Chapter 270 Transmissible Spongiform Encephalopathies
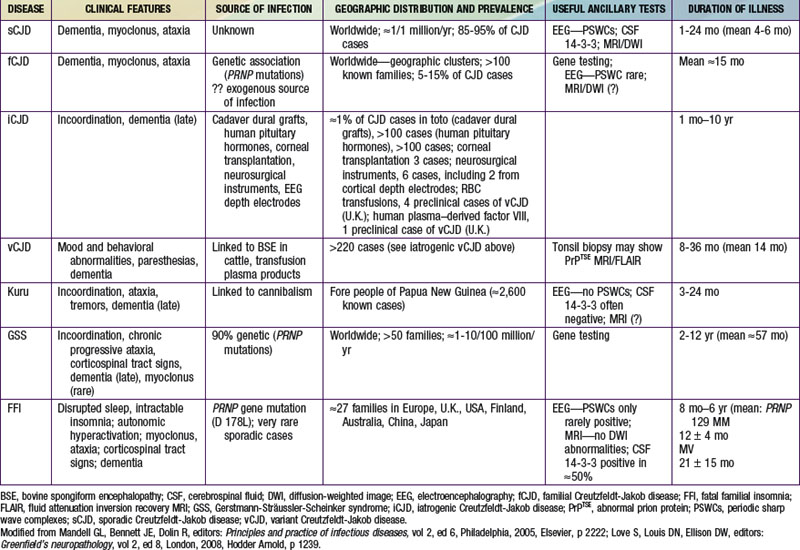
The transmissible spongiform encephalopathies (TSEs) are slow infections of the human nervous system, consisting of at least 4 diseases of humans (see Table 270-1  on the Nelson Textbook of Pediatrics website at www.expertconsult.com): kuru; Creutzfeldt-Jakob disease (CJD) with its variants—sporadic CJD (sCJD), familial CJD (fCJD), iatrogenic CJD (iCJD), and new-variant or variant CJD (vCJD); Gerstmann-Sträussler-Scheinker syndrome (GSS); and fatal familial insomnia (FFI), or the even more rare sporadic fatal insomnia syndrome. TSEs also affect animals; the most common and best-known TSEs of animals are scrapie in sheep, bovine spongiform encephalopathy (BSE or mad cow disease) in cattle, and a chronic wasting disease (CWD) of deer, elk, and moose found in parts of the USA and Canada. All TSEs have similar clinical manifestations and histopathology, and all are “slow” infections with very long asymptomatic incubation periods (often years), durations of several months or more, and overt disease affecting only the nervous system. The most striking neuropathologic change that occurs in each TSE, to a greater or lesser extent, is spongy degeneration of the cerebral cortical gray matter.
on the Nelson Textbook of Pediatrics website at www.expertconsult.com): kuru; Creutzfeldt-Jakob disease (CJD) with its variants—sporadic CJD (sCJD), familial CJD (fCJD), iatrogenic CJD (iCJD), and new-variant or variant CJD (vCJD); Gerstmann-Sträussler-Scheinker syndrome (GSS); and fatal familial insomnia (FFI), or the even more rare sporadic fatal insomnia syndrome. TSEs also affect animals; the most common and best-known TSEs of animals are scrapie in sheep, bovine spongiform encephalopathy (BSE or mad cow disease) in cattle, and a chronic wasting disease (CWD) of deer, elk, and moose found in parts of the USA and Canada. All TSEs have similar clinical manifestations and histopathology, and all are “slow” infections with very long asymptomatic incubation periods (often years), durations of several months or more, and overt disease affecting only the nervous system. The most striking neuropathologic change that occurs in each TSE, to a greater or lesser extent, is spongy degeneration of the cerebral cortical gray matter.
Etiology
The TSEs are transmissible to susceptible animals by inoculation of tissues from affected subjects. Although the infectious agents replicate in some cell cultures, they do not achieve the high titers of infectivity found in brain tissues or cause recognizable cytopathic effects in cultures. Most studies of TSE agents have used in vivo assays, relying on the transmission of typical neurologic disease to animals as evidence that the agent was present and intact. Inoculation of susceptible recipient animals with small amounts of infectious TSE agent results, months later, in the accumulation in tissues of large amounts of agent with the same physical and biologic properties as the original agent. The TSE agents display a spectrum of extreme resistance to inactivation by a variety of chemical and physical treatments that is unknown among conventional pathogens. This characteristic, as well as their partial sensitivity to protein-disrupting treatments and their consistent association with abnormal isoforms of a normal host-encoded protein (prion protein or PrP), stimulated the hypothesis that the TSE agents are probably subviral in size, composed of protein, and devoid of nucleic acid.
The term prion (for proteinaceous infectious agent) has been suggested as an appropriate name for such agents. The prion hypothesis proposes that the molecular mechanism by which the pathogen-specific information of TSE agents is propagated involves a self-replicating change in the folding host-encoded PrP associated with a transition from an α-helix–rich structure in the native protease-sensitive conformation (cellular PrP or PrPC) to a β-sheet–rich structure in the protease-resistant conformation associated with infectivity. The existence of a 2nd host-encoded protein—termed “protein X”—that participates in the transformation was also postulated to explain certain otherwise puzzling findings.
The prion hypothesis is not universally accepted; it relies on the postulated existence of a genome-like coding mechanism based on differences in protein folding that have not been satisfactorily explained at a molecular level. In addition, it has yet to account convincingly for the many biologic strains of TSE agent that have been observed, although strain-specific differences in the abnormal forms of the PrP have been found and proposed as providing a molecular basis for the coding. It fails to explain why pure PrP uncontaminated with nucleic acid from an infected host has not transmitted a convincingly typical spongiform encephalopathy associated with a serially self-propagating agent. Also troubling, in several experimental models, abnormal PrP and infectivity were not consistently associated. If the TSE agents ultimately prove to consist of protein and only protein, without any obligatory nucleic acid component, then the term prion will indeed be appropriate. If the agents are ultimately found to contain small nucleic acid genomes, then they might better be considered atypical viruses, for which the term virino has been suggested. Until the actual molecular structure of the infectious TSE pathogens and the presence or absence of a nucleic acid genome are rigorously established, it seems less contentious to continue calling them TSE agents, although many authorities use the term prion.
The 1st evidence that abnormal proteins are associated with the TSE was morphologic: scrapie-associated fibrils (SAFs) were found in extracts of tissues from patients and animals with spongiform encephalopathies but not in normal tissues. SAFs resemble but are distinguishable from the amyloid fibrils that accumulate in the brains of patients with Alzheimer disease. A group of antigenically-related protease-resistant proteins (PrPs) proved to be components of SAF and to be present in the amyloid plaques found in the brains of patients and animals with TSEs. The abnormal forms of PrP are variously designated PrPSc (scrapie-type PrP), PrP-res (protease-resistant PrP), PrPTSE (TSE-associated PrP), or PrPD (disease-associated PrP) by different authorities.
It remains unclear whether abnormal PrP constitutes the complete infectious particle of spongiform encephalopathies, is a component of those particles, or is a pathologic host protein not usually separated from the actual infectious entity by currently used techniques. The demonstration that PrP is encoded by a normal host gene seemed to favor the last possibility. Several studies have suggested that agent-specific pathogenic information can be transmitted and replicated by different conformations of a protein with the same primary amino acid sequence in the absence of agent-specific nucleic acids. Properties of 2 fungal proteins were found to be heritable without encoding in nucleic acid, although those properties have not been transmitted to recipient fungi as infectious elements. Whatever its relationship to the actual infectious TSE particles, PrP clearly plays a central role in susceptibility to infection, because the normal PrP must be expressed in mice and cattle if they are to acquire a TSE or to sustain replication of the infectious agents. Furthermore, inherited variations in PrP phenotype are associated with increased susceptibility to vCJD and with occurrence of fCJD.
PrPs are glycoproteins; protease-resistant PrPs have the physical properties of amyloid proteins. The PrPs of several species of animals are very similar in their amino acid sequences and antigenicity but are not identical in structure. The primary structure of PrP is encoded by the host and is not altered by the source of the infectious agent provoking its formation. The function of the ubiquitous protease-sensitive PrP precursor (designated PrPC or PrP-sen, for protease-sensitive PrP) in normal cells is unknown; it binds copper and may play some role in normal synaptic transmission, but it is not required for life or for relatively normal cerebral function in mice and cattle. As noted, expression of PrP is required both for development of scrapie disease and for replication of the transmissible scrapie agent in animals. The degree of homology between amino acid sequences of PrPs in different animal species may correlate with the “species barrier” that affects susceptibility of animals of 1 species to infection with a TSE agent adapted to grow in another species.
Attempts to find particles resembling those of viruses or virus-like agents in brain tissues of humans or animals with spongiform encephalopathies have been unsuccessful. Peculiar tubulovesicular structures reminiscent of some viruses have been seen in thin sections of TSE-infected brain tissues and cultured cells but not in normal cells. It has never been established that those structures are associated with infectivity.
It has been claimed that 2 other human diseases, familial Alzheimer disease of adults and Alpers disease of young children, may be caused by infections with agents similar to those causing the spongiform encephalopathies. The latter is a convulsive disorder associated with hemiatrophy and status spongiosus of the cerebral gray matter. Attempts to confirm these claims by transmission of disease to experimental animals failed.
Epidemiology
Kuru once affected many children ≥4 yr of age, adolescents, and young adults (mainly women) living in 1 limited area of Papua New Guinea. The complete disappearance of kuru among people born after 1957 suggests that the practice of ritual cannibalism (thought to have ended that year) was probably the only mechanism by which the infection was spread in Papua New Guinea.
CJD, the most common human spongiform encephalopathy, was formerly thought to occur only in older adults; however, iCJD and, much more rarely, sCJD have affected adolescents and young adults. GSS and the insomnia syndromes have not been diagnosed in children or adolescents. Variant CJD has a peculiar predilection for younger people; of 174 cases of CJD reported to date in the U.K., all except 23 were in people younger than 40 yr of age and 22 were under 20 yr of age. CJD has been recognized worldwide, at yearly rates of 0.25 to 2 cases/million population (not age-adjusted), with foci of considerably higher incidence among Libyan Jews in Israel, in isolated villages of Slovakia, and in other limited areas. Sporadic CJD has not been convincingly linked to any common exposure, and the source of infection remains unknown. Epidemiologic surveys have investigated several hypothetical mechanisms of spread of CJD. Person-to-person spread has been confirmed only for iatrogenic cases. Spouses and household contacts of patients are at very low risk of acquiring CJD, although 2 instances of conjugal CJD have been reported. However, medical personnel exposed to brains of patients with CJD may be at some increased risk; at least 20 health care workers have been recognized with the disease.
The striking resemblance of CJD to scrapie prompted a concern that infected sheep tissues might be a source of spongiform encephalopathy in humans. No reliable epidemiologic evidence suggests that exposure to potentially scrapie-contaminated animals, meat, meat products, or experimental preparations of the scrapie agent have transmitted a TSE to humans. The potential of the CWD agent to infect human beings has not been demonstrated but remains under investigation; deer, elk, and moose in 16 U.S. states and 2 Canadian provinces have been naturally infected and monkeys have been experimentally infected with the CWD agent. Exposure to contaminated meat, including venison from animals infected with the CWD agent, has not been implicated as a risk factor for sporadic CJD.
The outbreak of BSE among cattle (possibly infected by eating scrapie-agent–contaminated meat-and-bone meal added to feed) was 1st recognized in the United Kingdom in 1986, later reported in native cattle of 24 other countries, including Canada and the USA. The finding of a new TSE in ungulate and feline animals in British zoos and later in domestic cats raised a fear that some TSE agent (probably a strain of the scrapie agent), having crossed the species barrier from sheep to cattle, had acquired a broadened range of susceptible hosts, posing a potential danger for humans. That is the plausible explanation for the occurrence of vCJD, 1st described in adolescents in Britain in 1996 and as of December 2010 affecting at least 174 people in the U.K. (not counting several with evidence of “preclinical” vCJD infection), 25 in France, 5 in Spain, 4 in Ireland, 3 in the Netherlands, 2 each in Italy and Portugal, and single cases in Italy, Japan, and Saudi Arabia. Variant CJD has also occurred in former U.K. residents living in Canada (1 case) and the USA (2 cases); a 3rd case of vCJD in the USA was reported in a former resident of Saudi Arabia, a country that has not recognized BSE but might have imported contaminated meat products.
Iatrogenic transmissions of CJD have been recognized for >30 yr (Table 270-2). Such accidental transmissions of CJD have been attributed to use of contaminated neurosurgical instruments or operating facilities, use of cortical electrodes contaminated during epilepsy surgery, injections of human cadaveric pituitary growth hormone and gonadotropin, and transplantation of contaminated corneas and allografts of human dura mater used as a surgical patching material. Pharmaceuticals and tissue grafts derived from or contaminated with human neural tissues, particularly when obtained from unselected donors and large pools of donors, pose special risks.
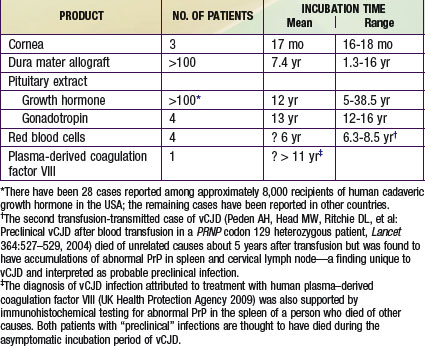
Studies of animals experimentally infected with TSE agents 1st suggested that blood and blood components from humans with preclinical CJD infections might pose a risk of transmitting disease to recipients, and since the 1980s such blood components have been withdrawn as a precaution when a donor was later found to have CJD and blood products were still in-date. While no epidemiologic study identified any subject exposed to such products obtained from donors later diagnosed with sporadic or familial CJD, a surveillance program in the U.K. has already reported vCJD in 3 recipients of red cells from donors later diagnosed with vCJD and evidence of a preclinical vCJD infection in a 4th red-cell recipient who died of another disease. Evidence of a preclinical vCJD infection was found at autopsy in a patient with hemophilia A treated with human plasma–derived coagulation factor VIII to which at least 1 vCJD-infected donor contributed. The coagulation factor involved is not licensed in the USA.
Pathogenesis and Pathology
The probable portal of entry for the TSE agent in kuru is thought to have been either through the gastrointestinal tract or lesions in the mouth or integument incidentally exposed to the agent during cannibalism. Subjects with vCJD (and animals with BSE and BSE-related TSEs) are thought to have been similarly infected with the BSE agent through exposure to a contaminated beef product, possibly through the intestinal tract. Except after direct introduction into the nervous system, the 1st site of replication of TSE agents appears to be in tissues of the reticuloendothelial system. TSE agents have been detected in low titers in blood of experimentally infected animals (mice, monkeys, hamsters, and sheep) and mainly associated with nucleated cells, although plasma contains a substantial portion of total infectivity in blood. Circulating lymphoid cells seem to be required to infect mice by peripheral routes. Limited evidence suggests that TSE agents also spread to the central nervous system (CNS) by ascending peripheral nerves. Several researchers claim to have developed tests that detected the CJD agent in human blood, although most attempts have failed. To date no blood-based test has been validated for antemortem testing of either humans or animals.
In human kuru, it seems probable that the only portal of exit of the agent from the body, at least in quantities sufficient to infect others, was through infected tissues exposed during cannibalism. In iatrogenically transmitted CJD, the brain and eyes of patients with CJD have been the probable sources of contamination. Kidney, liver, lung, lymph node, spleen, and cerebrospinal fluid (CSF) may also contain the CJD agent. At no time during the course of any TSE have antibodies or cell-mediated immunity to the infectious agents been convincingly demonstrated in either patients or animals. Mice must be immunologically competent to be infected with the scrapie agent by peripheral routes of inoculation.
Typical changes in TSE include vacuolation and loss of neurons with hypertrophy and proliferation of glial cells, most pronounced in the cerebral cortex in patients with CJD and in the cerebellum in those with kuru. The CNS lesions are usually most severe in or even confined to gray matter, at least early in the disease. Loss of myelin appears to be secondary to degeneration of neurons. There generally is no inflammation, but a marked increase in the number and size of astrocytes is usual. Spongiform changes are not a striking autopsy finding in patients with FFI, and neuronal degeneration and gliosis are largely restricted to thalamic nuclei.
Amyloid plaques are found in the brains of all patients with GSS and in at least 70% of those with kuru. These plaques are less common in patients with CJD. Amyloid plaques are most common in the cerebellum but occur elsewhere in the brain as well. In brains of patients with vCJD, plaques surrounded by halos of vacuoles (described as flower-like or florid plaques) have been a consistent finding. TSE amyloid plaques react with antiserum prepared against PrP. Even in the absence of plaques, extracellular PrP can be detected in the brain parenchyma by immunostaining.
Clinical Manifestations
Kuru is a progressive degenerative disease of the cerebellum and brainstem with less obvious involvement of the cerebral cortex. The 1st sign of kuru is usually cerebellar ataxia followed by progressive incoordination. Coarse, shivering tremors are characteristic. Variable abnormalities in cranial nerve function appear, frequently with impairment in conjugate gaze and swallowing. Patients die of inanition and pneumonia or of burns from cooking fires, usually 1 yr after onset. Although changes in mentation are common, there is no frank dementia or progression to coma, as in CJD. There are no signs of acute encephalitis such as fever, headaches, and convulsions.
CJD occurs throughout the world. Patients initially have either sensory disturbances (most often visual) or confusion and inappropriate behavior, with progression over weeks or months to frank dementia, akinetic mutism, and ultimately coma. Some patients have cerebellar ataxia early in disease, and most patients experience myoclonic jerking movements. Mean survival of patients with sCJD has been <1 yr from the earliest signs of illness, although about 10% live for 2 yr. Variant CJD (Table 270-3) differs from the more common sCJD: patients with vCJD are much younger at onset (as young as 12 yr) and more often present with complaints of dysesthesia and subtle behavioral changes, often mistaken for psychiatric illness. Severe mental deterioration occurs later in the course of vCJD. Patients with vCJD have survived substantially longer than those with sCJD.
Table 270-3 CLINICAL AND HISTOPATHOLOGIC FEATURES OF PATIENTS WITH VARIANT AND TYPICAL SPORADIC CREUTZFELDT-JAKOB DISEASE (CJD)
| FEATURE | VARIANT CJD (1st 10 PATIENTS) | SPORADIC CJD (185 PATIENTS) |
|---|---|---|
| Years of age at death* (range) | 29 (19-74) | 65 |
| Duration of illness, mo (range) | 12 (8-23) | 4 |
| Presenting signs | Abnormal behavior, dysesthesia | Dementia |
| Later signs | Dementia, ataxia, myoclonus | Ataxia, myoclonus |
| Periodic complexes on EEG | Rare | Most |
| PRNP 129 Met/Met | All tested (except 1 transfusion-transmitted case, 1 plasma-derivative transmitted case; 1 possible clinical case in U.K. where no tissue was available to confirm) | 83% |
| Histopathologic changes | Vacuolation, neuronal loss, astrocytosis, plaques (100%) | Vacuolation, neuronal loss, astrocytosis, plaques (≤15%) |
| Florid PrP plaques† | 100% | 0 |
| PrPTSE glycosylation pattern | BSE-like‡ | Not BSE-like |
BSE, bovine spongiform encephalopathy; EEG, electroencephalogram; Met, codon 129 of one PRNP gene encoding for methionine; PRNP, prion protein–encoding gene; PrP, prion protein.
* Median age and duration for variant CJD; averages for typical sporadic CJD.
† Dense plaques with a pale periphery of surrounding vacuolated cells.
‡ Characterized by an excess of high molecular mass band (diglycosylated) and 19 kd nonglycosylated band glycoform of PrP-res (Collinge J, Sidle KC, Meads J, et al: Molecular analysis of prion strain variation and the aetiology of “new variant” CJD, Nature 383:685–690, 1996).
Modified from Will RG, Ironside JW, Zeidler M, et al: A new variant of Creutzfeldt-Jakob disease in the UK, Lancet 347:921–925, 1996.
GSS is a familial disease resembling CJD but with more prominent cerebellar ataxia and amyloid plaques. Dementia may appear only late in the course, and the average duration of illness is longer than typical sCJD. Progressively severe insomnia and dysautonomia as well as ataxia, myoclonus, and other signs resembling those of CJD and GSS characterize FFI and sporadic fatal insomnia. Neither GSS nor an insomnia syndrome has been diagnosed in children or adolescents.
Diagnosis
Diagnosis of spongiform encephalopathies is most often determined on clinical grounds after excluding other diseases. The presence of 14-3-3 protein (see later) in CSF may aid in distinguishing between CJD and Alzheimer disease, although this is not a consideration in children. Elevations of 14-3-3 protein levels in CSF are not specific to TSEs and are common in viral encephalitis and other conditions causing rapid necrosis of brain tissue. Brain biopsy may be diagnostic of CJD, but it can be recommended only if a potentially treatable disease remains to be excluded or if there is some other compelling reason to make an antemortem diagnosis. Definitive diagnosis requires microscopic examination of brain tissue obtained at autopsy. The demonstration of protease-resistant PrP proteins in brain extracts can be useful to augment histopathologic diagnosis. Accumulation of the abnormal PrP in lymphoid tissues, even before the onset of neurologic signs, is typical of vCJD. Tonsil biopsy may prevent the need for brain biopsy when antemortem diagnosis of vCJD is indicated. Transmission of disease to susceptible animals by inoculation of brain suspension must be reserved for cases of special research interest.
Laboratory Findings
Virtually all patients with typical sporadic, iatrogenic, and familial forms of CJD have abnormal electroencephalograms (EEGs) as the disease progresses; the background becomes slow and irregular with diminished amplitude. A variety of paroxysmal discharges such as slow waves, sharp waves, spike-and-wave complexes may also appear, and these may be unilateral or focal or bilaterally synchronous. Paroxysmal discharges may be precipitated by loud noise. Many patients have typical periodic suppression-burst complexes of high-voltage slow activity on EEG at some time during the illness. Patients with vCJD have had only generalized slowing, without periodic bursts of high-voltage discharges on EEG. CT or MRI may show cortical atrophy and large ventricles late in the course of CJD. Many patients with vCJD have an increase in density of the pulvinar on MRI.
There may be modest elevation of CSF protein content in patients with TSE. Unusual protein spots were observed in CSF specimens after 2-dimensional separation in gels and silver staining; the spots were identified as 14-3-3 proteins, normal proteins (not related to PrP) abundant in neurons but not ordinarily detected in CSF. However, the finding of 14-3-3 protein in CSF has also been detected in CSF specimens from some patients with acute viral encephalitides and recent cerebral infarctions and thus is not specific to CJD. Finding the 14-3-3 protein in CSF is neither sensitive nor specific but has been of some help in confirming the diagnosis of vCJD, especially when accompanied by increases in other cellular proteins. Diagnosis usually rests on recognizing the typical constellation of clinical findings, clinical course, and testing (CSF examination, CT or MRI, EEG), confirmed by histopathology and detection of PrPTSE in tissues at autopsy (or, less often, tonsil or brain biopsy).
Treatment
No treatment has proven to be effective. Studies of cell cultures and rodents experimentally infected with TSE agents suggested that treatment with chlorpromazine, quinacrine, and tetracyclines might be of benefit, especially during the incubation period. Early reports of clinical trials based on those studies have been discouraging, and it seems unlikely that the severe brain damage found in late disease can be reversed by such treatment. Infusions with pentosan polysulfate directly into the cerebral ventricles appear to have delayed the progression of vCJD in a least 1 patient but did not reverse earlier brain damage. Appropriate supportive care should be provided to all CJD patients as for other progressive fatal neurologic diseases. On the basis of experimental studies in animals, several prophylactic postexposure treatment regimens have been suggested, but none have been widely accepted.
Genetic Counseling
TSE sometimes occurs in families in a pattern consistent with an autosomal dominant mode of inheritance. In patients with a family history of CJD, the clinical and histopathologic findings are similar to those seen in sporadic cases. In the USA, only about 10% of cases of CJD are familial. GSS and FFI are always familial. In some affected families, about 50% of siblings and children of a patient with a familial TSE eventually acquire the disease; in other families, the “penetrance” of illness may be less.
The gene coding for PrP is closely linked if not identical to that controlling the incubation periods of scrapie in sheep and both scrapie and CJD in mice. The gene encoding PrP in humans is designated the PRNP gene and is located on the short arm of chromosome 20. It has an open reading frame of about 759 nucleotides (253 codons), in which more than 20 different point mutations and a variety of inserted sequences encoding extra tandem-repeated octapeptides have been linked to the occurrence of spongiform encephalopathy in families with a pattern consistent with autosomal dominance of variable penetrance.
Although the interpretation of these findings in regard to the prion hypothesis is in dispute, in affected families with CJD or GSS, individuals who are heterozygous for linked mutations in the PRNP gene clearly have a high probability of eventually acquiring spongiform encephalopathy. The significance of mutations in the PRNP genes of individuals from families with no history of spongiform encephalopathy is not known. It seems wise to avoid alarming those who have miscellaneous mutations in the PRNP gene, because the implications are not yet clear.
The same nucleotide substitution at codon 178 of the PRNP gene associated with CJD in some families has been found in all patients with FFI. Homozygosity for valine and especially for methionine at codon 129 seems to increase susceptibility to iCJD and sCJD. Almost all patients with vCJD to be genotyped have been homozygous for methionine at codon 129 of the PRNP gene. A few probable preclinical vCJD infections and 1 clinically typical case of vCJD have been reported in persons with other genotypes.
Prognosis
The prognosis of all spongiform encephalopathies is uniformly poor. About 10% of patients may survive for >1 yr, but the quality of life is poor.
Family Support
The CJD Foundation, organized and maintained by family members and friends of patients with CJD and related disorders, working closely with the Centers for Disease Control and Prevention and with the National Prion Disease Pathology Surveillance Center, Cleveland, Ohio, is a useful source of information regarding available resources for those dealing with the diseases.
Prevention
Exposure to the BSE agent in meat products clearly poses a special danger. Authorities in Canada, the USA, and other countries have responded by implementing progressively more stringent agricultural and public health measures during the past 20 yr. No case of BSE in cattle has been recognized in the USA since 2006 (3 reported from the end of 2003 through 2006), but Canada has continued to find small numbers of affected cattle through 2010. In spite of encouraging epidemiological studies that failed to implicate exposure to scrapie or CWD agents in human TSEs, it seems prudent to avoid exposing children to meat and other products likely to be contaminated with any TSE agent.
The safety of human blood, blood components, and plasma derivatives in the USA and Canada is protected by deferring those donors with histories suggesting an increased risk of TSEs: persons treated with cadaveric pituitary hormones (no longer used) or dura mater allografts, patients with a family history of CJD (unless sequencing shows that they have no mutation in either PRNP gene), and patients spending substantial time in specified countries during years when BSE was prevalent. Persons transfused with blood in the U.K. and France after 1980 should be deferred from donating blood (similar deferral policies are in place for donors of human cells and tissues). U.K. authorities recently warned persons treated with U.K.-sourced pooled coagulation factor concentrates or antithrombin between 1989 and 2001 that they may be “at risk of vCJD for public health purposes” and that “special infection control precautions” apply to them.
In principle, it would be better to identify the few blood and tissue donors actually infected with a TSE rather than deferring all those at increased risk of exposure, most of whom are unlikely to have been infected. Accordingly, antemortem donor screening tests that might identify persons with preclinical TSE infections are currently under development but not validated. Another attractive approach would be to remove TSE agents from blood. Along these lines, a committee of expert advisors to the U.K. government recently recommended considering the use of an investigational device to filter red cells intended to transfuse children, because some unknown but possibly substantial number of U.K. blood donors might be incubating vCJD.
Standard precautions should be used to handle all human tissues, blood, and body fluids. Materials and surfaces contaminated with tissues or fluids from patients suspected of having CJD must be treated with great care. Whenever possible, discard contaminated instruments by careful packaging and incineration. Contaminated tissues and biologic products probably cannot be completely freed of infectivity without destroying their structural integrity and biologic activity; therefore, the medical and family histories of individual tissue donors should be carefully reviewed to exclude a diagnosis of TSE. Histopathologic examination of brain tissues of cadaveric donors and testing for abnormal PrP might be performed where feasible to provided an additional assurance of safety. Although no method of sterilization can be relied on to remove all infectivity from contaminated surfaces, exposures to moist heat, sodium hydroxide, chlorine bleach, concentrated formic acid, acidified detergent, and guanidine salts markedly reduced infectivity in experimental studies.
Asher DM. Kuru: memories of the NIH years. Philos Trans R Soc Lond B Biol Sci. 2008;363:3618-3625.
Asher DM. Slow viral infections: safe handling of the agents of subacute spongiform encephalopathies. In: Miller B, editor. Laboratory safety: principles and practices. Washington, DC: American Society for Microbiology; 1986:59-71.
Belay ED, Schonberger LB. The public health impact of prion diseases. Annu Rev Public Health. 2005;26:191-212.
Blossom DB, Maddox RA, Beavers SF, et al. A case of Creutzfeldt-Jakob disease associated with a dura mater graft in the United States. Infect Control Hosp Epidemiol. 2007;28:1396-1397.
Brandel JP, Heath CA, Head MW, et al. Variant Creutzfeldt-Jakob disease in France and the United Kingdom: evidence for the same agent strain. Ann Neurol. 2009;65:249-256.
Brown P, Gibbs CJr, Rodgers-Johnson P, et al. Human spongiform encephalopathy: the NIH series of 300 cases of experimentally transmitted disease. Ann Neurol. 1994;35:513-529.
Bruce ME, Will RG, Ironside JW, et al. Transmissions to mice indicate that ‘new variant’ CJD is caused by the BSE agent. Nature. 1997;389:498-501.
Büeler H, Aguzzi A, Sailer A, et al. Mice devoid of PrP are resistant to scrapie. Cell. 1993;73:1339-1347.
Centers for Disease Control and Prevention. BSE (bovine spongiform encephalopathy or mad cow disease). (website) www.cdc.gov/ncidod/dvrd/bse/ Accessed August 29, 2010
Centers for Disease Control and Prevention. Update: Creutzfeldt-Jakob disease associated with cadaveric dura mater grafts—Japan, 1978–2008. MMWR Morb Mortal Wkly Rep. 2008;57:1152-1154.
Croes EA, Roks G, Jansen GH, et al. Creutzfeldt-Jakob disease 38 years after diagnostic use of human growth hormone. J Neurol Neurosurg Psychiatry. 2002;72:792-793.
Deleault NR, Harris BT, Rees JR, et al. Formation of native prions from minimal components in vitro. Proc Natl Acad Sci U S A. 2007;104:9741-9746.
Dorsey K, Zou S, Schonberger LB, et al. Lack of evidence of transfusion transmission of Creutzfeldt-Jakob disease in a US surveillance study. Transfusion. 2009;49:977-984.
Gillies M, Chohan G, Llewelyn CA, et al. A retrospective case note review of deceased recipients of vCJD-implicated blood transfusions. Vox Sang. 2009;97:211-218.
Gregori L, Gurgel PV, Lathrop JT, et al. Reduction in infectivity of endogenous transmissible spongiform encephalopathies present in blood by adsorption to selective affinity resins. Lancet. 2006;368:2226-2230.
Gregori L, Lambert BC, Gurgel PV, et al. Reduction of transmissible spongiform encephalopathy infectivity from human red blood cells with prion protein affinity ligands. Transfusion. 2006;46:1152-1161.
Gregori L, McCombie N, Palmer D, et al. Effectiveness of leucoreduction for removal of infectivity of transmissible spongiform encephalopathies from blood. Lancet. 2004;364:529-531.
Kaski D, Mead S, Hyare H, et al. Variant CJD in an individual heterozygous for PRNP codon 129. Lancet. 2009;374:2128.
Manuelidis L. Transmissible encephalopathies: speculations and realities. Viral Immunol. 2003;16:123-139.
Manuelidis L, Yu ZX, Barquero N, et al. Cells infected with scrapie and Creutzfeldt-Jakob disease agents produce intracellular 25-nm virus-like particles. Proc Natl Acad Sci U S A. 2007;104:1965-1970.
Merz PA, Somerville RA, Wisniewski HM, et al. Abnormal fibrils from scrapie-infected brain. Acta Neuropathol. 1981;54:63-74.
Merz PA, Somerville RA, Wisniewski HM, et al. Scrapie-associated fibrils in Creutzfeldt-Jakob disease. Nature. 1983;306:474-476.
Mills JL, Schonberger LB, Wysowski DK, et al. Long-term mortality in the United States cohort of pituitary-derived growth hormone recipients. J Pediatr. 2004;144:430-436.
1991 . Occupational exposure to bloodborne pathogens; final rule. Fed Regist. 1991;56:64175-64182. www.osha.gov/pls/oshaweb/owadisp.show_document?p_table=STANDARDS&p_id=10051. Accessed August 29, 2010
Otto M, Wiltfang J, Cepek L, et al. Tau protein and 14–3-3 protein in the differential diagnosis of Creutzfeldt-Jakob disease. Neurology. 2002;58:192-197.
Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136-144.
Prusiner SB. Shattuck lecture—neurodegenerative diseases and prions. N Engl J Med. 2001;344:1516-1526.
Sehulster LM. Prion inactivation and medical instrument reprocessing: challenges facing healthcare facilities. Infect Control Hosp Epidemiol. 2004;25:276-279.
Spencer MD, Knight RS, Will RG. First hundred cases of variant Creutzfeldt-Jakob disease: retrospective case note review of early psychiatric and neurological features. BMJ. 2002;324:1479-1482.
Stewart LA, Rydzewska LH, Keogh GF, et al. Systematic review of therapeutic interventions in human prion disease. Neurology. 2008;70:1272-1281.
Stramer SL, Hollinger FB, Katz LM, et al. Emerging infectious disease agents and their potential threat to transfusion safety. Transfusion. 2009;49(Suppl 2):1S-29S.
Taylor D. Transmissible degenerative encephalopathies: inactivation of the unconventional causal agents. In: Fraise A, Lambert P, Maillard J-Y, editors. Russell, Hugo and Ayliffe’s principles and practice of disinfection, preservation and sterilization. ed 4. Malden, Massachusetts: Blackwell Publishing; 2004:324-341.
UK Health Protection Agency. vCJD abnormal prion protein found in a patient with haemophilia at post mortem. (press release February 17, 2009) www.hpa.org.uk/webw/HPAweb&HPAwebStandard/HPAweb_C/1234859690542?p=1231252394302 Accessed August 29, 2010
UK Health Protection Agency. vCJD-related abnormal prion protein in a person with hemophilia—an update. (press release February 17, 2009) www.hpa.org.uk/hpr/archives/2009/news2309.htm Accessed August 29, 2010
UK National CJD Surveillance Unit. CJD statistics. www.cjd.ed.ac.uk/figures.htm. Accessed December 7, 2009
UK National CJD Surveillance Unit. Variant Creutzfeldt-Jakob disease: current data (June 2010). www.cjd.ed.ac.uk/vcjdworld.htm. Accessed December 23, 2010
UK National CJD Surveillance Unit and Blood Services. Transfusion medicine epidemiology review (TMER) update Nov 2009. www.cjd.ed.ac.uk/TMER/TMER.htm. Accessed December 23, 2010
UK National CJD Surveillance Unit Edinburgh. Information on the new variant of CJD. www.cjd.ed.ac.uk/index.htm. Accessed December 23, 2010
Wilesmith JW, Wells GA, Cranwell MP, et al. Bovine spongiform encephalopathy: epidemiological studies. Vet Rec. 1988;123:638-644.
Will RG, Ironside JW, Zeidler M, et al. A new variant of Creutzfeldt-Jakob disease in the UK. Lancet. 1996;347:921-925.
Will RG, Zeidler M, Brown P, et al. Cerebrospinal-fluid test for new-variant Creutzfeldt-Jakob disease. Lancet. 1996;348:955.
World Health Organization. WHO guidelines on tissue infectivity distribution in transmissible spongiform encephalopathies (pdf). www.who.int/bloodproducts/TSEREPORT-LoRes.pdf. Accessed August 29, 2010
World Health Organization. WHO infection control guidelines for transmissible spongiform encephalopathies. Report of a WHO consultation Geneva, Switzerland, 23–26 March 1999 (pdf). www.who.int/csr/resources/publications/bse/whocdscsraph2003.pdf. Accessed August 29, 2010
Zou S, Fang CT, Schonberger LB. Transfusion transmission of human prion diseases. Transfus Med Rev. 2008;22:58-69.