Chapter 587 Conditions That Mimic Seizures
The misdiagnosis of epilepsy has been estimated to be as high as 5-40%, implying that many patients may be subjected to unnecessary therapy and tests. Often all that is needed to differentiate nonepileptic paroxysmal disorders from epilepsy is a careful history and thorough exam; but sometimes, more advanced testing may be necessary. Nonepileptic paroxysmal disorders can be classified according to the age of presentation and the clinical manifestations: (1) generalized paroxysms, (2) abnormal movements and postures, (3) oculomotor abnormalities, and (4) sleep-related disorders (see Table 587-1 on the Nelson Textbook of Pediatrics website at www.expertconsult.com).
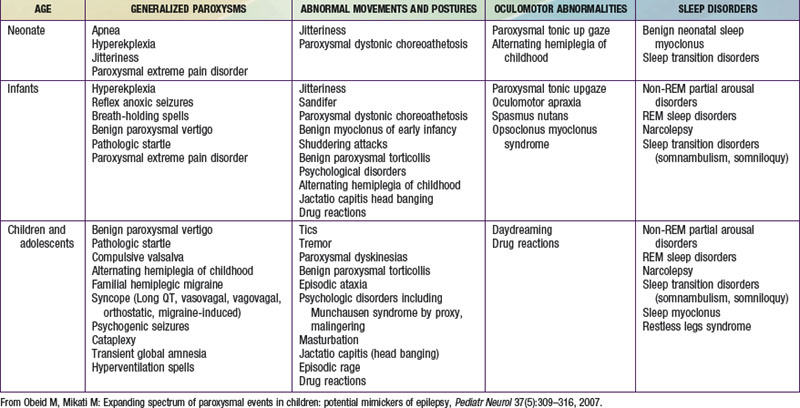
Generalized Paroxysms
Apnea
Apneic episodes in neonates are usually associated with bradycardia. In contrast, apnea associated with seizures is usually accompanied by tachycardia. Of note, exceptions do occur and severe apnea can also be followed by anoxic seizures.
Hyperekplexia and Pathologic Startles
Hyperekplexia is a rare, sporadic, or dominantly inherited disorder with neonatal onset of life-threatening episodes of tonic stiffening that precipitate apnea and convulsive hypoxic seizures. The genetic cause is a defect in the alpha or beta subunits of the strychnine-sensitive glycine receptors. It is characterized by a triad of generalized stiffness, nocturnal myoclonus, and later a pathologic startle reflex. A specific diagnostic sign can be elicited by tapping the nose, which produces a nonfatigable startle reflex with head retraction. Bathing, sudden awakening, and auditory, or tactile stimuli can induce attacks. Ictal electroencephalogram (EEG) reveals muscle artifact that can be confused with spikes. This diagnosis must not be missed, as hypoxic brain injury can result. Repeatedly flexing the baby at the neck and hips (the Vigevano maneuver) can abort the episodes. Clonazepam and sometimes other antiepileptics are used.
In other children after brain injury and in many patients with cerebral palsy an exaggerated startle reflex can occur. This is more common than hyperekplexia. In Tay-Sachs disease and similar gangliosidoses, exaggerated startle to sound occurs and has been, inappropriately, interpreted as hyperacusis.
Breath-Holding Spells
This term has been applied to 2 types of spells. The 1st is the pallid breath-holding spell, which is the vasovagal reflex described below. Often pallid breath-holding spells are made worse by iron deficiency anemia. The 2nd is the cyanotic or, “blue,” breath-holding spell. The term “breath-holding spells” is actually a misnomer, as these are not related to volition or behaviorally-mediated abnormalities. Prolonged expiratory apnea is responsible for the cyanotic episodes, which result from intrapulmonary shunting. On the other hand, reflex vagal-cardiac bradycardia is responsible for the pallid episodes. An episode starts with a cry (often a “silent” cry and marked pallor in the case of the pallid type), and progresses to apnea and cyanosis. Spells usually begin between 6 and 18 mo of age. Syncope, tonic posturing, and even reflex anoxic seizures may follow significant episodes, particularly in breath-holding spells of the pallid type. Injury, anger, and frustration, particularly with surprise, are common triggers. Education and reassurance of the parents is usually all that is needed, as these episodes are, as a rule, self-limited and outgrown within a few years. However, treatment of coexisting iron deficiency is needed if it is present. Education of the parents on how to handle more severe spells by first-aid measures is important. Anticholinergic drugs (e.g., atropine sulfate 0.01 mg/kg/24 hr in divided doses with a maximum daily dose of 0.4 mg), instructing parents in basic cardiopulmonary resuscitation (CPR), or antiepileptic drug therapy for anoxic seizures that are recurrent and not responding to other measures may, rarely, be needed. All parents should be taught not to provide secondary gain when the episodes occur, because this can reinforce the episodes. Also, preparation for unpleasant experiences (such as receiving a shot) rather than surprising the child with them can help limit the number of spells.
Compulsive Valsalva
In children with mental retardation, including Rett syndrome, syncopal convulsions may be self-induced by maneuvers like Valsalva. In this case, true breath-holding occurs, and it usually lasts for about 10 sec during inspiration. Some have advocated the use of naloxone in such cases.
Vagal Syncope
Syncope can present with drop attacks and can also lead to generalized convulsions, termed anoxic seizures. These convulsions, triggered by a sudden cutting off of oxygen to the brain, are clinically similar to and can be misdiagnosed as primary generalized seizures. Vasovagal (neurocardiogenic) syncope is one of the most common mimickers of generalized tonic clonic seizures and is usually triggered by dehydration, heat, standing for a long time without movement, hot showers, the sight of blood, pain, or sudden stress. History is usually the clue to distinguishing syncope from epileptic seizures. There is initially pallor and sweating followed by blurring of vision, dizziness, nausea, and then gradual collapse with loss of consciousness. However, these are not invariably present in syncope. Urinary incontinence and a brief period of convulsive jerks are not uncommon in vasovagal syncope. These occur with a frequency of 10% and 50%, respectively. Postictal confusion can also occur, though rarely. Abdominal pain, a common aura in temporal lobe epilepsy, occurs in vasovagal syncope, and can be a trigger or a consequence (intestinal vagal discharge). Most children with vasovagal syncope have an affected first-degree relative. EEG is normal and the tilt test has been used for diagnostic purposes. Although in most cases with typical history, it is not needed. Vagovagal syncope is triggered by swallowing or vomiting, and can progress to convulsive seizure if the asystole is sufficiently prolonged. Sudden cold exposure to the face or to the body can also trigger vagal syncope. Syncope has also been rarely reported to occur in association with cough, tight hair braiding, hair combing, extension of the neck while stretching due to compression of the vertebrals, and with flexion of the neck secondary to an abnormally prolonged stylomastoid process compressing the carotids. The latter 2 conditions require neuroimaging (CT, MRI) for proper diagnosis. Orthostatic hypotension and orthostatic intolerance manifest symptoms that develop during upright standing and can be relieved by recumbence. Postural tachycardia syndrome, the pathophysiology of which remains elusive, is a disease of adolescent females that is characterized by upright tachycardia and hypotension. Primary autonomic failure is rare in children, and familial dysautonomia is the only relatively common form. Familial dysautonomia is a disease common in Ashkenazi Jews, and is characterized by absence of overflow emotional tears, depressed patellar reflexes, and lack of a flare reaction following intradermal histamine. Dopamine beta-hydroxylase deficiency is a very rare cause of primary autonomic failure, and is characterized by impaired ejaculation, ptosis, nocturia, high palate, hyperflexible joints, and a complicated perinatal course (hypotension, hypotonia, hypothermia). Hypotension can also occur in adrenal insufficiency. Tilt test causes a drop in both blood pressure and heart rate in patients with classic vasovagal syncope. It results in a blood pressure drop with minimal change in heart rate in autonomic failure, and in blood pressure drop and an increase in heart rate in postural tachycardia syndrome.
Management of syncope centers on avoidance of precipitating factors (maintenance of hydration, avoidance of standing still, rising slowly from sitting, first aid measures, raise legs, positioning) and treatment of any accompanying or underlying medical conditions (anemia, adrenal insufficiency, cardiac, etc.). In addition, β-blockers (e.g., metoprolol starting dose 1-2 mg/kg once per day up to a maximum of 6 mg/kg/day), or flurohydrocortisone (0.05-0.1 mg/day) therapy may be needed in some selected cases.
Cardiac Syncope
Long QT syndromes (LQT) can cause life-threatening “pallid” or white syncope. Accompanying this are ventricular arrhythmias, usually torsades de pointes or even ventricular fibrillation. There are more than 10 types of prolonged QT syndromes. When accompanied by congenital deafness, it is part of the autosomal recessive Jervell and Lange-Nielson syndrome (type 1, LQT 1, associated with KvLQT1 potassium channel mutation). The Romano-Ward syndrome is an autosomal dominant syndrome with incomplete penetrance that is characterized by episodes of lying still like a dead body for several seconds before the anoxic convulsive episode (LQT 2 associated with an HERG potassium channel mutation). LQT 3 is associated with an SCN1A sodium channel mutation, type 4 ankyrin protein mutation, type 5 (milder form) with the minK KCNE1 mutation, type 6 with KCNE2 potassium gene mutations, type 9 with caveolin sodium channel–related protein mutations, and type 10 with SCN4B sodium channel mutations. Types 7 and 8 are of particular interest due to associated clinical and neurologic manifestations. Type 7 (Andersen-Tawil) syndrome is associated with periodic paralysis, skeletal developmental abnormalities, clinodactyly, low-set ears, and micrognathia (mutations in KCNJ2 gene). Type 8 or the Timothy syndrome (mutations in the calcium channel gene CACNA1c) manifests congenital heart disease, autism, syndactyly, and immune deficiency. All family members of an affected LQT syndrome individual should be investigated. Affected individuals need insertion of cardiac defibrillators, and their families should be taught CPR. As a rule children with new onset seizure disorder should get an electrocardiogram (ECG) to rule out LQT syndrome masquerading as seizure disorder. Cardiac syncope is usually sudden without the gradual onset and the symptoms that accompany vagal syncope. Aortic stenosis can cause sudden syncope at the height of the exercise (usually hypertrophic), or directly at the end (usually valvular) and, if suspected, warrants an echocardiogram.
Familial Hemiplegic Migraine
This is a rare type of migraine with a motor aura of weakness. Attacks begin as early as 5-7 yr of age. In a genetically susceptible person, attacks may be precipitated by head trauma, exertion, or emotional stress. Interictal cerebellar deficits (e.g., nystagmus, ataxia) may be present. Familial and sporadic cases are equally prevalent. The 3 genes implicated in the familial types are SCN1A (neuronal sodium channel subunit), CACNA1A (neuronal calcium channel subunit), and ATP1A2 (sodium potassium ATPase subunit). Several types of familial hemiplegic migraines can have coexisting seizures (e.g., in association with mitochondrial encephalopathy with lactic acidosis and strokelike episodes [MELAS], occipital epilepsy, and with episodic ataxia [Chapter 586.2]).
Benign Paroxysmal Vertigo
This is a common migraine equivalent that consists of episodes of brief imbalance during which the child appears frightened. Nystagmus, diaphoresis, nausea, and vomiting may be present. Episodes remit by 5 yr of age. MRI and EEG are normal, but caloric testing, if done, can show abnormal vestibular function. Patients can respond to diphenhydramine, 5 mg/kg/day (maximum of 300 mg/day) orally, intramuscularly, intravenously, or per rectum.
Cyclic Vomiting Syndrome
This syndrome is another related periodic migraine variant that can respond to antimigraine or antiepileptic drugs. This and other periodic syndromes have been associated with mutations that also can cause hemiplegic migraine.
The Alice in Wonderland Syndrome
This is the episodic experience of transient distortions of body image or visual images that, most often, constitute a migraine equivalent. It also can be an epileptic phenomenon.
Migraine Induced Syncope
Migraine, usually of the basilar variety, can trigger vasovagal syncope and, less commonly, epileptic seizures. Careful elicitation of the history of a migrainous prelude to the syncope helps in identifying these phenomena.
Psychologic Disorders
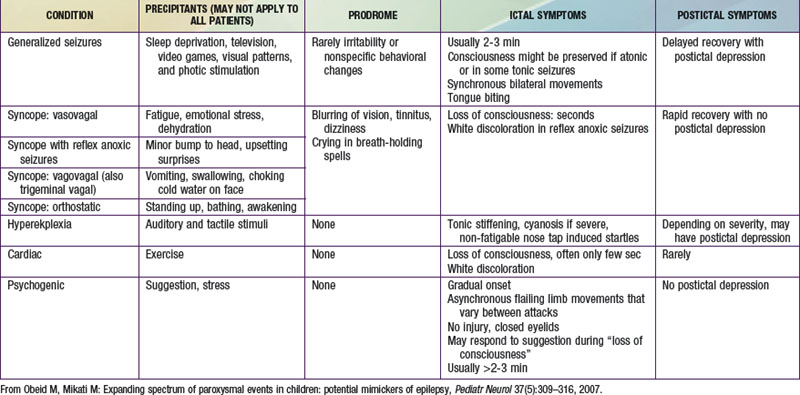
Psychogenic nonepileptic seizures are conversion reactions that are usually suspected clinically based on the characteristics of the spells (Table 587-2). The diagnosis can be confirmed by video EEG with capture of an episode to eliminate any residual doubts about their nature, as they can often occur in patients who also have epileptic seizures. Psychogenic seizures are also less likely than epileptic seizures to be associated with increased serum prolactin levels 15-120 min after the event. They are best managed acutely by reassurance about their relatively benign nature and by a supportive attitude. Psychiatric evaluation and follow-up are needed to uncover potential underlying psychopathology, particularly in adolescents and adults, and to establish continued support as psychogenic seizures can persist over long periods of time. Malingering and Munchausen syndrome by proxy are often difficult to diagnose but an approach similar to that for psychogenic seizures, including video-EEG monitoring, is often helpful.
Abnormal Movements and Postures
Neonatal Jitteriness and Clonus
Jitteriness consists of equal backward and forward movements of limbs, occurring spontaneously, or triggered by touch or loud sounds. Movement suppression by stimulus removal or by relaxing affected limbs, the lack of autonomic symptoms, and the clear difference from the two-phased (fast contraction, slow relaxation) clonic activity and the very quick myoclonic jerks, point to a nonepileptic event. Hypocalcemia, hypoglycemia, drug withdrawal, and hypoxic-ischemic encephalopathy are possible etiologies. Clonus due to corticospinal tract injury usually occurs in later infancy and childhood and can be stopped by change in position.
Paroxysmal Extreme Pain Disorder (Previously Familial Rectal Pain Syndrome)
This syndrome (caused by the SCN9A sodium channel gene mutation) usually starts in the neonatal period or infancy and persists throughout life. Autonomic manifestations predominate initially, with skin flushing in all and harlequin color change and tonic attacks in most. Dramatic syncope with bradycardia and sometimes asystole are common. Later, the disorder is characterized by attacks of excruciating deep burning pain often in the rectal, ocular, or jaw areas, but also diffusely in some. Attacks are triggered by defecation, cold, wind, eating, and emotion. Carbamazepine is used, but the response is often incomplete.
Benign Paroxysmal Torticollis of Infancy
This condition typically presents as morning episodes of painless retro-, and later, torticollis, often triggered by changes in posture. Attacks may start with abnormal ocular movements, and progress to stillness in an abnormal posture. This usually lasts for minutes or hours and, at times, days. Neurologic exam between attacks, EEG, and neuroimaging are normal. It affects girls more than boys (3 : 1), often begins before 3 mo of age, and spontaneously remits before the age of 5 yr. Medical therapy is not needed. It is considered to be a migraine equivalent and co-segregates with migraine in families.
Sandifer Syndrome
Gastroesophageal reflux in infants may cause paroxysmal episodes of generalized stiffening and opisthotonic posturing that may be accompanied by apnea, staring, and minimal jerking of the extremities. Episodes often occur 30 min after a feed.
Alternating Hemiplegia of Childhood
This consists of attacks of flaccid hemiplegia on 1 or both sides lasting minutes to days, starting in the 1st 18 mo of life. Earlier manifestations include paroxysmal nystagmus which is often monocular and ipsilateral to the hemiplegia. Dystonic and tonic spells often occur and can be confused with seizures and the hemiplegia with Todd paralysis. Usually sleep aborts the attacks and flunarizine 2.5-10 mg/day reduces them. Most children ultimately develop ataxia, developmental delay, and persistent choreoathetosis.
Paroxysmal Dyskinesias and Other Movement Disorders
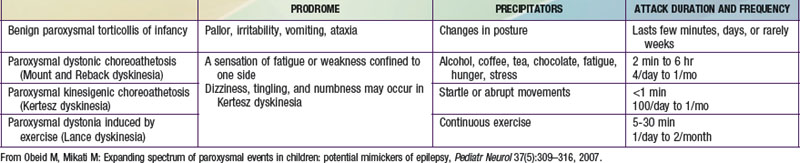
These disorders are characterized by sudden attacks that consist of choreic, dystonic, ballistic, or mixed movements (Table 587-3). A sensation of fatigue or weakness confined to 1 side may herald an attack. Consciousness is preserved and patients may be able perform a motor activity, like walking, despite the attack. The variability in the pattern of severity and localization between different attacks may also help in differentiating them from seizures. The frequency of attacks increases in adolescence, and steadily decreases in the third decade. Neurologic exam between attacks, EEG, laboratory investigations, and imaging studies are normal. These dyskinesias often respond to phenytoin, carbamazepine, clonazepam, or to antidopaminergic drugs such as haloperidol. Drug reactions can result in abnormal movements such as oculogyric crisis with many antiemetics, choreoathetosis with phenytoin, dystonia and facial dyskinesias with antidopaminergic drugs, and tics with carbamazepine. Strokes, focal brain lesions, connective tissue disorders (e.g., systemic lupus erythematosus), vasculitis, or metabolic and genetic disorders can also cause movement disorders. Mutations of glucose transporter 1 (GLUT1/SLC2AI) gene have been described in patients with exercise-induced dyskinesia.
Motor Tics
Tics are under partial control, are associated with an urge to do them and a subsequent relief, are usually exacerbated by emotions, and often change in character over time. In patients with tics who have Tourette syndrome, there is often a family history of tics and/or obsessive compulsive disorder or personality traits.
Episodic Ataxias
Episodic ataxia encompasses 7 clinically and genetically heterogeneous syndromes, only 2 of which (types 1 and 2) have been described in a large number of families. Type 1 is caused by mutations in the voltage-gated potassium channel Kv1.1. It consists of brief episodes (seconds to minutes) of cerebellar ataxia, and occasional partial seizures with clinical or electrophysiologic interictal myokymia as a main diagnostic feature. Type 2 is characterized by longer attacks (minutes to hours) and interictal cerebellar signs. It is caused by mutations in the voltage-gated calcium channel gene CACNA1A.
Benign Myoclonus of Early Infancy, Shuddering Attacks, and Chin Trembling
Benign myoclonus consists of myoclonic jerks of the extremities in wakefulness and sometimes also in sleep. It has been suggested by some that these attacks are in the same spectrum as shuddering attacks. Shuddering attacks are characterized by rapid tremor of the head, shoulder, and trunk, lasting a few seconds, often associated with eating, and recurring many times a day. Others have considered shuddering as an early manifestation of essential tremor as family history of essential tremor is often present. The clinical events in either of these can be mistaken for infantile spasms, but ictal and interictal EEG, MRI, and development are normal. Spontaneous remission occurs in both usually within a few months. Hereditary chin trembling at 3/sec precipitated by stress has also been reported.
Syndrome of Potassium Channel Autoantibodies
This syndrome is often associated with malignancy and with neurocognitive deterioration. It can manifest many seizure-like movement disorders that are responsive to immunotherapy rather than to antiepileptic drugs. These include choreoathetosis, dystonia, myoclonus, autonomic changes, and hallucinations. Epileptic seizures can also occur. It has been reported in older children and is more common in adults.
Autonomic Storms (Diencephalic Seizures)
Spells of hyperhidrosis, changes in blood pressure, temperature and autonomic instability occur in patients with severe diffuse brain injury or localized hypothalamic injury and have been termed autonomic storms. The term “diencephalic seizures” is discouraged as these are not truly seizures. Therapy is difficult and has included, with mixed results, clonidine, anticonvulsants, cyproheptadine, morphine, or sympathectomy.
Psychologic Disorders
Many psychologic disorders can be mistaken for epileptic seizures. Pleasurable behavior similar to masturbation may occur from infancy onward, and may consist of rhythmical rocking movement in a sitting or lying position, or rhythmic hip flexion and adduction. Masturbation may occur in girls 2-3 yr of age and is often associated with perspiration, irregular breathing, and grunting, but no loss of consciousness. Occasionally this is associated with child abuse or with other psychopathology. Stereotypies, or repetitive movements which are more complex than tics and do not change and wax and wane like tics (e.g., head banging, head rolling, body rocking, and hand flapping), usually occur in neurologically impaired children. Panic and anxiety attacks have been described in children; at times, these may be clinically indistinguishable from actual epileptic seizures, and therefore necessitate video-EEG monitoring. Rage attacks usually occur in patients with personality disorder and are not seizures. Hyperventilation spells can be precipitated by anxiety and are associated with dizziness, tingling, and, at times, carpopedal spasm. Transient global amnesia consists of isolated short term memory loss for minutes to hours that occurs mostly in the elderly. The etiology can by epileptic, vascular, or drug related.
Oculomotor Abnormalities
Paroxysmal Tonic Upgaze of Childhood
This usually starts before 3 mo of age, and consists of protracted attacks (hours to days) of continuous or episodic upward gaze deviation, during which horizontal eye movements are preserved. A down-beating nystagmus occurs on downward gaze. Symptoms are reduced or relieved by sleep, exacerbated by fatigue and infections, and spontaneously remit after a few years. Up to 50% of patients may have psychomotor and language delay. Imaging, laboratory, and neuropsychologic examinations are usually nonrevealing. Therapy with low dose levodopa/carbidopa may be helpful.
Oculomotor Apraxia and Saccadic Intrusions
Saccadic eye movements are impaired. Sudden head turns compensating for lateral gaze impairment mimic seizures. This disorder may be idiopathic (Cogan oculomotor apraxia) or may occur in the context of ataxia telangiectasia or lysosomal storage diseases. Genetic defects in DNA repair mechanisms have been implicated in at least 4 spinocerebellar ataxia disorders that are accompanied by oculomotor apraxia. A selective loss of Purkinje cells required to suppress omnipause neurons and initiate saccadic eye movement is believed to occur in those disorders. Saccadic intrusions are involuntary, sudden conjugate eye movements away from the desired eye position, which are not necessarily pathologic.
Spasmus Nutans
This disorder presents with a triad of nystagmus, head tilt, and head nodding. If diurnal fluctuation occurs, symptoms may look like epileptic seizures. Brain MRI should be performed, as the triad has been associated with masses in the optic chiasm and 3rd ventricle. Retinal disease should also be ruled out. Remission occurs before 5 yr of age.
Opsoclonus Myoclonus Syndrome
The so-called dancing eyes refers to continuous, random, irregular, and conjugate eye movements that may fluctuate in intensity. They usually accompany myoclonus and ataxia (“dancing feet”). Encephalitis and neuroblastoma are possible causes. Therapy is by treating the underlying etiology, but ACTH, corticosteroids, and clonazepam may be needed. Rituximab has been studied and looks in there preliminary trials to be effective as well.
Daydreaming and Behavioral Staring
Staring may be a manifestation of absence seizures, which should be differentiated from daydreaming, behavioral staring due to fatigue, or inattention. Episodes of staring only in certain settings (e.g., school) are unlikely to be seizures. In addition, responsiveness to touch and lack of interruption of playing characterizes nonepileptic staring.
Sleep Disorders
Paroxysmal nonepileptic sleep events are more common in epileptic patients than in the general population, which makes their diagnosis difficult. Semiology, timing of events, video-EEG and polysomnography help in distinguishing epileptic from non-epileptic events. Parasomnias typically occur less than once or twice a night; more frequent episodes suggest epileptic seizures.
Benign Sleep Myoclonus and Neonatal Sleep Myoclonus
Neonatal sleep myoclonus consists of repetitive, usually bilateral rhythmic jerks involving the upper and lower limbs during non-REM sleep, sometimes mimicking clonic seizures. A slow (1 Hz) rocking of the infant in a head to toe direction is a specific diagnostic test that reproduces the myoclonus. The lack of autonomic changes, occurrence only in sleep, and suppression by awakenings may help in differentiating these events from epileptic seizures. Remission is spontaneous at 2-3 mo of age. In older children and adults, sleep myoclonus consists of random myoclonic jerks of the limbs.
Non-REM Partial Arousal Disorders
Brief nocturnal confusional arousals occurring 1-2 hr after sleep in stage 4 sleep are normal in children. Such episodes can vary from chewing, sitting up, and mumbling to agitated sleep walking, and usually last for 10-15 min. Night terrors similarly occur a few hours after going to sleep in stage 3 or 4 of sleep, most often at 2-7 yr of age and more so in boys. The child screams; appears terrified; has dilated pupils, tachycardia, tachypnea, unresponsiveness, agitation, and thrashing that increase with attempts to be consoled; is difficult to arouse; and may have little or no vocalization. In older children with persistent night terrors an underlying psychologic etiology may be present. Diagnosis is based on the history. However, rarely, video EEG monitoring may be needed. At times, the use of bedtime diazepam (0.2-0.3 mg/kg) or clonazepam (0.01 mg/kg) may help control the problem while psychologic factors are being investigated. Restless leg syndrome can cause painful leg dysesthesias that cause nocturnal arousals and insomnia. It can be either genetic or associated with iron deficiency, systemic illness, or some drugs. Therapy depends upon treating the underlying cause and, if needed, on dopaminergic drugs such as levodopa/carbidopa, or antiepileptics like gabapentin.
REM Sleep Disorders
Nightmares and sleep paralysis are common disorders. Unlike night terrors, nightmares tend to occur later during the night and the child has a memory of the event.
Sleep Transition Disorders
Nocturnal head banging (jactatio capitis nocturna), rolling, or body rocking often occur in infants and toddlers as they are trying to fall asleep. These usually remit spontaneously by 5 yr of age. No specific therapy is needed.
Narcolepsy-Cataplexy Syndrome
Narcolepsy is characterized by excessive daytime sleepiness, cataplexy (sudden loss of tone), sleep paralysis, hypnogogic hallucinations, and disturbed nighttime sleep. Loss of tone occurs in response to strong emotions, and spreads from the face downwards. Consciousness is maintained in cataplexy. A selective loss of hypocretin-secreting neurons in the hypothalamus is at the origin of this disorder. The fact that DQB1*0602 is a predisposing HLA allele identified in 85-95% of patients with narcolepsy-cataplexy suggests an autoimmune mediated neuronal loss. Diagnosis is based on the multiple sleep latency test, and therapy relies on scheduled naps, amphetamines, methylphenidate, tricyclic antidepressants, and counseling about precautions in work and driving.
Bakker MJ, van Dijk JG, van den Maagdenberg AM, et al. Startle syndromes. Lancet Neurol. 2006;5:513-524.
Crompton DE, Berkovic SF. The borderland of epilepsy: clinical and molecular features of phenomena that mimic epileptic seizures. Lancet Neurol. 2009;8(4):370-381.
Dauvilliers Y, Arnulf I, Mignot E. Narcolepsy with cataplexy. Lancet. 2007;369:499-511.
DiMario FJJr. Paroxysmal nonepileptic events of childhood. Semin Pediatr Neurol. 2006;13:208-221.
Fertleman CR, Ferrie CD, Aicardi J, et al. Paroxysmal extreme pain disorder (previously familial rectal pain syndrome). Neurology. 2007;69:586-595.
Tan KM, Lennon VA, Klein CJ, et al. Clinical spectrum of voltage-gated potassium channel autoimmunity. Neurology. 2008;70:1883-1890.
Webster G, Berul CI. Congenital long-QT syndromes: a clinical and genetic update from infancy through adulthood. Trends Cardiovasc Med. 2008;18:216-224.