Chapter 458 Hemolytic Anemias Resulting from Extracellular Factors—Immune Hemolytic Anemias
Autoimmune Hemolytic Anemias
A number of extrinsic agents and disorders may lead to premature destruction of red blood cells (RBCs) (Table 458-1). Among the most clearly defined are antibodies associated with immune hemolytic anemias. The hallmark of this group of diseases is the positive result of the direct antiglobulin (Coombs) test, which detects a coating of immunoglobulin or components of complement on the RBC surface. The most important immune hemolytic disorder in pediatric practice is hemolytic disease of the newborn (erythroblastosis fetalis), caused by transplacental transfer of maternal antibody active against the RBCs of the fetus, that is, isoimmune hemolytic anemia (Chapter 97.2). Various other immune hemolytic anemias are autoimmune (see Table 458-1) and may be idiopathic or related to various infections (Epstein-Barr virus, rarely HIV, cytomegalovirus, and mycoplasma), immunologic diseases (systemic lupus erythematosus [SLE], rheumatoid arthritis), immunodeficiency diseases (agammaglobulinemia, autoimmune lymphoproliferative disorder, dysgammaglobulinemias), neoplasms (lymphoma, leukemia, and Hodgkin disease), or drugs (methyldopa, L-dopa). Other drugs (penicillins, cephalosporins) cause hemolysis by means of “drug-dependent antibodies—that is antibodies directed toward the drug and in some cases toward an RBC membrane antigen as well.
Table 458-1 DISEASES CHARACTERIZED BY IMMUNE-MEDIATED RED BLOOD CELL DESTRUCTION
AUTOIMMUNE HEMOLYTIC ANEMIA DUE TO WARM REACTIVE AUTOANTIBODIES
AUTOIMMUNE HEMOLYTIC ANEMIA DUE TO COLD REACTIVE AUTOANTIBODIES (CRYOPATHIC HEMOLYTIC SYNDROMES)
DRUG-INDUCED IMMUNE HEMOLYTIC ANEMIA (see Table 458-2)
Modified from Packman CH: Autoimmune hemolytic anemias. In Rakel R, editor: Conn’s current therapy, Philadelphia, 1995, WB Saunders, p 305.
Autoimmune Hemolytic Anemias Associated With “Warm” Antibodies
Etiology
In the autoimmune hemolytic anemias, abnormal antibodies are directed against RBC membrane antigens, but the pathogenesis of antibody induction is uncertain. The autoantibody may be produced as an inappropriate immune response to an RBC antigen or to another antigenic epitope similar to an RBC antigen, known as molecular mimicry. Alternatively, an infectious agent may alter the RBC membrane so that it becomes “foreign” or antigenic to the host. The antibodies usually react to epitopes (antigens) that are “public” or common to all human RBCs, such as Rh proteins.
In most instances of warm antibody hemolysis, no underlying cause can be found; this is the primary or idiopathic type (see Table 458-1). If the autoimmune hemolysis is associated with an underlying disease, such as a lymphoproliferative disorder, SLE, or immunodeficiency, it is secondary. In as many as 20% of cases of immune hemolysis, drugs may be implicated (Table 458-2).
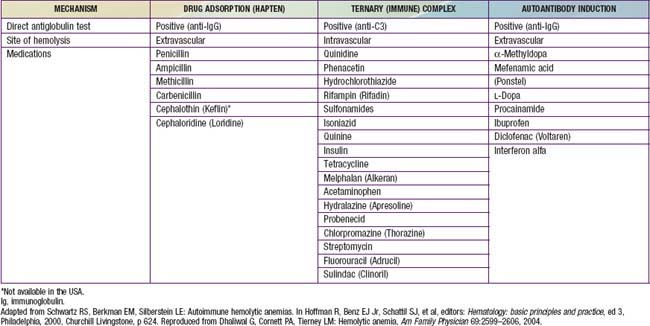
Drugs (penicillin or sometimes cephalosporins) that cause hemolysis via the “hapten” mechanism (immune but not autoimmune) bind tightly to the RBC membrane (see Table 458-1). Antibodies to the drug, either newly or previously formed, bind to the drug molecules on RBCs, mediating their destruction in the spleen. In other cases, certain drugs, such as quinine and quinidine, do not bind to RBCs but, rather, form part of a “ternary complex,” consisting of the drug, an RBC membrane antigen, and an antibody that recognizes both (see Table 458-1). Methyldopa and sometimes cephalosporins may, by unknown mechanisms, incite true autoantibodies to RBC membrane antigens, so that the presence of the drug is not required to cause hemolysis.
Clinical Manifestations
Autoimmune hemolytic anemias may occur in either of 2 general clinical patterns. The first, an acute transient type lasting 3-6 mo and occurring predominantly in children ages 2-12 yr, accounts for 70-80% of patients. It is frequently preceded by an infection, usually respiratory. Onset may be acute, with prostration, pallor, jaundice, fever, and hemoglobinuria, or more gradual, with primarily fatigue and pallor. The spleen is usually enlarged and is the primary site of destruction of immunoglobulin G (IgG)–coated RBCs. Underlying systemic disorders are unusual. A consistent response to glucocorticoid therapy, a low mortality rate, and full recovery are characteristic of the acute form. The other clinical pattern involves a prolonged and chronic course, which is more frequent in infants and in children >12 yr old. Hemolysis may continue for many months or years. Abnormalities involving other blood elements are common, and the response to glucocorticoids is variable and inconsistent. The mortality rate is approximately 10%, and death is often attributable to an underlying systemic disease.
Laboratory Findings
In many cases, anemia is profound, with hemoglobin levels <6 g/dL. Considerable spherocytosis and polychromasia (reflecting the reticulocyte response) are present. More than 50% of the circulating RBCs may be reticulocytes, and nucleated RBCs usually are present. In some cases, a low reticulocyte count may be found, particularly early in the episode. Leukocytosis is common. The platelet count is usually normal, but concomitant immune thrombocytopenic purpura sometimes occurs (Evans syndrome). The prognosis for patients with Evans syndrome is guarded, because many have or eventually have a chronic disease, including SLE, an immunodeficiency syndrome, or an autoimmune lymphoproliferative disorder.
Results of the direct antiglobulin test are strongly positive, and free antibody can sometimes be demonstrated in the serum (indirect Coombs test). These antibodies are active at 35-40°C (“warm” antibodies) and most often belong to the IgG class. They do not require complement for activity and are usually incomplete antibodies that do not produce agglutination in vitro. Antibodies from the serum and those eluted from the RBCs react with the RBCs of many persons in addition to those of the patient. They often have been regarded as nonspecific panagglutinins, but careful studies have revealed specificity for RBC antigens of the Rh system in 70% of patients (≈50% of adult patients). Complement, particularly fragments of C3b, may be detected on the RBCs in conjunction with IgG. The Coombs test result is rarely negative because of the limited sensitivity of the Coombs reaction. A minimum of 260-400 molecules of IgG per cell is necessary on the RBC membrane to produce a positive reaction. Special tests are required to detect the antibody in cases of “Coombs-negative” autoimmune hemolytic anemia. In warm antibody hemolytic anemia, the direct Coombs test may detect IgG alone, both IgG− and complement fragments, or solely complement fragments if the level of RBC-bound IgG is below the detection limit of the anti-IgG Coombs reagent.
Treatment
Transfusions may provide only transient benefit but may be lifesaving in cases of severe anemia by providing delivery of oxygen until the effect of other treatment is observed. In general, all tested units for transfusion are serologically incompatible. It is important to identify the patient’s ABO blood group in order to avoid a hemolytic transfusion reaction mediated by anti-A or anti-B. The blood bank should also test for the presence of an underlying allo-antibody, which could cause rapid hemolysis of transfused red cells. Patients who have neither been previously transfused nor pregnant are unlikely to harbor an alloantibody. Early consultation between the clinician and the blood bank physician is essential. Failure to transfuse a profoundly anemic infant or child may lead to serious morbidity and even death.
Patients with mild disease and compensated hemolysis may not require any treatment. If the hemolysis is severe and results in significant anemia or symptoms, treatment with glucocorticoids is initiated. Glucocorticoids decrease the rate of hemolysis by blocking macrophage function by down regulating Fcγ receptor expression, decreasing the production of the autoantibody, and perhaps enhancing the elution of antibody from the RBCs. Prednisone or its equivalent is administered at a dose of 2 mg/kg/24 hr. In some patients with severe hemolysis, doses of prednisone of up to 6 mg/kg/24 hr may be required to reduce the rate of hemolysis. Treatment should be continued until the rate of hemolysis decreases, and then the dose gradually reduced. If relapse occurs, resumption of the full dosage may be necessary. The disease tends to remit spontaneously within a few weeks or months. The Coombs test result may remain positive even after the hemoglobin level returns to normal. In general, it is safe to discontinue prednisone once the direct Coombs test result becomes negative. When hemolytic anemia remains severe despite glucocorticoid therapy, or if very large doses are necessary to maintain a reasonable hemoglobin level, IV immunoglobulin may be tried. Rituximab, a monoclonal antibody that targets B lymphocytes, the source of antibody production, has been useful in chronic cases refractory to conventional therapy. Plasmapheresis has been used in refractory cases but generally is not helpful. Splenectomy may be beneficial but is complicated by a heightened risk of infection with encapsulated organisms, particularly in patients <6 yr. Prophylaxis is indicated with appropriate vaccines (pneumococcal, meningococcal, and Haemophilus influenzae type b) before splenectomy and with oral penicillin after splenectomy.
Course and Prognosis
Acute idiopathic autoimmune hemolytic disease in childhood varies in severity but is self-limited; death from untreatable anemia is rare. Approximately 30% of patients have chronic hemolysis, often associated with an underlying disease, such as SLE, lymphoma, or leukemia. The presence of antiphospholipid antibodies in adult patients with immune hemolysis predisposes to thrombosis. Mortality in chronic cases depends on the primary disorder.
Autoimmune Hemolytic Anemias Associated with “Cold” Antibodies
“Cold” antibodies agglutinate RBCs at temperatures <37°C. They are primarily of the IgM class and require complement for hemolytic activity. The highest temperature at which RBC agglutination occurs is called the thermal amplitude. A higher thermal amplitude antibody—that is—one that can bind to RBCs at temperatures achievable in the body, results in hemolysis with exposure to a cold environment. High antibody titers are associated with a high thermal amplitude.
Cold Agglutinin Disease
Cold antibodies usually have specificity for the oligosaccharide antigens of the I/i system. They may occur in primary or idiopathic cold agglutinin disease, secondary to infections such as those from Mycoplasma pneumoniae and Epstein-Barr virus, or secondary to lymphoproliferative disorders. After M. pneumoniae infection, the anti-I levels may increase considerably, and occasionally, enormous increases may occur to titers ≥1/30,000. The antibody has specificity for the I antigen and thus reacts poorly with human cord RBCs, which possess the i antigen but exhibit low levels of I. Patients with infectious mononucleosis occasionally have cold agglutinin disease, and the antibodies in these patients often have anti-i specificity. This antibody causes less hemolysis in adults than in children because adults have fewer i molecules on their RBCs. Spontaneous RBC agglutination is observed in the cold and in vitro, and RBC aggregates are seen on the blood film. Mean corpuscular volume may be spuriously elevated because of RBC agglutination. The severity of the hemolysis is related to the thermal amplitude of the antibody, which itself partly depends on the IgM antibody titer.
When very high titers of cold antibodies are present and active near body temperature, severe intravascular hemolysis with hemoglobinemia and hemoglobinuria may occur and may be heightened on a patient’s exposure to cold (external temperature or ingested foods). Each IgM molecule has the potential to activate a C1 molecule so that large amounts of complement are found on the RBCs in cold agglutinin disease. These sensitized RBCs may undergo intravascular complement-mediated lysis or may be destroyed in the liver and spleen.
Cold agglutinin disease is less common in children than in adults and more frequently results in an acute, self-limited episode of hemolysis. Glucocorticoids are much less effective in cold agglutinin disease than in disease with warm antibodies. Patients should avoid exposure to cold and should be treated for underlying disease. In the uncommon patients with severe hemolytic disease, treatment includes immunosuppression and plasmapheresis. Successful treatment of cold agglutinin disease has been reported with the monoclonal antibody rituximab, which effectively depletes B lymphocytes. Splenectomy is not useful in cold agglutinin disease.
Paroxysmal Cold Hemoglobinuria
Paroxysmal cold hemoglobinuria is mediated by the Donath-Landsteiner hemolysin, which is an IgG cold-reactive autoantibody with anti-P specificity. This antibody fixes large amounts of complement in the cold, and the RBCs are lysed as the temperature is increased. Most reported cases are self-limited and are usually associated with nonspecific viral infections. They are now rarely found in association with congenital or acquired syphilis. This disorder may account for 30% of immune hemolytic episodes among children. Treatment includes transfusion for severe anemia and avoidance of cold ambient temperatures.
Berentsen S, Ulvestad E, Gjertsen BT, et al. Rituximab for primary cold agglutinin disease: a prospective study of 37 courses of therapy in 27 patients. Blood. 2004;103:2925-2928.
Flores G, Cunningham-Rundles C, Newland AC, et al. Efficacy of intravenous immunoglobulin in the treatment of autoimmune hemolytic anemia: results in 73 patients. Am J Hematol. 1993;44:237-242.
Gehrs BC, Friedberg RC. Autoimmune hemolytic anemia. Am J Hematol. 2002;69:258-271.
King KE, Ness PM. Treatment of hemolytic anemia. Semin Hematol. 2005;42:131-136.
Packman CH. Hemolytic anemia resulting from immune injury. In: Kanshansky K, Lichtman MA, Beutler E, et al, editors. Williams hematology. ed 8. New York: McGraw-Hill; 2010:777-798.
Petz L. Treatment of autoimmune hemolytic anemias. Curr Opin Hematol. 2001;8:411-416.
Sève P, Bourdillon L, Sarrot-Reynauld F, et al. Autoimmune hemolytic anemia and common variable immunodeficiency. Medicine. 2008;87:177-184.
Sparling TG, Andricevic M, Wass H. Remission of cold hemagglutinin disease induced by rituximab therapy. CMAJ. 2001;164:1405.
Zecca M, Nobili B, Ramenghi U, et al. Rituximab for the treatment of refractory autoimmune hemolytic anemia in children. Blood. 2003;101:3857.