Cannabinoids
The Endocannabinoid System
Two cannabinoid receptors (CB1 and CB2) have been identified to date (for review see Howlett et al 2002). Additional receptor subtypes (e.g., the G-protein receptor GPR55) that share little homology with CB1 and CB2 have also been recently postulated to represent novel cannabinoid receptors (Lauckner et al 2008).
In the early days of cannabinoid pharmacology it was hypothesized that lipophilic cannabinoids exerted their effects by perturbing neuronal membranes in a fashion similar to a theory proposed for general anesthetics. However, the demonstration of cannabinoid receptor binding sites (labeled with [3H]CP55,940) in the brain that possessed the characteristics of a G protein–coupled receptor (Devane et al 1988) established the existence of a cannabinoid transmitter system. This discovery was followed by cloning of the CB1 receptor from a rat cDNA library (Matsuda et al 1990). Rat CB1 has, in common with many other G protein–coupled receptors, seven transmembrane-spanning α-helices, a C-terminal that couples to G proteins, an extracellular N-terminal, and three potential glycosylation sites. It is 473 amino acids in length and has a molecular weight of 53 kDa, although variants of 59 and 64 kDa also exist. Cloning of human CB1 (hCB1) (472 amino acids; Gerard et al 1991) and mouse (473 amino acids; Chakrabarti et al 1995) homologues that share close sequence homology (>97%) with rat CB1 followed. Later, an immune cell human cannabinoid receptor (hCB2) was identified, initially in a human promyelocytic leukemia cell line that has 44% sequence homology (68% in the transmembrane regions) with hCB1 (Munro et al 1993). hCB2 is also a G protein–coupled receptor but is shorter than CB1 (360 amino acids, 40 kDa). Subsequently, the murine and rat homologues of CB2 have been cloned and found to have 82% (Shire et al 1995) and 81% (Griffin et al 2000) sequence homology with hCB2, respectively, and 90% homology between them. The presence of CB2 isoforms with different distributions in different tissues and species has also been documented; in the brain, CB2A has been reported to exist at levels of 0.1 or 1% of those expressed in the spleen (Liu et al 2009). Transgenic mice have been created in which the genes encoding CB1 (Ledent et al 1999, Zimmer et al 1999) or CB2 (Buckley et al 2000) have been disrupted.
Circumstantial evidence suggests the existence of further, hitherto uncharacterized, cannabinoid receptors predominantly based on residual pharmacological activity in cannabinoid receptor knockout mice or following the administration of receptor antagonists to naïve rodents (Breivogel et al 2001). An obvious strategy for identifying such receptors is to search databases for structural homology to CB1 or CB2. More recently, GPR55 has been identified as a putative third cannabinoid receptor, although it lacks the functional fingerprint of a cannabinoid receptor. GPR55 is highly sensitive to lysophosphatidylinositol (which does not bind cannabinoid receptors) and only some (e.g., tetrahydrocannabinol [THC], methanandamide, JWH015) cannabinoid and endocannabinoid ligands. Activation of GPR55 increases calcium and M currents, which engages signaling mechanisms distinct from CB1 and CB2 (Lauckner et al 2008). Intriguingly, this receptor may play a pro-nociceptive role in the nervous system inasmuch as mechanical hyperalgesia fails to develop in GPR55−/− mice following treatment with an inflammatory agent, complete Freund’s adjuvant (CFA), or traumatic nerve injury produced by partial sciatic nerve ligation (Staton et al 2008).
Molecular and Cellular Consequences of Cannabinoid Receptor Activation
CB1 receptors, via Gi/o proteins, influence several signal transduction pathways, including negative coupling to adenylate cyclase (AC) and activation of mitogen-activated protein kinase (MAPK) and immediate–early gene signaling pathways, including Krox-24 actions (see Howlett et al 2002). Additionally, CB1 receptors modulate the activity of a number of ion channels via a Gi/o protein also, including negative coupling to presynaptic voltage-dependent Ca2+ channels (particularly L, N, and P/Q types) (Mackie et al 1993) and positive coupling to inwardly rectifying K+ channels (Mackie et al 1995). Thus, the net consequence of the CB1 receptor is to augment membrane hyperpolarization and inhibit release of neurotransmitters. The conformational state of CB1 may determine which transduction mechanism will be operative (Howlett 1998). On binding to a ligand, the CB1 receptor–ligand complex rapidly internalizes, a process that may underlie the observed development of tolerance to cannabinoids (Coutts et al 2001, Kouznetsova et al 2002).
Activation of CB2 receptors engages multiple signaling mechanisms, including negative coupling to AC via Gi/o and activation of extracellular signal–regulated kinase (ERK) and voltage-dependent calcium channels (for review see Atwood et al 2012). Understanding these signaling mechanisms is further complicated by the fact that different agonists can traffic through distinct signaling mechanisms (Schatz et al 1997) and differ in their capacity to produce receptor internalization (Atwood et al 2012).
Tissue Expression of Cannabinoid Receptors
Expression of the CB1 receptor is largely restricted to neurons, and expression is high, similar to opioid receptors, at several loci important for nociceptive processing in the brain, spinal cord, and peripheral nervous system (Herkenham et al 1991, Tsou et al 1998, Egertova and Elphick 2000). Expression of CB2 is predominantly restricted to cells of the immune system, including glia, and is highly induced by insult or injury in the central nervous system (CNS) (for review see Guindon and Hohmann 2008, Atwood and Mackie 2010).
CB1 and the Brain
In the brain the existence of CB1 was first suggested by the results of in vitro ligand-binding studies (Devane et al 1992). The distribution of CB1 was subsequently mapped via both autoradiography (Herkenham et al 1991) and in situ hybridization (Mailleux et al 1992, Matsuda et al 1993). Expression of CB1 protein has been mapped comprehensively in the rat brain by immunohistochemistry (Tsou et al 1998, Egertová et al 2003). These studies have revealed a widespread distribution, with the cerebral cortex, hippocampus, olfactory bulb, basal ganglia, and cerebellum being particularly rich in CB1, a fact that is broadly in agreement with several of the known pharmacological effects of cannabinoids. Expression in the periaqueductal gray (PAG), rostral ventromedial medulla (RVM), and thalamus is particularly notable in the context of analgesia.
At a neuronal level, CB1 is expressed on axonal fibers, especially at their terminals. CB1 is expressed on presynaptic terminals of GABAergic (Katona et al 2000) and glutamatergic (Katona et al 2006) neurons. This presynaptic distribution, together with the presence of synthetic machinery residing post-synaptically, is consistent with the hypothesis that endocannabinoids act as short-range retrograde modulators of neurotransmitter release (Kreitzer and Regehr 2001, Ohno-Shosaku et al 2001, Wilson and Nicoll 2001, Katona and Freund 2008).
CB1 in the Spinal Cord
Following the initial reports of localization of CB1 in the spinal cord (Hohmann and Herkenham 1998, Hohmann et al 1999a, Sanudo-Pena et al 1999), detailed immunohistochemical mapping of CB1 in the rat spinal cord was undertaken (Farquhar-Smith et al 2000). Such mapping demonstrated expression of CB1 in areas important for nociceptive processing, particularly the dorsolateral funiculus, the superficial dorsal horn, and lamina X. In the dorsal horn, CB1 expression was evident as a bilayer in lamina I and III overlapping with the central terminals of the nerve growth factor (NGF)-dependent peptidergic class of primary afferent nociceptors. Evidence from both lesioning and electrophysiological studies suggests the existence of populations of CB1 in the spinal and medullary dorsal horn on both the central terminals of primary afferent neurons and intrinsic CNS neurons (Farquhar-Smith et al 2000), including those expressing protein kinase Cγ, γ-aminobutyric acid (GABA), and nitric oxide synthase (NOS). Recent electron microscopy studies have nonetheless detected CB1 receptors on Aδ- and C-fiber terminals (Nyilas et al 2009) via a sensitive CB1 antibody; this antibody was able to detect CB1 receptors on hippocampal glutamatergic axons (Katona et al 2006) in which CB1 protein levels were below the detection threshold for antibodies previously used in spinal cord studies (Farquhar-Smith et al 2000). Consistent with this interpretation, radioligand binding following extensive rhizotomy suggests that about 50% of spinal CB1 expression is on primary afferent neurons, although post-synaptic changes in response to such an extensive rhizotomy may contribute to the pattern of expression obtained (Hohmann et al 1999a). The precise nature of CB1-expressing neurons in the dorsolateral funiculus requires further investigation, but it is clear that this expression is on rostrocaudally coursing fibers, possibly concerned with descending control systems, or on local inhibitory networks (Farquhar-Smith et al 2000).
CB1 in the Peripheral Nervous System
CB1 is expressed by primary afferent neurons. Cannabinoid binding sites undergo axonal transport in peripheral nerves (Hohmann and Herkenham 1999b), and immunohistochemistry and flow cytometry have revealed the presence of CB1 in cultured neonatal dorsal root ganglion cells (Ross et al 2001). In situ hybridization (Hohmann and Herkenham 1999a, Bridges et al 2003) and immunohistochemistry (Bridges et al 2003) of lumbar dorsal root ganglia suggest that about 25% of dorsal root ganglion cells express CB1 and that most of these cells have a medium to large somal area. Approximately 75% of CB1-expressing dorsal root ganglia cells also display immunoreactivity for neurofilament 200 (a marker of myelinated neurons) and have a medium to large cell body area. This suggests that the majority of high–CB1-expressing dorsal root ganglion cells are not unmyelinated nociceptors, although some CB1-expressing cells in the NF200–co-expressing category could be Aδ or Aβ nociceptors. A smaller population of small-diameter (<30%) CB1-expressing dorsal root ganglion cells co-stain for markers of nociceptive neurons, such as calcitonin gene–related peptide (CGRP), isolectin B4 (IB4), tyrosine kinase receptor A (TrkA), and transient receptor potential vanilloid 1 (TRPV1). A very similar pattern of CB1 distribution has been observed with in situ hybridization in the trigeminal ganglion (Price et al 2003). A broadly similar pattern of CB1 expression has also been reported in dissociated rat dorsal root ganglion cells within a wide range of cell somal areas, with co-localization studies revealing that 48% of CB1-positive cells co-stain for RT-97 (labeling neurofilament 200) and 45% for CGRP (a marker of the peptidergic NGF-dependent subgroup of nociceptors). Functional effects of the synthetic cannabinoid CP55,940 (as measured by the depolarization-induced increase in intracellular [Ca2+]) occur primarily in large dorsal root ganglion cells (Khasabova et al 2002, 2004). Site-specific increases in both cannabinoid receptors and endocannabinoids (anandamide and 2-arachidonoylglycerol [2-AG]) have also been reported in dorsal root ganglia following nerve injury (Mitrirattanakul et al 2006), where co-localization with IB4, TRPV1, CGRP, and the NR2C/2D subunits of N-methyl-D-aspartate (NMDA) receptors are notably reported.
CB2 in Non-neuronal and Neuronal Cells
CB2 was originally identified in splenic macrophages (Munro et al 1993). Of the circulating human immune cells, B lymphocytes and natural killer cells are particularly rich in CB2 mRNA, with moderate expression in monocytes and minimal expression in polymorphonuclear leukocytes (Galiegue et al 1995). There is also evidence of CB2 expression on T-cell lines (Schatz et al 1997). Expression of CB2 by immune cells is dependent on their state of activation (Lee et al 2001). Functional CB2 (and CB1) has been identified in a mast cell line (Samson et al 2003). Finally, microglia express functional CB2 receptors (Walter et al 2001). With respect to nociceptive signaling, CB2 is induced in the spinal cord by injury in models of neuropathic pain, whereas levels of expression were below the detection threshold in naïve animals (Zhang et al 2003). CB2 is highly inducible but has also been reported in neurons (Van Sickle et al 2005) by immunohistochemistry, albeit at low levels relative to CB1. Primary afferent CB2 expression has been demonstrated via immunohistochemistry in TRPV1 (and CB1)-positive fibers (Anand et al 2008). Strikingly, CB2 expression is reportedly up-regulated in human neuromas and peripheral nerves following nerve injury (Anand et al 2008). Interestingly, a striking contralateral mirror-image pain that is interferon-γ dependent develops in CB2−/− mice following a neuropathic insult (Racz et al 2008a, 2008b); this phenotype is marked by bilateral hyperalgesia, allodynia, and microglial and astrocytic activation following a unilateral sciatic nerve injury, but similar levels of hypersensitivity ipsilateral to the injury. This latter effect is replicated in irradiated wild-type mice reconstituted with bone marrow cells from CB2−/− mice, thus suggesting that the CB2 expressed in hematopoietic cells is implicated in the development of neuropathic pain at the spinal cord.
Endocannabinoids
A number of endogenous ligands at cannabinoid receptors (“endocannabinoids”) have been identified, including anandamide (Devane et al 1992), 2-AG (Mechoulam et al 1995, Sugiura et al 1995), noladin ether (Hanus et al 2001), virodhamine (Porter et al 2002), and N-arachidonoyldopamine (Huang et al 2002). Anandamide and 2-AG remain the best studied of the endocannabinoids isolated to date. Endocannabinoid synthesis is not restricted to neurons; synthesis is also reported in immune cells, especially basophils, microglia, and macrophages (Bisogno et al 1997, De Petrocellis et al 2000, Walter et al 2003).
Anandamide
The fatty acid anandamide was first isolated from porcine brain (Devane et al 1992). Anandamide binds to both cannabinoid receptors; however, it has higher, but still weak affinity for CB1 (Ki ≈ 89nM) relative to CB2 (≈371 nM) but does not evoke CB2-mediated effects to a biologically significant degree. Anandamide gives rise to the classic “tetrad” (antinociception, catalepsy, hypothermia, and hypolocomotion) and evokes the cellular and molecular consequences of cannabinoid receptor activation, including inhibition of AC (Felder et al 1993, Bayewitch et al 1995); inhibition of N-, P/Q-, and L-type voltage-gated Ca2+ channels (Felder et al 1993; Mackie et al 1993, 1995); activation of inwardly rectifying K+ channels (Mackie et al 1995); and inhibition of neurotransmitter release (Shen et al 1996, Vaughan et al 2000). However, anandamide can produce effects in the opposing direction of the classic cannabinoids and antagonize the actions of Δ9-THC, perhaps by acting as a partial agonist or via its effects at other receptors.
Anandamide is unusual among endocannabinoids in that it also activates members of the TRP ion channel family, which includes thermal-sensing ion channels. Anandamide has weak affinity (Ki ≈ 10 μM) for and displays agonist activity at the TRPV1 noxious heat–gated ion channel both in vitro and in vivo (Smart et al 2000, Gauldie et al 2001). It has thus been hypothesized that CB1 represents the metabotropic receptor for anandamide whereas TRPV1 represents the ionotropic anandamide receptor. Consistent with this hypothesis, anandamide has been found to have a dual effect on cultured dorsal root ganglion cells and in vivo. There is also evidence indicating that anandamide, albeit indirectly via the formation of epoxyeicosatrienoic acids, activates another member of the TRP family, TRPV4, which senses innocuous heat (Watanabe et al 2003).
Anandamide Synthesis
It was initially thought that the major biosynthetic pathway for anandamide was the condensation of arachidonic acid and ethanolamine (Deutsch and Chin 1993). In vitro evidence has favored the model that membrane depolarization and consequent Ca2+ influx activate the intracellular cleavage of N-arachidonylphosphatidylethanolamine (NAPE) by phospholipase D (PLD) (Piomelli 2003), with anandamide being rapidly synthesized “on demand” rather than being stored in synaptic vesicles. However, NAPE-PLD−/− mice show no deficits in basal anandamide levels (Leung et al 2006), thus suggesting that this mechanism is not a major pathway for anandamide formation. This pathway may nonetheless contribute to anandamide formation under stimulated conditions or in the periphery.
Anandamide Degradation
The major degradation pathway for anandamide involves uptake into cells followed by hydrolysis catalyzed by the intracellular enzyme fatty acid amide hydrolase (FAAH) to arachidonic acid and ethanolamine. Uptake of anandamide by neurons is rapid (t1/2 = 2.5 minutes), temperature dependent, saturable, and selective, which suggests the presence of a specific transport process (Di Marzo et al 1994, Beltramo et al 1997). The existence of such a transporter has been controversial (Glaser et al 2003). Intracellular fatty-acid binding proteins that transport anandamide from the plasma membrane to sites of catabolism have recently been described (Kaczocha et al 2009).
FAAH was first identified as having the capability of hydrolyzing anandamide by Deutsch and Chin (1993) and was characterized and cloned by Cravatt and colleagues (1996). FAAH expression has best been characterized in neurons and glia. Expression of both FAAH mRNA and protein is widespread in the brain, particularly in the cerebellum, choroid plexus, hippocampus, and globus pallidus. In general, its pattern of expression is complementary to the CB1 receptor, but not exclusively so (Egertová et al 1998, 2003). In the spinal cord and periphery, FAAH expression has been identified and characterized in the cell bodies of primary afferent nociceptors in rat dorsal root ganglia and the superficial dorsal horn of the spinal cord, thus suggesting a potential analgesic drug target outside the CNS (Lever et al 2009). This study also demonstrated the phenotype of dorsal root ganglion neurons expressing changes in FAAH following peripheral nerve injury to also include cells that are not normally associated with nociceptive functions (Lever et al 2009).
FAAH “knockout” mice have been generated that have almost negligible levels of anandamide hydrolytic activity and 15-fold higher brain concentrations of anandamide than wild types do (Cravatt et al 2001). These mice also display evidence of cannabinoid receptor–dependent tonic analgesia to acute nociceptive and inflammatory stimuli, but not in a neuropathic pain model (presumably a phenomenon related to the increased concentration of anandamide), and significant augmentation of the CB1-mediated antinociceptive response to exogenous anandamide (Cravatt et al 2001, Lichtman et al 2004). The aberrant reduced pain sensitivity typical of FAAH−/− mice is not observed in mice lacking FAAH exclusively in the CNS, although these mice retained a cannabinoid receptor–independent anti-inflammatory phenotype (Cravatt et al 2004). Thus, both central and peripheral FAAH remains an analgesic target involving cannabinoid receptor–dependent and –independent mechanisms.
Therapeutic Potential of Inhibitors of Endocannabinoid Degradation
Inhibitors of endocannabinoid degradation are an alternative approach to receptor agonists in the drive to develop therapeutically useful cannabinoid-based drugs. Various inhibitors of FAAH have been described (Box 38-1) and include well-characterized selective ligands such as URB597 and OL-135. More recently, a peripherally restricted inhibitor of FAAH (e.g., URB937) has been characterized that elevates anandamide and other fatty acid amides exclusively outside the CNS (Clapper et al 2010). URB937 suppresses inflammatory and neuropathic pain and inflammation-evoked Fos protein expression in the spinal cord, consistent with suppression of nociceptive processing.
Intriguingly, certain non-steroidal analgesics, such as ibuprofen and ketorolac, in addition to their cyclooxygenase-inhibiting properties, inhibit FAAH (for review see Fowler et al 1997, Anand et al 2009). Furthermore, the antinociceptive actions of spinally administered indomethacin and flurbiprofen are antagonized by a CB1 receptor antagonist, and flurbiprofen inhibits the release of CGRP in the spinal cord via a CB1-mediated mechanism (Seidel et al 2003). Indomethacin is ineffective as an analgesic when administered intrathecally to CB1−/− mice (Guhring et al 2002, Ates et al 2003). These data could explain the non–cyclooxygenase-associated component of CNS-mediated analgesia with non-steroidal anti-inflammatory drugs.
Another approach is to explore inhibitors of endocannabinoid uptake, which may (or may not) be selective for anandamide. The mechanism of endocannabinoid transport remains incompletely understood. Drugs that engage in directed multi-targeting (e.g., inhibition of FAAH, MGL, cyclo-oxygenase, TRPV1) represent another potential approach to exploit the therapeutic properties of the endocannabinoid signaling system. Mechanisms for anandamide deactivation are summarized in Figure 38-1.
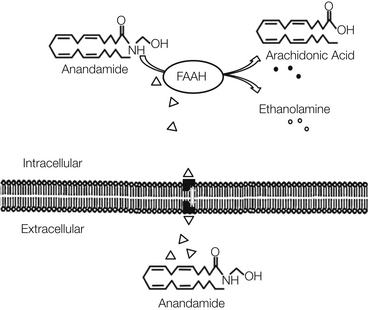
Figure 38-1 Mechanism of uptake and degradation of anandamide.
Anandamide is removed from the extracellular space by uptake into cells followed by metabolism. Intracellular fatty-acid binding proteins shuttle anandamide to FAAH, an intracellular enzyme localized to the endoplasmic reticulum. FAAH then catalyzes hydrolysis of anandamide into breakdown products arachidonic acid and glycerol.
2-Arachidonylglycerol
A second endocannabinoid, the monoacylglycerol 2-AG, was first isolated from canine gut and later from brain, where it is found in concentrations 170-fold greater than anandamide. This endocannabinoid is believed to be synthesized and degraded by an enzymatic reaction catalyzed by diacylglycerol lipase (DAGL or DGL) and monoacylglycerol lipase (MGL), respectively (for review see Guindon and Hohmann 2009). 2-AG binds to cannabinoid receptors and exhibits cannabimimetic effects (Mechoulam et al 1995, Sugiura et al 1995, Stella et al 1997). Appreciable concentrations of 2-AG have been measured in dorsal root ganglia and the spinal cord (Huang et al 1999), and neuropathic insults increase both 2-AG and anandamide in L5 dorsal root ganglion cells (Mitrirattanakul et al 2006). 2-AG is a full agonist at the CB1 receptor (Ki = 14 nM) and has been postulated to serve as the natural ligand at the CB2 receptor (Ki = 58 nM) (Sugiura et al 2000).
2-AG is the primary candidate for the role of retrograde endocannabinoid messenger (Gregg et al 2012). In vitro studies and studies in cell culture suggest that 2-AG is formed by a combined action of DAGL and phospholipase C (Jung et al 2005, 2007). Indeed, mice lacking the DAGL-α isoform show profound deficits in retrograde signaling, as well as in 2-AG accumulation, but also substantial changes in anandamide and other signaling lipids such as arachidonic acid (Gao et al 2010, Tanimura et al 2010). Both DAGL-α and DAGL-β isoforms exist, although deficits in retrograde signaling were associated predominantly with the DAGL-α isoform (Gao et al 2010). DAGL-α−/− mice exhibit 80% reductions in 2-AG and parallel reductions in arachidonic acid levels in the brain and spinal cord and deficits in neurogenesis. By contrast, DAGL-β−/− mice show 50% reductions in 2-AG in the brain and 90% reductions in the liver (Gao et al 2010). Alternative synthetic mechanisms are postulated to exist, such as hydrolysis of phosphatidylinositol by phospholipase A and subsequent hydrolysis by phospholipase C, but their roles have not been demonstrated in vivo.
Degradation of 2-AG is performed by a presynaptically localized enzyme, MGL (Dinh et al 2002, 2004; Hohmann et al 2005), distinct from FAAH. Approximately 85% of brain 2-AG hydrolase activity may be attributed to MGL, whereas the remaining 15% is mostly catalyzed by two less characterized enzymes, ABHD6 and ABHD12 (Blankman et al 2007). More work is necessary to determine whether ABHD6 or ABHD12 represent analgesic targets. Putative mechanisms for 2-AG synthesis and degradation are summarized in Figure 38-2.
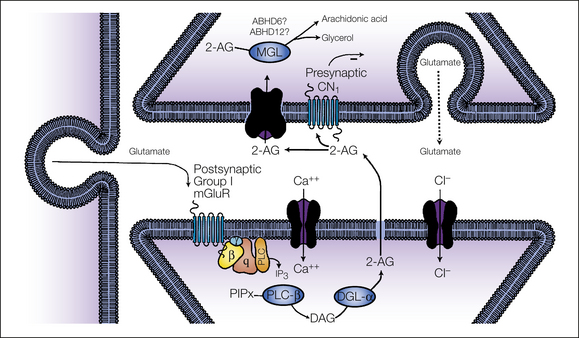
Figure 38-2 Putative mechanism for 2-arachidonoylglycerol (2-AG) synthesis and deactivation.
2-AG is formed by the consecutive activation of two enzymes: phospholipase C and diacylglycerol lipase. The 2-AG precursor diacylglycerol (DAG) is formed by phospholipase C–mediated hydrolysis of membrane phospholipid precursors. DAG is then hydrolyzed by DAG lipase (DGL-α) to generate 2-AG. 2-AG formed in the post-synaptic cell is then released and activates presynaptic CB1 receptors before deactivation by monoacylglycerol lipase (MGL), the presynaptic enzyme responsible for 2-AG deactivation. Minor 2-AG hydrolytic enzymes (ABHD6, ABHD12) have also been identified. (Adapted from Hohmann AG, Suplita RL 2006 Endocannabinoid mechanisms of pain modulation. AAPS Journal 8:E693–E708.)
Knockout mice lacking MGL exhibit reduced responses to the antinociceptive effects of cannabinoid agonists and impaired CB1-dependent forms of synaptic plasticity (Schlosburg et al 2010). Moreover, chronic and nearly complete blockade of MGL using the irreversible MGL inhibitor JZL184 is associated with profound desensitization and down-regulation of CB1 receptors (Schlosburg et al 2010). However, these observations do not necessarily preclude the use of MGL inhibitors as analgesics because antinociceptive tolerance to repeated administration of MGL inhibitors does not occur in conditions in which less complete suppression of MGL activity is observed (Sciolino et al 2011). Dual inhibition of MGL and FAAH has also received attention as a potential therapeutic strategy (Long et al 2009b, Cisneros et al 2012).
Physiological Function of Endocannabinoids
Global CB1 knockout mice (Ledent et al 1999, Zimmer et al 1999) display fairly subtle physiological deviations from the norm but are highly susceptible to seizures (Monory et al 2006). These observations suggest that endocannabinoids play an important, albeit modulatory, role in controlling neuronal excitability (Marsicano et al 2003). Cognitive effects (e.g., psychoactivity, memory impairment) remain the major impediment to the development of therapeutically useful cannabinoid-based analgesics.
At a cellular level, a major step forward in understanding the physiological role that endocannabinoids play in the brain was the revelation that endocannabinoids are synthesized “on demand” by post-synaptic neurons in response to membrane depolarization or receptor-stimulated cleavage of phospholipid precursors and are subsequently released into the synaptic cleft to activate presynaptic CB1 receptors. In addition to depolarization, activation of post-synaptic group I metabotropic glutamate type 5 (mGlu5) receptors can also trigger the rapid synthesis of endocannabinoids, probably 2-AG, in multiple brain regions (for review see Katona and Freund 2008). Activation of this mGlu5-DGL-α pathway can trigger CB1-dependent antinociceptive mechanisms and mobilize 2-AG in the PAG (Gregg et al 2012) and spinal cord (Nyilas et al 2009), a mechanism supported by results of pharmacological, electron microscopic, RNA silencing, and lipidomic analyses (Gregg et al 2012). Once released into the extracellular domain, endocannabinoids act as short-range (≈20 μm) retrograde modulators of neurotransmitter release, via inhibition of Ca2+ channels mediated by presynaptic CB1 receptors, before being removed by MGL. Anandamide, by contrast, is degraded by uptake into post-synaptic neurons—and perhaps glia—and subsequent hydrolysis by FAAH. The fact that endocannabinoids regulate activity in both excitatory and inhibitory neurons might explain why apparently contradictory observations have been made following the administration of exogenous cannabinoids to whole-animal models. Endocannabinoids are also molecular mediators of depolarization-induced suppression of inhibition (Ohno-Shosaku et al 2001, Wilson and Nicoll 2001, Yoshida et al 2002) and a similar process at excitatory synapses (depolarization-induced suppression of excitation) (Kreitzer and Regehr 2001; for review see Diana and Marty 2004, Katona and Freund 2008). Furthermore, endocannabinoids facilitate long-term potentiation (Carlson et al 2002) and long-term depression in various brain regions such as the striatum (Gerdeman et al 2002). Endocannabinoids suppress activity in systems that are fundamentally linked to the suppression of nociceptive activity in the brain stem, namely, by suppressing GABAergic activity in the RVM (Vaughan et al 1999) and both GABAergic (Vaughan et al 2000) and glutaminergic (Drew et al 2009) transmission in the PAG. Nonetheless, endocannabinoids at the spinal level have also paradoxically been shown to contribute to C-fiber–induced heterosynaptic pain sensitization through CB1 (Pernia-Andrade et al 2009). Inhibitors of 2-AG deactivation, by producing a major arachidonate precursor pool, can also generate prostaglandins that promote neuroinflammation through cannabinoid receptor–independent mechanisms (Nomura et al 2011), thus suggesting that the highly interactive nature of lipid signaling pathways may contribute to the observed phenotype.
Mobilization of endocannabinoids under physiological conditions is implicated in both stress-induced and fear-conditioned analgesia. Stress-induced analgesia is blocked by CB1 antagonists, attenuated in rats rendered tolerant of cannabinoids, enhanced by inhibitors of FAAH and MGL, and associated with mobilization of 2-AG and, subsequently, anandamide (Hohmann et al 2005). The time course of 2-AG mobilization in the PAG correlates well with stress antinociception, although endocannabinoid mobilization in other sites probably plays a role (RVM, spinal cord) (Hohmann et al 2005, Suplita et al 2005, Connell et al 2006, Hohmann and Suplita 2006, Nyilas et al 2009). Inhibitors of DAGL suppress stress antinociception following local administration under conditions in which 2-AG is selectively mobilized (Nyilas et al 2009; Gregg et al 2012). Fear-conditioned analgesia has recently been shown to involve endocannabinoid mobilization in the dorsolateral PAG (Olango et al 2012), thus suggesting considerable overlap with neural substrates subserving stress-induced analgesia (Hohmann et al 2005) and a primary importance of supraspinal sites of endocannabinoid analgesic action.
Plant-Derived and Synthetic Cannabinoids
Ligands used to probe the functions of the endocannabinoid signaling system are summarized in Box 38-1. This box includes agents showing efficacy in preclinical models of pathological pain (with an emphasis on neuropathic pain) and agents used clinically or currently under evaluation as analgesics.
Plant-Derived Cannabinoids
Herbal cannabis contains a complex and variable mixture of cannabinoids, with Δ9- and Δ8-THC, cannabidiol, and cannabinol being the major pharmacologically active molecules (Gaoni and Mechoulam 1964). The high lipophilicity of plant-derived cannabinoids is associated with limited gastrointestinal absorption and bioavailability (Gaoni and Mechoulam 1964), which represents a barrier to drug development. Inhalation provides the most predictable and titratable route of administration. Oral and rectal absorption of Δ9-THC and other plant-derived cannabinoids tends to be unpredictable and of limited bioavailability, but the rectal route does appear to have some advantage over the oral route, at least for the Δ9-THC prodrug THC hemisuccinate (Walker et al 1999b). An oromucosal spray containing Δ9-THC and cannabidiol (Sativex) has been shown clinically to have efficacy in relieving the pain caused by multiple sclerosis (for review see Rahn and Hohmann 2009). The lipid solubility of these compounds means that they are rapidly sequestered in fatty tissue, and the elimination half-life is considerable, from days to weeks. Δ9-THC is a substrate for the P450 mixed-function oxidases and is biotransformed in the liver; metabolites are excreted in feces and urine as glucuronide conjugates.
Synthetic Cannabinoids
Recent advances in medicinal chemistry have yielded a number of synthetic cannabinoids with different chemical backgrounds that are broadly classified as follows:
Classical Cannabinoids
These compounds are structurally related to plant-derived cannabinoids and are based on the tricyclic dibenzopyran ring, for example, HU210 (synthetic analogue of Δ8-THC) and HU243. Some classic cannabinoids have been described that have some selectivity for CB2 over CB1, such as L759,633; L759,656; and 1-deoxy-HU210 (JWH051) (Ross et al 1999, Huffman 2000).
Non-classical Cannabinoids
This term refers to bi- or tricyclic Δ9-THC analogues that lack a pyran ring, for example, CP55,940 and levonantradol. Radiolabeled CP55,940 has been widely used in radioligand-binding studies (e.g., Herkenham et al 1991, Hohmann et al 1999a), whereas levonantradol was administered to humans in early analgesic clinical trials but was associated with unacceptable adverse effects (Jain et al 1981). HU308 is a CB2 receptor agonist devoid of CB1-mediated effects that has analgesic properties (Hanus et al 1999).
Aminoalkylindoles
The best known example in this class is WIN55,212-2, which has affinity for both CB1 and CB2 receptors, with modest selectivity for CB2, and cannabinomimetic properties (Barth 1998). A number of CB2-selective ligands have been reported following investigation of the structure of WIN55,212-2, including indomethacin morpholinylamide; AM1241; JWH015; L768,242; and L759,787 (Gallant et al 1996).
Endocannabinoid Analogues
A number of endocannabinoid analogues with improved potency and metabolic stability, for example, methanandamide, have been synthesized (Abadji et al 1994). Arachidonyl-2-chloroethylamide (ACEA) and arachidonylcyclopropylamide (ACPA) and are CB1-selective analogs of anandamide (Hillard et al 1999). Arvanil is an anandamide analogue that behaves as a “hybrid” ligand at both CB1 and TRPV1, inhibits the anandamide transporter (Melck et al 1999), and has antinociceptive and antispastic properties in vivo (Brooks et al 2002).
Cannabinoid Receptor Antagonists (Inverse Agonists)
Sanofi developed the first widely available competitive antagonists at cannabinoid receptors. SR141716A (Rinaldi-Carmona et al 1994) and SR144528 (Rinaldi-Carmona et al 1998) are diarylpyrazoles that are competitive CB1 and CB2 receptor antagonists, respectively. These compounds behave as inverse agonists in that they can evoke signal transduction in the absence of agonists and are thus potentially capable of exerting biological effects by suppressing constitutive activity in cannabinoid receptors (Landsman et al 1997, Bouaboula et al 1999). SR141716A was used in phase I clinical trials that were subsequently discontinued because of concern about increases in suicidality in the subpopulation chronically taking the drug. Other CB1 receptor antagonists have also been reported (e.g., AM251 and AM281, which are analogues of SR141716A and LY320135, respectively, and act at CB1, and AM630, which acts at CB2; see Howlett et al 2002). Recently, AM6545, a peripherally restricted CB1 neutral antagonist, has been developed that improves cardiometabolic risk in rodent models of obesity (Tam et al 2010).
Mechanisms of Analgesia
In a striking parallel with opioids, a framework for cannabinoid-induced analgesia is recognized that has components at brain, spinal cord, and peripheral sites, and these involve both CB1 and CB2 receptors. These sites of action are not independent of each other, and there is a considerable degree of interaction. Both endocannabinoid (Guindon and Hohmann 2009) and cannabinoid CB2 mechanisms (Guindon and Hohmann 2008) of pain suppression have been reviewed recently.
Brain
The CB1 receptor is widely distributed in the brain (Herkenham et al 1991). Direct evidence implicates a role for the PAG, RVM, amygdala, thalamus, and noradrenergic A5 nucleus in CB1-mediated antinociception.
Early studies addressing how the brain mediates the antinociceptive actions of cannabinoids showed that intracerebroventricular administration of synthetic cannabinoids results in increased tail flick latency (Martin et al 1993). Microinjection experiments identified cannabinoid receptors in the PAG and dorsal raphe as the principal periventricular structures involved in cannabinoid-mediated antinociception (Martin et al 1995, Lichtman et al 1996). The nucleus reticularis gigantocellularis pars alpha of the RVM is also associated with CB1 receptor–dependent antinociception in the formalin test (Monhemius et al 2001). Microdialysis experiments subsequently revealed that anandamide concentrations are elevated in the PAG following local electrical stimulation or subcutaneous formalin injection (Walker et al 1999a). This elevation is observed in concert with signs of CB1 receptor–dependent antinociception. In vitro electrophysiological studies have revealed CB1-dependent modulation of excitatory post-synaptic currents in the PAG (Vaughan et al 2000). This modulation is effected by a decrease in the probability of release of neurotransmitters from the presynaptic terminal rather than by a direct effect on the post-synaptic membrane, as is the case for opioids. Glutamate spillover also modulates synaptic transmission at the level of the PAG through an endocannabinoid mechanism (Drew et al 2008). Here, orexin also produces antinociception through endocannabinoid-dependent mechanisms likely to involve 2-AG (Ho et al 2011). Microinjection of HU210 into the PAG also suppresses formalin-evoked Fos protein expression, consistent with suppression of nociceptive processing (Finn et al 2003).
A similar CB1-mediated effect has been shown in the RVM, where cannabinoids induce presynaptic inhibition of GABAergic neurotransmission (Vaughan et al 1999). Pharmacological inactivation of RVM neurons by microinjection of the GABAA agonist muscimol also eliminated cannabinoid antinociception (Meng et al 1998), an effect achieved by modulation of both “off” and “on” cell activity of RVM neurons. Consistent with the descending pain modulatory effects of cannabinoids, spinal transection reduces both the antinociceptive (Lichtman and Martin 1991b) and electrophysiological (on spinal nociceptive neuron) effects of cannabinoids (Hohmann et al 1999b).
Cannabinoid modulation of noxious stimulus–evoked activity in neurons of the ventroposterolateral nucleus of the thalamus correlates with the antinociceptive but not with the motor effects of cannabinoids (Martin et al 1996). Microinjection and lesion studies have also implicated roles for regions rostral to the brain stem (i.e., the amygdala, lateral posterior and submedial regions of the thalamus, superior colliculus, and noradrenergic A5 region) in cannabinoid antinociception (Martin et al 1999a). For example, bilateral lesions of the amygdala in primates reduce both opioid and cannabinoid antinociception (Manning et al 2001). Similarly, pharmacological (muscimol) lesions of the central nucleus of the amygdala reduce the ability of systemic WIN55,212-2 to produce antinociception (tail flick, formalin tests) and suppress the accompanying spinal Fos protein (Manning et al 2003). Endocannabinoid analgesic mechanisms were discussed previously.
Spinal Cord
Electrophysiological studies indicate that spinal CB1 receptors modulate the passage of nociceptive traffic. Walker’s group first showed that noxious thermal- and mechanical-evoked activity in spinal wide–dynamic range neurons is attenuated by systemically and intrathecally administered WIN55,212-2 (Hohmann 1995, 1998, 1999b), and systemic administration of WIN55,212-2 reduces the augmented activity (“wind-up”) in dorsal horn neurons evoked by a sustained noxious input (Strangman and Walker 1999). Chapman’s group also showed that cannabinoids, as well as endocannabinoid modulators, suppress evoked responses in spinal nociceptive neurons in rodent models of inflammation and neuropathic pain (for review see Walker and Hohmann 2005).
Evidence of spinal mechanisms of cannabinoid antinociception was first provided by intrathecal administration studies. Biodisposition studies also revealed that intrathecally administered cannabinoids tend to remain at the site of action and are not rapidly redistributed to the brain (Lichtman and Martin 1991b). Furthermore, intrathecal administration of the potent synthetic cannabinoid HU210 was more effective than morphine in attenuating formalin-induced pain behavior, and these effects could not be attributed to motor impairment (Guhring et al 2001). Intrathecal cannabinoids also reversed established mechanical hyperalgesia in the partial sciatic nerve ligation model of neuropathic pain (Fox et al 2001).
Systemic administration of Δ9-THC accompanied by the intrathecal administration of an α2-noradrenergic antagonist suggested that the antinociceptive effects of cannabinoids are mediated, at least in part, by a descending noradrenergic mechanism (Lichtman and Martin 1991a). Lesions of descending noradrenergic systems also attenuated the antinociceptive effects of systemically administered cannabinoids in animal models of acute and tonic nociception, as well as the accompanying changes in Fos protein expression (Gutierrez et al 2003).
Intrathecal administration of exogenous anandamide blocks the thermal hyperalgesia associated with intraplantar carrageenan at doses that did not alter basal nociceptive thresholds (Richardson et al 1998a). In vitro superfusion studies in the rat spinal cord revealed that anandamide prevents both capsaicin- and K+-evoked release of CGRP (Richardson et al 1998a) whereas the CB1 receptor antagonist SR141716A enhances capsaicin-evoked release of substance P, thus suggesting that release of this neuropeptide may be under tonic control by endocannabinoids (Lever and Malcangio 2002). However, the inverse agonist properties of SR141716A may also play a role.
Biochemical markers have also been used to detect the spinal cellular activity associated with central sensitization. Intrathecal administration of WIN55,212-2, which reversed mechanical allodynia following subcutaneous injection of CFA into the hindpaw, also prevented the associated appearance of Fos-like immunoreactivity in spinal cord neurons (Martin et al 1999b). Likewise, a reduction in dorsal horn Fos-like immunoreactivity by WIN55,212-2 and anandamide following intravesicular turpentine, injection of NGF (Farquhar-Smith et al 2002), intraplantar injection of carrageenan (Nackley et al 2003b), or subcutaneous formalin injection (Tsou et al 1996) has been demonstrated. The selective CB2 agonist AM1241 also reduces spinal Fos protein–like immunoreactivity following carrageenan-induced inflammation (Nackley et al 2003a).
Non-neuronal cells (e.g., microglia) also play a role in the CNS responses to inflammation and peripheral nerve injury that underlie persistent pain (DeLeo and Yezierski 2001, Watkins and Maier 2003). Microglia express functional CB2 receptors, synthesize endocannabinoids, and influence microglial migration (Walter et al 2003), and an increase in spinal CB2 expression is observed following peripheral nerve injury, but not following an inflammatory insult (Zhang et al 2003). Effects of endocannabinoids on microglial activity may be restricted to the process of migration because microglial proliferation, nitric oxide production, and phagocytosis are unaffected. Furthermore, cannabinoids block cytokine mRNA expression in cultured microglial cells through cannabinoid receptor–independent mechanisms (Puffenbarger et al 2000). Both the CB1- and CB2-mediated anti-inflammatory and neuroprotective effects of cannabinoids are also mediated by release of interleukin-1 receptor antagonist, which abrogates the effects of the key inflammatory cytokine interleukin-1 released from both glia and neurons (Molina-Holgado et al 2003). Nonetheless, spinally mediated cannabinoid antinociception is largely via CB1, although pharmacological specificity may be dependent on the model.
Peripheral Analgesic Effects
Peripheral cannabinoid antinociceptive mechanisms represent a promising target for analgesic drug development. Anti-hyperalgesic and anti-allodynic effects of locally delivered cannabinoids are observed at doses that are not systemically effective. Locally administered anandamide attenuated carrageenan-induced thermal hyperalgesia in a CB1-dependent manner, although a CB2-mediated component could not be excluded (Richardson et al 1998b). A peripheral CB2-mediated suppression of mechanical and heat hypersensitivity is an observed effect in carrageenan (Nackley et al 2003a, Quartilho et al 2003) and capsaicin models of inflammatory pain (Hohmann et al 2004). Suppression of noxious stimulus–evoked Fos protein expression (Nackley et al 2003a), as well as electrophysiological measures of spinal neuronal sensitization (Elmes et al 2004, Nackley et al 2004), accompanies the anti-allodynic effects of CB2 agonists. Local administration of various cannabinoids attenuated both phases of the behavioral response to subcutaneous formalin injection via CB1-dependent–mediated mechanisms (Calignano et al 1998). In the partial sciatic nerve ligation model of neuropathic pain, an infusion of WIN55,212-2 (30 μg) at the site of nerve injury prevented mechanical- and cold-induced hypersensitivity in a locally mediated effect that was a CB1 and, to a certain extent, CB2 receptor–dependent process (Lever et al 2007). Further evidence of a peripheral analgesic effect of cannabinoids comes from experiments in which the carrageenan model of cutaneous inflammatory hyperalgesia was examined (Nackley et al 2003a, 2003b). Locally administered WIN55,212-2 attenuated the behavioral signs of inflammatory hyperalgesia and the associated spinal Fos expression in an effect mediated by both CB1 and CB2 receptors. The same dose of WIN55,212-2 administered systemically or into the contralateral paw was ineffective (Nackley et al 2003a).
Evidence of functionally active CB1 receptors in the peripheral nervous system was provided by demonstrating the inhibitory effect of WIN55,212-2 on voltage-activated Ca2+ currents and antagonism of this effect by the CB1 antagonist SR141716A (Ross et al 2001). Cannabinoids are also known to inhibit the capsaicin-evoked influx of Ca2+ into cultured dorsal root ganglion cells (Millns et al 2001). Other authors have provided data that attenuation of the K+-evoked influx of Ca2+ in dissociated rat dorsal root ganglion cells by cannabinoids (in a CB1-dependent manner) occurs predominantly in intermediate-sized cells (somal area, 800–1500 μm2) (Khasabova et al 2002). This finding is in agreement with the expression of CB1 in medium to large dorsal root and trigeminal ganglion cells (Hohmann and Herkenham 1999a, Bridges et al 2003, Price et al 2003). Further evidence of a peripherally mediated cannabinoid effect comes from in vitro studies in which the synthetic cannabinoid CP55,940 attenuated the capsaicin-evoked release of CRGP in skin taken from normal and diabetic (streptozocin treated) rats in a CB1-dependent–mediated manner (Ellington et al 2002).
The literature supports the existence of both CB1- (Agarwal et al 2007) and CB2-mediated peripheral antinociception (for review see Guindon and Hohmann 2008). Genetic deletion of CB1 from nociceptive neurons attenuates the antinociception produced by systemic and local but not by intrathecal cannabinoids (Agarwal et al 2007), thus suggesting that peripheral CB1 receptors remain a viable analgesic target. Indeed, AZ11713908, a peripherally restricted CB1 agonist (with CB2 inverse agonist and partial agonist properties), produces CB1-mediated anti-hyperalgesic effects in a CFA model, and these effects are fully preserved in CB2−/− mice (Yu et al 2010). Since the majority of CB2 expression is on cells of immune origin, the periphery is an obvious site to investigate such effects. Agonist activity at CB2 generally down-regulates the activity of immune cells, and this effect contributes to the observed local anti-hyperalgesic effects of cannabinoids in inflammation and following peripheral nerve injury. For instance, the suppressive effect of locally administered anandamide on the enhanced, mechanically evoked responses observed in spinal dorsal horn neurons of rodents with carrageenan-induced inflammation of the hindpaw is mediated via peripheral CB2 receptors (Sokal et al 2003), as are the anti-hyperalgesic actions of AM1241 in inflammatory (Nackley et al 2003a, Quartilho et al 2003) and neuropathic pain (Ibrahim et al 2003). Both neuronal (via primary afferent) and non-neuronal (via mast cells, neutrophils, macrophages) mechanisms may contribute to these effects.
NGF contributes to the generation and maintenance of inflammatory hyperalgesia; NGF-induced mast cell degranulation amplifies the NGF “signal,” and cannabinoids may attenuate this effect (Rice 2001). Neutrophil migration is also a key component of the NGF-mediated component of inflammatory hyperalgesia (Bennett et al 1998). Palmitoylethanolamide (PEA), a fatty acid amide that is structurally related to anandamide but does not bind to cannabinoid receptors, decreases NGF-evoked cutaneous thermal hyperalgesia and neutrophil accumulation, as measured with the myeloperoxidase assay, by an SR144528-sensitive mechanism (Farquhar-Smith and Rice 2001). Efficacy of locally administered CB1-selective (Gutierrez et al 2007), CB2-selective (Quartilho et al 2003, Hohmann et al 2004, Nackley et al 2004), and mixed CB1/CB2 agonists (Nackley et al 2003b), as well as endocannabinoid modulators (e.g., MGL inhibitors; Guindon et al 2011), has been demonstrated (Table 38-1). A peripherally restricted FAAH inhibitor has also been shown to suppress inflammatory and neuropathic pain through CB1 mechanisms, whereas anti-inflammatory effects involve CB1 as well as CB2 receptors and peroxisome proliferator–associated receptor (PPAR-γ) (Clapper et al 2010).
Table 38-1
Modulators of the Cannabinoid Signaling System Showing Efficacy in Animal Models of Inflammatory Pain

Note: Pharmacological specificity, if demonstrated, is shown in parentheses.
ACEA, arachidonoyl-2-chloroethylamide; AEA, anandamide; 2-AG, 2-arachidonoylglycerol; CFA, complete Freund’s adjuvant; ECB, endocannabinoid; FAA, fatty acid amide; FAAH, fatty acid amide hydrolase; FLAT, FAAH-like anandamide transporter; MAFP, (5Z,8Z,11Z,14Z)-5,8,11,14-eicosatetraenyl-methyl ester phosphonofluoridic acid; MGL, monoacylglycerol lipase; NT, not tested; OEA, osteoarthritis; PEA, palmitoylethanolamine.
Adapted and updated from Guindon J, Beaulieu P, Hohmann AG, et al 2010 The pharmacology of the cannabinoid system. In: Beaulieu P, Lussier D, Porreca F, et al (eds) Pharmacology of pain. IASP Press, Seattle, p 111–138.
Studies in Animal Models of Pathological Pain
Cannabinoids produce antinociception in animal models of pathological pain that better mimic the neuronal sensitization characteristic of chronic pain states observed clinically. Cannabinoids appear to be efficacious in suppressing hyperalgesia and allodynia in virtually all animal models of pathological pain studied to date (Tables 38-1 and 38-2). This includes pain resulting from inflammation, traumatic nerve injury, metabolic challenges (e.g., streptozocin), toxic insults (e.g., chemotherapy), and disease states (cancer, human immunodeficiency virus [HIV], diabetes, herpes zoster). Although pharmacological specificity has not been established in all studies, we reviewed the literature to summarize the efficacy of synthetic cannabinoids targeting CB1 and/or CB2 receptors, as well as endocannabinoid modulators (FAAH inhibitors, MGL inhibitors, endocannabinoid uptake inhibitors) in suppressing inflammatory, neuropathic, and cancer pain. These data have recently been reviewed (Rice et al 2002; Guindon and Hohmann 2008, 2009, 2011; Rahn and Hohmann 2009). At the onset, however, it is important to emphasize that inhibitors of FAAH and MGL are not specific for the endocannabinoid system but also elevate levels of fatty acid amides and monoacylglycerols that do not bind to cannabinoid receptors. Thus, assessment of pharmacological specificity is critical to establish mediation by cannabinoid receptors. Cannabinoid receptor–independent effects may nonetheless contribute to the pattern of in vivo efficacy observed. For example, the fatty acid amide PEA, a product of FAAH inhibition, is an agonist at PPAR-γ receptors but does not bind to cannabinoid receptors. However, the in vivo effects of PEA are blocked by SR144528, a CB2 antagonist (Calignano et al 1998). Moreover, synthetic endocannabinoids—and specifically anandamide—like many endogenous compounds are not very good drugs because of their susceptibility to rapid degradation (e.g., by FAAH), low potency, and relatively low affinity for CB receptors. Finally, it is important to emphasize that the efficacy of endocannabinoid modulators may also vary with the level of endocannabinoid tone present in any specific model system.
Table 38-2
Modulators of the Cannabinoid Signaling System Showing Efficacy in Animal Models of Neuropathic Pain
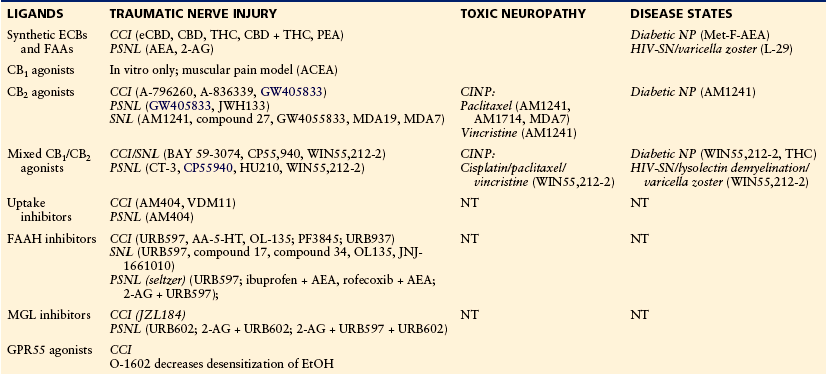
AA-5-HT, arachidonyl serotonin (N-[2-(5-Hydroxy-1H-indol-3-yl)ethyl]-5,8,11,14-eicosatetraenamide); ACEA, arachidonoyl-2-chloroethylamide; AEA, anandamide; 2-AG, 2-arachidonoylglycerol; CBD, cannabidiol; CCI, chronic constriction injury; CINP, chemotherapy-induced neuropathy; ECB, endocannabinoid; eCBD (cannabidiol with high CBD content); EtOH, ethanol; FAAH, fatty acid amide hydrolase; GPR55, G protein–coupled receptor GPR55; HIV-SN, human immunodeficiency virus sensory neuropathy (includes antiretroviral treatment [ddC], HIV-gp120, and HIV-gp120 + ddC models); MGL, monoacylglycerol lipase; NP, neuropathy; NT, not tested; PEA, palmitoylethanolamine; PSNL, partial sciatic nerve ligation; SNL, spinal nerve ligation; THC, Δ9-tetrahydrocannabinol.
Adapted and updated from Rahn EJ, Hohmann AG 2009 Cannabinoids as pharmacotherapies for neuropathic pain: from the bench to the bedside. Neurotherapeutics 6:713–737.
In models of inflammatory pain (formalin, carrageenan), anandamide produces antinociceptive effects through a CB1 mechanism, whereas the effects of 2-AG, when studied, are exclusively CB2 mediated (Guindon et al 2007). These observations are consistent with the greater affinity of 2-AG than anandamide for CB2. However, pro-nociceptive effects of anandamide (capsaicin evoked) and cannabinoid receptor–independent (TRPV1 mediated) effects, both in vitro and in vivo, are also observed. Unlike mixed cannabinoid agonists, which typically behave as CB1 agonists (at least following systemic administration), selective CB1 agonists (arachidonoyl-2-chloroethylamide [ACEA], methanandamide [MethAEA]) and CB2-selective agonists produce antinociceptive effects with the predicted pharmacological specificity (formalin, carrageenan, capsaicin, CFA). An exception to this generalization was observed in a model of osteoarthritic knee pain, where the CB2 agonist GW405833 was suggested to produce pro-nociceptive effects via a TRPV1-dependent mechanism (Schuelert et al 2010). Inhibitors of FAAH or endocannabinoid uptake, administered systemically or locally in the paw, produce antinociceptive effects mediated by either CB1 only or both CB1 and CB2 receptors (see Table 38-1). Similar results are observed with the peripherally restricted FAAH inhibitor URB937, which produces antinociceptive effects mediated by CB1 only but anti-edema effects mediated by CB1, CB2, and PPAR-γ (Clapper et al 2010). MGL inhibitors have been evaluated in capsaicin (local only), formalin, and carrageenan models (systemic, local), where they produce antinociceptive effects mediated by either CB1 or CB2 receptors (see Table 38-1). Strikingly, local inhibition of enzymes hydrolyzing 2-AG and AEA suppressed capsaicin-evoked behavioral sensitization with distinct patterns of pharmacological specificity and in a non-overlapping and modality-specific manner (Spradley et al 2010). In this study, JZL184 (local) suppressed capsaicin-evoked nocifensive behavior and thermal hyperalgesia through either CB1 or CB2 receptor mechanisms without altering mechanical allodynia. By contrast, URB597 (local) suppressed capsaicin-evoked mechanical allodynia through CB1 mechanisms only but did not alter capsaicin-evoked nocifensive behavior or heat hyperalgesia. An inhibitor of endocannabinoid uptake (VDM11) also mimicked the effects of the FAAH and MGL inhibitors in combination, with the predicted pattern of pharmacological specificity. Thus, local inhibition of endocannabinoid transport in the periphery was more effective than either FAAH or MGL alone in suppressing behavioral sensitization in the capsaicin model (Spradley et al 2010).
Cannabinoids are efficacious in suppressing nociception in preclinical models of neuropathic pain induced by diabetes (i.e., streptozocin), chemotherapy (i.e., paclitaxel, vincristine, cisplatin), HIV/retroviral treatment, demyelination disorders, and post-herpetic neuralgia (see Table 38-2). Of these, evaluations of CB2 agonists and mixed CB1/CB2 agonists, followed by inhibitors of FAAH and endocannabinoid uptake, have most permeated the literature, with models of surgically induced traumatic nerve injury being the most frequently studied. Both mixed cannabinoid agonists and CB2 agonists demonstrated efficacy in models of traumatic nerve injury (Herzberg et al 1997, Ibrahim et al 2003, Valenzano et al 2005) and toxic insults from chemotherapy (e.g.,Pascual et al 2005, Rahn et al 2007, Vera et al 2007, Rahn et al 2008). The anti-allodynic and anti-hyperalgesic effects of CB2 agonists are blocked by CB2 but not by CB1 antagonists, preserved in CB1−/− mice, and absent in CB2−/− mice, whereas mixed CB agonists (systemic) produce antinociceptive effects mediated largely by CB1 receptors, although CB2-mediated effects are unmasked following local administration of these agonists. Endocannabinoid modulators also show anti-hyperalgesic efficacy in models of neuropathic pain. The anti-allodynic effects of the MGL inhibitor JZL184, but not the FAAH inhibitor URB597, are preserved in FAAH−/− mice in a model of traumatic nerve injury (chronic constriction injury) (Kinsey et al 2009, 2010). These effects were mediated by CB1 (in the case of JZL184) or by both CB1 and CB2 (in the case of URB597).
Recently, neuropathic pain in disease states (streptozotocin-induced diabetic neuropathy, lysolecithin-induced demyelination, varicella zoster model of post-herpetic neuralgia) has been modeled in animals and shown to respond to mixed cannabinoid agonists and fatty acid amide analogues (the anandamide analogue Met-F-AEA and the PEA analogue L29). The mixed CB1 agonist WIN55,212-2, the CB2 agonist AM1241, and the CB1 agonist 2-methyl-2′-F anandamide suppress streptozotocin-induced mechanical allodynia following chronic administration, although pharmacological specificity was not assessed. These effects were greater in the presence of cyclooxygenase-1 and NOS inhibitors (Bujalska et al 2008), thus suggesting the presence of antinociceptive synergism between cyclooxygenase and cannabinoid mechanisms.
A mixed cannabinoid agonist suppresses mechanical allodynia in models of HIV neuropathy. Specifically, WIN55,212-2 reversed the mechanical hypersensitivity induced by either neurotoxic antiretroviral treatment, HIV-gp120 exposure, or a combination of these two neurotoxic insults (Wallace et al 2007a, 2007b). The demyelination-induced neuropathy produced by lysolecithin is resistant to the opioid analgesic Tyr-D-Ala-Gly-[NMePhe]-NH(CH2)2 (DAMGO) but is attenuated by the mixed cannabinoid agonist WIN55,212-2 in a CB1-dependent manner (Wallace et al 2003). The mechanical hypersensitivity produced by varicella-zoster virus in an animal model of post-herpetic neuralgia is also attenuated by both the PEA analogue L29 (through cannabinoid-independent mechanisms) and the cannabinoid agonist WIN55,212-2 (Hasnie et al 2007, Wallace et al 2007b).
Recent literature also suggests that the endocannabinoid signaling system can be targeted to suppress both the evolution and progression of cancer (i.e., breast, prostate, bone), as well as accompanying pain syndromes (for review see Guindon and Hohmann 2011). The anti-hyperalgesic effects of mixed cannabinoid agonists (WIN55,212-2, CP55,940) and CB1-selective agonists (ACEA), administered systemically, and synthetic endocannabinoids (AEA), administered locally, in the paw are CB1 mediated (for review see Guindon and Hohmann 2011). The anti-hyperalgesic/anti-allodynic effects of CB2 agonists (AM1241), administered either systemically, intrathecally, or peritumorally, are CB2 mediated (Curto-Reyes et al 2010). Changes in endocannabinoid signaling and the activity of endocannabinoid hydrolyzing enzymes have also been demonstrated in bone cancer models. Anandamide and FAAH inhibitors (local) produce CB1-mediated anti-hyperalgesic effects in a model of bone cancer produced by osteolytic fibrosarcoma; increased FAAH activity, FAAH mRNA, and CB1 expression are also observed in the dorsal root ganglia of tumor-bearing mice (Khasabova et al 2008). Most notably, locally administered 2-AG and JZL184 (MGL inhibitor) also attenuated hyperalgesia through CB2-specific mechanisms with an efficacy comparable to that of morphine (Khasabova et al 2011a). Moreover, CB1 and CB2 agonists produce synergistic anti-hyperalgesic effects (Khasabova et al 2011a, 2011b). Finally, in vitro studies suggest multiple antitumor effects of cannabinoids (inhibition of cell proliferation and migration, induction of apoptosis) that may contribute to a highly favorable therapeutic profile of the in vivo efficacy of cannabinoids (i.e., reduction of tumor growth). More work is necessary to determine whether cannabinoids exhibit sufficiently high therapeutic potential to reduce pain, promote antitumor effects, and improve quality of life in cancer patients.
Clinical Trials Testing the Efficacy of Cannabinoids
Since the last edition of this textbook was published, a number of high-quality randomized controlled trials (RCT) have been reported in which the analgesic efficacy of cannabinoids and related compounds was evaluated. The bulk of this evidence relates to neuropathic pain.
Efficacy
Of seven RCTs included in updates of a systematic review of cannabinoids for neuropathic pain, five examined patients with multiple sclerosis, including more than 1000 patient episodes of pain. These trials compared either various extracts of herbal cannabis or Δ9-THC with placebo. In four of these studies, pain relief was greater in patients randomized to cannabinoid than to placebo. Two RCTs reported responder rates (50% pain relief), and similar “numbers need to treat” (NNTs) of 3.5 (Svendsen et al 2004) and 3.7 (Rog et al 2005) were calculated.
For HIV-related peripheral neuropathy, a condition that does not respond to conventional antineuropathic pain agents such as tricyclic antidepressants (Phillips et al 2010), the evidence from RCTs also indicates that cannabis has analgesic properties. Two RCTs compared smoked cannabis with smoked placebo and demonstrated the clear efficacy of cannabis (Abrams et al 2007, Ellis et al 2009). Responder rates (NNTs [30% pain relief] of 3.6 [Abrams et al 2007] and 3.5 [Ellis et al 2009]) compared favorably with those of other agents for pain relief in this condition (Phillips et al 2010). Although smoking is unlikely to become a practical method of drug administration, these RCTs do provide encouragement for further examination of cannabinoids in patients with HIV neuropathy when administered by more acceptable methods.
For other neuropathic pain conditions, in two RCTs that evaluated 181 episodes in patients with peripheral neuropathic pain (brachial plexus avulsion and a mixture of conditions), no appreciable evidence of efficacy was found for cannabinoids (Rice and Hill 2006). Another more recently published study suggested a modest degree of analgesic efficacy when two concentrations of smoked cannabis were compared with a placebo preparation in subjects with a range of central and peripheral neuropathic pain conditions, mainly complex regional pain syndrome type 1 (Wilsey et al 2008). A small (23 participants) four-period crossover RCT examining four potencies of smoked cannabis for pain associated with peripheral nerve injury reported that only the highest potency tested (9.4% THC) had analgesic efficacy when compared with placebo, and this group also reported significant CNS-mediated adverse events (Ware et al 2010). Finally, a small RCT of oromucosal cannabis extract in patients with painful diabetic neuropathy did not demonstrate any efficacy of this preparation over placebo (Selvarajah et al 2010).
A small number of reports have assessed cannabinoids for efficacy in relieving postoperative pain; RCTs of oral THC for pain following pelvic surgery (Buggy et al 2003) and nabilone for pain after a variety of surgical procedures (Beaulieu 2006) did not demonstrate evidence of analgesic effects. One RCT examined the effect of oral cannabis extract in a complicated design of dose escalation of patient-controlled analgesia; even though there was perhaps some sign of efficacy, the study was terminated early because of a serious adverse event (Holdcroft et al 2006).
Another systematic review also identified reports of cannabis preparations evaluated in clinical trials of non-neuropathic chronic pain conditions, including cancer pain (six RCTs), rheumatoid arthritis (one), and fibromyalgia (one) (Martin-Sanchez et al 2009). However, although some evidence of modest efficacy was suggested, the review identified significant methodological flaws in the design of the studies included, and therefore further high-quality RCTs are required before evidence of efficacy can be concluded for these conditions.
In summary, there is good evidence of the analgesic efficacy of cannabinoids for multiple sclerosis and HIV-associated polyneuropathy, but not for other neuropathic pain conditions. There is little substantial evidence of cannabinoid analgesia for postoperative or non-neuropathic chronic pain. More work is necessary to determine whether adjunctive treatment with cannabinoids enhances the efficacy and reduces the side effects of other analgesics.
Harmful Effects
A systematic review assessed acute safety data in 31 reports in which cannabinoids were used for analgesia (Wang et al 2008). Using internationally accepted criteria for defining the severity of adverse events, the authors reported that although “non-serious” acute adverse events were associated with cannabinoid treatment (rate ratio, 1.86), “serious” adverse events were not associated with cannabinoid treatment in this limited, short-term data set (1.04). CNS-mediated adverse events such as dizziness (15.5%) were most frequently reported with cannabinoid therapy. The authors drew attention to the paucity of safety data related to the effects of long-term therapeutic exposure to cannabinoids. A further confounder is that many RCTs in this area included dosing regimens that permitted self-titration. Conversely, another systematic review of treatment of chronic pain with cannabis found evidence of frequent significant adverse events, particularly related to the CNS (Martin-Sanchez et al 2009). The NNH (number needed to harm) for perception-, motor-, and cognitive-related adverse events was 7, 5, and 8, respectively, which when considered in relation to the above NNTs for efficacy suggests a narrow therapeutic index. Furthermore, a small study of healthy volunteers that compared orally administered single doses of Δ9-THC (20 mg) with inactive placebo (5 mg diazepam) reported noteworthy evidence of CNS-mediated adverse effects associated with Δ9-THC treatment (Kaufmann et al 2010). Finally, implications for the development of cannabinoids as therapeutics do need to take into account the impact of cannabis intoxication on the cognitive skills required for driving motor vehicles (Asbridge et al 2012).
Implications of the Risk for Mental Illness with Cannabis Abuse
In any discussion of the evidence of the clinical potential of cannabinoids, the issue of potential long-term mental health risks associated with therapeutic cannabinoid administration requires serious consideration. There is a now a substantial and consistent epidemiological literature supporting a dose-related association between cannabis abuse and subsequent long-term risk for the development of psychotic illness or schizophrenia (see Rice 2008a, 2008b). A systematic review encompassing nine studies consistently identified such findings in studies with different methodology in different settings (Semple et al 2005). Cannabis users are two to three times more likely for serious psychotic illness to subsequently develop, including schizophrenia, than non-cannabis users are. Similar findings were reported in a subsequent systematic review (Moore et al 2010). This observation is strengthened by a clear dose-response relationship between cannabis use and schizophrenia or the development of psychotic symptoms (Zammit et al 2002, Henquet et al 2005, Di Forti et al 2009). Populations at enhanced risk can be identified; for instance, there is an 18.2% higher risk for cannabis-associated psychotic symptoms in persons who possess baseline risk factors for the development of psychosis (Henquet et al 2005). There is also a genetic element inherent in such risk: individuals who carry a functional polymorphism in the gene encoding the enzyme catechol O-methyltransferase are more likely to exhibit psychotic symptoms and to develop schizophrenia if they use cannabis (Caspi et al 2005). Polymorphisms in this same enzyme are associated with variations in pain sensitivity and risk for the development of chronic pain conditions such as temporomandibular joint dysfunction (Diatchenko et al 2005). It should be noted that many of these data have emerged largely from studies in adolescents, so the extent to which they can be generalized to the wider population and specifically to patients is unknown. Finally, a frequent criticism of such epidemiological studies is the “self-medication” hypothesis whereby individuals with early preclinical psychosis could self-medicate with cannabis—but this hypothesis has recently been refuted (Henquet et al 2005). The therapeutic trials of cannabinoids for neuropathic pain reported to date are insufficiently powered to detect such infrequent but significant adverse events. Furthermore, although some therapeutic trials have monitored patients for up to 1 year, this may be an insufficient period to detect the long-term adverse effects of cannabinoids. It is impossible to disregard the implications of these facts for cannabinoids being developed for chronic regular administration in patients with long-term conditions such as neuropathic pain.
Another important lesson regarding the danger of premature clinical introduction of therapeutic interventions that perturb the CNS endocannabinoid system is the increased risk for serious psychiatric adverse events that was revealed in the clinical development of CB1 receptor antagonists for the treatment of obesity and other indications (Christensen et al 2007, Hill and Gorzalka 2009, Topol et al 2010). These adverse events led to withdrawal from the market and termination of phase III clinical trials of these drugs. However, these data may reveal insight into the neurobiology of depression (Hill and Gorzalka 2009). More work is necessary to determine whether peripherally restricted modulators of the endocannabinoid system would produce a more circumscribed and beneficial spectrum of therapeutic efficacy in humans than brain-penetrant therapeutic agents would.
Conclusions for Clinical Studies
Justified by a strong basic science foundation, RCTs have been conducted that reveal evidence of analgesia for certain cannabinoids/cannabis in patients with multiple sclerosis and HIV-related polyneuropathy. The short-term adverse effect profiles of the cannabinoids evaluated in these RCTs are not dramatically different from those for other systemically administered neuropathic pain therapies. However, the implications of the strong and consistent epidemiological data associating cannabis misuse with a dose-dependent long-term risk for mental illness cannot be isolated from any discussion of the therapeutic use of potent cannabinoids, especially in the context of neuropathic pain, for which regular long-term therapy is required. The precise magnitude, relevance, and implications of this risk with therapeutic cannabinoid use are currently unknown. This should be borne in mind when taking informed consent in both RCT and therapeutic settings. It would seem prudent to exclude, both from clinical therapy and from RCTs, any patients with risk factors, including genetic ones, for psychosis or schizophrenia. Furthermore, arrangements for long-term follow-up of subjects who have been treated with cannabinoids should be made to ascertain whether such adverse events are revealed years after the intervention. From the foregoing discussion it would also seem sensible to avoid drugs that target brain CB1 receptors—at least until the issue of cannabis-induced risk for psychosis is resolved. Furthermore, preparations of centrally acting CB1 agonists or cannabis extracts will have significant misuse potential, which has implications for drug enforcement authorities and society. Encouragingly, there are some other avenues that could circumvent brain CB1 while retaining cannabinoid analgesia, including CB2 agonists (Malan et al 2003, Guindon and Hohmann 2008, Anand et al 2009), endocannabinoid-degrading enzyme inhibitors (Cravatt and Lichtman 2003, Hohmann 2007, Clapper et al 2010, Long et al 2009a), PEA analogues (LoVerme et al 2005, Wallace et al 2007b), and targeting CB1 in the peripheral nervous system (Agarwal et al 2007). All these targets are being keenly pursued in academia and the pharmaceutical industry. For example, there are preliminary case reports and open-label studies (Indraccolo and Barbieri 2010, Phan et al 2010) and unverified clinical trials (see Rice and Mackie 2001) suggesting an analgesic effect of PEA in humans. However, a recent report of a clinical trial investigating the CB2 agonist GW842166 indicated no superiority over placebo for this compound in relieving acute pain following third molar tooth extraction, and a number of other pain-related clinical trials, particularly for osteoarthritis, have been registered as completed (http://clinicaltrials.gov/ct2/results?term-GW842166 accessed March 5, 2012). Similarly, clinical trials of the FAAH inhibitor PF-04457845, which has shown promise in preclinical pain models of osteoarthritis (Ahn et al 2011), have been completed (http://clinicaltrials.gov/ct2/show/NCT00981357 [accessed March 5, 2012] and clinical trial gov/ct2/show/NCT00836082).” PF-04457845 failed to show analgesic efficacy in patients with pain due to osteoarthritis of the knee in the most recent reporting of a completed randomized placebo- controlled trial (Huggins et al 2012). A study of Sativex for the treatment of central pain due to multiple sclerosis has recently been completed (http://clinicaltrials.gov/ct2/show/NCT01604265?term=Sativex) and its evaluation in persistent pain due to cancer is ongoing (http://clinicaltrials.gov/ct2/show/NCT01604265?term=Sativex accessed September 2012). More work is necessary to determine whether adjunctive therapies that combine endocannabinoid modulators with exisiting analgesics may enhance the analgesic efficacy of available analgesics and reduce unwanted side-effects.
The references for this chapter can be found at www.expertconsult.com.
References
Abadji V., Lin S., Taha G., et al. (r)-Methanandamide: a chiral novel anandamide possessing higher potency and metabolic stability. Journal of Medicinal Chemistry. 1994;37:1889–1893.
Abrams D.I., Jay C.A., Shade S.B., et al. Cannabis in painful HIV-associated sensory neuropathy: a randomized placebo-controlled trial. Neurology. 2007;68:515–521.
Agarwal N., Pacher P., Tegeder I., et al. Cannabinoids mediate analgesia largely via peripheral type 1 cannabinoid receptors in nociceptors. Nature Neuroscience. 2007;10:870–879.
Ahn K., Smith S.E., Liimatta M.B., et al. Mechanistic and pharmacological characterization of PF-04457845: a highly potent and selective fatty acid amide hydrolase inhibitor that reduces inflammatory and noninflammatory pain. Journal of Pharmacology and Experimental Therapeutics. 2011;338:114–124.
Anand P., Whiteside G., Fowler C.J., et al. Targeting CB(2) receptors and the endocannabinoid system for the treatment of pain. Brain Research Reviews. 2009;60:255–266.
Anand U., Otto W.R., Sanchez-Herrera D., et al. Cannabinoid receptor CB2 localisation and agonist-mediated inhibition of capsaicin responses in human sensory neurons. Pain. 2008;138:667–680.
Asbridge M., Hayden J.A., Cartwright J.L. Acute cannabis consumption and motor vehicle collision risk: systematic review of observational studies and meta-analysis. British Medical Journal. 2012;344:e536.
Ates M., Hamza M., Seidel K., et al. Intrathecally applied flurbiprofen produces an endocannabinoid-dependent antinociception in the rat formalin test. European Journal of Neuroscience. 2003;17:597–604.
Atwood B.K., Mackie K. CB2: a cannabinoid receptor with an identity crisis. British Journal of Pharmacology. 2010;160:467–479.
Atwood B.K., Wager-Miller J., Haskins C., et al. Functional selectivity in CB2 cannabinoid receptor signaling and regulation: implications for the therapeutic potential of CB2 ligands. Molecular Pharmacology. 2012;81:250–263.
Barth F. Cannabinoid receptor agonists and antagonists. Expert Opinion on Therapeutic Patents. 1998;8:301–313.
Bayewitch M., Avidor Reiss T., Levy R., et al. The peripheral cannabinoid receptor: adenylate cyclase inhibition and G protein coupling. FEBS Letters. 1995;375:143–147.
Beaulieu P. Effects of nabilone, a synthetic cannabinoid, on postoperative pain. Canadian Journal of Anaesthesia/Journal Canadien d’Anesthesie. 2006;53:769–775.
Beltramo M., Stella N., Calignano A., et al. Functional role of high-affinity anandamide transport, as revealed by selective inhibition. Science. 1997;277:1094–1097.
Bennett G., al Rashed S., Hoult J.R., et al. Nerve growth factor induced hyperalgesia in the rat hind paw is dependent on circulating neutrophils. Pain. 1998;77:315–322.
Bisogno T., Maurelli S., Melck D., et al. Biosynthesis, uptake, and degradation of anandamide and palmitoylethanolamide in leukocytes. Journal of Biological Chemistry. 1997;272:3315–3323.
Blankman J.L., Simon G.M., Cravatt B.F. A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chemistry & Biology. 2007;14:1347–1356.
Bouaboula M., Desnoyer N., Carayon P., et al. Gi protein modulation induced by a selective inverse agonist for the peripheral cannabinoid receptor CB2: implication for intracellular signalization cross-regulation. Molecular Pharmacology. 1999;55:473–480.
Breivogel C.S., Griffin G., Di Marzo V., et al. Evidence for a new G protein–coupled cannabinoid receptor in mouse brain. Molecular Pharmacology. 2001;60:155–163.
Bridges D., Rice A.S., Egertová M., et al. Localisation of cannabinoid receptor 1 in rat dorsal root ganglion using in situ hybridisation and immunohistochemistry. Neuroscience. 2003;119:803–812.
Brooks J.W., Pryce G., Bisogno T., et al. Arvanil-induced inhibition of spasticity and persistent pain: evidence for therapeutic sites of action different from the vanilloid VR1 receptor and cannabinoid CB1/CB2 receptors. European Journal of Pharmacology. 2002;439:83–92.
Buckley N.E., McCoy K.L., Mezey E., et al. Immunomodulation by cannabinoids is absent in mice deficient for the cannabinoid CB(2) receptor. European Journal of Pharmacology. 2000;396:141–149.
Buggy D.J., Toogood L., Maric S., et al. Lack of analgesic efficacy of oral [delta]-9-tetrahydrocannabinol in postoperative pain. Pain. 2003;106:169–172.
Bujalska M., Tatarkiewicz J., de Cordé A., et al. Effect of cyclooxygenase and nitric oxide synthase inhibitors on streptozotocin-induced hyperalgesia in rats. Pharmacology. 2008;81:151–157.
Calignano A., La Rana G., Giuffrida A., et al. Control of pain initiation by endogenous cannabinoids. Nature. 1998;394:277–281.
Carlson G., Wang Y., Ali Z. Endocannabinoids facilitate the induction of LTP in the hippocampus. Nature Neuroscience. 2002;5:723–724.
Caspi A., Moffitt T.E., Cannon M., et al. Moderation of the effect of adolescent-onset cannabis use on adult psychosis by a functional polymorphism in the catechol-O-methyltransferase gene: longitudinal evidence of a gene X environment interaction. Biological Psychiatry. 2005;57:1117–1127.
Chakrabarti A., Onaivi E.S., Chaudhuri G. Cloning and sequencing of a cDNA encoding the mouse brain-type cannabinoid receptor protein. DNA Sequence: The Journal of DNA Sequencing and Mapping. 1995;5:385–388.
Christensen R., Kristensen P.K., Bartels E.M., et al. Efficacy and safety of the weight-loss drug rimonabant: a meta-analysis of randomised trials. Lancet. 2007;370:1706–1713.
Cisneros J.A., Bjorklund E., Gonzalez-Gil I., et al. Structure-activity relationship of a new series of reversible dual monoacylglycerol lipase/fatty acid amide hydrolase inhibitors. Journal of Medicinal Chemistry. 2012;55:824–836.
Clapper J.R., Moreno-Sanz G., Russo R., et al. Anandamide suppresses pain initiation through a peripheral endocannabinoid mechanism. Nature Neuroscience. 2010;13:1265–1270.
Connell K., Bolton N., Olsen D., et al. Role of the basolateral nucleus of the amygdala in endocannabinoid-mediated stress-induced analgesia. Neuroscience Letters. 2006;397:180–184.
Coutts A.A., Anavi-Goffer S., Ross R.A., et al. Agonist-induced internalisation and trafficking of cannabinoid CB1 receptors in hippocampal neurons. Journal of Neuroscience. 2001;21:2425–2433.
Cravatt B.F., Demarest K., Patricelli M.P., et al. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:9371–9376.
Cravatt B.F., Giang D.K., Mayfield S.P., et al. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature. 1996;384:83–87.
Cravatt B.F., Lichtman A.H. Fatty acid amide hydrolase: an emerging therapeutic target in the endocannabinoid system. Current Opinion in Chemical Biology. 2003;7:469–475.
Cravatt B.F., Saghatelian A., Hawkins E.G., et al. Functional disassociation of the central and peripheral fatty acid amide signaling systems. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:10821–10826.
Curto-Reyes V., Llames S., Hidalgo A., et al. Spinal and peripheral analgesic effects of the CB2 cannabinoid receptor agonist AM1241 in two models of bone cancer–induced pain. British Journal of Pharmacology. 2010;160:561–573.
DeLeo J.A., Yezierski R.P. The role of neuroinflammation and neuroimmune activation in persistent pain. Pain. 2001;90:1–6.
De Petrocellis L., Melck D., Bisogno T., et al. Endocannabinoids and fatty acid amides in cancer, inflammation and related disorders. Chemistry and Physics of Lipids. 2000;108:191–209.
Deutsch D.G., Chin S.A. Enzymatic synthesis and degradation of anandamide, a cannabinoid receptor agonist. Biochemical Pharmacology. 1993;46:791–796.
Devane W.A., Dysarz F.A., 3rd., Johnson M.R., et al. Determination and characterization of a cannabinoid receptor in rat brain. Molecular Pharmacology. 1988;34:605–613.
Devane W.A., Hanus L., Breuer A., et al. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949.
Diana M.A., Marty A. Endocannabinoid-mediated short-term synaptic plasticity: depolarization-induced suppression of inhibition (DSI) and depolarization-induced suppression of excitation (DSE). British Journal of Pharmacology. 2004;142:9–19.
Diatchenko L., Slade G.D., Nackley A.G., et al. Genetic basis for individual variations in pain perception and the development of a chronic pain condition. Human Molecular Genetics. 2005;14:135–143.
Di Forti M., Morgan C., Dazzan P., et al. High-potency cannabis and the risk of psychosis. British Journal of Psychiatry. 2009;195:488–491.
Di Marzo V., Fontana A., Cadas H., et al. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature. 1994;372:686–691.
Dinh T.P., Carpenter D., Leslie F.M., et al. Brain monoglyceride lipase participating in endocannabinoid inactivation. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:10819–10824.
Dinh T.P., Kathuria S., Piomelli D. RNA interference suggests a primary role for monoacylglycerol lipase in the degradation of the endocannabinoid 2-arachidonoylglycerol. Molecular Pharmacology. 2004;66:1260–1264.
Drew G.M., Lau B.K., Vaughan C.W. Substance P drives endocannabinoid-mediated disinhibition in a midbrain descending analgesic pathway. Journal of Neuroscience. 2009;29:7220–7229.
Drew G.M., Mitchell V.A., Vaughan C.W. Glutamate spillover modulates GABAergic synaptic transmission in the rat midbrain periaqueductal grey via metabotropic glutamate receptors and endocannabinoid signaling. Journal of Neuroscience. 2008;28:808–815.
Egertová M., Cravatt B.F., Elphick M.R. Comparative analysis of fatty acid amide hydrolase and cb(1) cannabinoid receptor expression in the mouse brain: evidence of a widespread role for fatty acid amide of endocannabinoid signaling. Neuroscience. 2003;119:481–496.
Egertova M., Elphick M.R. Localisation of cannabinoid receptors in the rat brain using antibodies to the intracellular C-terminal of CB 1. Journal of Comparative Neurology. 2000;422:159–171.
Egertová M., Giang D.K., Cravatt B.F., et al. A new perspective on cannabinoid signalling: complementary localization of fatty acid amide hydrolase and the CB1 receptor in rat brain. Proceedings of the Royal Society of London. Series B, Biological Sciences. 1998;265:2081–2085.
Ellington H.C., Cotter M.A., Cameron N.E., et al. The effect of cannabinoids on capsaicin-evoked calcitonin gene–related peptide (CGRP) release from the isolated paw skin of diabetic and non-diabetic rats. Neuropharmacology. 2002;42:966–975.
Ellis R.J., Toperoff W., Vaida F., et al. Smoked medicinal cannabis for neuropathic pain in HIV: a randomized, crossover clinical trial. Neuropsychopharmacology. 2009;34:672–680.
Elmes S.J., Jhaveri M.D., Smart D., et al. Cannabinoid CB2 receptor activation inhibits mechanically evoked responses of wide dynamic range dorsal horn neurons in naive rats and in rat models of inflammatory and neuropathic pain. European Journal of Neuroscience. 2004;20:2311–2320.
Farquhar-Smith W.P., Egertova M., Bradbury E.J., et al. Cannabinoid CB(1) receptor expression in rat spinal cord. Molecular and Cellular Neurosciences. 2000;15:510–521.
Farquhar-Smith W.P., Jaggar S.I., Rice A.S. Attenuation of nerve growth factor–induced visceral hyperalgesia via cannabinoid CB(1) and CB(2)-like receptors. Pain. 2002;97:11–21.
Farquhar-Smith W.P., Rice A.S.C. Palmitoylethanolamide (PEA) attenuates NGF-induced hyperalgesia by reducing neutrophil accumulation via cannabinoid CB2-like receptors. Society of Neuroscience Abstracts. 2001;27:510–512.
Felder C.C., Briley E.M., Axelrod J., et al. Anandamide, an endogenous cannabimimetic eicosanoid, binds to the cloned human cannabinoid receptor and stimulates receptor-mediated signal transduction. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:7656–7660.
Finn D.P., Jhaveri M.D., Beckett S.R., et al. Effects of direct periaqueductal grey administration of a cannabinoid receptor agonist on nociceptive and aversive responses in rats. Neuropharmacology. 2003;45:594–604.
Fowler C.J., Stenstrom A., Tiger G. Ibuprofen inhibits the metabolism of the endogenous cannabimimetic agent anandamide. Pharmacology & Toxicology. 1997;80:103–107.
Fox A., Kesingland A., Gentry C., et al. The role of central and peripheral cannabinoid1 receptors in the antihyperalgesic activity of cannabinoids in a model of neuropathic pain. Pain. 2001;92:91–100.
Galiegue S., Mary S., Marchand J., et al. Expression of central and peripheral cannabinoid receptors in human immune tissues and leukocyte subpopulations. European Journal of Biochemistry. 1995;232:54–61.
Gallant M., Dufresne C., Gareau Y., et al. New class of potent ligands for the human peripheral cannabinoid receptor. Bioorganic & Medicinal Chemistry Letters. 1996;6:2263–2268.
Gao Y., Vasilyev D.V., Goncalves M.B., et al. Loss of retrograde endocannabinoid signaling and reduced adult neurogenesis in diacylglycerol lipase knock-out mice. Journal of Neuroscience. 2010;30:2017–2024.
Gaoni Y., Mechoulam R. Isolation, structure and partial synthesis of an active constituent of hashish. Journal of the American Chemical Society. 1964;86:1946–1947.
Gauldie S.D., McQueen D.S., Pertwee R., et al. Anandamide activates peripheral nociceptors in normal and arthritic rat knee joints. British Journal of Pharmacology. 2001;132:617–621.
Gerard C.M., Mollereau C., Vassart G., et al. Molecular cloning of a human cannabinoid receptor which is also expressed in testis. Biochemical Journal. 1991;279:129–134.
Gerdeman G.L., Ronesi J., Lovinger D.M., et al. Postsynaptic endocannabinoid release is critical to long-term depression in the striatum. Nature Neuroscience. 2002;5:446–450.
Glaser S.T., Abumrad N.A., Fatade F., et al. Evidence against the presence of an anandamide transporter. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:4269–4274.
Gregg L.C., Jung K.M., Spradley J.M., et al. Activation of type-5 metabotropic glutamate receptors and diacylglycerol lipase-α initiates 2-arachidonoylglycerol formation and endocannabinoid-mediated analgesia. The Journal of Neuroscience. 2012;32:9457–9468.
Griffin G., Tao Q., Abood M.E. Cloning and pharmacological characterization of the rat CB(2) cannabinoid receptor. Journal of Pharmacology and Experimental Therapeutics. 2000;292:886–894.
Guhring H., Hamza M., Sergejeva M., et al. A role for endocannabinoids in indomethacin-induced spinal antinociception. European Journal of Pharmacology. 2002;454:153–163.
Guhring H., Schuster J., Hamza M., et al. HU-210 shows higher efficacy and potency than morphine after intrathecal administration in the mouse formalin test. Eur J Pharmacol. 2001;429:127–134.
Guindon J., Beaulieu P., Hohmann A.G. The pharmacology of the cannabinoid system. In: Beaulieu P., Lussier D., Porreca F., Dickenson A.H., eds. Pharmacology of Pain. Seattle WA: IASP Press; 2010:111–138.
Guindon J., Desroches J., Beaulieu P. The antinociceptive effects of intraplantar injections of 2-arachidonoyl glycerol are mediated by cannabinoid CB2 receptors. British Journal of Pharmacology. 2007;150:693–701.
Guindon J., Guijarro A., Piomelli D., et al. Peripheral antinociceptive effects of inhibitors of monoacylglycerol lipase in a rat model of inflammatory pain. British Journal of Pharmacology. 2011;163:1464–1478.
Guindon J., Hohmann A.G. Cannabinoid CB2 receptors: a therapeutic target for the treatment of inflammatory and neuropathic pain. British Journal of Pharmacology. 2008;153:319–334.
Guindon J., Hohmann A.G. The endocannabinoid system and pain. CNS & Neurological Disorders Drug Targets. 2009;8:403–421.
Guindon J., Hohmann A.G. The endocannabinoid system and cancer: therapeutic implications. British Journal of Pharmacology. 2011;163:1447–1463.
Gutierrez T., Farthing J.N., Zvonok A.M., et al. Activation of peripheral cannabinoid CB1 and CB2 receptors suppresses the maintenance of inflammatory nociception: a comparative analysis. British Journal of Pharmacology. 2007;150:153–163.
Gutierrez T., Nackley A.G., Neely M.H., et al. Effects of neurotoxic destruction of descending noradrenergic pathways on cannabinoid antinociception in models of acute and tonic nociception. Brain Research. 2003;987:176–185.
Hanus L., Abu-Lafi S., Fride E., et al. 2-arachidonyl glyceryl ether, an endogenous agonist of the cannabinoid CB1 receptor. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:3662–3665.
Hanus L., Breuer A., Tchilibon S., et al. HU 308: a specific agonist for CB(2), a peripheral cannabinoid receptor. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:14228–14233.
Hasnie F.S., Breuer J., Parker S., et al. Further characterization of a rat model of varicella zoster virus–associated pain: relationship between mechanical hypersensitivity and anxiety-related behavior, and the influence of analgesic drugs. Neuroscience. 2007;144:1495–1508.
Henquet C., Krabbendam L., Spauwen J., et al. Prospective cohort study of cannabis use, predisposition for psychosis, and psychotic symptoms in young people. British Medical Journal. 2005;330:11.
Herkenham M., Lynn A.B., Johnson M.R., et al. Characterization and localization of cannabinoid receptors in rat brain: a quantitative in vitro autoradiographic study. Journal of Neuroscience. 1991;11:563–583.
Herzberg U., Eliav E., Bennett G.J., et al. The analgesic effects of R(+)-WIN 55,212-2 mesylate, a high affinity cannabinoid agonist, in a rat model of neuropathic pain. Neuroscience Letters. 1997;221:157–160.
Hill M.N., Gorzalka B.B. Impairments in endocannabinoid signaling and depressive illness. JAMA: The Journal of the American Medical Association. 2009;301:1165–1166.
Hillard C.J., Manna S., Greenberg M.J. Synthesis and characterization of potent and selective agonists of the neuronal cannabinoid receptor (CB1). J Pharmacol Exp Ther. 1999;289:1427–1433.
Ho Y.C., Lee H.J., Tung L.W., et al. Activation of orexin 1 receptors in the periaqueductal gray of male rats leads to antinociception via retrograde endocannabinoid (2-arachidonoylglycerol)-induced disinhibition. Journal of Neuroscience. 2011;31:14600–14610.
Hohmann A.G. Inhibitors of monoacylglycerol lipase as novel analgesics. British Journal of Pharmacology. 2007;150:673–675.
Hohmann A.G., Briley E.M., Herkenham M. Pre- and postsynaptic distribution of cannabinoid and mu opioid receptors in rat spinal cord. Brain Research. 1999;822:17–25.
Hohmann A.G., Farthing J.N., Zvonok A.M., et al. Selective activation of cannabinoid CB2 receptors suppresses hyperalgesia evoked by intradermal capsaicin. Journal of Pharmacology and Experimental Therapeutics. 2004;308:446–453.
Hohmann A.G., Herkenham M. Regulation of cannabinoid and mu opioid receptor binding sites following neonatal capsaicin treatment. Neuroscience Letters. 1998;252:13–16.
Hohmann A.G., Herkenham M. Localization of central cannabinoid CB1 receptor messenger RNA in neuronal subpopulations of rat dorsal root ganglia: a double-label in situ hybridization study. Neuroscience. 1999;90:923–931.
Hohmann A.G., Herkenham M. Cannabinoid receptors undergo axonal flow in sensory nerves. Neuroscience. 1999;92:1171–1175.
Hohmann A.G., Martin W.J., Tsou K., et al. Inhibition of noxious stimulus–evoked activity of spinal cord dorsal horn neurons by the cannabinoid WIN 55,212-2. Life Sciences. 1995;56:2111–2118.
Hohmann A.G., Suplita R.L., 2nd. Endocannabinoid mechanisms of pain modulation. AAPS Journal. 2006;8:E693–E708.
Hohmann A.G., Suplita R.L., Bolton N.M., et al. An endocannabinoid mechanism for stress-induced analgesia. Nature. 2005;435:1108–1112.
Hohmann A.G., Tsou K., Walker J.M. Cannabinoid modulation of wide dynamic range neurons in the lumbar dorsal horn of the rat by spinally administered WIN55,212-2. Neuroscience Letters. 1998;257:119–122.
Hohmann A.G., Tsou K., Walker J.M. Cannabinoid suppression of noxious heat–evoked activity in wide dynamic range neurons in the lumbar dorsal horn of the rat. Journal of Neurophysiology. 1999;81:575–583.
Holdcroft A., Maze M., Dore C., et al. A multicenter dose-escalation study of the analgesic and adverse effects of an oral cannabis extract (Cannador) for postoperative pain management. Anesthesiology. 2006;104:1040–1046.
Howlett A.C. The CB1 cannabinoid receptor in the brain. Neurobiology of Disease. 1998;5:405–416.
Howlett A.C., Barth F., Bonner T.I., et al. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacological Reviews. 2002;54:161–202.
Huang S.M., Bisogno T., Trevisani M., et al. An endogenous capsaicin-like substance with high potency at recombinant and native vanilloid VR1 receptors. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:8400–8405.
Huang S.M., Strangman N.M., Walker J.M. Liquid chromatographic–mass spectrometric measurement of the endogenous cannabinoid 2-arachidonylglycerol in the spinal cord and peripheral nervous system. Acta Pharmacologica Sinica. 1999;20:1098–1102.
Huffman J.W. The search for selective ligands for the CB2 receptor. Current Pharmaceutical Design. 2000;6:1323–1337.
Huggins J.P., Smart T.S., Langman S., et al. An efficient randomised, placebo-controlled clinical trial with the irreversible fatty acid amide hydrolase-1 inhibitor PF-04457845, which modulates endocannabinoids but fails to induce effective analgesia in patients with pain due to osteoarthritis of the knee. Pain. 2012;153:1837–1846.
Ibrahim M.M., Deng H., Zvonok A., et al. Activation of CB2 cannabinoid receptors by AM1241 inhibits experimental neuropathic pain: pain inhibition by receptors not present in the CNS. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:10529–10533.
Indraccolo U., Barbieri F. Effect of palmitoylethanolamide-polydatin combination on chronic pelvic pain associated with endometriosis: preliminary observations. European Journal of Obstetrics, Gynecology, and Reproductive Biology. 2010;150:76–79.
Jain A.K., Ryan J.R., McMahon F.G., et al. Evaluation of intramuscular levonantradol and placebo in acute postoperative pain. Journal of Clinical Pharmacology. 1981;21(8–9 Suppl):320S–326S.
Jung K.M., Astarita G., Zhu C., et al. A key role for diacylglycerol lipase-alpha in metabotropic glutamate receptor–dependent endocannabinoid mobilization. Molecular Pharmacology. 2007;72:612–621.
Jung K.M., Mangieri R., Stapleton C., et al. Stimulation of endocannabinoid formation in brain slice cultures through activation of group I metabotropic glutamate receptors. Molecular Pharmacology. 2005;68:1196–1202.
Kaczocha M., Glaser S.T., Deutsh D.G. Identification of intracellular carriers for the endocannabinoid anandamide. Proceedings of the National Academy of Sciences USA. 2009;106:6375–6380.
Katona I., Freund T.F. Endocannabinoid signaling as a synaptic circuit breaker in neurological disease. Nature Medicine. 2008;14:923–930.
Katona I., Sperlagh B., Magloczky Z., et al. GABAergic interneurons are the targets of cannabinoid actions in the human hippocampus. Neuroscience. 2000;100:797–804.
Katona I., Urban G.M., Wallace M., et al. Molecular composition of the endocannabinoid system at glutamatergic synapses. Journal of Neuroscience. 2006;26:5628–5637.
Kaufmann R.M., Kraft B., Frey R., et al. Acute psychotropic effects of oral cannabis extract with a defined content of Δ9-tetrahydrocannabinol (THC) in healthy volunteers. Pharmacopsychiatry. 2010;43:24–32.
Khasabova I.A., Chandiramani A., Harding-Rose C., et al. Increasing 2-arachidonoyl glycerol signaling in the periphery attenuates mechanical hyperalgesia in a model of bone cancer pain. Pharmacological Research. 2011;64:60–67.
Khasabova I.A., Gielissen J., Chandiramani A., et al. CB1 and CB2 receptor agonists promote analgesia through synergy in a murine model of tumor pain. Behavioural Pharmacology. 2011;22:607–616.
Khasabova I.A., Harding-Rose C., Simone D.A., et al. Differential effects of CB1 and opioid agonists on two populations of adult rat dorsal root ganglion neurons. Journal of Neuroscience. 2004;24:1744–1753.
Khasabova I.A., Khasabov S.G., Harding-Rose C., et al. A decrease in anandamide signaling contributes to the maintenance of cutaneous mechanical hyperalgesia in a model of bone cancer pain. Journal of Neuroscience. 2008;28:11141–11152.
Khasabova I.A., Simone D.A., Seybold V.S. Cannabinoids attenuate depolarization-dependent Ca2+ influx in intermediate-size primary afferent neurons of adult rats. Neuroscience. 2002;115:613–625.
Kinsey S.G., Long J.Z., Cravatt B.F., et al. Fatty acid amide hydrolase and monoacylglycerol lipase inhibitors produce anti-allodynic effects in mice through distinct cannabinoid receptor mechanisms. Journal of Pain. 2010;11:1420–1428.
Kinsey S.G., Long J.Z., O’Neal S.T., et al. Blockade of endocannabinoid-degrading enzymes attenuates neuropathic pain. Journal of Pharmacology and Experimental Therapeutics. 2009;330:902–910.
Kouznetsova M., Kelley B., Shen M., et al. Desensitization of cannabinoid-mediated presynaptic inhibition of neurotransmission between rat hippocampal neurons in culture. Molecular Pharmacology. 2002;61:477–485.
Kreitzer A.C., Regehr W.G. Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron. 2001;29:717–727.
Landsman R.S., Burkey T.H., Consroe P., et al. SR141716A is an inverse agonist at the human cannabinoid CB1 receptor. European Journal of Pharmacology. 1997;334:R1–R2.
Lauckner J.E., Jensen J.B., Chen H.Y., et al. GPR55 is a cannabinoid receptor that increases intracellular calcium and inhibits M current. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:2699–2704.
Ledent C., Valverde O., Cossu G., et al. Unresponsiveness to cannabinoids and reduced addictive effects of opiates in CB1 receptor knockout mice. Science. 1999;283:401–404.
Lee S.F., Newton C., Widen R., et al. Differential expression of cannabinoid CB2 receptor mRNA in mouse immune cell subpopulations and following B cell stimulation. European Journal of Pharmacology. 2001;423:235–241.
Leung D., Saghatelian A., Simon G.M., et al. Inactivation of N-acyl phosphatidylethanolamine phospholipase D reveals multiple mechanisms for the biosynthesis of endocannabinoids. Biochemistry. 2006;45:4720–4726.
Lever I.J., Malcangio M. CB1 receptor antagonist SR141716A increases capsaicin-evoked release of substance P from the adult mouse spinal cord. British Journal of Pharmacology. 2002;135:21–24.
Lever I.J., Pheby T.M., Rice A.S. Continuous infusion of the cannabinoid WIN 55,212-2 to the site of a peripheral nerve injury reduces mechanical and cold hypersensitivity. British Journal of Pharmacology. 2007;151:292–302.
Lever I.J., Robinson M., Cibelli M., et al. Localization of the endocannabinoid-degrading enzyme fatty acid amide hydrolase in rat dorsal root ganglion cells and its regulation after peripheral nerve injury. Journal of Neuroscience. 2009;29:3766–3780.
Lichtman A.H., Cook S.A., Martin B.R. Investigation of brain sites mediating cannabinoid-induced antinociception in rats: evidence supporting periaqueductal gray involvement. Journal of Pharmacology and Experimental Therapeutics. 1996;276:585–593.
Lichtman A.H., Martin B.R. Cannabinoid-induced antinociception is mediated by a spinal α2-noradrenergic mechanism. Brain Research. 1991;559:309–314.
Lichtman A.H., Martin B.R. Spinal and supraspinal components of cannabinoid-induced antinociception. Journal of Pharmacology and Experimental Therapeutics. 1991;258:517–523.
Lichtman A.H., Shelton C.C., Advani T., et al. Mice lacking fatty acid amide hydrolase exhibit a cannabinoid receptor–mediated phenotypic hypoalgesia. Pain. 2004;109:319–327.
Liu Q.R., Pan C.H., Hishimoto A., et al. Species differences in cannabinoid receptor 2 (CNR2 gene): identification of novel human and rodent CB2 isoforms, differential tissue expression and regulation by cannabinoid receptor ligands. Genes, Brain, and Behavior. 2009;8:519–530.
Long J.Z., Li W., Booker L., et al. Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nature Chemical Biology. 2009;5:37–44.
Long J.Z., Nomura D.K., Vann R.E., et al. Dual blockade of FAAH and MAGL identifies behavioral processes regulated by endocannabinoid crosstalk in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:20270–20275.
LoVerme J., La Rana G., Russo R., et al. The search for the palmitoylethanolamide receptor. Life Sciences. 2005;77:1685–1698.
Mackie K., Devane W.A., Hille B. Anandamide, an endogenous cannabinoid, inhibits calcium currents as a partial agonist in N18 neuroblastoma cells. Molecular Pharmacology. 1993;44:498–503.
Mackie K., Lai Y., Westenbroek R., et al. Cannabinoids activate an inwardly rectifying potassium conductance and inhibit Q-type calcium currents in AtT20 cells transfected with rat brain cannabinoid receptor. Journal of Neuroscience. 1995;15:6552–6561.
Mailleux P., Parmentier M., Vanderhaeghen J.J. Distribution of cannabinoid receptor messenger RNA in the human brain: an in situ hybridization histochemistry with oligonucleotides. Neuroscience Letters. 1992;143:200–204.
Malan T.P., Jr., Ibrahim M.M., Lai J., et al. CB2 cannabinoid receptor agonists: pain relief without psychoactive effects? Current Opinion in Pharmacology. 2003;3:62–67.
Manning B.H., Martin W.J., Meng I.D. The rodent amygdala contributes to the production of cannabinoid-induced antinociception. Neuroscience. 2003;120:1157–1170.
Manning B.H., Merin N.M., Meng I.D., et al. Reduction in opioid- and cannabinoid-induced antinociception in rhesus monkeys after bilateral lesions of the amygdaloid complex. Journal of Neuroscience. 2001;21:8238–8246.
Marsicano G., Goodenough S., Monory K., et al. CB1 cannabinoid receptors and on-demand defense against excitotoxicity. Science. 2003;302:84–88.
Martin W.J., Coffin P.O., Attias E., et al. Anatomical basis for cannabinoid-induced antinociception as revealed by intracerebral microinjections. Brain Research. 1999;822:237–242.
Martin W.J., Gupta N.K., Loo C.M., et al. Differential effects of neurotoxic destruction of descending noradrenergic pathways on acute and persistent nociceptive processing. Pain. 1999;80:57–65.
Martin W.J., Hohmann A.G., Walker J.M. Suppression of noxious stimulus–evoked activity in the ventral posterolateral nucleus of the thalamus by a cannabinoid agonist: correlation between electrophysiological and antinociceptive effects. Journal of Neuroscience. 1996;16:6601–6611.
Martin W.J., Lai N.K., Patrick S.L., et al. Antinociceptive actions of cannabinoids following intraventricular administration in rats. Brain Research. 1993;629:300–304.
Martin W.J., Patrick S.L., Coffin P.O., et al. An examination of the central sites of action of cannabinoid-induced antinociception in the rat. Life Sciences. 1995;56:2103–2109.
Martin-Sanchez E., Furukawa T.A., Taylor J., et al. Systematic review and meta-analysis of cannabis treatment for chronic pain. Pain Medicine. 2009;10:1353–1368.
Matsuda L.A., Bonner T.I., Lolait S.J. Localization of cannabinoid receptor mRNA in rat brain. Journal of Comparative Neurology. 1993;327:535–550.
Matsuda L.A., Lolait S.J., Brownstein M.J., et al. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–564.
Mechoulam R., Ben Shabat S., Hanus L., et al. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochemical Pharmacology. 1995;50:83–90.
Melck D., Bisogno T., De Petrocellis L., et al. Unsaturated long chain N-acyl-vanillyl-amides (N-AVAMs): vanilloid receptor ligands that inhibit anandamide-facilitated transport and bind to CB 1 cannabinoid receptors. Biochemical and Biophysical Research Communications. 1999;262:275–284.
Meng I.D., Manning B.H., Martin W.J., et al. An analgesia circuit activated by cannabinoids. Nature. 1998;395:381–383.
Millns P.J., Chapman V., Kendall D.A. Cannabinoid inhibition of the capsaicin-induced calcium response in rat dorsal root ganglion neurones. British Journal of Pharmacology. 2001;132:969–971.
Mitrirattanakul S., Ramakul N., Guerrero A.V., et al. Site-specific increases in peripheral cannabinoid receptors and their endogenous ligands in a model of neuropathic pain. Pain. 2006;126:102–114.
Molina-Holgado F., Pinteaux E., Moore J.D., et al. Endogenous interleukin-1 receptor antagonist mediates anti-inflammatory and neuroprotective actions of cannabinoids in neurons and glia. Journal of Neuroscience. 2003;23:6470–6474.
Monhemius R., Azami J., Green D.L., et al. CB1 receptor mediated analgesia from the nucleus reticularis gigantocellularis pars alpha is activated in an animal model of neuropathic pain. Brain Research. 2001;908:67–74.
Monory K., Massa F., Egertova M., et al. The endocannabinoid system controls key epileptogenic circuits in the hippocampus. Neuron. 2006;51:455–466.
Moore R.A., Derry S., McQuay H.J., et al. Clinical effectiveness: an approach to clinical trial design more relevant to clinical practice, acknowledging the importance of individual differences. Pain. 2010;149:173–176.
Munro S., Thomas K.L., Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65.
Nackley A.G., Makriyannis A., Hohmann A.G. Selective activation of cannabinoid CB2 receptors suppresses spinal fos protein expression and pain behavior in a rat model of inflammation. Neuroscience. 2003;119:747–757.
Nackley A.G., Suplita I.I., Hohmann A.G. A peripheral cannabinoid mechanism suppresses spinal fos protein expression and pain behavior in a rat model of inflammation. Neuroscience. 2003;117:659–670.
Nackley A.G., Zvonok A.M., Makriyannis A., et al. Activation of cannabinoid CB2 receptors suppresses C-fiber responses and windup in spinal wide dynamic range neurons in the absence and presence of inflammation. Journal of Neurophysiology. 2004;92:3562–3574.
Nomura D.K., Morrison B.E., Blankman J.L., et al. Endocannabinoid hydrolysis generates brain prostaglandins that promote neuroinflammation. Science. 2011;334:809–813.
Nyilas R., Gregg L.C., Mackie K., et al. Molecular architecture of endocannabinoid signaling at nociceptive synapses mediating analgesia. European Journal of Neuroscience. 2009;29:1964–1978.
Ohno-Shosaku T., Maejima T., Kano M. Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to presynaptic terminals. Neuron. 2001;29:729–738.
Olango W.M., Roche M., Ford G.K., et al. The endocannabinoid system in the rat dorsolateral periaqueductal grey mediates fear-conditioned analgesia and controls fear expression in the presence of nociceptive tone. British Journal of Pharmacology. 2012;165:2549–2560.
Pascual D., Goicoechea C., Suardâiaz M., et al. A cannabinoid agonist, WIN 55,212-2, reduces neuropathic nociception induced by paclitaxel in rats. Pain. 2005;118:23–34.
Pernia-Andrade A.J., Kato A., Witschi R., et al. Spinal endocannabinoids and CB1 receptors mediate C-fiber–induced heterosynaptic pain sensitization. Science. 2009;325:760–764.
Phan N.Q., Siepmann D., Gralow I., et al. Adjuvant topical therapy with a cannabinoid receptor agonist in facial postherpetic neuralgia. Journal der Deutschen Dermatologischen Gesellschaft. 2010;8:88–91.
Phillips T.J.C., Cherry C.L., Cox S., et al. A metaanalysis of randomised controlled clinical trials assessing the effectiveness of pharmacological interventions for painful HIV-associated sensory neuropathy. European Journal of Pain Supplements. 4, 2010. 141–141
Piomelli D. The molecular logic of endocannabinoid signalling. Nature Reviews. Neuroscience. 2003;4:873–884.
Porter A.C., Sauer J.M., Knierman M.D., et al. Characterization of a novel endocannabinoid, virodhamine, with antagonist activity at the CB1 receptor. Journal of Pharmacology and Experimental Therapeutics. 2002;301:1020–1024.
Price T.J., Helesic G., Parghi D., et al. The neuronal distribution of cannabinoid receptor type 1 in the trigeminal ganglion of the rat. Neuroscience. 2003;120:155–162.
Puffenbarger R., Boothe A.C., Cabral G.A. Cannabinoids inhibit LPS-inducible cytokine mRNA expression in rat microglial cells. Glia. 2000;29:58–69.
Quartilho A., Mata H.P., Ibrahim M.M., et al. Inhibition of inflammatory hyperalgesia by activation of peripheral CB2 cannabinoid receptors. Anesthesiology. 2003;99:955–960.
Racz I., Nadal X., Alferink J., et al. Crucial role of CB(2) cannabinoid receptor in the regulation of central immune responses during neuropathic pain. Journal of Neuroscience. 2008;28:12125–12135.
Racz I., Nadal X., Alferink J., et al. Interferon-gamma is a critical modulator of CB(2) cannabinoid receptor signaling during neuropathic pain. Journal of Neuroscience. 2008;28:12136–12145.
Rahn E.J., Hohmann A.G. Cannabinoids as pharmacotherapies for neuropathic pain: from the bench to the bedside. Neurotherapeutics. 2009;6:713–737.
Rahn E.J., Makriyannis A., Hohmann A.G. Activation of cannabinoid CB1 and CB2 receptors suppresses neuropathic nociception evoked by the chemotherapeutic agent vincristine in rats. British Journal of Pharmacology. 2007;152:765–777.
Rahn E.J., Zvonok A.M., Thakur G.A., et al. Selective activation of cannabinoid CB2 receptors suppresses neuropathic nociception induced by treatment with the chemotherapeutic agent paclitaxel in rats. Journal of Pharmacology and Experimental Therapeutics. 2008;327:584–591.
Rice A.S., Farquhar-Smith W.P., Nagy I. Endocannabinoids and pain: spinal and peripheral analgesia in inflammation and neuropathy. Prostaglandins, Leukotrienes, and Essential Fatty Acids. 2002;66:243–256.
Rice A.S.C. Mechanisms of inflammatory pain: role of neurotrophins and cannabinoids. In: Soulsby L., Morton D., eds. Pain: its nature and management in man and animals. London: Royal Society of Medicine Press; 2001:35–45.
Rice A.S.C., Cannabinoids for neuropathic pain? Where next? Neuropathic Pain 2008;11:3–6. www neupsig.org
Rice A.S.C. Should cannabinoids be used as analgesics for neuropathic pain? Nature Clinical Practice Neurology. 2008;4:654–655.
Rice A.S.C., Mackie K. Analgesics: cannabinoids. In: Evers A.S., Maze M., eds. Anesthetic pharmacology: physiologic principles and clinical practise. St Louis: Harcourt Health Sciences, 2001.
Richardson J.D., Aanonsen L., Hargreaves K.M. Antihyperalgesic effects of spinal cannabinoids. European Journal of Pharmacology. 1998;345:145–153.
Richardson J.D., Kilo S., Hargreaves K.M. Cannabinoids reduce hyperalgesia and inflammation via interaction with peripheral CB1 receptors. Pain. 1998;75:111–119.
Rinaldi-Carmona M., Barth F., Heaulme M., et al. SR141716A, a potent and selective antagonist of the brain cannabinoid receptor. FEBS Letters. 1994;350:240–244.
Rinaldi-Carmona M., Barth F., Millan J., et al. SR 144528, the first potent and selective antagonist of the CB2 cannabinoid receptor. Journal of Pharmacology and Experimental Therapeutics. 1998;284:644–650.
Rog D.J., Nurmikko T.J., Friede T., et al. Randomized, controlled trial of cannabis-based medicine in central pain in multiple sclerosis. Neurology. 2005;65:812–819.
Ross R.A., Brockie H.C., Stevenson L.A., et al. Agonist–inverse agonist characterization at CB1 and CB2 cannabinoid receptors of L759633, L759656, and AM630. British Journal of Pharmacology. 1999;126:665–672.
Ross R.A., Coutt A.A., McFarlane S.M., et al. Actions of cannabinoid receptor ligands on rat cultured sensory neurones: implications for antinociception. Neuropharmacology. 2001;40:221–232.
Samson M.T., Small-Howard A., Shimoda L.M.N., et al. Differential roles of CB1 and CB2 cannabinoid receptors in mast cells. Journal of Immunology. 2003;170:4953–4962.
Sanudo-Pena M.C., Strangman N.M., Mackie K., et al. CB1 receptor localization in rat spinal cord and roots, dorsal root ganglion and peripheral nerve. Acta Pharmacologica Sinica. 1999;20:1115–1120.
Schatz A.R., Lee M., Condie R.B., et al. Cannabinoid receptors CB1 and CB2: a characterization of expression and adenylate cyclase modulation within the immune system. Toxicology and Applied Pharmacology. 1997;142:278–287.
Schlosburg J.E., Blankman J.L., Long J.Z., et al. Chronic monoacylglycerol lipase blockade causes functional antagonism of the endocannabinoid system. Nature Neuroscience. 2010;13:1113–1119.
Schuelert N., Zhang C., Mogg A.J., et al. Paradoxical effects of the cannabinoid CB2 receptor agonist GW405833 on rat osteoarthritic knee joint pain. Osteoarthritis and Cartilage. 2010;18:1536–1543.
Sciolino N.R., Zhou W., Hohmann A.G. Enhancement of endocannabinoid signaling with JZL184, an inhibitor of the 2-arachidonoylglycerol hydrolyzing enzyme monoacylglycerol lipase, produces anxiolytic effects under conditions of high environmental aversiveness in rats. Pharmacological Research. 2011;64:226–234.
Seidel K., Hamza M., Ates M., et al. Flurbiprofen inhibits capsaicin induced calcitonin gene related peptide release from rat spinal cord via an endocannabinoid dependent mechanism. Neuroscience Letters. 2003;338:99–102.
Selvarajah D., Gandhi R., Emery C.J., et al. Randomized placebo-controlled double-blind clinical trial of cannabis-based medicinal product (Sativex) in painful diabetic neuropathy. Diabetes Care. 2010;33:128–130.
Semple D.M., McIntosh A.M., Lawrie S.M. Cannabis as a risk factor for psychosis: systematic review. Journal of Psychopharmacology. 2005;19:187–194.
Shen M., Piser T.M., Seybold V.S., et al. Cannabinoid receptor agonists inhibit glutamatergic synaptic transmission in rat hippocampal cultures. Journal of Neuroscience. 1996;16:4322–4334.
Shire D., Carillon C., Kaghad M., et al, An amino-terminal variant of the central cannabinoid receptor resulting from alternative splicing. [published erratum appears in J Biol Chem 1996 Dec 27;271(52):33706]. Journal of Biological Chemistry. 1995;270:3726–3731.
Smart D., Gunthorpe M.J., Jerman J.C., et al. The endogenous lipid anandamide is a full agonist at the human vanilloid receptor (hVR1). British Journal of Pharmacology. 2000;129:227–230.
Sokal D.M., Elmes S.J.R., Kendall D.A., et al. Intraplantar injection of anandamide inhibits mechanically-evoked responses of spinal neurons via activation of CB2 receptors in anesthetized rats. Neuropharmacology. 2003;45:404–411.
Spradley J.M., Guindon J., Hohmann A.G. Inhibitors of monoacylglycerol lipase, fatty-acid amide hydrolase and endocannabinoid transport differentially suppress capsaicin-induced behavioral sensitization through peripheral endocannabinoid mechanisms. Pharmacological Research. 2010;62:249–258.
Staton P.C., Hatcher J.P., Walker D.J., et al. The putative cannabinoid receptor GPR55 plays a role in mechanical hyperalgesia associated with inflammatory and neuropathic pain. Pain. 2008;139:225–236.
Stella N., Schweitzer P., Piomelli D. A second endogenous cannabinoid that modulates long-term potentiation. Nature. 1997;388:773–778.
Strangman N.M., Walker J.M. The cannabinoid WIN 55,212-2 inhibits the activity-dependent facilitation of spinal nociceptive responses. Journal of Neurophysiology. 1999;81:472–477.
Sugiura T., Kondo S., Kishimoto S., et al. Evidence that 2-arachidonoylglycerol but not N-palmitoylethanolamine or anandamide is the physiological ligand for the cannabinoid CB2 receptor. Comparison of the agonistic activities of various cannabinoid receptor ligands in HL-60 cells. Journal of Biological Chemistry. 2000;275:605–612.
Sugiura T., Kondo S., Sukagawa A., et al. 2-Arachidonoylgylcerol: a possible endogenous cannabinoid receptor ligand in brain. Biochemical and Biophysical Research Communications. 1995;215:89–97.
Suplita R.L., 2nd., Farthing J.N., Gutierrez T., et al. Inhibition of fatty-acid amide hydrolase enhances cannabinoid stress-induced analgesia: sites of action in the dorsolateral periaqueductal gray and rostral ventromedial medulla. Neuropharmacology. 2005;49:1201–1209.
Svendsen K.B., Jensen T.S., Bach F.W. Does the cannabinoid dronabinol reduce central pain in multiple sclerosis? Randomised double blind placebo controlled crossover trial. British Medical Journal. 2004;329:253–257.
Tam J., Vemuri V.K., Liu J., et al. Peripheral CB1 cannabinoid receptor blockade improves cardiometabolic risk in mouse models of obesity. Journal of Clinical Investigation. 2010;120:2953–2966.
Tanimura A., Yamazaki M., Hashimotodani Y., et al. The endocannabinoid 2-arachidonoylglycerol produced by diacylglycerol lipase alpha mediates retrograde suppression of synaptic transmission. Neuron. 2010;65:320–327.
Topol E.J., Bousser M.G., Fox K.A., et al. Rimonabant for prevention of cardiovascular events (CRESCENDO): a randomised, multicentre, placebo-controlled trial. Lancet. 2010;376:517–523.
Tsou K., Brown S., Sañuedo-Peña M.C., et al. Immunohistochemical distribution of cannabinoid CB1 receptors in the rat central nervous system. Neuroscience. 1998;83:393–411.
Tsou K., Lowitz K.A., Hohmann A.G., et al. Suppression of noxious stimulus–evoked expression of FOS protein–like immunoreactivity in rat spinal cord by a selective cannabinoid agonist. Neuroscience. 1996;70:791–798.
Valenzano K.J., Tafesse L., Lee G., et al. Pharmacological and pharmacokinetic characterization of the cannabinoid receptor 2 agonist, GW405833, utilizing rodent models of acute and chronic pain, anxiety, ataxia and catalepsy. Neuropharmacology. 2005;48:658–672.
Van Sickle M.D., Duncan M., Kingsley P.J., et al. Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science. 2005;310:329–332.
Vaughan C.W., Connor M., Bagley E.E., et al. Actions of cannabinoids on membrane properties and synaptic transmission in rat periaqueductal gray neurons in vitro. Molecular Pharmacology. 2000;57:288–295.
Vaughan C.W., McGregor I.S., Christie M.J. Cannabinoid receptor activation inhibits GABAergic neurotransmission in rostral ventromedial medulla neurons in vitro. British Journal of Pharmacology. 1999;127:935–940.
Vera G., Chiarlone A., Cabezos P.A., et al. WIN 55,212-2 prevents mechanical allodynia but not alterations in feeding behaviour induced by chronic cisplatin in the rat. Life Sciences. 2007;81:468–479.
Walker J.M., Hohmann A.G. Cannabinoid mechanisms of pain suppression. Handbook of Experimental Pharmacology. 2005;168:509–554.
Walker J.M., Huang S.M., Strangman N.M., et al. Pain modulation by release of the endogenous cannabinoid anandamide. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:12198–12203.
Walker L.A., Harland E.C., Best A.M., et al. Delta 9 THC hemisuccinate in suppository form and an alternative to oral and smoked THC. In: Nahas G.G., Sutin K.M., Harvey D.J., et al, eds. Marihuana and medicine. Totowa, NJ: Humana Press; 1999:123–135.
Wallace V.C., Blackbeard J., Segerdahl A.R., et al. Characterization of rodent models of HIV-gp120 and anti-retroviral–associated neuropathic pain. Brain. 2007;130:2688–2702.
Wallace V.C., Cottrell D.F., Brophy P.J., et al. Focal lysolecithin-induced demyelination of peripheral afferents results in neuropathic pain behavior that is attenuated by cannabinoids. Journal of Neuroscience. 2003;23:3221–3233.
Wallace V.C., Segerdahl A.R., Lambert D.M., et al. The effect of the palmitoylethanolamide analogue, palmitoylallylamide (L-29) on pain behaviour in rodent models of neuropathy. British Journal of Pharmacology. 2007;151:1117–1128.
Walter L., Franklin A., Witting A., et al. Nonpsychotropic cannabinoid receptors regulate microglial cell migration. Journal of Neuroscience. 2003;23:398–1405.
Walter L.A., Franklin A., Myhre A., et al. Microglial cells produce endocannabinoids and express functional cannabinoid receptors. Society of Neuroscience Abstracts. 634:616, 2001.
Wang T., Collet J.-P., Shapiro S., et al. Adverse effects of medical cannabinoids: a systematic review. CMAJ: Canadian Medical Association Journal. 2008;178:1669–1678.
Ware M.A., Wang T., Shapiro S., et al. Smoked cannabis for chronic neuropathic pain: a randomized controlled trial. CMAJ: Canadian Medical Association Journal. 2010;182:E694–E701.
Watanabe H., Vriens J., Prenen J., et al. Anandamide and arachidonic acid use epoxyeicosatrienoic acids to activate TRPV4 channels. Nature. 2003;424:434–438.
Watkins L.R., Maier S.F. Glia: a novel drug discovery target for clinical pain. Nature Reviews Drug Discovery. 2003;2:973–985.
Wilsey B., Marcotte T., Tsodikov A., et al. A randomized, placebo-controlled, crossover trial of cannabis cigarettes in neuropathic pain. Journal of Pain. 2008;9:506–521.
Wilson R.I., Nicoll R.A. Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature. 2001;410:588–592.
Yoshida T., Hashimoto K., Zimmer A., et al. The cannabinoid CB1 receptor mediates retrograde signals for depolarisation-induced suppression of inhibition in cerebellar Purkinje cells. Journal of Neuroscience. 2002;22:1690–1697.
Yu X.H., Cao C.Q., Martino G., et al. A peripherally restricted cannabinoid receptor agonist produces robust anti-nociceptive effects in rodent models of inflammatory and neuropathic pain. Pain. 2010;151:337–344.
Zammit S., Allebeck P., Andreasson S., et al. Self reported cannabis use as a risk factor for schizophrenia in Swedish conscripts of 1969: historical cohort study. British Medical Journal. 2002;325:1199–2005.
Zhang J., Hoffert C., Vu H.K., et al. Induction of CB2 receptor expression in the rat spinal cord of neuropathic but not inflammatory chronic pain models. European Journal of Neuroscience. 2003;17:2750–2754.
Zimmer A., Zimmer A.M., Hohmann A.G., et al. Increased mortality, hypoactivity, and hypoalgesia in cannabinoid CB1 receptor knockout mice. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:5780–5785.
Abrams D.I., Jay C.A., Shade S.B., et al. Cannabis in painful HIV-associated sensory neuropathy: a randomized placebo-controlled trial. Neurology. 2007;68:515–521.
Agarwal N., Pacher P., Tegeder I., et al. Cannabinoids mediate analgesia largely via peripheral type 1 cannabinoid receptors in nociceptors. Nature Neuroscience. 2007;10:870–879.
Bridges D., Rice A.S., Egertová M., et al. Localisation of cannabinoid receptor 1 in rat dorsal root ganglion using in situ hybridisation and immunohistochemistry. Neuroscience. 2003;119:803–812.
Calignano A., La Rana G., Giuffrida A., et al. Control of pain initiation by endogenous cannabinoids. Nature. 1998;394:277–281.
Caspi A., Moffitt T.E., Cannon M., et al. Moderation of the effect of adolescent-onset cannabis use on adult psychosis by a functional polymorphism in the catechol-O-methyltransferase gene: longitudinal evidence of a gene X environment interaction. Biological Psychiatry. 2005;57:1117–1127.
Clapper J.R., Moreno-Sanz G., Russo R., et al. Anandamide suppresses pain initiation through a peripheral endocannabinoid mechanism. Nature Neuroscience. 2010;13:1265–1270.
Cravatt B.F., Demarest K., Patricelli M.P., et al. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:9371–9376.
Cravatt B.F., Saghatelian A., Hawkins E.G., et al. Functional disassociation of the central and peripheral fatty acid amide signaling systems. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:10821–10826.
Diatchenko L., Slade G.D., Nackley A.G., et al. Genetic basis for individual variations in pain perception and the development of a chronic pain condition. Human Molecular Genetics. 2005;14:135–143.
Dinh T.P., Carpenter D., Leslie F.M., et al. Brain monoglyceride lipase participating in endocannabinoid inactivation. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:10819–10824.
Fox A., Kesingland A., Gentry C., et al. The role of central and peripheral cannabinoid-1 receptors in the antihyperalgesic activity of cannabinoids in a model of neuropathic pain. Pain. 2001;92:91–100.
Gregg L.C., Jung K.M., Spradley J.M., et al. Activation of type-5 metabotropic glutamate receptors and diacylglycerol lipase-α initiates 2-arachidonoylglycerol formation and endocannabinoid-mediated analgesia. The Journal of Neuroscience. 2012;32:9457–9468.
Gao Y., Vasilyev D.V., Goncalves M.B., et al. Loss of retrograde endocannabinoid signaling and reduced adult neurogenesis in diacylglycerol lipase knock-out mice. Journal of Neuroscience. 2010;30:2017–2024.
Guindon J., Guijarro A., Piomelli D., et al. Peripheral antinociceptive effects of inhibitors of monoacylglycerol lipase in a rat model of inflammatory pain. British Journal of Pharmacology. 2011;163:1464–1478.
Guindon J., Hohmann A.G. Cannabinoid CB2 receptors: a therapeutic target for the treatment of inflammatory and neuropathic pain. British Journal of Pharmacology. 2008;153:319–334.
Guindon J., Hohmann A.G. The endocannabinoid system and pain. CNS & Neurological Disorders Drug Targets. 2009;8:403–421.
Guindon J., Hohmann A.G. The endocannabinoid system and cancer: therapeutic implications. British Journal of Pharmacology. 2011;163:1447–1463.
Hasnie F.S., Breuer J., Parker S., et al. Further characterization of a rat model of varicella zoster virus–associated pain: relationship between mechanical hypersensitivity and anxiety-related behavior, and the influence of analgesic drugs. Neuroscience. 2007;144:1495–1508.
Herkenham M., Lynn A.B., Johnson M.R., et al. Characterization and localization of cannabinoid receptors in rat brain: a quantitative in vitro autoradiographic study. Journal of Neuroscience. 1991;11:563–583.
Herzberg U., Eliav E., Bennett G.J., et al. The analgesic effects of R(+)-WIN 55,212-2 mesylate, a high affinity cannabinoid agonist, in a rat model of neuropathic pain. Neuroscience Letters. 1997;221:157–160.
Hill M.N., Gorzalka B.B. Impairments in endocannabinoid signaling and depressive illness. JAMA: The Journal of the American Medical Association. 2009;301:1165–1166.
Hohmann A.G., Suplita R.L., Bolton N.M., et al. An endocannabinoid mechanism for stress-induced analgesia. Nature. 2005;435:1108–1112.
Ibrahim M.M., Deng H., Zvonok A., et al. Activation of CB2 cannabinoid receptors by AM1241 inhibits experimental neuropathic pain: pain inhibition by receptors not present in the CNS. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:10529–10533.
Jung K.M., Astarita G., Zhu C., et al. A key role for diacylglycerol lipase-alpha in metabotropic glutamate receptor–dependent endocannabinoid mobilization. Molecular Pharmacology. 2007;72:612–621.
Katona I., Freund T.F. Endocannabinoid signaling as a synaptic circuit breaker in neurological disease. Nature Medicine. 2008;14:923–930.
Khasabova I.A., Khasabov S.G., Harding-Rose C., et al. A decrease in anandamide signaling contributes to the maintenance of cutaneous mechanical hyperalgesia in a model of bone cancer pain. Journal of Neuroscience. 2008;28:11141–11152.
Kinsey S.G., Long J.Z., Cravatt B.F., et al. Fatty acid amide hydrolase and monoacylglycerol lipase inhibitors produce anti-allodynic effects in mice through distinct cannabinoid receptor mechanisms. Journal of Pain. 2010;11:1420–1428.
Lever I.J., Robinson M., Cibelli M., et al. Localization of the endocannabinoid-degrading enzyme fatty acid amide hydrolase in rat dorsal root ganglion cells and its regulation after peripheral nerve injury. Journal of Neuroscience. 2009;29:3766–3780.
Martin W.J., Hohmann A.G., Walker J.M. Suppression of noxious stimulus–evoked activity in the ventral posterolateral nucleus of the thalamus by a cannabinoid agonist: correlation between electrophysiological and antinociceptive effects. Journal of Neuroscience. 1996;16:6601–6611.
Meng I.D., Manning B.H., Martin W.J., et al. An analgesia circuit activated by cannabinoids. Nature. 1998;395:381–383.
Mitrirattanakul S., Ramakul N., Guerrero A.V., et al. Site-specific increases in peripheral cannabinoid receptors and their endogenous ligands in a model of neuropathic pain. Pain. 2006;126:102–114.
Nackley A.G., Makriyannis A., Hohmann A.G. Selective activation of cannabinoid CB2 receptors suppresses spinal fos protein expression and pain behavior in a rat model of inflammation. Neuroscience. 2003;119:747–757.
Nyilas R., Gregg L.C., Mackie K., et al. Molecular architecture of endocannabinoid signaling at nociceptive synapses mediating analgesia. European Journal of Neuroscience. 2009;29:1964–1978.
Olango W.M., Roche M., Ford G.K., et al. The endocannabinoid system in the rat dorsolateral periaqueductal grey mediates fear-conditioned analgesia and controls fear expression in the presence of nociceptive tone. British Journal of Pharmacology. 2012;165:2549–2560.
Phillips T.J.C., Cherry C.L., Cox S., et al. A metaanalysis of randomised controlled clinical trials assessing the effectiveness of pharmacological interventions for painful HIV-associated sensory neuropathy. European Journal of Pain Supplements. 4, 2010. 141–141
Racz I., Nadal X., Alferink J., et al. Crucial role of CB(2) cannabinoid receptor in the regulation of central immune responses during neuropathic pain. Journal of Neuroscience. 2008;28:12125–12135.
Racz I., Nadal X., Alferink J., et al. Interferon-gamma is a critical modulator of CB(2) cannabinoid receptor signaling during neuropathic pain. Journal of Neuroscience. 2008;28:12136–12145.
Rahn E.J., Hohmann A.G. Cannabinoids as pharmacotherapies for neuropathic pain: from the bench to the bedside. Neurotherapeutics. 2009;6:713–737.
Rice A.S., Farquhar-Smith W.P., Nagy I. Endocannabinoids and pain: spinal and peripheral analgesia in inflammation and neuropathy. Prostaglandins, Leukotrienes, and Essential Fatty Acids. 2002;66:243–256.
Rog D.J., Nurmikko T.J., Friede T., et al. Randomized, controlled trial of cannabis-based medicine in central pain in multiple sclerosis. Neurology. 2005;65:812–819.
Schlosburg J.E., Blankman J.L., Long J.Z., et al. Chronic monoacylglycerol lipase blockade causes functional antagonism of the endocannabinoid system. Nature Neuroscience. 2010;13:1113–1119.
Spradley J.M., Guindon J., Hohmann A.G. Inhibitors of monoacylglycerol lipase, fatty-acid amide hydrolase and endocannabinoid transport differentially suppress capsaicin-induced behavioral sensitization through peripheral endocannabinoid mechanisms. Pharmacological Research. 2010;62:249–258.
Tanimura A., Yamazaki M., Hashimotodani Y., et al. The endocannabinoid 2-arachidonoylglycerol produced by diacylglycerol lipase alpha mediates retrograde suppression of synaptic transmission. Neuron. 2010;65:320–327.
Valenzano K.J., Tafesse L., Lee G., et al. Pharmacological and pharmacokinetic characterization of the cannabinoid receptor 2 agonist, GW405833, utilizing rodent models of acute and chronic pain, anxiety, ataxia and catalepsy. Neuropharmacology. 2005;48:658–672.
Wallace V.C., Cottrell D.F., Brophy P.J., et al. Focal lysolecithin-induced demyelination of peripheral afferents results in neuropathic pain behavior that is attenuated by cannabinoids. Journal of Neuroscience. 2003;23:3221–3233.
Ware M.A., Wang T., Shapiro S., et al. Smoked cannabis for chronic neuropathic pain: a randomized controlled trial. CMAJ: Canadian Medical Association Journal. 2010;182:E694–E701.
Yu X.H., Cao C.Q., Martino G., et al. A peripherally restricted cannabinoid receptor agonist produces robust anti-nociceptive effects in rodent models of inflammatory and neuropathic pain. Pain. 2010;151:337–344.
Zhang J., Hoffert C., Vu H.K., et al. Induction of CB2 receptor expression in the rat spinal cord of neuropathic but not inflammatory chronic pain models. European Journal of Neuroscience. 2003;17:2750–2754.
Zimmer A., Zimmer A.M., Hohmann A.G., et al. Increased mortality, hypoactivity, and hypoalgesia in cannabinoid CB1 receptor knockout mice. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:5780–5785.