26
Central Sympathetic Agents and Direct Vasodilators
Kazuomi Kario
Mechanism of Action
Central sympatholytics (e.g., methyldopa, guanabenz, guanfacine, clonidine, moxonidine, and rilmenidine) have a variety of antihypertensive actions1,2 that result in increased sodium excretion and decreases in the cardiac output, heart rate, total peripheral resistance, and renin release. Central sympatholytics cross the blood-brain barrier and stimulate the imidazoline 1 (I1) receptors and/or central postsynaptic alpha2 (α2) adrenoceptors in the brainstem’s sympathetic nervous control centers, the rostral ventrolateral medulla (RVLM) and the nucleus tractus solitarii (NTS). As shown in Fig. 26.1, the various central sympatholytics have differing affinities for these two types of receptors. Moxonidine and rilmenidine selectively stimulate the I1-imidazoline receptors. Methyldopa, guanabenz, and guanfacine selectively stimulate α2-adrenoceptors more than the I1-imidazoline receptors, and clonidine nonselectively stimulates both α2-adrenoceptors and I1-imidazoline receptors.
Treatment with one of the central sympatholytics that stimulate α2-adrenoceptors (e.g., methyldopa, clonidine, guanabenz, and guanfacine) is frequently accompanied by adverse effects such as dry mouth, decreased alertness, sedation, and depression. This is because α2-adrenoceptors are present not only in the RVLM but also in the NTS, nucleus coeruleus, and salivary grands. Treatment with a central sympatholytic that selectively stimulates only I1-imidazoline receptors (e.g., rilmenidine or moxonidine) results in central adverse effects much less frequently, because the I1-imidazoline receptors are located almost exclusively in the RVLM.
Perhaps the oldest agent (founded in the 1930s in India) that affects the sympathetic nervous system is reserpine. In contrast, the adrenergic uptake inhibitor reserpine depletes catecholamine storage in both the central and peripheral nervous systems and is associated with many dose-dependent side effects. It is no longer available in the United States.
Hemodynamic Effects
Stimulating the brainstem’s central I1-imidazoline receptors or α2-adrenoceptors results in several hemodynamic, neurohumoral, and adverse effects, because the stimulation directly inhibits the sympathetic outflow to the heart and blood vessels (Box 26.1).3 For the main pharmacodynamic effect of compounds in this class to occur, the drugs must pass the blood-brain barrier. As such, there is an implicit time lag between the plasma drug concentration that is achieved and the antihypertensive effect.
The blood pressure (BP)-lowering effect of the central sympatholytics is based on the following: a reduction in norepinephrine; decreased peripheral resistance and decreased cardiac output at rest and during exercise; reduced baroreflex to compensate for the decrease in BP, resulting in relative bradycardia with exaggerated hypotension on standing; decreased plasma levels of aldosterone, angiotensin II, and renin; and preserved glomerular filtration and renal blood flow despite the reduction in BP. However, central sympatholytics often cause fluid retention as overcompensation, and this limits their effectiveness.
Clinical Application
The significant overall effectiveness in reducing BP is a major advantage of the central sympatholytics.4 They are also useful for treating labile hypertensive patients with associated anxiety, especially when the anxiety is manifested by sympathetic hyperactivity. However, central sympatholytics are currently used less frequently because of their adverse effects, which can be significant. The centrally acting effects of the central sympatholytics(such as depression and sedation)are of particular concern.
Central sympatholytics are also not recommended for use as first-line or second-line monotherapy because they are often less effective in this role and have not been shown in clinical trials to reduce mortality. Moreover, most are associated with a dose-dependent rebound hypertension when abruptly stopped. A combination of a central sympatholytic with a thiazide-type diuretic is often used to manage resistant hypertension and occasionally hypertension in pregnant women, if beta-blockers are contraindicated. In resistant hypertension, these agents are added when combinations of three or more other antihypertensive drugs such as a calcium channel blocker, angiotensin-converting enzyme (ACE) inhibitor, angiotensin receptor blocker (ARB), or diuretic have failed to control blood pressure.
Central sympatholytics can be used safely in individuals with diabetes, with no significant loss of glycemic control. They can also be used safely for individuals with pulmonary diseases such as asthma. Intravenous preparations are available only in certain countries for clonidine and α-methyldopa, and clonidine is the only compound in this class that can be administered via a transdermal delivery system.
The central sympatholytics’ quick onset and long duration of action distinguish these drugs. The most rapid onset of action is seen in clonidine, at 30 to 60 minutes.
Adverse Effects
The most common adverse effects of the central sympatholytics that stimulate α2-adrenoceptors are sedation and dry mouth (40%), and these effects are the main reason that the use of central sympatholytics has declined. In addition, the sedative effects of drugs within this class are enhanced by other central nervous system (CNS) depressants such as antihistamines, benzodiazepines, sedative-hypnotics, and ethanol. The central sympatholytics that stimulate I1-imidazoline receptors do not cause as much sedation and dry mouth, and they are better tolerated by most patients. Dry mouth can be annoying, and the decreased level of saliva can increase an individual’s risk of dental caries and periodontal disease (Table 26.1).
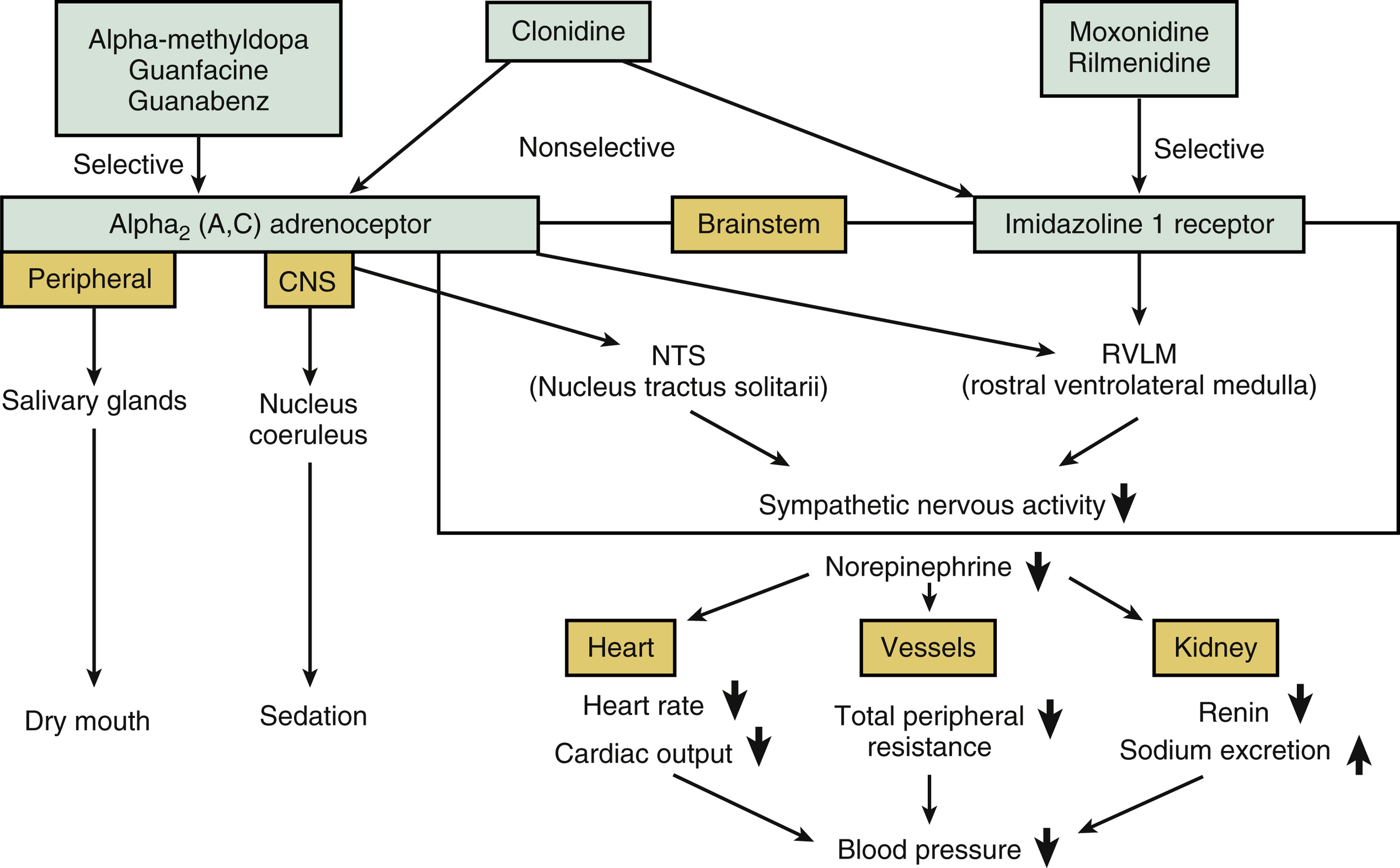
FIG. 26.1 Antihypertensive mechanisms of central sympatholytic agents. Central sympatholytic agents activate α2A and α2C, or imidazoline 1 receptors in the brainstem, resulting in decreases in heart rate, cardiac output, total peripheral resistance, and renin release, and an increase in sodium excretion.
The Central Sympatholytics
The pharmacodynamics, available preparations, daily dosages, contraindications, and adverse effects of the various central sympatholytics are listed in Table 26.1.
Clonidine
Clonidine, the most widely used central sympatholytic.2,5-8 The onset of action for oral clonidine is 30 to 60 minutes, which is advantageous for hypertensive urgencies. For primary hypertension, clonidine is recommended for use as the fourth or fifth line of therapy. Menopause-associated vasomotor syndrome symptoms such as hot flushes, and sympathetic hyperactivity-related hypertension with restless legs syndrome have all been treated successfully with clonidine. Clonidine has also been shown to be more effective in whites than in African Americans, and in older than in younger African Americans. However, in a blinded, randomized trial with a 2-by-2 factorial design study, it was shown that low-dose clonidine (0.2 mg per day) did not reduce the rate of the composite outcome (death or nonfatal myocardial infarction) and increase the risk of clinically important hypotension and nonfatal cardiac arrest in patients undergoing noncardiac surgery.9
Antihypertensive Effects
Clonidine is easily absorbed. The plasma levels of clonidine peak within 30 to 60 minutes after its oral administration, and its plasma half-life is 6 to 13 hours. The BP reduction peaks at 3 to 5 hours, and the BP-lowering effect lasts 8 to 12 hours. Oral clonidine preparations include 0.1-, 0.2-, and 0.3-mg dosages. Clonidine treatment is often initiated at 0.1 mg 2× per day and then gradually increased to a maximum dose of 2.4 mg per day.
Transdermal (patch) clonidine is particularly effective for treating labile hypertensive patients who need multiple medications, those who cannot take oral medications, and those with prominent early morning BP surges. Transdermal clonidine is available in three preparations: 2.5, 5.0, and 7.5 mg. The best absorption from a clonidine patch is obtained by placing the patch on the chest or upper arm. An optimal transdermal delivery system provides a constant clonidine dose for 7 days, and the peak effect is reached within days 1 to 2 days. The BP-lowering effect of transdermal clonidine lasts 8 to 24 hours after the patch is removed. However, compared with oral clonidine, at equivalent doses transdermal clonidine treatment is more likely to result in a dose-dependent retention of both water and salt.
Adverse Effects
Dry mouth and sedation, the most frequent adverse effects of clonidine treatment, are more common with clonidine than methyldopa, but clonidine does not present a risk of autoimmune hepatic damage as methyldopa does. Other potential adverse effects of clonidine are headache, impotence, and orthostatic hypotension. Known contraindications for clonidine are sick sinus syndrome and second-degree and third-degree atrioventricular (AV) block, because clonidine’s depression of sinus and atrioventricular nodal function may result in bradycardia.
TABLE 26.1
| Drug | Preparation | Pharmacodynamics | Daily Dosage | Adverse Effects | Contraindications |
| Clonidine: Oral | 0.1 mg 0.2 mg 0.3 mg | Onset: 0.5-1 hour Peak: 3-5 hours Plasma half-life: 12-16 hours Metabolism: liver | Initial: 0.1 mg Range: 0.2-1.2 mg Max.: 1.2 mg usually bid | Sedation, drowsiness, dry mouth, withdrawal syndrome, rebound hypertension (uncommon with doses <1.2 mg qd), headache, bradycardia, orthostatic hypotension, impotence (uncommon; 4%) | Sick sinus syndrome 2nd- and 3rd-degree atrioventricular block |
| Transdermal | 1 (containing 2.5 mg) 2 (containing 5.0 mg) 3 (containing 7.5 mg) | Duration of BP lowering: 1 week | 1, 2, 3 once weekly | ||
| Methyldopa | 125 mg 250 mg 500 mg | Onset: 2-3 hours Peak: 5 hours Plasma half-life: 12 hours Metabolism: renal | Avg.: 250-300 mg bid Max.: 3000 mg | Sedation, drowsiness, depression, dry mouth, positive Coombs test and anemia, lupuslike syndrome, withdrawal syndrome, rebound hypertension | Active hepatic disease |
| Guanabenz | 4 mg 8 mg | Onset: 1 hour Peak: 4 hours Plasma half-life 6 hours Metabolism: 75% Excretion: renal 80% | Average 16 mg Range 8-48 mg Maximum 48 mg | Sedation, drowsiness, dry mouth, withdrawal syndrome, rebound hypertension, impotence | Pregnancy |
| Guanfacine | 1 mg 2 mg | Onset: 1 hour Peak: 4 hours Plasma half-life: 12 hours Excretion: renal | 1 mg at bedtime Maximum 3 mg | Same as clonidine | Allergy to guanfacine |

Clonidine treatment is also subject to the risk of the occurrence of rebound syndrome and discontinuation syndrome. When treatment with any antihypertensive drug is abruptly stopped, the discontinuation syndrome may occur at different degrees of severity as follows: a rapid but asymptomatic return of the BP to the patient’s pretreatment level, a rebound of the BP with sympathetic hyperactivity symptoms, and the patient’s BP overshoots the pretreatment level. The central sympatholytic most frequently cited as resulting in discontinuation syndrome is clonidine (especially at ≥ 1.0 mg), caused by the rapid return of the catecholamine level, which is suppressed by clonidine treatment. Discontinuation syndrome is exacerbated in the presence of a β-blockade, but not in the presence of the α/β adrenergic antagonist labetalol or carvedilol. If discontinuation syndrome is detected in a patient who was treated with clonidine, the clonidine should be restarted, and the symptoms can be expected to quickly resolve.
Rebound hypertension may occur with clonidine transdermal treatment, but it is observed much less frequently than after clonidine oral administration. Skin hypersensitivity (e.g., allergic dermatitis) in response to a clonidine patch occurs in up to 20% of patients, most commonly in white and female patients.
Methyldopa
Methyldopa6-8,10 is used mainly to treat hypertension in pregnant women. It is not teratogenic and has been shown to produce no fetal adverse effects in utero. Methyldopa treatment maintains uterine perfusion and does not hinder the maternal cardiac output or renal or uterine blood flow. As the α-methylated derivative of dopa (the natural precursor of dopamine and norepinephrine), methyldopa is a suitable alternative to clonidine for patients for whom rebound hypertension or intolerable adverse effects preclude the use of clonidine.
Methyldopa is often used for hypertensive emergencies. It is available as an intravenous formulation (as the parent drug ester) at the typical intravenous dose range (for α-methyldopa) 20 to 40 mg per kg per day in divided doses every 6 hours.
Antihypertensive Effect
A relatively slow onset of action is a feature of methyldopa treatment, and the BP-lowering effect starts approximately 2 to 3 hours after dosing (cf. clonidine’s onset at 0.5 to 1.0 hour). At approximately 5 hours after an oral dose of methyldopa, the patient’s BP reaches its lowest point, and the effect persists for up to 24 hours. The commonly used initial dose of methyldopa is 250 mg 2× per day, titrated up to 3.0 g at most. In patients with renal insufficiency, this methyldopa dose should be halved.
Methyldopa also effectively reduces supine BP without producing orthostatic hypotension.
Adverse Effects
Methyldopa’s adverse effects include sedation and drowsiness, dry mouth, depression, postural hypotension, fluid retention, rebound hypertension, withdrawal syndrome, and various autoimmune reactions including flulike high fever, hepatitis, Coombs-positive hemolytic anemia, and lupuslike syndrome. Between 10% and 20% of patients who are treated with α-methyldopa (≥1 g/d) over a period of several months develop one or more of these adverse events. Occasionally, methyldopa treatment has resulted in drug-induced hepatitis with fever, eosinophilia, and increased transaminase values, but this is a self-limited process that resolves with discontinuation of the methyldopa. It is not necessary to stop methyldopa treatment in asymptomatic patients who become Coombs-positive but do not develop hemolytic anemia.
Guanabenz
The direct central α2-agonist guanabenz6-8 acts in the same manner as methyldopa and has similar adverse effects, but it has the advantage of not causing reactive fluid retention. Compared with clonidine, guanabenz treatment is less effective, but it results in rebound hypertension and orthostatic hypotension less frequently. The efficacy of guanabenz in reducing left ventricular hypertrophy in hypertensive patients and in attenuating morning hypertension when administered at nighttime has also been demonstrated.
Antihypertensive Effect
Guanabenz has a 1-hour onset of antihypertensive action. The most commonly used initial dose of guanabenz is 4 mg 2× per day, titrated to a maximum of 64 mg daily. It is eliminated predominantly via hepatic biotransformation. Thus, unlike clonidine, guanabenz dose adjustment is not required in patients with renal failure but is required in those with chronic liver diseases.
Guanabenz has also been shown to reduce total cholesterol levels by 10% to 20%.
Adverse Effects
The potential adverse effects of guanabenz include dry mouth, sedation, drowsiness, and impotence. Guanabenz treatment may also be followed by withdrawal syndrome and rebound hypertension. The adverse effects of guanabenz are essentially the same as those of clonidine.
Guanfacine
Unlike the other members of the class of central sympatholytics, the 17-hour duration of guanfacine’s action typically allows it to be dosed once daily.6-8 It is thought that compared with guanabenz, guanfacine enters the brain more slowly; its antihypertensive effect lasts longer than that of guanabenz. Evening dosing is preferable for guanfacine because its peak effect can be aligned with early-morning catecholamine and BP surges, and the potential sedating effect of guanfacine can play out during sleep. As with other central sympatholytics, the optimal effect of guanfacine can be achieved when it is coadministered with a low-dose diuretic, providing a BP-lowering effect with minimum CNS adverse effects. Patients who are intolerant to clonidine because of it strong sedation effect may benefit from treatment with guanfacine as an alternative.
Adverse Effects
Compared with clonidine, guanfacine has fewer CNS adverse effects and is much less likely to have withdrawal symptoms. The risk of adverse effects from guanfacine treatment increase significantly when doses greater than1 mg daily are administered.
Imidazoline Receptor Agonists
The imidazoline receptor agonists rilmenidine and moxonidine act on the RVLM’s imidazoline receptors.2 The α2-adrenergic receptors are less abundant in the RVLM. Moxonidine and rilmenidine effectively suppress sympathetic nervous activity without causing the adverse reactions observed with clonidine or methyldopa treatment such as sedation and dry mouth. In addition, the rebound syndrome caused by clonidine discontinuation has not been observed in cases of moxonidine or rilmenidine treatment.
Moxonidine
Moxonidine therapy effectively reduces BP although it does not reduce the heart rate as clonidine treatment can.7 Moxonidine’s plasma half-life is only 2 to 3 hours, and its extended duration of action suggests prolonged binding to central I1-imidazoline receptors. The dose of moxonidine must be adjusted according to the patient’s glomerular filtration rate (GFR), because moxonidine is extensively cleared by the kidneys.
For example, for patients with moderate renal impairment (i.e., a GFR 30 to 60 mL/min), single-dose moxonidine should not exceed 0.2 mg, and the daily dose should not go beyond 0.4 mg. For patients with severe renal impairment (i.e., a GFR < 30 mL/min), moxonidine should not be used. The same is true of patients with advanced heart failure; in a large cohort of New York Heart Association class II–IV heart failure patients with reduced ejection fraction, a sustained-release form of moxonidine that was force-titrated to 1.5 mg 2× per day was observed to be associated with early increases in morbidity and mortality.
Rilmenidine
For mild-to-moderate hypertension, oral rilmenidine (1 to 2 mg per day) is effective and well-tolerated, alone or in combination with another antihypertensive medication.7 The most favorable ratio of efficacy to tolerability has been observed with a 1-mg daily dose. Parasympathetic tone is increased by rilmenidine, which may account for its lack of an effect on the heart rate as it works to reduce BP.
Central and Peripheral Adrenergic Inhibitors
Reserpine
The only peripheral adrenergic inhibitor that is currently used is reserpine,5,8 which depletes norepinephrine by blocking the transport of norepinephrine into its storage granules at the site of postganglionic sympathetic nerve endings. Reserpine treatment decreases peripheral vascular resistance because the concentration of neurotransmitters is lower even when sympathetic nerves are stimulated. Catecholamines are also depleted by reserpine treatment, not only in peripheral sympathetic nerves, but also in the brain and other tissues. This CNS effect accounts for the adverse central reactions to reserpine, which include sedation, depression, and nasal congestion. In the myocardium, this may contribute to decreases in the heart rate and cardiac contractility. For drug-resistant hypertension, reserpine is now used as the fourth- or fifth-line drug in multiple-drug regimens.
Antihypertensive Effect
Although reserpine is extremely long-acting, only a relatively mild BP-lowering effect is obtained with reserpine monotherapy (mean BP reduction of 3/5 mm Hg). When administered with a diuretic, reserpine induces a significant regression of left ventricular hypertrophy.
Adverse Effects
When reserpine is used at a low dose, adverse effects are relatively infrequent. The minor adverse effects include nasal congestion. Clinically serious sedation and depression are rare side effects of reserpine treatment.
Direct Vasodilators
Direct vasodilators work by entering the vascular smooth muscle cells, whereas indirect vasodilators prevent the entry of calcium into the smooth muscle cells that initiate vasoconstriction (calcium channel blockers), or they inhibit hormonal vasoconstrictor mechanisms (e.g., ACE inhibitors and ARBs), or they block α-adrenergic receptor-mediated vasoconstriction (α1-blockers).
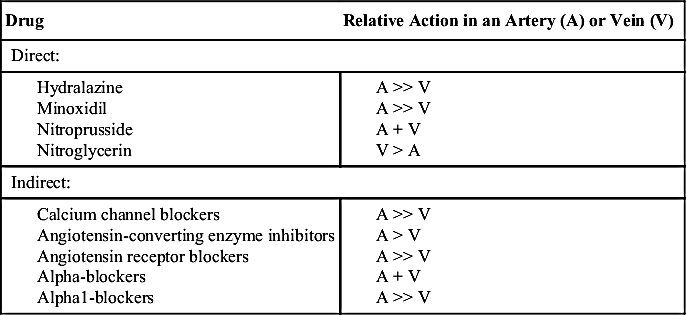
The vasodilator effect of the direct vasodilators (hydralazine, minoxidil, nitroprusside, and nitroglycerin) differs among large conduit arteries, small branch arteries, arterioles, and veins (Table 26.2). When conduit arteries are relaxed, their compliance increases and systolic and pulse pressures tend to become lower. When small arteries and arterioles are relaxed, wave reflection and systemic vascular resistance are reduced. When veins are relaxed, systemic capacitance is increased and the central venous pressure is lowered. The overall hemodynamic effect of the administration of vasodilators is affected by the balance of these effects of individual drugs, combined with the reflex neurohormonal response.
Minoxidil and hydralazine act by dilating resistance arterioles, thereby reducing peripheral resistance. Baroreflex-mediated venoconstriction occurs, resulting in an increase in the venous return to the heart and direct catecholamine-mediated positive inotropic and chronotropic stimulation of the heart (Fig. 26.2). These two drugs have no dilating effect on the venous side of the circulation.
The direct vasodilator sodium nitroprusside is used to lower BP in hypertensive crises and to treat severe left ventricular failure; it is particularly valuable when a patient’s survival is threatened by elevated pressure or severe left ventricular failure.
Nitrates are effective in producing sustained BP reductions when they are added to other antihypertensive regimens, but they are not yet used widely as antihypertensive agents. A recent systematic review demonstrated that when a nitrate and hydralazine were used together in some chronic heart failure trials, morbidity and mortality were reduced.
TABLE 26.2
Vasodilator Drugs for the Management of Hypertension
Hydralazine
The classic direct arteriolar dilator is hydralazine,11-13 which lowers the total peripheral resistance and BP levels by directly relaxing the smooth muscle cells in the peripheral resistance arteries more than in capacitance veins. Hydralazine’s vasodilating action may be mediated in part by its antioxidant action, which inhibits the vascular production of reactive oxygen species (ROS), thereby preventing the development of tolerance to exogenous nitrate, which serves as a source of nitrates.
Although hydralazine treatment significantly lowers BP, its use is limited by immunologic problems and the “pseudotolerance” phenomenon described later (Table 26.3). Hydralazine is currently used only infrequently to treat hypertension, and it is used as part of a multiple-drug regimen on top of other medications. It is usually combined with a sympathetic inhibitor to prevent the reflex activation of baroreflex, and it may be administered with a diuretic agent to prevent the sodium retention caused by reduced renal perfusion pressure. Moreover, given its short half-life of about 4 to 6 hours it must be given at least three times a day and preferably four times a day to maintain BP control over the 24-hour period.
Pregnancy-induced hypertension and eclampsia are the conditions for which hydralazine is most frequently used. Given during pregnancy, hydralazine is not toxic to the fetus.
Because its BP-lowering effect begins within a few minutes and its maximum effect occurs at 15 to 75 minutes after administration, hydralazine is also used for hypertensive emergencies. A parenteral dose of 20 to 40 mg hydralazine that can be repeated every 2 to 4 hours is usually administered. However, in hypertensive crises, hydralazine is not the best choice for patients with aortic dissection(because the hydralazine may increase the stroke volume and extend the dissection)or in patients with coexisting ischemic heart disease because it may worsen ischemia; see Chapter 46.
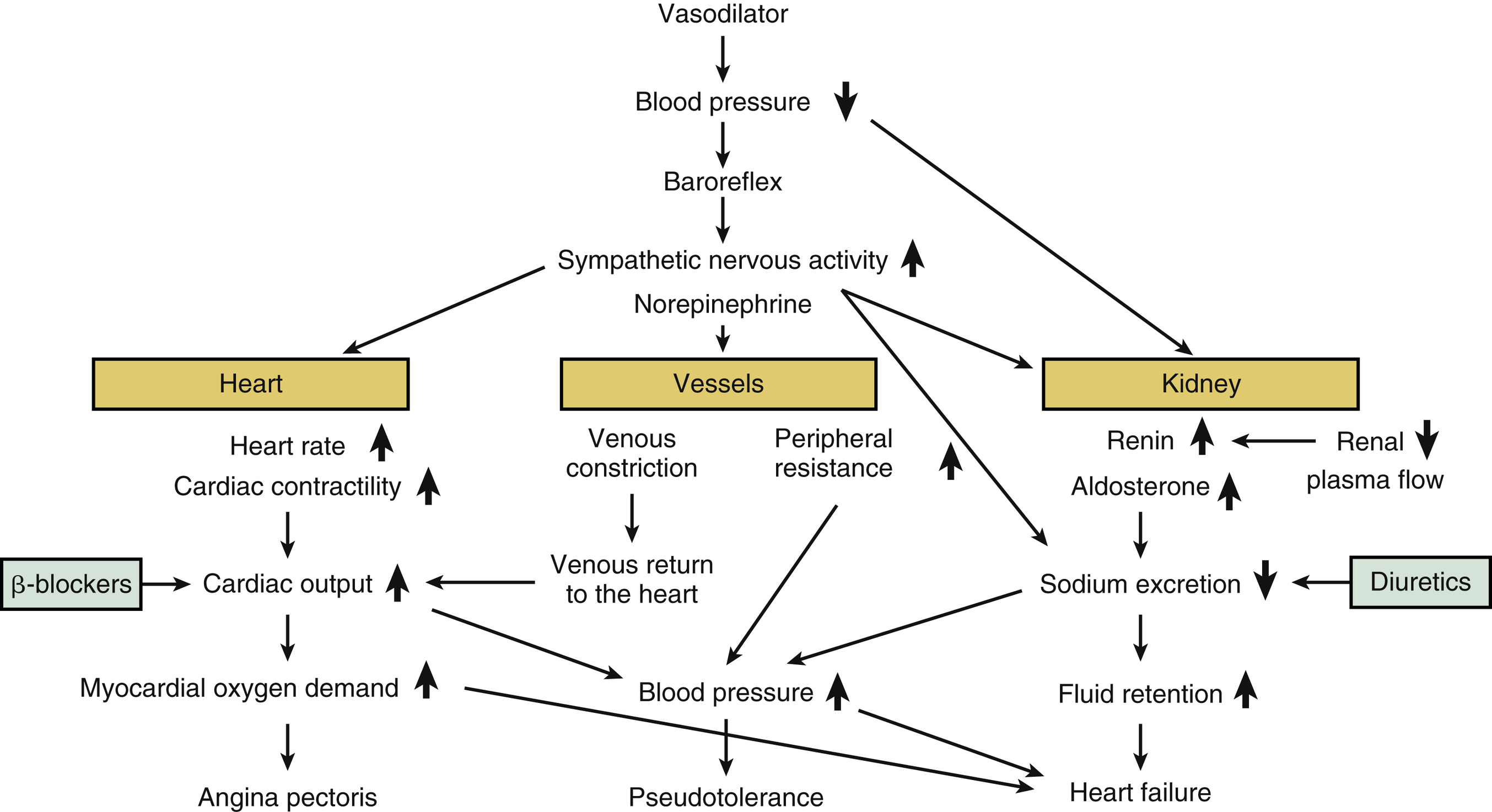
FIG. 26.2 Hemodynamic changes predisposing to pseudotolerance and adverse effects of vasodilators. Diuretics and β-blockers (shown in green) can counteract pseudotolerance when they are used concomitantly with vasodilators as the “standard triple therapy.”
Pseudotolerance
The BP-lowering effect of arterial vasodilators tends to wane over time, in a phenomenon called pseudotolerance. The prefix pseudo is used because the tolerance is attributed not to a loss of the drug’s direct BP-lowering effect, but rather to the compensatory mechanisms of BP regulation, that is, those of the renin-angiotensin-aldosterone system and the sympathetic nervous system, and fluid (sodium and water) retention (Fig. 26.2). Following peripheral vasodilation, to compensate for the hemodynamic changes, baroreflex-mediated sympathetic nervous activation increases the heart rate and cardiac output, and increased myocardial oxygen demand develops. Reduced blood flow, reduced renal perfusion pressure, and the sympathetic activation increase the secretion of renin, resulting in a compensatory retention of reactive sodium.
This compensatory sympathetic nervous activation along with the increased fluid retention (cardiac preload) and the increased heart rate may make the use of vasodilator monotherapy in patients with coronary artery disease (CAD) risky. These features may also trigger myocardial ischemia and CAD, and their occurrence helps explain the lesser regression of left ventricular hypertrophy. Hydralazine can be used with a diuretic and a β-blocker or other sympatholytic drug to block the pseudotolerance phenomenon and maintain the efficacy of the vasodilator, in a standard triple therapy. In fact, as a general principle, hydralazine should not be given to patients with cardiac ischemia until after they are on a beta-blocker and diuretic.
Antihypertensive Effect
Hydralazine’s plasma half-life is short (approximately 90 minutes) but its clinical effect far outlasts its presence in the blood. Hydralazine may thus be given effectively in a twice-a-day regimen. The most commonly used starting regimen is 10 to 25 mg 2× per day, which can be increased at weekly intervals to the maximum dose of 100 to 200 mg 2× per day. Further BP reduction is not provided by the higher doses, and the use of the higher doses increases the risk of lupuslike syndrome.
Hydralazine is metabolized primarily by N-acetylation in the liver. It also forms hydrazones (i.e., the acetone hydrazone and the pyruvic acid hydrazone), which may contribute to the BP-lowering effect. The rate of this N-acetylation step is determined genetically. This “acetylator status” determines the systemic bioavailability of orally administered hydralazine and, because the patient’s response is determined to a large extent by the level of the hydralazine in the blood, the acetylator status also determines the patient’s response to the hydralazine. The oral availability of hydralazine has been estimated to be 10% to 30%, depending on the patient’s acetylator status. Patients who are rapid acetylators require larger doses than slow acetylators to achieve an equivalent effect. Patients who develop lupuslike syndrome are likely to be slow acetylators and thus exposed to the drug longer.
Adverse Effects
In general, there are three types of adverse effects: (1) reflex sympathetic activation-related, (2) lupuslike syndrome-related, and (3) nonspecific adverse effects.
Adverse effects caused by reflex sympathetic activation include the anticipated tachycardia, palpitation, flashing, fluid retention, and headache, especially in the early days of therapy. Hydralazine may also trigger angina pectoris. However, these adverse effects can frequently be prevented by the concomitant use of a β-blocker. When a β-blocker is contraindicated, central sympatholytics are an alternative choice to reduce the pulse rate. Fluid retention causes not only edema but also pseudotolerance, and these can be prevented by a concomitant use of diuretics. Hydralazine should be avoided or used with caution in patients with a recent history of acute aortic dissection, stroke, coronary artery disease, or heart failure.
Similar to other drugs that are N-acetylated, high doses of hydralazine and long-term uses of hydralazine present a slight risk of lupuslike syndrome, with lupuslike symptoms such as a febrile reaction resembling that seen in systemic lupus erythematosus (SLE) and rheumatoid arthritis. The symptoms of lupuslike syndrome are: arthralgia, sometimes accompanied by pleural and pericardial effusion, splenomegaly, malaise, weight loss, and skin rash. These reactions were dose-dependent; the reactions did not occur in the patients given 50 mg daily, and they occurred in 5.4% of the patients given 100 mg daily and in 10.4% of those given 200 mg daily. The lupuslike reactions developed at approximately 6 to 24 months after the hydralazine therapy was initiated. The reactions were reversible, and when the drug was stopped or the dosage was lowered, a full recovery occurred within weeks.
In contrast to SLE, the hydralazine-induced lupuslike reaction is associated with antibodies directed against single-strand DNA (very high titers) rather than antibodies against the native double-strand DNA. A hydralazine-induced lupuslike reaction is also frequently accompanied by antibodies that are positive to histones, but glomerulonephritis rarely develops.
Other adverse reactions to hydralazine include gastrointestinal problems such as vomiting, nausea, diarrhea, and anorexia. Less common effects are muscle cramps, tremor, and paresthesia. Hydralazine treatment should be avoided in patients with liver damage, as fulminant hepatitis was reported in such patients.
Minoxidil
The direct vasodilator minoxidil12-15 was introduced in the early 1970s for the treatment of hypertension. Better known for its marketing as a hair restorer, minoxidil opens cardiovascular adenosine triphosphate (ATP)-sensitive potassium channels, which hyperpolarizes the smooth muscle membrane and inhibits the calcium influx through voltage-gated calcium channels. The cytosolic calcium concentration is thus reduced, producing smooth muscle relaxation. Minoxidil also dilates resistance vessels, with little or no action on the venous bed.
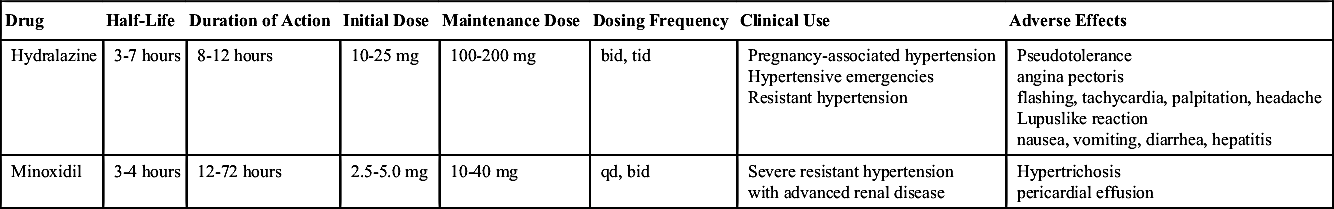
TABLE 26.3
| Drug | Half-Life | Duration of Action | Initial Dose | Maintenance Dose | Dosing Frequency | Clinical Use | Adverse Effects |
| Hydralazine | 3-7 hours | 8-12 hours | 10-25 mg | 100-200 mg | bid, tid | Pregnancy-associated hypertension Hypertensive emergencies Resistant hypertension | Pseudotolerance angina pectoris flashing, tachycardia, palpitation, headache Lupuslike reaction nausea, vomiting, diarrhea, hepatitis |
| Minoxidil | 3-4 hours | 12-72 hours | 2.5-5.0 mg | 10-40 mg | qd, bid | Severe resistant hypertension with advanced renal disease | Hypertrichosis pericardial effusion |
The vasodilatory action of minoxidil is stronger and lasts longer compared with that of hydralazine, but the potential adverse effects of minoxidil have limited its clinical use to hypertensive patients who are refractory to all other medications. The treatment of resistant hypertension,especially in patients with advanced renal disease,may be another option for minoxidil, the efficacy of which does not depend on the severity or etiology of the hypertension or the status of the patient’s renal function. Prolonged minoxidil treatment can stabilize or improve renal function after an initial decrease in the glomerular filtration rate.
Patients with acute or chronic hypertensive nephrosclerosis have been able to discontinue dialysis based on the sustained BP control they achieved with minoxidil treatment. This beneficial effect was caused primarily by minoxidil’s effective BP control rather than a specific renoprotective effect of minoxidil.
Minoxidil is usually administered with both a diuretic and a β-blocker, combined α/β-blocker or a central sympatholytic, because minoxidil increases sympathetic tone and causes significant sodium retention. For refractory edema, it may be necessary to apply a combination of thiazide-type and loop-type diuretics. The tachycardia caused by minoxidil treatment can aggravate myocardial ischemia and, if this is long-standing, left ventricular hypertrophy can develop.
Minoxidil’s safety in pregnancy has not been established, but minoxidil is excreted into breast milk and should thus not be used by breastfeeding mothers.
Antihypertensive Effect
Minoxidil for hypertension is usually administered at an initial dose of 2.5 mg to 5 mg, 2× per day or occasionally once daily. Although doses up to 100 mg have been used, the usual maximum daily dose is 50 mg.
The plasma half-life of minoxidil is 2.8 to 4.2 hours, and the plasma protein binding is negligible; its oral absorption is 100%. Minoxidil is metabolized extensively in the liver, along four pathways: glucuronidation (67%), hydroxylation (25%), sulphation, and conversion to an uncharacterized polar compound. The sulphated metabolite of minoxidil is pharmacologically active, and it probably accounts for much of the parent drug’s activity.
Adverse Effects
As the most common adverse effect of minoxidil, hirsutism is observed in nearly 80% of patients. The hirsutism begins with the development of fairly fine facial hair, progressing to coarse hair all over the body. The hair disappears gradually after the minoxidil treatment is stopped.
During the first few days of minoxidil treatment, electrocardiographic (ECG) changes are often observed; tachycardia as a result of reflex sympathetic activation may account for these ECG changes, which include T-wave inversion and ST depression but are not associated with cardiac enzyme elevation. In minoxidil-treated patients with ischemic heart disease, angina may be aggravated. Pericardial effusions appear in approximately 3% of minoxidil-treated patients attributed to potent fluid retention and are most common among those with advanced nephropathy or who are on dialysis.