Chapter 674 Arthrogryposis
Definition
Arthrogryposis multiplex congenita (AMC) is a congenital anomaly in the newborn involving multiple curved joints (see Fig. 674-1 on the Nelson Textbook of Pediatrics website at www.expertconsult.com  ). Arthrogryposis is a descriptive term and not an exact diagnosis, as there are as many as 300 possible underlying causes.
). Arthrogryposis is a descriptive term and not an exact diagnosis, as there are as many as 300 possible underlying causes.
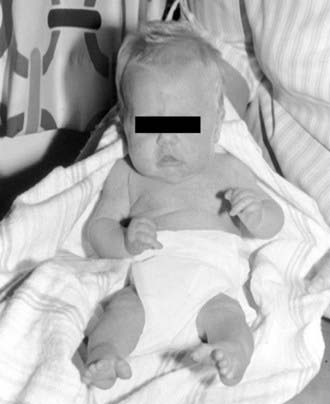
Figure 674-1 Arthrogryposis multiplex congenita. A 32-wk premature boy with severe multiple congenital contractures and severe congenital neuropathy.
(From Hosalkar HS, Moroz L, Drummond DS, et al: Neuromuscular disorders of infancy and childhood and arthrogryposis. In Dormans J, editor: Pediatric orthopedics: core knowledge in orthopedics, Philadelphia, 2005, Mosby, pp 454–482.)
The incidence of classic AMC is approximately 1/3,000 live births, and the three main groups include classic AMC, in which the limbs are primarily involved and the muscles are deficient or absent (amyoplasia) (Fig. 674-2); arthrogryposis in association with major neurogenic (brain, spinal cord, anterior horn cell, or peripheral nerve) or myopathic (congenital muscular dystrophy, myopathy, toxic myopathy) dysfunction; and arthrogryposis in association with other major anomalies and specific syndromes such as diastrophic dysplasia and craniocarpotarsal dystrophy (Table 674-1).
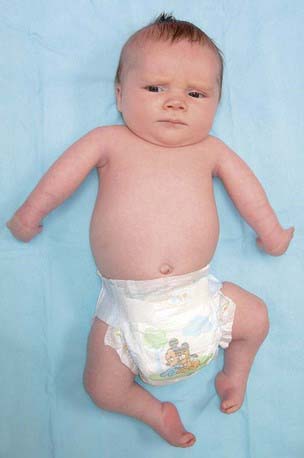
Figure 674-2 Child with amyoplasia. In the upper extremity, the shoulders are internally rotated, the elbows are extended, the wrists are flexed and ulnarly deviated, the fingers are stiff, and the thumbs are positioned in the palm. In the lower limbs, the hips may be dislocated, the knees are extended, and the feet have severe equinovarus contractures.
(From Bamshad M, Van Heest AE, Pleasure D: Arthrogryposis: a review and update, J Bone Joint Surg Am 91[Suppl 4]:40–46, 2009.)
Table 674-1 ASSOCIATED ETIOLOGIES OF ARTHROGRYPOSIS
ARTHROGRYPOSIS DUE TO NERVOUS SYSTEM DISORDERS
DISTAL ARTHROGRYPOSIS SYNDROMES
PTERYGIUM SYNDROMES
MYOPATHIES
ABNORMALITIES OF JOINTS AND CONTIGUOUS TISSUE
SKELETAL DISORDERS
INTRAUTERINE AND MATERNAL FACTORS
MISCELLANEOUS
SINGLE JOINT
Modified from Mennen U, Van Heest A, Ezaki MB: Arthrogryposis multiplex congenita, J Hand Surg [Br] 30:468–474, 2005.
Clinical Features
Multiple rigid joint deformities are present with defective muscles and normal sensation. There is rigidity of several joints in each case, resulting from both short tight muscles and capsular contractures. Pterygium may be present on the flexor aspects of contracted joints (Fig. 674-3). There is often an absence or fibrosis of muscles or muscle groups. There is normal intellectual development in most cases. All four limbs are involved in the classic form (AMC), but the condition can also occur in the upper or lower limbs. An autosomal dominant variant called distal arthrogryposis involves the hands and feet with severe deformation but with only minor contractures more proximally; scoliosis is a possible development. To date, 10 different distal arthrogryposes have been described. They are classified according to the proportion of features they share (Table 674-2). In addition to the multiple joint contractures, the lack of skin creases (cylindrical or tubular limbs) and deep dimples over the joints are very characteristic (Fig. 674-4). There is dislocation of joints, most commonly the hip but occasionally the knee; the trunk is rarely affected. Other congenital anomalies such as cryptorchidism, hernias, and gastroschisis can occur.

Figure 674-3 Fixed flexion of the knees in a boy with arthrogryposis multiplex congenita.
(From Hosalkar HS, Moroz L, Drummond DS, et al: Neuromuscular disorders of infancy and childhood and arthrogryposis. In Dormans J, editor: Pediatric orthopedics: core knowledge in orthopedics, Philadelphia, 2005, Mosby, pp 454–482.)
Table 674-2 CURRENT LABELS AND OMIM NUMBERS FOR THE DISTAL ARTHROGRYPOSIS SYNDROMES
| SYNDROME | NEW LABEL | OMIM NUMBER |
|---|---|---|
| Distal arthrogryposis type 1 | DA1 | 108120 |
| Distal arthrogryposis type 2A (Freeman-Sheldon syndrome) | DA2A | 193700 |
| Distal arthrogryposis type 2B (Sheldon-Hall syndrome) | DA2B | 601680 |
| Distal arthrogryposis type 3 (Gordon syndrome) | DA3 | 114300 |
| Distal arthrogryposis type 4 (scoliosis) | DA4 | 609128 |
| Distal arthrogryposis type 5 (ophthalmoplegia, ptosis) | DA5 | 108145 |
| Distal arthrogryposis type 6 (sensorineural hearing loss) | DA6 | 108200 |
| Distal arthrogryposis type 7 (trismus-pseudocamptodactyly) | DA7 | 158300 |
| Distal arthrogryposis type 8 (autosomal dominant multiple pterygium syndrome) | DA8 | 178110 |
| Distal arthrogryposis type 9 (congenital contractural arachnodactyly) | DA9 | 121050 |
| Distal arthrogryposis type 10 (congenital plantar contractures) | DA10 | 187370 |
OMIM, Online Mendelian Inheritance in Man.
From Bamshad M, Van Heest AE, Pleasure D: Arthrogryposis: a review and update, J Bone Joint Surg Am 91 Suppl 4:40–46, 2009.
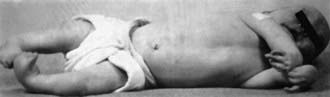
Figure 674-4 Characteristic lack of skin creases and tubular limbs.
(From Hosalkar HS, Moroz L, Drummond DS, et al: Neuromuscular disorders of infancy and childhood and arthrogryposis. In Dormans J, editor: Pediatric orthopedics: core knowledge in orthopedics, Philadelphia, 2005, Mosby, pp 454–482.)
Etiology
AMC is multifactorial in etiology. Factors liable to produce immobility of the fetus can contribute to congenital contractures. Some of these include structural abnormality of the uterus (bifid, large fibroids), oligohydramnios, increased intrauterine pressure, mechanical compression of the fetus, weak fetal movements, breech presentation, and prematurity. Inflammatory or infective etiology has also been postulated, including inflammation in the joint, muscle, spinal cord, or brain; rubella in early pregnancy; and infection with unknown viruses. A dominant theory is that the primary condition is patchy degeneration of the anterior horn cells occurring in the early months of gestation. A pregnant mother treated with curare for severe tetanus gave birth to an arthrogrypotic baby.
Diagnosis
Clinical examination remains the best modality for establishing the diagnosis of arthrogryposis. We have found a few factors that are often useful in making a diagnosis. Although not absolute criteria, they are helpful when considered in combination.
Unlike paralytic disorders, joint deformities of AMC are usually stiff or rigid from the beginning, with incomplete passive range of motion. Deformities of arthrogryposis tend to be symmetric.
The severity of contractures tends to increase as one reaches the periphery of the limb. The more proximal joints tend to be less involved, and the trunk is often spared. The most severe deformities tend to occur in the hands and feet.
The orthopedist, neurologist, geneticist, and pediatrician must participate in diagnosing and managing this condition. Radiographs of the involved extremities with joint involvement are recommended. These can demonstrate congenital bony abnormality and loss of subcutaneous fat and muscle. Radiographs of the whole spine identify any vertebral anomalies. CT and MRI of the brain and spine are useful in establishing or ruling out structural central nervous system (CNS) involvement. Electromyography and nerve conduction studies are of limited value and have been used to differentiate the peripheral neuropathic from the myopathic variants.
DNA diagnostic tests to distinguish the various congenital myopathies and/or dystrophies are not widely available, and thus skeletal muscle biopsy may be needed when a primary myopathic disorder is suspected. In such case, care must be taken to avoid malignant hyperthermia. Plasma creatine kinase may be estimated to exclude myopathic disorders. This is best checked on the 3rd day of life or after, once any transient initial increase in creatine kinase from the birth process has subsided. If a prenatal ultrasound scan detects an absence of fetal movement, especially in combination with polyhydramnios, the diagnosis of arthrogryposis can be suspected. Histologic analysis reveals a small muscle mass with fibrosis and fat between the muscle fibers. Myopathic and neuropathic features can overlap in the same specimen. The periarticular soft tissues are fibrotic. Genetic consultation, with chromosome analysis and collagen studies, should be considered in cases in which a distinct peripheral neuromuscular disorder is not readily apparent.
The differential diagnosis is shown in Table 674-3.
Prognosis
Most children with arthrogryposis require therapy; an estimated 80% of children with amyoplasia receive therapy services into their adolescence. Limb deformities that restrict function often require surgical treatment; in one series, 76% of children received surgery of the foot, >30% surgery of the knee, 25% surgeryof the elbow, and nearly 20% surgery of the hip. The clinician should be able to derive a general prognosis and treatment plan once the diagnosis of AMC is established. Vigorous occupational therapy and use of hand splints and serial casting can improve the range of motion and functional use of the hand in many cases of AMC in which the etiology is not a progressive disorder (amyoplasia). Surgery, in selected cases, is necessary to obtain a more neutral positioning of the wrist and fingers so the limited degree of strength can be used to optimal biomechanical advantage.
Corrected deformities are quite common and occur with growth in a limb in which the periarticular structures are incapable of stretching. Arthrogrypotic children have certain positive factors that must be used in their successful management.
Joint instability is not a problem in AMC, unlike other paralytic conditions.
With a coordinated and team approach in management, there often is little deterioration from the condition at birth. There is commonly central sparing and a relatively normal trunk.
The child with normal CNS findings can be expected to have reasonably normal intelligence and, with enough motivation, can contribute to successful management.
Principles of Orthopedic Management of Patients with Arthrogryposis and Multiple Congenital Contractures
Patients should be seen as early as possible, and treatment of the deformities should be started early because some respond remarkably well to physiotherapy, stretching, and splinting.
Muscle balance is usually less of a problem than in other paralytic neuromuscular conditions and is easier to achieve. Muscle balance should be possibly established if there are functioning muscles available for transfer. Recurrence of deformity is the rule because the dense, inelastic soft tissues about the joints do not properly elongate with growth. These structures are considered the key to successful management of arthrogryposis in the growing child. The maximal, safely obtainable correction should be achieved at the time of surgery. The use of wedging or corrective casts after surgery is of little additional benefit.
Range of Movement
Range of movement results from passive range of motion stretching varies. Aggressive treatment can result in a decrease in the already limited range of joint movement or have little impact. The arc of motion, however, can often be changed into a functionally better position.
Centrifugal Involvement
The severity of the deformity increases toward the periphery of the limb, and the more proximal joints tend to be less involved. Central sparing of the trunk is noted in most cases.
Aim of Management
The main aim of management is to achieve maximal functional gain for each patient. Minimal requirements are independent walking, self-care, and, ideally, the ability to eventually be gainfully employed.
Staging and Timing of Surgical Procedures
Maximal benefit from surgical reconstruction of a limb with arthrogryposis is achieved by careful staging of procedures. The disabling deformity and contractures should be corrected in the first stage. The major joints should be then put in a functional arc of motion most adapted to the patient’s needs.
In the final stage, tendon transfers are occasionally required to bring motor power to a joint that has been put to its optimal position. The elbow joint is well suited to tendon transfers and has been widely noted to give satisfactory results.
Lower Limbs
Foot Involvement
The most severe deformities in arthrogryposis are known to occur in the foot. The rigid foot deformity is usually the clubfoot or equinovarus deformity and, less commonly, a congenital vertical talus deformity. The goal of treatment is conversion of the rigid deformed foot into a rigid plantigrade foot.
Correction of the hindfoot takes precedence over the forefoot. Serial stretching (casting) sometimes produces a degree of correction. Once it is clear during the course of treatment that conservative treatment will not be successful, surgery should be considered, preferably when the child is ready to walk.
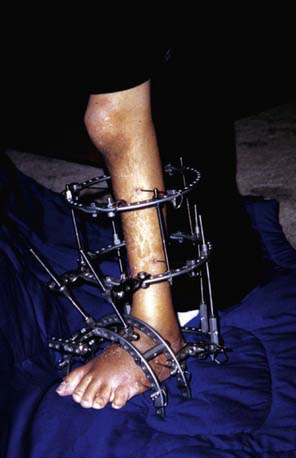
An extensive posteromedial and posterolateral release is recommended. If the foot fails to correct with even the most extensive soft-tissue release or relapses quickly within 2-3 yr, talectomy may be considered. The Ilizarov technique and apparatus do offer an opportunity to correct these deformities by gradual distraction and neohistiogenesis (Fig. 674-5). Applications of this fixator in the pediatric foot have been fairly successful and satisfactory. In the case of older children with neglected or relapsed equinovarus deformity, correction can be best obtained by triple arthrodesis. In rare cases of recurrence of the deformity after triple arthrodesis (at the level of the ankle), a pantalar arthrodesis may be offered by an easy conversion of the triple arthrodesis.
Knee Joint
Both common presentations of the knee deformity, fixed flexion and fixed extension, should be initially treated with repeated stretching and splinting. The goal is to get the knees straight and to keep them that way by bracing. Extension of the knee is considered the key to later walking. If a flexed knee is neglected, postural hip flexion contractures are likely to ensue. Combined contracture in both hips and knees is not compatible with good gait.
Nonsurgical Management
Manipulation and plaster casting in the younger child have been successful in a large population of our patients. Although the arc of motion is changed to a more extended position, the range is not increased. Even if complete extension cannot be obtained, the knee joints are usually stable and mild flexion deformities are quite compatible with a good gait pattern.
Surgical Management
Deformities not responding to soft-tissue stretching might need surgical intervention. It is advisable to plan the timing of knee surgery in keeping with the treatment plan for the foot. For example, if the foot requires immobilization with the knee flexed, then the knee flexion correction should be staged after the foot management. On the other hand, if the knee is in fixed extension, it is better to correct the knee extension before operation on the foot, so that the foot can be immobilized with a flexed knee.
Fixed hyperextension of the knee can respond reasonably well to serial stretching and casting. In severe cases that fail to respond to stretching, an extensive muscular release with quadricepsplasty may be necessary to correct the knee position. Splinting and knee support are likely to be required on a long-term basis. The Ilizarov fixator again provides a useful alternative to surgical release or in cases of failed surgery, especially in the older patient.
Hip Joint
A common finding at birth in arthrogrypotic patients is stiffness of the hips in flexion, abduction, and external rotation. The two most common involvements of the hips are fixed contractures and hip dislocation.
It is important to correct the knee deformity before attempting any surgical intervention and correction at the hip. With knee correction at an early stage, the results of hip deformity correction are encouraging. Full surgical correction of the growing child is not easily obtainable with soft-tissue release procedures. Recurrence is usually unavoidable with skeletal growth. Rather, if the child is able to ambulate with compensatory lordosis, it is best to wait until skeletal maturity and then hope for lasting correction with subtrochanteric osteotomy.
Arthrogryposis can lead to unilateral or bilateral dislocation of hips. Dislocations are usually stable and, if the pelvis is well balanced, are also consistent with a good gait. Treatment of hip dislocation is often not easy because closed reduction invariably fails and stiffness and persistent flexion deformity usually follow open reduction. Diagnosis can be difficult clinically because the marked stiffness may be a limiting factor for demonstrating the hip instability clinically. If the hips are dislocated, in most cases, they are not reducible on abduction and should not be splinted if irreducible. Splinting in such cases can lead to avascular necrosis. Bilateral dislocations tend to be high and stable, are usually symmetric, and tend to have a fairly balanced pelvis. This is often consistent with a good gait, and it may be advisable to leave them alone because it is often not possible to get a satisfactory result on both sides and in fact can lead to more stiffness with a high chance of redislocation. In cases of unilateral dislocation, there is a risk of progressive pelvic obliquity and secondary scoliosis. We therefore believe it is often worth reducing the dislocated hip, especially in the infant and the younger child. Open reduction should be done as soon as the child is healthy enough and knee flexion contractures have been controlled. Excessive delays make the procedure more technically demanding and the reduction more difficult to achieve.
Upper Limbs
Unlike management of lower limbs, where independent walking is the main goal, management of upper extremities in arthrogryposis requires considerable caution because the prognosis for successful treatment depends more on the extent of deformity and on the patient’s intelligence. The minimal requirements for the patient are ability to feed and attend to personal hygiene.
Again, in contrast to the lower limbs, where surgery cannot be postponed due to risk of delayed walking, operations on upper limbs can be postponed for several years. Interestingly, arthrogrypotic children develop a remarkable ability to get about well with their upper limbs in spite of the complexities of these deformities, developing a surprising amount of dexterity. Therefore, surgical intervention, if any, should be weighed very carefully in these cases.
Planning
Both limbs should be considered as a unit. The general principle that the arc of joint motion can be changed but not increased should be remembered. A reasonable expectation at the end of treatment is that the patient should ideally be able to move one hand to his or her mouth and the other to his or her anus, but still be capable of opposing both hands. This is important to consider because children with severe hand deformities and weakness depend on the integrated use of both hands (bimanual opposition) to perform any task that normal people do with one hand. In addition, he or she must be able to push him- or herself out of a chair.
Shoulder and elbow joints should be considered as a unit. The rotation of the shoulder is an important factor on which the axis of the elbow joint motion largely depends. Therefore, if elbow surgery is contemplated to enable the hand to reach the mouth, a severely fixed and internally rotated shoulder should also be simultaneously corrected.
Physiotherapy has a major contribution in obtaining motion in the stiff joints along with stretching and splinting. Detailed assessment by a physical therapist and occupational therapist is necessary before embarking on any surgical intervention, and surgery should be deferred until the age of 4 yr, if possible.
Shoulder Joint
The typical deformity is adduction and internal rotation. Although shoulder weakness is common, these children can develop remarkable trick movements, and therefore surgical intervention is rarely indicated. Adduction itself is not troublesome because usually there is enough passive abduction for self-care, and surgery is rarely required. Fixed internal rotation at the shoulder can in some cases jeopardize the axis of elbow motion. In severe cases, a simple external rotation osteotomy may be performed in the upper humerus to bring the forearm and hand into a more functional position.
Elbow Joint
The two common involvements of the elbow noted are fixed extension contracture and fixed flexion contracture. Both these conditions respond well to stretching and physical therapy and can be well controlled in infancy. Once the child is ambulating, activities such as crutch walking, toileting, and push-off from a chair or a seated position essentially need active elbow extension. Therefore, it is important to not inadvertently damage the active extension mechanism while intending to improve active flexion. Passive flexion can be surgically achieved by a posterior soft-tissue release of the elbow, lengthening of the triceps, and a posterior capsular and collateral ligament release. This can restore a very useful arc of motion, and further procedures might not be even necessary in most cases. In candidates in whom a value of increased active flexion can be established, active power could be provided in multiple ways including a Steindler flexorplasty and transfer of the triceps or pectoralis major.
Elbow and forearm soft tissue contractures can be successfully corrected using the Ilizarov fixator in many cases.
Wrist and Hand
The wrist is often involved in a flexion contracture, and the fingers may be curved and stiff as well. Thumb adduction deformity (thumb-in-palm) is also common. Manipulation, stretching, and splinting (especially early on) can be very important in establishing mobility and range. Interestingly, the flexed position of the wrist is very functional and might not need intervention. Surgical corrections involving partial or complete carpectomy have been described and often show high incidence of recurrence of deformity with growth. Finger stiffness is extremely difficult to correct in these cases, and surgery is rarely indicated. Most patients adapt extremely well to the finger stiffness and are impressive in their functioning abilities. Release of thumb adductors and web space enlargement are very useful in correcting the thumb deformity. Wrist arthrodesis for functional or cosmetic gain may be considered at or near skeletal maturity.
Spine
In consistency with the principle that the severity of the stiffness and deformity increases toward the periphery of limbs and that the more central or proximal areas are less involved, a straight, supple, and well-balanced trunk is an important asset. Scoliosis occurs commonly in arthrogryposis because of a high incidence of congenital curves (Chapter 671).
Idiopathic Scoliosis
Genetic or idiopathic scoliosis seems to occur as commonly in the arthrogrypotic population as in the general population. The progression and behavior of an idiopathic curve in arthrogryposis are possibly the same as in other children, although they may be more disabling because of the coexisting peripheral deformities.
Paralytic Scoliosis
Long paralytic scoliotic curves are sometimes seen in children with the typical neuropathic type of arthrogryposis. Some forms of congenital muscular dystrophy include a “rigid spine” component. The curve is typically seen before the 2nd yr of life and progresses to become long, severe, and eventually rigid. Well-controlled bracing is the treatment of choice in younger children, although surgical management may be necessary in progressive cases.
Congenital Scoliosis
These curves are common in arthrogryposis, perhaps owing to the other congenital anomalies common in arthrogryposis. Failure of vertebral formation and failure of segmentation have been noted. Klippel-Feil deformity is also observed. These curves must be observed closely, and aggressive treatment such as early surgical fusion may be indicated in children with asymmetric progressive curves.
Scoliosis Associated with Pelvic Obliquity
Scoliosis associated with pelvic obliquity is associated with neglected hip deformities that lead to femoropelvic obliquity and is potentially avoidable. Unilateral hip deformity should be particularly watched for and corrected. The trunk can then be kept supple and straight.
Alves PV, Zhao L, Patel PK, et al. Arthrogryposis: diagnosis and therapeutic planning for patients seeking orthodontic treatment or orthognathic surgery. J Craniofac Surg. 2007;18(4):838-843.
Bamshad M, Van Heest AE, Pleasure D. Arthrogryposis: a review and update. J Bone Joint Surg Am. 2009;91(Suppl 4):40-46.
Fixsen J. Arthrogryposis multiplex congenita. In: Benson MK, MF Macnicol MF, Parsch K, editors. Children’s orthopaedics and fractures. Philadelphia: Churchill Livingstone; 2002:293-298.
Fassier A, Wicart P, Dubousset J, et al. Arthrogryposis multiplex congenita. Long-term follow-up from birth until skeletal maturity. J Child Orthop. 2009;3(5):383-390.
Hall JG. Arthrogryposis multiplex congenita: etiology, genetics, classification, diagnostic approach, and general aspects. J Pediatr Orthop. 1997;6:159-166.
Hosalkar HS, Moroz L, Drummond DS, et al. Neuromuscular disorders of infancy and childhood and arthrogryposis. In: Dormans J, editor. Pediatric orthopedics: core knowledge in orthopedics. Philadelphia: Mosby; 2005:454-482.
Mennen U, Van Heest A, Ezaki MB. Arthrogryposis multiplex congenita. J Hand Surg [Br]. 2005;30:468-474.
Navti OB, Kinning E, Vasudevan P, et al. Review of perinatal management of arthrogryposis at a large UK teaching hospital serving a multiethnic population. Prenat Diagn. 2010;30(1):49-56.