Cancer Immunotherapy
The main role of the immune system is recognition of “foreign” proteins, including mutated or altered forms of self-proteins that arise during tumorigenesis, which are commonly referred to as tumor antigens (TAs). However, it is now apparent that powerful regulatory cells whose main function is to prevent rampant, uncontrolled immune responses also serve to block natural development of potent antitumor immune response. Therefore the role of immunotherapy in treatment of various cancers must take into account the ability to overcome these regulators in order to be successful.
It has long been known that the immune system is capable of controlling cancer. For example, it is well established that cancer incidence is increased in immunosuppressed individuals. Moreover, in some cases, spontaneous remission of tumors is observed without any therapeutic intervention, most likely attributed to a successful immune response. Biologically, tumor-specific T-cells are observed in the tumor tissue and tumor-draining lymph nodes, providing evidence that these cells have encountered and recognized the tumor cells as foreign. Finally, in some cases, there is the development of paraneoplastic autoimmunity, suggesting that the antitumor immune response has somehow gone unchecked by the regulator cells.
With the advancement and sophistication of techniques used to study the immune system comes the increased ability to precisely target the malignant cells while leaving normal tissues intact. One of the major goals of immunotherapy is to develop a potent, long-lasting antitumor immune response without producing untoward side effects. However, it is difficult to develop an immunotherapy that is not affected by tumor-induced immune tolerance or even the immune system itself. For example, therapy with the T-cell growth factor cytokine, interleukin-2 (IL-2), leads to expansion of regulatory T-cells, known for their powerful immunosuppressive abilities.1 In addition, when considering monoclonal antibody (MAb) immunotherapy in companion animals, one must account for the fact that the Fc region of the MAb, the part not involved in antigen recognition, is typically derived from mice or human protein sequences and thus is considered foreign by the immune system. Hence, repeated treatment with a non–canine-based antibody leads to a robust inactivation of the MAb, as well as systemic toxicity, thus eliminating its effectiveness over time.
Knowing more about the immune system and the way it is regulated will increase our ability to design better immunotherapies. In addition, immunotherapy has the potential to work in conjunction with and perhaps enhance the effectiveness of chemotherapies, radiation therapy, surgery, and other adjunct therapies. In this chapter, we will first discuss the role of the immune system in tumor development, then the various classes of immunotherapies both currently in use and those under investigation as part of clinical trials. We will discuss the biologic basis for the therapies, their use in human and companion animals, and their limitations.
Immune System Control of Tumor Development and Growth
Forty years ago, Thomas and Burnet, while studying how lymphocytes could respond to newly formed antigens on transformed cells, put forth the concept that the immune system could actively respond to and eliminate cancerous cells, an idea known as immune surveillance.2 In contrast, later studies3,4 showed that genetically manipulated immunodeficient (athymic) mice did not demonstrate an increased incidence of cancer—either spontaneously or carcinogen induced. Such observations were proposed to be due to residual immunity, leading to the immune surveillance concept falling out of favor.
Since the development of more sensitive and sophisticated technologies, many of the ideas behind the concept of the immune surveillance hypothesis are now more accepted, and currently, this modification of the original hypothesis is referred to as the immunoediting hypothesis.5 Pivotal studies by Robert Schreiber’s group showed that lymphocytes (T-cells) could directly or indirectly through production of a cytokine, interferon γ (IFN-γ), protect mice against the development of methylcholanthrene (MCA)-induced sarcomas.6,7 Moreover, they demonstrated that tumors from immunodeficient mice were more immunogenic than tumors from immunocompetent mice, thus leading to the immunoediting hypothesis.7,8 This hypothesis consists of three phases: (1) elimination: removal of the immunogenic tumor cells by the immune system; however, weakly immunogenic cells can survive; (2) equilibrium: tumor growth and immune destruction are equal; and (3) escape: tumor growth ensues as the result of decreased immunogenicity, immune suppression, and rapid tumor cell growth.5 However, despite recent data, there is still a controversy that remains around the immune surveillance hypothesis, discussed in a review by Schreiber et al.5
Mechanisms of Immune Evasion by Tumors
Given the fact that cancer can develop in immunocompetent individuals, some tumor cells are able to avoid recognition by the immune system.9 This is accomplished by various mechanisms (discussed later) that involve both changes in the tumor cells themselves and ways in which the tumor and the tumor stromal environment can manipulate the immune system and prevent antitumor immunity. These mechanisms of immune evasion pose a significant challenge to the development of effective immunotherapies. Figure 13-1 demonstrates some of these key mechanisms.
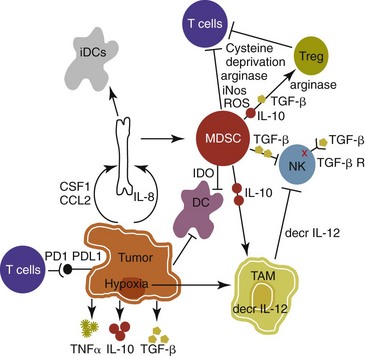
Figure 13-1 Mechanisms of tumor cell evasion via hijacking the immune system. DC, Dendritic cell; iDC, immature dendritic cell; MDSC, myeloid-derived suppressor cell; TAM, tumor-associated macrophage; Treg, regulatory T-cell.
Active Immune Suppression by Myeloid-Derived Suppressor Cells
One population of immune cells that play a major role in tumor immunosuppression are myeloid-derived suppressor cells (MDSCs). These cells consist of immature monocytes and granulocytes released from the bone marrow into the blood during pathologic conditions, including cancer. Sometimes included in the functional description of this group of cells are tumor-associated macrophages (TAMs), which have the same ability and use similar mechanisms as MDSCs to induce potent tumor immunosuppression.10,11 Numerous studies demonstrate increased numbers of MDSCs in humans with cancer12-14 and in mouse models of cancer.15,16 Furthermore, it has been shown that the presence of these cells correlates with clinical disease stage and metastatic tumor burden in humans with solid tumors.13 MDSCs are recruited to the tumor microenvironment through various chemoattractants,10 many of which are induced by the tumor during times of hypoxia and are directly released by the effects of hypoxia-inducible factor-1α (HIF-1α) production.17-20 Once at the tumor, MDSCs intercalate into the microenvironment and actively suppress the local antitumor immune response and promote tumor invasion and metastasis via the production of matrix metalloproteinases (MMPs) and various chemoattractants.21,22 Moreover, these cells, including the TAMs, contribute to increased tumor vessel growth, a process referred to as angiogenesis.10 Of note, although cell surface markers are used to identify these cells in mice and humans, similar markers have yet to be discovered for dogs, thus the role that MDSCs play in canine cancer is currently unknown.
The ability of MDSCs to suppress the antitumor response is the subject of many recent studies.23-25 Numerous mechanisms of suppression have been reported, and MDSCs have the ability to suppress not only T-cells, but also natural killer (NK) cells and dendritic cells (DCs) and they also are able to potentiate T-regulatory cells (Tregs), which are discussed later, and TAMs in the tumor (see Figure 13-1). Currently known mechanisms of immune suppression by MDSCs include suppression of T-cells through production of inducible nitric oxide (NO) species (iNOS), reactive oxygen species (ROS) and arginase, as well as cysteine deprivation.23 MDSCs can produce transforming growth factor-β (TGF-β) and IL-10, which stimulate Tregs and TAMs, and MDSCs can cause downregulation of the IL-12 production by TAMs, a cytokine involved in T-cell activation.24 MDSCs cause NK cell anergy (lack of function) also by this decreased IL-12 production and through membrane-bound TGF-β. 24,25 Thus, given the ability of these cells to use multiple pathways to induce tumor immunosuppression, the development of effective immunotherapies that can target these cells and either eliminate them or lead to their maturation, rather than ones that target specific pathways of suppression, is critical for that therapy’s success.
Induction of Regulatory T-Cells by Tumors
Another population of cells that are significantly increased in tumor-bearing humans and animals are Tregs. These cells are phenotypically defined by surface expression of CD4 and CD25 but are most specifically identified by the intracellular transcription factor, forkhead box P3 (foxp3).26,27 Other surface markers used to characterize these cells have been described, including cytotoxic T-lymphocyte antigen-4 (CTLA-4), glucocorticoid-induced tumor necrosis factor (TNF) receptor family-regulated gene (GITR), lymphocyte-activation gene 3 (Lag3), and folate receptor-4 (FR-4).28-31 This distinct subset of CD4+ T-cells is capable of directly suppressing tumor-specific CD4+ and CD8+ T-cells and NK cells and are enriched in the tumor microenvironment by conversion of CD4 T-cells to Tregs by DCs or TGF-β,32-35 proliferation of tumor-specific Tregs following antigen recognition, or recruitment of these cells via chemokine signaling (i.e., CCR5).36 Recent work has also suggested a role for the chemokine CCL-1 in specifically converting T-cells to Tregs and inducing their suppressive nature.37
Many studies demonstrate that increased numbers of Treg cells are correlated with a poor prognosis.38-41 Additionally, Tregs present in metastatic lymph nodes inhibit the ability of tumor-infiltrating lymphocytes to mount an effective antitumor response.42 Work in our laboratory demonstrated that canine Treg cells can also be identified via the expression of CD4 and foxp3.43 Moreover, we saw that cancer-bearing dogs had increased numbers of Tregs compared to healthy dogs and that this difference was greater in certain types of canine cancers.43,44 Therefore current therapies aimed at depleting Treg cells in humans could be applied to veterinary medicine. In particular, many studies have shown that the use of cyclophosphamide or anti–Treg-specific antibodies decreases the numbers of Tregs present in tumors and in circulation of tumor-bearing patients.45-49
Impaired Dendritic Cell Activation and Function
Another important mechanism of tumor suppression is through impairment of the potent antigen-presenting cells, dendritic cells (DCs). Numerous studies have denoted that overall numbers of DCs are decreased in various human cancers studied, including head and neck squamous cell carcinomas (HNSCCs),50 breast and prostate cancers, and malignant gliomas.51 A recent study showed that indoleamine 2,3-dioxygenase 1 (IDO1) expression in the tumor microenvironment led to increased DC apoptosis.52 Some tumor studies also demonstrated fewer circulating myeloid DCs and a concurrent increase in immature DCs (iDCs) that reduce presentation of antigens and stimulation of T-cells; thus they induce T-cell tolerance rather than activate T-cells.51,53,54 Thus the DCs present in the tumor tend to be immature and dysfunctional. Studies of DCs in numerous human cancers demonstrate minimal activation, decreased ability to stimulate in an alloreactive fashion, and decreased expression of co-stimulatory molecules.50,51,55-60 A similar study done in dogs with canine transmissible venereal tumors (CTVT) showed that the tumor environment caused downregulation of DC surface markers of activation and major histocompatibility (MHC), as well as decreased endocytic capabilities and decreased allogenic mixed lymphocyte reaction (MLR) responses.61 Possible mechanisms causing the DC dysfunction include the overexpression of the protein S100A9,62 accumulation of triglycerides in the DCs that leads to decreased capacity to present antigen,63 and downregulation of toll-like receptor 9 (TLR9).64 Moreover, factors such as IL-10 and vascular endothelial growth factor (VEGF) can negatively affect DC function and maturation.65,66 Finally, some DCs in the tumor are considered to be regulatory based on low expression of surface markers MHC II, CD86, and CD11c with high expression of co-stimulatory molecules CD80, CD40, CD106, and CD11b. These cells secrete regulatory factors such as IL-10 and NO and inhibit proliferation of naïve CD4+ T cells to antigen presented by mature, functional DCs.67 Overall, the microenvironment of the tumor leads to attraction of immature and regulatory DCs that due to their decreased activation and function can potently inhibit the development of antitumor T-cell responses even when copious amounts of antigen are present.
Production of Immunosuppressive Cytokines
In addition to the suppressive milieu established by tumor-infiltrating cells, the tumor cells themselves are capable of producing immunosuppressive cytokines.68 A few key cytokines produced by tumor cells are IL-10, TGF-β, and tumor necrosis factor-α (TNF-α).68,69 These cytokines act to suppress antitumor T-cell responses and inhibit DC function. IL-10 promotes Treg production and function70 and, in an autocrine and/or paracrine fashion, may potentially affect tumor cell proliferation and survival.71 In human cancer patients, increased levels of serum IL-10 is observed in patients with pancreatic carcinoma and non-Hodgkin’s lymphoma (NHL).72,73 In addition, elevated levels of IL-10 in diffuse large B-cell lymphoma in humans correlate with a poor prognosis.74 TGF-β acts similarly to IL-10 in that it is a potent immunosuppressive cytokine that can potentiate Treg proliferation and function.33,75-77 It can also enhance tumor progression; carcinomas can produce excess TGF-β, which in turn increases epithelial-to-mesenchymal transition, tumor invasion, and metastasis, and inhibit tumor-specific CD8+ T-cells.77 Moreover, tumor-produced TNF-α leads to promotion of tumor cell survival via induction of antiapoptotic proteins.78 Finally, TNF-α has been shown to promote tumor angiogenesis and metastasis and hamper cytotoxic T-cell and macrophage responses.79
One study in veterinary medicine examined a lymph node of a dog with metastatic melanoma. This study revealed an overexpression of IL-10 and TGF-β concurrent with a lack of expression of IL-2, IL-4, or IFN-γ—cytokines typically associated with antitumor immunity—thus demonstrating that tumor immunosuppression occurs in veterinary patients as well.80 For a review of cytokines relevant to tumor immunotherapy, see Table 13-1.
Table 13-1
Biologic Activities of Key Cytokines Relevant to Tumor Immunotherapy

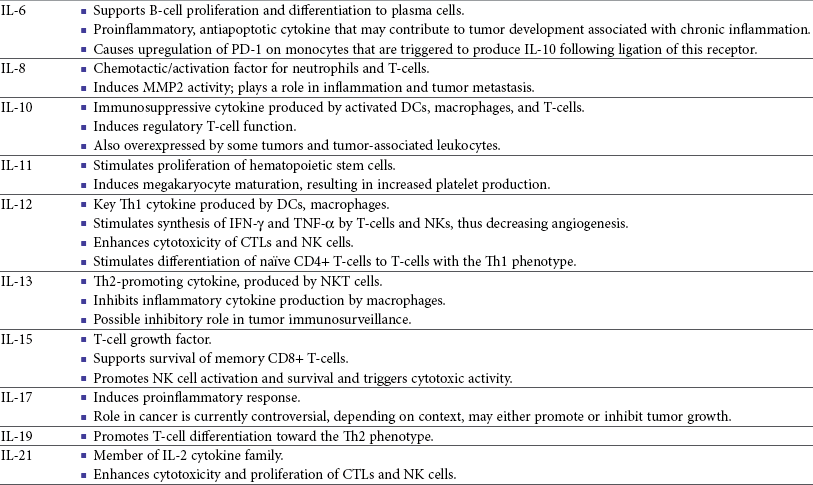

CTL, Cytotoxic T-lymphocyte; CSF-1, colony-stimulating factor-1; DCs, dendritic cells; FDA, Food and Drug Administration; G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte-macrophage colony-stimulating factor; IFN, interferon; IgE, immunoglobulin E; IgG, immunoglobulin G; IL, interleukin; LAK, lymphokine-activated killer; MHC, major histocompatibility; MMP, matrix metalloproteinase; NK, natural killer; NKT, natural killer T-cell; NO, nitric oxide; PD-1, programmed death-1; Th1, T-helper 1; Th2, T-helper 2; TGF, transforming growth factor; TILs, tumor-infiltrating lymphocytes; TNF, tumor necrosis factor.
Failure of Tumor Cells to Activate Immune System
Tumor cells are also capable of avoiding immune elimination by failing to be recognized by the immune system in the first place. For example, some tumor cells can downmodulate MHC surface expression to escape recognition by T-cells. MHC class I expression can be lost on tumor cells due to changes in protein synthesis, structure, or allelic loss.81,82 Moreover, defects in antigen processing and presentation can occur that can also lead to decreased MHC expression.81,82 A decrease in class II expression is also observed in certain human hematopoietic cancers, although it should be noted that most tumors are normally MHC class II negative.83,84 Reduced expression of MHC class II has been recently correlated with poor outcome in dogs with B-cell lymphoma.85
In addition, tumor cells can express co-inhibitory surface molecules, such as CD73 and/or PD-L1. CD73 is an ecto-5′-nucleotidase that catalyzes the breakdown of adenosine monophosphate (AMP) to adenosine. When expressed on tumors, this creates a local microenvironment rich in adenosine, which is immunosuppressive.52 The programmed death-1/programmed death ligand-1 (PD-1/PD-L1) axis also plays an immunosuppressive role in cancer. PD-L1 expression on tumors downmodulates antitumor T-cell function86-88 and NK cell89 activity via interaction through the PD-L1 expressed on these immune cells. Thus tumor cells themselves can actively and directly suppress antitumor T-cell responses through such mechanisms as decreased expression of MHC molecules and increased expression of inhibitory molecules.
Strategies to Control Tumor Growth through Immune Activation
Depletion of Immunosuppressive Myeloid-Derived Suppressor Cells to Allow for Effective Immunotherapy
In light of many recent studies, it has become clear that in order to develop an effective immunotherapy, it must be able to overcome, or be combined with other treatments that can overcome, the immunosuppression present in the tumor microenvironment. As mentioned previously, MDSCs are a key component of such immunosuppression. Box 13-1 lists the various potential ways in which MDSCs can be manipulated in order to enhance the effectiveness of immunotherapy.
Nonspecific Immune Activation to Generate Antitumor Activity Using Biologic-Response Modifiers
In the 1900s, William Coley observed that cancer patients who developed bacterial infections survived longer than those that did not (reviewed in Richardson and colleagues110). Building on these observations, Coley developed “Coley’s toxins,” which consisted of killed cultures of Streptococcus pyogenes and Serratia marcescens that he gave to patients with inoperable sarcomas. Although with this “vaccine,” Coley saw cure rates of approximately 15%, his therapy was discontinued because of its significant failure rate and intolerable side effects. However, this seminal work laid the foundation for further studies aimed at nonspecific, pan-immune activation to treat cancer through the use of biologic-response modifiers (BRMs).
Bacillus Calmette-Guérin and Corynebacterium parvum
One of the most well-known and clinically utilized BRMs is bacillus Calmette-Guérin (BCG), a live, attenuated strain of Mycobacterium bovis. Currently, in human medicine, BCG is intravesically instilled into the bladder where it is considered to be effective as a means to treat and prevent relapse of noninvasive transitional cell carcinoma (TCC).111,112 One proposed mechanism for its antitumor effects relates to the recruitment of neutrophils and their ability to promote urothelial cell turnover.113 This recruitment most likely relates to the ability of BCG to elicit T-helper 1 (Th1) inflammatory cytokines.114,115
The use of BCG in veterinary medicine is rather limited. Although its efficacy has been tested on numerous forms of canine cancer,116 its use as an immunotherapy in dogs is limited. BCG can be safely instilled into canine bladders,117 but the rate of true superficial bladder cancers is extremely low in dogs as compared to humans.118 Recent uses of BCG in canines include treatment of CTVT in conjunction with vincristine119 or, in combination with human chorionic gonadotropin (hCG; LDI-100), treatment of mast cell tumors (MCTs).120 In this study, response rates for grade I and II MCTs were comparable to single-agent vinblastine but without the myelosuppression.
Another BRM that has been studied in human and veterinary medicine is Corynebacterium parvum. In human and dog melanoma studies, C. parvum displayed antitumor activity as an adjunct to surgery.121,122 However, efficacy of C. parvum as an immunotherapy in other canine cancers has been disappointing.123
Salmonella
As a tumor grows, the core may become necrotic as the initial tumor cells are deprived of nutrients. Layered on this necrotic core are tumor cells that exist in an area of hypoxia, which puts them out of reach of blood vessels that can supply them with oxygen. These cells are able to remain viable and pose a challenge to most immunotherapies, chemotherapies, and even small molecule drugs due to their restricted location. Recently, researchers have begun to genetically modify facultative anaerobic bacteria that can penetrate and survive in these regions. In fact, it has been shown that several strains of Salmonella, including S. typhimurium and S. choleraesuis, target tumors following systemic administration. These bacteria penetrate the necrotic core and feed on the dead cells while also emitting natural toxins that will destroy surrounding, viable cells. Using a mouse melanoma model, treatment with VNP20009, an attenuated S. typhimurium, was able to slow tumor growth and specifically target primary tumor and metastatic lesions.124 Although this study showed that the effects were independent of B- and T-cells, possible indirect effects of the Salmonella include production of inflammatory cytokines, such as TNF-α.125 Recently, another proposed mechanism involves the ability of Salmonella to induce melanoma cells to express gap junctions that can interact with DCs and cause bits of tumor cell proteins to be loaded and expressed on the surface of these DCs for presentation to T cells.126 Unfortunately, in human trials, the bacteria failed to colonize some patients and did not provide any antitumor activity.127
Administration of VNP20009 in dogs resulted in a more positive outcome than in humans. In a phase I clinical trial, VNP20009 was administered to dogs with a variety of malignant tumors.128 In this study, 41 dogs received intravenous infusions of VNP20009 either weekly or biweekly at escalating doses. Fever and vomiting were reported as dose-limiting toxicities. Bacterial colonization was seen in approximately 40% of dogs, and significant clinical responses were observed in 15% of patients, with an overall rate of 37% of dogs experiencing either a transient response or stable disease. Thus the use of VNP20009 in specific dog tumors should be further investigated, perhaps in combination with modified Salmonella engineered to deliver tumor cytotoxic agents.
Superantigens
Bacteria, such as Staphylococcus aureus, produce enterotoxins known as superantigens (SAgs). Two of these S. aureus enterotoxins, referred to as SEA or SEB, stimulate T-cell proliferation and Th1 cytokine production (IL-2, TNF-α, and IFN-γ) via their ability to cross-link the T-cell receptor and MHC class II molecules. These activated T-cells are highly cytolytic and antitumorigenic in mouse models of cancer.129,130 Moreover, the potency of SAgs is increased when delivered with stimulatory cytokines.131,132 However, when these SAgs were injected into humans, toxic shock syndrome was elicited.133 Therefore genetically modified versions of the SAgs that maintain their immune potency have been evaluated in humans. In a study of patients with non–small-cell lung carcinoma using a modified SAg, stable disease occurred in 42% of patients with decreased tumor burdens of up to 50%. The dose-limiting side effect noted in this study was hypotension.134 SAgs and modified versions are still currently being evaluated for potential use as a human cancer therapeutic.135,136
In veterinary medicine, SAgs were evaluated for efficacy and safety in dogs with oral melanoma and soft tissue sarcoma (STS). Dow et al assessed the efficacy of intratumoral injection of lipid-complexed plasmid DNA that encoded for SEB and either granulocyte-macrophage colony-stimulating factor (GM-CSF) or IL-2.131 Complete or partial remission was seen in 46% of dogs and increased survival time for stage III diseased dogs was observed. There were no toxicities noted, and analysis of tissue sections revealed increased infiltration of tumors with T-cells and macrophages. Thamm et al assessed the efficacy of a lipid-DNA-SEA/IL-2 therapy in dogs with STS.137 In this study, dogs received once weekly intratumoral injections and surgery was done after the 12-week treatment. In the 25% of dogs that responded to the therapy (3 complete responders, 1 partial), a diffuse lymphoplasmacytic infiltrate was observed on histologic evaluation of the tumors. Thus SAgs show some promise for use in veterinary medicine.
Liposome-Encapsulated Muramyl Tripeptide
Similar to SAgs, bacterial cell components such as peptides derived from mycobacterial cell walls were evaluated for potential immunogenicity. One such product is muramyl tripeptide (MTP), which, when encapsulated in a phosphatidylethanolamine-based liposome (L-MTP-PE), can efficiently activate monocytes and macrophages to produce proinflammatory cytokines, such as IL-1α and β, IL-6, IL-7, IL-8, IL-12, and TNF-α.138 The use of L-MTP-PE as a therapeutic was assessed in phase I and II trials of people with osteosarcoma (OSA), renal carcinoma, and metastatic melanoma.138-140 Moreover, this drug has been approved for use in treating pediatric osteosarcoma in Europe under the name Mifamurtide.141
L-MTP-PE has been evaluated in veterinary medicine in a variety of studies.142-146 The survival benefit of L-MTP-PE therapy has been most clearly demonstrated in dogs with appendicular OSA.147 In this study, dogs receiving L-MTP-PE following limb amputation had a median survival time (MST) of 222 days, whereas dogs that received placebo had an MST of 77 days. However, since most of the dogs in both groups developed metastatic disease, further studies evaluated the efficacy of L-MTP-PE in conjunction with chemotherapy.142 In one study, dogs receiving L-MTP-PE after treatment with cisplatin had an MST of 14.4 months versus 9.8 months in dogs that received cisplatin only. Interestingly, only 73% of dogs receiving L-MTP-PE developed metastatic disease compared to 93% in the cisplatin only group. However, in a second trial, these investigators saw no significant survival advantage in dogs with OSA that received L-MTP-PE concurrently with cisplatin. The authors postulated that cisplatin obviated antimetastatic potential of L-MTP-PE due to impaired immune effectors. L-MTP-PE was also evaluated for efficacy in canine hemangiosarcoma (HSA).144 Dogs that received L-MTP-PE with chemotherapy following splenectomy had an MST of 9 months versus the 5.7 months seen with dogs receiving chemotherapy alone. In another study, only dogs with stage I oral melanoma that received L-MTP-PE had an increased survival over placebo-treated dogs.145 No differences were seen within the dogs with more advanced disease.
Liposome-DNA Complexes
Bacterial DNA can also stimulate the innate immune system via its CpG-oligonucleotides (CpG-ODNs), particularly when complexed with cationic liposomes, in a form known as cationic lipid-DNA complexes (CLDC).148 Complexing bacterial plasmid DNA to liposomes allows for more efficient delivery of the CpG DNA to the endosomal compartment of antigen-presenting cells such as DCs, in which it is released from the liposomes and binds to its receptor, TLR9.149,150 In mouse studies, CLDC stimulates the immune system largely through induction of NK cell activity and release of IFN-γ.148 Moreover, CLDC was also shown to stimulate the production of type I IFN151 and thus is a potent nonspecific immunostimulant.
The use of CLDC in dogs has been evaluated in metastatic OSA and in dogs with STS.152,153 Intravenous administration of a modified CLDC that encodes for IL-2 was performed in dogs with stage IV OSA.152 Dogs that received CLDC developed fevers and showed changes in their leukogram profile indicative of immune stimulation. Moreover, NK cell activity was observed, as assessed by target cell lysis, and monocytes showed increased expression of B7.2 on their surface, indicating activation. Treatment was associated with a significant increase in survival times compared to historic controls. Another study examined the use of CLDC in canine STS. Administration of CLDC intravenously once weekly for 6 weeks resulted in an objective response in 15% of the dogs and a decrease in tumor mean vessel density in half of the dogs receiving treatment.153 Thus CLDC has potential to be used as a stand-alone immunotherapeutic in veterinary medicine for a variety of cancer types.
Oncolytic Viruses
Oncolytic viruses are defined as viruses capable of replicating in and lysing tumor cells, thus making them a likely candidate for drug or gene delivery to tumors. A beneficial side effect of these viruses is that they can kill the tumor cell, thus providing release of TAs for processing by the immune system. Adenoviruses that have undergone genetic modification of their early genes, 1A (E1A) and 1B (E1B), preferentially target rapidly dividing tumor cells and have been used to target canine OSA cells.154-156 Canine distemper virus (CDV) has also been investigated as a treatment for B- and T-cell lymphoma in dogs.157 In vitro studies using fluorescently labeled, attenuated CDV and canine lymphoma cells demonstrated that CDV infected lymphoid cells via binding of the cell membrane protein CD150, which is overexpressed on malignant B cells, and induced cellular apoptosis.157
Nonspecific Tumor Immunotherapy Using Recombinant Cytokine Therapy
IL-2 is a cytokine that is released by T-cells following their activation via interactions of antigen-loaded MHC and co-stimulatory molecules expressed on the surface of antigen-presenting cells. Its function is to induce clonal expansion of T-cells in an antigen-specific fashion and activate DCs, macrophages, and B-cells, which in turn release proinflammatory cytokines. Moreover, IL-2 stimulates NK cells, thus playing an important role in inducing both the innate and adaptive arms of the immune system.
The therapeutic use of IL-2 in humans is fraught with toxicity.158-160 However, the use of IL-2 therapy in veterinary medicine holds some promise as a therapeutic. First, Helfand et al demonstrated that intravenously injected recombinant human IL-2 (rhIL-2) activates canine lymphocytes, causing only mild gastrointestinal toxicity, even at high doses for 4 consecutive days.161 Another study demonstrated the ability of rhIL-2 to induce canine lymphokine-activated killer (LAK) cells and incidentally showed that LAK cells from tumor-bearing dogs did not kill tumor cells as efficiently as compared to normal dogs.162 Further evaluation of toxicity and efficacy of rhIL-2 was done using dogs with primary lung cancer and with lung metastases in an aerosol formulation.163 In this study, complete regression was seen in two of the four dogs with pulmonary metastases, and these dogs remained disease free for at least 12 months after treatment. One of the two dogs with a primary lung tumor had disease stabilization for more than 8 months, whereas the other dog had progressive disease. Assessment of the lymphocytes obtained from bronchoalveolar lavage showed increased cytolytic activity following 15 days of IL-2 treatment. In addition, minimal toxicity was noted in this study. Finally, IL-2 gene therapy using viral vectors has been examined for treatment of feline fibrosarcomas and canine melanoma and was shown to be safe and effective.164-166 Therefore, given its low toxicity and promising effectiveness, rhIL-2 therapy is a plausible treatment for canine cancer.
Interleukin-12
IL-12, produced by antigen-stimulated DCs, macrophages, and B-cells, plays a role in stimulating the growth and function of T-cells and enhances the cytolytic activity of both T-cells and NK cells. Similar to IL-2, IL-12 therapy in humans leads to serious side effects; currently, it is not used clinically. Current investigation into the use of IL-12 in veterinary medicine revolves around recombinant gene therapy for treatment of canine head and neck tumors,167 with some in vitro work looking at its use in feline hyperthermia-induced gene therapy.168
Interleukin-15
IL-15 is structurally similar to and uses similar signaling molecules as IL-2. IL-15 plays a role in stimulation of NK cells and promoting proliferation of T-cells. However, from an immunotherapy standpoint, IL-15 holds more promise than IL-2 in that (1) it does not cause activation-induced cell death of CD4+ T-cells following prolonged periods of exposure; rather it sustains T cell proliferation,169 (2) IL-15 plays a critical role in CD8+ T-cell memory formation and maintenance,170 and (3) unlike IL-2, IL-15 does not appear to play a role in the development of Tregs.1
Clinical investigation of IL-15 will begin soon. An initial safety study in nonhuman primates was recently conducted.171 Twelve daily doses of clinical grade rhIL-15 revealed that neutropenia was the dose-limiting toxicity and documented an increase in circulating NK cells and memory CD8+ T-cells.
In veterinary medicine, one study used plasmid IL-15 in combination with plasmid IL-6 in beagles with CTVT.172 A threefold increase in the proportion of CD8+ T-cells that infiltrated the tumors and an enhancement of IFN-γ–producing cells and increased cytolytic activity against the tumor were observed. Thus IL-15 therapy shows promise as an effective immunotherapy in both human and veterinary medicine.
Interferons
IFNs are proteins produced by lymphocytes that play an important role in immune responses to pathogens and cancer. Broadly, they can influence cell proliferation, play a role in the induction of apoptosis, upregulate antigen presentation to T-cells, and enhance the ability of the adaptive immune system to mount a cytolytic immune response. Moreover, IFNs have antiangiogenic properties. The IFNs are typically classified as either type I (IFN-α, -β, and -ω) or type II (IFN-γ).
Interferon-α, Interferon-β, and Interferon-ω
Type I IFNs can affect cellular proliferation through various mechanisms, including interactions with cell cycle proteins (i.e., c-myc and retinoblastoma) and induction of apoptosis via Bcl-2/Bax and TNF/Fas interactions. Their antiangiogenic properties of downregulating VEGF and basic fibroblast growth factor (bFGF)173 make them attractive as immunotherapies, and they have been used successfully to treat pediatric hemangiomas.174
Clinical trials using type I IFNs have met limited success due to the high occurrence of severe toxicity in light of overall limited response rates. Nonetheless, their effectiveness was assessed in melanoma, multiple myeloma, renal cell carcinoma, leukemia, and other cancers, as well as in conjunction with chemotherapies. The best response, in terms of disease-free survival, was seen in renal cell carcinoma and melanoma when used as single agents.175,176
The use of type I IFNs in veterinary medicine is limited and mostly used for feline viral therapies.177 One study showed that recombinant feline IFN-ω was safe and easy to use for treating feline fibrosarcomas. As this was a safety study, the therapeutic effects of this treatment were not evaluated. Another recently published study also used recombinant feline IFN-ω with or without chemotherapy to study its effects in treating mammary tumors in vitro.178 This study reported that the antitumor cell effects of recombinant IFN and chemotherapy were additive and suggested further investigation into its clinical use as an adjuvant therapy.
Interferon-γ
IFN-γ plays an important role in stimulating the immune system. It is secreted mostly by NK cells, DCs, and antigen-activated T-cells and counteracts the effects of many of the immunosuppressive cytokines. It is a physiologic activator of macrophages, leading to increased antigen presentation and increased lysosomal function and NO production by macrophages. NO production by macrophages is an efficient mechanism of tumor cytolysis. IFN-γ can also cause increased MHC class I and II expression on a variety of cells, including tumors. Increased MHC expression has been confirmed to occur on in vitro IFN-γ–treated canine tumor cells lines179 and in vivo following treatment with INF-γ.180 Thus its role in antitumor immunity is characterized by increased tumor cell lysis and increased tumor antigen presentation to the adaptive immune response.
The use of IFN-γ in veterinary medicine is currently being investigated. A recently published study examined the use of IFN-γ in combination with a single injection of autologous, ex vivo activated DCs in dogs with various malignant or benign tumors.181 In the seven dogs enrolled in the study, the investigators noted four complete responses and two partial responses against malignant tumors and saw moderate partial responses against fast-growing benign tumors. Another study looked at the use of adenoviral IFN-γ gene transfer as an adjuvant therapy to treat a dog with astrocytoma.182 Following therapy and surgery, the dog was tumor free for greater than 450 days. Finally, a safety study was done in cats with fibrosarcomas using a triple-gene therapy that included IFN-γ along with IL-2 and GM-CSF.166 In this study, cats tolerated the therapy, although six of the eight cats developed local recurrence of disease within 1 year of treatment.
Specific Immunotherapy for Cancer: Tumor Vaccines
The development of a tumor vaccine that is safe, effective, and long-lasting is an ultimate goal of immunotherapy. Whereas the effects of traditional cancer treatments such as chemotherapy, surgery, and radiation therapy typically result in noticeable clinical responses within hours to days following treatment, cancer vaccine therapeutic responses typically take weeks to months to lead to an appreciable clinical response. This difference in response time, coupled with the lack of congruent and objective ways to measure efficacy, make it difficult to develop tumor vaccines.
In an attempt to alleviate the lack of objectivity in assessing responses to cancer therapy, the National Cancer Institute (NCI) created an objective way to measure clinical responses, termed response evaluation criteria in solid tumors (RECIST). Under RECIST criteria, a clinical response is defined as a 30% reduction in the total sum of the maximum diameter of all lesions concurrent with the lack of appearance of new lesions or progression of current lesions.183 The RECIST criteria were modified in 2009 to include changes to the number of lesions assessed, evaluation of pathologic lymph nodes, confirmation of response to treatments, clarification of disease progression, and incorporation of imaging modalities into the RECIST criteria.184
Nonetheless, despite the challenges to tumor vaccine development, there are many different varieties of tumor vaccines currently in use either clinically or as part of phase I, II, and III clinical trials. In fact, in April of 2010, the first therapeutic cancer vaccine for human prostate cancer was approved by the Food and Drug Administration (FDA). In this section, we will discuss only those vaccines showing success in human trials and those relevant to veterinary medicine.
Tumor Antigen Targets for Immunization
Mutations or differential expression of tumor-derived proteins are referred to as tumor antigens (TAs). These proteins include the broad categories of oncogenes, oncofetal proteins, and cancer testes antigens. Although TAs offer potential targets for vaccine development, their downside is that some of them tend to be individual or tumor type specific. Nonetheless, much work has been accomplished characterizing TAs for various forms of cancer, and a table of currently studied TAs can be found in a 2009 publication by a panel of experts organized by the NCI.185 Whereas numerous TAs exist, the use of these TAs in tumor vaccines is not trivial. As mentioned previously, the tumor is highly capable of inducing a potent, immunosuppressive microenvironment by various mechanisms, thus standard vaccine procedures using TAs can be rendered useless in this powerful environment. In fact, there are little data available showing a clear correlation between in vitro TA responses and prognosis. Success of most tumor vaccines has been limited to animal models of induced disease.186,187 However, through the use of better vaccine strategies and by combining therapies that can ultimately overcome tumor immunosuppression, more promising specific immunotherapies are being developed. Next, we will discuss the various platforms used to develop tumor vaccines.
Tumor Vaccine Approaches
Whole Tumor Cell and Tumor Cell Lysate Vaccines
One of the more simple approaches to tumor vaccine development is through the use of whole tumor cell or tumor cell lysate vaccines. These can either be made directly from the patient in the form of an autologous vaccine or from cell lines of similar tumor types from the same species as an allogeneic vaccine. Whole cell preparations are made by rendering tumor cells and/or tissues nonfunctional, typically via gamma irradiation. Tumor lysate vaccines, including membrane protein fraction vaccines, are made by mechanically disrupting the tumor cells and/or tissues. Both whole cell and tumor lysate vaccines are typically administered with some form of adjuvant to enhance the immune response. These polyvalent vaccines are superior to specific peptide or protein (subunit) vaccines in that they contain a heterogeneous population of TAs.
One study out of our laboratory assessed the use of an allogeneic HSA tumor lysate vaccine in combination with chemotherapy.188 In this phase I/II study, 28 dogs were evaluated and received eight immunizations of tumor lysate plus liposome-DNA adjuvant (see Liposome-DNA Complexes) over a 22-week period while concurrently receiving doxorubicin. The vaccine was well tolerated; side effects were limited to moderate diarrhea and anorexia. Tumor-specific antibody responses were detected in four to five of the six dogs tested, depending on which HSA cell-line they were screened against. Moreover, overall survival times of dogs receiving the combination treatment were significantly better than historic controls treated with only chemotherapy.
Whole cell and tumor lysate vaccines can also be modified to enhance their immunogenicity. Aside from different adjuvant strategies, combination of these vaccines with modifiers such as immunostimulatory cytokines has been examined. One clinical trial of 16 dogs with STS or melanoma assessed the use of an autologous, whole cell vaccine transfected with human GM-CSF.189 Three dogs in the study demonstrated objective tumor responses that included regression of primary and metastatic lesions. On histologic examination of tumor tissue in the dogs that received the vaccine, an impressive inflammatory response was noted. Another recent study using a similar human GM-CSF vaccine looked at its efficacy in treating dogs with B-cell lymphoma.190 Dogs in remission following a 19-week standard CHOP protocol (cyclophosphamide, hydroxydaunorubicin [doxorubicin], vincristine [Oncovin], prednisone) were randomized into placebo or vaccine treatment groups. Although no changes in median length of remission were seen, dogs receiving the vaccine demonstrated a significant increase in overall survival. Lastly, a recent study investigated the use of an allogeneic melanoma vaccine in combination with a xenogenic melanoma protein, human glycoprotein 100 (hgp100).191 In this phase II trial, the vaccine was well tolerated, and the researchers observed an overall response rate of 17% and a tumor control rate (including complete and partial responses as well as stable disease greater than 6 weeks duration) of 35%.
Immunization Against Defined Tumor Antigens Using Plasmid DNA
Vaccines that use specific gene sequences of TAs encoded in plasmid DNA have shown some clinical promise with their ability to invoke both cellular and humoral immunity. The ease of working with bacterial DNA and the ability to quickly produce large quantities of plasmid DNA make this an attractive vaccine platform. Moreover, the DNA sequences of a majority of TAs are known and can be easily inserted into the plasmid DNA and expressed under the control of a constitutively active bacterial promoter. Typically given intradermally or intramuscularly, the proteins expressed by transcription and translation of the plasmid are readily picked up by DCs, processed, and presented in the context of MHC class I and II, thus providing a more “natural” stimulation of the immune system. Moreover, the unmethylated dinucleotide-CpG residues, or CpG motifs, present in high frequency in the bacterial DNA, provide additional stimulation of DCs, triggering them to induce a Th1-type immune response.192
No DNA vaccines have been licensed for human use yet. However, many DNA vaccines have been tested in clinical trials, and results have thus far been disappointing for various reasons (see review in Liu193). Nonetheless, the first conditionally licensed (by the U.S. Department of Agriculture [USDA]) veterinary cancer vaccine is based off of the DNA plasmid technology.194 The ONCEPT vaccine (Merial, Duluth, GA) for canine malignant melanoma (CMM) uses xenogeneic DNA plasmids that contain the gene-encoding human tyrosinase (huTyr). Initial studies showed the development of an antibody-mediated immune response against the huTyr protein that cross-reacted to canine tyrosinase.195 Improved survival of dogs treated with this vaccine compared to historical control animals has been reported with no severe side effects noted.194,196 Further studies of this plasmid DNA technology demonstrated that the vaccine could induce antigen-specific IFN-γ+ T-cells in normal beagle dogs.197 Finally, the same group that developed the CMM vaccine has reportedly completed phase I trials of murine CD20 for treatment of canine B-cell lymphoma and is initiating a phase II trial soon.198
Tumor Vaccination Using Viral Vector Vaccines
As discussed previously, viruses have been used to target tumor cells, particularly those with innate oncolytic properties. However, viruses can also be used as vectors for expression of particular TAs. Typically, attenuated or replication-defective forms of the virus are used to allow for effective stimulation of the innate and adaptive immune responses without the risk of spreading and rapidly dividing within the host. The most commonly used viral platform for both human and veterinary studies is the Poxviridae family. The poxviruses are easy to work with, amenable to large amounts of foreign DNA, and highly immunogenic, allowing for strong immune responses against weak TAs, such as carcinoembryonic antigen (CEA).199 In humans, one of the most commonly used viral vaccine platforms is the canarypox virus ALVAC. Recent published human clinical trials using ALVAC include combining CEA-expressing ALVAC with chemotherapy for metastatic colorectal cancer,200 ALVAC-expressing human GM-CSF or IL-2 for treatment of melanoma or leiomyosarcoma,201 and intranodal injection of ALVAC-expressing gp100 in high-risk melanoma patients.202 Interestingly, although all of these studies reported that the vaccine was safe to use and that immunologic responses were observed, efficacy of these therapies is limited.203
Vaccination Against Tumor Antigens Using Dendritic Cells
Dendritic cells (DCs) possess very potent antigen-presenting abilities and are an attractive target for cancer vaccine strategies. Besides their role in vivo in processing and presenting TAs derived either naturally or from tumor vaccines, there are many clinical trials published that examine the use of ex vivo activated and expanded DCs injected back into the donor as a way of activating tumor-specific T-cells in vivo. The drawback to this method is that the ex vivo processing of DCs typically takes about 7 to 10 days, requires growth in a combination of cytokines, and can only be used autologously. Nonetheless, ex vivo prepared DCs have shown clinical efficacy, particularly in human patients with metastatic disease.204,205 Recently, it has been determined that the potency of the DCs produced ex vivo depends on the combination of cytokines used.206 DCs generated with GM-CSF and IFN-α or GM-CSF and IL-15 display potent priming of T-cell–mediated and CD8+ T-cell–mediated immune responses in vitro. Moreover, the use of mature DCs is better then immature DCs, since immature DCs actually induce immune tolerance via expansion of IL-10–secreting T-cells.207 However, the methods of maturation matter as well, with studies showing DCs activated with a mixture of IFN-α, polyinosinic-polycytidylic acid, IL-1β, TNF, and IFN-γ elicit many more anti-melanoma cytotoxic T-lymphocytes (CTLs) in vitro than the standard IL-1β, TNF, IL-6, and prostaglandin E2 (PGE2) cocktail.208 Finally, new methods of targeting antigens to DCs through anti-DC receptor (i.e., lectin receptors such as DEC-205, DC-SIGN, or DNGR-1) antibody-TA fusions, appropriate selection of adjuvants to deliver antigens to DCs, and combination therapies using chemotherapy and DC activation are being investigated.204,206
Of note, the first FDA-approved therapeutic cancer vaccine, Provenge (sipuleucel-T, Dendreon, Seattle) is derived from peripheral blood mononuclear cells (PBMCs) removed from patients with castration-refractory prostate cancer that are cultured and activated ex vivo with a fusion protein of human recombinant prostatic acid phosphatase (PAP) and human GM-CSF prior to transfusion back into the donor.209 This vaccine has demonstrated an increase in survival time that was a little over 4 months compared to the placebo group in a multicenter clinical trial of over 500 patients. Besides consisting of activated DCs, this vaccine also contains autologous lymphocytes that may be playing a role in its efficacy.210
DC vaccination in veterinary medicine has been and is still currently being explored. An initial study of three dogs with oral melanoma showed that bone marrow–derived DCs transduced with an adenovirus expressing human gp100 could safely be used. In this study, dogs received three subcutaneous vaccines over 4 months.211 One of the dogs that had a complete response with no evidence of disease 4 years later developed a robust CTL response against the gp100. Another dog that relapsed after 22 months had no evidence of anti-gp100 CTLs. A similar study performed in normal dogs was done to assess the immune response of DCs pulsed with canine melanoma cell (CMM2) lysates, in which a good delayed-type hypersensitivity response was seen against CMM2 following vaccination.212
Another study described previously saw success using ex vivo activated DCs and IFN-γ for treating canine solid tumors. Finally, a very recent study looked at the safety of using a DC-mammary tumor cell fusion hybrid vaccine.213 In this case, normal dog PBMCs were used to generate DCs that were subsequently fused to canine mammary tumor cells. Injection of normal Beagle dogs with this fusion plus CpG adjuvant resulted in a robust antibody response against the fusion partner tumor cell line, as well as three unrelated canine mammary tumor cells. However, no CTL responses were noted. Hence development of DC vaccines for use in veterinary medicine is currently being explored in various tumor models and by using various strategies to optimize the induced antitumor immune response.
Antibody Therapy for Cancer
The use of monoclonal antibody (MAb) therapy for cancer has been studied for over a quarter century, following the development of hybridoma technology by Kohler and Milstein in 1975.214 This technique consisted of antibody-producing cells fused with mouse myeloma cells, thus becoming immortalized and capable of continuously producing antibody that can be purified out of the culture media. Initially, the use of MAbs clinically was limited due to the responses mounted by the host against the foreign mouse proteins. However, recent technology allowed for “humanizing” these antibodies by genetically grafting the mouse hypervariable region of interest onto the human immunoglobulin, thus resulting in an antibody that is 95% human. Moreover, mice genetically rendered to express human immunoglobulins can successfully generate 100% human antibodies in response to various antigens.215 Using humanized antibodies improves antibody-dependent cell-mediated cytotoxicity (ADCC), improves antibody stability, and decreases immunogenicity of the antibody itself. The use of MAbs in human medicine has increased over the years. Several MAbs are approved by the FDA for use as human cancer treatments, and many are in active development and currently undergoing testing in human clinical trials.216 As a general guide, MAb names ending in -omab are murine based, -ximab and -zumab are chimeric, and -umab are humanized versions of the antibodies.
Unfortunately, the generation of canine versions of antibodies for use in veterinary medicine has yet to occur. Nonetheless, one recent study looked at the use of a murine (L243) and humanized form (IMMU-114) of an anti-HLA-DR MAb in normal dogs and dogs with B-cell lymphoma.217 In vitro studies demonstrated that both of these antibodies bound normal and malignant canine lymphocytes, inducing apoptosis in some. In vivo administration to normal dogs provided safety and pharmacokinetic data. A pilot study was then performed on seven dogs with either lymphoma or plasmacytomas; these dogs received 1 to 4 treatments with L243 MAb every 2 weeks. Myelosuppression was a cumulative-dose adverse effect, but some dogs displayed a transient response to the treatments. Thus future investigation of the use of this MAb in canine lymphoma is warranted.
Conjugated Monoclonal Antibodies
Another use of MAbs is linking them to potential toxins or radioisotopes. Initial studies involved linking chemical toxins to immunoglobulins to generate molecules called immunotoxins. Such chemicals tested were ricin and diphtheria toxins, but these conjugates were immunogenically and chemically unstable. Development of recombinant immunotoxins helped address this issue, although the current concern with immunotoxins is their ability to nonspecifically kill any cell expressing the antibody-specific receptor.
MAbs can also be linked to radionuclides. The concept behind these antibodies is that the antibody targets tumor tissue and the energy released by the radioisotope attached to the antibody can penetrate bulky solid tumors and may also kill surrounding tumor and stromal cells. Examples of radiolabeled MAbs in clinical therapeutic use in humans currently exist. The current use of radiolabeled MAbs in dogs is limited to imaging modalities rather than treatment of cancer.
Cancer Immunotherapy Using Adoptive Transfer of T-Cells
Adoptive T-cell transfer (ACT) is a technique in which cells are collected from a cancer patient, expanded and activated in culture, and then transferred back into the patient. Although this technique allows for the enhancement of tumor-specific T-cells, it is labor intensive and time consuming, thus its use is limited in both human and veterinary patients. Below, we will discuss a couple of historic methods used to generate these cells, as well as new techniques currently being investigated to improve this form of immunotherapy.
Transfer of Lymphokine-Activated Killer Cells
Initial T-cell transfer studies involved the generation of LAK cells. This was done by culturing PBMCs in high concentrations of IL-2, thus selecting for a population of cells with potent tumor cell-lysis ability. Clinical trials using this technique in humans were disappointing and unfeasible, despite promising mouse studies.218 Use of LAK cells in veterinary medicine is limited to studies of cats with feline leukemia virus (FeLV) or feline immunodeficiency virus (FIV).219
Transfer of Tumor-Infiltrating Lymphocytes
One source of potent antitumor T-cells is in the tumor itself. These cells, called tumor-infiltrating lymphocytes (TILs), when expanded using IL-2, exhibit potent cytolytic activity that is many times higher than LAK cells against tumors in both a specific and nonspecific way.220 Although they are considered the best source of T-cells for ACT,221,222 their use in human medicine is limited because of a few variables such as time of isolation, the tumor they were isolated from, and the functional state of the cells when isolated.218 Nonetheless, limited success has been observed in cases of treating human melanoma with TILs, particularly when combined with nonmyeloablative chemotherapy such as fludarabine and cyclophosphamide, which deplete lymphocytes but spare bone marrow stem cells.223 In one study, six of thirteen melanoma patients had significant tumor regression and four had a mixed response including regression of some lesions and growth of others.223 In a follow-up study involving a larger number of patients (34 in total), tumor regression was seen in 51% of the patients that received chemotherapy prior to the TIL transfer and IL-2 treatment.224 In addition to the use of nonmyeloablative treatments, recent studies have investigated the use of other forms of Th1 stimulation along with ACT. One group has investigated the use of adding CpG-ODNs to their TILs to increase their efficacy.225 In a study using ex vivo isolated human TILs, instillation of the activated TILs with CpG-ODN into athymic nude, tumor-bearing mice resulted in decreased tumor burden and prolonged survival. Regardless of the human clinical trials’ results, TILs have not been used in veterinary medicine, perhaps due to the lack of reliable efficacy across multiple tumor types.
New Approaches to Adoptive T-Cell Transfers
As mentioned at the beginning of this chapter, one limitation to most immunotherapies is the fact that tumors can orchestrate an immunosuppressive environment. Thus, even if one instilled thousands of activated, tumor-specific T-cells into a cancer patient, the majority of these cells will become inactivated on reaching the tumor, particularly when dealing with solid tumors.226 It is currently being recognized that strategies to overcome immunosuppression must be implemented in order to enhance the efficiency of tumor-specific T-cells in ACT studies. One technique to address this suppression is performing lymphodepletion prior to ACT.227-229 Recent studies have also suggested the isolation of CD4+ T-cells for ACT, rather than cytotoxic CD8+ T-cells because CD4+ T-cells are capable of activating both innate immune cells and CD8+ T-cells.230,231 However, CD4+ T-cells contain Treg cells, thus strategies to block the development of Treg cells during CD4+ T-cell ACT have also been investigated.230 In addition, the availability of TAs also appears to play a role in the strength of CD4+ ACT therapy.232 Similarly, the addition of cytokines and/or blocking antibodies against suppressor cells, along with ACTs, has been shown to enhance the effectiveness of this therapy.230,233-235
The Future of Cancer Immunotherapy
The use of immunotherapy for the treatment of cancer is an exciting and ever-evolving field of research and application. With the advancement of techniques used to assess immune responses to tumors; better ways of predicting responses, including development of RECIST; and an understanding that tumor responses to immunotherapies may be delayed as compared to conventional chemotherapy, radiation therapy, and surgery, one can more reliably assess the clinical efficacy and safety of novel immunotherapies. Moreover, a better understanding of the disease pathology in our veterinary patients has led to a movement toward using spontaneous canine and feline cancers as models for human disease, thus allowing for testing of novel immunotherapies in our small animal patients that will not only benefit them, but benefit human cancer patients as well.236,237
However, the development of a successful immunotherapy protocol is not without limitations. One of the main reasons for failure of many immunotherapies is due to the immunosuppressive microenvironment established by the tumor. Thus immunotherapies that are best able to overcome this suppression will prove the most successful.238 In addition, certain drugs and/or proteins that can deplete or inactivate the key players in immune suppression (i.e., MDSCs and Tregs) may be best used in concert with novel vaccines or other immunotherapies in order to optimize their effectiveness. Along those lines, the use of newer and more potent adjuvants such as various preparations of CpG motifs to stimulate the immune system will be a critical component of newer vaccines. It has now become clear that the most successful adjuvants are those that not only stimulate a strong primary response against the tumor, but also lead to the development of a robust central memory response.
One of the more successful categories of immunotherapies currently used in human medicine is MAbs. Advances in technology led to the development of humanized, nonimmunogenic forms of antibodies against key cellular receptors, either to activate key antitumor immune cells or lead to cytolytic activity against tumor cells. However, similar advances in treating dogs with MAbs are lacking, although some promising preliminary results have been seen using human or mouse antibodies to treat canine lymphoma.217
It should be understood that many, if not all, immunotherapies developed should work in concert and synergize with current cancer treatment modalities. Given the ability of tumor cells to become resistant to chemotherapy and radiation therapy and their ability to suppress the immune system, one would be naïve to think that a single-modality treatment is the most effective means of tumor control. While the immune system can be manipulated to mount an effective antitumor immune response, it is best utilized in cases of minimal residual and metastatic disease, in which radiation therapy, chemotherapy, and/or surgery are used to cytoreduce large tumors. Moreover, it is becoming very clear that the immune system is a key player involved in the tumor response to radiation and chemotherapy; thus finding ways to incorporate immunotherapy into current standards of care may actually enhance the effectiveness of these modalities. For example, we have observed that the use of liposomal clodronate therapy to eliminate MDSCs can enhance the tumor response to lomustine (CCNU) in dogs with malignant histiocytosis (unpublished observation). We hypothesize that the immunosuppressive cells present in the tumor microenvironment are capable of protecting tumor cells from the effects of chemotherapy, thus by removing these tumor cells we can enhance the effectiveness of the chemotherapy. We predict that the use of immunotherapy as part of a protocol to treat canine and feline diseases should soon become routine. By understanding the role of the immune system in cancer in our small animal patients, we can develop not only better immunotherapies that will benefit these patients, but also therapies that will be applicable to human medicine.
References
1. Antony, PA, Restifo, NP. CD4+CD25+ T regulatory cells, immunotherapy of cancer, and interleukin-2. J Immunother. 2005;28:120–128.
2. Burnet, M. Cancer; a biological approach. I. The processes of control. Br Med J. 1957;1:779–786.
3. Stutman, O. Tumor development after 3-methylcholanthrene in immunologically deficient athymic-nude mice. Science. 1974;183:534–536.
4. Rygaard, J, Povlsen, CO. The mouse mutant nude does not develop spontaneous tumours. An argument against immunological surveillance. Acta Pathol Microbiol Scand B Microbiol Immunol. 1974;82:99–106.
5. Schreiber, TH, Podack, ER. A critical analysis of the tumour immunosurveillance controversy for 3-MCA-induced sarcomas. Br J Cancer. 2009;101:381–386.
6. Kaplan, DH, Shankaran, V, Dighe, AS, et al. Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc Natl Acad Sci U S A. 1998;95:7556–7561.
7. Shankaran, V, Ikeda, H, Bruce, AT, et al. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410:1107–1111.
8. Dunn, GP, Bruce, AT, Ikeda, H, et al. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991–998.
9. Rabinovich, GA, Gabrilovich, D, Sotomayor, EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007;25:267–296.
10. Murdoch, C, Muthana, M, Coffelt, SB, et al. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer. 2008;8:618–631.
11. Qian, BZ, Pollard, JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39–51.
12. Almand, B, Clark, JI, Nikitina, E, et al. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol. 2001;166:678–689.
13. Diaz-Montero, CM, Salem, ML, Nishimura, MI, et al. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother. 2009;58:49–59.
14. Mandruzzato, S, Solito, S, Falisi, E, et al. IL4Ralpha+ myeloid-derived suppressor cell expansion in cancer patients. J Immunol. 2009;182:6562–6568.
15. Bunt, SK, Yang, L, Sinha, P, et al. Reduced inflammation in the tumor microenvironment delays the accumulation of myeloid-derived suppressor cells and limits tumor progression. Cancer Res. 2007;67:10019–10026.
16. Melani, C, Chiodoni, C, Forni, G, et al. Myeloid cell expansion elicited by the progression of spontaneous mammary carcinomas in c-erbB-2 transgenic BALB/c mice suppresses immune reactivity. Blood. 2003;102:2138–2145.
17. Bosco, MC, Puppo, M, Blengio, F, et al. Monocytes and dendritic cells in a hypoxic environment: Spotlights on chemotaxis and migration. Immunobiology. 2008;213:733–749.
18. Du, R, Lu, KV, Petritsch, C, et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell. 2008;13:206–220.
19. Sawanobori, Y, Ueha, S, Kurachi, M, et al. Chemokine-mediated rapid turnover of myeloid-derived suppressor cells in tumor-bearing mice. Blood. 2008;111:5457–5466.
20. Yang, L, Huang, J, Ren, X, et al. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell. 2008;13:23–35.
21. Ye, XZ, Yu, SC, Bian, XW. Contribution of myeloid-derived suppressor cells to tumor-induced immune suppression, angiogenesis, invasion and metastasis. J Genet Genomics. 2010;37:423–430.
22. Joyce, JA, Pollard, JW. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009;9:239–252.
23. Ostrand-Rosenberg, S, Sinha, P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol. 2009;182:4499–4506.
24. Sinha, P, Clements, VK, Bunt, SK, et al. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol. 2007;179:977–983.
25. Li, H, Han, Y, Guo, Q, et al. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta1. J Immunol. 2009;182:240–249.
26. Nomura, T, Sakaguchi, S. Naturally arising CD25+CD4+ regulatory T cells in tumor immunity. Curr Top Microbiol Immunol. 2005;293:287–302.
27. Fontenot, JD, Rudensky, AY. A well adapted regulatory contrivance: regulatory T cell development and the forkhead family transcription factor Foxp3. Nat Immunol. 2005;6:331–337.
28. Camisaschi, C, Casati, C, Rini, F, et al. LAG-3 expression defines a subset of CD4(+)CD25(high)Foxp3(+) regulatory T cells that are expanded at tumor sites. J Immunol. 2010;184:6545–6551.
29. Shimizu, J, Yamazaki, S, Takahashi, T, et al. Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self-tolerance. Nat Immunol. 2002;3:135–142.
30. Wing, K, Onishi, Y, Prieto-Martin, P, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;322:271–275.
31. Yamaguchi, T, Hirota, K, Nagahama, K, et al. Control of immune responses by antigen-specific regulatory T cells expressing the folate receptor. Immunity. 2007;27:145–159.
32. Qin, FX. Dynamic behavior and function of Foxp3+ regulatory T cells in tumor bearing host. Cell Mol Immunol. 2009;6:3–13.
33. Chen, W, Jin, W, Hardegen, N, et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–1886.
34. Chen, W, Wahl, SM. TGF-beta: the missing link in CD4+CD25+ regulatory T cell-mediated immunosuppression. Cytokine Growth Factor Rev. 2003;14:85–89.
35. Hawiger, D, Wan, YY, Eynon, EE, et al. The transcription cofactor Hopx is required for regulatory T cell function in dendritic cell-mediated peripheral T cell unresponsiveness. Nat Immunol. 2010;11:962–968.
36. Huehn, J, Hamann, A, Homing to suppress: address codes for Treg migration. Trends Immunol 2005;26:632–636.
37. Hoelzinger, DB, Smith, SE, Mirza, N, et al. Blockade of CCL1 inhibits T regulatory cell suppressive function enhancing tumor immunity without affecting T effector responses. J Immunol. 2010;184:6833–6842.
38. Curiel, TJ, Coukos, G, Zou, L, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–949.
39. Miller, AM, Lundberg, K, Ozenci, V, et al. CD4+CD25high T cells are enriched in the tumor and peripheral blood of prostate cancer patients. J Immunol. 2006;177:7398–7405.
40. Sasada, T, Kimura, M, Yoshida, Y, et al. CD4+CD25+ regulatory T cells in patients with gastrointestinal malignancies: possible involvement of regulatory T cells in disease progression. Cancer. 2003;98:1089–1099.
41. Turk, MJ, Guevara-Patino, JA, Rizzuto, GA, et al. Concomitant tumor immunity to a poorly immunogenic melanoma is prevented by regulatory T cells. J Exp Med. 2004;200:771–782.
42. Viguier, M, Lemaitre, F, Verola, O, et al. Foxp3 expressing CD4+CD25(high) regulatory T cells are overrepresented in human metastatic melanoma lymph nodes and inhibit the function of infiltrating T cells. J Immunol. 2004;173:1444–1453.
43. Biller, BJ, Elmslie, RE, Burnett, RC, et al. Use of FoxP3 expression to identify regulatory T cells in healthy dogs and dogs with cancer. Vet Immunol Immunopathol. 2007;116:69–78.
44. O’Neill, K, Guth, A, Biller, B, et al. Changes in regulatory T cells in dogs with cancer and associations with tumor type. J Vet Intern Med. 2009;23:875–881.
45. Teng, MW, Swann, JB, von Scheidt, B, et al. Multiple antitumor mechanisms downstream of prophylactic regulatory T-cell depletion. Cancer Res. 2010;70:2665–2674.
46. Piconese, S, Valzasina, B, Colombo, MP. OX40 triggering blocks suppression by regulatory T cells and facilitates tumor rejection. J Exp Med. 2008;205:825–839.
47. Berraondo, P, Nouze, C, Preville, X, et al. Eradication of large tumors in mice by a tritherapy targeting the innate, adaptive, and regulatory components of the immune system. Cancer Res. 2007;67:8847–8855.
48. Matar, P, Rozados, VR, Gonzalez, AD, et al. Mechanism of antimetastatic immunopotentiation by low-dose cyclophosphamide. Eur J Cancer. 2000;36:1060–1066.
49. Ghiringhelli, F, Menard, C, Puig, PE, et al. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol Immunother. 2007;56:641–648.
50. Hoffmann, TK, Muller-Berghaus, J, Ferris, RL, et al. Alterations in the frequency of dendritic cell subsets in the peripheral circulation of patients with squamous cell carcinomas of the head and neck. Clin Cancer Res. 2002;8:1787–1793.
51. Pinzon-Charry, A, Ho, CS, Laherty, R, et al. A population of HLA-DR+ immature cells accumulates in the blood dendritic cell compartment of patients with different types of cancer. Neoplasia. 2005;7:1112–1122.
52. Jin, D, Fan, J, Wang, L, et al. CD73 on tumor cells impairs antitumor T-cell responses: a novel mechanism of tumor-induced immune suppression. Cancer Res. 2010;70:2245–2255.
53. Steinman, RM, Hawiger, D, Nussenzweig, MC. Tolerogenic dendritic cells. Annu Rev Immunol. 2003;21:685–711.
54. Fuchs, EJ, Matzinger, P. Is cancer dangerous to the immune system? Semin Immunol. 1996;8:271–280.
55. Enk, AH, Jonuleit, H, Saloga, J, et al. Dendritic cells as mediators of tumor-induced tolerance in metastatic melanoma. Int J Cancer. 1997;73:309–316.
56. Nestle, FO, Burg, G, Fah, J, et al. Human sunlight-induced basal-cell-carcinoma-associated dendritic cells are deficient in T cell co-stimulatory molecules and are impaired as antigen-presenting cells. Am J Pathol. 1997;150:641–651.
57. Chaux, P, Favre, N, Martin, M, et al. Tumor-infiltrating dendritic cells are defective in their antigen-presenting function and inducible B7 expression in rats. Int J Cancer. 1997;72:619–624.
58. Ishida, T, Oyama, T, Carbone, DP, et al. Defective function of Langerhans cells in tumor-bearing animals is the result of defective maturation from hemopoietic progenitors. J Immunol. 1998;161:4842–4851.
59. Almand, B, Resser, JR, Lindman, B, et al. Clinical significance of defective dendritic cell differentiation in cancer. Clin Cancer Res. 2000;6:1755–1766.
60. Troy, AJ, Summers, KL, Davidson, PJ, et al. Minimal recruitment and activation of dendritic cells within renal cell carcinoma. Clin Cancer Res. 1998;4:585–593.
61. Liu, CC, Wang, YS, Lin, CY, et al. Transient downregulation of monocyte-derived dendritic-cell differentiation, function, and survival during tumoral progression and regression in an in vivo canine model of transmissible venereal tumor. Cancer Immunol Immunother. 2008;57:479–491.
62. Cheng, P, Corzo, CA, Luetteke, N, et al. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med. 2008;205:2235–2249.
63. Herber, DL, Cao, W, Nefedova, Y, et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat Med. 2010;16:880–886.
64. Hartmann, E, Wollenberg, B, Rothenfusser, S, et al. Identification and functional analysis of tumor-infiltrating plasmacytoid dendritic cells in head and neck cancer. Cancer Res. 2003;63:6478–6487.
65. Gerlini, G, Tun-Kyi, A, Dudli, C, et al. Metastatic melanoma secreted IL-10 down-regulates CD1 molecules on dendritic cells in metastatic tumor lesions. Am J Pathol. 2004;165:1853–1863.
66. Gabrilovich, DI, Chen, HL, Girgis, KR, et al. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat Med. 1996;2:1096–1103.
67. Zhang, M, Tang, H, Guo, Z, et al. Splenic stroma drives mature dendritic cells to differentiate into regulatory dendritic cells. Nat Immunol. 2004;5:1124–1133.
68. Lin, WW, Karin, M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest. 2007;117:1175–1183.
69. Ridge, J, Terle, DA, Dragunsky, E, et al. Effects of gamma-IFN and NGF on subpopulations in a human neuroblastoma cell line: flow cytometric and morphological analysis. In Vitro Cell Dev Biol Anim. 1996;32:238–248.
70. Maloy, KJ, Salaun, L, Cahill, R, et al. CD4+CD25+ T(R) cells suppress innate immune pathology through cytokine-dependent mechanisms. J Exp Med. 2003;197:111–119.
71. Sredni, B, Weil, M, Khomenok, G, et al. Ammonium trichloro(dioxoethylene-o,o’)tellurate (AS101) sensitizes tumors to chemotherapy by inhibiting the tumor interleukin 10 autocrine loop. Cancer Res. 2004;64:1843–1852.
72. Ebrahimi, B, Tucker, SL, Li, D, et al. Cytokines in pancreatic carcinoma: correlation with phenotypic characteristics and prognosis. Cancer. 2004;101:2727–2736.
73. Ozdemir, F, Aydin, F, Yilmaz, M, et al. The effects of IL-2, IL-6 and IL-10 levels on prognosis in patients with aggressive non-Hodgkin’s lymphoma (NHL). J Exp Clin Cancer Res. 2004;23:485–488.
74. Lech-Maranda, E, Bienvenu, J, Michallet, AS, et al. Elevated IL-10 plasma levels correlate with poor prognosis in diffuse large B-cell lymphoma. Eur Cytokine Netw. 2006;17:60–66.
75. Becker, C, Fantini, MC, Neurath, MF. TGF-beta as a T cell regulator in colitis and colon cancer. Cytokine Growth Factor Rev. 2006;17:97–106.
76. Ghiringhelli, F, Puig, PE, Roux, S, et al. Tumor cells convert immature myeloid dendritic cells into TGF-beta-secreting cells inducing CD4+CD25+ regulatory T cell proliferation. J Exp Med. 2005;202:919–929.
77. Derynck, R, Akhurst, RJ, Balmain, A. TGF-beta signaling in tumor suppression and cancer progression. Nat Genet. 2001;29:117–129.
78. Luo, JL, Maeda, S, Hsu, LC, et al. Inhibition of NF-kappaB in cancer cells converts inflammation- induced tumor growth mediated by TNFalpha to TRAIL-mediated tumor regression. Cancer Cell. 2004;6:297–305.
79. Elgert, KD, Alleva, DG, Mullins, DW. Tumor-induced immune dysfunction: the macrophage connection. J Leukoc Biol. 1998;64:275–290.
80. Catchpole, B, Gould, SM, Kellett-Gregory, LM, et al. Immunosuppressive cytokines in the regional lymph node of a dog suffering from oral malignant melanoma. J Small Anim Pract. 2002;43:464–467.
81. Reinis, M. Immunotherapy of MHC class I-deficient tumors. Future Oncology. 2010;6:1577–1589.
82. Garrido, F, Algarra, I, Garcia-Lora, AM. The escape of cancer from T lymphocytes: immunoselection of MHC class I loss variants harboring structural-irreversible “hard” lesions. Cancer Immunol Immunother. 2010;59:1601–1606.
83. Rimsza, LM, Farinha, P, Fuchs, DA, et al. HLA-DR protein status predicts survival in patients with diffuse large B-cell lymphoma treated on the MACOP-B chemotherapy regimen. Leuk Lymphoma. 2007;48:542–546.
84. Rimsza, LM, Roberts, RA, Miller, TP, et al. Loss of MHC class II gene and protein expression in diffuse large B-cell lymphoma is related to decreased tumor immunosurveillance and poor patient survival regardless of other prognostic factors: a follow-up study from the Leukemia and Lymphoma Molecular Profiling Project. Blood. 2004;103:4251–4258.
85. Rao, S, Lana, S, Eickhoff, J, et al. Class II major histocompatibility complex expression and cell size independently predict survival in canine B-cell lymphoma. J Vet Intern Med. 2011;25(5):1097–1105.
86. Shi, F, Shi, M, Zeng, Z, et al. PD-1 and PD-L1 upregulation promotes CD8(+) T-cell apoptosis and postoperative recurrence in hepatocellular carcinoma patients. Int J Cancer. 2011;128:887–896.
87. Mu, CY, Huang, JA, Chen, Y, et al. High expression of PD-L1 in lung cancer may contribute to poor prognosis and tumor cells immune escape through suppressing tumor infiltrating dendritic cells maturation. Med Oncol. 2010;28(3):682–688.
88. Nomi, T, Sho, M, Akahori, T, et al. Clinical significance and therapeutic potential of the programmed death-1 ligand/programmed death-1 pathway in human pancreatic cancer. Clin Cancer Res. 2007;13:2151–2157.
89. Benson, DM, Jr., Bakan, CE, Mishra, A, et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: a therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood. 2010;116:2286–2294.
90. Suzuki, E, Kapoor, V, Jassar, AS, et al. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res. 2005;11:6713–6721.
91. Vincent, J, Mignot, G, Chalmin, F, et al. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. 2010;70:3052–3061.
92. Ko, JS, Zea, AH, Rini, BI, et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res. 2009;15:2148–2157.
93. Kodumudi, KN, Woan, K, Gilvary, DL, et al. A novel chemoimmunomodulating property of docetaxel: suppression of myeloid-derived suppressor cells in tumor bearers. Clin Cancer Res. 2010;16:4583–4594.
94. Sinha, P, Clements, VK, Fulton, AM, et al. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res. 2007;67:4507–4513.
95. Pan, PY, Wang, GX, Yin, B, et al. Reversion of immune tolerance in advanced malignancy: modulation of myeloid-derived suppressor cell development by blockade of stem-cell factor function. Blood. 2008;111:219–228.
96. Lathers, DM, Clark, JI, Achille, NJ, et al. Phase 1B study to improve immune responses in head and neck cancer patients using escalating doses of 25-hydroxyvitamin D3. Cancer Immunol Immunother. 2004;53:422–430.
97. Shojaei, F, Singh, M, Thompson, JD, et al. Role of Bv8 in neutrophil-dependent angiogenesis in a transgenic model of cancer progression. Proc Natl Acad Sci U S A. 2008;105:2640–2645.
98. Melani, C, Sangaletti, S, Barazzetta, FM, et al. Amino-biphosphonate-mediated MMP-9 inhibition breaks the tumor-bone marrow axis responsible for myeloid-derived suppressor cell expansion and macrophage infiltration in tumor stroma. Cancer Res. 2007;67:11438–11446.
99. Mirza, N, Fishman, M, Fricke, I, et al. All-trans-retinoic acid improves differentiation of myeloid cells and immune response in cancer patients. Cancer Res. 2006;66:9299–9307.
100. Daurkin, I, Eruslanov, E, Vieweg, J, et al. Generation of antigen-presenting cells from tumor-infiltrated CD11b myeloid cells with DNA demethylating agent 5-aza-2′-deoxycytidine. Cancer Immunol Immunother. 2010;59:697–706.
101. Ko, HJ, Lee, JM, Kim, YJ, et al. Immunosuppressive myeloid-derived suppressor cells can be converted into immunogenic APCs with the help of activated NKT cells: an alternative cell-based antitumor vaccine. J Immunol. 2009;182:1818–1828.
102. Fernandez, A, Mesa, C, Marigo, I, et al. Inhibition of tumor-induced myeloid-derived suppressor cell function by a nanoparticulated adjuvant. J Immunol. 2011;186:264–274.
103. Priceman, SJ, Sung, JL, Shaposhnik, Z, et al. Targeting distinct tumor-infiltrating myeloid cells by inhibiting CSF-1 receptor: combating tumor evasion of antiangiogenic therapy. Blood. 2010;115:1461–1471.
104. Pan, PY, Ma, G, Weber, KJ, et al. Immune stimulatory receptor CD40 is required for T-cell suppression and T regulatory cell activation mediated by myeloid-derived suppressor cells in cancer. Cancer Res. 2010;70:99–108.
105. Curran, MA, Montalvo, W, Yagita, H, et al. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A. 2010;107:4275–4280.
106. De Santo, C, Serafini, P, Marigo, I, et al. Nitroaspirin corrects immune dysfunction in tumor-bearing hosts and promotes tumor eradication by cancer vaccination. Proc Natl Acad Sci U S A. 2005;102:4185–4190.
107. Serafini, P, Mgebroff, S, Noonan, K, et al. Myeloid-derived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer Res. 2008;68:5439–5449.
108. Nagaraj, S, Youn, JI, Weber, H, et al. Anti-inflammatory triterpenoid blocks immune suppressive function of MDSCs and improves immune response in cancer. Clin Cancer Res. 2010;16:1812–1823.
109. Serafini, P, Meckel, K, Kelso, M, et al. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J Exp Med. 2006;203:2691–2702.
110. Richardson, MA, Ramirez, T, Russell, NC, et al. Coley toxins immunotherapy: a retrospective review. Altern Ther Health Med. 1999;5:42–47.
111. Alexandroff, AB, Jackson, AM, O’Donnell, MA, et al. BCG immunotherapy of bladder cancer: 20 years on. Lancet. 1999;353:1689–1694.
112. van der Meijden, AP. Non-specific immunotherapy with bacille Calmette-Guerin (BCG). Clin Exp Immunol. 2001;123:179–180.
113. Vita, F, Siracusano, S, Abbate, R, et al. BCG prophylaxis in bladder cancer produces activation of recruited neutrophils. Can J Urol. 2011;18:5517–5523.
114. Ludwig, AT, Moore, JM, Luo, Y, et al. Tumor necrosis factor-related apoptosis-inducing ligand: a novel mechanism for Bacillus Calmette-Guerin-induced antitumor activity. Cancer Res. 2004;64:3386–3390.
115. Herr, HW, Morales, A. History of bacillus Calmette-Guerin and bladder cancer: an immunotherapy success story. J Urol. 2008;179:53–56.
116. Klein, WR, Rutten, VP, Steerenberg, PA, et al. The present status of BCG treatment in the veterinary practice. In Vivo. 1991;5:605–608.
117. Debruyne, FM, van der Meijden, AP, Schreinemachers, LM, et al. Intravesical and intradermal BCG-RIVM application: a toxicity study. Prog Clin Biol Res. 1985;185B:151–159.
118. Knapp, DW, Glickman, NW, Denicola, DB, et al. Naturally-occurring canine transitional cell carcinoma of the urinary bladder: a relevant model of human invasive bladder cancer. Urol Oncol. 2000;5:47–59.
119. Mukaratirwa, S, Chitanga, S, Chimatira, T, et al. Combination therapy using intratumoral bacillus Calmette-Guerin (BCG) and vincristine in dogs with transmissible venereal tumours: therapeutic efficacy and histological changes. J S Afr Vet Assoc. 2009;80:92–96.
120. Henry, CJ, Downing, S, Rosenthal, RC, et al. Evaluation of a novel immunomodulator composed of human chorionic gonadotropin and bacillus Calmette-Guerin for treatment of canine mast cell tumors in clinically affected dogs. Am J Vet Res. 2007;68:1246–1251.
121. Lipton, A, Harvey, HA, Balch, CM, et al. Corynebacterium parvum versus bacille Calmette-Guerin adjuvant immunotherapy of stage II malignant melanoma. J Clin Oncol. 1991;9:1151–1156.
122. MacEwen, EG, Patnaik, AK, Harvey, HJ, et al. Canine oral melanoma: comparison of surgery versus surgery plus Corynebacterium parvum. Cancer Invest. 1986;4:397–402.
123. Misdorp, W. Incomplete surgery, local immunostimulation, and recurrence of some tumour types in dogs and cats. Vet Q. 1987;9:279–286.
124. Luo, X, Li, Z, Lin, S, et al. Antitumor effect of VNP20009, an attenuated Salmonella, in murine tumor models. Oncol Res. 2001;12:501–508.
125. Leschner, S, Westphal, K, Dietrich, N, et al. Tumor invasion of Salmonella enterica serovar Typhimurium is accompanied by strong hemorrhage promoted by TNF-alpha. PLoS One. 2009;4:e6692.
126. Saccheri, F, Pozzi, C, Avogadri, F, et al. Bacteria-induced gap junctions in tumors favor antigen cross-presentation and antitumor immunity. Sci Transl Med. 2010;2:44–57.
127. Toso, JF, Gill, VJ, Hwu, P, et al. Phase I study of the intravenous administration of attenuated Salmonella typhimurium to patients with metastatic melanoma. J Clin Oncol. 2002;20:142–152.
128. Thamm, DH, Kurzman, ID, King, I, et al. Systemic administration of an attenuated, tumor-targeting Salmonella typhimurium to dogs with spontaneous neoplasia: phase I evaluation. Clin Cancer Res. 2005;11:4827–4834.
129. Ochi, A, Migita, K, Xu, J, et al. In vivo tumor immunotherapy by a bacterial superantigen. J Immunol. 1993;151:3180–3186.
130. Newell, KA, Ellenhorn, JD, Bruce, DS, et al. In vivo T-cell activation by staphylococcal enterotoxin B prevents outgrowth of a malignant tumor. Proc Natl Acad Sci U S A. 1991;88:1074–1078.
131. Dow, SW, Elmslie, RE, Willson, AP, et al. In vivo tumor transfection with superantigen plus cytokine genes induces tumor regression and prolongs survival in dogs with malignant melanoma. J Clin Invest. 1998;101:2406–2414.
132. Sun, D, Woodland, DL, Coleclough, C, et al. An MHC class II-expressing T cell clone presenting conventional antigen lacks the ability to present bacterial superantigen. Int Immunol. 1995;7:1079–1085.
133. Kotzin, BL, Leung, DY, Kappler, J, et al. Superantigens and their potential role in human disease. Adv Immunol. 1993;54:99–166.
134. Forsberg, G, Ohlsson, L, Brodin, T, et al. Therapy of human non-small-cell lung carcinoma using antibody targeting of a modified superantigen. Br J Cancer. 2001;85:129–136.
135. Xu, Q, Zhang, X, Yue, J, et al. Human TGFalpha-derived peptide TGFalphaL3 fused with superantigen for immunotherapy of EGFR-expressing tumours. BMC Biotechnol. 2010;10:91.
136. Imani Fooladi, AA, Sattari, M, Reza Nourani, M. Synergistic effects between staphylococcal enterotoxin type B and monophosphoryl lipid A against mouse fibrosarcoma. J BUON. 2010;15:340–347.
137. Thamm, DH, Kurzman, ID, Macewen, EG, et al. Intralesional lipid-complexed cytokine/superantigen immunogene therapy for spontaneous canine tumors. Cancer Immunol Immunother. 2003;52:473–480.
138. Kleinerman, ES, Jia, SF, Griffin, J, et al. Phase II study of liposomal muramyl tripeptide in osteosarcoma: the cytokine cascade and monocyte activation following administration. J Clin Oncol. 1992;10:1310–1316.
139. Asano, T, Kleinerman, ES. Liposome-encapsulated MTP-PE: a novel biologic agent for cancer therapy. J Immunother Emphasis Tumor Immunol. 1993;14:286–292.
140. Gianan, MA, Kleinerman, ES. Liposomal muramyl tripeptide (CGP 19835A lipid) therapy for resectable melanoma in patients who were at high risk for relapse: an update. Cancer Biother Radiopharm. 1998;13:363–368.
141. Anderson, PM, Tomaras, M, McConnell, K. Mifamurtide in osteosarcoma—a practical review. Drugs Today (Barc). 2010;46:327–337.
142. Kurzman, ID, MacEwen, EG, Rosenthal, RC, et al. Adjuvant therapy for osteosarcoma in dogs: results of randomized clinical trials using combined liposome-encapsulated muramyl tripeptide and cisplatin. Clin Cancer Res. 1995;1:1595–1601.
143. Fox, LE, King, RR, Shi, F, et al. Induction of serum tumor necrosis factor-alpha and interleukin-6 activity by liposome-encapsulated muramyl tripeptide-phosphatidylethanolamine (L-MTP-PE) in normal cats. Cancer Biother. 1994;9:329–340.
144. Vail, DM, MacEwen, EG, Kurzman, ID, et al. Liposome-encapsulated muramyl tripeptide phosphatidylethanolamine adjuvant immunotherapy for splenic hemangiosarcoma in the dog: a randomized multi-institutional clinical trial. Clin Cancer Res. 1995;1:1165–1170.
145. MacEwen, EG, Kurzman, ID, Vail, DM, et al. Adjuvant therapy for melanoma in dogs: results of randomized clinical trials using surgery, liposome-encapsulated muramyl tripeptide, and granulocyte macrophage colony-stimulating factor. Clin Cancer Res. 1999;5:4249–4258.
146. Teske, E, Rutteman, GR, vd Ingh, TS, et al. Liposome-encapsulated muramyl tripeptide phosphatidylethanolamine (L-MTP-PE): a randomized clinical trial in dogs with mammary carcinoma. Anticancer Res. 1998;18:1015–1019.
147. MacEwen, EG, Kurzman, ID, Rosenthal, RC, et al. Therapy for osteosarcoma in dogs with intravenous injection of liposome-encapsulated muramyl tripeptide. J Natl Cancer Inst. 1989;81:935–938.
148. Dow, SW, Fradkin, LG, Liggitt, DH, et al. Lipid-DNA complexes induce potent activation of innate immune responses and antitumor activity when administered intravenously. J Immunol. 1999;163:1552–1561.
149. Zaks, K, Jordan, M, Guth, A, et al. Efficient immunization and cross-priming by vaccine adjuvants containing TLR3 or TLR9 agonists complexed to cationic liposomes. J Immunol. 2006;176:7335–7345.
150. Hemmi, H, Takeuchi, O, Kawai, T, et al. A toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–745.
151. Sellins, K, Fradkin, L, Liggitt, D, et al. Type I interferons potently suppress gene expression following gene delivery using liposome(-)DNA complexes. Mol Ther. 2005;12:451–459.
152. Dow, S, Elmslie, R, Kurzman, I, et al. Phase I study of liposome-DNA complexes encoding the interleukin-2 gene in dogs with osteosarcoma lung metastases. Hum Gene Ther. 2005;16:937–946.
153. Kamstock, D, Guth, A, Elmslie, R, et al. Liposome-DNA complexes infused intravenously inhibit tumor angiogenesis and elicit antitumor activity in dogs with soft tissue sarcoma. Cancer Gene Ther. 2006;13:306–317.
154. Smith, BF, Curiel, DT, Ternovoi, VV, et al. Administration of a conditionally replicative oncolytic canine adenovirus in normal dogs. Cancer Biother Radiopharm. 2006;21:601–606.
155. Le, LP, Rivera, AA, Glasgow, JN, et al. Infectivity enhancement for adenoviral transduction of canine osteosarcoma cells. Gene Ther. 2006;13:389–399.
156. Hemminki, A, Kanerva, A, Kremer, EJ, et al. A canine conditionally replicating adenovirus for evaluating oncolytic virotherapy in a syngeneic animal model. Mol Ther. 2003;7:163–173.
157. Suter, SE, Chein, MB, von Messling, V, et al. In vitro canine distemper virus infection of canine lymphoid cells: a prelude to oncolytic therapy for lymphoma. Clin Cancer Res. 2005;11:1579–1587.
158. Siegel, JP, Puri, RK. Interleukin-2 toxicity. J Clin Oncol. 1991;9:694–704.
159. Vial, T, Descotes, J. Clinical toxicity of interleukin-2. Drug Saf. 1992;7:417–433.
160. Margolin, KA, Rayner, AA, Hawkins, MJ, et al. Interleukin-2 and lymphokine-activated killer cell therapy of solid tumors: analysis of toxicity and management guidelines. J Clin Oncol. 1989;7:486–498.
161. Helfand, SC, Soergel, SA, MacWilliams, PS, et al. Clinical and immunological effects of human recombinant interleukin-2 given by repetitive weekly infusion to normal dogs. Cancer Immunol Immunother. 1994;39:84–92.
162. Funk, J, Schmitz, G, Failing, K, et al. Natural killer (NK) and lymphokine-activated killer (LAK) cell functions from healthy dogs and 29 dogs with a variety of spontaneous neoplasms. Cancer Immunol Immunother. 2005;54:87–92.
163. Khanna, C, Anderson, PM, Hasz, DE, et al. Interleukin-2 liposome inhalation therapy is safe and effective for dogs with spontaneous pulmonary metastases. Cancer. 1997;79:1409–1421.
164. Jourdier, TM, Moste, C, Bonnet, MC, et al. Local immunotherapy of spontaneous feline fibrosarcomas using recombinant poxviruses expressing interleukin 2 (IL2). Gene Ther. 2003;10:2126–2132.
165. Quintin-Colonna, F, Devauchelle, P, Fradelizi, D, et al. Gene therapy of spontaneous canine melanoma and feline fibrosarcoma by intratumoral administration of histoincompatible cells expressing human interleukin-2. Gene Ther. 1996;3:1104–1112.
166. Jahnke, A, Hirschberger, J, Fischer, C, et al. Intra-tumoral gene delivery of feIL-2, feIFN-gamma and feGM-CSF using magnetofection as a neoadjuvant treatment option for feline fibrosarcomas: a phase-I study. J Vet Med A Physiol Pathol Clin Med. 2007;54:599–606.
167. Cutrera, J, Torrero, M, Shiomitsu, K, et al. Intratumoral bleomycin and IL-12 electrochemogenetherapy for treating head and neck tumors in dogs. Methods Mol Biol. 2008;423:319–325.
168. Siddiqui, F, Li, CY, Zhang, X, et al. Characterization of a recombinant adenovirus vector encoding heat-inducible feline interleukin-12 for use in hyperthermia-induced gene-therapy. Int J Hyperthermia. 2006;22:117–134.
169. Marks-Konczalik, J, Dubois, S, Losi, JM, et al. IL-2-induced activation-induced cell death is inhibited in IL-15 transgenic mice. Proc Natl Acad Sci U S A. 2000;97:11445–11450.
170. Zhang, X, Sun, S, Hwang, I, et al. Potent and selective stimulation of memory-phenotype CD8+ T cells in vivo by IL-15. Immunity. 1998;8:591–599.
171. Waldmann, TA, Lugli, E, Roederer, M, et al. Safety (toxicity), pharmacokinetics, immunogenicity, and impact on elements of the normal immune system of recombinant human IL-15 in rhesus macaques. Blood. 2011;117(18):4787–4795.
172. Chou, PC, Chuang, TF, Jan, TR, et al. Effects of immunotherapy of IL-6 and IL-15 plasmids on transmissible venereal tumor in beagles. Vet Immunol Immunopathol. 2009;130:25–34.
173. Streck, CJ, Zhang, Y, Miyamoto, R, et al. Restriction of neuroblastoma angiogenesis and growth by interferon-alpha/beta. Surgery. 2004;136:183–189.
174. Folkman, J. Successful treatment of an angiogenic disease. N Engl J Med. 1989;320:1211–1212.
175. Coates, A, Rallings, M, Hersey, P, et al. Phase-II study of recombinant alpha 2-interferon in advanced malignant melanoma. J Interferon Res. 1986;6:1–4.
176. Rosenthal, MA, Cox, K, Raghavan, D, et al. Phase II clinical trial of recombinant alpha-2 interferon for biopsy-proven metastatic or recurrent renal carcinoma. Br J Urol. 1992;69:491–494.
177. Zeidner, NS, Mathiason-DuBard, CK, Hoover, EA. Reversal of feline leukemia virus infection by adoptive transfer of activated T lymphocytes, interferon alpha, and zidovudine. Semin Vet Med Surg (Small Anim). 1995;10:256–266.
178. Penzo, C, Ross, M, Muirhead, R, et al. Effect of recombinant feline interferon-omega alone and in combination with chemotherapeutic agents on putative tumour-initiating cells and daughter cells derived from canine and feline mammary tumours. Vet Comp Oncol. 2009;7:222–229.
179. Whitley, EM, Bird, AC, Zucker, KE, et al. Modulation by canine interferon-gamma of major histocompatibility complex and tumor-associated antigen expression in canine mammary tumor and melanoma cell lines. Anticancer Res. 1995;15:923–929.
180. Hsiao, YW, Liao, KW, Chung, TF, et al. Interactions of host IL-6 and IFN-gamma and cancer-derived TGF-beta1 on MHC molecule expression during tumor spontaneous regression. Cancer Immunol Immunother. 2008;57:1091–1104.
181. Mito, K, Sugiura, K, Ueda, K, et al. IFNγ markedly cooperates with intratumoral dendritic cell vaccine in dog tumor models. Cancer Res. 2010;70:7093–7101.
182. Pluhar, GE, Grogan, PT, Seiler, C, et al. Anti-tumor immune response correlates with neurological symptoms in a dog with spontaneous astrocytoma treated by gene and vaccine therapy. Vaccine. 2010;28:3371–3378.
183. Duffaud, F, Therasse, P. [New guidelines to evaluate the response to treatment in solid tumors]. Bull Cancer. 2000;87:881–886.
184. Eisenhauer, EA, Therasse, P, Bogaerts, J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45:228–247.
185. Cheever, MA, Allison, JP, Ferris, AS, et al. The prioritization of cancer antigens: a National Cancer Institute pilot project for the acceleration of translational research. Clin Cancer Res. 2009;15:5323–5337.
186. Smyth, MJ, Dunn, GP, Schreiber, RD. Cancer immunosurveillance and immunoediting: the roles of immunity in suppressing tumor development and shaping tumor immunogenicity. Adv Immunol. 2006;90:1–50.
187. Whiteside, TL. Immune responses to malignancies. J Allergy Clin Immunol. 2010;125:S272–S283.
188. U’Ren, LW, Biller, BJ, Elmslie, RE, et al. Evaluation of a novel tumor vaccine in dogs with hemangiosarcoma. J Vet Intern Med. 2007;21:113–120.
189. Hogge, GS, Burkholder, JK, Culp, J, et al. Preclinical development of human granulocyte-macrophage colony-stimulating factor-transfected melanoma cell vaccine using established canine cell lines and normal dogs. Cancer Gene Ther. 1999;6:26–36.
190. Turek, MM, Thamm, DH, Mitzey, A, et al. Human granulocyte-macrophage colony-stimulating factor DNA cationic-lipid complexed autologous tumour cell vaccination in the treatment of canine B-cell multicentric lymphoma. Vet Comp Oncol. 2007;5:219–231.
191. Alexander, AN, Huelsmeyer, MK, Mitzey, A, et al. Development of an allogeneic whole-cell tumor vaccine expressing xenogeneic gp100 and its implementation in a phase II clinical trial in canine patients with malignant melanoma. Cancer Immunol Immunother. 2006;55:433–442.
192. Mutwiri, G, Pontarollo, R, Babiuk, S, et al. Biological activity of immunostimulatory CpG DNA motifs in domestic animals. Vet Immunol Immunopathol. 2003;91:89–103.
193. Liu, MA. DNA vaccines: an historical perspective and view to the future. Immunol Rev. 2011;239:62–84.
194. Bergman, PJ, Camps-Palau, MA, McKnight, JA, et al. Development of a xenogeneic DNA vaccine program for canine malignant melanoma at the Animal Medical Center. Vaccine. 2006;24:4582–4585.
195. Liao, JC, Gregor, P, Wolchok, JD, et al. Vaccination with human tyrosinase DNA induces antibody responses in dogs with advanced melanoma. Cancer Immun. 2006;6:8.
196. Bergman, PJ, McKnight, J, Novosad, A, et al. Long-term survival of dogs with advanced malignant melanoma after DNA vaccination with xenogeneic human tyrosinase: a phase I trial. Clin Cancer Res. 2003;9:1284–1290.
197. Goubier, A, Fuhrmann, L, Forest, L, et al. Superiority of needle-free transdermal plasmid delivery for the induction of antigen-specific IFNgamma T cell responses in the dog. Vaccine. 2008;26:2186–2190.
198. Bergman, PJ. Cancer immunotherapy. Vet Clin North Am Small Anim Pract. 2010;40:507–518.
199. von Mehren, M, Arlen, P, Tsang, KY, et al. Pilot study of a dual gene recombinant avipox vaccine containing both carcinoembryonic antigen (CEA) and B7.1 transgenes in patients with recurrent CEA-expressing adenocarcinomas. Clin Cancer Res. 2000;6:2219–2228.
200. Kaufman, HL, Lenz, HJ, Marshall, J, et al. Combination chemotherapy and ALVAC-CEA/B7.1 vaccine in patients with metastatic colorectal cancer. Clin Cancer Res. 2008;14:4843–4849.
201. Hofbauer, GF, Baur, T, Bonnet, MC, et al. Clinical phase I intratumoral administration of two recombinant ALVAC canarypox viruses expressing human granulocyte-macrophage colony-stimulating factor or interleukin-2: the transgene determines the composition of the inflammatory infiltrate. Melanoma Res. 2008;18:104–111.
202. Spaner, DE, Astsaturov, I, Vogel, T, et al. Enhanced viral and tumor immunity with intranodal injection of canary pox viruses expressing the melanoma antigen, gp100. Cancer. 2006;106:890–899.
203. Lech, PJ, Russell, SJ. Use of attenuated paramyxoviruses for cancer therapy. Expert Rev Vaccines. 2010;9:1275–1302.
204. Melief, CJ. Cancer immunotherapy by dendritic cells. Immunity. 2008;29:372–383.
205. Palucka, K, Ueno, H, Roberts, L, et al. Dendritic cells: are they clinically relevant? Cancer J. 2010;16:318–324.
206. Palucka, K, Ueno, H, Banchereau, J. Recent developments in cancer vaccines. J Immunol. 2011;186:1325–1331.
207. Slingluff, CL, Jr., Petroni, GR, Yamshchikov, GV, et al. Clinical and immunologic results of a randomized phase II trial of vaccination using four melanoma peptides either administered in granulocyte-macrophage colony-stimulating factor in adjuvant or pulsed on dendritic cells. J Clin Oncol. 2003;21:4016–4026.
208. Giermasz, AS, Urban, JA, Nakamura, Y, et al. Type-1 polarized dendritic cells primed for high IL-12 production show enhanced activity as cancer vaccines. Cancer Immunol Immunother. 2009;58:1329–1336.
209. Kantoff, PW, Higano, CS, Shore, ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411–422.
210. Carballido, E, Fishman, M. Sipuleucel-T: Prototype for development of anti-tumor vaccines. Curr Oncol Rep. 2011;13:112–119.
211. Gyorffy, S, Rodriguez-Lecompte, JC, Woods, JP, et al. Bone marrow-derived dendritic cell vaccination of dogs with naturally occurring melanoma by using human gp100 antigen. J Vet Intern Med. 2005;19:56–63.
212. Tamura, K, Yamada, M, Isotani, M, et al. Induction of dendritic cell-mediated immune responses against canine malignant melanoma cells. Vet J. 2008;175:126–129.
213. Bird, RC, Deinnocentes, P, Church Bird, AE, et al. An autologous dendritic cell canine mammary tumor hybrid-cell fusion vaccine. Cancer Immunol Immunother. 2011;60:87–97.
214. Kohler, G, Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256:495–497.
215. Osbourn, J, Jermutus, L, Duncan, A. Current methods for the generation of human antibodies for the treatment of autoimmune diseases. Drug Discov Today. 2003;8:845–851.
216. Abes, R, Teillaud, JL. Modulation of tumor immunity by therapeutic monoclonal antibodies. Cancer Metastasis Rev. 2011;30:111–124.
217. Stein, R, Balkman, C, Chen, S, et al. Evaluation of anti-human leukocyte antigen-DR monoclonal antibody therapy in spontaneous canine lymphoma. Leuk Lymphoma. 2011;52:273–284.
218. Yannelli, JR, Wroblewski, JM. On the road to a tumor cell vaccine: 20 years of cellular immunotherapy. Vaccine. 2004;23:97–113.
219. Blakeslee, J, Noll, G, Olsen, R, et al. Adoptive immunotherapy of feline leukemia virus infection using autologous lymph node lymphocytes. J Acquir Immune Defic Syndr Hum Retrovirol. 1998;18:1–6.
220. Yron, I, Wood, TA, Jr., Spiess, PJ, et al. In vitro growth of murine T cells. V. The isolation and growth of lymphoid cells infiltrating syngeneic solid tumors. J Immunol. 1980;125:238–245.
221. Rosenberg, SA, Restifo, NP, Yang, JC, et al. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008;8:299–308.
222. Dudley, ME, Rosenberg, SA. Adoptive-cell-transfer therapy for the treatment of patients with cancer. Nat Rev Cancer. 2003;3:666–675.
223. Dudley, ME, Wunderlich, JR, Yang, JC, et al. A phase I study of nonmyeloablative chemotherapy and adoptive transfer of autologous tumor antigen-specific T lymphocytes in patients with metastatic melanoma. J Immunother. 2002;25:243–251.
224. Rosenberg, SA, Dudley, ME. Cancer regression in patients with metastatic melanoma after the transfer of autologous antitumor lymphocytes. Proc Natl Acad Sci U S A. 2004;101(Suppl 2):14639–14645.
225. Xu, L, Wang, C, Wen, Z, et al. CpG oligodeoxynucleotides enhance the efficacy of adoptive cell transfer using tumor infiltrating lymphocytes by modifying the Th1 polarization and local infiltration of Th17 cells. Clin Dev Immunol. 2010;2010:410893.
226. Rosenberg, SA, Yang, JC, Restifo, NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10:909–915.
227. Gattinoni, L, Finkelstein, SE, Klebanoff, CA, et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med. 2005;202:907–912.
228. Paulos, CM, Wrzesinski, C, Kaiser, A, et al. Microbial translocation augments the function of adoptively transferred self/tumor-specific CD8+ T cells via TLR4 signaling. J Clin Invest. 2007;117:2197–2204.
229. Dudley, ME, Yang, JC, Sherry, R, et al. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol. 2008;26:5233–5239.
230. Quezada, SA, Simpson, TR, Peggs, KS, et al. Tumor-reactive CD4(+) T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J Exp Med. 2010;207:637–650.
231. Xie, Y, Akpinarli, A, Maris, C, et al. Naive tumor-specific CD4(+) T cells differentiated in vivo eradicate established melanoma. J Exp Med. 2010;207:651–667.
232. Corthay, A, Lorvik, KB, Bogen, B. Is secretion of tumour-specific antigen important for cancer eradication by CD4(+) T cells?—Implications for cancer immunotherapy by adoptive T cell transfer. Scand J Immunol. 2011;73(6):527–530.
233. Hanson, HL, Donermeyer, DL, Ikeda, H, et al. Eradication of established tumors by CD8+ T cell adoptive immunotherapy. Immunity. 2000;13:265–276.
234. Klebanoff, CA, Finkelstein, SE, Surman, DR, et al. IL-15 enhances the in vivo antitumor activity of tumor-reactive CD8+ T cells. Proc Natl Acad Sci U S A. 2004;101:1969–1974.
235. May, KF, Jr., Chen, L, Zheng, P, et al. Anti-4–1BB monoclonal antibody enhances rejection of large tumor burden by promoting survival but not clonal expansion of tumor-specific CD8+ T cells. Cancer Res. 2002;62:3459–3465.
236. Khanna, C, London, C, Vail, D, et al. Guiding the optimal translation of new cancer treatments from canine to human cancer patients. Clin Can Res. 2009;15:5671–5677.
237. Withrow, SJ, Khanna, C. Bridging the gap between experimental animals and humans in osteosarcoma. Cancer Treat Res. 2009;152:439–446.
238. Stewart, TJ, Smyth, MJ. Improving cancer immunotherapy by targeting tumor-induced immune suppression. Cancer Metastasis Rev. 2011;30:125–140.