Chapter 85 The Porphyrias
Porphyrias are metabolic diseases resulting from altered activities of specific enzymes of the heme biosynthetic pathway. These enzymes are most active in bone marrow and liver. Erythropoietic porphyrias, in which overproduction of heme pathway intermediates occurs primarily in bone marrow erythroid cells, usually present at birth or in early childhood with cutaneous photosensitivity, or in the case of congenital erythropoietic porphyria, even in utero as nonimmune hydrops. Most porphyrias are hepatic, with overproduction and initial accumulation of porphyrin precursors or porphyrins occurring 1st in the liver. Regulatory mechanisms for heme biosynthesis in liver are distinct from those in the bone marrow and appear to account for activation of hepatic porphyrias during adult life rather than childhood. Homozygous forms of the hepatic porphyrias may manifest clinically prior to puberty, and asymptomatic heterozygous children may present with nonspecific and unrelated symptoms. Parents often request advice about long-term prognosis and information about management of these disorders and drugs that can be taken safely to treat other common conditions.
The DNA sequences and chromosomal locations are established for the human genes of the enzymes in this pathway, and multiple disease-related mutations have been found for each porphyria. The inherited porphyrias display autosomal dominant or recessive inheritance, and recently an X-linked form of erythropoietic porphyria has been identified. Although initial diagnosis of porphyria by biochemical methods remains essential, it is especially important in children to confirm the diagnosis by demonstrating a specific gene mutation(s).
The Heme Biosynthetic Pathway
Heme is required for a variety of hemoproteins such as hemoglobin, myoglobin, respiratory cytochromes, and cytochrome P450 enzymes (CYPs). It is believed that the 8 enzymes in the pathway for heme biosynthesis are active in all tissues. Hemoglobin synthesis in erythroid precursor cells accounts for about 85% of daily heme synthesis in humans. Hepatocytes account for most of the rest, primarily for synthesis of CYPs, which are especially abundant in the liver endoplasmic reticulum (ER), and turn over more rapidly than many other hemoproteins, such as the mitochondrial respiratory cytochromes. As shown in Figure 85-1, pathway intermediates are the porphyrin precursors δ-aminolevulinic acid (ALA, also known as 5-aminolevulinic acid) and porphobilinogen (PBG), and porphyrins (mostly in their reduced forms, known as porphyrinogens). At least in humans, these intermediates do not accumulate in significant amounts under normal conditions or have important physiologic functions.
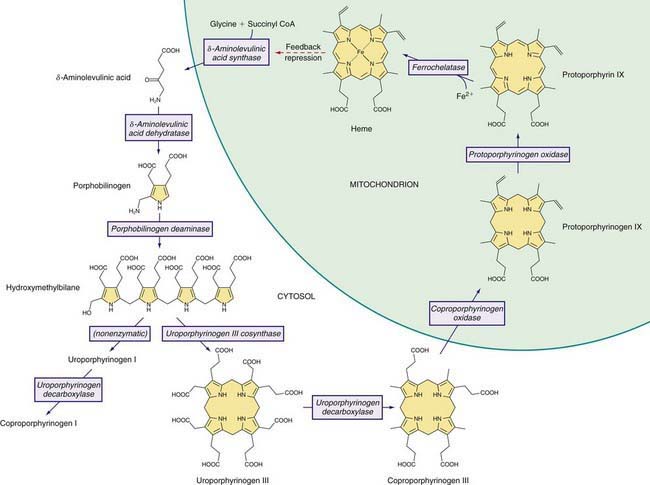
Figure 85-1 Enzymes and intermediates of the heme biosynthetic pathway. The pathway is regulated in the liver by the end product, heme, mainly by feedback repression (dashed arrow).
A deficiency of each enzyme in the pathway is associated with a specific porphyria (Table 85-1). The 1st enzyme, ALA synthase (ALAS), occurs in 2 forms. An erythroid specific form, termed ALAS2, is deficient in X-linked sideroblastic anemia, due to mutations of the ALAS2 gene on chromosome Xp11.2. Gain of function mutations of ALAS2 due to deletions in the last exon have been found in a variant form of erythropoietic protoporphyria (EPP). The housekeeping or ubiquitous form of this enzyme, termed ALAS1, is found in all tissues including liver, and its gene is located on chromosome 3p21.1. Disease-related mutations of ALAS1 have not been described.
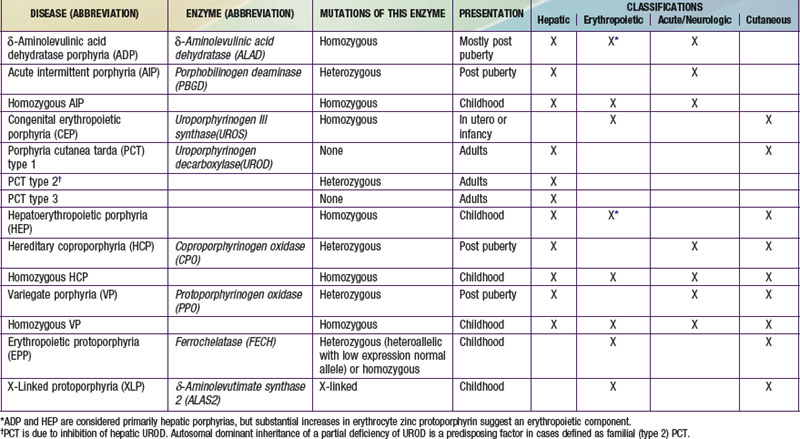
Table 85-1 THE HUMAN PORPHYRIAS: MUTATIONS, TIME OF PRESENTATION, AND TISSUE- AND SYMPTOM-BASED CLASSIFICATIONS
Regulation of heme synthesis differs in the 2 major heme-forming tissues. Liver heme biosynthesis is primary controlled by ALAS1. Synthesis of ALAS1 in liver is regulated by a “free” heme pool (see Fig. 85-1), which can be augmented by newly synthesized heme or by existing heme released from hemoproteins and destined for breakdown to biliverdin by heme oxygenase.
In the erythron, novel regulatory mechanisms allow for the production of the very large amounts of heme needed for hemoglobin synthesis. The response to stimuli for hemoglobin synthesis occurs during cell differentiation, leading to an increase in cell number. Also, unlike the liver, heme has a stimulatory role in hemoglobin formation, and the stimulation of heme synthesis in erythroid cells is accompanied by increases not only in ALAS2, but also by sequential induction of other heme biosynthetic enzymes. Separate erythroid-specific and nonerythroid or “housekeeping” transcripts are known for the 1st 4 enzymes in the pathway. The separate forms of ALAS are encoded by genes on different chromosomes, but for each of the other 3, erythroid and nonerythroid transcripts are transcribed by alternative promoters in the same gene. Heme also regulates the rate of its synthesis in erythroid cells by controlling the transport of iron into reticulocytes.
Intermediates of the heme biosynthetic pathway are efficiently converted to heme and, normally, only small amounts of the intermediates are excreted. Some may undergo chemical modifications before excretion. Whereas the porphyrin precursors ALA and PBG are colorless, nonfluorescent, and largely excreted unchanged in urine, PBG may degrade to colored products such as the brownish pigment called porphobilin or spontaneously polymerize to uroporphyrins. Porphyrins are red in color and display bright red fluorescence when exposed to long wavelength ultraviolet light. Porphyrinogens, which are colorless and nonfluorescent, are the reduced form of porphyrins, and when they accumulate are readily autoxidized to the corresponding porphyrins when outside the cell. Only the type III isomers of uroporphyrinogen and coproporphyrinogen are converted to heme (see Fig. 85-1).
ALA and PBG are excreted in urine. Excretion of porphyrins and porphyrinogens in urine or bile is determined by the number of carboxyl groups. Those with many carboxyl groups, such as uroporphyrin (octacarboxyl porphyrin) and heptacarboxyl porphyrin, are water soluble and readily excreted in urine. Those with fewer carboxyl groups, such as protoporphyrin (dicarboxyl porphyrin), are not water soluble and are excreted in bile and feces. Coproporphyrin (tetracarboxyl porphyrin) is excreted partly in urine and partly in bile. Because coproporphyrin I is more readily excreted in bile than is coproporphyrin III, impaired hepatobiliary function may increase total coproporphyrin excretion and the ratio of these isomers.
Classification and Diagnosis of Porphyrias
Two classification schemes reflect either the underlying pathophysiology or clinical features, and both are useful for diagnosis and treatment (see Table 85-1). In hepatic and erythropoietic porphyrias, the source of excess production of porphyrin precursors and porphyrins is the liver and bone marrow, respectively. Acute porphyrias cause neurologic symptoms that are associated with increases of 1 or both of the porphyrin precursors ALA and PBG. In the cutaneous porphyrias, photosensitivity results from transport of porphyrins in blood from the liver or bone marrow to the skin. Dual porphyria refers to the very rare cases of porphyria with deficiencies of 2 different heme pathway enzymes.
It is notable that acute intermittent porphyria (AIP), porphyria cutanea tarda (PCT), and erythropoietic protoporphyria (EPP), the 3 most common porphyrias, are very different in clinical presentation, precipitating factors, methods of diagnosis, and effective therapy (Table 85-2). Two of the 4 acute porphyrias, hereditary coproporphyria (HCP) and variegate porphyria (VP), can also cause lesions indistinguishable from PCT (see Table 85-1). Congenital erythropoietic porphyria (CEP) causes more severe blistering lesions, often with secondary infection and mutilation. EPP is distinct from the other cutaneous porphyrias in causing nonblistering photosensitivity that occurs acutely after sun exposure. EPP is also the most common porphyria to become manifest before puberty.
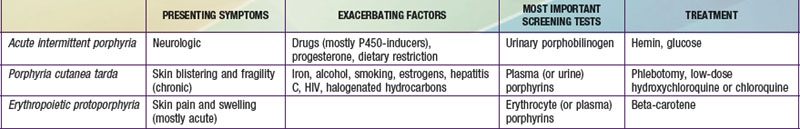
First-Line Laboratory Diagnostic Testing
A few sensitive and specific first-line laboratory tests should be obtained whenever symptoms or signs suggest the diagnosis of porphyria. If a first-line or screening test is significantly abnormal, more comprehensive testing should follow to establish the type of porphyria. Overuse of laboratory tests for screening can lead to unnecessary expense and even delay in diagnosis. In patients who present with a past diagnosis of porphyria, laboratory reports that were the basis for the original diagnosis must be reviewed, and if these were inadequate, further testing considered.
Acute porphyria should be suspected in patients with neurovisceral symptoms such as abdominal pain after puberty, when initial clinical evaluation does not suggest another cause, and urinary porphyrin precursors (ALA and PBG) should be measured. Urinary PBG is virtually always increased during acute attacks of AIP, HCP, and VP, and is not substantially increased in any other medical conditions. Therefore, this measurement is both sensitive and specific. A method for rapid, in-house testing for urinary PBG, such as the Trace PBG kit (Thermo Scientific, 1-800-640-0640), should be available in-house at all major medical facilities. Results from spot (single void) urine specimens are highly informative because very substantial increases are expected during acute attacks of porphyria. A 24 hr collection can unnecessarily delay diagnosis. The same spot urine specimen should be saved for quantitative determination of ALA and PBG, in order to confirm the qualitative PBG result, and also detect patients with ALA dehydratase porphyria. Urinary porphyrins may remain increased longer than porphyrin precursors in HCP and VP. Therefore, it is useful to measure total urinary porphyrins in the same sample, keeping in mind that urinary porphyrin increases are often nonspecific. Measurement of urinary porphyrins alone should be avoided for screening, because these are often increased in many disorders other than porphyrias, such as chronic liver disease, and misdiagnoses of porphyria can result from minimal increases in urinary porphyrins that have no diagnostic significance.
PBG is a colorless pyrrole that forms a violet pigment with Ehrlich reagent (p-dimethylaminobenzaldehyde). Other substances, principally urobilinogen, also react with Ehrlich aldehyde. A reliable quantitative method for both ALA and PBG, which uses small anion and cation exchange columns to separate interfering substances before adding Ehrlich reagent, has been available for many years. ALA is reacted to form a pyrrole, which is then also measured using Ehrlich reagent. The Trace PBG kit to detect increased PBG is based on this method.
Blistering Cutaneous Porphyrias
Blistering skin lesions due to porphyria are virtually always accompanied by increases in total plasma porphyrins. A fluorometric method is preferred, because the porphyrins in plasma in VP are mostly covalently linked to plasma proteins and may be less readily detected by high-pressure liquid chromatography (HPLC). The normal range for plasma porphyrins is somewhat increased in patients with end-stage renal disease. Urinary porphyrins are also increased in these porphyrias, as well as many other medical conditions.
Nonblistering Cutaneous Porphyria
Although a total plasma porphyrin determination will usually detect EPP, an erythrocyte protoporphyrin determination is more sensitive. Increases in erythrocyte protoporphyrin occur in many other conditions. Therefore, the diagnosis of EPP must be confirmed by showing a predominant increase in free protoporphyrin rather than zinc protoporphyrin. Interpretation of laboratory reports can be misleading, because the term “free erythrocyte protoporphyrin” sometimes actually represents zinc protoporphyrin.
Second-Line Testing
More extensive testing is well justified when a first-line test is positive. A substantial increase in PBG may be due to AIP, HCP, or VP. These acute porphyrias can be distinguished by measuring erythrocyte PBGD, urinary porphyrins (using the same spot urine sample), fecal porphyrins, and plasma porphyrins. The various porphyrias that cause blistering skin lesions are differentiated by measuring porphyrins in urine, feces, and plasma. Confirmation at the DNA level is important once the diagnosis is established by biochemical testing.
Testing for Subclinical Porphyria
It is often difficult to diagnose or “rule out” porphyria in patients who had suggestive symptoms months or years in the past, and in relatives of patients with acute porphyrias, because porphyrin precursors and porphyrins may be normal. More extensive testing and consultation with a specialist laboratory and physician may be needed. Before evaluating relatives, the diagnosis of porphyria should be firmly established in an index case, and the laboratory results reviewed to guide the choice of tests for the family members. The index case or another family member with confirmed porphyria should be retested if necessary. Identification of a disease-causing mutation in an index case greatly facilitates detection of additional gene carriers.
δ-Aminolevulinic Acid Dehydratase Porphyria (ADP)
This porphyria is sometimes termed Doss porphyria after the investigator who described the 1st cases. The term plumboporphyria emphasizes the similarity of this condition to lead poisoning, but incorrectly implies that it is due to lead exposure.
Etiology
This porphyria results from a deficiency of δ-aminolevulinic acid dehydratase (ALAD), which is inherited as an autosomal recessive trait. Only 6 cases have been confirmed by mutation analysis. The prevalence of heterozygous ALAD deficiency was estimated to be <1% in Germany and approximately 2% in Sweden.
Pathology and Pathogenesis
ALAD catalyzes the condensation of 2 molecules of ALA to form the pyrrole PBG (see Fig. 85-1). The enzyme is subject to inhibition by a number of exogenous and endogenous chemicals. ALAD is the principal lead-binding protein in erythrocytes, and lead can displace the zinc atoms of the enzyme. Inhibition of erythrocyte ALAD activity is a sensitive index of lead exposure.
To date, all ADP cases inherited a different ALAD mutation from each parent. Eleven abnormal ALAD alleles, most with point mutations, have been identified, some expressing partial activity, such that heme synthesis is partially preserved. The amount of residual enzyme activity may predict the phenotypic severity of this disease. Immunochemical studies in 3 cases demonstrated nonfunctional enzyme protein that cross-reacted with anti-ALAD antibodies. One late-onset case was associated with a myeloproliferative disorder and expansion of an affected clone of erythroid cells.
ADP is often classified as a hepatic porphyria, although the site of overproduction of ALA is not established. A patient with severe, early-onset disease underwent liver transplantation, without significant clinical or biochemical improvement, which might suggest that the excess intermediates did not originate in the liver. Excess urinary coproporphyrin III in ADP might originate from metabolism of ALA to porphyrinogens in a tissue other than the site of ALA overproduction. Administration of large doses of ALA to normal subjects also leads to substantial coproporphyrinuria. Increased erythrocyte protoporphyrin may, as in all other homozygous porphyrias, be explained by accumulation of earlier pathway intermediates in bone marrow erythroid cells during hemoglobin synthesis, followed by their transformation to protoporphyrin after hemoglobin synthesis is complete. The pathogenesis of the neurologic symptoms is poorly understood.
Clinical Manifestations
In most cases, symptoms resemble other acute porphyrias, including acute attacks of abdominal pain and neuropathy. Precipitating factors, such as exposure to harmful drugs, have not been evident in most cases. Four of the 6 reported cases were adolescent males. A Swedish infant had more severe disease, with neurologic impairment and failure to thrive. A 63 yr old man in Belgium developed an acute motor polyneuropathy concurrently with a myeloproliferative disorder.
Laboratory Findings
Urinary ALA, coproporphyrin III, and erythrocyte zinc protoporphyrin are substantially increased. Urinary PBG is normal or slightly increased. Erythrocyte ALAD activity is markedly reduced and both parents should have approximately half-normal activity of this enzyme and normal urinary ALA.
Diagnosis and Differential Diagnosis
The 3 other acute porphyrias are characterized by substantial increases in both ALA and PBG. In contrast, ALA but not PBG is substantially increased in ADP. A marked deficiency of erythrocyte ALAD and half-normal activity in the parents support the diagnosis. Other causes of ALAD deficiency, such as lead poisoning, must be excluded. Succinylacetone accumulates in hereditary tyrosinemia type 1 and is structurally similar to ALA, inhibits ALAD, and can cause increased urinary excretion of ALA and clinical manifestations that resemble acute porphyria. Idiopathic acquired ALAD deficiency has been reported. Unlike lead poisoning, the deficient ALAD activity is not restored by the in vitro addition of sulfhydryl reagents such as dithiothreitol. Even if no other cause of ALAD deficiency is found, it is essential to confirm the diagnosis of ADP by molecular studies.
Treatment
Treatment experience is limited but is similar to other acute porphyrias. Glucose seems not very effective but may be tried for mild symptoms. Hemin therapy was apparently effective for acute attacks in adolescent male cases, and weekly infusions prevented attacks in 1 of these cases. Hemin was not effective either biochemically or clinically in the Swedish child with severe disease, and produced a biochemical response but no clinical improvement in the Belgian man with a late-onset form, who had a peripheral neuropathy but no acute attacks. Hemin is also effective in treating porphyria-like symptoms associated with hereditary tyrosinemia, and can significantly reduce urinary ALA and coproporphyrin in lead poisoning. Avoidance of drugs that are harmful in other acute porphyrias is advisable. Liver transplantation was not effective in the child with severe disease.
Acute Intermittent Porphyria (AIP)
This disorder is also termed pyrroloporphyria, Swedish porphyria, and intermittent acute porphyria and is the most common type of acute porphyria in most countries.
Etiology
AIP results from the deficient activity of the housekeeping form of PBG deaminase (PBGD). This enzyme is also known as hydroxymethylbilane (HMB) synthase; the prior term uroporphyrinogen I synthase is obsolete. PBGD catalyzes the deamination and head-to-tail condensation of 4 PBG molecules to form the linear tetrapyrrole, HMB (also known as preuroporphyrinogen; see Fig. 85-1). A unique dipyrromethane cofactor binds the pyrrole intermediates at the catalytic site until 6 pyrroles (including the dipyrrole cofactor) are assembled in a linear fashion, after which the tetrapyrrole HMB is released. The apo-deaminase generates the dipyrrole cofactor to form the holo-deaminase, and this occurs more readily from HMB than from PBG. Indeed, high concentrations of PBG may inhibit formation of the holo-deaminase. The product HMB can cyclize nonenzymatically to form nonphysiologic uroporphyrinogen I, but in the presence of the next enzyme in the pathway is more rapidly cyclized to form uroporphyrinogen III.
Erythroid and housekeeping forms of the enzyme are encoded by a single gene on human chromosome 11 (11q24.1→q24.2), which contains 15 exons. The 2 isoenzymes are both monomeric proteins and differ only slightly in molecular weight (approximately 40 and 42 kd, respectively), and result from alternative splicing of 2 distinct mRNA transcripts arising from 2 promoters. The housekeeping promoter functions in all cell types, including erythroid cells.
The pattern of inheritance of AIP is autosomal dominant, with very rare homozygous cases that present in childhood. More than 300 PBGD mutations, including missense, nonsense, and splicing mutations and insertions and deletions have been identified in AIP, and in many population groups, including blacks. Most mutations are found in only 1 or a few families. But due to founder effects, some are more common in certain geographic areas such as northern Sweden (W198X), Holland (R116W), Argentina (G116R), Nova Scotia (R173W), and Switzerland (W283X). De novo mutations may be found in about 3% of cases. Chester porphyria was initially described as a variant form of acute porphyria in a large English family but was found to be due to a PBGD mutation. The nature of the PBGD mutation does not account for the severity of the clinical presentation, which varies markedly within families.
Most mutations lead to approximately half-normal activity of the housekeeping and erythroid isozymes and half-normal amounts of their respective enzyme proteins in all tissues of heterozygotes. In approximately 5% of unrelated AIP patients, the housekeeping isozyme is deficient, but the erythroid-specific isozyme is normal. Mutations causing this variant are usually found within exon 1 or its 5′ splice donor site or initiation of translation codon. Immunochemical methods can distinguish mutations that are CRIM-positive (i.e., having excess cross-reactive immunologic material [CRIM] relative to the mutant enzyme activity), whereas CRIM-negative mutations either do not synthesize a mutant enzyme protein, or the protein is not stable and not immunologically detectable using anti-PBGD antibodies. A child with homozygous AIP was found to have inherited a different CRIM-positive mutation from each parent.
Pathology and Pathogenesis
Induction of the rate-limiting hepatic enzyme ALAS1 is thought to underlie acute exacerbations of this and the other acute porphyrias. AIP remains latent (or asymptomatic) in the great majority of those who are heterozygous carriers of PBGD mutations, and this is almost always the case before puberty. In those with no history of acute symptoms, porphyrin precursor excretion is usually normal, suggesting that half-normal hepatic PBGD activity is sufficient and hepatic ALAS1 activity is not increased. Many nongenetic factors that lead to clinical expression of AIP, including certain drugs and steroid hormones, have the capacity to induce hepatic ALAS1 and CYPs. Under conditions in which heme synthesis is increased in the liver, half-normal PBGD activity may become limiting and ALA, PBG, and other heme pathway intermediates may accumulate. In addition, heme synthesis becomes impaired and heme-mediated repression of hepatic ALAS1 is less effective.
It is not proven, however, that hepatic PBGD remains constant at about 50% of normal activity during exacerbations and remission of AIP, as in erythrocytes. An early report suggested that the enzyme activity is considerably less than half-normal in the liver during an acute attack. Hepatic PBGD activity might be reduced further once AIP becomes activated if, as suggested, excess PBG interferes with assembly of the dipyrromethane cofactor for this enzyme. It also seems likely that currently unknown genetic factors play a contributing role in, for example, patients who continue to have attacks even when known precipitants are avoided.
The fact that AIP is almost always latent before puberty suggests that endocrine factors, and especially adult levels of steroid hormones, are important for clinical expression. Symptoms are more common in women suggesting a role for female hormones. Premenstrual attacks are probably due to endogenous progesterone. Acute porphyrias are sometimes exacerbated by exogenous steroids, including oral contraceptive preparations containing progestins. Surprisingly, pregnancy is usually well tolerated, suggesting that beneficial metabolic changes may ameliorate the effects of high levels of progesterone.
Drugs that are unsafe in acute porphyrias (Table 85-3) include those having the capacity to induce hepatic ALAS1, which is closely associated with induction of CYPs. Some chemicals (e.g., griseofulvin) can increase heme turnover by promoting the destruction of specific CYPs to form an inhibitor (e.g., N-methyl protoporphyrin) of ferrochelatase (FECH, the final enzyme in the pathway). Sulfonamide antibiotics are harmful but apparently not inducers of hepatic heme synthesis. Ethanol and other alcohols are inducers of ALAS1 and some CYPs.
Table 85-3 DRUGS REGARDED AS UNSAFE AND SAFE IN ACUTE PORPHYRIAS
| UNSAFE | SAFE |
|---|---|
| Barbiturates | Narcotic analgesics |
| Sulfonamide antibiotics* | Aspirin |
| Meprobamate* (also mebutamate,* tybutamate*) | Acetaminophen |
| Carisoprodol* | Phenothiazines |
| Glutethimide* | Penicillin and derivatives |
| Methyprylon | Streptomycin |
| Ethchlorvynol* | Glucocorticoids |
| Mephenytoin | Bromides |
| Phenytoin* | Insulin |
| Succinimides | Atropine |
| Carbamazepine* | Cimetidine |
| Clonazepam | Ranitidine† |
| Primidone* | Acetaminophen (paracetamol) |
| Valproic acid* | Acetazolamide |
| Pyrazolones (aminopyrine, antipyrine) | Allopurinol |
| Griseofulvin* | Amiloride |
| Ergots | Bethanidine |
| Metoclopramide* | Bumetanide |
| Rifampin* | Cimetidine |
| Pyrazinamide* | Coumarins |
| Diclofenac* | Fluoxetine |
| Progesterone and synthetic progestins* | Gabapentin |
| Danazol* | Gentamicin |
| Alcohol | Guanethidine |
| ACE inhibitors (especially enalapril) | Ofloxacin |
| Calcium channel blockers (especially nifedipine) | Propranolol |
| Ketoconazole | Succinylcholine |
| Rifampin | Tetracycline |
This partial listing does not include all available information about drug safety in acute porphyrias. Other sources should be consulted for drugs not listed here.
* Porphyria is listed as a contraindication, warning, precaution, or adverse effect in U.S. labeling for these drugs. Estrogens are also listed as harmful in porphyria, but have been implicated as harmful in acute porphyrias mostly based only on experience with estrogen-progestin combinations. While estrogens can exacerbate PCT, there is little evidence they are harmful in the acute porphyrias.
† Porphyria is listed as a precaution in U.S. labeling for this drug. However, this drug is regarded as safe by other sources.
Nutritional factors, principally reduced intake of calories and carbohydrates, as may occur with illness or attempts to lose weight, can increase porphyrin precursor excretion and induce attacks of porphyria. Increased carbohydrate intake may ameliorate attacks. Hepatic ALAS1 is modulated by the peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α), which is an important link between nutritional status and exacerbations of acute porphyria.
Other factors have been implicated. Chemicals in cigarette smoke, such as polycyclic aromatic hydrocarbons, can induce hepatic CYPs and heme synthesis. A survey of AIP patients found an association between smoking and repeated porphyric attacks. Attacks may result from metabolic stress and impaired nutrition associated with major illness, infection, or surgery.
The additive effect of multiple predisposing factors, including drugs, endogenous hormones, nutritional factors, and smoking, is suggested by clinical observations. Exposure to drugs and other precipitating factors is less likely to cause an attack in patients who have had no recent symptoms than in those with recent and frequent porphyric symptoms.
Neurologic Mechanisms
The mechanism of neural damage in acute porphyrias is poorly understood. Vasospasm resulting from decreased nitrous oxide production by nitrous oxide synthase (a hemoprotein) has been suggested to cause cerebral manifestations in AIP. The most favored hypothesis at present is that 1 or more heme precursors, or perhaps a derivative, are neurotoxic. Increased ALA in AIP, HCP, VP, ADP, plumbism, and hereditary tyrosinemia type 1, which have similar neurologic manifestations, suggests that this substance or a derivative may be neuropathic. Porphyrins derived from ALA after its uptake into cells may have toxic potential. ALA can also interact with γ-aminobutyric acid (GABA) receptors. Several reports that severe AIP improves markedly after allogeneic liver transplantation supports the hypothesis that heme precursors from the liver cause the neurologic manifestations.
Epidemiology
AIP occurs in all races and is the most common acute porphyria, with a roughly estimated prevalence in most countries of about 5/100,000. In Sweden, prevalence was estimated to be 7.7/100,000, including latent cases with normal porphyrin precursors. A much higher prevalence of 60-100/100,000 in northern Sweden is due to a common mutation and a founder effect. The combined prevalence of AIP and VP in Finland is approximately 3.4/100,000. A survey of chronic psychiatric patients in the USA using an erythrocyte PBGD determination found a high prevalence (210/100,000) of PBGD deficiency, but a study in Mexico found a similar prevalence in psychiatric patients and controls. Population screening by erythrocyte PBGD activity or DNA analysis revealed a prevalence of 200 heterozygotes per 100,000 in Finland, and 1 in about 1,675 (60/100,000) in France. Therefore, carriers of PBGD mutations that can cause AIP may be common.
Clinical Manifestations
Neurovisceral manifestations of acute porphyrias may appear any time after puberty, but rarely before. Very rare cases of homozygous AIP develop severe neurologic manifestations early in childhood.
In affected heterozygotes, acute attacks are characterized by a constellation of nonspecific symptoms, which may become severe and life-threatening. Abdominal pain occurs in 85-95% of cases, is usually severe, steady, and poorly localized, but sometimes cramping, and accompanied by signs of ileus, including abdominal distention and decreased bowel sounds. Nausea, vomiting, and constipation are common, and increased bowel sounds and diarrhea may occur. Bladder dysfunction may cause hesitancy and dysuria. Tachycardia, the most common physical sign, occurs in up to 80% of attacks. This is often accompanied by hypertension, restlessness, coarse or fine tremors, and excess sweating, which are attributed to sympathetic overactivity and increased catecholamines. Other common manifestations include mental symptoms; pain in the extremities, head, neck, or chest; muscle weakness; and sensory loss. Because all these manifestations are neurologic rather than inflammatory, there is little or no abdominal tenderness, fever, or leukocytosis.
Porphyric neuropathy is primarily motor and appears to result from axonal degeneration rather than demyelinization. Sensory involvement is indicated by pain in the extremities, which may be described as muscle or bone pain, and by numbness, paresthesias, and dysesthesias. Paresis may occur early in an attack, but is more often a late manifestation in an attack that is not recognized and adequately treated. Rarely, severe neuropathy develops when there is little or no abdominal pain. Motor weakness most commonly begins in the proximal muscles of the upper extremities and then progresses to the lower extremities and the periphery. It is usually symmetric, but occasionally asymmetric or focal. Initially, tendon reflexes may be little affected or hyperactive and become decreased or absent. Cranial nerves, most commonly X and VII, may be affected, and blindness from involvement of the optic nerves or occipital lobes has been reported. More common central nervous system manifestations include seizures, anxiety, insomnia, depression, disorientation, hallucinations, and paranoia. Seizures may result from hyponatremia, porphyria itself, or an unrelated cause. Chronic depression and other mental symptoms occur in some patients, but attribution to porphyria is often difficult.
Hyponatremia is common during acute attacks. Inappropriate antidiuretic hormone (ADH) secretion is often the most likely mechanism, but salt depletion from excess renal sodium loss, gastrointestinal loss, and poor intake have been suggested as causes of hyponatremia in some patients. Unexplained reductions in total blood and red blood cell volumes are sometimes found, and increased ADH secretion might then be an appropriate physiologic response. Other electrolyte abnormalities may include hypomagnesemia and hypercalcemia.
The attack usually resolves quite rapidly, unless treatment is delayed. Abdominal pain may resolve within a few hours and paresis within a few days. Even severe motor neuropathy can improve over months or several years, but may leave some residual weakness. Progression of neuropathy to respiratory and bulbar paralysis and death is uncommon with appropriate treatment and removal of harmful drugs. Sudden death may result from cardiac arrhythmia.
Laboratory Findings
Levels of ALA and PBG are substantially increased during acute attacks and these may decrease after an attack but usually remain increased unless the disease becomes asymptomatic for a prolonged period. A population-based study in Sweden indicated that symptoms suggestive of porphyria may occur in heterozygotes during childhood, in contrast to adults, even when urinary porphyria precursors are not elevated. This study lacked a comparison with the frequency of such nonspecific symptoms in a control group of children.
Porphyrins are also markedly increased, which accounts for reddish urine in AIP. These are predominantly uroporphyrins, which can form nonenzymatically from PBG. But because the increased urinary porphyrins in AIP are predominantly isomer III, their formation is likely to be largely enzymatic, which might occur if excess ALA produced in the liver enters cells in other tissues and is then converted to porphyrins via the heme biosynthetic pathway. Porphobilin, a degradation product of PBG, and dipyrrylmethenes appear to account for brownish urinary discoloration. Total fecal porphyrins and plasma porphyrins are normal or slightly increased in AIP. Erythrocyte protoporphyrin may be somewhat increased in patients with manifest AIP.
Erythrocyte PBGD activity is approximately half-normal in most patients (70-80%) with AIP. The normal range is wide and overlaps with the range for AIP heterozygotes. As noted, some PBGD gene mutations cause the enzyme to be deficient only in nonerythroid tissues. PBGD activity is also highly dependent on erythrocyte age, and an increase in erythropoiesis due to concurrent illness in an AIP patient may raise the activity into the normal range.
Diagnosis and Differential Diagnosis
An increased urinary PBG establishes that a patient has 1 of the 3 most common acute porphyrias (see Table 85-2). Measuring PBG in serum is preferred when there is coexistent severe renal disease but is less sensitive when renal function is normal. Measurement of urinary ALA is less sensitive than PBG and also less specific but will detect ADP, the fourth type of acute porphyria. Erythrocyte PBGD activity is decreased in most AIP patients and helps confirm the diagnosis in a patient with high PBG. A normal enzyme activity in erythrocytes does not exclude AIP.
Knowledge of the PBGD mutation in a family enables reliable identification of other gene carriers. PBGD deficiency can be documented in a fetus by measuring the enzyme activity in amniotic fluid cells, or more reliably by finding a PBGD mutation in these cells.
Complications
AIP and other acute porphyrias are commonly associated with mild abnormalities in liver function tests. The risk of more advanced liver disease and hepatocellular carcinoma is also increased during adult life, perhaps 60- to 70-fold, even in asymptomatic individuals who have increased porphyrins or porphyrin precursors. Few patients who developed this neoplasm had increases in serum α-fetoprotein. Patients with acute porphyrias, especially >50 yr old must be screened at least yearly by ultrasound or an alternative imaging method.
The risk of chronic hypertension and impaired renal function, most often with evidence of interstitial nephritis, is increased in AIP. A nephrotoxic effect of ALA may contribute. This may progress to severe renal failure and require renal transplantation.
Increased serum thyroxin levels due to increased thyroxin-binding globulin occur in some AIP patients. Hypercholesterolemia and elevated low-density lipoprotein cholesterol appear to be less common in this disorder than previously thought.
Treatment
Hemin
Intravenous hemin, combined with symptomatic and supportive measures, is the treatment of choice for most acute attacks of porphyria. There is a favorable biochemical and clinical response to early treatment with hemin, and less response if treatment is delayed. It is no longer recommended that therapy with hemin for a severe attack be started only after an unsuccessful trial of intravenous glucose for several days. Mild attacks, without severe manifestations such as paresis and hyponatremia, may be treated initially with intravenous glucose. After intravenous administration, hemin binds to hemopexin and albumin in plasma and is taken up primarily in hepatocytes. Hemin then enters and augments the regulatory heme pool in hepatocytes, represses the synthesis of hepatic ALAS1, and dramatically reduces porphyrin precursor overproduction.
Hemin* is available for intravenous administration in the United States as a lyophilized hematin preparation (Panhematin, Lundbeck). Degradation products begin to form as soon as the lyophilized product is reconstituted with sterile water, and these are responsible for phlebitis at the site of infusion and a transient anticoagulant effect. Loss of venous access due to phlebitis is common after repeated administration. Stabilization of lyophilized hematin by reconstitution with 30% human albumin can prevent these adverse effects, and is recommended, especially if a peripheral vein is used for the infusion. Uncommon side effects of hemin include fever, aching, malaise, hemolysis, anaphylaxis, and circulatory collapse. Heme arginate, a more stable hemin preparation, is available in Europe and South Africa.
Hemin treatment should be instituted only after a diagnosis of acute porphyria has been initially confirmed by a marked increase in urinary PBG (determined most rapidly using a kit). When prior documentation of the diagnosis is available for review, it is not essential to confirm an increase in PBG with every recurrent attack, if other causes of the symptoms are excluded clinically. The standard regimen of hemin for treatment of acute porphyric attacks is 3-4 mg/kg daily for 4 days. Lower doses have less effect on porphyrin precursor excretion and probably less clinical benefit.
General and Supportive Measures
Drugs that may exacerbate porphyrias (see Table 85-3) should be discontinued whenever possible, and other precipitating factors identified. Hospitalization is warranted, except for mild attacks, for treatment of severe pain, nausea, and vomiting; for administration of hemin and fluids; and for monitoring vital capacity, nutritional status, neurologic function, and electrolytes. Pain usually requires a narcotic analgesic; there is low risk for addiction after recovery from the acute attack. Ondansetron or a phenothiazine such as chlorpromazine is needed for nausea, vomiting, anxiety, and restlessness. Chloral hydrate or low doses of short-acting benzodiazepines can be given for restlessness or insomnia. β-Adrenergic blocking agents may be useful during acute attacks to control tachycardia and hypertension, but may be hazardous in patients with hypovolemia and incipient cardiac failure.
Carbohydrate Loading
The effects of carbohydrates on repressing hepatic ALAS1 and reducing porphyrin precursor excretion are weak compared to those of hemin. Therefore, only mild attacks (mild pain, no paresis or hyponatremia) are treated with carbohydrate loading. Glucose polymer solutions by mouth are sometimes tolerated. At least 300 g of intravenous glucose, usually given as a 10% solution, has been recommended for adults hospitalized with attacks of porphyria. Amounts up to 500 g daily may be more effective, but large volumes may favor development of hyponatremia.
Other Therapies
Liver transplantation was effective in several patients with severe AIP. Further evidence of efficacy is needed, however, before this can be recommended. Cimetidine, a well-known inhibitor of hepatic CYPs, can prevent experimental forms of porphyria induced by chemical agents that undergo activation by these enzymes, but these models are not highly relevant to human AIP. The drug’s use is based on uncontrolled observations.
Seizures and Other Complications
Seizures due to hyponatremia or other electrolyte imbalances may not require prolonged treatment with anticonvulsant drugs, most of which have at least some potential for exacerbating acute porphyrias. Bromides, gabapentin, and probably vigabatrin are safe. Clonazepam may be less harmful than phenytoin or barbiturates. Control of hypertension may help prevent chronic renal impairment, which can progress and require renal transplantation.
Safe and Unsafe Drugs
Patients often do well with avoidance of harmful drugs. Some drugs known or strongly suspected to be harmful or safe in the acute porphyrias are listed in Table 85-3. Updated and more extensive listings are available on an interactive website of the European Porphyria Initiative (www.porphyria-europe.com) and from the American Porphyria Foundation (www.porphyriafoundation.com). Information regarding safety is lacking for many drugs, especially for those recently introduced, and sometimes opinions are conflicting.
Exogenous progestins, usually in combination with estrogens, can induce attacks of porphyria. Estrogens are seldom reported to be harmful when given alone or in animal and hepatocyte culture systems. Synthetic steroids with an ethynyl substituent can cause a mechanism-based destruction of hepatic CYPs and should probably be avoided in patients with acute porphyria. Danazol is especially contraindicated.
Other Conditions
Major surgery can be carried out safely in patients with acute porphyria, especially if barbiturates are avoided. Halothane has been recommended as an inhalation agent and propofol and midazolam as intravenous induction agents.
Pregnancy is usually well tolerated, which is surprising, because levels of progesterone, a potent inducer of hepatic ALAS1, are considerably increased during pregnancy. Some women do experience continuing attacks during pregnancy. These sometimes result from reduced caloric intake or metoclopramide, a contraindicated drug sometimes used to treat hyperemesis gravidarum.
Diabetes mellitus and other endocrine conditions are not known to precipitate attacks of porphyria. In fact, the onset of diabetes mellitus and resulting high circulating glucose levels may decrease the frequency of attacks and lower porphyrin precursor levels in AIP.
Prognosis
The outlook for patients with acute porphyrias has improved markedly in the past several decades. In Finland, for example, 74% of patients with AIP or VP reported that they led normal lives, and less than 30% had recurrent attacks during several years of follow-up. In those presenting with acute symptoms, recurrent attacks were most likely within the next 1-3 yr. Moreover, only 6% of gene carriers who had never had attacks developed symptoms. The improved outlook may result from earlier detection, better treatment of acute attacks, and replacement of harmful drugs such as barbiturates and sulfonamides with safer drugs. A smaller number of patients continue to have recurrent attacks, chronic pain, and other symptoms even after avoiding known exacerbating factors.
Prevention
For prevention of attacks, it is important to identify multiple inciting factors and remove as many as possible. Drugs for concurrent medical conditions should be reviewed. Because dietary factors are often inapparent, consultation with a dietitian may be useful. A well-balanced diet that is somewhat high in carbohydrate (60-70% of total calories) and sufficient to maintain weight is recommended. There is little evidence that additional dietary carbohydrate helps further in preventing attacks, and it may lead to weight gain. Patients who wish to lose excess weight should do so gradually and when they are clinically stable. Rapid weight loss after bariatric surgery may exacerbate acute porphyrias. Iron deficiency, which can be detected by a low serum ferritin, should be corrected.
GnRH analogs, which reversibly suppress ovulation, can be dramatically effective for preventing frequently recurring luteal phase attacks, but baseline and continuing gynecologic evaluation and bone density measurements are important, and transdermal estrogen or a biphosphonate may be added to prevent bone loss. Hemin administered once or twice weekly can prevent frequent, noncyclic attacks of porphyria in some patients.
Genetic Counseling
Children with a family history of porphyria are often seen by pediatricians for evaluation and counseling. Information and laboratory results from a relative with proven porphyria must be reviewed in order to guide testing of the child, which is different depending on the type of acute porphyria. A mutation identified in the index case can be sought in the child. If the child is found to have inherited the mutation, counseling to avoid potentially harmful drugs is appropriate. Counseling should also emphasize that the great majority of those who inherit a PBGD mutation never develop symptoms, and the prognosis of those who do is favorable. Therefore, a normal, healthy life is expected, especially with avoidance of harmful drugs and other factors and prompt recognition and treatment of symptoms should they occur. Given the favorable outlook for most mutation carriers, even during pregnancy, having children is not precluded, and prenatal diagnosis of acute porphyrias is less important than it is for many other inherited diseases.
Congenital Erythropoietic Porphyria (CEP)
Also termed Günther disease, this rare disease usually presents with photosensitivity shortly after birth or in utero as nonimmune hydrops.
Etiology
CEP is an autosomal recessive disease due to a marked deficiency of uroporphyrinogen III synthase (UROS). Many UROS mutations have been identified among CEP families. Later-onset disease in adults is likely to be associated with myeloproliferative disorders and expansion of a clone of erythroblasts that carry a UROS mutation.
Pathology and Pathogenesis
UROS, which is markedly deficient in CEP, catalyzes inversion of pyrrole ring D of HMB (the pyrrole ring shown on the right end of the molecule in Fig. 85-1) and rapid cyclization of the linear tetrapyrrole to form uroporphyrinogen III. This enzyme is also termed uroporphyrinogen III cosynthase. The human enzyme is a monomer. The gene for the enzyme is found on chromosome 10q25.3→q26.3, and contains 10 exons. Erythroid and housekeeping transcripts are generated by alternative promoters but encode the same enzyme.
In CEP, HMB accumulates in erythroid cells during hemoglobin synthesis and cyclizes nonenzymatically to form uroporphyrinogen I, which is auto-oxidized to uroporphyrin I. Some of the uroporphyrinogen I that accumulates is metabolized to coproporphyrinogen I, which accumulates because it is not a substrate for coproporphyrinogen oxidase. Thus, both uroporphyrin I and coproporphyrin I accumulate in the bone marrow and are then found in circulating erythrocytes, plasma, urine, and feces.
A variety of UROS mutations have been identified in CEP, including missense and nonsense mutations, large and small deletions and insertions, splicing defects, and intronic branch point mutations. At least 4 mutations have been identified in the erythroid-specific promoter. Many patients inherited a different mutation from each parent, and most mutations have been detected in only 1 or a few families. An exception is a common mutation, C73R, which is at a mutational hotspot and was found in ≈33% of alleles. One child with CEP had a GATA1 mutation, with no UROS mutation.
Genotype-phenotype correlations have been based on the in vitro expression of various CEP mutations and the severity of associated phenotypic manifestations. The C73R allele, which is associated with a severe phenotype in homozygotes or in patients heteroallelic for C73R and another mutation expressing little residual activity, resulted in <1% of normal enzyme activity. Patients with the C73R allele and heteroallelic for other mutations expressing more residual activity have milder disease.
Hemolysis is a common feature of CEP. Excess porphyrins in circulating erythrocytes cause cell damage, perhaps by a phototoxic mechanism, leading to both intravascular hemolysis and increased splenic clearance of erythrocytes. Also important is ineffective erythropoiesis, with intramedullary destruction of porphyrin-laden erythroid cells and breakdown of heme. Expansion of the bone marrow due to erythroid hyperplasia may contribute to bone loss. Nutrient deficiencies sometimes cause erythroid hypoplasia. Despite the marked deficiency of UROS, heme production in the bone marrow is increased, due to hemolysis and a compensatory increase in hemoglobin production. This occurs, however, at the expense of marked accumulation of HMB, which is converted to porphyrinogens and porphyrins.
Clinical Manifestations
In severe cases, CEP can cause fetal loss, or be recognized in utero as intrauterine hemolytic anemia and nonimmune hydrops fetalis. CEP may be associated with neonatal hyperbilirubinemia, and phototherapy may unintentionally induce severe photosensitivity and scarring.
The most characteristic presentation is reddish urine or pink staining of diapers by urine or meconium shortly after birth (Fig. 85-2). With sun exposure, severe blistering lesions appear on exposed areas of skin on the face and hands, and have been termed hydroa aestivale because they are more severe with greater sunlight exposure during summer (Fig. 85-3). Vesicles and bullae, as well as friability, hypertrichosis, scarring, thickening, and areas of hypopigmentation and hyperpigmentation are very similar to those seen in PCT but usually much more severe. Infection and scarring sometimes cause loss of facial features and fingers and damage to the cornea, ears, and nails. Porphyrins are deposited in dentine and bone in utero. Reddish-brown teeth in normal light, an appearance termed erythrodontia, display reddish fluorescence under long-wave ultraviolet light (Fig. 85-4). An unaffected child born to a mother with CEP may have erythrodontia. Hemolysis and splenomegaly are common in CEP. Bone marrow compensation may be adequate, especially in milder cases. Patients with severe phenotypes, however, are often transfusion-dependent. Splenomegaly may contribute to the anemia and cause leukopenia and thrombocytopenia, which may be complicated by significant bleeding. Neuropathic symptoms are absent, and there is no sensitivity to drugs, hormones, and carbohydrate restriction. The liver may be damaged by iron overload or hepatitis acquired from blood transfusions.
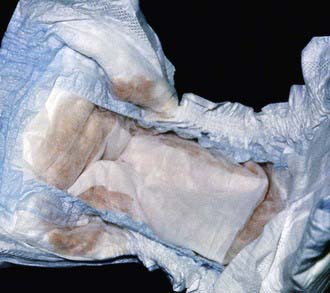
Figure 85-2 Congenital erythropoietic porphyria. The diaper of an affected baby demonstrates the red color of urine.
(From Paller AS, Macini AJ: Hurwitz clinical pediatric dermatology, ed 3, Philadelphia, 2006, Elsevier Saunders, p 517.)
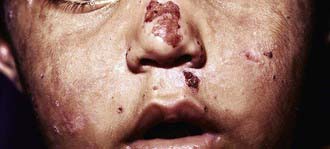
Figure 85-3 Congenital erythropoietic porphyria. Vesicles, bullae, and crusts on sun-exposed areas.
(From Paller AS, Macini AJ: Hurwitz clinical pediatric dermatology, ed 3, Philadelphia, 2006, Elsevier Saunders, p 517.)
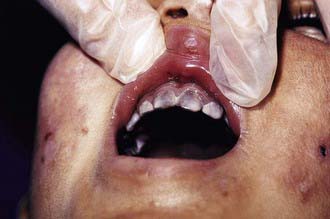
Figure 85-4 Congenital erythropoietic porphyria. Brownish teeth that fluoresce under Wood lamp examination.
(From Paller AS, Macini AJ: Hurwitz clinical pediatric dermatology, ed 3, Philadelphia, 2006, Elsevier Saunders, p 517.)
Milder cases of CEP with onset of symptoms in adult life and without erythrodontia may more closely mimic PCT. These late-onset cases are likely to be associated with myeloproliferative disorders, and expansion of a clone of cells carrying a UROS mutation.
Laboratory Findings
Urinary porphyrin excretion and circulating porphyrin levels in CEP are much higher than in almost all other porphyrias. Urinary porphyrin excretion can be as high as 50-100 mg daily, and consists mostly of uroporphyrin I and coproporphyrin I. ALA and PBG are normal. Fecal porphyrins are markedly increased, with a predominance of coproporphyrin I.
Marked increases in erythrocyte porphyrins in CEP consist mostly of uroporphyrin I and coproporphyrin I. These porphyrins are also increased in bone marrow, spleen, plasma, and, to a lesser extent, liver. The porphyrin pattern in erythrocytes is influenced by rates of erythropoiesis and erythroid maturation. A predominance of protoporphyrin has been noted in some CEP patients, and in 1 such patient, uroporphyrin and coproporphyrin increased when erythropoiesis was stimulated by blood removal.
Diagnosis and Differential Diagnosis
The diagnosis of CEP should be documented by full characterization of porphyrin patterns and identification of the underlying mutations. In later onset cases, an underlying myeloproliferative disorder and a UROS somatic mutation should be suspected and studied in detail.
The clinical picture in hepatoerythropoietic porphyria (HEP) may be very similar, but the porphyrin patterns in urine and feces in HEP resemble PCT. A predominant increase in erythrocyte protoporphyin is unusual in CEP but is characteristic of EPP, HEP, and rare homozygous cases of AIP, HCP, and VP. EPP is also distinguished by normal urinary porphyrins and by increases in erythrocyte free protoporphyrin, whereas the increased protoporphyrin in other conditions is complexed with zinc.
CEP should be suspected as a cause of nonimmune hydrops or hemolytic anemia in utero. With recognition of the disease at this stage, intrauterine transfusion can be considered, and severe, scarring photosensitivity from phototherapy for hyperbilirubinemia avoided. Prenatal diagnosis is feasible by finding red-brown discoloration and increased porphyrins in amniotic fluid, and measuring porphyrins in fetal erythrocytes and plasma. UROS activity can be measured in cultured amniotic fluid cells, or UROS gene mutations identified in chorionic villi or cultured amniotic cells.
Treatment
Protection from sunlight exposure, minimizing skin trauma, and prompt treatment of any cutaneous infections are highly important in managing CEP. Sunscreen lotions and beta-carotene are sometimes beneficial. Transfusions to achieve a level of hemoglobin sufficient to suppress erythropoiesis significantly can be quite effective in reducing porphyrin levels and photosensitivity. Concurrent deferoxamine to reduce iron overload, and hydroxyurea to suppress erythropoiesis further may provide additional benefit. Splenectomy reduces hemolysis and transfusion requirements in some patients. Oral charcoal may increase fecal loss of porphyrins, but may contribute little in more severe cases. Intravenous hemin may be somewhat effective, but has not been extensively studied and seems unlikely to provide long-term benefit.
The most effective treatment is bone marrow or stem cell transplantation in early childhood, which has markedly reduced porphyrin levels and photosensitivity and increased long-term survival.
Porphyria Cutanea Tarda (PCT)
This is the most common and readily treated human porphyria (see Table 85-2). It occurs in mid or late adult life, and is rare in children. Previous terms include symptomatic porphyria, PCT symptomatica, and idiosyncratic porphyria. The underlying cause is a liver-specific, acquired deficiency of uroporphyrinogen decarboxylase (UROD) with contributions by several types of genetic factors. UROD mutations are found in familial PCT. HEP, the homozygous form of familial PCT, usually has a more severe presentation in childhood, resembling CEP clinically.
Etiology
PCT is due to a reduction of hepatic UROD to 20% of normal activity or less. An inhibitor of hepatic UROD has been characterized as uroporphomethene, which is derived from partial oxidation of the enzyme substrate uroporphyrinogen. CYPs such as CYP1A2, as well as iron, are involved in its formation (Fig. 85-5). Although the enzyme is inhibited, the amount of hepatic enzyme protein measured immunochemically remains at its genetically determined level.
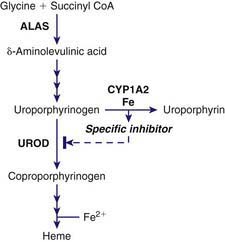
Figure 85-5 Formation of a specific inhibitor of uroporphyrinogen decarboxylase in the liver in porphyria cutanea tarda. ALAS, δ-aminolevulinic acid synthase; CYP1A2, cytochrome P450 1A2; UROD, uroporphyrinogen decarboxylase.
UROD catalyzes the decarboxylation of the 4 acetic acid side chains of uroporphyrinogen (an octacarboxyl porphyrinogen) to form coproporphyrinogen (a tetracarboxyl porphyrinogen) (see Fig. 85-1). The enzyme reaction occurs in a sequential, clockwise fashion, with the intermediate formation of hepta-, hexa-, and pentacarboxyl porphyrinogens. Uroporphyrinogen III, as compared with other uroporphyrinogen isomers, is the preferred substrate. Human UROD is a dimer with the 2 active site clefts juxtaposed. The UROD gene is on chromosome 1p34 and contains 10 exons, with only 1 promoter. Therefore, the gene is transcribed as a single mRNA in all tissues.
The majority of PCT patients (i.e., ≈80%) have no UROD mutations and are said to have sporadic (type 1) disease. Some are heterozygous for UROD mutations and are said to have familial (type 2) PCT. Described mutations include missense, nonsense, and splice site mutations, several small and large deletions, and small insertions, with only a few identified in more than 1 family. A few of these mutations may be located near the active site cleft, but most appear to involve regions with important structural roles. Being heterozygous for a UROD mutation is insufficient to cause PCT unless a UROD inhibitor is also generated. Because penetrance of the genetic trait is low, many patients with familial PCT have no family history of the disease.
Induction of hepatic ALAS1 is not a prominent feature in PCT, although alcohol may increase this enzyme slightly. Iron and estrogens are also not potent inducers of ALAS1 and drugs that are potent inducers of ALAS1 and CYPs are much less commonly implicated in PCT than in acute porphyrias.
Blistering skin lesions result from porphyrins that are released from the liver. Sunlight exposure leads to generation of reactive oxygen species in the skin, complement activation, and lysosomal damage.
Epidemiology
Different prevalences probably relate to geographic variations in susceptibility factors such as hepatitis C and ethanol use. The yearly incidence in the United Kingdom was estimated at 2-5/1,000,000, and the prevalence in the USA and Czechoslovakia was estimated at approximately 1/25,000 and 1/5,000, respectively. The disease was reported to be prevalent in the Bantus of South Africa in association with iron overload. PCT is more common in males, possible due to greater alcohol intake, and in women it is commonly associated with estrogen use.
A massive outbreak of PCT occurred in eastern Turkey in the 1950s. Wheat intended for planting and treated with hexachlorobenzene as a fungicide was consumed by many at a time of food shortage. Cases and small outbreaks of PCT after exposure to other chemicals including di- and trichlorophenols and 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD, dioxin) have been reported. The manifestations improved in most cases when the exposure was stopped. There are reported cases of delayed onset many years after chemical exposure.
Pathology and Pathogenesis
PCT is currently classified into 3 clinically similar types. Generation of a UROD inhibitor in the liver plays an important role in all 3 types. The 80% of patients with type 1 (sporadic) PCT have no UROD mutations, and UROD activity is normal in nonhepatic tissues such as erythrocytes. In familial (type 2) PCT, a heterozygous UROD mutation results in a partial (about 50%) deficiency of UROD in nonhepatic tissues from birth, and the disease becomes active in some heterozygotes when other susceptibility factors are present and a UROD inhibitor is generated. HEP results from inheritance of a UROD mutation from each parent and typically causes severe photosensitivity, resembling CEP, starting in early childhood. Some compound heterozygotes have developed symptoms in childhood more typical of PCT. Type 3 is rare, and describes PCT with normal erythrocyte UROD activity occurring in more than 1 family member. UROD mutations or another genetic basis have not been identified in type 3, and familial occurrence is the only feature that distinguishes it from type 1.
CYPs, especially CYP1a2, can catalyze the oxidation of uroporphyrinogen to uroporphyrin. This uroporphyrinogen oxidase (URO-OX) activity is enhanced by iron, and leads to formation of a UROD inhibitor (see Fig. 85-5). CYP1a2 seems essential for development of uroporphyria in rodents, because experimental uroporphyria does not develop in CYP1a2 knockout mice.
Susceptibility Factors
The following factors are implicated in the development of PCT, and these occur in various combinations in individual patients.
Iron
A normal or increased amount of iron in the liver is essential for developing PCT, and treatment by phlebotomy to reduce hepatic iron leads to remission. Serum ferritin levels are usually in the upper part of the normal range or moderately increased and liver histology commonly shows increased iron staining. Prevalence of the C282Y mutation of the HFE gene, which is the major cause of hemochromatosis in people of northern European ancestry, is increased in both type 1 and type 2 PCT, and 10% of patients are C282Y homozygotes. In southern Europe, the H63D mutation is more commonly associated. PCT may occur with secondary iron overload. Reduced hepatic expression of the hormone hepcidin occurs in hemochromatosis and also in PCT, regardless of HFE genotype, which may explain hepatic siderosis in this condition.
Hepatitis C
This viral infection is highly prevalent in PCT in most geographic locations; in the United States, for example, it is present in 56-74% of cases, which is similar to the rate cited in earlier reports from southern Europe. Prevalence of hepatitis C in PCT is lower in northern Europe (<20%). Steatosis and oxidative stress in hepatitis C may favor iron-mediated generation of reactive oxygen species and a UROD inhibitor. Dysregulation of hepcidin occurs in hepatitis C as well as hemochromatosis and may lead to increased iron absorption.
HIV
Many reports suggest that HIV infection can contribute to the development of PCT, although less commonly than does hepatitis C.
Ethanol
The long-recognized association between alcohol and PCT may be explained by the generation of active oxygen species, which may cause oxidative damage, mitochondrial injury, depletion of reduced glutathione and other antioxidant defenses, increased production of endotoxin, and activation of Kupffer cells.
Smoking and Cytochrome P450 Enzymes
Smoking has not been extensively studied as a susceptibility factor but is commonly associated with alcohol use in PCT. It may act to induce hepatic CYPs and oxidative stress. Hepatic CYPs are thought to be important in oxidizing uroporphyrinogen and generating a UROD inhibitor (see Fig. 85-5). Genetic polymorphisms of CYP1a2 and 1A1 have been implicated in human PCT. The frequency of an inducible CYP1a2 genotype was more common in PCT patients than in controls in several studies.
Antioxidant Status
Ascorbic acid deficiency contributes to uroporphyria in laboratory models and perhaps in human PCT. In 1 series, plasma ascorbate levels were substantially reduced in 84% of patients with PCT. Low levels of serum carotenoids were also described, further suggesting that oxidant stress in hepatocytes is important in PCT.
Clinical Manifestations
Cutaneous Manifestations
PCT is readily recognized by blistering and crusted skin lesions on the backs of the hands, which are the most sun-exposed areas of the body, and somewhat less commonly on the forearms, face, ears, neck, legs, and feet. The fluid-filled vesicles commonly rupture and become crusted or denuded areas, heal slowly, and are subject to infection. The skin on the backs of the hands is characteristically friable, and minor trauma may cause blisters or denudation of skin. Small white plaques, termed milia, may precede or follow vesicle formation. Facial hypertrichosis and hyperpigmentation are also common. Severe scarring and thickening of sun-exposed skin may resemble scleroderma. Skin biopsy findings include subepidermal blistering and deposition of PAS-positive material around blood vessels and fine fibrillar material at the dermoepithelial junction, which may relate to excessive skin fragility. IgG, other immunoglobulins, and complement are also deposited at the dermoepithelial junction and around dermal blood vessels. The skin lesions and histologic changes are not specific for PCT. The same findings occur in VP and HCP, and resemble those of CEP and HEP but are usually less severe. PCT usually develops in mid or late adult life. Earlier onset may be seen in those with UROD or HFE mutations. Childhood onset is rare, and suggests heterozygosity or even compound heterozygosity for UROD mutations.
Liver Abnormalities
PCT is almost always associated with nonspecific liver abnormalities, especially increased serum transaminases and γ-glutamyltranspeptidase, even in the absence of heavy alcohol intake or hepatitis C. Most histologic findings, such as necrosis, inflammation, increased iron, and increased fat, are nonspecific. Specific findings include red fluorescence of liver tissue, and fluorescent, birefringent, needlelike inclusions presumably consisting of porphyrins. Electron microscopy shows these inclusions are in lysosomes, and paracrystalline inclusions are found in mitochondria. Distorted lobular architecture and cirrhosis are more common with long-standing disease.
The risk of developing hepatocellular carcinoma is increased, with reported incidences ranging from 4 to 47% in PCT. These tumors seldom contain large amounts of porphyrins.
Other Findings and Associations
Mild or moderate erythrocytosis in some adult patients is not well understood, but chronic lung disease due to smoking may contribute. An earlier onset of symptoms may be noted in patients with genetic predisposing factors, such as an inherited partial deficiency of UROD or the C282Y/C282Y HFE genotype. Iron overload secondary to conditions such as myelofibrosis and end-stage renal disease may be associated with PCT. The disease can be especially severe in patients with end-stage renal disease, because the lack of urinary excretion leads to much higher concentrations of porphyrins in plasma, and the excess porphyrins are poorly dialyzable. PCT occurs more frequently in patients with systemic lupus erythematosis and other immunologic disorders than would have been expected by chance.
Laboratory Findings
Porphyrin accumulates in the liver mostly as the oxidized porphyrins rather than porphyrinogens in PCT, as indicated by the immediate red fluorescence observed in liver tissue. This develops in weeks or months, and then porphyrins appear in plasma and are transported to the skin, causing photosensitivity. In contrast to the acute hepatic porphyrias, only a very small increase in synthesis of heme pathway intermediates and little or no increase in hepatic ALAS1 are required to account for the excess porphyrins excreted in PCT.
Hepatic UROD deficiency leads to a complex pattern of excess porphyrins, which initially accumulate as porphyrinogens, and then undergo nonenzymatic oxidation to the corresponding porphyrins (uro-, hepta-, hexa-, and pentacarboxyl porphyrins, and isocoproporphyrins). Uroporphyrin and heptacarboxyl porphyrin predominate in urine, with lesser amounts of coproporphyrin and penta- and hexacarboxyl porphyrin. A normally minor pathway is accentuated by UROD deficiency, whereby pentacarboxyl porphyrinogen is oxidized by coproporphyrinogen oxidase (CPO; the next enzyme in the pathway), forming isocoproporphyrinogen, an atypical tetracarboxyl porphyrinogen. Relative to normal values, urinary porphyrins are increased to a greater extent than fecal porphyrins. However, the total amount of porphyrins excreted in feces in PCT exceeds that in urine, and total excretion of type III isomers (including isocoproporphyrins, which are mostly derived from the type III series) exceeds that of type I isomers. Perhaps because uroporphyrinogen III is the preferred substrate for UROD, more uroporphyrinogen I than III accumulates and is excreted in PCT. Hepta- and hexacarboxyl porphyrin are mostly isomer III; and pentacarboxyl porphyrin and coproporphyrin are approximately equal mixtures of isomers I and III.
Diagnosis and Differential Diagnosis
Plasma porphyrins are always increased in clinically manifest PCT, and a total plasma porphyrin determination is most useful for screening. A normal value rules out PCT and other porphyrias that produce blistering skin lesions. If increased, it is useful to determine the plasma fluorescence emission maximum at neutral pH, because a maximum near 619 nm is characteristic of PCT (as well as CEP and HCP) and, most important, excludes VP, which has a distinctly different fluorescence maximum. Increased urinary porphyrins, with a predominance of uroporphyrin and heptacarboxyl porphyrin, is confirmatory. Urine porphyrins are less useful for initial screening because nonspecific increases, especially of coproporphyrin, occur in liver disease and other medical conditions. Urinary ALA may be increased slightly, and PBG is normal.
Familial (type 2) can be distinguished from sporadic (type 1) PCT by finding decreased erythrocyte UROD activity (in type 2), or more reliably by finding a disease-related UROD mutation. Type 3 is distinguished from type 1 only by occurrence of PCT in a relative. Biochemical findings in HEP are similar to those in PCT, but with an additional marked increase in erythrocyte zinc protoporphyrin.
Pseudoporphyria (also known as pseudo-PCT) presents with skin lesions that closely resemble PCT, but without significant increases in plasma porphyrins. A photosensitizing drug such as a nonsteroidal antiinflammatory agent is sometimes implicated. Both PCT and pseudoporphyria may occur in patients with end-stage renal disease.
Complications
Cutaneous blisters may rupture and become infected, sometimes leading to cellulitis. In more severe disease in patients with end-stage renal disease, repeated infections can be mutilating, as in CEP. Pseudoscleroderma, with scarring, contraction, and calcification of skin and subcutaneous tissue, is a rare complication. Other complications include advanced liver disease and hepatocellular carcinoma.
Treatment
Two specific and effective forms of treatment, namely phlebotomy or low-dose hydroxychloroquine, are available. Susceptibility factors should be removed when possible. The diagnosis of PCT must be firmly established, because conditions that produce identical cutaneous lesions do not respond to these treatments. Treatment can usually be started after demonstrating an increase in plasma total porphyrins and excluding VP by analysis of the fluorescence spectrum at neutral pH, while urine and fecal studies are still pending. Use of alcohol, estrogens (in women), and smoking should be stopped, and patients tested for hepatitis C, HIV, and HFE mutations. Some susceptibility factors and the degree of iron overload as assessed by the serum ferritin concentration, influence the choice of treatment.
Phlebotomy is considered standard therapy, and is effective both in children and adults with PCT because it reduces hepatic iron content. Treatment is guided by plasma (or serum) ferritin and porphyrin levels. Hemoglobin or hematocrit levels should be followed to prevent symptomatic anemia. For adults, a unit of blood (≈450 mL) is removed at about 2 wk intervals until a target serum ferritin near the lower limit of normal (≈15 ng/mL) is achieved. A total of 6 to 8 phlebotomies is often sufficient. After this, plasma porphyrin concentrations continue to fall from pretreatment levels (generally 10-25 µg/dL) to below the upper limit of normal (≈1 µg/dL), usually after several more weeks. This is followed by gradual clearing of skin lesions, sometimes including pseudoscleroderma. Liver function abnormalities may improve, and hepatic siderosis, needle-like inclusions, and red fluorescence of liver tissue will disappear. Although remission usually persists even if ferritin levels later return to normal, it is advisable to follow porphyrin levels and reinstitute phlebotomies if these begin to rise. Infusions of deferoxamine, an iron chelator, may be used when phlebotomy is contraindicated.
An alternative when phlebotomy is contraindicated or poorly tolerated is a low-dose regimen of hydroxychloroquine (or chloroquine). Normal doses of these 4-aminoquinoline antimalarials increase plasma and urinary porphyrin levels and increase photosensitivity in PCT, reflecting an outpouring of porphyrins from the liver. This is accompanied by acute hepatocellular damage, with fever, malaise, nausea, and increased serum transaminases, but is followed by complete remission of the porphyria. These adverse consequences of normal doses are largely avoided by a low-dose regimen (hydroxychloroquine 100 mg or chloroquine 125 mg, 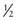 of a normal tablet, twice weekly), which can be continued until plasma or urine porphyrins are normalized. There is at least some risk of retinopathy, which may be lower with hydroxychloroquine. Prospective treatment trials comparing this treatment with phlebotomy are lacking. Low-dose chloroquine was reportedly not effective in patients homozygous for the C282Y mutation in the HFE gene. The mechanism of action of 4-aminoquinolines in PCT is not known but is quite specific, because these drugs are not useful in other porphyrias.
of a normal tablet, twice weekly), which can be continued until plasma or urine porphyrins are normalized. There is at least some risk of retinopathy, which may be lower with hydroxychloroquine. Prospective treatment trials comparing this treatment with phlebotomy are lacking. Low-dose chloroquine was reportedly not effective in patients homozygous for the C282Y mutation in the HFE gene. The mechanism of action of 4-aminoquinolines in PCT is not known but is quite specific, because these drugs are not useful in other porphyrias.
In patients with PCT and hepatitis C, PCT should be treated 1st because this condition is more symptomatic and can be treated more quickly and effectively. Treatment of PCT by phlebotomy may not be possible once interferon-ribavirin treatment is complicated by anemia. Moreover, treatment of hepatitis C may be more effective after iron reduction.
PCT in patients with end-stage renal disease is often more severe and difficult to treat. However, erythropoietin administration can correct anemia, mobilize iron, and support phlebotomy in many cases. Improvement after renal transplantation may be due in part to resumption of endogenous erythropoietic production.
Liver imaging and a serum α-fetoprotein determination may be advisable in all PCT patients, perhaps at 6-12 mo intervals for early detection of hepatocellular carcinoma. Finding low erythrocyte UROD activity or a UROD mutation identifies those with an underlying genetic predisposition, which does not alter treatment but is useful for genetic counseling (see later).
Prognosis
PCT is the most readily treated form of porphyria, and complete remission is expected with treatment either by phlebotomy or low-dose hydroxychloroquine. There is little information on rates of recurrence and long-term outlook. Risk for hepatocellular carcinoma is increased, and some susceptibility factors such as hepatitis C can lead to complications even after PCT is in remission.
Prevention and Genetic Counseling
Patients with PCT may have concerns about risk to other family members. A heritable UROD mutation can usually be detected or excluded by measuring erythrocyte UROD activity, although DNA studies are more sensitive. Relatives of patients with UROD mutations have an increased risk for developing PCT, and may have increased motivation to avoid adverse behaviors such as ethanol and tobacco use and exposures to hepatitis C and HIV. Such counseling would be given to anyone, however. The finding of HFE mutations, and especially C282Y, should prompt screening of relatives, some of whom may be C282Y homozygotes and warrant lifelong monitoring of serum ferritin.
Hepatoerythropoietic Porphyria
Hepatoerythropoietic porphyria (HEP), which is the homozygous form of familial (type 2) PCT, resembles CEP clinically. Excess porphyrins originate mostly from liver, with a pattern consistent with severe UROD deficiency. This rare disorder has no particular racial predominance.
Etiology
HEP is an autosomal recessive disorder, although most patients have inherited a different mutation from unrelated parents. In contrast to most mutations in familial (type 2) PCT, most causing HEP are associated with expression of some residual enzyme activity. At least 1 genotype is associated with the predominant excretion of pentacarboxyl porphyrin.
Pathology and Pathogenesis
Excess porphyrins originate primarily from the liver in HEP, although the substantial increase in erythrocyte zinc protoporphyrin indicates that the heme biosynthetic pathway is also impaired in bone marrow erythroid cells. Apparently, porphyrinogens accumulate in the marrow while hemoglobin synthesis is most active, and are metabolized to protoporphyrin after hemoglobin synthesis is complete. The cutaneous lesions are due to photoactivation of porphyrins in skin, as in other cutaneous porphyrias.
Clinical Manifestations
Like CEP, this disease usually presents with blistering skin lesions, hypertrichosis, scarring, and red urine in infancy or childhood. Sclerodermoid skin changes are sometimes prominent. Unusually mild cases have been described. Concurrent conditions that affect liver function can alter disease severity. For example, the disease became manifest due to hepatitis A in a 2 yr old child, and then improved with recovery of liver function.
Laboratory Findings
Biochemical findings resemble those in PCT with accumulation and excretion of uroporphyrin, heptacarboxyl porphyrin, and isocoproporphyrin. But in addition, erythrocyte zinc protoporphyrin is substantially increased.
Diagnosis and Differential Diagnosis
HEP is distinguished from CEP by increases in both uroporphyrin and heptacarboxyl porphyrin, and isocoproporphyrins. In CEP the excess erythrocyte porphyrins are predominantly uroporphyrin and coproporphyrin rather than protoporphyrin. Blistering skin lesions are unusual in EPP, the excess erythrocyte protoporphyrin in that disease is free and not complexed with zinc, and urinary porphyrins are normal.
Treatment and Prognosis
Avoiding sunlight exposure is most important in managing this disease, as in CEP. Oral charcoal was helpful in a severe case associated with dyserythropoiesis. Phlebotomy has shown little or no benefit. The outlook depends on the severity of the enzyme deficiency and may be favorable if sunlight can be avoided. Retrovirus-mediated gene transfer corrected porphyria in transduced lymphoblastoid cells from HEP patients, suggesting that gene therapy may eventually be developed.
Hereditary Coproporphyria (HCP)
This autosomal dominant hepatic porphyria is due to a deficiency of coproporphyrinogen oxidase (CPO). The disease presents with acute attacks, as in AIP. Cutaneous photosensitivity may occur, but much less commonly than in VP. Rare homozygous cases present in childhood.
Etiology
A partial (50%) deficiency in CPO activity has been found in all cells studied from patients with HCP. A much more profound deficiency is found in homozygous cases. Human CPO is a homodimer composed of 39-kd subunits, and contains no metals or prosthetic groups. The enzyme requires molecular oxygen, and is localized in the mitochondrial intermembrane space. A single active site on the enzyme catalyzes the oxidative decarboxylation of 2 of the 4 proprionic acid groups of coproporphyrinogen III to form the 2 vinyl groups at positions 2 and 4, on rings A and B, respectively, of protoporphyrinogen IX (see Fig. 85-1). Most of the intermediate tricarboxyl porphyrinogen, termed harderoporphyrinogen, is not released before undergoing the 2nd decarboxylation to protoporphyrinogen IX. Coproporphyrinogen I is not a substrate for this enzyme.
The human CPO gene contains 7 exons and is located on chromosome 3q12.1. A single promoter contains elements for both housekeeping and erythroid-specific expression. A variety of CPO gene mutations have been described in HCP, with a predominance of missense mutations and no genotype-phenotype correlations. Harderoporphria, an autosomal recessive biochemical variant form of HCP, is due to CPO mutations that impair substrate binding, leading to premature release of harderoporphyrinogen.
Epidemiology
HCP is less common than AIP and VP, but its prevalence has not been carefully estimated. There is no obvious racial predominance. Homozygous HCP is rare and presents during childhood. Harderoporphyria, a biochemically distinguishable variant of HCP, has been recognized in heteroallelic and homoallelic forms.
Pathology and Pathogenesis
Increased ALA and PBG during acute attacks of HCP may be explained by induction of ALAS1 and by the normally relatively low activity of PBGD in the liver. Hepatic ALAS1 is increased during acute attacks, but is normal when the disease is latent and porphyrin precursor excretion is normal. Because coproporphyrinogen III concentration in the liver is probably less than the Km for CPO, the reaction rate is likely to be determined in part by substrate concentration. The substrate coproporphyrinogen appears to be lost more readily from the liver cell than, for example, uroporphyrinogen, especially when heme synthesis is stimulated. Coproporphyrin and coproporphyrinogen are both transported into bile and excreted in urine, and do not appear to accumulate in the liver in HCP.
Clinical Manifestations
Symptoms are identical to those of AIP except that attacks are generally milder, and cutaneous lesions that resemble those in PCT develop occasionally. Severe motor neuropathy and respiratory paralysis can occur. Like other acute porphyrias, HCP is almost always latent before puberty, and symptoms are most common in adult women. Attacks are precipitated by the same factors that cause attacks in AIP, including fasting, oral contraceptive steroids, and hormone increases during the luteal phase of the menstrual cycle. Concomitant liver diseases may increase porphyrin retention and photosensitivity. The risk of hepatocellular carcinoma is increased, as in other acute porphyrias.
The clinical features of homozygous HCP or harderoporphria, which begin in early childhood, may include jaundice, hemolytic anemia, hepatosplenomegaly, and skin photosensitivity. These symptoms are generally quite distinct from those seen in heterozygotes.
Laboratory Findings
The porphyrin precursors ALA and PBG are increased during acute attacks, but may decrease more rapidly than in AIP. Marked increases in coproporphyrin III in urine and feces are more persistent. In homozygous cases, porphyrin excretion may be more markedly increased and is accompanied by substantial increases in erythrocyte zinc protoporphyrin. Harderoporphyria is characterized by a marked increase in fecal excretion of harderoporphyrin (tricarboxyl porphyrin) as well as coproporphyrin. Plasma porphyrins are usually normal or only slightly increased.
Diagnosis and Differential Diagnosis
The diagnosis of HCP is readily established in patients with clinically manifest disease, although urinary ALA, PBG, and uroporphyrin may revert to normal more quickly than in AIP. Urinary coproporphyrin III is increased. Urinary porphyrins, especially coproporphyrin, can be increased in many medical conditions such as liver disease, and small increases may not be clinically significant and lead to an incorrect diagnosis of HCP. Fecal porphyrins are mostly coproporphyrin (isomer III) in HCP, whereas in VP, coproporphyrin III and protoporphyrin are often increased approximately equally. Plasma porphyrins are usually normal in HCP and increased in VP.
The ratio of fecal coproporphyrin III to coproporphyrin I is especially sensitive for detecting latent heterozygotes (especially adults). Assays for CPO, a mitochondrial enzyme, require cells such as lymphocytes and are not widely available. Identification of a CPO mutation in an index case greatly facilitates screening family members.
Treatment and Prognosis
Acute attacks of HCP are treated as in AIP, which includes intravenous hemin and identifying and avoiding precipitating factors. Cholestyramine may be of some value for photosensitivity occurring with liver dysfunction. Phlebotomy and chloroquine are not effective. GnRH analogues can be effective for prevention of cyclic attacks. The prognosis is generally better than in AIP.
Variegate Porphyria (VP)
This hepatic porphyria is due to a deficiency of protoporphyrinogen oxidase (PPO), which is inherited as an autosomal dominant trait. The disorder is termed variegate because it can present with neurologic or cutaneous manifestations. Other terms have included porphyria variegata, protocoproporphyria, and South African genetic porphyria. Rare cases of homozygous VP are symptomatic in childhood.
Etiology
PPO is approximately half normal in all cells studied in patients with VP. The enzyme is more markedly deficient in rare cases of homozygous VP, with approximately half-normal enzyme activity in parents.
Human PPO is a homodimer that contains FAD and is localized to the cytosolic side of the inner mitochondrial membrane. Membrane-binding domains may be docked onto human FECH, the next enzyme in the pathway, which is embedded in the opposite side of the membrane. PPO catalyzes the oxidation of protoporphyrinogen IX to protoporphyrin IX by the removal of 6 hydrogen atoms (see Fig. 85-1). The enzyme requires molecular oxygen. The substrate is readily oxidized nonenzymatically to protoporphyrin under aerobic conditions, or if exported into the cytosol. PPO is highly specific for protoporphyrinogen IX, and is inhibited by tetrapyrroles such as heme, biliverdin, and bilirubin and by certain herbicides that cause protoporphyrin to accumulate and induce phototoxicity in plants. Inhibition by bilirubin may account for decreased PPO activity in Gilbert disease.
The human PPO gene on chromosome 1q22-q23 consists of 1 noncoding and 12 coding exons. A single PPO transcript is produced in a variety of tissues, but putative transcriptional element binding sequences may allow for erythroid-specific expression. Many PPO mutations have been reported in VP families. A missense mutation, R59W, is prevalent in South Africa. No convincing genotype-phenotype correlations have been identified. Mutations in homozygous cases of VP are more likely to encode enzyme proteins with residual activity.
Epidemiology
VP is less common than AIP in most countries. The R59W mutation is highly prevalent in South African whites (3/1,000). This example of genetic drift or “founder effect” has been traced to a man or his wife who emigrated from Holland to South Africa in 1688. In Finland, the prevalence is 1.3/100,000 and is about as common as AIP.
Pathology and Pathogenesis
Acute attacks develop in a minority (about 25%) of heterozygotes for PPO deficiency, and are often attributable to drugs, steroids, and nutritional factors that play a role in other acute porphyrias. Protoporphyrinogen IX accumulates and undergoes auto-oxidation to protoporphyrin IX. Coproporphyrinogen III may accumulate due to a close functional association between PPO in the inner mitochondrial membrane and CPO in the intermembrane space. Liver porphyrin content is not increased. The increased porphyrin content in plasma consists of porphyrin-peptide conjugates, which may be formed from protoporphyrinogen. Increased ALA and PBG during acute attacks may be explained, as in HCP, by induction of ALAS1 by exacerbating factors, and by the normally relatively low activity of PBGD in liver. Furthermore, PBGD is inhibited by protoporphyrinogen, the substrate for PPO.
Clinical Manifestations
Symptoms develop in some heterozygotes after puberty. Neurovisceral symptoms occurring as acute attacks are identical to AIP but are generally milder and less often fatal. Drugs, steroids, and nutritional alterations such as fasting, which are harmful in AIP, can also induce attacks of VP. Attacks occur equally in males and females, at least in South Africa. Cutaneous fragility, vesicles, bullae, hyperpigmentation, and hypertrichosis of sun-exposed areas are much more common than in HCP. They are likely to occur apart from and be more long lasting than the neurovisceral symptoms. Oral contraceptives can precipitate cutaneous manifestations. Acute attacks have become less common, and skin manifestations are more frequently the initial presentation; this may be due to earlier diagnosis and counseling. The risk of hepatocellular carcinoma is increased.
Symptoms of homozygous VP begin in infancy or childhood. These children generally have severe photosensitivity, neurologic symptoms, convulsions, developmental disturbances, and sometimes growth retardation, but do not have acute attacks.
Laboratory Findings
Urinary ALA, PBG, and uroporphyrin are increased during acute attacks but often less so than in AIP, and may be normal or only slightly increased during remission. Plasma porphyrins, urinary coproporphyrin III, and fecal coproporphyrin III and protoporphyrin are more persistently increased between attacks. Erythrocyte zinc protoporphyrin levels are markedly increased in homozygous VP, and may be modestly increased in heterozygous cases.
Diagnosis and Differential Diagnosis
VP is readily distinguished from AIP and HCP, which also present with acute attacks and increases in PBG. Plasma porphyrin analysis is especially useful, because the plasma porphyrins in VP are tightly protein bound, resulting in a characteristic fluorescence emission spectrum at neutral pH. Fecal porphyrins are increased, with approximately equal amounts of coproporphyrin III and protoporphyrin. Fluorometric detection of plasma porphyrins is more sensitive than stool porphyrin analysis in asymptomatic VP. PPO assays using cells that contain mitochondria, such as lymphocytes, are sensitive for identifying asymptomatic carriers but are not widely available. Knowing the PPO mutation in an index case enables the identification of relatives who carry the same mutation.
Treatment
Acute attacks are treated as in AIP. Hemin is beneficial for acute attacks but not for cutaneous symptoms. Light protection is important in patients with skin manifestations, using long-sleeved clothing, gloves, a broad-brimmed hat, and opaque sunscreen preparations. Exposure to short-wavelength ultraviolet light, which does not excite porphyrins, may increase skin pigmentation and provide some protection. Phlebotomy and chloroquine are not effective. Surprisingly, oral activated charcoal was reported to increase porphyrin levels and worsen skin manifestations.
Prognosis and Prevention
The outlook of patients with VP has improved, which may be attributed to improved treatment, earlier diagnosis, and detection of latent cases. Cyclic acute attacks in women can be prevented with a GnRH analog, as in AIP. A diagnosis of VP or any other acute porphyria should not lead to difficulty obtaining insurance, because the prognosis is usually good once the diagnosis is established.
Erythropoietic Protoporphyria (EPP)
In this autosomal recessive disorder, protoporphyrin accumulates due to a marked deficiency of FECH, the last enzyme in the heme biosynthetic pathway. EPP is sometimes termed protoporphyria or erythrohepatic protoporphyria, although the liver does not contribute substantially to production of excess protoporphyrin in uncomplicated cases.
Etiology
FECH, the enzyme that is deficient in EPP, catalyzes the final step in heme synthesis, which is insertion of ferrous iron (Fe2+) into protoporphyrin IX (see Fig. 85-1). The enzyme is also termed heme synthetase or protoheme ferrolyase. The human enzyme is a dimer, and each homodimer contains a [2Fe-2S] cluster, which may have a role in bridging homodimers. FECH is found in the mitochondrial inner membrane where its active site faces the mitochondrial matrix. It may be associated with complex I of the mitochondrial electron transport chain, and the ferrous iron substrate may be produced upon nicotinamide adenine dinucleotide (NADH) oxidation. FECH is specific for the reduced form of iron, but can utilize other metals such as Zn2+ and Co2+ and other dicarboxyl porphyrins. Accumulation of free protoporphyrin rather than zinc protoporphyrin in EPP indicates that formation of the latter is dependent on FECH activity in vivo.
The human FECH gene is located on chromosome 18q21.3, has a single promoter sequence, and contains 11 exons. Two mRNAs of 1.6 and 2.5 kb were described, which may be explained by the use of 2 alternative polyadenylation signals. The larger transcript is more abundant in murine erythroid cells, suggesting erythroid-specific regulation of FECH. A variety of FECH mutations have been reported in EPP, including missense, nonsense, and splicing mutations, small and large deletions, and an insertion.
The inheritance of 2 alleles associated with reduced FECH activity is required for disease expression. This is consistent with FECH activities as low as 15-25% of normal in EPP patients. In most patients, a disabling mutation on 1 FECH allele is combined with a common polymorphism affecting the other allele, which produces less than normal amounts of enzyme. This intronic single nucleotide polymorphism, IVS3-48T>C, results in the expression of an aberrantly spliced mRNA that is degraded by a nonsense-mediated RNA decay mechanism, which decreases the steady-state level of FECH mRNA. The IVS3-48T>C polymorphism by itself does not cause disease, even when homozygous. In a few families, 2 severe FECH mutations have been found. EPP with autosomal recessive inheritance occurs naturally in cattle and in mouse models.
A variant clinically indistinguishable form of EPP has been associated with gain of function deletions in the last exon of ALAS2. These lesions delete the last 10-20 amino acids of the ALAS2 polypeptide and apparently make the enzyme more stable. Free protoporphyrin predominates in erythrocytes in these cases, but because FECH activity is normal the proportion of zinc protoporphyrin is greater than in classic EPP.
EPP is sometimes associated with myelodysplastic syndromes and expansion of a clone of hematopoietic cells with deletion of 1 FECH allele. In such cases, there is late onset of the disease.
Epidemiology
EPP is the 3rd most common porphyria, although its prevalence is not precisely known (see Table 85-2). It is described mostly in white people, but occurs in other races. The IVS3-48T>C splice variant is common in whites and East Asians but rare in Africans, which would be predictive of a lower disease prevalence in populations of African origin. EPP is the most common porphyria in children, but is often not diagnosed until adult life.
Pathology and Pathogenesis
FECH is deficient in all tissues in EPP, but bone marrow reticulocytes are thought to be the primary source of the excess protoporphyrin, some of which enters plasma and circulates to the skin. Circulating erythrocytes are no longer synthesizing heme and hemoglobin, but they contain excess free protoporphyrin, which also contributes. In the variant form of EPP due to terminal deletions in exon 11 of ALAS2, all intermediates of the heme pathway are overproduced and ultimately accumulate in bone marrow erythroblasts as protoporphyrin. FECH is not deficient in the variant form, and this enzyme chelates some of the excess protoporphyrin with zinc. An aberrantly spliced mitoferrin transcript, which limits iron transport into mitochondria, has also been described in this condition. The liver functions as an excretory organ for excess protoporphyrin rather than a major source. But FECH deficiency in the skin and liver may be important, as tissue transplantation studies in mice suggest that skin photosensitivity and liver damage occur only when FECH is deficient in these tissues.
Patients with EPP are maximally sensitive to light in the 400 nm range, which corresponds to the so-called Soret band (the narrow peak absorption maximum that is characteristic for protoporphyrin and other porphyrins). Having absorbed light, porphyrins enter an excited energy state and release energy as fluorescence, singlet oxygen, and other reactive oxygen species. Tissue damage is accompanied by lipid peroxidation, oxidation of amino acids, cross linking of proteins in cell membranes, and damage to capillary endothelial cells. Such damage may be mediated by photoactivation of the complement system and release of histamine, kinins, and chemotactic factors. Repeated acute damage leads to thickening of the vessel walls and perivascular deposits from accumulation of serum components. Deposition of amorphous material containing immunoglobulin, complement components, glycoproteins, acid glycosaminoglycans, and lipids around blood vessels occurs in the upper dermis.
There is little evidence for impaired erythropoiesis or hemolysis in EPP. However, mild anemia with microcytosis, hypochromia and reticulocytosis is common. Iron accumulation in erythroblasts and ring sideroblasts have been noted in bone marrow in some patients. Decreased transferrin saturation may suggest iron deficiency, but serum ferritin and transferrin receptor levels are usually normal. Iron status should be carefully evaluated in EPP patients, keeping in mind that iron deficiency may lead to further increases in protoporphyrin and increase the risk for cholestasis.
Liver damage that develops in a small proportion of EPP patients is attributed to excess protoporphyrin, which is cholestatic, insoluble in water and excreted only by hepatic uptake and biliary excretion. Some may be reabsorbed by the intestine and undergo enterohepatic circulation. With cholestasis the excess protoporphyrin that accumulates in the liver can form crystalline structures in hepatocytes, and impair mitochondrial function.
Clinical Manifestations
Symptoms of cutaneous photosensitivity begin in childhood, and consist of pain, redness, and itching occurring within minutes of sunlight exposure. Swelling may resemble angioneurotic edema, and is termed solar urticaria. Symptoms are usually worse in the spring and summer. Petechiae and purpuric lesions may be seen, but blisters are usually absent. Chronic changes may include lichenification, leathery pseudovesicles, labial grooving, and nail changes, but changes in pigmentation and pronounced scarring are unusual. Although physical findings in EPP may not be impressive, the symptoms significantly impair quality of life and to a greater extent than in PCT and VP. An association between autosomal recessive EPP and seasonal palmar keratoderma is unexplained. Neuropathy develops only in some patients with severe hepatic decompensation.
Unless hepatic or other complications develop, protoporphyrin levels and symptoms of photosensitivity remain remarkably stable for many years in most patients. Factors that exacerbate hepatic porphyrias play little or no role in EPP. Mild, unexplained hypertriglyceridemia has been described. Erythrocyte protoporphyrin levels may decrease and sunlight tolerance may improve during pregnancy, which is unexplained.
Laboratory Findings
Protoporphyrin is substantially increased in circulating erythrocytes in EPP, and consists almost entirely of free protoporphyrin. In a variant form of EPP due to ALAS2 exon 11 deletions, zinc protoporphyrin as well as free protoporphyrin are increased, although the latter still predominates. Protoporphyrin is also increased in bone marrow, plasma, bile, and feces. Other porphyrins and porphyrin precursors are normal in uncomplicated EPP.
Diagnosis and Differential Diagnosis
A diagnosis of EPP is confirmed primarily by finding a substantially elevated concentration of erythrocyte protoporphyrin, which is predominantly free and not complexed with zinc. Erythrocyte zinc protoporphyrin concentration is increased in some homozygous porphyrias, iron deficiency, lead poisoning, anemia of chronic disease, hemolytic conditions, and many other erythrocytic disorders. Because many assays for erythrocyte protoporphyrin or “free erythrocyte protoporphyrin” measure either total or zinc protoporphyrin, and specific assays for metal-free protoporphyrin are less widely available, reports of increased erythrocyte protoporphyrin must be interpreted with care.
Plasma total porphyrin concentration is often less increased in EPP than in other cutaneous porphyrias, and may be normal. Great care must be taken to avoid light exposure during sample processing, because plasma porphyrins in EPP are particularly subject to photodegradation. Urinary porphyrin precursors and porphyrins are not increased.
Measurement of FECH activity requires cells containing mitochondria and is not widely available. A greater than expected proportion of zinc protoporphyrin (more than 15% of the total) in erythrocytes is important in identifying variant EPP. DNA studies are increasingly important for confirming FECH or ALAS2 mutations and for genetic counseling.
Life-threatening protoporphyric hepatopathy is heralded by increasingly abnormal liver function tests, increasing erythrocyte and plasma protoporphyrin levels, and worsening photosensitivity. Increases in urinary porphyrins, especially coproporphyrin, in this setting are attributable to liver dysfunction.
Complications
Biliary stones containing protoporphyrin are sometimes symptomatic and require cholecystectomy. Liver disease occurs in less than 5% of EPP patients, including children, and may be chronic or progress rapidly to death from liver failure. Liver disease is sometimes the major presenting feature of EPP. Upper abdominal pain may suggest biliary obstruction, and unnecessary laparotomy to exclude this possibility can be detrimental. Concurrent conditions that impair liver function, such as viral hepatitis, alcohol intake, iron deficiency, fasting, or oral contraceptive steroids, may contribute. Liver histology shows marked deposition of protoporphyrin in liver cells and bile canaliculi. Patients with protoporphyric liver failure, which represents a severe phenotype, most often have “null mutations” and the IVS3–48T>C hypoexpression allele, but some may have 2 severe mutant alleles. Patients with EPP due to ALAS2 exon 11 deletions are also at risk for liver failure. The bone marrow is probably the major source of protoporphyrin, even in EPP patients with hepatic failure.
Treatment
Exposure to sunlight should be avoided, which is aided by wearing closely woven clothing. Oral beta-carotene leads to clinical improvement and greater tolerance to light in some patients, usually 1 to 3 mo after starting treatment. In most adults, doses of 120-180 mg daily will maintain serum carotene levels in the recommended range of 600-800 mg/dL, but doses up to 300 mg daily may be needed. Mild skin discoloration due to carotenemia is expected. The recommended product is Solatene (Tishcon), which was initially developed as a drug for treating this disease, rather than nutritional products that are less standardized. Beta-carotene may quench singlet oxygen or free radicals, but does not substantially alter circulating porphyrin levels. Better tolerance of sunlight may result in tanning, which provides additional protection. Oral cysteine may also quench excited oxygen species and was found to increase tolerance to sunlight in EPP.
Measures to darken the skin may also be helpful. This may be accomplished by narrow-band UV-B phototherapy or with topical products such as dihydroxyacetone and lawsone (naphthoquinone). A synthetic analogue of melanocyte stimulating hormone shows promise for increasing sunlight tolerance in EPP. Caloric restriction and drugs or hormone preparations that impair hepatic excretory function should be avoided, and iron deficiency should be corrected if present. Vitamin D supplementation and hepatitis A and B vaccination are recommended.
Treatment of protoporphyric liver disease must be individualized and results are unpredictable. Ursodeoxycholic acid may be of some value in early stages. Cholestyramine or activated charcoal may interrupt the enterohepatic circulation of protoporphyrin, promote its fecal excretion, and reduce liver protoporphyrin content. Splenectomy may be beneficial when EPP is complicated by hemolysis and splenomegaly. Spontaneous resolution may occur, especially if another reversible cause of liver dysfunction, such as viral hepatitis or alcohol abuse, is contributing. In patients with severe hepatic decompensation, combined treatment with plasmapheresis, transfusion to suppress erythropoiesis, intravenous hemin to suppress erythroid and hepatic protoporphyrin production, ursodeoxycholic acid, vitamin E, and cholestyramine may be beneficial.
Motor neuropathy resembling that seen in acute porphyrias sometimes develops in EPP patients with liver disease after transfusion or liver transplantation and is sometimes reversible. Artificial lights, such as operating room lights during liver transplantation or other surgery, may cause severe photosensitivity, with extensive burns of the skin and peritoneum and photodamage of circulating erythrocytes.
With continued progression of liver disease, liver transplantation may be considered. Although liver disease may recur in the transplanted liver due to continued bone marrow production of excess protoporphyrin, outcomes are comparable to transplantation for other types of liver disease. Bone marrow transplantation (BMT) should also be considered after liver transplantation if a suitable donor is available.
Prognosis
Typical EPP patients have lifelong photosensitivity but can otherwise expect normal longevity. Protoporphyric liver disease is often life-threatening; however, the incidence is low.
Prevention
Symptoms can be prevented by avoiding sunlight. Avoiding agents that may cause liver damage may help prevent liver complications.
Genetic Counseling
DNA studies to identify FECH mutations, the common IVS3–48T>C FECH hypoexpressing mutant allele, or ALAS2 exon 11 deletions are increasingly important for genetic counseling. EPP may improve during pregnancy. In classic EPP, DNA studies in both parents can predict the risk for EPP occurring in an offspring.
Dual Porphyria
Dual porphyria refers to patients with porphyria who have deficiencies of more than 1 enzyme of the heme biosynthetic pathway. An unusual pattern of porphyrin precursors and porphyrins may suggest the presence of two enzyme deficiencies. Mutations of 2 heme pathway enzymes have been documented in only 2 patients with porphyria. One presented with acute porphyria and had heterozygous mutations in both the CPO and ALAD genes. The other had symptoms of AIP and PCT and was documented to have both PBGD and UROD mutations. In other reported cases, 1 or both enzyme deficiencies were based on enzyme measurements.
Porphyria due to Tumors
Very rarely, hepatocellular tumors contain and presumably produce excess porphyrins, but such cases have not been studied carefully. Hepatocellular carcinomas complicating PCT and acute hepatic porphyrias usually are not described as containing large amounts of porphyrins. Erythropoietic porphyrias can develop late in life due to clonal expansion of erythroid cells containing a specific enzyme deficiency in patients who have developed myelodysplastic or myeloproliferative syndromes.
Anderson KE, Sassa S, Bishop DF, et al. Disorders of heme biosynthesis: X-linked sideroblastic anemias and the porphyrias, ed 8. Scriver CR, Beaudet AL, Sly WS, et al, editors. The metabolic and molecular basis of inherited disease, vol II. McGraw-Hill, New York, 2001;2991-3062.
Puy H, Gouya L, Deybach JC. Porphyrias. Lancet. 2010;375:924-934.
Sarkany RP. Making sense of the porphyrias. Photodermatol Photoimmunol Photomed. 2008;24:102-108.
Seth AK, Badminton MN, Mirza D, et al. Liver transplantation for porphyria: who, when, and how? Liver Transpl. 2007;13:1219-1227.
Akagi R, Kato N, Inoue R, et al. δ-Aminolevulinate dehydratase (ALAD) porphyria: the first case in North America with two novel ALAD mutations. Mol Genet Metab. 2006;87:329-336.
Anderson KE, Bloomer JR, Bonkovsky HL, et al. Recommendations for the diagnosis and treatment of the acute porphyrias. Ann Intern Med. 2005;142:439-450.
Solis C, Martinez-Bermejo A, Naidich TP, et al. Acute intermittent porphyria: studies of the severe homozygous dominant disease provide insights into the neurologic attacks in acute porphyrias. Arch Neurol. 2004;61:1764-1770.
Congenital Erythropoietic Porphyria
Dupuis-Girod S, Akkari V, Ged C, et al. Successful match-unrelated donor bone marrow transplantation for congenital erythropoietic porphyria (Gunther disease). Eur J Pediatr. 2005;164:104-107.
Hallai N, Anstey A, Mendelsohn S, et al. Pregnancy in a patient with congenital erythropoietic porphyria. N Engl J Med. 2007;357:622-623.
Phillips JD, Steensma DP, Pulsipher MA, et al. Congenital erythropoietic porphyria due to a mutation in GATA1: the first trans-acting mutation causative for a human porphyria. Blood. 2007;109:2618-2621.
Ajioka RS, Phillips JD, Weiss RB, et al. Down-regulation of hepcidin in porphyria cutanea tarda. Blood. 2008;112:4723-4728.
Phillips JD, Bergonia HA, Reilly CA, et al. A porphomethene inhibitor of uroporphyrinogen decarboxylase causes porphyria cutanea tarda. Proc Natl Acad Sci U S A. 2007;104:5079-5084.
Remenyik E, Lecha M, Badenas C, et al. Childhood-onset mild cutaneous porphyria with compound heterozygotic mutations in the uroporphyrinogen decarboxylase gene. Clin Exp Dermatol. 2008;33:602-605.
Goodwin RG, Kell WJ, Laidler P, et al. Photosensitivity and acute liver injury in myeloproliferative disorder secondary to late-onset protoporphyria caused by deletion of a ferrochelatase gene in hematopoietic cells. Blood. 2006;107:60-62.
Harms J, Lautenschlager S, Minder CE, et al. An alpha-melanocyte-stimulating hormone analogue in erythropoietic protoporphyria. N Engl J Med. 2009;360:306-307.
Holme SA, Anstey AV, Finlay AY, et al. Erythropoietic protoporphyria in the U.K.: clinical features and effect on quality of life. Br J Dermatol. 2006;155:574-581.
Holme SA, Whatley SD, Roberts AG, et al. Seasonal palmar keratoderma in erythropoietic protoporphyria indicates autosomal recessive inheritance. J Invest Dermatol. 2009;129:599-605.
Holme SA, Worwood M, Anstey AV, et al. Erythropoiesis and iron metabolism in dominant erythropoietic protoporphyria. Blood. 2007;110:4108-4110.
Minder EI, Schneider-Yin X, Steurer J, et al. A systematic review of treatment options for dermal photosensitivity in erythropoietic protoporphyria. Cell Mol Biol (Noisy-le-grand). 2009;55:84-97.
Rand EB, Bunin N, Cochran W, et al. Sequential liver and bone marrow transplantation for treatment of erythropoietic protoporphyria. Pediatrics. 2006;118:e1896-1899.
Whatley SD, Ducamp S, Gouya L, et al. C-terminal deletions in the ALAS2 gene lead to gain of function and cause X-linked dominant protoporphyria without anemia or iron overload. Am J Hum Genet. 2008;83:408-414.