Chapter 691 Disorders for Which Defects Are Poorly Understood or Unknown
There are many chondrodysplasias, or chondrodysplasia clinical phenotypes, for which the genetic cause or basic mechanism is poorly understood or not known. Many illustrate features not found in other disorders and have historical significance in the evolution of chondrodysplasia nomenclature and classification.
Ellis–Van Creveld Syndrome
The Ellis-van Creveld syndrome (OMIM 225500), also known as chondroectodermal dysplasia, is a skeletal and an ectodermal dysplasia. The skeletal dysplasia presents at birth with short limbs, especially the middle and distal segments, accompanied by postaxial polydactyly of the hands and sometimes of the feet. Nail dysplasia and dental anomalies (including neonatal, absent, and premature loss of teeth and upper lip defects) constitute the ectodermal dysplasia. Common manifestations also include atrial septal defects and other congenital heart defects.
Skeletal radiographs reveal short tubular bones with clubbed ends, especially the proximal tibia and ulna (Fig. 691-1). Carpal bones display extra ossification centers and fusion; cone-shaped epiphyses are evident in the hands. A bony spur is often noted above the medial aspect of the acetabulum.

Figure 691-1 Radiograph of lower extremities in Ellis-van Creveld syndrome. Tubular bones are short, and proximal fibula is short. Ossification is retarded in lateral tibia epiphyses, causing a knock-knee deformity.
Ellis-van Creveld syndrome is an autosomal recessive trait that occurs most often in the Amish. Mutations have been identified in one of two genes, EVC (EVC1) and EVC2, which map in a head-to-head configuration to chromosome 4p. The functions of the gene products are unknown. About 30% of patients die of cardiac or respiratory problems during infancy. Life span is otherwise normal; adult heights range from 119 to 161 cm.
Asphyxiating Thoracic Dystrophy
Asphyxiating thoracic dystrophy (OMIM 208500), or Jeune syndrome, is an autosomal recessive chondrodysplasia that resembles Ellis-van Creveld syndrome. Newborn infants present with a long, narrow thorax and respiratory insufficiency associated with pulmonary hypoplasia. Neonates often die. Other neonatal manifestations include slightly short limbs and postaxial polydactyly. Mutations have been identified in the gene encoding cytoplasmic dynein 2 heavy chain 1 (DYNC2H1), which maps to chromosome 11q14.3-q23.1.
Skeletal radiographs show very short ribs with anterior expansion. Tubular limb bones are short with bulbous ends; cone-shaped epiphyses occur in hand bones. The iliac bones are short and square with a spur above the medial aspect of the acetabulum.
If infants survive the neonatal period, respiratory function usually improves as the rib cage grows. Surgery that produces lateral thoracic expansion improves rib growth and enhances chest wall dimensions. Progressive renal dysfunction often develops during childhood. Intestinal malabsorption and hepatic dysfunction have also been reported.
Short-Rib Polydactyly Syndromes
Four types of short-rib polydactyly syndrome (types I-IV) (OMIM 263530, 263520, 263510, 269860) have been described. All are lethal in the newborn period. Neonates present with respiratory distress, an extremely small thorax, very short extremities, polydactyly, and a variety of nonskeletal defects. Radiographs demonstrate very short ribs and tubular bones with changes characteristic for each type. All four types are autosomal recessive traits. Mutations of DYNC2H1 have been detected in short-rib polydactyly syndrome type III, making it allelic to asphyxiating thoracic dystrophy.
Cartilage-Hair Hypoplasia
Cartilage-hair hypoplasia (CHH) (OMIM 250250) is also known as metaphyseal chondrodysplasia—McKusick type. It is recognized during the 2nd year because of growth deficiency affecting the limbs, accompanied by flaring of the lower rib cage, a prominent sternum, and bowing of the legs. The hands and feet are short, and the fingers are very short with extreme ligamentous laxity. The hair is thin, sparse, and light colored, and nails are hypoplastic. The skin is hypopigmented.
Radiographs show short tubular bones with flared, irregularly mineralized, and cupped metaphyses (Fig. 691-2). The knees are more affected than are the hips, and the fibula is disproportionately longer than the tibia. The metacarpals and phalanges are short and broad. Spinal radiographs reveal mild platyspondyly.
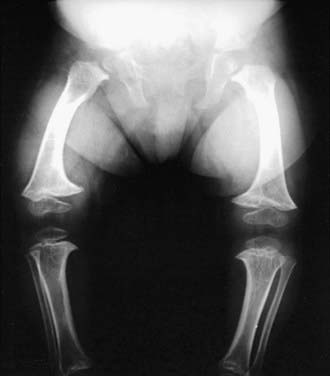
Figure 691-2 Radiograph of lower extremities in cartilage-hair hypoplasia. The tubular bones are short and the metaphyses are flared and irregular. The fibula is disproportionately long compared with the tibia. The femoral necks are short.
Nonskeletal manifestations associated with CHH include immunodeficiency (T-cell abnormalities, neutropenia, leukopenia, and susceptibility to chickenpox; children also may have complications from smallpox and polio vaccinations), malabsorption, celiac disease, and Hirschsprung disease. Adults are at risk for malignancy, especially non-Hodgkin lymphoma and skin tumors. Adults reach heights of 107-157 cm.
CHH shows autosomal recessive inheritance. Although rare, its highest prevalence is in the Amish and Finnish populations. It results from mutations of a gene coding for a large untranslated RNA component of an enzyme complex involved in processing mitochondrial RNA (RMRP). Loss of this gene product interferes with processing of both mRNA and rRNA. Loss of rRNA processing correlates with the extent of bone dysplasia, whereas loss of mRNA processing correlates with degree of hair hypoplasia, immunodeficiency, and hematologic abnormality. RMRP mutations are occasionally detected in patients with mild metaphyseal dysplasia lacking the extraskeletal features characteristic of CHH. Prenatal diagnosis is available if the mutation is identified either in the patient or parents.
Metatropic Dysplasia
There are two recognized forms of metatropic dysplasia (OMIM 156530, 250600), an autosomal dominant and an autosomal recessive form, although the existence of a recessive form has been questioned. Heterozygous mutations of TRPV4 (transient receptor potential vanilloid family 4), which encodes a calcium-permeable cation channel, have been identified in the dominant form.
Newborn infants present with a long narrow trunk and short extremities. A tail-like appendage sometimes extends from the base of the spine. Odontoid hypoplasia is common and may be associated with cervical instability. Kyphoscoliosis appears in late infancy and progresses through childhood, often becoming severe enough to compromise cardiopulmonary function. The joints are large and become progressively restricted in mobility, except in the hands. Contractures often develop in the hips and knees during childhood. Although severely affected infants can die at a young age from respiratory failure, patients usually survive, although they can become disabled as adults from the progressive musculoskeletal deformities. Adult heights range from 110 to 120 cm.
Skeletal radiographs show characteristic changes dominated by severe platyspondyly and short tubular bones with expanded and deformed metaphyses that exhibit a dumbbell appearance (Fig. 691-3). The pelvic bones are hypoplastic and exhibit a halberd appearance because of a small sacrosciatic notch and a notch above the lateral margin of the acetabulum.
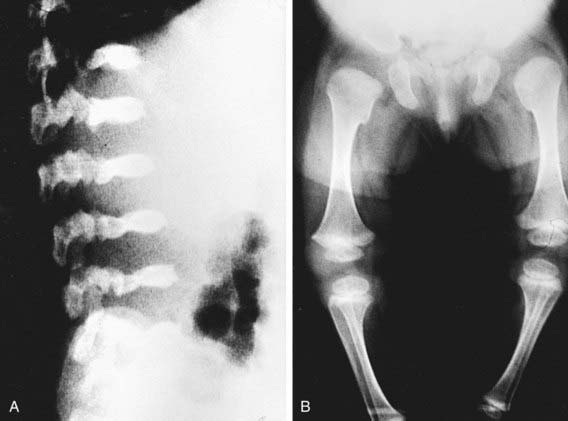
Spondylometaphyseal Dysplasia, Kozlowski Type
The Kozlowski type of spondylometaphyseal dysplasia (SMDK) (OMIM 184252) is an allelic disorder to autosomal dominant metatropic dysplasia as mutations of TRPV4 have been detected. Mutations of TRPV4 have also been identified in autosomal dominant brachyolmia, whose phenotype is dominated by progressive scoliosis and platyspondyly on x-rays.
SMDK manifests in early childhood with mild short stature involving mostly the trunk and a waddling gait. The hands and feet may be short and stubby. Radiographs show flattening of vertebral bodies. The metaphyses of tubular bones are widened and irregularly mineralized, especially at the proximal femur. The pelvic bones manifest mild hypoplasia.
Scoliosis can develop during adolescence. The disorder is otherwise uncomplicated, and manifestations are limited to the skeleton. Adults reach heights of 130-150 cm. The Kozlowski type of spondylometaphyseal dysplasia is an autosomal dominant trait.
Disorders Involving Filamins
Mutations of genes encoding filamin A and filamin B proteins have been detected in diverse disorders of skeletal development: filamin A mutations in otopalatodigital syndromes type 1 and 2 frontometaphyseal dysplasia and Melnick-Needles syndrome (OMIM 311300, 304120, 305620, 309350) and filamin B mutations in Larsen syndrome and perinatal lethal ateosteogenesis types I and III (OMIM 150250, 108720, 108721). Filamins functionally connect extracellular to intracellular structural proteins, thereby linking cells to their local microenvironment, which is essential for skeletal development and growth.
Juvenile Osteochondroses
The juvenile osteochondroses are a heterogeneous group of disorders in which regional disturbances in bone growth cause noninflammatory arthropathies. They are summarized in Table 691-1. Some have localized pain and tenderness (Freiberg disease, Osgood-Schlatter disease, osteochondritis dissecans), whereas others present with painless limitation of joint movement (Legg-Calvé-Perthes disease, Scheuermann disease). Bone growth may be disrupted, leading to deformities. The diagnosis is usually confirmed radiographically, and treatment is symptomatic. The pathogenesis of these disorders is believed to involve ischemic necrosis of primary and secondary ossification centers. Although familial forms have been reported, these disorders usually occur sporadically.
TABLE 691-1 JUVENILE OSTEOCHONDROSES
| EPONYM | AFFECTED REGION | AGE AT PRESENTATION |
|---|---|---|
| Legg-Calvé-Perthes disease | Capital femoral epiphysis | 3-12 yr |
| Osgood-Schlatter disease | Tibial tubercle | 10-16 yr |
| Sever disease | Os calcaneus | 6-10 yr |
| Freiberg disease | Head of second metatarsal | 10-14 yr |
| Scheuermann disease | Vertebral bodies | Adolescence |
| Blount disease | Medial aspect of proximal tibial epiphysis | Infancy or adolescence |
| Osteochondritis dissecans | Subchondral regions of knee, hip, elbow, and ankle | Adolescence |
Caffey Disease (Infantile Cortical Hyperostosis)
This is a rare disorder of unknown etiology characterized by cortical hyperostosis with inflammation of the contiguous fascia and muscle. It is often sporadic, but both autosomal dominant and autosomal recessive inheritance have been reported. In three unrelated families with autosomal dominant inheritance, a linkage to mutations of the COL1A1 gene (codes for the α1 chain of type I collagen) has been reported.
Prenatal and more often postnatal onset have been described. Prenatal onset may be mild (autosomal dominant) or severe (autosomal recessive). Severe prenatal disease is characterized by typical bone lesions, polyhydramnios, hydrops fetalis, severe respiratory distress, prematurity, and high mortality. Onset in infancy (<6 mo; average, 10 wk) is most common; manifestations include the sudden onset of irritability, swelling of contiguous soft tissue that precedes the cortical thickening of the underlying bones, fever, and anorexia. The swelling is painful with a woodlike induration but with minimal warmth or redness; suppuration is absent. There are unpredictable remissions and relapses; an episode can last 2 wk to 3 mo. The most common bones involved include the mandible (75%) (Fig. 691-4), the clavicle, and the ulna. If swelling is not prominent or visible, the diagnosis might not be evident.
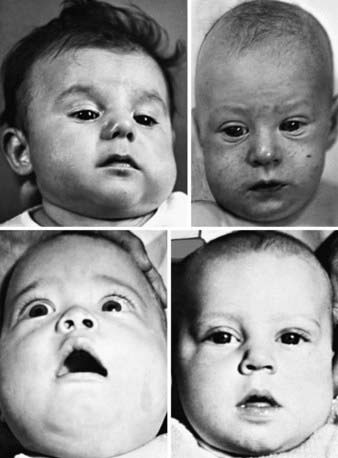
Figure 691-4 Facies in infantile cortical hyperostosis. In almost all cases, the changes have appeared before the 5th month of life. Unilateral swelling of the left cheek and left side of the jaw in an infant 12 wk of age.
(From Kuhn JP, Slovis TL, Haller JO: Caffey’s pediatric diagnostic imaging, ed 10, vol 2, Philadelphia, 2004, Mosby.)
Laboratory features include elevated erythrocyte sedimentation rate and serum alkaline phosphatase as well as, in some patients, increased serum prostaglandin E levels. There may be thrombocytosis and anemia. The radiographic features include soft-tissue swelling and calcification and cortical hyperostosis (Fig. 691-5). All bones may be affected except the phalanges or vertebral bodies. The differential diagnosis includes other causes of hyperostosis such as chronic vitamin A intoxication, prolonged prostaglandin E infusion in children with ductal dependent congenital heart disease, primary bone tumors, and scurvy.

Figure 691-5 Residual bony bridges between each radius and ulna in infantile cortical hyperostosis. A, Massive cortical thickenings of the radii and ulnas at  mo of age. Pressure from the external thickenings has forced the radial heads laterad out of the elbows. B, At
mo of age. Pressure from the external thickenings has forced the radial heads laterad out of the elbows. B, At 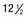 mo, 9 mo after onset, all affected bones are still greatly swollen, owing largely to expansion of the medullary cavities, although there are still residues of the earlier cortical thickening. The radial heads are still dislocated, and the radial diaphyses are now anchored in this ectopic position by solid bony bridges between them and the ulnar diaphyses, a single bridge on the right and three on the left. At 32 mo, these bridges were still intact, although they had diminished slightly in caliber. It is possible that these bony bridges represent ossification of parts of the interosseous membrane.
mo, 9 mo after onset, all affected bones are still greatly swollen, owing largely to expansion of the medullary cavities, although there are still residues of the earlier cortical thickening. The radial heads are still dislocated, and the radial diaphyses are now anchored in this ectopic position by solid bony bridges between them and the ulnar diaphyses, a single bridge on the right and three on the left. At 32 mo, these bridges were still intact, although they had diminished slightly in caliber. It is possible that these bony bridges represent ossification of parts of the interosseous membrane.
(From Kuhn JP, Slovis TL, Haller JO: Caffey’s pediatric diagnostic imaging, ed 10, vol 2, Philadelphia, 2004, Mosby.)
Complications are unusual but include pseudoparalysis with limb or scapula involvement, pleural effusions (rib), torticollis (clavicle), mandibular asymmetry, bone fusion (ribs or ulna and radius), and bone angulation deformities (common with severe prenatal onset).
Treatment includes indomethacin and prednisone if there is a poor response to indomethacin.
Baujat G, Le Merrer M. Ellis-van Creveld syndrome. Orphanet J Rare Dis. 2007;2:27.
Dagoneau N, Goulet M, Genevieve D, et al. DYNC2H1 mutations cause asphyxiating thoracic dystrophy and short rib-polydactyly syndrome, type III. Am J Hum Genet. 2009;84:706-711.
de Vries J, Yntema JL, van Die CE, et al. Jeune syndrome: description of 13 cases and a proposal for follow-up protocol. Eur J Pediatr. 2010;169(1):77-88.
Genevieve D, Le Merrer M, Feingold J, et al. Revisiting metatropic dysplasia: presentation of a series of 19 novel patients and review of the literature. Am J Med Genet A. 2008;146:992-996.
Glorieux FH. Caffey disease: an unlikely collagenopathy. J Clin Invest. 2005;115:1142-1144.
Kamoun-Goldrat A, le Merrer M. Infantile cortical hyperostosis (Caffey disease): a review. J Oral Maxillofac Surg. 2008;66:2145-2150.
Krakow D, Robertson SP, King LM, et al. Mutations in the gene encoding filamin b disrupt vertebral segmentation, joint formation and skeletogenesis. Nat Genet. 2004;36:405-410.
Makitie O, Sulisalo T, de la Chapelle A, et al. Cartilage-hair hypoplasia. J Med Genet. 1995;32:39-43.
Restrepo S, Sanchez AM, Palacios E. Infantile cortical hyperostosis of the mandible. Ear Nose Throat J. 2004;83:454-455.
Ruiz-Perez VL, Ide SE, Strom TM, et al. Mutations in a new gene in Ellis-van Creveld syndrome and weyers acrodental dysostosis. Nat Genet. 2000;24:283-286.
Sharrard WJW. Abnormalities of the epiphyses and limb inequality. In: Sharrard WJW, editor. Paediatric orthopaedics and fracture. Oxford, UK: Blackwell Scientific Publications, 1993.
Taskinen M, Ranki A, Pukkala E, et al. Extended follow-up of the Finnish cartilage-hair hypoplasia cohort confirms high incidence of non-Hodgkin lymphoma and basal cell carcinoma. Am J Med Genet A. 2008;146:2370-2375.
Thiel CT, Mortier G, Kaitila I, et al. Type and level of rmrp functional impairment predicts phenotype in the cartilage hair hypoplasia-anauxetic dysplasia spectrum. Am J Hum Genet. 2007;81:519-529.