Chapter 84 Progeria
The Hutchinson-Gilford progeria syndrome (HGPS) is a rare, fatal, sporadic, autosomal dominant disorder with an incidence of ≈1/4,000,000 live births. HGPS is regarded as the most prominent of the senile-like appearance syndromes. Sexual maturation is incomplete and these patients do not reproduce. Parent to child transmission has not occurred. The most prominent features of HGPS are changes that simulate accelerated aging, the recognition of which establishes the diagnosis. Genetic diagnosis is available; HGPS is caused by a single base mutation in LMNA, which results in the production of a mutant lamin A, progerin. Progerin is found in increased concentration in fibroblasts of normal older compared to younger individuals, suggesting a role in normal aging. The mean age of survival of children with HGPS is 13 yr, with a range of 5 to 20 yr, such that there are approximately 40 patients world wide at any point in time. A study of 15 patients, about 40% of the world’s population, revealed that all were heterozygous for the G608G mutation.
Clinical Manifestations
Children with progeria usually appear normal at birth, but occasionally, findings such as circumoral cyanosis, pinched nose, and tight skin suggest the diagnosis. Shortly after birth profound changes in growth parameters, skin, hair, dentition, ophthalmologic status, hearing, bone development, and blood vessels occur.
Birthweight is normal, but by 2 mo of age, weight is below the 3rd percentile. Between 2 and 10 yr of age, normal children gain 1.80 kg/yr whereas children with HGPS gain 0.44 to 0.65 kg/yr. Linear growth, while severely retarded, is less affected than weight. Height falls below the 3rd percentile at the age of 16 mo; because the majority of children with HGPS have developed knee contractures by that time, height measurements are probably an underestimate. The disparity of weight and height delay, associated with the loss of subcutaneous fat, results in the emaciated appearance characteristic of HGPS. These abnormalities of growth cannot be accounted for by inadequate caloric intake. Growth hormone is normal, and insulin resistance is usually only mild. Abnormalities in skin development result in thin, taut, pigmented skin with prominent veins and scleroderma-like changes. Alopecia, loss of eyebrows and eyelashes, and diminution of subcutaneous fat, including earlobes, are characteristic.
The abnormalities of bone development are unique to HGPS. Recession of the mandible with the development of an accentuated vertical angle gives rise to prominent micrognathia and contributes to crowded dentition. The secondary incisors erupt on the lingual and palatal surfaces of the mandibular and maxillary alveolar ridges, rather than in place of the primary incisors. These dental changes are specific for HGPS. The mandibular changes, in association with the skull finding of persistently patent fontanels, result in the appearance of a large head relative to face and prominent eyes. Acro-osteolysis, progressive distal radiolucency, and resorption of bone are the earliest characteristics of long bone changes. These changes begin as early as 3 mo of age and involve the digits, clavicles, and ribs. There is resultant shortening of distal phalanges, resorption of the clavicle, and thinning as well as tapering of the ribs. The rib changes give rise to the pyramidal or pyriform thorax seen in HGPS. Long bone remodeling of the femoral head-neck axis following knee and ankle contractures results in coxa valga, which is straightening of the femoral head-neck axis to >125 degrees. The bony pelvis is normal and these changes give rise to the “horse riding” stance of HGPS. Other long bone changes include flaring of the humeral and femoral metaphyses and constriction of the radial neck. Unlike the bone and joint changes seen in normal aging, these changes do not result in fractures; arthritis is not a feature of HGPS. Growth plates and bone age are normal. The progressive bone changes of HGPS resemble a skeletal dysplasia and may be due to a microvasculopathy.
Ophthalmological changes seen in HGPS include dry eye syndrome, hyperopia, and keratitis.
Orobuccal difficulties include abnormalities of chewing, limitation and weakness of lingual movement, dysarthria, and lisping, all of which are in part related to mandibular maldevelopment. Auditory comprehension and expressive language skills are normal.
Hearing abnormalities are quite consistent and the majority of HGPS patients have conductive hearing loss. High-frequency sensorial hearing loss is also described.
Laboratory abnormalities include thrombophilia, prolonged prothrombin time, and elevated serum phosphorus. Some children have elevated serum triglycerides, total cholesterol, and low density lipoproteins, but have reduced high density lipoproteins. Thyroid and immune studies are normal. Glucose tolerance tests have occasionally revealed elevated glucose and insulin levels with normal free fatty acid levels that decreased appropriately with insulin elevation.
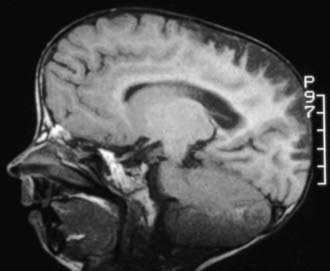
Progressive vascular changes are a major feature of HGPS. Medial smooth muscle cells are lost, vascular remodeling is defective resulting in intimal thickening, disrupted elastin, and deposition of extracellular matrix. Sclerotic plaques deposited in the aorta, coronary, and cerebral blood vessels lead to stenosis (Fig. 84-1). Neurologic abnormalities include transient ischemic attacks, stroke, and seizures, but the major cause of death is cardiovascular compromise. In the absence of neurologic events, motor and mental developments are usually normal.
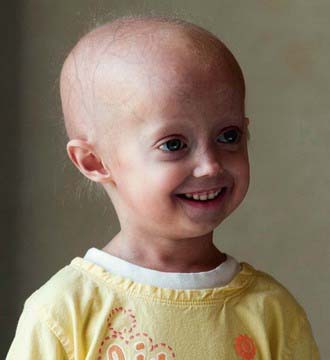
The constellation of bone, hair, subcutaneous fat, and skin changes results in the marked physical resemblance among patients with HGPS (Fig. 84-2).
Molecular Pathogenesis
The nuclear lamina is a protein-containing layer attached to the inner nuclear membrane and composed of a family of polypeptides, a major component of which is lamin A. CAAX is the motif cysteine (C), 2 aliphatic amino acids (AA), and any amino acid (X). Farnesyl groups linked to cysteines of C-terminal CAAX boxes tether the normal and mutant lamin A (progerin) to the nuclear membrane. Normal lamin A is released by cleavage of the 15 C-terminal amino acids, but progerin, lacking a cleavage site, remains anchored to the membrane, binding other proteins, causing blebbing of the nucleus, disrupting mitosis, and altering gene expression. Progerin acts in a dominant negative fashion.
A growing list of other diseases is associated with different mutations in LMNA that are known as laminopathies. These include Emery-Dreifuss muscular dystrophy type 2, familial dilated cardiomyopathy and conduction system defects, Dunnigan-type familial partial lipodystrophy, limb girdle muscular dystrophy 1B, Charcot-Marie-Tooth disease 2B1, mandibuloacral dysplasia, and atypical Werner syndrome.
Differential Diagnosis
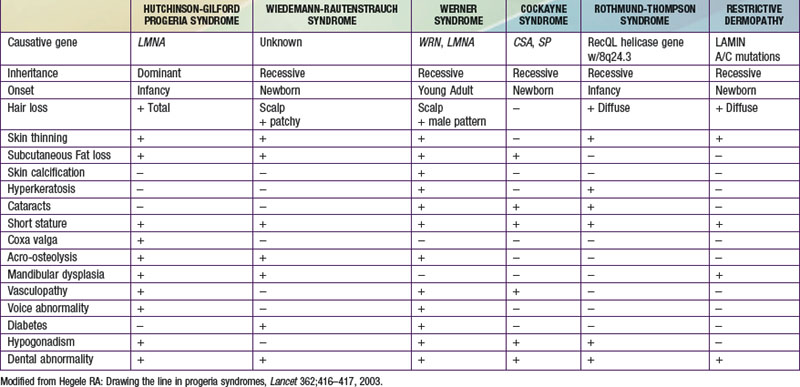
The disorders that resemble HGPS are those grouped as the senile-like syndromes and include Wiedmann-Rautenstrauch syndrome, Werner syndrome, Cockayne syndrome, Rothmund-Thomson syndrome, and restrictive dermopathy (Table 84-1).
Prognosis and Treatment
Children with progeria usually have severe atherosclerosis, and death occurs as a result of complications of cardiac or cerebrovascular disease, generally between age 5 and 20 yr, with a median life span of 13 yr. Cataracts and tumors have infrequently been noted, but many changes associated with normal aging in adults, such as presbyopia, arcus senilis, osteoarthritis, senile personality changes, or Alzheimer disease, are not found.
No specific treatment for this condition exists. There is interpatient variation in weight gain, but the projected weight gain over time in individual patients is constant and very predictable. Growth hormone has resulted in increased rate of weight gain, but still well below that seen in normal children. Growth hormone therapy is no longer used. A promising therapy appears to be inhibition of farnesyl transferase to prevent the anchoring of progerin to the nuclear membrane. This treatment has normalized morphology in fibroblasts from HGPS patients. An open label clinical trial of farnesyl transferase inhibition using weight gain as the outcome variable has been initiated.
The Progeria Foundation (www.progeriaresearch.org) maintains a progeria registry to help with diagnosis and to clearly define the incidence and molecular basis of the disorder. The Foundation website is an excellent source of current information on HGPS for families of children with the disorder.
Brown WT, Gordon LB, Collis FS. Hutchinson-Gilford progeria syndrome. In GeneReviews at GeneTests: Medical Genetics Information Rescurce (website) http://www.ncbi.nlm.nih.gov/sites/GeneTests/review/disease/progeria?db=genetests&search_param=contains Accessed February 17, 2011
Capell BC, Erdos MR, Madigan JP, et al. Inhibiting farnesylation of progerin prevents the characteristic nuclear blebbing of Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2005;102:12879-12884.
Gordon L, McCarten K, Giobbie-Hurder A, et al. Disease progression in Hutchinson-Gilford progeria syndrome: impact on growth and development. Pediatrics. 2007;120:824-833.
Meridith M, Gordon L, Clauss S, et al. Phenotype and course of Hutchinson-Gilford progeria syndrome. N Engl J Med. 2008;358:592-604.