Chapter 64 Peripheral Vascular Anomalies, Malformations, and Vascular Tumors
Nonmalignant vascular anomalies can be functionally divided into two groups: proliferative vascular lesions and static vascular malformations. Unfortunately, this distinction is not universally appreciated, and diagnoses are often incorrect in the literature and clinical practice because of knowledge gaps and lack of clarity. Box 64-1A delineates a classification initially proposed by Mulliken and Glowaki,1 and Box 64-1B is an updated version published by the International Society for the Study of Vascular Anomalies (access at www.issva.org/). Functional classification helps guide management and prognosis. This chapter discusses peripheral (i.e., not central nervous system [CNS] or cardiac) vascular anomalies, including vascular tumors and syndromic vascular disorders, and offers new genetic information and insights into putative signaling pathways implicated in their development.
 Box 64-1A Functional Classification of Vascular Anomalies
Box 64-1A Functional Classification of Vascular Anomalies
Proliferative Nonmalignant* Vascular Lesions and Tumors
* Mitotic figures absent or rare.
Box 64-1B Updated ISSVA Classification of Vascular Anomalies
Vascular Tumors
Tufted angioma (± Kasabach-Merritt’s syndrome)
Kaposiform hemangioendothelioma (± Kasabach-Merritt’s syndrome)
Spindle cell hemangioendothelioma
Other rare hemangioendotheliomas (epithelioid, composite, retiform, polymorphous, Dabska tumor, lymphangioendotheliomatosis, etc.)
Acquired vascular tumors (pyogenic granuloma, targetoid hemangioma, glomeruloid hemangioma, microvenular hemangioma, etc.)
Vascular Malformations
Familial cutaneous and mucosal venous malformation (VMCM)
Fast-flow vascular malformations:
Complex-combined vascular malformations: CVM, CLM, LVM, CLVM, AVM-LM, CM-AVM
A, arterial; AVF, arteriovenous fistula; AV, arteriovenous; C, capillary; G, glomovenous; ISSVA, International Society for the Study of Vascular Anomalies; L, lymphatic; M, malformation; NICH, noninvoluting congenital hemangioma; RICH, rapidly involuting congenital hemangioma; V, venous.
From Enjolras O, Wassef M, Chapot R: Color atlas of vascular tumors and vascular malformations, Cambridge, 2007, Cambridge University Press.
Proliferative Vascular Anomalies and Tumors
Hemangiomas are considered the most common tumors of childhood. They are benign growths of endothelial cells (ECs), with a unique natural history characterized by a rapid growth phase usually beginning in the first weeks of life and continuing until 9 to 12 months of age (Fig. 64-1). The majority of hemangiomas subsequently undergo spontaneous gradual (but extensive) involution. Histological correlation with the growth phase demonstrates involution and is characterized by increased connective tissue in the dermis and fat in the subcutaneous tissues.2 An important exception to this growth/regression pattern is the group of rapidly involuting congenital hemangiomas (RICH), which are generally present in full at birth (or even detected prenatally), and noninvoluting congenital hemangiomas (NICH), which do not change size postnatally.3 Growth curves for these hemangiomas are illustrated in Figure 64-2. A subset of patients with RICH may have high-flow lesions with prenatal or postnatal high-flow characteristics and/or transient coagulopathy4,5 (Fig. 64-3). Congenital nonprogressive hemangiomas have been shown by North et al. to be histologically and immunophenotypically distinct from classical hemangiomas of infancy and are speculated to have a differing pathogenesis.6 NICH-type lesions were found to have high flow clinically (as assessed by Doppler), and inferred histologically, in that small arteries were seen shunting into lobular vessels or abnormal veins.7 Another subtype of hemangiomas are those with minimal or arrested growth, presenting as areas of telangiectasia with peripheral bulkiness. In one series, the majority of this type of hemangioma was present on the lower extremities.8
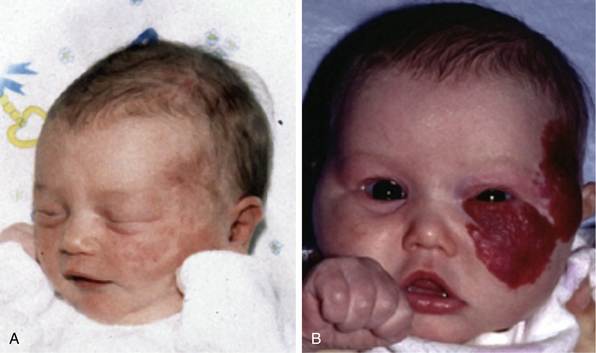
Figure 64-1 Hemangioma of infancy.
A-B, Sequential photos of infant who developed aggressive proliferative hemangioma with ophthalmological as well as cosmetic issues. In early phases (A), this lesion is not easily differentiated from a capillary malformation.
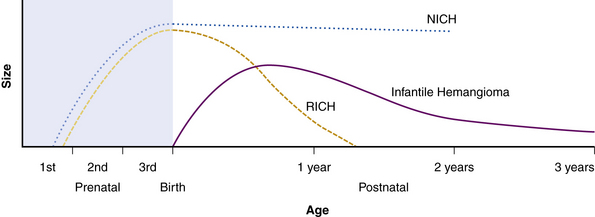
Figure 64-2 Growth curves for infantile hemangioma, rapidly involuting congenital hemangioma (RICH), and noninvoluting congenital hemangioma (NICH).
(From Mulliken J, Enjolras O: Congenital hemangiomas and infantile hemangioma: missing links. J Am Acad Dermatol 50:875–882, 2004; used with permission.)3
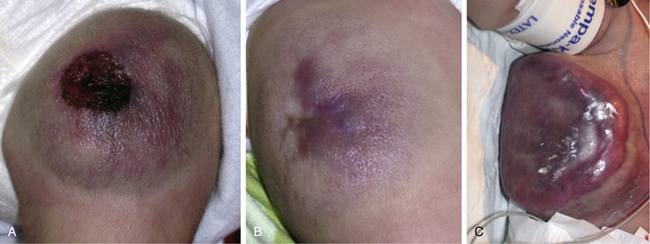
Figure 64-3 Rapidly involuting congenital hemangioma (RICH).
RICH of thigh with ulceration (A) and during natural involution (B). C. RICH of chest wall will high-flow component necessitating inotropic agents, intubation, and embolization. Infant had transient coagulopathy. Notice circumferential halo in both cases.
Typical hemangiomas are known to be most common in females, premature infants, and in the facial region. Results of the multicenter Hemangioma of Infancy Study of over 1000 children with hemangiomas showed an increased incidence in white non-Hispanic infants, multiple gestations, infants born to older mothers, and in association with placenta previa and/or preeclampsia.9 Other studies have shown (1) a threefold increased risk of hemangiomas in infants born to mothers who had transcervical chorionic villous sampling compared to amniocentesis (the incidence of hemangiomas in the amniocentesis group was equivalent to the incidence of hemangiomas in the general population)10,11 and (2) a correlation with placental anomalies with abnormal uteroplacental circulation.12,13 Waner et al. noted a nonrandom distribution of facial hemangiomas and found two patterns of growth: focal lesions (in 76.3% of the 205 patients assessed) and diffuse lesions (in 23.7%). The focal hemangiomas correlated to 22 sites of occurrence, all near lines of mesenchymal or mesenchymal-ectodermal embryonic fusion. The diffuse hemangiomas were in a segmental distribution and were specified as frontonasal (27%), maxillary (35%), or mandibular (38%). There was a threefold increased incidence of ulceration in patients with diffuse hemangiomas compared to that in patients with focal hemangiomas.14 Haggstrom et al. expanded the observation of nonrandom distribution, designating four primary segments (Seg1-Seg4) to correspond with cutaneous location15 (Fig. 64-4). Large hemangioma size, facial location, and/or segmental hemangiomas were more likely to require medical intervention.16 Segmental hemangiomas can be associated with a higher incidence of PHACE(S) syndrome, visceral hemangiomas, and underlying lumbosacral anomalies (e.g., occult spinal dysraphism, including lipomyelomeningocele with tethered cord).17–20PHACE(S) Association is an acronym for posterior fossa structural malformations, hemangiomas (segmental), arterial anomalies, cardiac defects, eye abnormalities, (and sternal and other midline deformities)21 (Fig. 64-5). A patient with a segmental hemangioma and one or more of these criteria has PHACES. In one series, approximately one third of patients with facial segmental hemangiomas were found to have PHACES, those at higher risk having large hemangiomas involving more than one anatomical segment, and in the frontonasal or frontotemporal distribution. Of those with PHACES, most (90%) had more than one extracutaneous finding (most commonly CNS arteriopathy or cardiac anomaly).22 Similarly, Oza et al. observed that patients with large facial segmental cutaneous (Seg1-Seg4) hemangiomas were especially at risk of CNS structural and cerebrovascular anomalies, those with Seg1 distribution hemangiomas had a higher incidence of ocular anomalies, and those with Seg3 distribution had airway, ventral, and cardiac anomalies. In this series, all patients with CNS structural anomalies had concomitant CNS arteriopathies. Also identified were supratentorial CNS anomalies (cortical dysgenesis and migration abnormalities). Arteriopathies are most commonly dysplastic vessels with an aberrant course involving the internal cerebral artery and its embryonic branches ipsilateral to the side of the cutaneous hemangioma.23 Hypoplasia, agenesis, or absence of normal arteries can also occur. In one review, some 20% of patients had arterial occlusions and stenoses.24 Progressive changes can lead to aneurysm formation.23
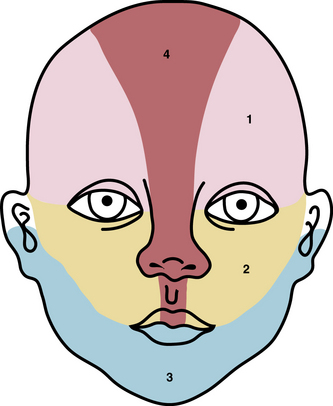
Figure 64-4 Distribution patterns of facial segmental hemangiomas.
(From Haggstrom AN, Lammer EJ, Schneider RA, et al: Patterns of infantile hemangiomas: new clues to hemangioma pathogenesis and embryonic facial development. Pediatrics 117:698–703, 2006.) Reproduced with permission, copyright by the AAP.
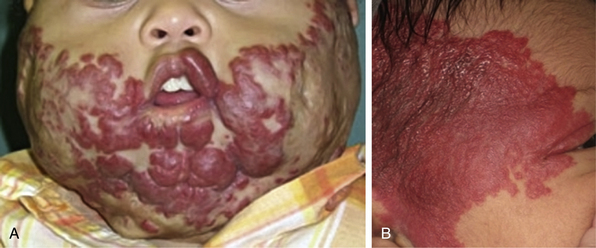
Figure 64-5 Segmental hemangiomas.
Note beard distribution and Seg3 distribution (A). Patient also had subglottic hemangioma. B, Partial Seg1&2 hemangioma that extended to neck and back. Patient was also found to have PHACE (posterior fossa structural malformations, hemangiomas [segmental], arterial anomalies, cardiac defects, eye abnormalities) arteriopathy.
Most hemangiomas are asymptomatic and require no therapy. Despite this clinical course, hemangiomas nonetheless may be the source of significant psychosocial morbidity (although this has not been well studied). Early intervention may be considered to prevent morbidity and/or preclude the need for future surgery. Hemangiomas may cause complications requiring medical therapy to catalyze the involution phase. These complications may include obstruction of the upper airway, ophthalmological disturbances, ulceration or bleeding, persistent soft-tissue deformity, cerebral vasculopathy, and/or high-output congestive heart failure (CHF); all are discussed below.
Kasabach-Merritt Phenomenon, Kaposiform Hemangioendothelioma, and Tufted Angioma
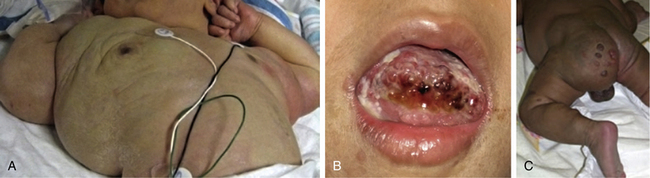
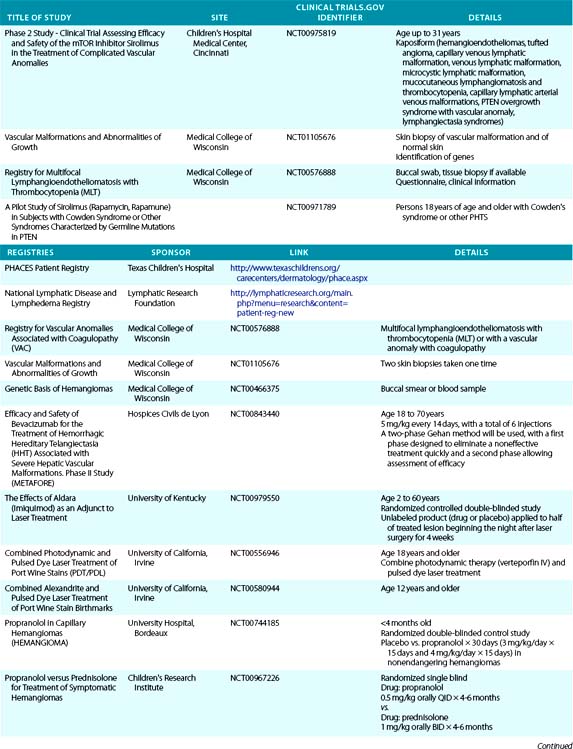
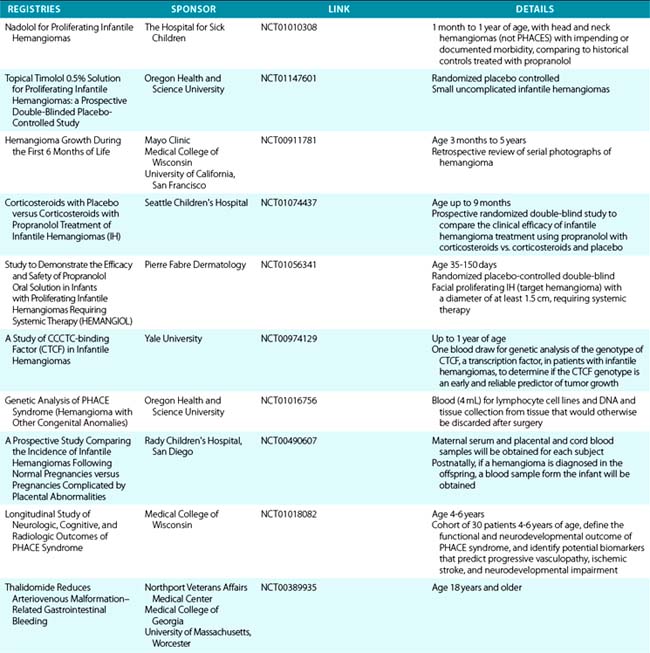
Trapping of platelets and other blood elements (Kasabach-Merritt phenomenon) has been known to occur in association with a subset of vascular anomalies since it was first described in 1940.25 This is an extremely important diagnosis because early detection and rapid evaluation and treatment (if clinically symptomatic) are essential. Kasabach-Merritt phenomenon is not associated with common hemangiomas of infancy, but with kaposiform hemangioendothelioma (KHE) or tufted angiomas.26,27 On examination, the lesion is often edematous, boggy, and ecchymotic (Fig. 64-6). Anatomical predilection is for the chest wall and shoulder, groin extending down the leg, retroperitoneum, or face. Gender distribution tends to be equal. Hematological features of Kasabach-Merritt phenomenon include thrombocytopenia, hypofibrinogenemia, elevated fibrin degradation products, and D-dimers. Radiological hallmarks of KHE are cutaneous thickening, diffuse enhancement with ill-defined margins, small feeding/draining vessels, stranding, and hemosiderin deposits. The histological features of KHE are spindled ECs resembling Kaposi sarcoma (but not associated with human immunodeficiency virus [HIV] infection), abnormal lymphatic-like vessels, microthrombi, hemosiderin, and decreased mast cells and pericytes (which are often seen in hemangiomas). There may be residual tumor after resolution of hematological abnormalities, and radiological studies often demonstrate persistent vascular tumors. Residua of KHE-associated tumors may be dormant vascular tumors rather than scars. Clinically as well as histologically, they differ considerably from involuted hemangiomas. A subset of patients with KHE do not have an associated coagulopathy.28 Treatment of KHE is not standardized but depends on the morbidity, location, and radiological features. Multimodal therapy may include steroids, chemotherapy (most commonly vincristine), interferon (IFN), antifibrinolytic agents, antiplatelet agents, and/or embolization. Diffuse intramuscular involvement often makes surgery not an option. Treatment with Rapamune (sirolimus) has been reported in one case2,29 and is currently being studied in a clinical trial (http://clinicaltrials.gov/ct2/show/NCT00975819?term=sirolimus&rank=82; see Table 64-4).
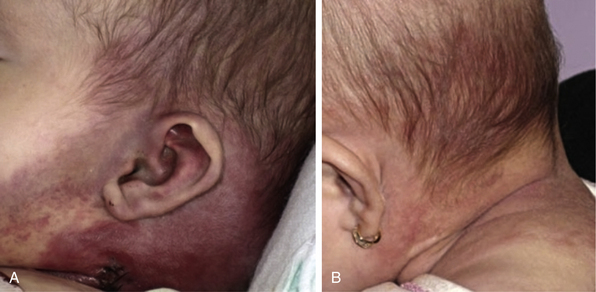
Figure 64-6 A, Kaposiform heman- gioendothelioma presenting with boggy diffuse mass, thrombocytopenia, and coagulopathy. B, After several courses of vincristine therapy.
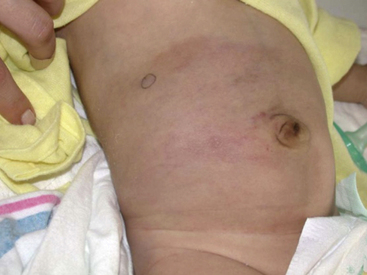
Tufted angioma, first described in the late 1980s, is a benign vascular tumor typified by tufts of capillaries in the dermis. The clinical appearance ranges from erythematous indurated annular nodules to plaques, with or without hypertrichosis (Fig. 64-7). They commonly occur on the trunk and extremities, and they may be associated with Kasabach-Merritt phenomenon. Chu et al. suggest that KHE and tufted angioma may represent a continuum; they report a case of transformation between both tumors within a single patient.30
Pyogenic Granuloma
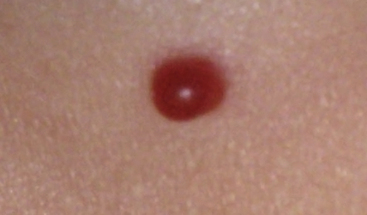
Pyogenic granuloma (also termed lobular capillary hemangioma) is an acquired vascular lesion of the skin and mucous membranes seen in pediatric patients (Fig. 64-8). The lesions have a cervicofacial propensity but can also be located on the trunk or extremities. The majority occur on the skin, and less frequently the mucous membranes (oral cavity and conjunctivae). These lesions are small and papular and tend to bleed. Treatment includes: (1) excision and linear closure, (2) shave excision, (3) cauterization, (4) cryotherapy, (5) carbon dioxide (CO2) or pulsed dye laser, or (6) sclerotherapy.31
Kaposi Sarcoma
Kaposi sarcoma is a neoplasm commonly but not exclusively seen in patients with acquired immunodeficiency syndrome (AIDS).32,33 It is an unusual vascular neoplasm originally described in 1872. The clinical appearance begins as violaceous macules that progress to plaques and papules and then nodules. Kaposi sarcoma is thought to be multifocal rather than metastatic, with multiple lesions occurring simultaneously at different anatomical locations. Histological features include spindle cells and ECs with rare mitotic figures. Evidence indicates that Kaposi sarcoma is monoclonal, although these data are conflicting. A novel human herpesvirus known as Kaposi sarcoma–associated herpesvirus (KSHV), or human herpesvirus type 8 (HHV8), has been identified in Kaposi sarcoma tissue, supporting a viral etiology. Growth factors and cytokines are also believed to be involved in Kaposi sarcoma development. Therapies directed against Kaposi sarcoma include antiviral agents, antiangiogenic drugs, and immunosuppressive agents. Recent studies show the effectiveness of antiretroviral therapy suppressing HIV/AIDS-associated Kaposi sarcoma growth.34
Vascular Malformations
Vascular malformations are present at birth and grow in parallel with the rate of growth of the child, with no propensity to spontaneous involution. They are due to developmental anomalies of the vasculature and may involve one or several types of vessels (arteries, veins, capillaries, or lymphatics). Vascular malformations are properly described according to the affected anomalous vascular channel. They can range from capillary malformations (commonly referred to as port-wine stains; Fig. 64-9) to large bulky growths that can distort the normal structures of the body and potentially lead to a high-output cardiac state (arterial malformations). Studies suggest that capillary malformations may be a result of abnormal innervation of discrete capillary beds causing chronic focal vascular ectasia.35,36 In one study, nerve density was significantly decreased in biopsies of capillary malformations, as compared to uninvolved skin.37
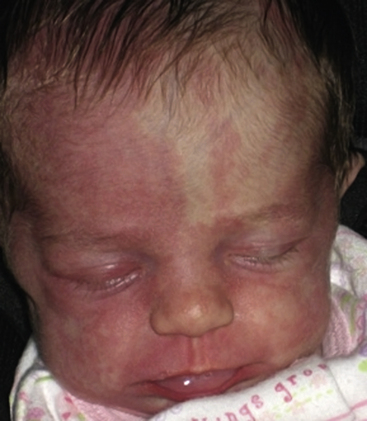
Figure 64-9 Capillary malformations.
Patient has facial capillary malformation and Sturge-Weber’s syndrome.
Lymphatic malformations may cause focal or generalized lymphedema, depending on the magnitude of aberrant lymphatics (Fig. 64-10). Abnormal growth of lymphatic circulation encompasses overdevelopment (in lymphangiodysplasias, lymphangiomas, and lymphangiomatosis), underdevelopment of lymphatic vasculature, or both. Disorders of the lymphatic circulation are common, diverse, and often devastating in their functional consequences. Clinical issues common to lymphatic anomalies reflect the tendency of these malformations to develop: (1) local and systemic infections/cellulitis (infectious and aseptic); (2) leakage (e.g., superficial blebs, chylous ascites, chylothorax, peritonitis, pleural effusions); (3) malabsorption syndromes with significant metabolic consequences; (4) craniofacial distortion interfering with swallowing, airway, or causing significant visceral dysfunction; (5) recurrences or complications after surgery; and (6) swelling of the affected anatomy, with functional limitation.38
Syndromic Vascular Anomalies
There is a spectrum of vascular malformations with dysregulated skeletal/adipose/soft-tissue growth (Table 64-1). Klippel-Trénaunay’s syndrome (capillary-lymphatic-venous malformation with ipsilateral limb enlargement or hypoplasia, venous varicosities or developmental anomalies, or both ) is one of the most common peripheral vascular malformation syndromes (Fig. 64-11). Males and females are affected in equal proportion, and the lower limb is the most frequent site of the anomaly. In severe cases, there may be an accompanying bleeding diathesis characterized by a normal to slightly decreased platelet count, decrease in fibrinogen, and increased D-dimers and fibrin degradation products39 (Fig. 64-12).
Table 64-1 Syndromic Vascular Anomalies and Genetic Information
| Name | Features | Omim |
|---|---|---|
| Blue rubber bleb nevus syndrome Bean syndrome |
Multiple small soft venous malformations on skin, gastrointestinal tract, elsewhere | 112200 |
| CLOVES syndrome | Congenital lipomatous overgrowth, vascular malformations, and epidermal nevi, skeletal/spinal anomalies | 612918 |
| Gorham’s syndrome Gorham-Stout’s syndrome Cystic angiomatosis of bone, diffuse disappearing bone disease |
Lymphangiomatosis, bony destruction | 123880 |
| Klippel-Trénaunay’s syndrome | Capillary, venous, ± lymphatic malformation, hypertrophy of the related bones and soft tissues ± atretic deep venous system of affected extremity | 149000 |
| Maffucci’s syndrome (osteochondromatosis/ dyschondroplasia with vascular lesions) | Enchondromatosis and subcutaneous spindle cell hemangioendotheliomas, risk of chondrosarcoma, other malignancies including CNS | 166000 |
| Proteus’ syndrome | Gigantism (partial) of hands and feet, nevi, asymmetrical and disproportionate overgrowth, hemihypertrophy, macrocephaly, dysregulated adipose tissue, vascular malformations | 176920 |
| CM-AVM; CMC1 5q13-22 RASA-1 (RAS p21 protein activator 1) loss of function |
Multifocal small macular CMs + AVM | 608354 |
| Venous malformations, multiple cutaneous and mucosal; VMCM 9p21 TIE2/TEK gain of function AD (most are sporadic) |
Focal venous dilation with sparse vascular smooth muscle cells, cutaneous, mucosal, ± underlying areas | 600195 |
| Hennekam syndrome 18q21.32 CCBE1 Collagen and calcium-binding EGF domain–containing protein 1 |
Intestinal lymphangiectasia, severe lymphedema, mental retardation | 235510 |
| Hypotrichosis-lymphedema-telangiectasia syndrome HLTS 20q13.33 SOX18 |
Alopecia and/or areas of sparse hair, transparent skin, lymphedema, telangiectasia | 607823 |
| Lymphedema-distichiasis syndrome 16q24.3 AD or de novo FOXC2 loss of function |
Limb edema and double rows of eyelashes (distichiasis) ± other associated anomalies including cardiac, renal, vascular, CNS gene mutation | 153400 |
| Milroy’s disease 5q35.3 AD, AR, or de novo FLT4 vascular endothelial growth factor receptor 3; VEGFR3 loss of function |
Primary congenital hereditary lymphedema type Ia | 153100 |
| Lymphedema praecox Meige’s disease Late-onset lymphedema |
Hereditary lymphedema type II Peripubertal onset |
153200 |
| Lymphangioleiomyomatosis LAM 16p13.3, 9q34 |
Pulmonary (and extrapulmonary) lymphangiomyomatosis; female predominance, adult onset | 606690 |
| HHT Osler-Weber-Rendu AD Loss of function HHT type I 9q34.1 Endoglin (131195) Part of TGF-β receptor complex HHT type 2 12q11-q14 ALK1 Activin A receptor, type II-like kinase-1; ACVRLK1 cell surface receptor for TGF-β superfamily HHT type 3 5q31.3-q32 HHT type 4 7p14 Juvenile polyposis/HHT syndrome; JPHT 18q21.1 SMAD4 tumor suppressor; mutations affect TGF-β signaling |
Cutaneous, mucosal and visceral telangiectasias and AVMs, epistaxis, and gastrointestinal bleeding, ± pulmonary AV fistulas, hepatic, CNS, spinal AVM HHT1: cerebral AVMs > pulmonary AVMs HHT2: hepatic AVMs more common |
187300 600376 601101 610655 175050 |
| Cutis marmorata telangiectatica congenita CMTC Macrocephaly-cutis marmorata telangiectatica congenita |
Cutaneous reticulated mottling, telangiectasia, and phlebectasia, undergrowth or overgrowth of an involved extremity ± other anomalies | 219250 |
| Glomovenous malformation GVM AD 1p22-p21 Glomulin (601749) FKBP (FK506 binding proteins)-associated protein, 48-KD; FAP48 |
Glomovenous malformations Cutaneous venous malformations with glomus cells surrounding distended vein-like channels |
138000 |
| PHACES Association | Posterior fossa brain malformations Segmental facial hemangiomas Arterial anomalies Cardiac anomalies Eye abnormalities Sternal or midline anomalies |
606519 |
| Bannayan-Riley-Ruvalcaba 10q23.31 PTEN Phosphatase and tensin homolog; tumor suppressor |
Macrocephaly, multiple lipomas, vascular anomalies, pigmented macules of the penis | 153480 |
| Cowden’s syndrome 10q23.31 AD PTEN Phosphatase and tensin homolog; tumor suppressor PHTS |
Macrocephaly, multiple hamartomas, cutaneous verrucous lesions, gingival/buccal papules, facial trichilemmomas, risk of breast/ thyroid/renal/endometrial malignancies, cerebelloparenchymal disorder VI (Lhermitte-Duclos’ disease) | 158350 |
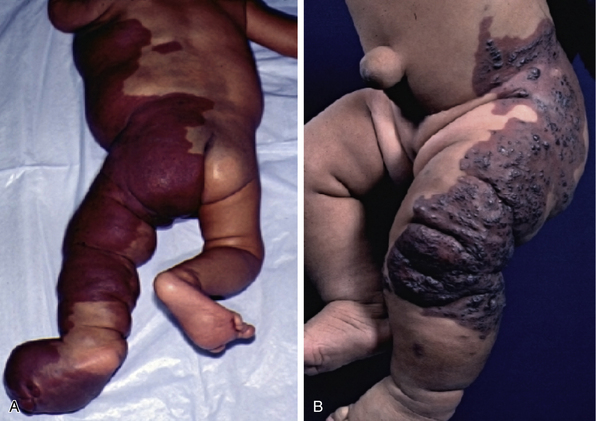
Figure 64-11 A, Patient with Klippel-Trénaunay’s vascular malformation syndrome complicated by leg-length discrepancy, asymmetrical foot size requiring custom orthotics, lymphopenia, and frequent septic episodes due to abnormal lymphatic communications. B, Patient with Klippel-Trénaunay’s vascular malformation syndrome, leg length discrepancy with cutaneous capillary malformation, and blebs prone to bleeding.
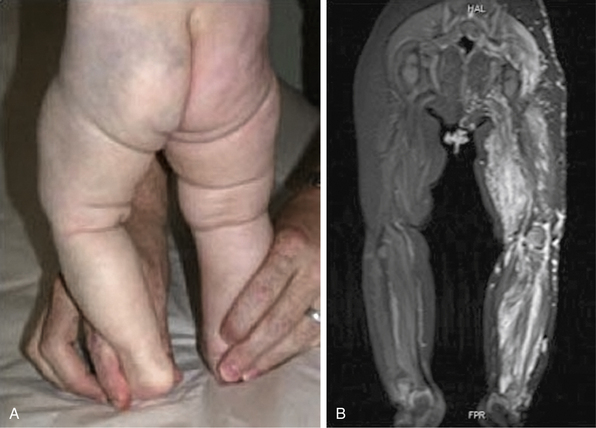
Figure 64-12 Patient with venous vascular malformation of left leg (A), with leg length discrepancy and extensive involvement, as noted on magnetic resonance imaging (MR) (B).
Patient later developed knee contractures, pain, and coagulopathy.
Sturge-Weber’s syndrome includes a capillary malformation in the trigeminal distribution, intracranial angiomatosis and dysplasia, seizures, and glaucoma (see Fig. 64-9). Other examples of dysmorphic syndromes associated with vascular malformation are Turner’s and Noonan’s syndromes, Parkes Weber’s syndrome, hereditary hemorrhagic telangiectasia (HHT), blue rubber bleb nevus syndrome (Fig. 64-13), Maffucci’s syndrome, CLOVES syndrome (congenital lipomatous overgrowth, vascular malformations, epidermal nevi, spinal/skeletal anomalies or scoliosis), Proteus’s syndrome, Bannayan-Riley-Ruvalcaba’s syndrome, and Cowden’s syndrome (see Table 64-1). Syndromes noted for CNS vascular anomalies include von Hippel-Lindau, ataxia-telangiectasia, Sturge-Weber, and tuberous sclerosis; however, CNS and spinal arterial or venous anomalies are now known to occur in association with a number of vascular anomalies.23,40–45
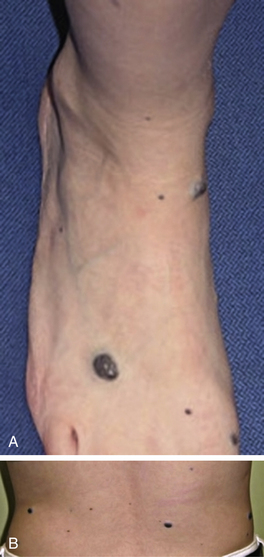
Figure 64-13 Patient with blue rubber bleb nevus syndrome.
Patient has large vascular malformation of neck and history of severe gastrointestinal bleeding due to similar vascular malformations in gastrointestinal tract.
Dysmorphic syndromes associated with hemangiomas are predominantly associated with superficial segmental hemangiomas such as PHACES Association or sacral and/or genitourinary defects, associated with hemangiomas in the lumbar area.19,46
PTEN-Associated Hamartoma Syndromes
PTEN (phosphatase and tensin homolog protein) is a tumor suppressor gene. Patients with a PTEN mutation are susceptible to cancers and warrant early and regular screening. Some patients with vascular anomalies (arteriovenous, lymphatic, venous) have the PTEN mutation, such as those with Cowden’s and Bannayan-Riley-Ruvalcaba’s syndromes.47 A family history or presence of lipomas, thyroid disorders, tricholemmas, macrocephaly, and penile lentigines may point to a PTEN mutation. Consultation with a geneticist and family screening for mutations is indicated, and early screening for thyroid, breast, brain, gynecological, and other cancers should be initiated for all individuals with the PTEN mutation.47,48 Tan et al. recommend screening for PTEN mutations in patients with vascular malformations and the described findings and/or multiple vascular anomalies with a characteristic angiographic appearance, adipose-containing intramuscular lesions, and multiple intracranial developmental venous anomalies.40
Patients with Cowden’s syndrome have typical skin growths that may resemble small, uniform, cutaneous and mucosal warts or skin tags, as well as macrocephaly and cognitive delay. Malignancies seen in patients with Cowden’s syndrome are usually breast, thyroid, or endometrium. Additional findings are benign tumors, thyroid nodules, breast masses, and Lhermitte-Duclos’ disease, a benign noncancerous brain tumor, which is pathognomonic. Often the suspicion of Cowden’s syndrome begins with a family history of thyroid nodules, lipomas (benign fatty lumps), and/or cancers. If the family history and clinical spectrum (macrocephaly, hamartomas, skin tag–appearing lesions) are present, the patient and family should be referred to a genetics specialist for further discussion and blood testing for the presence of the PTEN mutation. Since not all the mutations are available for testing, more sophisticated tests may be required if the initial test is negative and the patient/family fulfills the criteria for the disorder. Upon the suspicion or diagnosis of Cowden’s syndrome, individuals should be placed in a cancer surveillance program to facilitate early detection and prompt referral for further evaluation and treatment.
Bannayan-Riley-Ruvalcaba’s syndrome is characterized by macrocephaly, noncancerous fatty masses (lipomas), vascular malformations, intestinal polyps, thyroid disorders, pectus excavatum, hyperextensible joints, proximal muscle abnormalities, and predisposition to breast and thyroid cancers. Male patients have penile lentigines. Bannayan-Riley-Ruvalcaba’s syndrome, which is often diagnosed in childhood, is also associated with mutations of the PTEN gene, thus the same guidelines hold true for patients suspected of having this disorder, as well as their family members.
Prenatal Diagnosis of Vascular Anomalies
With the availability of improved techniques in fetal ultrasound and magnetic resonance imaging (MRI), prenatally diagnosed vascular anomalies are becoming increasingly recognized. Most prenatally diagnosed vascular lesions are vascular malformations. Vascular lesions detected prenatally are generally identified by asymmetrical limbs and/or high-flow lesions (e.g., arteriovenous malformations [AVMs]or high-flow RICH-type lesions).49 If symptomatic in utero, such as high-flow vascular lesions compromising fetal hemodynamic status, prenatal therapy with maternal steroids or digoxin can be instituted. Maternal steroid therapy may be helpful in the management of fetal hemangiomas.50,51
Etiology of Hemangiomas and Vascular Malformations
Why do vascular anomalies occur? The simple answer is that they are due to many causes—mechanical, environmental, hormonal, and genetic—although no single etiology is thematic. Within the last several years, major research breakthroughs are unraveling potential etiological factors leading to formation of vascular anomalies, as detailed in excellent reviews.52–55
As subtypes of hemangiomas with segmental cutaneous distribution and associated visceral anomalies became evident, researchers speculated involvement of neural crest–derived cells, further supported by identification of neural crest cell markers (neurotrophin receptor p75) in proliferating hemangioma tissue.56 Several studies demonstrated markers for progenitor mesodermal stem cells (brachyury, GATA) or endothelial and hematopoietic cells (platelet endothelial adhesion molecule [PECAM]-1 [CD31]), intracellular adhesion molecule (ICAM)-3, bcl-2 gene expression, KDR+, CD133+, CD34+, endothelial precursor cells, lymphatic endothelial hyaluronan receptor-1, von Willebrand factor (vWF), and Snrk-1 in hemangioma tissue.57–60 Constitutive activation of the endothelial tie-2 receptor and vascular endothelial growth factor receptor (VEGFR)-2-related signaling pathways have been identified in human hemangiomas of infancy.52,54,61
Clonality of ECs was demonstrated,62,63 and the potential role of ECs in hemangioma development elucidated.64–66 Bischoff et al. isolated hemangioma-derived stem cells, which unlike other precursor cells, grew in vitro and differentiated in vivo into cells with properties of hemangiomas, including the eventual presence of adipocytes, as seen in involuting hemangiomas.2 Hemangiomas and placental vessels express common proteins including glucose transporter (GLUT)-1.67 This discovery is of diagnostic utility and spearheaded insights into placenta-based hypotheses. For example, Mihm and Nelson proposes a metastatic niche theory for hemangioma development, suggesting the placenta prepares hemangioma precursor cells that “home” to sites of hemangioma growth68 (Fig. 64-14). Proliferating hemangiomas have been shown to express VEGF-A as well as genes involved with nuclear factor (NF)-κB-related pathways.69,70
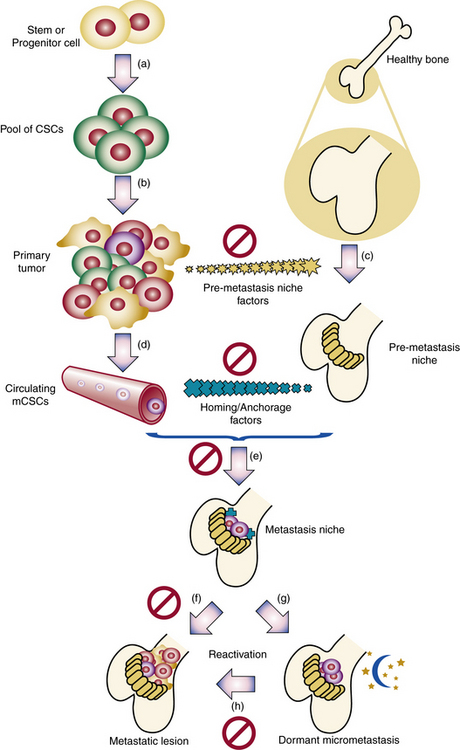
Figure 64-14 Metastatic niche theory of hemangioma development.
(From Mihm MC, Nelson JS: Hypothesis: the metastatic niche theory can elucidate infantile hemangioma development. J Cutan Pathol 37:83–87, 2010; used with permission.)
In addition, proapoptotic factors and appearance of adipocytes during the involution phase support a role for inflammation and immunoregulation in this process71 (Fig. 64-15). The vast majority of hemangiomas appear to be sporadic; however, familial cases harboring germline mutations of angiogenesis-related genes (VEGF2 and tumor endothelial matrix marker [TEM8]) have been identified.72 A secondary somatic event appears to be necessary for hemangioma development. Box 64-2 summarizes features of hemangioma endothelial cells.
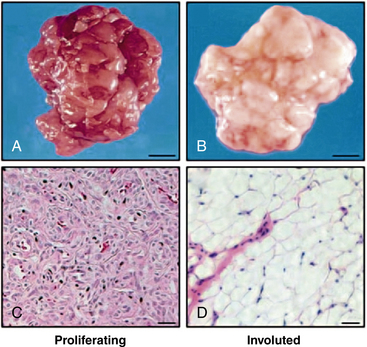
Figure 64-15 Adipocytes in hemangioma involution.
Photographs and hematoxylin & eosin–stained frozen sections of resected proliferating hemangioma from 3-month-old child (A, C) and involuted hemangioma of 7-year-old child (B, D). Note presence of adipocytes in involuted hemangioma.
(From Yu Y, Fuhr, J, Boye, E, Gyorffy, S, Soker, S, Atalia, A, Mulliken, J, Bischoff. Mesenchymal Stem Cells and Adipogenesis in Hemangioma Involution. Stem Cells 2006;24:1605–1612.2 used with permission.)
Box 64-2 Summary of Properties of Hemangioma Endothelial Cells*
Express
Glut-1, vascular endothelial growth factor (VEGF) receptors, CD 31, CD34, VEGF-A, β-FGF, IGF-2, HIF-1α, Snrk-1, Tie-2, angiopoietin-2
Markers for progenitor mesodermal stem cell (brachyury, GATA)
Endothelial and hematopoietic cell markers (platelet endothelial adhesion molecule [PECAM]-1; CD31), intracellular adhesion molecule-2 (ICAM-3), bcl-2 gene expression, KDR+, CD133+, CD34+
Lymphatic endothelial hyaluronan receptor-1 (LYVE-1)
Angiotensin-converting enzyme (ACE) and angiotensin receptor 2 (ATR2)
Partially differentiated, resembling fetal endothelial cells in culture
Intrinsic Vascular Endothelial Growth Factor Signaling
Calcitonin gene-related peptide (CGRP)–containing nerve fibers
Increased TIE-2 expression/enhanced response to angiopoietin-1
Increased pericytes during involution
Increased mast cells (produce tissue inhibitors of metalloproteinases [TIMPs], interferon, transforming growth factor [TGF]-β during involution)
Endothelial Progenitor Cells or Stem Cells
* Summary of several reviews and manuscripts.
Genetics and Vascular Malformations
Newly identified genetic mutations are present in many hereditary vascular anomalies, as are well-defined genetic mutations and inheritance patterns (see Table 64-1). Although modes of inheritance are not consistently straightforward, genetic counselors have an increasing role consulting with patients and family members. Furthermore, identification of genes associated with vascular anomalies has greatly contributed to the windfall of basic research studying the signaling mechanisms involved with these disorders.
The gene for hereditary lymphedema has been linked to distal chromosome 5q, an area where a VEGF-C receptor (FLT4) has been mapped. The FLT4 gene is a marker for lymphatic endothelium during development, and VEGF-C receptor has been detected in lymphatic vasculature. Distichiasis is the presence of a second row of eyelashes arising from the meibomian glands of the eyelids. This can be inherited alone or as a component of lymphedema-distichiasis syndrome. Mutations in the FOXC2 gene (a forkhead, or Fox-box gene, coding for winged helix transcription factors) have been identified in the lymphedema-distichiasis syndrome.65 Additionally, Brooks et al.73 identified the same mutation in patients with isolated distichiasis, suggesting that hereditary distichiasis and lymphedema-distichiasis may represent the same disorder with different phenotypic expression. Levinson et al. reported genotype-phenotype correlations, with FLT4 mutations associated with congenital lymphedema and FOXC2 mutations in pubertal-onset lymphedema. Both groups had similar male and female penetrance.74
Mutations that are passed through the germline and predispose family members to hemangioma development may also be involved in sporadic hemangiomas. Consistent with this speculation, two recent studies indicate clonality, demonstrating nonrandom X-inactivation and loss of heterozygosity.62,63 Furthermore, Walter et al. identified two unique somatic mutations of the VEGFR genes, VEGFR2 (FLK1/KDR) and VEGFR3 (FLT4) in hemangioma specimens.63
Capillary malformations–arteriovenous malformations (CM-AVM) have been correlated with RASA-1 mutations, characterized by AVMs in the brain, limbs, or spine and evolving cutaneous capillary malformations44,75,76 (Fig. 64-16). Mutations in the angiopoietin receptor TIE2/TEK, causing up-regulation of Tie-2, are associated with multifocal cutaneomucosal venous malformation (VMCM) with phenotypic heterogeneity.77–79
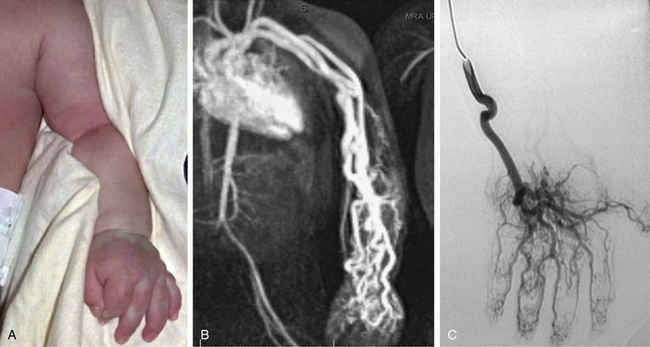
Figure 64-16 Arteriovenous malformation (AVM) with RASA1 mutation.
A, AVM of arm noted at birth. Patient had thrill and bruit of right arm and high-output cardiac failure. B, Magnetic resonance image (MRI) of patient. C, Angiogram. Patient was later found to have scattered multiple, small, macular, cutaneous red lesions, and RASA1 mutation was identified.
Clinical Issues
Clinical issues warranting evaluation and treatment of hemangiomas and vascular malformations can be found in Tables 64-2 and 64-3, and clinical trials are listed in Table 64-4.
Table 64-2 Significant Clinical Issues Warranting Evaluation of Hemangiomas
| Clinical Finding | Recommended Evaluation |
|---|---|
| Hemangiomatosis—multiple small cutaneous hemangiomas | Evaluate for parenchymal hemangiomas, especially hepatic/CNS/gastrointestinal |
| Cutaneous hemangiomas in beard distribution | Evaluate for airway hemangioma, especially if presenting with stridor |
| Facial hemangioma involving significant area of face | Evaluate for PHACES MRI ± contrast for orbital hemangioma ± posterior fossa malformation MRA brain, neck to thoracic aorta Cardiac, ophthalmological evaluation Evaluate for midline abnormality—supraumbilical raphe, sternal atresia, cleft palate, thyroid abnormality Evaluate thyroid function MRA evaluation of craniocervical vessels for anomaly |
| Periocular hemangioma | MRI ± contrast of orbit Ophthalmological evaluation |
| Paraspinal midline vascular lesion | Ultrasound (if <6 months of age) or MRI to evaluate for occult spinal dysraphism ± underlying vascular lesion |
| Thrill or bruit, or both, associated with hemangioma | Cardiac evaluation and echo to rule out diastolic reversal of flow of aorta MRI/Doppler of vascular lesion to evaluate flow characteristics |
| Large hemangioma, especially hepatic | Ultrasound with Doppler flow MRI ± contrast Thyroid function studies |
| Preferential position (e.g., torticollis, flexure contracture) | Consider physical therapy evaluation |
| Delayed milestones | Consider side effect of corticosteroids (myopathy, weight related) or interferon (especially spastic diplegia), CNS issue, hearing assessment |
CNS, central nervous system; MRA, magnetic resonance angiography; MRI, magnetic resonance imaging.
Table 64-3 Clinical Findings and Treatment of Hemangiomas and Vascular Malformations
| Clinical Finding | Recommended Treatment |
|---|---|
| Hemangiomas | |
| Segmental Distribution | |
| Severe ulceration/maceration | Encourage cleansing regimen twice daily Sterile saline soaks/air drying/nonstick gauze ± Flashlamp-pulsed dye laser ± Oral propranolol ± Metronidazole gel Analgesics |
| Bleeding (not Kasabach-Merritt’s phenomenon) | Gelfoam (Pharmacia Pfizer, New York)/Surgifoam (Johnson & Johnson, Somerset, NJ) Compression therapy Kids QR Powder (Biolife Inc., Sarasota, FL) ± Embolization |
| Hemangioma with ophthalmological sequelae | Patching therapy as directed by ophthalmologist Topical timolol maleate gel-forming solution (Merck & Co., Whitehouse Station, NJ) vs. oral propranolol vs. surgery |
| Subglottic hemangioma | Propranolol ± KTP laser ± surgery Tracheotomy if required |
| Kasabach-Merritt’s phenomenon | Corticosteroids, aminocaproic acid, vincristine, ± chemotherapy, interferon alfa ± Embolization |
| High-flow hepatic hemangioma | Propranolol or corticosteroids ± embolization ± chemotherapy, ± surgery, ± Synthroid Transplantation |
| Vascular Malformations | |
| Swelling | If airway, consider tracheotomy vs. surgery Massage, compression therapy Look for source of infection and treat |
| Phlebolith | Antiinflammatory agent |
| Limb-length discrepancy | Shoe insert vs. epiphysiodesis vs. serial observation |
| Shoe-size discrepancy | Wear two different shoe sizes Epiphysiodesis vs. ray resection |
| Pain | Evaluate for phlebolith or deep venous thrombosis Analgesics Anticoagulation if thrombosis ± Nerve block, ± sclerotherapy (if not thrombosis) |
| Recurrent infections/swelling | Local hygienic care Rotating oral antibiotic prophylaxis |
| Chylous ascites | Drainage/low-fat diet/parenteral nutrition/albumin ± Immunoglobulin replacement |
Facial Port-Wine Stains
When an infant has a macular vascular stain covering the trigeminal distribution, the diagnosis may not initially be apparent. If the lesion remains static, the diagnosis is capillary malformation, and the child is at risk for Sturge-Weber’s syndrome (dysmorphogenesis of cephalic neuroectoderm) and must be followed for development of glaucoma, seizures, and developmental delay. The risk of ophthalmological sequelae is highest in patients with lesions located in the ophthalmic (or V1 trigeminal) cutaneous area. In one study, port-wine stains of the eyelids, bilateral distribution of the birthmark, and unilateral port-wine stains involving all three branches of the trigeminal nerve were associated with a significantly higher likelihood of having eye or CNS complications, or both.80 Open studies assessing imaging, laser, epilepsy, and other assessments are listed at http://www.sturge-weber.org/research/current-studies-seeking-participants.html.
Patients with segmental facial and upper trunk hemangiomas, especially of plaque-like quality, should be evaluated for PHACES Association, with (1) cervicofacial MRI evaluation for assessment of structural and vascular abnormalities, (2) magnetic resonance angiography (MRA) of brain, neck, and upper chest for identification of arteriopathies, (3) cardiac evaluation, (4) ophthalmological evaluation, (5) thyroid function studies, and (6) thorough examination to assess for clefting or other anomalies (e.g., sternum, palate, midline supraumbilical raphe).
Airway Symptoms
Recurrent stridor with progressive worsening of symptoms (in an infant with or without cutaneous hemangiomas) should alert the physician to the possibility of a subglottic hemangioma. Definitive diagnosis is made by bronchoscopy with direct visualization of the airway. Orlow et al. reported increased risk of symptomatic airway hemangiomas in association with a distinctive cutaneous beard hemangioma distribution.81 However, even in the absence of cutaneous signs, one should also entertain this diagnosis. Controversies in management of subglottic hemangiomas include surgery (submucous resection); laser (CO2 vs. potassium titanyl phosphate [KTP]); oral propranolol; steroids (intralesional vs. systemic); and interferon alpha (IFN-a) or tracheotomy, or both. Carbon dioxide or KTP laser may be helpful for noncircumferential subglottic hemangiomas.82
Periocular Vascular Anomalies
Periocular lesions present unique management problems. What is visible externally is the tip of the iceberg. Thus early radiological evaluation by MRI, ophthalmological evaluation, and follow-up are essential, since vascular lesions put patients at risk for blepharoptosis, amblyopia, strabismus, proptosis, optic nerve compression, and anisotropia. Early intervention is essential to minimize ocular sequelae. Patients with PHACES or Sturge-Weber’s syndrome must be monitored for glaucoma. PHACES patients may also have retinopathies and other anomalies. Periocular hemangiomas belong to the class of hemangiomas that warrant close evaluation and early, active treatment because some have the potential to threaten or permanently compromise vision. Failure to do so can lead to severe and permanent visual disturbances by occluding the visual axis, compressing the globe, or expanding into the retrobulbar space.
Complications such as amblyopia, significant refractive errors, and strabismus are seen in up to 80% of patients with untreated periocular hemangiomas. Hemangioma size (>1 cm in largest diameter) and diffuse segmental hemangiomas were associated with amblyopia in one study,83 and prompt treatment resulted in reversal of ophthalmological sequelae.84 Thus, all children with periocular hemangiomas warrant early evaluation and serial follow-up by regular serial cycloplegic refractions performed by a physician skilled in retinoscopy of preverbal children. Therapies include patching of the contralateral eye, topical or systemic pharmacotherapy, and/or surgery. Unique risks of intralesional steroids for periocular hemangiomas include central retinal artery occlusion or iris depigmentation.85,86
Hepatic Hemangiomas
Hepatic hemangiomas represent a special category. Although many hepatic hemangiomas are asymptomatic, a subset carries a high morbidity and mortality rate. They may be solitary or multiple and may be seen in association with cutaneous hemangiomatosis or be an isolated finding. Even if radiologically extensive, the clinical spectrum ranges from asymptomatic to life threatening, with high-output CHF or profound consumptive hypothyroidism due to elaboration of type 3 iodothyronine deiodinase by the mass.87 Therapies include steroids, propranolol, interferon, embolization, antifibrinolytic therapies, surgical resection, and liver transplantation, with inconsistent results.88
Hemangiomatosis
The child with multiple hemangiomas may have diffuse neonatal hemangiomatosis (DNH), a dermatosis with a graver prognosis, or benign neonatal hemangiomatosis. A subset of babies with numerous (small) cutaneous hemangiomas is predisposed to parenchymal hemangiomas, especially of the liver (also CNS, eye, pancreas, gastrointestinal tract, lungs, or other organs).
Ulcerating Lesions
Hemangiomas in mucosal (perineum, lip) or intertriginous areas or at pressure points (e.g., back) are prone to ulceration, generally during the proliferative phase.89 Local wound care may be adequate (e.g., metronidazole or other antibiotic cream, Vaseline gauze, hydrocolloid gels). If infected, topical or systemic antibiotics, or both, are indicated. Other required therapies may be intralesional or systemic steroids or flashlamp pulsed dye laser. Topical Imiquimod90,91 and platelet-derived growth factor (PDGF)92,93 have been reported as successful therapies for ulcerated hemangiomas, but the latter carries a black box warning. Topical eosin was reported to be efficacious for ulcerated hemangiomas.94 Pain management can be achieved with topical and oral analgesics. Simple but helpful measures to comfort the infant with a painful ulcerating hemangioma include twice-daily sitz baths, air drying, and construction of foam rubber cushions with custom-designed cutout areas to relieve direct pressure on the painful area.
Bleeding Associated with Coagulation and Other Abnormalities in Patients with Vascular Anomalies
As noted earlier, bleeding due to Kasabach-Merritt phenomenon (thrombocytopenia, hypofibrinogenemia, and increased fibrinolysis) is often associated with KHE or tufted angioma. In addition to therapy directed toward the primary tumor, antifibrinolytic agents, antiplatelet agents, and heparin are helpful. A transient coagulopathy may be seen in a subgroup of RICH-type lesions.5 Bleeding may occur with ulcerated hemangiomas. Topical hemostasis may be achieved with QR Powder, Surgifoam, or Gelfoam. Consensus guidelines have been established for management of hereditary hemorrhagic telangiectasia, including bleeding problems.95
Hemodynamic Sequelae
Rarely, hemangiomas may demonstrate transient high-flow functionally (until they have undergone significant involution), mimicking AVMs. Hemangiomas with high flow are most frequently located in the liver. These lesions can lead to significant morbidity with high-output cardiac failure. Nonhepatic hemangiomas, prone to develop a high-flow component, include those involving the parotid gland, upper arm, chest wall, scalp, and (rarely) upper lip. These lesions appear to behave as transiently arterialized hemangiomas. During this time, patients may have a failure to thrive–type picture, hyperdynamic precordium, tachycardia, bounding pulses with a widened pulse pressure, and a thrill/bruit over the hemangioma. These findings should alert the treating physician to monitor the hemodynamic status of these patients by careful physical examination and frequent follow-up evaluation. RICH-type hemangiomas may have arterial flow diagnosed pre- or postnatally. Overall, a minority of patients develop high cardiac output states requiring intervention, including diuretics, inotropic agents, or an embolization procedure.
Orthopedic Concerns
Orthopedic issues associated with vascular anomalies involve those relating to limb-dimension discrepancies (e.g., limb length, hypertrophy, atrophy, macrodactyly, polydactyly, gigantism), scoliosis, and other less common orthopedic problems (e.g., foot and hand deformities, joint abnormalities). Limb-length discrepancies may be associated with quadriceps fatigue, hip and lower back pain, or secondary scoliosis. Serial assessment of limb-length data and bone ages at regular intervals is recommended. Interventions include shoe lifts or epiphyseodesis (surgical growth plate closure) for more modest discrepancies; however, for discrepancies predicted to be greater than 5 cm, or in patients who have already reached skeletal maturity, limb shortening and lengthening are the only options to equalize limb lengths.96 Macrodactyly and gigantism may cause functional problems and difficulty with shoe fit. Therapeutic options include custom shoes, ray or digital resection for macrodactyly of the fingers or toes, debulking procedures, and amputation for severe and otherwise unmanageable cases of hypertrophy.96 These procedures include removal of subcutaneous fat and ray resection, removal of one or more metatarsals and the associated phalanges. Patients with vascular anomalies can also develop joint contractures due to a mass effect from the lesion. Physical therapy with stretching exercises may be adequate to relieve symptoms; however, direct sclerotherapy plus or minus surgical excision may be required.
Gynecological Issues in Patients with Vascular Malformations
Some women with vascular anomalies have such severe menorrhagia that they undergo hysterectomy. Furthermore, pregnancy is not often seen as an option for women with severe vascular anomalies of the lower extremities, owing to exacerbation of leg swelling, pain, and bleeding from the increased pressure of a gravid uterus. The normal physiological changes of pregnancy include increased plasma volume and cardiac output, increased venous pressure, leg edema, and venous stasis. Additionally, during pregnancy there is a 5.5 times increased risk of thromboembolism; this risk is augmented in patients with vascular anomalies, who already have an increased prothrombotic risk. Increased risk of thrombosis with oral contraceptives limits these patients’ choices of contraception. This is also an issue when oral contraceptives are considered to treat dysmenorrhea or other gynecological problems.97
Pregnancy for women with vascular anomalies, especially those in the lower extremities, may cause unique problems related to hormonal changes and compression of venous structures by the enlarging uterus. Preliminary data suggest that the risk of obstetric complications, especially preeclampsia and thrombotic events, is higher in women with vascular anomalies of the lower extremities.83 It is recommended that management of pregnancy be under the direction of an obstetrician who is aware of these risks. Therapy with daily injections of low-molecular-weight heparin (LMWH) during pregnancy may prevent some prothrombotic complications.97 In addition to pregnancy, other hormonal changes, such as those associated with puberty (in males or females) or the menstrual cycle, may present an increased risk of thrombosis within vascular lesions, necessitating medical intervention with anticoagulants.
Psychosocial Issues
Despite the benign clinical course of infantile hemangiomas in the majority of patients, and the tendency of these lesions to naturally involute, families of patients frequently undergo stress related to social interactions and medical care.98,99 Tanner et al. conducted interviews of parents of 25 children (5 months to 8 years of age) with facial hemangiomas.100 They found great variability in parental emotion regarding the lesion. However, support from extended family appeared to be an important factor in coping. Interactions with strangers were a major stress in the majority of cases. Oster studied mother-infant interactions, comparing facial expressions in infants with facial anomalies (including vascular anomalies) and controls, showing that affected infants were capable of effective emotional communication by showing a wide range of facial expressions.101 Earlier studies by this researcher recognized the role of maternal emotion on affective communication.
Contact with stable familiars (e.g., family members, friends, preschool) appeared to be the least stressful. Many families were dissatisfied with medical care for two reasons: (1) imprecise treatment plans, which are inherent with the nature of many hemangiomas, and (2) what the parents perceived as insensitivity on the part of physicians. Williams et al. assessed the psychological profile of children with hemangiomas and their families in a survey distributed to parents of children with hemangiomas. The results suggested that the families, rather than the infants, experienced emotional and psychological distress.102
Contact with other families who are going through or have gone through the same experience enables families to see the light at the end of the tunnel of this curable disorder. In this sense, local family support groups organized at some medical centers, as well as national support networks and meetings, are increasingly providing the necessary stability for families and patients.
Psychosocial stress related to lymphedema is reviewed by Ridner.103 Although the focus of this review is primarily patients with cancer-related secondary lymphedema, the issues apply to patients with vascular anomalies and lymphedema as well. The Internet has played an enormous role in assisting the exchange of information, as well as enabling families and physicians to connect with one another (Table 64-5). As the field becomes more familiar, older patients who had hemangiomas as infants and children are becoming role models, publishing their experiences and speaking at meetings—further enforcing the optimistic outcome. Furthermore, adult patients with vascular malformations are networking with younger patients.
Table 64-5 Web Resources for Patients and Physicians
| Program | Website |
|---|---|
| Medline Plus | www.medlineplus.gov |
| Genetics Home Reference | http://ghr.nlm.nih.gov/ |
| About Face | http://www.aboutfaceinternational.org |
| Arkansas Children’s Hospital Vascular Anomalies Program | http://www.birthmarks.org |
| Boston Children’s Hospital Vascular Anomalies Program | http://web1.tch.harvard.edu |
| Children’s Hospital of Wisconsin | http://www.chw.org |
| Cincinnati Children’s Hospital Vascular Anomalies Program |
http://www.cincinnatichildrens.org/svc/prog/vascular
http://ghr.nlm.nih.gov |
| Hereditary Hemorrhagic Telangiectasia (HHT) Foundation International | www.hht.org (includes link to: International Guidelines for the Diagnosis and Management of HHT J Med Genet 2009)) |
| Klippel-Trénaunay Foundation | http://www.ktfoundation.com/ |
| Klippel-Trénaunay Support Group | http://www.k-t.org/ |
| Lymphatic Research Foundation | http://www.lymphaticresearch.org/ |
| Lymphatic Disorders | http://www.littleleakers.com/ |
| National Foundation for Facial Reconstruction | http://www.nffr.org/ |
| National Lymphedema Network | http://www.lymphnet.org/ |
| National Organization for Rare Diseases | http://www.rarediseases.org |
| National Organization of Vascular Anomalies | http://www.novanews.org |
| Proteus Syndrome | http://www.proteus-syndrome.org |
| Sturge-Weber Foundation | http://www.sturge-weber.com |
| UCSF Vascular Anomalies Program | http://dermatology.medschool.ucsf.edu/ |
| Vascular Birthmarks Foundation | http://www.birthmark.org |
Malignancies
Treatment-related secondary malignancies due to radiation or chemotherapy may arise in patients with vascular anomalies. Thyroid adenoma, thyroid carcinoma, angiosarcoma, or breast cancer may arise in patients who received radiation therapy (an outdated treatment) for cutaneous hemangiomas of infancy.104–106 Acute myeloid leukemia occurred in one case of severe hemangiomatosis treated with multiple medications including chemotherapy.107
Malignancies such as infantile fibrosarcoma, hemangiopericytoma, rhabdomyosarcoma, glioma, neurofibroma, neuroblastoma, leukemia, and lymphoma in infants may mimic benign hemangiomas of infancy.108–117An atypical history and/or physical examination and/or a fixed nonmotile firm mass should alert the practitioner to obtain histological confirmation of the diagnosis.
Concomitant cutaneous hemangiomatosis with hepatic type 2 infantile hepatic hemangioendothelioma (angiosarcoma) has been reported.118 Additional reports include cases of angiosarcoma in adulthood arising at sites of hemangiomas or vascular malformations,119 as well as a case of metastatic hepatic lymphangiosarcoma in a child with multiple benign cutaneous and visceral capillary-lymphatic-venous malformations.108 A higher rate of malignancies can also be seen in young adulthood in patients with PTEN hamartoma syndromes and Maffucci’s syndrome.
Treatment of Hemangiomas
Cautious observation is recommended for the majority of hemangiomas, providing there is no impending danger associated with the lesion. Various reviews and guidelines for treatment are available in the medical literature.86,120 Flashlamp pulsed dye laser is a therapeutic option for some cutaneous hemangiomas, especially in the early phase. Via selective photothermolysis, flashlamp pulsed dye laser selectively destroys superficial dermal vessels while sparing surrounding tissue.121–125 Pulsed dye laser therapy may also be an effective means of treating ulcerated hemangiomas, although this remains controversial. Further information regarding laser therapy of subglottic hemangiomas is discussed later.
Previously, corticosteroids were the most common medication for proliferating hemangiomas necessitating medical intervention. Along with reports supporting their use126,127 are concerns of undue side effects such as hypertension, pseudotumor cerebri, and Pneumocystis carinii pneumonia,128–130 as well as iris depigmentation, failure to thrive, adrenal suppression, cellulitis, and retinal artery occlusion with intralesional use.85,131–135
Steroid therapy has been supplanted by oral propranolol, a nonselective β-blocker. In 2008, Leaute-Labreze et al. serendipitously noted a dramatic effect of propranolol as a treatment for proliferating infantile hemangioma.136 Two patients who were treated with oral corticosteroids developed cardiac issues necessitating treatment with propranolol. An immediate improvement in the hemangiomas led to a pilot study that confirmed this perceptive observation. This has revolutionized therapy for infantile hemangioma warranting treatment, and since then several papers have been published, the majority documenting its efficacy.137–141 Topical β-blockers have also been shown to catalyze involution of superficial hemangiomas.142,143
Side effects are cool extremities, gastrointestinal symptoms, hypotension, and bradycardia, with rare but significant reports of hypoglycemia.144–146 The medication should be held during intercurrent illnesses associated with diminished oral intake and/or respiratory symptoms, as well as prior to any procedures where the child will be fasting.147
The mechanism of propranolol-induced involution is under investigation. Inhibition of (1) proliferation (via G0/G1 cell cycle arrest) and chemotactic mobility and differentiation of cultured endothelial cells, and (2) VEGF-induced phosphorylation of VEGFR-2 and other angiogenesis-related pathways were demonstrated by Lamy et al.148 Itinteang et al. identified angiotensin-converting enzyme (ACE) and angiotensin receptor 2 (ATII) in proliferating hemangioma cells, and suggest a propranolol-induced effect on the renin-angiotensin system of proliferating infantile hemangiomas,149 as shown in Figure 64-17. Other putative mechanisms involve vasoconstriction resulting from decreased nitric oxide (NO) release, cyclic adenosine monophosphate (cAMP)-induced inhibition of VEGF- and fibroblast growth factor (FGF)-β-induced EC proliferation, or inducing apoptosis.150,151
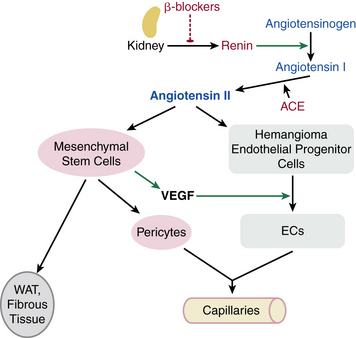
Figure 64-17 Proposed model for mechanism of action of propranolol via effect on renin-angiotensin system on hemangioma endothelial cell (EC) proliferation and differentiation.
Proliferating hemangiomas were shown to express angiotensin-converting enzyme (ACE) and angiotensin receptor 2 (ATR2). Premature infants, Caucasians, and females (groups with predilection to hemangiomas) have high renin levels, contributing to ATII production. Propranolol, by inhibiting renal renin production, blocks this process. EC, endothelial cell; VEGF, vascular endothelial growth factor.
(From Itinteang T, Brasch HD, Tan ST, Day DJ: Expression of components of the renin-angiotensin system in proliferating infantile haemangioma may account for the propranolol-induced accelerated involution. J Plast Reconstr Aesthet Surg 64:759–765, 2011; used with permission.)
Ulcerated hemangiomas may respond to oral propranolol152; however, local management with various medications has been used. Recombinant PDGF has been effective for ulcerated hemangiomas, but a black box warning asserting a higher incidence of cancer fatality in adult patients who used three or more tubes of this medication limits its use (http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0001057).92,93 Various combinations of local and systemic therapies for ulcerated hemangiomas are included in the reviews noted earlier. Topical or systemic antibiotics may be warranted for superinfected ulcerated hemangiomas. Topical and/or systemic analgesics, as well as hemostatic agents (e.g., Kids QR Powder, Biolife Inc., Sarasota, Fla.) for bleeding, may also be needed. A study by Lapidoth et al. demonstrated topical application of eosin, a triphenylmethane dye traditionally used as a topical antibiotic and for treatment of diaper dermatitis, hastened healing of ulcerated hemangioma. In vitro studies demonstrated angiotensin (Ang-2) messenger ribonucleic acid (mRNA) suppression in cultured ECs.94 Other agents include topical Imiquimod,90,91 IFN, and vincristine. Interferon alpha was used in the 1990s, with improvement of endangering hemangiomas, but use of this agent has fallen out of favor, primarily because of concerns about potential neurotoxicity.153–155 Vincristine has been used for problematic hemangiomas, but the necessity for indwelling intravenous (IV) access and potential toxicities limit its use for straightforward hemangiomas.156
Recently, Lazaridou et al. reported treatment of six patients with superficial vascular lesions with topical tacrolimus or pimecrolimus, calcineurin inhibitors.157 Although the cases are listed as hemangiomas based on the age of the patients, results, and photographs, some of these patients may have had vascular malformations. Calcineurin activates NFATCA (nuclear factor of activated T cell, cytoplasmic), leading to interleukin (IL)-2 expression and T-cell activation. Calcineurin inhibitors block this activation pathway. The Down syndrome critical region 1 (DSCR1) gene provides endogenous feedback inhibition of the calcineurin/VEGF-mediated NFAT signaling pathway in ECs.158,159
Surgery
Indications for and timing of surgery for hemangiomas remain controversial. Some surgeons prefer to defer surgery until the hemangioma has undergone substantial involution, with the rationale that the surgery will be less complex and esthetically more favorable. Other surgeons advocate early intervention to possibly prevent medical complications or avert psychological stresses on the patient and/or family. In any case, a well-planned strategy with medical, laser, and surgical management decisions discussed amongst multidisciplinary physicians can provide excellent results.160–171 Surgical techniques are not discussed in this chapter.
Treatment of Vascular Malformations
Because vascular malformations represent a more chronic condition and fewer specific therapies are available for it, a more supportive approach is often taken (see Tables 64-2 and 64-3). The general rule is, “If it’s not broken, don’t fix it.” If there are no clinical symptoms, alert observation is often adequate. Patients with venous vascular malformations (see Fig. 64-12) often experience painful episodes—this is often in conjunction with a phlebolith, or local clot. Therapy with ibuprofen usually suffices. Bleeding from cutaneous blebs may respond to laser cauterization. More severe bleeding from the genitourinary tract may require a combined approach with angiography-guided embolization, laser, or sclerotherapy. Large symptomatic thromboses may require anticoagulation, as might the coagulopathy associated with severe cases. Marimastat, a matrix metalloproteinase (MMP) inhibitor, provided symptomatic relief and reversed bony destruction in a child with intraosseous AVM.172 The clinical trial Safety and Efficacy Study of Sirolimus in Complicated Vascular Anomalies is assessing the effectiveness and safety of the mammalian target of rapamycin (mTOR) inhibitor, rapamycin, in the treatment of children and young adults diagnosed with complicated vascular anomalies (http://clinicaltrials.gov/ct2/show/NCT00975819).
Patients with arterial and venous abnormalities must be cautioned about triggers. Quiescent vascular anomalies may become more problematic secondary to local trauma, infections, or hormonal fluctuations such as puberty, menstruation, and pregnancy. These changes usually manifest as increased fullness of the malformation as well as pain. The etiology of these difficulties is not clearly understood, but in the hormonally mediated settings it is likely related to hormonal stimulation of EC surface receptors. Use of hormonal birth control can elicit undesirable thromboses and/or pain, thus alternative means of contraception are advised.
Lymphatic malformations are among the most frustrating cases encountered. Evaluation of patients with lymphedema involves physical examination and radiological studies including lymphoscintigraphy for extremity involvement. Lymphatic abnormalities involving the mouth and gastrointestinal tract are prone to infection, so patients with lymphatic malformation in these sites benefit from diligent oral hygiene and, occasionally, rotating prophylactic antibiotic regimens. Microcystic lymphatic malformations of the head and neck may respond well to sclerotherapy (doxycycline, bleomycin, picibanil) or surgery, or a combined approach.173 Picibanil (OK-432) is a sclerosing agent derived from a low-virulence strain of Streptococcus pyogenes. A multicenter prospective nonrandomized trial demonstrated the efficacy of picibanil in the treatment of macrocystic cervicofacial lymphatic malformations, and the results were corroborated by further reports.174–176 Breakthroughs in the embryology and biology of lymphatic vasculature provide insights into the therapeutic potential for lymphangiogenesis with lymphangiogenic growth factors (e.g., VEGF-C).177,178
Therapy is generally supportive, with hygienic skin care, sclerotherapy when possible, complete decompressive physiotherapy with compression bandaging, and intermittent pneumatic compression (IPC) therapy.179,180 Rockson and Mayrovitz provide detailed reviews of these therapies.181,182 To date, pharmacological intervention (other than antibiotics when indicated) has not been fruitful. Liposuction for lymphedema has been successful in selected patients with recalcitrant disease.183 Future molecular-based therapies may evolve from laboratory studies, such as those that demonstrated VEGF-C, a lymphatic growth factor, improved lymphedema in a mouse model.184
Acknowledgments
The author extends gratitude to the patients with vascular anomalies and their families, as well as the staff of the Vascular Birthmark Institute of New York, the Institute for Reconstructive Surgery, and the Stephen D. Hassenfeld Children’s Center.
1 Mulliken J.B., Glowacki J. Classification of pediatric vascular lesions. Plast Reconstr Surg. 1982;70(1):120–121.
2 Yu Y., Fuhr J., Boye E., et al. Mesenchymal stem cells and adipogenesis in hemangioma involution. Stem Cells. 2006;24(6):1605–1612.
3 Mulliken J.B., Enjolras O. Congenital hemangiomas and infantile hemangioma: missing links. J Am Acad Dermatol. 2004;50(6):875–882.
4 Konez O., Burrows P.E., Mulliken J.B., et al. Angiographic features of rapidly involuting congenital hemangioma (RICH). Pediatr Radiol. 2003;33(1):15–19.
5 Baselga E., Cordisco M.R., Garzon M., et al. Rapidly involuting congenital haemangioma associated with transient thrombocytopenia and coagulopathy: a case series. Br J Dermatol. 2008;158(6):1363–1370.
6 North P.E., Waner M., James C.A., et al. Congenital nonprogressive hemangioma: a distinct clinicopathologic entity unlike infantile hemangioma. Arch Dermatol. 2001;137(12):1607–1620.
7 Enjolras O., Mulliken J.B., Boon L.M., et al. Noninvoluting congenital hemangioma: a rare cutaneous vascular anomaly. Plast Reconstr Surg. 2001;107(7):1647–1654.
8 Suh K.Y., Frieden I.J. Infantile hemangiomas with minimal or arrested growth: a retrospective case series. Arch Dermatol. 2010;146(9):971–976.
9 Haggstrom A.N., Drolet B.A., Baselga E., et al. Prospective study of infantile hemangiomas: demographic, prenatal, and perinatal characteristics. J Pediatr. 2007;150(3):291–294.
10 Burton B.K., Schulz C.J., Angle B., et al. An increased incidence of haemangiomas in infants born following chorionic villus sampling (CVS). Prenat Diagn. 1995;15(3):209–214.
11 Bauland C.G., Smit J.M., Bartelink L.R., et al. Hemangioma in the newborn: increased incidence after chorionic villus sampling. Prenat Diagn. 2010;30(10):913–917.
12 Lopez Gutierrez J.C., Avila L.F., Sosa G., et al. Placental anomalies in children with infantile hemangioma. Pediatr Dermatol. 2007;24(4):353–355.
13 Colonna V., Resta L., Napoli A., et al. Placental hypoxia and neonatal haemangioma: clinical and histological observations. Br J Dermatol. 2010;162(1):208–209.
14 Waner M., North P.E., Scherer K.A., et al. The nonrandom distribution of facial hemangiomas. Arch Dermatol. 2003;139(7):869–875.
15 Haggstrom A.N., Lammer E.J., Schneider R.A., et al. Patterns of infantile hemangiomas: new clues to hemangioma pathogenesis and embryonic facial development. Pediatrics. 2006;117(3):698–703.
16 Haggstrom A.N., Drolet B.A., Baselga E., et al. Prospective study of infantile hemangiomas: clinical characteristics predicting complications and treatment. Pediatrics. 2006;118(3):882–887.
17 Metry D.W., Hawrot A., Altman C., et al. Association of solitary, segmental hemangiomas of the skin with visceral hemangiomatosis. Arch Dermatol. 2004;140(5):591–596.
18 Metry D.W. Potential complications of segmental hemangiomas of infancy. Semin Cutan Med Surg. 2004;23(2):107–115.
19 Stockman A., Boralevi F., Taieb A., et al. SACRAL syndrome: spinal dysraphism, anogenital, cutaneous, renal and urologic anomalies, associated with an angioma of lumbosacral localization. Dermatology. 2007;214(1):40–45.
20 Drolet B., Garzon M. SACRAL syndrome. Dermatology. 2007;215(4):360. author reply -1
21 Metry D.W., Haggstrom A.N., Drolet B.A., et al. A prospective study of PHACE syndrome in infantile hemangiomas: demographic features, clinical findings, and complications. Am J Med Genet A. 2006;140(9):975–986.
22 Haggstrom A.N., Garzon M.C., Baselga E., et al. Risk for PHACE syndrome in infants with large facial hemangiomas. Pediatrics. 2010;126(2):e418–e426.
23 Oza V.S., Wang E., Berenstein A., et al. PHACES association: a neuroradiologic review of 17 patients. AJNR Am J Neuroradiol. 2008;29(4):807–813.
24 Heyer G.L., Dowling M.M., Licht D.J., et al. The cerebral vasculopathy of PHACES syndrome. Stroke. 2008;39(2):308–316.
25 Kasabach H., Merritt K. Capillary hemangioma with extensive purpura. Am J Dis Child. 1940;59:1063.
26 Sarkar M., Mulliken J.B., Kozakewich H.P., et al. Thrombocytopenic coagulopathy (Kasabach-Merritt phenomenon) is associated with kaposiform hemangioendothelioma and not with common infantile hemangioma. Plast Reconstr Surg. 1997;100(6):1377–1386.
27 Enjolras O., Wassef M., Mazoyer E., et al. Infants with Kasabach-Merritt syndrome do not have “true” hemangiomas. J Pediatr. 1997;130(4):631–640.
28 Gruman A., Liang M.G., Mulliken J.B., et al. Kaposiform hemangioendothelioma without Kasabach-Merritt phenomenon. J Am Acad Dermatol. 2005;52(4):616–622.
29 Blatt J., Stavas J., Moats-Staats B., et al. Treatment of childhood kaposiform hemangioendothelioma with sirolimus. Pediatr Blood Cancer. 2010;55(7):1396–1398.
30 Chu C.Y., Hsiao C.H., Chiu H.C. Transformation between kaposiform hemangioendothelioma and tufted angioma. Dermatology. 2003;206(4):334–337.
31 Gilmore A., Kelsberg G., Safranek S. Clinical inquiries. What’s the best treatment for pyogenic granuloma. J Fam Pract. 2010;59(1):40–42.
32 Greene W., Kuhne K., Ye F., et al. Molecular biology of KSHV in relation to AIDS-associated oncogenesis. Cancer Treat Res. 2007;133:69–127. PMCID: 2798888
33 Patrikidou A., Vahtsevanos K., Charalambidou M., et al. Non-AIDS Kaposi’s sarcoma in the head and neck area. Head Neck. 2009;31(2):260–268.
34 Barbaro G., Barbarini G. HIV infection and cancer in the era of highly active antiretroviral therapy (Review). Oncol Rep. 2007;17(5):1121–1126.
35 Rosen S., Smoller B.R. Port-wine stains: a new hypothesis. J Am Acad Dermatol. 1987;17(1):164–166.
36 Smoller B.R., Rosen S. Port-wine stains. A disease of altered neural modulation of blood vessels? Arch Dermatol. 1986;122(2):177–179.
37 Chang C.J., Yu J.S., Nelson J.S. Confocal microscopy study of neurovascular distribution in facial port wine stains (capillary malformation). J Formos Med Assoc. 2008;107(7):559–566.
38 Blei F. Congenital lymphatic malformations. Ann N Y Acad Sci. 2008;1131:185–194.
39 Mazoyer E., Enjolras O., Bisdorff A., et al. Coagulation disorders in patients with venous malformation of the limbs and trunk: a case series of 118 patients. Arch Dermatol. 2008;144(7):861–867.
40 Tan W.H., Baris H.N., Burrows P.E., et al. The spectrum of vascular anomalies in patients with PTEN mutations: implications for diagnosis and management. J Med Genet. 2007;44(9):594–602. PMCID: 2597949
41 Bisdorff A., Mulliken J.B., Carrico J., et al. Intracranial vascular anomalies in patients with periorbital lymphatic and lymphaticovenous malformations. AJNR Am J Neuroradiol. 2007;28(2):335–341.
42 Viswanathan V., Smith E.R., Mulliken J.B., et al. Infantile hemangiomas involving the neuraxis: clinical and imaging findings. AJNR Am J Neuroradiol. 2009;30(5):1005–1013.
43 Hess C.P., Fullerton H.J., Metry D.W., et al. Cervical and intracranial arterial anomalies in 70 patients with PHACE syndrome. AJNR Am J Neuroradiol. 2010;31(10):1980–1986.
44 Thiex R., Mulliken J.B., Revencu N., et al. A novel association between RASA1 mutations and spinal arteriovenous anomalies. AJNR Am J Neuroradiol. 2010;31(4):775–779.
45 Pascual-Castroviejo I., Alvarez-Linera J., Coya J., et al. Pascual-Castroviejo type II syndrome (P-CIIS). Importance of the presence of persistent embryonic arteries. Childs Nerv Syst. 2011;27(4):617–625.
46 Metry D.W., Garzon M.C., Drolet B.A., et al. PHACE syndrome: current knowledge, future directions. Pediatr Dermatol. 2009;26(4):381–398.
47 Hobert J.A., Eng C. PTEN hamartoma tumor syndrome: an overview. Genet Med. 2009;11(10):687–694.
48 Blumenthal G.M., Dennis P.A. PTEN hamartoma tumor syndromes. Eur J Hum Genet. 2008;16(11):1289–1300.
49 Elia D., Garel C., Enjolras O., et al. Prenatal imaging findings in rapidly involuting congenital hemangioma of the skull. Ultrasound Obstet Gynecol. 2008;31(5):572–575.
50 Morris J., Abbott J., Burrows P., et al. Antenatal diagnosis of fetal hepatic hemangioma treated with maternal corticosteroids. Obstet Gynecol. 1999;94(5 Pt 2):813–815.
51 Schmitz R., Heinig J., Klockenbusch W., et al. Antenatal diagnosis of a giant fetal hepatic hemangioma and treatment with maternal corticosteroid. Ultraschall Med. 2009;30(3):223–226.
52 Arbiser J.L., Bonner M.Y., Berrios R.L. Hemangiomas, angiosarcomas, and vascular malformations represent the signaling abnormalities of pathogenic angiogenesis. Curr Mol Med. 2009;9(8):929–934.
53 Bischoff J. Progenitor cells in infantile hemangioma. J Craniofac Surg. 2009;20(Suppl 1):695–697. PMCID: 2810465
54 Boye E., Olsen B.R. Signaling mechanisms in infantile hemangioma. Curr Opin Hematol. 2009;16(3):202–208. PMCID: 2895461
55 Jinnin M., Ishihara T., Boye E., et al. Recent progress in studies of infantile hemangioma. J Dermatol. 2010;37(4):283–298.
56 Itinteang T., Tan S.T., Brasch H., et al. Primitive mesodermal cells with a neural crest stem cell phenotype predominate proliferating infantile haemangioma. J Clin Pathol. 2010;63(9):771–776.
57 Dadras S.S., North P.E., Bertoncini J., et al. Infantile hemangiomas are arrested in an early developmental vascular differentiation state. Mod Pathol. 2004;17(9):1068–1079.
58 Nguyen V.A., Kutzner H., Furhapter C., et al. Infantile hemangioma is a proliferation of LYVE-1-negative blood endothelial cells without lymphatic competence. Mod Pathol. 2006;19(2):291–298.
59 Khan Z.A., Boscolo E., Picard A., et al. Multipotential stem cells recapitulate human infantile hemangioma in immunodeficient mice. J Clin Invest. 2008;118(7):2592–2599. PMCID: 2413184
60 Itinteang T., Tan S.T., Brasch H., et al. Haemogenic endothelium in infantile haemangioma. J Clin Pathol. 2010;63(11):982–986.
61 Perry B.N., Govindarajan B., Bhandarkar S.S., et al. Pharmacologic blockade of angiopoietin-2 is efficacious against model hemangiomas in mice. J Invest Dermatol. 2006;126(10):2316–2322.
62 Boye E., Yu Y., Paranya G., et al. Clonality and altered behavior of endothelial cells from hemangiomas. J Clin Invest. 2001;107(6):745–752. PMCID: 208946
63 Walter J.W., North P.E., Waner M., et al. Somatic mutation of vascular endothelial growth factor receptors in juvenile hemangioma. Genes Chromosomes Cancer. 2002;33(3):295–303.
64 Yu Y., Flint A.F., Mulliken J.B., et al. Endothelial progenitor cells in infantile hemangioma. Blood. 2004;103(4):1373–1375.
65 Khan Z.A., Melero-Martin J.M., Wu X., et al. Endothelial progenitor cells from infantile hemangioma and umbilical cord blood display unique cellular responses to endostatin. Blood. 2006;108(3):915–921. PMCID: 1895853
66 Kleinman M.E., Blei F., Gurtner G.C. Circulating endothelial progenitor cells and vascular anomalies. Lymphat Res Biol. 2005;3(4):234–239.
67 North P.E., Waner M., Mizeracki A., et al. A unique microvascular phenotype shared by juvenile hemangiomas and human placenta. Arch Dermatol. 2001;137(5):559–570.
68 Mihm M.C., Nelson J.S. Hypothesis: the metastatic niche theory can elucidate infantile hemangioma development. J Cutan Pathol. 2010;37:83–87.
69 Greenberger S., Adini I., Boscolo E., et al. Targeting NF-kappaB in infantile hemangioma-derived stem cells reduces VEGF-A expression. Angiogenesis. 2010;13(4):327–335.
70 Greenberger S., Boscolo E., Adini I., et al. Corticosteroid suppression of VEGF-A in infantile hemangioma-derived stem cells. N Engl J Med. 2010;362(11):1005–1013. PMCID: 2845924
71 Sun Z.J., Zhao Y.F., Zhang W.F. Immune response: a possible role in the pathophysiology of hemangioma. Med Hypotheses. 2007;68(2):353–355.
72 Jinnin M., Medici D., Park L., et al. Suppressed NFAT-dependent VEGFR1 expression and constitutive VEGFR2 signaling in infantile hemangioma. Nat Med. 2008;14(11):1236–1246. PMCID: 2593632
73 Brooks B.P., Dagenais S.L., Nelson C.C., et al. Mutation of the FOXC2 gene in familial distichiasis. J AAPOS. 2003;7(5):354–357.
74 Levinson K.L., Feingold E., Ferrell R.E., et al. Age of onset in hereditary lymphedema. J Pediatr. 2003;142(6):704–708.
75 Eerola I., Boon L.M., Mulliken J.B., et al. Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am J Hum Genet. 2003;73(6):1240–1249. PMCID: 1180390
76 Boon L.M., Mulliken J.B., Vikkula M. RASA1: variable phenotype with capillary and arteriovenous malformations. Curr Opin Genet Dev. 2005;15(3):265–269.
77 Wouters V., Limaye N., Uebelhoer M., et al. Hereditary cutaneomucosal venous malformations are caused by TIE2 mutations with widely variable hyper-phosphorylating effects. Eur J Hum Genet. 2010;18(4):414–420. PMCID: 2841708
78 Hershkovitz D., Bercovich D., Sprecher E., et al. RASA1 mutations may cause hereditary capillary malformations without arteriovenous malformations. Br J Dermatol. 2008;158(5):1035–1040.
79 Hershkovitz D., Bergman R., Sprecher E. A novel mutation in RASA1 causes capillary malformation and limb enlargement. Arch Dermatol Res. 2008;300(7):385–388.
80 Comi A.M. Update on Sturge-Weber syndrome: diagnosis, treatment, quantitative measures, and controversies. Lymphat Res Biol. 2007;5(4):257–264.
81 Orlow S.J., Isakoff M.S., Blei F. Increased risk of symptomatic hemangiomas of the airway in association with cutaneous hemangiomas in a “beard” distribution. J Pediatr. 1997;131(4):643–646.
82 Balakrishnan K., Perkins J.A. Management of airway hemangiomas. Expert Rev Respir Med. 2010;4(4):455–462.
83 Schwartz S.R., Blei F., Ceisler E., et al. Risk factors for amblyopia in children with capillary hemangiomas of the eyelids and orbit. J AAPOS. 2006;10(3):262–268.
84 Schwartz S.R., Kodsi S.R., Blei F., et al. Treatment of capillary hemangiomas causing refractive and occlusional amblyopia. J AAPOS. 2007;11(6):577–583.
85 Al-Mahdi H. Iris depigmentation: an unusual complication of intralesional corticosteroid injection for capillary hemangioma. Middle East Afr J Ophthalmol. 2010;17(1):100–102. PMCID: 2880367
86 Maguiness S.M., Frieden I.J. Current management of infantile hemangiomas. Semin Cutan Med Surg. 2010;29(2):106–114.
87 Huang S.A., Tu H.M., Harney J.W., et al. Severe hypothyroidism caused by type 3 iodothyronine deiodinase in infantile hemangiomas. N Engl J Med. 2000;343(3):185–189.
88 Christison-Lagay E.R., Burrows P.E., Alomari A., et al. Hepatic hemangiomas: subtype classification and development of a clinical practice algorithm and registry. J Pediatr Surg. 2007;42(1):62–67. discussion 7–8
89 Chamlin S.L., Haggstrom A.N., Drolet B.A., et al. Multicenter prospective study of ulcerated hemangiomas. J Pediatr. 2007;151(6):684–689. 9 e1
90 Ho N.T., Lansang P., Pope E. Topical imiquimod in the treatment of infantile hemangiomas: a retrospective study. J Am Acad Dermatol. 2007;56(1):63–68.
91 McCuaig C.C., Dubois J., Powell J., et al. A phase II, open-label study of the efficacy and safety of imiquimod in the treatment of superficial and mixed infantile hemangioma. Pediatr Dermatol. 2009;26(2):203–212.
92 Sugarman J.L., Mauro T.M., Frieden I.J. Treatment of an ulcerated hemangioma with recombinant platelet-derived growth factor. Arch Dermatol. 2002;138(3):314–316.
93 Metz B.J., Rubenstein M.C., Levy M.L., et al. Response of ulcerated perineal hemangiomas of infancy to becaplermin gel, a recombinant human platelet-derived growth factor. Arch Dermatol. 2004;140(7):867–870.
94 Lapidoth M., Ben-Amitai D., Bhandarkar S., et al. Efficacy of topical application of eosin for ulcerated hemangiomas. J Am Acad Dermatol. 2009;60(2):350–351.
95 Faughnan M.E., Palda V.A., Garcia-Tsao G., et al. International guidelines for the diagnosis and management of hereditary hemorrhagic telangiectasia. J Med Genet. 2011;48(2):73–87.
96 Scher D.M. Orthopedic issues in patients with vascular anomalies. Lymphat Res Biol. 2004;2(1):51–55.
97 Rebarber A., Roman A.S., Roshan D., et al. Obstetric management of Klippel-Trenaunay syndrome. Obstet Gynecol. 2004;104(5 Pt 2):1205–1208.
98 Cohen S.G. Hemangiomas in infancy and childhood: psychosocial issues require close attention. Adv Nurse Pract. 2005;13(11):41–44.
99 Lande R.G., Crawford P.M., Ramsey B.J. Psychosocial impact of vascular birthmarks. Facial Plast Surg Clin North Am. 2001;9(4):561–567.
100 Tanner J.L., Dechert M.P., Frieden I.J. Growing up with a facial hemangioma: parent and child coping and adaptation. Pediatrics. 1998;101(3 Pt 1):446–452.
101 Oster H. Emotion in the infant’s face: insights from the study of infants with facial anomalies. Ann N Y Acad Sci. 2003;1000:197–204.
102 Williams E.F.3rd, Hochman M., Rodgers B.J., et al. A psychological profile of children with hemangiomas and their families. Arch Facial Plast Surg. 2003;5(3):229–234.
103 Ridner S.H. The psycho-social impact of lymphedema. Lymphat Res Biol. 2009;7(2):109–112. PMCID: 2904185
104 Cabo H., Cohen E.S., Casas G.J., et al. Cutaneous angiosarcoma arising on the radiation site of a congenital facial hemangioma. Int J Dermatol. 1998;37(8):638–639.
105 Haddy N., Andriamboavonjy T., Paoletti C., et al. Thyroid adenomas and carcinomas following radiotherapy for a hemangioma during infancy. Radiother Oncol. 2009;93(2):377–382.
106 Haddy N., Dondon M.G., Paoletti C., et al. Breast cancer following radiotherapy for a hemangioma during childhood. Cancer Causes Control. 2010;21(11):1807–1816.
107 Sovinz P., Urban C., Hausegger K. Life-threatening hemangiomatosis of the liver in an infant: multimodal therapy including cyclophosphamide and secondary acute myeloid leukemia. Pediatr Blood Cancer. 2006;47(7):972–973.
108 Al Dhaybi R., Agoumi M., Powell J., et al. Lymphangiosarcoma complicating extensive congenital mixed vascular malformations. Lymphat Res Biol. 2010;8(3):175–179.
109 Al-Mubarak L., Al-Khenaizan S. A wolf in sheep’s disguise: rhabdomyosarcoma misdiagnosed as infantile hemangioma. J Cutan Med Surg. 2009;13(5):276–279.
110 Assen Y.J., Madern G.C., de Laat P.C., et al. Rhabdoid tumor mimicking hemangioma. Pediatr Dermatol. 2011;28(3):295–298.
111 Boon L.M., Fishman S.J., Lund D.P., et al. Congenital fibrosarcoma masquerading as congenital hemangioma: report of two cases. J Pediatr Surg. 1995;30(9):1378–1381.
112 Hassanein A., Fishman S., Mulliken J., et al. Metastatic neuroblastoma mimicking infantile hemangioma. J Pediatr Surg. 2010;45(10):2045–2049.
113 Hayward P.G., Orgill D.P., Mulliken J.B., et al. Congenital fibrosarcoma masquerading as lymphatic malformation: report of two cases. J Pediatr Surg. 1995;30(1):84–88.
114 Martinez-Perez D., Fein N.A., Boon L.M., et al. Not all hemangiomas look like strawberries: uncommon presentations of the most common tumor of infancy. Pediatr Dermatol. 1995;12(1):1–6.
115 Megarbane H., Doz F., Manach Y., et al. Neonatal rhabdomyosarcoma misdiagnosed as a congenital hemangioma. Pediatr Dermatol. 2011;28(3):299–301.
116 Requena C., Miranda L., Canete A., et al. Congenital fibrosarcoma simulating congenital hemangioma. Pediatr Dermatol. 2008;25(1):141–144.
117 Yan A.C., Chamlin S.L., Liang M.G., et al. Congenital infantile fibrosarcoma: a masquerader of ulcerated hemangioma. Pediatr Dermatol. 2006;23(4):330–334.
118 Nord K.M., Kandel J., Lefkowitch J.H., et al. Multiple cutaneous infantile hemangiomas associated with hepatic angiosarcoma: case report and review of the literature. Pediatrics. 2006;118(3):e907–e913.
119 Rossi S., Fletcher C.D. Angiosarcoma arising in hemangioma/vascular malformation: report of four cases and review of the literature. Am J Surg Pathol. 2002;26(10):1319–1329.
120 Buckmiller L., Richter G., Suen J. Diagnosis and management of hemangiomas and vascular malformations of the head and neck. Oral Dis. 2010;16(5):405–418.
121 Stier M.F., Glick S.A., Hirsch R.J. Laser treatment of pediatric vascular lesions: port wine stains and hemangiomas. J Am Acad Dermatol. 2008;58(2):261–285.
122 Rizzo C., Brightman L., Chapas A.M., et al. Outcomes of childhood hemangiomas treated with the pulsed-dye laser with dynamic cooling: a retrospective chart analysis. Dermatol Surg. 2009;35(12):1947–1954.
123 Hunzeker C.M., Geronemus R.G. Treatment of superficial infantile hemangiomas of the eyelid using the 595-nm pulsed dye laser. Dermatol Surg. 2010;36(5):590–597.
124 Li D.N., Gold M.H., Sun Z.S., et al. Treatment of infantile hemangioma with optimal pulse technology. J Cosmet Laser Ther. 2010;12(3):145–150.
125 Frieden I.J. Commentary: early laser treatment of periorbital infantile hemangiomas may work, but is it really the best treatment option? Dermatol Surg. 2010;36(5):598–601.
126 Greene A.K. Corticosteroid treatment for problematic infantile hemangioma: evidence does not support an increased risk for cerebral palsy. Pediatrics. 2008;121(6):1251–1252.
127 Greene A.K. Systemic corticosteroid is effective and safe treatment for problematic infantile hemangioma. Pediatr Dermatol. 2010;27(3):322–323.
128 Ray W.Z., Lee A., Blackburn S.L., et al. Pseudotumor cerebri following tapered corticosteroid treatment in an 8-month-old infant. J Neurosurg Pediatr. 2008;1(1):88–90.
129 Maronn M.L., Corden T., Drolet B.A. Pneumocystis carinii pneumonia in infant treated with oral steroids for hemangioma. Arch Dermatol. 2007;143(9):1224–1225.
130 Aviles R., Boyce T.G., Thompson D.M. Pneumocystis carinii pneumonia in a 3-month-old infant receiving high-dose corticosteroid therapy for airway hemangiomas. Mayo Clin Proc. 2004;79(2):243–245.
131 Deboer M.D., Boston B.A. Failure-to-thrive in an infant following injection of capillary hemangioma with triamcinolone acetonide. Clin Pediatr (Phila). 2008;47(3):296–299.
132 Goyal R., Watts P., Lane C.M., et al. Adrenal suppression and failure to thrive after steroid injections for periocular hemangioma. Ophthalmology. 2004;111(2):389–395.
133 Kushner B.J., Lemke B.N. Bilateral retinal embolization associated with intralesional corticosteroid injection for capillary hemangioma of infancy. J Pediatr Ophthalmol Strabismus. 1993;30(6):397–399.
134 Ang L.P., Lee M.W., Seah L.L., et al. Orbital cellulitis following intralesional corticosteroid injection for periocular capillary haemangioma. Eye. 2007;21(7):999–1001.
135 Buckmiller L.M., Francis C.L., Glade R.S. Intralesional steroid injection for proliferative parotid hemangiomas. Int J Pediatr Otorhinolaryngol. 2008;72(1):81–87.
136 Leaute-Labreze C., Dumas de la Roque E., Hubiche T., et al. Propranolol for severe hemangiomas of infancy. N Engl J Med. 2008;358(24):2649–2651.
137 Leboulanger N., Fayoux P., Teissier N., et al. Propranolol in the therapeutic strategy of infantile laryngotracheal hemangioma: a preliminary retrospective study of French experience. Int J Pediatr Otorhinolaryngol. 2010;74(11):1254–1257.
138 Haider K.M., Plager D.A., Neely D.E., et al. Outpatient treatment of periocular infantile hemangiomas with oral propranolol. J AAPOS. 2010;14(3):251–256.
139 Rosbe K.W., Suh K.Y., Meyer A.K., et al. Propranolol in the management of airway infantile hemangiomas. Arch Otolaryngol Head Neck Surg. 2010;136(7):658–665.
140 Tan S.T., Itinteang T., Leadbitter P. Low-dose propranolol for infantile haemangioma. J Plast Reconstr Aesthet Surg. 64(2929), 2011.
141 Mazereeuw-Hautier J., Hoeger P.H., Benlahrech S., et al. Efficacy of propranolol in hepatic infantile hemangiomas with diffuse neonatal hemangiomatosis. J Pediatr. 2010;157(2):340–342.
142 Pope E., Chakkittakandiyil A. Topical timolol gel for infantile hemangiomas: a pilot study. Arch Dermatol. 2010;146(5):564–565.
143 Guo S., Ni N. Topical treatment for capillary hemangioma of the eyelid using beta-blocker solution. Arch Ophthalmol. 2010;128(2):255–256.
144 Bonifazi E., Acquafredda A., Milano A., et al. Severe hypoglycemia during successful treatment of diffuse hemangiomatosis with propranolol. Pediatr Dermatol. 2010;27(2):195–196.
145 Breur J.M., de Graaf M., Breugem C.C., et al. Hypoglycemia as a result of propranolol during treatment of infantile hemangioma: a case report. Pediatr Dermatol. 2011;28(2):169–171.
146 Holland K.E., Frieden I.J., Frommelt P.C., et al. Hypoglycemia in children taking propranolol for the treatment of infantile hemangioma. Arch Dermatol. 2010;146(7):775–778.
147 Allford M.A., Brown J.L. Case report: intraoperative hypoglycaemia in a child treated with propranolol following a short preoperative fast. Eur J Anaesthesiol. 2011;28(1):71–72.
148 Lamy S., Lachambre M.P., Lord-Dufour S., et al. Propranolol suppresses angiogenesis in vitro: inhibition of proliferation, migration, and differentiation of endothelial cells. Vascul Pharmacol. 2010;53(5–6):200–208.
149 Itinteang T., Brasch H.D., Tan S.T., et al. Expression of components of the renin-angiotensin system in proliferating infantile haemangioma may account for the propranolol-induced accelerated involution. J Plast Reconstr Aesthet Surg. 2011;64(6):759–765.
150 Sans V., de la Roque E.D., Berge J., et al. Propranolol for severe infantile hemangiomas: follow-up report. Pediatrics. 2009;124(3):e423–e431.
151 Storch C.H., Hoeger P.H. Propranolol for infantile haemangiomas—insights into the molecular mechanisms of action. Br J Dermatol. 2010;163(2):269–274.
152 Naouri M., Schill T., Maruani A., et al. Successful treatment of ulcerated haemangioma with propranolol. J Eur Acad Dermatol Venereol. 2010;24(9):1109–1112.
153 Barlow C.F., Priebe C.J., Mulliken J.B., et al. Spastic diplegia as a complication of interferon alfa-2a treatment of hemangiomas of infancy. J Pediatr. 1998;132(3 Pt 1):527–530.
154 Michaud A.P., Bauman N.M., Burke D.K., et al. Spastic diplegia and other motor disturbances in infants receiving interferon-alpha. Laryngoscope. 2004;114(7):1231–1236.
155 Chao Y.H., Liang D.C., Chen S.H., et al. Interferon-alpha for alarming hemangiomas in infants: experience of a single institution. Pediatr Int. 2009;51(4):469–473.
156 Perez-Valle S., Peinador M., Herraiz P., et al. Vincristine, an efficacious alternative for diffuse neonatal haemangiomatosis. Acta Paediatr. 2010;99(2):311–315.
157 Lazaridou E., Giannopoulou C., Apalla Z., et al. Calcineurin inhibitors in the treatment of cutaneous infantile haemangiomas. J Eur Acad Dermatol Venereol. 2010;24(5):614–615.
158 Yao Y.G., Duh E.J. VEGF selectively induces Down syndrome critical region 1 gene expression in endothelial cells: a mechanism for feedback regulation of angiogenesis? Biochem Biophys Res Commun. 2004;321(3):648–656.
159 Gollogly L.K., Ryeom S.W., Yoon S.S. Down syndrome candidate region 1-like 1 (DSCR1-L1) mimics the inhibitory effects of DSCR1 on calcineurin signaling in endothelial cells and inhibits angiogenesis. J Surg Res. 2007;142(1):129–136. PMCID: 1995402
160 Serena T. Wound closure and gradual involution of an infantile hemangioma using a noncontact, low-frequency ultrasound therapy. Ostomy Wound Manage. 2008;54(2):68–71.
161 Spector J.A., Blei F., Zide B.M. Early surgical intervention for proliferating hemangiomas of the scalp: indications and outcomes. Plast Reconstr Surg. 2008;122(2):457–462.
162 Waner M., Kastenbaum J., Scherer K. Hemangiomas of the nose: surgical management using a modified subunit approach. Arch Facial Plast Surg. 2008;10(5):329–334.
163 Warren S.M., Longaker M.T., Zide B.M. The subunit approach to nasal tip hemangiomas. Plast Reconstr Surg. 2002;109(1):25–30.
164 Watanabe S., Takagi S., Sato Y., et al. Early surgical intervention for Japanese children with infantile hemangioma of the craniofacial region. J Craniofac Surg. 2009;20(Suppl 1):707–709.
165 Wu J.K., Rohde C.H. Purse-string closure of hemangiomas: early results of a follow-up study. Ann Plast Surg. 2009;62(5):581–585.
166 Zide B.M., Glat P.M., Stile F.L., et al. Vascular lip enlargement: part I. Hemangiomas--tenets of therapy. Plast Reconstr Surg. 1997;100(7):1664–1673.
167 Canavese F., Soo B.C., Chia S.K., et al. Surgical outcome in patients treated for hemangioma during infancy, childhood, and adolescence: a retrospective review of 44 consecutive patients. J Pediatr Orthop. 2008;28(3):381–386.
168 Levi M., Schwartz S., Blei F., et al. Surgical treatment of capillary hemangiomas causing amblyopia. J AAPOS. 2007;11(3):230–234.
169 Mandrekas A.D., Zambacos G.J., Hapsas D.A. Pediatric nasal reconstruction for nasal tip hemangioma. Plast Reconstr Surg. 2010;125(5):1571–1572. author reply 3
170 Mehta M., Waner M., Fay A. Amniotic membrane grafting in the management of conjunctival vascular malformations. Ophthal Plast Reconstr Surg. 2009;25(5):371–375.
171 Mulliken J.B., Rogers G.F., Marler J.J. Circular excision of hemangioma and purse-string closure: the smallest possible scar. Plast Reconstr Surg. 2002;109(5):1544–1554. discussion 55
172 Burrows P.E., Mulliken J.B., Fishman S.J., et al. Pharmacological treatment of a diffuse arteriovenous malformation of the upper extremity in a child. J Craniofac Surg. 2009;20(Suppl 1):597–602.
173 Niramis R., Watanatittan S., Rattanasuwan T. Treatment of cystic hygroma by intralesional bleomycin injection: experience in 70 patients. Eur J Pediatr Surg. 2010;20(3):178–182.
174 Smith M.C., Zimmerman M.B., Burke D.K., et al. Efficacy and safety of OK-432 immunotherapy of lymphatic malformations. Laryngoscope. 2009;119(1):107–115.
175 Narkio-Makela M., Makela T., Saarinen P., et al. Treatment of lymphatic malformations of head and neck with OK-432 sclerotherapy induce systemic inflammatory response. Eur Arch Otorhinolaryngol. 2011;268:123–1219.
176 Greene A.K., Burrows P.E., Smith L., et al. Periorbital lymphatic malformation: clinical course and management in 42 patients. Plast Reconstr Surg. 2005;115(1):22–30.
177 Oliver G., Srinivasan R.S. Lymphatic vasculature development: current concepts. Ann N Y Acad Sci. 2008;1131:75–81.
178 Oliver G., Srinivasan R.S. Endothelial cell plasticity: how to become and remain a lymphatic endothelial cell. Development. 2010;137(3):363–372. PMCID: 2858906
179 Perkins J.A., Manning S.C., Tempero R.M., et al. Lymphatic malformations: review of current treatment. Otolaryngol Head Neck Surg. 2010;142(6):795–803. e1
180 Perkins J.A., Manning S.C., Tempero R.M., et al. Lymphatic malformations: current cellular and clinical investigations. Otolaryngol Head Neck Surg. 2010;142(6):789–794.
181 Rockson S.G. Current concepts and future directions in the diagnosis and management of lymphatic vascular disease. Vasc Med. 2010;15(3):223–231.
182 Mayrovitz H.N. The standard of care for lymphedema: current concepts and physiological considerations. Lymphat Res Biol. 2009;7(2):101–108.
183 Brorson H. From lymph to fat: complete reduction of lymphoedema. Phlebology. 2010;25(Suppl 1):52–63.
184 Rockson S.G. Diagnosis and management of lymphatic vascular disease. J Am Coll Cardiol. 2008;52(10):799–806.