Chapter 605 Hereditary Motor-Sensory Neuropathies
The hereditary motor-sensory neuropathies (HMSNs) are a group of progressive diseases of peripheral nerves. Motor components generally dominate the clinical picture, but sensory and autonomic involvement is expressed later.
605.1 Peroneal Muscular Atrophy (Charcot-Marie-Tooth Disease; HMSN Type I)
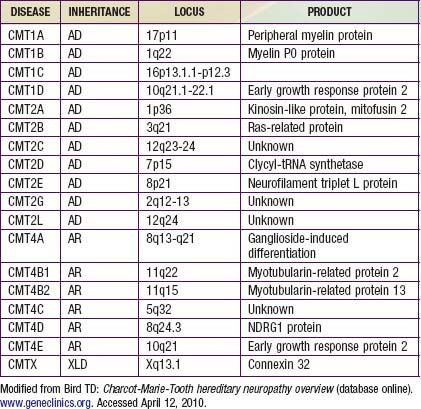
Charcot-Marie-Tooth disease is the most common genetically determined neuropathy and has an overall prevalence of 3.8/100,000. It is transmitted as an autosomal dominant trait with 83% expressivity; the 17p11.2 locus is the site of the abnormal gene. Autosomal recessive transmission also is described but is rarer. The gene product is peripheral myelin protein 22 (PMP22). A much rarer X-linked HMSN type I results from a defect at the Xq13.l locus, causing mutations in the gap junction protein connexin-32. Other forms have been reported (Table 605-1).
Clinical Manifestations
Most patients are asymptomatic until late childhood or early adolescence, but young children sometimes manifest gait disturbance as early as the 2nd year of life. The peroneal and tibial nerves are the earliest and most severely affected. Children with the disorder are often described as being clumsy, falling easily, or tripping over their own feet. The onset of symptoms may be delayed until after the 5th decade.
Muscles of the anterior compartment of the lower legs become wasted, and the legs have a characteristic stork-like contour. The muscular atrophy is accompanied by progressive weakness of dorsiflexion of the ankle and eventual footdrop. The process is bilateral but may be slightly asymmetric. Pes cavus deformities invariably develop due to denervation of intrinsic foot muscles, further destabilizing the gait. Atrophy of muscles of the forearms and hands is usually not as severe as that of the lower extremities, but in advanced cases contractures of the wrists and fingers produce a claw hand. Proximal muscle weakness is a late manifestation and is usually mild. Axial muscles are not involved.
The disease is slowly progressive throughout life, but patients occasionally show accelerated deterioration of function over a few years. Most patients remain ambulatory and have normal longevity, although orthotic appliances are required to stabilize the ankles.
Sensory involvement mainly affects large myelinated nerve fibers that convey proprioceptive information and vibratory sense, but the threshold for pain and temperature can also increase. Some children complain of tingling or burning sensations of the feet, but pain is rare. Because the muscle mass is reduced, the nerves are more vulnerable to trauma or compression. Autonomic manifestations may be expressed as poor vasomotor control with blotching or pallor of the skin of the feet and inappropriately cold feet.
Nerves often become palpably enlarged. Tendon stretch reflexes are lost distally. Cranial nerves are not affected. Sphincter control remains well preserved. Autonomic neuropathy does not affect the heart, gastrointestinal tract, or bladder. Intelligence is normal. A unique point mutation in PMP22 causes progressive auditory nerve deafness in addition, but this is usually later in onset than the peripheral neuropathy.
Davidenkow syndrome is a variant of HMSN type I with a scapuloperoneal distribution.
Laboratory Findings and Diagnosis
Motor and sensory nerve conduction velocities are greatly reduced, sometimes as slow as 20% of normal conduction time. In new cases without a family history, both parents should be examined, and nerve conduction studies should be performed.
Electromyography (EMG) and muscle biopsy are not usually required for diagnosis, but they show evidence of many cycles of denervation and reinnervation. Serum creatine kinase level is normal. Cerebrospinal fluid (CSF) protein may be elevated, but no cells appear in the CSF.
Sural nerve biopsy is diagnostic. Large- and medium-sized myelinated fibers are reduced in number, collagen is increased, and characteristic onion bulb formations of proliferated Schwann cell cytoplasm surround axons. This pathologic finding is called interstitial hypertrophic neuropathy. Extensive segmental demyelination and remyelination also occur.
The definitive molecular genetic diagnosis may be made in blood.
Treatment
Stabilization of the ankles is a primary concern. In early stages, stiff boots that extend to the mid-calf often suffice, particularly when patients walk on uneven surfaces such as ice and snow or stones. As the dorsiflexors of the ankles weaken further, lightweight plastic splints may be custom made to extend beneath the foot and around the back of the ankle. They are worn inside the socks and are not visible, reducing self-consciousness. External short-leg braces may be required when footdrop becomes complete. Surgical fusion of the ankle may be considered in some cases.
The leg should be protected from traumatic injury. In advanced cases, compression neuropathy during sleep may be prevented by placing soft pillows beneath or between the lower legs. Burning paresthesias of the feet are not common but are often abolished by phenytoin or carbamazepine. No medical treatment is available to arrest or slow the progression.
605.2 Peroneal Muscular Atrophy (Axonal Type)
Peroneal muscular atrophy is clinically similar to HMSN type I, but the rate of progression is slower and the disability is less. EMG shows denervation of muscle. Sural nerve biopsy reveals axonal degeneration rather than the demyelination and whorls of Schwann cell processes typical in type I. The locus is on chromosome 1 at 1p35-p36; this is a different disease than HMSN type I, although both are transmitted as autosomal dominant traits. An autosomal recessive infantile motor axonal neuropathy can closely mimic infantile spinal muscular atrophy.
605.3 Déjerine-Sottas Disease (HMSN Type III)
Déjerine-Sottas disease is an interstitial hypertrophic neuropathy of autosomal dominant transmission. It is similar to HMSN type I but is more severe. Symptoms develop in early infancy and are rapidly progressive. Pupillary abnormalities, such as lack of reaction to light and Argyll Robertson pupil, are common. Kyphoscoliosis and pes cavus deformities complicate about 35% of cases. Nerves become palpably enlarged at an early age.
The onion-bulb formations seen in the sural nerve biopsy specimen are more pronounced. Hypomyelination also occurs.
The genetic locus of 17p11.2 is identical to that of HMSN type I or Charcot-Marie-Tooth disease. The clinical and pathologic differences may be phenotypical variants of the same disease, analogous to the situation in Duchenne and Becker muscular dystrophies. An autosomal recessive form of Déjerine-Sottas disease is also described but is incompletely documented.
605.4 Roussy-Lévy Syndrome
Roussy-Lévy syndrome is defined as a combination of HMSN type I and cerebellar deficit resembling Friedreich ataxia, but it does not have cardiomyopathy.
605.5 Refsum Disease
(See Chapter 80.2.)
Refsum disease is a rare autosomal recessive disease caused by an enzymatic block in β-oxidation of phytanic acid to pristanic acid. Phytanic acid is a branched-chain fatty acid that is derived mainly from dietary sources: spinach, nuts, and coffee. Levels of phytanic acid are greatly elevated in plasma, CSF, and brain tissue. The CSF shows an albuminocytologic dissociation, with a protein concentration of 100-600 mg/dL. Genetic linkage studies identify 2 distinct loci at 10p13 and 6q22-q24 with PHYH and PEX7 genetic mutations, respectively.
Clinical onset is usually between 4 and 7 yr of age, with intermittent motor and sensory neuropathy. Ataxia, progressive neurosensory hearing loss, retinitis pigmentosa with loss of night vision, ichthyosis, and liver dysfunction also develop in various degrees. Skeletal malformations from birth and cardiac findings of conduction disturbances and cardiomyopathy appear in the majority. Motor and sensory nerve conduction velocities are delayed. Sural nerve biopsy shows loss of myelinated axons. Treatment is by dietary management and periodic plasma exchange. With careful management, life expectancy can be normal.
Refsum disease should not be confused with another autosomal recessive disease, infantile Refsum disease, in which phytanic acid accumulation is only secondary to a primary peroxisomal disorder.
605.6 Fabry Disease
(See Chapter 80.4.)
Fabry disease, a rare X-linked recessive trait, results in storage of ceramide trihexose due to deficiency of the enzyme ceramide trihexosidase, which cleaves the terminal galactose from ceramide trihexose (ceramide-glucose-galactose-galactose), resulting in tissue accumulation of this trihexose lipid in central nervous system (CNS) neurons, Schwann cells and perineurial cells, ganglion cells of the myenteric plexus, skin, kidneys, blood vessel endothelial and smooth muscle cells, heart, sweat glands, cornea, and bone marrow. It results from a missense mutation disrupting the crystallographic structure of α-galactosidase A.
Clinical Manifestations
The presentation is in late childhood or adolescence, with recurrent episodes of burning pain and paresthesias of the feet and lower legs so severe that patients are unable to walk. These episodes are often precipitated by fever or by physical activity. Objective sensory and motor deficits are not demonstrated on neurologic examination, and reflexes are preserved. Characteristic skin lesions are seen in the perineal region, scrotum, buttocks, and periumbilical zone as flat or raised red-black telangiectases known as angiokeratoma corporis diffusum. Hypohidrosis may be present. Corneal opacities, cataracts, and necrosis of the femoral heads are inconstant features. Tortuosity of retinal vessels and of the vertebral and basilar arteries can occur. The disease is progressive. Hypertension and renal failure are usually delayed until early adult life. Recurrent strokes result from vascular wall involvement. Death often occurs in the 5th decade owing to cerebral infarction or renal insufficiency, but a significant morbidity already occurs in childhood despite the absence of major organ failure. Heterozygous female carriers may be asymptomatic or, rarely, are as affected as males; corneal opacities involve 70-80%, though cataracts are rare.
Laboratory Findings
Motor and sensory nerve conduction velocities are normal to only mildly slow, showing preservation of large myelinated nerve fibers. CSF protein is normal. Proteinuria is present early in the course.
Pathologic features are usually first detected in skin or sural nerve biopsy specimens. Crystalline glycosphingolipids appear as zebra bodies in lysosomes of endothelial cells, in smooth myocytes of arterioles, and in Schwann cells, best demonstrated by electron microscopy. Nerves show a selective loss of small myelinated fibers and relative preservation of large and medium-sized axons, contrasting to most axonal neuropathies in which large myelinated fibers are most involved.
Assay for the deficient enzyme, α-galactosidase-A, may be performed from skin fibroblasts, leukocytes, and other tissues. This test permits detection of the female carrier state and provides a reliable means of prenatal diagnosis.
Treatment
(See Chapter 80.4 for specific therapy of Fabry disease, including enzyme replacement.)
Medical therapy of painful neuropathies includes management of the initiating disease and therapy directed to the neuropathic pain independent of etiology. Pain may be burning or associated with paresthesias, hyperalgesia (abnormal response to noxious stimuli), or allodynia (induced by non-noxious stimuli; Chapter 71). Neuropathic pain is often successfully managed by tricyclic antidepressants; selective serotonin reuptake inhibitors are less effective. Anticonvulsants (carbamazepine, phenytoin, gabapentin, lamotrigine) are effective, as are narcotic and non-narcotic analgesics. Enzyme replacement therapy has improved the short and long term prognosis.
605.7 Giant Axonal Neuropathy
Giant axonal neuropathy is a rare autosomal recessive disease with onset in early childhood. It is a progressive mixed peripheral neuropathy and degeneration of central white matter, similar to the leukodystrophies. Ataxia and nystagmus are accompanied by signs of progressive peripheral neuropathy. A large majority of affected children have frizzy hair, which microscopically shows variation in diameter of the shaft and twisting, similar to that in Menkes disease; hence, microscopic examination of a few scalp hairs provides a simple screening tool in suspected cases. Focal axonal enlargements are seen in both the peripheral nervous system and the CNS, but the myelin sheath is intact. The disease is a general proliferation of intermediate filaments, including neurofilaments in axons, glial filaments (i.e., Rosenthal fibers) in brain, cytokeratin in hair, and vimentin in Schwann cells and fibroblasts.
Nonsense and missense mutations or deletions occur in the GAN gene, with allelic heterogeneity, at 16q24. These mutations are responsible for defective synthesis of the protein gygaxonin, a member of the cytoskeletal BTB/kelch superfamily, crucial to linkage between intermediate proteins and the cell membrane. MRI shows white matter lesions of the brain similar to leukodystrophies, and magnetic resonance spectroscopy (MRS) demonstrates increased ratios of choline : creatine and myoinositol : creatine, with a normally preserved ratio of N-acetyl aspartate : creatine, indicating demyelination and glial proliferation without axonal loss. Gygaxonin is expressed in a wide variety of neuronal cell types and is localized to the Golgi apparatus and endoplasmic reticulum.
The diagnosis is established by microscopy of scalp hair and by MRI and MRS of the brain; it is confirmed by sural nerve biopsy and/or by genetic studies, if available, of the GAN gene.
605.8 Congenital Hypomyelinating Neuropathy
Congenital hypomyelinating neuropathy is a lack of normal myelination of motor and sensory peripheral nerves but not of CNS white matter. It is not a degeneration or loss of previously formed myelin, thus differentiating it from a leukodystrophy. Schwann cells are preserved, and axons are normal. Cases in siblings suggest autosomal recessive inheritance. Mutations in the MTMR2, PMP22, EGR2, and MPZ genes have been demonstrated in various children with this neuropathy; hence, it is a syndrome rather than a single disease.
The condition is present from birth; hypotonia and developmental delay are the hallmark clinical findings. Many patients present clinically as having congenital insensitivity to pain. Cranial nerves are inconsistently involved, and respiratory distress and dysphagia are rare complications. Tendon reflexes are absent. Arthrogryposis is present at birth in at least half the cases. It is uncertain whether the condition is progressive; myelination of nerves proceeds at a slow rate and remains incomplete. Motor and sensory nerve conduction velocities are slow. The diagnosis is confirmed by sural nerve biopsy, which shows lack of myelination of large and small fibers and sometimes interstitial hypertrophic reactive changes. It is useful in distinguishing this condition from several hereditary demyelinating neuropathies without resorting to batteries of expensive genetic markers. Muscle biopsy can show mild neurogenic atrophy but not the characteristic alterations of spinal muscular atrophy. No inflammation is demonstrated in muscle or nerve. Treatment is supportive.
605.9 Tomaculous (Hypermyelinating) Neuropathy; Hereditary Neuropathy with Liability to Pressure Palsies
This hereditary neuropathy is characterized by redundant overproduction of myelin around each axon in an irregular segmental fashion so that tomaculous (sausage-shaped) bulges occur in the individual myelinated nerve fibers. Other sections of the same nerve can show loss of myelin. Such nerves are particularly prone to pressure palsies, and patients, usually beginning in adolescence, present with recurrent or intermittent mononeuropathies secondary to minor trauma or entrapment neuropathies, such as carpal tunnel syndrome, peroneal palsies, and even “writer’s cramp.” It is transmitted as an autosomal dominant trait, with loci identified at 17p11.2 and 17p12, deletion of exons in the PMP22 gene. Duplication of the same 17p12 locus leads to Charcot-Marie-Tooth disease type 1A, myelin protein zero (MPZ) gene mutation. Sural nerve biopsy is diagnostic, but special teased fiber preparations should be made to demonstrate the myelin abnormalities most clearly. Skin or conjunctival biopsies also may be diagnostic. Electrophysiologic nerve conduction studies are abnormal but nonspecific. Genetic studies are definitive.
Treatment is supportive and includes avoiding trauma and prolonged nerve compression, including postures when sitting or lying. Surgical release of entrapped nerves is indicated at times.
605.10 Leukodystrophies
Several hereditary degenerative diseases of white matter of the CNS also cause peripheral neuropathy. The most important are Krabbe disease (globoid cell leukodystrophy), metachromatic leukodystrophy, and adrenoleukodystrophy (see Chapters 80 and 592).
Bruno C, Bertini E, Federico A, et al. Clinical and molecular findings in patients with giant axonal neuropathy (GAN). Neurology. 2004;62:13-16.
Chance PF. Inherited focal, episodic neuropathies: hereditary neuropathy with liability to pressure palsies and hereditary neuralgic amyotrophy. Neuromolec Med. 2006;8:159-174.
Chance PF, Alderson MK, Leppig KA, et al. DNA deletion associated with hereditary neuropathy with liability to pressure palsies. Cell. 1993;72:143-151.
Evgrafov OV, Mersiyanova I, Irobi J, et al. Mutant small heat-shock protein 27 causes axonal Charcot-Marie-Tooth disease and distal hereditary motor neuropathy. Nat Genet. 2004;36:602-606.
Gordon N. Giant axonal neuropathy. Dev Med Child Neurol. 2004;46:717-719.
Houlden H, Blake J, Reilly MM. Hereditary sensory neuropathies. Curr Opin Neurol. 2004;17:569-577.
Kochanski A, Drac H, Kabzinska D, et al. A novel MPZ gene mutation in congenital neuropathy with hypomyelination. Neurology. 2004;62:2122-2123.
Kovach MJ, Lin JP, Boyadjiev S, et al. A unique point mutation in the PMP22 gene is associated with Charcot-Marie-Tooth disease and deafness. Am J Hum Genet. 1999;64:1580-1593.
Landouré G, Zdebik AA, Martinez TL, et al. Mutations in TRPV4 cause Charcot-Marie-tooth disease type 2C. Nat Genet. 2010;42(2):170-174.
Lupski JR, Reid JG, Gonzaga-Jauregui C, et al. Whole-genome sequencing in a patient with Charcot-Marine-tooth neuropathy. N Engl J Med. 2010;362(13):1181-1190.
Mendell JR, Sahenk Z. Painful sensory neuropathy. N Engl J Med. 2003;348:1243-1255.
Pleasure D. New treatments for denervating diseases. J Child Neurol. 2005;20:258-262.
Ries M, Gupta S, Moore DF, et al. Pediatric Fabry disease. Pediatrics. 2005;115:344-355.
Seithi PK, Khandelwal D, Sethi NK, et al. Hereditary infantile motor axonal neuropathy mimicking SMA: a case report. J Pediatr Neurol. 2009;7:175-179.
Shy ME. Charcot-Marie-Tooth disease: An update. Curr Opin Neurol. 2004;17:579-585.