Chapter 641 Ectodermal Dysplasias
Ectodermal dysplasia (ED) is a heterogeneous group of disorders characterized by a constellation of findings involving defects of 2 or more of the following: teeth, skin, and appendageal structures including hair, nails, and eccrine and sebaceous glands. Although more than 150 ectodermal dysplasias have been described, the majority are rare and most have not been genetically defined.
Anhidrotic (Hypohidrotic) Ectodermal Dysplasia
The syndrome known as anhidrotic ectodermal dysplasia manifests as a triad of defects: partial absence (hypohidrosis) or complete absence of sweat glands, anomalous dentition, and hypotrichosis. There are 4 recognized types of anhidrotic ectodermal dysplasia (Table 641-1). The X-linked form is most common.
Table 641-1 FOUR RECOGNIZED TYPES OF ANHIDROTIC ECTODERMAL DYSPLASIA
| TYPE | INHERITANCE | GENE DEFECT |
|---|---|---|
| ED-1 | X-linked recessive | Ectodysplasin A (EDA) |
| ED-anhidrotic | Autosomal recessive | Ectodyplasin A anhidrotic receptor (EDAR) |
| EDAR-associated death gene (EDARADD) | ||
| ED-3 | Autosomal dominant | EDAR |
| ED-anhidrotic with immune deficiency |
In ED-1, affected males are unable to sweat and may experience episodes of high fever in warm environments, which may be mistakenly considered to be fevers of unknown origin. This error is particularly common in infancy, when the facial changes are not easily appreciated. Diagnosis at this time may be made using the starch-iodine test or palmar or scalp biopsy. Scalp biopsy is the most sensitive and is 100% specific. The typical facies is characterized by frontal bossing; malar hypoplasia; a flattened nasal bridge; recessed columella; thick, everted lips; wrinkled, hyperpigmented periorbital skin; and prominent, low-set ears (Fig. 641-1). The skin over the entire body is dry, finely wrinkled, and hypopigmented, often with a prominent venous pattern. Extensive peeling of the skin is a clinical clue to diagnosis in the newborn period. The paucity of sebaceous glands may account for the dry skin. The scalp hair is sparse, fine, and lightly pigmented, and eyebrows and lashes are sparse or absent. Other body hair is also sparse or absent. Sexual hair growth is normal. Anodontia or hypodontia with widely spaced, conical teeth is a consistent feature (see Fig. 641-1). Otolaryngeal and ophthalmologic abnormalities secondary to decreased saliva and tear production are seen. The incidence of atopic diseases in children with ED-1 is high. Gastroesophageal reflux is common and may play a role in failure to thrive, which is seen in 20% of cases. Sexual development is usually normal. Historically, the infant mortality rate has been 30%. Carrier females have no or variable clinical manifestations.
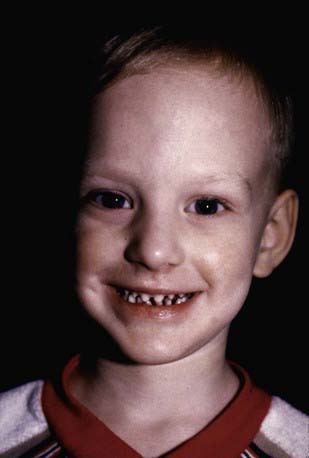
Figure 641-1 Hypohidrotic ectodermal dysplasia is characterized by pointed ears, fine hair, periorbital hyperpigmentation, midfacial hypoplasia, and pegged teeth.
(Courtesy of the Fitzsimons Army Medical Center teaching file.)
The clinical findings in autosomal recessive anhidrotic ectodermal dysplasia are identical to those of the X-linked recessive disorder, except that females are affected to the same degree as males. The clinical findings in the autosomal dominant form are also seen in both sexes and are similar to the X-linked recessive form but much milder. Hypohidrotic ED with immune deficiencies causes similar findings in sweating and hair and nail development, in association with a dysgammaglobulinemia. Significant mortality is seen from recurrent infections.
Treatment of children with hypohidrotic ED includes protecting them from exposure to high ambient temperatures. Early dental evaluation is necessary so that prostheses can be provided for cosmetic reasons and for adequate nutrition. The use of artificial tears prevents damage to the cornea in patients with defective lacrimation. Alopecia may necessitate the wearing of a wig to improve appearance.
Hidrotic Ectodermal Dysplasia (Clouston Syndrome)
The salient features of the autosomal dominant disorder hidrotic ED are dystrophic, hypoplastic, or absent nails; sparse hair; and hyperkeratosis of the palms and soles. Conjunctivitis and blepharitis are common. The dentition and sweating are always normal. Absence of eyebrows and eyelashes and hyperpigmentation over the knees, elbows, and knuckles have been noted in some affected individuals. Mutations in the GJB6 gene encoding the gap junction protein connexin 30 are responsible for this disorder. A similar disorder associated with deafness has been described with mutations in the GJB2 gene encoding the connexin 26 protein.
Baris HN, Zlotogorski A, Peretz-Amit G, et al. A novel GJB6 mutation in hidrotic ectodermal dysplasia 2 (Clouston syndrome) broadens its genotypic basis. Br J Dermatol. 2008;159:1373-1376.
Gunadi, M K, Ohta M, et al. Two novel mutations in the ED1 gene in Japanese families with X-linked hypohidrotic ectodermal dysplasia. Pediatr Res. 2009;65:453-457.
Lu PD, Schaffer JV. Hypohidrotic ectodermal dysplasia. Dermat Online J. 2008;14:22.