Molecular/Targeted Therapy of Cancer
 Section A
Section A
Gene Therapy for Cancer
Since its development, recombinant DNA technology has been vigorously applied to the advancement of medicine. New molecular techniques have been used to study the role of specific genes and their products in disease, to improve diagnosis, and to produce novel therapeutics. Gene therapy, in its simplest definition, is the introduction of genes into cells in vivo to treat a disease.1 Although one may recognize this as one of the newest areas of medicine, the actual concept of gene therapy is not a new idea. In the late 1960s, the idea of gene therapy was hypothesized by many working in the field of molecular biology, in particular the use of gene therapy to deliver a normal copy of a gene into a patient with a single gene defect, such as hemophilia. However, the technology to manipulate genes and be able to deliver them safely to patients was not available until very recently. Even now and despite improvements in technologies involving gene manipulation and delivery, there are still many technical hurdles to overcome before gene therapy can become accepted clinical practice.1,2
Efficient Gene Delivery: The Major Hurdle to Clinical Benefits
In simple terms, gene therapy is the introduction of nucleic acid into a cell to ameliorate a disease process. For this to be effective, the gene has to be delivered to sufficient numbers of target cells in the body, and this requires a vehicle or vector for delivery. In addition, the gene has to be expressed at a sufficient level and for a length of time appropriate for the disease.2
Early studies on how viruses were able to cause tumors by delivery of their own DNA to foreign cells made them ideal candidates for vectors for gene therapy (i.e., the vehicles by which we could transfer therapeutic genes to patients). However, it was not until the 1980s when work on the retroviral life cycle started to revolutionize the development of gene therapy. In these early studies, it was demonstrated that retroviruses could transfer DNA to cells and this DNA could be stably integrated into the host cell’s genome. Since these early experiments, a number of viruses have been manipulated to act as vehicles for gene delivery. In addition, because of concerns over safety of viral vectors for gene delivery, a number of workers have also explored the possibility of using naked DNA (a therapeutic gene delivered in a bacterial plasmid or lipid/DNA complexes) with some success. The vehicles used for gene delivery and their associated problems are shown in Table 14-1.
The ideal vector would efficiently deliver the gene of interest (transgene) specifically to the cancer cell. It would be easy and cheap to manufacture and would be nonimmunogenic. Obviously the ideal vector does not exist, but there have been great advances in both viral and nonviral delivery systems.2
Viral Vectors
The great advantage to viral vectors for gene delivery is their ability to infect cells and our ability to exploit their replicative machinery. The majority of systems utilize replicative-defective viruses to overcome concerns that recombination within the host may lead to the production of wild-type virus with pathogenic potential. The common systems rely on oncogenic retroviruses (e.g., murine leukemia virus [MuLV]) or adenoviruses (e.g., human adenovirus type 5 [AD5]), but great strides are also being made with lentiviral vectors (particularly human immunodeficiency virus-1 [HIV-1]).3-8 Most of the systems described involve the local delivery of virus to tumor deposits (e.g., by intratumoral injection). Systemic delivery is hindered by rapid clearance of viruses from the body by the immune and complement systems. To overcome this, work is in progress to explore cellular delivery of viruses by the systemic route. In this delivery system, viral producer cells are delivered to the patient, and virus production is triggered when the cells reach the tumor. Endothelial cell cultures lend themselves well to this technology because they specifically home in to areas of neoangiogenesis. However, T-cells, macrophages, and dendritic cells are also being explored as potential cell delivery systems. The advantage of this system is that virus could potentially be delivered to metastatic disease, as well as primary tumors.9-13 Recent evidence suggests that mesenchymal stem cells also have homing ability to tumors and could be used to deliver “suicide” genes (see later).
Nonviral Gene Delivery
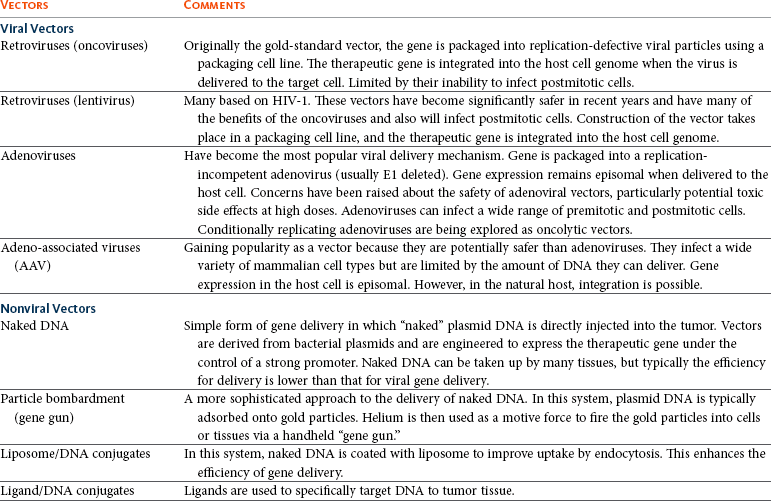
Concerns relating to virus safety and an inability to produce high enough viral titers have led to the development of nonviral delivery systems for gene therapy.14 Liposomes have been used to safely and efficiently deliver genes to tumor cells through direct injection.15,16 Further, naked DNA delivery (the delivery of plasmid DNA alone containing the gene of interest) has been shown to be taken up by tumor cells and antigen-presenting cells after simple direct injection. A modification of this is particle-mediated gene delivery using a “gene gun.” In this approach, DNA is adsorbed on to gold particles and fired into tissues under high pressure (using helium as the motive force).17,18 However, the majority of these techniques are still inefficient vehicles for delivery and are not able to be given systemically (Figure 14-1; see Table 14-1).
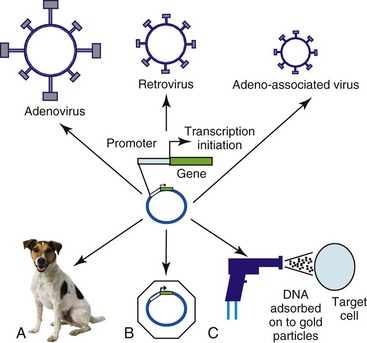
Figure 14-1 Viral and nonviral gene delivery. Adenoviral vectors are produced in “producer cell lines.” They enter the cell by transduction, and their genetic material is transported to the nucleus. In contrast to retroviruses, the DNA is not integrated into the host genome—gene expression is achieved episomally. Retroviral vectors are also produced in specialized producer cell lines. They enter the cell by transduction, but their RNA genome is reverse transcribed into proviral DNA. This integrates into the host genome in which expression of the transgene takes place. DNA plasmid vectors contain a gene cassette that incorporates the therapeutic transgene under the control of a promoter. The plasmid can be delivered by direct injection as naked or liposome-encapsulated DNA (A), by direct injection or systemically wrapped in nanomedicine particles (B), or by direct injection utilizing a helium-driven “biolistic” gene gun (C).
Targeted Gene Delivery
One of the major barriers to gene therapy becoming accepted in clinical practice is the ability to give vectors systemically and to ensure that therapeutic transgenes are not expressed in normal cells. Numerous strategies have been attempted to provide levels of targeting to spare normal tissue. In a later section, we describe the use of conditionally replicating viruses, which is one method of targeting. Surface modification of viruses (transductional targeting) is also being explored. An example of this is the use of modified fibers on the surface of adenoviruses that only allow the virus to enter cells with specific receptors (Figure 14-2).
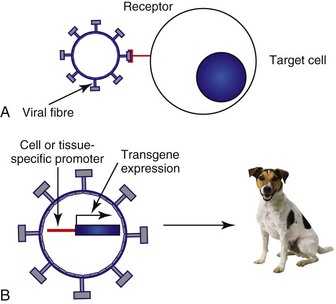
Figure 14-2 Vector targeting. The specificity of viral vectors can be improved utilizing either transductional targeting (A) where the viral surface proteins are modified so they will only enter the cell of interest or transcriptional targeting (B) where the expression of the therapeutic transgene is under the control of a tissue or cell-specific promoter. Transcriptional targeting can also be employed in nonviral vectors and directly delivered to the patient.
A further targeting strategy is transcriptional targeting once the vector has entered the cell.19-26 Although every gene is represented in every cell of the body, expression of any one gene requires specific transcription factors that may be unique to a particular cell or tissue type. Certain genes have been identified that are expressed in cancer cells but are not expressed in normal cells (e.g., telomerase) or are only expressed in a specific tissue type (e.g. prostate-specific antigen [PSA]). By using the promoter sequences for these genes to drive transgene expression, targeted expression in cancer cells only (e.g., using the promoter for telomerase) or to a specific tissue type (e.g., to the prostate using the promoter for PSA) can be achieved (see Figure 14-2).
Gene Therapy Strategies for Cancer
Despite advances in surgical techniques and the use of radiotherapy and chemotherapy, cancer still remains a disease of high mortality in both human and veterinary medicine, warranting the investigation of alternative treatments. Gene therapy has the potential to play a major role in the development of new cancer therapeutic agents, and there are four broad approaches that can be applied, as follows.
Rescue of the Cancer Cell Through Gene Replacement Technologies
The increased understanding of the molecular events in cancer has made possible the identification of defective genes involved in the cancer phenotype. One of the most studied genes in cancer development has been the tumor suppressor gene p53. p53 acts as a genomic guardian for the cell; it is switched on when a cell’s DNA is damaged. The product of this gene causes the cell to either stop dividing or become apoptotic (programmed cell death), depending on the degree of damage. In many cancers, this gene is defective; thus damaged cells fail to stop dividing and can accumulate further damaging events, which can allow selection for a malignant phenotype. A number of studies have addressed this by attempting to replace the defective p53 gene with its normal counterpart.27 However, problems associated with this approach include the following:
• The inability of our current technology to be able to efficiently deliver a normal p53 gene to every cancer cell in a tumor mass.
• Cancer is a multigenetic abnormality, and the delivery of one correct gene to a tumor cell may still not have the desired phenotypic effect.
A more promising approach has been to use the lack of a normal functioning p53 gene to target viruses to kill cells. The use of E1b-deleted adenoviruses to specifically cause oncolysis in p53 null cells is described later and has proved successful in some human clinical models.28 p53 mutations in domestic species such as the dog and cat have also been well characterized, and these may provide targets for therapy, particularly for diseases such as canine osteosarcoma (OSA) and feline vaccine-associated sarcomas (FVAS).
Destruction of Cancer Cells Through Delivery of “Suicide Genes”
Typically, the “suicide gene” approach involves the delivery of a gene (usually an enzyme) to cancer cells that has the ability to convert a relatively nontoxic prodrug to an active compound within the cancer cell (gene-directed enzyme prodrug therapy [GDEPT]) (Figure 14-3). At the clinical level, the gene would be delivered to the patient’s tumor and the enzyme activity would be confined to the cancer cells.29 The patient would then be given a prodrug systemically. In the cancer cells, this novel enzyme can convert the prodrug to a more active compound that has the ability to kill the cancer cell (see Figure 14-3). A number of successful approaches have been developed based on this system. For example, the Escherichia coli nitroreductase gene has been used in preclinical models to cause reduction of an inactive prodrug (CB1954, a weak alkylating agent) to promote cell killing in cancer cells.29 However, due to the low efficiency of existing vectors, the success of this therapy will largely depend on the extent of the bystander effect. In this, the activation of the prodrug in the cell causes cell death and also leakage of toxic metabolites to neighboring cells. Consequently, it is estimated that only a small fraction of the cells need receive the gene for there to be a dramatic effect on tumor volume. Further, in mouse models, a distant bystander effect on tumor metastases has been demonstrated that is mediated through the patient’s immune system.22 The in-situ destruction of tumor cells is mediated through necrosis rather than apoptosis, creating an ideal inflammatory environment for the exposure and presentation of tumor antigens to the immune system. This allows the patient’s immune system to recognize tumor metastases and has caused regression in a number of preclinical model systems. These systems have been combined with transcriptionally targeted vectors (described previously) to improve the eventual therapeutic index.
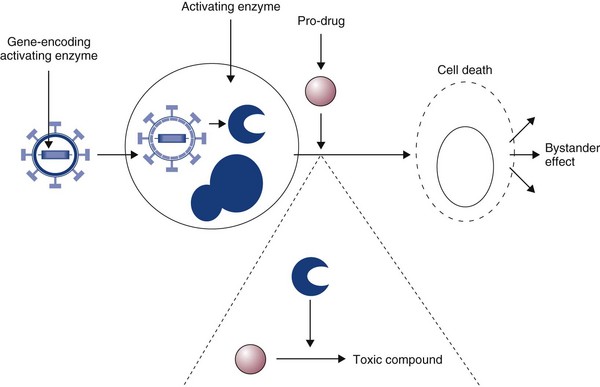
Figure 14-3 In gene-directed enzyme prodrug therapy (GDEPT), an activating gene is delivered to the cancer cells. A relatively inactive prodrug is then given to the patient systemically. In cells processing the activating gene, the prodrug is converted to a highly toxic drug, which can kill the cancer cell. The advantage of this system is evidence of the bystander effect. In this, only a small proportion of cancer cells need to receive the activating gene, as toxic metabolites leak across gap junctions and kill surrounding cancer cells.
Utilizing Stem Cells to Deliver “Suicide Genes”
The attractiveness of prodrug cancer gene therapy has been described earlier, but it does rely on the ability to specifically target prodrugs to tumors. This can be achieved using transcriptional targeting of vectors (i.e., the use of cell-specific promoters to drive prodrug gene expression). However, this targeting needs an effective and efficient delivery vehicle, and viruses demonstrate some severe deficiencies in this role. The use of stem cells to target tumors can avoid systemic toxicity. Tumor stroma is composed of a variety of cells, including proliferating tumor cells, cancer stem cells (CSCs), tumor fibroblasts, endothelial cells, lymphocytes, and other cells. Many therapeutic strategies are now geared toward killing both tumor cells and CSCs. Prodrug cancer gene therapy driven by mesenchymal stem cells (MSCs) has been suggested as a treatment modality that could achieve this.30 It represents an attractive tool for activating the prodrug directly within the tumor mass, thus avoiding systemic toxicity. In addition, MSCs lack major histocompatibility complex (MHC)-II and show only minimal MHC-I expression.31-33 Thanks to their immunosuppressive properties, allogeneic MSCs can substitute for autologous stem cells in delivering the therapeutic agent in targeted tumor therapy. Stem cell–driven cancer gene therapy is based on the tumor-trophic property of MSCs. Tumor-homing ability of MSCs holds therapeutic advantages compared to vehicles such as proteins, antibodies, nanoparticles, and to some extent viruses. The success of an enzyme-prodrug gene therapy depends on several factors. The catalytic activity of the enzyme encoded by a suicide gene, a suitable prodrug-enzyme combination, ability of the vector to target tumor cells, sufficient transgene expression, and, importantly, the extent of the bystander effect are the main indicators of effective and successful suicide gene therapy. Various combinations of suicide genes, prodrugs, and gene transfer technologies have been investigated in order to find the most suitable and effective system in combating the otherwise incurable tumors. The failures of the present suicide gene therapies were mainly caused by the inability of vectors carrying the suicide gene to reach invasive tumor cells distant from the tumor bulk, as well as inefficient spread of the vectors within the tumor. Therefore the stem cell–based suicide gene therapy based on the inherent and privileged tumor-trophic nature of MSCs holds great potential for moving suicide gene therapy closer to translation (Figure 14-4).
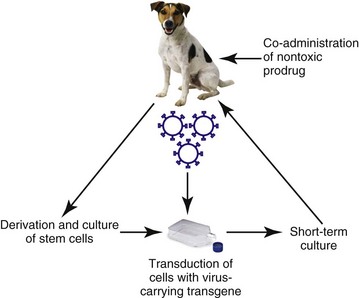
Figure 14-4 Mesenchymal stem cell (MSC)–targeted cancer gene therapy. MSCs are isolated from patients. Bone marrow-MSCs (BM-MSCs) are isolated from mononuclear fraction of bone marrow obtained by Percoll density gradient centrifugation. Obtained cells are plated in a plastic dish and expanded. Stem cells are transduced with virus containing a suicide gene and gene encoding resistance to antibiotic G418. Transduced cells are exposed to selective medium containing antibiotic G418. The population of selected therapeutic stem cells is expanded and used for systemic or intratumoral injections. Therapeutic cells migrate to the tumor site. Subsequently, nontoxic prodrug is administered and converted at the tumor site to toxic drug killing tumor cells and therapeutic cells as well. No adverse systemic toxicities have been observed.
Gene-Directed Immunotherapy
The search for an effective cancer vaccine over the past 150 years has led to extensive studies of the immune response of cancer patients. These studies have suggested that cell-mediated immune responses are important components of the antitumor immune response. Cytokines are small glycoprotein molecules that orchestrate the immune response, tissue repair, and hemopoiesis, and it has been demonstrated that the relative amounts of individual cytokines can direct the immune system toward either a mainly humoral or a mainly cell-mediated response. In particular, cytokines such as interleukin-2 (IL-2), interferon-γ (IFN-γ), IL-12, and IL-18 have the ability to promote cell-mediated responses. Further, evidence derived from animal models suggested that local production of cytokines around a tumor mass can lead to production of an antitumor immune response and a reversal of T-cell anergy (nonresponsiveness).34,35 Thus there appears a rationale for using cytokine molecules in cancer patients to improve the antitumor immune response to tumors that present weakly antigenic epitopes or epitopes that evade immune recognition. In the 1980s and 1990s, a number of clinical studies were undertaken using recombinant cytokine proteins to improve the survival of human cancer patients. However, cytokines tend to be autocrine or paracrine in nature and the levels of protein required to demonstrate a biologic effect were often too toxic for the patient to withstand. However, a more promising approach has been to deliver the actual cytokine genes to cancer cells rather than delivery of the protein to the whole patient.36-38 This approach has also been adopted in a number of small-scale veterinary studies, including one on canine malignant melanoma, which used cells to deliver IL-2 to tumors.39 These studies have had encouraging results and warrant further larger scale trials.
In the preceding section, we described how the in-situ destruction of tumor cells by enzymes can lead to a distant bystander effect mediated by the immune system. Many trials have now combined this approach with gene-directed immunotherapy. In this approach, cytokine genes, as well as the prodrug-activating gene, are delivered to cancer cells. The conversion of an inactive prodrug by the activating gene leads to destruction of the cancer cells by necrosis. The co-delivery of cytokines that enhance cell-mediated immune responses such as IL-2, IFN-γ, IL-12, and IL-18 enhances the antitumor response and may potentially improve the distant bystander effect against micrometastatic disease.1,2
Delivery of Chemoprotective Genes
An alternative approach to gene therapy for cancer involves the delivery of genes to normal cells of the bone marrow to protect them against the cytotoxic effects of conventional chemotherapeutic drugs. In particular, the multidrug resistance (MDR) gene has been cloned and delivered to normal bone marrow cells. When patients are given high doses of chemotherapy, the normal cells with the MDR gene are able to export the toxic drugs across their membranes and reduce potential side effects.40-42 However, this approach does not protect gastrointestinal cells, which limit this approach. Further, there is also a danger that the MDR gene could transfer to malignant cells, rendering them insensitive to the effects of standard drugs.
The Use of Replication-Competent Viral Vectors
Progress has been made in the development of replication-competent viruses that conditionally replicate in cancer cells.43-48 As an example, the Onyx 015 vector is an E1b-deleted adenovirus that conditionally replicates in cells with a nonfunctional p53 gene. p53 protein has the potential to shut down cell cycling when infected with wild-type adenovirus but is prevented from doing so through the actions of the product of viral E1b. E1b-deficient viruses cannot replicate in normal cells with p53 intact. However, in cells that have no functional p53 protein, viral replication can proceed and cause cell lysis (Figure 14-5). Many other conditionally replicating viruses are being developed that rely on specific cancer cell defects (e.g., Reo viruses that conditionally replicate in cells with intact Ras signalling pathways) or are transcriptionally targeted. p53 mutations in domestic species such as the dog and cat have also been well characterized, and this may provide targets for therapy, particularly for diseases such as canine OSA and feline vaccine-associated sarcomas. In one study, researchers have utilized the osteocalcin promoter for restricting the replication of a canine adenovirus to dog OSA cells.47 This has shown promise in preclinical evaluations and has been shown to yield a therapeutic benefit in vivo. A cautionary note, however, is that the majority of dogs in the United States and Europe are vaccinated against canine adenovirus, and these vectors may not be able to overcome host immunity.
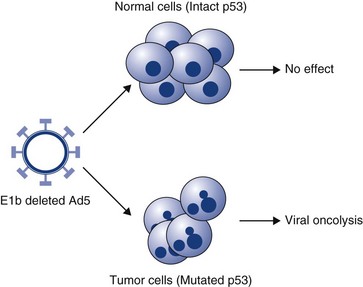
Figure 14-5 Conditionally replicating adenovirus. The Onyx 015 vector is an E1b-deleted adenovirus that conditionally replicates in cells with a nonfunctional p53 gene. p53 protein has the potential to shut down cell cycling when infected with wild-type adenovirus but is prevented from doing so through the actions of the product of viral E1b. E1b-deficient viruses cannot replicate in normal cells with p53 intact. However, in cells that have no functional p53 protein, viral replication can proceed and cause cell lysis.
Miscellaneous Approaches to Cancer Gene Therapy
The multigenetic abnormalities of cancer lend themselves to multiple gene therapy approaches. In addition to those approaches described previously, there is vigorous exploration of the following:
• Gene delivery of sodium iodide symporter genes to tumors to allow them to concentrate radioactive iodine.1
• Delivering proapoptotic genes to cancer cells.1
• Delivering antiangiogenesis genes to cancers to inhibit growth of their blood supply.1
It is highly likely that one individual approach to cancer will be insufficient to cure or control particular cancers. However, it is possible that a combination of treatments, with or without conventional therapies, will prove to provide the best therapeutic solution.
Safety Considerations in Gene Therapy
One of the major considerations in gene therapy revolves around issues of safety, in particular the safety of the vectors used for gene delivery. In 1999, gene therapy suffered a major setback with the death of a patient as a direct result of adenovirus gene therapy. Problems associated with vector delivery include inappropriate inflammatory responses caused by vector delivery (e.g., adenoviruses), the generation of replication-competent viruses (although this is unlikely with new generation vectors), and insertional mutagenesis caused by integrating viruses (e.g., retroviruses).
Until recently, many gene therapy trials had utilized retroviral vectors for gene delivery. There are many advantages to using retroviruses as outlined in Table 14-1. However, retroviruses are also associated with serious diseases of domestic animals and the use of these in gene therapy poses a risk of insertional mutagenesis or the production of replication-competent viruses during the manufacturing process. Realistically, insertional mutagenesis leading to a malignant transformation is an unlikely event because cancer is a multistep process. In fact, there may be a greater risk of malignant transformation from external beam radiation than from the use of retroviruses to treat cancer. The production of replication-competent retroviruses during the production process would also be unlikely because of the rigorous testing that is required prior to clinical application. Many of these issues are resolving with the development of new generation vectors.1 As an example, in the use of retroviruses and to prevent insertional mutagenesis in normal tissues, one group recently described the use of zinc finger nucleases (engineered DNA-editing enzymes) that allows the insertion of DNA to a site of choice within the genome.49 This adds a further level of safety in a high-risk procedure.
In recent years, there has been a shift from using retroviruses in gene therapy to using viruses such as adenovirus and the human adeno-associated viruses (AAV). Adenoviruses may also pose some risks, including inappropriate inflammatory responses leading to serious clinical complications. However, a great deal of work is currently underway to improve viral vectors by removing most of their genetic material to produce “ghost vectors.” These would be potentially less toxic and offer a brighter future for virally mediated gene transfer.
One might imagine that the delivery of naked DNA may offer a safer alternative. However, all of the potential safety issues using this technology are still not fully answered. These include potential risks of autoimmunity and also the actual fate of the DNA when it has been delivered to the patient; in the case of the former, however, there would appear to be no evidence of autoimmunity being a problem in preclinical models.
One of the most exciting developments in cancer gene therapy is the use of conditionally replicating viruses. However, the safety of these vector systems needs special consideration as many of them in their native form could pose a risk to both human and animal health.
New Horizons
Gene therapy promises a completely new approach to the treatment of cancer and represents the newest area of pharmacology. It has suffered over the past 10 years as clinical trials in human medicine have not delivered what they had originally promised. However, one has to put this into context in that many of these studies were conducted in patients with high-grade or end-stage disease and many studies were conducted prematurely without refining the delivery technologies. Clearly, there are a number of technical issues such as safety issues surrounding the delivery and efficiency of the vectors that need to be resolved before gene therapy becomes established clinical practice. Despite gene therapy being very much in its infancy, the field is advancing at a rapid rate. A number of clinical trials have begun in companion animals, and products are in development for clinical application. However, although these treatments would appear to be powerful in preclinical models, it is likely that their greatest benefit will be in the management of patients with minimal disease states. Thus gene therapy will probably have its greatest advantage not as a stand-alone treatment but as an adjunct to more conventional therapies such as surgery, radiation, or chemotherapy.
References
1. Argyle, DJ. Gene therapy in veterinary medicine. Vet Rec. 1999;144:369–376.
2. Harris, J, Sikora, K. Gene therapy in the clinic. (1993). Aspects Med. 1993;14:251–546.
3. Bartosch, B, Cosset, FL. Strategies for retargeted gene delivery using vectors derived from lentiviruses. Curr Gene Ther. 2004;4(4):427–443.
4. Tomanin, R, Scarpa, M. Why do we need new gene therapy viral vectors? Characteristics, limitations and future perspectives of viral vector transduction. Curr Gene Ther. 2004;4(4):357–372.
5. Lachmann, RH. Herpes simplex virus-based vectors. Int J Exp Path. 2004;85(4):177–190.
6. Buning, H, Braun-Falco, M, Hallek, M. Progress in the use of adeno-associated viral vectors for gene therapy. Cells Tissues Organs. 2004;177(3):139–150.
7. Mah, C, Byrne, BJ, Flotte, TR. Virus-based gene delivery systems. Clin Pharmacokinet. 2002;41(12):901–911.
8. Dornburg, R. The history and principles of retroviral vectors. Front Biosci. 2003;8:D818–D835.
9. Culver, KW, Ram, Z, Wallbridge, S, et al. In vivo gene transfer with retroviral vector-producer cells for treatment of experimental brain tumors. Science. 1992;256:1550–1552.
10. Gomez Navarro, J, Contreras, JL, Arafat, W, et al. Genetically modified CD34+ cells as cellular vehicles for gene delivery into areas of angiogenesis in a rhesus model. Gene Ther. 2000;7:43–52. [1].
11. Harrington, K, Alvarez-Vallina, L, Crittenden, M, et al. Cells as vehicles for cancer gene therapy: the missing link between targeted vectors and systemic delivery? Hum Gene Ther. 2002;13(11):1263–1280.
12. Muta, M, Matsumoto, G, Hiruma, K, et al. Study of cancer gene therapy using IL-12-secreting endothelial progenitor cells in a rat solid tumor model. Oncol Rep. 2003;10(6):1765–1769.
13. Pereboeva, L, Komarova, S, Mikheeva, G, et al. Approaches to utilize mesenchymal progenitor cells as cellular vehicles. Stem Cells. 2003;21:389–404. [4].
14. Annier, AK, Shea, LD. Controlled release systems for DNA delivery. Mol Ther. 2004;10(1):19–26.
15. Tranchant, I, Thompson, B, Nicolazzi, C, et al. Physicochemical optimisation of plasmid delivery by cationic lipids. J Gene Med. 2004;6:S24–S35.
16. Hirko, A, Tang, FX, Hughes, JA. Cationic lipid vectors for plasmid DNA delivery. Curr Med Chem. 2003;10(14):1185–1193.
17. Yang, N, Sun, WH. Gene and non-viral approaches to cancer gene therapy. Nat Med. 1995;1:481–483.
18. Keller, ET, Burkholder, JK, Shi Pugh, TD, et al. In-vivo particle mediated cytokine gene transfer into canine oral mucosa and epidermis. Cancer Gene Therapy. 1996;3:186–191.
19. Dachs, GU, Dougherty, GJ, Stratford, IJ, et al. Targeting gene therapy to cancer. Oncol Res. 1997;9:313–325.
20. Scanlon, KJ. Cancer gene therapy: Challenges and opportunities. Anticancer Res. 2004;24(2A):501–504.
21. Blackwood, L, Onions, DE, Argyle, DJ. The feline thyroglobulin promoter: towards targeted gene therapy of hyperthyroidism. Domest Anim Endocrinol. 2001;20(3):185–201.
22. Vile, RG, Hart, IR. In-vitro and in-vivo targeting of gene expression to melanoma cells. Cancer Res. 1993;53(5):962–967.
23. Gu, R, Fang, BL. Telomerase promoter-driven cancer gene therapy. Cancer Biol Ther. 2003;2(4):S64–S70.
24. Song, JS. Adenovirus-mediated suicide SCLC gene therapy using the increased activity of the hTERT promoter by the MMRE and SV40 enhancer. Biosci Biotechnol Biochem. 2005;69(1):56–62.
25. Helder, MN, Wiseman, GBA, van der Zee, AGJ. Telomerase and telomeres: From basic biology to cancer treatment. Cancer Invest. 2002;20(1):82–101.
26. Fullerton, NE, Boyd, M, Mairs, RJ, et al. Combining a targeted radiotherapy and gene therapy approach for adenocarcinoma of prostate. Prostate Cancer Prostatic Dis. 2004;7(4):355–363.
27. Edelman, J, Edelman, J, Nemunaitis, J. Adenoviral p53 gene therapy in squamous cell cancer of the head and neck region. Curr Opin Mol Ther. 2003;5(6):611–617.
28. Bortolanza, S, Hernandez-Alcoceba, R, Kramer, G, et al. Evaluation of the tumor specificity of a conditionally replicative adenovirus controlled by a modified human core telomerase promoter. Mol Ther. 2004;9:S375.
29. Blackwood, L, O’Shaughnessy, PJ, Reid, SJ, et al. E. coli Nitroreductase/CB1954: In vitro studies into a potential system for feline cancer gene therapy? Vet J. 2001;161:269–279.
30. Cihova, M, Altanerova, V, Altaner, C. Stem cell based cancer gene therapy. Mol Pharma. 2011;8(5):1480–1487.
31. Le Blanc, K. Immunomodulatory effects of fetal and adult mesenchymal stem cells. Cytotherapy. 2003;5:485–489.
32. Koppula, PR, Chelluri, LK, Polisetti, N, et al. Histocompatibility testing of cultivated human bone marrow stromal cells—a promising step towards pre-clinical screening for allogeneic stem cell therapy. Cell Immunol. 2009;259:61–66.
33. Griffin, MD, Ritter, T, Mahon, BP. Immunological aspects of allogeneic mesenchymal stem cell therapies. Hum Gene Ther. 2010;21:1641–1655.
34. Lasek, W, Basak, G, Switaj, T, et al. Complete tumour regressions induced by vaccination with IL-12 gene-transduced tumour cells in combination with IL-15 in a melanoma model in mice. Cancer Immunol Immunother. 2004;53(4):363–372.
35. Yamazaki, M, Straus, FH, Messina, M, et al. Adenovirus-mediated tumor-specific combined gene therapy using Herpes simplex virus thymidine/ganciclovir system and murine interleukin-12 induces effective antitumor activity against medullary thyroid carcinoma. Cancer Gene Ther. 2004;11(1):8–15.
36. Nagayama, Y, Nakao, K, Mizuguchi, H, et al. Enhanced antitumor effect of combined replicative adenovirus and nonreplicative adenovirus expressing interleukin-12 in an immunocompetent mouse model. Gene Ther. 2003;10(16):1400–1403.
37. Liu, YQ, Huang, H, Saxena, A, et al. Intratumoral co-injection of two adenoviral vectors expressing functional interleukin-18 and inducible protein-10, respectively, synergizes to facilitate regression of established tumors. Cancer Gene Therapy. 2002;9(6):533–542.
38. Goto, H, Osaki, T, Nishino, K, et al. Construction and analysis of new vector systems with improved interleukin-18 secretion in a xenogeneic human tumor model. J Immunother. 2002;25:S35–S41.
39. Quintin-Colonna, F, Devauchelle, P, Fradelizi, D, et al. Gene therapy of spontaneous canine melanoma and feline fibrosarcoma by intratumoral administration of histoincompatible cells expressing human interleukin-2. Gene Ther. 1996;3(12):1104–1112.
40. Carpinteiro, A, Peinert, S, Ostertag, W, et al. Genetic protection of repopulating hematopoietic cells with an improved MDR1-retrovirus allows administration of intensified chemotherapy following stem cell transplantation in mice. Int J Cancer. 2002;98(5):785–792.
41. Schiedlmeier, B, Schilz, AJ, Kuhlcke, K, et al. Multidrug resistance 1 gene transfer can confer chemoprotection to human peripheral blood progenitor cells engrafted in immunodeficient mice. Hum Gene Ther. 2002;13(2):233–242.
42. Fairbairn, LJ, Rafferty, JA, Lashford, LS. Engineering drug resistance in human cells. Bone Marrow Transplant. 2000;25:S110–S113.
43. Chiocca, EA, Abbed, KM, Tatter, S, et al. A phase I open-label, dose-escalation, multi-institutional trial of injection with an E1B-attenuated adenovirus, ONYX-015, into the peritumoral region of recurrent malignant gliomas, in the adjuvant setting. Mol Ther. 2004;10(5):958–966.
44. Post, DE, Fulci, G, Chiocca, EA, et al. Replicative oncolytic herpes simplex viruses in combination cancer therapies. Curr Gene Ther. 2004;4(1):41–51.
45. Shah, AC, Benos, D, Gillespie, GY, et al. Oncolytic viruses: clinical applications as vectors for the treatment of malignant gliomas. J Neuroncol. 2003;65(3):203–226.
46. Dirven, CMF, van Beusechem, VW, Lamfers, MLM, et al. Oncolytic adenoviruses for treatment of brain tumours. Exp Opin Biol Ther. 2002;2(8):943–952.
47. Hemminki, A, Kanerva, A, Kremer, EJ, et al. A canine conditionally replicating adenovirus for evaluating oncolytic virotherapy in a syngeneic animal model. Mol Ther. 2003;7(2):163–173.
48. Zhan, JH, Gao, Y, Wang, WS, et al. Tumor-specific intravenous gene delivery using oncolytic adenoviruses. Cancer Gene Therapy. 2005;12(1):19–25.
49. Lombardo, A, Genovese, P, Beausejour, CM, et al. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat Biotechnol. 2007;25:1298–1306.
Section B
Signal Transduction and Cancer
In normal cells, signals are generated that begin at the outside of the cell and transmit through the cytoplasm to the nucleus, regulating cell growth, differentiation, survival, and death. Over the past few years, researchers have recognized that many of the components of the signal transduction pathways are dysregulated in cancer cells, leading to uncontrolled cell growth and thereby contributing to tumorigenesis. Because many tumors have similar alterations in signal transduction components, these have become promising targets for therapeutic intervention. This section primarily focuses on the role of a particular group of signal transducers called protein kinases, their role in normal cells, the mechanisms by which they contribute to tumorigenesis, and the use of agents designed to inhibit them when they become dysfunctional.
Protein Kinases and Normal Cells
Protein kinases play critical roles in normal cell signal transduction, acting to tightly regulate critical cellular processes such as growth and differentiation. These proteins work through phosphorylation; that is, they bind adenosine triphosphate (ATP) and use it to add phosphate groups to key residues on themselves (a process called autophosphorylation) and on other molecules, thereby stimulating a downstream signal inside the cell. This process typically occurs in response to external signals generated by growth factors (GFs) or other stimuli that initiate the cascade. Protein kinases are classified as tyrosine kinases (TKs) if they phosphorylate proteins on tyrosine residues or serine/threonine kinases if they phosphorylate proteins on serine and threonine residues. In some cases the kinases perform both functions (i.e., dual function kinases). These kinases can be expressed on the cell surface, in the cytoplasm, and in the nucleus. The human genome encodes approximately 518 kinases, of which 90 are classified as TKs.1
TKs on the cell surface that are activated through binding of GFs are called receptor TKs (RTKs). Of the 90 identified TKs, 58 are known to be RTKs. Each RTK contains an extracellular domain that binds the GF, a transmembrane domain, and a cytoplasmic kinase domain that positively and negatively regulates phosphorylation of the RTK (Figure 14-6).2-4 Most RTKs are monomers on the cell surface and are dimerized through the act of GF binding; this changes the three-dimensional structure of the receptor, permitting ATP to bind and autophosphorylation to occur, resulting in generation of a downstream signal through subsequent binding of adaptor proteins and nonreceptor kinases.2 Dysregulation of RTKs resulting in pathway activation/uncontrolled signaling is known to contribute to several human cancers, and work is ongoing to characterize such abnormalities in canine and feline cancers. Examples of RTKs known to play prominent roles in specific cancers include KIT, MET, epidermal growth factor receptor (EGFR), and anaplastic lymphoma kinase (ALK), which can be activated by overexpression, mutation, and chromosomal translocation.5-9
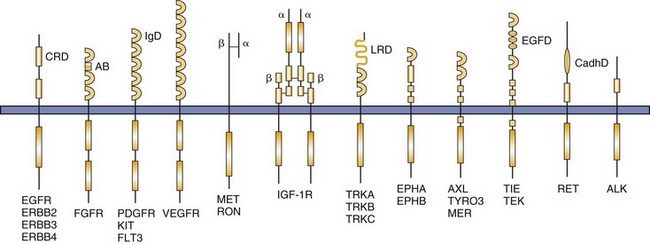
Figure 14-6 The structures of receptor tyrosine kinase (RTK) families implicated in a variety of malignancies are shown. The symbols α and β indicate specific RTK subunits. AB, Acid box; ALK, anaplastic lymphoma kinase; CadhD, cadherin-like domain; CRD, cysteine-rich domain; EGFD, epidermal growth factor-like domain; EGFR, epidermal growth factor receptor; EPH, member of ephrin receptor family; FGFR, fibroblast growth factor receptor; IgD, immunoglobulin-like domain; IGF-1R, insulin-like growth factor receptor 1; LRD, leucine-rich domain; PDGFR, platelet-derived growth factor receptor; Tie, tyrosine kinase receptor on endothelial cells; TRK, member of nerve growth factor receptor family; VEGFR, vascular endothelial growth factor receptor. (From London CA: Kinase inhibitors in cancer therapy, Vet Comp Oncol 2:177–193, 2004. Reprinted with permission from Blackwell Publishing.)
RKT signaling is critical for regulating typical cell functions and is also an important regulator of angiogenesis, a process known to be essential for continued tumor cell growth. The RTKs involved in angiogenesis include vascular endothelial growth factor receptor (VEGFR), platelet-derived growth factor receptor (PDGFR), fibroblast growth factor receptor (FGFR), and Tie-1 and Tie-2 (receptors for angiopoietin).10-13 VEGFRs are expressed on vascular endothelium, and VEGFR signaling drives endothelial migration and proliferation.10 PDGFR is expressed on stroma and pericytes that are critical for the maintenance of newly formed blood vessels. It also supports angiogenesis by inducing VEGF transcription and secretion.12,13 FGFR is expressed on vascular endothelium and works with VEGFR to promote increased expression of VEGF.12 Tie-1 and Tie-2 are expressed on blood vessels in tumors and are important in the recruitment of pericytes and smooth muscle cells to the newly forming vascular channels.14
Kinases in the cytoplasm act as a bridge, conducting signals generated by RTKs to the nucleus through a series of intermediates that become phosphorylated.15 The cytoplasmic kinases may be directly on the inside of the cell membrane or free in the cytoplasm. With respect to tumor cell biology, two particular cytoplasmic pathways are often dysregulated in a number of cancers. The first includes members of the RAS-RAF-MEK-ERK/p38/JNK families (Figure 14-7).16,17 Most of these are serine/threonine kinases, and their activation leads to ERK phosphorylation, translocation into the nucleus, and subsequent alteration of transcription factors and nuclear kinase activity important for controlling the cell cycle. Examples of dysregulation in human cancers include RAS mutations in lung cancer, colon cancer, and several hematologic malignancies and BRAF mutations in cutaneous melanomas and papillary thyroid carcinomas.18-20
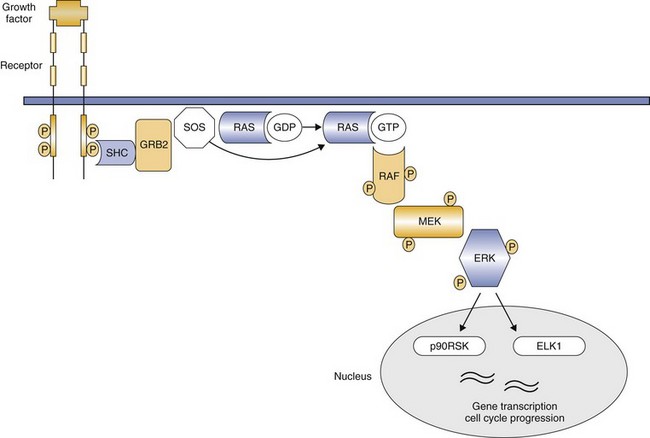
Figure 14-7 RAS signal transduction. Activated receptor tyrosine kinases recruit SOS to the plasma membrane through binding of SHC and GRB2. SOS replaces bound GDP with GTP, thereby activating RAS. The downstream target RAF is then phosphorylated by RAS, leading to subsequent activation of MEK, then ERK. ERK has several substrates both in the nucleus and in the cytoplasm, including ETS transcription factors such as ELK1 and RSK, which regulate cell cycle progression. (From London CA: Kinase inhibitors in cancer therapy, Vet Comp Oncol 2:177–193, 2004. Reprinted with permission from Blackwell Publishing.)
The second cytoplasmic pathway includes phosphatidyl inositol-3 kinase (PI3K) and its associated downstream signal transducers AKT, nuclear factor κB (NFκB), and mTOR, among others (Figure 14-8).21,22 PI3K is activated by RTKs and in turn activates AKT, which alters several additional proteins involved in the regulation of cell survival, cycling, and growth.23 AKT phosphorylates targets that promote apoptosis (BAD, procaspase-9, and Forkhead transcription factors) and activates NFκB, a transcription factor that has antiapoptotic activity.21-23 AKT also phosphorylates other proteins such as mTOR, p21, p27, and glycogen synthase kinase 3 (GSK3). This leads to redistribution of these proteins either in or out of the nucleus, ultimately inhibiting apoptosis while promoting cell cycling.21-23 Abnormalities of PI3K resulting in pathway activation are commonly found in human cancers, including mutations (breast and colorectal cancers and glioblastoma) and gene amplification (gastric, lung, ovarian cancers).24 This pathway may also become dysregulated through loss of activity of PTEN, a phosphatase that normally acts to dephosphorylate AKT and terminate signaling.21,25,26 PTEN mutations and/or decreased PTEN expression are found in several human cancers (e.g., glioblastoma and prostate cancer)24,25 and have been documented in canine cancers (OSA, melanoma).27-29
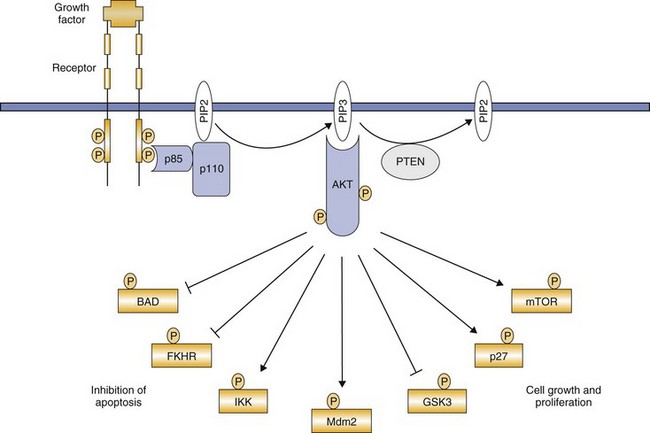
Figure 14-8 Phosphatidyl inositol-3 kinase (PI3K) signal transduction. Following receptor tyrosine kinase (RTK) activation, PI3K is recruited to the phosphorylated receptor through binding of the p85 adaptor subunit, leading to activation of the catalytic subunit (p110). This activation results in the generation of the second messenger phosphatidyl inositol-3,4,5-triphosphate (PIP3). PIP3 recruits AKT to the membrane and following its phosphorylation, several downstream targets are subsequently phosphorylated, leading to either their activation or inhibition. The cumulative effect results in cell survival, growth, and proliferation. (From London CA: Kinase inhibitors in cancer therapy, Vet Comp Oncol 2:177–193, 2004. Reprinted with permission from Blackwell Publishing.)
RTK-induced signaling ultimately influences cellular events by affecting transcription and the proteins that control cell cycling. The cyclins and their kinase partners (cyclin-dependent kinases [CDKs]) act to regulate the progression of cells through various phases of the cell cycle (Figure 14-9).30-32 The cyclins comprise several families. Cyclins D and E control restriction point passage by activating their respective CDKs (CDK4 and CDK6 for cyclin D and CDK2 for cyclin E). Coordinated function of cyclins D and E is required for cells to progress from G1 into S phase (see Figure 14-9). In many cases, RTK-generated signals induce expression of cyclin D, which complexes with CDK4 and CDK6, resulting in phosphorylation of the tumor suppressor Rb, partially repressing its function.31,32 Functional cyclin D/CDK complexes induce transcription of cyclin E, and active cyclin E/CDK complexes further reduce Rb activity through phosphorylation. This in turn initiates the process of DNA replication important for cell cycling. Dysregulation of the cyclins and CDKs is common in human cancers; for example, overexpression of cyclins D and E is often present in breast, pancreas, and head and neck carcinomas.32
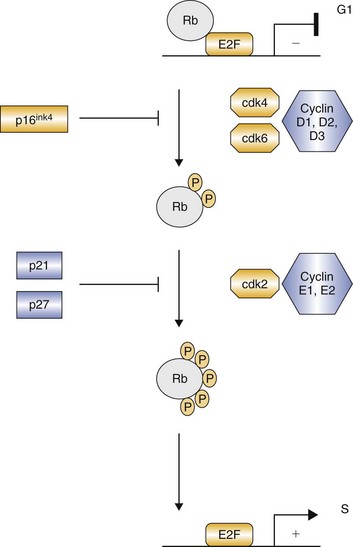
Figure 14-9 Cyclin and cyclin-dependent kinase (CDK) regulation of G1-S transition. CDK inhibitors such as p16ink4 and p21 restrict the activity of cyclin D- and cyclin E-dependent kinases. Progressive Rb phosphorylation by the cyclins results in liberation of E2F and the resultant transcription of S-phase genes. (From London CA: Kinase inhibitors in cancer therapy, Vet Comp Oncol 2:177–193, 2004. Reprinted with permission from Blackwell Publishing.)
Protein Kinases and Cancer Cells
Dysfunction of protein kinases is now recognized to be a common event in tumors. Although this has been best characterized in humans, recent data indicate that dog and cat cancers experience similar dysregulation (Table 14-2). Kinases may be dysregulated through a variety of mechanisms, including mutation, overexpression, fusion proteins, or autocrine loops. In the case of mutations, these may result in phosphorylation of the kinase in the absence of an appropriate signal. Such mutations can consist of a single amino acid change through a point mutation, deletion of amino acids, or insertion of amino acids, usually in the form of an internal tandem duplication (ITD). For example, a point mutation occurs in the BRAF gene (V600E, exon 15) in approximately 60% of human cutaneous melanomas.18,33,34 This amino acid change causes a conformation change in BRAF that mimics its activated form, thereby inducing constitutive downstream ERK signaling and abnormal promotion of cell growth and survival.35,36 RAS is another kinase that is dysregulated through point mutation in several hematopoietic neoplasms (multiple myeloma, juvenile chronic myelogenous leukemia [CML], acute myelogenous leukemia [AML], and chronic myelomonocytic leukemia [CMML]) and in lung cancer, colon cancer, and many others.17,37,38
Table 14-2
Receptor Tyrosine Kinases Associated with Cancer
| Tyrosine Kinase | Cancer Association |
| EGFR family | Breast, ovary, lung, stomach, colon, glioblastoma |
| Insulin receptor family | Sarcomas, cervix, kidney |
| PDGFR family | Glioblastoma, ovary, CMML, GIST |
| KIT | AML, GIST, seminoma, MCT, melanoma |
| Flt3 | AML |
| VEGFR family | Angiogenesis, Kaposi’s sarcoma, hemangiosarcoma, melanoma |
| FGFR family | AML, lymphoma, breast, prostate, multiple myeloma, TCC |
| NGFR family | Thyroid cancer, neuroblastoma, fibrosarcoma, AML |
| Met/Ron | Thyroid cancer, osteosarcoma, rhabdomyosarcoma, liver, kidney, colon |
| EPHR family | Melanoma, stomach, colon, breast, esophagus |
| AXL | AML |
| Tie family | Angiogenesis, stomach, hemangioblastoma |
| RET family | Thyroid cancer, multiple endocrine neoplasia |
| ALK | Non-Hodgkin’s lymphoma, lung |
ALK, Anaplastic lymphoma kinase; AML, acute myelogenous leukemia; CMML, chronic myelomonocytic leukemia; EGFR, epidermal growth factor receptor; EPHR, ephrin receptor; FGFR, fibroblast growth factor receptor; GIST, gastrointestinal stromal tumors; MCT, mast cell tumors; NGFR, nerve growth factor receptor; PDGFR, platelet-derived growth factor receptor; TCC, transitional cell carcinoma; Tie, tyrosine kinase receptor on endothelial cells; VEGFR, vascular endothelial growth factor receptor.
Another example of a mutation involves KIT, an RTK that normally is expressed on hematopoietic stem cells, on melanocytes, in the central nervous system, and on mast cells.39 In approximately 25% to 30% of canine grade 2 and grade 3 mast cell tumors (MCTs), mutations consisting of ITDs are found in the juxtamembrane domain of KIT, resulting in constitutive activation in the absence of ligand binding. These mutations are associated with a higher risk of local recurrence and metastasis.40-42 KIT mutations consisting of deletions in the juxtamembrane domain are also found in approximately 50% of human patients with gastrointestinal stromal tumors (GISTs) and are also found in GISTs in dogs.43-45 There are now several well-characterized mutations involving RTKs in human cancers, including FLT3 ITDs in AML,46-49 EGFR point mutations in lung carcinomas,50,51 and PI3K mutations in several types of carcinomas.24
Overexpression of kinases usually involves the RTKs and may result in enhanced response of the cancer cells to normal levels of growth factor; or, if the levels are high enough, the kinase may become activated through spontaneous dimerization in the absence of signal/growth factor. In humans, the RTK human epidermal growth factor receptor 2 (HER2; also known as ErbB2, a member of the EGFR family) is overexpressed in both breast and ovarian carcinomas and often correlates with a more aggressive phenotype.4,52,53 EGFR is also overexpressed in human lung, bladder, cervical, ovarian, renal, and pancreatic cancers, and some tumors have as many as 60 copies of the gene per cell.7,54,55 As with HER2, such overexpression is linked to a worse outcome in affected patients.7
Fusion proteins are generated when a portion of the kinase becomes attached to another gene through chromosomal rearrangement and the normal mechanisms that control protein function are disrupted. One of the best characterized fusion proteins is BCR-ABL, which is found in 90% of patients with CML.56-59 ABL is a cytoplasmic tyrosine kinase that, when fused to BCR, results in dysregulation of ABL, inappropriate activity of the protein, and resultant malignant transformation. Other examples include TEL-PDGFRβ in CMML, FIP1-PDGFRα in hypereosinophilic syndrome with mastocytosis, and (EML4)-ALK in non–small-cell lung cancer (NSCLC).60
Autocrine loops of activation primarily occur when the tumor cell expresses both the RTK and the growth factor; in most cases, one or the other usually is also overexpressed, resulting in constitutive activation of the RTK. Examples include coexpression of transforming growth factor α (TGFα) and EGFR in glioblastoma and squamous cell carcinoma, insulin-like growth factor (IGF) and its ligand, IGF-1R, in breast and colorectal cancer, and VEGF and VEGFR in melanoma.4,61-63 In canine cancers, possible autocrine loops have been documented in OSA (co-expression of MET and its ligand, HGF) and hemangiosarcoma (HSA; co-expression of KIT and its ligand, SCF).64-66
Inhibition of Kinases
Given the detailed molecular characterization of signal transducer dysregulation in cancer cells, significant efforts have been directed at developing strategies to inhibit those transducers that participate in tumorigenesis through direct effects on the cancer cell or through modulation of tumor microenvironment (stroma and neovasculature). The two most successful approaches to date have been monoclonal antibodies (MAbs) and small molecule inhibitors.
Several antibodies have been developed to target the extracellular domain of RTKs known to be important in a variety of tumors. These antibodies may prevent the growth factor from binding, may promote internalization of the RTK and subsequent degradation, or may induce an immune response against the cancer cell. One of the most successful examples is a humanized MAb called trastuzumab (Herceptin). This antibody targets HER2, which as previously discussed is overexpressed in approximately 30% of breast cancers, as well as other cancers, including prostate cancer, ovarian cancer, and NSCLC.67 In initial clinical trials, trastuzumab treatment of HER2-positive breast cancer resulted in a response rate of approximately 25% in patients with metastatic disease.68 The response rate approached 50% when trastuzumab was combined with chemotherapy.69 When used in the adjuvant setting, multiple studies have demonstrated that trastuzumab markedly improves survival rates of women with HER2-positive disease; trastuzumab is now part of the routine standard of care for this disease.70,71 Other examples of MAbs that have demonstrated significant activity in human cancers include rituximab (Rituxan) that targets CD20 expressed in a number of B-cell lymphomas72,73 and cetuximab (Erbitux) that targets ERBB1/HER1 EGFR known to be overexpressed in several carcinomas.7,67,74
Small molecule inhibitors work primarily by blocking the ATP-binding site of kinases, essentially acting as competitive inhibitors; a smaller number of these inhibitors work by preventing necessary protein-protein interactions (allosteric inhibition).75 In the absence of ATP, the kinase is unable to phosphorylate itself or downstream signaling elements, thereby interrupting a survival/growth signal essential to the tumor cell, ultimately resulting in cell death. As the molecular characterization of tumors has improved, the development and application of small molecule inhibitors have rapidly expanded in human oncology and their use is markedly altering how cancers are managed. Such inhibitors often are easy to synthesize in large quantities, frequently orally bioavailable, and can readily enter cells to bind the intended target.
The first small molecule inhibitor to be approved for human use was imatinib (Gleevec), an orally administered drug that binds the ATP pocket of ABL, as well as the RTKs KIT and PDGFRα.76 As previously discussed, BCR-ABL fusion proteins are present in 90% of human patients with CML, making ABL a good target for therapeutic intervention. The application of imatinib to CML has been transformative, with significant biologic activity demonstrated in several clinical trials, resulting in the approval of imatinib for up-front care of affected individuals.77-82 In the chronic phase of CML, imatinib induces a remission rate of close to 95%, and most patients remain in remission for longer than 1 year. Unfortunately, the remission rate is much lower for patients in blast crisis (20% to 50%), often lasting less than 10 months. Resistance to imatinib has been well characterized and is primarily due to the development of mutations in ABL that preclude drug binding, although gene amplification has also been documented.83,84 Imatinib also has clinical activity against human GIST in which 60% to 80% of the tumors have point mutations or deletions in the juxtamembrane domain of KIT, resulting in constitutive activation.85,86 Response rates of 50% to 70% have been reported with imatinib, far better than the 5% response rate seen with standard chemotherapy.87,88 A small number of GISTs have activating mutations in PDGFRα instead of KIT mutations; these patients also respond to imatinib.89
There are now several small molecule inhibitors approved for the treatment of human cancers that possess specific mutations in kinases known to drive tumor growth and survival. A subset of patients with NSCLC have tumors with activating mutations in EGFR that respond to erlotinib (Tarceva) or gefitinib (Iressa), small molecule inhibitors of EGFR.90 Response rates in patients with EGFR mutations can be as high as 80% compared to less than 10% to 20% for those without, demonstrating that efficacy of targeted therapies often depends on the presence of a known activated signaling element. A small number of patients with NSCLC also exhibit activation of the RTK ALK through its fusion to EML4.91 A small molecule inhibitor of ALK, crizotinib (Xalkori), has demonstrated significant activity against lung cancer patients whose tumors express the EML4-ALK translocation. In a recent phase II study, an objective response rate of 56% was achieved in this subset of patients, with another 31% of patients experiencing stable disease.91,92 As expected with a targeted therapeutic that disrupts key cell-signaling events, responses were rapid with most occurring by 8 weeks of treatment.
Vemurafenib (Zelboraf) is a small molecule inhibitor of BRAF that has shown significant activity against cutaneous malignant melanomas that possess activating mutations in BRAF. Most patients treated with vemurafenib experienced tumor shrinkage, with close to 50% meeting the criteria for objective response.93 This compares to an objective response rate of only 5% in patients treated with dacarbazine. Inhibition of mTOR has become of interest in several cancers given the activation of the PI3K pathway and the critical role of mTOR in mediating its effects. Rapamycin, a drug used for many years as an immunosuppressive agent, is the prototypical mTOR inhibitor.94,95 Temsirolimus and everolimus, two rapamycin analogs, have already been approved for use in patients with metastatic renal carcinoma and other mTOR inhibitors are currently under investigation for their potential utility in treating soft tissue sarcomas and bone sarcomas.94,95
Flavopiridol, a partly synthetic flavonoid derived from an indigenous plant (rohitukine) found in India, sits in the ATP-binding pocket of CDK2, acting as a competitive inhibitor.89,90 This compound has been shown to inhibit most of the CDKs evaluated, although some less potently than others.91,92 Although flavopiridol initially failed to demonstrate significant efficacy when used as a single agent,89 recent pharmacokinetic data demonstrated activity in some patients, particularly those with chronic lymphocytic leukemia (CLL), when an alternative dosing schedule that enhanced the area under the curve was employed.96
The inhibitors discussed previously tend to affect a restricted set of kinases, although other drugs exhibit much more broadly targeted inhibition. Sunitinib (Sutent) is a small molecule inhibitor of several RTKs, including VEGFR1, VEGFR2, PDGFRα/β, KIT, FLT3, receptor of colony-stimulating factor 1 (CSFR1), and rearranged during transfection receptor (RET).97 The multitargeted nature of this inhibitor may be responsible for its observed activity in several types of cancer, including GIST, renal cell carcinoma, thyroid carcinoma, and insulinoma, among others.97 Although such agents often have significant clinical activity, they are typically associated with a broader range of toxicities that may limit their use.
Kinase Inhibitors in Veterinary Medicine
There are now two small molecule inhibitors approved for use in dogs in veterinary medicine. Toceranib (Palladia) is a multitargeted inhibitor closely related to sunitinib that exhibits a similar target profile, including VEGFR, PDGFR, KIT, FLT3, and CSF1R. Toceranib has demonstrated activity against MCT, as well as sarcomas and carcinomas. In the original phase I study, 28% of dogs experienced objective responses to treatment, with an additional 26% experiencing stable disease for an overall biologic activity of 54%.98 A pivotal study of toceranib was subsequently conducted in dogs with recurrent or metastatic intermediate or high grade MCT resulting in an objective response rate of 42.8% (21 complete responses, 42 partial responses), with an additional 16 dogs experiencing stable disease for an overall biologic activity of 60%.99 Dogs whose MCT harbored activating mutations in KIT were roughly twice as likely to respond to toceranib than those without mutation (69% versus 37%). Following approval of toceranib in 2009, it has been used to treat a number of different solid tumors.100 Preliminary observations of biologic activity were reported in dogs with anal sac adenocarcinoma, thyroid carcinoma, head and neck carcinoma, nasal carcinoma, and OSA. Several studies are ongoing to more clearly define the role of toceranib in the treatment of canine and feline cancer.
Masitinib (Kinavet) is a small molecule inhibitor of KIT, PDGFRα/β, and Lyn. In dogs with MCTs, masitinib significantly improved time to progression compared to placebo, and outcome was improved in dogs with MCT harboring KIT mutations.101 Subsequent follow-up of patients treated with long-term masitinib identified an increased number of patients with long-term disease control compared to those treated with placebo (40% versus 15% alive at 2 years).102 Finally, small studies have evaluated the efficacy of imatinib for the treatment of canine and feline MCT.103-105 Imatinib was well tolerated, and objective antitumor responses were observed in dogs with both mutant and wild-type KIT. Responses have also been observed in cats with MCT.106,107
Conclusion
With the advent of molecular techniques, the characterization of signal transduction pathways that are dysfunctional in cancer cells has become commonplace. Advances in computer modeling and small molecule engineering have led to the rapid development of inhibitors capable of blocking specific pathways critical for cancer cell survival. The success of inhibitors such as imatinib and crizotinib indicate that the application of this therapeutic strategy can markedly improve clinical outcome. Perhaps the greatest challenges will be determining how these novel therapeutics can be effectively combined with standard treatment regimens such as chemotherapy and radiation therapy to provide optimal anticancer efficacy without enhancing toxicity and identifying strategies to use these therapeutics in ways that are less likely to result in drug resistance.
References
1. Manning, G, Whyte, DB, Martinez, R, et al. The protein kinase complement of the human genome. Science. 2002;298:1912–1934.
2. Lemmon, MA, Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117–1134.
3. Madhusudan, S, Ganesan, TS. Tyrosine kinase inhibitors in cancer therapy. Clin Biochem. 2004;37:618–635.
4. Zwick, E, Bange, J, Ullrich, A. Receptor tyrosine kinases as targets for anticancer drugs. Trends Mol Med. 2002;8:17–23.
5. Barreca, A, Lasorsa, E, Riera, L, et al. Anaplastic lymphoma kinase in human cancer. J Mol Endocrinol. 2011;47:R11–R23.
6. Fletcher, JA. Role of KIT and platelet-derived growth factor receptors as oncoproteins. Semin Oncol. 2004;31:4–11.
7. Laskin, JJ, Sandler, AB. Epidermal growth factor receptor: a promising target in solid tumours. Cancer Treat Rev. 2004;30:1–17.
8. Ma, PC, Jagadeeswaran, R, Jagadeesh, S, et al. Functional expression and mutations of c-Met and its therapeutic inhibition with SU11274 and small interfering RNA in non-small cell lung cancer. Cancer Res. 2005;65:1479–1488.
9. Ma, PC, Maulik, G, Christensen, J, et al. c-Met: structure, functions and potential for therapeutic inhibition. Cancer Metastasis Rev. 2003;22:309–325.
10. Thurston, G, Gale, NW. Vascular endothelial growth factor and other signaling pathways in developmental and pathologic angiogenesis. Int J Hematol. 2004;80:7–20.
11. Eskens, FA. Angiogenesis inhibitors in clinical development; where are we now and where are we going? Br J. Cancer. 2004;90:1–7.
12. Cherrington, JM, Strawn, LM, Shawver, LK. New paradigms for the treatment of cancer: the role of anti-angiogenesis agents. Adv Cancer Res. 2000;79:1–38.
13. McCarty, MF, Liu, W, Fan, F, et al. Promises and pitfalls of anti-angiogenic therapy in clinical trials. Trends Mol Med. 2003;9:53–58.
14. Thurston, G. Role of Angiopoietins and Tie receptor tyrosine kinases in angiogenesis and lymphangiogenesis. Cell Tissue Res. 2003;314:61–68.
15. Blume-Jensen, P, Hunter, T. Oncogenic kinase signalling. Nature. 2001;411:355–365.
16. Johnson, GL, Lapadat, R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912.
17. Downward, J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11–22.
18. Davies, H, Bignell, GR, Cox, C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954.
19. Kumar, R, Angelini, S, Snellman, E, et al. BRAF mutations are common somatic events in melanocytic nevi. J Invest Dermatol. 2004;122:342–348.
20. Mercer, KE, Pritchard, CA. Raf proteins and cancer: B-Raf is identified as a mutational target. Biochim Biophys Acta. 2003;1653:25–40.
21. Fresno Vara, JA, Casado, E, de Castro, J, et al. PI3K/Akt signalling pathway and cancer. Cancer Treat Rev. 2004;30:193–204.
22. Franke, TF, Hornik, CP, Segev, L, et al. PI3K/Akt and apoptosis: size matters. Oncogene. 2003;22:8983–8998.
23. Mitsiades, CS, Mitsiades, N, Koutsilieris, M. The Akt pathway: molecular targets for anti-cancer drug development. Curr Cancer Drug Targets. 2004;4:235–256.
24. Markman, B, Atzori, F, Perez-Garcia, J, et al. Status of PI3K inhibition and biomarker development in cancer therapeutics. Ann Oncol. 2010;21:683–691.
25. Simpson, L, Parsons, R. PTEN: Life as a tumor suppressor. Exp Cell Res. 2001;264:29–41.
26. Weng, LP, Smith, WM, Dahia, PL, et al. PTEN suppresses breast cancer cell growth by phosphatase activity-dependent G1 arrest followed by cell death. Cancer Res. 1999;59:5808–5814.
27. Kanae, Y, Endoh, D, Yokota, H, et al. Expression of the PTEN tumor suppressor gene in malignant mammary gland tumors of dogs. Am J Vet Res. 2006;67:127–133.
28. Koenig, A, Bianco, SR, Fosmire, S, et al. Expression and significance of p53, rb, p21/waf-1, p16/ink-4a, and PTEN tumor suppressors in canine melanoma. Vet Pathol. 2002;39:458–472.
29. Levine, RA, Forest, T, Smith, C. Tumor suppressor PTEN is mutated in canine osteosarcoma cell lines and tumors. Vet Pathol. 2002;39:372–378.
30. Swanton, C. Cell-cycle targeted therapies. Lancet Oncol. 2004;5:27–36.
31. Ortega, S, Malumbres, M, Barbacid, M. Cyclin D-dependent kinases, INK4 inhibitors and cancer. Biochim Biophys Acta. 2002;1602:73–87.
32. Malumbres, M, Barbacid, M. To cycle or not to cycle: a critical decision in cancer. Nat Rev Cancer. 2001;1:222–231.
33. Wellbrock, C, Ogilvie, L, Hedley, D, et al. V599EB-RAF is an oncogene in melanocytes. Cancer Res. 2004;64:2338–2342.
34. Pollock, PM, Meltzer, PS. A genome-based strategy uncovers frequent BRAF mutations in melanoma. Cancer Cell. 2002;2:5–7.
35. Wan, PT, Garnett, MJ, Roe, SM, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116:855–867.
36. Dhillon, AS, Kolch, W. Oncogenic B-Raf mutations: crystal clear at last. Cancer Cell. 2004;5:303–304.
37. Brose, MS, Volpe, P, Feldman, M, et al. BRAF and RAS mutations in human lung cancer and melanoma. Cancer Res. 2002;62:6997–7000.
38. Malumbres, M, Barbacid, M. RAS oncogenes: the first 30 years. Nat Rev Cancer. 2003;3:459–465.
39. Galli, SJ, Zsebo, KM, Geissler, EN. The kit ligand, stem cell factor. Adv Immunol. 1994;55:1–95.
40. Downing, S, Chien, MB, Kass, PH, et al. Prevalence and importance of internal tandem duplications in exons 11 and 12 of c-kit in mast cell tumors of dogs. Am J Vet Res. 2002;63:1718–1723.
41. London, CA, Galli, SJ, Yuuki, T, et al. Spontaneous canine mast cell tumors express tandem duplications in the proto-oncogene c-kit. Exp Hematol. 1999;27:689–697.
42. Zemke, D, Yamini, B, Yuzbasiyan-Gurkan, V. Mutations in the juxtamembrane domain of c-KIT are associated with higher grade mast cell tumors in dogs. Vet Pathol. 2002;39:529–535.
43. Demetri, GD. Targeting the molecular pathophysiology of gastrointestinal stromal tumors with imatinib. Mechanisms, successes, and challenges to rational drug development. Hematol Oncol Clin North Am. 2002;16:1115–1124.
44. Demetri, GD. Differential properties of current tyrosine kinase inhibitors in gastrointestinal stromal tumors. Semin Oncol. 2011;38(Suppl 1):S10–S19.
45. Frost, D, Lasota, J, Miettinen, M. Gastrointestinal stromal tumors and leiomyomas in the dog: a histopathologic, immunohistochemical, and molecular genetic study of 50 cases. Vet Pathol. 2003;40:42–54.
46. Kondo, M, Horibe, K, Takahashi, Y, et al. Prognostic value of internal tandem duplication of the FLT3 gene in childhood acute myelogenous leukemia. Med Pediatr Oncol. 1999;33:525–529.
47. Nakoa, M, Yokota, S, Iwai, T, et al. Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia. 1996;10:1911–1918.
48. Yokota, S, Kiyoi, H, Nakao, M, et al. Internal tandem duplication of the FLT3 gene is preferentially seen in acute myeloid leukemia and myelodysplastic syndrome among various hematological malignancies. A study on a large series of patients and cell lines. Leukemia. 1997;11:1605–1609.
49. Iwai, T, Yokota, S, Nakao, M, et al. Internal tandem duplication of the FLT3 gene and clinical evaluation in childhood acute myeloid leukemia. The Children’s Cancer and Leukemia Study Group, Japan. Leukemia. 1999;13:38–43.
50. Pao, W, Chmielecki, J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat Rev Cancer. 2010;10:760–774.
51. Wen, J, Fu, J, Zhang, W, et al. Genetic and epigenetic changes in lung carcinoma and their clinical implications. Mod Pathol. 2011;24:932–943.
52. Paik, S, Hazan, R, Fisher, ER, et al. Pathologic findings from the National Surgical Adjuvant Breast and Bowel Project: prognostic significance of erbB-2 protein overexpression in primary breast cancer. J Clin Oncol. 1990;8:103–112.
53. Slamon, DJ, Clark, GM, Wong, SG, et al. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–182.
54. Libermann, TA, Nusbaum, HR, Razon, N, et al. Amplification, enhanced expression and possible rearrangement of EGF receptor gene in primary human brain tumours of glial origin. Nature. 1985;313:144–147.
55. Libermann, TA, Nusbaum, HR, Razon, N, et al. Amplification and overexpression of the EGF receptor gene in primary human glioblastomas. J Cell Sci Suppl. 1985;3:161–172.
56. Golub, TR, Barker, GF, Lovett, M, et al. Fusion of PDGF receptor beta to a novel ets-like gene, tel, in chronic myelomonocytic leukemia with t(5;12) chromosomal translocation. Cell. 1994;77:307–316.
57. Gotlib, J, Cools, J, Malone, JM, 3rd., et al. The FIP1L1-PDGFRalpha fusion tyrosine kinase in hypereosinophilic syndrome and chronic eosinophilic leukemia: implications for diagnosis, classification, and management. Blood. 2004;103:2879–2891.
58. Melo, JV, Hughes, TP, Apperley, JF. Chronic myeloid leukemia. Hematology (Am Soc Hematol Educ Program). 2003:132–152. [2003].
59. Van Etten, RA. Mechanisms of transformation by the BCR-ABL oncogene: new perspectives in the post-imatinib era. Leuk Res. 2004;28(Suppl 1):S21–S28. [2004].
60. Medves, S, Demoulin, JB. Tyrosine kinase gene fusions in cancer: Translating mechanisms into targeted therapies. J Cell Mol Med. 2012;16(2):237–248.
61. Sciacca, L, Costantino, A, Pandini, G, et al. Insulin receptor activation by IGF-II in breast cancers: evidence for a new autocrine/paracrine mechanism. Oncogene. 1999;18:2471–2479.
62. Ekstrand, AJ, James, CD, Cavenee, WK, et al. Genes for epidermal growth factor receptor, transforming growth factor alpha, and epidermal growth factor and their expression in human gliomas in vivo. Cancer Res. 1991;51:2164–2172.
63. Graeven, U, Fiedler, W, Karpinski, S, et al. Melanoma-associated expression of vascular endothelial growth factor and its receptors FLT-1 and KDR. J Cancer Res Clin Oncol. 1999;125:621–629.
64. Fosmire, SP, Dickerson, EB, Scott, AM, et al. Canine malignant hemangiosarcoma as a model of primitive angiogenic endothelium. Lab Invest. 2004;84:562–572.
65. MacEwen, EG, Kutzke, J, Carew, J, et al. c-Met tyrosine kinase receptor expression and function in human and canine osteosarcoma cells. Clin Exp Metastasis. 2003;20:421–430.
66. Ferracini, R, Angelini, P, Cagliero, E, et al. MET oncogene aberrant expression in canine osteosarcoma. J Orthop Res. 2000;18:253–256.
67. Harris, M. Monoclonal antibodies as therapeutic agents for cancer. Lancet Oncol. 2004;5:292–302.
68. Vogel, CL, Cobleigh, MA, Tripathy, D, et al. First-line Herceptin monotherapy in metastatic breast cancer. Oncology. 2001;61(Suppl 2):37–42.
69. Slamon, DJ, Leyland-Jones, B, Shak, S, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;2001(344):783–792.
70. Arteaga, CL, Sliwkowski, MX, Osborne, CK, et al. Treatment of HER2-positive breast cancer: current status and future perspectives. Nat Rev Clin Oncol. 2011;9(1):16–32.
71. Mukai, H. Treatment strategy for HER2-positive breast cancer. Int J Clin Oncol. 2010;15:335–340.
72. Cabanillas, F. Front-line management of diffuse large B cell lymphoma. Curr Opin Oncol. 2010;22:642–645.
73. Vidal, L, Gafter-Gvili, A, Salles, G, et al. Rituximab maintenance for the treatment of patients with follicular lymphoma: an updated systematic review and meta-analysis of randomized trials. J Natl Cancer Inst. 2001;103:1799–1806.
74. Brand, TM, Iida, M, Wheeler, DL. Molecular mechanisms of resistance to the EGFR monoclonal antibody cetuximab. Cancer Biol Ther. 2011;11:777–792.
75. Zhang, J, Yang, PL, Gray, NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009;9:28–39.
76. de Kogel, CE, Schellens, JH. Imatinib. Oncologist. 2007;12:1390–1394.
77. Mauro, MJ, Druker, BJ. STI571: targeting BCR-ABL as therapy for CML. Oncologist. 2001;6:233–238.
78. Kantarjian, H, Sawyers, C, Hochhaus, A, et al. Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. N Engl J Med. 2002;346:645–652.
79. Beham-Schmid, C, Apfelbeck, U, Sill, H, et al. Treatment of chronic myelogenous leukemia with the tyrosine kinase inhibitor STI571 results in marked regression of bone marrow fibrosis. Blood. 2002;99:381–383.
80. Druker, BJ, Talpaz, M, Resta, DJ, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031–1037.
81. Druker, BJ, Sawyers, CL, Kantarjian, H, et al. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med. 2001;344:1038–1042.
82. Sawyers, CL. Rational therapeutic intervention in cancer: Kinases as drug targets. Curr Opin Genet Dev. 2002;12:111–115.
83. Weisberg, E, Griffin, JD. Resistance to imatinib (Glivec): update on clinical mechanisms. Drug Resist Updat. 2003;6:231–238.
84. Nardi, V, Azam, M, Daley, GQ. Mechanisms and implications of imatinib resistance mutations in BCR-ABL. Curr Opin Hematol. 2004;11:35–43.
85. Duffaud, F, Blay, JY. Gastrointestinal stromal tumors: Biology and treatment. Oncology. 2003;65:187–197.
86. Heinrich, MC, Rubin, BP, Longley, BJ, et al. Biology and genetic aspects of gastrointestinal stromal tumors: KIT activation and cytogenetic alterations. Hum Pathol. 2002;33:484–495.
87. Miettinen, M, Sarlomo-Rikala, M, Lasota, J. Gastrointestinal stromal tumors: recent advances in understanding of their biology. Hum Pathol. 1999;30:1213–1220.
88. Miettinen, M, Sarlomo-Rikala, M, Lasota, J. Gastrointestinal stromal tumours. Ann Chir Gynaecol. 1998;87:278–281.
89. Heinrich, MC, Corless, CL, Duensing, A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299:708–710.
90. Peled, N, Yoshida, K, Wynes, MW, et al. Predictive and prognostic markers for epidermal growth factor receptor inhibitor therapy in non-small cell lung cancer. Ther Adv Med Oncol. 2009;1:137–144.
91. Bang, YJ. The potential for crizotinib in non-small cell lung cancer: A perspective review. Ther Adv Med Oncol. 2011;3:279–291.
92. Shaw, AT, Yeap, BY, Solomon, BJ, et al. Effect of crizotinib on overall survival in patients with advanced non-small-cell lung cancer harbouring ALK gene rearrangement: a retrospective analysis. Lancet Oncol. 2011;12:1004–1012.
93. Chapman, PB, Hauschild, A, Robert, C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–2516.
94. Markman, B, Dienstmann, R, Tabernero, J. Targeting the PI3K/Akt/mTOR pathway–beyond rapalogs. Oncotarget. 2010;1:530–543.
95. Vilar, E, Perez-Garcia, J, Tabernero, J. Pushing the envelope in the mTOR pathway: the second generation of inhibitors. Mol Cancer Ther. 2011;10:395–403.
96. Christian, BA, Grever, MR, Byrd, JC, et al. Flavopiridol in the treatment of chronic lymphocytic leukemia. Curr Opin Oncol. 2007;19:573–578.
97. Papaetis, GS, Syrigos, KN. Sunitinib: a multitargeted receptor tyrosine kinase inhibitor in the era of molecular cancer therapies. BioDrugs. 2009;23:377–389.
98. London, CA, Hannah, AL, Zadovoskaya, R, et al. Phase I dose-escalating study of SU11654, a small molecule receptor tyrosine kinase inhibitor, in dogs with spontaneous malignancies. Clin Cancer Res. 2003;9:2755–2768.
99. London, CA, Malpas, PB, Wood-Follis, SL, et al. Multi-center, placebo-controlled, double-blind, randomized study of oral toceranib phosphate (SU11654), a receptor tyrosine kinase inhibitor, for the treatment of dogs with recurrent (either local or distant) mast cell tumor following surgical excision. Clin Cancer Res. 2009;15:3856–3865.
100. London, C, Mathie, T, Stingle, N, et al. Preliminary evidence for biologic activity of toceranib phosphate (Palladia) in solid tumours. Vet Comp Oncol. 2011. [Epub ahead of print June 1].
101. Hahn, KA, Ogilvie, G, Rusk, T, et al. Masitinib is safe and effective for the treatment of canine mast cell tumors. J Vet Intern Med. 2008;22:1301–1309.
102. Hahn, KA, Legendre, AM, Shaw, NG, et al. Evaluation of 12- and 24-month survival rates after treatment with masitinib in dogs with nonresectable mast cell tumors. Am J Vet Res. 2010;71:1354–1361.
103. Isotani, M, Ishida, N, Tominaga, M, et al. Effect of tyrosine kinase inhibition by imatinib mesylate on mast cell tumors in dogs. J Vet Intern Med. 2008;22(4):985–988.
104. Marconato, L, Bettini, G, Giacoboni, C, et al. Clinicopathological features and outcome for dogs with mast cell tumors and bone marrow involvement. J Vet Intern Med. 2008;22(4):1001–1007.
105. Yamada, O, Kobayashi, M, Sugisaki, O, et al. Imatinib elicited a favorable response in a dog with a mast cell tumor carrying a c-kit c.1523A>T mutation via suppression of constitutive KIT activation. Vet Immunol Immunopathol. 2011;142:101–106.
106. Isotani, M, Tamura, K, Yagihara, H, et al. Identification of a c-kit exon 8 internal tandem duplication in a feline mast cell tumor case and its favorable response to the tyrosine kinase inhibitor imatinib mesylate. Vet Immunol Immunopathol. 2006;114:168–172.
107. Isotani, M, Yamada, O, Lachowicz, JL, et al. Mutations in the fifth immunoglobulin-like domain of kit are common and potentially sensitive to imatinib mesylate in feline mast cell tumours. Br J Haematol. 2009;148:144–153.
Section C
Antiangiogenic and Metronomic Therapy
Tumor Angiogenesis
Angiogenesis has emerged as a validated therapeutic target in oncology over the last several years, particularly with the widespread approval of drugs that target the proangiogenic vascular endothelial growth factor (VEGF) signaling pathway.1-5 Drugs such as bevacizumab (Avastin), the humanized anti-VEGF monoclonal antibody, and small molecule RTK inhibitors (RTKIs) such as sunitinib (Sutent) and sorafenib (Nexavar) in human oncology and toceranib (Palladia) in veterinary medicine are all considered to be efficacious, at least in part, because of an antiangiogenic mechanism of action.6,7 The guiding principle of tumor angiogenesis states that in order for a solid tumor to grow beyond a few millimeters in size, it must recruit its own blood supply to provide adequate nutrients and oxygen to the dividing cell mass, as well as the removal of waste products.8,9 Inducing angiogenesis is considered a hallmark of cancer progression,10 yet angiogenesis is a prominent aspect of normal physiology and transient pathology (e.g., in wound healing) and is tightly regulated. A large number of proangiogenic growth factors and signaling pathways promote the process of blood vessel growth.8 Similarly, endogenous inhibitors of angiogenesis are temporally expressed to suppress vessel expansion and maintain angiogenic balance.11 The net effect depends on this relative balance, which is tipped toward promoting angiogenesis during tumor growth. Therapeutic interventions aim to tip the scales back toward inhibition of tumor angiogenesis.12
Angiogenesis occurs through the sprouting of new vessels from existing vasculature, whereas the term vasculogenesis is generally used to describe the de novo formation of new blood vessels from bone marrow–derived progenitor cell populations that respond to locally produced proangiogenic signals.13,14 In addition, although controversial, circulating endothelial progenitor cells (CEPs) from the bone marrow may contribute to tumor angiogenesis by travelling to tumor sites and incorporating into existing vessel walls.15-18 Finally, mature endothelial cells originating from the growing vasculature itself may be shed into the bloodstream, and are referred to as circulating endothelial cells (CECs).19 These CECs may represent a byproduct of local angiogenesis rather than an indicator of bone marrow involvement. At the present time, there is still much to learn regarding the morphology, surface marker expression, and relative contribution circulating cell populations make to tumor blood vessel growth and maintenance.20,21
Despite the principles of angiogenesis finding extensive application in the field of solid tumor growth, blood vessels are also a normal component of the bone marrow microenvironment, and angiogenesis is a prominent feature of malignant bone marrow disorders. Many of the same growth factor and inhibitory pathway components described in solid tumor angiogenesis contribute to the biology of myelodysplasia, leukemias, myeloma, and lymphoma, among other “liquid” tumors.22 As a result, targeting angiogenesis is being investigated for the treatment of blood and bone marrow neoplasia and, indeed, one such agent with demonstrated antiangiogenic effects, thalidomide, has been approved for treatment of human multiple myeloma.23
In addition to blood vessels, lymphatics are an important component of the tumor microenvironment and a prominent feature of tissue homeostasis, immunosurveillance, and a gateway to metastatic spread.24 The regulation of lymphatic vessel growth is physiologically similar in principle to that of blood vessels, although the contribution of cells from the bone marrow to the formation and maintenance of lymphatic channels may be equally controversial.25 Similar to angiogenesis, targeting lymphangiogenesis is an attractive prospect in clinical oncology that is receiving an increased research focus.26
Antiangiogenic Therapy
There are multiple theories to explain the potential antitumor effects that result from antiangiogenic therapies.3 The first and most intuitive is vascular collapse resulting in impaired oxygen delivery to the tumor, leading to nutrient starvation, hypoxia, and death of cancer cells that cannot survive in this oxygen-deprived environment. Conversely, a second mechanism actually involves more efficient delivery of oxygen, nutrients, and indeed other drugs (such as chemotherapeutics) to tumor cells by a process termed vessel normalization.27,28 Normalization occurs when smaller, tortuous, inefficient, and leaky tumor vessels are selectively destroyed, resulting in improved blood flow through the tumor as a whole.29 Vascular normalization has been demonstrated clinically using functional magnetic resonance imaging (fMRI) methods in patients undergoing treatment with VEGF pathway inhibitors.30,31 The most common clinical approach to targeting tumor angiogenesis has been either inhibition of overexpressed proangiogenic stimuli or supplementation of factors that inhibit angiogenesis. Targeting vasculogenesis through neutralization of CEPs is also a potential treatment strategy.32
Inhibition of Proangiogenic Factors—Receptor Tyrosine Kinase Inhibitors
Currently, most RTKIs target numerous receptors to a varying degree.7,33 Toceranib is an example of an RTKI approved for veterinary use that complements its direct effects on tumor cells (e.g., via mutated c-kit inhibition) with angiogenesis inhibition because it directly targets VEGF receptors (VEGFR).34 Binding to VEGFR results in blockade of the most powerful endothelial growth and survival factor. Additionally, through inhibition of platelet-derived growth factor receptors (PDGFR), toceranib and another veterinary approved RTKI, masitinib (Masivet, Kinavet), disrupt signaling pathways for blood vessel support structures, such as the stromal pericyte component of larger vessels.35,36 The relative antiangiogenic and direct antitumor effect of these RTKIs is made more complicated by the fact that some angiogenesis receptors such as VEGFR and PDGFR may also be concomitantly expressed by certain cancer cell types, resulting in an autocrine growth factor loop.37,38 The end result is that in certain cases targeting angiogenic pathways may have concurrent antitumor cell and antiangiogenic effects.
Significant cross-talk exists between growth signaling pathways, and as a result there is strong interaction between tumor cell–based oncogene or tumor suppressor gene expression and regulation of blood vessel growth via factors such as VEGF. Therefore many drugs designed to target oncogenes have demonstrated antiangiogenic “off-target” effects as a byproduct of VEGF inhibition.39-41 For example, the anti-HER2 (ErbB-2) human MAb trastuzumab (Herceptin) and anti-EGFR antibodies such as cetuximab (Erbitux) are examples of drugs that indirectly suppress angiogenesis because neutralization of their oncogenic targets leads to a dramatic reduction in tumor cell VEGF production, which increases again at the time of targeted drug resistance.42,43 Through reduction in VEGF or other growth factor levels, there is an antiangiogenic component inherent in many forms of cancer treatment, including not only targeted inhibitors but also cytotoxic chemotherapy and radiation therapy. As a result, antiangiogenic mechanisms are not necessarily restricted to treatment modalities that target known angiogenesis pathways directly.
Angiogenic Inhibitor Supplementation
Since the discovery of endogenous proteins and protein fragments that inhibit blood vessel growth such as angiostatin, endostatin, and thrombospondins, there has been clinical interest in using angiogenesis inhibitors as cancer therapeutics.11,44 In veterinary medicine, thrombospondin-1 mimetic peptides have been evaluated as an antiangiogenic strategy in dogs with multiple tumor types. Treatment with the mimetic peptides ABT-526 and ABT-510 in a prospective clinical trial of 242 dogs with multiple tumor types showed an objective response or substantially stabilized disease in 42 dogs and a lack of dose-limiting toxicity.45 Interestingly, most objective responses were recorded after 60 days of continuous drug treatment. A prospective randomized placebo-controlled clinical trial has also been reported in dogs with multicentric lymphoma treated at first relapse.46 Results revealed a significant improvement in time to tumor progression and remission duration, but not remission rate, when ABT-526 was used in combination with lomustine, compared to dogs treated with lomustine alone, with no ABT-526–specific toxicities noted. In addition to thrombospondin-1, preliminary studies documenting detection of the endogenous inhibitors angiostatin and endostatin in normal and tumor-bearing dogs have been reported.47,48 In a preliminary study of endostatin as a therapeutic agent, 13 dogs with soft tissue sarcomas were treated with the canine endostatin gene delivered via liposome-DNA complexes.49 Although endostatin gene expression was not detected in the tumors following treatment, objective responses were documented in 2 dogs, and 8 dogs experienced stable disease, suggesting potential nonspecific antitumor activity.
Direct Targeting of Tumor Endothelial Cell Surface Markers
Tumor endothelial cells differ from normal endothelia in their genetic and protein composition.50 These differences may represent an opportunity for a favorable therapeutic index when drugs are designed to bind differentially expressed targets on tumor-specific endothelium. If not naturally destructive to these cells, drugs can be designed to carry a payload to induce local cytotoxicity, resulting in an antiangiogenic effect. A phage vector delivering tumor necrosis factor (RGD-A-TNF) to αV integrins on tumor endothelium is one example of such a strategy that has undergone evaluation in dogs.51 Through serial biopsy, this dose escalation trial was able to demonstrate selective targeting of tumor endothelium via αV integrin-targeted expression of TNF, and treatment of a cohort of tumor-bearing dogs with the maximum tolerated dose (MTD) resulting in partial remission in 2/14 and stable disease in 6/14 dogs.
Other Antiangiogenic Agents
A vast array of drugs not necessarily designed as anticancer therapeutics may derive at least a portion of their activity through inhibition of angiogenesis. Two examples investigated in a veterinary setting include inhibitors of matrix metalloproteinase (MMP) and cyclooxygenase (COX). The MMPs are a family of enzymes that degrade the extracellular matrix and basement membrane, thereby mediating tumor invasion, angiogenesis, and metastasis.52 Unfortunately, clinical evaluation of compounds that inhibit these enzymes has to date been largely unrewarding,53,54 including results from a large randomized trial of 303 dogs with OSA, in which treatment with the MMP inhibitor Bay12-9566 or placebo after doxorubicin chemotherapy did not improve overall survival.55 Inhibition of the proinflammatory COX enzymes has been commonly studied in numerous tumor types in veterinary oncology.56-58 The effects of COX inhibition on reducing angiogenesis specifically have been evaluated with piroxicam treatment of canine transitional cell carcinoma (TCC). In a study of 18 dogs, piroxicam treatment was associated with reduced urinary concentrations of the proangiogenic growth factor basic fibroblast growth factor (bFGF) and induction of apoptosis.59 In a subsequent study evaluating these parameters in 12 dogs treated with piroxicam in combination with cisplatin, reductions in urinary bFGF and VEGF were associated with response to the combination regimen.60
Metronomic Chemotherapy
Conventional cytotoxic chemotherapy agents that have been in clinical use for many decades also achieve at least a portion of their therapeutic efficacy by inhibition of tumor angiogenesis.61,62 The basis for this notion likely originated with the observation that tumor endothelial cell populations are much more proliferative than the quiescent endothelium found elsewhere in the body. These rapidly dividing endothelial cells would be targets of chemotherapy in the same way that other proliferative cells such as intestinal epithelia and bone marrow precursors are damaged. The key to maximizing the antiangiogenic potential of chemotherapeutic drugs was considered to be through reduction or elimination of the break period between doses. It is during this break period that repair and repopulation of endothelial cells were likely to occur, as they do for other cell populations.61 However, conventional chemotherapeutic protocols are largely based on the principle of MTD, which originated through models of optimized tumor cell kill that require a break period to allow recovery of normal cell populations.63,64 Despite the success of MTD chemotherapy protocols for the treatment and outright cure of certain cancers, critics of such models point out that the preclinical experiments were performed in exponential growth phase cell culture models, and the exposure time of the cells to chemotherapy was not an included variable.65 More continuous exposure to chemotherapy, with reduction or elimination of the break period between each dose, has come to be known as “metronomic” delivery.66 Not surprisingly, dose reduction is required to provide a more continuous treatment schedule. Generally speaking, metronomic chemotherapy protocols are well tolerated. The low toxicity profile, combined with ease of administration when oral drugs are used, and often lower cost make metronomic chemotherapy protocols appealing in veterinary oncology. However, mechanistic and clinical evaluation in veterinary patients is still at an early stage.
Mechanisms of Treatment
When different types of dividing cells in culture are exposed to continuous ultra-low doses of chemotherapy, endothelial cell populations display an exquisite sensitivity compared to other cell types, including tumor cells in many instances.67,68 Although the reasons for this selectivity are not entirely clear, the explanation likely goes beyond mere targeting of rapidly dividing cells. Experimental evidence has suggested that upregulation of the endogenous angiogenesis inhibitor thrombospondin-1 occurs during treatment with metronomic cyclophosphamide and possibly other agents.69,70 The resulting angiogenic suppression is likely reinforced by a decrease in tumor cell production of proangiogenic growth factors such as VEGF. Decreased VEGF production may be accomplished through reduced tumor cell mass as a result of direct cancer cell killing by chemotherapy. This explains why the antiangiogenic actions of metronomic chemotherapy may be enhanced when there is concomitant tumor cell kill; however, in theory, antiangiogenic targeting of chemotherapy drugs does not require drug sensitivity from the tumor cell population. Targeting the genetically stable endothelial component of the tumor microenvironment instead of the genetically unstable tumor cell population was promoted early on as a potential advantage of antiangiogenic treatment strategies.71
Other components of the tumor microenvironment may also be targeted by metronomic delivery of certain cytotoxic chemotherapy drugs. As mentioned earlier, tumor angiogenesis may involve recruitment of CEPs.17 These cells, if they play a significant role in the growth of tumor blood vessels, may be most influential after the acute damage that would occur in the early break period of MTD chemotherapy schedules.72 Preclinical models have demonstrated that CEPs acutely decrease with MTD treatment, only to rapidly rebound during the break period, when they may contribute to endothelial cell repopulation.73 In contrast, metronomic chemotherapy delivery does not appear to be associated with this CEP surge, leading instead to a sustained antiangiogenic effect.73-75
Other cells also compose a significant component of the tumor microenvironment. Immune effector cells such as lymphocytes and macrophages influence tumor biology and may be affected by chemotherapy treatment. As such, the potential immunomodulatory antitumor mechanisms of metronomic chemotherapy, particularly with the alkylating agent cyclophosphamide, are receiving significant investigation. Low doses of cyclophosphamide influence the T-lymphocyte subset population by decreasing levels of CD4+CD25+ regulatory T-cells.76 Regulatory T-cells (Tregs) inhibit the immune response, thereby suppressing tumor immunosurveillance. In dogs with various malignancies, regulatory T-cells have been quantified by flow cytometry77,78 and have been shown to decrease in blood samples taken from dogs with soft tissue sarcoma treated with low-dose cyclophosphamide chemotherapy.79 Further effects of metronomic cyclophosphamide on tumor immunology remain to be determined, and the nature of potential immunomodulatory mechanisms for other chemotherapeutics delivered metronomically is currently largely unexplored.
Clinical Trial Evaluation
Recent small retrospective and phase II clinical trials have been reported in human and veterinary oncology with a variety of chemotherapy drugs and combinations.80-86 Most clinical trial evaluations to date have paired conventional chemotherapy drugs with noncytotoxic drugs that target angiogenesis directly or indirectly. Preclinical results demonstrated over a decade ago that such combinations resulted in superior outcomes, likely due to more robust neutralization of the prosurvival functions of VEGF in the tumor endothelium.87,88 As a result, most clinical trials have evaluated combination approaches without necessarily any clear prior documentation of single-agent clinical activity for each drug. Bevacizumab has been a popular choice to pair with human metronomic chemotherapy schedules and is FDA-approved for multiple indications.83,89-92 Other, more readily available “targeted” drugs, shown to have at least indirect antiangiogenic effects, are also commonly utilized. Examples include COX inhibitors,81,93-97 tetracyclines, thalidomide,23,98 and others.99 Generally speaking, in contrast to MTD chemotherapy protocols, targeted drugs are often given for longer periods of time at their optimal biologic dose (OBD). As a result, targeted drug combinations with metronomic chemotherapy may be an effective treatment strategy, as each follows a similar dose and scheduling principle. In addition, it is possible that there may be a role for targeted antiangiogenic treatment or metronomic chemotherapy in combination with MTD chemotherapy schedules to neutralize rebound angiogenic responses that occur during the break period. This approach has been demonstrated successfully in preclinical studies.72,100 Ultimately, rigorous clinical trials are necessary to identify and optimize successful drug combinations and schedules of antiangiogenic drugs and chemotherapy.
Veterinary Trials with Cyclophosphamide
Cyclophosphamide has been the most common drug by far to be studied in low-dose metronomic treatment protocols in both human and veterinary oncology. At this time, metronomic chemotherapy protocols have only been evaluated in small retrospective studies and phase I and II veterinary clinical trials (Table 14-3).78,100-104 A protocol of low-dose cyclophosphamide (12.5 to 25 mg/m2 orally daily) alternating with etoposide (50 mg/m2 orally daily) and paired with continuous piroxicam treatment has been evaluated for adjuvant treatment of splenic HSA in dogs.105 The survival time of 9 dogs treated with this regimen was not different from a historic control group of 24 dogs treated with doxorubicin alone. The metronomic protocol was well tolerated over a 6-month period. Pharmacokinetic analysis of etoposide in 3 dogs revealed detectable drug levels. Another report of etoposide administration in dogs demonstrated a low and variable oral bioavailability with this drug.101

Low-dose continuous cyclophosphamide and piroxicam were also studied for the adjuvant treatment of canine incompletely resected soft tissue sarcoma.102 Unlike the HSA study, in this trial the dose of cyclophosphamide was 10 mg/m2 daily or every other day. The disease-free interval was significantly prolonged in the treated group of 30 dogs compared to an age, site, and grade-matched contemporary control group of 55 dogs treated with surgery alone. Again, the protocol was well tolerated, although 40% of dogs experienced mild toxicity at some point of the treatment, and 1 dog experienced grade 4 sterile hemorrhagic cystitis. The every-other-day dosing regimen was better tolerated than daily cyclophosphamide dosing.
Metronomic cyclophosphamide treatment was also recently reported for first-line therapy of metastatic canine tumors of varying histologies.106 Fifteen dogs were treated with daily oral cyclophosphamide at 25 mg/m2 combined with the COX-2 inhibitor celecoxib at 2 mg/kg. One dog had a complete response, and five dogs had stable disease with a treatment protocol that was devoid of toxicity. All dogs were reported to have improved quality of life scores. Most of the tumors treated were carcinomas, and as with many other trials it is not apparent what the relative contribution of each drug may have been to the overall response rate, as single-agent responses to COX inhibitors have been documented in cancer-bearing dogs.58 Interestingly, the authors measured circulating VEGF and IL-6 and observed that pretreatment plasma VEGF levels were significantly lower in responders than nonresponders,106 as has been observed in other clinical trials of metronomic cyclophosphamide.103
Other Alkylating Agents
Chlorambucil has been used in many veterinary treatment protocols, such as first-line treatment of canine chronic lymphocytic leukemia (CLL), low-grade lymphoma, MCTs, and as a cyclophosphamide replacement for cases that develop sterile hemorrhagic cystitis.107 A recent prospective clinical trial of daily single-agent metronomic chlorambucil therapy at a dose of 4 mg/m2 in 36 dogs with tumors of varying histologies and prior therapies revealed a complete response over 35 weeks duration in 3 dogs, a partial response in 1 dog, and stable disease in 17 dogs.104 The median progression-free interval was 61 days, and there were no grade 3 or 4 toxicities noted.
Lomustine (CCNU) has also been evaluated in a metronomic protocol at a dose of 2.84 mg/m2 by mouth, which corresponds to the conversion of a 60 mg/m2 every 3 week dose into a daily schedule.108 A total of 81 dogs with various primary and metastatic tumors were treated with this single-agent regimen for a median of 98 days. Twenty-two dogs had treatment discontinued for adverse events of gastrointestinal, bone marrow, hepatic, or renal origin, although the authors concluded that the treatment was generally well tolerated.
Platinum Compounds
While not designed to be administered in a metronomic fashion per se, the various slow-release and oral formulations of platinum compounds that have been evaluated in veterinary oncology may represent metronomic application of these drugs. Slow-release cisplatin using delivery systems such as the open-cell polylactic acid polymer impregnated with drug (OPLA-Pt) has been studied in a variety of scenarios, including treatment of canine OSA, soft tissue sarcoma, and nasal tumors with desirable efficacy and manageable toxicity.109-111 A dose escalation and pharmacokinetic study of satraplatin, the first orally bioavailable platinum agent, has recently been reported.112 The aim of this study was to determine the MTD; however, the fact that this drug is administered orally makes possible the future investigation of satraplatin in a metronomic-dosing schedule. These studies with platinum compounds raise the general question of whether forms of slow-release chemotherapeutic delivery such as liposome encapsulation represent a form of metronomic drug delivery.
Biomarkers for Antiangiogenic and Metronomic Therapy
The identification and application of biomarkers is becoming increasingly important as the field of oncology moves further into clinical application of targeted therapeutics.113 The aims of validated biomarkers are to (1) predict patient populations that will or will not respond to treatment, (2) monitor response to therapy on a cellular or molecular level, or (3) determine the proper therapeutic dose for targeted agents that often possess optimal biologic activity at doses well below the traditionally defined MTD. The use of biomarkers for antiangiogenic agents is valid in all three categories.
Tumor tissue expression and/or blood-based circulating growth factor levels have been the most popular approaches to predicting response to angiogenesis inhibitors, with VEGF being the most studied molecule.114-116 For example, in the study of metronomic cyclophosphamide and celecoxib by Marchetti and colleagues, low baseline plasma VEGF was predictive of response.106 In many cases it may be through comparison of pretreatment to posttreatment levels that insight will be gained from this surrogate marker, as was demonstrated with urinary bFGF and VEGF with piroxicam treatment of canine TCC.59,60 The most important biomarker in angiogenesis may be one that defines tumor response because inhibition of angiogenesis does not necessarily correspond to a reduction in tumor volume. This static response sits in sharp contrast to MTD chemotherapy approaches that result in measureable tumor shrinkage. Systems for clinical evaluation of tumor response to therapy are often formally based on World Health Organization (WHO) criteria or response evaluation criteria in solid tumors (RECIST), which equate tumor shrinkage with positive outcome. Therefore there may be a need for both recognition of sustained stable disease as a desirable clinical trial endpoint and biomarkers to evaluate angiogenic function during treatment. Imaging modalities that can provide information about vascular function are a potentially valuable tool for monitoring tumor angiogenesis and the effects of antiangiogenic therapy.114 Dynamic contrast-enhanced MRI (DCE-MRI) has been studied for its ability to quantitatively assess vascular parameters.117 A study by MacLeod and colleagues demonstrated blood volume and permeability measurements in canine intracranial masses using this modality.118
The area of dose optimization also represents an unmet need for biomarkers in antiangiogenic and metronomic chemotherapy. Tregs are providing insight in this regard. Recent results from Burton and coworkers demonstrated a dose-dependent reduction in Tregs with metronomic cyclophosphamide treatment, observed at 15 mg/m2 but not 12.5 mg/m2 doses, leading the investigators to conclude that 15 mg/m2 may be a more appropriate dose for future clinical trials.79 In addition to Treg levels, temporal evaluation of other markers has been studied. Assessment of CECs and/or CEPs has been applied for biomarker analysis of dosing antiangiogenic and metronomic therapy.75,97,119 For example, in the study by Rusk and colleagues, decreasing CEC levels in dogs treated with the thrombospondin-1 mimetic peptide ABT-526 may have indicated adequate exposure to the antiangiogenic drug, which in this study was utilized at a single dose.45
Side Effects (Adverse Events)
Despite the therapeutic index achieved by treating activated versus dormant vasculature, there is still potential risk that drug effects on vessels have unwanted consequences. For bevacizumab, potential side effects of a vascular etiology include hypertension, edema, hemorrhage, thromboembolism, proteinuria, intestinal perforation, and impaired wound healing.120 Through further clinical evaluation, we will learn the relative risks for different antiangiogenic drugs applied to a number of tumor settings. Resistance to antiangiogenic therapy has also become evident from early clinical data. Given the genetically stable nature of the blood vessel/endothelial cell target compared to the mutationally prone cancer cell population, antiangiogenic treatment was originally postulated to be less likely to demonstrate drug resistance.71 Proposed mechanisms of resistance to antiangiogenic therapies involve both tumor and host-mediated pathways that can be intrinsic or induced by treatment.121 One alarming consequence of antiangiogenic treatment that has emerged from recent preclinical studies is the potential that certain drugs may alter the host microenvironment, leading to metastatic conditioning.122,123 This phenomenon of increased invasion and metastasis as a result of antiangiogenic drug treatment, despite successful suppression of primary tumor growth, has been demonstrated in preclinical models with certain antiangiogenic RTKIs. The drugs studied include sunitinib, an RTKI with a similar target spectrum to toceranib.123 Further validation and investigation into potential explanations for these provocative results are ongoing.
Finally, it is worth noting that similar to most other drugs, antiangiogenic agents are not innocuous and may have a unique side-effect profile that is independent of their effects on blood vessels or the tumor microenvironment. Side effects for toceranib include dose-dependent gastrointestinal upset, including bleeding, myelosuppression, azotemia, anemia, lethargy, or lameness.124-126 Masitinib’s side effects include gastrointestinal upset, regenerative or nonregenerative anemia, and protein-losing nephropathy.35,124 Cyclophosphamide has become a popular drug for metronomic scheduling but is associated with the potential for sterile hemorrhagic cystitis.127 It is currently unknown whether cystitis is more frequent in metronomic protocols; however, close urinary monitoring is strongly recommended for these patients. Similarly, chlorambucil has been clinically evaluated for metronomic use104 and can be used as an alternative to cyclophosphamide, although it is still not known whether drug efficacy will be similar for different drugs applied across multiple tumor types.
References
1. Potente, M, Gerhardt, H, Carmeliet, P. Basic and therapeutic aspects of angiogenesis. Cell. 2011;146:873–887.
2. Kerbel, RS. Tumor angiogenesis. N Engl J Med. 2008;358:2039–2049.
3. Kerbel, RS. Antiangiogenic therapy: A universal chemosensitization strategy for cancer? Science. 2006;312:1171–1175.
4. Khosravi, SP, Fernandez, PI. Tumoral angiogenesis: review of the literature. Cancer Invest. 2008;26:104–108.
5. Carmeliet, P, Jain, RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307.
6. Motzer, RJ, Hoosen, S, Bello, CL, et al. Sunitinib malate for the treatment of solid tumours: a review of current clinical data. Expert Opin Investig Drugs. 2006;15:553–561.
7. Ivy, SP, Wick, JY, Kaufman, BM. An overview of small-molecule inhibitors of VEGFR signaling. Nat Rev Clin Oncol. 2009;6:569–579.
8. Folkman, J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995;1:27–31.
9. Folkman, J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182–1186.
10. Hanahan, D, Weinberg, RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674.
11. Folkman, J. Endogenous angiogenesis inhibitors. APMIS. 2004;112:496–507.
12. Ferrara, N, Kerbel, RS. Angiogenesis as a therapeutic target. Nature. 2005;438:967–974.
13. Patel-Hett, S, D’Amore, PA. Signal transduction in vasculogenesis and developmental angiogenesis. Int J Dev Biol. 2011;55:353–363.
14. Stewart, KS, Kleinerman, ES. Tumor Vessel Development and Expansion in Ewing’s Sarcoma: A Review of the Vasculogenesis Process and Clinical Trials with Vascular-Targeting Agents. Sarcoma. 2011;2011:165837.
15. Asahara, T, Murohara, T, Sullivan, A, et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275:964–967.
16. Purhonen, S, Palm, J, Rossi, D, et al. Bone marrow-derived circulating endothelial precursors do not contribute to vascular endothelium and are not needed for tumor growth. Proc Natl Acad Sci U S A. 2008;105:6620–6625.
17. Shaked, Y, Ciarrocchi, A, Franco, M, et al. Therapy-induced acute recruitment of circulating endothelial progenitor cells to tumors. Science. 2006;313:1785–1787.
18. Kerbel, RS, Benezra, R, Lyden, DC, et al. Endothelial progenitor cells are cellular hubs essential for neoangiogenesis of certain aggressive adenocarcinomas and metastatic transition but not adenomas. Proc Natl Acad Sci USA. 2008;105:E54.
19. Bertolini, F, Mancuso, P, Braidotti, P, et al. The multiple personality disorder phenotype(s) of circulating endothelial cells in cancer. Biochim Biophys Acta. 2009;1796:27–32.
20. Bertolini, F, Shaked, Y, Mancuso, P, et al. The multifaceted circulating endothelial cell in cancer: towards marker and target identification. Nat Rev Cancer. 2006;6:835–845.
21. Yoder, MC, Ingram, DA. Endothelial progenitor cell: Ongoing controversy for defining these cells and their role in neoangiogenesis in the murine system. Curr Opin Hematol. 2009;16:269–273.
22. Vacca, A, Ribatti, D. Angiogenesis and vasculogenesis in multiple myeloma: role of inflammatory cells. Recent Results Cancer Res. 2011;183:87–95.
23. Suvannasankha, A, Fausel, C, Juliar, BE, et al. Final report of toxicity and efficacy of a phase II study of oral cyclophosphamide, thalidomide, and prednisone for patients with relapsed or refractory multiple myeloma: A Hoosier Oncology Group Trial, HEM01-21. Oncologist. 2007;12:99–106.
24. Tammela, T, Alitalo, K. Lymphangiogenesis: Molecular mechanisms and future promise. Cell. 2010;140:460–476.
25. Adams, RH, Alitalo, K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol. 2007;8:464–478.
26. Holopainen, T, Bry, M, Alitalo, K, et al. Perspectives on lymphangiogenesis and angiogenesis in cancer. J Surg Oncol. 2011;103:484–488.
27. Jain, RK. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science. 2005;307:58–62.
28. Carmeliet, P, Jain, RK. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov. 2011;10:417–427.
29. Goel, S, Duda, DG, Xu, L, et al. Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev. 2011;91:1071–1121.
30. Sorensen, AG, Emblem, KE, Polaskova, P, et al. Increased survival of glioblastoma patients who respond to antiangiogenic therapy with elevated blood perfusion. Cancer Res. 2012;72(2):402–407.
31. Batchelor, TT, Sorensen, AG, di, TE, et al. AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell. 2007;11:83–95.
32. Shaked, Y, Henke, E, Roodhart, JM, et al. Rapid chemotherapy-induced acute endothelial progenitor cell mobilization: implications for antiangiogenic drugs as chemosensitizing agents. Cancer Cell. 2008;14:263–273.
33. Fabian, MA, Biggs, WH, III., Treiber, DK, et al. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat Biotechnol. 2005;23:329–336.
34. London, CA, Malpas, PB, Wood-Follis, SL, et al. Multi-center, placebo-controlled, double-blind, randomized study of oral toceranib phosphate (SU11654), a receptor tyrosine kinase inhibitor, for the treatment of dogs with recurrent (either local or distant) mast cell tumor following surgical excision. Clin Cancer Res. 2009;15:3856–3865.
35. Hahn, KA, Ogilvie, G, Rusk, T, Devauchelle, P, Leblanc, A, Legendre, A, et al. Masitinib is safe and effective for the treatment of canine mast cell tumors. J Vet Intern Med. 2008;22:1301–1309.
36. Pietras, K, Hanahan, D. A multitargeted, metronomic, and maximum-tolerated dose “chemo-switch” regimen is antiangiogenic, producing objective responses and survival benefit in a mouse model of cancer. J Clin Oncol. 2005;23:939–952.
37. Mentlein, R, Forstreuter, F, Mehdorn, HM, et al. Functional significance of vascular endothelial growth factor receptor expression on human glioma cells. J Neurooncol. 2004;67:9–18.
38. Jackson, MW, Roberts, JS, Heckford, SE, et al. A potential autocrine role for vascular endothelial growth factor in prostate cancer. Cancer Res. 2002;62:854–859.
39. Kerbel, RS, Viloria-Petit, A, Klement, G, et al. “Accidental” anti-angiogenic drugs: anti-oncogene directed signal transduction inhibitors and conventional chemotherapeutic agents as examples. Eur J Cancer. 2000;36:1248–1257.
40. Lopez-Ocejo, O, Viloria-Petit, A, Bequet-Romero, M, et al. Oncogenes and tumor angiogenesis: the HPV-16 E6 oncoprotein activates the vascular endothelial growth factor (VEGF) gene promoter in a p53 independent manner. Oncogene. 2000;19:4611–4620.
41. Ebos, JM, Tran, J, Master, Z, et al. Imatinib mesylate (STI-571) reduces Bcr-Abl-mediated vascular endothelial growth factor secretion in chronic myelogenous leukemia. Mol Cancer Res. 2002;1:89–95.
42. du Manoir, JM, Francia, G, Man, S, et al. Strategies for delaying or treating in vivo acquired resistance to trastuzumab in human breast cancer xenografts. Clin Cancer Res. 2006;12:904–916.
43. Viloria-Petit, A, Crombet, T, Jothy, S, et al. Acquired resistance to the antitumor effect of epidermal growth factor receptor-blocking antibodies in vivo: a role for altered tumor angiogenesis. Cancer Res. 2001;61:5090–5101.
44. O’Reilly, MS, Boehm, T, Shing, Y, et al. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell. 1997;88:277–285.
45. Rusk, A, McKeegan, E, Haviv, F, et al. Preclinical evaluation of antiangiogenic thrombospondin-1 peptide mimetics, ABT-526 and ABT-510, in companion dogs with naturally occurring cancers. Clin Cancer Res. 2006;12:7444–7455.
46. Rusk, A, Cozzi, E, Stebbins, M, et al. Cooperative activity of cytotoxic chemotherapy with antiangiogenic thrombospondin-I peptides, ABT-526 in pet dogs with relapsed lymphoma. Clin Cancer Res. 2006;12:7456–7464.
47. Pirie-Shepherd, SR, Coffman, KT, Resnick, D, et al. The role of angiostatin in the spontaneous bone and prostate cancers of pet dogs. Biochem Biophys Res Commun. 2002;292:886–891.
48. Troy, GC, Huckle, WR, Rossmeisl, JH, et al. Endostatin and vascular endothelial growth factor concentrations in healthy dogs, dogs with selected neoplasia, and dogs with nonneoplastic diseases. J Vet Intern Med. 2006;20:144–150.
49. Kamstock, D, Guth, A, Elmslie, R, et al. Liposome-DNA complexes infused intravenously inhibit tumor angiogenesis and elicit antitumor activity in dogs with soft tissue sarcoma. Cancer Gene Ther. 2006;13:306–317.
50. St, CB, Rago, C, Velculescu, V, Traverso, G, Romans, KE, Montgomery, E, et al. Genes expressed in human tumor endothelium. Science. 2000;289:1197–1202.
51. Paoloni, MC, Tandle, A, Mazcko, C, et al. Launching a novel preclinical infrastructure: comparative oncology trials consortium directed therapeutic targeting of TNFalpha to cancer vasculature. PLoS One. 2009;4:e4972.
52. Hua, H, Li, M, Luo, T, et al. Matrix metalloproteinases in tumorigenesis: an evolving paradigm. Cell Mol Life Sci. 2011;68:3853–3868.
53. Hirte, H, Vergote, IB, Jeffrey, JR, et al. A phase III randomized trial of BAY 12-9566 (tanomastat) as maintenance therapy in patients with advanced ovarian cancer responsive to primary surgery and paclitaxel/platinum containing chemotherapy: A National Cancer Institute of Canada Clinical Trials Group Study. Gynecol Oncol. 2006;102:300–308.
54. Moore, MJ, Hamm, J, Dancey, J, et al. Comparison of gemcitabine versus the matrix metalloproteinase inhibitor BAY 12-9566 in patients with advanced or metastatic adenocarcinoma of the pancreas: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2003;21:3296–3302.
55. Moore, AS, Dernell, WS, Ogilvie, GK, et al. Doxorubicin and BAY 12-9566 for the treatment of osteosarcoma in dogs: a randomized, double-blind, placebo-controlled study. J Vet Intern Med. 2007;21:783–790.
56. Mohammed, SI, Khan, KN, Sellers, RS, et al. Expression of cyclooxygenase-1 and 2 in naturally-occurring canine cancer. Prostaglandins Leukot Essent Fatty Acids. 2004;70:479–483.
57. Knapp, DW, Richardson, RC, Chan, TC, et al. Piroxicam therapy in 34 dogs with transitional cell carcinoma of the urinary bladder. J Vet Intern Med. 1994;8:273–278.
58. Knapp, DW, Richardson, RC, Bottoms, GD, et al. Phase I trial of piroxicam in 62 dogs bearing naturally occurring tumors. Cancer Chemother Pharmacol. 1992;29:214–218.
59. Mohammed, SI, Bennett, PF, Craig, BA, et al. Effects of the cyclooxygenase inhibitor, piroxicam, on tumor response, apoptosis, and angiogenesis in a canine model of human invasive urinary bladder cancer. Cancer Res. 2002;62:356–358.
60. Mohammed, SI, Craig, BA, Mutsaers, AJ, et al. Effects of the cyclooxygenase inhibitor, piroxicam, in combination with chemotherapy on tumor response, apoptosis, and angiogenesis in a canine model of human invasive urinary bladder cancer. Mol Cancer Ther. 2003;2:183–188.
61. Kerbel, RS, Kamen, BA. The anti-angiogenic basis of metronomic chemotherapy. Nat Rev Cancer. 2004;4:423–436.
62. Pasquier, E, Kavallaris, M, Andre, N. Metronomic chemotherapy: new rationale for new directions. Nat Rev Clin Oncol. 2010;7:455–465.
63. Skipper, HE, Schabel, FM, Jr., Mellett, LB, et al. Implications of biochemical, cytokinetic, pharmacologic, and toxicologic relationships in the design of optimal therapeutic schedules. Cancer Chemother Rep. 1970;54:431–450.
64. Skipper, HE, Perry, S. Kinetics of normal and leukemic leukocyte populations and relevance to chemotherapy. Cancer Res. 1970;30:1883–1897.
65. Kamen, BA, Glod, J, Cole, PD. Metronomic therapy from a pharmacologist’s view. J Pediatr Hematol Oncol. 2006;28:325–327.
66. Hanahan, D, Bergers, G, Bergsland, E. Less is more, regularly: metronomic dosing of cytotoxic drugs can target tumor angiogenesis in mice. J Clin Invest. 2000;105:1045–1047.
67. Drevs, J, Fakler, J, Eisele, S, et al. Antiangiogenic potency of various chemotherapeutic drugs for metronomic chemotherapy. Anticancer Res. 2004;24:1759–1763.
68. Bocci, G, Nicolaou, KC, Kerbel, RS. Protracted low-dose effects on human endothelial cell proliferation and survival in vitro reveal a selective antiangiogenic window for various chemotherapeutic drugs. Cancer Res. 2002;62:6938–6943.
69. Bocci, G, Francia, G, Man, S, et al. Thrombospondin 1, a mediator of the antiangiogenic effects of low-dose metronomic chemotherapy. Proc Natl Acad Sci U S A. 2003;100:12917–12922.
70. Hamano, Y, Sugimoto, H, Soubasakos, MA, et al. Thrombospondin-1 associated with tumor microenvironment contributes to low-dose cyclophosphamide-mediated endothelial cell apoptosis and tumor growth suppression. Cancer Res. 2004;64:1570–1574.
71. Kerbel, RS. A cancer therapy resistant to resistance. Nature. 1997;390:335–336.
72. Shaked, Y, Kerbel, RS. Antiangiogenic strategies on defense: on the possibility of blocking rebounds by the tumor vasculature after chemotherapy. Cancer Res. 2007;67:7055–7058.
73. Bertolini, F, Paul, S, Mancuso, P, et al. Maximum tolerable dose and low-dose metronomic chemotherapy have opposite effects on the mobilization and viability of circulating endothelial progenitor cells. Cancer Res. 2003;63:4342–4346.
74. Daenen, LG, Shaked, Y, Man, S, et al. Low-dose metronomic cyclophosphamide combined with vascular disrupting therapy induces potent antitumor activity in preclinical human tumor xenograft models. Mol Cancer Ther. 2009;8:2872–2881.
75. Shaked, Y, Emmenegger, U, Man, S, et al. Optimal biologic dose of metronomic chemotherapy regimens is associated with maximum antiangiogenic activity. Blood. 2005;106:3058–3061.
76. Ghiringhelli, F, Menard, C, Puig, PE, et al. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol Immunother. 2007;56:641–648.
77. Biller, BJ, Elmslie, RE, Burnett, RC, et al. Use of FoxP3 expression to identify regulatory T cells in healthy dogs and dogs with cancer. Vet Immunol Immunopathol. 2007;116:69–78.
78. O’Neill, K, Guth, A, Biller, B, et al. Changes in regulatory T cells in dogs with cancer and associations with tumor type. J Vet Intern Med. 2009;23:875–881.
79. Burton, JH, Mitchell, L, Thamm, DH, et al. Low-dose cyclophosphamide selectively decreases regulatory T cells and inhibits angiogenesis in dogs with soft tissue sarcoma. J Vet Intern Med. 2011;25:920–926.
80. Sanborn, SL, Cooney, MM, Dowlati, A, et al. Phase I trial of docetaxel and thalidomide: a regimen based on metronomic therapeutic principles. Invest New Drugs. 2008;26:355–362.
81. Bhatt, RS, Merchan, J, Parker, R, et al. A phase 2 pilot trial of low-dose, continuous infusion, or “metronomic” paclitaxel and oral celecoxib in patients with metastatic melanoma. Cancer. 2010;116:1751–1756.
82. Wong, NS, Buckman, RA, Clemons, M, et al. Phase I/II trial of metronomic chemotherapy with daily dalteparin and cyclophosphamide, twice-weekly methotrexate, and daily prednisone as therapy for metastatic breast cancer using vascular endothelial growth factor and soluble vascular endothelial growth factor receptor levels as markers of response. J Clin Oncol. 2010;28:723–730.
83. Garcia, AA, Hirte, H, Fleming, G, et al. Phase II clinical trial of bevacizumab and low-dose metronomic oral cyclophosphamide in recurrent ovarian cancer: a trial of the California, Chicago, and Princess Margaret Hospital phase II consortia. J Clin Oncol. 2008;26:76–82.
84. Gorn, M, Habermann, CR, Anige, M, et al. A pilot study of docetaxel and trofosfamide as second-line ‘metronomic’ chemotherapy in the treatment of metastatic non-small cell lung cancer (NSCLC). Onkologie. 2008;31:185–189.
85. Bottini, A, Generali, D, Brizzi, MP, et al. Randomized phase II trial of letrozole and letrozole plus low-dose metronomic oral cyclophosphamide as primary systemic treatment in elderly breast cancer patients. J Clin Oncol. 2006;24:3623–3628.
86. Kieran, MW, Turner, CD, Rubin, JB, et al. A feasibility trial of antiangiogenic (metronomic) chemotherapy in pediatric patients with recurrent or progressive cancer. J Pediatr Hematol Oncol. 2005;27:573–581.
87. Klement, G, Huang, P, Mayer, B, et al. Differences in therapeutic indexes of combination metronomic chemotherapy and an anti-VEGFR-2 antibody in multidrug-resistant human breast cancer xenografts. Clin Cancer Res. 2002;8:221–232.
88. Browder, T, Butterfield, CE, Kraling, BM, et al. Antiangiogenic scheduling of chemotherapy improves efficacy against experimental drug-resistant cancer. Cancer Res. 2000;60:1878–1886.
89. Reardon, DA, Desjardins, A, Vredenburgh, JJ, et al. Metronomic chemotherapy with daily, oral etoposide plus bevacizumab for recurrent malignant glioma: a phase II study. Br J Cancer. 2009;101(12):1986–1994.
90. Dellapasqua, S, Bertolini, F, Bagnardi, V, et al. Metronomic cyclophosphamide and capecitabine combined with bevacizumab in advanced breast cancer. J Clin Oncol. 2008;26:4899–4905.
91. Garcia-Saenz, JA, Martin, M, Calles, A, et al. Bevacizumab in combination with metronomic chemotherapy in patients with anthracycline- and taxane-refractory breast cancer. J Chemother. 2008;20:632–639.
92. Jurado, JM, Sanchez, A, Pajares, B, et al. Combined oral cyclophosphamide and bevacizumab in heavily pre-treated ovarian cancer. Clin Transl Oncol. 2008;10:583–586.
93. Buckstein, R, Kerbel, RS, Shaked, Y, et al. High-Dose celecoxib and metronomic “low-dose” cyclophosphamide is an effective and safe therapy in patients with relapsed and refractory aggressive histology non-Hodgkin’s lymphoma. Clin Cancer Res. 2006;12:5190–5198.
94. Andre, N, Rome, A, Coze, C, et al. Metronomic etoposide/cyclophosphamide/celecoxib regimen given to children and adolescents with refractory cancer: a preliminary monocentric study. Clin Ther. 2008;30:1336–1340.
95. Krzyzanowska, MK, Tannock, IF, Lockwood, G, et al. A phase II trial of continuous low-dose oral cyclophosphamide and celecoxib in patients with renal cell carcinoma. Cancer Chemother Pharmacol. 2007;60:135–141.
96. Stempak, D, Gammon, J, Halton, J, et al. A pilot pharmacokinetic and antiangiogenic biomarker study of celecoxib and low-dose metronomic vinblastine or cyclophosphamide in pediatric recurrent solid tumors. J Pediatr Hematol Oncol. 2006;28:720–728.
97. Twardowski, PW, Smith-Powell, L, Carroll, M, et al. Biologic markers of angiogenesis: circulating endothelial cells in patients with advanced malignancies treated on phase I protocol with metronomic chemotherapy and celecoxib. Cancer Invest. 2008;26:53–59.
98. D’Amato, RJ, Loughnan, MS, Flynn, E, et al. Thalidomide is an inhibitor of angiogenesis. Proc Natl Acad Sci U S A. 1994;91:4082–4085.
99. Hafner, C, Reichle, A, Vogt, T. New indications for established drugs: combined tumor-stroma-targeted cancer therapy with PPARgamma agonists, COX-2 inhibitors, mTOR antagonists and metronomic chemotherapy. Curr Cancer Drug Targets. 2005;5:393–419.
100. Shaked, Y, Emmenegger, U, Francia, G, et al. Low-dose metronomic combined with intermittent bolus-dose cyclophosphamide is an effective long-term chemotherapy treatment strategy. Cancer Res. 2005;65:7045–7051.
101. Flory, AB, Rassnick, KM, Balkman, CE, et al. Oral bioavailability of etoposide after administration of a single dose to tumor-bearing dogs. Am J Vet Res. 2008;69:1316–1322.
102. Elmslie, RE, Glawe, P, Dow, SW. Metronomic therapy with cyclophosphamide and piroxicam effectively delays tumor recurrence in dogs with incompletely resected soft tissue sarcomas. J Vet Intern Med. 2008;22:1373–1379.
103. Calleri, A, Bono, A, Bagnardi, V, et al. Predictive potential of angiogenic growth factors and circulating endothelial cells in breast cancer patients receiving metronomic chemotherapy plus bevacizumab. Clin Cancer Res. 2009;15:7652–7657.
104. Leach, TN, Childress, MO, Greene, SN, et al. Prospective trial of metronomic chlorambucil chemotherapy in dogs with naturally occurring cancer. Vet Comp Oncol. 2011;10(2):102–112.
105. Lana, S, U’ren, L, Plaza, S, et al. Continuous low-dose oral chemotherapy for adjuvant therapy of splenic hemangiosarcoma in dogs. J Vet Intern Med. 2007;21:764–769.
106. Marchetti, V, Giorgi, M, Fioravanti, A, et al. First-line metronomic chemotherapy in a metastatic model of spontaneous canine tumours: a pilot study. Invest New Drugs. 2011.
107. Taylor, F, Gear, R, Hoather, T, et al. Chlorambucil and prednisolone chemotherapy for dogs with inoperable mast cell tumours: 21 cases. J Small Anim Pract. 2009;50:284–289.
108. Tripp, CD, Fidel, J, Anderson, CL, et al. Tolerability of metronomic administration of lomustine in dogs with cancer. J Vet Intern Med. 2011;25:278–284.
109. Withrow, SJ, Liptak, JM, Straw, RC, et al. Biodegradable cisplatin polymer in limb-sparing surgery for canine osteosarcoma. Ann Surg Oncol. 2004;11:705–713.
110. Lana, SE, Dernell, WS, Lafferty, MH, et al. Use of radiation and a slow-release cisplatin formulation for treatment of canine nasal tumors. Vet Radiol Ultrasound. 2004;45:577–581.
111. Havlicek, M, Straw, RS, Langova, V, et al. Intra-operative cisplatin for the treatment of canine extremity soft tissue sarcomas. Vet Comp Oncol. 2009;7(2):122–129.
112. Selting, KA, Wang, X, Gustafson, DL, et al. Evaluation of satraplatin in dogs with spontaneously occurring malignant tumors. J Vet Intern Med. 2011;25:909–915.
113. Park, JW, Kerbel, RS, Kelloff, GJ, et al. Rationale for biomarkers and surrogate end points in mechanism-driven oncology drug development. Clin Cancer Res. 2004;10:3885–3896.
114. Drevs, J, Schneider, V. The use of vascular biomarkers and imaging studies in the early clinical development of anti-tumour agents targeting angiogenesis. J Intern Med. 2006;260:517–529.
115. Sandri, MT, Johansson, HA, Zorzino, L, et al. Serum EGFR and serum HER-2/neu are useful predictive and prognostic markers in metastatic breast cancer patients treated with metronomic chemotherapy. Cancer. 2007;110:509–517.
116. Lindauer, A, Di, GP, Kanefendt, F, et al. Pharmacokinetic/Pharmacodynamic modeling of biomarker response to sunitinib in healthy volunteers. Clin Pharmacol Ther. 2010;87:601–608.
117. Morgan, B, Thomas, AL, Drevs, J, et al. Dynamic contrast-enhanced magnetic resonance imaging as a biomarker for the pharmacological response of PTK787/ZK 222584, an inhibitor of the vascular endothelial growth factor receptor tyrosine kinases, in patients with advanced colorectal cancer and liver metastases: results from two phase I studies. J Clin Oncol. 2003;21:3955–3964.
118. MacLeod, AG, Dickinson, PJ, LeCouteur, RA, et al. Quantitative assessment of blood volume and permeability in cerebral mass lesions using dynamic contrast-enhanced computed tomography in the dog. Acad Radiol. 2009;16:1187–1195.
119. Bertolini, F, Mancuso, P, Shaked, Y, et al. Molecular and cellular biomarkers for angiogenesis in clinical oncology. Drug Discov Today. 2007;12:806–812.
120. Shih, T, Lindley, C. Bevacizumab: an angiogenesis inhibitor for the treatment of solid malignancies. Clin Ther. 2006;28:1779–1802.
121. Ebos, JM, Lee, CR, Kerbel, RS. Tumor and host-mediated pathways of resistance and disease progression in response to antiangiogenic therapy. Clin Cancer Res. 2009;15:5020–5025.
122. Paez-Ribes, M, Allen, E, Hudock, J, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15:220–231.
123. Ebos, JM, Lee, CR, Cruz-Munoz, W, et al. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell. 2009;15:232–239.
124. London, CA. Tyrosine kinase inhibitors in veterinary medicine. Top Companion Anim Med. 2009;24:106–112.
125. London, CA, Hannah, AL, Zadovoskaya, R, et al. Phase I dose-escalating study of SU11654, a small molecule receptor tyrosine kinase inhibitor, in dogs with spontaneous malignancies. Clin Cancer Res. 2003;9:2755–2768.
126. London, CA, Malpas, PB, Wood-Follis, SL, et al. Multi-center, placebo-controlled, double-blind, randomized study of oral toceranib phosphate (SU11654), a receptor tyrosine kinase inhibitor, for the treatment of dogs with recurrent (either local or distant) mast cell tumor following surgical excision. Clin Cancer Res. 2009;15:3856–3865.
127. Charney, SC, Bergman, PJ, Hohenhaus, AE, et al. Risk factors for sterile hemorrhagic cystitis in dogs with lymphoma receiving cyclophosphamide with or without concurrent administration of furosemide: 216 cases (1990-1996). J Am Vet Med Assoc. 2003;222:1388–1393.
Section D
Novel and Emerging Therapeutic Targets
Douglas H. Thamm and David J. Argyle
The recent explosion in available cancer bioinformatics, rational and combinatorial drug design, and high-throughput drug screening has resulted in a massive increase in potential therapeutic targets and anticancer treatment strategies. An exhaustive survey of all potential novel targets for cancer therapy would be impossible; thus this review is designed to present a brief overview of some of the more promising and well-developed “druggable” targets that have been discovered recently and, when applicable, their potential application to veterinary oncology.
DNA Methylation
In addition to the information encoded within the genome sequence, it has been shown that epigenetic changes are of great importance in the modification and maintenance of gene expression. These changes take place through a number of mechanisms, including polymerase enzyme modulation, chromatin condensation, and DNA methylation. Mammalian DNA is methylated at cytosines within cytosine-guanine (CpG) dinucleotide sequences. During tissue differentiation, the methylation pattern is one governor of tissue-specific gene expression and thus phenotype.1-3
Two different methylation-related phenomena have been identified in cancer. First, tumor cell DNA has been shown in both dogs and other mammals to be globally hypomethylated,4,5 specifically in pericentromeric satellite sequences. This may lead to decreased genome stability and an increase in the incidence of oncogenic chromosome defects. Indeed, the purposeful induction of genomic hypomethylation by reduction in germline DNA methyltransferase-1 (DNMT1) levels in genetically engineered mice is associated with a high incidence of T-cell lymphomas displaying trisomy 15.6 Second, cancer cells also acquire sequence-specific promoter hypermethylation and transcriptional repression in normally unmethylated regions, several of which have been shown to be associated with known tumor suppressor genes, including Rb, p16, p73, and the von Hippel-Lindau protein (VHL),1,2,7-9 or other important tumor-associated genes such as E-cadherin and estrogen and retinoic acid receptors.10
The methylation of DNA is controlled by four known DNMTs, of which DNMT1 may be the most important in cancer.1-3 A variety of agents can inhibit DNMT function. The two best studied are 5-azacytidine (Vidaza) and 5-aza-deoxycitidine (decitabine, Dacogen), which are nucleoside analogs that incorporate into DNA and inhibit DNMT activity but allow replication to proceed. A large number of single-agent clinical trials with these agents have been reported, and significant activity has been demonstrated in hematopoietic neoplasia, leading to the Food and Drug Administration (FDA) approval of 5-azacytidine and decitabine for the treatment of myelodysplastic syndrome.11,12 Encouraging response rates to the nucleoside analog decitabine have also been seen in human patients with imatinib-refractory CML.13,14 Results in advanced solid tumors have been generally disappointing15,16; however, studies in combination with standard antineoplastic therapy and other targeted agents (e.g., histone deacetylase inhibitors) are ongoing.17 One drawback of these drugs is the requirement of DNA replication for activity. There are various other drugs that have DNMT inhibitory activity in nonreplicating cells, including green tea polyphenols1 and marine products of the psammaplin class,18 a synthetic derivative of which concurrently inhibits histone deacetylase (see later) and is in human clinical trials.19 Interestingly, the commonly used cardiac medications procainamide and hydralazine also possess demethylating activity,10,20 and clinical trials have demonstrated alterations in promoter methylation and reactivation of silenced genes following administration of well-tolerated doses of hydralazine to human cervical cancer patients.21 Hydralazine-valproic acid combinations have demonstrated activity in myelodysplastic syndrome in early human trials.22 Procainamide and hydralazine have a long track record of use in veterinary medicine and as such could serve as interesting and readily available drugs for the initial evaluation of methylation inhibition in veterinary cancer patients.
Two potential problems exist regarding the wide clinical implementation of DNMT inhibitors for cancer treatment. As discussed previously, induction of long-term genome-wide hypomethylation could decrease chromosome stability leading to potentially tumorigenic chromosome rearrangements.3 Demethylation could also trigger the reactivation of genes promoting a more aggressive or metastatic phenotype.3 In support of this theory, treatment of nonmetastatic breast cancer cells with 5-azacytidine was shown to upregulate expression of urokinase-like plasminogen activator, an enzyme important in tumor invasion and metastasis, leading to enhanced metastatic potential.23
Histone Deacetylase
Another critical determinant of gene expression is the condensation of chromatin in the form of heterochromatin, which results in transcriptional silencing and enhanced genome stability. This is accomplished by a number of pathways, one of which is the acetylation and deacetylation of histones, controlled by histone acetyltransferases and histone deacetylases (HDACs).24 The HDACs specifically maintain chromatin in a condensed form, and can associate with specific transcription factors resulting in transcription repression. The acetylation of histones may be key in regulating the expression of genes associated with cellular proliferation, differentiation, and survival, both in development and carcinogenesis.25,26 Studies suggest that histone acetylation reduces electrostatic charge interactions between histones, leading to chromatin decondensation, and more recent work has suggested that HDAC enzymes’ effects may also be indirect through modulation of other proteins important in chromatin condensation, including DNMT1.27 Induction of HDAC expression, leading to transcriptional repression, is a common feature in human cancers such as colon cancer28 and negatively regulates the expression of several tumor suppressor genes, including p53 and VHL.29 Differential expression of certain HDAC isoforms has been associated with outcome in a variety of human tumors, with HDAC2 and 6 studied most completely.30
Pharmacologic inhibition of HDAC can affect multiple facets of the malignant phenotype. HDAC inhibition inhibits colon carcinogenesis in the adenomatosis polyposis coli (APC) mouse model.28 Angiogenesis can be inhibited through upregulation of VHL and subsequent inhibition of hypoxia-inducible factor-1α (HIF-1α) function and VEGF production29,31,32; decreased expression of other proangiogenic factors such as bFGF, angiopoietin-2, and Tie231,32; inhibition of endothelial nitric oxide (NO) synthase and endothelial cell proliferation and tube formation33,34; and inhibition of the commitment of endothelial progenitor cells to the endothelial lineage.35 Inhibition of HDAC can enhance apoptosis in tumor and endothelial cells32,36-38 and directly inhibit tumor cell proliferation.37,39-41 Consistent with its role as a transcription repressor, inhibition of HDAC has been shown to induce differentiation in thyroid and prostate cancers, neuroblastoma, and the leukemias.42-46 HDAC inhibition dramatically enhances the in vitro and in vivo efficacy of multiple standard cytotoxic therapies, as well as novel antibodies and small molecules.27,32,38,47-54
Two HDAC inhibitors, vorinostat (suberoylanilide hydroxamic acid [SAHA], Zolinza) and romidepsin (FK228, Istodax), have recently been approved by the FDA for the treatment of cutaneous T-cell lymphoma (CTCL),55-57 and a large number of additional agents are in intermediate to late-stage clinical trials. A number of studies are being conducted evaluating HDAC inhibitors for various other hematologic and solid tumors, and early studies combining these drugs with other targeted and cytotoxic therapies have been reported.58-61
A recent in vitro study demonstrated potent induction of histone acetylation, growth inhibition, and induction of apoptosis in a variety of canine tumor cell lines treated with either vorinostat or the novel HDAC inhibitor OSU-HDAC42 (AR42).62 The commercially available anticonvulsant drug valproic acid (VPA) can function as an HDAC inhibitor and is capable of inhibiting tumor cell invasion, P-glycoprotein expression, proliferation, and angiogenesis and enhancing chemosensitivity.24,27,43 VPA was recently shown to enhance the antiproliferative and proapoptotic effects of doxorubicin in canine OSA cell lines in vitro and to synergize with doxorubicin in a canine OSA xenograft.53
There is a single case report describing administration of vorinostat to a dog with microscopic HSA.63 Although apparently well tolerated, the dose was empirically chosen, there was no pharmacokinetic analysis or demonstration of target inhibition, and the drug’s impact on the course of the disease could not be unequivocally determined. A recent study demonstrated that VPA could be administered prior to a standard dose of doxorubicin in tumor-bearing dogs, at dosages sufficient to enhance histone acetylation in tumor and peripheral blood mononuclear cells, but was not associated with toxicity or apparent potentiation of doxorubicin’s adverse effects.54
The Proteasome
The abundance of cellular proteins is tightly controlled at the levels of both production and destruction. Protein production can be modified at the transcriptional and posttranscriptional level, and therapies based on these approaches are relatively abundant. Until recently, relatively little attention has been paid to the manipulation of protein degradation as a therapeutic modality. The ubiquitin-proteasome pathway (UPP) is responsible for the degradation of the majority of intracellular proteins and for the regulation of many proteins with key roles in cancer.
The 26S proteasome is a large multiprotein complex containing ubiquitin recognition domains, which bind ubiquitinated proteins tagged for degradation, and proteolytic domains with trypsin-like, chymotrypsin-like, and caspase-like activity, which degrade proteins into short polypeptide sequences.64 It is responsible for the degradation of a variety of proteins responsible for cell-cycle regulation, angiogenesis, apoptosis, and chemotherapy and radiation sensitivity (Table 14-4).64-66
Table 14-4
Molecular Targets and Consequences of Proteasome Inhibition
| Process | Proteins Degraded by Proteasome | Cellular Consequences |
| Nuclear factor-κB (NF-κB) activation | IκB | Accumulation of IκB inhibits nuclear translocation and activity of NF-κB, leading to decreased proliferation, survival, invasion, angiogenesis |
| Apoptosis | p53, Bax, tBid, Smac, JNK, Noxa | Accumulation of these proteins directly or indirectly promotes apoptosis through various pathways. |
| Cell cycle regulation | p21, p27, other cyclin-dependent kinase (CDK) inhibitors, cyclins, p53 | Accumulation of CDK inhibitors can cause cell-cycle arrest and apoptosis. The dysregulated elevation of multiple cyclins can send contradictory signals to the cell resulting in apoptosis. |
| Signal transduction | Mitogen-activated protein (MAP) kinase (MKP-1) phosphatase | Accumulation dephosphorylates p44/42 MAP kinase, leading to decreased MAP kinase pathway signaling, proliferation, survival, ± angiogenesis. |
| Oncogenic transformation | c-Fos, c-jun, c-myc, N-myc | Unclear how overabundance of these proteins exerts an antitumor effect. |
| Unfolded protein response | Various damaged/misfolded proteins | Accumulation of damaged proteins leads to endoplasmic reticulum stress and apoptosis. |
| Chemo/radiation sensitivity | IκB, P-glycoprotein, topoisomerase IIα, DNA damage repair enzyme downregulation | NF-κB is induced in response to DNA damage; normal proteasome function is required for correct folding of P-glycoprotein; downregulation of topoisomerase IIα may reduce sensitivity to doxorubicin. |
Adapted from Adams J: The development of proteasome inhibitors as anticancer drugs, Cancer Cell 5:417–421, 2004; Rajkumar SV, Richardson PG, Hideshima T, et al: Proteasome inhibition as a novel therapeutic target in human cancer, J Clin Oncol 23:630–639, 2005; and Voorhees PM, Dees EC, O’Neil B, et al: The proteasome as a target for cancer therapy, Clin Cancer Res 9:6316–6325, 2003.
Tumor cells are generally more sensitive to the effects of proteasome inhibition than are normal cells. Various studies have demonstrated a threefold to fortyfold increase in susceptibility to proteasome inhibitor–associated apoptosis when comparing tumor cells to corresponding normal tissues.66-70 The mechanisms for this differential sensitivity are unclear, but proliferating cells generally appear more sensitive than do quiescent cells.64,66 Additionally, dysregulation of UPP function appears to occur in many cancer cells, thus potentially rendering them more sensitive to inhibition.66,71 Recent studies implicate profound upregulation of the proapoptotic factor Noxa as being a key mediator of proteasome inhibitor–induced tumor cell apoptosis.72,73
Although a number of chemicals appear capable of proteasome inhibition in vitro, only the boronic acid derivatives appear suitable for clinical use and only one drug, bortezomib (Velcade, PS-341), has received FDA approval for the treatment of multiple myeloma.74-77 Several additional proteasome inhibitors are in clinical development. Meaningful antitumor activity has been observed in patients with other hematopoietic neoplasms,78-80 but less activity has been seen in solid tumors to date.81-84 Although toxicology studies with bortezomib have been performed in dogs,85 there is no information available to date regarding the safety or biologic effect of proteasome inhibitors in veterinary patients or tumor cells.
Heat Shock Protein 90
Given the complex nature of cancer and the multiple pathways that can be subjugated to contribute to the malignant phenotype, an optimal cancer drug might target a variety of oncogenic pathways simultaneously. One molecular target that has the potential to interrupt a wide variety of pathways important in cancer is heat shock protein 90 (HSP90), a molecular chaperone responsible for the conformational maturation of many proteins involved in diverse oncogenic activities such as cell adhesion/migration/invasion, signal transduction, cell cycle progression, angiogenesis, and survival (Table 14-5).86-111 HSP90 and other chaperones are responsible for ensuring the correct folding and prevention of aggregation of their client proteins.112 Misfolding and aggregation of proteins lead to ubiquitination and proteasomal destruction, resulting in proteins with diminished function and greatly shortened half-lives.113 Although several classes of compound are capable of inhibiting HSP90 chaperone function,97,98,103 the best studied are ansamycin antibiotics of the geldanamycin class.
Table 14-5
Molecules Targeted by Heat Shock Protein 90 (HSP90) Inhibition
| Process | Targets | References |
| Invasion and migration | Urokinase-like plasminogen activator,* focal adhesion kinase (FAK) phosphorylation | 86-88 |
| Cell cycle progression | Cyclin D3, cyclin-dependent kinase 4 (CDK4) | 89 |
| Signal transduction | AKT, KIT, RAF-1, EGFR, HER2, Jun, Lyn, Src, IGF-1R, PDGFR, Met, Bcr-Abl, ILK, androgen receptor, progesterone receptor, glucocorticoid receptor | 89-96 |
| Hypoxic response/angiogenesis | HIF-1, VEGF, Glut-1, nitric oxide synthase | 90, 97-102 |
| Antiapoptosis | Wild-type and mutant p53, survivin | 103-109 |
| Cell senescence | Telomerase | 110, 111 |
EGFR, Epidermal growth factor receptor; HIF-1, hypoxia-inducible factor-1; IGF-1R, insulin-like growth factor receptor 1; ILK, integrin-linked kinase; PDGFR, platelet-derived growth factor receptor; VEGF, vascular endothelial growth factor.
*Urokinase-like plasminogen activator activity appears to be inhibited by geldanamycin class drugs through a mechanism other than heat shock protein 90 (HSP90) inhibition.
Many HSP90 inhibitors appear to demonstrate significant preferential activity against malignant versus normal somatic cells. The HSP90 derived from most tumor cells has a binding affinity for 17-allylaminogeldanamycin (17-AAG) approximately 100-fold higher than HSP90 derived from normal cells. This may occur as a result of the overaccumulation of mutated, misfolded, and overexpressed signaling proteins in tumor cells, leading to increased HSP90 chaperone activity and a greater proportion of the molecule in the bound, active, and 17-AAG–sensitive state.114
Tumor cells display considerable variation in sensitivities to HSP90 inhibition. Although the mechanisms underlying this differential sensitivity are incompletely characterized, some important characteristics include reliance on certain kinase cascades, expression of apoptotic and cell-cycle regulators, and p-glycoprotein expression.90
Many RTKs targeted by the geldanamycins may have an important role in canine and feline tumors. For example, they are capable of inhibiting the function of mutant and wild-type KIT,91 important in canine mast cell neoplasia115; Met,92 expressed in multiple canine tumor types116,117; PDGFR,118 expressed in FVAS and OSA119,120; and IGF-1R,91 which is expressed and functional in canine OSA and melanoma.121-123 The geldanamycins are likewise able to attenuate the function of the HIF-1α protein, a key transcription factor responsible for sensing and responding to hypoxia and activating the angiogenic switch.97,99,100 They are additionally able to deplete key antiapoptotic proteins such as mutant p53 and survivin,104-107 contributing to enhanced in vitro sensitivity to standard cytotoxic therapies such as radiation and chemotherapy when used in combination.93,94,101,124-126
Under certain circumstances, HSP90 inhibitors may have undesirable effects from the standpoint of cancer therapy. For example, 17-AAG has been shown to protect colon carcinoma cells from cisplatin-mediated toxicity,95 whereas it has additive or synergistic activity when combined with cisplatin against human neuroblastoma and OSA cells.124 Additionally, 17-AAG inhibited primary tumor formation, although it potentiated bone-specific mammary carcinoma metastasis by enhancing osteoclastogenesis in one murine model.127
The impressive preclinical data generated with compounds such as HSP90 inhibitors have led to phase I human clinical trials of multiple agents,128-132 including some early combinatorial studies.130-132 Evidence of biologic effect in the form of upregulation of HSP70 chaperone expression in peripheral blood cells has been observed. There is in vitro evidence of antitumor activity of HSP90 inhibitors in canine OSA and mast cell tumor cell lines133,134; however, no clinical evaluation of these agents has been reported to date.
Poly Adenosine Diphosphate Ribose Polymerase and Poly Adenosine Diphosphate Ribose Glycohydrolase
Poly adenosine diphosphate (ADP)-ribose polymerase (PARP) is a “nick-sensor” that signals the presence of DNA damage and facilitates DNA repair.135 The first PARP enzyme was discovered by Chambon et al and is now recognized as a superfamily of 18 members,136 although only PARP-1 and PARP-2 are known to act in DNA damage.137 The PARP family is also involved in the regulation of several transcription factors such as NFκB in modulating the expression of chemokines, adhesion molecules, inflammatory cytokines, and mediators.135 Poly ADP-ribose glycohydrolase (PARG) is the main enzyme in catabolizing poly ADP-ribose to ADP-ribose. To date, only one single PARG gene has been detected in mammals, encoding for three complementary DNAs (cDNA), which generate three isoforms.135
PARP has multiple intracellular functions, including signaling DNA damage, and recognizing and binding to DNA strand breaks generated by DNA-damaging agents (cytotoxic drugs and ionizing radiation).138 Activation of PARP is one of the earliest DNA damage responses. PARP is also a modulator of DNA base excision repair (BER), which constitutes a major mechanism for genomic stability. There is increasing evidence demonstrating that both PARP and PARG repair DNA.136 When PARP binds to DNA strand breaks, it activates an enzyme causing shuttling of PARP and, subsequently, opening of the chromatin. PARG enters the nucleus, it moves to the PARP substrate, and DNA strand breaks are repaired. Due to excessive PARG, poly ADP-ribose decreases and thus chromatin reverts back to its original structure.
PARP inhibition has been suggested as an important approach in sensitizing cancer cells to conventional cancer therapy, leading to early clinical trials with PARP inhibitors.138 PARP inhibitors have been shown to be lethal in BRCA-deficient cells due to persistence of DNA lesions that would normally be repaired in a BRCA-dependent fashion,139 suggesting that PARP inhibitors might be an effective monotherapy in these cancers. However, one might expect that their major benefit would be to enhance conventional cytotoxic drug treatment or radiation therapy, and studies have demonstrated that PARP inhibition potentiates the cytotoxicity of anticancer drugs and ionizing radiation through inhibition of DNA repair in cancer cells. PARG inhibition could also be one of the pathways selected for cancer management due to its effects on increased sensitivity to both radiation and chemotherapy.
Although PARG inhibitors have lagged behind PARP inhibitors, a number of molecules have been developed that target these pathways, with varying degrees of specificity. In experimental mouse models, these have shown promise in breast, colon, lung, and brain tumors and in melanoma, either as monotherapy or combined with conventional drugs or radiation.140-144 Only PARP inhibitors have been used in early human clinical trials (phase I). It is too early to pass judgment on these early trials, and the next 5 years will demonstrate whether PARP inhibitors will have a place in the drug arsenal for cancer treatment.
Carbonic Anhydrase
Hypoxia in cancer tissues is emerging as a key negative prognostic marker in cancer survival. Carbonic anhydrase functions to interconvert carbon dioxide and bicarbonate to maintain acid-base balance in tissues and may play important roles in hypoxic states. In humans, there are 13 active isoenzymes of carbonic anhydrase (CA), and the transmembrane isoform CA IX has been linked with carcinogenesis.145 High levels of CA IX expression have been demonstrated in human epithelial tumors such as carcinomas of the cervix, uterus, kidney, lung, esophagus, breast, and colon. Normally, this isoenzyme has restricted expression to the epithelia of the gastrointestinal tract, which is in contrast to studies in cancer where ectopic expression has been demonstrated in hypoxic tumors, participating in tumor cell environment acidosis and contributing to malignant progression and poor treatment outcome.145 Modulation of extracellular tumor pH via inhibition of CA IX activity has been suggested as a promising approach to novel anticancer therapies. Much attention has recently been paid to the CA IX inhibitors’ drug design, and efforts have been made to obtain isozyme IX inhibitors, with putative applications as antitumor drugs or diagnostic agents. A large number of selective CA IX inhibitors have been developed in the past 5 years in the sulfonamide, sulfamide, and sulfamate series.146 Some of these compounds can constitute interesting leads for further development. Furthermore, new classes of prodrug have emerged in the design of new anticancer compounds, which can be activated under hypoxia. As yet, there is little clinical information regarding the efficacy of any of these compounds, but one may anticipate that these drugs could be beneficial in large hypoxic tumors that have failed conventional treatments or as possible sensitizers to radiation damage.145,147
References
1. Brueckner, B, Lyko, F. DNA methyltransferase inhibitors: old and new drugs for an epigenetic cancer therapy. Trends Pharmacol Sci. 2004;25:551–554.
2. Herman, JG, Baylin, SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349:2042–2054.
3. Szyf, M. DNA methylation and cancer therapy. Drug Resist Update. 2003;6:341–353.
4. Pelham, JT, Irwin, PJ, Kay, PH. Genomic hypomethylation in neoplastic cells from dogs with malignant lymphoproliferative disorders. Res Vet Sci. 2003;74:101–104.
5. Ehrlich, M. DNA methylation in cancer: too much, but also too little. Oncogene. 2002;21:5400–5413.
6. Gaudet, F, Hodgson, JG, Eden, A, et al. Induction of tumors in mice by genomic hypomethylation. Science. 2003;300:489–492.
7. Catto, JW, Azzouzi, AR, Rehman, I, et al. Promoter hypermethylation is associated with tumor location, stage, and subsequent progression in transitional cell carcinoma. J Clin Oncol. 2005;23:2903–2910.
8. Baylin, SB, Herman, JG. DNA hypermethylation in tumorigenesis: epigenetics joins genetics. Trends Genet. 2000;16:168–174.
9. van Doorn, R, Zoutman, WH, Dijkman, R, et al. Epigenetic profiling of cutaneous T-cell lymphoma: promoter hypermethylation of multiple tumor suppressor genes including BCL7a, PTPRG, and p73. J Clin Oncol. 2005;23:3886–3896.
10. Segura-Pacheco, B, Trejo-Becerril, C, Perez-Cardenas, E, et al. Reactivation of tumor suppressor genes by the cardiovascular drugs hydralazine and procainamide and their potential use in cancer therapy. Clin Cancer Res. 2003;9:1596–1603.
11. Silverman, LR, Demakos, EP, Peterson, BL, et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J Clin Oncol. 2002;20:2429–2440.
12. Kantarjian, H, Issa, JP, Rosenfeld, CS, et al. Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study. Cancer. 2006;106:1794–1803.
13. Issa, JP, Gharibyan, V, Cortes, J, et al. Phase II study of low-dose decitabine in patients with chronic myelogenous leukemia resistant to imatinib mesylate. J Clin Oncol. 2005;23:3948–3956.
14. Cashen, AF, Schiller, GJ, O’Donnell, MR, et al. Multicenter, phase II study of decitabine for the first-line treatment of older patients with acute myeloid leukemia. J Clin Oncol. 2010;28:556–561.
15. Aparicio, A, Eads, CA, Leong, LA, et al. Phase I trial of continuous infusion 5-aza-2’-deoxycytidine. Cancer Chemother Pharmacol. 2003;51:231–239.
16. Momparler, RL, Bouffard, DY, Momparler, LF, et al. Pilot phase I-II study on 5-aza-2’-deoxycytidine (Decitabine) in patients with metastatic lung cancer. Anticancer Drugs. 1997;8:358–368.
17. Pohlmann, P, DiLeone, LP, Cancella, AI, et al. Phase II trial of cisplatin plus decitabine, a new DNA hypomethylating agent, in patients with advanced squamous cell carcinoma of the cervix. Am J Clin Oncol. 2002;25:496–501.
18. Pina, IC, Gautschi, JT, Wang, GY, et al. Psammaplins from the sponge Pseudoceratina purpurea: inhibition of both histone deacetylase and DNA methyltransferase. J Org Chem. 2003;68:3866–3873.
19. Remiszewski, SW. The discovery of NVP-LAQ824: from concept to clinic. Curr Med Chem. 2003;10:2393–2402.
20. Villar-Garea, A, Fraga, MF, Espada, J, et al. Procaine is a DNA-demethylating agent with growth-inhibitory effects in human cancer cells. Cancer Res. 2003;63:4984–4989.
21. Zambrano, P, Segura-Pacheco, B, Perez-Cardenas, E, et al. A phase I study of hydralazine to demethylate and reactivate the expression of tumor suppressor genes. BMC Cancer. 2005;5:44.
22. Candelaria, M, Herrera, A, Labardini, J, et al. Hydralazine and magnesium valproate as epigenetic treatment for myelodysplastic syndrome. Preliminary results of a phase-II trial. Ann Hematol. 2011;90:379–387.
23. Guo, Y, Pakneshan, P, Gladu, J, et al. Regulation of DNA methylation in human breast cancer. Effect on the urokinase-type plasminogen activator gene production and tumor invasion. J Biol Chem. 2002;277:41571–41579.
24. Blaheta, RA, Michaelis, M, Driever, PH, et al. Evolving anticancer drug valproic acid: insights into the mechanism and clinical studies. Med Res Rev. 2005;25:383–397.
25. Berger, SL. Histone modifications in transcriptional regulation. Curr Opin Genet Dev. 2002;12:142–148.
26. Jenuwein, T, Allis, CD. Translating the histone code. Science. 2001;293:1074–1080.
27. Marchion, DC, Bicaku, E, Daud, AI, et al. Valproic acid alters chromatin structure by regulation of chromatin modulation proteins. Cancer Res. 2005;65:3815–3822.
28. Zhu, P, Martin, E, Mengwasser, J, et al. Induction of HDAC2 expression upon loss of APC in colorectal tumorigenesis. Cancer Cell. 2004;5:455–463.
29. Kim, MS, Kwon, HJ, Lee, YM, et al. Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nat Med. 2001;7:437–443.
30. Weichert, W. HDAC expression and clinical prognosis in human malignancies. Cancer Lett. 2009;280:168–176.
31. Zgouras, D, Becker, U, Loitsch, S, et al. Modulation of angiogenesis-related protein synthesis by valproic acid. Biochem Biophys Res Commun. 2004;316:693–697.
32. Qian, DZ, Wang, X, Kachhap, SK, et al. The histone deacetylase inhibitor NVP-LAQ824 inhibits angiogenesis and has a greater antitumor effect in combination with the vascular endothelial growth factor receptor tyrosine kinase inhibitor PTK787/ZK222584. Cancer Res. 2004;64:6626–6634.
33. Michaelis, M, Michaelis, UR, Fleming, I, et al. Valproic acid inhibits angiogenesis in vitro and in vivo. Mol Pharmacol. 2004;65:520–527.
34. Rossig, L, Li, H, Fisslthaler, B, et al. Inhibitors of histone deacetylation downregulate the expression of endothelial nitric oxide synthase and compromise endothelial cell function in vasorelaxation and angiogenesis. Circ Res. 2002;91:837–844.
35. Rossig, L, Urbich, C, Bruhl, T, et al. Histone deacetylase activity is essential for the expression of HoxA9 and for endothelial commitment of progenitor cells. J Exp Med. 2005;201(11):1825–1835.
36. Phillips, A, Bullock, T, Plant, N. Sodium valproate induces apoptosis in the rat hepatoma cell line, FaO. Toxicology. 2003;192:219–227.
37. Tang, R, Faussat, AM, Majdak, P, et al. Valproic acid inhibits proliferation and induces apoptosis in acute myeloid leukemia cells expressing P-gp and MRP1. Leukemia. 2004;18:1246–1251.
38. Roh, MS, Kim, CW, Park, BS, et al. Mechanism of histone deacetylase inhibitor Trichostatin A induced apoptosis in human osteosarcoma cells. Apoptosis. 2004;9:583–589.
39. Maeda, T, Nagaoka, Y, Kawai, Y, et al. Inhibitory effects of cancer cell proliferation by novel histone deacetylase inhibitors involve p21/WAF1 induction and G2/M arrest. Biol Pharm Bull. 2005;28:849–853.
40. Takai, N, Desmond, JC, Kumagai, T, et al. Histone deacetylase inhibitors have a profound antigrowth activity in endometrial cancer cells. Clin Cancer Res. 2004;10:1141–1149.
41. Olsen, CM, Meussen-Elholm, ET, Roste, LS, et al. Antiepileptic drugs inhibit cell growth in the human breast cancer cell line MCF7. Mol Cell Endocrinol. 2004;213:173–179.
42. Fortunati, N, Catalano, MG, Arena, K, et al. Valproic acid induces the expression of the Na+/I- symporter and iodine uptake in poorly differentiated thyroid cancer cells. J Clin Endocrinol Metab. 2004;89:1006–1009.
43. Gottlicher, M. Valproic acid: an old drug newly discovered as inhibitor of histone deacetylases. Ann Hematol. 2004;83(suppl 1):S91–S92.
44. Gottlicher, M, Minucci, S, Zhu, P, et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001;20:6969–6978.
45. Stockhausen, MT, Sjolund, J, Manetopoulos, C, et al. Effects of the histone deacetylase inhibitor valproic acid on Notch signalling in human neuroblastoma cells. Br J Cancer. 2005;92:751–759.
46. Thelen, P, Schweyer, S, Hemmerlein, B, et al. Expressional changes after histone deacetylase inhibition by valproic acid in LNCaP human prostate cancer cells. Int J Oncol. 2004;24:25–31.
47. Chobanian, NH, Greenberg, VL, Gass, JM, et al. Histone deacetylase inhibitors enhance paclitaxel-induced cell death in ovarian cancer cell lines independent of p53 status. Anticancer Res. 2004;24:539–545.
48. Fuino, L, Bali, P, Wittmann, S, et al. Histone deacetylase inhibitor LAQ824 down-regulates Her-2 and sensitizes human breast cancer cells to trastuzumab, Taxotere, gemcitabine, and epothilone B. Mol Cancer Ther. 2003;2:971–984.
49. Kim, MS, Blake, M, Baek, JH, et al. Inhibition of histone deacetylase increases cytotoxicity to anticancer drugs targeting DNA. Cancer Res. 2003;63:7291–7300.
50. Maggio, SC, Rosato, RR, Kramer, LB, et al. The histone deacetylase inhibitor MS-275 interacts synergistically with fludarabine to induce apoptosis in human leukemia cells. Cancer Res. 2004;64:2590–2600.
51. Watanabe, K, Okamoto, K, Yonehara, S. Sensitization of osteosarcoma cells to death receptor-mediated apoptosis by HDAC inhibitors through downregulation of cellular FLIP. Cell Death Differ. 2005;12:10–18.
52. Chinnaiyan, P, Vallabhaneni, G, Armstrong, E, et al. Modulation of radiation response by histone deacetylase inhibition. Int J Radiat Oncol Biol Phys. 2005;62:223–229.
53. Wittenburg, LA, Bisson, L, Rose, BJ, et al. The histone deacetylase inhibitor valproic acid sensitizes human and canine osteosarcoma to doxorubicin. Cancer Chemother Pharmacol. 2011;67:83–92.
54. Wittenburg, LA, Gustafson, DL, Thamm, DH. Phase I pharmacokinetic and pharmacodynamic evaluation of combined valproic acid/doxorubicin treatment in dogs with spontaneous cancer. Clin Cancer Res. 2010;16:4832–4842.
55. Whittaker, SJ, Demierre, MF, Kim, EJ, et al. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J Clin Oncol. 2010;28:4485–4491.
56. Duvic, M, Talpur, R, Ni, X, et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood. 2007;109:31–39.
57. Olsen, EA, Kim, YH, Kuzel, TM, et al. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol. 2007;25:3109–3115.
58. Munster, PN, Thurn, KT, Thomas, S, et al. A phase II study of the histone deacetylase inhibitor vorinostat combined with tamoxifen for the treatment of patients with hormone therapy-resistant breast cancer. Br J Cancer. 2011;104:1828–1835.
59. Kirschbaum, M, Frankel, P, Popplewell, L, et al. Phase II study of vorinostat for treatment of relapsed or refractory indolent non-Hodgkin’s lymphoma and mantle cell lymphoma. J Clin Oncol. 2011;29:1198–1203.
60. Otterson, GA, Hodgson, L, Pang, H, et al. Phase II study of the histone deacetylase inhibitor Romidepsin in relapsed small cell lung cancer (Cancer and Leukemia Group B 30304). J Thor Oncol. 2010;5:1644–1648.
61. Stathis, A, Hotte, SJ, Chen, EX, et al. Phase I study of decitabine in combination with vorinostat in patients with advanced solid tumors and non-Hodgkin’s lymphomas. Clin Cancer Res. 2011;17:1582–1590.
62. Kisseberth, WC, Murahari, S, London, CA, et al. Evaluation of the effects of histone deacetylase inhibitors on cells from canine cancer cell lines. Am J Vet Res. 2008;69:938–945.
63. Cohen, LA, Powers, B, Amin, S, et al. Treatment of canine haemangiosarcoma with suberoylanilide hydroxamic acid, a histone deacetylase inhibitor. Vet Compar Oncol. 2004;2:243–248.
64. Adams, J. The development of proteasome inhibitors as anticancer drugs. Cancer Cell. 2004;5:417–421.
65. Rajkumar, SV, Richardson, PG, Hideshima, T, et al. Proteasome inhibition as a novel therapeutic target in human cancer. J Clin Oncol. 2005;23:630–639.
66. Voorhees, PM, Dees, EC, O’Neil, B, et al. The proteasome as a target for cancer therapy. Clin Cancer Res. 2003;9:6316–6325.
67. Hideshima, T, Richardson, P, Chauhan, D, et al. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 2001;61:3071–3076.
68. Masdehors, P, Omura, S, Merle-Beral, H, et al. Increased sensitivity of CLL-derived lymphocytes to apoptotic death activation by the proteasome-specific inhibitor lactacystin. Br J Haematol. 1999;105:752–757.
69. Orlowski, RZ, Eswara, JR, Lafond-Walker, A, et al. Tumor growth inhibition induced in a murine model of human Burkitt’s lymphoma by a proteasome inhibitor. Cancer Res. 1998;58:4342–4348.
70. Soligo, D, Servida, F, Delia, D, et al. The apoptogenic response of human myeloid leukaemia cell lines and of normal and malignant haematopoietic progenitor cells to the proteasome inhibitor PSI. Br J Haematol. 2001;113:126–135.
71. Masdehors, P, Merle-Beral, H, Maloum, K, et al. Deregulation of the ubiquitin system and p53 proteolysis modify the apoptotic response in B-CLL lymphocytes. Blood. 2000;96:269–274.
72. Fernandez, Y, Verhaegen, M, Miller, TP, et al. Differential regulation of noxa in normal melanocytes and melanoma cells by proteasome inhibition: therapeutic implications. Cancer Res. 2005;65:6294–6304.
73. Qin, JZ, Ziffra, J, Stennett, L, et al. Proteasome inhibitors trigger NOXA-mediated apoptosis in melanoma and myeloma cells. Cancer Res. 2005;65:6282–6293.
74. Jagannath, S, Barlogie, B, Berenson, J, et al. A phase 2 study of two doses of bortezomib in relapsed or refractory myeloma. Br J Haematol. 2004;127:165–172.
75. Jagannath, S, Durie, BG, Wolf, J, et al. Bortezomib therapy alone and in combination with dexamethasone for previously untreated symptomatic multiple myeloma. Br J Haematol. 2005;129:776–783.
76. Richardson, PG, Sonneveld, P, Schuster, MW, et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med. 2005;352:2487–2498.
77. Richardson, PG, Barlogie, B, Berenson, J, et al. A phase 2 study of bortezomib in relapsed, refractory myeloma. N Engl J Med. 2003;348:2609–2617.
78. O’Connor, OA, Wright, J, Moskowitz, C, et al. Phase II clinical experience with the novel proteasome inhibitor bortezomib in patients with indolent non-Hodgkin’s lymphoma and mantle cell lymphoma. J Clin Oncol. 2005;23:676–684.
79. Goy, A, Younes, A, McLaughlin, P, et al. Phase II study of proteasome inhibitor bortezomib in relapsed or refractory B-cell non-Hodgkin’s lymphoma. J Clin Oncol. 2005;23:667–675.
80. Cortes, J, Thomas, D, Koller, C, et al. Phase I study of bortezomib in refractory or relapsed acute leukemias. Clin Cancer Res. 2004;10:3371–3376.
81. Davis, NB, Taber, DA, Ansari, RH, et al. Phase II trial of PS-341 in patients with renal cell cancer: a University of Chicago phase II consortium study. J Clin Oncol. 2004;22:115–119.
82. Maki, RG, Kraft, AS, Scheu, K, et al. A multicenter Phase II study of bortezomib in recurrent or metastatic sarcomas. Cancer. 2005;103:1431–1438.
83. Markovic, SN, Geyer, SM, Dawkins, F, et al. A phase II study of bortezomib in the treatment of metastatic malignant melanoma. Cancer. 2005;103:2584–2589.
84. Shah, MH, Young, D, Kindler, HL, et al. Phase II study of the proteasome inhibitor bortezomib (PS-341) in patients with metastatic neuroendocrine tumors. Clin Cancer Res. 2004;10:6111–6118.
85. Bouchard, PR, Juedes, MJ, Nix, D, et al, Nonclinical discovery and development of Bortezomib (PS-341, VELCADE), a proteasome inhibitor for the treatment of cancer. In Proceedings of the 55th Annual Meeting of the American College of Veterinary Pathology, Orlando, FL 2004.
86. Xie, Q, Gao, CF, Shinomiya, N, et al. Geldanamycins exquisitely inhibit HGF/SF-mediated tumor cell invasion. Oncogene. 2005;24:3697–3707.
87. Zagzag, D, Nomura, M, Friedlander, DR, et al. Geldanamycin inhibits migration of glioma cells in vitro: a potential role for hypoxia-inducible factor (HIF-1alpha) in glioma cell invasion. J Cell Physiol. 2003;196:394–402.
88. Masson-Gadais, B, Houle, F, Laferriere, J, et al. Integrin alphavbeta3, requirement for VEGFR2-mediated activation of SAPK2/p38 and for Hsp90-dependent phosphorylation of focal adhesion kinase in endothelial cells activated by VEGF. Cell Stress Chaperones. 2003;8:37–52.
89. Smith, V, Sausville, EA, Camalier, RF, et al. Comparison of 17-dimethylaminoethylamino-17-demethoxy-geldanamycin (17DMAG) and 17-allylamino-17-demethoxygeldanamycin (17AAG) in vitro: effects on Hsp90 and client proteins in melanoma models. Cancer Chemother Pharmacol. 2005;56:126–137.
90. Maloney, A, Clarke, PA, Workman, P. Genes and proteins governing the cellular sensitivity to HSP90 inhibitors: a mechanistic perspective. Curr Cancer Drug Targets. 2003;3:331–341.
91. Fumo, G, Akin, C, Metcalfe, DD, et al. 17-Allylamino-17-demethoxygeldanamycin (17-AAG) is effective in down-regulating mutated, constitutively activated KIT protein in human mast cells. Blood. 2004;103:1078–1084.
92. Maulik, G, Kijima, T, Ma, PC, et al. Modulation of the c-Met/hepatocyte growth factor pathway in small cell lung cancer. Clin Cancer Res. 2002;8:620–627.
93. Machida, H, Matsumoto, Y, Shirai, M, et al. Geldanamycin, an inhibitor of Hsp90, sensitizes human tumour cells to radiation. Int J Radiat Biol. 2003;79:973–980.
94. Solit, DB, Basso, AD, Olshen, AB, et al. Inhibition of heat shock protein 90 function down-regulates Akt kinase and sensitizes tumors to Taxol. Cancer Res. 2003;63:2139–2144.
95. Vasilevskaya, IA, Rakitina, TV, O’Dwyer, PJ. Geldanamycin and its 17-allylamino-17-demethoxy analogue antagonize the action of cisplatin in human colon adenocarcinoma cells: differential caspase activation as a basis for interaction. Cancer Res. 2003;63:3241–3246.
96. Aoyagi, Y, Fujita, N, Tsuruo, T. Stabilization of integrin-linked kinase by binding to Hsp90. Biochem Biophys Res Commun. 2005;331:1061–1068.
97. Kurebayashi, J, Otsuki, T, Kurosumi, M, et al. A radicicol derivative, KF58333, inhibits expression of hypoxia-inducible factor-1alpha and vascular endothelial growth factor, angiogenesis and growth of human breast cancer xenografts. Jpn J Cancer Res. 2001;92:1342–1351.
98. Osada, M, Imaoka, S, Funae, Y. Apigenin suppresses the expression of VEGF, an important factor for angiogenesis, in endothelial cells via degradation of HIF-1alpha protein. FEBS Lett. 2004;575:59–63.
99. Mabjeesh, NJ, Post, DE, Willard, MT, et al. Geldanamycin induces degradation of hypoxia-inducible factor 1alpha protein via the proteosome pathway in prostate cancer cells. Cancer Res. 2002;62:2478–2482.
100. Isaacs, JS, Jung, YJ, Mimnaugh, EG, et al. Hsp90 regulates a von Hippel Lindau-independent hypoxia-inducible factor-1 alpha-degradative pathway. J Biol Chem. 2002;277:29936–29944.
101. Bisht, KS, Bradbury, CM, Mattson, D, et al. Geldanamycin and 17-allylamino-17-demethoxygeldanamycin potentiate the in vitro and in vivo radiation response of cervical tumor cells via the heat shock protein 90-mediated intracellular signaling and cytotoxicity. Cancer Res. 2003;63:8984–8995.
102. Kaur, G, Belotti, D, Burger, AM, et al. Antiangiogenic properties of 17-(dimethylaminoethyl amino)-17-demethoxygeldanamycin: An orally bioavailable heat shock protein 90 modulator. Clin Cancer Res. 2004;10:4813–4821.
103. Plescia, J, Salz, W, Xia, F, et al. Rational design of shepherdin, a novel anticancer agent. Cancer Cell. 2005;7:457–468.
104. Muller, L, Schaupp, A, Walerych, D, et al. Hsp90 regulates the activity of wild type p53 under physiological and elevated temperatures. J Biol Chem. 2004;279:48846–48854.
105. Muller, P, Ceskova, P, Vojtesek, B. Hsp90 is essential for restoring cellular functions of temperature-sensitive p53 mutant protein but not for stabilization and activation of wild-type p53: implications for cancer therapy. J Biol Chem. 2005;280:6682–6691.
106. Walerych, D, Kudla, G, Gutkowska, M, et al. Hsp90 chaperones wild-type p53 tumor suppressor protein. J Biol Chem. 2004;279:48836–48845.
107. Fortugno, P, Beltrami, E, Plescia, J, et al. Regulation of survivin function by Hsp90. Proc Natl Acad Sci U S A. 2003;100:13791–13796.
108. Park, JW, Yeh, MW, Wong, MG, et al. The heat shock protein 90-binding geldanamycin inhibits cancer cell proliferation, down-regulates oncoproteins, and inhibits epidermal growth factor-induced invasion in thyroid cancer cell lines. J Clin Endocrinol Metab. 2003;88:3346–3353.
109. Hawkins, LM, Jayanthan, AA, Narendran, A. Effects of 17-allylamino-17-demethoxygeldanamycin (17-AAG) on pediatric acute lymphoblastic leukemia (ALL) with respect to Bcr-Abl status and imatinib mesylate sensitivity. Pediatr Res. 2005;57:430–437.
110. Villa, R, Folini, M, Porta, CD, et al. Inhibition of telomerase activity by geldanamycin and 17-allylamino, 17-demethoxygeldanamycin in human melanoma cells. Carcinogenesis. 2003;24:851–859.
111. Haendeler, J, Hoffmann, J, Rahman, S, et al. Regulation of telomerase activity and anti-apoptotic function by protein-protein interaction and phosphorylation. FEBS Lett. 2003;536:180–186.
112. Neckers, L. Hsp90 inhibitors as novel cancer chemotherapeutic agents. Trends Mol Med. 2002;8:S55–S61.
113. Isaacs, JS, Xu, W, Neckers, L. Heat shock protein 90 as a molecular target for cancer therapeutics. Cancer Cell. 2003;3:213–217.
114. Kamal, A, Thao, L, Sensintaffar, J, et al. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature. 2003;425:407–410.
115. Downing, S, Chien, MB, Kass, PH, et al. Prevalence and importance of internal tandem duplications in exons 11 and 12 of c-kit in mast cell tumors of dogs. Am J Vet Res. 2002;63:1718–1723.
116. Liao, AT, McMahon, M, London, CA. Characterization, expression and function of c-Met in canine spontaneous cancers. Vet Compar Oncol. 2005;3:61–72.
117. MacEwen, EG, Kutzke, J, Carew, J, et al. c-Met tyrosine kinase receptor expression and function in human and canine osteosarcoma cells. Clin Exp Metastasis. 2003;20:421–430.
118. Sakagami, M, Morrison, P, Welch, WJ. Benzoquinoid ansamycins (herbimycin A and geldanamycin) interfere with the maturation of growth factor receptor tyrosine kinases. Cell Stress Chaperones. 1999;4:19–28.
119. Katayama, R, Huelsmeyer, MK, Marr, AK, et al. Imatinib mesylate inhibits platelet-derived growth factor activity and increases chemosensitivity in feline vaccine-associated sarcoma. Cancer Chemother Pharmacol. 2004;54:25–33.
120. Levine, RA. Overexpression of the sis oncogene in a canine osteosarcoma cell line. Vet Pathol. 2002;39:411–412.
121. MacEwen, EG, Pastor, J, Kutzke, J, et al. IGF-1 receptor contributes to the malignant phenotype in human and canine osteosarcoma. J Cell Biochem. 2004;92:77–91.
122. Serra, M, Pastor, J, Domenzain, C, et al. Effect of transforming growth factor-beta1, insulin-like growth factor-I, and hepatocyte growth factor on proteoglycan production and regulation in canine melanoma cell lines. Am J Vet Res. 2002;63:1151–1158.
123. Thamm, DH, Huelsmeyer, MK, Mitzey, AM, et al. RT-PCR-based tyrosine kinase display profiling of canine melanoma: IGF-1 receptor as a potential therapeutic target. Melanoma Res. 2010;20:35–42.
124. Bagatell, R, Beliakoff, J, David, CL, et al. Hsp90 inhibitors deplete key anti-apoptotic proteins in pediatric solid tumor cells and demonstrate synergistic anticancer activity with cisplatin. Int J Cancer. 2005;113:179–188.
125. Jones, DT, Addison, E, North, JM, et al. Geldanamycin and herbimycin A induce apoptotic killing of B chronic lymphocytic leukemia cells and augment the cells’ sensitivity to cytotoxic drugs. Blood. 2004;103:1855–1861.
126. Munster, PN, Basso, A, Solit, D, et al. Modulation of Hsp90 function by ansamycins sensitizes breast cancer cells to chemotherapy-induced apoptosis in an RB- and schedule-dependent manner. Clin Cancer Res. 2001;7:2228–2236.
127. Price, JT, Quinn, JMW, Sims, NA, et al. The heat shock protein 90 inhibitor, 17-allylamino-17-demethoxygeldanamycin, enhances osteoclast formation and potentiates bone metastasis of a human breast cancer cell line. Cancer Res. 2005;65:4929–4938.
128. Goetz, MP, Toft, D, Reid, J, et al. Phase I trial of 17-allylamino-17-demethoxygeldanamycin in patients with advanced cancer. J Clin Oncol. 2005;23:1078–1087.
129. Grem, JL, Morrison, G, Guo, XD, et al. Phase I and pharmacologic study of 17-(allylamino)-17-demethoxygeldanamycin in adult patients with solid tumors. J Clin Oncol. 2005;23:1885–1893.
130. Pacey, S, Wilson, RH, Walton, M, et al. A phase I study of the heat shock protein 90 inhibitor alvespimycin (17-DMAG) given intravenously to patients with advanced solid tumors. Clin Cancer Res. 2011;17:1561–1570.
131. Richardson, PG, Chanan-Khan, AA, Alsina, M, et al. Tanespimycin monotherapy in relapsed multiple myeloma: results of a phase 1 dose-escalation study. Br J Haematol. 2010;150:438–445.
132. Ramanathan, RK, Egorin, MJ, Erlichman, C, et al. Phase I pharmacokinetic and pharmacodynamic study of 17-dimethylaminoethylamino-17-demethoxygeldanamycin, an inhibitor of heat-shock protein 90, in patients with advanced solid tumors. J Clin Oncol. 2010;28:1520–1526.
133. Lin, TY, Bear, M, Du, Z, et al. The novel HSP90 inhibitor STA-9090 exhibits activity against Kit-dependent and -independent malignant mast cell tumors. Exper Hematol. 2008;36:1266–1277.
134. McCleese, JK, Bear, MD, Fossey, SL, et al. The novel HSP90 inhibitor STA-1474 exhibits biologic activity against osteosarcoma cell lines. Int J Cancer. 2009;125:2792–2801.
135. Fauzee, NJ, Pan, J, Wang, YL. PARP and PARG inhibitors—new therapeutic targets in cancer treatment. Pathol Oncol Res. 2010;16:469–478.
136. D’Amours, D, Desnoyers, S, D’Silva, I, et al. Poly (ADP-ribosyl) ation reactions in the regulation of nuclear functions. Biochem J. 1999;342(Pt 2):249–268.
137. Hochegger, H, Dejsuphong, D, Fukushima, T, et al. Parp-1 protects homologous recombination from interference by Ku and Ligase IV in vertebrate cells. EMBO J. 2006;25:1305–1314.
138. Plummer, R, Jones, C, Middleton, M, et al. Phase I study of the poly (ADP-ribose) polymerase inhibitor, AG014699, in combination with temozolomide in patients with advanced solid tumors. Clin Cancer Res. 2008;14:7917–7923.
139. De Soto, JA, Wang, X, Tominaga, Y, et al. The inhibition and treatment of breast cancer with poly (ADP-ribose) polymerase (PARP-1) inhibitors. Int J Biol Sci. 2006;2:179–185.
140. Albert, JM, Cao, C, Kim, KW, et al. Inhibition of poly (ADP-ribose) polymerase enhances cell death and improves tumor growth delay in irradiated lung cancer models. Clin Cancer Res. 2007;13:3033–3042.
141. Donawho, CK, Luo, Y, Penning, TD, et al. ABT-888, an orally active poly (ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin Cancer Res. 2007;13:2728–2737.
142. Li, M, Threadgill, MD, Wang, Y, et al. Poly(ADP-ribose) polymerase inhibition down-regulates expression of metastasis-related genes in CT26 colon carcinoma cells. Pathobiology. 2009;76:108–116.
143. Tentori, L, Leonetti, C, Scarsella, M, et al. Systemic administration of GPI 15427, a novel poly(ADP-ribose) polymerase-1 inhibitor, increases the antitumor activity of temozolomide against intracranial melanoma, glioma, lymphoma. Clin Cancer Res. 2003;9:5370–5379.
144. Dungey, FA, Caldecott, KW, Chalmers, AJ. Enhanced radiosensitization of human glioma cells by combining inhibition of poly(ADP-ribose) polymerase with inhibition of heat shock protein 90. Mol Cancer Ther. 2009;8:2243–2254.
145. Winum, JY, Scozzafava, A, Montero, JL, et al. Inhibition of carbonic anhydrase IX: a new strategy against cancer. Anticancer Agents Med Chem. 2009;9:693–702.
146. El-Sayed, NS, El-Bendary, ER, El-Ashry, SM, et al. Synthesis and antitumor activity of new sulfonamide derivatives of thiadiazolo[3, 2-a]pyrimidines. Eur J Med Chem. 2011;46:3714–3720.
147. Muller, V, Riethdorf, S, Rack, B, et al. Prospective evaluation of serum tissue inhibitor of metalloproteinase 1 and carbonic anhydrase IX in correlation to circulating tumor cells in patients with metastatic breast cancer. Breast Can Res. 2011;13:R71.