Chapter 490 Lymphoma*
Lymphoma is the third most common cancer among U.S. children (age ≤14 yr), with an annual incidence of 15 cases per million children. It is the most common cancer in adolescents, accounting for >25% of newly diagnosed cancers in persons 15 to 19 yr old. The two broad categories of lymphoma, Hodgkin lymphoma (HL) and non-Hodgkin lymphoma (NHL), have different clinical manifestations and treatments.
490.1 Hodgkin Lymphoma
HL is a malignant process of the lymphoreticular system that accounts for 6% of childhood cancers. In the USA, HL accounts for about 5% of cancers in persons ≤14 yr of age and for about 15% of cancers in adolescents, making HL the most common malignancy in this age group (15-19 yr). It is rare in children <10 yr of age.
Epidemiology
The worldwide incidence of HL is approximately 2-4 new cases/100,000 population/year; there is a bimodal age distribution, with peaks at 15-35 yr of age and again after 50 yr. In developing countries, the early peak tends to occur prior to adolescence. A male:female predominance is found among young children but lessens with age. Several studies suggest that infectious agents may be involved, such as human herpes virus 6, cytomegalovirus, and Epstein-Barr virus (EBV). The role of EBV is supported by prospective serologic studies and confers a fourfold higher risk of developing HL and may precede the diagnosis by years. EBV antigens have been demonstrated in HL tissues, although EBV status is not prognostic of outcome.
Pathogenesis
The Reed-Sternberg (RS) cell, a pathognomonic feature of HL, is a large cell (15-45 µm in diameter) with multiple or multilobulated nuclei. This cell type is considered the hallmark of HL, although similar cells are seen in infectious mononucleosis, NHL, and other conditions. The Reed-Sternberg cell is clonal in origin and arises from the germinal center B cells. HL is characterized by a variable number of RS cells surrounded by an inflammatory infiltrate of lymphocytes, plasma cells, and eosinophils in different proportions, depending on the HL histologic subtype. Other features that distinguish the histologic subtypes include various degrees of fibrosis and the presence of collagen bands, necrosis, or malignant reticular cells (Fig. 490-1). The distribution of subtypes varies with age (Fig. 490-2).
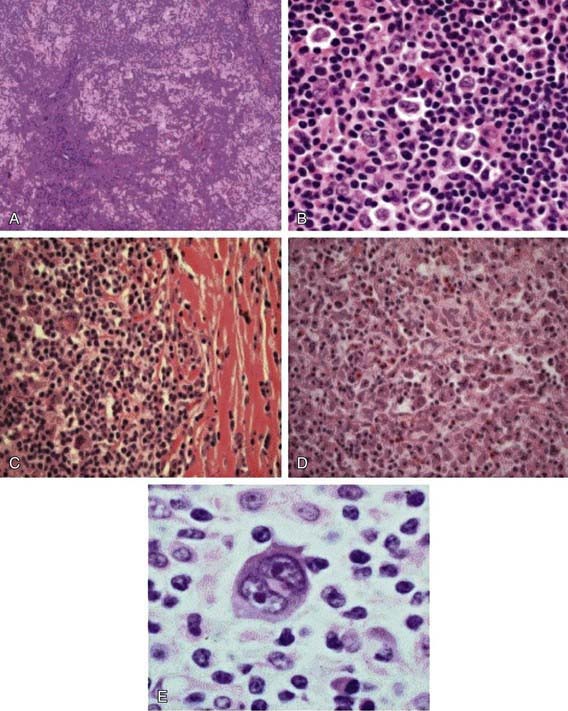
Figure 490-1 Histologic subtypes of Hodgkin lymphoma. A, Hematoxylin & eosin stains of nodular lymphocyte-predominant Hodgkin lymphoma (NLPHL) demonstrating a nodular proliferation with a moth-eaten appearance. B, High-power view demonstrating the neoplastic L and H cells found in NLPHL. C, Classic Hodgkin lymphoma, nodular sclerosis subtype. Large mononuclear and binucleate Reed-Sternberg cells are seen admixed in the inflammatory cell background. D, Classic Hodgkin lymphoma, mixed cellularity subtype, demonstrating increased numbers of Reed-Sternberg cells in a mixed inflammatory background without sclerotic changes. E, High-power view of a classic Reed-Sternberg cell showing binucleate cells with prominent eosinophilic nucleoli and relatively abundant cytoplasm.
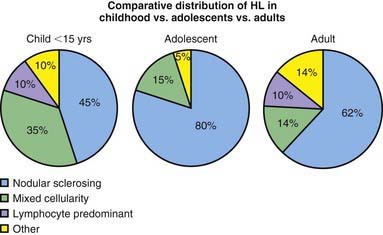
Figure 490-2 Comparative distribution of Hodgkin lymphoma in children, adolescents, and adults.
(Adapted from Hochberg J, Waxman IM, Kelly KM, et al: Adolescent non-Hodgkin lymphoma and Hodgkin lymphoma: state of the science, Br J Haematol 144:24–40, 2008.)
The Revised World Health Organization (WHO) Classification of Lymphoid Neoplasms (Table 490-1) includes two modifications of the older Rye system. HL appears to arise in lymphoid tissue and spreads to adjacent lymph node areas in a relatively orderly fashion. Hematogenous spread also occurs, leading to involvement of the liver, spleen, bone, bone marrow, or brain, and is usually associated with systemic symptoms.
Table 490-1 NEW WORLD HEALTH ORGANIZATION/REVISED EUROPEAN-AMERICAN CLASSIFICATION OF LYMPHOID NEOPLASMS CLASSIFICATION SYSTEM FOR HODGKIN LYMPHOMA
From Harris NL, Jaffe ES, Diebold J, et al: The World Health Organization classification of neoplastic diseases of the haematopoietic and lymphoid tissues: report of the Clinical Advisory Committee Meeting, Airlie House, Virginia, November 1997, Histopathology 36:69–87, 2000.
Clinical Manifestations
Patients commonly present with painless, nontender, firm, rubbery, cervical or supraclavicular lymphadenopathy and usually some degree of mediastinal involvement. Clinically detectable hepatosplenomegaly is rarely encountered. Depending on the extent and location of nodal and extranodal disease, patients may present with symptoms and signs of airway obstruction (dyspnea, hypoxia, cough), pleural or pericardial effusion, hepatocellular dysfunction, or bone marrow infiltration (anemia, neutropenia, or thrombocytopenia). Disease manifesting below the diaphragm is rare and occurs in approximately 3% of all cases. Systemic symptoms, classified as B symptoms that are considered important in staging, are unexplained fever >39°C, weight loss >10% total body weight over 3 mo, and drenching night sweats. Less common and not considered of prognostic significance are symptoms of pruritus, lethargy, anorexia, or pain that worsens after ingestion of alcohol. Patients also exhibit immune system abnormalities that often persist during and after therapy.
Diagnosis
Any patient with persistent, unexplained lymphadenopathy unassociated with an obvious underlying inflammatory or infectious process should undergo chest radiography to identify the presence of a large mediastinal mass before undergoing lymph node biopsy. Formal excisional biopsy is preferred over needle biopsy to ensure that adequate tissue is obtained, both for light microscopy and for appropriate immunohistochemical and molecular studies. Once the diagnosis of HL is established, extent of disease (stage) should be determined to allow selection of appropriate therapy (Table 490-2). Evaluation includes history, physical examination, and imaging studies, including chest radiograph; CT scans of the chest, abdomen, and pelvis; and either gallium scan or positron emission tomography (PET) scan. Laboratory studies should include a complete blood cell count (CBC) to identify abnormalities that might suggest marrow involvement; erythrocyte sedimentation rate (ESR); and measurement of serum ferritin, which is of some prognostic significance and, if abnormal at diagnosis, serves as a baseline to evaluate the effects of treatment. A chest radiograph is particularly important for measuring the size of the mediastinal mass in relation to the maximal diameter of the thorax (Fig. 490-3). This determines “bulk” disease and becomes prognostically significant. Chest CT more clearly defines the extent of a mediastinal mass if present and identifies hilar nodes and pulmonary parenchymal involvement, which may not be evident on chest radiographs (Fig. 490-4). Bone marrow aspiration and biopsy should be performed to rule out advanced disease. Bone scans are performed in patients with bone pain and/or elevation of alkaline phosphatase. Gallium scan can be particularly helpful in identifying areas of increased uptake, which can then be re-evaluated at the end of treatment. Fluorodeoxyglucose (FDG)–PET imaging has advantages over gallium scanning, as it is a 1-day procedure with higher resolution, better dosimetry, less intestinal activity, and the potential to quantify disease. PET scans are being evaluated as a prognostic tool in HL enabling therapy to be reduced in those predicted to have a good outcome.
Table 490-2 ANN ARBOR STAGING CLASSIFICATION FOR HODGKIN LYMPHOMA*
| STAGE | DEFINITION |
|---|---|
| I | Involvement of a single lymph node (I) or of a single extralymphatic organ or site (IE) |
| II | Involvement of two or more lymph node regions on the same side of the diaphragm (II) or localized involvement of an extralymphatic organ or site and one or more lymph node regions on the same side of the diaphragm (IIE) |
| III | Involvement of lymph node regions on both sides of the diaphragm (III), which may be accompanied by involvement of the spleen (IIIS) or by localized involvement of an extralymphatic organ or site (IIIE) or both (IIISE) |
| IV | Diffuse or disseminated involvement of one or more extralymphatic organs or tissues with or without associated lymph node involvement |
* The absence or presence of fever >38°C for 3 consecutive days, drenching night sweats, or unexplained loss of ≥10% of body weight in the 6 months preceding admission are to be denoted in all cases by the suffix letter A or B, respectively.
From Lister TA, Crowther D, Sutcliffe SB, et al: Report of a committee convened to discuss the evaluation and staging of patients with Hodgkin’s disease: Cotswolds meeting, J Clin Oncol 7:1630–1636, 1989.
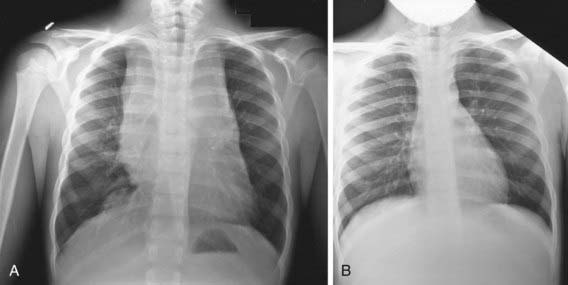
Figure 490-3 A, Anterior mediastinal mass in a patient with Hodgkin disease before therapy. B, After 2 mo of chemotherapy, the mediastinal mass has disappeared.
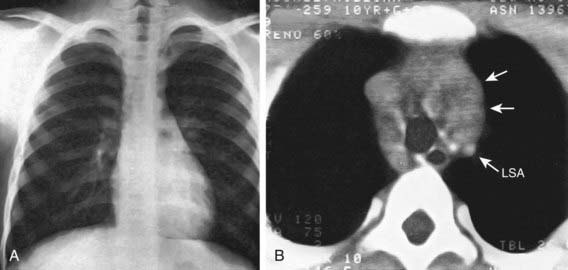
Figure 490-4 CT for the diagnosis of mediastinal disease in Hodgkin lymphoma. A, Chest radiograph, findings of which were interpreted as normal. B, Contrast-enhanced CT scan reveals marked widening of the mediastinum by an irregular enhancing mass (arrows) that is displacing the great vessels posteriorly. Right-sided paratracheal adenopathy is also present posterior to the superior vena cava. LSA, left subclavian artery.
(From Slovis TL, editor: Caffey’s pediatric diagnostic imaging, ed 11, vol 1, Philadelphia, 2008, Mosby/Elsevier.)
The staging classification currently used for HL was adopted at the Ann Arbor Conference in 1971 and was revised in 1989 (see Table 490-2). HL can be subclassified into A or B categories: A is used to identify asymptomatic patients and B is for patients who exhibit any B symptoms. Extralymphatic disease resulting from direct extension of an involved lymph node region is designated by category E. A complete response in HL is defined as the complete resolution of disease on clinical examination and imaging studies or at least 70-80% reduction of disease and a change from initial positivity to negativity on either gallium or PET scanning because residual fibrosis is common.
Treatment
Multiple agents allow different mechanisms of action to have non-overlapping toxicities so that full doses can be given to each patient. Chemotherapy and radiation therapy are both effective in the treatment of HL. Current treatment of HL in pediatric patients is risk adapted and involves the use of combined chemotherapy with or without low-dose involved-field radiation therapy based on response. Treatment is determined largely by disease stage, presence or absence of B symptoms, and the presence of bulky nodal disease. Radiation therapy alone, once given at higher doses, initially resulted in prolonged remission and cure rates in patients with low-stage HL. However, this treatment approach also caused significant long-term morbidity in pediatric patients, including growth retardation, thyroid dysfunction, and cardiac and pulmonary toxicity. The development of effective multiagent combination chemotherapy was a major milestone in the treatment of HL resulting in a complete response rate of 70-80% and cure rate of 40-50% in patients with advanced stage disease. However, as with radiation therapy, this regimen also led to significant acute and long-term toxicity. The desire to reduce side effects and morbidity has stimulated attempts to reduce the intensity of chemotherapy as well as radiation dose and volume. Newer combinations of chemotherapy have reduced the risk of secondary cancers. Also, current radiation therapy utilizes lower amounts of overall radiation in addition to narrowing the radiation treatment field to either involved-field or even involved-node irradiation. The current Children’s Oncology Group trials are investigating whether radiation therapy can be eliminated altogether in patients who have a very good rapid early response to pre-radiation induction chemotherapy.
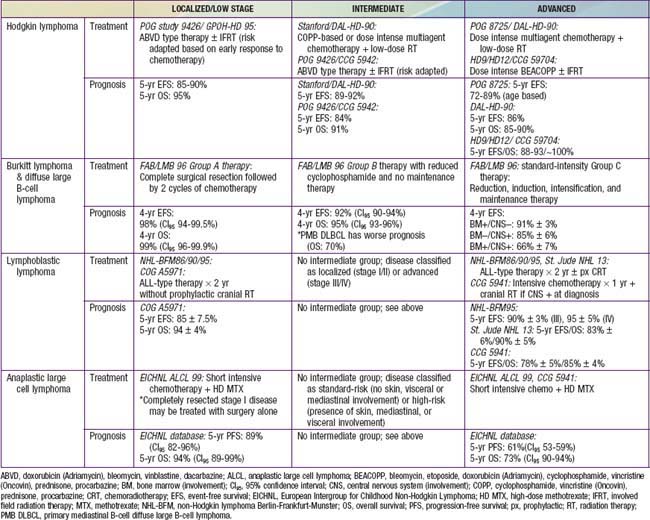
Chemotherapy agents commonly used to treat children and adolescents with HL include cyclophosphamide, procarbazine, vincristine or vinblastine, prednisone or dexamethasone, doxorubicin, bleomycin, dacarbazine, etoposide, methotrexate, and cytosine arabinoside. The combination chemotherapy regimens in current use are based on COPP (cyclophosphamide, vincristine [Oncovin], procarbazine, and prednisone) or ABVD (doxorubicin [Adriamycin], bleomycin, vinblastine, and dacarbazine), with the addition of prednisone, cyclophosphamide and etoposide (ABVE-PC and BEACOPP) in various combinations for intermediate- and high-risk groups (Table 490-3). “Risk-adapted” protocols are based on staging criteria as well as rapidity of response to initial chemotherapy. The aim is to reduce total drug doses and treatment duration and to eliminate radiation therapy if possible.
Table 490-3 CHEMOTHERAPY REGIMENS COMMONLY USED FOR CHILDREN, ADOLESCENTS, AND YOUNG ADULTS WITH HODGKIN LYMPHOMA
| CHEMOTHERAPY REGIMEN | CORRESPONDING AGENTS |
|---|---|
| ABVD | Doxorubicin (Adriamycin), bleomycin, vinblastine, dacarbazine |
| ABVE (DBVE) | Doxorubicin (Adriamycin), bleomycin, vincristine, etoposide |
| VAMP | Vincristine, doxorubicin (Adriamycin), methotrexate, prednisone |
| OPPA ± COPP (females) | Vincristine (Oncovin), prednisone, procarbazine, doxorubicin (Adriamycin), cyclophosphamide, vincristine (Oncovin), prednisone, procarbazine |
| OEPA ± COPP (males) | Vincristine (Oncovin), etoposide, prednisone, doxorubicin (Adriamycin), cyclophosphamide, vincristine (Oncovin), prednisone, procarbazine |
| COPP/ABV | Cyclophosphamide, vincristine (Oncovin), prednisone, procarbazine, doxorubicin (Adriamycin), bleomycin, vinblastine |
| BEACOPP (advanced stage) | Bleomycin, etoposide, doxorubicin (Adriamycin), cyclophosphamide, vincristine (Oncovin), prednisone, procarbazine |
| COPP | Cyclophosphamide, vincristine (Oncovin), prednisone, procarbazine |
| CHOP | Cyclophosphamide, doxorubicin (Adriamycin), vincristine (Oncovin), prednisone |
| ABVE-PC (DBVE-PC) | Doxorubicin (Adriamycin), bleomycin, vincristine, etoposide, prednisone, cyclophosphamide |
Newer agents such as those that disrupt the nuclear factor-κB (NF-κB) pathway or monoclonal antibodies that target RS tumor cells as well as the benign reactive cells that surround them are currently being investigated. Ongoing clinical trials report encouraging results with the use of anti-CD20 antibody (rituximab). In addition, anti-CD30 agents are being used that are targeted to the RS cells themselves, where CD30 is abundantly expressed. EBV-specific cytotoxic lymphocytes (CTLs) can be generated from patients with advanced HL. In clinical trials, these show promising results, with enhanced antiviral activity and stabilization of disease even though all patients have continued to have persistent disease. Still, this approach represents an exciting direction in adoptive cellular tumor immunology, and success may come with CTLs that have improved cytotoxicity that can overcome inhibitory signals.
Relapse
Most relapses occur within the first 3 yr after diagnosis, but relapses as late as 10 yr have been reported. Relapse cannot be predicted accurately with this disease. Poor prognostic features include tumor bulk, stage at diagnosis, and presence of B symptoms. Patients who never achieve remission or suffer relapse <12 mo after initiation of therapy are candidates for myeloablative chemotherapy and autologous stem cell transplantation with or without the addition of radiation therapy. This treatment is most successful in patients with chemoresponsive disease. Myeloablative allogeneic stem cell transplantation reduces the relapse rate in patients with high-risk relapsed or refractory HL. There was no improvement in overall survival in the studies reported, owing to a high transplant-related mortality. These results do suggest a graft versus HL effect. The use of nonmyeloablative, nontoxic regimens to reduce regimen-related morbidity and mortality associated with myeloablative allogeneic stem cell transplantation but still achieve a graft versus HL effect is under investigation.
Prognosis
With the use of current therapeutic regimens, patients with favorable prognostic factors and early-stage disease have an event-free survival (EFS) of 85-90% and an overall survival (OS) at 5 yr >95%. Patients with advanced stage disease have slightly lower EFS (80-85%) and OS o (90%), respectively, although OS has approached 100% with dose-intense chemotherapy (Table 490-4). Prognosis after relapse depends on the time from completion of treatment to recurrence, site of relapse (nodal vs. extranodal), and presence of B symptoms at relapse. Patients whose disease relapses >12 mo after chemotherapy alone or combined-modality therapy have the best prognosis, and their relapses usually respond to additional standard therapy, resulting in a long-term survival of 60-70%. A myeloablative autologous stem cell transplantation in patients with refractory disease or relapse within 12 mo of therapy results in a long-term survival rate of 40-50%.
Armitage JO. Early-stage Hodgkin’s lymphoma. N Engl J Med. 2010;363(7):653-662.
Bartlett NL, Younes A, Carabasi MH, et al. A phase 1 multidose study of SGN-30 immunotherapy in patients with refractory or recurrent CD30+ hematologic malignancies. Blood. 2008;111:1848-1854.
NIH Pub No. 06-5767. Bleyer WA, O’Leary M, Barr R, et al, editors. Cancer epidemiology in older adolescents and young adults 15–29 years of age, including seer incidence and survival, 1975–2000. Bethesda, MD: National Cancer Institute, 2006.
Bollard CM, Aguilar L, Straathof KC, et al. Cytotoxic T lymphocyte therapy for Epstein-Barr virus + Hodgkin’s disease. J Exp Med. 2004;200:1623-1633.
Diehl V. Hodgkin’s disease—from pathology specimen to cure. N Engl J Med. 2007;357:1968-1971.
Engert A, Plütschow A, Eich HT, et al. Reduced treatment intensity in patients with early-stage Hodgkin’s lymphoma. N Engl J Med. 2010;363(7):640-652.
Ferme C, Eghbali H, Meerwaldt JH, et al. Chemotherapy plus involved-field radiation in early-stage Hodgkin’s disease. N Engl J Med. 2007;357:1916-1927.
Hudson MM, Donaldson SS. Hodgkin disease. In: Pizzo PA, Poplack DG, editors. Principles and practice of pediatric oncology. ed 4. Philadelphia: Lippincott Williams & Wilkins; 2002:637-660.
Kapatai G, Murray P. Contribution of the Epstein Barr virus to the molecular pathogenesis of Hodgkin lymphoma. J Clin Pathol. 2007;60:1342-1349.
Oeffinger KC, Mertens AC, Sklar CA, et al. Chronic health conditions in adult survivors of childhood cancer. N Engl J Med. 2006;355:1572-1582.
Oki Y, Pro B, Fayad LE, et al. Phase 2 study of gemcitabine in combination with rituximab in patients with recurrent or refractory Hodgkin lymphoma. Cancer. 2008;112:831-836.
Peggs KS, Hunter A, Chopra R, et al. Clinical evidence of a graft-versus-Hodgkin’s-lymphoma effect after reduced-intensity allogeneic transplantation. Lancet. 2005;365:1934-1940.
Steidl C, Lee T, Shah SP, et al. Tumor-associated macrophages and survival in classic Hodgkin’s lymphoma. N Engl J Med. 2010;362:875-885.
490.2 Non-Hodgkin Lymphoma
NHL accounts for approximately 60% of all lymphomas in children and adolescents. It represents 8-10% of all malignancies in children between 5 and 19 yr of age, with an annual incidence in the USA of 750-800 cases/yr in children ≤19 yr of age. Although >70% of patients present with advanced disease at diagnosis, the prognosis has improved dramatically, with survival rates of 90-95% for localized disease and 60-90% with advanced disease.
Epidemiology
Although most children and adolescents with NHL present with de novo disease, a small number of patients have NHL secondary to specific etiologies, including inherited or acquired immune deficiencies (e.g., severe combined immunodeficiency syndrome, Wiskott-Aldrich syndrome), viruses (e.g., HIV, EBV), and as part of genetic syndromes (e.g., ataxia-telangiectasia, Bloom syndrome). Most children in whom NHL develops have no obvious genetic or environmental etiology.
Pathogenesis
The four major pathologic subtypes of childhood and adolescent NHL are Burkitt lymphoma (BL), lymphoblastic lymphoma (LL), diffuse large B-cell lymphoma (DLBCL), and anaplastic large cell lymphoma (ALCL; Figs. 490-5 and 490-6). DLBCL is further divided into several subtypes: the germinal center B-cell like (GCB), which carries a favorable prognosis and accounts for the vast majority of pediatric cases of DLBCL, and the subtypes with poorer prognosis, activated B cell–like (ABC) and primary mediastinal B-cell (PMB) subtypes. Pediatric NHL is usually high grade and very aggressive, whereas adult NHL is generally less aggressive or indolent. Almost all forms of BL and DLBCL are of B-cell origin; 80% cases of LL are of T-cell origin, and 20% of B-cell origin; and 70% of cases of ALCL are of T-cell origin, 20% of null-cell origin, and 10% of B-cell origin. Some pathologic subtypes have specific cytogenetic aberrations. Children with BL commonly have a t(8;14) translocation (90%) or, less commonly, a t(2;8) or t(8;22) translocation (10%), whereas those with DLBCL may have a t(8;14) translocation (30%) and often have a complex (80%) and aneuploid (80%) karyotype. Patients with ALCL commonly have a t(2;5) translocation (90%), which results in the formation of a fusion gene encoding the constitutively active NPM-ALK tyrosine kinase. Variant ALK translocations, all with a breakpoint at 2p23, have also been reported. T-cell LBL harbors many of the same cytogenetic abnormalities as T-cell acute lymphoblastic leukemia (ALL), including rearrangements with breakpoints at 14q11.2 involving the T-cell receptor, and t(5;14) translocation (20%), which does not involve the 14q11.2 breakpoint.
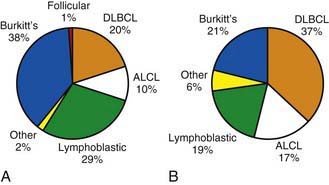
Figure 490-5 Incidence of non-Hodgkin lymphoma subtypes in (A) 0-14 yr age group and (B) 15-19 yr age group. ALCL, anaplastic large cell lymphoma; DLBCL, diffuse large B-cell lymphoma.
(Adapted from Hochberg J, Waxman IM, Kelly KM, et al: Adolescent non-Hodgkin lymphoma and Hodgkin lymphoma: state of the science, Br J Haematol 144:24–40, 2008.)
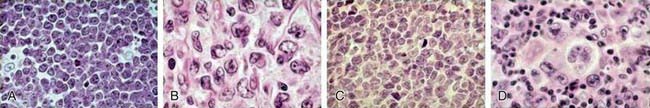
Figure 490-6 Distribution of childhood and adolescent non-Hodgkin lymphoma. Hematoxylin & eosin stains showing morphology of Burkitt lymphoma (A, high power), diffuse large B-cell lymphoma (B, high power), precursor T-lymphoblastic lymphoma (C, high power), and anaplastic large cell lymphoma (D, high power).
(From Cairo MS, Raetz E, Lim MS, et al: Childhood and adolescent non-Hodgkin lymphoma: new insights in biology and critical challenges for the future, Pediatr Blood Cancer 45:753–769, 2005.)
Clinical Manifestations
The clinical manifestations of childhood and adolescent NHL depend primarily on pathologic subtype and primary and secondary sites of involvement. Tumors grow rapidly and can cause symptoms based on size and location. Approximately 70% of patients with NHL present with advanced disease, at stage III or IV (Table 490-5), including extranodal disease with gastrointestinal, bone marrow, and central nervous system (CNS) involvement. BL commonly manifests as abdominal (sporadic type) or head and neck (endemic type) disease with involvement of the bone marrow or CNS. LL commonly manifests as an intrathoracic or mediastinal supradiaphragmatic mass and also has a predilection for spreading to the bone marrow and CNS. DLBCL commonly manifests as either an abdominal or mediastinal (PMB subtype) primary and, rarely, dissemination to the bone marrow or CNS. ALCL manifests either as a primary cutaneous manifestation (10%) or as systemic disease (fever, weight loss) with dissemination to liver, spleen, lung, mediastinum, or skin; spread to the bone marrow or CNS is rare. Patients have also been staged according to risk classification in pediatric international cooperative group trials Table 490-6.
Table 490-5 ST. JUDE STAGING SYSTEM FOR CHILDHOOD NON-HODGKIN LYMPHOMA
| STAGE | DESCRIPTION |
|---|---|
| I | A single tumor (extranodal) or single anatomic area (nodal), with the exclusion of mediastinum or abdomen |
| II | A single tumor (extranodal) with regional node involvement |
| Two or more nodal areas on the same side of the diaphragm | |
| Two single (extranodal) tumors with or without regional node involvement on the same side of the diaphragm | |
| A primary gastrointestinal tract tumor, usually in the ileocecal area, with or without involvement of associated mesenteric nodes only, which must be grossly (>90%) resected | |
| III | Two single tumors (extranodal) on opposite sides of the diaphragm |
| Two or more nodal areas above and below the diaphragm | |
| Any primary intrathoracic tumor (mediastinal, pleural, or thymic) | |
| Any extensive primary intra-abdominal disease | |
| IV | Any of the above, with initial involvement of central nervous system or bone marrow at time of diagnosis |
From Murphy SB: Classification, staging and end results of treatment of childhood non-Hodgkin’s lymphomas: dissimilarities from lymphomas in adults, Semin Oncol 7:332–339, 1980.
Table 490-6 RISK STRATIFICATION GROUPS FOR PEDIATRIC B-CELL NHL
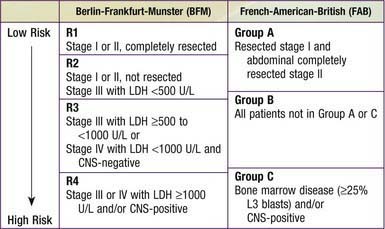
CNS, central nervous system; LDH, lactate dehydrogenase.
Site-specific manifestations include painless, rapid lymph node enlargement; cough, superior mediastinal syndrome (SMS), dyspnea with thoracic involvement; abdominal (massive and rapidly enlarging) mass, intestinal obstruction, intussusception-like symptoms, ascites with abdominal involvement; nasal stuffiness, earache, hearing loss, tonsil enlargement with Waldeyer ring involvement; and localized bone pain (primary or metastatic).
Three clinical manifestations that require special alternative treatment strategies are SMS symptoms secondary to a large mediastinal mass obstructing blood flow or respiratory airways; acute paraplegias secondary to spinal cord or CNS compression from neighboring tumor; and tumor lysis syndrome (TLS) secondary to severe metabolic abnormalities, including hyperuricemia, hyperphosphatemia, hyperkalemia, and hypocalcemia from massive tumor cell lysis.
Laboratory Findings
Recommended laboratory and radiologic testing includes: CBC; measurements of electrolytes, uric acid, calcium, phosphorus, blood urea nitrogen, creatinine, bilirubin, alanine aminotransferase, and aspartate aminotransferase; bone marrow aspiration and biopsy; lumbar puncture with cerebrospinal fluid (CSF) cytology, cell count and protein; chest radiographs; and neck, chest, abdominal, and pelvic CT scans (head CT for suspicion of CNS disease), and PET scan. Tumor tissue (i.e., biopsy, bone marrow, CSF, or pleurocentesis/paracentesis fluid) should be tested by flow cytometry for immunophenotypic origin (T, B, or null) and cytogenetics (karyotype). Additional tests might include fluorescent in situ hybridization (FISH) or quantitative reverse transcription polymerase chain reaction (RT-PCR) for specific genetic translocations, T- and B-cell gene rearrangement studies, and molecular profiling by oligonucleotide microarray.
Treatment
The primary modality of treatment for childhood and adolescent NHL is multiagent systemic chemotherapy with intrathecal chemotherapy. Surgery is used mainly for diagnosis and staging. Radiation therapy is used only in special circumstances, such as when there is CNS involvement in LL or, occasionally, BL and in the presence of acute SMS and acute paraplegias. Newly diagnosed patients and those otherwise at risk for TLS (usually advanced/bulky BL or LL) require vigorous hydration and either a xanthine oxidase inhibitor (allopurinol, 10 mg/kg/day PO divided tid) or, more often, recombinant urate oxidase (rasburicase, 0.2 mg/kg/day IV once daily up to 5 days).
Specific treatment for localized and advanced disease is similar for BL and DLBCL (see Table 490-4). Localized BL or DLBCL requires 6 wk to 6 mo of multiagent chemotherapy. Complete surgical resection followed by 2 cycles of COPAD (cyclophosphamide, vincristine, prednisone, and doxorubicin) led to a 4-yr overall survival (OS) in the international FAB/LMB 96 (French-American-British Lymphoma, mature B cell) trial. Advanced disease is usually treated with a 4- to 6-mo regimen of multiagent chemotherapy, such as FAB/LMB 96 protocol therapy or NHL-BFM (Berlin-Frankfurt-Munich) 95 protocol therapy.
Localized or advanced LL usually requires almost 24 mo of therapy. The best results in advanced LL have been obtained using the NHL-BFM90 protocol, which uses therapeutic approaches similar to those for childhood acute leukemia, including induction, consolidation, interim maintenance, and re-induction (advanced disease only) phases as well as a year-long maintenance phase with 6-mercaptopurine and methotrexate.
Localized ALCL may require only cutaneous excision or more aggressive therapy similar to that for advanced ALCL. Advanced ALCL is commonly treated with NHL-BFM90 protocol–type therapy.
Intrathecal chemotherapy is administered in the presence of moderate to advanced disease in all subtypes of childhood and adolescent NHL and may include methotrexate, hydrocortisone, or Ara-C.
Patients with NHL in whom progressive or relapsed disease develops require re-induction chemotherapy and either allogeneic or autologous stem cell transplantation. The specific re-induction regimen or transplant type depends on the pathologic subtype, previous therapy, site or reoccurrence, and stem cell donor availability.
Complications
Patients receiving multiagent chemotherapy for advanced disease are at acute risk for serious mucositis, infections, cytopenias that require red blood cell and platelet blood product transfusions, electrolyte imbalances, and poor nutrition. Long-term complications may include growth retardation, cardiac toxicity, gonadal toxicity with infertility, and secondary malignancies.
Prognosis
The prognosis is excellent for most forms of childhood and adolescent NHL (see Table 490-4). Patients with localized disease have a 90-100% chance of survival, and those with advanced disease have a 60-95% chance of survival. The variation in survival depends on pathologic subtype, tumor burden at diagnosis as reflected in serum lactate dehydrogenase (LDH) level, presence or absence of CNS disease, and specific sites of metastatic spread. Specific cytogenetic and molecular genetic subtyping may also be important in predicting outcome and influencing specific therapeutic strategies.
Abromowitch M, Sposto R, Perkins S, et al. Shortened intensified multi-agent chemotherapy and non-cross resistant maintenance therapy for advanced lymphoblastic lymphoma in children and adolescents: report from the Children’s Oncology Group. Br J Haematol. 2008;143:261-267.
Brugieres L, Le Deley M-C, Rosolen A, et al. Impact of the methotrexate administration dose on the need for intrathecal treatment in children and adolescents with anaplastic large-cell lymphoma: results of a randomized trial of the EICHNL group. J Clin Oncol. 2009;27:897-903.
Burkhardt B, Woessmann W, Zimmermann M, et al. Impact of cranial radiotherapy on central nervous system prophylaxis in children and adolescents with central nervous system-negative stage III or IV lymphoblastic lymphoma. J Clin Oncol. 2006;24:491-499.
Cairo MS, Sposto R, Perkins SL, et al. Burkitt’s and Burkitt-like lymphoma in children and adolescents: a review of the Children’s Cancer Group experience. Br J Haematol. 2003;120:660-670.
Cairo MS, Gerrard M, Sposto R, et al. Results of a randomized study of high-risk central nervous system B non-Hodgkin lymphoma and B acute lymphoblastic leukemia in children and adolescents. Blood. 2007;109:2736-2743.
Coiffier B, Altman A, Pui CH, et al. Guidelines for the management of pediatric and adult tumor lysis syndrome: an evidence-based review. J Clin Oncol. 2008;26:2767-2778.
Czuczman MS, Gregory SA. The future of CD20 monoclonal antibody therapy in B-cell malignancies. Leuk Lymphoma. 2010;51(6):983-994.
Gerrard M, Cairo MS, Weston C, et al. Excellent survival following two courses of COPAD chemotherapy in children and adolescents with resected localized B-cell non-Hodgkin’s lymphoma: results of the FAB/LMB 96 international study. Br J Hematol. 2008;141:840-847.
Hochberg J, Waxman IM, Kelly KM, et al. Adolescent non-Hodgkin lymphoma and Hodgkin lymphoma: state of the science. Br J Haematol. 2009;144:24-40.
Lenz G, Staudt LM. Aggressive lymphomas. N Engl J Med. 2010;362:1417-1428.
Meinhardt A, Burkhardt B, Zimmermann M, et al. Phase II window study on rituximab in newly diagnosed pediatric mature B-cell non-Hodgkin’s lymphoma and Burkitt leukemia. J Clin Oncol. 2010;28(19):3115-3121.
Oeffinger KC, Mertens AC, Sklar CA, et al. Chronic health conditions in adult survivors of childhood cancer. N Engl J Med. 2006;355:1572-1582.
Patte C, Auperin A, Gerrard M, et al. Results of the randomized international FAB/LMB96 trial for intermediate risk B-cell non-Hodgkin lymphoma in children and adolescents: it is possible to reduce treatment for the early responding patients. Blood. 2007:2773-2780.
Tai E, Pollack LA, Townsend J, et al. Differences in non-Hodgkin lymphoma survival between young adults and children. Arch Pediatr Adolesc Med. 2010;164:218-224.
Woessmann W, Seidemann K, Mann G, et al. The impact of the methotrexate administration schedule and dose in the treatment of children and adolescents with B-cell neoplasms: a report of the BFM Group Study NHL-BFM95. Blood. 2005;105:948-958.
490.3 Late Effects in Children and Adolescents with Lymphoma
Ian M. Waxman, Jessica Hochberg, and Mitchell S. Cairo
The majority of patients with newly diagnosed HL and NHL have overall survival rates well above 90%. However, this survival has often been achieved at the expense of an increased relative risk of long-term complications, including solid tumors, leukemia, cardiac disease, pulmonary complications, thyroid disease, and infertility. An analysis of >1,000 long-term childhood NHL survivors found increased rates of death >20 yr after treatment. A review of Surveillance, Epidemiology and End Results (SEER) data over a 25-yr follow-up period has demonstrated that the relative survival curves do not plateau after 10 yr following diagnosis of HL but, rather, accelerate. This finding highlights the importance of late morbidity and mortality among survivors of lymphoma. The first Childhood Cancer Survivor Study, a retrospective cohort study of 10,397 cancer survivors, showed that 62.3% of survivors report at least one chronic condition; with 27.5% reporting severe or life-threatening conditions. The survivor’s adjusted relative risk of a severe or life-threatening chronic condition, compared with that of a sibling, was 8.2 (95% confidence interval [CI], 6.9- 9.7). When disease-specific health outcomes were looked at, both HL and NHL were found to be associated with a cumulative incidence of chronic health conditions approaching 70-80%, with severe conditions being reported in close to 50% of HL survivors (Fig. 490-7). Newer treatment strategies continue to be investigated to address the morbidity and consequences survivors of childhood lymphoma must face when undergoing treatment.
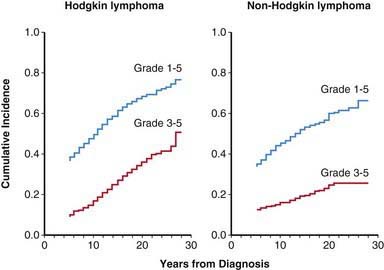
Figure 490-7 Incidence of chronic health conditions in adult survivors of pediatric cancer. Among the survivors of Hodgkin lymphoma and non-Hodgkin lymphoma, the severity of subsequent health conditions was scored according to the National Cancer Institute’s Common Terminology Criteria for Adverse Events (version 3) as either mild (grade 1), moderate (grade 2), severe (grade 3), life-threatening or disabling (grade 4), or fatal (grade 5).
(Adapted from Oeffinger KC, Mertens AC, Sklar CA, et al: Chronic health conditions in adult survivors of childhood cancer, N Engl J Med 355:1572–1582, 2006.)