Chapter 619 Abnormalities of the Cornea
Megalocornea
This is a nonprogressive symmetric condition characterized by an enlarged cornea (>12 mm in diameter) and an anterior segment in which there is no evidence of previous or concurrent ocular hypertension. High myopia is frequently present and may lead to reduced vision. A frequent complication is the development of lens opacities in adult life. All modes of inheritance have been described, although X-linked recessive is the most common; therefore, this disorder more commonly affects males. Systemic abnormalities that may be associated with megalocornea include Marfan syndrome, craniosynostosis, and Alport syndrome. The cause of the enlargement of the cornea and the anterior segment is unknown, but possible explanations include a defect in the growth of the optic cup and an arrest of congenital glaucoma. The region on the X chromosome responsible for this disorder has been identified.
Pathologic corneal enlargement caused by glaucoma is to be differentiated from this anomaly. Any progressive increase in the size of the cornea, especially when accompanied by photophobia, lacrimation, or haziness of the cornea, requires prompt ophthalmologic evaluation.
Microcornea
Microcornea, or anterior microphthalmia, is an abnormally small cornea in an otherwise relatively normal eye. It may be familial, with transmission being dominant more often than recessive. More commonly, a small cornea is just 1 feature of an otherwise developmentally abnormal or microphthalmic eye; associated defects include colobomas, microphakia, congenital cataract, glaucoma, and aniridia.
Keratoconus
This is a disease of unclear pathogenesis characterized by progressive thinning and bulging of the central cornea, which becomes cone shaped. Although familial cases are known, most cases are sporadic. Eye rubbing and contact lens wear have been implicated as pathogenic, but the evidence to support this is equivocal. The incidence is increased in individuals with atopy, Down syndrome, Marfan syndrome, and retinitis pigmentosa.
Most cases are bilateral, but involvement may be asymmetric. The disorder usually presents and progresses rapidly during adolescence; progression slows and stabilizes when patients reach full growth. Desçemet membrane may occasionally be stretched beyond its elastic breaking point, causing an acute rupture in the membrane with resultant sudden and marked corneal edema (acute hydrops) and decrease in vision. The corneal edema resolves as endothelial cells cover the defective area. Some degree of corneal scarring occurs, but the visual acuity is often better than before the initial incident. Signs of keratoconus include Munson sign (bulging of the lower eyelid on looking downward) and the presence of a Fleischer ring (a deposit of iron in the epithelium at the base of the cone). Corneal transplantation is indicated if satisfactory visual acuity cannot be attained with the use of contact lenses.
Neonatal Corneal Opacities
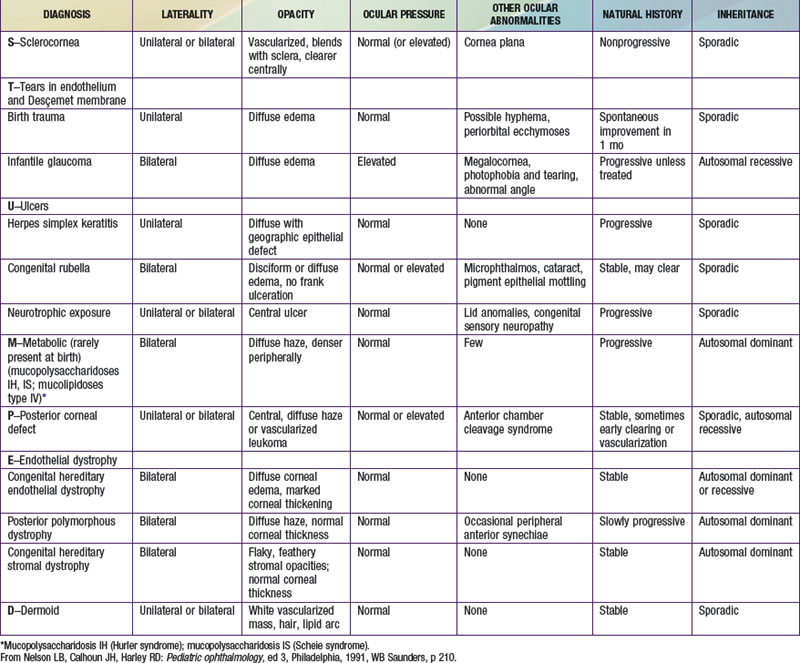
Loss of the normal transparency of the cornea in neonates may occur secondary to either intrinsic hereditary or extrinsic environmental causes (Table 619-1).
Sclerocornea
In sclerocornea, the normal translucent cornea is replaced by sclera-like tissue. Instead of a clearly demarcated cornea, white, feathery, often ill-defined and vascularized tissue develops in the peripheral cornea, appearing to blend with and extend from the sclera. The central cornea is usually clearer, but total replacement of the cornea with sclera may occur. The curvature of the cornea is often flatter, similar to the sclera. Potentially coexisting abnormalities include a shallow anterior chamber, iris abnormalities, and microphthalmos. This condition is usually bilateral. In approximately 50% of cases, a dominant or recessive inheritance has been described. Sclerocornea has been reported in association with numerous systemic abnormalities including limb deformities, craniofacial defects, and genitourinary disorders. In generalized sclerocornea, early keratoplasty should be considered in an effort to provide vision.
Peters Anomaly
Peters anomaly is a central corneal opacity (leukoma) that is present at birth (Fig. 619-1). It is often associated with iridocorneal adhesions that extend from the iris collarette to the border of the corneal opacity. Approximately half of patients have other ocular abnormalities, which may include cataracts, glaucoma, and microcornea. As many as 80% of cases may be bilateral, and 60% are associated with systemic malformations that may affect any major organ system. Some investigators have divided Peters anomaly into 2 types: a mesodermal or neuroectodermal form (type I), which shows no associated lens changes, and a surface ectodermal form (type II), which does. Histologic findings include a focal absence of Desçemet membrane and corneal endothelium in the region of the opacity. Peters anomaly may be caused by incomplete migration and differentiation of the precursor cells of the central corneal endothelium and Desçemet membrane or a defective separation between the primitive lens and cornea during embryogenesis.
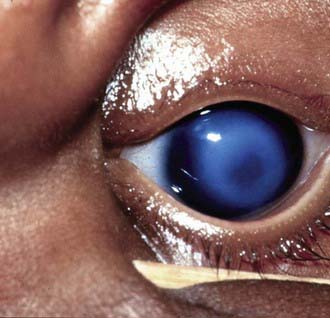
Dermoids
Epibulbar dermoids are choristomas. They are often present at birth and may increase in size with age. They occur most frequently in the lower temporal quadrant. They most commonly straddle the limbus and extend into the peripheral cornea. Rarely, they may be confined entirely to the cornea or conjunctiva. Epibulbar dermoids may cause visual disturbance by encroaching on the visual axis or by contributing to the development of astigmatism, which may lead to amblyopia.
A dermoid usually appears as a well-circumscribed rounded or oval, gray or pinkish-yellow mass with a dry surface from which short hairs may protrude. It may affect only the superficial layers of the cornea, although full-thickness involvement is common. Associated ocular anomalies include eyelid and iris colobomas, microphthalmos, and retinal and choroidal defects. A total of 30% of dermoids are associated with systemic abnormalities. Many of the associated anomalies involve developmental defects of the 1st branchial arch (vertebral anomalies, dysostosis of the facial bones and dental anomalies, and Goldenhar syndrome). Epibulbar dermoids are found in 75% of cases of Goldenhar syndrome.
Dendritic Keratitis
Infection of the cornea with the herpes simplex virus produces a characteristic lesion of the corneal epithelium, referred to as a dendrite; it has a branching treelike pattern that can be demonstrated by fluorescein staining. The acute episode is accompanied by pain, photophobia, tearing, blepharospasm, and conjunctival injection. Specific treatment may include mechanical debridement of the involved corneal epithelium to remove the source of infection and eliminate an antigenic stimulus to inflammation in the adjacent stroma. Medical treatment involves the use of trifluridine or systemic acyclovir. In addition, a cycloplegic agent is useful to relieve pain from spasm of the ciliary muscle. Overly aggressive topical antiviral treatment itself can be toxic to the cornea and should be avoided. Recurrent infection and deep stromal involvement can lead to corneal scarring and loss of vision.
Topical use of corticosteroids causes exacerbation of superficial herpetic disease of the eye and may lead to corneal perforation; eyedrops combining steroids and antibiotics are therefore to be avoided in treatment of red eye unless there are clear-cut indications for their use and close supervision during therapy.
Infants born to mothers infected with herpes simplex virus should be examined carefully for signs of ocular involvement. Intravenous acyclovir is required for treatment of ocular herpes in newborns.
Corneal Ulcers
The usual signs and symptoms are focal or diffuse corneal haze, hyperemia, lid edema, pain, photophobia, tearing, and blepharospasm. Hypopyon (pus in the anterior chamber) is common. Corneal ulcers require prompt treatment. They result most frequently from traumatic lesions that become secondarily infected. Many organisms are capable of infecting the cornea. One of the most serious is Pseudomonas aeruginosa; it can rapidly destroy stromal tissue and lead to corneal perforation. Neisseria gonorrhoeae also is particularly damaging to the cornea. Indolent ulcers may be caused by fungi, often in association with the use of contact lenses. In each case, scrapings of the cornea must be studied in an effort to identify the infectious agent and to determine the best therapy. Although aggressive local treatment is generally needed to save the eye, systemic treatment may be necessary in some cases as well. Perforation or scarring resulting from corneal ulceration is an important cause of blindness throughout the world and is estimated to be responsible for 10% of blindness in the USA.
Unexplained corneal ulcers in infants and young children should raise the question of a sensory defect, as in Riley-Day or Goldenhar-Gorlin syndrome, or of a metabolic disorder such as tyrosinemia.
Phlyctenules
These are small, yellowish, slightly elevated lesions usually located at the corneal limbus; they may encroach on the cornea and extend centrally. A small corneal ulcer is often found at the head of the advancing lesion, with a fascicle of blood vessels behind the head of the lesion. Although once thought to represent a sign of systemic tuberculin infection, phlyctenular keratoconjunctivitis is now accepted as a morphologic expression of delayed hypersensitivity to diverse antigens. In children, it commonly occurs as a result of a hypersensitivity reaction of the conjunctiva or cornea to bacterial products. Treatment usually consists of eliminating the underlying disorder, usually staphylococcal blepharitis or meibomianitis, and suppressing the immune response with the use of topical corticosteroid therapy. A superficial stromal pannus and scarring sometimes remain after treatment.
Interstitial Keratitis
This denotes inflammation of the corneal stroma. The most common cause is syphilis, interstitial keratitis being one of the characteristic late manifestations of congenital syphilis. The corneal changes in congenital syphilis occur in 2 phases. The acute phase presents between the ages of 5 and 10 yr, with an intense keratitis that may last for several months and causes a severe reduction in vision. The acute effects of syphilis are mainly due to the host immune response, such as mononuclear cell infiltrates, proliferative vascular changes, and occasionally granuloma formation. The deep inflammation produces pain, photophobia, tearing, circumcorneal injection, and corneal haze. The acute episode is followed by a chronic stage with significant regression in the corneal findings along with a parallel improvement in visual acuity. Although the corneal findings may regress with time, “ghost vessels,” which represent the previous vascular changes, and patchy corneal scarring remain and serve as permanent stigmata of the disease.
Cogan syndrome is a nonluetic interstitial keratitis associated with hearing loss and vestibular symptoms. Although its cause is unknown, a systemic vasculitis is suspected. Prompt treatment is required to avoid permanent hearing loss. Both the corneal changes and the auditory involvement may respond to the use of immunosuppressive agents.
Less frequently, interstitial keratitis is caused by other infectious diseases, such as tuberculosis or leprosy.
Corneal Manifestations of Systemic Disease
Several metabolic diseases produce distinctive corneal changes in childhood. Refractile polychromatic crystals are deposited throughout the cornea in cystinosis (Chapter 79.4). Corneal deposits producing various degrees of corneal haze also occur in certain types of mucopolysaccharidosis (MPS; Chapter 82), particularly MPS IH (Hurler), MPS IS (Scheie), MPS I H/S (Hurler-Scheie compound), MPS IV (Morquio), MPS VI (Maroteaux-Lamy), and sometimes MPS VII (Sly). Corneal deposits may develop in patients with GM1 (generalized) gangliosidosis (Chapter 80.4). In Fabry disease, fine opacities radiating in a whorl or fanlike pattern occur, and corneal changes can be important in identifying the carrier state (Chapter 80.4). A spraylike pattern of corneal opacities may also be seen in the Bloch-Sulzberger syndrome. In Wilson disease, the distinctive corneal sign is the Kayser-Fleischer ring, a golden brown ring in the peripheral cornea resulting from changes in Desçemet membrane (Chapter 349.2). Pigmented corneal rings may develop in neonates with cholestatic liver disease. Corneal changes may occur in autoimmune hypoparathyroidism and band keratopathy in patients with hypercalcemia. Transient keratitis may occur with rubeola and sometimes with rubella.
Beauchamp GR, Gillette TE, Friendly DS. Phlyctenular keratoconjunctivitis. J Pediatr Ophthalmol Strabismus. 1981;18:22-28.
Chang DC, Grant GB, O’Donnell K, et al. Multistate outbreak of Fusarium keratitis associated with use of a contact lens solution. JAMA. 2006;296:953-963.
Comer RM, Daya SM, O’Keefe M. Penetrating keratoplasty in infants. J AAPOS. 2001;5:285-290.
Dana MR, Moyes AL, Gomes JA, et al. The indications for and outcome in pediatric keratoplasty. A multicenter study. Ophthalmology. 1995;102:1129-1138.
Garg P, Krishna PV, Stratis AK, et al. The value of corneal transplantation in reducing blindness. Eye (Lond). 2005;19:1106-1114.
Mackey DA, Buttery RG, Wise GM, et al. Description of X-linked megalocornea with identification of the gene locus. Arch Ophthalmol. 1991;109:829-833.
Michaeli A, Markovich A, Rootman DS. Corneal transplants for the treatment of congenital corneal opacities. J Pediatr Ophthalmol Strabismus. 2005;42:34-44.
Mohandessan MM, Romano PE. Neuroparalytic keratitis in Goldenhar-Gorlin syndrome. Am J Ophthalmol. 1978;85:111-113.
Reidy JJ. Congenital corneal opacities. Ophthalmol Clin North Am. 1996;2:199-213.
Traboulsi EI, Maumenee IH. Peters’ anomaly and associated congenital malformations. Arch Ophthalmol. 1992;110:1739-1742.
Yang LL, Lambert SR, Fernhoff PM, et al. Peters’ anomaly: associated congenital malformations and etiology. Invest Ophthalmol Vis Sci. 1995;36:S41.
Yang LL, Lambert SR, Lynn MJ, et al. Long-term results of corneal graft survival in infants and children with Peters anomaly. Ophthalmology. 1999;106:833-848.