Chapter 599 Evaluation and Investigation
The term neuromuscular disease defines disorders of the motor unit and excludes influences on muscular function from the brain, such as spasticity. The motor unit has 4 components: a motor neuron in the brainstem or ventral horn of the spinal cord; its axon, which, together with other axons, forms the peripheral nerve; the neuromuscular junction; and all muscle fibers innervated by a single motor neuron. The size of the motor unit varies among different muscles and with the precision of muscular function required. In large muscles, such as the glutei and quadriceps femoris, hundreds of muscle fibers are innervated by a single motor neuron; in small, finely tuned muscles, such as the stapedius or the extraocular muscles, a 1 : 1 ratio can prevail. The motor unit is influenced by suprasegmental or upper motor neuron control that alters properties of muscle tone, precision of movement, reciprocal inhibition of antagonistic muscles during movement, and sequencing of muscle contractions to achieve smooth, coordinated movements. Suprasegmental impulses also augment or inhibit the monosynaptic stretch reflex; the corticospinal tract is inhibitory upon this reflex.
Diseases of the motor unit are common in children. These neuromuscular diseases may be genetically determined, congenital or acquired, acute or chronic, and progressive or static. Because specific therapy is available for many diseases and because of genetic and prognostic implications, precise diagnosis is important; laboratory confirmation is required for most diseases because of overlapping clinical manifestations.
Many chromosomal loci have been identified with specific neuromuscular diseases as a result of genetic linkage studies and the isolation and cloning of a few specific genes. In some cases, such as Duchenne muscular dystrophy, the genetic defect has been shown to be a deletion of nucleotide sequences and is associated with a defective protein product, dystrophin; in other cases, such as myotonic muscular dystrophy, the genetic defect is an expansion or repetition, rather than a deletion, in a codon (a set of three consecutive nucleotide repeats that encodes for a single amino acid), with many copies of a particular codon, in this example also associated with abnormal mRNA. Some diseases manifest as autosomal dominant and autosomal recessive traits in different pedigrees; these distinct mendelian genotypes can result from different genetic mutations on different chromosomes (nemaline rod myopathy) or may be small differences in the same gene at the same chromosomal locus (myotonia congenita), despite many common phenotypic features and shared histopathologic findings in a muscle biopsy specimen. Among the several clinically defined mitochondrial myopathies, specific mtDNA deletions and tRNA point mutations are recognized. The inheritance patterns and chromosomal and mitochondrial loci of common neuromuscular diseases affecting infants and children are summarized in Table 600-1.
Clinical Manifestations
Examination of the neuromuscular system includes an assessment of muscle bulk, tone, and strength. Tone and strength should not be confused: Passive tone is range of motion around a joint; active tone is physiologic resistance to movement. Head lag when an infant is pulled to a sitting position from supine is a sign of weakness, not of low tone. Hypotonia may be associated with normal strength or with weakness; enlarged muscles may be weak or strong; thin, wasted muscles may be weak or have unexpectedly normal strength. The distribution of these components is of diagnostic importance. In general, myopathies follow a proximal distribution of weakness and muscle wasting (with the notable exception of myotonic muscular dystrophy); neuropathies are generally distal in distribution (with the notable exception of juvenile spinal muscular atrophy; Table 599-1). Involvement of the face, tongue, palate, and extraocular muscles provides an important distinction in the differential diagnosis. Tendon stretch reflexes are generally lost in neuropathies and in motor neuron diseases and are diminished but preserved in myopathies (see Table 599-1). A few specific clinical features are important in the diagnosis of some neuromuscular diseases. Fasciculations of muscle, which are often best seen in the tongue, are a sign of denervation. Sensory abnormalities indicate neuropathy. Fatigable weakness is characteristic of neuromuscular junctional disorders. Myotonia is specific for a few myopathies.
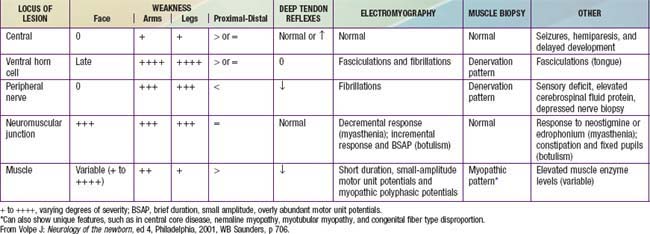
Some features do not distinguish myopathy from neuropathy. Muscle pain or myalgias are associated with acute disease of either myopathic or neurogenic origin. Acute dermatomyositis and acute polyneuropathy (Guillain-Barré syndrome) are characterized by myalgias. Muscular dystrophies and spinal muscular atrophies are not associated with muscle pain. Myalgias also occur in several metabolic diseases of muscle and in ischemic myopathy, including vascular diseases such as dermatomyositis. Myalgias denote the acuity, rather than the nature, of the process, so that progressive but chronic diseases, such as muscular dystrophy and spinal muscular atrophy, are not painful, but acute stages of inflammatory myopathies and acute denervation of muscle often do present muscular pain and tenderness to palpation. Contractures of muscles, whether present at birth or developing later in the course of an illness, occur in both myopathic and neurogenic diseases.
Infant boys who are weak in late fetal life and in the neonatal period often have undescended testes. The testes are actively pulled into the scrotum from the anterior abdominal wall by a pair of cords that consist of smooth and striated muscle called the gubernaculum. The gubernaculum is weakened in many congenital neuromuscular diseases, including spinal muscular atrophy, myotonic muscular dystrophy, and many congenital myopathies.
The thorax of infants with congenital neuromuscular disease often has a funnel shape, and the ribs are thin and radiolucent as a result of intercostal muscle weakness during intrauterine growth. This phenomenon is characteristically found in infantile spinal muscular atrophy but also occurs in myotubular myopathy, neonatal myotonic dystrophy, and other disorders (Fig. 599-1). Because of the small muscle mass, birth weight may be low for gestational age.
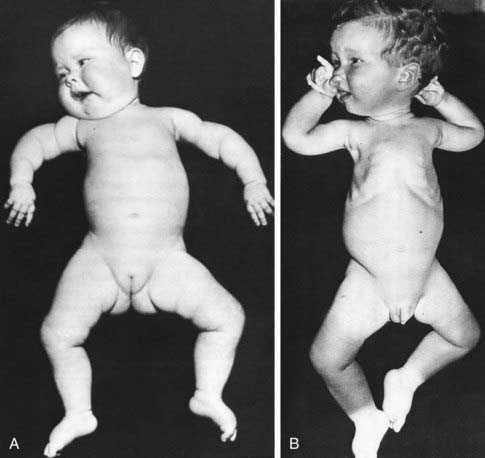
Figure 599-1 Type 1 spinal muscular atrophy (Werdnig-Hoffmann disease). Characteristic postures in 6 wk old (A) and 1 yr old (B) infants with severe weakness and hypotonia from birth. Note the frog-leg posture of the lower limbs and internal rotation (“jug handle”) (A) or external rotation (B) at the shoulders. Note also intercostal recession, especially evident in B, and normal facial expressions.
(From Volpe J: Neurology of the newborn, ed 4, Philadelphia, 2001, WB Saunders, p 645.)
Generalized hypotonia and motor developmental delay are the most common presenting manifestations of neuromuscular disease in infants and young children (Table 599-2). These features can also be expressions of neurologic disease, endocrine and systemic metabolic diseases, and Down syndrome, or they may be nonspecific neuromuscular expressions of malnutrition or chronic systemic illness (Table 599-3). A prenatal history of decreased fetal movements and intrauterine growth retardation is often found in patients who are symptomatic at birth. Developmental disorders tend to be of slow onset and are progressive. Acute flaccid paralysis in older infants and children has a different differential diagnosis (Table 599-4).
Table 599-2 PATTERN OF WEAKNESS AND LOCALIZATION IN THE FLOPPY INFANT
| ANATOMIC REGION OF HYPOTONIA | CORRESPONDING DISORDERS | PATTERN OF WEAKNESS AND INVOLVEMENT |
|---|---|---|
| Central nervous system | ||
| Motor neuron | Spinal muscular atrophy | Generalized weakness; often spares the diaphragm, facial muscles, pelvis, and sphincters |
| Nerve | Peripheral neuropathies | |
| Neuromuscular junction | Bulbar, oculomotor muscles exhibit greater degree of involvement | |
| Muscle |
CMD, congenital muscular dystrophy.
From Prasad AN, Prasad C: The floppy infant: contribution of genetic and metabolic disorders, Brain Dev 27:457–476, 2003.
Table 599-3 DIFFERENTIAL DIAGNOSIS OF INFANTILE HYPOTONIA
From Fenichel GM: The hypotonic infant. In Clinical pediatric neurology: a signs and symptoms approach, ed 5, Philadelphia, 2005, Saunders, p. 150.
Laboratory Findings
Serum Enzymes
Several lysosomal enzymes are released by damaged or degenerating muscle fibers and may be measured in serum. The most useful of these enzymes is creatine kinase (CK), which is found in only 3 organs and may be separated into corresponding isozymes: MM for skeletal muscle, MB for cardiac muscle, and BB for brain. Serum CK determination is not a universal screening test for neuromuscular disease because many diseases of the motor unit are not associated with elevated enzymes. The CK level is characteristically elevated in certain diseases, such as Duchenne muscular dystrophy, and the magnitude of increase is characteristic for particular diseases.
Molecular Genetic Markers
Many DNA markers of hereditary myopathies and neuropathies are available from blood samples. If the clinical manifestations suggest a particular disease, these tests can provide a definitive diagnosis and not subject the child to more-invasive procedures, such as muscle biopsy. Other molecular markers are available only in muscle biopsy tissue.
Nerve Conduction Velocity
Motor and sensory nerve conduction velocity (NCV) may be measured electrophysiologically by using surface electrodes. Neuropathies of various types are detected by decreased conduction. The site of a traumatic nerve injury may also be localized. The nerve conduction at birth is about half of the mature value achieved by age 2 yr. Tables are available for normal values at various ages in infancy, including for preterm infants. Because the NCV study measures only the fastest conducting fibers in a nerve, 80% of the total nerve fibers must be involved before slowing in conduction is detected.
Electromyography
Electromyography (EMG) requires insertion of a needle into the belly of a muscle and recording the electric potentials in various states of contraction. It is less useful in pediatrics than in adult medicine, in part because of technical difficulties in recording these potentials in young children and in part because the best results require the patient’s cooperation for full relaxation and maximal voluntary contraction of a muscle. Many children are too frightened to provide such cooperation. Characteristic EMG patterns distinguish denervation from myopathic involvement. The specific type of myopathy is not usually definitively diagnosed, but certain specialized myopathic conditions, such as myotonia, may be demonstrated. An EMG can transiently raise the serum CK level.
EMG combined with repetitive electrical stimulation of a motor nerve supplying a muscle to produce tetany is useful in demonstrating myasthenic decremental responses. Small muscles, such as the abductor digiti quinti of the hypothenar eminence, are used for such studies.
Imaging of Muscle
Ultrasonography, CT scans, and, more often, MRI are used to image muscle in many neuromuscular diseases. Although these methods are not always definitively diagnostic, in experienced hands, they provide a supplementary means of following the progression of disease over time. MRI is quite useful in identifying inflammatory myopathies of immune (dermatomyositis) or infectious (viral, bacterial, parasitic) origin. MRI is the study of choice to image the spinal cord or nerve roots and plexus (e.g., brachial plexus).
Muscle Biopsy
The muscle biopsy is the most important and specific diagnostic study of most neuromuscular disorders, if the definitive diagnosis of a hereditary disease is not provided by molecular genetic testing in blood. Not only are neurogenic and myopathic processes distinguished, but also the type of myopathy and specific enzymatic deficiencies may be determined. The vastus lateralis (quadriceps femoris) is the muscle that is most commonly sampled. The deltoid muscle should be avoided in most cases because it normally has a 60-80% predominance of type I fibers so that the distribution patterns of fiber types are difficult to recognize. Muscle biopsy is a simple outpatient procedure that may be performed under local anesthesia with or without femoral nerve block. Needle biopsies are preferred in some centers, but are not percutaneous and require an incision in the skin similar to open biopsy; numerous samples must be taken to conduct an adequate examination of the tissue, and they provide inferior specimens. The volume of tissue from a needle biopsy is usually not adequate for all required studies, including supplementary biochemical studies, such as mitochondrial respiratory chain enzymes; a small, clean, open biopsy is therefore advantageous.
Histochemical studies of frozen sections of the muscle are obligatory in all pediatric muscle biopsies because many congenital and metabolic myopathies cannot be diagnosed from paraffin sections using conventional histologic stains. Immunohistochemistry is a useful supplement in some cases, such as for demonstrating dystrophin in suspected Duchenne muscular dystrophy or merosin in congenital muscular dystrophy. A portion of the biopsy specimen should be fixed for potential electron microscopy, but ultrastructure has additional diagnostic value only in selected cases. Interpretation of muscle biopsy samples is complex and should be performed by an experienced pathologist. A portion of frozen muscle tissue should also be routinely saved for possible biochemical analysis (mitochondrial cytopathies, carnitine palmityltransferase, acid maltase).
Nerve Biopsy
The most commonly sampled nerve is the sural nerve, a pure sensory nerve that supplies a small area of skin on the lateral surface of the foot. Whole or fascicular biopsy specimens of this nerve may be taken. When the sural nerve is severed behind the lateral malleolus of the ankle, regeneration of the nerve occurs in >90% of cases, so that permanent sensory loss is not experienced. The sural nerve is often involved in many neuropathies whose clinical manifestations are predominantly motor.
Electron microscopy is performed on most nerve biopsy specimens because many morphologic alterations cannot be appreciated at the resolution of a light microscope. Teased fiber preparations are sometimes useful in demonstrating segmental demyelination, axonal swellings, and other specific abnormalities, but this time-consuming procedure is not done routinely. Special stains may be applied to ordinary frozen or paraffin sections of nerve biopsy material to demonstrate myelin, axoplasm, and metabolic products.
Electrocardiography
Cardiac evaluation is important if myopathy is suspected because of involvement of the heart in muscular dystrophies and in inflammatory and metabolic myopathies. Electrocardiography (ECG) often detects early cardiomyopathy or conduction defects that are clinically asymptomatic. At times, a more complete cardiac work-up, including echocardiography and consultation with a pediatric cardiologist, is indicated. Serial pulmonary function tests also should be performed in muscular dystrophies and in other chronic or progressive diseases of the motor unit.