Chapter 504 Isolated Glomerular Diseases with Recurrent Gross Hematuria
Approximately 10% of children with gross hematuria have an acute or a chronic form of glomerulonephritis that may be associated with a systemic illness. The gross hematuria, which is usually characterized by brown or cola-colored urine, may be painless or associated with vague flank or abdominal pain. Presentation with gross hematuria is common within 1-2 days after the onset of an apparent viral upper respiratory tract infection in immunoglobulin A (IgA) nephropathy, and typically resolves within 5 days. This relatively short period contrasts to a latency period of 7-21 days occurring between the onset of a streptococcal pharyngitis or impetiginous skin infection and the development of poststreptococcal acute glomerulonephritis. Gross hematuria in these circumstances can last as long as 4-6 wk. Gross hematuria can also be seen in children with glomerular basement membrane (GBM) disorders such as hereditary nephritis (Alport syndrome [AS]) and thin GBM disease. These glomerular diseases can also manifest as microscopic hematuria and/or proteinuria without gross hematuria.
504.1 Immunoglobulin A Nephropathy (Berger Nephropathy)
IgA nephropathy is the most common chronic glomerular disease. It is characterized by a predominance of IgA immunoglobulin within mesangial glomerular deposits in the absence of systemic disease (e.g., symptomatic systemic lupus erythematosus or Henoch-Schönlein purpura). Diagnosis requires renal biopsy, which is performed when clinical features warrant confirmation of the diagnosis or characterization of the histologic severity, which might affect therapeutic decisions.
Pathology and Pathologic Diagnosis
Focal and segmental mesangial proliferation and increased mesangial matrix are seen in the glomerulus (Fig. 504-1). Renal histology demonstrates mesangial proliferation that may be associated with epithelial cell crescent formation and sclerosis. IgA deposits in the mesangium are often accompanied by C3 complement (Fig. 504-2).
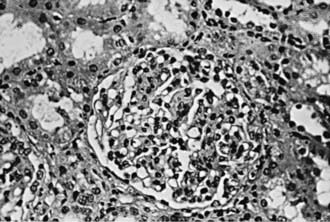
Figure 504-1 Light microscopy of immunoglobulin A nephropathy demonstrating segmental mesangial proliferation and increased matrix (×180).
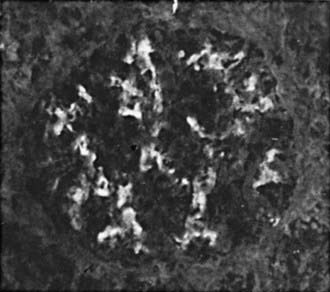
Figure 504-2 Immunofluorescence microscopy of the biopsy specimen from a child with episodes of gross hematuria demonstrating mesangial deposition of immunoglobulin A (×150).
IgA nephropathy is an immune complex disease that appears to be caused by abnormalities in the IgA immune system. The abnormalities identified in the IgA immunoglobulin system have also been observed in patients with Henoch-Schönlein purpura and lends support to the hypothesis that these two diseases are part of the same disease spectrum. Familial clustering of IgA nephropathy cases suggests the importance of genetic factors. Genome-wide linkage analysis suggests the linkage of IgA nephropathy to 6q22-23 in multiplex IgA nephropathy kindreds.
Clinical and Laboratory Manifestations
IgA nephropathy is seen more often in male than in female patients. Though there are rare cases of rapidly progressive forms of the disease, the clinical presentation of childhood IgA nephropathy is often benign in comparison to that of adults. IgA nephropathy is an uncommon cause of end-stage renal failure during childhood. A majority of children with IgA nephropathy in the USA and Europe present with gross hematuria, whereas microscopic hematuria and/or proteinuria is a more common presentation in Japan. Other types of presentation include acute nephritic syndrome, nephrotic syndrome, or a combined nephritic-nephrotic picture. Gross hematuria often occurs within 1-2 days of onset of an upper respiratory or gastrointestinal infection, in contrast to the longer latency period observed in acute postinfectious glomerulonephritis, and may be associated with loin pain. Proteinuria is often <1000 mg/24 hr in patients with asymptomatic microscopic hematuria. Mild to moderate hypertension is most often seen in patients with nephritic or nephrotic syndrome but is rarely severe enough to result in hypertensive emergencies. Normal serum levels of C3 in IgA nephropathy help to distinguish this disorder from poststreptococcal glomerulonephritis. Serum IgA levels have no diagnostic value because they are elevated in only 15% of pediatric patients.
Prognosis and Treatment
Although IgA nephropathy does not lead to significant kidney damage in most children, progressive disease develops in 20-30% of patients 15-20 yr after disease onset. Therefore, most children with IgA nephropathy do not display progressive renal dysfunction until adulthood, prompting the need for careful long-term follow-up. Poor prognostic indicators at presentation or follow-up include persistent hypertension, diminished renal function, and heavy or prolonged proteinuria. A more-severe prognosis is correlated with histologic evidence of diffuse mesangial proliferation, extensive glomerular crescents, glomerulosclerosis, and diffuse tubulointerstitial changes, including inflammation and fibrosis.
The primary treatment of IgA nephropathy is appropriate blood pressure control. Fish oil, which contains anti-inflammatory omega-3 polyunsaturated fatty acids, decreases the rate of disease progression in adults. Immunosuppressive therapy with corticosteroids or more intensive multidrug regimens may be beneficial in some patients. Angiotensin-converting enzyme inhibitors and angiotensin II receptor antagonists are effective in reducing proteinuria and retarding the rate of disease progression when used as single agents or in combination. Tonsillectomy has been used as treatment for IgA nephropathy in many countries including Japan, but demonstration of its efficacy will require prospective controlled trials. Patients with IgA nephropathy may undergo successful kidney transplantation. Although recurrent disease is frequent, allograft loss caused by IgA nephropathy occurs in only 15-30% of patients.
Delos Santos NM, Wyatt RJ. Pediatric IgA nephropathies: clinical aspects and therapeutic approaches. Semin Nephrol. 2004;24:269-286.
Dillon JJ. Angiotensin-converting enzyme inhibitors and angiotensin receptor blockers for IgA nephropathy. Semin Nephrol. 2004;24:218-224.
Donadio JV, Grande JP. The role of fish oil/omega-3 fatty acids in the treatment of IgA nephropathy. Semin Nephrol. 2004;3:225-243.
Gharavi AG, Yan Y, Scolari F, et al. IgA nephropathy, the most common cause of glomerulonephritis is linked to 6q22-23. Nat Genet. 2000;26:354-357.
Naganishi K, Yoshikawa N. Immunoglobulin A nephropathy. In: Avner ED, Harmon WE, Niaudet P, et al, editors. Pediatric nephrology. ed 6. Heidelberg, Germany: Springer-Verlag; 2009:757-782.
Sanders JT, Wyatt RJ. IgA nephropathy and Henoch-Schönlein purpura nephritis. Curr Opin Pediatr. 2008;20:163-170.
Wang AY, Lai FM, Yu AW, et al. Recurrent IgA nephropathy in renal transplant allografts. Am J Kidney Dis. 2001;38:588-596.
504.2 Alport Syndrome
AS, hereditary nephritis, is a genetically heterogeneous disease caused by mutations in the genes coding for type IV collagen, a major component of basement membranes. These genetic alterations are associated with marked variability in clinical presentation, natural history, and histologic abnormalities.
Genetics
Approximately 85% of patients have X-linked disease caused by a mutation in the COL4A5 gene encoding the α5 chain of type IV collagen. Patients with a subtype of X-linked AS and diffuse leiomyomatosis demonstrate a contiguous mutation within the COL4A5 and COL4A6 genes that encodes the α5 and α6 chains, respectively, of type IV collagen. Autosomal recessive forms of AS are caused by mutations in the COL4A3 and COL4A4 genes on chromosome 2 encoding the α3 and α4 chains, respectively, of type IV collagen. An autosomal dominant form of AS linked to the COL4A3-COL4A4 gene locus occurs in 5% of cases.
Pathology
Kidney biopsy specimens during the 1st decade of life might show few changes on light microscopy. Later, the glomeruli can develop mesangial proliferation and capillary wall thickening, leading to progressive glomerular sclerosis. Tubular atrophy, interstitial inflammation and fibrosis, and lipid-containing tubular or interstitial cells, called foam cells, develop as the disease progresses. Immunopathologic studies are usually nondiagnostic.
In most patients, electron microscopy reveals diffuse thickening, thinning, splitting, and layering of the glomerular and tubular basement membranes (Fig. 504-3). Ultrastructural analysis of the GBM in all genetic forms of AS may be completely normal, display nonspecific alterations, or demonstrate only uniform thinning.
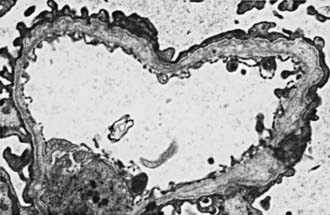
Clinical Manifestations
All patients with AS have asymptomatic microscopic hematuria, which may be intermittent in girls and younger boys. Single or recurrent episodes of gross hematuria commonly occurring 1-2 days after an upper respiratory infection are seen in approximately 50% of patients. Proteinuria is often seen in boys but may be absent, mild, or intermittent in girls. Progressive proteinuria, often exceeding 1 g/24 hr, is common by the 2nd decade of life and can be severe enough to cause nephrotic syndrome.
Bilateral sensorineural hearing loss, which is never congenital, occurs in 90% of hemizygous males with X-linked AS, 10% of heterozygous females with X-linked AS, and 67% of patients with autosomal recessive AS. This deficit begins in the high-frequency range but progresses to involve conversational speech, prompting the need for hearing aids. Ocular abnormalities, which occur in 30-40% of patients with X-linked AS, include anterior lenticonus (extrusion of the central portion of the lens into the anterior chamber), macular flecks, and corneal erosions. Leiomyomatosis of the esophagus, tracheobronchial tree, and female genitals in association with platelet abnormalities is rare.
Diagnosis
A careful family history, a screening urinalysis of first-degree relatives, an audiogram, and an ophthalmologic examination are critical in making the diagnosis of AS. The presence of anterior lenticonus is pathognomonic. AS is highly likely in the patient who has hematuria and at least 2 of the following characteristic clinical features: macular flecks, recurrent corneal erosions, GBM thickening and thinning, or sensorineural deafness. Absence of epidermal basement membrane staining for the α5 chain of type IV collagen in male hemizygotes and discontinuous epidermal basement membrane staining in female heterozygotes is pathognomonic for X-linked AS and can preclude diagnostic renal biopsy. Mutation screening or linkage analysis is not readily available for routine clinical use. Prenatal diagnosis is available for families with members who have X-linked AS and who carry an identified mutation.
Prognosis and Treatment
The risk of progressive renal dysfunction leading to end-stage renal disease (ESRD) is highest among hemizygotes and autosomal recessive homozygotes. ESRD occurs before age 30 yr in approximately 75% of hemizygotes with X-linked AS. The risk of ESRD in X-linked heterozygotes is 12% by age 40 yr and 30% by age 60 yr. Risk factors for progression are gross hematuria during childhood, nephrotic syndrome, and prominent GBM thickening. Intrafamilial variation in phenotypic expression results in significant differences in the age of ESRD among family members. No specific therapy is available to treat AS, although angiotensin-converting enzyme inhibitors can slow the rate of progression. Careful management of renal failure complications such as hypertension, anemia, and electrolyte imbalance is critical. Patients with ESRD are treated with dialysis and kidney transplantation (Chapter 529). Approximately 5% of kidney transplant recipients develop anti-GBM nephritis, which occurs primarily in males with X-linked AS who develop ESRD before age 30 yr.
Heidet L, Gubler MC. The renal lesions of Alport syndrome. J Am Soc Nephrol. 2009;20:1210-1215.
Jais JP, Knebelmann B, Giatras I, et al. X-linked Alport syndrome: natural history and genotype-phenotype correlations in girls and women belonging to 195 families: a “European Community Alport Syndrome Concerted Action” study. J Am Soc Nephrol. 2003;14:2603-2610.
Kashtan CE. Familial hematuria due to type IV collagen mutations: Alport syndrome and thin basement membrane nephropathy. Curr Opin Pediatr. 2004;16:177-181.
Kashtan CE, Gubler MC. Inherited glomerular diseases. In: Avner ED, Harmon WE, Niaudet P, et al, editors. Pediatric nephrology. ed 6. Heidelberg, Germany: Springer-Verlag; 2009:621-642.
504.3 Thin Basement Membrane Disease
Thin basement membrane disease (TBMD) is defined by the presence of persistent microscopic hematuria and isolated thinning of the glomerular basement membrane (GBM) (and occasionally tubular basement membranes) on electron microscopy. Microscopic hematuria is often initially observed during childhood and may be intermittent. Episodic gross hematuria can also be present, particularly after a respiratory illness. Isolated hematuria in multiple family members without renal dysfunction is referred to as benign familial hematuria. Although most of these patients will not undergo renal biopsy, it is often presumed that the underlying pathology is TBMD.
TBMD may be sporadic or transmitted as an autosomal dominant trait. Heterozygous mutations in the COL4A3 and COL4A4 genes, which encode the α3 and α4 chains of type IV collagen present in the GBM, result in TBMD. Rare cases of TBMD progress, and such patients develop significant proteinuria, hypertension, or renal insufficiency. Homozygous mutations in these same genes result in autosomal recessive Alport syndrome. Therefore, in these rare cases, the absence of a positive family history for renal insufficiency or deafness would not necessarily predict a benign outcome. Therefore, monitoring patients with benign familial hematuria for progressive proteinuria, hypertension, or renal insufficiency is important through childhood and young adulthood.