Chapter 614 Abnormalities of Pupil and Iris
Aniridia
The term aniridia is a misnomer because iris tissue is usually present, although it is hypoplastic (Fig. 614-1). Two thirds of the cases are dominantly transmitted with a high degree of penetrance. The other 30% of cases are sporadic and are considered to be new mutations. The condition is bilateral in 98% of all patients, regardless of the means of transmission, and is found in approximately 1/50,000 persons.
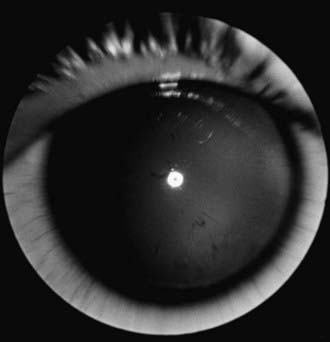
Figure 614-1 Aniridia with minimal iris tissue.
(From Nelson LB, Spaeth GL, Nowinski TS, et al: Aniridia: a review, Surv Ophthalmol 28:621–642, 1984.)
Aniridia is a panocular disorder and should not be thought of as an isolated iris defect. Macular and optic nerve hypoplasias are commonly present and lead to decreased vision and sensory nystagmus. The visual acuity is measured as 20/200 in most patients, although the vision occasionally is better. Other ocular deformities are common and can involve the lens and cornea. The cornea may be small, and a cellular infiltrate (pannus) occasionally develops in the superficial layers of the peripheral cornea. Clinically, this appears as a gray opacification. The pannus results from a stem cell deficiency and therefore must be treated with keratolimbal stem cell transplantation rather than cornea transplantation. Lens abnormalities include cataract formation and partial or total lens dislocation. Glaucoma develops in as many as 75% of patients with aniridia.
Aniridia is caused by a defect in the PAX6 gene on chromosome 11p13. The PAX6 gene is the master control gene for eye morphogenesis. Aniridia can be sporadic or familial. The familial form is autosomal dominant with complete penetrance but variable expressivity. Sporadic aniridia is associated with Wilms tumor in as many as 30% of cases (Chapter 493.1). The combination of aniridia and Wilms tumor represents a contiguous gene syndrome in which the adjacent PAX6 and Wilms tumor (WT1) genes are both deleted. Some deletions create the WAGR complex of Wilms tumor, aniridia, genitourinary malformations, and mental retardation. All children with sporadic aniridia should undergo chromosomal deletional analysis to exclude the possibility of Wilms tumor formation. Children who test positive for the deletion should undergo repeated abdominal ultrasonographic and clinical examinations. Wilms tumor has also been reported in patients with familial aniridia. Therefore, these patients should also undergo chromosomal analysis.
Coloboma of the Iris
Coloboma of the iris is a developmental defect that can occur as a defect in a sector of the iris, a hole in the substance of the iris, or a notch in the pupil’s margin. Simple colobomas are often transmitted as an autosomal dominant trait and can occur alone or in association with other anomalies. A coloboma is formed when the embryonic fissure fails to close completely. Because of the anatomic location of the embryonic fissure, an iris coloboma is always located inferiorly, giving the iris a keyhole appearance. An iris coloboma may be the only externally visible part of an extensive malclosure of the embryonic fissure that also involves the fundus and optic nerve. When this occurs, vision is likely to be severely affected. Therefore, all children with an iris coloboma should undergo a full ophthalmologic examination.
Microcoria
Microcoria (congenital miosis) appears as a small pupil that does not react to light or accommodation and that dilates poorly, if at all, with medication. The condition may be unilateral or bilateral. In bilateral cases, the degree of miosis may be different in each eye. The eye may be otherwise normal or may demonstrate other abnormalities of the anterior segment. Congenital microcoria is usually transmitted as an autosomal dominant trait, although it can occur sporadically.
Congenital Mydriasis
In congenital mydriasis, the pupils appear dilated, do not constrict significantly to light or near gaze, and respond minimally to miotic agents. The iris is otherwise normal, and affected children are usually healthy. Trauma, pharmacologic mydriasis, and neurologic disorders should be considered. Many apparent cases of congenital mydriasis show abnormalities of the central iris structures and may be considered a form of aniridia.
Dyscoria and Corectopia
Dyscoria is abnormal shape of the pupil, and corectopia is abnormal position of the pupil. They can occur together or independently as congenital or acquired anomalies.
Congenital corectopia is usually bilateral and symmetric and rarely occurs as an isolated anomaly; it is usually accompanied by dislocation of the lens (ectopia lentis et pupillae), and the lens and pupil are commonly dislocated in opposite directions. Ectopia lentis et pupillae is transmitted as an autosomal recessive disorder; consanguinity is common.
When these are acquired, distortion and displacement of the pupil are often a result of trauma or intraocular inflammation. Prolapse of the iris after perforating injuries of the eye leads to peaking of the pupil in the direction of the perforation. Posterior synechiae (adhesions of the iris to the lens) are commonly seen when inflammation due to any cause occurs in the anterior segment.
Anisocoria
This is inequality of the pupils. The difference in size may be due to local or neurologic disorders. As a rule, if the inequality is more pronounced in the presence of bright focal illumination or on near gaze, there is a defect in pupillary constriction and the larger pupil is abnormal. If the anisocoria is worse in reduced illumination, a defect in dilation exists and the smaller pupil is abnormal. Neurologic causes of anisocoria (parasympathetic or sympathetic lesions) must be differentiated from local causes such as synechiae (adhesions), congenital iris defects (colobomas, aniridia), and pharmacologic effects. Simple central anisocoria can occur in otherwise healthy children. The combination of anisocoria and ptosis may be seen in Horner syndrome.
Horner Syndrome
The principal signs of oculosympathetic paresis (Horner syndrome) are homolateral miosis, mild ptosis, and apparent enophthalmos with slight elevation of the lower lid. Patients may also have decreased facial sweating, increased amplitude of accommodation, and transient decrease in intraocular pressure. If paralysis of the ocular sympathetic fibers occurs before the age of 2 yr, iris heterochromia with hypopigmentation of the iris can occur on the affected side but can take time to develop.
Oculosympathetic paralysis may be caused by a lesion in the midbrain, brainstem, upper spinal cord, neck, middle fossa, or orbit. Congenital oculosympathetic paresis, often as part of Klumpke brachial palsy, is common, although the ocular signs, particularly the anisocoria, can pass undetected for years. Horner syndrome is also seen in some children after thoracic surgery, such as for congenital heart disease. Congenital Horner syndrome can occur in association with vertebral anomalies and with enterogenous cysts. In some infants and children, Horner syndrome is the presenting sign of tumor in the mediastinal or cervical region, particularly neuroblastoma.
The diagnosis of Horner syndrome can be confirmed with the use of topical cocaine or apraclonidine drops. A normal pupil dilates within 20-45 min after instillation of 1 or 2 drops of 4% cocaine, whereas the miotic pupil of an oculosympathetic paresis dilates poorly, if at all, with cocaine. Apraclonidine causes reversal of the anisocoria with dilation of the affected (smaller) pupil and no effect on the normal pupil. It should be used in caution in young children because it can cause excessive sedation owing to its CNS side effects. Pharmacologic testing might not be needed in the presence of typical clinical findings.
Horner’s syndrome in children can result from trauma, surgery, or the presence of neuroblastoma affecting the sympathetic chain in the chest. Evaluation of acquired Horner syndrome in a child without a history of trauma or surgery that could explain the anisocoria should include imaging studies of the brain, neck, and chest. Examining old photographs and old records is sometimes helpful in establishing the age at onset of Horner syndrome.
Dilated Fixed Pupil
Differential diagnosis of a dilated unreactive pupil includes internal ophthalmoplegia caused by a central or peripheral lesion, Hutchinson pupil of transtentorial herniation, tonic pupil, pharmacologic blockade, and iridoplegia secondary to ocular trauma.
The most common cause of a dilated unreactive pupil is purposeful or accidental instillation of a cycloplegic agent, particularly atropine and related substances. Central nervous system lesions, such as a pinealoma, can cause internal ophthalmoplegia in children. Because the external surface of the oculomotor nerve carries the fibers responsible for pupillary constriction, compression of the nerve along its intracranial course may be associated with internal ophthalmoplegia, even before the development of ptosis or an ocular motility deficit. Although ophthalmoplegic migraine is a common cause of a 3rd nerve palsy with pupillary involvement in children, an intracranial aneurysm must also be considered in the differential diagnosis. The blown pupil of transtentorial herniation, occurring with increasing intracranial pressure, is generally unilateral, and patients usually are obviously ill. The pilocarpine test can help differentiate neurologic iridoplegia from pharmacologic blockade. In the case of neurologic iridoplegia, the dilated pupil constricts within minutes after instillation of 1 or 2 drops of 0.5-1% pilocarpine; if the pupil has been dilated with atropine, pilocarpine has no effect. Because pilocarpine is a long-acting drug, this test is not to be used in acute situations in which pupillary signs must be carefully monitored. Because of the consensual pupil response to light, even complete uniocular blindness does not cause a unilaterally dilated pupil.
Tonic Pupil
Tonic pupil is typically a large pupil that reacts poorly to light (the reaction may be very slow or essentially nil), reacts poorly and slowly to accommodation, and redilates in a slow, tonic manner. The features of tonic pupil are explained by cholinergic supersensitivity of the sphincter after peripheral (postganglionic) denervation and imperfect reinnervation. A distinctive feature of a tonic pupil is its sensitivity to dilute cholinergic agents. Instillation of 0.125% pilocarpine causes significant constriction of the involved pupil and has little or no effect on the unaffected side. The condition is usually unilateral.
Tonic pupil can develop after the acute stage of a partial or complete iridoplegia. It can be seen after trauma to the eye or orbit and can occur in association with toxic or infectious conditions. For those in the pediatric age group, tonic pupil is uncommon. Infectious processes (primarily viral syndromes) and trauma are the primary causes. Features of tonic pupil may also be seen in infants and children with familial dysautonomia (Riley-Day syndrome), although the significance of these findings has been questioned. Tonic pupil has also been reported in young children with Charcot-Marie-Tooth disease. The occurrence of tonic pupil in association with decreased deep tendon reflexes in young women is referred to as Adie syndrome.
Marcus Gunn Pupil
This relative afferent pupillary defect indicates an asymmetric, prechiasmatic, afferent conduction defect. It is best demonstrated by the swinging flashlight test; this allows comparison of the direct and consensual pupillary responses in both eyes. With patients fixing on a distant target (to control accommodation), a bright focal light is directed alternately into each eye in turn. In the presence of an afferent lesion, both the direct response to light in the affected eye and the consensual response in the other eye are subnormal. Swinging the light to the better or normal eye causes both pupils to react (constrict) normally. Swinging the light back to the affected eye causes both pupils to redilate to some degree, reflecting the defective conduction. This is a very sensitive and useful test for detecting and confirming optic nerve and retinal disease. This test is only abnormal if there is a “relative” difference in the conduction properties of the optic nerves. Therefore, patients with bilateral and symmetric optic nerve disease do not demonstrate an afferent pupillary defect. A subtle relative afferent defect is found in some children with amblyopia.
Paradoxical Pupil Reaction
Some children exhibit paradoxical constriction of the pupils to darkness. An initial brisk constriction of the pupils occurs when the light is turned off, followed by slow redilatation of the pupils. The response to direct light stimulation and the near response are normal. The mechanism is not clear, but paradoxical constriction of the pupils in reduced light can be a sign of retinal or optic nerve abnormalities. The phenomenon has been observed in children with congenital stationary night blindness, albinism, retinitis pigmentosa, Leber congenital retinal amaurosis, and Best disease. It has also been observed in those with optic nerve anomalies, optic neuritis, optic atrophy, and possibly amblyopia.
Persistent Pupillary Membrane
Involution of the pupillary membrane and anterior vascular capsule of the lens is usually completed during the 5th-6th mo of fetal development. It is common to see some remnants of the pupillary membrane in newborns, particularly in premature infants. These membranes are nonpigmented strands of obliterated vessels that cross the pupil and can secondarily attach to the lens or cornea. The remnants tend to atrophy in time and usually present no problem. In some children, however, significant remnants that remain obscure the pupil and interfere with vision. Rarely, there is patency of the vascular elements; hyphema can result from rupture of persistent vessels.
Intervention must be considered to minimize amblyopia in infants with extensive persistent pupillary membrane of sufficient degree to interfere with vision in the early months of life. In some cases, mydriatics and occlusion therapy are effective, but in others, surgery is needed to provide an adequate pupillary aperture.
Heterochromia
In heterochromia, the two irides are of different color (heterochromia iridium) or a portion of an iris differs in color from the remainder (heterochromia iridis). Simple heterochromia can occur as an autosomal dominant characteristic. Congenital heterochromia is also a feature of Waardenburg syndrome, an autosomal dominant condition characterized principally by lateral displacement of the inner canthi and puncta, pigmentary disturbances (usually a median white forelock and patches of hypopigmentation of the skin), and defective hearing. The color of the iris can change as a result of trauma, hemorrhage, intraocular inflammation (iridocyclitis, uveitis), intraocular tumor (especially retinoblastoma), intraocular foreign body, glaucoma, iris atrophy, oculosympathetic palsy (Horner syndrome), melanosis oculi, previous intraocular surgery, and some glaucoma medications.
Other Iris Lesions
Discrete nodules of the iris, referred to as Lisch nodules, are commonly seen in patients with neurofibromatosis. Lisch nodules represent melanocytic hamartomas of the iris and vary from slightly elevated pigmented areas to distinct ball-like excrescences. Lisch nodules are found in 92-100% of patients >5 yr of age who have neurofibromatosis. Slit-lamp identification of these nodules can help to fulfill the criteria required to confirm the diagnosis of neurofibromatosis.
In leukemia, there may be infiltration of the iris, sometimes with hypopyon, an accumulation of white blood cells in the anterior chamber, which can herald relapse or involvement of the central nervous system.
The lesion of juvenile xanthogranuloma (nevoxanthoendothelioma) can occur in the eye as a yellowish fleshy mass or plaque of the iris. Spontaneous hyphema (blood in the anterior chamber), glaucoma, or a red eye with signs of uveitis may be associated. A search for the skin lesions of xanthogranuloma (Chapter 80.3) should be made in any infant or young child with spontaneous hyphema. In many cases, the ocular lesion responds to topical corticosteroid therapy.
Leukocoria
Leukocoria includes any white pupillary reflex, also called cat eye reflex. Primary diagnostic considerations in any child with leukocoria are cataract, persistent hyperplastic primary vitreous, cicatricial retinopathy of prematurity, retinal detachment and retinoschisis, larval granulomatosis, and retinoblastoma (Fig. 614-2). Also to be considered are endophthalmitis, organized vitreous hemorrhage, leukemic ophthalmopathy, exudative retinopathy (as in Coats disease), and less-common conditions such as medulloepithelioma, massive retinal gliosis, the retinal pseudotumor of Norrie disease, the pseudoglioma of the Bloch-Sulzberger syndrome, retinal dysplasia, and the retinal lesions of the phakomatoses. A white reflex might also be seen with fundus coloboma, large atrophic chorioretinal scars, and ectopic medullation of retinal nerve fibers. Leukocoria is an indication for prompt and thorough evaluation.

The diagnosis can often be made by direct examination of the eye by ophthalmoscopy and biomicroscopy. Ultrasonographic and radiologic examinations are often helpful. In some cases, the final diagnosis rests with a pathologist.
Frank JW, Kushner BJ, France TD. Paradoxic pupillary phenomenon: a review of patients with pupillary constriction to darkness. Arch Ophthalmol. 1988;106:1564-1566.
Greenwald MJ, Folk ER. Afferent pupillary defects in amblyopia. J Pediatr Ophthalmol Strabismus. 1983;20:63-67.
Ivanov I, Shuper A, Shohat M, et al. Aniridia: recent achievements in paediatric practice. Eur J Pediatr. 1995;154:795-800.
Jaffe N, Cassady JR, Filler RM, et al. Heterochromia and Horner syndrome associated with cervical and mediastinal neuroblastoma. J Pediatr. 1975;87:75-77.
Jeffery AR, Ellis FJ, Repka MX, et al. Pediatric Horner syndrome. J Am Assoc Pediatr Ophthalmol Strabismus. 1998;2:159-167.
Loewenfeld IE. “Simple, central” anisocoria: a common condition seldom recognized. Trans Am Acad Ophthalmol Otolaryngol. 1977;83:832.
Mahoney NR, Liu GT, Menacker SJ, et al. Pediatric horner syndrome: etiologies and roles of imaging and urine studies to detect neuroblastoma and other responsible mass lesions. Am J Ophthalmol. 2006;142:651-659.
Thompson HS. Segmental palsy of the iris sphincter in Adie’s syndrome. Arch Ophthalmol. 1978;96:1615-1620.