New England Journal of Medicine. 2005;352:1071–1080.
Sudano I., Flammer A.J., Périat D., et al. Acetaminophen increases blood pressure in patients with coronary artery disease. Circulation. 2010;122:1789–1796.
Suzuki K., Araki H., Mizoguchi H., et al. Prostaglandin E inhibits indomethacin-induced gastric lesions through EP-1 receptors. Digestion. 2001;63:92–101.
Svensson C.I., Zattoni M., Serhan C.N. Lipoxins and aspirin-triggered lipoxin inhibit inflammatory pain processing. Journal of Experimental Medicine. 2007;204:245–252.
Takeuchi K., Ukawa H., Kato S., et al. Impaired duodenal bicarbonate secretion and mucosal integrity in mice lacking prostaglandin E–receptor subtype EP(3). Gastroenterology. 1999;117:1128–1135.
Tegeder I., Lotsch J., Krebs S., et al. Comparison of inhibitory effects of meloxicam and diclofenac on human thromboxane biosynthesis after single doses and at steady state. Clinical Pharmacology and Therapeutics. 1999;65:533–544.
Tilley S.L., Hartney J.M., Erikson C.J., et al. Receptors and pathways mediating the effects of prostaglandin E2 on airway tone. American Journal of Physiology. Lung Cellular and Molecular Physiology. 2003;284:L599–606.
Ushikubi F., Segi E., Sugimoto Y., et al. Impaired febrile response in mice lacking the prostaglandin E receptor subtype EP3. Nature. 1998;395:281–284.
Vane J.R. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nature New Biology. 1971;231:232–235.
Vane J.R., Mitchell J.A., Appleton I., et al. Inducible isoforms of cyclooxygenase and nitric-oxide synthase in inflammation. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:2046–2050.
Vardeh D., Wang D., Costigan M., et al. COX2 in CNS neural cells mediates mechanical inflammatory pain hypersensitivity in mice. The Journal of Clinical Investigation. 2009;119:287–294.
Warner T.D., Mitchell J.A. COX-2 selectivity alone does not define the cardiovascular risks associated with non-steroidal anti-inflammatory drugs. Lancet. 2008;371:270–273.
Werner U., Werner D., Rau T., et al. Celecoxib inhibits metabolism of cytochrome P450 2D6 substrate metoprolol in humans. Clinical Pharmacology and Therapeutics. 2003;74:130–137.
White W.B., Campbell P. Blood pressure destabilization on nonsteroidal antiinflammatory agents: acetaminophen exposed? Circulation. 2010;122:1779–1781.
Wolhoff H., Altrogge G., Pola W., et al. [Metamizol—acute overdose with suicidal intent] Deutsche Medizinische Wochenschrift. 1983;108:1761–1764.
Woodward D.F., Liang Y., Krauss A.H. Prostamides (prostaglandin-ethanolamides) and their pharmacology. British Journal of Pharmacology. 2008;153:410–419.
Yamagata K., Andreasson K.I., Kaufmann W.E., et al. Expression of a mitogen-inducible cyclooxygenase in brain neurons: regulation by synaptic activity and glucocorticoids. Neuron. 1993;11:371–386.
Yu M., Ives D., Ramesha C.S. Synthesis of prostaglandin E2 ethanolamide from anandamide by cyclooxygenase-2. Journal of Biological Chemistry. 1997;272:21181–21186.
Yue H.Y., Fujita T., Kawasaki Y., et al. AM404 enhances the spontaneous release of l-glutamate in a manner sensitive to capsazepine in adult rat substantia gelatinosa neurones. Brain Research. 2004;1018:283–287.