Chapter 46 Vitamin B Complex Deficiency and Excess
Vitamin B complex includes a number of water soluble nutrients, including thiamine (B1), riboflavin (B2), niacin (B3), pyridoxine (B6), folate, cobalamin (B12), biotin, and pantothenic acid. Choline and inositol are also considered part of the B complex and are important for normal body functions, but specific deficiency syndromes have not been attributed to a lack of these factors in the diet.
B-complex vitamins serve as coenzymes in many metabolic pathways that are functionally closely related. Consequently, a lack of one of the vitamins has the potential to interrupt a chain of chemical processes, including reactions that are dependent on other vitamins, and ultimately can produce diverse clinical manifestations. Because diets deficient in any one of the B-complex vitamins are often poor sources of other B vitamins, manifestations of several vitamin B deficiencies usually can be observed in the same person. It is therefore a general practice in a patient who has evidence of deficiency of a specific B vitamin to treat with the entire B-complex group of vitamins.
46.1 Thiamine (Vitamin B1)
Thiamine (vitamin B1) consists of thiazole and pyrimidine rings joined by a methylene bridge. Thiamine diphosphate, the active form of thiamine, serves as a cofactor for several enzymes involved in carbohydrate catabolism such as pyruvate dehydrogenase, transketolase, and α-ketoglutarate. These enzymes also play a role in the hexose monophosphate shunt that generates nicotinamide adenine dinucleotide phosphate (NADP) and pentose for nucleic acid synthesis. Thiamine is also required for the synthesis of acetylcholine and gamma-aminobutyric acid (GABA), which have important roles in nerve conduction. Thiamine is absorbed efficiently in the gastrointestinal (GI) tract, and may be deficient in persons with GI or liver disease. The requirement of thiamine is increased when carbohydrates are taken in large amounts and during periods of increased metabolism, for example, fever, muscular activity, hyperthyroidism, and pregnancy and lactation. Alcohol affects various aspects of thiamine transport and uptake, contributing to the deficiency in alcoholics.
Pork (especially lean), fish, and poultry are good nonvegetarian dietary sources of thiamine. Main sources of thiamine for vegetarians are rice, oat, wheat, and legumes. Most ready-to-eat breakfast cereals are enriched with thiamine. Thiamine is water soluble and heat-labile; most of the vitamin is lost when the rice is repeatedly washed and the cooking water is discarded. The breast milk of a well-nourished mother provides adequate thiamine; breast-fed infants of thiamine-deficient mothers are at risk for deficiency. Most infants and older children consuming a balanced diet obtain an adequate intake of thiamine from food and do not require supplements.
Deficiency
Deficiency of thiamine is associated with severely malnourished states, including malignancy and following surgery. The disorder (or spectrum of disorders) is classically associated with a diet consisting largely of polished rice (oriental beriberi), it can also arise if highly refined wheat flour forms a major part of the diet, in alcoholics, and in food faddists (occidental beriberi). Thiamine deficiency has often been reported from inhabitants of refugee camps consuming the polished rice–based monotonous diets. Thiamine-responsive megaloblastic anemia (TRMA) syndrome is a rare autosomal recessive disorder characterized by megaloblastic anemia, diabetes mellitus, and sensorineural deafness, responding in varying degrees to thiamine treatment. The syndrome occurs because of mutations in the SLC19A2 gene, encoding a thiamine transporter protein, leading to abnormal thiamine transportation and vitamin deficiency in the cells. Thiamine and related vitamins can improve the outcome in children with Leigh encephalomyelopathy and type 1 diabetes mellitus.
Clinical Manifestations
Thiamine deficiency can develop within 2-3 mo of a deficient intake. Early symptoms of thiamine deficiency are nonspecific such as fatigue, apathy, irritability, depression, drowsiness, poor mental concentration, anorexia, nausea, and abdominal discomfort. As the condition progresses, more-specific manifestations of beriberi such as peripheral neuritis (manifesting as tingling, burning, paresthesias of the toes and feet), decreased deep tendon reflexes, loss of vibration sense, tenderness and cramping of the leg muscles, congestive heart failure, and psychic disturbances develop. Patients can have ptosis of the eyelids and atrophy of the optic nerve. Hoarseness or aphonia caused by paralysis of the laryngeal nerve is a characteristic sign. Muscle atrophy and tenderness of the nerve trunks are followed by ataxia, loss of coordination, and loss of deep sensation. Later signs include increased intracranial pressure, meningismus, and coma. The clinical picture of thiamine deficiency is usually divided into a dry (neuritic) type and a wet (cardiac) type. The disease is wet or dry depending on the amount of fluid that accumulates in the body due to factors such as cardiac and renal dysfunction, even though the exact cause for this edema has not been explained. Many cases of thiamine deficiency show a mixture of the 2 main features and are more properly termed thiamine deficiency with cardiopathy and peripheral neuropathy.
The classic clinical triad of Wernicke encephalopathy (mental status changes, ocular signs, ataxia) is rarely reported in infants and young children with severe deficiency secondary to malignancies or feeding of defective formula. An epidemic of life-threatening thiamine deficiency was seen in infants fed a defective soy-based formula that had undetectable thiamine levels. Manifestations included emesis, lethargy, restlessness, ophthalmoplegia, abdominal distention, developmental delay, failure to thrive, lactic acidosis, nystagmus, diarrhea, apnea, and seizures. Intercurrent illnesses that resembled Wernicke encephalopathy often precipitated the symptoms.
Death from thiamine deficiency usually is secondary to cardiac involvement. The initial signs are slight cyanosis and dyspnea, but tachycardia, enlargement of the liver, loss of consciousness, and convulsions can develop rapidly. The heart, especially the right side, is enlarged. The electrocardiogram shows an increased Q-T interval, inverted T waves, and low voltage. These changes as well as the cardiomegaly rapidly revert to normal with treatment, but without prompt treatment, cardiac failure can develop rapidly and result in death. In fatal cases of beriberi, lesions are located principally in the heart, peripheral nerves, subcutaneous tissue, and serous cavities. The heart is dilated, and fatty degeneration of the myocardium is common. Generalized edema or edema of the legs, serous effusions, and venous engorgement are often present. Degeneration of myelin and axon cylinders of the peripheral nerves, with wallerian degeneration beginning in the distal locations, also is common, particularly in the lower extremities. Lesions in the brain include vascular dilation and hemorrhage.
Diagnosis
The diagnosis is often suspected on the basis of clinical setting and compatible symptoms. Objective biochemical tests of thiamine status include measurement of erythrocyte transketolase activity (ETKA) and the thiamine pyrophosphate effect (TPPE). The biochemical diagnostic criteria of thiamine deficiency consist of low ETKA and high TPPE (normal range, 0-14%). Urinary excretion of thiamine or its metabolites (thiazole or pyrimidine) after an oral loading dose of thiamine may also be measured to help identify the deficiency state. MRI changes of thiamine deficiency in infants are characterized by bilateral symmetric hyperintensities of the frontal lobes and basal ganglia, in addition to the lesions in the periaqueductal region, thalami, and the mammillary bodies described in adults.
Prevention
A maternal diet containing sufficient amounts of thiamine prevents thiamine deficiency in breast-fed infants, and infant formulas marketed in all developed countries provide recommended levels of intake. During complementary feeding, adequate thiamine intake can be achieved with a varied diet that includes meat and enriched or whole-grain cereals. When the staple cereal is polished rice, special efforts need to be made to include legumes and/or nuts in the ration. Thiamine and other vitamins can be retained in rice by parboiling, a process of steaming the rice in the husk before milling. Improvement in cooking techniques, such as not discarding the water used for cooking, minimal washing of grains, and reduction of cooking time help to minimize the thiamine losses during the preparation of food.
Treatment
In the absence of GI disturbances, oral administration of thiamine is effective. Children with cardiac failure, convulsions, or coma should be given 10 mg of thiamine intramuscularly or intravenously daily for the 1st week. This treatment should then be followed by 3-5 mg of thiamine per day orally for at least 6 wk. The response is dramatic in infants and in those having predominantly cardiovascular manifestations, whereas the neurologic response is slow and often incomplete. Patients with beriberi often have other B-complex vitamin deficiencies; therefore, all other B-complex vitamins should also be administered. Treatment of TRMA and other dependency states require higher dosages (100-200 mg/day). The anemia responds well to thiamine administration, and insulin for associated diabetes mellitus can also be discontinued in many cases of TRMA.
Toxicity
There are no reports of adverse effects from consumption of excess thiamine by ingestion of food or supplements. A few isolated cases of pruritus and anaphylaxis have been reported in patients after parenteral administration of the vitamin.
Boonsiri P, Tangrassameeprasert R, Panthongviriyakul C, Yongvanit P. A preliminary study of thiamine status in northeastern Thai children with acute diarrhea. Southeast Asian J Trop Med Public Health. 2007;38:1120-1125.
Fattal-Valevski A, Kesler A, Sela BA, et al. Outbreak of life-threatening thiamine deficiency in infants in Israel caused by a defective soy-based formula. Pediatrics. 2005;115:e233-e238.
Kornreich L, Bron-Harlev E, Hoffmann C, et al. Thiamine deficiency in infants: MR findings in the brain. Am J Neuroradiol. 2005;26:1668-1674.
Ricketts CJ, Minton JA, Samuel J, et al. Thiamine-responsive megaloblastic anaemia syndrome: long-term follow-up and mutation analysis of seven families. Acta Paediatr. 2006;95:99-104.
46.2 Riboflavin (Vitamin B2)
Riboflavin is part of the structure of the coenzymes flavin adenine dinucleotide (FAD) and flavin mononucleotide, which participate in oxidation-reduction reactions in numerous metabolic pathways and in energy production via the mitochondrial respiratory chain. Riboflavin is stable to heat but is destroyed by light.
Milk, eggs, organ meats, legumes, and mushrooms are rich dietary sources of riboflavin. Most commercial cereals, flours, and breads are enriched with riboflavin.
Deficiency
The causes of riboflavin deficiency are mainly related to malnourished and malabsorptive states, including GI infections. Treatment with some drugs, such as probenecid, phenothiazine, or oral contraceptives, can also cause the deficiency. The side chain of the vitamin is photochemically destroyed during phototherapy for hyperbilirubinemia, as it is involved in the photosensitized oxidation of bilirubin to more polar excretable compounds. Isolated complex II deficiency, a rare mitochondrial disease manifesting in infancy and childhood, responds favorably to riboflavin supplementation and thus can be termed a dependency state.
Clinical Manifestations
Clinical features of riboflavin deficiency include cheilosis, glossitis, keratitis, conjunctivitis, photophobia, lacrimation, corneal vascularization, and seborrheic dermatitis. Cheilosis begins with pallor at the angles of the mouth and progresses to thinning and maceration of the epithelium, leading to fissures extending radially into the skin (Fig. 46-1). In glossitis, the tongue becomes smooth, with loss of papillary structure (Fig. 46-2). Normochromic, normocytic anemia may also be seen because of the impaired erythropoiesis. A low riboflavin content of the maternal diet has been linked to congenital heart defects, but the evidence is weak.
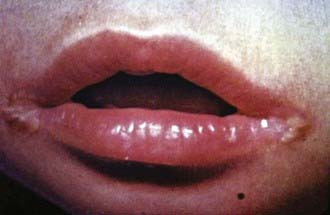
Figure 46-1 Angular cheilosis with ulceration and crusting.
(Courtesy of National Institute of Nutrition, Indian Council of Medical Research, Hyderabad, India.)

Figure 46-2 Glossitis as seen in riboflavin deficiency.
(From Zappe HA, Nuss S, Becker K, et al: Riboflavin deficiency in baltistan (website). www.rzuser.uni-heidelberg.de/%7Ecn6/baltista/ribofl_e.htm. Accessed May 23, 2010.)
Diagnosis
Most often, the diagnosis is based on the clinical features of angular cheilosis in a malnourished child, which responds promptly to riboflavin supplementation. A functional test of riboflavin status is done by measuring the activity of erythrocyte glutathione reductase (EGR), with and without the addition of FAD. An EGR activity coefficient (ratio of EGR activity with added FAD to EGR activity without FAD) of >1.4 is used as an indicator of deficiency. Urinary excretion of riboflavin <30 µg/24 hr also suggests low intakes.
Prevention
The recommended daily allowance (RDA) of riboflavin for infants, children and adolescents is presented in Table 46-1. Adequate consumption of milk, milk products, and eggs prevents riboflavin deficiency. Fortification of cereal products is helpful for those who follow vegan diets or are consuming inadequate amounts of milk products because of other reasons.
Toxicity
No adverse effects associated with riboflavin intakes from food or supplements have been reported, and the upper safe limit for consumption has not been established. Though the photosensitizing property of this vitamin raises the possibility for some potential risks, limited absorption in high-intake situations precludes such concerns.
Bugiani M, Lamantea E, Invernizzi F, et al. Effects of riboflavin in children with complex II deficiency. Brain Dev. 2006;28:576-581.
Rohner F, Zimmermann MB, Wegmueller R, et al. Mild riboflavin deficiency is highly prevalent in school-age children but does not increase risk for anaemia in Côte d’Ivoire. Br J Nutr. 2007;97:970-976.
Shaw NS, Wang JL, Pan WH, et al. Thiamin and riboflavin status of Taiwanese elementary schoolchildren. Asia Pac J Clin Nutr. 2007;16(Suppl 2):564-571.
Smedts HP, Rakhshandehroo M, Verkleij-Hagoort AC, et al. Maternal intake of fat, riboflavin and nicotinamide and the risk of having offspring with congenital heart defects. Eur J Nutr. 2008;47:357-365.
46.3 Niacin (Vitamin B3)
Niacin (nicotinamide or nicotinic acid) forms part of 2 cofactors, nicotinamide adenine dinucleotide (NAD) and NADP, which are important in several biologic reactions, including the respiratory chain, fatty acid and steroid synthesis, cell differentiation, and DNA processing. Niacin is rapidly absorbed from the stomach and the intestines and can also be synthesized from tryptophan in the diet.
Major dietary sources of niacin are meat, fish, and poultry for nonvegetarians and cereals, legumes, and green leafy vegetables for vegetarians. Enriched and fortified cereal products and legumes also are major contributors to niacin intake. Milk and eggs contain little niacin but are good sources of tryptophan, which can be converted to NAD (60 mg tryptophan = 1 mg niacin).
Deficiency
Pellagra, the classic niacin deficiency disease, occurs chiefly in populations where corn (maize), a poor source of tryptophan, is the major foodstuff. A severe dietary imbalance such as in anorexia nervosa and in war or famine conditions also can cause pellagra. Pellagra can also develop in conditions associated with disturbed tryptophan metabolism such as carcinoid syndrome and Hartnup’s disease.
Clinical Manifestations
The early symptoms of pellagra are vague: anorexia, lassitude, weakness, burning sensations, numbness, and dizziness. After a long period of deficiency, the classic triad of dermatitis, diarrhea, and dementia appears. Dermatitis, the most characteristic manifestation of pellagra, can develop suddenly or insidiously and may be initiated by irritants, including intense sunlight. The lesions first appear as symmetric areas of erythema on exposed surfaces, resembling sunburn, and might go unrecognized. The lesions are usually sharply demarcated from the surrounding healthy skin, and their distribution can change frequently. The lesions on the hands and feet often have the appearance of a glove or stocking (Fig. 46-3). Similar demarcations can also occur around the neck (Casal necklace) (see Fig. 46-3). In some cases, vesicles and bullae develop (wet type). In others, there may be suppuration beneath the scaly, crusted epidermis, and in still others, the swelling can disappear after a short time, followed by desquamation (Fig. 46-4). The healed parts of the skin might remain pigmented. The cutaneous lesions may be preceded by or accompanied by stomatitis, glossitis, vomiting, and/or diarrhea. Swelling and redness of the tip of the tongue and its lateral margins is often followed by intense redness, even ulceration, of the entire tongue and the papillae. Nervous symptoms include depression, disorientation, insomnia, and delirium.
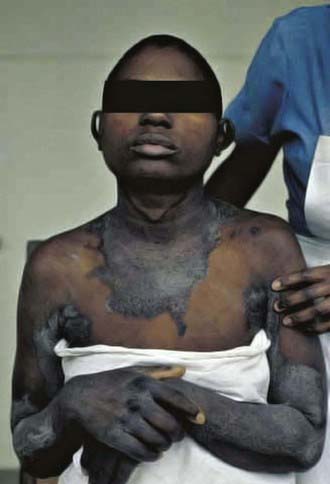
Figure 46-3 Characteristic skin lesions of pellagra on hands and lesions on the neck (Casal necklace).
(Courtesy of Dr. J.D. MacLean, McGill Centre for Tropical Diseases, Montreal, Canada.)

Figure 46-4 Clinical manifestations of niacin deficiency before (A) and after (B) therapy.
(From Weinsier RL, Morgan SL: Fundamentals of clinical nutrition, St Louis, 1993, Mosby, p 99.)
The classic symptoms of pellagra usually are not well developed in infants and young children, but anorexia, irritability, anxiety, and apathy are common. Young patients might also have sore tongues and lips and usually have dry and scaly skin. Diarrhea and constipation can alternate, and anemia can occur. Children who have pellagra often have evidence of other nutritional deficiency diseases.
Diagnosis
Because of lack of a good functional test to evaluate niacin status, the diagnosis of deficiency is usually made from the physical signs of glossitis, GI symptoms, and a symmetric dermatitis. Rapid clinical response to niacin is an important confirmatory test. A decrease in the concentration and/or a change in the proportion of the niacin metabolites N1-methyl-nicotinamide (1-mn) and 2-pyridone (2-pyr) in the urine provide biochemical evidence of deficiency and can be seen before the appearance of overt signs of deficiency. Serum levels of NAD and NADP do not correlate well with clinical deficiency.
Prevention
Adequate intakes of niacin are easily met by consumption of a diet that consists of a variety of foods and includes meat, eggs, milk, and enriched or fortified cereal products. The dietary reference intake (DRI) is expressed in mg niacin equivalents (NE) in which 1 mg NE = 1 mg niacin or 60 mg tryptophan. An intake of 2 mg of niacin is considered adequate for infants 0-6 mo of age; and 4 mg is adequate for infants 7-12 mo of age. For older children, the recommended intakes are 6 mg for 1-3 yr of age, 8 mg for 4-8 yr of age, 12 mg for 9-13 yr of age, and 14-16 mg for 14-18 yr of age.
Treatment
Children usually respond rapidly to treatment. A liberal and varied diet should be supplemented with 50-300 mg/day of niacin; in severe cases or in patients with poor intestinal absorption, 100 mg may be given intravenously. The diet should also be supplemented with other vitamins, especially other B-complex vitamins. Sun exposure should be avoided during the active phase of pellagra, and the skin lesions may be covered with soothing applications. Other coexisting nutrient deficiencies such as iron deficiency anemia should be treated. Even after successful treatment, the diet should continue to be monitored to prevent recurrence.
Toxicity
There are no toxic effects associated with the intake of naturally occurring niacin in foods. Shortly after the ingestion of large doses of nicotinic acid taken as a supplement or a pharmacologic agent, a person often experiences a burning, tingling, and itching sensation as well as flushing on the face, arms, and chest. Large doses of niacin also can have nonspecific GI effects and can cause cholestatic jaundice or hepatotoxicity. Tolerable upper intake levels for children are approximately double the recommended dietary allowance.
Birjmohun RS, Hutten BA, Kastelein JJ, Stroes ES. Efficacy and safety of high-density lipoprotein cholesterol–increasing compounds: a meta-analysis of randomized controlled trials. J Am Coll Cardiol. 2005;45:185-197.
Creeke PI, Dibari F, Cheung E, et al. Whole blood NAD and NADP concentrations are not depressed in subjects with clinical pellagra. J Nutr. 2007;137:2013-2017.
Jagielska G, Tomaszewicz-Libudzic EC, Brzozowska A. Pellagra: a rare complication of anorexia nervosa. Eur Child Adolesc Psychiatry. 2007;16:417-420.
Seal AJ, Creeke PI, Dibari F, et al. Low and deficient niacin status and pellagra are endemic in postwar Angola. Am J Clin Nutr. 2007;85:218-224.
46.4 Vitamin B6 (Pyridoxine)
Vitamin B6 includes a group of closely related compounds: pyridoxine, pyridoxal, pyridoxamine, and their phosphorylated derivatives. Pyridoxal 5′-phosphate (PLP) and, to a lesser extent, pyridoxamine phosphate function as coenzymes for many enzymes involved in amino acid metabolism, neurotransmitter synthesis, glycogen metabolism, and steroid action. If vitamin B6 is lacking, glycine metabolism can lead to oxaluria. The major excretory product in the urine is 4-pyridoxic acid.
The vitamin B6 content of human milk and infant formulas is adequate. Good food sources of the vitamin include fortified ready-to-eat cereals, meat, fish, poultry, liver, bananas, rice, and certain vegetables. Large losses of the vitamin can occur during high-temperature processing of foods or milling of cereals, whereas parboiling of rice prevents its loss.
Deficiency
Because of the importance of vitamin B6 in amino acid metabolism, high protein intakes can increase the requirement for the vitamin; the RDAs are sufficient to cover the expected range of protein intake in the population. The risk of deficiency is increased in persons taking medications that inhibit the activity of vitamin B6 (isoniazid, penicillamine, corticosteroids, anticonvulsants), in young women taking oral progesterone-estrogen contraceptives, and in patients receiving maintenance dialysis.
Clinical Manifestations
The deficiency symptoms seen in infants are listlessness, irritability, seizures, vomiting, and failure to thrive. Peripheral neuritis is a feature of deficiency in adults but is not usually seen in children. Electroencephalogram (EEG) abnormalities have been reported in infants as well as in young adult subjects in controlled depletion studies. Skin lesions include cheilosis, glossitis, and seborrheic dermatitis around the eyes, nose, and mouth. Microcytic anemia can occur in infants but is not common. Oxaluria, oxalic acid bladder stones, hyperglycinemia, lymphopenia, decreased antibody formation, and infections also have been associated with vitamin B6 deficiency.
Several types of vitamin B6 dependence syndromes, presumably due to errors in enzyme structure or function, respond to very large amounts of pyridoxine (see Table 46-1). These syndromes include pyridoxine-dependent seizures, a vitamin B6–responsive anemia, xanthurenic aciduria, cystathioninuria, and homocystinuria (Chapters 79, 448, and 586).
Diagnosis
The activity of the erythrocyte transaminases glutamic oxaloacetic transaminase and glutamic pyruvic transaminase is low in vitamin B6 deficiency; tests measuring the activity of these enzymes before and after the addition of PLP may be useful as indicators of vitamin B6 status. Abnormally high xanthurenic acid excretion after tryptophan ingestion also provides evidence of deficiency. Plasma PLP assays are being used more often, but factors other than deficiency can influence the results. Vitamin B6 deficiency or dependence should be suspected in all infants with seizures. If more common causes of infantile seizures have been eliminated, 100 mg of pyridoxine can be injected, with EEG monitoring if possible. If the seizure stops, vitamin B6 deficiency should be suspected. In older children, 100 mg of pyridoxine may be injected intramuscularly while the EEG is being recorded; a favorable response of the EEG suggests pyridoxine deficiency.
Prevention
Deficiency is unlikely in children consuming diets that meet their energy needs and contain a variety of foods. Parboiling of rice prevents the loss of vitamin B6 from the grains. The DRIs for vitamin B6 are 0.1 mg/day for infants up to 6 mo of age; 0.3 mg/day for ages 6 mo to 1 yr; 0.5 mg/day for ages 1-3 yr; 0.6 mg/day for ages 4-8 yr; 1.0 mg/day for ages 9-13 yr; and 1.2-1.3 mg/day for ages 14-18 yr. Infants whose mothers have received large doses of pyridoxine during pregnancy are at increased risk for seizures from pyridoxine dependence, and supplements during the 1st few weeks of life should be considered. Any child receiving a pyridoxine antagonist, such as isoniazid, should be carefully observed for neurologic manifestations; if these develop, vitamin B6 should be administered or the dose of the antagonist should be decreased.
Treatment
Intramuscular or intravenous administration of 100 mg of pyridoxine is used to treat convulsions due to vitamin B6 deficiency. One dose should be sufficient if adequate dietary intake follows. For pyridoxine-dependent children, daily doses of 2-10 mg intramuscularly or 10-100 mg orally may be necessary. Occasionally vitamin B6 has been used in large doses along with magnesium in children said to have “autism”; the functional benefit of such intervention is minimal.
Toxicity
Adverse effects have not been associated with high intakes of vitamin B6 from food sources. However, ataxia and sensory neuropathy have been reported with dosages as low as 100 mg/day in adults taking vitamin B6 supplements for several months.
Been JV, Bok LA, Andriessen P, Renier WO. Epidemiology of pyridoxine dependent seizures in the Netherlands. Arch Dis Child. 2005;90:1293-1296.
Nye C, Brice A: Combined vitamin B6-magnesium treatment in autism spectrum disorder, Cochrane Database Syst Rev (19):CD003497, 2005.
Teune LK, vd Hoeven JH, Maurits NM, et al. Pyridoxine induces non-specific EEG alterations in infants with therapy resistant seizures. Seizure. 2007;16:459-464.
46.5 Biotin
Biotin functions as a cofactor for enzymes involved in carboxylation reactions within and outside mitochondria. These biotin-dependent carboxylases catalyze key reactions in gluconeogenesis, fatty acid metabolism, and amino acid catabolism.
There is limited information on the biotin content of foods; it is believed to be widely distributed, thus making a deficiency unlikely. Avidin found in raw egg whites acts as a biotin antagonist. Signs of biotin deficiency have been demonstrated in persons who consume large amounts of raw egg whites over long periods. Deficiency also has been described in infants and children receiving enteral and parenteral nutrition infusates that lack biotin. Treatment with valproic acid may result in a low biotinidase activity and/or biotin deficiency.
The clinical findings of biotin deficiency include scaly periorificial dermatitis, conjunctivitis, thinning of hair, and alopecia. Central nervous system abnormalities seen with biotin deficiency are lethargy, hypotonia, and withdrawn behavior. Biotin deficiency can be successfully treated using 1-10 mg of biotin orally daily. The adequate dietary intake values for biotin are 5 µg/day for 0-6 mo, 6 µg/day for 7-12 mo, 8 µg/day for ages 1-3 yr, 12 µg/day for ages 4-8 yr, 20 µg/day for ages 9-13 yr, and 25 µg/day for ages 14-18 yr. No toxic effects have been reported with very high doses. Conditions involving deficiencies in the enzymes holocarboxylase synthetase and biotinidase that respond to treatment with biotin are described in Chapter 79.6.
46.6 Folate
Folate exists in a number of different chemical forms. Folic acid (pteroylglutamic acid) is the synthetic form used in fortified foods and supplements. Naturally occurring folates in foods retain the core chemical structure of pteroylglutamic acid but vary in their state of reduction, the single carbon moiety they bear, or the length of the glutamate chain. These polyglutamates are broken down and reduced in the small intestine to dihydro- and tetrahydrofolates, which are involved as coenzymes in amino acid and nucleotide metabolism as acceptors and donors of 1-carbon units.
Rice and cereals are rich dietary sources of folate, especially if enriched. Beans, leafy vegetables, and fruits such as oranges and papaya are good sources, too. The vitamin is readily absorbed from the small intestine and is broken down to monoglutamate derivatives by mucosal polyglutamate hydrolases. A high-affinity proton-coupled folate transporter (PCFT) seems to be essential for absorption of folate in intestine and in various cell types at low pH. The vitamin is also synthesized by the colonic bacteria, and the half-life of the vitamin is prolonged by enterohepatic recirculation.
Deficiency
Because of its role in protein, DNA, and RNA synthesis, the risk of deficiency is increased during periods of rapid growth or increased cellular metabolism. Folate deficiency can result from poor nutrient content in diet, inadequate absorption (celiac disease, inflammatory bowel disease), increased requirement (sickle cell anemia, psoriasis, malignancies, periods of rapid growth as in infancy and adolescence), or inadequate utilization (long-term treatment with high-dose nonsteroidal anti-inflammatory drugs, anticonvulsants such as phenytoin and phenobarbital, and methotrexate). Rare causes of deficiency are hereditary folate malabsorption, inborn errors of folate metabolism (methylene tetrahydrofolate reductase, methionine synthase reductase, and glutamate formiminotransferase deficiencies), and cerebral folate deficiency. A loss-of-function mutation in the gene coding for proton-coupled folate transporter (PCFT) is the molecular basis for hereditary folate malabsorption. A high-affinity blocking autoantibody against the membrane-bound folate receptor in the choroid plexus preventing its transport across the blood-brain barrier is the likely cause of the infantile cerebral folate deficiency.
Clinical Manifestations
Folic acid deficiency results in megaloblastic anemia and hypersegmentation of neutrophils. Nonhematologic manifestations include glossitis, listlessness, and growth retardation not related to anemia. There is an association between low maternal folic acid status and neural tube defects, primarily spina bifida and anencephaly, and the role of periconceptional folic acid in their prevention is well established.
Hereditary folate malabsorption manifests at 1-3 mo of age with recurrent or chronic diarrhea, failure to thrive, oral ulcerations, neurologic deterioration, megaloblastic anemia, and opportunistic infections. Cerebral folate deficiency manifests at 4-6 mo of age with irritability, microcephaly, developmental delay, cerebellar ataxia, pyramidal tract signs, choreoathetosis, ballismus, seizures, and blindness due to optic atrophy. 5-Methyltetrahydrofolate levels are normal in serum and red blood cells (RBCs) but are markedly depressed in the cerebrospinal fluid (CSF).
Diagnosis
The diagnosis of folic acid deficiency anemia is made in the presence of macrocytosis along with low folate levels in serum and/or RBCs. Normal serum folic acid levels are 5-20 ng/mL; with deficiency, serum folic acid levels are <3 ng/mL. Levels of RBC folate are a better indicator of chronic deficiency. The normal RBC folate level is 150-600 ng/mL of packed cells. The bone marrow is hypercellular because of erythroid hyperplasia, and megaloblastic changes are prominent. Large, abnormal neutrophilic forms (giant metamyelocytes) with cytoplasmic vacuolation also are seen.
Cerebral folate deficiency is associated with low levels of 5-methyltetrahydrofolate in the CSF and normal folate levels in the plasma and red blood cells. Mutations in the PCFT gene are demonstrated in the hereditary folate malabsorption.
Prevention
Breast-fed infants have better folate nutriture than non–breast-fed infants throughout infancy. Consumption of folate-rich foods and food-fortification programs are important to ensure adequate intake in children and in women of childbearing age. The DRIs for folate are 65 µg of dietary folate equivalent (DFE) for infants 0-6 mo and 80 µg of DFE for infants aged between 6 and 12 mo. (1 DFE = 1 µg food folate = 0.6 µg of folate from fortified food or as a supplement consumed with food = 0.5 µg of a supplement taken on an empty stomach.) For older children, the DRIs are 150 µg of DFE for ages 1-3 yr; 200 µg of DFE for ages 4-8 yr; 300 µg of DFE for ages 9-13 yr; and 400 µg of DFE for ages 14-18 yr. Providing iron and folic acid tablets for prevention of anemia in children and pregnant women is a routine strategy in at-risk populations. Health-education programs increase women’s knowledge and use of folate supplements to prevent birth defects.
Treatment
When the diagnosis of folate deficiency is established, folic acid may be administered orally or parenterally at 0.5-1.0 mg/day. Folic acid therapy should be continued for 3-4 wk or until a definite hematologic response has occurred. Maintenance therapy with 0.2 mg of folate is adequate. Prolonged treatment with oral folinic acid is required in cerebral folate deficiency, and the response may be incomplete. Treatment of hereditary folate malabsorption may be possible with intramuscular folinic acid; some patients may respond to high-dose oral folinic acid therapy.
Toxicity
No adverse effects have been associated with consumption of the amounts of folate normally found in fortified foods. Excessive intake of folate supplements might obscure and potentially delay the diagnosis of vitamin B12 deficiency. Massive doses given by injection have the potential to cause neurotoxicity.
Allen LH. Causes of vitamin B12 and folate deficiency. Food Nutr Bull. 2008;29(2 Suppl):S20-S34.
Gordon N. Cerebral folate deficiency. Dev Med Child Neurol. 2009;51:180-182.
Qiu A, Jansen M, Sakaris A, et al. Identification of an intestinal folate transporter and the molecular basis for hereditary folate malabsorption. Cell. 2006;127:917-928.
Taneja S, Bhandari N, Strand TA, et al. Cobalamin and folate status in infants and young children in a low-to-middle income community in India. Am J Clin Nutr. 2007;86:1302-1309.
Wilson RD, Johnson JA, Wyatt P, et al. Pre-conceptional vitamin/folic acid supplementation 2007: the use of folic acid in combination with a multivitamin supplement for the prevention of neural tube defects and other congenital anomalies. J Obstet Gynaecol Can. 2007;29:1003-1026.
46.7 Vitamin B12 (Cobalamin)
Vitamin B12 in the form of deoxyadenosylcobalamin functions as a cofactor for isomerization of methylmalonyl-CoA to succinyl-CoA, an essential reaction in lipid and carbohydrate metabolism. Methylcobalamin is another circulating form of vitamin B12 and is essential for methyl group transfer during the conversion of homocysteine to methionine. This reaction also requires a folic acid cofactor and is important for protein and nucleic acid biosynthesis.
Dietary sources of vitamin B12 are almost exclusively from animal foods. Organ meats, muscle meats, sea foods (mollusks, oysters, fish), poultry, and egg yolk are rich sources. Fortified ready-to-eat cereals and milk and their products are the important sources of the vitamin for vegetarians. Human milk is an adequate source for breast-feeding infants if the maternal serum B12 levels are adequate. The vitamin is absorbed from ileum at alkaline pH after binding with intrinsic factor. Enterohepatic circulation, direct absorption, and synthesis by intestinal bacteria are additional mechanisms helping to maintain the vitamin B12 nutriture.
Deficiency
Vitamin B12 deficiency due to inadequate dietary intake occurs primarily in persons consuming strict vegetarian or vegan diets. Prevalence of vitamin B12 deficiency is high in predominantly vegetarian or lacto-vegetarian populations. Malabsorption of B12 occurs in pernicious anemia due to intrinsic factor deficiency, and in ileal resections and Crohn disease. Breast-feeding infants of B12-deficient mothers are also at risk for significant deficiency. Newborn metabolic screening can detect high levels of methylmalonic acid in the neonate’s blood spot, which suggests maternal and neonatal B12 deficiencies.
Clinical Manifestations
The hematologic manifestations of vitamin B12 deficiency are similar to manifestations of folate deficiency and are discussed in Chapter 448.2. Irritability, hypotonia, developmental delay, developmental regression, and involuntary movements are the common neurologic symptoms in infants and children, whereas sensory deficits, paresthesias, and peripheral neuritis are seen in adults. Hyperpigmentation of the knuckles and palms is another common observation with B12 deficiency in children.
Treatment
The hematologic symptoms respond promptly to parenteral administration of 1,000 µg vitamin B12. Oral administration has also been found to be equally effective in achieving hematologic and neurologic responses in adults, but the data are inadequate in children.
Prevention
The DRIs are 0.4 µg/day at age 0-6 mo, 0.5 µg/day at age 6-12 mo, 0.9 µg/day at age 1-3 yr, 1.2 µg/day at age 4-8 yr, 1.8 µg/day at age 9-13 yr, 2.4 µg/day at age 14-18 yr and in adults, 2.6 µg/day in pregnancy, and 2.8 µg/day in lactation. Pregnant and breastfeeding women should ensure an adequate consumption of animal products to prevent the deficiency in infants. Food fortification with the vitamin helps to prevent deficiency in predominantly vegetarian populations.
Andrès E, Dali-Youcef N, Vogel T, et al. Oral cobalamin (vitamin B12) treatment. An update. Int J Lab Hematol. 2009;31:1-8.
Bjørke-Monsen AL, Torsvik I, Saetran H, et al. Common metabolic profile in infants indicating impaired cobalamin status responds to cobalamin supplementation. Pediatrics. 2008;122:83-91.
Coelho D, Suormala T, Stucki M, et al. Gene identification for the cb1D defect of vitamin B12 metabolism. N Engl J Med. 2008;358:1454-1464.
Dror DK, Allen LH. Effect of vitamin B12 deficiency on neurodevelopment in infants: current knowledge and possible mechanisms. Nutr Rev. 2008;66:250-255.
Hudson B. Vitamin B-12 deficiency. BMJ. 2010;340:c2305.
Marble M, Copeland S, Khanfar N, Rosenblatt DS. Neonatal vitamin B12 deficiency secondary to maternal subclinical pernicious anemia: identification by expanded newborn screening. J Pediatr. 2008;152:731-733.
Marsch AF, Shashidhar H, D’Orazio JA. B12 deficient megaloblastic anemia in a toddler with a history of gastroschisis. J Pediatr. 2011;158:512.
McLean E, de Benoist B, Allen LH. Review of the magnitude of folate and vitamin B12 deficiencies worldwide. Food Nutr Bull. 2008;29(2 Suppl):S38-S51.
Molloy AM, Kirke PN, Brody LC, et al. Effects of folate and vitamin B12 deficiencies during pregnancy on fetal, infant, and child development. Food Nutr Bull. 2008;29(2 Suppl):S101-S111.
Vidal-Alaball J, Butler CC, Cannings-John R, et al: Oral vitamin B12 versus intramuscular vitamin B12 for vitamin B12 deficiency, Cochrane Database Syst Rev (3):CD004655, 2005.