Chapter 688 Disorders Involving Ion Transporters
In order of decreasing severity, the disorders involving ion transporters include achondrogenesis type 1B, atelosteogenesis type II, and diastrophic dysplasia. They result from the functional loss of the sulfate ion transporter called diastrophic dysplasia sulfate transporter (DTDST), which is also referred to as SLC26A2 (solute carrier family 26, member 2). This protein transports sulfate ions into cells and is important for cartilage cells that add sulfate moieties to newly synthesized proteoglycans destined for cartilage extracellular matrix. Matrix proteoglycans are responsible for many of the properties of cartilage that allow it to serve as a template for skeletal development. The clinical manifestations result from defective sulfation of cartilage proteoglycans.
A number of mutant alleles have been found for the DTDST gene; they variably disturb transporter function. The disorders are recessive traits requiring the presence of 2 mutant alleles. The phenotype is determined by the combination of mutant alleles; some alleles are present in more than one disorder.
Diastrophic Dysplasia
Diastrophic dysplasia (OMIM 22600) is a well-characterized disorder recognized at birth by the presence of very short extremities, clubfoot, and short hands, with proximal displacement of the thumb producing a hitchhiker appearance (Fig. 688-1). The hands are usually deviated in an ulnar direction. Bony fusion of the metacarpophalangeal joints (symphalangism) is common, as is restricted movement of many joints, including hips, knees, and elbows. The external ears often become inflamed soon after birth. The inflammation resolves spontaneously but leaves the ears fibrotic and contracted (cauliflower ear deformity). Many newborns have a cleft palate.
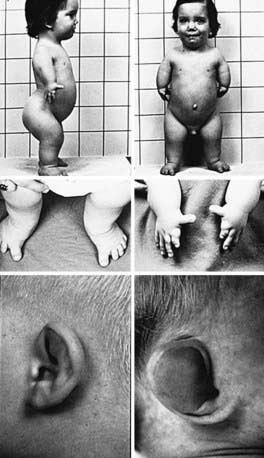
Figure 688-1 Child with diastrophic dysplasia. The extremities are dramatically shortened (top). Clubfoot is commonly observed (middle left). The fingers are short, especially the index finger; the thumb characteristically is proximally placed and has a hitchhiker appearance (middle right). The upper helix of the ears becomes swollen 3-4 wk postnatally (lower left), and this inflammation spontaneously resolves, leaving a cauliflower deformity of the pinnae (lower right).
Radiographs reveal short and broad tubular bones with flared metaphyses and flat, irregular epiphyses (Fig. 688-2).
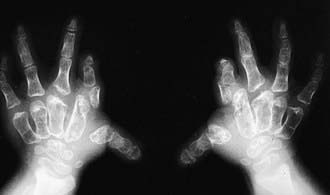
Figure 688-2 Radiograph of hands in diastrophic dysplasia. The metacarpals and phalanges are irregular and short. The first metacarpal is ovoid.
The capital femoral epiphyses are hypoplastic, and the femoral heads are broad. The ulnas and fibulas are disproportionately short. Carpal centers may be developmentally advanced; the first metacarpal is typically ovoid, and the metatarsals are twisted medially. There may be vertebral abnormalities, including clefts of cervical vertebral lamina and narrowing of the interpedicular distances in the lumbar spine.
Complications are primarily orthopedic and tend to be severe and progressive. The clubfoot deformity in the newborn resists usual treatments, and multiple corrective surgeries are common. Scoliosis typically develops during early childhood. It often requires multiple surgical procedures to control, and it sometimes compromises respiratory function in older children. Despite the orthopedic problems, patients typically have a normal life span and reach adult heights in the 105-130 cm range, depending on the severity of scoliosis. Growth curves are available for diastrophic dysplasia.
Some patients are mildly affected and exhibit slight short stature and joint contractures, no clubfoot or cleft palate, and correspondingly mild radiographic changes. The mild phenotype tends to recur within families. The recurrence risk of this autosomal recessive condition is 25%. Ultrasonographic examination can be employed for prenatal diagnosis, but if DTDST mutations can be identified in the patients or parents, molecular genetic diagnosis is possible.
Achondrogenesis Type 1b and Atelosteogenesis Type Ii
Achondrogenesis type 1B (OMIM 600972) and atelosteogenesis type II (OMIM 256050) are rare recessive lethal chondrodysplasias. The most serious is achondrogenesis type 1B, which demonstrates a severe lack of skeletal development usually detected in utero or after a miscarriage. The limbs are extremely short, and the head is soft. Skeletal radiographs show poor to missing ossification of skull bones, vertebral bodies, fibulas, and ankle bones. The pelvis is hypoplastic, and the ribs are short. The femurs are short and exhibit a trapezoid shape with irregular metaphyses.
Infants with atelosteogenesis type II are stillborn or die soon after birth; prematurity is common. They exhibit very short limbs, especially the proximal segments. Clubfoot and dislocations of the elbows and knees may be detected. Hypoplasia of vertebral bodies, especially in the cervical and lumbar spine, is found on radiographs. The femora and humeri are hypoplastic and display a club-shaped appearance. The distal limb bones, including the ulna and fibula, are poorly ossified.
Both disorders carry a 25% recurrence risk and are potentially detectable in utero by mutation analysis if the mutant alleles are identified in the parents. Prenatal diagnosis is possible with fetal imaging.
Hall BD. Diastrophic dysplasia: Extreme variability within a sibship. Am J Med Genet. 1996;63:28-33.
Makitie O, Kaitila I. Growth in diastrophic dysplasia. J Pediatr. 1997;130:641-646.
Newbury-Ecob R. Atelosteogenesis type 2. J Med Genet. 1998;35:49-53.
Rossi A, Superti-Furga A. Mutations in the diastrophic dysplasia sulfate transporter (DTDST) gene (SLC26A2): 22 novel mutations, mutation review, associated skeletal phenotypes, and diagnostic relevance. Hum Mutat. 2001;17:159-171.
Superti-Furga A. Skeletal dysplasias related to defects in sulfate metabolism. In: Royce PM, Steinmann B, editors. Connective tissue and its heritable disorders. New York: Wiley-Liss; 2002:939-960.