Chapter 496 Retinoblastoma
Retinoblastoma is an embryonal malignancy of the retina and is the most common intraocular tumor in children. Although the survival rate of children in the USA and developed countries with retinoblastoma is extremely high, retinoblastoma progresses to metastatic disease and death in over 50% of children worldwide. Furthermore, the associated loss of vision and side effects of therapy are significant problems that remain to be addressed.
Epidemiology
Approximately 250-350 new cases of retinoblastoma are diagnosed each year in the USA, with no known racial or gender predilection. The cumulative lifetime incidence of retinoblastoma is approximately 1:20,000 live births, and retinoblastoma accounts for 4% of all pediatric malignancies. The median age at diagnosis is approximately 2 yr, and over 90% of cases are diagnosed in children under 5 yr of age. Overall, about two thirds to three quarters of children with retinoblastoma have unilateral tumors, with the remainder having bilateral retinoblastoma. Bilateral involvement is more common in younger children, particularly in those diagnosed under the age of 1 yr.
Retinoblastoma can be either hereditary or sporadic. Hereditary cases usually are diagnosed at a younger age and are multifocal and bilateral, while sporadic cases are usually diagnosed in older children who tend to have unilateral, unifocal involvement. The hereditary form is associated with loss of function of the retinoblastoma gene (RB1) via gene mutation or deletion. The RB1 gene is located on chromosome 13q14 and encodes the retinoblastoma protein (Rb), a tumor suppressor protein that controls cell-cycle phase transition and has roles in apoptosis and cell differentiation. Many different causative mutations have been identified, including translocations, deletions, insertions, point mutations, and epigenetic modifications such as gene methylation. The nature of the predisposing mutation can affect the penetrance and expressivity of retinoblastoma development.
According to Knudson’s “two-hit” model of oncogenesis, two mutational events are required for retinoblastoma tumor development (Chapter 486). In the hereditary form of retinoblastoma, the first mutation in the RB1 gene is inherited through germinal cells and a second mutation occurs subsequently in somatic retinal cells. Second mutations that lead to retinoblastoma often result in the loss of the normal allele and concomitant loss of heterozygosity. Most children with hereditary retinoblastoma have spontaneous new germinal mutations, and both parents have wild-type retinoblastoma genes. In the sporadic form of retinoblastoma, the two mutations occur in somatic retinal cells. Heterozygous carriers of oncogenic RB1 mutations demonstrate variable phenotypic expression.
Pathogenesis
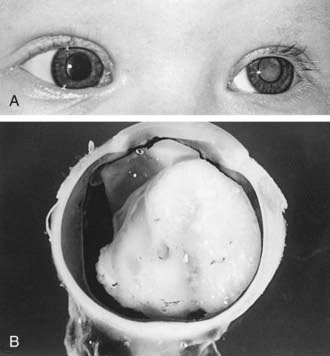
Histologically, retinoblastoma appears as a small round blue cell tumor with rosette formation (Flexner-Wintersteiner rosettes). It may arise in any of the nucleated layers of the retina and exhibit various degrees of differentiation. Retinoblastoma tumors tend to outgrow their blood supply, resulting in necrosis and calcification.
Endophytic tumors arise from the inner surface of the retina and grow into the vitreous, and can also grow as tumors suspended within the vitreous itself, known as vitreous seeding. Exophytic tumors grow from the outer retinal layer and can cause retinal detachment. Tumors can also be both endophytic and exophytic. These tumors can also spread by direct extension to the choroid or along the optic nerve beyond the lamina cribrosa, or by hematogenous or lymphatic spread to distant sites.
Clinical Manifestations
Retinoblastoma classically presents with leukocoria, a white pupillary reflex (Fig. 496-1), which often is first noticed when a red reflex is not present at a routine newborn or well child examination or in a flash photograph of the child. Strabismus often is an initial presenting complaint. Orbital inflammation, hyphema, and pupil irregularity can occur with advancing disease. Pain can occur if secondary glaucoma is present. Only about 10% of retinoblastoma cases are detected by routine ophthalmologic screening in the context of a positive family history.
Diagnosis
The diagnosis is established by the characteristic ophthalmologic findings. Imaging studies are not diagnostic, and biopsies are contraindicated. Indirect ophthalmoscopy with slit-lamp evaluation can detect retinoblastoma tumors, but a complete evaluation requires an examination under general anesthesia by an experienced ophthalmologist to obtain complete visualization of both eyes, which also facilitates photographing and mapping of the tumors. Retinal detachment or vitreous hemorrhage can complicate the evaluation.
Orbital ultrasonography, CT, or MRI are used to evaluate the extent of intraocular disease and extraocular spread. Occasionally, a pineal area tumor is detected in a child with hereditary retinoblastoma, a phenomenon known as trilateral retinoblastoma. MRI allows for better evaluation of optic nerve involvement. Evaluation of the cerebrospinal fluid and bone marrow for tumor metastasis is required only if indicated by other clinical, laboratory, or imaging findings.
The differential diagnosis of retinoblastoma includes other causes of leukocoria, including persistent hyperplastic primary vitreous, Coats disease, cataract, endophthalmitis from Toxocara canis, choroidal coloboma, and retinopathy of prematurity.
Treatment
Treatment is determined by the size and location of the tumors and whether the child has hereditary or sporadic disease. The primary goal of treatment is always cure; the secondary goals include preserving vision and the eye itself. As newer modalities for local control of intraocular tumors and more effective systemic chemotherapy have emerged, primary enucleation is being performed less often.
Most unilateral disease presents with a solitary, large tumor. Enucleation is performed if there is no potential for the salvage of useful vision. With bilateral disease, chemoreduction in combination with focal therapy (laser photocoagulation or cryotherapy) has replaced the traditional approach of enucleation of the more severely affected eye and irradiation of the remaining eye. If feasible, small tumors can be treated with focal therapy with careful follow-up for recurrence or new tumor growth. Larger tumors often respond to multiagent chemotherapy including carboplatin, vincristine, and etoposide. If this approach fails, external-beam irradiation should be considered, although this approach may result in significant orbital deformity and increased incidence of second malignancies in patients with germ line RB1 mutations. Brachytherapy, or episcleral plaque radiotherapy, is an alternative with less morbidity. Enucleation may be required for unresponsive or recurrent tumors. Alternative treatment options currently under investigation include other systemic chemotherapy agents such as topotecan and other sites of chemotherapy administration, including periocular and ophthalmic artery infusions.
All first-degree relatives of children with known or suspected hereditary retinoblastoma should have retinal examinations to identify retinomas or retinal scars, which may suggest hereditary retinoblastoma even though malignant retinoblastoma did not develop.
Prognosis
Approximately 95% of children with retinoblastoma are cured with modern treatment in the USA. Current efforts using chemotherapy in combination with focal therapy are intended to preserve useful vision and avoid external-beam radiation or enucleation. Routine ophthalmologic examinations should continue until children are over age 7 yr. Unfortunately the diagnosis of retinoblastoma in many children from third-world countries is delayed, resulting in spread of the tumor outside of the orbit. The prognosis for these children with retinoblastoma that has spread outside of the eye is poor.
Children with germ line RB1 mutations are at significant risk for development of second malignancies, especially osteosarcoma and also soft tissue sarcomas and malignant melanoma. The risk of second malignancies is further increased by the use of radiation therapy. Other radiation-related late adverse effects include cataracts, orbital growth deformities, lacrimal dysfunction, and late retinal vascular injury.
Abramson DH, Schefler AC. Update on retinoblastoma. Retina. 2004;24:828-848.
Classon M, Harlow E. The retinoblastoma tumour suppressor in development and cancer. Nat Rev Cancer. 2002;2:910-917.
Kleinerman R, Tucker MA, Tarone RF, et al. Risk of new cancers after radiotherapy in long-term survivors of retinoblastoma: an extended follow-up. J Clin Oncol. 2005;23:2272-2279.
Lohman DR, Gallie BL. Retinoblastoma: Revisiting the model prototype of inherited cancer. Am J Med Genet C Semin Med Genet. 2004;129:23-28.
MacCarthy A, Bayne AM, Draper GJ, et al. Non-ocular tumours following retinoblastoma in Great Britain 1951 to 2004. Br J Opthalmol. 2009;93:1159-1162.
Richter S, Vandezande K, Chen N, et al. Sensitive and efficient detection of RB1 gene mutations enhances care of families with retinoblastoma. Am J Hum Genet. 2003;72:253-269.
Rodriguez-Galindo C, Chantada G, Wilson MW, et al. Treatment of retinoblastoma: current status and future perspectives. Curr Treat Options Neurol. 2007;9:294-307.
Woo KI, Harbour W. Review of 676 second primary tumors in patients with retinoblastoma. Arch Ophthalmol. 2010;128(7):865-870.