Chapter 157 Hereditary Periodic Fever Syndromes
Hereditary periodic fever syndromes are a group of autoinflammatory diseases caused by an inborn error in the innate immune system. They are characterized by recurrent short episodes of fever that are self-limited and occur in the absence of infection or autoimmune reaction, such as high titer autoantibodies or autoreactive T cells. The innate immune system provides the first immunologic line of defense against many microbes and uses pattern recognition receptors (PRRs) such as Toll-like receptors (TLRs) to recognize a limited number of widely expressed viral and bacterial molecular structures known as pathogen-associated molecular patterns (PAMPs). These pattern recognition receptors stimulate inflammation by activating intracellular proteins (also known as intracellular sensors), which mediate the regulation of nuclear factor-κB (NF-κB), cell apoptosis, and interleukin-1β (IL-1β) through cross-regulated and common signaling pathways. Mutations in these intracellular proteins lead to increased production and secretion of IL-1β, resulting in clinical signs and symptoms.
The most common hereditary periodic fever disorders are familial Mediterranean fever (FMF), tumor necrosis factor (TNF) receptor–associated periodic syndrome (TRAPS), and hyperimmunoglobulinemia D syndrome (HIDS) (Table 157-1). The cryopyrin-associated periodic syndromes (CAPS) include Muckle-Wells syndrome (MWS), familial cold autoinflammatory syndrome (FCAS) (also known as familial cold urticaria [FCU]), and chronic infantile neurologic cutaneous and articular (CINCA) disease (also known as neonatal-onset multisystem inflammatory disease [NOMID]). A syndrome called pyogenic arthritis, pyoderma gangrenosum, and acne (PAPA) and Blau syndrome (known also as familial juvenile systemic granulomatosis) have now been added to this group. Secondary amyloidosis (AA amyloidosis) is a complication in all of these periodic fever disorders, although it is less commonly reported with HIDS. FMF and HIDS are autosomal recessive diseases, whereas TRAPS, PAPA, and Blau syndrome are autosomal dominant conditions. The diagnosis of each of these entities depends on the clinical features and the genetic confirmation (see Table 157-1). Another periodic fever syndrome is periodic fever, aphthous stomatitis, pharyngitis, and cervical adenitis (PFAPA), but it is not clear yet whether PFAPA is an autoinflammatory syndrome (see Table 157-1). Among the conditions that are not in the category of periodic fever and that have been classified as autoinflammatory diseases are Crohn disease, Behçet diseases, early-onset childhood sarcoidosis, systemic juvenile idiopathic arthritis (JIA), and chronic recurrent multifocal osteomyelitis (known also as Majeed syndrome) (Table 157-2).
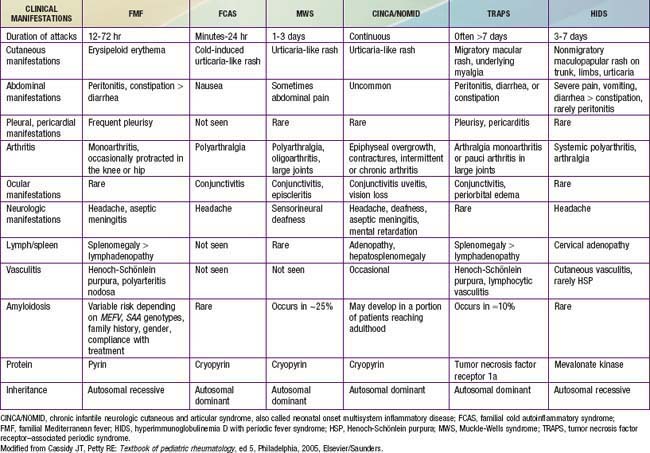
Table 157-2 RECURRENT OR PERIODIC FEVER SYNDROMES IN CHILDREN
INFECTIOUS DISEASES
RHEUMATIC DISEASES
HEREDITARY AUTOINFLAMMATORY SYNDROMES
CYCLIC HEMATOPOIESIS
IDIOPATHIC CONDITIONS
Periodic fever with aphthous stomatitis, pharyngitis, and adenitis (PFAPA)
From Cassidy JT, Petty RE: Textbook of pediatric rheumatology, ed 5, Philadelphia, 2005, Elsevier/Saunders.
Familial Mediterranean Fever
FMF is an autosomal recessive disorder characterized by brief, acute, self-limited episodes of fever and polyserositis that recur at irregular intervals and are associated with development of AA amyloidosis (Chapter 158).
Etiology
The gene responsible for FMF is mapped to a small interval on the short arm of chromosome 16p13.3. It is designated MEFV (ME for Mediterranean and FV for fever) and is a member of the RoRet gene family. It has 10 exons that express a 15-kb transcript encoding a 781–amino acid protein known as pyrin (from pyrus, the Greek word for “fever”), or marenostrin (Latin word for “our sea”), which is expressed in myeloid cells. Exon 10 and exon 2 carry most FMF-associated mutations. To date, more than 70 mutations have been discovered, mostly missense mutations. It is unclear whether all are truly disease-related mutations. The 5 most common mutations (M694V, V726A, M694I, M680I, E148Q) are found in more than two thirds of Mediterranean patients with FMF. Haplotypes and mutational analyses show ancestral relationships among carrier chromosomes that have been separated for centuries.
Approximately 70% of patients with clinical manifestations of FMF are heterozygous and have one of the two mutations that are identifiable by genetic analysis. The most common missense mutation is M694V (substitution of methionine with valine at codon 694), which occurs in 20-67% of cases and is associated with full penetrance. Homozygosity for M694V is associated with a greater disease severity and a higher incidence of amyloidosis. It is also associated with increased risk for onset at an early age. The V726A mutation occurs in 7-35% of cases and is associated with milder disease and a lower incidence of amyloidosis. The E148Q mutation is associated with low penetrance and very mild phenotype. These findings suggest that phenotypic differences may reflect different mutations. As with other recessive diseases, it is likely that some heterozygous patients may show attenuated clinical symptoms, with or without increased levels of acute phase reactants.
Epidemiology
FMF occurs primarily among ethnic groups of Mediterranean origin, mainly Sephardic Jews, Turks, Armenians, and individuals of Arab descent. In these populations, the carrier frequency is estimated to be as high as 1 in 5 persons, suggesting a carrier advantage for heterozygotes. Greeks, Hispanics, and Italians are less commonly affected. In addition, cases of FMF are found among non-Mediterranean persons. It is seen rarely among Ashkenazi Jews, Germans, and Anglo-Saxons.
Pathogenesis
The exact pathogenesis of the acute episodes of FMF is unknown. Between episodes, patients with FMF have increased serum levels of interferon-γ and enhanced production of other proinflammatory cytokines, such as TNF-α, IL-1β, IL-6, and IL-8, in circulating leukocytes. Pyrin/marenostrin is a member of the death domain superfamily and consists of 4 different functional domains that interact with other proteins. Of particular interest is the domain known as the pyrin domain (PYD), a 92–amino acid N-terminal domain shared by several proteins that are involved in the regulation of the inflammatory response and apoptosis. Pyrin acts as an anti-inflammatory factor by inhibiting processing of pro–IL-1β cytokine to the active form. This inhibition normally takes place through interactions with a caspase recruitment domain (ASC) and NF-κB. It has been suggested that normally pyrin inhibits the binding of ASC to caspase-1 in a competitive manner. The C-terminal domain of the pyrin molecule interacts with caspase-1, leading to inhibition of IL-1β production. It is speculated that the defective (or mutated) pyrin found in patients with FMF is functionally inactive, allowing binding of ASC to caspase-1 to take place. As a consequence, stimulation of IL-1β processing and secretion occur, resulting in increased IL-1β levels that are responsible for the uncontrolled inflammation (Fig. 157-1). Another possibility that was previously more popular is based on the finding of C5a inhibitor (inactivating enzyme) deficiency in peritoneal and synovial fluids of patients with FMF. C5a is a fragment of complement, an anaphylatoxin, and a potent chemotactic agent (Chapter 127). Normally, C5a inhibitor neutralizes the small amounts of C5a released into serosal cavities before they precipitate overt inflammation. The hypothesis is that a deficiency of C5a inhibitor, which is a consequence of pyrin/marenostrin dysfunction in patients with FMF, allows further accumulation of C5a, leading to the acute attack. Further understanding of pyrin/marenostrin functions will shed light on aspects of FMF pathogenesis that are not yet fully understood.
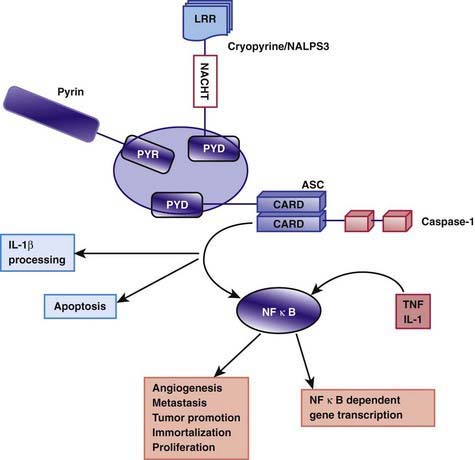
Figure 157-1 Proteins containing pyrin domain (PYD) regulate inflammation through their interaction with apoptotic speck protein (ASC). The assembly of cryopyrin and ASC induces interleukin-1 (IL-1) processing through caspase-1, whereas pyrin may act as an inhibitor. Loss of function by mutations in the pyrin could potentially lead to autoinflammation by reducing the pyrin inhibitory role. Alternatively, gain-of-function mutations in cryopyrin, as found in patients with Muckle-Wells syndrome/familial cold urticaria/neonatal-onset multisystem inflammatory disease, could activate this pathway. ASC participates in apoptosis and activation of nuclear factor–κB (NF-κB), a transcription factor involved in both initiation and resolution of the inflammatory response. LRR, leucine-rich repeat(s); TNF, tumor necrosis factor.
(From Padeh S: Periodic fever syndromes, Pediatr Clin North Am 52:577–609, 2005.)
Clinical Manifestations
The onset of clinical manifestations occurs before 5 yr of age in 65% of cases and before 20 yr of age in 90% of cases. Onset may be as early as 6 mo of age. Exercise, emotional stress, infection, menses, and surgery may precipitate acute episodes. The typical acute episode lasts 1-4 days and includes fever and 1 or more symptoms of sterile peritonitis, manifested as abdominal pain (90%), arthritis or arthralgia (85%), or pleuritis manifested by chest pain (20%). Other serosal tissues, such as the pericardium and tunica vaginalis testis (acute scrotum), are rarely affected. Some patients experience prolonged and protracted episodes of fever and myalgia in upper and lower extremities that may last up to 6 wk. Erysipelas-like rash, myalgia, splenomegaly, scrotal involvement in boys, neurologic involvement, Henoch-Schönlein purpura, and hypothyroidism are other, less common clinical manifestations.
Diagnosis
Genetic testing for the FMF gene confirms the diagnosis of FMF, which is especially important in areas where the disease is rare and less familiar to physicians. Genetic screening using polymerase chain reaction (PCR) and restriction analysis is available in some commercial clinical genetics laboratories. However, genetics laboratories usually screen for only the 10 to 15 most common mutations, and thus, rare mutations will be missed. Therefore, the diagnosis of FMF is still based on clinical manifestations, with genetic testing used as a confirmatory test.
Treatment
Attacks of FMF can be prevented by prophylactic colchicine (0.02-0.03 mg/kg/day; maximum 2 mg/day) in 1 to 2 divided doses. In general, the initial dose should be 0.5 mg/day for children <5 yr of age, 1 mg/day for children 5-10 yr, and 1.5 mg/day for those >10 yr. Approximately 65% of patients experience remission of attacks, 20-30% experience improvement with significant reduction in the number and severity of the episodes, and 5-10% show no response. Colchicine therapy reduces the frequency of acute attacks and also greatly decreases the probability of development of amyloidosis; it may produce partial regression of existing amyloidosis. Poor compliance is common, owing to gastrointestinal side effects and may contribute to treatment failure. Toxic effects (acute myopathy and bone marrow hypoplasia) can be seen with doses >0.1 mg/kg, resulting in lethality at a dose ≥0.8 mg/kg. Colchicine therapy for FMF during pregnancy has not been reported to harm either the mother or her fetus. Prolonged colchicine use seems to have no effects on male or female fertility, pregnancy, fetal development, or development after birth. It has also been shown that biologic treatments, especially the IL-1 inhibitor anakinra, produce a beneficial response in cases of FMF that do not respond to colchicine.
Complications and Prognosis
In 30-50% of untreated children and in 75% of adults with FMF, a form of renal amyloidosis develops in which the amyloid derives from a normal serum protein and an acute-phase reactant, serum amyloid A (SAA), resulting in AA amyloidosis. Renal disease manifests as proteinuria that progresses to nephrotic syndrome and renal failure over a period of months to several years. Transplantation may be required for renal failure. Amyloidosis is common among Sephardic Jews and Turks and less common in Armenians. Homozygosity for M694V is associated with a greater disease severity and a higher incidence of amyloidosis. Armenians living in Armenia are reported to have a significantly higher incidence of amyloidosis than their counterparts in North America, suggesting that environmental factors may also play a role. The country of residence rather than the MEFV genotype has been playing the major role in the development of amyloidosis. Mortality from FMF usually results from complications of renal failure and amyloidosis, such as infection, thromboembolism, and uremia. Other rare complications are joint contractures, abdominal adhesions, and impairment in social development, although patients are capable of physical activity with some limitations due to their illness.
Hyperimmunoglobulinemia D Syndrome
HIDS, known also as Dutch fever, is an inherited periodic fever syndrome with an autosomal recessive mode of transmission. This condition is reported primarily among families of European descent, especially Dutch and French, and is caused by mutations in the mevalonate kinase (MVK) gene found on chromosome 12 at 12q24. Mevalonate kinase is an enzyme that enhances the metabolism of mevalonic acid, an intermediary product of cholesterol and isoprenoid synthesis pathways (Chapter 80). Cells from patients with HIDS still contain residual MVK enzyme activity (1-8%). A complete deficiency of this enzyme causes a distinct disorder known as mevalonic aciduria, which is associated with severe mental retardation, ataxia, myopathy, cataracts, and failure to thrive. In these patients, MVK enzyme activity is below the detection level. It is speculated that shortage of isoprenoid end products contributes to increased secretion of IL-1β, which subsequently leads to overt inflammation and fever.
More than 100 different mutations in the MVK gene have been reported so far. Some variants are strongly associated with a severe mevalonic aciduria phenotype. The most common mutation is V377I, likely of Dutch origin, which is exclusively associated with a mild phenotype. These mutations are associated with decreased activity of mevalonate kinase in lymphocytes, leading to increased plasma levels of mevalonic acid, which is excreted in large amounts in the urine. The majority of patients have onset within the 1st yr of life. The manifestations include recurrent, short episodes of fever lasting 3-7 days, with abdominal pain that is often accompanied by diarrhea, nausea, and vomiting. Other clinical manifestations include cervical lymphadenopathy, rash, aphthous ulcers, symmetric polyarthritis/arthralgia or oligoarthralgia/arthritis, and occasional splenomegaly. In some patients, the attacks may last several weeks. During the attacks, leukocytosis and increased serum levels of acute-phase reactants and proinflammatory cytokines are commonly present.
HIDS is a difficult diagnosis to make, and diagnosis may be delayed by as long as 10 yr from the onset of symptoms. The finding of elevated serum values of immunoglobulin (Ig) D (>100 mU/mL) is present in ≈80% of patients and strongly supports the diagnosis of HIDS, but it is not diagnostic. In particular, IgD levels may be increased in other autoinflammatory diseases. The symptoms of HIDS may persist for years but tend to become less prominent with time. Unlike in patients with FMF or TRAPS, the incidence of AA amyloidosis in patients with HIDS is remarkably low (3 out of 103 in an international study). The low susceptibility to amyloidosis in HIDS is not fully understood. Other rare complications include joint contractures and abdominal adhesions. There is no known therapy for this condition, although treatment with glucocorticoids may be associated with dramatic or partial relief. Antagonists of IL-1 receptor (anakinra) and TNF-α (etanercept) are effective in case reports of patients with HIDS. A trial of simvastatin showed a beneficial clinical effect in 5 of 6 patients with HIDS. Bone marrow transplantation has been effective in one reported patient.
Tumor Necrosis Factor Receptor–Associated Periodic Syndrome
TRAPS is an autosomal-dominant periodic fever syndrome caused by mutation of the soluble TNF receptor superfamily 1A gene, TNFRSF1A. This syndrome was previously known by other names, including familial Hibernian fever, familial periodic fever, and autosomal dominant recurrent fever. TRAPS is a rare disorder that was initially reported in a few families of Irish and Scottish ancestry, although other ethnic groups, including African-Americans, Japanese, Puerto Rican, and Finnish, may be affected. The TNFRSF1A gene is on chromosome 12 at 12p13 and encodes the type 1A TNF receptor protein (TNFR1). In TRAPS a mutation in the TNFRSF1A gene leads to a defective TNFR1 molecule on the cell surface that is unable to neutralize TNF-α. More than 50 different disease-associated mutations in TNFRSF1A have been reported. Phenotype-genotype correlations have demonstrated that mutations at cysteine residues have a higher penetrance and are associated with a severe disease course and an increased risk of secondary AA amyloidosis.
Patients with TRAPS usually have brief, intermittent febrile episodes, typically lasting 4-6 days and associated with severe abdominal pain, nausea, and vomiting. Oligoarthritis, myalgias, rash, conjunctivitis, and unilateral periorbital edema are universally present in patients with TRAPS (Fig. 157-2). Arthralgias are less common. The acute attacks of TRAPS are slightly longer than the episodes of FMF and may persist for up to 3 wk. AA amyloidosis develops in up to 25% of patients with TRAPS, depending on the specific gene mutation and the duration of attacks. Amyloidosis may affect various organs but commonly involves the kidneys and liver, leading to renal and/or hepatic failure. Increased levels of acute-phase reactants may be seen, with the most specific findings being low serum levels of soluble type 1A TNF receptor and increased serum levels of TNF.
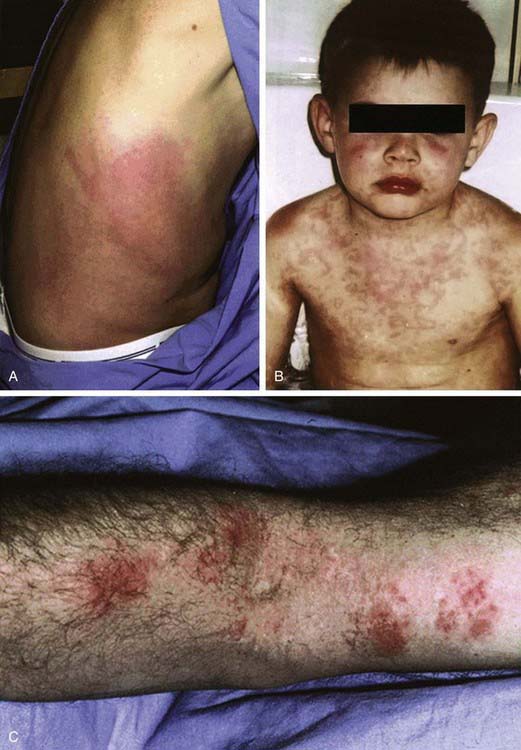
Figure 157-2 Cutaneous manifestations of tumor necrosis factor receptor–associated periodic syndrome. A, Right flank of a patient with the T50M mutation. B, Serpiginous rash involving the face, neck, torso, and upper extremities of a child with the C30S mutation. C, Erythematous, macular patches with crusting on the flexor surface of the right arm of a patient with the T50M mutation.
Colchicine has no effect on the acute attacks or on the development of amyloidosis in patients with TRAPS. Prednisone (1 mg/kg; maximum dose 20 mg) may be helpful and can attenuate the length and severity of the attacks. Although the use of etanercept appears promising and may reverse AA amyloidosis, not all patients show a response to this agent. In addition, IL-1 receptor antagonist (anakinra) has been shown to be effective in a case report of TRAPS.
Muckle-Wells Syndrome, Familial Cold Autoinflammatory Syndrome, and Chronic Infantile Neurologic Cutaneous and Articular Disease
The 3 separate clinical entities NWS, FCAS, and CINCA disease, also known as cryopyrin-associated periodic syndromes, are autosomal dominant disorders. They represent a clinical spectrum that ranges from mild symptoms in FCAS to severe symptoms in CINCA disease. These three entities are associated with mutations in the cold-induced anti-inflammatory syndrome gene, CIAS1, located on chromosome 1 at 1q44. The CIAS1 gene encodes the protein cryopyrin, which shares homology in several regions (see Fig. 157-1). The term cryopyrin was designated because of the association with cold urticaria. Approximately 50 mutations in the CIAS1 gene that have different effects on cryopyrin expression have been described. Like pyrin, cryopyrin is expressed in polymorphonuclear leukocytes and monocytes and activates an adaptor protein known as ASC. As with FMF, mutations in cryopyrin lead to increased production of IL-1β (a common underlying mechanism), which eventually causes these diverse disorders. All 3 entities are characterized by periodic febrile attacks with an urticarial rash. The skin rash can be distinguished from classic urticaria on the basis of histopathologic findings, which include perivascular infiltrates of polymorphonuclear leucocytes rather than mast cells. Other characteristics include arthralgia and arthritis, ocular involvement, and the development of AA amyloidosis. In FCAS, autoinflammatory attacks start within 8 hours of generalized cold exposure. Typically, localized cold exposure does not trigger the episodes. The joint symptoms consist of polyarthralgias (hands, knees, and ankles) in more than 90% of patients. Both MWS and CINCA disease are typically associated with progressive sensorineural hearing loss, optic nerve involvement, and chronic aseptic meningitis. CINCA disease is a more severe entity, typically having a neonatal onset and being associated with dysmorphic features, rash, neurologic disease with mental retardation, and destructive arthropathy, mainly of the knees, which may lead to major malformation and disability (Fig. 157-3). No definitive therapy exists for these conditions, although treatment with colchicine, nonsteroidal anti-inflammatory drugs (NSAIDs), and glucocorticoids may provide some relief. Remarkable responses to anakinra (IL-1 receptor antagonist) in 3 family members with MWS and 18 patients with CINCA disease have been reported. Anakinra seems to improve visual and hearing impairment and in some cases induces amelioration of amyloidosis within 6 mo of treatment. Treatment with rilonacept, an IL-1 trap given by weekly subcutaneous injections, can markedly reduce symptoms and inflammatory markers. In addition, canakinumab, an anti-interleukin-1β monoclonal antibody, has demonstrated efficacy in cryopyrin associated periodic fever syndromes.
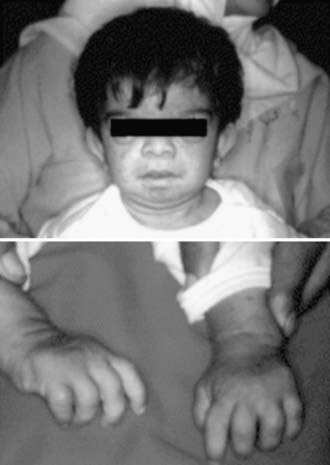
Figure 157-3 A 3 yr old girl with neonatal-onset multisystem inflammatory disease (NOMID)/chronic infantile neurologic cutaneous and articular (CINCA) disease. Note the markedly deformed hands, rash, frontal bossing, and large head.
(From Padeh S: Periodic fever syndromes, Pediatr Clin North Am 52:577–560, 2005.)
Deficiency of the interleukin-1 receptor antagonist produces an autoinflammatory syndrome characterized by inflammation and a pustulosis rash, sterile multifocal osteomyelitis, widened ribs, periosteal elevation, osteopenia, and onset before 1 yr of age. Anakinra is the treatment of choice.
Pyogenic Arthritis, Pyoderma Gangrenosum, and Acne and Blau Syndrome
Further understanding of pyrin functions, especially the interactions with other proteins, has led to the discovery of two other entities, PAPA and Blau syndromes. PAPA syndrome is an autosomal dominant disorder with mutations in the gene encoding the adaptor protein proline serine threonine phosphatase–interacting protein (PSTPIP1) located on chromosome 15 at 15q24. Pyoderma gangrenosum and severe cystic acne associated with skin ulcerations are usually seen on the extremities and are triggered by trauma. Typically, the arthritis is sterile and the synovial fluid is rich in neutrophils. Blau syndrome is a rare autosomal dominant disorder that manifests as early-onset granulomatous arthritis, uveitis, rash, and flexion contractures at the fingers associated with mutations in the gene encoding CARD15 (caspase recruitment domain 15 protein), also known as NOD2 (nucleotide-binding oligomerization domain 2 protein), located on chromosome 16 at 16q12. Although fever is not a major symptom in PAPA and Blau syndromes, these conditions represent additional rare members of the hereditary periodic fever syndromes family.
Periodic Fever, Aphthous Stomatitis, Pharyngitis, and Adenitis
Another distinct periodic fever syndrome, PFAPA, also known as Marshall syndrome, manifests as episodes of periodic fever, aphthous stomatitis, pharyngitis, and adenitis. PFAPA occurs sporadically and has no ethnic predilection. Symptoms begin around 2-5 yr of age and include recurring fever, malaise, exudative-appearing tonsillitis with negative throat culture results, cervical lymphadenopathy, oral aphthae ulceration, and, less commonly, headache, abdominal pain, and arthralgia. The episodes last 4-6 days, regardless of antipyretic or antibiotic treatment, and occur at a frequency of 8-12 episodes/yr. Findings during the episodes may include mild hepatosplenomegaly, mild leukocytosis, and elevated acute-phase reactants. Both the frequency and intensity of the episodes diminish over time.
The etiology and the pathogenesis of PFAPA remain unknown. It is not clear whether this syndrome represents an infectious or immunogenetic dysregulation entity. Clinical experience suggests that NSAIDs and antipyretics such as acetaminophen are ineffective in controlling the clinical manifestations of PFAPA. The majority of patients show dramatic response to a single dose of prednisone (1-2 mg/kg) or betamethasone (0.3 mg/kg) with prompt resolution of symptoms within 24 hr. In addition, cimetidine in 3-4 divided doses of 20 mg to 40 mg/kg/day has been reported to be effective in inducing sustained remission after 6 mo of therapy. Complete resolution has also been reported after tonsillectomy in some but not all patients with this disorder. Affected children grow normally and have spontaneous resolution within 4-8 yr with no long-term sequelae. One patient with PFAPA demonstrated TRAPS at age 22 yr.
Adachi M, Watanabe A, Nishiyama A, et al. Familial cases of periodic fever with aphthous stomatitis, pharyngitis, and cervical adenitis syndrome. J Pediatr. 2011;158:155-159.
Alksentijevich I, Masters SL, Ferguson PJ, et al. An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N Engl J Med. 2009;360:2426-2436.
Belkhir R, Moulonguet-Doleris L, Hachulla F, et al. Treatment of familial Mediterranean fever with anakinra. Ann Intern Med. 2007;146:825-826.
Bodar EJ, Drenth JPH, Van der Meer JWM, et al. Dysregulation of innate immunity: hereditary periodic fever syndromes. Brit J Haematol. 2008;144:279-302.
Caudy AA, Reddy ST, Chatila T, et al. CD25 deficiency causes an immune dysregulation, polyendocrinopathy, enteropathy, X-linked-like syndrome and defective IL-10 expression from CD4 lymphocytes. J Allergy Clin Immunol. 2007;119:482-487.
Chitkara P, Stojanov S, Kastner DL. The hereditary autoinflammatory syndromes. Pediatr Infect Dis J. 2007;26:353-354.
Church LD, McDermott MF. Rilonacept in cryopyrin-associated periodic syndromes: the beginning of longer-acting interleukin-1 antagonism. Nat Clin Pract Rheumatol. 2009;5:14-15.
Garavello W, Romagnoli M, Gaini RM. Effectiveness of adenotonsillectomy in PFAPA syndrome: a randomized study. J Pediatr. 2009;155:250-253.
Gattorno M, Caorsi R, Meini A, et al. Differentiating PFAPA syndrome from monogenic periodic fevers. Pediatrics. 2009;124(4):e721-e728.
Goldbach-Mansky R, Dailey NJ, Canna SW, et al. Neonatal-onset multisystem inflammatory disease responsive to interleukin-1β inhibition. N Engl J Med. 2006;355:581-592.
Hawkins PN, Lachmann HJ, Aganna E, et al. Spectrum of clinical features in Muckle-Wells syndrome and response to anakinra. Arthritis Rheum. 2004;50:607-612.
Hoffman HM, Throne ML, Amar NJ, et al. Efficacy and safety of rilonacept (interleukin-1 TRAP) in patients with cryopyrin-associated periodic syndromes: results from two sequential placebo-controlled studies. Arthritis Rheum. 2008;58:2443-2452.
Hull KM, Drewe E, Aksentijevich I, et al. The TNF receptor-associated periodic syndrome (TRAPS): emerging concepts of an autoinflammatory disorder. Medicine. 2002;81:349-368.
Jacobs Z, Ciaccio CE. Periodic fever syndromes. Curr Allergy Asthma Rep. 2010;10(6):398-404.
Kallinich T, Haffner D, Niehues T, et al. Colchicine use in children and adolescents with familial Mediterranean fever: literature review and consensus statement. Pediatrics. 2007;119:e474-e483.
Kalyoncu U, Eker A, Oguz KK, et al. Familial Mediterranean fever and central nervous system involvement. Medicine. 2010;89:75-84.
Lachmann HJ, Kone-Paut I, Kuemmerle-Deschner JB, et al. Use of canakinumab in the cryopyrin-associated periodic syndrome. N Engl J Med. 2009;360:2416-2425.
Lepore L, Paloni G, Caorsi R, et al. Follow-up and quality of life of patients with cryopyrin-associated periodic syndromes treated with anakinra. J Pediatr. 2010;157:310-315.
Nevien B, Valayannopoulos V, Quartier P, et al. Allogenic bone marrow transplantation in mevalonic aciduria. N Engl J Med. 2007;356:2700-2703.
Obici L, Manno C, Muda AO, et al. First report of systemic reactive (AA) amyloidosis in a patient with the hyperimmunoglobulinemia D with periodic fever syndrome. Arthritis Rheum. 2004;50:2966-2969.
Padeh S, Livneh A, Pras E, et al. Familial Mediterranean fever in the first two years of life: a unique phenotype of disease in evolution. J Pediatr. 2010;156:985-989.
Reddy S, Jia S, Geoffrey R, et al. An autoinflammatory disease due to homozygous deletion of the IL1RN locus. N Engl J Med. 2009;360:2438-2444.
Renko M, Salo E, Putto-Laurila A, et al. A randomized, controlled trial of tonsillectomy in periodic fever, aphthous stomatitis, pharyngitis, and adenitis syndrome. J Pediatr. 2007;151:289-292.
Ryan JG, Goldbach-Mansky R. The spectrum of autoinflammatory diseases: recent bench to bedside observations. Curr Opin Rheumatol. 2008;20:66-75.
Steichen O, van der Hilst J, Simon A, et al. A clinical criterion to exclude the hyperimmunoglobulin D syndrome (mild mevalonate kinase deficiency) in patients with recurrent fever. J Rheumatol. 2009;36:1677-1681.
Tasher D, Somekh E, Dalal I. PFAPA syndrome: new clinical aspects disclosed. Arch Dis Child. 2006;91:981-984.
Tunca M, Akar S, F Onen. Turkish FMF Study Group: The familial Mediterranean fever (FMF) in Turkey. Medicine. 2005;84:1-11.
Van der Hilst JCH, Bodar EJ, Barron KS, et al. Long-term follow-up, clinical features, and quality of life in a series of 103 patients with hyperimmunoglobulinemia D syndrome. Medicine. 2008;87:301-310.
Zhan H, Sinclair J, Adams S, et al. Immune reconstruction and recovery of FOXP3 (forkhead box P3)-expressing T cells after transplantation for IPEX (immune dysregulation, polyendocrinopathy, enteropathy, X-linked) syndrome. Pediatrics. 2008;121:e998-e1002.