Inflammatory Conditions
Acute Appendicitis
Appendicitis, inflammation of the vermiform appendix (blind sac at the end of the cecum), is the most common cause of emergency abdominal surgery in childhood. In the United States 100,000 cases are diagnosed each year (Aiken & Oldham, 2016a). The peak incidence of appendicitis is between 12 and 18 years, with boys affected slightly more often than girls (Aiken & Oldham, 2016a). Classically, the first symptom of appendicitis is periumbilical pain, followed by nausea, right lower quadrant pain, and, later, vomiting with fever (Rentea, Peter, & Snyder, 2017). Perforation occurs in up to 82% of children under 5 years of age, likely due to an inability to verbalize their symptoms (Aiken & Oldham, 2016a). Perforation of the appendix can occur within approximately 48 hours of the initial complaint of pain (Aiken & Oldham, 2016a). Complications from appendiceal perforation include major abscess, phlegmon, enterocutaneous fistula, peritonitis, and partial bowel obstruction. A phlegmon is an acute suppurative inflammation of subcutaneous connective tissue that spreads.
Etiology
The cause of appendicitis is obstruction of the lumen of the appendix, usually by hardened fecal material (fecalith). Swollen lymphoid tissue, frequently occurring after a viral infection, can also obstruct the appendix. Another rare cause of obstruction is a parasite such as Enterobius vermicularis, or pinworms, which can obstruct the appendiceal lumen.
Pathophysiology
With acute obstruction, the outflow of mucus secretions is blocked and pressure builds within the lumen, resulting in compression of blood vessels. The resulting ischemia is followed by ulceration of the epithelial lining and bacterial invasion. Subsequent necrosis causes perforation or rupture with fecal and bacterial contamination of the peritoneal cavity. The resulting inflammation spreads rapidly throughout the abdomen (peritonitis), especially in young children, who are unable to localize infection. Progressive peritoneal inflammation results in functional intestinal obstruction of the small bowel (ileus) because intense GI reflexes severely inhibit bowel motility. Because the peritoneum represents a major portion of total body surface, the loss of extracellular fluid to the peritoneal cavity leads to electrolyte imbalance and hypovolemic shock.
Clinical Manifestations.
The first symptom of appendicitis is usually colicky, cramping, abdominal pain located around the umbilicus (Box 25.12). Referred pain is the term used for this vague periumbilical localization. The midgut shares the same T10 dermatome, so pain is often perceived to be coming from this area. Generally, this pain progresses and becomes constant. The most important physical finding is focal abdominal tenderness. As the inflammation progresses to involve the serosa of the appendix and the peritoneum of the abdominal wall, the pain may shift to the right lower quadrant. The McBurney point, located two-thirds the distance along a line between the umbilicus and the anterosuperior iliac spine, is the most common point of tenderness. Localized peritoneal signs may occur with gentle percussion or maneuvers such as heel strike or shaking the bed. Other helpful findings are Rovsing sign, tenderness in the right lower quadrant that occurs during palpation or percussion of other abdominal quadrants; obturator sign, pain with flexion and internal rotation of the right hip; psoas sign, pain with left side with right hip extension; and Dunphy sign, pain with coughing (Rentea, Peter, & Snyder, 2017). Rebound tenderness—pain on deep palpation with sudden release—may be present, but it is not a finding specific to appendicitis (Aiken & Oldham, 2016a). Nausea, vomiting, and anorexia typically occur after the pain starts. Diarrhea, as well as other common signs of childhood illness such as upper respiratory tract congestion, poor feeding, lethargy, or irritability, may accompany appendicitis.
The child may not be able to walk well and may complain of pain in the right hip caused by inflammation in the psoas or iliopsoas muscles. Low-grade fever (38°C [100.4°F]) may occur with the initial presentation; however, the absence of fever does not exclude appendicitis. Because of the great variability in the presentation and location of appendicitis, any child with focal tenderness, regardless of the location, should be considered to potentially have acute appendicitis (see Community and Home Health Considerations box).
 Community and Home Health Considerations
Community and Home Health Considerations
Acute Appendicitis
Abdominal pain is a common complaint among school-age children, but in some cases it may indicate acute appendicitis. School nurses and nurse practitioners in school-based clinics should become familiar with the “typical” pattern of symptoms in acute appendicitis and how to assess and evaluate an acute abdomen. School nurses also need to impress on teachers and coaches the importance of early referral to the health suite for further assessment. Early referral to the health suite and an alert school nurse or nurse practitioner may make the difference between an uncomplicated appendectomy and a delayed diagnosis of a perforated appendix with peritonitis.
 Nursing Alert
Nursing Alert
Signs of peritonitis in addition to fever usually include sudden relief from pain after perforation; subsequent increase in pain, which is usually diffuse and accompanied by rigid guarding of the abdomen; progressive abdominal distention; tachycardia; rapid, shallow breathing as the child refrains from using abdominal muscles; pallor; chills; irritability; and restlessness.
Diagnostic Evaluation
Diagnosis is not always straightforward. Fever, vomiting, abdominal pain, and an elevated white blood cell count are associated with appendicitis but are also seen in IBD, pelvic inflammatory disease, gastroenteritis, urinary tract infection, right lower lobe pneumonia, mesenteric adenitis, Meckel diverticulum, and intussusception. Prolonged symptoms and delayed diagnosis often occur in younger children, in whom the risk of perforation is greatest because of their inability to verbalize their complaints.
The diagnosis is based primarily on the history and physical examination (see Box 25.12). Pain, the cardinal feature, is initially generalized (usually periumbilical). However, it usually descends to the lower right quadrant. The most intense site of pain may be at the McBurney point. Rebound tenderness is not a reliable sign and is extremely painful to the child. Referred pain, elicited by light percussion around the perimeter of the abdomen, indicates peritoneal irritation. Movement, such as riding over bumps in an automobile or gurney, aggravates the pain. In addition to pain, significant clinical manifestations include fever, a change in behavior, anorexia, and vomiting.
Laboratory studies usually include a complete blood count (CBC); urinalysis (to rule out a urinary tract infection); and, in adolescent females, serum human chorionic gonadotropin (to rule out an ectopic pregnancy). A white blood cell count greater than 10,000/mm3 and an elevated C-reactive protein (CRP) are common but are not necessarily specific for appendicitis. An elevated percentage of bands (often referred to as “a left shift”) may indicate an inflammatory process. CRP is an acute-phase reactant that rises within 12 hours of the onset of infection.
Ultrasound is the imaging technique of choice in diagnosing appendicitis, although a computed tomography (CT) scan may be used. Ultrasound is considered positive in the presence of enlarged appendiceal diameter; appendiceal wall thickening; and periappendiceal inflammatory changes, including fat streaks, phlegmon, fluid collection, and extraluminal gas (Aiken & Oldham, 2016a). The accuracy of imaging for diagnosing appendicitis is 95% (Rentea, Peter, & Snyder, 2017).
Therapeutic Management
The treatment for appendicitis before perforation is surgical removal of the appendix (appendectomy). Usually antibiotics are administered preoperatively. IV fluids and electrolytes are often required before surgery, especially if the child is dehydrated as a result of the marked anorexia characteristic of appendicitis.
The operation is usually performed through a right lower quadrant incision (open appendectomy). Laparoscopic surgery is commonly used to treat nonperforated acute appendicitis in pediatric patients. Three cannulas are inserted in the abdomen: one in the umbilicus, one in the left lower abdominal quadrant, and one in the suprapubic area. A small telescope is inserted through the left lower quadrant cannula, and an endoscopic stapler is inserted through the umbilical cannula. The appendix is ligated with the stapler and removed through the umbilical cannula. Advantages of laparoscopic appendectomy include reduced time in surgery and under anesthesia and reduced risk of postoperative wound infection (Aiken & Oldham, 2016b).
Ruptured Appendix.
Management of the child diagnosed with peritonitis caused by a ruptured appendix often begins preoperatively with IV administration of fluid and electrolytes, systemic antibiotics, and NG suction. Postoperative management includes IV fluids, continued administration of antibiotics, and NG suction for abdominal decompression until intestinal activity returns. Sometimes surgeons close the wound after irrigation of the peritoneal cavity. Other times, they leave the wound open (delayed closure) to prevent wound infection.
The treatment of a localized perforation with an appendiceal abscess is controversial. Some surgeons prefer to treat these children with antibiotics and IV fluids and allow the abscess to drain spontaneously. An elective appendectomy is then performed 2 to 3 months later.
Prognosis.
Complications are uncommon after a simple appendectomy, and recovery is usually rapid and complete. The mortality rate from perforating appendicitis has improved from nearly certain death a century ago to less than 1% at the present time (Rentea, Peter, & Snyder, 2017). Complications, however, including wound infection and intraabdominal abscess, are not uncommon. Early recognition of the illness is important to prevent complications.
Nursing Alert
In any instance in which severe abdominal pain is observed, the nurse must be aware of the danger of administering laxatives or enemas. Such measures stimulate bowel motility and increase the risk of perforation.
Nursing Care Management
Because successful treatment of appendicitis is based on prompt recognition of the disorder, an important nursing objective is to assist in establishing a diagnosis. Because abdominal pain is a common childhood complaint, the nurse needs to make some preliminary assessment of the severity of the pain. (See Chapter 5.) One of the most reliable estimates is the degree of change in behavior. A child who stays home from school and voluntarily lies down or refuses to play is much more likely to have considerable pain than a child who is absent from school but plays contentedly at home. Younger, nonverbal children will assume a rigid, side-lying position with the knees flexed and have decreased range of motion of the right hip.
For nurses involved in primary ambulatory care, the responsibility of recognizing a possible case of appendicitis and prompt medical or surgical referral is particularly important. The importance of a detailed history and thorough abdominal examination cannot be overemphasized. Palpating the abdomen should be delayed until all other assessments have been made. Instruct the child to point with one finger to the site of the abdominal pain. Rebound tenderness may be present but is not always a sufficiently reliable test in children. Light palpation will satisfactorily elicit pain without causing excessive trauma (see Atraumatic Care box). Ask the child with mild pain to lift the heels and drop them to the floor two or three times, to hop on one foot, or to “puff out” or “pull in” the abdomen to check for tenderness without more painful probing. Chapter 4 discusses other techniques for assessment of the abdomen.
Physical preparation of the child with appendicitis is similar to that for any child undergoing surgery. (See Chapter 22.) In situations in which medical treatment is required to correct problems associated with peritonitis, the nurse must anticipate procedures and set up equipment as quickly as possible to avoid any delay in preparing the child for surgery. Psychologic preparation of the child and parents is similar to that used in other emergency situations. (See Chapter 22.)
Postoperative care for the nonperforated appendix is the same as for most abdominal operations. Care of the child with a ruptured appendix and peritonitis is more complex. The child may need to remain in the hospital for several days or may be discharged with home care services to provide IV antibiotics and dressing changes.
Postoperatively the child is maintained on IV fluids and antibiotics and is allowed nothing by mouth (NPO). The child also remains on low, intermittent gastric decompression until there is evidence of return of intestinal motility. Listening for bowel sounds and observing for other signs of bowel activity (such as passage of stool) are part of the routine assessment.
A drain may be placed in the wound during surgery, and frequent dressing changes with meticulous skin care are essential to prevent excoriation of the surgery area. If the wound is left open, moist dressings (usually saline-soaked gauze) and wound irrigations with antibacterial solution are used to provide an optimum healing environment.
Pain management is an essential part of the child's care. Not only is the incision painful, but the repeated dressing changes and irrigations also cause considerable distress. Because pain is continuous during the first few postoperative days, analgesics are given regularly to control pain. Procedures are performed when the analgesics are at peak effect. (See Chapter 5.)
Psychosocial care after surgery is also important. Sudden, acute illnesses cause unique stress because there is little time for preparation or planning. Parents and older children need an opportunity to express their feelings and concerns regarding the events surrounding the illness and hospitalization. The nurse can provide important education and psychosocial support to promote adequate coping, with alleviation of anxiety for both the child and the family (see Nursing Care Plan box).
 Nursing Care Plan
Nursing Care Plan
The Child With Appendicitis
Case Study
Lisa is a 10-year-old girl who has a 2-day history of generalized periumbilical pain and anorexia. Today she developed a fever and vomiting, so her parents took her to her pediatrician. On examination, Lisa was febrile with abdominal pain midway between the anterior superior iliac crest and umbilicus. The pain intensifies with any activity or deep breathing. Blood work was performed and a complete blood count (CBC) with differential shows a white blood cell (WBC) count of 21,000/mm3, 79% bands, 14% lymphocytes, 6% eosinophils, and a normal hemoglobin and platelet count. With Lisa's history and physical findings, she was referred to a local emergency department.
Assessment
Based on Lisa's history, what are the most important signs and symptoms that you need to be aware of?
Appendicitis Defining Characteristics
- History of abdominal pain for 2 days that started around the umbilicus and has now progressed to the lower right abdomen (McBurney point)
- Fever
- Anorexia
- Nausea and vomiting
- Elevated WBC count (>10,000/mm3) along with a high percentage of bands (left shift)
- Elevated C-reactive protein (CRP)
Nursing Interventions
| Nursing Interventions | Rationale |
|---|---|
| Close monitoring of the patient's status. Follow clinical and laboratory findings. Blood studies included CBC, CRP, and electrolytes. | To identify infection, signs of inflammation, changes in fluid and electrolyte status which require additional treatment |
| Close monitoring of diagnostic evaluation studies (i.e., computed tomography [CT] scan and/or ultrasound). | To confirm diagnosis of appendicitis |
| Administer intravenous (IV) fluids. | To correct fluid deficit and electrolyte imbalances |
| Administer analgesics as ordered. | To reduce pain |
| Administer antiemetics as ordered. | To reduce nausea and alleviate vomiting |
| Monitor temperature and vital signs. | To observe for signs of infection |
| Administer antipyretic medication as indicated. | To reduce fever |
| Administer antibiotics as ordered. | To treat infection |
| Maintain nothing-by-mouth (NPO) status. | To keep stomach empty in anticipation of possible surgery |
| Identify patient and family stressors that may accompany a diagnosis of appendicitis. | Providing financial and emotion support for family can help decrease some of the stressors associated with this condition |
| Review disease, medication, dietary restrictions. | Understanding the medical condition and therapies allows family to make informed decisions about care |
Expected Outcomes
- Lisa will exhibit decreased pain.
- Lisa will exhibit no evidence of nausea or vomiting.
- The temperature will remain within normal limits.
- Sufficient fluid and electrolytes are maintained.
- Patient/family indicate understanding of appendicitis and treatment.
Case Study (Continued)
Results of the CT scan demonstrate a ruptured appendix. Lisa is now being prepared for surgery. The nurse performing the assessment finds Lisa's temperature to be elevated. Lisa reports the pain had initially resolved but she now reports increasing pain (rated 9 out of 10) and nausea.
Assessment
- What concerns you most based on the scenario?
- What immediate steps should be taken to further evaluate Lisa's status?
- The following laboratory results have returned from Lisa's blood work:
Nursing Interventions
| Nursing Interventions | Rationale |
|---|---|
| Administer antibiotics as ordered. IV antibiotics are given for a minimum of 3 days postoperatively in children with complicated appendicitis then transitioned to oral antibiotics at discharge. | To treat infection |
| Administer analgesics as ordered. | To reduce pain |
| Administer antiemetics as ordered. | To reduce nausea and alleviate vomiting |
| Monitor temperature and vital signs. | To observe for signs of infection and shock |
| Administer IV fluids and monitor electrolytes. | To correct fluid deficit and electrolyte imbalances |
| Follow laboratory findings. Blood studies including CBC, CRP, and intraoperative cultures if obtained. | To identify infection, and signs of inflammation |
| Advance diet as tolerated postoperatively. | To maintain nutritional status |
Expected Outcome
- Lisa will exhibit no signs of infection.
- Pain is controlled initially with IV analgesics then transitioned to oral analgesics.
- Lisa will exhibit no evidence of nausea or vomiting and tolerate a regular diet.
- The temperature is within normal limits.
- Sufficient fluid and electrolytes are maintained.
Case Study (Continued)
Lisa's parents are anxious and upset with the urgent need for surgery and hospitalization. You are concerned that they do not understand what is happening to their daughter.
Assessment
Family's Knowledge of Illness-Defining Characteristics
- Understands definition of appendicitis and ruptured appendix
- Describes rationale for urgent surgery
- Describes rationale for subsequent hospitalization and need for IV antibiotics
- Expresses fears and concerns
- Shows appropriate reactions to child's illness
Nursing Interventions
| Nursing Interventions | Rationale |
|---|---|
| Review disease and treatment before surgery. | Understanding the medical condition and therapies allow families to make informed decisions about care |
| Review disease and treatment after surgery. | To increase knowledge and compliance with treatment plan to control pain, treat infection, maintain adequate fluid and electrolyte balance, and maximize nutrition |
| Arrange for social worker to meet with family to assess emotional and financial needs. | To identify and modify stressors associated with urgent and prolonged hospitalization |
| As child nears discharge, arrange for discussions with parents to discuss home care. | Family must be aware of necessary treatment and monitoring to be compliant with care |
Expected Outcome
Meckel Diverticulum
Meckel diverticulum is a remnant of the fetal omphalomesenteric duct, which connects the yolk sac with the primitive midgut during fetal life (Kennedy & Liacouras, 2016a). Normally the structure is obliterated between the fifth and seventh week of gestation, when the placenta replaces the yolk sac as the source of nutrition for the fetus. Failure of obliteration may result in an omphalomesenteric fistula (a fibrous band connecting the small intestine to the umbilicus), umbilical cyst, vitelline duct remnant, mesodiverticular bands, and Meckel diverticula (Bagade & Khanna, 2015).
Meckel diverticulum is a true diverticulum because it arises from the antimesenteric border of the small intestine and includes all layers of the intestinal wall. Meckel diverticulum is often referred to by the “rule of 2s” because it occurs in 2% of the population, has a 2 : 1 male-to-female ratio, is located within 2 feet of the ileocecal valve, is commonly 2 cm in diameter and 2 inches in length, contains 2 types of ectopic tissue (pancreatic and gastric), and is more common before age 2 (Kennedy & Liacouras, 2016a).
Pathophysiology
Bleeding, obstruction, or inflammation causes the symptomatic complications of Meckel diverticulum (Lin, Huang, Bao, et al., 2017). Bleeding, which is the most common problem in children, is caused by peptic ulceration or perforation because of the unbuffered acidic secretion. Several mechanisms may cause obstruction such as intussusception or entanglement of the small intestine.
Clinical Manifestations
Signs and symptoms are based on the specific pathologic process, such as inflammation, bleeding, or intestinal obstruction (Box 25.13). The most common clinical presentation is rectal bleeding caused by ulceration at the junction of the ectopic gastric mucosa and normal ileal mucosa. The bleeding is usually painless and may be dramatic and occur as bright red or currant jelly–like stools, or it may occur intermittently and appear as tarry stools. The bleeding may be significant enough to cause hypotension. Volvulus and intussusception are common obstructive mechanisms in children with Meckel diverticulum, and these children present with symptoms of abdominal pain, distention, nausea, and vomiting (Kennedy & Liacouras, 2016a).
Diagnostic Evaluation
Diagnosis is usually based on the history, physical examination, and radiographic studies. Meckel diverticulum is often a diagnostic challenge. A technetium-99 pertechnetate scan (Meckel scan) is the most effective diagnostic testing, especially for a bleeding diverticulum, with sensitivity ranging from 80% to 90% and a specificity of 95% (Lin, Huang, Bao, et al., 2017). Laboratory studies such as a CBC and a basic metabolic panel are usually part of the general workup to rule out any bleeding disorder and to evaluate for dehydration.
Therapeutic Management
The standard treatment for symptomatic Meckel diverticulum is surgical removal. In instances in which severe hemorrhage increases the surgical risk, medical intervention to correct hypovolemic shock (e.g., blood replacement, IV fluids, and oxygen) may be necessary. Antibiotics may be used preoperatively to control infection. If intestinal obstruction has occurred, appropriate preoperative measures are used to correct fluid and electrolyte imbalances and prevent abdominal distention.
Prognosis.
If symptomatic Meckel diverticulum is diagnosed and treated early, full recovery is likely. Because of the potential for surgical complications, resection of asymptomatic Meckel diverticulum remains controversial.
Nursing Care Management
Nursing objectives are the same as for any child undergoing surgery. (See Chapter 22.) When intestinal bleeding is present, specific preoperative considerations include frequent monitoring of vital signs and blood pressure, keeping the child on bed rest, and recording the approximate amount of blood lost in stools.
Postoperatively the child requires IV fluids and an NG tube for decompression and evacuation of gastric secretions. Because the onset of illness is usually rapid, psychologic support is important, as in other acute conditions, such as appendicitis. It is important to remember that massive rectal bleeding is usually traumatic to both the child and the parents and may significantly affect their emotional reaction to hospitalization and surgery.
Inflammatory Bowel Disease
Inflammatory bowel disease (IBD) should not be confused with IBS. IBD is a term used to refer to three major forms of chronic intestinal inflammation: Crohn disease (CD), ulcerative colitis (UC), and inflammatory bowel disease unspecified (IBDU). CD and UC have similar epidemiologic, immunologic, and clinical features, but they are distinct disorders. The diagnosis of IBDU is used for patients with colonic disease, but their features are not specific to UC or CD; it is very rare (Conrad & Rosh, 2017).
Approximately 70,000 children in the United States have IBD (Rosen, Dhawan, & Saeed, 2017). Over the past 30 years the incidence of CD has risen, whereas the incidence of UC in children has remained stable (Grossman & Baldassano, 2016). Both CD and UC tend to be more aggressive if the onset occurs in childhood (Conrad & Rosh, 2017). Exacerbations and remissions without complete resolution of symptoms are also characteristics of IBD.
Etiology
Despite decades of research, the etiology of IBD is not completely understood, and there is no known cure. There is evidence to indicate a multifactorial etiology. Genetic, environmental, and microbial factors are associated with IBD, and research focuses on genetic associations and theories of defective immunoregulation of the inflammatory response to bacteria or viruses in the GI tract (Rosen, Dhawan, & Saeed, 2017). Genome-wide studies have confirmed at least 150 genes that increase the risk for IBD in individuals (Rosen, Dhawan, & Saeed, 2017). Furthermore, children who immigrate from developing countries to Western countries show an incidence of IBD similar to that of Western populations, confirming an environmental factor with the disease (Rosen, Dhawan, & Saeed, 2017). Finally, most individuals have 10 trillion bacteria and fungi in their intestinal microbiome, but children and adults with IBD have small diversity of intestinal bacterial species with an overrepresentation and underrepresentation of some species (Rosen, Dhawan, & Saeed, 2017).
Pathophysiology.
The inflammation found with UC is limited to the colon and rectum, with the distal colon and rectum the most severely affected. Inflammation affects the mucosa and submucosa and involves continuous segments along the length of the bowel with varying degrees of ulceration, bleeding, and edema. Thickening of the bowel wall and fibrosis are unusual, but long-standing disease can result in shortening of the colon and strictures. Toxic megacolon is the most dangerous form of severe colitis.
The chronic inflammatory process of CD involves any part of the GI tract from the mouth to the anus but most often affects the terminal ileum. The disease involves all layers of the bowel wall (transmural) in a discontinuous fashion, meaning that between areas of intact mucosa, there are areas of affected mucosa (skip lesions). The inflammation may result in ulcerations; fibrosis; adhesions; stiffening of the bowel wall; stricture formation; and fistulas to other loops of bowel, bladder, vagina, or skin.
Clinical Signs and Symptoms
Children with UC may experience mild, moderate, or severe symptoms, depending on the extent of mucosal inflammation and systemic symptoms. UC often manifests with the insidious onset of diarrhea, possibly with hematochezia, and usually without fever or weight loss. The course of the disease may remain mild with intermittent exacerbations. Some children and adolescents are seen with grossly bloody diarrhea, cramps, urgency with defecation, mild anemia, fever, anorexia, weight loss, and moderate signs of systemic illness. Severe UC is characterized by frequent bloody stools, abdominal pain, significant anemia, fever, and weight loss. Extraintestinal manifestations are not common in UC. Enlarged lymph nodes (lymphadenopathy), arthritis, and the skin lesions of erythema nodosum may be present.
Common presenting manifestations of CD include diarrhea, abdominal pain with cramps, fever, and weight loss. Extraintestinal manifestations, including aphthous ulcers, peripheral arthritis, erythema nodosum, digital clubbing, renal stones, and gallstones, are more common with CD than UC (Grossman & Baldassano, 2016). Growth failure and delayed sexual maturation are often present for several years before overt GI symptoms are present (Conrad & Rosh, 2017). Both malabsorption and anorexia are factors that contribute to the growth problems that are prevalent in CD. Children with CD may have perianal disease, including tags, fissures, fistulas, or abscesses (Rosen, Dhawan, & Saeed, 2017). The effects of UC and CD are listed in Fig. 25.4. Table 25.6 provides a comparison of UC and CD.
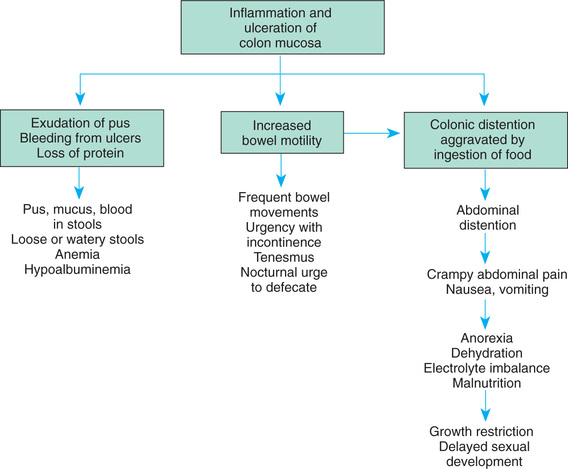
TABLE 25.6
Diagnostic Evaluation
The diagnosis of UC and CD comes from the history, physical examination, laboratory evaluation, and other diagnostic procedures. Laboratory tests include a CBC to evaluate anemia and an erythrocyte sedimentation rate (ESR) or CRP to assess the systemic reaction to the inflammatory process. The ESR or CRP may be elevated, indicating a systemic response to an inflammatory process. Levels of total protein, albumin, iron, zinc, magnesium, vitamin B12, and fat-soluble vitamins may be low in children with CD. Stools are examined for blood, leukocytes, and infectious organisms. A serologic panel is often used in combination with clinical findings to diagnose IBD and to differentiate between CD and UC.
In patients with CD, an upper GI series with small bowel follow-through assists in assessing the existence, location, and extent of disease. Upper endoscopy and colonoscopy with biopsies are an integral part of diagnosing IBD (Rosen, Dhawan, & Saeed, 2017). Endoscopy allows direct visualization of the surface of the GI tract so that the extent of inflammation and narrowing can be evaluated. CT and ultrasound also may be used to identify bowel wall inflammation, intraabdominal abscesses, and fistulas. Colonoscopy can confirm the diagnosis and evaluate the extent of the disease. Discrete ulcers are commonly seen in patients with CD, whereas microulcers and diffuse abnormalities and inflammation are seen in patients with UC (Grossman & Baldassano, 2016). CD lesions may pierce the walls of the small intestine and colon, creating tracts called fistulas between the intestine and adjacent structures such as the bladder, anus, vagina, or skin.
Therapeutic Management
The natural history of the disease continues to be unpredictable and characterized by recurrent flare-ups that can severely impair patients' physical and social functioning (Grossman & Baldassano, 2016). The goals of therapy are to control the inflammatory process to reduce or eliminate the symptoms, obtain long-term remission, promote normal growth and development, and allow as normal a lifestyle as possible. Treatment is individualized and managed according to the type and the severity of the disease, its location, and the response to therapy. CD is more disabling, has more serious complications, and is often less amenable to medical and surgical treatment than is UC. Because UC is confined to the colon, a colectomy may cure UC.
Medical Treatment.
The goal of any treatment regimen is first to induce remission of acute symptoms and then to maintain remission over time. 5-Aminosalicylates (5-ASAs) are effective in the induction and maintenance of remission in mild to moderate UC. Mesalamine, olsalazine, and balsalazide are preferred over sulfasalazine because of reduced side effects (e.g., headache, nausea, vomiting, neutropenia, and oligospermia). Suppository and enema preparations of mesalamine are used to treat left-sided colitis. These drugs decrease inflammation by inhibiting prostaglandin synthesis. 5-ASAs can be used to induce remission in mild CD.
Corticosteroids, such as prednisone and prednisolone, are indicated in induction therapy in children with moderate to severe UC and CD. These drugs inhibit the production of adhesion molecules, cytokines, and leukotrienes. Although these drugs reduce the acute symptoms of IBD, they are not commonly used for maintenance therapy because of their long-term side effects including growth suppression (adrenal suppression), weight gain, and decreased bone density (Rosen, Dhawan, & Saeed, 2017). High doses of IV corticosteroids may be administered in acute episodes and tapered according to clinical response. Budesonide, a synthetic corticosteroid, is designed for controlled release in the ileum and is indicated for ileal and right-sided colitis; budesonide has fewer side effects than prednisone and prednisolone but is also less effective (Rosen, Dhawan, & Saeed, 2017).
Immunomodulators, such as azathioprine and its metabolite 6-mercaptopurine (6-MP), are used to induce and maintain remission in children with IBD who are steroid resistant or steroid dependent and in treating chronic draining fistulas. They block the synthesis of purine, thus inhibiting the ability of deoxyribonucleic acid (DNA) and ribonucleic acid (RNA) to hinder lymphocyte function, especially that of T cells. Side effects include infection, pancreatitis, hepatitis, bone marrow toxicity, arthralgia, and malignancy. Methotrexate is also useful in inducing and maintaining remission in CD patients who are unresponsive to standard therapies. Cyclosporine and tacrolimus have both been effective in inducing remission in severe steroid-dependent UC. 6-MP or azathioprine is then used to maintain remission. Patients on immunomodulating medications require regular monitoring of their CBC and differential to assess for changes that reflect suppression of the immune system because many of the side effects can be prevented or managed by dose reduction or discontinuation of medication.
Antibiotics, such as metronidazole and ciprofloxacin, may be used as an adjunctive therapy to treat complications such as perianal disease or small bowel bacterial overgrowth in CD. Side effects of these drugs are peripheral neuropathy, nausea, and a metallic taste.
Biologic therapies act to regulate inflammatory and anti-inflammatory cytokines. The use of anti–tumor necrosis factor-α (TNF-α) agents such as infliximab and adalimumab decrease active inflammation and are effective in healing the intestinal mucosal lining and perianal fistulas, and have even improved linear growth in children (Grossman & Baldassano, 2016; Rosen, Dhawan, & Saeed, 2017). These agents are now being used as front-line therapy in children with CD with severe deep mucosal ulcerations, perianal fistulas, or growth failure (Rosen, Dhawan, & Saeed, 2017).
Nutritional Support.
Nutritional support is important in the treatment of IBD. Growth failure is a common serious complication, especially in CD. Growth failure is characterized by weight loss, alteration in body composition, retarded height, and delayed sexual maturation. Malnutrition causes the growth failure, and its etiology is multifactorial. Malnutrition occurs as a result of inadequate dietary intake, excessive GI losses, malabsorption, drug-nutrient interaction, and increased nutritional requirements. Inadequate dietary intake occurs with anorexia and episodes of increased disease activity. Excessive loss of nutrients (e.g., protein, blood, electrolytes, and minerals) occur secondary to intestinal inflammation and diarrhea. Carbohydrate, lactose, fat, vitamin, and mineral malabsorption, as well as vitamin B12 and folic acid deficiencies, occur with disease episodes and with drug administration and when the terminal ileum is resected. Finally, nutritional requirements are increased with inflammation, fever, fistulas, and periods of rapid growth (e.g., adolescence).
The goals of nutritional support include correction of nutrient deficits and replacement of ongoing losses, provision of adequate energy and protein for healing, and provision of adequate nutrients to promote normal growth. Nutritional support includes both enteral and parenteral nutrition. A well-balanced, high-protein, high-calorie diet is recommended for children whose symptoms do not prohibit an adequate oral intake. There is little evidence that avoiding specific foods influences the severity of the disease. Supplementation with multivitamins, iron, and folic acid is recommended.
Special enteral formulas, given either by mouth or continuous NG infusion (often at night), may be required. Elemental formulas are completely absorbed in the small intestine with almost no residue. A diet consisting only of elemental formula not only improves nutritional status but also induces disease remission, either without steroids or with a diminished dosage of steroids required. An elemental diet is a safe and potentially effective primary therapy for patients with CD. Unfortunately, remission is not sustained when NG feedings are discontinued unless maintenance medications are added to the treatment regimen.
Total parenteral nutrition (TPN) has also improved nutritional status in patients with IBD. Short-term remissions have been achieved after TPN, although complete bowel rest has not reduced inflammation or added to the benefits of improved nutrition by TPN. Nutritional support is less likely to induce a remission in UC than in CD. Improvement of nutritional status is important, however, in preventing deterioration of the patient's health status and in preparing the patient for surgery.
Surgical Treatment.
Surgery is indicated for UC when medical and nutritional therapies fail to prevent complications. Surgical options include a subtotal colectomy and ileostomy that leaves a rectal stump as a blind pouch. A reservoir pouch is created in the configuration of a J or an S to help improve continence postoperatively. An ileoanal pull-through preserves the normal pathway for defecation. Pouchitis, an inflammation of the surgically created pouch, is the most common late complication of this procedure. In many cases UC can be cured with a total colectomy.
Surgery may be required in children with CD when complications cannot be controlled by medical and nutritional therapy. Segmental intestinal resections are performed for small bowel obstructions, strictures, or fistulas. Diversion of the fecal stream, such as a colostomy, allows the colon to be less active and causes the disease to become dormant but on reconnection of the colon, disease often reoccurs (Grossman & Baldassano, 2016).
Prognosis.
IBD is a chronic disease. Relatively long periods of quiescent disease may follow exacerbations. The outcome is influenced by the regions and severity of involvement, as well as by appropriate therapeutic management. Malnutrition, growth failure, and bleeding are serious complications. The overall prognosis for UC is good.
The development of colorectal cancer (CRC) is a long-term complication of IBD. Because the risk for CRC occurs 8 to 10 years after diagnosis, surveillance colonoscopy with multiple biopsies should begin approximately 7 to 10 years after diagnosis of UC or CD (Rosen, Dhawan, & Saeed, 2017). In CD, surgical removal of the affected colon does not prevent cancer from developing elsewhere in the GI tract.
Nursing Care Management
The nursing considerations in the management of IBD extend beyond the immediate period of hospitalization. These interventions involve continued guidance of families in terms of (1) managing diet; (2) coping with factors that increase stress and emotional lability; (3) adjusting to a disease of remissions and exacerbations; and (4) when indicated, preparing the child and parents for the possibility of diversionary bowel surgery. (See Quality Patient Outcomes box.)
Because nutritional support is an essential part of therapy, encouraging the anorexic child to consume sufficient quantities of food is often a challenge. Successful interventions include involving the child in meal planning; encouraging small, frequent meals or snacks rather than three large meals a day; serving meals around medication schedules when diarrhea, mouth pain, and intestinal spasm are controlled; and preparing high-protein, high-calorie foods such as eggnog, milkshakes, cream soups, puddings, or custard (if lactose is tolerated). (See Feeding the Sick Child, Chapter 22.) Using bran or a high-fiber diet for active IBD is questionable. Bran, even in small amounts, has been shown to worsen the patient's condition. Occasionally the occurrence of aphthous stomatitis further complicates adherence to dietary management. Mouth care before eating and the selection of bland foods help relieve the discomfort of mouth sores.
When NG feedings or TPN is indicated, nurses play an important role in explaining the purpose and the expected outcomes of this therapy. The nurse should acknowledge the anxieties of the child and family members and give them adequate time to demonstrate the skills necessary to continue the therapy at home, if needed (see Critical Thinking Case Study box).
 Critical Thinking Case Study
Critical Thinking Case Study
Inflammatory Bowel Disease
Susan, a 13-year-old girl, was admitted to the hospital because of bloody diarrhea, abdominal pain, and weight loss. After a thorough evaluation, including laboratory tests, radiographic studies, and gastrointestinal endoscopy procedures, the diagnosis of Crohn disease (CD) was made. Medical treatment, including corticosteroid drugs and nutritional support, was implemented during this hospitalization.
Susan has improved considerably and is to be discharged home this week. Enteral formula administered by continuous nighttime nasogastric (NG) tube infusion will be continued at home, and both Susan and her family are eager to learn how to perform these feedings. You are the nurse responsible for Susan's discharge planning. Which interventions relating to these feedings should you include in Susan's preparations for discharge?
- 1. Evidence—Are there sufficient data to formulate any specific interventions for discharge?
- 2. Assumptions—Describe some underlying assumptions about the following:
- 3. What are the priorities for discharge planning at this time?
- 4. Does the evidence support your conclusion?
Answers are available at http://evolve.elsevier.com/wong/ncic.
The importance of continued drug therapy despite remission of symptoms must be stressed to the child and family members. Failure to adhere to the pharmacologic regimen can result in exacerbation of the disease. (See Compliance, Chapter 22.) Unfortunately, exacerbation of IBD can occur even if the child and family are compliant with the treatment regimen; this is difficult for the child and family to cope with.
Emotional Support.
The nurse should attend to the emotional components of the disease and assess any sources of stress. Frequently, the nurse can help children adjust to problems of growth retardation, delayed sexual maturation, dietary restrictions, feelings of being “different” or “sickly,” inability to compete with peers, and necessary absence from school during exacerbations of the illness.
If a permanent colectomy-ileostomy is required, the nurse can teach the child and family how to care for the ileostomy. The nurse can also emphasize the positive aspects of the surgery, particularly accelerated growth and sexual development, permanent recovery, and the normality of life despite bowel diversion. Introducing the child and parents to other ostomy patients, especially those who are the same age, is effective in fostering eventual acceptance. Whenever possible, offer continent ostomies as options to the child, although they are not performed in all centers in the United States.
Because of the chronic and often lifelong nature of the disease, families benefit from the educational services provided by organizations such as the Crohn's and Colitis Foundation of America (CCFA).* If diversionary bowel surgery is indicated, United Ostomy Associations of America† and the Wound, Ostomy and Continence Nurses Society‡ are available to assist with ileostomy care and provide important psychologic support through their self-help groups. Adolescents often benefit by participating in peer-support groups, which are sponsored by the CCFA.
Peptic Ulcer Disease
Peptic ulcer disease (PUD) is a chronic condition that affects the stomach or duodenum. Ulcers are described as gastric or duodenal and as primary or secondary. A gastric ulcer involves the mucosa of the stomach; a duodenal ulcer involves the pylorus or duodenum. Most primary ulcers are idiopathic or associated with Helicobacter pylori infection and tend to be chronic, occurring more frequently in the duodenum (Blanchard & Czinn, 2016). Secondary ulcers result from the stress of a severe underlying disease or injury (e.g., severe burns, sepsis, increased intracranial pressure, severe trauma, multisystem organ failure) and are more frequently gastric with an acute onset (Blanchard & Czinn, 2016).
Etiology
The exact cause of PUD is unknown, although infectious, genetic, and environmental factors are important. There is an increased familial incidence, likely due to H. pylori, which is known to cluster in families (Blanchard & Czinn, 2016). H. pylori is a microaerophilic, gram-negative, slow-growing, spiral-shaped, and flagellated bacterium known to colonize the gastric mucosa in about half of the population of the world (Blanchard & Czinn, 2016). H. pylori synthesizes the enzyme urease, which hydrolyses urea to form ammonia and carbon dioxide. Ammonia then absorbs acid to form ammonium, thus raising the gastric pH. H. pylori may cause ulcers by weakening the gastric mucosal barrier and allowing acid to damage the mucosa. It is believed that it is acquired via the fecal-oral route, and this hypothesis is supported by finding viable H. pylori in feces.
In addition to ulcerogenic drugs, both alcohol and smoking contribute to ulcer formation. There is no conclusive evidence to implicate particular foods, such as caffeine-containing beverages or spicy foods, but polyunsaturated fats and fiber may play a role in ulcer formation. Psychologic factors may play a role in the development of PUD, and stressful life events, dependency, passiveness, and hostility have all been implicated as contributing factors.
Pathophysiology
Most likely, the pathology is due to an imbalance between the destructive (cytotoxic) factors and defensive (cytoprotective) factors in the GI tract. The toxic mechanisms include acid, pepsin, medications such as aspirin and nonsteroidal antiinflammatory drugs (NSAIDs), bile acids, and infection with H. pylori. The defensive factors include the mucus layer, local bicarbonate secretion, epithelial cell renewal, and mucosal blood flow. Prostaglandins play a role in mucosal defense because they stimulate both mucus and alkali secretion. The primary mechanism that prevents the development of peptic ulcer is the secretion of mucus by the epithelial and mucus glands throughout the stomach. The thick mucus layer acts to diffuse acid from the lumen to the gastric mucosal surface, thus protecting the gastric epithelium. The stomach and the duodenum produce bicarbonate, decreasing acidity on the epithelial cells and thereby minimizing the effects of the low pH. When abnormalities in the protective barrier exist, the mucosa is vulnerable to damage by acid and pepsin. Exogenous factors, such as aspirin and NSAIDs, cause gastric ulcers by inhibition of prostaglandin synthesis.
Zollinger-Ellison syndrome is rare but may occur in children who have multiple, large, or recurrent ulcers. This syndrome is characterized by hypersecretion of gastric acid, intractable ulcer disease, and intestinal malabsorption caused by a gastrin-secreting tumor of the pancreas. The pathogenesis, manifestations, and complications of PUD are outlined in Fig. 25.5.
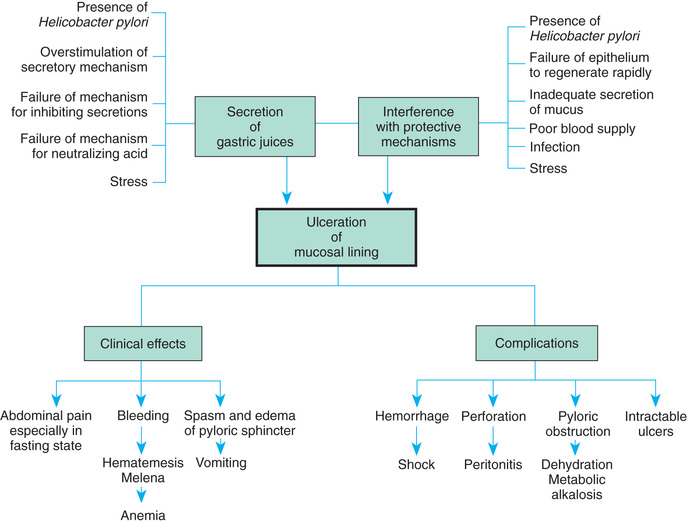
Clinical Manifestations
The clinical manifestations of PUD vary according to the child's age and the ulcer's location. Common clinical manifestations include chronic abdominal pain, especially when the stomach is empty, such as during the night or early morning; recurrent vomiting; hematemesis; melena; chronic anemia; and abdominal tenderness (Box 25.14).
Diagnostic Evaluation
Diagnosis is based on the history of symptoms, physical examination, and diagnostic testing. The focus is on symptoms such as epigastric abdominal pain, nocturnal pain, oral regurgitation, heartburn, weight loss, hematemesis, and melena. History should include questions relating to the use of potentially causative substances such as NSAIDs, corticosteroids, alcohol, and tobacco. Frequently a history of epigastric and periumbilical pain accompanies PUD. However, children often find it difficult to describe the location of their pain and frequently indicate the location by moving their hand in a circular movement all around the stomach area. Asking the child to take one finger and point to the area where it hurts the most often helps identify the location of the pain. Pain may also be elicited during the examination with palpation.
Laboratory studies may include a CBC to detect anemia, stool analysis for occult blood, liver function tests (LFTs), ESR, or CRP to evaluate IBD; amylase and lipase to evaluate pancreatitis; and gastric acid measurements to identify hypersecretion. Stool analysis is performed to rule out infection. Polyclonal and monoclonal stool antigen tests are an accurate, noninvasive method both for the initial diagnosis of H. pylori and for the confirmation of its eradication after treatment (Yang, 2016). A C13 urea breath test measures bacterial colonization in the gastric mucosa and can be used as an additional noninvasive test to determine the presence of antibodies to H. pylori.
An upper GI series is the most reliable way to detect and diagnose PUD in children (Blanchard & Czinn, 2016). Direct visualization of the gastric and duodenal mucosa helps identify specific lesions, and biopsy specimens can determine the presence of H. pylori.
Therapeutic Management
The major goals of therapy for children with PUD are to relieve discomfort, promote healing, prevent complications, and prevent recurrence. Management is primarily medical and consists of administration of medications to treat the infection and to reduce or neutralize gastric acid secretion. Antacids are beneficial medications to neutralize gastric acid. Histamine (H2) receptor antagonists (antisecretory drugs) act to suppress gastric acid production. These medications have few side effects. PPIs, such as omeprazole, lansoprazole, pantoprazole, and esomeprazole, act to inhibit the hydrogen ion pump in the parietal cells, thus blocking the production of acid. Although these drugs have not been well studied in children, they are used in clinical practice to treat ulcers, GER, esophagitis, and gastritis and appear to be well tolerated with infrequent side effects (e.g., headache, diarrhea, nausea) (Blanchard & Czinn, 2016).
Mucosal protective agents, such as sucralfate and bismuth-containing preparations, may be prescribed for PUD. Sucralfate is an aluminum-containing agent that forms a barrier over ulcerated mucosa to protect against acid and pepsin. Bismuth compounds are sometimes prescribed for the relief of ulcers, but they are used less frequently than PPIs. Although these compounds inhibit the growth of microorganisms, the mechanism of their activity is poorly understood. In combination with antibiotics, bismuth is effective against H. pylori. Although concern has been expressed about the use of bismuth salts in children because of potential side effects, none of these side effects has been reported when these compounds have been used in the treatment of H. pylori infection. These agents are available in both pill and liquid forms. Because they block the absorption of other medications, they should be given separately from other medications.
Triple-drug therapy is the standard first-line treatment regimen for H. pylori and has demonstrated 90% efficacy in the eradication of H. pylori (Kalach, Bontems, & Cadranel, 2015). Examples of drug combinations used in triple therapy are (1) bismuth, clarithromycin, and metronidazole; (2) lansoprazole, amoxicillin, and clarithromycin; and (3) metronidazole, clarithromycin, and omeprazole. Common side effects of medications include diarrhea, nausea, and vomiting.
In addition to medications, the child with PUD should have a nutritious diet and avoid caffeine. Warn adolescents about gastric irritation associated with alcohol use and smoking.
Children with an acute ulcer who have developed complications, such as massive hemorrhage, require emergency care. The administration of IV fluids, blood, or plasma depends on the amount of blood loss. Replacement with whole blood or packed cells may be necessary for significant loss.
Surgical intervention may be required for complications such as hemorrhage, perforation, or gastric outlet obstruction. Ligation of the source of bleeding or closure of a perforation is performed. A vagotomy and pyloroplasty may be indicated in children with bleeding ulcers despite aggressive medical treatment (Patel, Bommayya, Choudhry, et al., 2015).
Prognosis.
The long-term prognosis for PUD is variable. Many ulcers are successfully treated with medical therapy; however, primary duodenal peptic ulcers often recur. Complications such as GI bleeding can occur and extend into adult life. The effect of maintenance drug therapy on long-term morbidity remains to be established with further studies.
Nursing Care Management
The primary nursing goal is to promote healing of the ulcer through compliance with the medication regimen. If an analgesic-antipyretic is needed, acetaminophen, not aspirin or NSAIDs, is used. Critically ill neonates, infants, and children in intensive care units should receive H2 blockers to prevent stress ulcers.
 Drug Alert
Drug Alert
H2 Blockers
Critically ill children receiving IV H2 blockers should have their gastric pH values checked at frequent intervals.
For nonhospitalized children with chronic illnesses, consider the role stress plays. In children, many ulcers occur secondary to other conditions, and the nurse should be aware of family and environmental conditions that may aggravate or precipitate ulcers. Children may benefit from psychologic counseling and from learning how to cope constructively with stress.
Obstructive Disorders
Obstruction in the GI tract occurs when the passage of nutrients and secretions is impeded by a constricted or occluded lumen or when there is impaired motility (paralytic ileus). Obstructions may be congenital or acquired. Congenital obstructions, such as esophageal or intestinal atresias, imperforate anus, and meconium ileus, usually appear in the neonatal period. Other obstructions of congenital etiology (e.g., malrotation, HD, pyloric stenosis, volvulus, incarcerated hernia, and Meckel diverticulum) appear after the first few weeks of life. Intestinal obstruction from acquired causes such as intussusception and tumors may occur in infancy or childhood.
Acute intestinal obstruction is commonly characterized by abdominal pain, nausea, vomiting, abdominal distention, and a change in stooling patterns (Box 25.15). Pain is caused by intermittent muscular contractions proximal to the obstruction as the bowel attempts to move luminal contents along the normal path. It may also be due to severe abdominal distention, which results from accumulation of gas and fluid above the level of the obstruction. As abdominal distention progresses, the abdomen may become extremely tender, rigid, and firm.
When abdominal contents continue to accumulate, nausea and vomiting occur. Vomiting of gastric contents is often the first sign of a high obstruction, such as obstruction of the pylorus, and vomiting of bile-stained material is a sign of obstruction of the small intestine. Persistent vomiting can lead to dehydration and electrolyte disturbances. Constipation and obstipation (prolonged absence of defecation) are early signs of low obstructions and later signs of higher obstructions. In acute conditions such as intussusception, the clinical manifestations are apparent within a few hours of the onset of the disorder. In other conditions such as hypertrophic pyloric stenosis the signs and symptoms may have a more gradual onset. Bowel sounds may initially be hyperactive, then diminish or cease. Respiratory distress may occur when the diaphragm is pushed up into the pleural cavity as a result of severe abdominal distention.
Hypertrophic Pyloric Stenosis
Hypertrophic pyloric stenosis (HPS) occurs when the circumferential muscle of the pyloric sphincter becomes thickened, resulting in elongation and narrowing of the pyloric canal. This produces an outlet obstruction and compensatory dilation, hypertrophy, and hyperperistalsis of the stomach. This condition usually develops in the first few weeks of life, causing nonbilious vomiting, which occurs after a feeding; projectile vomiting may develop and the infant is fussy and hungry after vomiting. If the condition is not diagnosed early, dehydration, metabolic alkalosis, and failure to thrive may occur. The precise etiology of HPS is not known. Boys are affected four to six times more frequently than girls (Hunter & Liacouras, 2016). It is more common in white infants and is seen less frequently in African American and Asian infants (Hunter & Liacouras, 2016).
Pathophysiology
The circular muscle of the pylorus thickens as a result of hypertrophy. This produces severe narrowing of the pyloric canal between the stomach and the duodenum. Consequently, the lumen at this point is partially obstructed. Over time, inflammation and edema further reduce the size of the opening, resulting in complete obstruction. The hypertrophied pylorus may be palpable as an olive-like mass in the upper abdomen (Fig. 25.6).
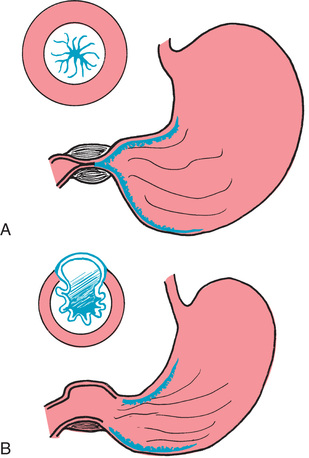
Pyloric stenosis is not a congenital disorder. It is believed that local innervation may be involved in the pathogenesis. In most cases, HPS is an isolated lesion; however, it may be associated with intestinal malrotation, esophageal and duodenal atresia, and anorectal anomalies.
Clinical Manifestations
Infants with HPS have nonbilious vomiting in the early stages (Box 25.16). Vomiting usually begins at 3 weeks of age but can start as early as 1 week and as late as 5 months. Vomiting usually occurs 30 to 60 minutes after feeding and becomes projectile as the obstruction progresses. Initially the infant is hungry and irritable, but prolonged vomiting may lead to dehydration, weight loss, and failure to thrive. Gastric peristalsis may be visible on examination, and the olive-shaped mass in the epigastrium just to the right of the umbilicus may be palpated (see Fig. 25.6, A). Indirect (unconjugated) hyperbilirubinemia may be present in a small percentage of affected infants; this usually resolves with surgical correction and is reported to occur as a result of a decreased level of glucuronyl transferase (see also Chapter 8).
Diagnostic Evaluation
The diagnosis of HPS is often made after the history and physical examination. The olive-like mass is most easily palpated when the stomach is empty, the infant is quiet, and the abdominal muscles are relaxed. If the diagnosis is inconclusive from the history and physical examination, ultrasonography will demonstrate an elongated mass surrounding a long pyloric canal. If ultrasonography does not demonstrate a hypertrophied pylorus, upper GI radiography should be done to rule out other causes of vomiting.
If the condition is not diagnosed early, laboratory findings reflect the metabolic alterations created by moderate to severe depletion of both water and electrolytes from extensive and prolonged vomiting. There are decreased serum levels of both sodium and potassium, although these may be masked by the hemoconcentration from extracellular fluid depletion. Of greater diagnostic value are a decrease in serum chloride levels and increases in pH and bicarbonate (carbon dioxide content), indicative of metabolic alkalosis. The blood urea nitrogen will be elevated as evidence of dehydration. However, in those cases diagnosed early, laboratory findings may not be significant.
Therapeutic Management
Surgical relief of the pyloric obstruction by pyloromyotomy is the standard therapy for this disorder. Preoperatively the infant must be rehydrated and metabolic alkalosis corrected with parenteral fluid and electrolyte administration. Replacement fluid therapy usually delays surgery for 24 to 48 hours. The stomach is decompressed with an NG tube if the infant continues with vomiting. In infants with no evidence of fluid and electrolyte imbalance, surgery is performed without delay.
The surgical procedure is often performed by laparoscope and consists of a longitudinal incision through the circular muscle fibers of the pylorus down to, but not including, the submucosa (pyloromyotomy, or the Fredet-Ramstedt operative procedure) (see Fig. 25.6, B). The procedure has a high success rate. Laparoscopic surgery may result in a shorter surgical time, more rapid postoperative feeding, and shorter hospital stay (Hunter & Liacouras, 2016).
Feedings are usually begun 4 to 6 hours postoperatively, beginning with small, frequent feedings of water or electrolyte solution. If clear fluids are retained, about 24 hours after surgery formula is started in the same small increments. The amount and the interval between feedings are gradually increased until a full feeding schedule is reinstated, which usually takes about 48 hours.
Prognosis.
The prognosis for infants and small children with HPS is excellent when the diagnosis is confirmed early, and the mortality rate is low (0% to 0.5%). A small percentage of children with HPS will have GER.
Nursing Care Management
Nursing care involves primarily observation for clinical features that help establish the diagnosis, careful regulation of fluid therapy, and reestablishment of normal feeding patterns. Nurses must be alert to signs of HPS in infants and refer them for medical evaluation. HPS should be considered a possibility in the very young infant who appears alert but fails to gain weight and has a history of vomiting after feedings. Assessment is based on observation of eating behaviors and evidence of other characteristic clinical manifestations, hydration, and nutritional status.
Preoperatively, the emphasis is on restoring hydration and electrolyte balance. The infant is kept NPO and given IV fluids with glucose and electrolytes based on serum electrolyte values and clinical appearance. Careful monitoring of the IV fluids and strict monitoring of intake and output are important. Record accurate description of any vomiting and the number and character of stools.
Observations include assessment of vital signs, particularly those that indicate fluid or electrolyte imbalances. These infants are especially prone to metabolic alkalosis from loss of hydrogen ions and depletion of potassium, sodium, and chloride, all of which are contained in gastric secretions. Assess the skin and mucous membranes for alterations in hydration status. (See Chapter 23 for manifestations of fluid and electrolyte disturbances.)
If stomach decompression and gastric lavage are part of preoperative management, the nurse is responsible for ensuring that the NG tube is patent and functioning properly and for measuring and recording the type and amount of drainage. Encourage parents to visit and become involved in the child's care. Most parents need support and reassurance that the condition is caused by a structural problem and is not a reflection of their parenting skills and capacities.
Postoperative vomiting is common, and most infants, even with successful surgery, exhibit some vomiting during the first 24 to 48 hours. IV fluids are administered until the infant is taking and retaining adequate amounts by mouth. Much of the same care that was instituted before surgery is continued postoperatively, including observation of vital signs, monitoring of IV fluids, and careful monitoring of intake and output. In addition, the infant is observed for responses to the stress of surgery and for evidence of pain. Appropriate analgesics should be given around the clock because pain is continuous. The surgical incision(s) is inspected for drainage or erythema, and any signs of infection are reported to the surgeon. A surgical adhesive may be used for incision closure, and parents are instructed regarding the care of the incision and any dressings before discharge.
Feedings are usually instituted within 12 to 24 hours postoperatively, beginning with clear liquids. They are offered in small quantities at frequent intervals. If the infant has been breastfed, breast milk expressed by the mother may be given by bottle when the infant is able to tolerate feedings, or the mother is instructed to limit nursing time and gradually increase the time to previous patterns. Observation and recording of feedings and the infant's responses to feedings are a vital part of postoperative care. Care of the operative site consists of observation for any drainage or signs of inflammation and care of the incision.
Intussusception
Intussusception is the most common cause of intestinal obstruction in children between 3 months and 6 years old (Carroll, Kavanagh, Ni Leidhin, et al., 2017). Intussusception is more common in males than in females and is more common in children younger than 2 years old. Although specific intestinal lesions occur in a small percentage of the children, generally the cause is not known. Only 12.5% to 25% of intussusception cases have a pathologic lead point, such as a polyp, lymphoma, or Meckel diverticulum (Carroll, Kavanagh, Ni Leidhin, et al., 2017). The idiopathic cases may be caused by hypertrophy of intestinal lymphoid tissue secondary to viral infection.
Pathophysiology
Intussusception occurs when a proximal segment of the bowel telescopes into a more distal segment, pulling the mesentery with it. The mesentery is compressed and angled, resulting in lymphatic and venous obstruction. As the edema from the obstruction increases, pressure within the area of intussusception increases. When the pressure equals the arterial pressure, arterial blood flow stops, resulting in ischemia and the pouring of mucus into the intestine. Venous engorgement also leads to leaking of blood and mucus into the intestinal lumen, forming the classic currant jelly–like stools. The most common site is the ileocecal valve (ileocolic), where the ileum invaginates into the cecum and then further into the colon (Fig. 25.7). Other forms include ileoileal (i.e., one part of the ileum invaginates into another section of the ileum) and colocolic (i.e., one part of the colon invaginates into another area of the colon) intussusceptions, usually in the area of the hepatic or splenic flexure or at some point along the transverse colon.
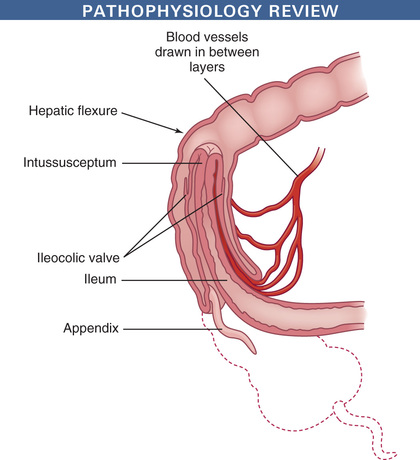
Clinical Manifestations
Intussusception usually manifests with the sudden onset of crampy abdominal pain, inconsolable crying, and a drawing up of the knees to the chest in an otherwise healthy child (Box 25.17). Between episodes the child appears normal. As the obstruction progresses, bilious vomiting may occur and lethargy increases. The classic triad of intussusception symptoms (abdominal pain, abdominal mass, bloody stools) is present in less than 30% of children (Kennedy & Liacouras, 2016b). A more chronic case may be presented, characterized by diarrhea, anorexia, weight loss, occasional vomiting, and periodic pain. Because intussusception is potentially life threatening, be aware of such signs, and closely observe and refer these children for further medical evaluation. With atypical cases, lethargy may be the primary symptom. If the distal bowel remains distended, necrosis and perforation are possible.
Diagnostic Evaluation
Frequently, subjective findings lead to the diagnosis. However, definitive diagnosis is based on ultrasonography that reveals a characteristic heterogenous mass and a “bull's-eye.” A rectal examination reveals mucus, blood, and occasionally a low intussusception itself.
Therapeutic Management
Conservative treatment consists of radiologist-guided pneumoenema (gas enema) or ultrasound-guided hydrostatic enema, the advantage of the latter being that no ionizing radiation is needed (Kennedy & Liacouras, 2016b). Recurrence of intussusception after conservative treatment is rare; however, this procedure should not be attempted with prolonged intussusception, signs of shock, peritoneal irritation, or intestinal perforation (Kennedy & Liacouras, 2016b).
IV fluids, NG decompression, and antibiotic therapy may be used before hydrostatic reduction is attempted. If these procedures are not successful, the child may require surgical intervention. Surgery involves manually reducing the invagination and, when indicated, resecting any nonviable intestine.
Prognosis.
Nonoperative reduction is successful in the majority of stable cases. Gas enema is slightly more successful with reduction compared to a hydrostatic enema (83% versus 70%, respectively) (Carroll, Kavanagh, Ni Leidhin, et al., 2017). Surgery is required for patients in whom the reduction is unsuccessful or for patients who are unstable. With early diagnosis and treatment, serious complications and death are uncommon.
Nursing Care Management
The nurse can help establish a diagnosis by listening to the parent's description of the child's physical and behavioral symptoms. It is not unusual for parents to state that they thought something was seriously wrong before others shared their concerns. The description of the child's severe colicky abdominal pain combined with vomiting is a significant sign of intussusception.
As soon as a possible diagnosis of intussusception is made, the nurse prepares the parents for the immediate need for hospitalization, the nonsurgical technique of hydrostatic reduction, and the possibility of surgery. It is important to explain the basic defect of intussusception. The nurse can easily demonstrate this by creating a model of the defect. Use the example of a telescoping rod, or push the end of a finger on a rubber glove back into itself. Then demonstrate the principle of reduction by hydrostatic pressure by filling the glove with water, which pushes the “finger” into a fully extended position.
Physical care of the child does not differ from that for any child undergoing abdominal surgery. Even though nonsurgical intervention may be successful, the usual preoperative procedures, such as maintenance of NPO status, routine laboratory testing (CBC and urinalysis), signed parental consent, and preanesthetic sedation, are performed. Children with perforation will require IV fluids, systemic antibiotics, and bowel decompression before undergoing surgery. Fluid volume replacement and restoration of electrolytes may be required in such children before surgery. Before surgery the nurse monitors all stools.
Nursing Alert
Passage of a normal brown stool usually indicates that the intussusception has reduced itself. This is immediately reported to the practitioner, who may choose to alter the diagnostic and therapeutic care plan.
Postprocedural care includes observations of vital signs, blood pressure, intact sutures and dressing, and the return of bowel sounds. After spontaneous or hydrostatic reduction, the nurse observes for passage of water-soluble contrast material (if used) and the stool patterns because the intussusception may recur. Children may be admitted to the hospital or monitored on an outpatient basis. A recurrence of intussusception is treated with the conservative reduction techniques described previously, but a laparotomy is considered for multiple recurrences.
Malrotation and Volvulus
Malrotation of the intestine is caused by the abnormal rotation of the intestine around the superior mesenteric artery during embryologic development. Malrotation may manifest in utero or at any age, but the majority of patients (80%) present in the first month of life (Carroll, Kavanagh, Ni Leidhin, et al., 2017). Infants may have intermittent bilious vomiting, recurrent abdominal pain, distention, or lower GI bleeding. Malrotation is the most serious type of intestinal obstruction because if the intestine undergoes complete volvulus (i.e., the intestine twisting around itself), compromise of the blood supply will result in intestinal necrosis, peritonitis, perforation, and death.
Diagnostic Evaluation
It is imperative that malrotation and volvulus be diagnosed promptly and surgical treatment instituted quickly. In addition to a history and physical, a plain abdominal radiograph and lateral decubitus view are obtained; bowel distention will be present proximal to the distention on plain radiograph, and a lateral view will demonstrate air-fluid levels in the distended bowel (Bales & Liacouras, 2016). An upper GI series is the most accurate imaging study (Carroll, Kavanagh, Ni Leidhin, et al., 2017).
Therapeutic Management
Surgery is indicated to remove the affected area. Because of the extensive nature of some lesions, short-bowel syndrome (SBS) is a postoperative complication.
Nursing Care Management
Preoperatively the nursing care is the same as that provided to an infant or child with intestinal obstruction. IV fluids, NG decompression, and systemic antibiotics are implemented. In the rapidly deteriorating infant, fluid volume resuscitation and vasopressors may be required for preoperative stabilization. Postoperatively, the nursing care is similar to that provided to the infant or child who has undergone abdominal surgery.
Malabsorption Syndromes
Chronic diarrhea and malabsorption of nutrients characterize malabsorption syndromes. An important complication of malabsorption syndromes in children is failure to thrive. Most cases are classified according to the location of the supposed anatomic or biochemical defect. The term celiac disease is often used to describe a symptom complex with four characteristics: (1) steatorrhea (fatty, foul, frothy, bulky stools), (2) general malnutrition, (3) abdominal distention, and (4) secondary vitamin deficiencies.
Digestive defects are conditions in which the enzymes necessary for digestion are diminished or absent, such as (1) cystic fibrosis, in which pancreatic enzymes are absent; (2) biliary or liver disease, in which bile flow is affected; or (3) lactase deficiency, in which there is congenital or secondary lactose intolerance.
Absorptive defects are conditions in which the intestinal mucosal transport system is impaired. This may occur because of a primary defect (e.g., celiac disease) or secondary to inflammatory disease of the bowel that results in impaired absorption because bowel motility is accelerated (e.g., ulcerative colitis). Obstructive disorders (e.g., Hirschsprung disease) also cause secondary malabsorption from enterocolitis.
Anatomic defects, such as extensive resection of the bowel or SBS, affect digestion by decreasing the transit time of substances and affect absorption by severely compromising the absorptive surface.
Celiac Disease (Gluten-Sensitive Enteropathy)
Celiac disease, also known as gluten-induced enteropathy, gluten-sensitive enteropathy, and celiac sprue, is an autoimmune disorder triggered by the ingestion of gluten in genetically susceptible individuals (Fok, Holland, Gil-Zaragozano, et al., 2016). The disorder results in permanent intestinal intolerance to dietary gluten, a protein present in wheat, barley, and rye that causes damage to the villi in the small intestine. Children with unexplained iron deficiency anemia, recurrent aphthous stomatitis, dental enamel defects, type 1 diabetes, Down syndrome, selective immunoglobulin A deficiency, autoimmune thyroid disease, Turner syndrome, or Williams syndrome are more susceptible to being diagnosed with the disease (Paul, McVeigh, Gil-Zaragozano, et al., 2016). The disease is seen more frequently in Europe and the United States in approximately 1% of these populations, and it is rarely reported in Asians or African Americans (Branski, Troncone, & Fasano, 2016).
Pathophysiology
Celiac disease is characterized by villous atrophy in the small intestine in response to the protein gluten. When individuals are unable to digest the gliadin component of gluten, an accumulation of a toxic substance occurs that is damaging to the mucosal cells. Damage to the mucosa of the small intestine leads to villous atrophy, hyperplasia of the crypts, and infiltration of the epithelial cells with lymphocytes. Villous atrophy leads to malabsorption due to the reduced absorptive surface area (see Fig. 25.1).
Genetic predisposition is an essential factor in the development of celiac disease. Membrane receptors involved in preferential antigen presentation to CD4+ T cells play a crucial role in the immune response characteristic of celiac disease. Children with genetic susceptibilities, namely HLA-DQ2 or HLA-DQ8, are more susceptible to being diagnosed with celiac disease (Lebwohl, Sanders, & Green, 2017).
Clinical Manifestations
Symptoms of celiac disease appear when solid foods such as beans and pasta are introduced into the child's diet between the ages of 1 and 5 years (Box 25.18). There is usually an interval of several months between the introduction of gluten into the diet and the onset of symptoms. Intestinal symptoms are common in children diagnosed within the first 2 years of life. Other symptoms include failure to thrive, chronic diarrhea, abdominal distention and pain, muscle wasting, aphthous ulcers, and fatigue.
Diagnostic Evaluation
Gluten should not be excluded from the diet until the diagnostic evaluation is complete so proper identification can occur. The first step is a serologic blood test for tissue transglutaminase and antiendomyseal antibodies in children 18 months of age or older (Branski, Troncone, & Fasano, 2016). Positive serologic markers should be followed by an upper GI endoscopy with biopsy. The diagnosis of celiac disease is based on a biopsy of the small intestine demonstrating the characteristic changes of mucosal inflammation, crypt hyperplasia, and villous atrophy (Branski, Troncone, & Fasano, 2016).
Therapeutic Management
Treatment of celiac disease consists primarily of dietary management. Although a gluten-free diet is prescribed, it is actually low in gluten because it is impossible to remove every source of this protein. Because gluten is found primarily in wheat and rye, but also in smaller quantities in barley and oats, these four foods are eliminated. Corn, rice, and millet are substitute grain foods.
Children with untreated celiac disease may have lactose intolerance, especially if their mucosal lesions are extensive. Lactose intolerance usually improves as the mucosa heals with gluten withdrawal. Specific nutritional deficiencies, such as iron, folic acid, and fat-soluble vitamin deficiencies, are treated with appropriate supplements.
Prognosis.
Celiac disease is regarded as a chronic disease; its severity varies greatly among children. The most severe symptoms usually occur in early childhood and again in adult life. Most children who comply with dietary management are healthy and remain free of symptoms and complications; however, children should be evaluated annually for nutritional deficiencies, impaired growth, delayed puberty, and reduced bone mineral density (Fok, Holland, Gil-Zaragozano, et al., 2016).
Nursing Care Management
The main nursing consideration is helping the child adhere to the dietary regimen. Considerable time is involved in explaining the disease process to the child and parents, the specific role of gluten in aggravating the disorder, and those foods that must be restricted. It is difficult to maintain a diet indefinitely when the child has no symptoms and temporary transgressions result in no difficulties. However, the majority of individuals who relax their diet will experience a relapse of their disease.
Although the chief source of gluten is cereal and baked goods, grains are frequently added to processed foods as thickeners or fillers. To compound the difficulty, gluten is added to many foods as hydrolyzed vegetable protein, which is derived from cereal grains. The nurse must advise parents of the necessity of reading all label ingredients carefully to avoid hidden sources of gluten.
Many of children's favorite foods contain gluten, including bread, cake, cookies, crackers, donuts, pies, spaghetti, pizza, prepared soups, hot dogs, luncheon meats, and some prepared hamburgers. Many of these products can be eliminated from the infant's or young child's diet fairly easily, but monitoring the diet of the school-age child or adolescent is more difficult. Luncheon preparation away from home is particularly difficult because bread, luncheon meats, and instant soups are not allowed. For families on restricted food budgets, the diet adds an additional financial burden because many inexpensive or convenient foods cannot be used.
In addition to restricting gluten, other dietary alterations may be necessary. For example, in some children who have more severe mucosal damage, the digestion of disaccharides is impaired, especially in relation to lactose. Therefore these children often need a temporarily lactose-free diet, which necessitates eliminating all milk products. In general, dietary management includes a diet high in calories and proteins with simple carbohydrates such as fruits and vegetables, but low in fats. Because the bowel is inflamed as a result of the pathologic processes in absorption, the child must avoid high-fiber foods such as nuts, raisins, raw vegetables, and raw fruits with skin until inflammation has subsided.
It is important to stress long-range complications and to remind parents of the child's physical status before dietary treatment and the dramatic improvement after treatment. The nurse can be instrumental in allowing the child to express concerns and frustration while focusing on ways in which the child can still feel normal. Encourage the child and parents to find new recipes using suitable ingredients, such as Mexican or Chinese dishes that use corn or rice. Consult a registered dietitian to provide children and their families with detailed dietary instructions and education.
Several resources are available to assist children and parents in all aspects of coping with celiac disease. The National Celiac Association* provides support and guidance to families and supplies educational materials concerning a gluten-free diet, food sources, recipes, and travel information.
Short-Bowel Syndrome
Short-bowel syndrome (SBS) is a malabsorptive disorder that occurs as a result of decreased mucosal surface area, usually because of extensive resection of the small intestine. Malabsorption may be exacerbated by other factors, such as bacterial overgrowth and dysmotility. The most common congenital causes of SBS in children are short-bowel syndrome, multiple atresias, and gastroschisis; other causes resulting in bowel resection include necrotizing volvulus, meconium peritonitis, Crohn disease, and trauma (Vanderhoof & Branski, 2016).
The definition of SBS includes two important findings: (1) decreased intestinal surface area for absorption of fluid, electrolytes, and nutrients; and (2) a need for parenteral nutrition (PN) (Martin, Ladd, Werts, et al., 2017). The prognosis for infants with SBS has improved dramatically, but the mortality rate within 5 years after diagnosis remains at 27% to 37% (Martin, Ladd, Werts, et al., 2017).
Therapeutic Management
The goals of therapy for infants and children with SBS include (1) preserving as much length of bowel as possible during surgery; (2) maintaining optimum nutritional status, growth, and development while intestinal adaptation occurs; (3) stimulating intestinal adaptation with enteral feeding; and (4) minimizing complications related to the disease process and therapy (Vanderhoof & Branski, 2016).
Nutritional Support.
Nutritional support is the long-term focus of care for children with SBS. The initial phase of therapy includes PN as the primary source of nutrition. The second phase is the introduction of enteral feeding, which usually begins as soon as possible after surgery. Elemental formulas containing glucose, sucrose and glucose polymers, hydrolyzed proteins, and medium-chain triglycerides facilitate absorption. Usually these formulas are given by continuous infusion through an NG or gastrostomy tube. As the enteral feedings are advanced, the PN solution is decreased in terms of calories, amount of fluid, and total hours of infusion per day. If enteral feedings are tolerated, oral feedings should be attempted to minimize oral aversion and preserve oral skills (Vanderhoof & Branski, 2016).
The final phase of nutritional support occurs when growth and development are sustained. When PN is discontinued, there is a risk of nutritional deficiency secondary to malabsorption of fat-soluble vitamins (A, D, E, and K) and trace minerals (iron, selenium, zinc). Serum vitamin and mineral levels should be monitored closely and supplemented enterally, if needed. Pharmacologic agents have been used to reduce secretory losses. H2 blockers, PPIs, and octreotide inhibit gastric or pancreatic secretion. Cholestyramine is often prescribed to improve diarrhea that is associated with bile salt malabsorption.
Numerous complications are associated with SBS and long-term PN. Infectious, metabolic, and technical complications can occur. Sepsis can occur after improper care of the catheter. The GI tract can also be a source of microbial seeding of the catheter. Bowel atrophy may foster increased intestinal permeability of bacteria. A lack of adequate sites for central lines may become a significant problem for the child in need of long-term PN. Hepatic dysfunction and cholestasis may also occur (Cohran, Prozialeck, & Cole, 2017).
Bacterial overgrowth is likely to occur when the ileocecal valve is absent or when stasis exists as a result of a partial obstruction or a dilated segment of bowel with poor motility. Alternating cycles of broad-spectrum antibiotics are used to reduce bacterial overgrowth. This treatment may also decrease the risk of bacterial translocation and subsequent central venous catheter infections. A high-fat and low-carbohydrate diet may be helpful in reducing bacterial overgrowth (Vanderhoof & Branski, 2016). Other complications of bacterial overgrowth and malabsorption include metabolic acidosis and gastric hypersecretion.
Many surgical interventions, including intestinal valves, tapering enteroplasty or stricturoplasty, intestinal lengthening, and interposed segments, have been used to slow intestinal transit, reduce bacterial overgrowth, or increase mucosal surface area. Intestinal transplantation has been performed successfully in children. Children with a permanent dependence on PN or severe complications of long-term PN are candidates for transplantation.
Prognosis.
The prognosis for infants with SBS has improved with advances in PN and with the understanding of the importance of intraluminal nutrition. Improved supportive care for the management of therapy-related problems and the development of more specific immunosuppressive medications for transplantation have all contributed to improved management. The prognosis depends in part on the length of the residual small intestine. An intact ileocecal valve also improves the prognosis. Infants and children with SBS die from PN-related problems, such as fulminant sepsis or severe PN cholestasis.
Nursing Care Management
The most important components of nursing care are administration and monitoring of nutritional therapy. During PN therapy, care must be taken to minimize the risk of complications related to the central venous access device (i.e., catheter infections, occlusions, dislodgment, or accidental removal). Care of the enteral feeding tubes and monitoring of enteral feeding tolerance are also important nursing responsibilities.
When hospitalization is prolonged, the child's developmental and emotional needs must be met. This often requires special planning to promote normal family adjustment and adaptation of the hospital routines. Family members require psychosocial support and education to cope successfully with SBS.
Many infants with SBS have an intestinal ostomy performed at the time of the initial bowel resection. Routine ostomy care is another important nursing responsibility. Because infants and children with SBS have chronic diarrhea, perineal skin irritation is often a problem after ostomy closure. Frequent diaper changes, gentle perineal cleansing, and protective skin ointments help prevent skin breakdown. (See Diaper Dermatitis, Chapter 32.)
Home Care.
When long-term PN is required, preparation of the family for home care of the child is a major nursing responsibility. Preparation for home nutritional support begins as early as possible to prevent lengthy hospitalizations with subsequent problems such as developmental delays and family stresses. Many infants and children can be successfully cared for at home with enteral and parenteral nutrition if the family is thoroughly prepared and provided with adequate support services. Most families benefit from home nursing care to assist with and supervise therapy. Careful follow-up care by a multidisciplinary nutritional support service is essential. Most home health agencies now provide portable enteral and parenteral equipment, which enables the child and family to maintain a more normal and active lifestyle. The nurse plays an active and important role in the success of a home nutrition program. Home infusion companies provide portable equipment, which enables the child and family to maintain a more normal lifestyle.
Gastrointestinal Bleeding
GI bleeding in infants and children is an uncommon but potentially serious problem (Casciani, Nardo, Chin, et al., 2017). Most actual or apparent instances of GI bleeding cause great anxiety for the parents or caregivers. Blood may be vomited or passed per rectum, but the origin of the blood may not be the GI tract. In the newborn, swallowed maternal blood at the time of delivery may account for some episodes of apparent GI bleeding. A bleeding site on the nipple of a nursing mother may lead to heme-positive stools in the breastfed infant. Finally, blood can be swallowed during epistaxis and then passed as hematemesis or melena.
Once it has been established that the cause of bleeding is from a source in the GI tract, further investigation for the source and cause is undertaken. Upper GI bleeding is defined as bleeding from a site above the ligament of Treitz, which is attached to the duodenum at its junction with the jejunum. Lower GI bleeding comes from a source distal to the ligament of Treitz. Diagnostic studies such as endoscopy, scintigraphy, and angiography have improved the ability to localize the site of bleeding.
Etiology
The esophagus is a common site of upper GI bleeding. Esophagitis caused by GER may lead to chronic and often occult blood loss. Esophageal varices secondary to portal hypertension may cause massive bleeding. Peptic inflammation (gastritis and duodenitis) or ulceration is the most common cause of upper GI bleeding in children. Hemorrhagic gastritis may occur in the newborn infant after a difficult delivery or asphyxia. In this circumstance gastric perforation is a serious complication that requires emergent treatment. Less common causes of upper GI bleeding include bleeding disorders, vascular malformations, GI duplications, Mallory-Weiss syndrome (an esophageal tear caused by protracted vomiting), and hematobilia (bleeding into biliary passages).
In lower GI bleeding, small amounts of bright red blood in the stool of a healthy child may be due to an anal fissure. Colonic polyps are another cause of passage of bright red blood per rectum in toddlers and older children. Bleeding associated with diarrhea may indicate a serious problem. Enteric infections remain the leading cause, but the nurse should consider necrotizing enterocolitis, hemolytic uremic syndrome, IBD, and food allergy. Other causes are intussusception with the passage of blood per rectum (see earlier in this chapter) or Meckel diverticulum with the painless passage of currant jelly–like stools (see earlier in this chapter).
Pathophysiology
The GI tract has an extensive surface area and a rich vascular supply. Bleeding can occur anywhere along the GI tract from a vein, artery, or vascular malformation. In an otherwise healthy newborn infant, hemophilia or inherited coagulation-factor deficits are rarely accompanied by bleeding unless other conditions are superimposed. Children with liver disease may also have deficient coagulation factors because of poor synthesis and malabsorption of vitamin K, which is a risk factor for GI bleeding.
Portal hypertension may lead to GI bleeding because the formation of portosystemic shunts can result in dilated venous channels in vulnerable locations such as the esophagus and stomach. These dilated venous channels (varices) may bleed, causing severe GI hemorrhage.
Diagnostic Evaluation
The diagnosis of GI bleeding is often made on the basis of the history and physical examination. Hematemesis is the vomiting of bright red blood or denatured blood that looks like coffee grounds, usually representing an upper GI source of bleeding. Hematochezia is the passage of bright red blood per rectum, indicating lower GI bleeding. This blood may precede or follow a bowel movement or be mixed with or coat the stool. Bright red blood that coats the stool may be due to a hard bowel movement, hemorrhoids, or anal fissures. Blood mixed with stool indicates a bleeding source proximal to the rectum. Blood passed alone after a bowel movement is most likely due to bleeding in the perianal or rectal area, possibly caused by a polyp. Blood with mucus in the stool indicates an inflammatory or infectious condition, and currant jelly–like stools indicate vascular compromise, such as intussusception. Melena is the passage of black, tarry stools that contain denatured (digested) blood and suggests an upper GI source of bleeding. Occasionally, bright red blood may be passed per rectum from an upper GI source of bleeding when the bleeding is massive. It is important to test emesis or stool for occult blood to differentiate true bleeding from the ingestion of food containing food coloring. In older children, false-positive stool tests for occult blood can also occur with the ingestion of red meats and iron preparations.
Laboratory studies are determined on the basis of the history and physical examination. In many instances, a CBC with platelet quantification, prothrombin, partial thromboplastin, and coagulation studies will be done. Children who have acute illness, fever, and joint pain in addition to GI bleeding need an ESR and stool studies with culture to evaluate for enteric pathogens, ova and parasites, and C. difficile. When IBD is suspected, a metabolic panel to determine total protein and albumin may be added to a CBC, ESR, and LFTs. The child with massive painless rectal bleeding may require a nuclear medicine scan to rule out Meckel diverticulum. If there is evidence of portal hypertension or chronic liver disease, LFTs, liver imaging studies, and a liver biopsy may be necessary. A barium enema is performed if intussusception is suspected.
Imaging studies help differentiate among several suspected diagnoses. CT of the sinuses helps localize bleeding that is coming from the nasopharynx or sinuses. A chest radiograph may distinguish hemoptysis related to cystic fibrosis, bronchiectasis, or other chronic lung conditions from hematemesis. Angiography can be used to identify the source of bleeding and to allow embolization or vasopressin infusion for treatment. Endoscopy is the diagnostic method chosen when the source of bleeding is thought to be secondary to gastritis, esophagitis, PUD, colitis, or polyps. Endoscopic examinations also permit visualization of the intestinal mucosa and collection of biopsy specimens and cultures.
Therapeutic Management
Treatment of GI bleeding in children depends on its severity and cause. The first step in management of acute GI bleeding is to assess the magnitude of blood loss and restore the child's hemodynamic stability. Severe bleeding necessitates hospitalization. IV fluids (normal saline or lactated Ringer solution) are administered rapidly. Oxygen therapy is indicated if the bleeding is severe. Transfusion of blood products may be required if the blood loss is significant, and any existing coagulopathy should be corrected.
Upper GI mucosal lesions are usually treated with H2-receptor antagonists (e.g., cimetidine, ranitidine, or famotidine) or PPIs (e.g., omeprazole, lansoprazole, pantoprazole, or esomeprazole) and antacids to reduce acidity and promote mucosal healing. Variceal hemorrhage can be treated with peripheral vasopressin infusion and endoscopic sclerotherapy to hasten tissue fibrosis. Balloon tamponade to place pressure on the bleeding area may be performed as a temporary measure until endoscopic sclerotherapy can be done.
Therapy for lower GI bleeding is directed toward the primary underlying condition. The treatment may include medical or surgical management. Surgery may be required if the bleeding is severe despite aggressive medical intervention.
Nursing Care Management
The infant or child with acute and severe GI bleeding requires emergency care. Initial management includes assessment of the magnitude of bleeding and hemodynamic status and assistance with resuscitation efforts (see Critical Thinking Case Study box).
Critical Thinking Case Study
Hematemesis
A 6-month-old infant is seen in the emergency department. The parents brought the infant to the hospital because he spit up formula with blood streaks. In the emergency department the infant has tachypnea, tachycardia, and a fever of 39°C (102.2°F). A chest x-ray film shows pneumonia. The infant is admitted to the hospital to receive antibiotics and for observation. Several hours after admission to the inpatient unit, the mother calls the nurse when the infant vomits a large amount of bright red blood. The infant is pale and lethargic. Which of the following should not be included in the initial nursing actions?
- 1. Call for assistance and estimate the amount of blood loss.
- 2. Obtain vital signs and monitor capillary refill, skin color, and behavior.
- 3. Prepare to pass a nasogastric tube, obtain blood for laboratory analyses, and start an intravenous line.
- 4. Test stool for blood (Hematest or Hemoccult).
Questions
- 1. Evidence—Are there sufficient data to support your decision?
- 2. Assumptions—Describe some underlying assumptions about the following:
- 3. What are the priorities for this child at this time?
- 4. What are the nursing actions that need to be implemented?
Answers are available at http://evolve.elsevier.com/wong/ncic.
Nursing Alert
Monitor closely for signs of shock: restlessness; increased respiratory and heart rate; poor capillary refill; pallor; cool, clammy extremities; and decreased blood pressure (a late sign). Call for assistance immediately if these signs are observed.
Make certain oxygen and suction equipment is available. An IV line should be inserted and preparation made for the administration of IV fluids, usually normal saline or lactated Ringer solution. Draw blood for laboratory analysis, including hemoglobin, hematocrit, blood urea nitrogen, creatinine, coagulation studies, and type and crossmatch. The nurse should be prepared to insert an NG tube to help locate the site of bleeding and to lavage the stomach with normal saline at room temperature if upper GI bleeding is suspected. Avoid taking rectal temperatures to prevent further irritation or damage to the rectal mucosa of a child suspected of having rectal bleeding or fissures. After the child is stabilized, ongoing monitoring in an intensive care setting may be indicated.
In cases of mild or chronic bleeding, there is more time for a thorough history and diagnostic evaluation, often in an outpatient setting. Important nursing responsibilities include assisting with the history and physical examination, diagnostic procedures, and education regarding the therapeutic plan.
The parents or caregivers of a child with GI bleeding may be extremely anxious and panic stricken. They need reassurance that most instances of bleeding are self-limiting and can be treated successfully. In life-threatening situations, special emotional support is required. Keep the family informed about the source, cause, and treatment of the bleeding.
Hepatic Disorders
The liver is a vital organ whose functions can be divided into several groups: (1) vascular functions of storing and filtering blood; (2) secretory function of producing bile; (3) metabolism of carbohydrate, protein, and fat; (4) synthesis of blood-clotting components and storage of iron and vitamins (A, D, B12, and K); and (5) detoxification and excretion of certain drugs and metabolic substances. Many disorders, including biliary atresia, hepatitis, and cirrhosis, can cause liver dysfunction in children.
Acute Hepatitis
Hepatitis is an acute or chronic inflammation of the liver that can result from infectious or noninfectious reasons. Viruses such as hepatitis viruses, Epstein-Barr virus (EBV), and cytomegalovirus (CMV) are common causes of many types of hepatitis. Other causes of hepatitis are nonviral (e.g., abscess, amebiasis), autoimmune, metabolic, drug induced, anatomic (e.g., choledochal duct cyst and biliary atresia), hemodynamic (e.g., shock, congestive heart failure), and idiopathic (e.g., sclerosing cholangitis and Reye syndrome). Determining the cause of acute or chronic hepatitis is important in determining the treatment and prognosis for the child. Epidemiologic features and serologic testing are used to differentiate the causes. Table 25.7 compares the features of hepatitis A, B, and C viruses.
TABLE 25.7
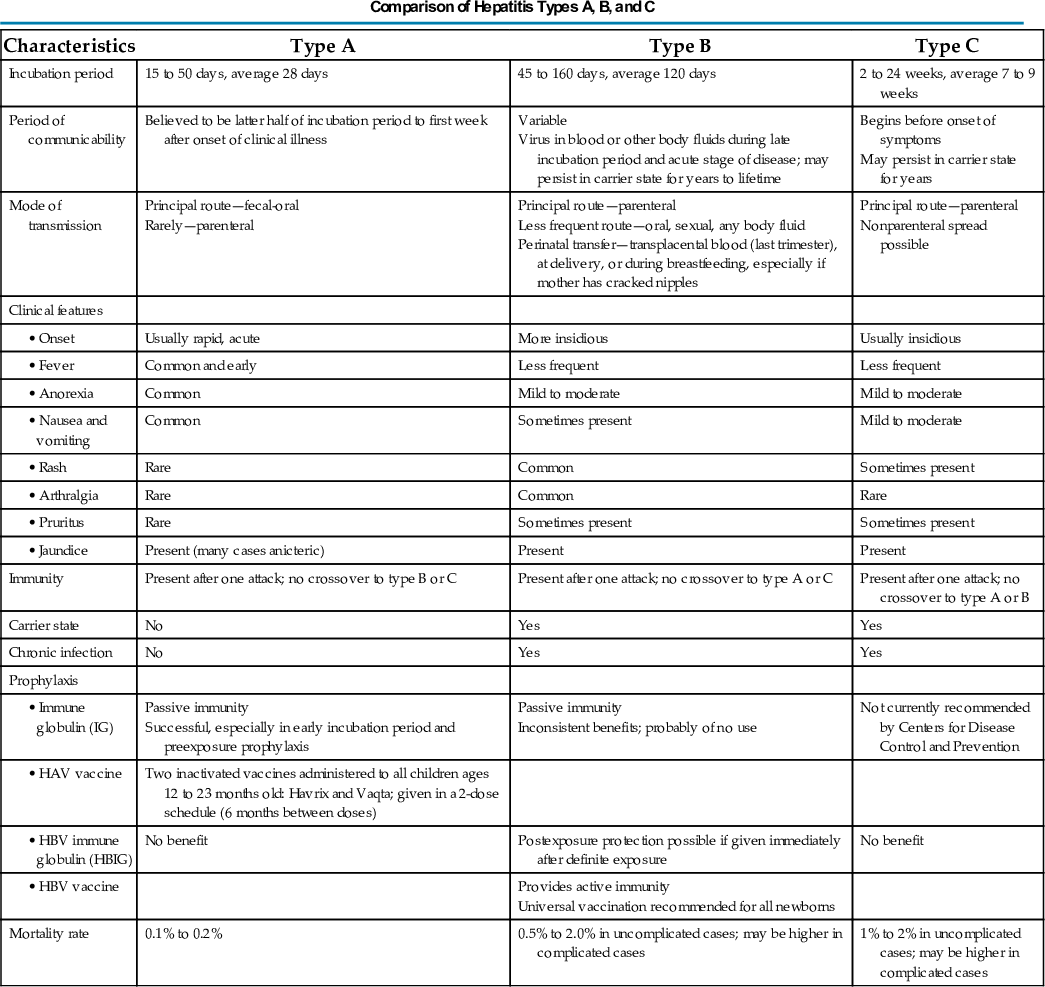
HAV, Hepatitis A virus; HBV, hepatitis B virus.
Hepatitis A Virus
Hepatitis A virus (HAV) incidence in the United States has declined since the introduction of a vaccine in 1995. There were approximately 1390 new cases in the United States in 2015 (Centers for Disease Control and Prevention, 2017). The virus is spread directly or indirectly by the fecal-oral route by ingestion of contaminated foods, direct exposure to infected fecal material, or close contact with an infected person. The virus is particularly prevalent in developing countries with poor living conditions, inadequate sanitation, crowding, and poor personal hygiene practices. The spread of HAV has been associated with improper food handling and high-risk areas such as households with infected persons, residential centers for the disabled, and day care centers. The average incubation period is about 21 days (Jensen & Balistreri, 2016). Fecal shedding of the virus can occur for 2 weeks before and for 1 week after the onset of jaundice. During this time, although the individual is asymptomatic, the virus is most likely to be transmitted. Infants with HAV infection are likely to be asymptomatic (anicteric hepatitis). Children often have diarrhea, and their symptoms are frequently attributed to gastroenteritis. Younger children rarely develop jaundice; however, 70% of older children and adults infected with HAV develop clinical signs with icteric hepatitis that typically lasts 7 to 14 days (Jensen & Balistreri, 2016). The prognosis of HAV infection is usually good, and complications are rare.
Hepatitis B Virus
Although the incidence of hepatitis B virus (HBV) is declining after the introduction of a universal immunization program, approximately 1.25 million people in the United States are infected with HBV, with 400 million HBV cases worldwide (Jensen & Balistreri, 2016). HBV can be an acute or chronic infection, ranging from an asymptomatic, limited infection to fatal, fulminant (rapid and severe) hepatitis (Jensen & Balistreri, 2016). There are no environmental or animal reservoirs for HBV. Humans are the main source of infections. HBV may be transmitted parenterally, percutaneously, or transmucosally. Hepatitis B surface antigen (HBsAg) has been found in all body fluids, including feces, bile, breast milk, sweat, tears, vaginal secretions, and urine, but only blood, semen, and saliva have been found to contain infectious HBV particles. HBV infection from human bites has been documented, but transmission from feces has not. HBV has been acquired after blood transfusion, but the likelihood of this has been reduced through blood product–screening procedures. Adults whose occupations are associated with considerable exposure to blood or blood products, such as health care workers, are at an increased risk of contracting HBV.
Most HBV infections in children are acquired perinatally. Transmission from mother to infant during the perinatal period (i.e., blood exposure during delivery) results in chronic infection in up to 90% of infants if the mother is positive for HBsAg and HBeAg (Jensen & Balistreri, 2016). HBsAg has been inconsistently detected in breast milk, but no increased risk of transmission has been found, and breastfeeding is currently recommended after infant immunization (Jensen & Balistreri, 2016). Infants and children who are not infected during the perinatal period remain at high risk for acquiring person-to-person transmission from their mother.
HBV infection occurs in children and adolescents in the following specific high-risk groups: (1) individuals with hemophilia or other disorders who have received multiple transfusions, (2) children and adolescents involved in IV drug abuse, (3) institutionalized children, (4) preschool children in endemic areas, and (5) individuals engaged in sexual activity with an infected partner. The incubation period for HBV infection ranges from 45 to 160 days, with an average of 120 days (Jensen & Balistreri, 2016). HBV infection can cause a carrier state and lead to chronic hepatitis with eventual cirrhosis or hepatocellular carcinoma in adulthood.
Hepatitis C Virus
Hepatitis C virus (HCV) is the most common cause of chronic liver disease, with an estimated 4 million people in the United States and 170 million people worldwide infected (Jensen & Balistreri, 2016). HCV is transmitted parenterally through exposure to blood and blood products from HCV-infected persons, whereas perinatal transmission is the most common mode of transmission among children (Jensen & Balistreri, 2016). Recent improvements in donor screening and inactivation procedures for blood products, such as the factor concentrates used for hemophilia patients, have significantly reduced the risk of transmission through blood products. Currently the most common cause of HCV infection is illegal drug use with exposure to blood or blood products from an HCV-infected individual, and sexual transmission is the second most common cause of HCV infection (Jensen & Balistreri, 2016).
The clinical course is variable. The incubation period for HCV ranges from 2 to 24 weeks, with an average of 7 to 9 weeks (Jensen & Balistreri, 2016). The natural history of the disease in children is not well defined. Some children may be asymptomatic, but HCV can become a chronic condition and can cause cirrhosis and hepatocellular carcinoma. About 85% of individuals infected with HCV develop chronic disease (Jensen & Balistreri, 2016).
Hepatitis D Virus
Hepatitis D virus (HDV) rarely occurs in children and must occur in individuals already infected with HBV (Jensen & Balistreri, 2016). HDV is a defective RNA virus that requires the helper function of HBV. The incubation period is 2 to 8 weeks, but with coinfection of HBV the incubation period is similar to an HBV infection (Jensen & Balistreri, 2016). HDV infection occurs through blood and sexual contact and commonly occurs among drug abusers, individuals with hemophilia, and persons immigrating from endemic areas.
Hepatitis E Virus
Hepatitis E virus (HEV) was formerly known as non-A, non-B hepatitis. Transmission may occur through the fecal-oral route or from contaminated water. The incubation period ranges from 15 to 60 days, with an average of 40 days (Jensen & Balistreri, 2016). This illness is uncommon in children, does not cause chronic liver disease, is not a chronic condition, and has no carrier state. However, it can be a devastating disease among pregnant women, with an unusually high fatality rate.
Pathophysiology
Pathologic changes occur primarily in the parenchymal cells of the liver and result in variable degrees of swelling; infiltration of liver cells by mononuclear cells; and subsequent degeneration, necrosis, and fibrosis. Structural changes within the hepatocyte account for altered liver functions, such as impaired bile excretion, elevated transaminase levels, and decreased albumin synthesis. The disorder may be self-limiting, with regeneration of liver cells without scarring, leading to a complete recovery. However, some forms of hepatitis do not result in complete return of liver function. These include fulminant hepatitis, which is characterized by a severe, acute course with massive destruction of the liver tissue causing liver failure and high mortality risk within 1 to 2 weeks; and subacute or chronic active hepatitis, which is characterized by progressive liver destruction, uncertain regeneration, scarring, and potential cirrhosis.
The progression of liver disease is characterized pathologically by four stages: (1) stage one is characterized by mononuclear inflammatory cells surrounding small bile ducts, (2) in stage two there is proliferation of small bile ductules, (3) stage three is characterized by fibrosis or scarring, and (4) stage four is cirrhosis.
Clinical Manifestations
The clinical manifestations and course of uncomplicated acute viral hepatitis are similar for most of the hepatitis viruses. Usually the prodromal, or anicteric, phase (absence of jaundice) lasts 5 to 7 days. Anorexia, malaise, lethargy, and easy fatigability are the most common symptoms. Fever may be present, especially in adolescents. Nausea, vomiting, and epigastric or right upper quadrant abdominal pain or tenderness may occur. Arthralgia and skin rashes may occur and are more likely in children with hepatitis B than those with hepatitis A. The transaminases, rather than the bilirubin, are often be elevated in acute hepatitis, and hepatomegaly may be present. Some mild cases of acute viral hepatitis do not cause symptoms or can be mistaken for influenza.
In young children most of the prodromal symptoms disappear with the onset of jaundice, or the icteric phase. Many children with acute viral hepatitis, however, never develop jaundice. If jaundice occurs, it is often accompanied by dark urine and pale stools. Pruritus may accompany jaundice and can be bothersome for children.
Children with chronic active hepatitis may be asymptomatic but more commonly have nonspecific symptoms of malaise, fatigue, lethargy, weight loss, or vague abdominal pain. Hepatomegaly may be present, and the transaminases are often very high, with mild to severe hyperbilirubinemia.
Fulminant hepatitis is due primarily to HBV or HCV. Many children with fulminant hepatitis develop characteristic clinical symptoms and rapidly develop manifestations of liver failure, including encephalopathy, coagulation defects, ascites, deepening jaundice, and an increasing white blood cell count. Changes in mental status or personality indicate impending liver failure. Although children with acute hepatitis may have hepatomegaly, a rapid decrease in the size of the liver (indicating loss of tissue due to necrosis) is a serious sign of fulminant hepatitis. Complications of fulminant hepatitis include GI bleeding, sepsis, renal failure, and disseminated coagulopathy.
Diagnostic Evaluation
Diagnosis is based on the history, physical examination, and serologic markers for hepatitis A, B, and C. No LFT is specific for hepatitis, but serum aspartate aminotransferase (AST) and serum alanine aminotransferase (ALT) levels are markedly elevated. Serum bilirubin levels peak 5 to 10 days after clinical jaundice appears. Histologic evidence from liver biopsy may be required to establish the diagnosis and to assess the severity of the liver disease. Serologic markers indicate the antibodies or antigens formed in response to the specific virus and confirm the diagnosis. Serum immunologic tests are not available to detect HAV antigen, but there are two HAV antibody tests: anti-HAV immunoglobulin G (IgG) and immunoglobulin M (IgM). Anti-HAV antibodies are present at the onset of the disease and persist for life. A positive anti-HAV antibody test can indicate acute infection, immunity from past infection, passive antibody acquisition (e.g., from transfusion, serum immunoglobulin infusion), or immunization. To diagnose an acute or recent HAV infection, a positive anti-HAV IgM test that is present with the onset of the disease and that persists for only 2 or 3 days is required.
Diagnosis of hepatitis B is confirmed by the detection of various hepatitis virus antigens and the antibodies that are produced in response to the infection. These antibodies and antigens and their significance include the following:
- HBsAg—Hepatitis B surface antigen (found on the surface of the virus), indicating ongoing infection or carrier state
- Anti-HBs—Antibody to surface antigen HbsAg, indicating resolving or past infection
- HBcAg—Hepatitis B core antigen (found on the inner core of the virus), detected only in the liver
- Anti-HBc—Antibody to core antigen HbcAg, indicating ongoing or past infection
- HBeAg—Hepatitis B antigen (another component of the HBV core), indicating active infection
- Anti-HBe—Antibody to HbeAg, indicating resolving or past infection
- IgM anti-HBc—IgM antibody to core antigen
Tests are available for detection of all the HBV antigens and antibodies except HBcAg. HBsAg is detectable during acute infection. Presence of HBsAg indicates that the individual has been infected with the hepatitis virus. If the infection is self-limiting, HBsAg disappears in most patients before serum anti-HBs can be detected (termed the window phase of infection). IgM anti-HBc is highly specific in establishing the diagnosis of acute infection, as well as during the window phase in older children and adults. However, IgM anti-HBc usually is not present in perinatal HBV infection. Clinical improvement is usually associated with a decrease in or disappearance of these antigens, followed by the appearance of their antibodies. For example, anti-HBc of the IgM class often occurs early in the disease, followed by a rise in anti-HBc of the IgG class. Because the antibodies persist indefinitely, they are used to identify the carrier state (i.e., individuals with HBV who have no clinical disease but are able to transmit the organism). Persons with chronic HBV infection have circulating HBsAg and anti-HBc, and on rare occasions anti-HBsAg is present. Both anti-HBs and anti-HBc are detected in persons with resolved infection, but anti-HBs alone are present in individuals who have been immunized with the HBV vaccine.
HCV-RNA is the earliest serologic marker for HCV. HCV-RNA can be detected during the incubation period before symptoms of HCV disease are expressed. A positive HCV-RNA result indicates active infection, and persistence of HCV-RNA indicates chronic infection. A negative test correlates with resolution of the disease. HCV-RNA is also used to determine patient response to antiviral therapy for HCV.
The history of all patients should include questions to seek evidence of (1) contact with a person known to have hepatitis, especially a family member; (2) unsafe sanitation practices, such as contaminated drinking water; (3) ingestion of certain foods, such as clams or oysters (especially from polluted water); (4) multiple blood transfusions; (5) ingestion of hepatotoxic drugs, such as salicylates, sulfonamides, antineoplastic agents, acetaminophen, and anticonvulsants; and (6) parenteral administration of illicit drugs or sexual contact with a person who uses these drugs.
Therapeutic Management
The goals of management include early detection, support and monitoring of the disease, recognition of chronic liver disease, and prevention of spread of the disease. Special high-protein, high-carbohydrate, low-fat diets are generally not of value. The use of corticosteroids alone or with immunosuppressive drugs is not advocated in the treatment of chronic viral hepatitis. However, steroids have been used to treat chronic autoimmune hepatitis. Hospitalization is required in the event of coagulopathy or fulminant hepatitis.
Therapy for hepatitis depends on the severity of inflammation and the cause of the disorder. HAV is treated primarily with supportive care. The US Food and Drug Administration approved several medications for treatment of children with HBV and HCV. Human interferon-alpha is being used successfully in the treatment of chronic hepatitis B and C in children. Lamivudine is used for the treatment of HBV. It is well tolerated with no significant side effects and is approved for children older than 2 years of age (Jensen & Balistreri, 2016). Combined therapy with lamivudine and interferon-alpha does not improve response rates (Jensen & Balistreri, 2016). Adefovir is used to treat HBV in children older than 12 years of age. Entecavir and tenofovirhave recently been approved for HBV treatment in adolescents ages 16 years or older (Jensen & Balistreri, 2016). Peginterferon-α2b has not been approved for use in children in the United States but has been used to treat HBV in people in other countries (Jensen & Balistreri, 2016).
Prevention.
Proper hand washing and Standard Precautions prevent the spread of viral hepatitis. Prophylactic use of standard immune globulin is effective in preventing hepatitis A in situations of preexposure (such as anticipated travel to areas where HAV is prevalent) or within 2 weeks of exposure.
Hepatitis B immune globulin (HBIG) is effective in preventing HBV infection after one-time exposures such as accidental needle punctures or other contact of contaminated material with mucous membranes and should be given to newborns whose mothers are HbsAg positive. HBIG is prepared from plasma that contains high titers of antibodies against HBV. HBIG should be given within 72 hours of exposure.
Vaccines have been developed to prevent HAV and HBV infection (see Table 25.7). HBV vaccination is recommended for all newborns and children who did not receive the vaccination as a newborn. (See Immunizations, Chapter 6.) Because HDV cannot be transmitted in the absence of HBV infection, it is possible to prevent HDV infection by preventing HBV infection. Routine serologic testing for anti-HCV of children older than 12 months of age who were born to women previously identified as being infected with HCV is also recommended (Jensen & Balistreri, 2016).
Prognosis.
The prognosis for children with hepatitis is variable and depends on the type of virus and the child's age and immunocompetency. Hepatitis A and E are usually mild, brief illnesses with no carrier state. Hepatitis B can cause a wide spectrum of acute and chronic illness. Infants are more likely than older children to develop chronic hepatitis. Hepatocellular carcinoma during adulthood is a potentially fatal complication of chronic HBV infection. Hepatitis C frequently becomes chronic, and cirrhosis may develop in these children.
Nursing Care Management
Nursing objectives depend largely on the severity of the hepatitis, the medical treatment, and factors influencing the control and transmission of the disease. Because children with mild viral hepatitis are frequently cared for at home, it is often the nurse's responsibility to explain any medical therapies and infection control measures. When further assistance is needed for parents to comply with instructions, a public health nursing referral is necessary.
Encourage a well-balanced diet and a schedule of rest and activity adjusted to the child's condition. Because the child with HAV is not infectious within 1 week after the onset of jaundice, the child may feel well enough to resume school shortly thereafter. Caution parents about administering any medication to the child because normal doses of many drugs may become dangerous because of the liver's inability to detoxify and excrete them.
Standard Precautions are followed when children are hospitalized. However, these children are not usually isolated in a separate room unless they are fecally incontinent or their toys and other personal items are likely to become contaminated with feces. Discourage children from sharing their toys.
Hand washing is the single most effective measure in prevention and control of hepatitis in any setting. Parents and children need an explanation of the usual ways in which hepatitis is spread (fecal-oral route and parenteral route). Parents should also be aware of the recommendation for universal vaccination against HBV for newborns and adolescents. (See Chapter 6.)
In young people with HBV infection who have a known or suspected history of illicit drug use, the nurse has the responsibility of helping them realize the associated dangers of drug abuse, stressing the parenteral mode of transmission of hepatitis, and encouraging them to seek counseling through a drug program.
Biliary Atresia
Biliary atresia (BA), or extrahepatic biliary atresia (EHBA), is a progressive inflammatory process that causes both intrahepatic and extrahepatic bile duct fibrosis, resulting in eventual ductal obstruction. The incidence of BA is approximately 1 in 10,000 to 15,000 live births (Hassan & Balistreri, 2016). Associated malformations include polysplenia and malrotation of the intestine. BA, if untreated, usually leads to cirrhosis, liver failure, and death.
Pathophysiology
The exact cause of BA is unknown, although immune- or infection-mediated mechanisms may be responsible for the progressive process that results in complete obliteration of the bile ducts. BA is not seen in the fetus or stillborn or newborn infant. This suggests that BA is acquired late in gestation or in the perinatal period and is manifested a few weeks after birth.
Congenital infections have been implicated as a cause of hepatocellular damage leading to BA, yet no specific agent is identified in every case. Immune-mediated bile duct injury from viral exposure and immaturity of the neonatal immune system may play a role in the destruction of bile ducts and development of EHBA. Other potential causes include an early first trimester insult to the developing bile ducts or a postnatal viral insult (Hassan & Balistreri, 2016). Early in the course of the disease, the intrahepatic ducts are patent from the interlobular ductules to the porta hepatis. The size of these structures is variable and is correlated with the infant's age and with bile excretion after surgical treatment. These structures are present in most affected infants under 2 months of age but gradually disappear over the next few months and by 4 months are completely replaced by fibrous tissue.
The degree of involvement of the extrahepatic biliary ducts is also variable. The majority of cases of BA (85%) have a complete obliteration of the extrahepatic biliary tree at or above the porta hepatis (Hassan & Balistreri, 2016). But some infants have a patent proximal portion of the extrahepatic duct or patency of the gallbladder, cystic duct, and common bile duct. Microscopic examination of the liver tissue reveals cholestasis with absent or diminished bile duct proliferation and fibrosis.
Clinical Manifestations
Many infants with BA are full term and appear healthy at birth. If jaundice persists beyond 2 weeks of age, especially if the direct (conjugated) serum bilirubin is elevated, the nurse should suspect BA. The urine may be dark, and the stools often become progressively acholic or gray, indicating absence of bile pigment. Hepatomegaly is present early in the course of the disease, and the liver is firm on palpation.
Diagnostic Evaluation
Early diagnosis is critical to the child with EHBA; the outcome in children surgically treated before 2 months of age is much better than in patients with delayed treatment. The diagnosis of BA is suspected on the basis of the history, physical findings, and laboratory studies. Laboratory tests include a CBC, bilirubin levels, and liver function studies. Additional laboratory analyses, including α1-antitrypsin level, TORCH titers and other intrauterine infections (see Maternal Infections, Chapter 9), hepatitis serology, and urine cytomegalovirus, may be indicated to rule out other conditions that cause cholestasis and jaundice. An abdominal ultrasound is usually performed to identify potential causes of extrahepatic obstruction, such as a choledochal cyst. The patency of the extrahepatic biliary system is demonstrated by a nuclear scintiscan using technetium 99m iminodiacetic acid (99mTc-IDA, or HIDA; HIDA scan). If there is no evidence of radioactive material excreted into the duodenum, BA is the most probable diagnosis. Because the nuclear scan may take up to 5 days for the results, a percutaneous liver biopsy is probably the most useful method of diagnosing BA (Govindarajan, 2016). The definitive diagnosis of BA is further established during an exploratory laparotomy and an intraoperative cholangiogram that demonstrates complete obstruction at some level of the biliary tree.
Therapeutic Management
Medical management of BA is primarily supportive. It includes nutritional support with infant formulas that contain medium-chain triglycerides and essential fatty acids. Supplementation with fat-soluble vitamins (A, D, E, and K); a multivitamin; and minerals, including iron, zinc, and selenium, is usually required. Aggressive nutritional support in the form of continuous gastrostomy feedings or TPN may be indicated for moderate to severe growth failure; the enteral solution should be low in sodium. Phenobarbital may be prescribed after hepatic portoenterostomy to stimulate bile flow, and ursodeoxycholic acid may be used to decrease cholestasis and the intense pruritus from jaundice. In cases of advanced liver dysfunction, management is the same as in infants with cirrhosis.
The primary surgical treatment of BA is hepatic portoenterostomy (Kasai procedure), which a segment of intestine is anastomosed to the resected porta hepatis to attempt bile drainage (Fig. 25.8). A Roux-en-Y jejunal limb is then anastomosed to the porta hepatis (a Y-shaped anastomosis performed to provide bile drainage without reflux). Complications after portoenterostomy include ascending cholangitis, cirrhosis, portal hypertension, and GI bleeding. Prophylactic antibiotics are given after the Kasai procedure to minimize the risk of ascending cholangitis. After the Kasai procedure approximately one-third of infants become jaundice free and regain normal liver function. Another one-third of infants demonstrate liver damage; however, they may be supported by medical and nutritional interventions. A final third require liver transplantation. Liver transplantation is required for children who cannot regain bile flow and for those with end-stage liver disease or severe portal hypertension. Complications after liver transplantation include obstruction and bile leaks at the biliary anastomosis, portal hypertension, hemorrhage, infection, and rejection. Immunosuppressive drugs are required after transplantation.
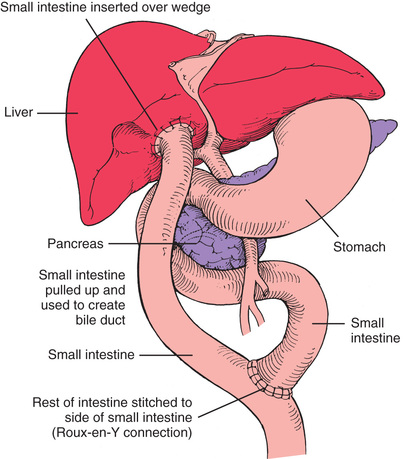
Prognosis.
Untreated BA results in progressive cirrhosis and death in most children by 3 years of age (Govindarajan, 2016). The Kasai procedure improves the prognosis but is not a cure. There is only 20% survival in patients 20 years after the Kasai procedure and only 10% survival in patients 30 years after the procedure (Govindarajan, 2016).
Biliary drainage can often be achieved if the surgery is performed before the intrahepatic bile ducts are destroyed; the success rate is much higher, up to 90%, if surgery is performed in an infant younger than 2 months of age (Hassan & Balistreri, 2016). Long-term survival rates of 64% to 92% have been noted in children after portoenterostomy (Govindarajan, 2016). However, even with successful bile drainage, many children ultimately develop liver failure and require liver transplantation.
The advances in surgical techniques for liver transplantation and the development of immunosuppressive and antifungal drugs have significantly improved the success of transplantation. Surgical techniques and immunosuppression have contributed to survival rates of 83% to 91% in children who underwent transplant (Yanagi, Matsuura, Hayashida, et al., 2017). The major obstacle remains the shortage of suitable infant donors.
Liver disease progresses in infants with delayed diagnosis or in children in whom surgery has failed to provide adequate bile drainage. Cirrhosis and splenomegaly occur with hypoalbuminemia, ascites, and coagulopathy. Malabsorption of fat and fat-soluble vitamins and malnutrition result in severe growth failure. Retained bile salts and cholesterol further contribute to pruritus (itching) and xanthomas, often requiring the administration of ursodeoxycholic acid. The severity of pruritus intensifies as the jaundice progresses as the result of disease advancement.
Nursing Care Management
There are many important nursing interventions for the child with BA. The nurse should educate family members regarding all aspects of the treatment plan and the rationale for therapy. Immediately after a hepatic portoenterostomy, nursing care is similar to that after any major abdominal surgery. If an interrupted jejunal conduit has been performed, the family needs to learn how to care for the two stomas and how to refeed the bile after feedings. Teaching includes the proper administration of medications. Administration of nutritional therapy, including special formulas, vitamin and mineral supplements, gastrostomy feedings, or parenteral nutrition, is an essential nursing responsibility. Growth failure in such infants is common, and increased metabolic needs combined with ascites, pruritus, and nutritional anorexia constitute a challenge for care. The nurse teaches caregivers how to monitor and administer nutritional therapy in the home. Pruritus may be a significant problem that is addressed by drug therapy or comfort measures such as baths in colloidal oatmeal compounds and trimming of fingernails. The risk of complications of BA, such as cholangitis, portal hypertension, GI bleeding, and ascites, should be explained to the caregivers.
These children and their families require special psychosocial support. The uncertain prognosis, discomfort, and waiting for transplantation can produce considerable stress. (See Cirrhosis, next.) In addition, extended hospitalizations, as well as pharmacologic and nutritional therapy, can impose significant financial burdens on the family, as with any chronic condition. The expertise of a multidisciplinary health care team, including surgeons, gastroenterologists, pediatricians, nurses, nutritionists, pharmacists, child life specialists, and social workers, is often necessary. Parent support groups can be beneficial as well. The Children's Liver Association for Support Services (C.L.A.S.S.)* and the American Liver Foundation† provide educational materials, programs, and support systems for parents of children with liver disease.
Cirrhosis
Cirrhosis occurs as an end stage of many chronic liver diseases, including BA and chronic hepatitis. Infectious, autoimmune, toxic injury, and chronic diseases such as hemophilia and cystic fibrosis can cause severe liver damage. A cirrhotic liver is irreversibly damaged.
Pathophysiology
Cirrhosis occurs as a result of hepatocyte injury with necrosis, fibrosis, regeneration, and eventual degeneration. The diminished parenchymal cell mass causes regeneration of tissue with nodular areas of proliferating hepatocytes that stretch the surrounding connective tissue. Hepatocytes respond to injury with deposition of collagen that forms fibrous connective tissue. This scar tissue and nodular areas of regeneration impair the intrahepatic blood flow. Ongoing necrosis and self-perpetuation of this pathologic process are the result of cirrhosis.
Failure of hepatocellular function and portal hypertension occur and often lead to complications, including ascites, severe cholestasis, encephalopathy (hepatic coma), and GI bleeding.
Clinical Manifestations
Clinical manifestations of cirrhosis include jaundice, poor growth, anorexia, muscle weakness, and lethargy. Ascites, edema, GI bleeding, anemia, and abdominal pain may be present in children with impaired intrahepatic blood flow. Pulmonary function may be impaired because of pressure against the diaphragm due to hepatosplenomegaly and ascites. Dyspnea and cyanosis may occur, especially on exertion. Intrapulmonary arteriovenous shunts may develop, which can also cause hypoxemia. Spider angiomas and prominent blood vessels on the upper torso are often present.
Diagnostic Evaluation
The diagnosis of cirrhosis is based on (1) the history, especially in regard to prior liver disease, such as hepatitis; (2) physical examination, particularly hepatosplenomegaly; (3) laboratory evaluation, especially LFTs, such as bilirubin and transaminases, ammonia, albumin, cholesterol, and prothrombin time; and (4) liver biopsy for characteristic changes. Doppler ultrasonography of the liver and spleen is useful to confirm ascites, to evaluate the blood flow through the liver and spleen, and to determine the patency and size of the portal vein if liver transplantation is considered.
Nursing Alert
The most common complication from percutaneous liver biopsy is internal bleeding. Vital signs and laboratory values, especially hematocrit, should be monitored for evidence of hemorrhage and shock.
Therapeutic Management
Unfortunately, there is no successful treatment to arrest the progression of cirrhosis. The goals of management include monitoring liver function and managing specific complications such as esophageal varices and malnutrition. Assessment of the child's degree of liver dysfunction is important so that the child can be evaluated for transplantation at the appropriate time.
Liver transplantation has improved the prognosis substantially for many children with cirrhosis. Pediatric liver transplantation is one of the most successful solid organ transplants with a 1-year survival rate of 83% to 91%, depending on the age at transplant (Yanagi, Matsuura, Hayashida, et al., 2017). The policy governing the allocation of livers for transplantation by the United Network for Organ Sharing allows pediatric patients less than 12 years of age, those with acute fulminant liver failure, or those with chronic liver disease to be placed at the top of the network's transplantation lists (United Network for Organ Sharing, 2017). Although this change has benefited many pediatric patients, the shortage of available donors for children continues to dictate transplantation decisions, and many children continue to die while waiting for a suitable donor.
Nutritional support is an important therapy for children with cirrhosis and malnutrition. Supplements of fat-soluble vitamins are often required, and mineral supplements may be indicated. In some instances aggressive nutritional support in the form of enteral feeding or PN may be necessary.
Esophageal and gastric varices can be a life-threatening complication of portal hypertension. Acute hemorrhage is managed with IV fluids; blood products; vitamin K, if needed to correct coagulopathy; vasopressin or somatostatin; and gastric lavage. If acute hemorrhage persists, the most common secondary approach is endoscopic sclerotherapy or endoscopic banding ligation (Choudhary, Puri, Saigal, et al., 2016). Balloon tamponade with a Sengstaken-Blakemore tube may be indicated for the unstable patient with acute hemorrhage (Choudhary, Puri, Saigal, et al., 2016). Ascites is managed by sodium and fluid restrictions and diuretics. Severe ascites with respiratory compromise is managed with albumin infusions or by paracentesis.
Although the full mechanism of hepatic encephalopathy is unknown, failure of the damaged liver to remove endogenous toxins, such as ammonia, plays a role. Treatment is directed at limiting the ammonia formation and absorption that occur in the bowel, especially with the drugs neomycin and lactulose. Because ammonia is formed in the bowel by the action of bacteria on ingested protein, neomycin reduces the number of intestinal bacteria so less ammonia is produced. The fermentation of lactulose by colonic bacteria produces short-chain fatty acids, which lower the colonic pH, thereby inhibiting bacterial metabolism. This decreases the formation of ammonia from bacterial metabolism of protein.
Prognosis.
The success of liver transplantation has revolutionized the approach to liver cirrhosis. Liver failure and cirrhosis are indications for transplantation. Careful monitoring of the child's condition and quality of life is necessary to evaluate the need for and timing of transplantation.
Nursing Care Management
Several factors influence nursing care of the child with cirrhosis, including the cause of the cirrhosis, the severity of complications, and the prognosis. The prognosis is often poor unless successful liver transplantation occurs. Therefore nursing care of this child is similar to that for any child with a life-threatening illness. (See Chapter 19.) Hospitalization is required when complications such as hemorrhage, severe malnutrition, or hepatic failure occur. Nursing assessments are directed at monitoring the child's condition, and interventions are aimed at treatment of specific complications. If liver transplantation is an option, the family needs support and assistance to cope (see Family-Centered Care box).
 Family-Centered Care
Family-Centered Care
End-Stage Liver Disease
In many cases the child with liver disease and the family must cope with an uncertain progression of the disease. The only hope for long-term survival may be liver transplantation. Transplantation can be successful, but the waiting period may be long because there are many more children in need of organs than there are donors. The procedure is expensive and is only performed at designated medical centers, which are often far from the family's home. The nurse should recognize the unique stresses of coping with end-stage liver disease and waiting for transplantation, and should offer support and assistance to the family in coping with these stressors. The assistance of social workers and support from other parents can also be beneficial.
Structural Defects
Congenital defects of the GI tract can involve any portion from the mouth to the anus. Most are apparent at birth or shortly thereafter and are anomalies in which normal growth ceased at a crucial stage of embryonic development, leaving the structure in an embryonic form or only partially completed. The result may be atresia, malposition, nonclosure, or any number of variations.
Atresia is absence of a normal opening or normally patent lumen. Atresia at any point along the length of the GI tract creates an obstruction to the normal progress of nutrients and secretions. The most common anomalies requiring surgical intervention are atresias of the esophagus, intestine, and anus. The congenital defects considered in this chapter include abnormalities of the lip, palate, trachea, esophagus, and anus.
Esophageal Atresia and Tracheoesophageal Fistula
Congenital esophageal atresia (EA) and tracheoesophageal fistula (TEF) are rare malformations that represent a failure of the esophagus to develop as a continuous passage and a failure of the trachea and esophagus to separate into distinct structures. These defects may occur as separate entities or in combination, and without early diagnosis and treatment they pose a serious threat to the infant's well-being.
The incidence of EA is estimated to be approximately 1 in 3000 neonates (Wu, Kuang, Lv, et al., 2017). There appears to be a slightly higher incidence in males, and the birth weight of most affected infants is significantly lower than average, with an unusually high incidence of preterm birth in infants with EA and a subsequent increase in mortality. A history of maternal polyhydramnios is common.
Approximately 50% of the cases of EA/TEF are a component of VATER or VACTERL association, acronyms used to describe associated anomalies (VATER for Vertebral defects, imperforate Anus, Tracheoesophageal fistula, and Radial and Renal dysplasia; and VACTERL for Vertebral, Anal, Cardiac, Tracheal, Esophageal, Renal, and Limb) (Khan & Orenstein, 2016c). Cardiac anomalies may also occur with EA/TEF; therefore all patients should undergo a workup for associated anomalies.
Pathophysiology
The esophagus develops from the first segment of the embryonic gut. During the fourth and fifth weeks of gestation, the foregut normally lengthens and separates longitudinally. Each longitudinal portion fuses to form two parallel channels (the esophagus and the trachea) that are joined only at the larynx. Anomalies involving the trachea and esophagus are caused by defective separation, incomplete fusion of the tracheal folds after this separation, or altered cellular growth during embryonic development.
The most commonly encountered form of EA and TEF (80% to 90% of cases) is one in which the proximal esophageal segment terminates in a blind pouch and the distal segment is connected to the trachea or primary bronchus by a short fistula at or near the tracheal bifurcation (Fig. 25.9, C). The second most common type (7% to 8%) consists of a blind pouch at each end, widely separated and with no communication to the trachea (Fig. 25.9, A). An H-type EA refers to an otherwise normal trachea and esophagus connected by a fistula (4% to 5%) (Fig. 25.9, E). Extremely rare anomalies involve a fistula from the trachea to the upper esophageal segment (0.8%) (Fig. 25.9, B) or to both the upper and lower segments (0.7% to 6%) (Fig. 25.9, D).
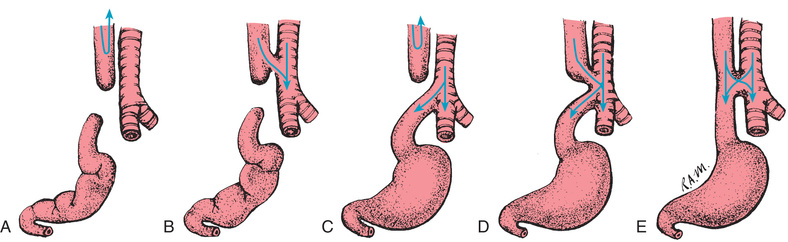
Clinical Manifestations
The presence of EA is suspected in a newborn with frothy saliva in the mouth and nose, drooling, choking, and coughing. Respiratory distress may be mild or significant, depending on the type of defect and the infant's gestational age. If fed, the infant may swallow normally but suddenly cough and gag, with return of fluid through the nose and mouth. The infant may become cyanotic and apneic because of aspiration of breastmilk or saliva.
In the infant who has EA with a distal TEF (type C), the stomach becomes distended with air, and thoracic and abdominal compressions (especially during crying) cause the gastric contents to be regurgitated through the fistula and into the trachea, producing a chemical pneumonitis. When the upper segment of the esophagus opens directly into the trachea (types B and D), the infant is in danger of aspirating any swallowed material. Cyanosis or choking during feeding may be the only symptom of type H fistula (see Fig. 25.9, E). The child with this type of EA may not manifest symptoms until later in life when he or she shows signs of chronic respiratory problems, recurrent pneumonia, and signs of GER (Khan & Orenstein, 2016c).
Diagnostic Evaluation
Although the diagnosis is established on the basis of clinical signs and symptoms, the exact type of anomaly is determined by radiographic studies. A radiopaque catheter is inserted into the hypopharynx and advanced until it encounters an obstruction. Chest radiographs are taken to ascertain esophageal patency or the presence and level of a blind pouch. Films that show air in the stomach indicate a connection between the trachea and the distal esophagus in types C, D, and E. Complete absence of air in the stomach is seen in types A and B. Occasionally, fistulas are not patent, which makes their presence more difficult to diagnose. A careful bronchoscopic examination may be performed in an attempt to visualize the fistula.
The presence of polyhydramnios (accumulation of 2000 ml of amniotic fluid) prenatally is a clue to the possibility of EA in the unborn infant, especially with defect type A, B, or C. With these types of EA/TEF, amniotic fluid normally swallowed by the fetus is unable to reach the GI tract to be absorbed and excreted by the kidneys. The result is an abnormal accumulation of amniotic fluid, or polyhydramnios.
Therapeutic Management
The treatment of EA and TEF includes maintenance of a patent airway, prevention of pneumonia, gastric or blind pouch decompression, supportive therapy, and surgical repair of the anomaly.
When EA with a TEF is suspected, the infant is immediately deprived of oral intake, IV fluids are initiated, and the infant is positioned to facilitate drainage of secretions and decrease the likelihood of aspiration. Accumulated secretions are suctioned frequently from the mouth and pharynx. A double-lumen catheter should be placed into the upper esophageal pouch and attached to intermittent or continuous low suction. The infant's head is kept upright to facilitate removal of fluid collected in the pouch and to prevent aspiration of gastric contents. Broad-spectrum antibiotic therapy is often instituted if there is a concern about aspiration of gastric contents.
Most malformations can be corrected surgically in one operation or in two or more staged procedures. The success depends on early diagnosis before complications occur and on the presence and severity of associated anomalies and illness factors, including preterm birth. With measures instituted to prevent aspiration pneumonia and to ensure adequate hydration and nutrition, surgery may be postponed to allow for more effective treatment of pneumonia and physiologic stabilization so that the infant can better withstand the complex surgery. The delay also offers an opportunity for further evaluation and assessment to rule out any associated anomalies and to optimize respiratory support.
Thoracoscopic repair of EA/TEF is being used successfully, thus negating the need for a thoracotomy and minimizing associated postoperative complications and morbidities (Khan & Orenstein, 2016c). The surgery consists of a thoracotomy with division and ligation of the TEF and an end-to-end or end-to-side anastomosis of the esophagus. A chest tube may be inserted to drain intrapleural air and fluid. For infants who are not stable enough to undergo definitive repair or those with a lengthy gap (>3 to 4 cm) between the proximal and distal esophagus, a staged operation is preferred that involves gastrostomy, ligation of the TEF, and constant drainage of the esophageal pouch (Khan & Orenstein, 2016c). A delayed esophageal anastomosis is usually attempted after several weeks to months.
A primary anastomosis may be impossible because of insufficient length of the two segments of esophagus. This occurs if the distance between the two segments is 3 to 4 cm (1.2 to 1.6 inches) (approximately 3 vertebral bodies) or greater; this is often referred to as long-gap EA (Khan & Orenstein, 2016c). In these cases an esophageal replacement procedure using a part of the colon or gastric tube interposition may be necessary to bridge the missing esophageal segment. Further surgical techniques may be performed later to facilitate esophageal lengthening.
Tracheomalacia may occur as a result of weakness in the tracheal wall that exists when a dilated proximal pouch compresses the trachea early in fetal life. It may also occur as a result of inadequate intratracheal pressure causing abnormal tracheal development. Clinical signs of tracheomalacia include barking cough, stridor, wheezing, recurrent respiratory tract infections, cyanosis, and sometimes apnea.
Prognosis.
The survival rate is nearly 100% in otherwise healthy children. Most deaths are the result of extreme prematurity or other lethal associated anomalies.
Potential complications after the surgical repair of EA and TEF depend on the type of defect and surgical correction. Complications of repair include an anastomotic leak, strictures caused by tension or ischemia, esophageal motility disorders causing dysphagia, respiratory compromise, scoliosis, chest wall deformity, and GER. Anastomotic esophageal strictures may cause dysphagia, choking, and respiratory distress. The strictures are often treated with routine esophageal dilation. Feeding difficulties are often present for months or years after surgery, and the infant must be monitored closely to ensure adequate weight gain, growth, and development. In some cases laparoscopic fundoplication may be required. At times the infant must be fed via gastrostomy or jejunostomy to provide adequate caloric intake.
Nursing Care Management
Nursing responsibility for detection of this serious malformation begins immediately after birth. For the infant with the classic signs and symptoms of EA (see Nursing Alert) the major concern is the establishment of a patent airway and prevention of further respiratory compromise. Cyanosis is usually a result of laryngeal spasm caused by overflow of saliva into the larynx from the proximal esophageal pouch or aspiration; it normally resolves after removal of the secretions from the oropharynx by suctioning. The passage of a small-gauge orogastric (OG) feeding tube via the mouth into the stomach during the initial nursing physical assessment is helpful to determine the presence of EA or other obstructive defects.
Nursing Alert
Any infant who has an excessive amount of frothy saliva in the mouth or difficulty with secretions and unexplained episodes of apnea, cyanosis, or oxygen desaturation should be suspected of having an EA/TEF and referred immediately for medical evaluation.
Preoperative Care.
The nurse carefully suctions the mouth and nasopharynx and places the infant in an optimum position to facilitate drainage and avoid aspiration. The most desirable position for a newborn who is suspected of having the typical EA with a TEF (e.g., type C) is supine (or sometimes prone) with the head elevated on an inclined plane of at least 30 degrees. This positioning minimizes the reflux of gastric secretions at the distal esophagus into the trachea and bronchi, especially when intraabdominal pressure is elevated.
It is imperative to immediately remove any secretions that can be aspirated. Until surgery the blind pouch is kept empty by intermittent or continuous suction through an indwelling double-lumen catheter passed orally or nasally to the end of the pouch. In some cases a percutaneous gastrostomy tube is inserted and left open so that any air entering the stomach through the fistula can escape, thus minimizing the danger of gastric contents being regurgitated into the trachea. The gastrostomy tube is emptied by gravity drainage. Feedings through the gastrostomy tube and irrigations with fluid are contraindicated before surgery in the infant with a distal TEF.
Nursing interventions include respiratory assessment, airway management, thermoregulation, fluid and electrolyte management, and parenteral nutritional support.
Often the infant must be transferred to a hospital with a specialized care unit and pediatric surgical team. The nurse advises the parents of the infant's condition and provides them with necessary support and information.
Postoperative Care.
Postoperative care for these infants is the same as for any high-risk newborn. Adequate thermoregulation is provided, the double-lumen NG catheter is attached to low-suction or gravity drainage, parenteral nutrition is provided, and the gastrostomy tube (if applicable) is returned to gravity drainage until feedings are tolerated. If a thoracotomy is performed and a chest tube is inserted, attention to the appropriate function of the closed drainage system is imperative. Pain management in the postoperative period is important even if only a thoracoscopic approach is used. In the first 24 to 36 hours the nurse should provide pain management for the neonate just as for an adult undergoing a similar procedure. (See Pain in Neonates, Chapter 5.) Tracheal suction should only be done using a premeasured catheter and with extreme caution to avoid injury to the suture line.
If tolerated, gastrostomy feedings may be initiated and continued until the esophageal anastomosis is healed. Before oral feedings are initiated and the chest tube (if applicable) is removed, a contrast study or esophagram will verify the integrity of the esophageal anastomosis.
The nurse must carefully observe the initial attempt at oral feeding to make certain the infant is able to swallow without choking. Oral feedings are begun with sterile water, followed by frequent small feedings of breast milk or formula. Until the infant is able to take a sufficient amount by mouth, oral intake may need to be supplemented by bolus or continuous gastrostomy feedings. Ordinarily infants are not discharged until they can take oral fluids well. The gastrostomy tube may be removed before discharge or maintained for supplemental feedings at home.
Special Problems.
Upper respiratory tract complications are a threat to life in both the preoperative and postoperative periods. In addition to pneumonia, the infants are in constant danger of respiratory distress resulting from atelectasis, pneumothorax, and laryngeal edema. Any persistent respiratory difficulty after removal of secretions must be reported to the surgeon immediately. The infant should be monitored for anastomotic leaks and signs of infection such as purulent chest tube drainage, an increased white blood cell count, and temperature instability.
For the infant who requires esophageal replacement, nonnutritive sucking should be provided with a pacifier. Infants who are on NPO status for an extended period and have not received oral stimulation frequently have difficulty eating by mouth after corrective surgery and may develop oral hypersensitivity and feeding aversion. They require patient, firm guidance in learning the techniques of taking food into the mouth and swallowing after repair. A referral to a multidisciplinary feeding behavior team may be necessary.
One of the difficulties in TEF is the immediate transfer of the sick infant to the intensive care unit and sometimes lengthy hospitalization. Parent-infant bonding is facilitated by encouraging parents to visit the infant, participate in his or her care when appropriate, and express their feelings regarding the infant's condition. The nurse in the intensive care unit should assume responsibility for ensuring that the parents are fully informed of the infant's progress.
Family Support, Discharge Planning, and Home Care.
Some infants with EA/TEF may require periodic esophageal dilations on an outpatient basis. Discharge education should include instructions about feeding techniques in the child with a repaired esophagus, including a semiupright feeding position, small feedings, and observation for adequacy of swallowing (e.g., regurgitation, cyanosis, choking). Tracheomalacia is often a complication; parents are educated about the signs and symptoms of this condition, which include a barking cough, stridor, wheezing, recurrent respiratory tract infections, cyanosis, and sometimes apnea. GER may also occur when feedings resume and may contribute to reactive airway disease with wheezing and labored respirations as the prominent clinical manifestations. Problems with thriving and gaining weight may occur in the first 5 years of life in the child with EA/TEF, especially if the infant is born preterm. The nurse should be alert to the achievement of developmental milestones that indicate a need for early intervention and multidisciplinary referral.
Preparing parents for discharge of their infant involves teaching the techniques that will be continued at home. The parents learn signs of respiratory difficulty and of esophageal stricture (e.g., poor feeding, choking, dysphagia, drooling, regurgitating undigested food) and GER.
Parents must be aware of feeding restrictions. Remind parents that it is particularly important to guard against the infant swallowing foreign objects. They should cut solid food into small pieces, teach the child to chew thoroughly, give frequent sips of liquid to help swallow food, and avoid foods such as whole hot dogs or large pieces of meat that may become lodged in the esophagus. (See Safety Promotion and Injury Prevention, Chapter 10.)
Discharge planning should include attainment of needed equipment and home nursing services to assist with ongoing assessment of the child and continuity of care.
Abdominal Wall Defects
Gastroschisis and omphalocele are two of the more common forms of congenital abdominal wall defects. Gastroschisis occurs in varying incidences worldwide from about 3 to 4 in 10,000 births, and omphalocele occurs in approximately 1 to 2 in 10,000 live births (St. Louis, Kim, Browne, et al., 2017). Numerous reports cite an increase in the incidence of gastroschisis, although the cause of this increased incidence is unknown (St. Louis, Kim, Browne, et al., 2017). An omphalocele occurs when the abdominal contents herniate through the umbilical ring (hernia of the umbilical cord), usually with an intact peritoneal sac, whereas gastroschisis occurs when the herniation of intestine is lateral to the umbilical ring. This herniation is usually to the right of the umbilicus, and a peritoneal sac is not present.
Omphalocele
Omphalocele is related to a true failure of embryonic development. It occurs when there is failure of the caudal or lateral in folding of the abdominal wall at approximately the third week of gestation. With the deficiency in the abdominal wall, the bowel is unable to complete its return to the abdomen between the tenth and twelfth weeks of gestation.
The omphalocele is usually covered only by a translucent peritoneal sac (Fig. 25.10). The sac may contain only a small portion of the bowel or most of the bowel and other abdominal viscera, such as the liver. If the sac ruptures, the abdominal contents become exposed. Omphalocele often is associated with other anomalies (50% to 70% incidence of anomalies), including cardiac, neurologic, skeletal, and genitourinary (GU) anomalies; imperforate anus; ileal atresia; and bladder exstrophy. Omphalocele is also associated with trisomies 18, Beckwith-Wiedemann syndrome, congenital heart disease, Meckel diverticulum, inguinal hernias, renal or limb deformities, and closed gastroschisis (Watanabe, Suzuki, Hara, et al., 2017).
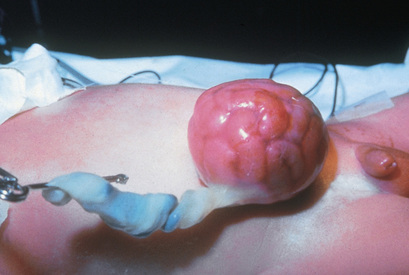
A small omphalocele may go undetected at first glance and appear as a bulge in the umbilical cord. It is therefore imperative to inspect an unusually large umbilical cord for omphalocele before clamping to prevent possible damage to bowel tissue. (See Care of the Umbilicus, Chapter 7.)
With the increasing frequency of and improvements in prenatal ultrasonography, many abdominal wall defects are being diagnosed prenatally. The benefits of prenatal ultrasonographic diagnosis include the ability to transfer the mother to a tertiary care center, where pediatric surgeons and a neonatal intensive care unit are available to assist with care after delivery.
Initial management after delivery includes inspection of the defect and any associated anomalies. If the bowel covering is intact, a nonadherent dressing is placed over the defect to prevent injury; if the bowel is exposed, the exposed abdominal contents and membranes are covered with a bowel bag or moist dressings and a plastic drape to prevent excessive fluid loss, drying, and temperature instability. IV fluids and antibiotics are administered, and a further evaluation for other associated anomalies is completed. Placement of a Silastic double-lumen catheter (NG-OG) is performed to accomplish gastric bowel decompression.
After initial medical management and stabilization, several surgical options may be carried out, depending on the size of the defect, associated medical problems, and surgeon preference. Primary closure of the omphalocele is one option if the defect is small. The sac is resected, contents are reduced into the abdominal cavity, and an attempt is made to close the abdominal fascia with sutures. The abdominal wall may need to be stretched. If an intestinal atresia exists, a bowel resection may be performed, possibly involving a diverting stoma.
When primary closure of the defect is not possible because of the small size of the abdominal cavity or an extremely large omphalocele, staged reduction is accomplished. One nonoperative approach with a large omphalocele is to treat the omphalocele sac with a topical substance such as silver sulfadiazine to enhance epithelialization of the membrane (Ledbetter, 2012). Additional reduction of the defect may be used by applying a compression dressing or elastic bandage. This process may take up to 12 months and the infant can be started on regular feedings; parents can be taught to apply the topical ointment or dressings at home (Ledbetter, 2012). Another approach involves closure with skin flaps from the lateral abdominal wall. In the event that the sac has been disrupted, a silo mesh may be used to house the omphalocele as described in the care of the gastroschesis.
Postoperatively these infants may require mechanical ventilation and parenteral nutrition. Intraabdominal compression may prevent effective respiration and restrict blood flow to the lower extremities and abdominal organs. Feedings may resume once adequate bowel function is established. Postoperatively the infant is monitored for complications seen with abdominal surgery, including infection, evisceration, intestinal volvulus, obstruction, and a ventral hernia.
Long-term complications include GER, failure to thrive (growth failure), ventral hernia, and feeding issues if the infant has been on NPO status for a lengthy period. Additional complications may occur related to the presence of associated conditions such as Beckwith-Wiedemann syndrome.
Gastroschisis
Gastroschisis occurs when the bowel herniates through a defect in the abdominal wall to the right of the umbilical cord and through the rectus muscle (Fig. 25.11). There is no membrane covering the exposed bowel. Controversy exists regarding the etiology of gastroschisis. It has been suggested that at some point between the bowel's stay in the umbilical cord and the completion of fixation, a tear occurs at the base of the umbilical cord, allowing the intestine to herniate. The gap between the cord and the tear is filled in by skin, giving the appearance of a defect in the abdominal wall to the right of the umbilical cord. The base of the defect is narrow, and the lack of membranes results in thickening and foreshortening of the bowel. Gastroschisis is usually not associated with other major congenital anomalies (10% to 20% incidence of associated anomalies); however, jejunoileal atresia, ischemic enteritis, and malrotation may occur as a result of the defect itself. Gastroschisis has been classified as simple and complicated; those within the latter category may involve bowel atresia, perforation, ischemia, or necrosis (Lakshminarayanan & Lakhoo, 2014). Prenatal management of gastroschesis is evolving with emphasis on protection of the bowel from the effects of amniotic fluid, but there is no evidence-based consensus on prenatal management at this time.
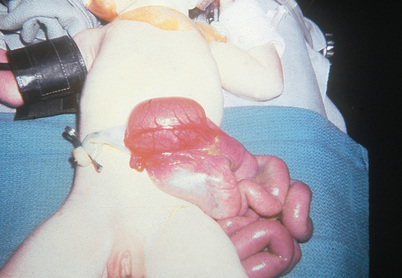
Initial management involves covering the exposed bowel with a transparent plastic bowel bag or loose, moist dressings. If the opening in the abdominal cavity through which the bowel is protruding is small and strangulation of the bowel is possible, the abdominal opening is enlarged at the bedside. IV fluids and antibiotics are administered, and a double-lumen NG tube is inserted for bowel decompression. Fluid replacement for gastroschisis is increased twofold to threefold because of large losses from the exposed viscera.
Adequate thermoregulation and fluid management are extremely important for both omphalocele and gastroschisis. During surgery the abdominal wall is stretched and the mass of bowel is replaced in the abdomen. If primary closure is not possible, a prefabricated, spring-loaded Silastic silo is placed over the unprotected bowel in labor and delivery or in the neonatal intensive care unit shortly after birth to protect the bowel and decrease fluid loss; primary surgical closure is attempted at a later date once the bowel is reduced. The silo is reduced over several days or weeks, at which time it is removed surgically and the defect is closed. Infection is a concern during this period.
Infants with gastroschisis have traditionally been operated on within 24 hours of birth because of temperature instability, risk of infection in the unprotected bowel, and fluid loss. Studies have shown that outcomes vary in regards to early surgical closure versus silo management and later surgical closure; some outcomes are heavily dependent on the amount of bowel to be replaced into the abdominal cavity and subsequent intraabdominal pressure with primary closure. However, simple gastroschisis when the bowel is in good condition is usually treated with immediate closure (Lakshminarayanan & Lakhoo, 2014).
Postoperatively most infants require mechanical ventilation because of respiratory distress secondary to increased abdominal pressure. Pain management is imperative, especially in the first 72 hours. Morphine and fentanyl are effective opioid analgesics. Many infants also require prolonged nutritional support (parenteral and enteral) because of poor bowel function. Prolonged parenteral nutrition may cause liver failure. Exposure of the bowel to amniotic fluid in utero predisposes the infant to prolonged paralytic ileus and hypomotility. Other complications include infection, transient renal impairment, intestinal obstruction, vena cava compression, and a subsequent decrease in blood flow to the lower extremities.
Prognosis
Advanced surgical techniques, improved parenteral nutrition delivery systems, and better medical management have improved the prognosis for the newborn with an abdominal wall defect. Survival estimates for infants with gastroschisis range from 96% for simple gastroschisis to 89% for complex gastroschisis (Lakshminarayanan & Lakhoo, 2014). Because many newborns with omphalocele often have serious associated congenital anomalies, the prognosis for survival of such infants is often not as predictable or as positive as it is for those with gastroschisis (Watanabe, Suzuki, Hara, et al., 2017).
Nursing Care Management
Nursing care is similar to that for any high-risk infant. Infection is a constant threat before surgery, and careful positioning and handling are necessary to prevent rupture of the omphalocele sac or herniated bowel, or disturbance of the Silastic material used for gradual silo reduction. Viscera should be protected with moist dressings or a silo as described previously. Heat and fluid loss from the exposed viscera are major concerns in the preoperative period. Therefore thermoregulation and attention to adequate fluid volume are critical. Fluid replacement is vital and must compensate for losses. The GI tract is decompressed via an NG tube before surgery to aid in bowel reduction.
Postoperative care includes monitoring for signs of complications and assessment of bowel function; pain management with an opioid is also important in the recovery of the infant. Parenteral nutritional support may be necessary when ileus persists. It may require several days or weeks for normal bowel function to return and before full feedings can be achieved. Infants with a prolonged bowel recovery phase are prime candidates for the development of feeding resistance; therefore consultation with a feeding specialist in the early postoperative period is recommended to enhance feeding success. Associated long-term problems with gastroschisis include bowel adhesions, bowel obstruction, necrotizing enterocolitis, parenteral nutrition–related cholestasis, and poor weight gain (Lakshminarayanan & Lakhoo, 2014).
Family Support, Discharge Planning, and Home Care.
Because these abdominal defects are visible and may be shocking to parents, immediate emotional support at the time of birth is essential. The family needs a brief explanation of the defect and reassurance that their child is in no immediate danger (unless circumstances are different). After the parents have had time to interact with their newborn, inform them about the surgical treatment and postoperative care. At the time of discharge from the hospital, many of these infants are receiving oral feedings, but extended parenteral nutrition may be required if malabsorption and poor bowel function occur. The nurse can ensure continuity of care by referral to a home health care agency, especially if long-term nutritional support is required.
Hernias
A hernia is a protrusion of a portion of an organ or organs through an abnormal opening. The danger of herniation arises when the protrusion is constricted, impairing circulation, or when the protrusion interferes with the function or development of other structures. The herniations discussed in this section are those that protrude through the diaphragm, the abdominal wall, or the inguinal canal.
Umbilical Hernia
The umbilical hernia is a common hernia observed in infants. It occurs when fusion of the umbilical ring is incomplete at the point where the umbilical vessels exit the abdominal wall. It affects low-birth-weight and preterm infants more often than full-term infants. An umbilical hernia usually is an isolated defect, but it may be associated with other congenital anomalies, such as Down syndrome (trisomy 21) and trisomies 13 and 18. The size of the defect is variable, and the protrusion is more prominent when the infant is crying (Fig. 25.12). Incarceration, in which the hernia is constricted and cannot be reduced manually, is rare. Hernias usually resolve spontaneously by 3 to 5 years of age. If the hernia persists beyond this age, it is usually surgically corrected on an elective basis.
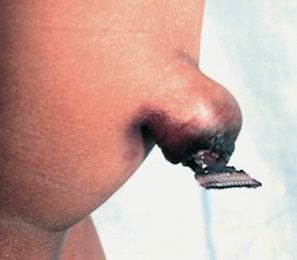
Nursing Care Management
The appearance of an umbilical hernia may be disconcerting to parents. Therefore they need reassurance that the defect usually is not harmful. Taping or strapping the abdomen to flatten the protrusion does not aid in resolution and can produce skin irritation.
Nursing care of the child with an umbilical hernia repair is essentially the same as that for other minor GI surgery. The procedure may be performed on an outpatient basis. Observe the child for complications related to a hematoma or infection. The child may resume a normal diet and activity postoperatively; however, strenuous activity or play is restricted for 2 to 3 weeks.
Inguinal Hernia
Inguinal hernias account for approximately 80% of all childhood hernias and occur more frequently in boys than in girls (approximately 6 : 1). An incidence of 0.8% to 5% is reported in term newborns and up to 30% in low-birth-weight and preterm infants (Abdulhai, Glenn, & Ponsky, 2017).
Pathophysiology
Inguinal hernia comes from persistence of all or part of the processus vaginalis, the tube of peritoneum that precedes the testicle through the inguinal canal into the scrotum (in boys), or the round ligament into the labia (in girls), during the eighth month of gestation. After descent of the testicle, the proximal portion of the processus vaginalis normally atrophies and closes, whereas the distal portion forms the tunica vaginalis, which envelops the testicle in the scrotum. When the upper portion fails to atrophy, the abdominal fluid or an abdominal structure (e.g., bowel, ovary, fallopian tubes) can be forced into it, creating a palpable bulge or mass. The persistent sac may end at any point along the inguinal canal; it may stop at the inguinal ring or extend all the way into the scrotum or labia (Fig. 25.13).
Clinical Manifestations
This common defect is usually asymptomatic unless the abdominal contents are forced into the patent sac. Most often it appears as a painless inguinal swelling that varies in size. It disappears during periods of rest or is reducible by gentle compression. It appears when the infant cries or strains or when the older child strains, coughs, or stands for a long time. The defect can be palpated as a thickening of the cord in the groin, and the silk glove sign can be elicited by rubbing together the sides of the empty hernial sac.
Sometimes the herniated loop of intestine becomes partially obstructed, producing variable symptoms that may include irritability, tenderness, anorexia, abdominal distention, and difficulty defecating. Occasionally the loop of bowel becomes incarcerated (irreducible) or strangulated (loss of blood supply), with symptoms of complete intestinal obstruction that, left untreated, will progress to strangulation and necrotic bowel. Incarceration occurs more often in infants under 12 months of age. The incidence of incarceration is reported to be 3% to 16% but as high as 30% in premature infants (Abdulhai, Glenn, & Ponsky, 2017).
Therapeutic Management
The treatment for hernias is prompt, elective surgical repair in the healthy child as soon as the defect is diagnosed. However, an incarcerated hernia requires emergent surgical care. Because there was believed to be a significant incidence of bilateral involvement, many surgeons advocated exploration of both sides; however, this practice has gained disfavor due to complications occurring with open exploration (Aiken & Oldham, 2016b). Laparoscopic exploration of the contralateral side may be performed without risk of injury to the vas deferens (Aiken & Oldham, 2016b).
Nursing Care Management
Prompt recognition of an inguinal hernia is imperative. The hernia may first be noticed when the infant is crying or straining to stool (Valsalva maneuver). Nursing care of the infant or child with an inguinal hernia involves preoperative preparation of the infant and appropriate explanation to the parents of the child's expected postoperative status. Most hernia repairs can be managed on an outpatient basis. The preterm infant usually has hernia repair several days before discharge. The former preterm infant diagnosed after discharge is admitted the day of surgery and, after repair, is observed for 12 to 24 hours for apnea and bradycardia.
Postoperatively the incision is kept clean and dry, and the infant's pain is managed appropriately. In infants and small children who are not yet toilet trained, the wound may be covered with an occlusive dressing or left without a dressing. Changing diapers as soon as they become damp helps reduce the chance of irritation or infection of the incision.
No restrictions are placed on activity for the infant or toddler, but older children are cautioned against lifting, pushing, wrestling or fighting, bicycle riding, and participation in sporting events for about 3 weeks.
If surgery is postponed, parents need to learn the signs of incarcerated hernia, simple measures to reduce it (e.g., a warm bath, avoidance of upright positioning, and comfort measures to reduce crying), and where to call for assistance if relief is not obtained in a reasonably short time.
Femoral Hernia
Femoral hernias are rare in children, with a reported incidence of less than 1% (Abdulhai, Glenn, & Ponsky, 2017). The incidence is higher in girls than in boys. The hernia may manifest as a recurrent hernia after inguinal hernia repair. Initial symptoms are swelling in the groin area associated with severe abdominal pain and cramping. Treatment and management are the same as for inguinal hernia. Incarceration and strangulation are frequent complications.
Anorectal Malformations
Anorectal malformations (ARMs) are among the more common congenital malformations caused by abnormal development, with an incidence of approximately 1 in 3000 births (Akay & Klein, 2016). These malformations may range from simple imperforate anus to other associated complex anomalies of GU and pelvic organs, which may require extensive treatment for fecal, urinary, and sexual function. Anorectal malformations may occur in isolation or as a part of the VACTERL association (see earlier in this chapter). These anomalies are classified according to the newborn's gender and abnormal anatomic features, including GU defects (Box 25.19). More than half of all ARMs are associated with other anomalies.
Rectal atresia and stenosis occur when the anal opening appears normal, there is a midline intergluteal groove, and usually no fistula exists between the rectum and urinary tract. Rectal atresia is a complete obstruction (inability to pass stool) and requires immediate surgical intervention. Rectal stenosis may not become apparent until later in infancy when the infant has a history of difficult stooling, abdominal distention, and ribbonlike stools. A persistent cloaca is a complex anorectal malformation in which the rectum, vagina, and urethra drain into a common channel opening into the perineum (Fig. 25.14, A).
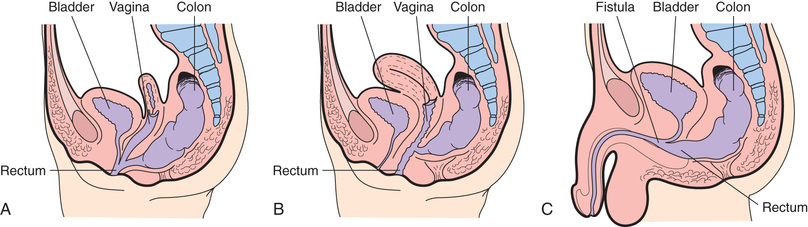
Imperforate anus includes several forms of malformation without an obvious opening (Fig. 25.15). Frequently a fistula (an abnormal communication) leads from the distal rectum to the perineum or GU system (see Fig. 25.14, B and C). The fistula may be evidenced when meconium is evacuated through the vaginal opening, the perineum below the vagina, the male urethra, or the perineum under the scrotum. The presence of meconium on the perineum does not indicate anal patency. A fistula may not be apparent at birth, but as peristalsis increases, meconium is forced through the fistula into the urethra or onto the newborn's perineum.
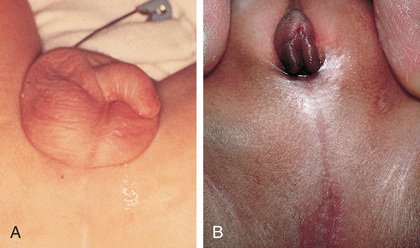
Pathophysiology
During embryonic development the cloaca becomes the common channel for the developing urinary, genital, and rectal systems. The cloaca is divided at the sixth week of gestation into an anterior urogenital sinus and a posterior intestinal channel by the urorectal septum. After the lateral folds join the urorectal septum, separation of the urinary and rectal segments takes place. Further differentiation results in the anterior GU system and the posterior anorectal channel. An interruption of this development leads to incomplete migration of the rectum to its normal perineal position.
Diagnostic Evaluation
The diagnosis of an anorectal malformation is based on the physical finding of an absent anal opening. Other symptoms may include abdominal distention, vomiting, absence of meconium passage, or presence of meconium in the urine. Additional physical findings with an anorectal malformation are a flat perineum and the absence of a midline intergluteal groove. The appearance of the perineum alone does not accurately predict the extent of the defect and associated anomalies. GU and spinal-vertebral anomalies associated with anorectal malformations should be considered when an anomaly is noted. EA with or without TEF, cardiac defects, and spinal or vertebral anomalies may occur in association with anorectal malformations, and the infant should be carefully evaluated for the presence of these and other anomalies. Although rare, some ARMs may not be diagnosed until later in infancy or early childhood.
A perineal fistula (see Box 25.19) may be diagnosed by clinical observation. The presence of a prominent anal dimple and a band of skin tissue commonly known as a bucket handle is indicative of a perineal fistula. Abdominal and pelvic ultrasonography is performed to further evaluate the infant's anatomic malformation. An IV pyelogram and a voiding cystourethrogram are performed to evaluate associated anomalies involving the urinary tract. Other diagnostic examinations that may be performed include pelvic magnetic resonance imaging, radiography, ultrasound, and fluoroscopic examination of pelvic anatomic contents and lower spinal anatomy.
Therapeutic Management
The primary management of anorectal malformations is surgical. Once the defect is identified, take steps to rule out associated life-threatening defects, which need immediate surgical intervention. Provided no immediate life-threatening problems exist, the newborn is stabilized and kept NPO for further evaluation. IV fluids are provided to maintain glucose and fluid and electrolyte balance. Current recommendation is that surgery be delayed at least 24 hours to properly evaluate for the presence of a fistula and possibly other anomalies (Akay & Klein, 2016).
The surgical treatment of anorectal malformations varies according to the defect but usually involves one, or possibly a combination of several, of the following procedures: anoplasty, colostomy, posterior sagittal anorectoplasty (PSARP) or other pull-through with colostomy, and colostomy (take-down) closure. The Nursing Care Management discussion below outlines some aspects of preoperative and postoperative care.
A primary laparoscopic repair (without colostomy) of some anorectal malformations is being is being used successfully for the treatment of ARMs. This minimizes surgical risks, associated morbidity, and postoperative pain management with no significant risk for rectal prolapse, anal stenosis, or anorectal manometry (Han, Xia, Guo, et al., 2017).
Prognosis.
The long-term prognosis depends on such factors as the type of defect, anatomy of the sacrum and vertebrae, quality of muscles, and the success of the surgery.
The presence of a flat or “rocker” bottom and no midline groove usually carries a poor prognosis for bowel continence because of associated neurologic, muscular, and anatomic problems. When the internal anal sphincter is absent, incontinence is a common long-term problem. These children may achieve socially acceptable continence over time with the aid of a bowel management program. Other potential complications after surgical treatment of anorectal anomalies include strictures, recurrent rectourinary fistula, mucosal prolapse, and constipation. Long-term outcomes in adults with surgically treated ARM are reported to be positive, depending on the type and severity of the defect, as well as associated anomalies (Gangopadhyay & Pandey, 2015). Constipation, fecal soiling, and fecal incontinence are problems that such children and adults must deal with on a regular basis, but the functional outcome is reported to be excellent (Gangopadhyay & Pandey, 2015).
Nursing Care Management
The first nursing responsibility is assisting in identification of anorectal malformations. A newborn who does not pass stool within 24 hours after birth or has meconium that appears at a location other than the anal opening requires further assessment. Preoperative care includes diagnostic evaluation, GI decompression, bowel preparation, and IV fluids.
For the newborn with a perineal fistula, an anoplasty is performed, which involves moving the fistula opening to the center of the sphincter and enlarging the rectal opening. Postoperative nursing care after anoplasty is primarily directed toward healing the surgical site without other complications. A program of anal dilations is usually initiated when the child returns for the 2-week checkup. Feedings are started soon after surgical repair, and breastfeeding is encouraged because it causes less constipation.
In neonates with anomalies such as cloaca (female), rectourethral prostatic fistula (male), and vestibular fistula (female), a descending colostomy is performed to allow fecal elimination and avoid fecal contamination of the distal imperforate section and subsequent urinary tract infection in infants with urorectal fistulas. With a colostomy, postoperative nursing care is directed toward maintaining appropriate skin care at the stoma sites (both distal and proximal), managing postoperative pain, and administering IV fluids and antibiotics. Postoperative NG decompression may be required with laparotomy, and nursing care focuses on maintenance of appropriate drainage. (See Chapter 22 for colostomy care.)
The PSARP is a common surgical procedure for the repair of anorectal malformations in infants approximately 1 to 2 months after the initial colostomy. Preoperative PSARP care often involves irrigation of the distal stoma to prevent fecal contamination of the operative site. During this time parents must be given accurate yet simple information regarding the infant's appearance postoperatively and expectations as to their level of involvement in the child's care.
In the PSARP procedure the repair is made via a posterior midline sacral approach to dissect the different muscle groups involved without damaging strategic innervation of pelvic structures so optimum postoperative bowel continence is achieved. A laparotomy may be required if the rectum is unidentifiable by the posterior approach. Additional management after successful repair involves a program of anal dilations, colostomy closure, and a bowel management program.
Parents are instructed in perineal and wound care or care of the colostomy as needed. Anal dilations may be necessary for some infants. Parents should observe stooling patterns and observe for signs of anal stricture or complications. Information on dietary modifications and administration of medications is included in counseling. Nurses have a vital role in helping families of a child with anorectal malformations provide optimum care so bowel management is successful and quality of life enhanced for the child and family.
Family Support, Discharge Planning, and Home Care.
Long-term follow-up care is essential for children with complex malformations. Parents need reassurance when a colostomy is performed regarding the child's appearance and their ability to care for the child at home. With much patience and reassurance, parents learn how to provide optimum care of the skin and the appliance, while maintaining an appropriate bond with the child.
After the definitive pull-through procedure, toilet training may be delayed. Complete continence is seldom achieved at the usual age of 2 to 3 years. Bowel habit training, bowel management irrigation programs, diet modification, and administration of stool softeners or fiber help children improve bowel function and social continence. Some children never achieve bowel continence and must rely on daily bowel irrigations. Support and reassurance during the slow progression to normal, socially acceptable function are essential.
NCLEX Review Questions
1. A 16-month-old has a history of diarrhea for 3 days with poor oral intake. He received intravenous fluids, has tolerated some oral fluids in the emergency department, and is being discharged home. Instructions for diet for this child should include:
A. BRAT diet (bananas, rice, applesauce, and toast) for 24 hours, then a soft diet as tolerated
B. Chicken or beef broth for 24 hours, then resume a soft diet
C. Offer a regular diet as child's appetite warrants
D. Keep on clear liquids and toast for 24 hours
2. A 5-month-old infant is seen in the well-child clinic for a complaint of vomiting and failure to grow. His birth weight was 7 lb, and he now weighs 8 lb, 10 oz. The infant's mother reports that he is taking 4 to 7 oz of formula every 4 to 5 hours, but he “spits up a lot after eating and then is hungry again.” The child is noted to be alert but appears malnourished. The mother reports that his stools are brown in color, and he has 1 to 2 bowel movements every day. Based on these findings, the nurse anticipates the infant has:
B. Hypertrophic pyloric stenosis
3. Because children with celiac disease must limit their intake of products containing gluten in wheat, rye, oats, and barley, they are at risk for which of the following nutritional deficiencies? Select all that apply.
D. Vitamin A, D, E, and K deficiency
4. A formerly preterm infant who had surgery for necrotizing enterocolitis is now 6 months old and has short-bowel syndrome. He is unable to absorb most nutrients taken by mouth and is totally dependent on parenteral nutrition, which he receives via a central venous catheter. The clinic nurse following this infant is aware that this infant should be closely observed for the development of:
5. The nurse caring for a 4-month-old infant with biliary atresia and significant urticaria can anticipate administering:
B. Ursodiol (ursodeoxycholic acid)
6. Hepatitis A virus is transmitted by which of the following? Select all that apply.
A. Breast milk from mother with HAV
B. Ingestion of contaminated food
D. Casual contact with infected person
Correct Answers
1. C; 2. B; 3. A, B, D; 4. C; 5. B; 6. B, C