Chapter 446 Congenital Dyserythropoietic Anemias
The congenital dyserythropoietic anemias (CDA) are a heterogeneous class of genetic disorders characterized by unique morphologic abnormalities in marrow erythroblasts: multinuclearity, abnormal nuclear fragments, and intrachromatin bridges between cells, associated with ineffective erythropoiesis. Three major types of CDA (types I, II, and III) are defined (see  Table 446-1 on the Nelson Textbook of Pediatrics website at www.expertconsult.com), although additional subgroups and variants have also been identified. These rare disorders are characterized by variable degrees of anemia, increased marrow erythroid activity (ineffective erythropoiesis), and secondary hemochromatosis.
Table 446-1 on the Nelson Textbook of Pediatrics website at www.expertconsult.com), although additional subgroups and variants have also been identified. These rare disorders are characterized by variable degrees of anemia, increased marrow erythroid activity (ineffective erythropoiesis), and secondary hemochromatosis.
Type I Congenital Dyserythropoietic Anemia
Pathogenesis
Type I CDA is an autosomal recessive disorder. A causative gene (CDAN1) has been localized to chromosome 15q15, and several different mutations have been identified. This gene encodes codanin-1, a protein that has been identified as a cell cycle–dependent nuclear protein whose synthesis is promoted by the transcription factor E2F1. Its exact cellular role remains to be elucidated. Notably, not all CDA-1 cases are linked to chromosome 15q15, implying that at least one other locus must exist.
Clinical Manifestations
The onset of macrocytic anemia and/or jaundice may be noted at any age, although most cases are first recognized during childhood or adolescence. Rarely, type 1 CDA is diagnosed in utero. Symptoms can include splenomegaly, hepatomegaly, gallstones, and mild jaundice. Type I CDA has been associated with skeletal (primarily syndactyly) and other dysmorphologies. Symptoms of iron overload and, in more severe cases, evidence of extramedullary hematopoiesis in frontal or parietal bones of the skull, may be present.
Laboratory Findings
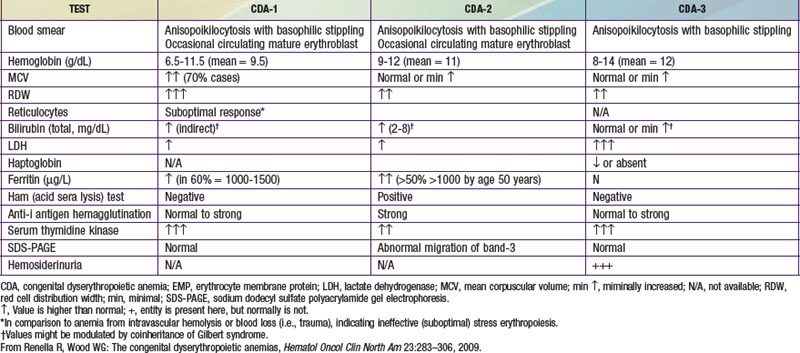
There is usually a moderate hemolytic anemia, abnormal or slightly elevated (but inadequate) reticulocyte count, macrocytosis, and high mean corpuscular volume (MCV). Anisopoikilocytosis is also appreciated on the peripheral blood smear. In some cases, normoblasts and basophilic stippling of red blood cells (RBCs) may be seen. White blood cells and platelets are normal. Laboratory evidence of iron overload may be present. The marrow exhibits erythroid hyperplasia and megaloblastic erythroblasts. A small number of erythroblasts manifest dyserythropoietic features with interchromatin bridges between cells that are diagnostic for type I CDA. Electron microscopy is the gold standard for diagnosis, revealing erythroblasts with a characteristic heterochromatin pattern.
Treatment
Treatment of this disorder is primarily supportive. The most important long-term complication is hemosiderosis caused by increased intestinal absorption of iron and ineffective erythropoiesis. Transfusions should be avoided where possible so as to prevent further iron loading. Chelation therapy should be employed when repeated ferritin levels exceed 1000 µg/L. Interferon-α has effectively raised the hemoglobin concentration and improved liver iron overload in many cases of documented CDA type I. Patients do not respond to erythropoietin, and splenectomy is not helpful. Transplantation with HLA-matched stem cells has been used in severe cases.
Type II Congenital Dyserythropoietic Anemia
Pathogenesis
CDA II is the most common form of CDA and is also an autosomal recessive disorder. Genome-wide linkage analysis identified a region of chromosome 20q11.2 as the location of the candidate CDAN2 gene, but the responsible gene remains elusive at present. There are cases of CDA II that are not linked to chromosome 20q11.2, implying genetic heterogeneity in this subtype as well. Defective glycosylation, affecting erythroid precursors, liver cells, and transferrin, is an associated biochemical feature of CDA II. However, CDA II is not a distinct glycosylation disorder but is rather caused by defective Golgi processing in erythroblasts.
Clinical Manifestations
In contrast to CDA I, this diagnosis is usually made later in life because symptoms can be mild. Characteristic findings include normocytic anemia, jaundice, splenomegaly, or hepatomegaly. Signs of iron overload may also be present.
Laboratory Findings
The anemia is normocytic and is generally mild. The reticulocyte count is inadequate and there is anisopoikilocytosis on the peripheral smear. Occasional basophilic stippling may be found as well. The bone marrow is normoblastic but hypercellular, with erythroid hyperplasia. Many more late marrow erythroblasts (up to 50%) may be abnormal, as manifested by binuclearity, multinuclearity, and abnormal lobulation. Pseudo-Gaucher cells may be present. The pathognomonic findings in type II CDA are that the patient’s RBCs are lysed by acidified serum. Type II CDA is known by the acronym HEMPAS (hereditary erythroblastic multinuclearity with a positive acidified serum test) because it features both erythroblast multinuclearity and circulating RBCs that are sensitive to lysis by acidified normal serum. RBCs also agglutinate strongly in sera containing anti-i antigens. Red cell membrane band 3 (anion exchange protein-1) and band 4.5 (glucose transporter-1) show thinner and faster migration on sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) due to defective glycosylation.
Treatment
In contrast to type I CDA, splenectomy is useful in patients with type II CDA. As in hereditary spherocytosis, RBC survival is normal following surgery, even though abnormal RBCs continue to circulate. Splenectomy does not prevent further iron overloading, even in patients whose hemoglobin is normalized, presumably because of persistent ineffective erythropoiesis in the bone marrow. Patients with severe anemia require blood transfusions. Iron overload may be present, and iron chelation therapy should be considered as clinically indicated. Transplantation has been curative. Most patients can lead a normal life and have a normal life expectancy if complications and consequences are managed appropriately.
Type III Congenital Dyserythropoietic Anemia
Type III CDA is the least common type of CDA and is a mild to moderate macrocytic anemia. It is inherited in an autosomal dominant fashion, although there have been cases that might represent de novo mutations or other inheritance patterns. The gene for type III CDA has been mapped to chromosome 15q22 near the CDAN1 gene. In contrast to other CDA types, iron overload is absent (probably because hemolysis is predominantly intravascular) and spleen size is generally normal. Patients can present with angioid streaks with macular degeneration. The blood smear shows macrocytes, anisopoikilocytosis, and occasional basophilic stippling. The bone marrow is notable for giant erythroid precursors that are often multinucleated, containing up to 12 nuclei per cell. Such multinucleated erythroblasts can also be seen in myelodysplasia and erythroleukemia. Transfusions usually are not required.
Chrobak L. Successful treatment of iron overload with phlebotomies in two siblings with congenital dyserythropoietic anemias-type II (CDA II). Acta Medica (Hradee Krabue). 2006;49:193-195.
Heimpel H. Congenital dyserythropoietic anemias: epidemiology, clinical significance, and progress in understanding their pathogenesis. Ann Hematol. 2004;83:613-621.
Heimpel H, Wilts H, Hirschmann WD, et al. Aplastic crisis as a complication of congenital dyserythropoietic anemia type II. Acta Haematol. 2006;117:115-118.
Iolascon A, Delaunay J. Close to unraveling the secrets of congenital dyserythropoietic anemias types I and II. Hematologica. 2009;94:599-602.
Noy-Lotan S, Dgany O, Lahmi R, et al. Codanin-1, the protein encoded by the gene mutated in congenital dyserythropoietic anemia type I (CDAN1) is cell cycle regulated. Hematologica. 2009;94:629-637.
Renella R, Wood WG. The congenital dyserythropoietic anemias. Hematol Oncol Clin N Am. 2009;23:283-306.
Wickramasinghe SN, Wood WB. Advances in the understanding of the congenital dyserythropoietic anaemias. Br J Haematol. 2005;131:431-446.