Painful Peripheral Neuropathies
Introduction
Peripheral nerves may be affected in a number of different ways by a great variety of diseases. Motor, autonomic, or sensory fibers may be preferentially affected, but in most neuropathies all components of the peripheral nervous system are involved, which leads to various patterns of sensorimotor deficit and autonomic dysfunction. Neuropathies affecting motor or sensory neurons are frequently accompanied by positive sensory symptoms that usually take the form of fasciculations or paresthesias, which are not particularly troublesome and are overshadowed by the symptoms of sensory or motor deficits. This reflects the common clinical experience that most lesions of the peripheral nervous system do not produce chronic pain. However, there are neuropathies in which pain and severe paresthesias are typical and troublesome features and in which pain rather than the neurological deficit is the predominant clinical complaint. In these neuropathies, ongoing and stimulus-evoked pain may be the initial and most severe continuing symptoms. Conditions in which damage to the nervous system causes pain may seem paradoxical in that impairment of nerve fibers carrying nociceptive information in the peripheral or central nervous system should result in a decrease in pain sensibility (hypo- or analgesia). Thus, the presence of pain after neural injury implies qualitative changes in the neurobiological mechanisms encoding pain. In fact, it is one of the puzzles of pain that lesions in the peripheral and central pathways normally signaling pain rather than those subserving non-nociceptive functions are the culprits of neuropathic pain. This chapter is concerned with these neuropathies, which include many poly- and mononeuropathies.
Pain in Peripheral Neuropathy
Definition of Neuropathic Pain
Neuropathic pain is never a complete diagnosis in its own right but is always a symptom of an underlying neurological disease. From a historical perspective, it is interesting to reflect that the terms neuralgia and neuropathic pain were coined to describe pain of peripheral nerve origin. However, it has long been recognized that these definitions are often inappropriately used, a sentiment expressed by Robert Wartenberg (1958) more than 5 decades ago: “The indiscriminate use of the term neuralgia to designate almost every undeterminable, and often not neurogenic, painful affection is a menace to exact medical diagnosis.” Recently, a redefinition of neuropathic pain has been endorsed by the International Association for the Study of Pain (IASP) (Jensen 2011). In the new definition, neuropathic pain is defined as “pain caused by a lesion or disease of the somatosensory system.” This supersedes previous IASP definitions and, importantly, removes it from the ill-defined term of “dysfunction of the nervous system.” The new, more restrictive definition of neuropathic pain provides a welcomed increase in diagnostic stringency. However, its major weakness is the associated grading system of diagnostic certainty. Under this grading system, classic trigeminal neuralgia does not fulfill the criteria for neuropathic pain, whereas many non-neuropathic conditions could still be categorized as neuropathic pain by virtue of an altered von Frey hair detection threshold in the skin (Treede et al 2008).
It is common clinical experience that patients with neuropathic pain have significant co-morbid conditions and that these conditions have an important impact on the global pain experience. Psychological factors such as changes in mood, anxiety, and altered sleep patterns have all been identified as significant adjuncts of painful neuropathies, and in addition there may be social isolation and reduced employment status (Meyer-Rosberg et al 2001). Approximately 60% of patients report at least discomfort as a result of difficulty sleeping, and moderate to severe depression is present in a third of patients and anxiety in a quarter (Fig. 65-1). Newly referred neurology outpatients with pain have a high prevalence of depression, pain is more likely to persist in patients with depression, and depression is more likely to persist in those with co-existent pain (Williams et al 2004).
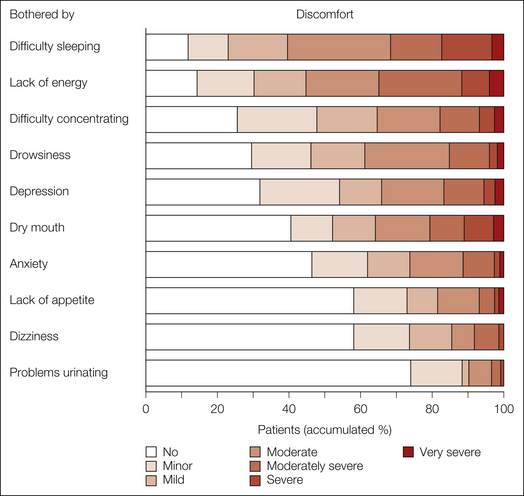
Figure 65-1 Frequency of the extent to which patients had been bothered by symptoms related to pain or the adverse effects of treatment during the previous 7 days in categories of increasing discomfort (from 1, no discomfort, to 7, very severe discomfort).
Eighty-eight percent of the patients reported some degree of discomfort. (Reproduced from Meyer-Rosberg K, Kvarnstrom A, Kinnman E, et al 2001 Peripheral neuropathic pain—a multidimensional burden for patients. European Journal of Pain 5:379–389. Copyright 2001 International Association for the Study of Pain.)
Diseases of the nervous system commonly cause a variety of secondary consequences such as skeletal deformity, arthropathy, and musculoskeletal changes, all of which may be painful. In these circumstances the pain is likely to be nociceptive in type. Finally, the disease causing the neuropathy may, as in the case of diabetes mellitus, lead to a host of non-neuropathic types of pain, particularly those of vascular, joint, and skin origin. In individual patients it may be clinically difficult to differentiate between the relative contribution of these secondary factors to the overall burden of pain. This underlines the need for careful clinical assessment of all patients with pain and a neuropathy; the pain may not be of a neuropathic type and will demand appropriate investigations and treatment.
A number of questionnaires and pain scales have been devised to assist in the recognition and diagnosis of neuropathic pain, including the Neuropathic Pain Scale (NPS) (Galer and Jensen 1997), the Leeds Assessment of Neuropathic Symptoms and Signs (LANSS) (Bennett, 2001), the Neuropathic Pain Questionnaire (NPQ) (Krause and Backonja 2003), the Douleur Neuropathique en 4 questions (DN4) (Bouhassira et al 2005), painDETECT (Freynhagen et al 2006), and ID-Pain (Portenoy 2006). These instruments can be useful in formalizing the assessment of symptoms and signs and may be a helpful alert for the non-specialist to consider the possibility of a condition causing neuropathic pain in the differential diagnosis of a pain state. Although these instruments are often hailed as being “diagnostic,” they are in fact not and can even be positively misleading in non-painful neuropathies such as Charcot–Marie–Tooth (CMT) disease type 1, where the pain is often of musculoskeletal origin and can frequently co-exist in regions of reported numbness, mild painless paresthesias, and hypoesthesia to touch or pinprick. Therefore, questionnaires are not a shortcut and no substitute for the traditional approach to neurological diagnosis. A positive score on a questionnaire is therefore a potential starting point and not the end of the diagnostic process. In other words, if a pain is thought to be neuropathic in type, a neurological diagnosis must be sought.
Classification of Painful Neuropathies
There are different classification schemes for painful peripheral neuropathies. The anatomical distribution pattern of the affected nerves provides valuable differential diagnostic clues to possible underlying causes. It is therefore common clinical practice to group painful neuropathies into symmetrical polyneuropathies, diseases affecting many nerves simultaneously, typically in a length-related glove-and-stocking distribution; asymmetrical neuropathies with a mono- or multiplex distribution; or processes affecting the brachial or lumbosacral plexuses. Neuropathies commonly associated with pain are listed in Box 65-1.
The diversity of clinical conditions known to produce peripheral neuropathic pain is astonishing, and it has been difficult to identify a common denominator for the pain in conditions as heterogeneous as post-traumatic and diabetic neuropathy.
This has led to the development of a symptom-orientated diagnostic approach to neuropathic pain conditions that supplements the etiology-based classification scheme. It recognizes the fact that neuropathic pain is usually manifested as a composite of several distinct pain symptoms (Koltzenburg 1996, Woolf et al 1998). A symptom-orientated approach does not negate the fact that distinct neuropathies behave differently clinically and that some neuropathic disease states may predispose to certain constellations of pain symptoms (e.g., touch-evoked pain in post-herpetic neuralgia [PHN]). The rationale of this approach recognizes several principles:
A symptom-based approach to painful neuropathies can be useful for dissecting the underlying neural mechanisms, and this knowledge may eventually be harnessed for the development of novel analgesic drugs that differentially target these mechanisms. However, a purely descriptive symptomatic approach to neuropathic pain that is not accompanied by a rigorous clinical differential neurological diagnostic assessment and appropriate investigations is negligent.
Epidemiology of Neuropathic Pain in Peripheral Neuropathy
Several studies have investigated the prevalence of neuropathies, but from these investigations it is often not possible to obtain information about the prevalence of pain. The difficulties surrounding the definition of neuropathic pain have compounded epidemiological surveys. Furthermore, the outcomes of epidemiological surveys obviously depend on the tools (questionnaires, clinical examination, outcome of appropriate investigations) that are used to make a diagnosis. Neuropathies represent one of the most common neurological disorders, with a prevalence of 2.4% in the general population that rises to 8% with age (Martyn and Hughes 1998). Estimates of the point prevalence of neuropathic pain in the general population are as high as 5–7% (Daousi et al 2004, Bouhassira 2008, Institute of Medicine 2011 http://download.nap.edu/catalog.php?record_id=13172).
A complicating factor is that even within an etiologic entity such as diabetic polyneuropathy, chronic neuropathic pain develops in only a minority of patients. Thus, the point prevalence of chronic painful peripheral polyneuropathy is on the order of 10–20% of patients with diabetes mellitus (Daousi et al 2004). Moreover, it is still unclear why the symptoms within an etiologically defined population of patients can be extremely diverse. In fact, it is one of the challenges of the field to understand the factors that determine why painful symptoms develop in some individuals only with apparently identical conditions and what determines the range of the initial symptoms.
Range of Painful Symptoms and Signs in Peripheral Neuropathies
No single symptom or sign is pathognomonic for neuropathic pain. The symptoms may be divided into those that are unprovoked (stimulus independent) and those that are provoked by maneuvers (stimulus induced) such as skin stimulation, pressure over affected nerves, or changes in temperature. The term deafferentation syndrome is often used for diseases characterized by extensive and complete disconnection of peripheral nerves from their target, such as with amputations or plexus lesions. In these situations the pain is stimulus independent. In contrast, when the connections in the periphery are partially retained or are at the borders of a completely deafferentated zone, the pain often has stimulus-independent and stimulus-induced components.
Many terms may be used by patients with neuropathies to describe their painful neuropathic sensations. Because some neuropathic pain symptoms are not experienced in normal situations, patients sometimes use bizarre and ornate verbal descriptors. The most commonly described spontaneous symptoms are deep aching in the extremities and a superficial burning, stinging, or prickling pain. Verbal descriptors from the McGill Pain Questionnaire that are used significantly more frequently by patients with neuropathic pain than by those with non-neuropathic pain include “electric shocks,” “burning,” “tingling,” “itching,” or “prickling,” whereas descriptions such as “dull,” “heavy,” and “tiring” are more often reported by patients with non-neuropathic pain (Boureau et al 1990). Patients also report paroxysmal, shock-like lancinating pain, sometimes radiating through an entire limb. Signs of cutaneous hypersensitivity such as touch-evoked pain are more common in neuropathic pain states than in non-neuropathic pain states (Bennett 2001, Rasmussen et al 2004). In addition, investigations of patients suffering from different neuropathies demonstrate patterns of sensory abnormalities. Table 65-1 shows that the symptoms and signs can be similar in neuropathic pain conditions, such as painful polyneuropathy and PHN, but that there can also be substantial differences when comparing these conditions with non-freezing cold injury (NFCI; also known as trench foot neuropathy) or complex regional pain syndrome (CRPS). However, the diagnosis of neuropathic pain on the basis of the patient’s description of symptoms in the absence of confirmatory findings on clinical examination and investigations is often not possible. One study of patients who were referred to a tertiary neurological center with the suspected diagnosis of neuropathic pain concluded that superficial ongoing pain and brush-evoked pain, but not cold or pinprick hyperalgesia, were more frequently found in patients with definitive and probable neuropathic pain than in patients unlikely to have neuropathic pain (Rasmussen et al 2004). In the following descriptions of the different neuropathies, the major painful complaints typical of each condition are given, but it should be emphasized that within a single etiological or pathological diagnostic category, considerable variation in symptoms occurs in different individuals.
Table 65-1
Symptoms and Signs of Neuropathic Pain
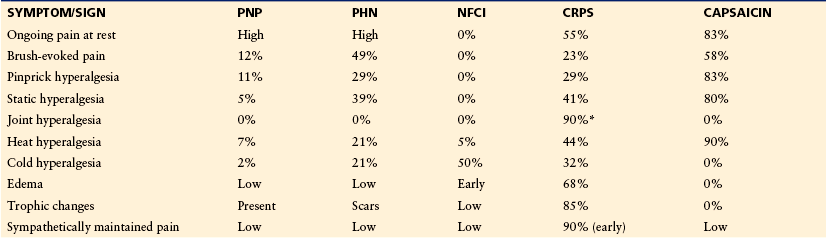
CRPS, complex regional pain syndrome; NFCI, non-freezing cold injury; PHN, post-herpetic neuralgia; PNP, peripheral neuropathic pain.
Data from Gierthmuhlen J, Maier C, Baron R, et al 2012 Sensory signs in complex regional pain syndrome and peripheral nerve injury. Pain 153:765–774; Liu M, Max MB, Robinovitz E, et al 1998 The human capsaicin model of allodynia and hyperalgesia: sources of variability and methods for reduction. Journal of Pain and Symptom Management 16:10–20; Maier C, Baron R, Tölle T R, et al 2010 Quantitative sensory testing in the German Research Network on Neuropathic Pain (DFNS): somatosensory abnormalities in 1236 patients with different neuropathic pain syndromes. Pain 150:439–450; and unpublished data from Jørum (NFCI) and Baron (CRPS).
Type of Fibers Affected in Painful Neuropathies
The relationship of painful symptoms to morphological and electrophysiological changes in peripheral nerves has been a subject of interest for many decades, particularly since the introduction of nerve biopsy and the development of clinical electrophysiological techniques (Dyck and Thomas 2005). Overall, the human evidence converges on several principles:
Neuropathies involving small fibers, with or without large-fiber involvement, are often painful. Many studies converge to suggest that axonal injury involving a portion of the nociceptive fibers in a peripheral nerve is the single most important causal factor for neuropathic pain:
Pathophysiological Findings in Patients with Neuropathic Pain
Although it is clear that changes in central nervous system properties are crucial for the development of painful symptoms in peripheral neuropathy, several lines of independent evidence converge to indicate that changes in the excitability of primary nociceptive afferents are the single most important factor in the generation and maintenance of chronic neuropathic pain in humans. These experimental studies in patients are entirely in agreement with the neuropathological observations mentioned earlier that implicate small-diameter primary afferent fibers as the primary culprits in peripheral neuropathic pain:
The evidence from microneurographic investigations generally supports the notion of an abnormal sensitivity of primary sensory neurons in patients with painful nerve lesions by demonstrating abnormal activity and reduced thresholds in cutaneous afferents (Ochoa et al 1982, Nordin et al 1984, Cline et al 1989, Ochs et al 1989, Burchiel and Baumann 2004). Abnormal ectopic activity in myelinated mechanosensitive fibers has been recorded in patients with traumatic nerve lesions, entrapment neuropathies, and radiculopathies (Fig. 65-2). A classic study by Nyström and Hagbarth (1981) provided evidence that ectopic excitation can occur at multiple sites in damaged sensory neurons. Ongoing activity and mechanical sensitivity were recorded proximal to a nerve neuroma in an amputee with phantom limb pain. Following local anesthetic blockade of the nerve distal to the recording site, impulses evoked by mechanical stimulation of the neuroma were abolished, but ongoing activity at the recording site continued, thus suggesting that this residual activity arose from the dorsal root ganglion (DRG) (Fig. 65-3).
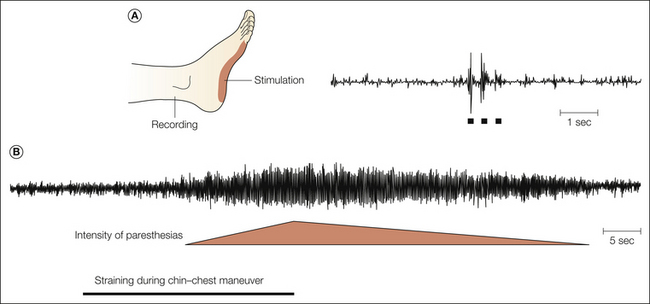
Figure 65-2 Microneurographic multiunit recording from the sural nerve in a patient with compression of the S1 spinal root by a herniated disc.
A, Excitation of mechanosensitive units in the receptive field by tactile stimulation (bars). B, Straining- and chin–chest maneuver–provoked paresthesias and ectopic discharge of afferent fibers originating from the compressed root. (After Nordin M, Nyström B, Wallin U, et al 1984 Ectopic sensory discharges and paresthesiae in patients with disorders of peripheral nerves, dorsal roots and dorsal columns. Pain 20:231–245. Copyright 1984 Elsevier Ltd.)
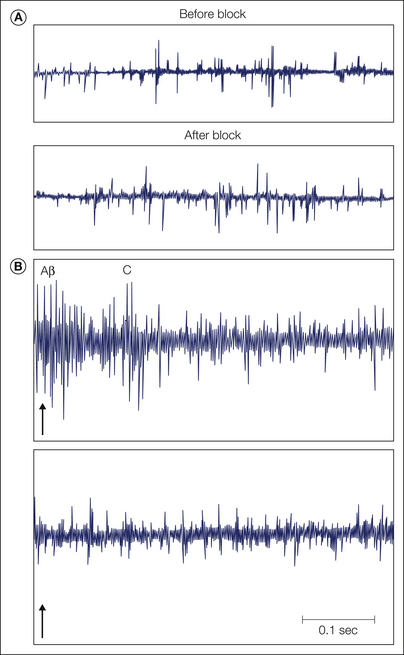
Figure 65-3 Microneurographic multi-unit recording from a skin fascicle of the median nerve at the wrist in a patient with a hand amputation and phantom pain in the hand and fingers that was accentuated by taps on a stump neuroma.
A, Single sweeps showing spontaneous impulse activity (left) that is uninfluenced by local anesthetic block of the neuroma (right). Sustained phantom pain was also uninfluenced. B, Superimposed neural responses evoked by taps (arrows). Units with Aβ- and C-fiber latency were excited by tapping the painful neuroma. This response (arrow) was eliminated by the local anesthetic block (right), which also abolished tap-induced accentuation of the phantom pain. (After Nyström B, Hagbarth KE 1981 Microelectrode recordings from transected nerves in amputees with phantom limb pain. Neuroscience Letters 27:211–216. Copyright 1981 Elsevier.)
Microneurographic studies of unmyelinated nociceptors in patients with polyneuropathy have shown that individuals with pain have a significantly higher percentage of ongoing activity than do those without pain (Kleggetveit 2012). This difference was particularly pronounced in the population of mechanically insensitive nociceptors, which are thought to be important for central sensitization. Furthermore, nociceptors with ongoing activity were much more likely to discharge multiple times after a single electrical stimulation, a response that is unusual in healthy subjects or patients with painless neuropathies (Serra et al 2012, Schmidt et al 2012). This abnormal spiking with natural stimulation provides an explanation for the hyperalgesia in these patients.
Abnormal nociceptors have also been recorded in patients suffering from erythermalgia (also known as erythromelalgia), a condition characterized by painful, red, hot extremities. Nociceptors displayed ongoing activity that is not normally observed in nociceptors, and there was evidence of sensitization of mechanically insensitive afferents to non-painful tactile stimuli (Ørstavik et al 2003) (Fig. 65-4). The inherited form is caused by a gain-of-function mutation in the gene encoding the voltage-gated sodium channel Nav1.7 (Yang et al 2004, Dib-Hajj et al 2010). In heterologous expression systems the mutations can produce a hyperpolarizing shift in activation and a slowing of deactivation (Cummins et al 2004) (Fig. 65-5).
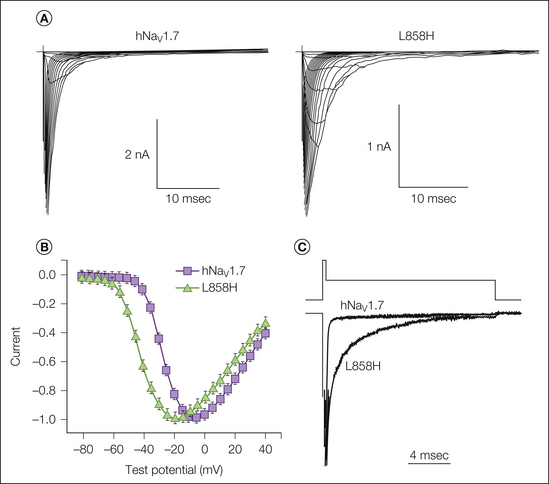
Figure 65-4 The L858H mutation of human Nav1.7 (hNav1.7) alters activation and deactivation properties.
A, Current traces recorded in whole-cell patch-clamp recordings from HEK293 cells expressing either wild-type hNav1.7 or the mutant channel L858H. Cells were held at −100 mV, and currents were elicited with 50-msec test pulses to potentials ranging from −80 to 40 mV. B, Normalized peak current–voltage relationship for wild-type (squares) and L858H (triangles) channels. C, Representative tail currents of wild-type and L858H channels. Cells were held at −100 mV and depolarized to −20 mV for 0.5 msec, followed by repolarization to −50 mV to elicit tail currents. (Adapted with permission from Cummins TR, Dib-Hajj SD, Waxman SG 2004 Electrophysiological properties of mutant Nav1.7 sodium channels in a painful inherited neuropathy. Journal of Neuroscience 24:8232–8236. Copyright 2004 by the Society for Neuroscience.)
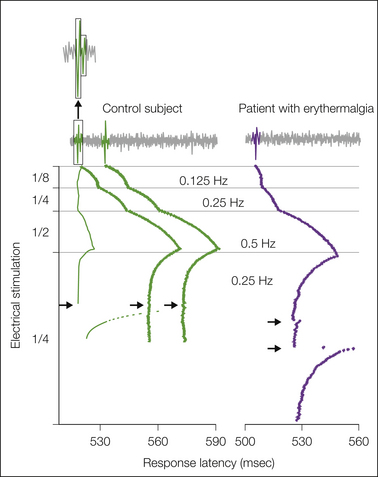
Figure 65-5 Microneurographic C-fiber recordings from the peroneal nerves of a normal volunteer (left) and a patient suffering from erythermalgia (right).
The first trace shows the original nerve signal. The subsequent recordings are a falling leaf display in which each action potential is symbolized by a series of dots. Each row of dots represents the latency of its receptive field in the skin at different electrical stimulation frequencies of the fiber. Simultaneous recording of three afferent C fibers in a control subject shows two units with activity-dependent increases in latency at low-frequency stimulation and one unit that displays increases in latency when the excitation frequency increases to 1 impulse every 2 seconds. These biophysical features are characteristic of mechanically insensitive and mechanically sensitive nociceptive C fibers, respectively. Stimulation of the receptive field with a 750-mN von Frey hair (arrow) excited only the mechanosensitive C nociceptor, as evidenced by a strong shift in latency with electrical stimulation. Mechanically insensitive afferents are not activated by this stimulus. The right panel shows a C-fiber recording from a patient with erythermalgia. The unit had the biophysical properties of a mechanically insensitive C fiber but responded reproducibly to mechanical stimulation (arrows). This is consistent with the hypothesis that mechanically insensitive afferents become sensitized in erythermalgia. An alternative explanation would be that mechanically sensitive nociceptors start to display a different pattern of activity-dependent slowing of latency in this disease. (After Ørstavik K, Weidner C, Schmidt R, et al 2003 Pathological C-fibres in patients with a chronic painful condition. Brain 126:567–578. Copyright 2003 Oxford University Press.)
Stimulus-Induced Pain (Hyperalgesia)
Stimulus-induced pain is common in individuals with neuropathic pain. Most often patients report mechanical hyperalgesia followed by hyperalgesia to heat and cold. In neuropathic conditions the distinction between primary and secondary areas is less clearly defined than in the classic studies on tissue injury but probably corresponds to the tissue supplied by damaged nerves and the area outside this innervation territory. The mechanisms for the different symptoms appear to be distinct (Table 65-2).
Table 65-2
Mechanisms of Stimulus-Independent (Ongoing) and Stimulus-Induced Pains (Hyperalgesia) in Painful Neuropathies

Ongoing or ectopic activity in nociceptors will initiate central sensitization, which must therefore be considered part of the mechanism, and it in turn will initiate touch-evoked pain. There is the theoretical possibility that after tissue or nerve injury, non-nociceptive primary afferent fibers (Aβ mechanoreceptors, sensitive cold receptors) that physiologically have ongoing activity can add to the magnitude of pain when central sensitization is present.
Hyperalgesia to Heat
Hyperalgesia to heat is a hallmark of tissue injury, and this symptom occurs only occasionally in neuropathic conditions. The hyperalgesia to heat persists during differential nerve blocks of myelinated fibers, and microneurographic investigations have demonstrated chronic sensitization of nociceptors to heat (Cline et al 1989, Torebjörk 1990).
Sensitization to Catecholamines
Sensitization of nociceptors may prevail in sympathetically maintained pain, and although no hard epidemiological data are available, the number of patients with a predominant sympathetically maintained component of their pain is probably small. The importance of the sympathetic nervous system in the generation of pain has been the focus of a long, if controversial, debate. An explanation that is consistent with many experimental findings is that nociceptors acquire a sensitivity to catecholamines that permits abnormal excitation by either noradrenaline or circulating catecholamines. Two lines of evidence suggest that the ongoing pain can be caused or maintained by the sympathetic nervous system in selected patients. First, sympatholytic therapy can abolish pain and hyperalgesia (Loh and Nathan 1978, Bonica 1990, Campbell et al 1992). Second, in patients in whom sympatholytic therapy had provided pain relief, intracutaneous injection of adrenoceptor agonists can under certain conditions rekindle the ongoing pain and hyperalgesia (Davis et al 1991, Torebjörk et al 1995). Furthermore, injections of catecholamines around a stump neuroma can precipitate attacks of pain in humans (Chabal et al 1992). Since noradrenalin-induced pain occurs during a differential blockade of myelinated fibers, unmyelinated fibers appear to signal sympathetically maintained pain (Torebjörk et al 1995). This has been corroborated by direct microneurographic recordings of C fibers in a patient with sympathetically maintained pain, in whom activation of sympathetic efferents or injection of noradrenaline led to excitation of mechanically insensitive nociceptors (Jørum 2007).
Mechanical Hyperalgesia
The signs and symptoms of mechanical hyperalgesia in neuropathy are diverse, and at least three distinct types have been described in patients with neuropathic pain: (1) brush-evoked pain, (2) pinprick hyperalgesia, and (3) hyperalgesia to blunt pressure (Koltzenburg 1996). Even in an etiologically defined group of patients such as those with PHN, these symptoms can exist in various degrees (Pappagallo et al 2000) (see Table 65-1).
Touch-Evoked Pain: There is consensus that the touch-evoked pain in neuropathic conditions is signaled out of the skin by sensitive mechanoreceptors with large myelinated axons that normally encode non-painful tactile events (see Table 65-2). First, differential blockade of large myelinated non-nociceptive afferents abolishes brush-evoked pain (Campbell et al 1988, Gracely et al 1992, Koltzenburg et al 1994). Second, electrical stimulation (Price et al 1989, Torebjörk 1990) of these afferents causes painful dysesthesias. Third, reaction time measurements indicate that brush-evoked pain is signaled by fast conducting myelinated fibers (Fruhstorfer and Lindblom 1984). Finally, light punctate mechanical stimuli that can activate only sensitive mechanoreceptors are often called painful in patients with neuralgia (Price et al 1992, Koltzenburg et al 1994). The central sensitization that promotes brush-evoked pain in painful neuropathies requires ongoing excitation of nociceptors (Gracely et al 1992, Koltzenburg et al 1994) and involves N-methyl-D-aspartate (NMDA) receptors (Eide et al 1994).
Pinprick Hyperalgesia: Hyperalgesia to pinprick stimuli—typically elicited by probing the skin with a stiff von Frey hair—can be found in patients suffering from neuropathy (Pappagallo et al 2000). It is distinct from brush-evoked pain because of its different spatial and temporal profile and the fact that it is signaled by non-sensitized heat-insensitive Aδ nociceptors (LaMotte et al 1991, Koltzenburg et al 1992, Kilo et al 1994, Ziegler et al 1999). Although excitation of mechanically insensitive unmyelinated afferents is important for the initiation of this type of central sensitization, it is not required for sustaining it (LaMotte et al 1991, Kilo et al 1994).
Hyperalgesia to Blunt Pressure: Hyperalgesia to blunt pressure has also been described in patients with neuropathic pain (Ochs et al 1989, Price et al 1992, Ochoa and Yarnitsky 1993), and differential nerve block experiments suggest that this type of hyperalgesia is signaled by nociceptors in humans. One possible explanation for its generation is spatial summation of nociceptive input, which could be brought about by recruitment of mechanically insensitive nociceptors (Schmidt et al 2000) or expansion of the receptive field of mechanosensitive nociceptors (Schmelz et al 1994).
Hypersensitivity to Cold
Hypersensitivity to cold is particularly prominent after traumatic nerve injury (Wahren and Torebjörk 1992), but it can also be present in painful polyneuropathic conditions (Ochoa and Yarnitsky 1994) or in PHN (Pappagallo et al 2000). Cold hypersensitivity has been recognized as one of the major painful chronic sequelae of the so-called trench foot neuropathy, which is brought about by NFCI in the extremities (Thomas and Holdorff 1993, Irwin 1996). Another clinically important condition of acute cold intolerance is related to administration of the chemotherapeutic agent oxaliplatin, which is associated with aggravation of paresthesias and dysesthesias by cold (Ibrahim et al 2004, Park et al 2011).
Currently, three hypotheses have been advanced to explain the generation of cold hyperalgesia in neuropathic pain in humans.
Central Disinhibition
Because cold stimulation, through excitation of cold-sensitive thermoreceptive afferents, normally suppresses noxious stimuli on a central level (Craig 2003), selective loss or dysfunction of these afferents shifts the cold pain threshold to warmer temperatures (Wahren et al 1989, Yarnitsky and Ochoa 1990, Ochoa and Yarnitsky 1994).
Peripheral Sensitization
Psychophysical studies of human volunteers suggest that sensitization of cold-sensitive nociceptors can produce cold hyperalgesia (Wasner et al 2004) in normal volunteers. Microneurographic studies have shown that sensitization can occur in some patients (Serra et al 2009). Peripheral sensitization also appears to occur in the acute oxaliplatin-induced peripheral neuropathy (Lehky et al 2004, Park et al 2011).
Central Sensitization
Reaction time measurements (Fruhstorfer and Lindblom 1984, Lindblom 1994) and differential nerve block experiments (Torebjörk et al 1995) suggest that thin myelinated cold-sensitive afferents signal pain in some patients with neuropathy. The qualitative switch from signaling of innocuous cool sensations to cold pain by cold receptors could be analogous to the mechanisms that mediate brush-evoked pain signaled by large myelinated non-nociceptive fibers and involve NMDA receptor signaling (Jørum et al 2003).
Clinical Features and Investigations of Peripheral Neuropathies
The cause of some neuropathies is often quickly apparent clinically, supplemented by a few simple tests, and there may be no need for specialized investigation. Nevertheless, even after extensive investigation the cause of a substantial minority of neuropathies remains uncertain, and detailed discussion of the clinical diagnostic aspects of peripheral nerve disease and of specialized investigative techniques can be found elsewhere (Kimura 2001, Dyck and Thomas 2005). The brief account that follows provides an overview of the main currently available diagnostic procedures that complement the history and clinical examination. In addition, the diagnostic work-up of patients with a neuropathy often includes a range of appropriate investigations, including chemical pathology, cerebrospinal fluid analysis, and genetic testing.
Clinical Features
The cardinal clinical features of a peripheral neuropathy are weakness or wasting of the affected muscles, hypoesthesia, loss of or attenuated tendon reflexes, and impaired autonomic functions. It is often possible to demonstrate this with a careful neurological examination of patients with neuropathic pain. However, none of these findings is specific for peripheral nerve disease, and consequently the neurological examination is only the initial step in a diagnostic process that makes use of a range of appropriate investigations. Pain or hyperalgesia in the absence of neurological symptoms or signs can be caused by peripheral nerve disease, but such diagnosis has to be treated with considerable suspicion until investigations provide definitive confirmatory evidence of nerve involvement.
Nerve Conduction Studies, Electromyography, and Evoked Potentials
Clinical neurophysiological investigations are some of the most important investigations to confirm the diagnosis of a neuropathy (Kimura 2001). Nerve conduction studies in combination with electromyography (EMG) can make the important differentiation between demyelinating or axonal neuropathies and the presence or absence of conduction blocks. However, it is important to realize the limitations of these techniques. In routine practice, nerve conduction studies are restricted to the distal branches of a few major nerves in the extremities. Somatosensory evoked potentials (SEPs) or magnetic evoked potentials (MEPs) can be helpful in the diagnosis of proximal lesions. The main drawback of all these techniques is that they are in principle restricted to the assessment of large myelinated fibers, which are not the culprit in neuropathic pain. Laser-evoked potentials (LEPs) (Treede et al 2003), contact heat–evoked potentials (CHEPs) (Chen 2001, Chao 2008), and pain-related electrically evoked potentials (PREPs) (Katsarava et al 2006) are capable of testing the function of Aδ fibers. These techniques are often not available outside tertiary referral centers and do not allow topographical differentiation between peripheral and central lesions. Even though EMG is a sensitive technique for the detection of axonal lesions that can be applied to virtually all skeletal muscles, it provides information only about the motor system. Therefore, abnormal findings on neurophysiological examination can provide positive evidence of nerve disease, whereas normal findings on examination can exclude major nerve damage but cannot fully exclude minor nerve damage or selective damage to thin myelinated or unmyelinated sensory nerve fibers, which are the main source of peripheral neuropathic pain. Microneurography, the only technique that permits direct assessment of the function of small fibers in humans, is generally available just in the research setting.
Nerve Biopsy
Nerve biopsy is an important diagnostic tool to establish the cause of a neuropathy. However, the invasive nature and the suitability of only a few nerves in routine practice are the major limitations of this technique. The two major pathological processes in peripheral neuropathy are axonal degeneration and segmental demyelination. Division of polyneuropathies into either of these pathological categories is somewhat artificial since both processes are usually present, albeit in varying proportions. The findings of axonal degeneration, affecting distal parts of the axon, the cell body, or both, the details of segmental demyelination, remyelination, and onion bulb formation, and the pathological reactions of unmyelinated fibers in different neuropathies are described extensively elsewhere (Dyck and Thomas 2005). Some problems with morphological studies of standard nerve biopsy specimens are that examination of transverse sections by light microscopy will fail to recognize segmentally demyelinated axons and thus will underestimate the myelinated fiber population. An increase in the density of small fibers does not necessarily imply a selective loss of large fibers since regeneration will increase the population of small fibers. By relating axonal diameter to myelin sheath thickness by electron microscopic examination and by using certain criteria for differentiating the sprouts of myelinated and unmyelinated fibers, this problem can be overcome to some extent (Ochoa 1970). Unfortunately, not all studies of nerve specimens include electron microscopic examination, and this essentially precludes the analysis of unmyelinated neurons.
Very few electrophysiological and pharmacological studies have been performed on isolated human nerves, which have essentially remained research investigations. Lambert and Dyck (1993) took long multifascicular biopsy specimens of the sural nerve and compared compound action potentials with morphological changes in normal volunteers and those with various neuropathies. In Friedreich’s ataxia, a substantial reduction in Aβ potential with preservation of Aδ and C potential correlated well with the morphological decrease in the large myelinated fiber population. Conversely, in dominantly inherited amyloidosis, the absent C-fiber potential, the greatly reduced Aδ potential, and the only moderately reduced Aβ potential correlated well with a near absence of C fibers and reduced small myelinated fiber population on electron microscopy. Similar good correlations were found in two types of hereditary sensory neuropathies and in uremic neuropathy, but not in chronic relapsing inflammatory neuropathy. This was thought to be due to extensive segmental demyelination and remyelination and the resultant dispersion of large-fiber action potentials. In aggregate, these observations established that reasonable predictions about fiber population could be made from physiological observations, except when segmental demyelination was a prominent feature. Grafe and colleagues showed that in unmyelinated axons of the human sural nerve, the action potential propagation of many C fibers was accomplished by tetrodotoxin (TTX)-resistant sodium currents (Fig. 65-6)(Grosskreutz et al 1996). Furthermore, there was a good correlation between the expression of TTX-resistant sodium channels and voltage-dependent calcium channels on nociceptive fibers expressing the capsaicin receptor transient receptor potential vanilloid 1 (TRPV1) (Grosskreutz et al 1996).
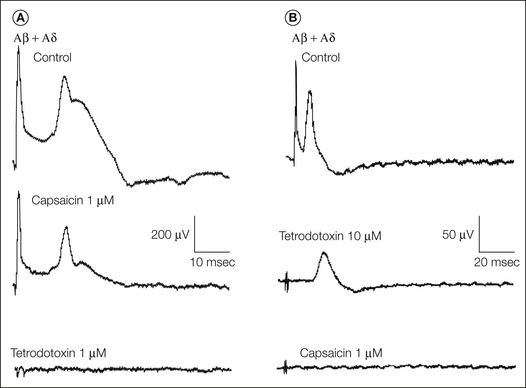
Figure 65-6 Compound action potential recordings from a human sural nerve in vitro.
Capsaicin blocks the tetrodotoxin-resistant sodium spike. A, After the application of capsaicin, A-fiber potentials were only slightly reduced, whereas a capsaicin-resistant component of the C-fiber potential remained. Both components were blocked by tetrodotoxin. B, Tetrodotoxin abolishes A-fiber potentials but blocks only a portion of the C-fiber potential. This tetrodotoxin-resistant C-fiber potential is blocked by capsaicin. (Reprinted with permission from Grosskreutz J, Quasthoff S, Kuhn M, et al 1996 Capsaicin blocks tetrodotoxin-resistant sodium potentials and calcium potentials in unmyelinated C fibres of biopsied human sural nerve in vitro. Neuroscience Letters 208:49–52. Copyright 1996 Elsevier Ltd.)
Magnetic Resonance Imaging
In the diagnostic work-up of peripheral nerve disease, imaging studies are often used to exclude focal mass lesions or external compression and to visualize muscle atrophy. Recently, it has been recognized that magnetic resonance imaging (MRI) can identify changes in peripheral nerves and secondary neurogenic alterations in skeletal muscle (Koltzenburg and Bendszus 2004). Prolongation of the T2 relaxation time and gadolinium enhancement of denervated muscle develop in parallel with the development of pathological spontaneous activity on EMG that is a feature of axonal damage. Axonal nerve lesions cause a hyperintense signal on T2-weighted MRI of the affected nerve at and distal to the lesion site, which correlates with wallerian degeneration and nerve edema (Fig. 65-7). Although changes on MRI do not distinguish between painful and painless nerve lesions, they supplement the differential diagnosis of peripheral nerve disease.
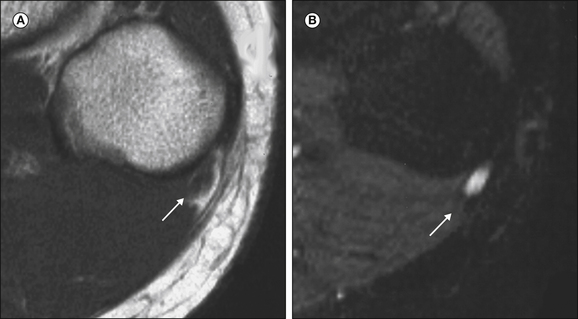
Figure 65-7 Axial T1-weighted (A) and corresponding turbo inversion recovery magnitude (B) images of the region of the fibular head with a peroneal nerve lesion.
The arrow points to a hyperintense nerve. FH, fibular head. (Reprinted from Koltzenburg M, Bendszus M 2004 Imaging of peripheral nerve lesions. Current Opinion in Neurology 17:621–626. Copyright 2004 Lippincott, Williams & Wilkins.)
Quantitative Sensory Testing
The quantitative somatosensory thermotest (QST) assesses the function of different classes of unmyelinated and thin myelinated afferent fibers. Ramps of ascending or descending temperature are applied to the skin through a Peltier contact thermode, and detection thresholds are recorded. The method of limits is most frequently used, and the subject operates a switch when a particular sensation is reached. It is generally thought that the warm detection threshold requires signaling through unmyelinated fibers whereas cold detection is signaled by thin myelinated afferents, and these two modalities are thus commonly tested. Abnormal findings on the QST may occur in patients with normal large-fiber function, as assessed by clinical examination and neurophysiological investigations, and hypoesthesia for cold and warm may dissociate (Verdugo and Ochoa 1992). The QST permits additional documentation of thermal hyperalgesia, and several different patterns (Fig. 65-8) have been described in different subgroups of patients with painful neuropathy (Verdugo and Ochoa 1992, Koltzenburg et al 1994). This psychophysical test does not specify the location in the somatosensory pathway where hypoesthesia or hyperalgesia is generated. Because of this non-specificity, the relatively large intraindividual variability, and the obvious potential for change that patients can introduce when they are biased against a particular outcome, the usefulness of this technique is limited (Zaslansky and Yarnitsky 1998). It is therefore perceived as a relatively weak diagnostic tool when compared with other electrodiagnostic procedures. The diagnostic efficiency of thermal threshold testing is inferior to measurements of ENF density in skin biopsy specimens (Devigili et al 2008).
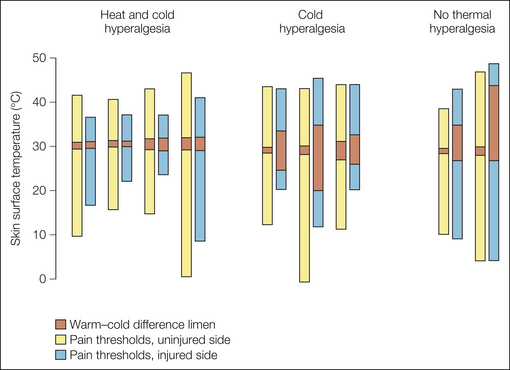
Figure 65-8 Results of quantitative thermal threshold testing with a Peltier device in patients after unilateral painful traumatic nerve injury.
The ends of the bars show the pain thresholds for cold or heat stimuli on the symptomatic and uninjured sides. The dark columns illustrate the cold–warm difference limen. Hyperalgesia for heat is present only in subjects with a normal cold–warm difference limen; such individuals have relatively little damage to C and Aδ fibers. (From Koltzenburg M, Lundberg LER, Torebjörk HE 1992 Dynamic and static components of mechanical hyperalgesia in human hairy skin. Pain 51:207–219.)
Indirect Test of Unmyelinated Fiber Function
A number of investigations are available that use effector responses as an indirect means of studying the function of unmyelinated fibers. Studies of the axon reflex (neurogenic flare) by visual inspection, thermography, or laser Doppler recordings in response to a chemical stimulus that activates cutaneous C fibers are commonly used. Though independent of patient cooperation, a major drawback of this technique is the dependence of a host of other factors that affect the effector response (Low 1993). Autonomic tests can be used to study the function of postganglionic sympathetic neurons. This includes various measures of sudomotor function, such as sweat testing, sympathetic skin response, or quantitative sudomotor axon reflex testing (Low 1993). Except for the latter, it is difficult to define the location of the dysfunction with abnormal test results.
Skin Biopsy
Quantification of ENF density has emerged as an objective specific and sensitive tool for the investigation of unmyelinated nerve fibers in humans (Griffin et al 2001). Skin punch biopsy specimens are immunostained for the pan-neuronal marker UCHL1, which is used to identify the terminals of unmyelinated nociceptors terminating in the epidermis (Fig. 65-9). It is also possible to visualize unmyelinated fibers innervating blood vessels and sweat glands. Skin biopsy can be performed in multiple sites and can be repeated over time so that a spatiotemporal profile of epidermal innervation can be constructed. This approach is currently the best technology to assess the progression of fiber loss in disease and the progression of regeneration and re-innervation with treatment (Devigili et al 2008, Haanpää et al 2011). Small-fiber loss is an important feature in idiopathic small-fiber neuropathy (Singer et al 2004) and in the early stages of diabetes mellitus or in individuals with impaired glucose tolerance (Polydefkis et al 2003). Many patients with restless legs syndrome (RLS) harbor a small-fiber neuropathy as well. RLS is characterized by a desire to move the extremities and is often associated with paresthesias or dysesthesias, motor restlessness, worsening of symptoms with rest and relief by activity, and worsening of symptoms in the evening or night. One group of RLS is triggered by painful dysesthesias associated with small-fiber loss and has a later onset and no family history. It is clinically distinct from another RLS group without pain, no small-fiber loss, a positive family history, and earlier onset (Polydefkis et al 2000). Skin biopsy can be helpful in the differential diagnosis of other painful states (Herrmann et al 2010), but thus far no unique histological features have been identified that distinguish painful from painless neuropathy (Kalliomaki et al 2011).
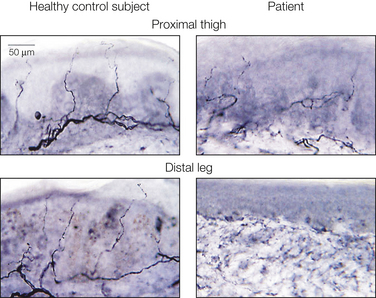
Figure 65-9 Representative skin biopsy samples from a healthy control subject and a patient with a diabetic small-fiber neuropathy.
Epidermal nerve fibers are visualized by immunohistochemical staining of cutaneous punch biopsy specimens with a polyclonal antibody against the pan-neuronal marker UCHL1. (Reprinted from Polydefkis M, Allen RP, Hauer P, et al 2000 Subclinical sensory neuropathy in late-onset restless legs syndrome. Neurology 55:1115–1121. Copyright 2000 Lippincott, Williams & Wilkins.)
Polyneuropathies
We have already mentioned that most neuropathies are accompanied by positive sensory symptoms that are not particularly bothersome. Furthermore, secondary changes such as joint lesions or tissue injury can be the cause of pain in peripheral neuropathy. What follows is an account of neuropathies that are typically painful and in which the primary source of the pain is thought to be the main consequence of the nerve disease.
It is clinically convenient to subdivide neuropathies on the basis of whether they are symmetrical polyneuropathies or asymmetrical neuropathies. Because neurophysiological investigations can positively diagnose large-fiber involvement, this provides a further important differential diagnostic clue to the underlying disease process, even though it may not be an essential pathophysiological mechanism in the generation of pain. Box 65-2 lists neuropathies important to the present discussion, divided on the basis of painfulness, their topographical distribution, and fiber size involvement.
Polyneuropathies with Selective Loss of Pain Sensation
Congenital analgesia comprises an exceedingly rare heterogeneous group of inherited disorders in which insensitivity to pain is evident from an early age and can be explained on the basis of an abnormality in peripheral sensory neurons. It is important to distinguish these conditions from severe generalized peripheral neuropathy or disorders in which the peripheral and central nervous systems appear to be intact, where the problem appears to be lack of recognition of pain, indifference, or asymbolia (Schilder et al 1931). In the latter group, patients are able to identify noxious stimuli and sensory thresholds are normal, but they do not react behaviorally or physiologically in the expected way, and the peripheral nerves, spinal cord, and thalamus are all thought to be normal (Baxter and Olszewski 1960). An important caveat is that some patients who have been described in the literature as having an indifference to pain may in fact have abnormal peripheral nociceptor function, such as a loss-of-function mutation in SCN9A (see below).
HSANs are causes of congenital insensitivity to pain (Table 65-3). Some of these disorders appear to be caused by defective signaling of nerve growth factor (NGF), which is critically involved in embryonic development of the peripheral nociceptive system. Patients with HSAN 4 (also called CIPA, Online Mendelian Inheritance in Man [OMIM] 256800) have a mutation of the gene encoding the tyrosine kinase A (TrkA) receptor, the high-affinity receptor for NGF. Patients lack superficial and deep pain sensitivity, and thermosensation is severely impaired or absent, which frequently leads to massive injuries, particularly of the joints, early in life. All patients lack sweating and have episodes of unexplained hyperpyrexia. Mental retardation and self-mutilation are present in more than 90% of cases. The results of routine electrophysiological investigations are usually normal, save for the generally absent sympathetic skin response and absent histamine flare (Shatzky et al 2000). HSAN 4 shows absent ENFs and severe loss of unmyelinated nerve fibers in nerve biopsy specimens and massively reduced tract of Lissauer on autopsy (Houlden et al 2004). It is distinct from the even rarer HSAN 5 (OMIM 608654), which is characterized by a prevalent loss of thin myelinated fibers and relative sparing of unmyelinated fibers. At least some of the patients with HSAN 5 appear to be allelic to HSAN 4 and have a mutation in TrkA, whereas others appear to have a mutation of the β subunit of NGF (Houlden et al 2004). Patients with HSAN 3, familial dysautonomia (also called Riley–Day syndrome), also have impairment in their pain sensation, though less complete than in patients with HSAN 4 or 5 (Axelrod 2002). Patients with HSAN 2 show severe loss of small-fiber function in the distal ends of extremities that characteristically leads to severe joint deformities (Houlden et al 2004).
Table 65-3
Hereditary Sensory and Autonomic Neuropathies

AD, autosomal dominant; AR, autosomal recessive; CMT, Charcot–Marie–Tooth disease; GTPase, guanosine triphosphatase; HSAN, hereditary sensory and autonomic neuropathy; HSN, hereditary sensory neuropathy.
Loss-of-function mutations in the gene SCN9A encoding the TTX-sensitive voltage-gated sodium channel Nav1.7 also cause insensitivity to pain (Cox et al 2006, Goldberg et al 2007). These patients are otherwise almost completely normal neurologically, save for anosmia, and in the past, indifference to pain may have been diagnosed in some of these patients. In these patients neurogenic flare is retained following stimulation of cutaneous nociceptors or purinoceptors by capsaicin or histamine, thus suggesting that initiation and peripheral spread of action potentials are intact but that either a deficit in centripetal propagation of action potentials or a defect in synaptic transmission in the dorsal horn of the spinal cord is present.
Because there have been reports of several patients with congenital analgesia, including those with the ill-defined congenital autonomic dysfunction with universal pain loss (Axelrod 2002), who do not fall neatly into any of the HSAN categories or in whom mutations in the genes encoding for Nav1.7, TrkA, or NGF have been excluded, it is likely that there are several other distinct disorders with congenital insensitivity to pain whose pathophysiological and genetic basis has thus far remained elusive.
Tangier Disease (OMIM 205400)
Tangier disease, or familial high-density lipoprotein deficiency, was first described in a kindred living on Tangier island in Chesapeake Bay and is an extremely rare lipid disorder in which a neuropathy occurs in at least half of those affected (Dyck and Thomas 2005). Patients can have a remarkable dissociated sensory loss of pain and temperature sensation over most of the body, and radial nerve biopsy shows that small myelinated fibers are selectively lost and unmyelinated fibers are virtually absent (Kocen et al 1973). The disease is caused by a loss-of-function mutation in the gene encoding the adenosine triphosphate (ATP)-binding cassette transporter ABC1 (Brooks-Wilson et al 1999), but it has remained unclear how this relates to the development of a neuropathy.
Painful Polyneuropathies with Prevalent Loss of Large Fibers
Pain as a feature of polyneuropathies in which there is profound large-fiber loss occurs in some neuropathies. In many instances, such as isoniazid neuropathy, there is also appreciable small-fiber involvement. Many pathological studies in which selective loss of large fibers has been described have not investigated small-fiber function by electron microscopy or skin biopsy, and thus it is difficult to assess the relative proportion of fiber loss in the myelinated or unmyelinated fiber group.
Isoniazid Neuropathy
In isoniazid neuropathy, the initial symptoms are distal numbness and tingling paresthesias, which are later accompanied by pain that may be felt as a deep ache or burning sensation. The calf muscles are often painful and tender, and exacerbation of the symptoms by walking may prevent the patient from walking. The spontaneous pain and paresthesias may be particularly troublesome at night. Examination shows signs of a sensorimotor neuropathy, often confined to the legs. Cutaneous hyperesthesia is a frequent finding. Ochoa (1970) examined sural nerve biopsy specimens from nine patients and reported primary axonal degeneration in myelinated fibers with evidence of degeneration in unmyelinated fibers and regeneration of both types, together with degeneration of regenerated myelinated fibers. By using several ultrastructural criteria it was possible to distinguish as yet unmyelinated sprouts of myelinated fibers from unmyelinated fibers, as well as make an accurate assessment of differential myelinated fiber damage, through which it was found that large fibers were preferentially lost.
Pellagra Neuropathy
Peripheral neuropathy is one of the many neurological manifestations of pellagra, which is due to a deficiency of niacin (Spillane 1947). A predominant feature of the sensorimotor neuropathy is spontaneous pain in the feet and lower part of the legs, tenderness in the calf muscles, and cutaneous hyperesthesia of the feet. There have been no recent pathological studies of this neuropathy and no ultrastructural study, and thus the degree of small-fiber loss is unclear. The early light microscopic investigations showed a decreased density of myelinated fibers, with a preferential loss of larger fibers. In the spinal cord, extensive degeneration was found in the dorsal and lateral tracts and in the posterior columns. These changes in the spinal cord suggest that this is an example of a central–peripheral distal axonopathy.
Hypothyroid Neuropathy
Pollard and colleagues (1982) reported the pathological changes in sural nerve biopsy specimens from two patients with untreated hypothyroidism. One had a long history of pain in the feet and progressive difficulty walking; the other had pain and paresthesias in the hands. Both had signs of a sensorimotor neuropathy. The nerve specimens showed mainly axonal degeneration with occasional segmental demyelination. In both patients, myelinated fiber density was decreased with a relative loss of large fibers, but there were regenerating myelinated fibers that may have contributed to the small-fiber bias, though probably not to a significant extent. Unmyelinated fiber densities were increased because of small-diameter regenerating axons. Electrophysiological studies of sural nerve specimens studied in vitro also found reduced myelinated fiber density in two hypothyroid patients associated with reduced Aβ potentials in vitro, together with relatively normal C-fiber potentials (Lambert and Dyck 1993). However, over half of patients with overt hypothyroidism appear to also have a small-fiber neuropathy (Magri et al 2012).
Painless Polyneuropathies with Prevalent Large-Fiber Loss
In contrast to the polyneuropathies described above, two conditions are associated with neuropathies in which the selective loss of large fibers is not generally accompanied by painful symptoms. These conditions are Friedreich’s ataxia and chronic renal failure.
Friedreich’s Ataxia (OMIM 229300)
Friedreich’s ataxia, an autosomal recessive neurodegenerative disease, is the most common of the inherited ataxias and is caused in most cases by a GAA trinucleotide repeat expansion in intron 1 of the FRDA gene and hence reduced levels of the protein frataxin. This may result in an excess of free radicals, which then leads to cellular damage and death (Delatycki et al 2000). Pain is an unusual complaint in this condition and is only occasionally severe. Friedreich himself mentioned it, although others with a large experience in patients with the condition do not report pain as an important feature (Dyck and Thomas 2005). It is worth emphasizing that the selective loss of myelinated fibers takes place only in the earlier stages of the disorder and that loss of all fiber sizes eventually occurs (Dyck and Thomas 2005).
Chronic Renal Failure Neuropathy
Chronic renal failure of any cause may be associated with a neuropathy in which selective loss of large fibers occurs, but it is rarely painful. A complaint of restless legs is an early symptom, followed by distal numbness and paresthesias, with the distal weakness usually being confined to the legs. The rate of progression and the eventual extent of the disability are extremely variable, and painful symptoms are uncommon (Thomas et al 1971), although some studies have mentioned it (Asbury et al 1963). Extensive pathological studies have been performed on patients at autopsy and have shown axonal degeneration in distal parts of the lower limb nerves, and in neuropathy of long duration, myelin degenerative changes were observed in the cervical dorsal columns, thus suggesting that this may be a central–peripheral distal axonopathy (Asbury et al 1963). Although demyelination and remyelination are noted in teased fiber preparations, the main pathology is primary axonal degeneration (Thomas et al 1971).
Painful Polyneuropathies with Prevalent Loss of Small Fibers
There are several examples of painful small-fiber polyneuropathies, including some patients with diabetic polyneuropathy, amyloid, Fabry’s disease, certain hereditary neuropathies, and idiopathic small-fiber neuropathy.
Diabetes Mellitus
Diabetes mellitus is associated with several types of polyneuropathies, the most common of which is a symmetrical sensory polyneuropathy (Dyck and Thomas 1999). Evidence is emerging that the causes of neuropathy differ in type 1 and type 2 diabetes mellitus (Callaghan et al 2012). Numbness and paresthesias are common initial complaints. In addition, about a quarter to a third of patients complain of a spontaneous deep aching, burning, or lightning pain (Daousi et al 2006, Abbott et al 2011), and it is frequently troublesome, even when the sensory and motor deficits are mild (Dyck and Thomas 1999). Severe sensory neuropathy in diabetes with loss of protective sensitivity may lead to painless perforating foot ulcers, and in such patients the upper limbs may also be involved and there may be an associated autonomic neuropathy. The characteristic slowing demonstrated in routine nerve conduction studies in most cases of diabetic neuropathy suggests that demyelination is present, but it is also clear that at times extensive demyelination may occur as a result of pathological processes primarily affecting axons.
Brown and co-authors (1976) reported the clinical and pathological findings in three patients with severe pain secondary to diabetic polyneuropathy and distal sensory impairment but preserved tendon reflexes. Nerve biopsies suggested a predominant axonal degeneration affecting mainly small myelinated and unmyelinated fibers.
Britland and colleagues (1992) reported a morphometric study of sural nerve biopsy specimens from six diabetic patients, four with active acute painful neuropathy and two with recent remission from this type of neuropathy. Myelinated and unmyelinated fiber degeneration and regeneration were present in all the nerves, the only discernible differences between the nerves from patients with and without pain being that those with remission from the pain had a less abnormal axon–to–Schwann cell caliber ratio, more successful myelinated fiber regeneration, and less active myelinated fiber regeneration. However, these were all differences in severity, and the authors emphasized the similarity of the pathological changes in the two groups. These observations indicate that small fibers are affected early and large fibers later in diabetic polyneuropathy.
With the advent of ENF measurements in skin biopsy samples it has become clear that unmyelinated fibers are involved early in diabetic neuropathy (Polydefkis et al 2003). Several studies have shown small-fiber loss in patients with early diabetes or impaired glucose tolerance. More than 80% of patients with abnormal ENF density also had an increased warm detection threshold (Shun et al 2004). Furthermore, the ability of C fibers to regenerate after an experimental challenge with topical capsaicin is reduced relative to normal subjects (Polydefkis et al 2004). Over and above the common structural and physiological alterations, two other factors may be of importance in diabetic neuropathy. Hyperglycemia in diabetics may itself be an important factor in acute exacerbations of pain (Dyck and Thomas 1999), and changes in blood flow have also been implicated (Archer et al 1984). Microneurographic investigations have shown that in diabetic patients the ratio of mechanically responsive to mechanically insensitive nociceptors is much lower than in controls (Ørstavik 2006). This was explained by the presence of mechanically responsive nociceptors that had lost their mechanical and heat responsiveness. These finding could explain the loss of heat and pinprick sensitivity in these patients. Afferent fibers with spontaneous activity or mechanical sensitization were found in patients with and without pain, and there was no obvious neurophysiological marker for patients in pain. However, the findings suggested that there was a higher rate of ongoing activity in the subpopulations of mechanically insensitive C fibers in patients with painful neuropathy.
Amyloid Neuropathy
Another example of a painful small-fiber neuropathy is that caused by amyloid, both the inherited and the sporadic varieties (Dyck and Thomas 2005). Patients typically have distal sensory loss that initially affects pain and thermal sensations, frequently with autonomic involvement. It is common experience that this type of polyneuropathy is often very painful, the pain usually having a deep aching quality, sometimes with superimposed shooting pain. As the neuropathy progresses, all modalities are affected, reflexes are lost, and motor involvement ensues. The physiological and morphological findings showed that small myelinated and unmyelinated fibers (Fig. 65-10) are selectively lost (Thomas and King 1974).
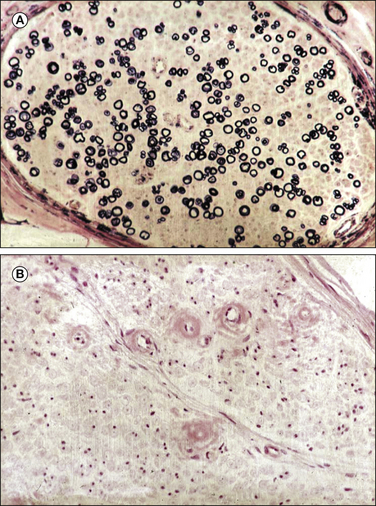
Figure 65-10 Light microscopic analysis of a sural nerve fascicle in a patient with amyloid neuropathy.
A, Kulchitsky stain showing relative preservation of myelinated fibers. B, Congo red stain showing the characteristic endoneurial red amyloid deposits. (With permission of Thomas PK, King RH 1974 Peripheral nerve changes in amyloid neuropathy. Brain 97:395–406.)
Fabry’s Disease
Fabry’s disease, angiokeratoma corporis diffusum, is a rare lipid storage disorder in which a painful peripheral neuropathy is the usual initial feature. The dermatological manifestation is telangiectasia with proliferation of keratin and epidermal cells, and most tissues, including those in the heart, kidneys, and lungs, may be involved (Dyck and Thomas 2005). Fabry’s disease can lead to renal failure, which in turn may complicate the neuropathy. There is a deficiency of ceramide trihexosidase in this sex-linked recessive disease that leads to accumulation of ceramide trihexoside in tissues. Typically, boys or young men have tenderness of the feet and spontaneous burning pain in the legs, which may be extremely severe. The accompanying sensorimotor deficit is often mild. A rash is usually present early, and this should always suggest the diagnosis of Fabry’s disease in a young man. In heterozygous carrier females, symptoms occasionally develop later in life (Dyck and Thomas 2005). DRG cells are variably affected, but in peripheral nerves there is a selective loss of small myelinated fibers and a decrease in unmyelinated axons, particularly those of larger diameter. On electron microscopy the accumulated lipid appears to be lamellated, often with concentric inclusions known as zebra bodies (Kocen and Thomas 1970). The results of routine nerve conduction studies and large-fiber quantitation by sural nerve biopsy are normal in patients with Fabry’s disease. An elevated cold detection threshold is frequently present (Morgan et al 1990), and involvement of small cutaneous fibers in these patients was shown by a profound reduction in ENFs in skin biopsy specimens taken from the ankle, with less severe loss at the thigh. The nerve damage in patients with Fabry’s disease and preserved renal function involves exclusively small myelinated and unmyelinated fibers (Scott et al 1999), and this underscores the finding that damage to small and unmyelinated fibers can be a primary cause of neuropathic pain.
Enzyme replacement therapy with recombinant human α-galactosidase A had limited transient effects on neuropathic pain (Schiffmann 2009) and no effect on ENF density in the lower extremities (Schiffmann et al 2006, Üçeyler et al 2011).
Painful Hereditary Neuropathies
Hereditary neuropathies can affect peripheral sensory, motor, and autonomic neurons. Depending on the leading initial phenotype, they are classified as CMT diseases (also known as HMSN) or HSAN. These clinical classification schemes are somewhat arbitrary in view of the motor involvement in some patients with HSAN, the dominant sensory fiber involvement in patients with CMT, and variable autonomic features. Of the different types of hereditary and autonomic sensory neuropathies it is the subtype 1 that is typically accompanied by lancinating pain (Houlden et al 2004). Patients are usually first seen in their thirties with dense distal sensory loss, a history of painless injuries, chronic skin ulceration, and a high incidence of lancinating pain. There is an intriguing combination of stimulus-independent pain and frequent accidental injuries caused by the absence of protective sensibility in these patients. This can progress to bone deformities and osteomyelitis. A characteristic finding on neurological examination is the relative preservation of vibration sense over the severe impairment of other large-fiber modalities such as touch or joint position sense. Distal weakness can be present in some patients, and electrophysiological investigations often reveal sensory and motor involvement. In a combined morphological and in vitro electrophysiological study, a preferential reduction in Aδ- and C-fiber potentials was associated with a prevalent loss of unmyelinated and small myelinated fibers, although there was also a considerable reduction in larger myelinated fibers (Lambert and Dyck 1993). Evidence is increasing that the group of autosomal dominantly inherited neuropathies in which sensory loss and pain are the leading complaints is heterogeneous (Rotthier et al 2012). HSAN 1 is caused by a mutation in the gene encoding serine palmitoyltransferase, long-chain base subunit 1 (SPTLC1) or subunit 2 (SPTLC2), which catalyzes the initial step in the de novo synthesis of sphingolipids. Evidence is emerging that this causes the accumulation of neurotoxic atypical deoxysphingoid bases (Penno et al 2010). Another hereditary neuropathy, CMT 2B, shares many features with HSAN 1, including the presence of lancinating pain. CMT 2B is caused by mutations in the gene encoding small guanosine triphosphatase (GTPase) late endosomal protein RAB7 (RAB7). Understanding how the diverse mutations in hereditary and sensory neuropathy lead to the clinical phenotypes remains a challenge (Rotthier et al 2012). Some mutations affect well-known signaling cascades such as TrkA (HSAN 4) and NGF (HSAN 5), and others affect proteins involved in axonal transport (CMT 2B, HSAN 2C, HSAN 3). However, in the majority of cases the pathophysiological mechanisms have been incompletely understood as well as, indeed, whether the pain is a consequence of the specific mutation or the sequela of general degenerative changes.
Idiopathic Small-Fiber Neuropathy
We mentioned previously that investigations of unselected cohorts of patients with neuropathy will reveal a high percentage of subjects with impaired glucose tolerance or diabetes mellitus as a probable cause of their small-fiber neuropathy (Polydefkis et al 2003). Even when this and other known causes of small-fiber neuropathy are excluded, a considerable number of patients will remain and are grouped together as having cryptogenic or idiopathic small-fiber neuropathy. A number of these distal and generalized small-fiber neuropathies involve somatic and autonomic neurons, notably distal small-fiber neuropathy, neuropathic postural tachycardia syndrome, and idiopathic autonomic neuropathy (Singer et al 2004). Pain is generally a feature only when somatic unmyelinated fibers are involved.
Idiopathic small-fiber neuropathy often affects elderly patients and is a frequent cause of the so-called burning feet syndrome. This neuropathy is ubiquitous in practice, and its main features are a burning sensation, dysesthesia and paresthesia in the feet, lancinating pain, and minimal or no distal weakness (Gorson and Ropper 1995). A detailed study of more than 30 patients showed morphological abnormalities in epidermal skin innervation in all, but astonishingly, muscle strength was normal, as were proprioception, tendon reflexes, and the results of nerve conduction studies. Two clinical patterns were apparent based on the natural history and spatial distribution of cutaneous denervation. More than 80% of the patients had neuropathic pain initially restricted to the feet and toes but extending more proximally to involve the legs and hands with time, whereas a minority had an abrupt onset of generalized cutaneous burning pain and hyperesthesia. In these patients, intraepidermal nerve fiber (IENF) density was reduced in skin from both proximal and distal sites. Of the six patients who underwent sural nerve biopsy, four were found to have selective loss of small myelinated or unmyelinated axons and two had normal histology and fiber density despite reduced IENF density in skin biopsy specimens (Holland et al 1998). This elegant study emphasizes that small-fiber involvement with sparing of large fibers can be the sole feature of a peripheral neuropathy and, when it occurs, can be the cause of pain. Evidence is forthcoming that mutations in Nav1.7 can be the cause of a structural neuropathy. A recent study of patients with biopsy-proven “idiopathic” small-fiber neuropathy in whom other causes had been excluded showed that a quarter harbored a gain-of-function mutation that rendered DRG neurons hyperexcitable. These results extend the phenotype spectrum of diseases and suggest that the gain-of-function mutant sodium channels in small-diameter peripheral axons may cause these fibers to degenerate (Faber et al 2012, Han et al 2012).
Painful Polyneuropathies with Non-selective Fiber Loss
Two commonly painful polyneuropathies are associated with non-selective fiber loss: alcoholic and myeloma neuropathy. They are discussed separately from the miscellaneous group of painful neuropathies that follow since they have been extensively studied pathologically, particularly with regard to the question of differential fiber involvement.
Alcoholic Neuropathy
The incidence of neuropathy in chronic alcoholics is in the region of 10%, including asymptomatic patients. Together with diabetes mellitus it constitutes the main cause of peripheral neuropathy. Of symptomatic patients, approximately one-quarter complain of pain or paresthesias as the first symptom (Dyck and Thomas 2005). Burning pain and tenderness of the feet and legs are the characteristic complaints, the upper limbs being involved only rarely. Examination reveals a sensorimotor neuropathy, and the occurrence of painful symptoms is not related to the severity of the deficit. In a pathological study, Walsh and McLeod (1970) examined sural nerve biopsy specimens from 11 patients who were divided into those with acute and those with chronic neuropathies, and in addition, their diet was assessed. Myelinated fiber density was decreased in all the biopsy specimens. Fiber size histograms of five specimens showed a reduction in all fiber sizes in three, but in two there was a relative excess of small-diameter fibers. Teased-fiber preparations showed that these were regenerating sprouts. Patients with an acute neuropathy and a poor diet had active axonal degeneration, whereas those with chronic neuropathy and a better diet had less degeneration, and regeneration was present. However, Walsh and McLeod (1970) did not relate this to painfulness in their patients.
In the early stages of alcoholic neuropathy in some patients, clinical evidence of large sensory fiber involvement is slight or even absent, and abnormal thermal thresholds indicate preferential involvement of unmyelinated afferent fibers. This is thus another example of a painful neuropathy with selective small-fiber involvement. Treatment of alcoholic neuropathy consists of stopping drinking and ensuring an adequate diet. Poor diet is a major contributory factor to development of the neuropathy, and there are obvious clinical similarities to the neuropathies caused by specific vitamin deficiencies; pellagra has already been discussed and some further examples are considered below.
Myeloma
Both multiple and solitary myeloma may be associated with a peripheral sensorimotor neuropathy (Walsh 1971). The neuropathy is extremely variable in severity and rate of progression and ranges from mild, predominantly sensory neuropathy to complete tetraplegia. Bone pain is of course a common symptom, but in addition the pain can be attributed to the associated neuropathy, and its incidence may be as high as 60% in those with a neuropathy (Davis and Drachman 1972). In five sural nerve biopsy specimens from patients with neuropathies but without painful symptoms, loss of myelinated fibers of all sizes was found, and in teased fibers the appearance suggested a primary axonal degeneration. Unmyelinated axon counts showed a substantial decrease in number, without regeneration. Amyloid has only occasionally been found in the nerves of patients with myeloma neuropathy and is not the cause of the neuropathy. The neuropathy responds well to myeloma chemotherapy or radiotherapy for solitary myeloma, and this includes resolution of the painful symptoms. Neuropathies can also occur with a monoclonal gammopathy, which may be the sentinel event in malignant disease, but it is mostly a “monoclonal gammopathy of undetermined significance” (MGUS). Sensory symptoms predominate and consist of paresthesias, numbness, imbalance, and gait ataxia, but rarely pain (Gorson 1999).
Other Painful Polyneuropathies
Some further polyneuropathies not included in the aforementioned categories that may be painful are considered here. They have been excluded from the foregoing sections either because of insufficient morphological data concerning differential fiber damage and loss or because a primary demyelinating pathology precludes accurate assessment of differential fiber loss.
Acute Inflammatory Polyneuropathy
In acute inflammatory polyneuropathy (AIP) of the Guillain–Barré type, pain is a common early symptom and often precedes sensory impairment or weakness. It may be manifested in a distal distribution as generalized muscular pain or as root pain, which sometimes leads to diagnostic difficulty. It has been suggested that such pain may be due to local inflammation in roots, mediated by the nervi nervorum, rather than neuropathic pain from the pathological processes involving the root fibers themselves. The pathology of AIP is demyelination, usually predominantly affecting the roots, and in the great majority of patients full recovery occurs. The pain with AIP may be severe but is usually transient. Persisting pain is more often in a distal distribution. Guillain–Barré syndrome (GBS) has traditionally been considered to be a large-fiber neuropathy, and in more than 50% of cases there is evidence of small-fiber involvement based on reduced ENF density and pathological evidence of active nerve degeneration in the dermis: fragmentation of subepidermal nerve plexuses and a beaded appearance of dermal nerves. Correspondingly, patients with GBS have significantly elevated thermal thresholds (Pan et al 2003).
Chronic Inflammatory Demyelinating Polyneuropathy
Severe pain is not generally a common feature of chronic inflammatory demyelinating polyneuropathy (CIDP) because of the predominantly large-fiber involvement in this condition. However, one prospective study revealed that pain is a symptom in up to 42% of patients, although it was not clear how severe the pain was nor whether it was related to the neuropathic process (Gorson et al 1997). A more recent study involving 46 patients reported pain in 7% (Nasu 2012).
POEMS Syndrome
POEMS syndrome is a rare paraneoplastic disorder caused by plasma cell dyscrasia. The acronym stands for polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, and skin changes. The disorder is often mistaken for CIDP because both are initially manifested as subacute length-dependent sensorimotor polyneuropathy. However, POEMS results in more axonal loss than CIDP does (Mauermann et al 2012), and this may explain why pain affects more than three-quarters of patients (Nasu et al 2012).
Neuropathy Associated with Sjögren’s Syndrome
Sjögren’s syndrome is an autoimmune attack on exocrine glands. The disease affects mainly women and is typically characterized by xerostomia and keratoconjunctivitis sicca, although other organs, including the peripheral nervous system, may be involved. One study reported that burning pain in the feet was present in 80% of patients with a neuropathy. A high proportion showed either reduced or abnormal morphology of ENFs. Loss of IENFs was frequently not length dependent, thus suggesting that patients with this disorder commonly have a small-fiber sensory neuronopathy rather than a “dying-back” axonopathy (Chai et al 2005).
Nutritional Neuropathies
In addition to niacin deficiency neuropathy and nutritional factors in the pathogenesis of alcoholic neuropathy already discussed, several other painful neuropathies attributed to specific nutritional deficiencies have been described (Dyck and Thomas 2005). Many accounts in the literature provide descriptions of clinical features but lack biochemical or neuropathological investigation. Other problems of etiological differentiation are that nutritional deficiency of a single vitamin seldom occurs and that in some cases, such as alcoholic neuropathy, a known neurotoxin is also involved.
Beriberi Neuropathy
In beriberi, a painful sensorimotor polyneuropathy is very common. There is spontaneous pain in the feet and sometimes the hands, often with a burning character. The calf muscles may be particularly painful, and although sensory thresholds are raised, skin stimulation may produce extremely unpleasant paresthesias. It is likely, though not completely proved, that thiamine deficiency is the cause of this neuropathy and the associated cardiac disorder, and improvement with thiamine is well recorded (Dyck and Thomas 2005). Pathological studies suggest that beriberi neuropathy is primary axonal degeneration of the central–peripheral distal type.
Strachan’s Syndrome (Jamaican Neuropathy)
In 1897, Strachan described 510 cases of a neuropathy observed in Jamaica. Patients had pain, paresthesias, and sensory impairment in the feet and hands, together with pain proximally around the shoulder and hip girdles, visual impairment, deafness, and orogenital dermatitis, in some cases resembling the lesions of pellagra. Many of the patients were ataxic, although whether this was peripheral or central in origin is not clear. More recent studies have shown that the Jamaican neuropathy is accompanied by features of spinal cord disease (Montgomery et al 1964) and that a pure peripheral disorder of the type described by Strachan is not seen now. There is no evidence that the patients described by Strachan (1897) had a specific nutritional deficiency, although nutritional or toxic factors may have been important in some patients included in this clinically heterogeneous group.
Burning Feet Syndrome
The so-called burning feet syndrome is now often used to describe the painful symptoms of small-fiber neuropathies. However, this term was first introduced to describe a syndrome seen in many prisoners of war during the Second World War (Cruickshank 1946), and it is distinct from what is currently described. The symptoms were severe aching and burning pain with unpleasant paresthesias, starting on the soles of the feet and sometimes spreading up the legs. The symptoms were often worse at night and were relieved by cold. Objective signs of neuropathy were not always present in these patients. Hyperhidrosis of the feet was sometimes a prominent feature. A single nutritional deficiency was not identified in this condition, and most patients responded to an improvement in general diet and vitamin B–rich foods.
Cuban Neuropathy
A very large epidemic of painful sensory neuropathy and bilateral optic atrophy occurred in western Cuba between 1991 and 1993, with more than 45,000 people reported to have been affected. Clinical features included bilateral optic neuropathy with centrocecal scotomas or a predominantly sensory polyneuropathy, sometimes associated with deafness, or a combination of optic and peripheral neuropathy (Thomas et al 1995). There are clearly similarities between the Cuban disease and Strachan’s syndrome. It was found that smoking, particularly cigars, was associated with increased risk for optic neuropathy and the risk was lower in patients with higher dietary intake of methionine, vitamin B12, riboflavin, and niacin and higher serum concentrations of antioxidant carotenoids (Cuba Neuropathy Field Investigation Team 1995). In support of a nutritional basis for Cuban neuropathy is the fact that the numbers of new cases began to decrease after vitamin supplementation was initiated in the population.
Tanzanian Neuropathy
A syndrome similar to Strachan’s syndrome and Cuban neuropathy has also been observed in coastal Tanzania since 1988. An optic neuropathy, with primary retinal involvement in some patients, was associated with a dysesthetic peripheral neuropathy. It was suspected but not proved that the condition had a nutritional basis (Plant and Perry 1990).
Infectious Neuropathies
Several infectious diseases, as well as anti-infectious drugs, can cause painful neuropathies. Sometimes, as in human immunodeficiency virus (HIV)-related neuropathies, it is difficult to differentiate the neurotoxic action of the infectious disease from that of the drug.
Human Immunodeficiency Virus– and Highly Active Antiretroviral Therapy–Associated Neuropathies
Neuropathies of various types are very commonly associated with HIV infection, and they are frequently painful. In one large series of ambulant patients with acquired immunodeficiency syndrome (AIDS), 28% had pain related to peripheral neuropathies (Hewitt et al 1997). By far the most frequent painful neuropathy is AIDS-associated sensory polyneuropathy, in which neuropathic pain and hyperalgesia, usually confined to the feet and lower part of the legs, are leading symptoms in at least 30% of patients, and there may be associated features of autonomic neuropathy (Manji 2000). Nerve pathology in this painful sensory polyneuropathy includes axonal atrophy and endoneurial capillary thickening. Cytomegalovirus infection leads to a characteristically painful, rapidly progressive lumbosacral polyradiculopathy that can result in paraplegia. Nutritional neuropathies, occasionally painful, have been described with HIV infection, and because herpes zoster infection is so frequent, PNH is well recognized in patients with AIDS (Pardo et al 2001). Finally, highly active antiretroviral therapy (HAART) with ddI (2′,3′-dideoxyinosine), ddC (2′,3′-dideoxycytidine), and d4t (stavudine) can all lead to the development of painful neuropathies (Luciano et al 2003). Based on the clinical findings it is not usually possible to distinguish whether the neuropathy is the consequence of the viral disease or the antiretroviral therapy (Pardo et al 2001). In untreated patients the painful sensory polyneuropathy is often a manifestation of later-stage disease, whereas the toxic neuropathy has an increased prevalence in Western countries and is found in patients with relatively little immune system compromise. Several studies have shown that hypoesthesia to warm and cold, as well as elevated thresholds to heat pain, are found in HIV-related neuropathy (Bouhassira et al 1999, Martin et al 2003), and this correlated with reduced ENF density (Polydefkis et al 2002). ENF density was inversely correlated with pain (Polydefkis et al 2002), and sensory impairment on the QST was higher in patients with a painful neuropathy (Martin et al 2003), thus suggesting that more profound damage to unmyelinated fibers was contributing to the pain. In longitudinal studies, the emergence of abnormal small sensory fiber function was associated with a transition to symptomatic distal sensory neuropathy 6 to 12 months later (Herrmann et al 2006).
Neuroborreliosis
Infection with Borrelia burgdorferi may lead to multifocal neurological lesions, both central and peripheral. The condition with its oligoarthritic manifestation became known as Lyme disease following an epidemic in Lyme, Connecticut, in the 1970s, and it was recognized then that B. burgdorferi was the infecting agent and that Borrelia was also associated with a variety of neurological findings. In Europe, tick-borne lymphocytic meningopolyneuritis, or Garin–Bujadoux–Bannwarth syndrome, had been described since the 1920s (Halperin 2003).
Stage 1 of the disease, which occurs up to 1 month after a tick bite, is characterized by fever and erythema migrans. In stage 2, the erythema migrans persists and is associated with neurological or cardiac features. The former include lymphocytic meningitis, cranial polyneuritis, and polyradiculoneuritis. A painful thoracic sensory radiculitis is well recognized, and brachial and lumbar plexopathies and the peripheral neuropathy frequently cause neuropathic pain. These manifestations usually resolve over a period of up to 6 months without specific treatment. Stage 3 of the disease, which occurs months to years after the primary infection, is characterized by arthritis, central nervous system manifestations (encephalopathy, cerebellar ataxia, spastic paraparesis, and occasionally other lesions), and a peripheral polyneuropathy that is often painful. In most patients the neuropathy resolves in several months with antibiotic treatment.
Parenteral antibiotic treatment is indicated in the initial stages of the disease. The precise indications for corticosteroids remain uncertain, and a specific role in the treatment of painful polyradiculoneuropathy is unproven.
Erythromelalgia (OMIM 133020) and Paroxysmal Extreme Pain Disorder (OMIM 167400)
Erythromelalgia (also called erythermalgia) is a rare condition characterized by painful, red, hot extremities. Patients can suffer from an acquired form of the disease that is sometimes associated with polycythemia rubra vera or from a hereditary form of the disease that is caused by a mutations in SCN9A, the gene encoding the sodium channel Nav1.7 (Yang et al 2004) (see earlier). The characteristic symptoms are aggravated by heat, and patients obtain relief of pain by cooling the affected limb. In more than 80% of cases symptoms involve the feet and in approximately a quarter involve the hands (Davis et al 2000). Characteristic changes in the sodium channel Nav1.7 are a shift in activation to hyperpolarized membrane potentials, slow deactivation, and an enhanced ramp response (Dib-Hajj et al 2010). There is evidence that many patients also have a structural small-fiber neuropathy (Ørstavik et al 2003, Davis et al 2006).
Paroxysmal extreme pain disorder (PEPD, also previously known as familial rectal pain syndrome) is caused by a gain-of-function mutation in Nav1.7. The distinctive features of PEPD are paroxysmal episodes of burning pain in the rectal, ocular, and mandibular areas accompanied by skin flushing. The symptoms are often brought about by defecation and lower body movement. Functional analysis in heterologous expression systems showed a severe reduction in the fast inactivation of the these mutant Nav1.7 channels, which leads to persistent sodium currents (Fertleman et al 2006).
Genotype–phenotype correlations have suggested that certain mutations predispose to either erythromelalgia or PEPD, but for some mutations there appears to be considerable phenotypic diversity, even in the same family (Estacion et al 2011).
TRPA1-Related Channelopathy
Individuals of a kindred with a gain-of-function mutation in the gene encoding the transient receptor potential (TRP) channel A1 (Kremeyer et al 2010), which is expressed by nociceptors, have episodes of upper body pain starting in infancy that are usually triggered by a combination of factors such as fasting, fatigue, illness, cold temperature, and physical exertion. These episodes of intense pain last for approximately 90 minutes and terminate in a period of exhaustion and deep sleep. The period of intense pain is accompanied by difficulty breathing and autonomic symptoms such as tachycardia, sweating, and generalized pallor. Affected individuals report no altered pain sensitivity outside the episodes and have normal results on neurological examination. Patients had normal thermal thresholds and no structural small-fiber neuropathy. Heterologous expression studies show increases in current flow through the activated channel at more negative potentials than in normal controls, and this could explain the increased flare response and hyperalgesia after mustard oil application in these patients.
Pain as a Manifestation of Potassium Channel Complex Autoimmunity
More recently it has been recognized that autoantibodies directed against voltage-gated potassium channels may cause pain. Autoantibodies associated with voltage-gated potassium channels have been found in rare disorders of motor nerve hyperexcitability such as neuromyotonia (also called Isaacs’ or Mertens–Zschocke syndrome) or Morvan’s syndrome (neuromyotonia, dysautonomia, and encephalopathy) (Irani et al 2012). It is becoming clear that the autoantibodies are directed proteins that are complexed with potassium channels and include contactin-associated protein 2 (CASPAR2) or leucine-rich glioma inactivated 1 (LGI1). In a large series of several hundred of these patients, half of them had pain, and intriguingly, a quarter of them had no other neurological manifestations (Klein et al 2012). The pain had a subacute onset, was chronic and affected the extremities or the entire body, and was frequently described as burning (Irani et al 2012, Klein et al 2012). In many patients antibodies against CASPAR2 were found, but because the majority had no detectable antibodies to this or to LIG1, it is possible that the pain is associated with antibodies against other antigens.
Non-freezing Cold Injury (Trench Foot)
NFCI (trench foot) is caused by prolonged exposure of one or more limbs to wet conditions at non-freezing cold temperatures (0–15°C). NFCI usually occurs in the feet and legs but may affect the upper limbs. The symptoms and signs are due to altered vascular reactivity and direct tissue damage and are always maximal in the distal parts of the affected limbs. In the majority, the acute symptoms and signs resolve within weeks, but chronic pain develops in a substantial minority.
The acute features of NFCI result from both prolonged vasoconstriction and direct cellular damage, including damage to peripheral nerves. Clinically, four stages have been recognized (Ungley and Blackwood 1942), but the symptoms and signs often overlap and the time course of each stage is variable. In the first stage, the affected limb becomes cold and numb during exposure to cold. Sensory loss may be limited to cutaneous modalities, but loss of proprioception may cause unsteadiness of gait. Peripheral pulses are reduced or absent. With warming, blue mottling of the skin occurs, but the limb remains numb and cold; this second stage is often short-lived. In the third stage, the hyperemic phase, which lasts from 2 weeks to 3 months, the limb becomes swollen and warm, with dry skin. Pulses return, but the microcirculation remains poor. The numbness may persist but is usually overshadowed by pain and tingling paresthesias. In the fourth stage, when the signs of hyperemia have resolved, pain and cold allodynia develop as a result of a small-fiber neuropathy, which may persist in more than 70% of cases (Francis and Oakley 1996), and large-fiber function is restored. Abnormal cold hyperalgesia persists in a large percentage of these patients (Rosen et al 1991, Namer 2008). Reliable data from extended longitudinal studies are lacking, but in a proportion of those with NFCI, chronic neuropathic pain and cold hyperalgesia persist for years and are sometimes lifelong. In some patients there appears to be a depletion in ENF density (Irwin et al 1997).
Toxic Neuropathies Caused by Metals and Other Poisons
Although many heavy metals and some other poisons frequently cause peripheral neuropathies, the majority is not typically painful. Of those causing pain, arsenic is the most common (see Box 65-1), and a small-fiber neuropathy has been described in patients with thallium intoxication (Lu et al 2007).
Toxic Neuropathies Caused by Drugs
Many drugs may cause peripheral neuropathies, but pain is not generally a prominent feature. The typically painful neuropathies secondary to isoniazid and antiretroviral medication have already been described. Other less common causes are listed in Box 65-1.
Carcinomatous (Paraneoplastic) Neuropathies
Mild sensorimotor neuropathies are relatively common in patients with cancer, particularly when associated with substantial loss of body weight. True paraneoplastic neuropathies are rare. Of the various paraneoplastic neuropathies described (Grisold and Drlicek 1999), only the most frequently occurring, subacute sensory neuronopathy, is commonly a cause of pain (Posner 1995).
Typically, patients have burning pain and paresthesias, often asymmetrical at the onset and frequently more severe in the upper limbs, and sensory loss of all modalities. The underlying tumor is small cell lung cancer in about 90% of cases, with other rare causes including lymphoma and breast, gastric, esophageal, prostate, and uterine cancer.
Although there are some strong autoantibody associations with the paraneoplastic neuropathies, their role in pathogenesis is uncertain.
Central–Peripheral Distal Axonopathies
It is now well established that that some pathological processes may have differential effects on the peripheral and central axons. Several references have already been made to the process of dying back or distal axonopathy, and it is likely that this is a common pattern in peripheral neuropathies in which axonal degeneration is the primary pathology. The process of distal axonopathy has been observed in a number of experimental neuropathies, for example, those caused by triorthocresyl phosphate and acrylamide (LoPachin et al 2003). In humans, anatomical information on the central processes of primary sensory neurons is rather more difficult to obtain than information on peripheral axons, but there is physiological evidence that central processes may be selectively involved in some diseases, such as in subacute myelo-optic neuropathy (SMON) secondary to clioquinol poisoning in Japan and in pure hereditary spastic paraplegia (Thomas 1982). A more common pattern in distal axonopathies is probably involvement of both central and distal axons, but differential recovery of function in peripheral and central axons may occur, as is probably the case in SMON, where there is poor resolution of the painful symptoms after removal of the toxin, and similarly in patients with vitamin E deficiency, in whom differential electrophysiological changes can occur. The implications of differential vulnerability and recovery in relation to pain in neuropathies other than SMON are not clear.
Mononeuropathies and Multiple Mononeuropathies
Clinical and pathological observations on mononeuropathies have drawn attention to several potentially important mechanisms underlying the production of painful symptoms, which may also be relevant to pain production in some polyneuropathies. Nerve transection with neuroma formation and other types of nerve traumas are discussed elsewhere in this book, but brief reference is made here to entrapment neuropathies.
Post-herpetic Neuralgia
PHN is one of the most common conditions seen in pain clinics. There is no generally agreed definition of PHN. An often underappreciated fact is that the severity of PHN tends to diminish with time and it may cease to be troublesome many months or even years later. There is generally no qualitative change in the pain of acute herpes and PHN. Furthermore, there is not a distinct point in the natural history of the disease when the pain following healing of the rash ends, but a tail distribution of the pain is observed. Thus, the time for distinguishing the different phases of the disease is somewhat arbitrary. After 3 months following healing of the acute herpes rash the pain is considered to be PHN (Watson et al 1988), and most studies of antiviral therapy now choose this time point (Dworkin et al 1997).
The most common sites for herpes zoster and PHN are the mid-thoracic dermatomes and the ophthalmic division of the trigeminal nerve, but they may occur in any dermatome. Women are more often affected than men in a ratio of approximately 3:2 (Watson et al 1988).
It is unusual for shingles to be entirely painless in middle-aged or older people. Pre-eruptive pain for up to 3 weeks has been described, although pain for more than 2 days before the rash is uncommon. Acute herpetic neuralgia is often severe (Fig. 65-11). No features of the pain are unique to the acute neuralgia, and it merges into PHN without distinct qualitative change. The pain is most often of two types: an ongoing pain described as burning, raw, severe aching, or tearing and a superimposed paroxysmal pain, stabbing or electric shock–like. Both the ongoing and paroxysmal pain may be present throughout the entire affected dermatome, but the pain commonly becomes concentrated in one part of the dermatome, particularly after a period of more than 6 months.
Figure 65-11 A, Rash of acute herpes zoster on the trunk. B, Scars of the first division of the trigeminal nerve following zoster.
The pain is frequently accompanied by a very unpleasant sensitivity of the skin, which again is often most severe in part of the dermatome. The scars themselves tend to be hypoesthetic, but elsewhere there is hyperesthesia and various forms of stimulus-induced pains (Watson et al 1988). Mechanical stimuli frequently exacerbate the underlying ongoing pain, and most patients can distinguish stimulus-induced pain by its quality from the relentless ongoing stimulus-independent pain. These evoked sensations constitute the most unbearable part of PHN for many patients. At least three forms of mechanical hyperalgesia have been described: touch-evoked pain, pinprick hyperalgesia, and pressure hyperalgesia. Even though touch-evoked pain is most prevalent, all three types of hypersensitivities can be found to various degrees in individual patients (Pappagallo et al 2000). In this group of patients, pain is usually produced by contact with clothes and stretching of the skin with movement. Although mechanical hyperalgesia is also found with other neuropathic pain, it is a particularly prominent feature of PHN (see Table 65-1).
Incidence and Natural History
Recent studies have shown that the risk for neuralgia after herpes zoster may be lower than previously estimated. The incidence of zoster in a general immune-competent population is approximately 0.2% in persons younger than 50 years but increases to 1% in those older than 80 years (Kost and Straus 1996). The risk for PHN also increases with age (Kost and Straus 1996), and data collected from patients in the placebo arm of antiviral trials of herpes zoster show that pain is present in approximately 10% of individuals at 3–6 months (Jackson et al 1997). Prospective studies have shown that the incidence of post-herpetic pain continuously decreases to low numbers over the first year after herpes zoster. In one study conducted in more than 400 patients (Helgason et al 2000), pain was present at 3 months in less than 2% of those younger than 60 years but in 10% of older individuals. After 1 year none of the patients in the younger age group reported severe pain, and the percentage had dropped to 3% in the older patient group. In one study of over 100 patients (Haanpää et al 2000), more than 90% reported pain during the acute skin eruption, but pain and hyperalgesia were present in 25% of the patients at 3 months and in 12% at 6 months. Another study showed that only 2% of patients with herpes zoster had pain of 30% intensity or greater on a visual analog scale at 6 months (Thyregod et al 2007). Good or bad outcome did not depend on age, sex, or affected dermatome. Another study of the prognosis of PHN found that patients with a longer duration of PHN at initial evaluation tended to do worse and that some patients were identified whose pain appeared to gradually worsen despite all attempts at relief of the pain (Watson et al 1991b). However, in patients with PHN defined as pain persisting for more than 3 months after healing of the rash, it was found that more than 50% either had no pain or had pain that had decreased to a level of no longer being troublesome at a median of 3 years of follow-up (Watson et al 1991b). Patients in whom persistent pain developed after healing of the rash had significantly more impairment in detecting warmth and especially cold and a larger area of altered sensation (Petersen and Robotham 2010). More than half of these patients had had PHN for longer than 1 year at the time of first evaluation. Given the progressive reduction in pain after herpes zoster, it has been difficult to evaluate the effect of treatment on prevention of PHN.
Pathology and Pathogenesis of Pain
There have been few pathological studies of PHN. Head and Campbell (1900) were the first to document changes in the DRGs and sensory roots, but of 21 patients only 1 was reported to have PHN. Lhermitte and Nicholas (1924) studied a patient with acute zoster myelitis who had acute hemorrhagic inflammatory changes in the DRGs, posterior roots, and peripheral nerves that consisted of demyelination and axonal degeneration. Denny-Brown and colleagues (1944) showed marked lymphocytic infiltration at the sites of inflammation. In the peripheral nerves of patients who underwent biopsy early and late after the acute eruption, wallerian degeneration, followed by marked fibrotic change leading to severe depletion of myelinated fibers and preservation of small unmyelinated fibers, was found in only some patients (Zachs et al 1964).
In an autopsy study of five patients, Watson and co-workers (1991a) examined the spinal cord, DRGs, dorsal roots, and peripheral nerves from five patients, three of whom had severe persistent PHN and two of whom had no pain. Dorsal horn atrophy was found only in patients with PHN. In all patients the peripheral nerves showed severe loss of myelin and axons. In these nerves there was a relative loss of larger myelinated fibers. The dorsal roots in all patients with PHN and in one patient without pain showed loss of myelin and axons, but in the other pain-free patient the dorsal root was normal. Substance P and calcitonin gene–related peptide (CGRP) levels were measured in two patients with PHN. Staining was absent in the affected DRG but apparently normal in the dorsal horn. Quantitative study of the unmyelinated fibers was not performed in either study, so the degree to which unmyelinated afferents were affected in these patients remains uncertain. An additional finding of interest in one patient who had PHN for 22 months before death was marked inflammatory change with lymphocytic infiltration bilaterally in the DRGs of four adjacent segments and in the respective peripheral nerves, thus suggesting that an ongoing inflammatory process may result following acute zoster in some patients. The persistent inflammation found in one patient at a longer interval after acute zoster might indicate continuing low-grade infection and could be a feature of those whose pain gradually worsens. As yet, pathological studies have not unequivocally revealed a specific change present in patients with PHN and absent in patients without pain that might offer a clue to the mechanism of the pain. Preferential loss of larger myelinated fibers appears to be a feature common to patients both with and without PHN.
It is clear that changes in the central and peripheral nervous systems contribute to pain, and several lines of evidence indicate that the mechanisms causing pain in PHN are heterogeneous. Based on psychophysical experiments, assessment of C-fiber function, and skin biopsy, several subtypes have been recognized, although many components of these different pathologies may co-exist in individuals (Fields et al 1998). One group of patients with PHN appears to have chronic abnormal sensitization of unmyelinated cutaneous nociceptors (irritable nociceptors). Such patients characteristically have minimal sensory loss, a normal flare response, strong pain following the application of a capsaicin patch to the affected dermatome, and good pain relief during peripheral local anesthetic blocks. Many of the patients also have strong touch-evoked pain and relatively mild loss of cutaneous innervation as assessed in skin biopsy specimens. Other patients have pain associated with substantial loss of small-fiber function and at least partially preserved large-fiber function. In such patients temperature sensation is profoundly impaired, but light moving mechanical stimuli can produce pain. It is presently unclear how pain is generated in these patients. One possibility is that it is maintained by ectopically discharging nociceptors that have lost their connection to the skin. If the deafferentation is even more pronounced and associated with profound and extensive loss of small and large fibers, there may be ongoing pain without hyperalgesia. In this group the pain may possibly be due to increased excitability of deafferentated central neurons.
Diabetic Mononeuropathy and Amyotrophy
Mononeuropathies occur more frequently in diabetics than in the normal population and in particular affect the motor nerves to the extraocular muscles but also single peripheral nerves, including the median, ulnar, peroneal, femoral, and lateral cutaneous nerve of the thigh. It is particularly interesting that approximately half the patients with acute lesions of the third, fourth, and sixth cranial nerves have pain that may precede the ocular palsy by a few days (Zorilla and Kozak 1967). Third nerve palsy is the most common and usually spares the pupil. Pain is felt around or behind the eye and may be severe. Since there are no somatosensory fibers in these nerves, where is the pain arising? One possibility is that the lesions involve the nervi nervorum and produce pain, and in animals the cranial motor nerves contain unmyelinated sensory fibers, presumably from the trigeminal nerve, that supply the ocular muscles with nociceptors and use the motor nerve as a convenient route. This is suggestive of pain arising as a result of activity in the nervi nervorum, also termed nerve trunk pain (Asbury and Fields 1984).
Postmortem studies of the pathology of diabetic mononeuropathy show a swollen nerve retro-orbitally, degenerative changes in the central parts of the nerve, and wallerian degeneration distally. This lesion was considered to have a vascular cause on the basis of the arteriolar changes observed, although no vessel occlusion was seen. One patient who underwent autopsy a month after the onset of a third nerve palsy had suffered a contralateral transient third nerve palsy 3 years earlier (Asbury et al 1970). The changes in the acutely affected nerve were maximal in the central parts of the nerve and consisted mainly of demyelination. The contralateral previously affected nerve was structurally normal.
Diabetic amyotrophy, also now known as painful proximal diabetic neuropathy, is an asymmetrical predominantly motor neuropathy, sometimes related to poor diabetic control, that is found in middle-aged and elderly diabetics (Dyck and Thomas 1999, Said et al 2003). In a clinical and pathological study, inflammatory changes and ischemic lesions associated with vasculitis of epineurial and perineurial vessels were found. There was an associated mixed axonal and demyelinating neuropathy with degeneration of unmyelinated axons, together with evidence of regeneration. Endoneurial and perineurial inflammatory infiltrates were common. The findings were considered to be in keeping with an ischemic mechanism secondary to occlusion of blood vessels (Said et al 1997). In view of the marked inflammatory component, it is likely that the pain may in part be another example of pain mediated by the nervi nervorum, or nerve trunk pain.
Entrapment Neuropathies
Entrapment neuropathies of sensory or mixed nerves, such as carpal tunnel syndrome (CTS), are usually characterized in the early stages by paresthesias and pain (Mumenthaler et al 1998). Entrapment of nerves often occurs at physiologically narrowed passages (Stewart 1999). Entrapment of spinal roots or DRGs can also occur as a result of a herniated disc. Morphologically, entrapment lesions cause damage to myelinated fibers in the first instance (Dyck and Thomas 2005), with a near absence of myelinated fibers only in severe lesions, in which there is preservation of only C fibers. The local pain and tenderness at the site of nerve entrapment in many patients is likely to be nerve trunk pain mediated by the nervi nervorum, as discussed in relation to diabetic mononeuropathy. Paresthesias of the non-painful type with entrapment neuropathies presumably reflect activity in damaged myelinated fibers (Nordin et al 1984).
The explanation for pain in entrapment neuropathies (other than nerve trunk pain) is more difficult. Severe pain, sometimes with a burning quality, is frequently a symptom, albeit often in short-lived episodes in electrophysiologically mild CTS, in which myelinated fiber conduction is relatively mildly affected, and thus it is difficult to imagine that C fibers are damaged. In CTS the average pain correlates inversely with warmth detection on the index finger but not on the little finger, thus suggesting that lesions of unmyelinated fibers could be involved (Lang et al 1995).
Morton’s neuralgia is an example of a frequently histologically severe entrapment neuropathy in which a plantar digital nerve becomes compressed in the region of the metatarsal heads in the foot. In badly affected nerves, many myelinated fibers may be disrupted within the region of compression and produce a nerve that is populated almost exclusively by C fibers. However, explanations of pain pathogenesis based on these findings has to take account of the fact that similar changes are often found in control nerves from subjects who never suffered this type of neuralgia.
The sciatic nerve is vulnerable during surgical procedures on the hip joint. Severe persistent neuropathic pain may result from surgical trauma to the nerve, even when the neurological deficit resulting from the injury is very mild. Clinically severe sciatic lesions, usually compressive in nature and seen in emaciated drug users and occasionally as critical illness compression neuropathies, are characteristically painful.
Blood vessels can also be the cause of irritation and compression of peripheral nerves and give rise to episodic pain under provocation maneuvers. Such conditions can include thoracic outlet syndrome, fistulas or varicosities (Fig. 65-12), or venous thrombosis compressing the sciatic nerve (Bendszus et al 2003). MRI is the investigation of choice for definitive diagnosis of these conditions (Koltzenburg and Bendszus 2004).
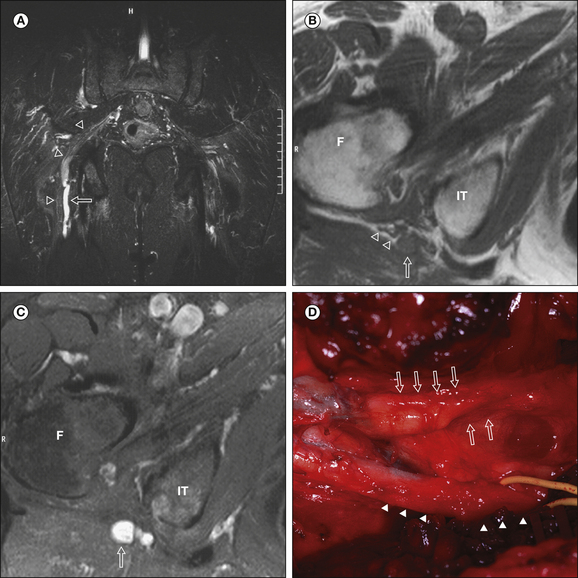
Figure 65-12 Painful vascular compression of the sciatic nerve.
A, Coronal, fat-suppressed, T2-weighted magnetic resonance image of the pelvis and proximal part of the thigh demonstrating a hyperintense tubular structure with focal notches (arrow) in immediate contact medial to the sciatic nerve (arrowheads). An axial, unenhanced, T1-weighted image (B) and a contrast-enhanced, fat-saturated image (C) reveal intense contrast enhancement of the tubular structure (arrow) posterior and medial to the sciatic nerve (arrowheads in B) at the level of the piriform (F, femur; IT, ischial tuberosity). D, Intraoperative view showing the close contact between the varicose vein (elevated by a loop, arrowheads) and the sciatic nerve (four large arrows) causing a focal flattening of the nerve (two large arrows). Decompression surgery resulted in relief of the pain. (Reprinted from Bendszus M, Rieckmann P, Perez J, et al 2003. Painful vascular compression syndrome of the sciatic nerve caused by gluteal varicosities. Neurology 61:985–987. Copyright 2003 Lippincott, Williams & Wilkins.)
Ischemic and Vasculitic Neuropathies
Vasculitic neuropathies can be divided into those in which the nerves are affected as part of the systemic disease and into non-systemic vasculitic neuropathies (Collins and Periquet 2004). Of the vasculitic diseases, periarteritis nodosa and Churg–Strauss syndrome are characteristically painful, which probably has an ischemic microangiopathic basis (Seo et al 2004), and some appear to have a small-fiber neuropathy (Said 1999).
Clinical signs of sensory neuropathy are present in more than three-fourths of patients undergoing amputation for major vessel atheromatous disease, and loss of myelinated fibers with segmental demyelination, remyelination, and wallerian degeneration is observed in nerves from these patients (Eames and Lange 1967). Ischemia involving the nerves of a limb may, of course, be associated with ischemia of non-neural structures, which may be the source of pain rather than the neuropathic changes.
Neuralgic Amyotrophy
Neuralgic amyotrophy (also called cryptogenic brachial plexus neuropathy or Parsonage–Turner syndrome) is a condition characterized by an acute onset of severe pain around the shoulder girdle or in the arms, often in a root distribution, followed within 2 weeks by weakness in the limb. It occurs at an incidence of approximately 1–2 per 100,000 (Rubin 2001). This condition is most frequently distributed around the shoulder girdle and upper limb but may involve distal muscle groups. A minority of patients report a minor, possibly viral preceding illness. The ensuing paralysis is extremely variable in severity and duration, but good recovery is usual within 2 years. Little is known about the pathology of the condition or even whether the site of the initial lesion is in the roots or more distally in the brachial plexus, although the patchy distribution favors involvement of the plexus and its branches rather than the roots in the majority of patients (Rubin 2001). The time course of milder lesions suggests demyelination without axonal disruption, whereas that of severe lesions indicates that axonal degeneration probably occurs. It has been suggested that in some patients the swollen roots may become entrapped in the cervical exit foramina and that this is the cause of the radicular pain. There are rare familial forms of this disorder. Hereditary neuralgic amyotrophy is an autosomal dominant form of recurrent focal neuropathy characterized clinically by acute, recurrent episodes of brachial plexus neuropathy with muscle weakness and atrophy preceded by severe pain in the affected arm. In some individuals the condition is caused by a mutation in Septin 9 (Kuhlenbaumer et al 2005), a member of the family of GTPases that interacts with the cytoskeleton, including microtubules. There is evidence of genetic heterogeneity since some forms map to the gene locus 17q25 whereas other do not (Kuhlenbaumer et al 2001).
Carcinomatous and Radiation Plexopathy
Cancer and cancer therapy can cause a variety of peripheral neurological complications (Posner 1995). By far the most common tumors causing metastatic spread to the brachial plexus are lung and breast cancer. Less common tumors include lymphoma, melanoma, and sarcoma. The lower trunks and divisions are preferentially affected because of the proximity of this part of the plexus to the lateral group of axillary lymph nodes. This contrasts with radiation plexopathy, in which the upper part of the plexus (C5 and C6 innervation) is usually more severely affected, possibly because of the shorter length of the lower trunks in the field of radiation and a protective effect of the clavicle. Horner’s syndrome is common with tumor invasion and unusual with radiation damage.
Pain is virtually always the initial symptom of brachial plexus carcinomatous invasion and is generally experienced diffusely around the shoulder and in the elbow and medial aspect of the forearm and hand. It is often severe and proves refractory to treatment. Swelling of the arm as a result of lymphedema is mild or absent, again in contrast to radiation plexopathy, in which swelling is almost always present. Pain is usually a less prominent symptom in radiation plexopathy, but differentiation on the basis of symptoms and signs can be difficult, and the same applies to lumbosacral plexopathy (Jaeckle et al 1985). Radiation plexopathy and plexopathy from tumor spread or recurrence may co-exist in individual patients.
Radiation plexopathy is uncommon if a dose of less than 60 Gy has been given. The interval between completion of radiotherapy and the onset of symptoms ranges from 3 months to 26 years (median 6 years).
Electrophysiological changes may be helpful in distinguishing the two types of pathologies: myokymic discharges on EMG are common in radiation plexopathy (Kimura 2001). Combined investigation with MRI and positron emission tomography is also helpful, although imaging does not always yield a definitive diagnosis and a few patients will require biopsy to differentiate carcinomatous invasion from radiation damage.
Lumbosacral Plexus
Tumor invasion of the lumbosacral plexus is due to direct spread of primary pelvic malignancies in about 75% of cases. The onset is between 1 and 31 years following radiotherapy, with a median of 5 years. Three common patterns of plexus involvement are recognized—upper, lower, and pan-plexus—but several unusual and distinctive variants have been described (Chad 2003). As with malignant brachial plexopathy, pain is often the earliest symptom, followed by sensory and motor deficits and swelling of the limb.
Imaging usually establishes the diagnosis. Because spread of tumor to the sacrum and thence to the epidural space is very common, imaging should include the lumbosacral spine in suspected cases.
The prognosis is worse than with malignant brachial plexus invasion, with a median survival of only about 6 months from the time of diagnosis.
Radiation lumbosacral plexopathy is commonly painless at the onset, with the findings usually being progressive distal weakness initially predominantly in an L5–S1 distribution, later extending proximally, and in most patients becoming bilateral but remaining asymmetrical (Chad and Recht 1991).
Imaging is often necessary to make sure that the deficit is not due to malignant invasion, and EMG characteristically demonstrates myokymic discharges in the affected leg muscles and fibrillation potentials in the paraspinal muscles because of the extent of the field of radiation. Both changes are helpful in differentiating radiation damage from malignant invasion.
Pain Caused by Tumors of Peripheral Nerves
Benign tumors of peripheral nerves, such as schwannomas and neurofibromas, are sometimes painful, with both local pain and tenderness associated with swelling, as well as pain and paresthesias radiating in the distribution of the nerve. The diagnosis is not difficult to make in the case of superficial nerves, but benign tumors affecting deeper nerves sometimes occasion considerable diagnostic problems and delay. Neurinomas are characteristically painful with a segmental projection and can produce signs of spinal cord or cauda equina compression (Fig. 65-13).
Figure 65-13 Sagittal (A) and axial (B) magnetic resonance images of a typical dumbbell-shaped neurinoma (arrows) in the lumbar region of a patient with progressive painful wasting of the quadriceps muscle and radiating leg pain.
Malignant peripheral nerve tumors are nearly always of Schwann cell origin. They are exceptionally rare except in patients with neurofibromatosis, in whom the incidence is approximately 4%. Pain and rapidly increasing swelling, together with a deficit in the distribution of the nerve concerned, are the initial features. The prognosis is poor.
Conclusion
Despite considerable progress in our understanding of the neurobiology of pain and peripheral nerve disease, the crucial mechanisms in the generation of pain in neuropathies have remained obscure. Although experimental observations in animal models of neuropathic pain continue to provide important insight into their importance, applicability to humans is far from being clear. Studies in humans have begun to shed light on distinct mechanisms that have not been predicted from animal work. Thus, molecular genetic studies have identified mutations in hereditary painful neuropathies, and one example is the identification of mutations in the sodium channel Nav1.7, which is a crucial ion channel of the pain pathway. Refinement of morphological techniques to study unmyelinated fibers in skin biopsy specimens has revealed their involvement in many neuropathies. Microneurographic evidence is forthcoming that there are functionally distinct subtypes of nociceptive C fibers, and data are emerging to suggest that sensitization of mechanically insensitive afferents may be responsible for the generation of pain and mechanical hyperalgesia in some neuropathic conditions. Knowledge from investigation of patients with neuropathic pain is a prerequisite for the establishment of rational and specific treatment. Clinicians who regularly deal with patients suffering from pain resulting from peripheral nerve disease are the first to admit the current inadequacies and unpredictability of the present range of treatments that can be offered to this unfortunate group of patients.
The references for this chapter can be found at www.expertconsult.com.
References
Abbott C.A., Malik R.A., Van Ross E.R., et al. Prevalence and characteristics of painful diabetic neuropathy in a large community-based diabetic population in the U.K. Diabetes Care. 2011;34:2220–2224.
Archer A.G., Roberts V.C., Watkins P.J. Blood flow patterns in painful diabetic neuropathy. Diabetologia. 1984;27:563–567.
Arnér S., Lindblom U., Meyerson B.A., et al. Prolonged relief of neuralgia after regional anesthetic blocks. A call for further experimental and systematic clinical studies. Pain. 1990;43:287–297.
Asbury A.K., Aldredge H., Hershberg R., et al. Oculomotor palsy in diabetes mellitus: a clinico-pathological study. Brain. 1970;93:555–566.
Asbury A.K., Fields H.L. Pain due to peripheral nerve damage: an hypothesis. Neurology. 1984;34:1587–1590.
Asbury A.K., Victor M., Dams R.D. Uremic polyneuropathy. Archives of Neurology. 1963;8:413–428.
Axelrod F.B. Hereditary sensory and autonomic neuropathies. Familial dysautonomia and other HSANs. Clinical Autonomic Research. 2002;12(Suppl 1):I/2–I/14.
Baxter D.W., Olszewski J. Congenital universal insensitivity to pain. Brain. 1960;83:381–393.
Bendszus M., Rieckmann P., Perez J., et al. Painful vascular compression syndrome of the sciatic nerve caused by gluteal varicosities. Neurology. 2003;61:985–987.
Bennett M. The LANSS Pain Scale: the Leeds assessment of neuropathic symptoms and signs. Pain. 2001;92:147–157.
Bonica J.J. Causalgia and other sympathetic reflex dystrophies. In: Bonica J.J., ed. The management of pain. Philadelphia: Lea & Febiger; 1990:220–243.
Bouhassira D., Attal N., Alchaar H., et al. Comparison of pain syndromes associated with nervous or somatic lesions and development of a new neuropathic pain diagnostic questionnaire (DN4). Pain. 2005;114:29–36.
Bouhassira D., Attal N., Willer J.C., et al. Painful and painless peripheral sensory neuropathies due to HIV infection: a comparison using quantitative sensory evaluation. Pain. 1999;80:265–272.
Bouhassira D., Lanteri-Minet M., Attal N., et al. Prevalence of chronic pain with neuropathic characteristics in the general population. Pain. 2008;136:380–387.
Boureau F., Doubrere J.F., Luu M. Study of verbal description in neuropathic pain. Pain. 1990;42:145–152.
Britland S.T., Young R.J., Sharma A.K., et al. Acute and remitting painful diabetic polyneuropathy: a comparison of peripheral nerve fiber pathology. Pain. 1992;48:361–370.
Brooks-Wilson A., Morcil M., Clee S.M., et al. Mutations in ABC1 in Tangier disease and familial high-density lipoprotein deficiency. Nature Genetics. 1999;22:336–345.
Brown M.J., Martin J.R., Asbury A.K. Painful diabetic neuropathy. A morphometric study. Archives of Neurology. 1976;33:164–171.
Burchiel K.J., Baumann T.K. Pathophysiology of trigeminal neuralgia: new evidence from a trigeminal ganglion intraoperative microneurographic recording. Case report. Journal of Neurosurgery. 2004;101:872–873.
Callaghan B.C., Hur J., Feldman E.L. Diabetic neuropathy: one disease or two? Current Opinion in Neurology. 2012;25:536–541.
Campbell J.N., Meyer R.A., Raja S.N. Is nociceptor activation by alpha-1 adrenoreceptors the culprit in sympathetically maintained pain. APS Journal. 1992;13:344–350.
Campbell J.N., Raja S.N., Meyer R.A., et al. Myelinated afferents signal the hyperalgesia associated with nerve injury. Pain. 1988;32:89–94.
Chabal C., Jacobson L., Russell L.C., et al. Pain response to perineuronal injection of normal saline, epinephrine, and lidocaine in humans. Pain. 1992;49:9–12.
Chad D.A. Disorders of nerve root and plexuses. In: Bradley W.G., Daroff R.B., Fenichel G.M., et al, eds. Neurology in clinical practice. Boston: Butterworth-Heinemann; 2003:2267–2298.
Chad D.A., Recht L.D. Neuromuscular complications of systemic cancer. Neurologic Clinics. 1991;9:901–918.
Chai J., Herrmann D.N., Stanton M., et al. Painful small-fiber neuropathy in Sjögren syndrome. Neurology. 2005;65:925–927.
Chao C.C., Hsieh S.T., Chiu M.J., et al. Effects of aging on contact heat-evoked potentials: the physiological assessment of thermal perception. Muscle Nerve. 2007;36(1):30–38.
Chen A.C., Niddam D.M., Arendt-Nielsen L. Contact heat evoked potentials as a valid means to study nociceptive pathways in human subjects. Neurosci Lett. 2001;316(2):79–82.
Cline M.A., Ochoa J., Torebjörk H.E. Chronic hyperalgesia and skin warming caused by sensitized C nociceptors. Brain. 1989;112:621–647.
Collins M.P., Periquet M.I. Non-systemic vasculitic neuropathy. Current Opinion in Neurology. 2004;17:587–598.
Cox J.J., Reimann F., Nicholas A.K., et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature. Dec 14 2006;444(7121):894–898.
Craig A.D. Pain mechanisms: labeled lines versus convergence in central processing. Annual Review of Neuroscience. 2003;26:1–30.
Cruickshank E.K. Painful feet in prisoners of war in the Far East. Lancet. 1946;2:369–381.
Cummins T.R., Dib-Hajj S.D., Waxman S.G. Electrophysiological properties of mutant Nav1.7 sodium channels in a painful inherited neuropathy. Journal of Neuroscience. 2004;24:8232–8236.
Daousi C., Benbow S.J., Woodward A., et al. The natural history of chronic painful peripheral neuropathy in a community diabetes population. Diabetic Medicine. 2006;23:1021–1024.
Daousi C., MacFarlane I.A., Woodward A., et al. Chronic painful peripheral neuropathy in an urban community: a controlled comparison of people with and without diabetes. Diabetic Medicine. 2004;21:976–982.
Davis K.D., Treede R.D., Raja S.N., et al. Topical application of clonidine relieves hyperalgesia in patients with sympathetically maintained pain. Pain. 1991;47:309–317.
Davis L.E., Drachman D.B. Myeloma neuropathy. Successful treatment of two patients and review of cases. Archives of Neurology. 1972;27:507–511.
Davis M.D., O’Fallon W.M., Rogers RS, III., et al. Natural history of erythromelalgia: presentation and outcome in 168 patients. Archives of Dermatology. 2000;136:330–336.
Davis M.D., Weenig R.H., Genebriera J., et al. Histopathologic findings in primary erythromelalgia are nonspecific: special studies show a decrease in small nerve fiber density. Journal of the American Academy of Dermatology. 2006;55:519–522.
Delatycki M.B., Williamson R., Forrest S.M. Friedreich ataxia: an overview. Journal of Medical Genetics. 2000;37:1–8.
Denny-Brown D., Adams R.D., Fitzgerald P.J. Pathologic features of herpes zoster: a note on “geniculate herpes,”. Archives of Neurology and Psychiatry. 1944;77:337–349.
Devigili G., Tugnoli V., Penza P., et al. The diagnostic criteria for small fiber neuropathy: from symptoms to neuropathology. Brain. 2008;131:1912–1925.
Dib-Hajj S.D., Cummins T.R., Black J.A., et al. Sodium channels in normal and pathological pain. Annual Review of Neuroscience. 2010;33:325–347.
Dworkin R.H., Carrington D., Cunningham A., et al. Assessment of pain in herpes zoster: lessons learned from antiviral trials. Antiviral Research. 1997;33:73–85.
Dyck P.J., Lambert E.H., O’Brien P.C. Pain in peripheral neuropathy related to rate and kind of fiber degeneration. Neurology. 1976;26:466–471.
Dyck P.J., Thomas P.K. Peripheral neuropathy. Philadelphia: Saunders; 2005.
Dyck P.J., Thomas P.K. Diabetic neuropathy. Philadelphia: Saunders; 1999.
Eames R.A., Lange L.S. Clinical and pathological study of ischaemic neuropathy. Journal of Neurology, Neurosurgery, and Psychiatry. 1967;30:215–226.
Eide P.K., Jorum E., Stubhaug A., et al. Relief of post-herpetic neuralgia with the N-methyl-D-aspartic acid receptor antagonist ketamine: a double-blind, cross-over comparison with morphine and placebo. Pain. 1994;58:347–354.
Estacion M., Han C., Choi J.S., et al. Intra- and interfamily phenotypic diversity in pain syndromes associated with a gain-of-function variant of NaV1.7. Molecular Pain. 2011;7:92.
Faber C.G., Hoeijmakers J.G., Ahn H.S., et al. Gain of function Na(V) 1.7 mutations in idiopathic small fiber neuropathy. Annals of Neurology. 2012;71:26–39.
Fertleman C.R., Baker M.D., Parker K.A., et al. SCN9A mutations in paroxysmal extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes. Neuron. 2006;52:767–774.
Fields H.L., Rowbotham M., Baron R. Postherpetic neuralgia: irritable nociceptors and deafferentation. Neurobiology of Disease. 1998;5:209–227.
Fields H.L., Rowbotham M.C., Devor M. Excitability blockers: anticonvulsants and low concentration local anesthetics in the treatment of chronic pain. In: Dickenson A., Besson J.M., eds. The pharmacology of pain. Berlin: Springer Verlag; 1997:93–116.
Francis T.J.R., Oakley E.H.N. Cold injury. In: Tooke J.E., Lowe G.D.O., eds. A textbook of vascular medicine. London: Arnold; 1996:353–370.
Freynhagen R., Baron R., Gockel U., et al. painDETECT: a new screening questionnaire to identify neuropathic components in patients with back pain. Current Medical Research and Opinion. 2006;22:1911–1920.
Fruhstorfer H., Lindblom U. Sensibility abnormalities in neuralgic patients studied by thermal and tactile pulse stimulation. In: von Euler U., Franzén O., Lindblom U., et al, eds. Somatosensory mechanisms. London: Macmillan; 1984:353–361.
Galer B.S., Jensen M.P. Development and preliminary validation of a pain measure specific to neuropathic pain: the Neuropathic Pain Scale. Neurology. 1997;48:332–338.
Gierthmuhlen J., Maier C., Baron R., et al. Sensory signs in complex regional pain syndrome and peripheral nerve injury. Pain. 2012;153:765–774.
Goldberg Y.P., MacFarlane J., MacDonald M.L., et al. Loss-of-function mutations in the Na(v)1.7 gene underlie congenital indifference to pain in multiple human populations. Clinical Genetics. 2007;71:311–319.
Gorson K.C. Clinical features, evaluation, and treatment of patients with polyneuropathy associated with monoclonal gammopathy of undetermined significance (MGUS). Journal of Clinical Apheresis. 1999;14:149–153.
Gorson K.C., Allam G., Ropper A.H. Chronic inflammatory demyelinating polyneuropathy: clinical features and response to treatment in 67 consecutive patients with and without a monoclonal gammopathy. Neurology. 1997;48:321–328.
Gorson K.C., Ropper A.H. Idiopathic distal small fiber neuropathy. Acta Neurologica Scandinavica. 1995;92:376–382.
Gracely R.H., Lynch S.A., Bennett G.J. Painful neuropathy: altered central processing, maintained dynamically by peripheral input. Pain. 1992;51:175–194.
Griffin J.W., McArthur J.C., Polydefkis M. Assessment of cutaneous innervation by skin biopsies. Current Opinion in Neurology. 2001;14:655–659.
Grisold W., Drlicek M. Paraneoplastic neuropathy. Current Opinion in Neurology. 1999;12:617–625.
Grosskreutz J., Quasthoff S., Kuhn M., et al. Capsaicin blocks tetrodotoxin-resistant sodium potentials and calcium potentials in unmyelinated C fibers of biopsied human sural nerve in vitro. Neuroscience Letters. 1996;208:49–52.
Haanpää M., Attal N., Backonja M., et al. NeuPSIG guidelines on neuropathic pain assessment. Pain. 2011;152:14–27.
Haanpää M., Laippala P., Nurmikko T. Allodynia and pinprick hypesthesia in acute herpes zoster, and the development of postherpetic neuralgia. Journal of Pain and Symptom Management. 2000;20:50–58.
Halperin J.J. Lyme disease and the peripheral nervous system. Muscle & Nerve. 2003;28:133–143.
Han C., Hoeijmakers J.G., Ahn H.S., et al. Nav1.7-related small fiber neuropathy: impaired slow-inactivation and DRG neuron hyperexcitability. Neurology. 2012;78:1635–1643.
Head H., Campbell A.W. The pathology of herpes zoster and its bearing on sensory localisation. Brain. 1900;23:353–523.
Helgason S., Petursson G., Gudmundsson S., et al. Prevalence of postherpetic neuralgia after a first episode of herpes zoster: prospective study with long term follow up. British Medical Journal. 2000;321:794–796.
Herrmann D.N., McDermott M.P., Sowden J.E., et al. Is skin biopsy a predictor of transition to symptomatic HIV neuropathy? A longitudinal study. Neurology. 2006;66:857–861.
Herrmann D.N., O’Connor A.B., Schwid S.R., et al. Broadening the spectrum of controls for skin biopsy in painful neuropathies. Muscle & Nerve. 2010;42:436–438.
Hewitt D.J., McDonald M., Portenoy R.K., et al. Pain syndromes and etiologies in ambulatory AIDS patients. Pain. 1997;70:117–123.
Hoeijmakers J.G., Faber C.G., Lauria G., et al. Small-fiber neuropathies—advances in diagnosis, pathophysiology and management. Nature Reviews. Neurology. 2012;8:369–379.
Holland N.R., Crawford T.O., Hauer P., et al. Small-fiber sensory neuropathies: clinical course and neuropathology of idiopathic cases. Annals of Neurology. 1998;44:47–59.
Houlden H., Blake J., Reilly M.M. Hereditary sensory neuropathies. Current Opinion in Neurology. 2004;17:569–577.
Ibrahim A., Hirschfeld S., Cohen M.H., et al. FDA drug approval summaries: oxaliplatin. Oncologist. 2004;9:8–12.
Irani S.R., Pettingill P., Kleopa K.A., et al. Morvan syndrome: clinical and serological observations in 29 cases. Annals of Neurology. 2012;72:241–255.
Irwin M.S. Nature and mechanism of peripheral nerve damage in an experimental model of non-freezing cold injury. Annals of the Royal College of Surgeons of England. 1996;78:372–379.
Irwin M.S., Sanders R., Green C.J., et al. Neuropathy in non-freezing cold injury (trench foot). Journal of the Royal Society of Medicine. 1997;90:433–438.
Jackson J.L., Gibbons R., Meyer G., et al. The effect of treating herpes zoster with oral acyclovir in preventing postherpetic neuralgia. A meta-analysis. Archives of Internal Medicine. 1997;157:909–912.
Jaeckle K.A., Young D.F., Foley K.M. The natural history of lumbosacral plexopathy in cancer. Neurology. 1985;35:8–15.
Jensen T.S., Baron R., Haanpää M., et al, A new definition of neuropathic pain. Pain. 2011;152;10:2204220–2204225. 10.1016/j.pain.2011.06.017. Epub 2011 Jul 18
Jørum E., Ørstavik K., Schmidt R., et al. Catecholamine-induced excitation of nociceptors in sympathetically maintained pain. Pain. Feb 2007;127(3):296–301.
Jørum E., Warncke T., Stubhaug A. Cold allodynia and hyperalgesia in neuropathic pain: the effect of N-methyl-D-aspartate (NMDA) receptor antagonist ketamine—a double-blind, cross-over comparison with alfentanil and placebo. Pain. 2003;101:229–235.
Kalliomaki M., Kieseritzky J.V., Schmidt R., et al. Structural and functional differences between neuropathy with and without pain? Experimental Neurology. 2011;231:199–206.
Katsarava Z., Yaldizli O., Voulkoudis C., et al. Pain related potentials by electrical stimulation of skin for detection of small-fiber neuropathy in HIV. Journal of Neurology. 2006;253:1581–1584.
Kilo S., Schmelz M., Koltzenburg M., et al. Different patterns of hyperalgesia induced by experimental inflammation in human skin. Brain. 1994;117:385–396.
Kimura J. Electrodiagnosis in diseases of nerve and muscle. Oxford: Oxford University Press; 2001.
Kleggetveit I.P., Namer B., Schmidt R., et al. High spontaneous activity of C-nociceptors in painful polyneuropathy. Pain. 2012;153:2040–2047.
Klein C.J., Lennon V.A., Aston P.A., et al. Chronic pain as a manifestation of potassium channel–complex autoimmunity. Neurology. 2012;79:1136–1144.
Kocen R.S., King R.H., Thomas P.K., et al. Nerve biopsy findings in two cases of Tangier disease. Acta Neuropathologica. 1973;26:317–327.
Kocen R.S., Thomas P.K. Peripheral nerve involvement in Fabry’s disease. Archives of Neurology. 1970;22:81–88.
Koltzenburg M. Afferent mechanisms mediating pain and hyperalgesia in neuralgia. In: Jänig W., Stanton-Hicks M., eds. Reflex sympathetic dystrophy: a reappraisal. Seattle: IASP Press; 1996:123–150.
Koltzenburg M., Bendszus M. Imaging of peripheral nerve lesions. Current Opinion in Neurology. 2004;17:621–626.
Koltzenburg M., Lundberg L.E.R., Torebjörk H.E. Dynamic and static components of mechanical hyperalgesia in human hairy skin. Pain. 1992;51:207–219.
Koltzenburg M., Torebjork H.E., Wahren L.K. Nociceptor modulated central sensitization causes mechanical hyperalgesia in acute chemogenic and chronic neuropathic pain. Brain. 1994;117:579–591.
Kost R.G., Straus S.E. Postherpetic neuralgia—pathogenesis, treatment, and prevention. New England Journal of Medicine. 1996;335:32–42.
Krause S.J., Backonja M.M. Development of a neuropathic pain questionnaire. Clinical Journal of Pain. 2003;19:306–314.
Kremeyer B., Lopera F., Cox J.J., et al. A gain-of-function mutation in TRPA1 causes familial episodic pain syndrome. Neuron. 2010;66:671–680.
Kuhlenbaumer G., Meuleman J., De Jonghe P., et al. Hereditary neuralgic amyotrophy (HNA) is genetically heterogeneous. Journal of Neurology. 2001;248:861–865.
Kuhlenbaumer G., Hannibal M.C., Nelis E., et al. Mutations in SEPT9 cause hereditary neuralgic amyotrophy. Nature Genetics. 2005;37:1044–1046.
Lambert E.H., Dyck P.J. Compound action potentials of sural nerve in vitro in peripheral neuropathy. In: Dyck P.J., Thomas P.K., Griffin J.W., et al, eds. Peripheral neuropathy. Philadelphia: Saunders; 1993:672–684.
LaMotte R.H., Shain C.N., Simone D.A., et al. Neurogenic hyperalgesia: psychophysical studies of underlying mechanisms. Journal of Neurophysiology. 1991;66:190–211.
Lang E., Claus D., Neundorfer B., et al. Parameters of thick and thin nerve-fiber functions as predictors of pain in carpal tunnel syndrome. Pain. 1995;60:295–302.
Lehky T.J., Leonard G.D., Wilson R.H., et al. Oxaliplatin-induced neurotoxicity: acute hyperexcitability and chronic neuropathy. Muscle & Nerve. 2004;29:387–392.
Lhermitte J., Nicholas M. Les lesions spinales du zona. La myelite zosterienne. Revue Neurologique. 1924;2:361–364.
Lindblom U. Analysis of abnormal touch, pain, and temperature sensation in patients. In: Boivie J., Hansson P., Lindblom U., eds. Touch, temperature, and pain in health and disease: mechanisms and assessments. Seattle: IASP Press; 1994:63–84.
Liu M., Max M.B., Robinovitz E., et al. The human capsaicin model of allodynia and hyperalgesia: sources of variability and methods for reduction. Journal of Pain and Symptom Management. 1998;16:10–20.
Loh L., Nathan P.W. Painful peripheral states and sympathetic blocks. Journal of Neurology, Neurosurgery, and Psychiatry. 1978;41:664–671.
LoPachin R.M., Balaban C.D., Ross J.F. Acrylamide axonopathy revisited. Toxicology and Applied Pharmacology. 2003;188:135–153.
Low P.A. Clinical autonomic disorders. Boston: Little, Brown; 1993.
Lu C.I., Huang C.C., Chang Y.C., et al. Short-term thallium intoxication: dermatological findings correlated with thallium concentration. Archives of Dermatology. 2007;143:93–98.
Luciano C.A., Pardo C.A., McArthur J.C. Recent developments in the HIV neuropathies. Current Opinion in Neurology. 2003;16:403–409.
Magri F., Buonocore M., Camera A., et al, Improvement of intra-epidermal nerve fiber density in hypothyroidism after L-thyroxine therapy. Clin Endocrinol (Oxf). 2013;78;1:152–153. 10.1111/j.1365-2265.2012.04447.x.
Maier C., Baron R., Tölle T.R., et al. Quantitative sensory testing in the German Research Network on Neuropathic Pain (DFNS): somatosensory abnormalities in 1236 patients with different neuropathic pain syndromes. Pain. 2010;150:439–450.
Manji H. Neuropathy in HIV infection. Current Opinion in Neurology. 2000;13:589–592.
Martin C., Solders G., Sonnerborg A., et al. Painful and non-painful neuropathy in HIV-infected patients: an analysis of somatosensory nerve function. European Journal of Pain. 2003;7:23–31.
Martyn C., Hughes R.A.C. Peripheral neuropathies. In: Martyn C.N., Hughes R.A.C., eds. The epidemiology of neurological disorders. London: BMJ Books; 1998:96–117.
Mauermann M.L., Sorenson E.J., Dispenzieri A., et al. Uniform demyelination and more severe axonal loss distinguish POEMS syndrome from CIDP. Journal of Neurology, Neurosurgery, and Psychiatry. 2012;83:480–486.
Meyer-Rosberg K., Kvarnstrom A., Kinnman E., et al. Peripheral neuropathic pain—a multidimensional burden for patients. European Journal of Pain. 2001;5:379–389.
Montgomery R.D., Cruickshank E.K., Robertson W.B., et al. Clinical and pathological observations on Jamaican neuropathy; a report on 206 cases. Brain. 1964;87:425–462.
Morgan S.H., Rudge P., Smith S.J., et al. The neurological complications of Anderson-Fabry disease (alpha-galactosidase A deficiency)—investigation of symptomatic and presymptomatic patients. Quarterly Journal of Medicine. 1990;75:491–507.
Mumenthaler M., Schliack H., Stöhr M. Läsionen peripherer Nerven und radikuläre Syndrome. Stuttgart: Thieme; 1998.
Namer B., Kleggetveit I.P., Handwerker H., et al, Role of TRPM8 and TRPA1 for cold allodynia in patients with cold injury. Pain. 2008;139;1:63–72. 10.1016/j.pain.2008.03.007. Epub 2008 Apr 25
Nasu S., Misawa S., Sekiguchi Y., et al. Different neurological and physiological profiles in POEMS syndrome and chronic inflammatory demyelinating polyneuropathy. Journal of Neurology, Neurosurgery, and Psychiatry. 2012;83:476–479.
Nordin M., Nyström B., Wallin U., et al. Ectopic sensory discharges and paresthesiae in patients with disorders of peripheral nerves, dorsal roots and dorsal columns. Pain. 1984;20:231–245.
Nyström B., Hagbarth K.E. Microelectrode recordings from transected nerves in amputees with phantom limb pain. Neuroscience Letters. 1981;27:211–216.
Ochoa J. Isoniazid neuropathy in man: quantitative electron microscope study. Brain. 1970;93:831–850.
Ochoa J., Torebjörk H.E., Culp W.J., et al. Abnormal spontaneous activity in single sensory nerve fibers in humans. Muscle & Nerve. 1982;5:S74–S77.
Ochoa J.L., Yarnitsky D. Mechanical hyperalgesias in neuropathic pain patients: dynamic and static subtypes. Annals of Neurology. 1993;33:465–472.
Ochoa J.L., Yarnitsky D. The triple cold syndrome: cold hyperalgesia, cold hypoaesthesia and cold skin in peripheral nerve disease. Brain. 1994;117:185–197.
Ochs G., Schenk M., Struppler A. Painful dysaesthesias following peripheral nerve injury: a clinical and electrophysiological study. Brain Research. 1989;496:228–240.
Ørstavik K., Namer B., Schmidt R., et al. Abnormal function of C-fibers in patients with diabetic neuropathy. Journal of Neuroscience. 2006;26:11287–11294.
Ørstavik K., Weidner C., Schmidt R., et al. Pathological C-fibers in patients with a chronic painful condition. Brain. 2003;126:567–578.
Pan C.L., Tseng T.J., Lin Y.H., et al. Cutaneous innervation in Guillain-Barré syndrome: pathology and clinical correlations. Brain. 2003;126:386–397.
Pappagallo M., Oaklander A.L., Quatrano-Piacentini A.L., et al. Heterogenous patterns of sensory dysfunction in postherpetic neuralgia suggest multiple pathophysiologic mechanisms. Anesthesiology. 2000;92:691–698.
Pardo C.A., McArthur J.C., Griffin J.W. HIV neuropathy: insights in the pathology of HIV peripheral nerve disease. Journal of the Peripheral Nervous System. 2001;6:21–27.
Park S.B., Lin C.S., Krishnan A.V., et al. Dose effects of oxaliplatin on persistent and transient Na+ conductances and the development of neurotoxicity. PLoS One. 2011;6:e18469.
Penno A., Reilly M.M., Houlden H., et al. Hereditary sensory neuropathy type 1 is caused by the accumulation of two neurotoxic sphingolipids. Journal of Biological Chemistry. 2010;285:11178–11187.
Petersen K.L., Rowbotham M.C. Natural history of sensory function after herpes zoster. Pain. 2010;150:83–92.
Plant G.T., Perry V.H. The anatomical basis of the caecocentral scotoma. New observations and a review. Brain. 1990;113:1441–1457.
Pollard J.D., Mcleod J.G., Honnibal T.G., et al. Hypothyroid polyneuropathy. Clinical, electrophysiological and nerve biopsy findings in two cases. Journal of the Neurological Sciences. 1982;53:461–471.
Polydefkis M., Allen R.P., Hauer P., et al. Subclinical sensory neuropathy in late-onset restless legs syndrome. Neurology. 2000;55:1115–1121.
Polydefkis M., Griffin J.W., McArthur J. New insights into diabetic polyneuropathy. JAMA: Journal of the American Medical Association. 2003;290:1371–1376.
Polydefkis M., Hauer P., Sheth S., et al. The time course of epidermal nerve fiber regeneration: studies in normal controls and in people with diabetes, with and without neuropathy. Brain. 2004;127:1606–1615.
Polydefkis M., Yiannoutsos C.T., Cohen B.A., et al. Reduced intraepidermal nerve fiber density in HIV-associated sensory neuropathy. Neurology. 2002;58:115–119.
Portenoy R. Development and testing of a neuropathic pain screening questionnaire: ID pain. Current Medical Research and Opinion. 2006;22:1555–1565.
Posner J.B. Neurological complications of cancer. Philadelphia: Davis; 1995.
Price D.D., Bennett G.J., Rafii A. Psychophysical observations on patients with neuropathic pain relieved by a sympathetic block. Pain. 1989;36:273–288.
Price D.D., Long S., Huitt C. Sensory testing of pathophysiological mechanisms of pain in patients with reflex sympathetic dystrophy. Pain. 1992;49:163–173.
Rasmussen P.V., Sindrup S.H., Jensen T.S., et al. Symptoms and signs in patients with suspected neuropathic pain. Pain. 2004;110:461–469.
Rosen L., Eltvik L., Arvesen A., et al. Local cold injuries sustained during military service in the Norwegian Army. Arctic Medical Research. 1991;50:159–165.
Rotthier A., Baets J., Timmerman V., et al. Mechanisms of disease in hereditary sensory and autonomic neuropathies. Nature Reviews. Neurology. 2012;8:73–85.
Rowbotham M.C., Davies P.S., Fields H.L. Topical lidocaine gel relieves postherpetic neuralgia. Annals of Neurology. 1995;37:246–253.
Rubin D.I. Neuralgic amyotrophy: clinical features and diagnostic evaluation. Neurologist. 2001;7:350–356.
Said G. Vasculitic neuropathy. Current Opinion in Neurology. 1999;12:627–629.
Said G., Elgrably F., Lacroix C., et al. Painful proximal diabetic neuropathy: inflammatory nerve lesions and spontaneous favorable outcome. Annals of Neurology. 1997;41:762–770.
Said G., Lacroix C., Lozeron P., et al. Inflammatory vasculopathy in multifocal diabetic neuropathy. Brain. 2003;126:376–385.
Schiffmann R. Fabry disease. Pharmacology & Therapeutics. 2009;122:65–77.
Schiffmann R., Hauer P., Freeman B., et al. Enzyme replacement therapy and intraepidermal innervation density in Fabry disease. Muscle & Nerve. 2006;34:53–56.
Schilder P., Schmidt B.J., Leon L. Asymbolia for pain. Archives of Neurology and Psychiatry. 1931;25:598–600.
Schmelz M., Schmidt R., Ringkamp M., et al. Sensitization of insensitive branches of nociceptors in human skin. Journal of Physiology. 1994;480:389–394.
Schmidt R., Kleggetveit I.P., Namer B., et al. Double spikes to single electrical stimulation correlates to spontaneous activity of nociceptors in painful neuropathy patients. Pain. 2012;153:391–398.
Schmidt R., Schmelz M., Torebjork H.E., et al. Mechano-insensitive nociceptors encode pain evoked by tonic pressure to human skin. Neuroscience. 2000;98:793–800.
Scott L.J., Griffin J.W., Luciano C., et al. Quantitative analysis of epidermal innervation in Fabry disease. Neurology. 1999;52:1249–1254.
Seo J.H., Ryan H.F., Claussen G.C., et al. Sensory neuropathy in vasculitis: a clinical, pathologic, and electrophysiologic study. Neurology. 2004;63:874–878.
Serra J., Bostock H., Solà R., Aleu J., et al, Microneurographic identification of spontaneous activity in C-nociceptors in neuropathic pain states in humans and rats. Pain. 2012;153;1:42–55. 10.1016/j.pain.2011.08.015. Epub 2011 Oct 10
Serra J., Sola R., Quiles C., et al. C-nociceptors sensitized to cold in a patient with small-fiber neuropathy and cold allodynia. Pain. 2009;147:46–53.
Shatzky S., Moses S., Levy J., et al. Congenital insensitivity to pain with anhidrosis (CIPA) in Israeli-Bedouins: genetic heterogeneity, novel mutations in the TRKA/NGF receptor gene, clinical findings, and results of nerve conduction studies. American Journal of Medical Genetics. 2000;92:353–360.
Shun C.T., Chang Y.C., Wu H.P., et al. Skin denervation in type 2 diabetes: correlations with diabetic duration and functional impairments. Brain. 2004;127:1593–1605.
Singer W., Spies J.M., McArthur J., et al. Prospective evaluation of somatic and autonomic small fibers in selected autonomic neuropathies. Neurology. 2004;62:612–618.
Spillane J.D. Nutritional disorders of the nervous system. Baltimore: Williams & Wilkins; 1947.
Strachan H. On a form of multiple neuritis prevalent in the West Indies. Practitioner. 1897;59:477–484.
The Cuba Neuropathy Field Investigation Team. Epidemic optic neuropathy in Cuba—clinical characterization and risk factors. New England Journal of Medicine. 1995;333:1176–1182.
Thomas P.K. Selective vulnerability of the centrifugal and centripetal axons of primary sensory neurons. Muscle & Nerve. 1982;5:S117–S121.
Thomas P.K. The anatomical substratum of pain evidence derived from morphometric studies on peripheral nerve. Canadian Journal of Neurological Sciences. 1974;1:92–97.
Thomas P.K., Holdorff B. Neuropathy due to physical agents. In: Dyck P.J., Thomas P.K., Griffin J.W., et al, eds. Peripheral neuropathy. Philadelphia: Saunders; 1993:990–1013.
Thomas P.K., Hollinrake K., Lascelles R.G., et al. The polyneuropathy of chronic renal failure. Brain. 1971;94:761–780.
Thomas P.K., King R.H. Peripheral nerve changes in amyloid neuropathy. Brain. 1974;97:395–406.
Thomas P.K., Plant G.T., Baxter P., et al. An epidemic of optic neuropathy and painful sensory neuropathy in Cuba: clinical aspects. Journal of Neurology. 1995;242:629–638.
Thyregod H.G., Rowbotham M.C., Peters M., et al. Natural history of pain following herpes zoster. Pain. 2007;128:148–156.
Torebjörk E. Clinical and neurophysiological observations relating to pathophysiological mechanisms in reflex sympathetic dystrophy. In: Stanton-Hicks M., Jänig W., Boas R.A., eds. Reflex sympathetic dystrophy. Boston: Kluwer; 1990:71–80.
Torebjörk E., Wahren L., Wallin G., et al. Noradrenaline-evoked pain in neuralgia. Pain. 1995;63:11–20.
Treede R.D., Jensen T.S., Campbell J.N., et al. Neuropathic pain: redefinition and a grading system for clinical and research purposes. Neurology. 2008;70:1630–1635.
Treede R.D., Lorenz J., Baumgartner U. Clinical usefulness of laser-evoked potentials. Neurophysiologie Clinique. 2003;33:303–314.
Üçeyler N., He L., Schonfeld D., et al. Small fibers in Fabry disease: baseline and follow-up data under enzyme replacement therapy. Journal of the Peripheral Nervous System. 2011;16:304–314.
Ungley C.C., Blackwood W. Peripheral vasoneuropathy after chilling. Lancet. 1942;240:447–451.
Verdugo R., Ochoa J.L. Quantitative somatosensory thermotest. A key method for functional evaluation of small calibre afferent channels. Brain. 1992;115:893–913.
Wahren L.K., Torebjörk E. Quantitative sensory tests in patients with neuralgia 11 to 25 years after injury. Pain. 1992;48:237–244.
Wahren L.K., Torebjörk E., Jørum E. Central suppression of cold-induced C fiber pain by myelinated fiber input. Pain. 1989;38:313–319.
Walsh J.C. The neuropathy of multiple myeloma. An electrophysiological and histological study. Archives of Neurology. 1971;25:404–414.
Walsh J.C., Mcleod J.G. Alcoholic neuropathy. An electrophysiological and histological study. Journal of the Neurological Sciences. 1970;10:457–469.
Wartenberg R. Neuritis, sensory neuritis, neuralgia. New York: Oxford University Press; 1958.
Wasner G., Schwarz K., Schattschneider J., et al. Interaction between histamine-induced itch and experimental muscle pain. European Journal of Pain. 2004;8:179–185.
Watson C.P.N., Deck J.H., Morshead C., et al. Post-herpetic neuralgia: further post-mortem studies of cases with and without pain. Pain. 1991;44:105–117.
Watson C.P.N., Evans R.J., Watt V.R., et al. Post-herpetic neuralgia: 208 cases. Pain. 1988;35:289–297.
Watson C.P.N., Watt V.R., Chipman M., et al. The prognosis with postherpetic neuralgia. Pain. 1991;46:195–199.
Williams L.S., Jones W.J., Shen J., et al. Outcomes of newly referred neurology outpatients with depression and pain. Neurology. 2004;63:674–677.
Woolf C.J., Bennett G.J., Doherty M., et al. Towards a mechanism-based classification of pain? Pain. 1998;77:227–229.
Yang Y., Wang Y., Li S., et al. Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. Journal of Medical Genetics. 2004;41:171–174.
Yarnitsky D., Ochoa J.L. Release of cold-induced burning pain by block of cold-specific afferent input. Brain. 1990;113:893–902.
Zachs S.I., Langfit T.W., Elliot F.A. Herpetic neuritis: a light and electron microscopic study. Neurology. 1964;14:644–750.
Zaslansky R., Yarnitsky D. Clinical applications of quantitative sensory testing (QST). Journal of the Neurological Sciences. 1998;153:215–238.
Ziegler E.A., Magerl W., Meyer R.A., et al. Secondary hyperalgesia to punctate mechanical stimuli. Central sensitization to A-fiber nociceptor input. Brain. 1999;122:2245–2257.
Zorilla E., Kozak G.P. Ophthalmoplegia in diabetes mellitus. Annals of Internal Medicine. 1967;67:968–976.