Chapter 598 Spinal Cord Disorders
598.1 Tethered Cord
Beyond infancy the spinal cord in humans ends in the conus medullaris at about the level of L1. The position of the conus below L2 is consistent with a congenital tethered spinal cord. For normal humans as the spine flexes and extends, the spinal cord is free to move up and down within the spinal canal. If the spinal cord is fixed at any point, this movement is restricted and the spinal cord and associated nerve roots become stretched. This fixing of the spinal cord, regardless of the underlying cause of the fixation, is called a tethered cord. When severe pain or neurologic deterioration occurs in response to the fixation, it is called the tethered cord syndrome.
In its simplest form the tethered cord syndrome results from a thickened filum terminale, which normally extends as a thin, very mobile structure from the tip of the conus to the sacrococcygeal region where it attaches. When this structure is thickened and shortened, the conus is found to end at levels below L2. This stretching between 2 points is likely to cause symptoms later in life. Fatty infiltration is often seen in the thickened filum (Fig. 598-1).
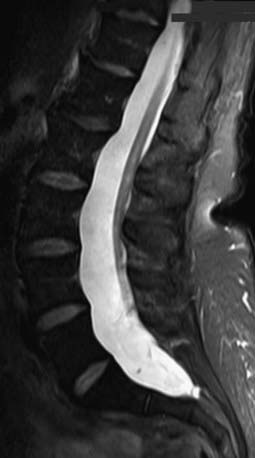
Figure 598-1 Sagittal MRI showing thickening of the filum terminale in a patient with a symptomatic tethered spinal cord.
(Used with permission from Barrow Neurological Institute.)
Any condition that fixes the spinal cord can be the cause of the tethered cord syndrome. Conditions that are well established to cause symptomatic tethering include various forms of occult dysraphism such as lipomyelomeningocele, myelocystocele, and diastematomyelia. These conditions are associated with cutaneous manifestations such as midline lipomas often with asymmetry of the gluteal fold (Fig. 598-2), and hairy patches called hypertrichosis (Fig. 598-3). Probably the most common type of symptomatic tethered cord involves patients who had previously undergone closure of an open myelomeningocele and later become symptomatic with pain or neurologic deterioration. Tethered cord syndrome can also be associated with attachment of the spinal cord in patients who undergo surgical procedures that disrupt the pial surface of the spinal cord.
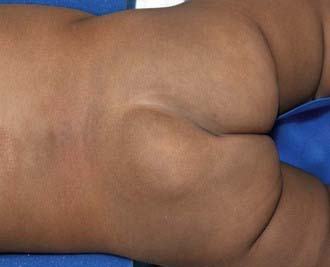
Figure 598-2 Child with a lipomyelomeningocele demonstrating an extraspinal mass and an asymmetry of the gluteal fold indicative of underlying occult dysraphism.
(Used with permission from Barrow Neurological Institute.)
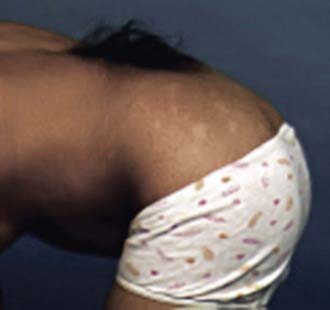
Figure 598-3 Hairy patch or hypertrichosis usually associated with diastematomyelia.
(Used with permission from Barrow Neurological Institute.)
Clinical Manifestations
Patients at risk for the subsequent development of the tethered cord syndrome can often be identified at birth by the presence of an open myelomeningocele or by cutaneous manifestations of dysraphism. It is important to examine the back of the newborn for cutaneous midline lesions (lipoma, dermal sinus, tail, hair patch, hemangioma, port-wine stain) that may signal an underlying form of occult dysraphism. Dermal sinuses are usually located above the gluteal fold. Cutaneous abnormalities are not found in patients with an isolated thickened filum terminale. Patients who become symptomatic later in life often exhibit an asymmetry of the feet (i.e., one is smaller than the other). The smaller foot will show a high arch and clawing of the toes (Fig. 598-4). Characteristically, there is no ankle jerk on the involved side and the calf is atrophied. This condition is termed the neuro-orthopedic syndrome.
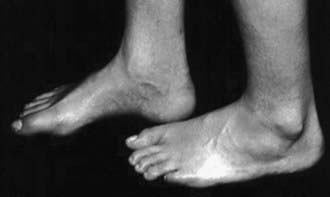
Figure 598-4 Example of the neuro-orthopedic syndrome involving a larger left foot than right foot, a high arch, and absent ankle jerk frequently associated with tethered cord regardless of the etiology.
(Used with permission from Barrow Neurological Institute.)
Three clinical syndromes can occur at the time of deterioration. The most likely clinical presentation is increasing urinary urgency and, finally, incontinence. Deterioration of motor and sensory function in the lower extremities is a compelling reason for intervention. Finally, severe generalized back pain, often radiating into the lower extremities, can occur, particularly in older adolescents and adults.
Diagnostic Evaluation
When patients present with symptoms related to the tethered cord syndrome, a thorough motor and sensory examination of the patient must be documented. Assessment of bladder function with an ultrasound of the bladder and urodynamic studies is useful in analyzing bladder innervation. MRI is the diagnostic study of choice to reflect the anatomy of the tethering lesion and to provide information about the risks of surgical intervention.
Treatment
There are no nonsurgical options for the management of tethered cord syndrome. Because the presence of tethering is most likely to be at least suspected in the newborn, prophylactic surgery to prevent late deterioration has been advocated by some neurosurgeons. This strategy remains controversial and depends to some extent on a careful assessment of the risks compared to the benefits. If surgical intervention is chosen, microsurgical dissection with release of the spinal cord attachment to the overlying dura is the goal of treatment.
Outcome
The outcome of releasing a thickened filum terminale or detethering of patients with diastematomyelia is routinely good, and the chance of recurrent symptoms is very low. Patients with symptomatic tethered cord who undergo repair of a myelomeningocele or a lipomyelomeningocele have a significant possibility of recurrent tethering and recurrent symptoms.
598.2 Diastematomyelia
Diastematomyelia: Split Cord Malformation
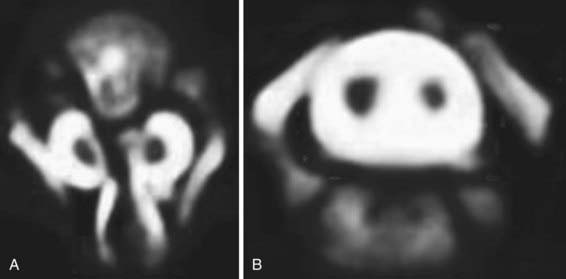
Diastematomyelia is a relatively rare form of occult dysraphism in which the spinal cord is divided into 2 halves. In type 1 split cord malformation, there are 2 spinal cords, each in its own dural tube and separated by a spicule of bone and cartilage (Fig. 598-5A). In a type 2 split cord malformation, the 2 spinal cords are enclosed in a single dural sac with a fibrous septum between the 2 spinal segments (Fig. 598-5B). In both cases the anatomy of the outer half of the spinal cord is essentially normal while the medial half is extremely underdeveloped. Undeveloped nerve roots and dentate ligaments terminate medially into the medial dural tube in type 1 cases and terminate in the membranous septum in type 2 cases. Both types have an associated defect in the bony spinal segment. In the case of type 2 lesions, this defect can be quite subtle.
Clinical Manifestations
Patients with both type 1 and type 2 split cord malformations may have subtle signs of neurologic involvement such as unilateral calf atrophy and a high arch to one or both feet early in life, but they are more likely to be neurologically normal. These patients are tethered by the adherence of the spinal cord to the median membrane or dural sac. Later they may develop progressive loss of bowel and bladder function and sensory and motor difficulties in the lower extremities. Back pain is a common symptom in adolescents and adults with split cord malformation but is uncommon in small children.
Cutaneous manifestations of dysraphism are present in 90% of patients with split cord malformations. Large hairy midline patches called hypertrichosis, the most common cutaneous manifestations, are present in about 60% of the cases.
Diagnostic Evaluation
MRI, the study of choice, shows the 2 spinal cords. The frequent association of bony abnormalities in this condition may require further evaluation with radiography or computed tomography.
Treatment
The treatment of split cord malformations is surgical. This abnormality is a form of tethered cord syndrome, and its treatment is to release the spinal cord to move freely with movement of the spine. In type 1 split cord malformations, the 2 half cords are in separate dural sacs with medial attachment to the dura and bony septum. In this case the dura needs to be opened, the bony septum removed, the medial attachments to the dura lysed, and a single dural tube created. For type 2 lesions, the membranous septum should be lysed. An attachment of this membrane to the anterior dura should be explored and lysed as well. Retethering of this type is rare as there is no reason to disrupt the pial layer of the spinal cord.
598.3 Syringomyelia
Syringomyelia is a cystic distension of the spinal cord caused by obstruction of the flow of spinal fluid from within the spinal cord to its point of absorption. There are 3 recognized forms of syringomyelia depending on the underlying cause. Communicating syringomyelia implies that cerebrospinal fluid (CSF) from within the ventricles communicates with the fluid within the spinal cord and is assumed to be the source of the CSF that distends the spinal cord. Noncommunicating syringomyelia implies that ventricular CSF does not communicate with the fluid within the spinal cord. It primarily occurs in the context of intramedullary tumors and obstructive lesions. In the final form of syringomyelia, that is, post-traumatic syringomyelia, spinal cord injury results in damage and subsequent softening of the spinal cord. This softening, combined with the scarring of the surrounding spinal cord tissue, results in progressive distension of the cyst.
Clinical Manifestations
Signs and symptoms of syringomyelia develop insidiously over years or decades. The classic presentation is the central cord syndrome. In this situation the patient develops numbness beginning in the shoulder in a capelike distribution followed by the development of atrophy and weakness in the upper extremities. Trophic ulcers of the hands are characteristic of advanced cases. The central cord syndrome results from damage to the central spinal cord and the orientation of spinal tracts from proximal to distal leading to selective involvement of the upper rather than the lower extremities.
Other forms of presentation include scoliosis that may be rapidly progressive and often can be presumed from the absence of superficial abdominal reflexes. Urgency and bladder dysfunction as well as lower extremity spasticity also may be part of the presentation.
In patients with syringomyelia related to spinal cord injury, the presentation is usually severe pain in the area of the spinal cord distension above the level of the initial injury. There is also an ascending level of motor and sensory dysfunction.
Diagnostic Evaluation
MRI is the radiologic study of choice (Figs. 598-6 and 598-7). The study should include the entire spine and should include gadolinium-enhanced sequences. Specific attention should be paid to the craniovertebral junction due to the frequent association of syringomyelia with Chiari I and II malformations. Obstruction to the flow of CSF from the 4th ventricle can cause syringomyelia; therefore, most patients also should undergo imaging of the brain.
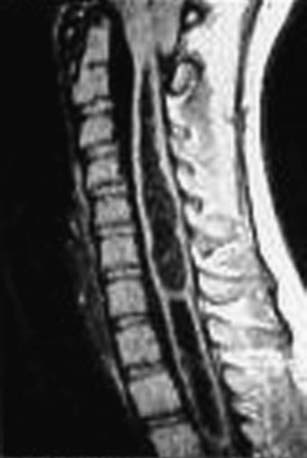
Treatment
The treatment of syringomyelia should be tailored to the underlying cause. If that cause can be removed or ameliorated, the syrinx should improve. Traumatic syrinxes are treated by preventing distension of the spinal cord by transecting the spinal cord in cases of complete spinal cord injury. Doing so drains the fluid from the spinal cord. In cases of incomplete spinal cord injury, functioning neurologic elements must be protected. Microscopic lysis of the scar surrounding the spinal cord at the point of injury allows the spinal cord to collapse and prevents it from being distorted by a hydrostatic column.
Communicating syringomyelia is most frequently seen in the context of abnormalities at the craniovertebral junction caused by inflammatory conditions such as chronic meningitis as seen in tuberculosis or meningeal carcinomatosis. However, it is most often associated with hindbrain herniation as in Chiari malformations. In such cases decompression of the craniovertebral junction is usually effective in the management of the syringomyelia. In the context of the Chiari II malformation associated with spina bifida, syringomyelia usually results from an insidious failure of the shunt used to treat the hydrocephalus. This distension of the spinal cord results in a rapid development of scoliosis and occasionally spasticity in the lower extremities. Repair of the shunt is effective treatment.
Noncommunicating syringomyelia results from blocking the flow of spinal cord extracellular fluid or CSF within the central canal by an intramedullary spinal cord tumor or severe external compression of the spinal cord. In such cases management should be directed to tumor resection or to decompression of constricting elements.
Drainage procedures can result in symptomatic and radiographic improvement. Syrinx-to-subarachnoid shunting with a small piece of shunt tubing is 1 form of treatment. Syrinx-to-pleural or syrinx-to-peritoneal shunting is more likely to result in improvement in the radiographic appearance of the syrinx. In patients with syringomyelia that extends to the conus medullaris, remnants of the central canal can be found in the filum terminale. Lysis of this structure near the conus can provide effective drainage.
598.4 Spinal Cord Tumors
Tumors of the spine and spinal cord are rare in children. Different types of tumors have different relationships with the spinal cord, meninges, and bony elements of the spine (Fig. 598-8). Intramedullary spinal cord tumors arise within the substance of the spinal cord itself (Fig. 598-9). They represent between 5% and 15% of primary central nervous system tumors. This percentage may well reflect the total volume of spinal cord as opposed to brain. About 10% of intramedullary spinal cord tumors are malignant astrocytic tumors, but most are World Health Organization grade I or II tumors of glial or ependymal origin. In children, low-grade astrocytomas and gangliogliomas represent the most common tumor types with ependymomas being less common than in adults. Ependymomas in children are frequently associated with neurofibromatosis (NF 2).
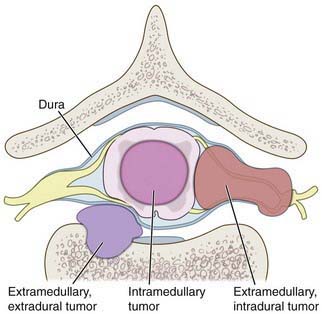
Figure 598-8 Diagram of the relationship of various tumors to the spine, nerve roots, and spinal cord.
(Used with permission from Barrow Neurological Institute.)
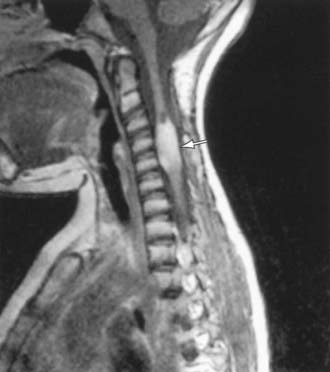
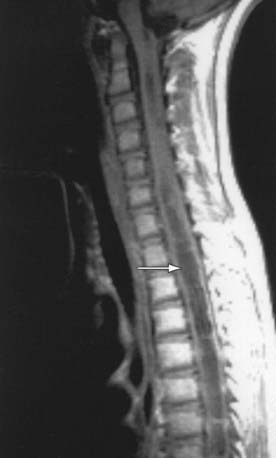
Figure 598-9 T1-weighted MRI scan of a spinal cord tumor (arrow). The fusiform expansion of the cervical cord enhances after intravenous gadolinium injection.
Except in the context of NF 1 and NF 2, intradural extramedullary tumors are extremely rare in children. Most are nerve sheath tumors, either schwannomas or, in the case of NF 2, neurofibromas. Intraspinal meningiomas in children are essentially found only in patients with NF 2. The intradural extramedullary compartment is also a site for metastatic tumors from primary cancers such as leukemia or primitive neuroectodermal tumors.
Extradural spinal tumors characteristically begin in the bones of the spine. Primary tumors in this location include aneurysmal bone cysts, Langerhans cell histiocytosis (formerly called eosinophilic granuloma), and giant cell tumors. In infants the extradural space is often the site of neuroblastomas or ganglioneuroblastomas, which tend to be present in the epidural space and in the paraspinous tissue through the intervertebral foramen. In older patients the bones of the spine may be the site of multiple myeloma and metastases from common malignant tumors.
Clinical Manifestations
With the exception of the uncommon malignant glial tumors of the spinal cord, which tend to present precipitously, intramedullary spinal cord tumors present in a very insidious manner. Back pain related to the level of the tumor is a common presenting complaint. It is likely that this pain will awaken the child from sleep and improve as the day progresses. Before the use of MRI became routine, the time from the 1st onset of symptoms to diagnosis of the tumor could be as long as 9 yr. Weakness, gait disturbance, and sensory deficits are usually minor and are often found when formal neurologic examinations are performed. Scoliosis, urinary urgency, and incontinence may be the presenting complaints associated with intramedullary spinal cord tumors.
Extramedullary extradural tumors have a propensity to cause an acute block of the CSF pathways owing to rapid growth within a confined space. Such children present with a flaccid paraplegia, urinary retention, and a patulous anus. Some extramedullary tumors produce the Brown-Séquard syndrome, which consists of ipsilateral weakness, spasticity, and ataxia, with contralateral loss of pain and temperature sensation. Papilledema is observed in a few patients, usually in association with markedly elevated CSF protein levels that presumably interfere with normal CSF flow dynamics.
Nerve sheath tumors primarily arise from the sensory rootlet of the exiting spinal nerve. They are very slow-growing tumors and present with symptoms and signs relative to the nerve root involved. Pain in a bandlike distribution around the chest or into an extremity is the most common presenting complaint. Tumor growth eventually leads to spinal cord compression and involvement of adjacent nerve roots.
Tumors rarely arise in the fat of the epidural space; most epidural tumors arise in the bony compartment of the spine. They can present abruptly with severe pain and neurologic deficit at the time of pathologic fracture of the vertebral body. Benign tumors such as giant cell tumors and aneurysmal bone cysts present more insidiously as the tumor slowly grows and begins to compress neural structures.
Diagnostic Evaluation
MRI with and without gadolinium enhancement of the spinal cord is the diagnostic study of choice and is essential in the diagnosis of spinal cord tumors, especially intramedullary spinal cord tumors. Most astrocytic tumors of the spinal cord and most ependymomas show diffuse enhancement and will distend the spinal cord focally. These tumors may involve the entire length of the spinal cord (holocord astrocytomas). MRI also shows the relationship between the normal spinal cord and tumor embedded within spinal cord tissue. These tumors are frequently associated with a syrinx, which is usually distal to the tumor. Nerve sheath tumors characteristically enhance and are focal. They may exit through the neural foramen and distend the canal as can be seen on MRI. They also may be visualized on plain radiographs of the affected area of the spine.
Plain radiographs of the spine are helpful in defining the relationship of extradural tumors to the bony spine and in documenting evidence of instability in the case of pathologic compression fractures. When a pathologic fracture occurs, CT is essential to determine the effect of the tumor on the bone. Because many of these tumors occur as metastatic lesions, a general staging of the extent of disease is essential. In the case of Langerhans cell histiocytosis, a thorough bone survey should be conducted to look for other lesions. Radionucleotide bone scanning is also useful in determining the extent of the disease.
Treatment
The primary treatment of both intramedullary and extramedullary intradural tumors is surgical removal. For both low-grade astrocytomas and ependymomas, microsurgical removal with the intent of total removal is the treatment of choice. This goal should be attainable in all patients with ependymomas and in most patients with low-grade astrocytomas and gangliogliomas. Adjunctive treatment of these tumors is unwarranted in patients treated with adequate surgical resection. Likewise, schwannomas should be resectable. Occasionally, however, the nerve root must be resected. Doing so is of no consequence in the thoracic spinal cord, but an attempt to remove the tumor while salvaging the motor root in the cervical and thoracic region is critical to preserve movement. Malignant astrocytic tumors cannot be resected without major morbidity and, in any case care, carry an extremely poor prognosis. In the case of grade III and IV astrocytomas of the spinal cord, decompression and biopsy followed by radiation therapy and possibly chemotherapy are utilized.
The diagnosis and treatment of extramedullary spinal cord tumors must be individualized. Patients with distension of the vertebral body or with unstable pathologic fractures benefit from extensive resection of the involved vertebral bodies and will likely need fusion. For extramedullary tumors with soft tissue components such as neuroblastomas, treatment is determined by the nature of the tumor and degree of spinal cord compression, and directed following needle biopsy of the lesion. In the absence of significant neurologic compression, surgical intervention is rarely indicated.
Outcome
The prognosis for patients with benign intramedullary spinal cord tumors depends, to some extent, on the patient’s condition at the time of surgical intervention. It is very unlikely that nonambulatory patients will improve after surgery. If, however, patients are ambulatory at the time of surgery, they may experience increased weakness after surgery. They are likely to recover at least their preoperative level of function. Malignant spinal cord tumors are usually lethal with death resulting from diffuse metastases via the CSF pathways. Successful resection of nerve sheath tumors should be curative. In the context of neurofibromatosis, however, many more tumors can be found at other levels or can be expected to develop later in life. Surgical intervention in the context of neurofibromatoses should be performed only on clearly symptomatic lesions.
The outcome of treatment of extramedullary tumors depends on the cell type and, in most cases, on the efficacy of nonsurgical, adjunctive therapies. For aneurysmal bone cysts and giant cell tumors, resection of the tumor and fusion of the spine are the treatments of choice.
McGirt MJ, Chaichana KL, Atiba A, et al. Resection of intramedullary spinal cord tumors in children: assessment of long-term motor and sensory deficits. J Neurosurg Pediatr. 2008;1:63-67.
Wilne S, Walker D. Spine and spinal cord tumours in children: a diagnostic and therapeutic challenge to healthcare. Arch Dis Child Educ Pract Ed. 2010;95:47-54.
598.5 Spinal Cord Injuries in Children
Spine and spinal cord injuries are very rare in children, particularly in young children. The spine of a small child is very mobile, and fractures of the spine are exceedingly rare. This increased mobility is not always a positive feature. Transfer of energy leading to spinal distortion can maintain the structural integrity of the spine but lead to significant injuries of the spinal cord. Spinal cord injury without radiographic bone (vertebral) abnormalities, called SCIWORA, is more common in children than adults. There seem to be 2 distinct forms. The infantile form involves severe injury of the cervical or thoracic spine. These patients have a poor likelihood of complete recovery. In older children and adolescents, SCIWORA is more likely to cause a less severe injury and the likelihood of complete recovery over time is high. The adolescent form is assumed to be a spinal cord concussion or mild contusion as opposed to the severe spinal cord injury related to the mobility of the spine in small children.
Although the mechanisms of spinal cord injury in children include birth trauma, falls, and child abuse, the major cause of morbidity and mortality remains motor vehicle injuries. While the mechanisms of injury and diagnosis are distinct in very small children, adolescents incur spinal cord injuries with epidemiology similar to that of adults, including significant male predominance and a high likelihood of fracture dislocations of the lower cervical spine or thoracolumbar region. In infants and children under the age of 5 yr, fractures and mechanical disruption of spinal elements are limited to the upper cervical spine between the occiput and C3.
Clinical Manifestations
One in three patients with significant trauma to the spine and spinal cord will have a concomitant severe head injury, which makes early diagnosis challenging. For these patients clinical evaluation may be difficult. They need to be maintained in a protective environment such as a collar until the appropriate radiographs can be obtained. A careful neurologic examination is necessary for infants with suspected spinal cord injuries. Complete spinal cord injury will lead to spinal shock with early areflexia. Severe cervical spinal cord injuries will usually lead to paradoxical respiration in patients who are breathing spontaneously. Paradoxical respiration occurs when the diaphragm functions because the phrenic nerves from C3, C4, and C5 are functioning normally but the intercostal musculature innervated by the thoracic spinal cord is paralyzed. In this situation, inspiration fails to expand of the chest wall but distends the abdomen.
The mildest injury to the spinal cord is transient quadriparesis evident for seconds or minutes with complete recovery in 24 hr. This injury follows a concussion of the cord.
A transverse injury in the high cervical cord level (C1-C2) causes respiratory arrest and death in the absence of ventilatory support. Fracture dislocations at the C5-C6 level resulting in spinal cord injuries are characterized by flaccid quadriparesis, loss of sphincter function, and a sensory level corresponding to the upper sternum. Fractures or dislocations in the low thoracic (T12-L1) region may produce the conus medullaris syndrome, which includes a loss of urinary and rectal sphincter control, flaccid weakness, and sensory disturbances of the legs. A central cord lesion may result from contusion and hemorrhage and typically involves the upper extremities to a greater degree than the legs. There are lower motor neuron signs in the upper extremities and upper motor neuron signs in the legs, bladder dysfunction, and loss of sensation caudal to the lesion. There may be considerable recovery, particularly in the lower extremities.
Thoracolumbar injuries are usually fracture-dislocations such as occur in severe motor vehicle accidents when children are wearing lap belts but not shoulder harnesses. These injuries lead to a conus medullaris syndrome. These patients exhibit a loss of bowel and bladder function and lower motor neuron injuries involving the innervation of the lower extremities.
Treatment
The initial management of spine and spinal cord injuries in children is similar to that in adults. The cervical spine should be immobilized in the field by the emergency medical technicians. In cases of acute spinal cord injury, some data support the acute infusion of a bolus of high-dose (30 mg/kg) methylprednisolone followed by a 23-hr infusion (5.4 mg/kg/hr). The data for this treatment in children are controversial. Imaging studies, including lateral cervical spine x-rays, should be performed in the emergency room. If instability is recognized, precautions such as fixation with a collar or halo device should be instituted. In patients with documented neurologic dysfunction, MRI should be performed, including imaging of soft tissue to look for ligamentous instability.
Surgical management of unstable spinal injuries must be tailored to the patient’s age. For occipitocervical dislocations, early surgery with fusion from the occiput to C2 or C3 should be performed, even in babies older than 6 mo. Fixation of the subaxial spine must be tailored to the size of the pedicles and other osseus structures of the developing axial skeleton.
Prevention
The most important aspect of the care of spinal cord injuries in children relates to injury prevention. In this regard, the use of appropriate child restraints in automobiles is the most important precaution. In older children and adolescents, rules against spear tackling in football and the “Feet First, First Time Program” from the Think First Foundation aimed at adolescents diving into swimming pools and natural water areas are important ways to help prevent severe cervical spinal cord injuries.
Garton HJ, Hammer MR. Detection of pediatric cervical spine injury. Neurosurgery. 2008;62:700-708.
Lavy C, James A, Wilson-MacDonald J, et al. Cauda equina syndrome. BMJ. 2009;338:881-884.
Polk-Williams A, Carr BG, Blinman TA, et al. Cervical spine injury in young children: a National Trauma Data Bank review. J Pediatr Surg. 2008;43:1718-1721.
598.6 Transverse Myelitis
Transverse myelitis is a condition characterized by rapid development of both motor and sensory deficits (Table 598-1). It has multiple causes and tends to occur in 2 distinct contexts. Small children, 3 yr of age and younger, develop spinal cord dysfunction over hours to a few days. They have a history of an infectious disease, usually of viral origin, or of an immunization within the few weeks preceding the 1st development of their neurologic difficulties. The clinical loss of function is often severe and may seem complete. Although a slow recovery is common in these cases, it is likely to be incomplete. The likelihood of independent ambulation in these small children is about 40%. The pathologic findings of perivascular infiltration with mononuclear cells imply an infectious or inflammatory basis. Overt necrosis of spinal cord can be seen.
Table 598-1 DIAGNOSTIC CRITERIA FOR TRANSVERSE MYELITIS*
* Clinical events that are consistent with transverse myelitis but that are not associated with cerebrospinal fluid abnormalities or abnormalities detected on MRI and that have no identifiable underlying cause are categorized as possible idiopathic transverse myelitis.
† The IgG index is a measure of intrathecal synthesis of immunoglobulin and is calculated with the use of the following formula: (CSF IgG ÷ serum IgG) ÷ (CSF albumin ÷ serum albumin), where CSF denotes cerebrospinal fluid.
From Frohman EM, Wingerchuk DM: Transverse myelitis, N Engl J Med 363:564–572, 2010, Table 1, p 565.
In older children, the syndrome is somewhat different. Although the onset is also rapid with a nadir in neurologic function occurring between 2 days and 2 wk, recovery is more rapid and more likely to be complete. Pathologic or imaging examination shows acute demyelination.
Clinical Manifestations
In both forms the patient shows or complains of discomfort or overt pain in the neck or back, depending on the level of the lesion. Depending on its severity, the condition progresses to numbness, anesthesia, and weakness in the truncal and appendicular musculature. Paralysis begins as flaccidity, but over a few weeks spasticity develops. Urinary retention is an early finding; incontinence occurs later in the course.
The differential diagnosis includes demyelination disorders, overt meningitis, spinal cord infarction, or mass lesions such as bony distortion, abscess, and spine and spinal cord tumors.
Diagnostic Evaluation
MRI with and without contrast enhancement is essential to rule out a mass lesion requiring neurosurgical intervention. In both conditions, T1 weighted images of the spine at the anatomic level of involvement may be normal or may show distension of the spinal cord. In the infantile form, T2 weighted images show high signal intensity that extends over multiple segments. In the adolescent form, the high signal intensity will likely be limited to 1 or 2 segments. A limited degree of contrast enhancement after the administration of gadolinium is expected, especially in the infantile form, and denotes an inflammatory condition. MRI of the brain is also indicated and shows evidence of other foci of demyelination in at least 30% of patients similar to the adult population.
After a mass lesion associated with spinal cord compression or complete subarachnoid column block from spinal cord swelling have been ruled out, a lumbar puncture is indicated. In both forms of disease, the number of mononuclear cells is usually elevated minimally. The level of CSF protein is elevated mildly. CSF should be analyzed for myelin basic protein and immunoglobulin levels, which are usually elevated in transverse myelitis. The presence of inflammatory cells is essential for the diagnosis of transverse myelitis.
Because one of the most important possibilities for this condition is neuromyelitis optica (NMO; Devic syndrome) the serum of all patients should be analyzed for the NMO antigen. This test is positive in 60% of patients with Devic syndrome which, as opposed to a presenting syndrome for multiple sclerosis, is likely to be a monophasic condition. As in adults with transverse myelitis, older children with the condition should have serum studies sent for autoimmune disorders, especially systemic lupus erythematosus.
Treatment
There are no standards for the treatment of transverse myelitis. Available evidence suggests that modulation of the immune response may be effective in decreasing the severity and length of the condition. In the absence of open clinical trials, the use of high-dose steroids, particularly methylprednisolone, is a reasonable approach to treatment of both the early and late childhood forms of this condition.
Brinar VV, Habek M, Brinar M, et al. The differential diagnosis of acute transverse myelitis. Clin Neurol Neurosurg. 2006;108:278-282.
Frohman EM, Wingerchuk DM. Transverse myelitis. N Engl J Med. 2010;363:564-572.
Krishnan C, Kaplin AI, Deshpande DM, et al. Transverse myelitis: pathogenesis, diagnosis and treatment. Front Biosci. 2004;9:1483-1499.
Pawate S, Sriram S. Isolated longitudinal myelitis: a report of six cases. Spinal Cord. 2009;47:257-261.
Pidcock FS, Krishnan C, Crawford TO, et al. Acute transverse myelitis in childhood. Neurology. 2007;68:1474-1480.
598.7 Spinal Arteriovenous Malformations
Arteriovenous malformations of the spinal cord are rare lesions in children. Only about 60 patients under the age of 18 yr are treated in the USA each year. These lesions are complex. Despite their rarity there are multiple subtypes, which require different treatment strategies. Patients commonly present with back or neck pain, depending on the segments of the spinal cord involved, and they may experience the insidious onset of motor and sensory disturbances. Sudden onset of paraplegia secondary to hemorrhage has been reported. Occasionally, patients present with subarachnoid hemorrhage without overt neurologic deficits, similar to the presentation associated with cerebral aneurysms. In some cases, bruits are audible upon auscultation over the bony spine.
Diagnostic Evaluation
When a spinal arteriovenous malformation is suspected, MRI of the spinal cord is 1st needed to make the diagnosis and to obtain a general idea of the location of the lesion. MR angiography or CT angiography may provide further information, but formal catheter angiography of the spinal cord is needed to obtain an adequate understanding of the complex anatomy of the lesion and to plan the intervention.
Treatment
Open microsurgery had been the mainstay of treatment for spinal cord arteriovenous fistulae and arteriovenous malformations. With the rapid development of interventional techniques, the percentage of patients undergoing microsurgery has decreased from 70% to about 30%. Stereotactic radiosurgery may be used adjunctively. Treatment of these complex lesions requires the commitment of an organized neurovascular treatment program.