Chapter 593 Demyelinating Disorders of the CNS
Demyelinating disorders of the central nervous system (CNS) cause acute or relapsing-remitting encephalopathy and other multifocal signs of brain, brainstem, and spinal cord dysfunction. They affect white matter, which is formed by myelin contained within oligodendrocytes, providing electrical insulation for neurons and neuronal connections. In contrast to genetically determined leukodystrophies (sometimes called dysmyelinating disorders) that also produce disrupted white matter, demyelinating disorders generally target normally formed white matter through immune-mediated mechanisms. Major demyelinating disorders in childhood include multiple sclerosis (MS) and acute disseminated encephalomyelitis (ADEM). The rare macrophage activating syndromes and isolated angiitis of the CNS can sometimes be confused with ADEM (Table 593-1).
Table 593-1 SUMMARY OF CONSENSUS DEFINITIONS FOR PEDIATRIC CENTRAL NERVOUS SYSTEM INFLAMMATORY DEMYELINATION
| MONOPHASIC CNS INFLAMMATORY DEMYELINATION | |
| ADEM | Clinical event must include encephalopathy (behavioral change and/or altered consciousness) |
| New symptoms or signs within 3 months are considered part of same ADEM event | |
| CIS | Clinical event that can be monofocal (e.g., isolated optic neuritis) or polyfocal, but cannot include encephalopathy |
| NMO | Must have optic neuritis and transverse myelitis as major criteria. Must have spinal MRI lesion extending over 3 or more segments or be NMO-IgG positive |
| RELAPSING CNS INFLAMMATORY DEMYELINATION | |
| Recurrent ADEM | New event of ADEM (must have encephalopathy) with recurrence of initial ADEM symptoms and signs 3 or more months after initial event and not related to withdrawal of steroids |
| Multiphasic ADEM | ADEM followed by new clinical event also meeting criteria for ADEM, but involving new CNS lesions (clinically and radiologically) |
| Relapsing NMO | Relapse of NMO as described above. Additional clinical and radiologic features extrinsic to optic nerve and spinal cord are well described and acceptable for diagnosis |
| Pediatric MS | Two or more events separated in time (4 or more weeks) and space. First episode cannot be ADEM. If 1st episode is ADEM, 2 or more non-ADEM events are required for diagnosis of MS. New MRI lesions 3 months or longer after the initial clinical event can be used to satisfy criteria for dissemination in time |
All events must include compatible MRI features of CNS inflammatory demyelination and exclude alternative causes.
ADEM, acute disseminated encephalomyelitis; CIS, clinically isolated syndrome; CNS, central nervous system; MS, multiple sclerosis; NMO, neuromyelitis optica.
(Data from Krupp LB, Banwell B, Tenembaum S: Consensus definitions proposed for pediatric multiple sclerosis and related disorders, Neurology 68[16 Suppl 2]:S7–S12, 2007.)
593.1 Multiple Sclerosis
Multiple sclerosis (MS) is a chronic demyelinating disorder of the brain, spinal cord and optic nerves characterized by a relapsing-remitting course of neurologic episodes separated in time and space.
Epidemiology
Pediatric MS is rare, with an estimated 2-5% of MS patients experiencing their 1st symptoms before age 18 yr. Pediatric MS has a slight male predominance when disease onset is before age 6 yr, but by age 12 yr, females outnumber males 2 : 1.
Pathogenesis
A complex interplay of environmental, infectious, and genetic factors influence MS susceptibility. Immune system dysregulation involving T and B lymphocytes triggers inflammation, axonal demyelination, axonal loss, and regeneration within both white and gray matter. Inflammatory infiltrates within actively demyelinating lesions of relapsing-remitting MS are targets for disease modifying therapy (DMT). Neurodegenerative changes predominate in progressive forms of MS.
Clinical Manifestations
Presenting symptoms in pediatric MS include hemiparesis or paraparesis, unilateral or bilateral optic neuritis, focal sensory loss, ataxia, diplopia, dysarthria, or bowel/bladder dysfunction (Table 593-2). Polyregional symptoms are reported in 30% of patients. Encephalopathy is less common and suggests consideration of acute disseminated encephalomyelitis (ADEM).
Table 593-2 SYMPTOMS AND SIGNS OF MULTIPLE SCLEROSIS BY SITE
| SYMPTOMS | SIGNS | |
|---|---|---|
| Cerebrum | Cognitive impairment | Deficits in attention, reasoning, and executive function (early); dementia (late) |
| Hemisensory and motor | Upper motor neuron signs | |
| Affective (mainly depression) | ||
| Epilepsy (rare) | ||
| Focal cortical deficits (rare) | ||
| Optic nerve | Unilateral painful loss of vision | Scotoma, reduced visual acuity, color vision, and relative afferent papillary defect |
| Cerebellum and cerebellar pathways | Tremor | Postural and action tremor, dysarthria |
| Clumsiness and poor balance | Limb incoordination and gait ataxia | |
| Brainstem | Diplopia, oscillopsia | Nystagmus, internuclear and other complex ophthalmoplegias |
| Vertigo | ||
| Impaired swallowing | Dysarthria | |
| Impaired speech and emotional lability | Pseudobulbar palsy | |
| Paroxysmal symptoms | ||
| Spinal cord | Weakness | Upper motor neuron signs |
| Stiffness and painful spasms | Spasticity | |
| Bladder dysfunction | ||
| Erectile impotence | ||
| Constipation | ||
| Other | Pain | |
| Fatigue | ||
| Temperature sensitivity and exercise intolerance |
Modified from Compston A, Coles A: Multiple sclerosis, Lancet 372:1502–1517, 2008, p 1503.
Laboratory Findings
Cranial MRI exhibits discrete T2 lesions in cerebral white matter, particularly periventricular regions as well as brainstem, cerebellum, and juxtacortical and deep gray matter. Alternatively, tumefactive T2 lesions are also seen. Spine MRI typically shows partial-width cord lesions restricted to 1-2 spine segments. CSF may be normal or exhibit mild pleocytosis, particularly in younger children. Abnormal MS profiles (increased IgG index, and/or CSF oligoclonal bands) increase likelihood of MS but may be negative in 10-60% of pediatric MS patients, particularly prepubertal children (Fig. 593-1). Abnormal evoked potential studies can localize disruptions in visual, auditory, or somatosensory pathways.
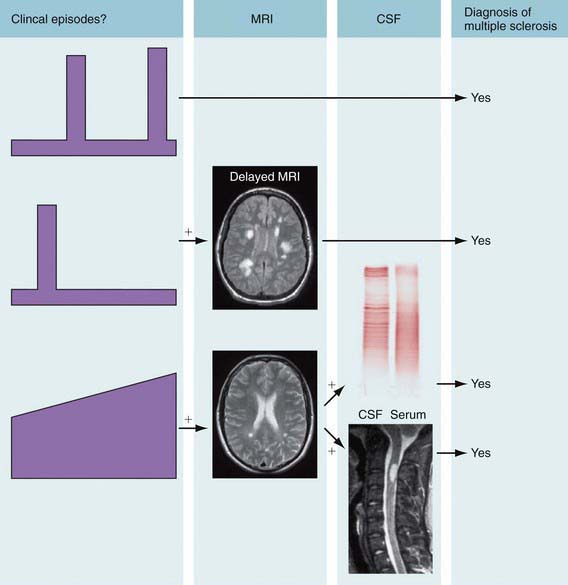
Figure 593-1 Criteria for the diagnosis of multiple sclerosis (MS). The principle is to establish dissemination in time and place of lesions, meaning that episodes affecting separate sites within the central nervous system have occurred at least 30 days apart. MRI can substitute for 1 of these clinical episodes. Dissemination in time of magnetic resonance lesions requires: 1 gadolinium-enhancing lesion at least 3 mo after the onset of the clinical event; or a new T2 lesion compared with a reference scan done at least 30 days after onset of the clinical event. In the case of recurrent stereotyped clinical episodes at the same neurologic site, criteria for MRI definition of dissemination in space are 3 features from the following: (1) 1 gadolinium-enhancing lesion or 9 T2 MRI lesions; (2) 1 or more infratentorial lesions; (3) 1 or more juxtacortical lesions; or (4) 3 or more periventricular lesions (a spinal cord lesion can replace some of these brain lesions). Primary progressive MS can be diagnosed after 1 yr of a progressive deficit and 2 of the following: (1) a positive brain MRI; (2) a positive spinal cord MRI; and (3) positive oligoclonal bands. Patients having an appropriate clinical presentation but who do not meet all of the diagnostic criteria can be classified as having possible MS. CSF, cerebrospinal fluid.
(From Compston A, Coles A: Multiple sclerosis, Lancet 372:1502–1517, 2008, Fig 1, p 1504.)
Diagnosis and Differential Diagnosis
Like adult MS, pediatric MS can be diagnosed following 2 demyelinating episodes localizing to distinct CNS regions, lasting >24 hr and separated by >30 days, provided no other plausible explanation exists. Alternatively, accumulation of T2 or gadolinium-enhancing lesions in the brain or spine >3 mo later can demonstrate dissemination in time, enabling MS diagnosis after the 1st event. Challenges arise in distinguishing pediatric MS from other demyelinating syndromes such as acute disseminated encephalomyelitis (ADEM) or neuromyelitis optica (NMO), ADEM is a self-limited syndrome characterized by encephalopathy, polyregional neurologic deficits, and diffuse multifocal MRI T2 abnormalities followed by subsequent clinical improvement and resolution of MRI T2 lesions (see Tables 593-1 and 593-3). However, a subset of pediatric MS patients (10-25%) present with an ADEM phenotype and then experience multiple relapses with accumulation of MRI T2 lesions. NMO, traditionally a combined myelitis and optic neuritis with normal brain MRI, now has a broader phenotype with the recent identification of the NMO antibody against the CNS water channel aquaporin-4. The NMO spectrum now includes isolated bilateral ON or longitudinally extensive transverse myelitis even in the presence of brain MRI abnormalities or encephalopathy.
Table 593-3 CLINICAL AND MRI FEATURES THAT MAY DISTINGUISH ADEM FROM FIRST ATTACK OF MS
| ADEM | MS | |
|---|---|---|
| Age | <10 yr | >10 yr |
| Stupor/coma | + | − |
| Fever/vomiting | + | − |
| Family history | No | 20% |
| Sensory complaints | + | + |
| Optic neuritis | Bilateral | Unilateral |
| Manifestations | Polysymptomatic | Monosymptomatic |
| MRI imaging | Widespread lesions: basal ganglia, thalamus, cortical gray-white junction | Isolated lesions: periventricular white matter, corpus callosum |
| CSF | Pleocytosis (lymphocytosis) | Oligoclonal bands |
| Response to steroids | + | + |
| Follow-up | No new lesions | New lesions |
Some features that may help distinguish an initial acute episode of demyelination from a 1st attack of MS in children. Final diagnosis of MS is based on follow-up evaluation and possibly MRI.
ADEM, acute disseminated encephalomyelitis; CSF, cerebrospinal fluid; MS, multiple sclerosis; +, more likely to be present; –, less likely to be present.
Complications
Similar to adults with MS, pediatric MS patients can acquire fixed neurologic deficits affecting vision and other cranial nerves, motor and sensory function, balance, and bowel/bladder function. Cognitive impairment can impede academic achievement.
Treatment
Relapses causing functional disability may be treated with methylprednisolone, 20-30 mg/kg/day (maximum 1,000 mg/day) for 3-5 days, with or without prednisone taper. Injectable DMT (interferon-beta1α or interferon-beta1β; glatiramer acetate) reduces relapse frequency and lowers MRI T2 lesion load in adult MS, especially with early institution of therapy. Although not yet approved by the U.S. Food and Drug Administration for pediatric MS, injectable DMT use has been reported in >1,000 children and adolescents with MS. Pediatric experience is limited with intravenously infused DMT, natalizumab, and mitoxantrone. Drugs that target lymphocyte subtypes (cladribine, alemtuzumab) or interfere with lymphocyte myelin interaction (fingolimod, natalizumab) are promising agents in trials in adult patients with MS. Natalizumab therapy has been associated with the risk of developing progressive multifocal encephalopathy (CNS infection with human polyomavirus JC).
Prognosis
Retrospective studies of patients diagnosed with MS prior to widespread DMT use suggest slower disease progression in pediatric MS patients compared to adults. Despite a longer time to irreversible disability (20-30 yr), pediatric MS patients acquire irreversible disability at a younger age than adults.
Banwell B, Kennedy J, Sadovnick D, et al. Incidence of acquired demyelination of the CNS in Canadian children. Neurology. 2009;72:232-239.
Callen DJ, Shroff MM, Branson HM, et al. MRI in the diagnosis of pediatric multiple sclerosis. Neurology. 2009;72:961-967.
The CAMMS223 Trial Investigators. Alemtuzumab vs. interferon beta-1a in early multiple sclerosis. N Engl J Med. 2008;359:1786-1801.
Cohen JA, Barkhof F, Comi G, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. 2010;362:402-414.
Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372:1502-1517.
Franklin RJM. Stem cell treatments and multiple sclerosis. BMJ. 2010;340:985-986.
Gajofatto A, Monaco S, Fiorini M, et al. Assessment of outcome predictors in first-episode acute myelitis. Arch Neurol. 2010;67(6):724-730.
Giovannoni G, Comi G, Cook S, et al. A placebo-controlled trial of oral cladribine for relapsing multiple sclerosis. N Engl J Med. 2010;362:416-426.
Goodman AD, Brown TR, Edwards KR, et al. A phase 3 trial of extended release oral dalfampridine in multiple sclerosis. Ann Neurol. 2010;68:494-502.
Kalluri SR, Illes Z, Srivastava R, et al. Quantification and functional characterization of antibodies to native aquaporin 4 in neuromyelitis optica. Arch Neurol. 2010;67(10):1201-1208.
Krupp LB, Banwell B, Tenembaum S, et al. Consensus definitions proposed for pediatric multiple sclerosis and related disorders. Neurology. 2007;68:S7-S12.
Linda H, von Heijne A, Major EO, et al. Progressive multifocal leukoencephalopathy after natalizumab monotherapy. N Engl J Med. 2009;361:1081-1087.
The Medical Letter. Interferon beta-lb (extavia) for multiple sclerosis. Med Lett. 2010;52:86-87.
The Medical Letter. Oral fingolimod (gilenya) for multiple sclerosis. Med Lett. 2010;52:98-100.
Pohl D, Waubant E, Banwell B, et al. Treatment of pediatric multiple sclerosis and variants. Neurology. 2007;68:S54-S65.
Thompson A, Polman C. Improving function: a new treatment era for multiple sclerosis? Lancet. 2009;373:697-698.
Weng WC, Yang CC, Yu TW, et al. Multiple sclerosis with childhood onset: report of 21 cases in Taiwan. Pediatr Neurol. 2006;35:327-334.
Venkateswaran S, Banwell B. Pediatric multiple sclerosis. Neurologist. 2010;16:92-105.
Yeh EA, Chitnis T, Krupp L, et al. US Network of Pediatric Multiple Sclerosis Centers of Excellence. Pediatric multiple sclerosis. Nat Rev Neurol. 2009;5:621-631.
593.2 Neuromyelitis Optica
Neuromyelitis optica (NMO) (Devic disease) is a demyelinating disorder characterized by monophasic or polyphasic episodes of optic neuritis and/or transverse myelitis. It was once thought that NMO was a variant of MS; most authorities believe that NMO is a distinct disorder.
Epidemiology
NMO has an age of onset of 31.2 ± 11 yr. In 1 study, in monophasic patients, the range of age of onset was 1-54 yr; in polyphasic patients, the range was 6-72 yr. NMO is more common in women and girls than in men and boys; 65% of monophasic and 80-85% of polyphasic NMO patients are female. It is also more common in Asians than in blacks or whites and appears to have a higher mortality rate in individuals of African descent than in others.
Pathogenesis
NMO is associated with IgG antibodies to the aquaporin-4 water channel. It is not clear why the attack on and depletion of spinal cord and optic nerve aquaporin-4 results in disruption of the myelin sheath in these parts of the CNS, but pathologic examination of autopsy specimens reveals both anti–aquaporin-4 IgG deposits and B cells in the spinal cord and optic nerves of NMO patients. Although most cases of NMO are idiopathic and only occasional familial cases have been reported, there have been reports of postinfectious NMO. HIV, syphilis, chlamydia, varicella, cytomegalovirus, and Epstein-Barr virus have been associated with subsequent development of NMO.
Clinical Manifestations
NMO presents with either optic neuritis or transverse myelitis or both. Visual field defects and color desaturation are common. The symptoms and signs of transverse myelitis depend on the spinal level and completeness of the inflammatory changes. NMO differs from MS in that other parts of the nervous system are generally not involved either symptomatically or on imaging studies; recovery of visual and spinal cord function is generally not as complete after each episode; optic neuritis is more frequently bilateral in NMO than in MS; and NMO is more frequently fatal than MS.
Laboratory Findings
CSF in patients with NMO often has 50 or more WBC/µL. Unlike MS, it is devoid of oligoclonal bands. Serum positivity for anti–aquaporin-4 antibodies (so-called NMO antibodies) has a sensitivity of 73% and a specificity of 91% for NMO. Neuroimaging studies may show small, asymptomatic lesions in the white matter of the brainstem or hemispheres, but not the large (>3 mm), oval lesions in the periventricular white matter seen in MS.
Diagnosis and Differential Diagnosis
Clinical criteria for the diagnosis of NMO include finding at least 2 of the following: normal brain MRI, spinal cord widening and cavitation involving at least 3 spinal segments, decreased serum/CSF albumin ratio with normal IgG synthesis rate and the absence of oligoclonal bands, and acute episode(s) of spinal cord and/or optic nerve involvement separated by months or years without any other systemic or neurologic features. The differential diagnosis includes MS; ADEM (Chapter 593.3); rheumatologic etiologies of transverse myelitis and/or optic neuritis including systemic lupus erythematosus, Behçet disease, and neurosarcoidosis (usually accompanied by other nonneurologic manifestations); idiopathic transverse myelitis, tropical spastic paraparesis, and viral encephalomyelitis (none of which have NMO antibodies in the serum or CSF); and metabolic and idiopathic causes of isolated optic neuritis or other acute monocular or binocular visual loss (Chapter 623).
Complications
Similar to adults with NMO, pediatric NMO patients often are left with fixed neurologic deficits affecting visual acuity, visual fields, color vision, motor and sensory function, balance, and bowel/bladder function.
Treatment
Initial episodes and relapses may be treated acutely with methylprednisolone, 20-30 mg/kg/day (maximum 1,000 mg/day) for 3-5 days, followed by a slow prednisone taper. It is not yet known whether injectable DMT (interferon-beta1α or interferon-beta1β; glatiramer acetate) reduces relapse frequency in NMO. However, agents like rituximab that reduce B cell number and function have shown promise in this regard in early studies.
593.3 Acute Disseminated Encephalomyelitis (ADEM)
ADEM is an initial inflammatory, demyelinating event with multifocal neurologic deficits, typically accompanied by encephalopathy.
Epidemiology
ADEM can occur at any age but most series report a mean age between 5 and 8 yr with a slight male predominance. Reported incidence ranges from 0.07-0.4 per 100,000 per yr in the pediatric population.
Pathogenesis
Molecular mimicry induced by infectious exposure or vaccine may trigger production of CNS autoantigens. Many patients experience a transient febrile illness in the month prior to ADEM onset. Preceding infections associated with ADEM include influenza, Epstein-Barr virus, cytomegalovirus, varicella, enterovirus, measles, mumps, rubella, herpes simplex, and Mycoplasma pneumoniae. Postvaccination ADEM has been reported following immunizations for rabies, smallpox, measles, mumps, rubella, Japanese encephalitis B, pertussis, diphtheria-polio-tetanus, and influenza.
Clinical Manifestations
Initial symptoms of ADEM may include lethargy, fever, headache, vomiting, meningeal signs, and seizure, including status epilepticus. Encephalopathy is a hallmark of ADEM, ranging from ongoing confusion to persistent irritability to coma. Focal neurologic deficits can be difficult to ascertain in the obtunded or very young child but common neurologic signs in ADEM include visual loss, cranial neuropathies, ataxia, motor and sensory deficits, plus bladder/bowel dysfunction with concurrent spinal cord demyelination.
Neuroimaging
Head CT may be normal or show hypodense regions. Cranial MRI, the imaging study of choice, typically exhibits large, multifocal and sometimes confluent or tumefactive T2 lesions with variable enhancement within white and often gray matter of the cerebral hemispheres, cerebellum, and brainstem (Fig. 593-2). Deep gray matter structures (thalami, basal ganglia) are often involved although this may not be specific to ADEM. Spinal cord may have abnormal T2 signal or enhancement, with or without clinical signs of myelitis. MRI lesions of ADEM typically appear to be of similar age but their evolution may lag behind the clinical presentation. Serial MRI imaging 3-12 mo following ADEM shows improvement and often complete resolution of T2 abnormalities although residual gliosis may remain.
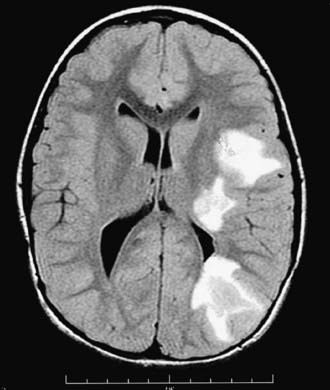
Figure 593-2 Axial T2-weighted FLAIR MRI of the brain in a child with acute disseminated encephalomyelitis (ADEM). High signal (white) lesions in the T2-weighted image reflect areas of demyelination and edema in deep subcortical and periventricular white matter as well as the basal ganglia and thalamus on the left side.
Laboratory Findings
There is no biologic marker for ADEM and laboratory findings can vary widely. CSF studies often exhibit pleocytosis with lymphocytic or monocytic predominance. CSF protein can be elevated, especially in repeat studies. Up to 10% of ADEM have oligoclonal bands in the CSF and/or elevated CSF immune globulin production. Electroencephalograms (EEG) often show generalized slowing, consistent with encephalopathy, although polyregional demyelination of ADEM can also cause focal slowing or epileptiform discharges.
Differential Diagnosis
The differential diagnosis for ADEM is broad but can be narrowed by careful history, appropriate laboratory evaluations, and MRI. Empirical antibiotic and antiviral treatment should be considered while infectious evaluations are pending. Follow-up MRI examinations 3-12 mo after ADEM should show improvement; new or enlarging T2 lesions should prompt re-evaluation for other etiologies such as MS, leukodystrophies, tumor, vasculitis, or mitochondrial, metabolic, or rheumatologic disorders (see Table 593-3).
Treatment
Although there are no randomized controlled trials to compare acute treatments for ADEM or other demyelinating disorders of childhood, high dose intravenous steroids are commonly employed (typically methylprednisolone 20-30 mg/kg per day for 5 days with a maximum dose of 1,000 mg per day). An oral prednisone taper over 1 mo may prevent relapse. Other treatment options include intravenous immune globulin (IVIG; usually 2 g/kg administered over 2-5 days) or plasmapheresis (typically 5-7 exchanges administered every other day). There is no consensus about timing of these treatments for ADEM.
Prognosis
Many children experience full recovery after ADEM but some are left with residual motor and/or cognitive deficits. ADEM is usually a monophasic illness but demyelinating symptoms can fluctuate for several months. Repeated bouts of demyelination more than 3 mo after ADEM later raise the question of MS versus repeated ADEM.
Dale RC, Brilot F, Banwell B. Pediatric central nervous system inflammatory demyelination: acute disseminated encephalomyelitis, clinically isolated syndromes, neuromyelitis optica, and multiple sclerosis. Curr Opin Neurol. 2009;22:233-240.
Huynh W, Cordato DJ, Kehdi E, et al. Post-vaccination encephalomyelitis: literature review and illustrative case. J Clin Neurosci. 2008;15:1315-1322.
Tenembaum S, Chitnis T, Ness J, et al. Acute disseminated encephalomyelitis. Neurology. 2007;68:S23-S36.
Yiu EM, Kornberg AJ, Ryan MM, et al. Acute transverse myelitis and acute disseminated encephalomyelitis in childhood: spectrum or separate entities? J Child Neurol. 2009;24:287-296.