Chapter 9 Pathophysiology of Vasculitis
The pathophysiological mechanisms that underlie vasculitis include elements of virtually all effector limbs of host defenses, including innate and adaptive immunity. Various classification schemes separate the vasculitides into primary and secondary families, and categorize them by size of the afflicted vessel. Classification of vasculitides is currently in ferment, with the classical American College of Rheumatology classification and the Chapel Hill Consensus Conference criteria under reconsideration from several fronts and for various reasons.1–3 Epidemiological studies and clinical trials, for example, require standardized definitions of these diseases, and these may overlap considerably. The confusion that reigns in this field reflects in part an incomplete understanding of the fundamentals of the pathogenesis of human vasculitides—an endeavor still in considerable flux. Whereas Chapter 41 provides a detailed classification of the vasculitides, this chapter focuses on the primary vasculitides and, for pedagogical purposes, considers the pathophysiological mechanisms of vasculitis in two broad categories: (1) mechanisms that underlie small-vessel vasculitis, and (2) mechanisms involved in medium- and large-sized arteritides (Fig. 9-1). Although this is an oversimplified categorization, it provides an organizational framework for discussing elements of humoral immunity involved chiefly in primary small-vessel vasculitides, and cellular immunity, likely the central mechanism underlying vasculitides of medium- and large-sized arteries (Table 9-1).
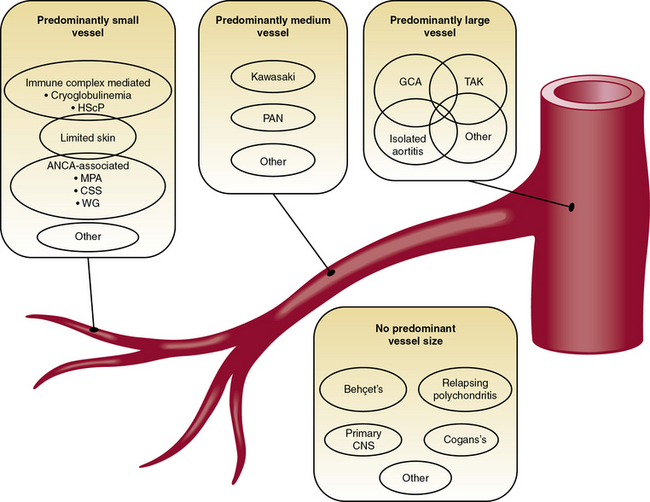
Figure 9-1 Classification scheme for the primary vasculitides.
CNS, central nervous system; CSS, Churg-Strauss syndrome; GCA, giant-cell arteritis; HScP, Henoch-Schönlein purpura; MPA, microscopic polyangiitis; PAN, polyarterits nodosa; TAK, Takayasu arteritis; WG, Wegener granulomatosis.
(Modified from Suppiah R, Basu N, Watts RA, et al: Advances in the classification of primary systemic vasculitis. Int J Adv Rheumatol 7:10, 2009. Reproduced with permission from Watts RA, Suppiah R, Merkel PA, et al: Systemic vasculitis—Is it time to reclassify? Rheumatology 50:643, 2011.)
Table 9-1 Pathophysiological Mechanisms of Some Primary Vasculitides
| Putative Pathogenic Effectors | |
|---|---|
| Large-Artery Arteritides | |
| Takayasu arteritis | γδ T cells |
| Giant-cell arteritis | TH1 and TH17 helper T cells |
| Small-Artery Arteritides | |
| Microscopic polyangiitis | Anti-MPO ANCA > Anti-PR3 ANCA |
| Wegener granulomatosis | Anti-PR3 ANCA > Anti-MPO ANCA |
| Henoch-Schönlein purpura | Viral infections, food allergies (?) |
| Churg-Strauss’ syndrome | Eosinophils, CD95 |
| Cryoglobulinemia | IgM > IgG, hepatitis C virus infection |
ANCA, antineutrophil cytoplasmic antibody; Ig, immunoglobulin; MPO, myeloperoxidase; PR3, proteinase-3; TH, helper T cell.
Pathophysiology of Small-Vessel Vasculitis
Small-vessel vasculitides include Wegener granulomatosis, Churg-Strauss’ syndrome, microscopic polyangiitis, Henoch-Schönlein purpura, and essential cryoglobulinemic vasculitis.2,3 Recognition that antineutrophil cytoplasmic antibodies (ANCAs) associate with many (but not all) small-vessel vasculitides has advanced understanding of their pathophysiology. In particular, Wegener granulomatosis, microscopic polyangiitis, and Churg-Strauss’ syndrome associate strongly with ANCA. Many of the ANCA-positive small-vessel vasculitides involve the kidney.4
The principal antigens recognized by ANCA are the neutrophil enzymes myeloperoxidase (MPO) and proteinase-3 (PR3) (see Table 9-1); some ANCA may recognize human neutrophil elastase as well. Anti-PR3 antibodies also may recognize plasminogen.5 Antigens recognized by ANCA usually localize within polymorphonuclear (PMNs) leukocytes. When primed by stimuli such as tumor necrosis factor (TNF)-α or when undergoing apoptosis or NETosis (release of chromatin fibers called neutrophil extracellular traps (NETs) that trap and kill microbes extracellularly), PMN leukocytes can release these antigens. These antigens in turn can bind back to the cell surface—in the case of PR3, through CD1776—or decorate NETs.7 Binding of ANCAs to cell surface–associated MPO and PR3 on intact neutrophils leads to further activation of these leukocytes (i.e., generation of reactive oxygen species [ROS], release of lytic enzymes, binding of cells to the endothelium), as does engagement of the surface-bound Fc-portion of immunoglobulin (Ig)G in immune complexes with FcγRs on neighboring cells. Uptake of neutrophil-released MPO and PR3 by endothelial cells (ECs) also may impair the viability and vasomotor responses of ECs.8 These events together aggravate the local inflammatory response. As opposed to secondary vasculitides, which characteristically have substantial immune complex deposition upon histological examination, lesions of ANCA-associated conditions show modest Ig deposits.9 When released in soluble form, proteinases such as PR3 and neutrophil elastase readily bind to widely distributed and abundant antiproteinases that may mask their recognition by ANCA. Circulating ANCA can also complex with these antigens, but such complexes form preferentially when proteinase antigens remain associated with the neutrophil cell surface (Fig. 9-2).
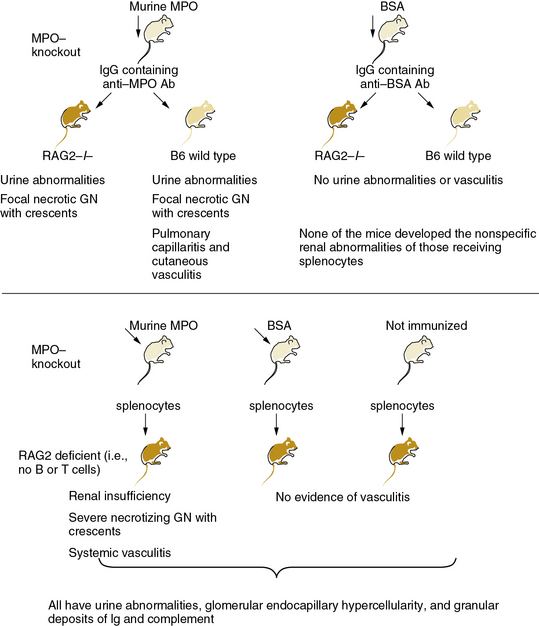
Figure 9-2 MPO-ANCA mice cause small-vessel arteritis.
ANCA, antineutrophil cytoplasmic antibody; BSA, bovine serum albumin; GN, glomerulonephritis; IgG, immunoglobulin G; MPO, myeloperoxidase; RAG2, recombination activating gene 2.
(Data from Xiao H, Herringa P, Hu P, et al: Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. J Clin Invest 110:955, 2002. Reproduced with permission from Day CJ, Hewins P, Savage CO: New directions in the pathogenesis of ANCA-associated vasculitis. Clin Exp Rheumatol 21:S35, 2003.)
Individuals who express primarily MPO-directed ANCA (vs. PR3-directed ANCA) may have distinct clinical courses.10 Microscopic polyangiitis associates particularly with anti-MPO ANCA, while Wegener granulomatosis typically associates with anti-PR3 ANCA (see Table 9-1). The possible clinical dichotomy between these patient categories may relate to the functions of target antigens. For example, binding to ANCA may protect MPO from clearance and inactivation by ceruloplasmin, increasing the ability of this enzyme to produce the highly oxidant species, hypochlorous acid (HOCl). Hypochlorous acid has many properties that may contribute to the pathophysiology of vasculitis, including stimulation of endothelial apoptosis.11
Not all patients with small-vessel vasculitis have ANCA-positive serology, indicating that some small-vessel vasculitides may involve other mechanisms or have low titer antibodies. Additionally, “atypical” ANCA directed against antigens other than MPO or PR3 may participate in the pathogenesis of vasculitis. Recent studies have implicated lysosomal-associated membrane protein-2 (LAMP-2) as a novel autoantigen in vasculitis.6,12,13 In addition to neutrophils, endothelial and other cells express LAMP-2, a recognition target for ANCA.
Antineutrophil cytoplasmic antibodies have proven unequivocally pathogenic in mice. In a landmark investigation, Xiao et al. immunized mice lacking endogenous MPO, owing to targeted gene inactivation, with exogenous mouse MPO.14 Transfer of splenocytes from these MPO-immunized mice into immunodeficient mice caused severe necrotizing crescentic glomerulonephritis. In some cases, a systemic necrotizing and granulomatous vasculitis affected lung capillaries as well as the renal microvasculature (see Fig. 9-2). Purified anti-MPO IgG isolated from the MPO-immunized mice caused renal, pulmonary, and cutaneous small-vessel vasculitis. Experimental depletion of neutrophils abrogated formation of glomerular lesions in anti-MPO IgG-treated mice, thus implicating granulocytes in the pathogenesis of ANCA-induced angiitis.15 Moreover, studies in bone marrow chimeric mice (transplantation of MPO wild-type or MPO-deficient bone marrow into MPO-immunized MPO-null animals) suggest that leukocytes are targets of anti-MPO ANCA.16
Immunization of PR3-deficient mice with PR3 yielded circulating anti-PR3 antibodies and modest renal and pulmonary vasculitis.17 These mice with anti-PR3 antibodies developed cutaneous vasculitis at sites of TNF-α injection. While not directly comparable to the passive/adoptive transfer studies in MPO-deficient mice, these results support different pathogenic capabilities of these two major classes of ANCA in terms of severity and localization of vasculitis, at least in mice. Plasma exchange causes improvement in patients with ANCA-associated disease exacerbation, supporting the causal role of antibody in these conditions.6
Antineutrophil cytoplasmic antibodies may provoke vasculitis in several ways (Figs. 9-3 and 9-4). These autoantibodies may increase activation and adherence of neutrophils to ECs.18 When neutrophils “primed” by exposure to TNF-α encounter MPO-ANCA, a respiratory burst can ensue and produce ROS such as superoxide anion and hydrogen peroxide—proinflammatory mediators that can injure ECs and activate smooth muscle cells (SMCs).19,20 Antineutrophil cytoplasmic antibodies promote neutrophil degranulation, and can activate intracellular signaling pathways and heighten sensitivity of PMN leukocytes to classic stimulants, such as formyl peptides.21 The mechanism of vascular damage in immune complex disease also involves complement activation (see Fig. 9-4).6 Antigen-antibody complexes (immune complexes) containing IgM or IgG can bind to complement factor 1 (C1), lead to assembly of C3 convertase, and yield activation of C3, C4, and C5. Ultimately, assembly of the membrane attack complex (MAC, composed of oligomers of C9 and other terminal complement components) can damage ECs by forming pores in their plasma membranes. Circulating immune complexes can sequester in subendothelial basement membranes at sites where interendothelial separation has occurred. These trapped immune complexes then activate complement, and can engage neutrophils and monocytes via their Fc and complement receptors. Anaphylatoxins (fragments of C3a, C4a, and C5a) generated during activation of the classical complement pathway can recruit granulocytes and monocytes and activate mast cells at sites of immune complex deposition in vessels. These leukocytes can amplify local inflammation and aggravate and perpetuate the vasculitic response.
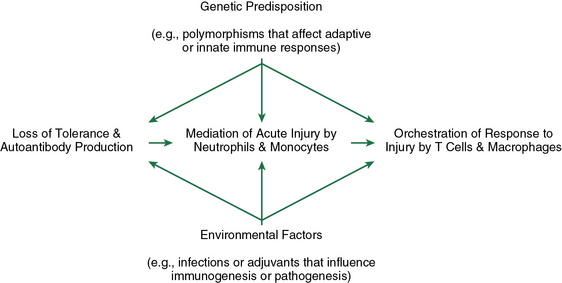
Figure 9-3 Pathogenic events during the evolution of ANCA disease.
Production of pathogenic amounts of ANCA requires loss of tolerance. ANCAs activate neutrophils and monocytes, which results in acute inflammation and necrosis, eliciting a response orchestrated by macrophages and T cells. Multiple environmental and genetic factors modulate each pathogenic step from onset to outcome. ANCA, antineutrophil cytoplasmic antibody.
(Reproduced with permission from Jennette JC, Xiao H, Falk RJ. Pathogenesis of vascular inflammation by anti-neutrophil cytoplasmic antibodies. J Am Soc Nephrol 17:1235, 2006.)
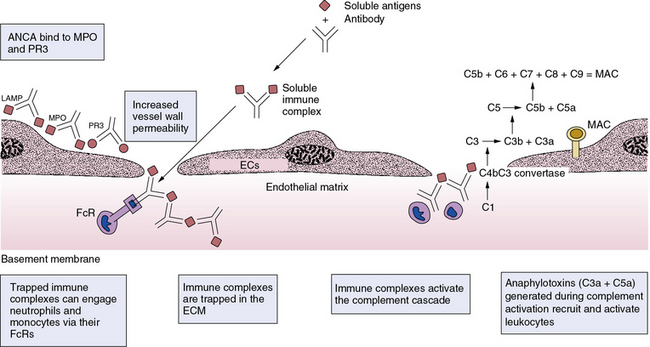
Figure 9-4 Some mechanisms of small-vessel vasculitides.
In resting polymorphonuclear (PMN) leukocytes, antigens such as myeloperoxidase (MPO) or proteinase-3 (PR3) remain localized within cells in millimolar quantities and hidden from the immune system. On priming or activation, neutrophils can release or exteriorize MPO or PR3. In addition, dying PMN leukocytes can furnish externally disposed MPO, PR3, or lysosomal-associated membrane protein-2 (LAMP-2) to the immune system. Binding of antibodies known as antineutrophil cytoplasmic antibodies (ANCA) can activate PMN leukocytes, enhancing their adhesion to endothelial cells, (ECs) (middle EC). Activated PMN leukocytes can undergo an oxidative burst, producing high levels of reactive oxygen species (ROS) such as superoxide anion or hypochlorous acid (HOCl), which can injure ECs. Release of neutrophil elastase and other hydrolases can digest basement membrane, leading to the classic picture of a necrotizing vasculitis affecting small vessels of the glomerulus, lung, or skin. Formation of immune complexes can directly activate cells by binding to Fc receptors (FCRs), and can unleash complement which, in turn, can bind complement receptors. Activation of the complement pathway can also generate anaphylotoxins, which can recruit and activate additional leukocytes. Similar mechanisms are involved in the secondary vasculitides (e.g., those associated with endocarditis and serum sickness), systemic lupus erythematosus, and rheumatoid arthritis. ECM, extracellular matrix; MAC, membrane attack complex.
Some pathogenic schemes for vasculitis invoke the participation of viruses—in some cases well substantiated, such as the association of hepatitis C and cryoglobulinemia. Given the ubiquity of viral infections and antigen-specific antibody responses, circulation of viral antigen-antibody complexes probably occurs frequently. Yet, most common viral infections do not produce clinically evident or sustained vasculitis. The relative rarity of symptomatic or sustained vasculitis in common viral infections probably relates to tight control of the complement system—which, like many protease cascades, undergoes intricate regulation by endogenous inhibitors.
The ANCA-positive vasculitides have a solid experimental and clinical evidence base that supports a pathogenic role for ANCA in the development of vasculitis. These findings illustrate the importance of humoral immunity in the pathogenesis of this category of vasculitides, but the cellular immune response may regulate aspects of the primarily humoral immune pathogenesis of the small-vessel vasculitides. For example, the balance between type 1 helper T cell (TH1 cell, interferon [IFN]-γ predominant) responses and type 2 helper T cell (TH2 cell, interleukin [IL]-4 predominant) slanted reactions may modulate expression of small-vessel vasculitides; a TH1 response may associate with the localized variant of Wegener granulomatosis rather than with the generalized form.22 Indeed, T cells appear to modulate the consequences of the antibody-induced disease.6,23 In later stages of ANCA-induced vasculitides, T cells and their mediators may promote granulomatous features of lesions that are more necrotizing and destructive of elastic fibers in their earlier phases. Some even speculate that such granulomatous lesions may constitute a local lymphoid tissue that can perpetuate the disease through augmented production of autoantibodies and by presenting antigens regionally via abundant dendritic cells.23,24
Churg-Strauss’ syndrome, which characteristically occurs in individuals with asthma, comprises a special case of small-vessel vasculitis. Tissue accumulation of eosinophils and peripheral blood eosinophilia characterize this disease. This form of small-vessel vasculitis appears to result from prolonged eosinophil survival due to an excess of soluble CD95, which antagonizes apoptosis usually mediated by engagement of CD95 on the eosinophil surface—a mechanism that usually holds the eosinophil population tonically in check. Indeed, patients with Churg-Strauss’ syndrome have elevated levels of soluble CD95, and CD95 ligation appears important in regulating eosinophil apoptosis.25,26
Observations in some secondary vasculitides further support the involvement of antibodies in vasculitis. In particular, the cryoglobulinemic vasculitis associated with hepatitis C virus infection frequently involves a polyclonal elevation of IgG and IgM antibodies, perhaps in response to polyclonal activation of B cells. In some cases, the mixed cryoglobulins in patients with chronic hepatitis C virus infection may contain ANCA, illustrating the overlap between primary and secondary vasculitides.27
Mechanisms that give rise to ANCA (primum movens) remain speculative. The association of Staphylococcus aureus nasal infections and serological evidence of exposure to Ross River virus and other viruses with Wegener granulomatosis suggests that infection may trigger ANCA formation. Antigenic mimicry with such pathogens might explain selective induction of ANCA.12,28 A novel complex pathogenic model invokes autoantigenic complementarity rather than strict antigenic mimicry: PR3-ANCA patients have antibodies against a product of PR3 antisense ribonucleic acid (RNA) with sequence similarity to microbial and viral proteins. About half of ANCA-positive patients transcribe the complementary protein’s antisense RNA. This protein product elicits antibodies that in turn provoke an antiidiotype immune response. Antiidiotypic antibodies bind the autoantigen, raising the possibility that autoantigen complement, rather than the autoantigen itself, initiates the autoimmune response.29 Mouse experiments have shown important influences of genetic background on expression of ANCA-related vasculitis, suggesting that modifier genes or immunogenetic mechanisms modulate individual responses to ANCA—mechanisms that might explain the daunting clinical heterogeneity of these conditions in terms of severity and organ involvement.4
Pathogenesis of Medium- and Large-Sized Arterial Vasculitides
The pathological hallmark of medium- and large-sized artery vasculitides—fibroproliferation with or without granuloma formation—implicates a chronic cellular immune response in their pathogenesis. In contrast to the ANCA-associated small-vessel arteritides considered previously, current evidence implicates a primary role for cellular immunity rather than humoral immunity in large-vessel arteritis (see Table 9-1).
The stimuli driving arteritis in Takayasu’s disease, Kawasaki disease, and polyarteritis nodosa remain unknown. Among the many mechanisms proposed for Takayasu’s disease, evidence has accumulated for T-cell involvement.30 Recent work has implicated heat shock protein 60 (hsp60)-reactive γδ T cells in the pathogenesis of Takayasu’s disease.31 Other studies have pointed to antiendothelial cell antibodies in Takayasu arteritis.32 In this and other arterial diseases, whether such antibodies cause or result from the disease remains unknown.
Understanding of the pathogenesis of giant-cell arteritis as T cell–mediated disease has continued to evolve. Weyand et al. have provided strong experimental evidence for the critical involvement of CD4-positive T lymphocytes in the pathogenesis of giant-cell arteritis. Their approach involved grafting human temporal artery specimens from patients with giant-cell arteritis into immunodeficient mice. Ablation of the human T lymphocytes by administering anti–T-cell antibodies to these mice halts the inflammatory process in the xenografted human arterial specimen.33
Analysis of T-cell populations in human peripheral blood has shown high levels of TH1 and TH17 lymphocytes in untreated patients with active giant-cell arteritis.34 These T-cell subtypes produce IFN-γ, a cytokine implicated in many aspects of arterial pathophysiology that might promote vasculitis.35,36 Using the human-mouse chimera preparation, glucocorticoid treatment could inhibit IL-17 but not IFN-γ production. These results have clinical implications for the management of giant-cell arteritis.
The initiating stimulus for medium- and large-vessel arteritis remains uncertain. The predisposition of certain populations, particularly those of Northern European descent, suggests a genetic component. As in other diseases of obscure origin, many have hypothesized that infectious processes trigger medium- and large-artery vasculitides, but recovery of infectious agents from lesions has not proven reproducible. Some experimental results support the presence of antigens that can stimulate T cells in extracts of lesions of giant-cell or Takayasu arteritis, such as members of the hsp60 family.
The inflammatory process that initiates medium- and large-sized artery vasculitis may originate in the adventitia. Normal arterial adventitia contains resting dendritic cells. When activated, dendritic cells function importantly as antigen-presenting cells (Fig. 9-5); they patrol their environment, engulfing and presenting antigens to T helper cells that typically bear the CD4 marker. Unactivated dendritic cells usually inhibit T-cell stimulation, a process known as tolerization, but activated dendritic cells can trigger the cellular immune response. Normal arterial tunica media appears “immunoprivileged”—it usually does not support afferent signaling to the cellular immune system. The “break” in medial immunoprivilege may result from local expression of the characteristic TH1 cytokine, IFN-γ.37
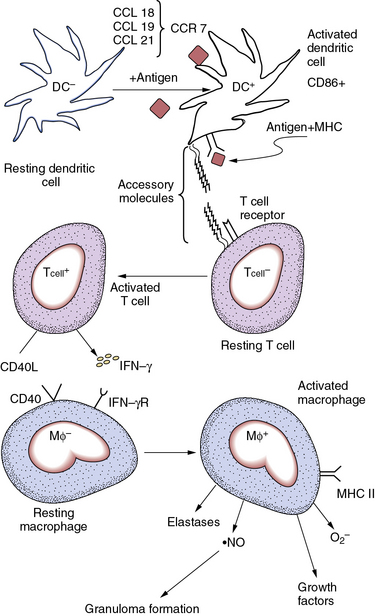
Figure 9-5 Some mechanisms of large-vessel vasculitides.
Resting dendritic cells (DC−) can exert an inhibitory influence on T cells. DC− localized in adventitia may provide a check on T-cell activation in the normal arterial wall. When appropriately stimulated, activated dendritic cells (DC+) express markers such as CD86. Expression of chemokine receptor-7 (CCR-7) allows recruitment of DC+ by the chemokine ligands CCL-18, -19, and -21. High levels of expression of class II histocompatibility antigens facilitate presentation of foreign peptides to T lymphocytes. When activated, the CD4-positive T cell (T cell+) augments production of interferon-γ (IFN-γ) and expresses high levels of CD40 ligand on its surface. These and other stimuli can activate the resting macrophage (Mφ−) by engaging cognate receptors on its surface. The stimulated macrophage (Mφ+) can secrete multiple mediators of the fibroproliferative response involved in intimal expansion characteristic of large- and medium-cell arteritides, including growth factors and cytokines, reactive oxygen species (ROS) such as superoxide anion (O2−), or reactive nitrogen species such as nitric oxide ( NO). The activated macrophage also augments expression of class II major histocompatibility antigens (MHC) that enhance the cell type’s antigen-presenting function. Elastolytic and other proteinases secreted by activated macrophages can contribute to arterial remodeling in inflamed vessels. Coalescence of activated macrophages creates the multinucleated giant cell characteristic of granulomata that form in these arteritides. These mechanisms participate in the pathogenesis of giant-cell arteritis and other medium- and large-vessel arteritides.
NO). The activated macrophage also augments expression of class II major histocompatibility antigens (MHC) that enhance the cell type’s antigen-presenting function. Elastolytic and other proteinases secreted by activated macrophages can contribute to arterial remodeling in inflamed vessels. Coalescence of activated macrophages creates the multinucleated giant cell characteristic of granulomata that form in these arteritides. These mechanisms participate in the pathogenesis of giant-cell arteritis and other medium- and large-vessel arteritides.
In arteries affected by giant-cell arteritis, dendritic cells become activated, as disclosed by their expression of the markers CD86 and CD83. Dendritic cells can accumulate in the media and intima of the artery. Activated dendritic cells express the chemokine receptor CCR-7, which can bind a trio of chemokine ligands, CCL-18, -19, and -21. These chemoattractant cytokines, overexpressed in the inflamed artery wall, can attract dendritic cells into the nascent vasculitic lesion. Recruited and activated dendritic cells can now effectively present antigen to T lymphocytes and stimulate the afferent limb of cellular immunity.
When activated, T cells can secrete cytokines such as IFN-γ and express CD40 ligand on their surface. These effectors of the activated T lymphocyte act as strong stimuli for macrophage activation; activated macrophages then can form granulomas. Accumulating macrophages can fuse into multinucleated giant cells, one of the histological hallmarks of granuloma formation. T cell–driven macrophage activation classically instigates the delayed-type hypersensitivity (DTH) response characteristic of granulomatous disease. For example, the cutaneous reaction to injected purified protein derivative from tubercle bacilli elicits a T cell–driven macrophage response believed to replicate the initial steps in formation of granulomas (e.g., those characteristic of infection with the tubercle bacillus).
Activated macrophages can release multiple mediators that participate in the fibroproliferative process characteristic of medium- and large-artery vasculitis. Production of mediators such as transforming growth factor (TGF)-β can stimulate collagen synthesis, as well as that of other constituents of the extracellular matrix (ECM).38 This fibrogenic response contributes to expansion of intimal volume, characteristic of these arteritides. Mitogenic and chemoattractant factors released from activated macrophages can also stimulate SMC migration and proliferation. For example, activated macrophages can produce platelet-derived growth factor (PDGF), fibroblast growth factor (FGF) family members, and other stimulators of SMC multiplication and directed migration.39 Activated macrophages also produce ROS and lipid hydroperoxides that can injure ECs and further amplify arterial injury.
The elastic laminae in medium- and large-sized arteries affected by giant-cell arteritis typically do not show severe dissolution, but fragmentation of the internal elastic lamina can occur. Elastolysis mediated by elastolytic enzymes of the matrix metalloproteinase (MMP) family (e.g., MMP-9, MMP-12), or by elastolytic cathepsins secreted by macrophages stimulated by CD40 ligand or other proinflammatory cytokines, contributes to elastic fiber fragmentation. This elastolysis can promote migration of SMCs and remodeling characteristic of intima expansion. Arterial ectasia or aneurysm formation sometimes occur in advanced giant-cell arteritis, or frequently as a chronic complication of Kawasaki disease. In this case, elastin destruction and digestion of other ECM macromolecules likely pave the way for arterial expansion. Compared with small-vessel vasculitides, medium- and large-sized arteritides do not involve extensive necrosis. A concentric fibroproliferative expansion of the intima with or without granuloma formation, rather than cell death, predominates.
Summary of Pathogenic Mechanisms in Vasculitides
This chapter has dichotomized the pathophysiological mechanisms of vasculitis—those that affect primarily small vessels, and those that characteristically involve medium- and large-sized arteries (see Table 9-1). Although exceptions and overlap clearly exist, the small-vessel vasculitides appear more driven by antibodies and activated PMN leukocytes than by cells of the adaptive immune response, such as T cells and dendritic cells involved in the initiation of medium- and large-sized arteritis. Activated PMN leukocytes appear to act as the major effector in small-vessel vasculitides. In contrast, the macrophage, recruited in response to dendritic cell–T lymphocyte control, likely accounts for much of the tissue response in medium- and large-vessel arteritides. The immunoprivilege of the arterial media provides a fertile field for dendritic cells to survey for, and to present, antigens to incite the cellular immune response. Antigens that drive small-vessel vasculitides include MPO and PR3, common stimuli for ANCA development. The antigenic drivers of medium- and large-sized arteritides remain unknown.
Necrosis, the histological hallmark of small-vessel vasculitides, occurs in all lesions, whether they affect glomerular capillaries, pulmonary microvasculature, or small vessels in the skin. In contrast, rapid formation of concentric intimal lesions, characterized by accumulation of ECM and SMCs, occurs during the formation of medium- and large-sized arteritides. The histological hallmark of these arteritides, the granuloma, usually does not involve necrosis or widespread elastolysis, but instead involves formation of giant cells derived from macrophages. Pathogenesis of both types of vasculitis appears to involve oxidative stress, as exemplified by the production of ROS including superoxide anion and MPO-derived HOCl. But elastolysis in small-vessel vasculitides probably involves neutrophil elastase, rather than the metalloelastases or cysteinyl elastases characteristically elaborated by mononuclear phagocytes stimulated by TH1 cytokines.
Future goals of the investigation in this field include delineation of the antigens involved in instigating medium- and large-sized arteritides. We should aim to achieve a greater understanding of the differences between giant-cell arteritis, polymyalgia rheumatica (which in some ways resembles a forme fruste of giant-cell arteritis), and Takayasu arteritis. Immunogenetic components may participate in these distinct manifestations of arteritis. The current practicality of high-throughput genome sequencing may facilitate identification of susceptibility and modifier genes. Yet low numbers of patients, heterogeneity and overlap of the vasculitic syndromes, and attendant nosological controversies will likely hamper this effort.
Individuals of Northern European descent display the greatest susceptibility to giant-cell arteritis, whereas Asian and Hispanic populations seem most at risk for Takayasu arteritis. As we understand better the specific triggers for inflammatory processes involved in these various forms of vasculitis, we should strive to target therapies more precisely and develop finer diagnostic tools. Ultimately, deeper insight into pathogenesis may guide clinical trials, inform management guidelines, and enable us to prevent and effectively treat these often serious and debilitating diseases of the vasculature.40,41
Acknowledgments
I thank Dr. Tanya Mayadas for her critical reading of this manuscript and helpful comments and suggestions, and Ms. Sara Karwacki for her editorial expertise.
1 Watts R., Lane S., Hanslik T., et al. Development and validation of a consensus methodology for the classification of the ANCA-associated vasculitides and polyarteritis nodosa for epidemiological studies. Ann Rheum Dis. 2007;66(2):222–227.
2 Watts R.A., Suppiah R., Merkel P.A., et al. Systemic vasculitis--is it time to reclassify? Rheumatology (Oxford). 2010.
3 Basu N., Watts R., Bajema I., et al. EULAR points to consider in the development of classification and diagnostic criteria in systemic vasculitis. Ann Rheum Dis. 2010;69(10):1744–1750.
4 Jennette J.C, Xiao H., Falk R., et al. Experimental models of vasculitis and glomerulonephritis induced by antineutrophil cytoplasmic autoantibodies. Contrib Nephrol. 2011;169:211–220.
5 Bautz D.J., Preston G.A., Lionaki S., et al. Antibodies with dual reactivity to plasminogen and complementary PR3 in PR3-ANCA vasculitis. J Am Soc Nephrol. 2008;19(12):2421–2429.
6 Chen M., Kallenberg C.G. New advances in the pathogenesis of ANCA-associated vasculitides. Clin Exp Rheumatol. 2009;27(1 Suppl 52):S108–S114.
7 Kessenbrock K., Krumbholz M., Schonermarck U., et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med. 2009;15(6):623–625.
8 Preston G.A., Yang J.J., Xiao H., et al. Understanding the pathogenesis of ANCA: where are we today? Cleve Clin J Med. 2002;69(Suppl 2):SII51–SII54.
9 Jennette J.C., Xiao H., Falk R.J. Pathogenesis of vascular inflammation by anti-neutrophil cytoplasmic antibodies. J Am Soc Nephrol. 2006;17(5):1235–1242.
10 Franssen C.F., Stegeman C.A., Kallenberg C.G, et al. Antiproteinase 3- and vasculitis. Kidney Int. 2000;57(6):2195–2206.
11 Sugiyama S., Kugiyama K., Aikawa M., et al. Hypochlorous acid, a macrophage product, induces endothelial apoptosis and tissue factor expression: involvement of myeloperoxidase-mediated oxidant in plaque erosion and thrombogenesis. Arterioscler Thromb Vasc Biol. 2004;24(7):1309–1314.
12 Kain R., Exner M., Brandes R., et al. Molecular mimicry in pauci-immune focal necrotizing glomerulonephritis. Nat Med. 2008;14(10):1088–1096.
13 Wilde B., Thewissen M., Damoiseaux J., et al. T cells in ANCA-associated vasculitis: what can we learn from lesional versus circulating T cells? Arthritis Res Ther. 2010;12(1):204.
14 Xiao H., Heeringa P., Hu P., et al. Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. J Clin Invest. 2002;110(7):955–963.
15 Xiao H., Heeringa P., Liu Z., et al. The role of neutrophils in the induction of glomerulonephritis by anti-myeloperoxidase antibodies. Am J Pathol. 2005;167(1):39–45.
16 Schreiber A., Xiao H., Falk R.J, et al. Bone marrow-derived cells are sufficient and necessary targets to mediate glomerulonephritis and vasculitis induced by anti-myeloperoxidase antibodies. J Am Soc Nephrol. 2006;17(12):3355–3364.
17 Pfister H., Ollert M., Frohlich L.F., et al. Antineutrophil cytoplasmic autoantibodies against the murine homolog of proteinase 3 (Wegener autoantigen) are pathogenic in vivo. Blood. 2004;104(5):1411–1418.
18 Nolan S.L., Kalia N., Nash G.B., et al. Mechanisms of ANCA-mediated leukocyte-endothelial cell interactions in vivo. J Am Soc Nephrol. 2008;19(5):973–984.
19 Little M.A., Savage C.O. The role of the endothelium in systemic small vessel vasculitis. Clin Exp Rheumatol. 2008;26(3 Suppl 49):S135–S140.
20 Guilpain P., Servettaz A., Goulvestre C., et al. Pathogenic effects of antimyeloperoxidase antibodies in patients with microscopic polyangiitis. Arthritis Rheum. 2007;56(7):2455–2463.
21 Hattar K., Sibelius U., Bickenbach A., et al. Subthreshold concentrations of anti-proteinase 3 antibodies (c-ANCA) specifically prime human neutrophils for fMLP-induced leukotriene synthesis and chemotaxis. J Leukoc Biol. 2001;69(1):89–97.
22 Mueller A., Holl-Ulrich K., Feller A.C, et al. Immune phenomena in localized and generalized Wegener’s granulomatosis. Clin Exp Rheumatol. 2003;21(6 Suppl 32):S49–S54.
23 Tervaert J.W. Translational mini-review series on immunology of vascular disease: accelerated atherosclerosis in vasculitis. Clin Exp Immunol. 2009;156(3):377–385.
24 Voswinkel J., Muller A., Lamprecht P. Is PR3-ANCA formation initiated in Wegener’s granulomatosis lesions? Granulomas as potential lymphoid tissue maintaining autoantibody production. Ann N Y Acad Sci. 2005;1051:12–19.
25 Muschen M., Warskulat U., Perniok A., et al. Involvement of soluble CD95 in Churg-Strauss syndrome. Am J Pathol. 1999;155(3):915–925.
26 Hellmich B., Ehlers S., Csernok E., et al. Update on the pathogenesis of Churg-Strauss syndrome. Clin Exp Rheumatol. 2003;21(6 Suppl 32):S69–S77.
27 Lamprecht P., Gutzeit O., Csernok E., et al. Prevalence of ANCA in mixed cryoglobulinemia and chronic hepatitis C virus infection. Clin Exp Rheumatol. 2003;21(6 Suppl 32):S89–S94.
28 Pendergraft W.F.3rd, Pressler B.M., Jennette J.C., et al. Autoantigen complementarity: a new theory implicating complementary proteins as initiators of autoimmune disease. J Mol Med. 2005;83(1):12–25.
29 Pendergraft W.F., Alcorta D.A., Segelmark M., et al. ANCA antigens, proteinase 3 and myeloperoxidase, are not expressed in endothelial cells. Kidney Int. 2000;57(5):1981–1990.
30 Seko Y. Giant cell and Takayasu arteritis. Curr Opin Rheumatol. 2007;19(1):39–43.
31 Chauhan S.K., Singh M., Nityanand S. Reactivity of gamma/delta T cells to human 60-kd heat-shock protein and their cytotoxicity to aortic endothelial cells in Takayasu arteritis. Arthritis Rheum. 2007;56(8):2798–2802.
32 Chauhan S.K., Tripathy N.K., Nityanand S. Antigenic targets and pathogenicity of anti-aortic endothelial cell antibodies in Takayasu arteritis. Arthritis Rheum. 2006;54(7):2326–2333.
33 Brack A., Geisler A., Martinez-Taboada V.M., et al. Giant cell vasculitis is a T cell-dependent disease. Mol Med. 1997;3(8):530–543.
34 Deng J., Younge B.R., Olshen R.A., et al. Th17 and Th1 T-cell responses in giant cell arteritis. Circulation. 2010;121(7):906–915.
35 Nagano H., Mitchell R.N., Taylor M.K., et al. Interferon-gamma deficiency prevents coronary arteriosclerosis but not myocardial rejection in transplanted mouse hearts. J Clin Invest. 1997;100(3):550–557.
36 Tellides G., Tereb D.A., Kirkiles-Smith N.C., et al. Interferon-gamma elicits arteriosclerosis in the absence of leukocytes. Nature. 2000;403(6766):207–211.
37 Dal Canto A.J., Swanson P.E., O’Guin A.K., et al. IFN-gamma action in the media of the great elastic arteries, a novel immunoprivileged site. J Clin Invest. 2001;107(2):R15–R22.
38 Amento E.P., Ehsani N., Palmer H., et al. Cytokines and growth factors positively and negatively regulate intersitial collagen gene expression in human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 1991;11:1223–1230.
39 Raines E., Rosenfeld M., Libby P. The role of macrophages. In: Fuster V., Topol E.J., Nabel E.G. Atherothrombosis and coronary artery disease. ed 2. Philadelphia: Lippincott Williams & Wilkins; 2004:505–520.
40 Jones R.B., Tervaert J.W., Hauser T., et al. Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis. N Engl J Med. 2010;363(3):211–220.
41 JCS Joint Working Group. Guideline for Management of Vasculitis Syndrome (JCS 2008). Circ J. 2011;75(2):474–503.