2: Cell Injury, Cell Death, and Adaptations
Introduction to Pathology (p. 33)
Pathology is the study of the structural, biochemical, and functional causes of human disease.
Pathology is traditionally divided into general and systemic pathology. General pathology focuses on the typical reactions of cells and tissues anywhere in the body in response to injury; systemic pathology examines the alterations and underlying mechanisms in diseases of particular organ systems. The first nine chapters will describe principles of general pathology and the subsequent chapters will involve specific disease processes as they affect different organs.
Four aspects of a disease process form the core of pathology:
Overview of Cellular Responses to Stress and Noxious Stimuli (p. 34)
Normal cell function requires a balance between physiologic demands and the constraints of cell structure and metabolic capacity; the result is a steady state, or homeostasis. Cells can fine-tune their functional state in response to modest stress to maintain the steady state. More excessive physiologic stresses, or adverse pathologic stimuli (injury), result in (1) adaptation, (2) reversible injury, or (3) irreversible injury and cell death (Fig. 2.1).
- • Adaptation occurs when physiologic or pathologic stressors induce a new state that changes the cell but otherwise preserves its viability in the face of the exogenous stimuli. These changes are reversible (if the stressors recede) and include:
- • Cell injury occurs when damaging insults, nutrient withdrawal, or mutations that compromise cellular functions exceed adaptive responses (see also following).
-
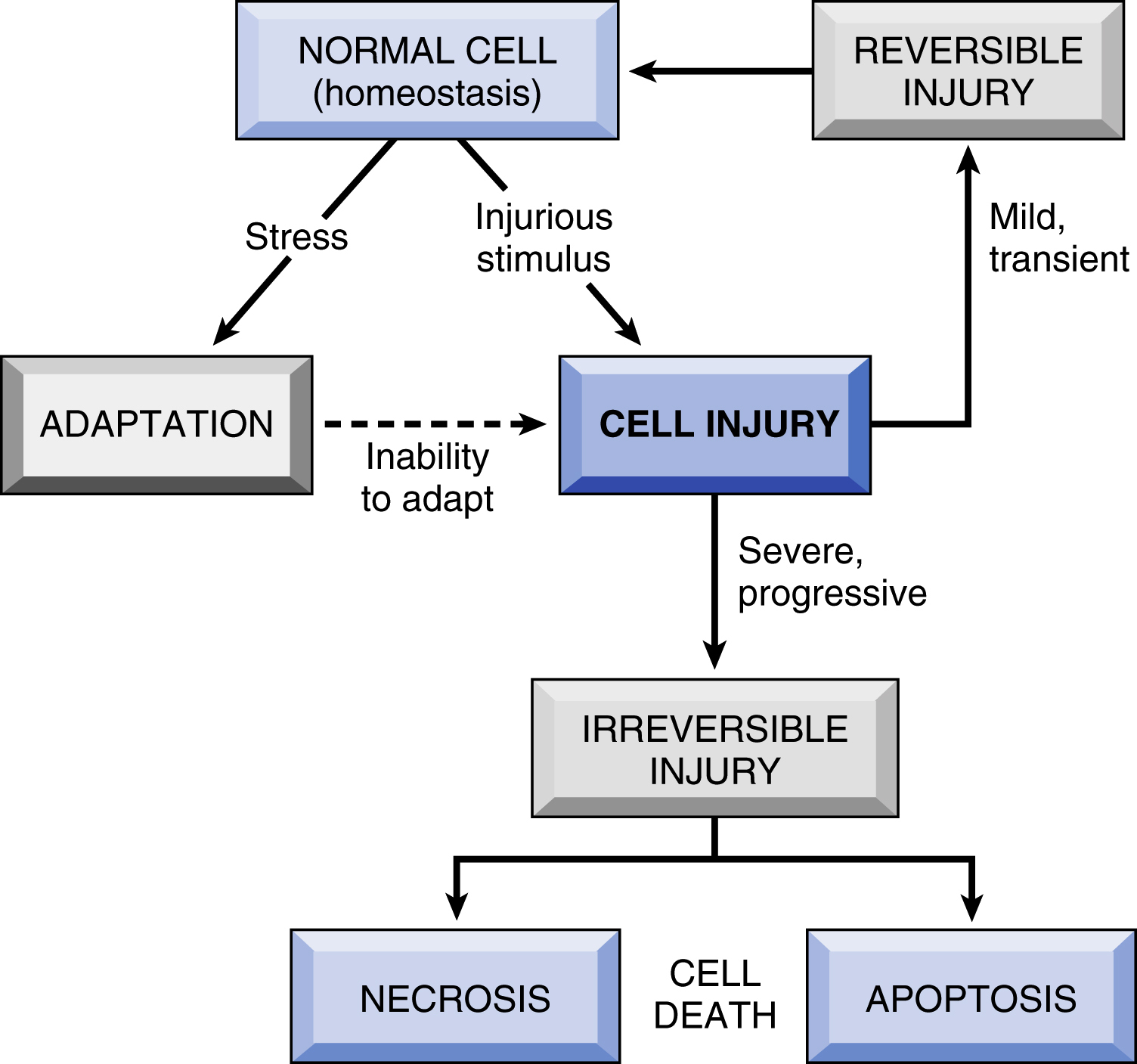
Fig. 2.1 Stages of the cellular response to stress and injurious stimuli. - • Reversible injury denotes pathologic cell changes that can be restored to normalcy if the stimulus is removed or if the cause of injury is mild.
- • Irreversible injury occurs when stressors exceed the capacity of the cell to adapt (beyond a point of no return) causing pathologic changes that lead to cell death.
- • Cell death occurs primarily through two morphologic and mechanistic patterns, necrosis and apoptosis (Table 2.1 and Fig. 2.2). Although necrosis always represents a pathologic process, apoptosis may also occur in several normal physiologic situations (e.g., in embryogenesis) and is not necessarily associated with cell injury. Nutrient deprivation triggers an adaptive cellular response called autophagy, that can also culminate in cell death.
Causes of Cell Injury (p. 36)
- • Oxygen deprivation (hypoxia) affects aerobic respiration and therefore the ability to generate adenosine triphosphate (ATP). Depending on the severity of the hypoxia, cells may either adapt (e.g., atrophy) or experience injury and die. Hypoxia occurs due to:
- • Physical agents, including trauma, heat, cold, radiation, and electric shock (see Chapter 9)
- • Chemical agents and drugs, including therapeutic drugs, poisons, environmental pollutants, and “social stimuli” (alcohol and narcotics) (see Chapter 9)
-
Features of Necrosis and Apoptosis Feature Necrosis Apoptosis Cell size Enlarged (swelling) Reduced (shrinkage) Nucleus Pyknosis, karyorrhexis, karyolysis Fragmentation into nucleosome-size fragments Plasma membrane Disrupted Intact; altered structure, especially orientation of lipids Cellular contents Enzymatic digestion; may leak out of cell Intact; may be released in apoptotic bodies Adjacent inflammation Frequent No Physiologic or pathologic role Usually pathologic (culmination of irreversible cell injury) Often physiologic, means of eliminating unwanted cells; may be pathologic after some forms of cell injury, especially DNA damage - • Infectious agents, including viruses, bacteria, fungi, and parasites (see Chapter 8)
- • Immunologic reactions, including autoimmune diseases (see Chapter 6) and cell injury following responses to infection (see Chapter 3)
- • Genetic derangements, such as chromosomal alterations and specific gene mutations (see Chapter 5)
- • Nutritional imbalances, including protein-calorie deficiency or lack of specific vitamins, as well as nutritional excesses (see Chapter 9)
The Progression of Cell Injury and Death (p. 36)
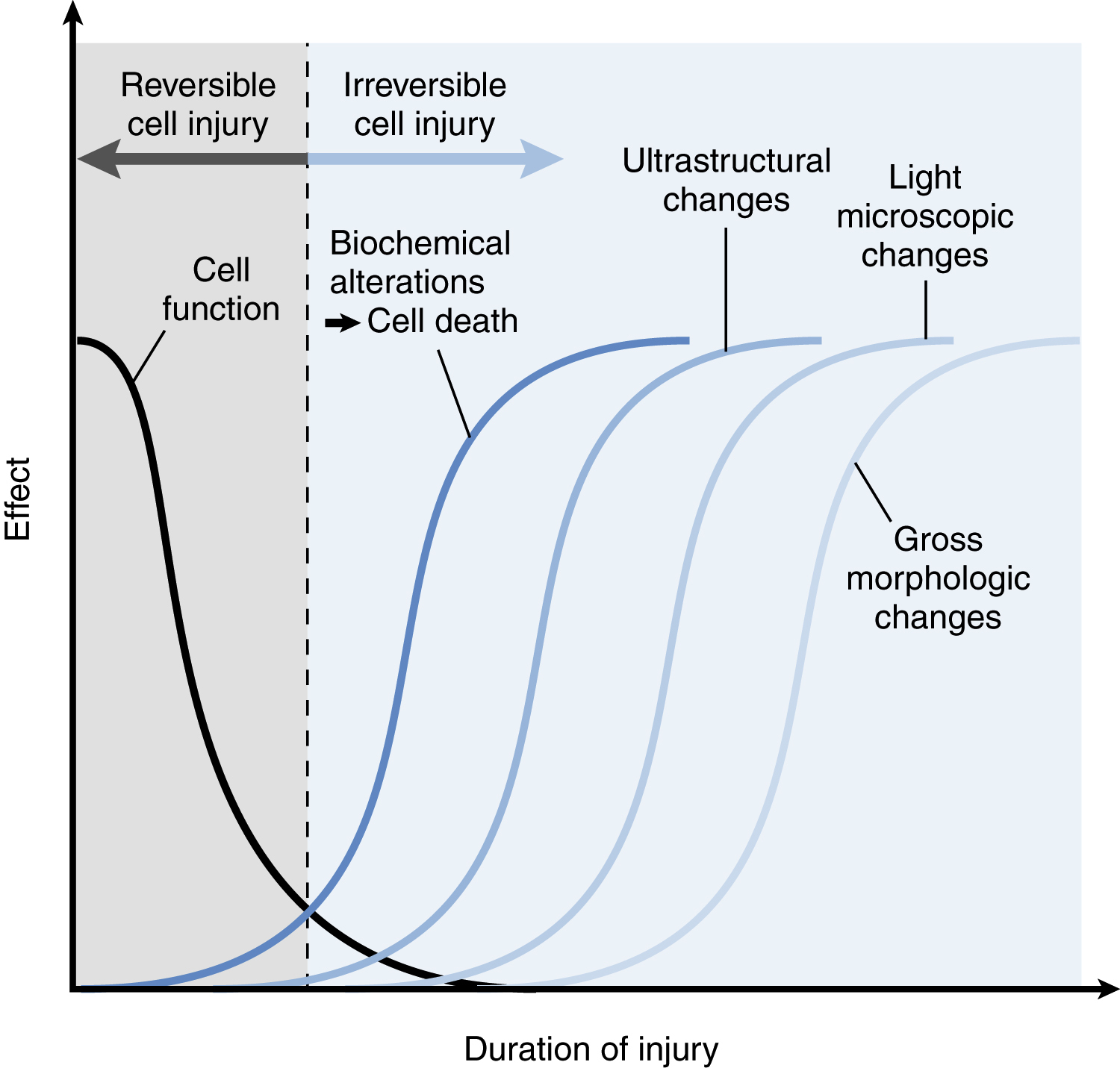
Injury leads to loss of cell function long before damage is morphologically recognizable. Morphologic changes become apparent only sometime after a critical biochemical system within the cell has been deranged; the interval between injury and morphologic change depends on the method of detection (Fig. 2.3).
Reversible Cell Injury (p. 37)
- • Cell swelling appears whenever cells cannot maintain ionic and fluid homeostasis. This is usually caused by failure of the adenosine triphosphate (ATP)-dependent Na+-K+ plasma membrane pump due to depletion of ATP due to hypoxia or mitochondrial damage or dysfunction.
- • Fatty change is manifested by cytoplasmic lipid vacuoles, principally encountered in cells involved in or dependent on fat metabolism (e.g., hepatocytes and myocardial cells).
Cell Death (p. 37)
There are two principal types of cell death, necrosis and apoptosis (Table 2.1, Fig. 2.2
). Necrosis is traditionally described as “accidental” cell death, reflecting severe injury and irreparable damage; necrosis also elicits an inflammatory response that clears the “scene
of the accident.” In contrast, apoptosis is “regulated” cell death without inflammation. While it is useful to describe these principal mechanism (and histologic features) separately, the differences are not always so distinct; some forms of necrosis are genetically controlled through a defined molecular pathway, called “necroptosis” (discussed later), and in some cases, dying cells may show morphologic features of both apoptosis and necrosis.
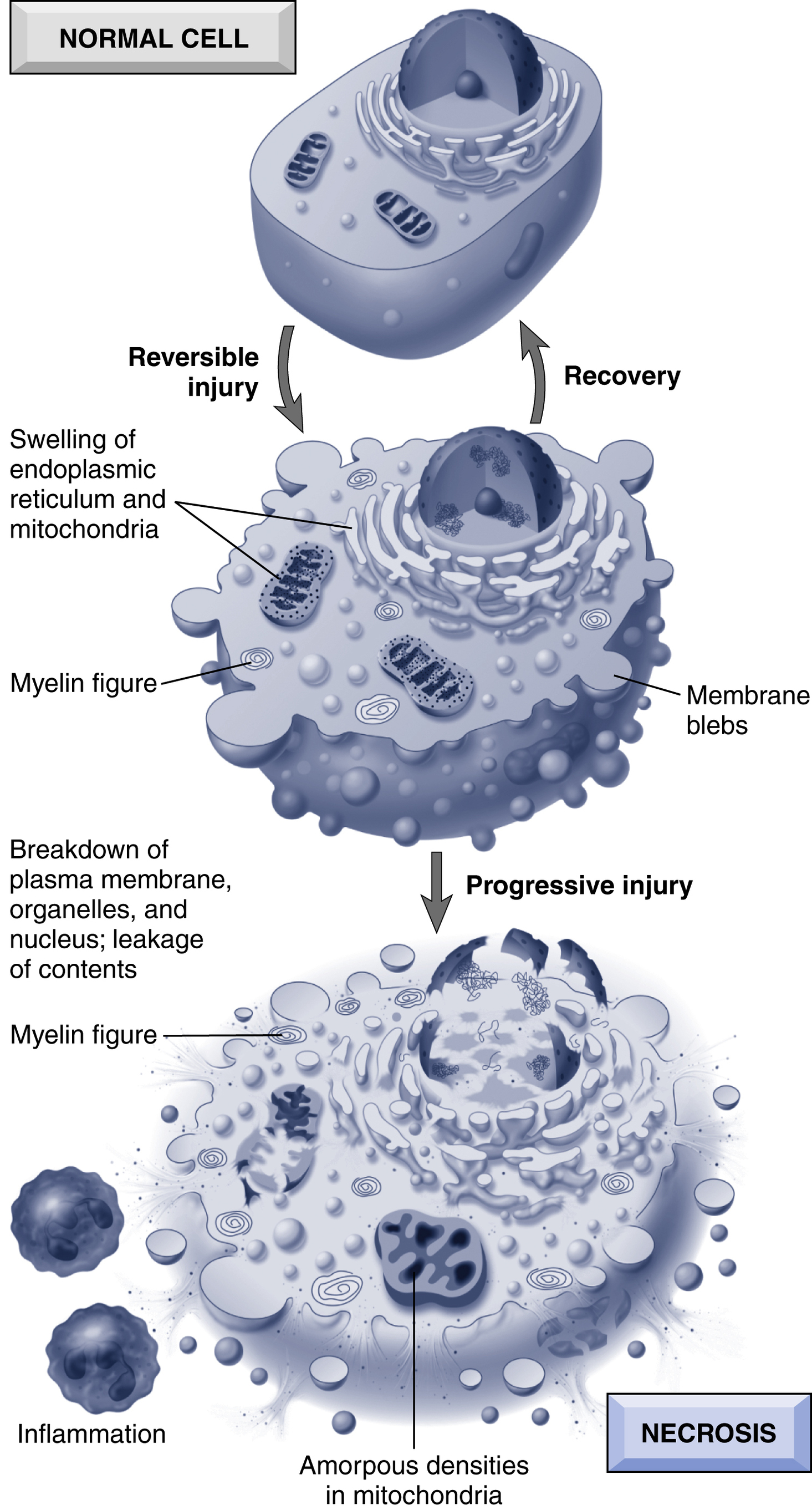
Necrosis (p. 39)
Necrosis is the consequence of severe irreversible injury; it is characterized by:
Some of the leaked cell contents comprise damage-associated molecular patterns (DAMPs); these include ATP and uric acid from nucleotide catabolism. The DAMPs bind to specific receptors on cells of innate immunity (e.g., macrophages), triggering debris phagocytosis, as well as cytokine production that elicits a secondary inflammatory response. The leaked proteins also include intracellular molecules that can be detected in the blood and used as markers of tissue-specific injury.
Morphologically, necrotic cells have lost basophilic nucleic acids, and have more denatured cytoplasmic proteins, making them more eosinophilic (pink) than viable cells by standard hematoxylin and eosin (H&E) staining. They appear “glassy,” owing to glycogen loss, and may be vacuolated due to organellar degradation; cell membranes are fragmented. Necrotic cells may attract calcium salts; this is particularly true of necrotic fat cells (forming fatty soaps). Nuclear changes secondary to DNA breakdown include pyknosis (small, dense nucleus), karyolysis (faint, dissolved nucleus), and karyorrhexis (fragmented nucleus).
Patterns of Tissue Necrosis (p. 40)
- • Coagulative necrosis (p. 40) is the most common pattern, predominated by protein denaturation with preservation of the cell and tissue framework. This pattern is characteristic of ischemic/hypoxic death in all tissues except the brain (see as follows). Necrotic tissue undergoes either heterolysis (digestion by lysosomal enzymes of invading leukocytes) or autolysis (digestion by its own lysosomal enzymes). A localized area of coagulative necrosis is called an infarct.
- • Liquefactive necrosis (p. 40) occurs when autolysis or heterolysis predominates over protein denaturation. The necrotic area is soft and filled with fluid. This type of necrosis is most frequently seen in localized bacterial infections (abscesses) and in the brain.
- • Gangrenous necrosis (p. 41) is not a specific pattern but is rather just coagulative necrosis, typically as applied to an ischemic limb; superimposed bacterial infection makes for a more liquefactive pattern called wet gangrene.
- • Caseous necrosis (p. 41) is characteristic of tuberculous lesions (Chapter 8); it appears grossly as soft, friable, “cheesy” material and microscopically as amorphous eosinophilic material with cell debris, usually associated with collections of activated macrophages (granuloma).
- • Fat necrosis (p. 41) is seen in adipose tissue; lipase activation (e.g., from injured pancreatic cells or macrophages) releases fatty acids from triglycerides, which then complex with calcium to create soaps. Grossly these are white, chalky areas (fat saponification); histologically there are vague cell outlines, with basophilic calcium deposition, and a variable inflammatory response.
- • Fibrinoid necrosis (p. 41 and Chapter 11) is a pathologic pattern due to antigen-antibody (immune complex) deposition in blood vessels. Microscopically there is bright pink amorphous material (protein deposition) in arterial walls, often with associated inflammation and thrombosis.
Apoptosis (p. 42)
Apoptosis occurs when a cell dies through activation of a tightly regulated internal suicide program controlled by the action of a small number of genes. The function of apoptosis is to selectively eliminate unwanted cells, with minimal disturbance to surrounding cells and tissues. Apoptosis is a generic term for this pattern of cell death, while programmed cell death refers only to apoptosis occurring during normal development.
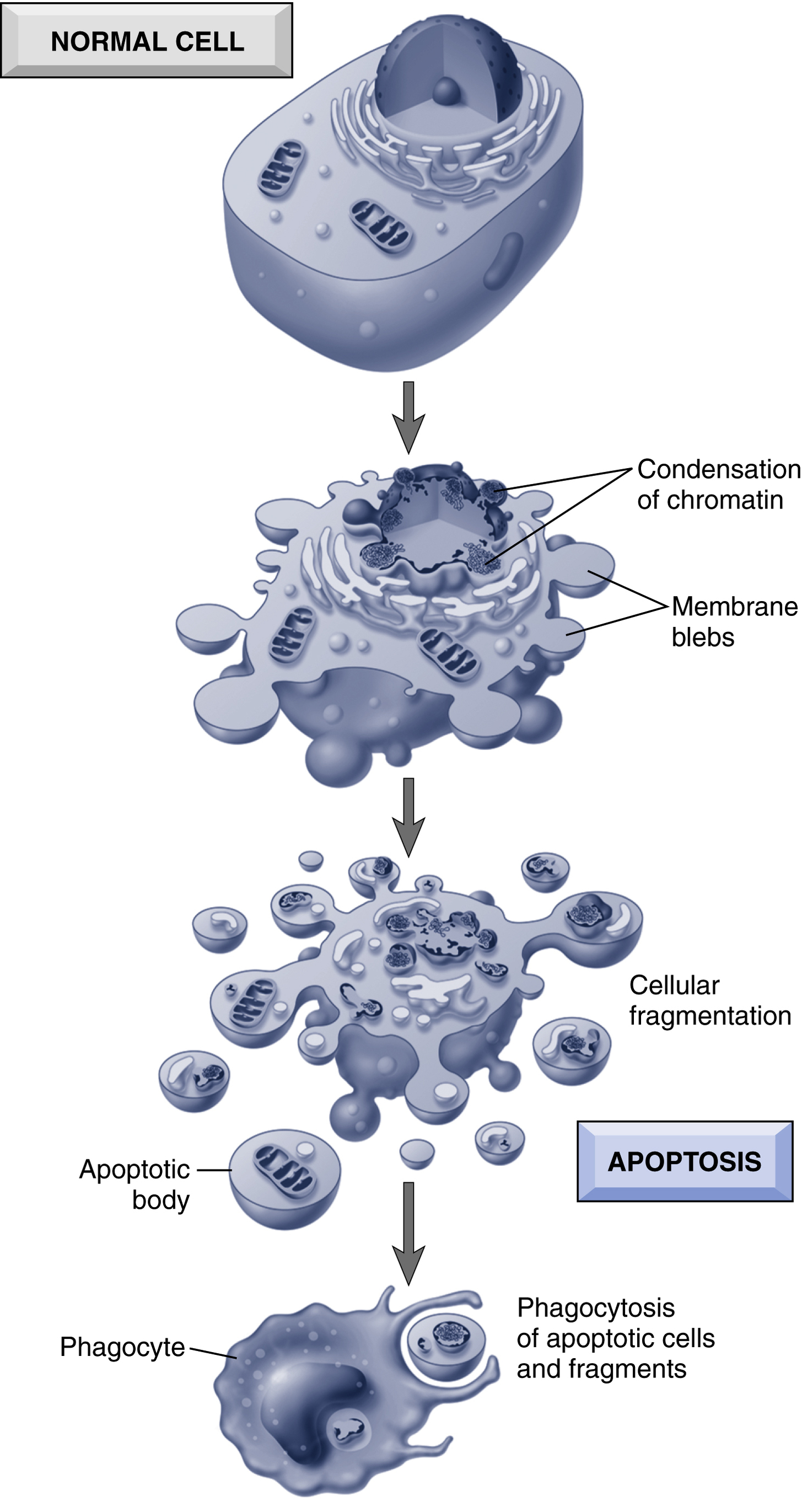
In apoptosis, the plasma membrane remains intact, but its structure is altered so that the cell fragments into smaller apoptotic bodies; those express “find me” and “eat me” signals so that they become avid targets for phagocytosis before any contents have leaked out. Importantly, cell death by this pathway also does not elicit an inflammatory response. Thus apoptosis is fundamentally different from necrosis, which is characterized by loss of membrane integrity, enzymatic digestion of cells, and frequently a host reaction (Fig. 2.2; Table 2.1). Nevertheless, apoptosis and necrosis sometimes coexist, and they may share some common features and mechanisms.
Causes of Apoptosis (p. 42)
Apoptosis can be physiologic or pathologic.
- Physiologic Causes (p. 42)—when cells are deprived of necessary survival signals (growth factors or matrix interactions) or receive proapoptotic signals.
-
- • Programmed cell death during embryogenesis
- • Hormone-dependent involution of tissues in the adult organisms (e.g., endometrium during the menstrual cycle, regression of the lactating breast after weaning)
- • Cell deletion in proliferating cell populations (e.g., intestinal epithelium) to maintain a constant cell number
- • Death of cells that have served their useful purpose (e.g., neutrophils following an acute inflammatory response)
- • Deletion of potentially harmful self-reactive lymphocyte
- Pathologic Causes (p. 42)—when cells are damaged beyond repair, but without eliciting collateral damage.
-
- • DNA damage (e.g., due to hypoxia, radiation, or cytotoxic drugs). If repair mechanisms cannot correct the damage, cells will undergo apoptosis rather than risk mutations that could result in malignant transformation.
- • Accumulation of misfolded proteins (e.g., due to inherited defects or due to free radical damage).
- • Cell death in certain viral infections (e.g., hepatitis), either caused directly by the infection or by cytotoxic T cells (CTL).
- • CTL may also be a cause of apoptotic cell death in tumors and in the rejection of transplanted tissues.
- • Pathologic atrophy in parenchymal organs after duct obstruction (e.g., pancreas).
Morphologic and Biochemical Changes in Apoptosis (p. 43)
Morphologic features of apoptosis (Table 2.1, Fig. 2.2) include cell shrinkage, chromatin condensation and fragmentation, cellular blebbing and fragmentation into apoptotic bodies, and phagocytosis of apoptotic bodies by adjacent healthy cells or macrophages. Lack of inflammation makes it difficult to detect apoptosis histologically.
Mechanisms of Apoptosis (p. 44) (Fig. 2.4)
Protein breakdown in apoptosis occurs through a family of proteases called caspases (they have an active site
c
ysteine and cleave at
asp
artate residues). Caspases are synthesized as inactive proenzymes and are activated by proteolytic cleavage; the demonstration of active caspases is a marker that identifies cells undergoing apoptosis.
Apoptosis is a cascade of molecular events that are initiated by a variety of triggers. The process of apoptosis is divided into an initiation phase, when caspases become active, and an execution phase, when the enzymes cause cell death. Initiation of apoptosis occurs through two distinct but converging pathways: the intrinsic mitochondrial pathway and the extrinsic death receptor-mediated pathway.
Intrinsic (Mitochondrial) Pathway of Apoptosis (p. 44) (Fig. 2.5)
The mitochondrial pathway is responsible for apoptosis in most physiologic and pathologic circumstances. Increased mitochondrial permeability leads to the release of cytochrome c (and other proapoptotic molecules) into the cytoplasm. Mitochondrial permeability is regulated by a host of BCL proteins—so-named because the first of the family was a protein product of the BCL2 gene which was overexpressed in certain B cell lymphomas. There are over 20 members of the BCL family, with three different roles:
- • Antiapoptotic. BCL2, BCL-XL, and MCL1 are the principal antiapoptotic proteins; they reside in the outer mitochondrial membrane and are responsible for maintaining membrane impermeability. As you might expect, the synthesis of these proteins is regulated; in many cases, their transcription relies on the presence of survival signals. They all have four BCL-2-homology (BH) domains.
-
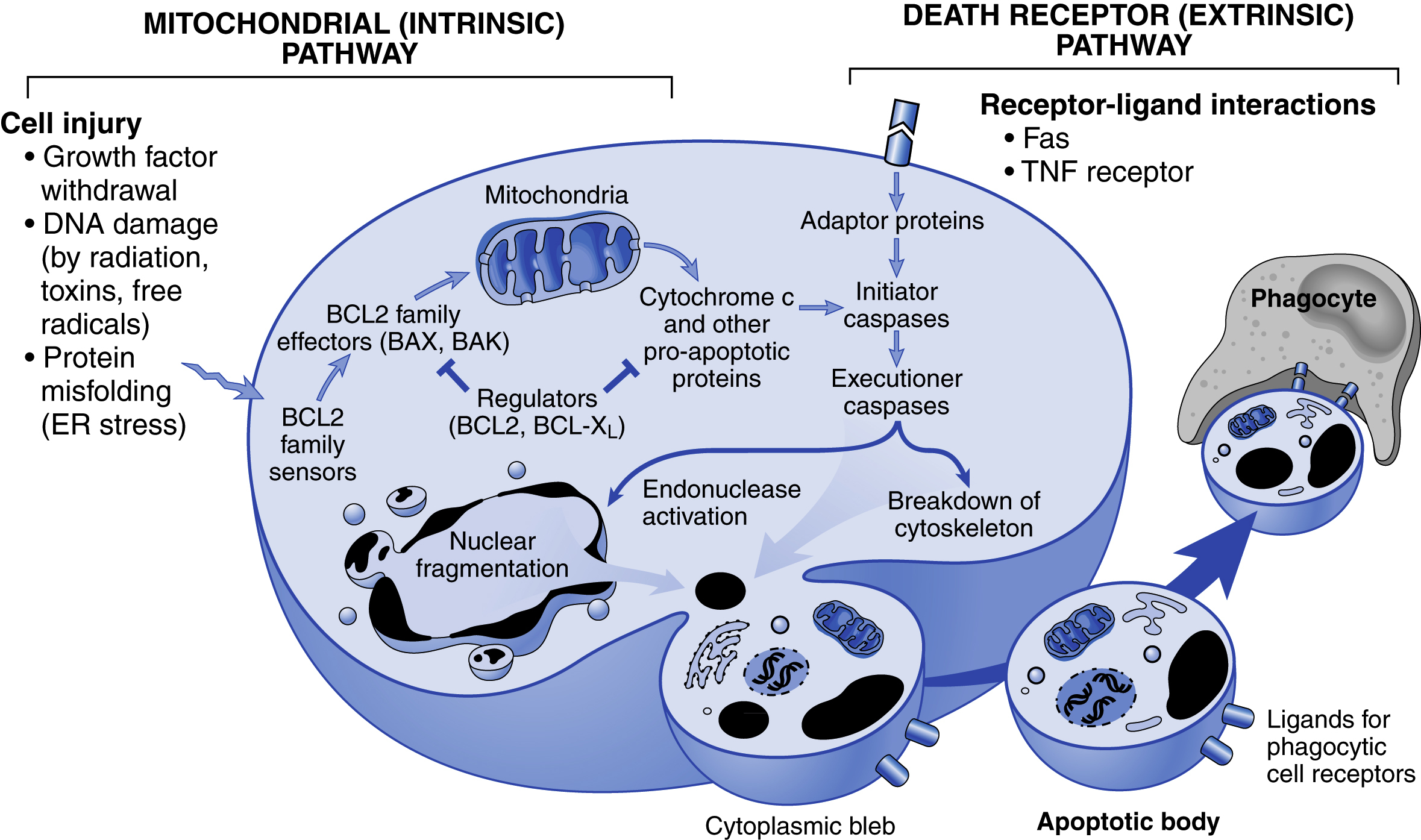
Fig. 2.4 Mechanisms of apoptosis.Although the two pathways of apoptosis differ in their induction and regulation, they both culminate in the activation of caspases. In the mitochondrial pathway, proteins of the BCL2 family, which regulate mitochondrial permeability, become imbalanced such that the ratio of proapoptotic versus antiapoptotic proteins results in the leakage of various substances from mitochondria that lead to caspase activation. In the death receptor pathway, signals from plasma membrane receptors lead to the assembly of adaptor proteins into a “death-inducing signaling complex,” which activates caspases, and the end result is the same. 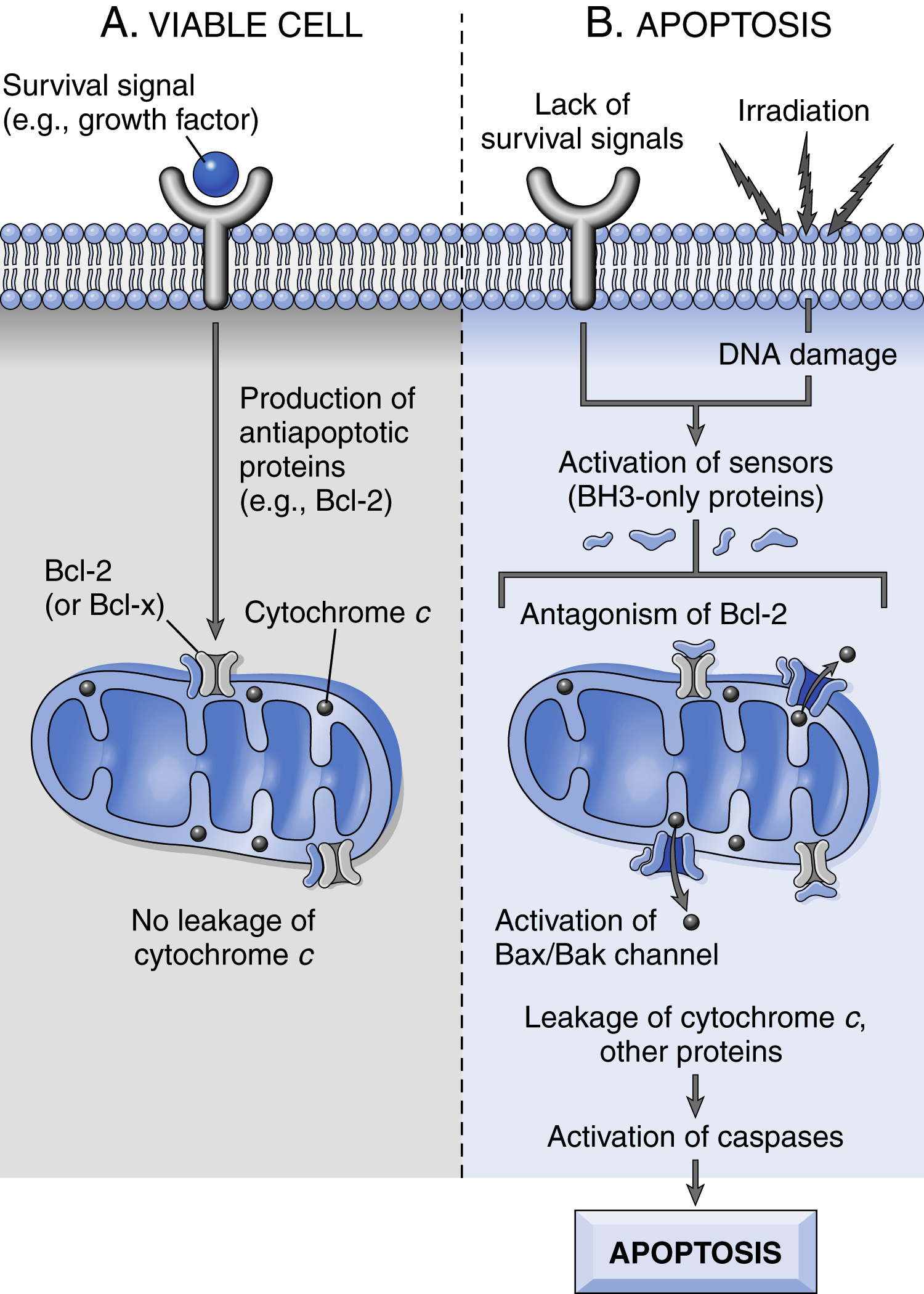
Fig. 2.5 The intrinsic (mitochondrial) pathway of apoptosis. A, Cell viability is maintained by the induction of antiapoptotic proteins such as BCL2 by survival signals. These proteins maintain the integrity of mitochondrial membranes and prevent leakage of mitochondrial proteins. B, Loss of survival signals, DNA damage, and other insults activate sensors that antagonize the antiapoptotic proteins and activate the proapoptotic proteins BAX and BAK, which form channels in the mitochondrial membrane. The subsequent leakage of cytochrome c (and other proteins, not shown) leads to caspase activation and apoptosis. - • Proapoptotic. BAX and BAK are the two prototypical proapoptotic proteins; these form oligomers that insert into the mitochondrial membrane and create permeability channels. They contain the first three (of four) BH domains (BH1–3).
- • Regulated apoptosis initiators. Cellular stressors (e.g., misfolded proteins, DNA damage) or loss of survival signals is sensed by these BCL members (e.g., BIM, BID, and BAD); when upregulated and/or activated (e.g., by phosphorylation) they can drive apoptosis. These have a single BH domain, the third of the four, and are therefore also called BH3-only proteins. BH3-only proteins also bind to and block the antiapoptotic activities of BCL2, etc.
The net result of Bax-Bak activation coupled with declining Bcl-2/Bcl-x levels is an increased mitochondrial membrane permeability, leaking out several proteins that can activate caspases and/or neutralize cytoplasmic proteins that normally inhibit apoptosis. Thus released cytochrome c binds to apoptosis activating factor-1 (Apaf-1) to form a large multimeric apoptosome complex that triggers the
autocatalytic cleavage activation of caspase-9 (an initiator caspase). This, in turn, leads to a cascade of proteolytic cleavage activation of other procaspases (e.g., caspase-3), eventually culminating in cell death. Other mitochondrial proteins such as Smac/DIABLO neutralize the physiologic inhibitors of apoptosis (IAPs). The essence of the intrinsic pathway is a balance between pro- and antiapoptotic molecules that regulate mitochondrial permeability.
Extrinsic (Death Receptor-Initiated) Pathway of Apoptosis (p. 45)
This pathway is initiated by engagement of plasma membrane death receptors. These are members of the tumor necrosis factor (TNF) receptor family (e.g., type 1 TNF receptor and Fas), with a cytoplasmic death domain involved in protein-protein interactions. Cross-linking of these receptors by external ligands, such as TNF or Fas ligand (FasL) causes them to trimerize to form binding sites for adapter proteins (Fas-associated death domain or FADD). FADD serves to bring multiple inactive caspase-8 molecules into close proximity, culminating in their autocatalytic cleavage and activation, and initiating the same executioner caspase cascade as for the intrinsic pathway (Fig. 2.6). This enzymatic pathway can be blocked by a protein called FLIP; viruses and normal cells can produce FLIP to protect themselves against Fas-mediated death.
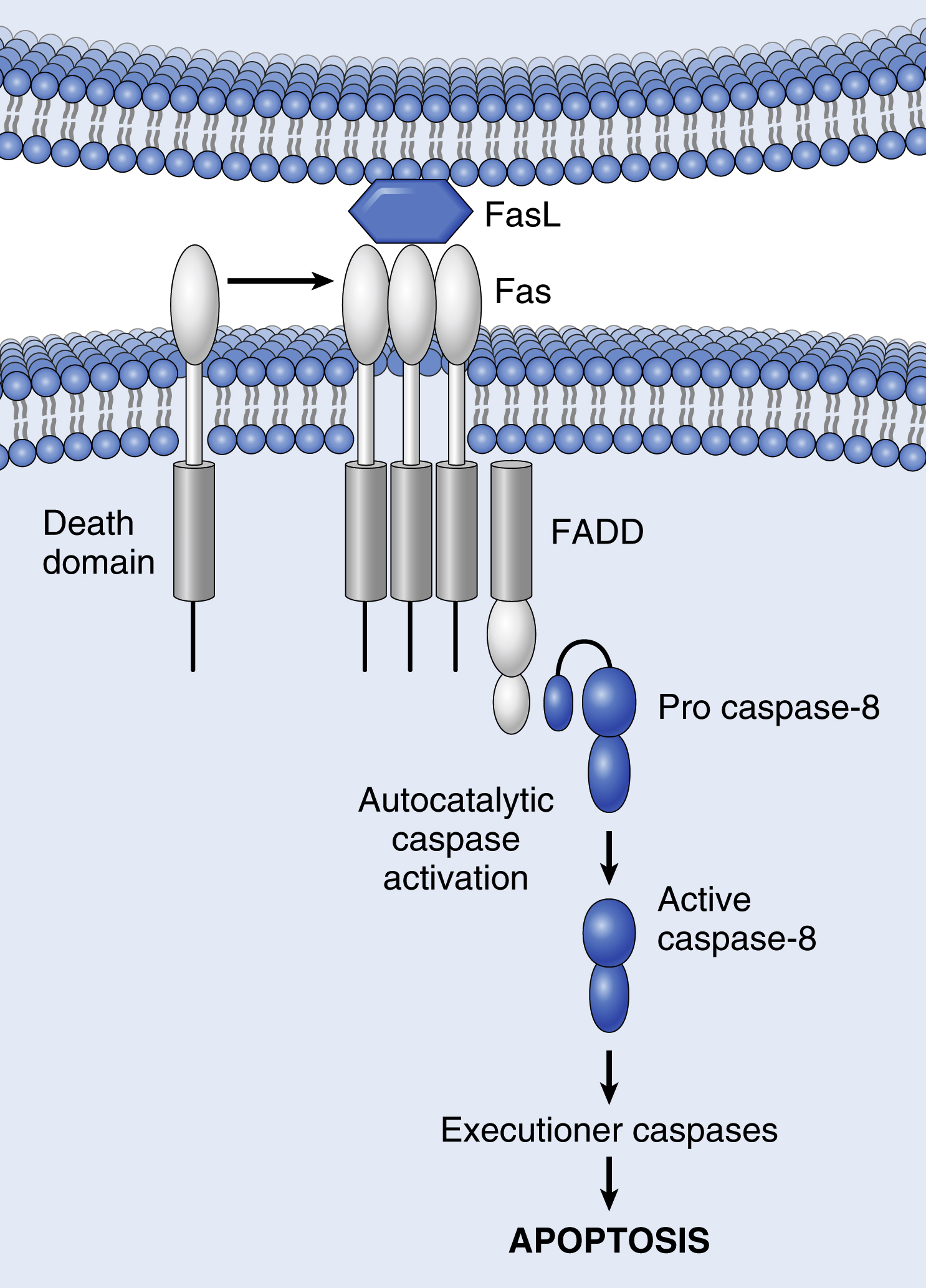
Execution Phase (p. 46)
Caspases occur as inactive proenzymes that are activated through proteolytic cleavage; the cleavage sites can be hydrolyzed by other caspases or autocatalytically. Initiator caspases (e.g., caspase-8 and -9) are activated early in the sequence and induce the cleavage of the executioner caspases (e.g., caspase-3 and -6) that do the bulk of the intracellular proteolytic degradation. Once an initiator caspase is activated, the death program is set in motion by rapid and sequential activation of other caspases. Executioner caspases act on many cell components; they cleave cytoskeletal and nuclear matrix proteins, disrupting the cytoskeleton and leading to nuclear breakdown. In the nucleus, caspases cleave proteins involved in transcription, DNA replication, and DNA repair; in particular, caspase-3 activates a cytoplasmic DNase, resulting in the characteristic cleavage between nucleosomes.
Removal of Dead Cells (p. 46)
The apoptotic process results in the formation of apoptotic bodies—”bite-sized” fragments that are engulfed by phagocytes (in a process called efferocytosis). In apoptotic cells, phosphatidylserine that is normally present only on the inner leaflet of the plasma membrane “flips” to be expressed on the outer plasma membrane, where it is recognized by several macrophage receptors. Apoptotic bodies can also become coated with natural antibodies and proteins of the complement system, which are also recognized by phagocytes. Finally, ingestion of ingested apoptotic bodies reduces the macrophage production of proinflammatory cytokines, thus limiting inflammatory reactions, even in the setting of extensive apoptosis.
Other Mechanisms of Cell Death (p. 47)
While necrosis and apoptosis are the best-defined mechanisms of cell death, there are other ways for cells to die; students should at least be aware of these:
- • Necroptosis, a hybrid of both necrosis and apoptosis that morphologically and biochemically resembles necrosis (loss of ATP, cell swelling, lysosomal enzyme release, and plasma membrane rupture), but is triggered by signal transduction pathways akin to apoptosis. Notably, the signals leading to necroptosis do not result in caspase activation, but rather involve two kinases called receptor-interacting protein kinase 1 and 3 (RIPK1 and RIPK3; Fig. 2.7). Thus ligation of TNFR1 (for example) recruits these kinases into a multiprotein complex, leading to phosphorylation and oligomerization of the cytoplasmic protein MLKL and causing a plasma membrane disruption reminiscent of necrosis. Physiologic necroptosis occurs during the formation of the mammalian bone growth plate, and can be part of the host defense against certain viruses that encode caspase inhibitors (e.g., cytomegalovirus). Pathologic necroptosis is associated with cell death in steatohepatitis, acute pancreatitis, ischemia-reperfusion injury, and neurodegenerative diseases such as Parkinson disease.
- • Pyroptosis is a form of apoptosis associated with the release of the fever-inducing cytokine IL-1 (pyro refers to fever). This pathway is induced by cytoplasmic immune receptors that recognize intracellular microbial products and activate a multiprotein complex called the inflammasome (Chapter 6); inflammasomes then activate caspase-1 to cleave a precursor of interleukin-1 (IL-1) to its biologically active form. Caspase-1 (and the closely related caspases-4 and -5) also induce cell death.
-

Fig. 2.7 Molecular mechanism of TNF-mediated necroptosis.Cross-linking of TNFR1 by TNF initiates the illustrated series of downstream events, which ultimately lead to plasma membrane disruption, cell death, and inflammation. See text for details. Modified from Galluzi L, et al: Programmed necrosis from molecules to health and disease, Int Rev Cell Molec Biol 289:1, 2011. - • Ferroptosis is a distinct form of cell death triggered when the levels of intracellular iron or reactive oxygen species overwhelm glutathione-dependent antioxidant defenses and cause membrane lipid peroxidation. This disrupts many aspects of membrane function, including fluidity, lipid-protein interactions, ion and nutrient transport, and signaling pathways, and leads to cell death that resembles necrosis. Unlike necrosis, ferroptosis is regulated by specific signals, and can be prevented by reducing iron levels.
Autophagy (p. 48)
Autophagy is a process by which a cell consumes its own contents (Greek: auto, self; phagy, eating), delivering cytoplasmic materials to the lysosome for degradation. It is a survival mechanism during states of nutrient deprivation whereby cells can endure by cannibalizing themselves; it occurs in physiologic states (e.g., aging and exercise), as well as pathologic processes. The mechanism involves (Fig. 2.8):

- • Formation of an isolation membrane (phagophore) derived from the endoplasmic reticulum
- • Formation of an autophagosome vesicle with intracellular organelles and cytosolic structures within the isolation membrane
- • Fusion of the autophagosome with lysosomes, to deliver digestive enzymes for degradation of the contents
The process is regulated by protein products of autophagy-related genes (Atgs). Nutrient deprivation or depletion of growth factors activate a complex of four proteins that recruit Atgs to nucleate the phagophore, which then elongates and captures its cytosolic cargo before closing to form the autophagosome. The elongation and closure steps require covalent linkage of cytosolic microtubule-associated protein light chain 3 (LC3) to membrane phosphatidylethanolamine.
Autophagy is not a random process that engulfs cytosolic contents indiscriminately; cargo-loading is selective, with LC3 allowing the targeting and clearance of protein aggregates that accumulate during aging and stress, as well as senescent organelles. Autophagy can also precipitate cell death if stressors are too great, with a cell death pathway distinct from necrosis and apoptosis. Autophagy plays a role in both cancer growth and defense (Chapter 7), neurodegenerative disorders, infectious diseases (where many pathogens are degraded by autophagic pathways), and inflammatory bowel disease (Chapter 17).
Mechanisms of Cell Injury (p. 49)
The biochemical pathways in cell injury can be organized around a few general principles:
- • Responses to injurious stimuli depend on the type of injury, duration, and severity.
- • The consequences of injury depend on the type, state, and adaptability of the injured cell.
- • Cell injury results from perturbations in:
-
- • ATP production (mostly through effects on mitochondrial aerobic respiration)
- • Mitochondrial integrity independent of ATP production
- • Plasma membrane integrity, responsible for ionic and osmotic homeostasis
- • Protein synthesis, folding, degradation, and refolding
- • Integrity of the genetic apparatus
The intracellular mechanisms of cell injury fall into a small handful of general pathways (Fig. 2.9). Structural and biochemical elements of the cell are so closely interrelated that regardless of the locus of initial injury, secondary effects rapidly propagate through other elements.
Mitochondrial Damage (p. 49; Fig. 2.10)
Mitochondrial damage and dysfunction can occur due to heritable mutations (Chapter 5), hypoxia or toxins, or as a consequence of increased cytosolic Ca2+ or reactive oxygen species (ROS). There are three major consequences:
- • ATP depletion due to mitochondrial damage (toxins) or reduced supply of oxygen and nutrients (ischemia and/or hypoxia). Since ATP is critical for membrane transport, maintenance of ionic gradients (particularly Na+, K+, and Ca2+), and protein synthesis, reduced ATP synthesis will dramatically impact those pathways. ATP is generated through glycolysis (anaerobic, inefficient) and oxidative phosphorylation (aerobic, efficient). Hypoxia will lead to increased anaerobic glycolysis with glycogen depletion, increased lactic acid production, and intracellular acidosis. Mitochondrial damage can also result in formation of a high-conductance channel in the mitochondrial membrane (mitochondrial permeability transition pore; see Fig. 2.10). This causes loss of the mitochondrial membrane potential, with progressive ATP depletion.
-

Fig. 2.9 The principal forms and sites of damage in cell injury. ATP, Adenosine triphosphate; ROS, reactive oxygen species. -

Fig. 2.10 Role of mitochondria in cell injury and death.Mitochondria are affected by a variety of injurious stimuli, and their abnormalities lead to necrosis or apoptosis. ATP, Adenosine triphosphate; ROS, reactive oxygen species. - • Incomplete oxidative phosphorylation also leads to the formation of ROS (see later).
- • Leakage of mitochondrial proteins due to channel formation by proapoptotic BAX and BAK is the initial step in apoptosis by the intrinsic pathway.
Membrane Damage (p. 51; Fig. 2.11)
Plasma membrane damage or dysfunction can affect the integrity and functions of all cellular membranes, e.g., through the loss of osmotic balance and the influx of fluids and ions. If cells leak metabolites (e.g., glycolytic intermediates) that are vital for the reconstitution of ATP, energy stores can be further depleted. As noted previously mitochondrial membrane damage that opens mitochondrial permeability transition pores will decrease ATP generation and release proteins that drive apoptosis. Injury to lysosomal membranes results in enzyme leakage (e.g., of RNases, DNases, proteases, phosphatases, and glucosidases) and can cause necrosis.
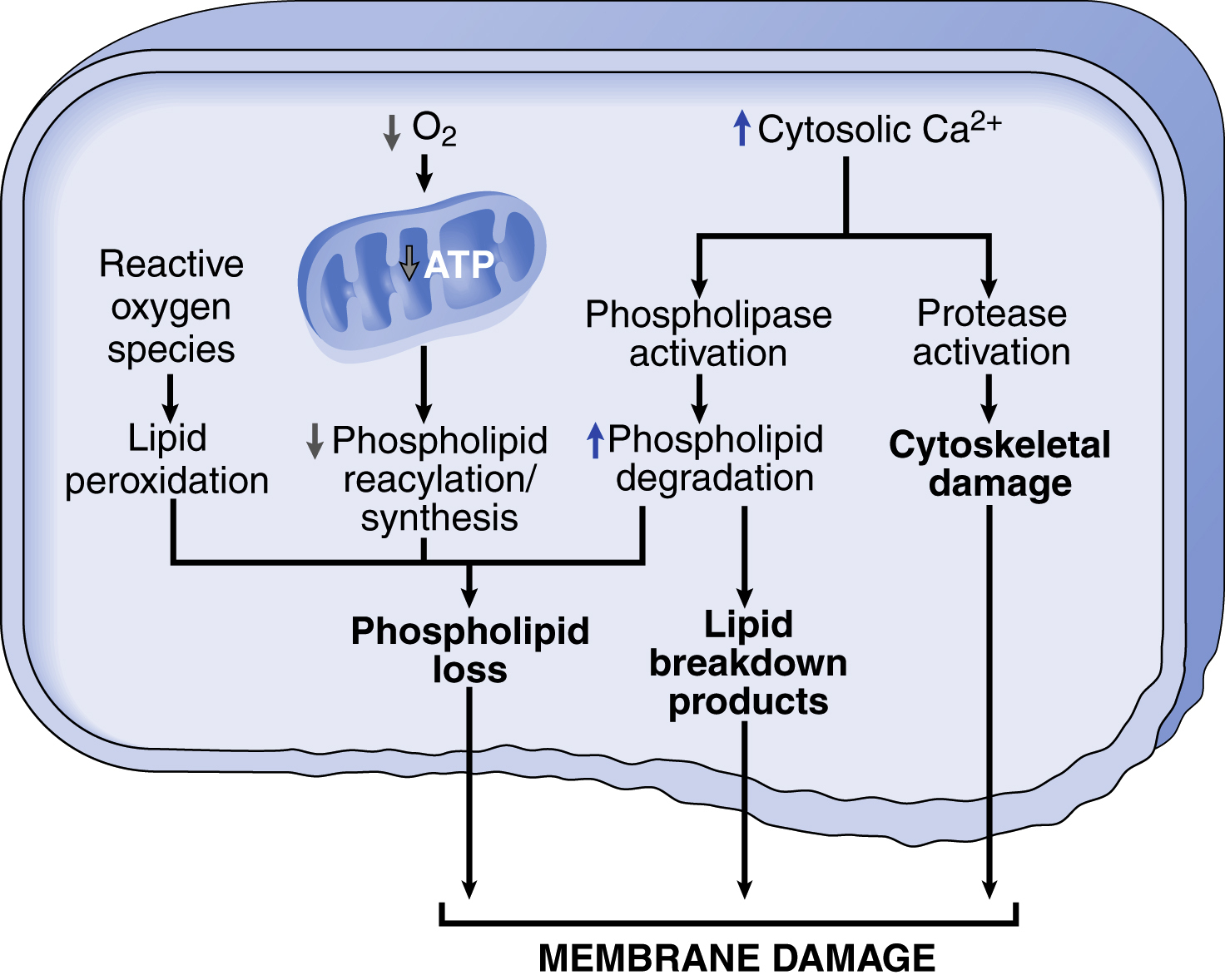
Membrane defects can result from ATP depletion, calcium-mediated activation of phospholipases, bacterial toxins, viral proteins, lytic complement components, and a variety of physical and chemical agents.
- • ROS cause injury to cell membranes by lipid peroxidation (see later).
- • Decreased phospholipid synthesis due to diminished mitochondrial function impacting energy-dependent biosynthetic pathways.
- • Increased phospholipid breakdown due to activation of calcium-dependent phospholipases; accumulation of lipid breakdown products has a detergent effect on membranes, and can cause permeability and electrophysiologic alterations.
- • Cytoskeletal abnormalities due to proteolysis activated by cytosolic Ca2+; detachment of the cell membrane from the cytoskeleton renders it susceptible to stretching and rupture.
Damage to DNA (p. 51)
Damage to nuclear DNA triggers p53-dependent pathways to stop cell proliferation while repair attempts are underway (Chapter 7). If repair is unsuccessful, p53 triggers apoptosis by the intrinsic mitochondrial pathway, so that the cell dies rather than mutates. Predictably, mutations in p53 that interfere with its function are associated with malignancy (Chapter 7). DNA damage can occur secondary to radiation, chemotherapeutic (anticancer) drugs, and ROS, or can occur spontaneously as a part of aging (due to deamination of cytosine residues to become uracil residues).
Oxidative Stress: Accumulation of Oxygen-Derived Free Radicals (p. 52)
Cell injury induced by free radicals is an important damage mechanism underlying chemical and radiation injury, ischemia-reperfusion injury, cellular aging, and microbial killing. Free radicals are chemical species that have a single unpaired electron in an
outer orbit, which is highly reactive and avidly modifies proteins, lipids, carbohydrates, and nucleic acids. These reactions are typically autocatalytic, meaning that molecules that react with free radicals are themselves converted into free radicals, thus propagating the chain of damage (see Table 2.2). ROS are produced at low levels during normal mitochondrial respiration, but are efficiently degraded by intracellular ROS scavengers. However, increased production or decreased scavenging of ROS may lead to an excess of free radicals—called oxidative stress. ROS are also produced in large amounts by activated leukocytes, particularly neutrophils and macrophages, during inflammatory reactions (Chapter 3).
Generation of Free Radicals (p. 52) (Fig. 2.12)
-
• Reduction-oxidation reactions that occur during normal respiration. As molecular O2 is converted to water along the electron transport chain, small amounts of partially reduced intermediates are produced; these include superoxide anion (
 ), hydrogen peroxide (H2O2), and hydroxyl radicals (˙OH).
), hydrogen peroxide (H2O2), and hydroxyl radicals (˙OH). - • Absorption of radiant energy. Ionizing radiation (e.g., x-rays) can hydrolyze water into ˙OH and hydrogen (H) free radicals.
- • Inflammation. Activated leukocytes use a plasma membrane multiprotein complex including NADPH oxidase to generate ROS to kill phagocytized pathogens (Chapter 3).
- • Enzymatic metabolism of exogenous chemicals or drugs. Although these are not ROS they will have similar effects (e.g., CCl4 can generate ˙CCl3)
- • Transition metals (e.g., iron and copper) donate or accept free electrons during intracellular reactions and catalyze free radical formation; the Fenton reaction is an example: H2O2 + Fe2+ → Fe3+ + ˙OH + OH−.
- • Nitric oxide (NO), generated by endothelial cells, macrophages, neurons, and other cell types (Chapter 3), can act as a free radical and be converted to highly reactive peroxynitrite anion (ONOO − ) as well as NO2 and NO3 − (see Table 2.2)
Removal of Free Radicals (p. 53)
Free radicals are inherently unstable and generally rapidly decay. Thus
spontaneously dismutates to O2 and H2O2 in the presence of water. Additional mechanisms to eliminate free radicals include (Fig. 2.12):
spontaneously dismutates to O2 and H2O2 in the presence of water. Additional mechanisms to eliminate free radicals include (Fig. 2.12):- • Antioxidants can block free radical formation or scavenge free radicals. These include vitamins E and A, as well as ascorbic acid and glutathione.
- • The reactivity of transition metals (e.g., iron and copper) is minimized by binding to storage and transport proteins (e.g., transferrin, ferritin, lactoferrin, and ceruloplasmin).
-
• Several enzymes break down H2O2 and
:
-
- • Catalase, present in peroxisomes, converts 2H2O2 → O2 + 2H2O.
-
• Superoxidase dismutases (SODs) convert
 + 2H → H2O2 + O2.
+ 2H → H2O2 + O2. - • Glutathione peroxidase removes H2O2 or ˙OH by catalyzing the formation of disulfide-linked glutathione homodimers (GSSG) from the reduced glutathione tripeptide (γ-glutamyl-cysteine-glycine; GSH): H2O2 + 2GSH → GSSG + 2H2O, or 2˙OH + 2GSH → GSSG + 2H2O. The intracellular ratio of oxidized GSSG to reduced GSH is an important indicator of the cell’s ability to detoxify ROS.


Pathologic Effects of Free Radicals (p. 53, Fig. 2.12)
Free radical damage can lead to necrosis as well as trigger apoptosis.
- • Membrane lipid peroxidation. Damage occurs when double bonds in membrane unsaturated fatty acids are attacked by ROS. The lipid–free radical interactions yield peroxides, which are unstable and reactive, causing an autocatalytic chain reaction (propagation) resulting in extensive membrane damage.
- • Protein oxidative modification. Free radicals promote oxidation of amino acid side chains, formation of covalent protein-protein cross-links (e.g., disulfide bonds), and oxidation of the protein backbone. These can damage enzyme active sites, disrupt structural proteins, and engender proteasomal degradation.
- • DNA damage. Free radicals cause single- and double-strand DNA breaks, cross-link DNA strands, and form new adducts. These are implicated in cell aging and malignant transformation (Chapter 7).
Disturbance in Calcium Homeostasis (p. 54)
Calcium ions normally serve as second messengers in several signaling pathways, but disproportionate levels of cytosolic calcium are an important cause of injury. Cytosolic free Ca2+ is normally maintained at very low concentrations (∼0.1 μmol) compared with extracellular levels of 1.3 mmol; most intracellular Ca2+ is sequestered in mitochondria and the ER. Cell injury (e.g., ischemia, toxins) initially cause Ca2+ release from these intracellular stores, followed by influx across the plasma membrane (Fig. 2.13). The resulting excessive intracellular Ca2+ causes further cell injury by:
- • Mitochondrial Ca2+ accumulation opens the mitochondrial permeability transition pore with failure of ATP generation.
- • Increased cytosolic Ca2+ activates enzymes including phospholipases (membrane damage), proteases (protein catabolism), endonucleases (DNA and chromatin fragmentation), and ATPases (hastening ATP depletion).
Endoplasmic Reticulum (ER) Stress: The Unfolded Protein Response (p. 54; Fig. 2.14)
ER chaperones control the proper folding of newly synthesized proteins; misfolded polypeptides are shuttled into the cytoplasm and are targeted for proteasome degradation (Chapter 1). However, if misfolded proteins accumulate in the ER, they trigger the unfolded protein response (UPR). Misfolded proteins can accumulate due to:
- • reduced ability to repair or eliminate them
- • deleterious mutations
- • synthesis of viral proteins at a level that overwhelms the folding quality control system
- • increased demand for secretory proteins such as insulin in insulin-resistant states
- • changes in intracellular pH and redox state
- • insufficient ATP for the “foldases” to function
The UPR increases chaperone production, enhances proteasomal degradation, and slows protein translation (Fig. 2.14). If this cytoprotective response is insufficient, the cell activates caspases and will undergo apoptosis, so-called ER stress. Protein misfolding is likely causal in a number of disorders (Table 2.3, p. 25).

Examples of Cell Injury and Death (p. 55)
Hypoxia and Ischemia (p. 55)
Ischemia and hypoxic injury are the most common forms of cell injury in clinical medicine. Hypoxia is reduced oxygen-carrying capacity; ischemia (which also clearly causes hypoxia) is due to reduced blood flow. Hypoxia alone allows continued delivery of substrates for glycolysis and removal of accumulated wastes (e.g., lactic acid); ischemia does neither and therefore tends to injure tissues faster than hypoxia alone.
Mechanisms of Ischemic Cell Injury (p. 55; Fig. 2.15)
As intracellular oxygen tension falls with ischemia or hypoxia, oxidative phosphorylation fails, and ATP generation decreases. As described in the Mitochondrial Damage section, loss of ATP results initially in reversible cell injury (cell and organelle swelling) and later in cell death by necrosis. Responses to protect against hypoxic stress include induction of the transcription factor hypoxia-inducible
factor-1 (HIF-1); it promotes new blood vessel formation, stimulates cell survival pathways, and enhances glycolysis.

Ischemia-Reperfusion Injury (p. 56)
Restoration of blood flow to ischemic tissues can result in recovery of reversibly injured cells, but can paradoxically also exacerbate cell injury and even cause cell death. Called ischemia-reperfusion injury, the process is clinically important in myocardial and cerebral infarction following therapies that restore blood flow. Potential mechanisms include:
- • Oxidative stress. New damage may occur during reoxygenation by increased generation of ROS from parenchymal and endothelial cells and from infiltrating leukocytes. ROS in reperfused tissue result from incomplete reduction of oxygen by damaged mitochondria or because of the normal action of oxidases from tissue cells or invading inflammatory cells. Antioxidant defense mechanisms may also be compromised, favoring radical accumulation.
- • Intracellular calcium overload. This occurs due to cell membrane damage and ROS-mediated injury to sarcoplasmic reticulum. Calcium overload drives mitochondrial permeability transition pore opening and subsequent ATP depletion.
- • Inflammation. Ischemic injury recruits circulating inflammatory cells (see Chapter 3) through release of “danger signals,” as well as enhanced cytokine and adhesion molecule expression by hypoxic parenchymal and endothelial cells. The ensuing inflammation causes additional injury. By restoring blood flow, reperfusion may actually increase local inflammatory cell infiltration.
-
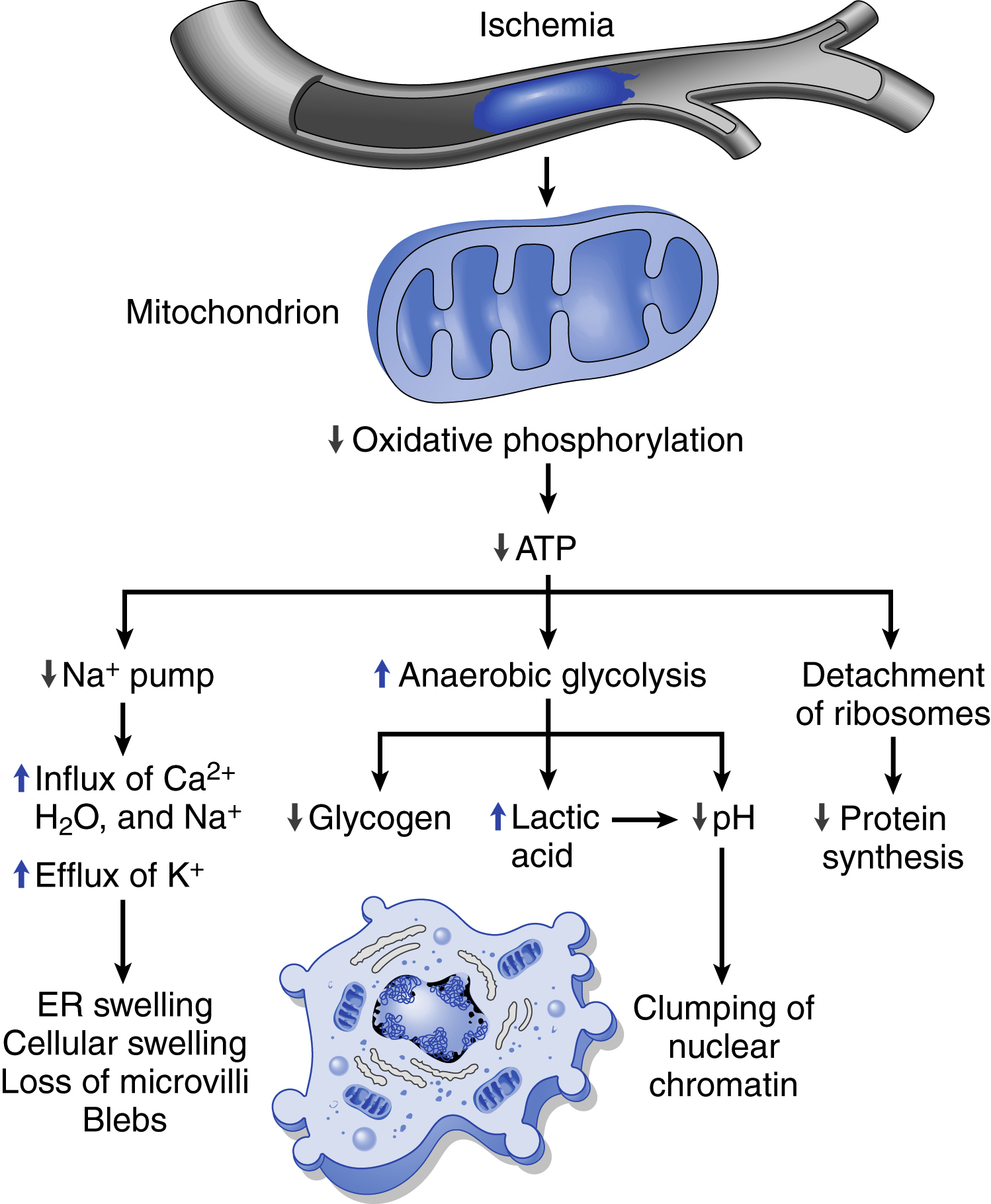
Fig. 2.15 Functional and morphologic consequences of decreased intracellular adenosine triphosphate (ATP) in ischemic cell injury.The morphologic changes shown here are indicative of reversible cell injury. Further depletion of ATP results in cell death, typically by necrosis. ER, Endoplasmic reticulum. - • Complement activation. Immunoglobulin M (IgM) antibodies can deposit in ischemic tissues; when blood flow is resumed, complement proteins are activated by binding to the antibodies, resulting in further cell injury and inflammation.
Chemical (Toxic) Injury (p. 56)
Chemical injury occurs by two general mechanisms:
- • Direct toxicity, by binding to some critical molecular component. Thus mercuric chloride binds to cell membrane protein sulfhydryl groups, inhibiting ATPase-dependent transport, and causing increased permeability, and cyanide poisons mitochondrial cytochrome oxidase and inhibits oxidative phosphorylation.
- • Conversion to toxic metabolites. Most chemicals that ultimately exhibit toxicity are not injurious in their native form; they must be converted to reactive toxic metabolites that act on target molecules. This is usually accomplished by the cytochrome P-450 mixed-function oxidases in the smooth ER of the liver and other organs. The resulting metabolites can then cause membrane damage and cell injury by formation of free radicals and subsequent lipid peroxidation; CCl4 and acetaminophen are examples of compounds that cause such indirect injury.
Adaptations of Cellular Growth and Differentiation (p. 57)
Hypertrophy (p. 57)
Hypertrophy is an increase in the size of cells; it typically results in an increase in the size of the affected organ. The hypertrophied organ has no new cells, just cells that are larger due to the synthesis of additional intracellular structural components. Cells capable of division may respond to stress by undergoing both hyperplasia (increased cell number; below) and hypertrophy, whereas nondividing cells (e.g., myocardial fibers) can only hypertrophy.
Physiologic hypertrophy occurs with increased functional demand or stimulation by hormones and growth factors, e.g., uterine hypertrophy stimulated by estrogenic signaling during pregnancy.
Pathologic hypertrophy occurs, for example, when the left ventricular myocardium experiences increased pressure due to aortic stenosis or systemic hypertension; the myocytes respond by synthesizing more protein and increasing the number of myofilaments per cell, which increases the amount of force each myocyte can generate. Initially, cardiac hypertrophy improves function, but there is a point of diminishing returns, and the hypertrophy can become maladaptive, setting the stage for heart failure (Chapter 12).
Mechanisms of Hypertrophy (p. 57)
Cardiac hypertrophy results from a combination of growth factors and direct mechanical effects (Fig. 2.16):
- • Cellular mechanical sensors (in myocyes, fibroblasts, and endothelial cells) detect an increased load
- • Downstream signaling is activated, including phosphoinositide 3-kinase (PI3K)/AKT and G-protein-coupled receptor pathways
- • Signaling pathways stimulate growth factor production, and the release of vasoactive molecules (e.g., α-adrenergic agonists, endothelin-1, and angiotensin II)
- • Transcription factors are produced (e.g., GATA4, nuclear factor of activated T cells, and myocyte enhancer factor 2), increasing the expression of genes including those that encode muscle proteins.
Cardiac hypertrophy is also associated with a switch in gene expression from adult-type contractile proteins to distinct fetal isoforms of the same proteins. For example, the α isoform of myosin heavy chain is replaced by the β isoform, which has a slower, more energetically economical contraction. Genes that participate in the cellular response to stress are also increased in hypertrophic myocytes.
Hyperplasia (p. 59)
Hyperplasia is an increase in the number of cells in an organ or tissue. Although hyperplasia and hypertrophy are distinct processes, they frequently occur together, and can be triggered by the same external stimuli. Hyperplasia results from growth factor-driven proliferation of mature cells and/or increased output of new cells from stem cells (Chapter 3). Hyperplasia can only take place if the tissue contains cells capable of dividing, and can be physiologic or pathologic.
- • Physiologic hyperplasia occurs when there is a need to increase functional capacity of hormone-sensitive organs, or when there is need for compensatory increase after damage or resection. Hormonal hyperplasia is well-illustrated by the proliferation of the glandular epithelium of the female breast at puberty and during pregnancy. Liver regeneration is a good example of compensatory hyperplasia; individuals who donate a liver lobe for transplantation will experience cell proliferation that restores the organ to its original size.
-

Fig. 2.16 Biochemical mechanisms of myocardial hypertrophy.The major known signaling pathways and their functional effects are shown. Mechanical sensors appear to be the major triggers for physiologic hypertrophy, and agonists and growth factors may be more important in pathologic states. ANF, Atrial natriuretic factor; GATA4, transcription factor that binds to DNA sequence GATA; IGF1, insulin-like growth factor; NFAT, nuclear factor–activated T cells; MEF2, myocardial enhancing factor 2. - • Pathologic hyperplasia is typically caused by excessive or inappropriate response of target cells to hormones or growth factors. Benign prostatic hyperplasia is a good example of pathologic hyperplasia, resulting from hormonal stimulation by androgens. Although such pathologic hyperplasia is abnormal, the process remains controlled and the hyperplasia can either regress or stabilize if the hormonal stimulation is eliminated. Hyperplasia is also a characteristic response to certain viral infections, such as papillomaviruses; skin warts and mucosal lesions composed of masses of hyperplastic epithelium arise when viruses make factors that interfere with the host proteins that regulate cell proliferation.
Increased cell division associated with hyperplasia elevates the risk of acquiring genetic aberrations that drive unrestrained proliferation and cancer (Chapter 7).
Atrophy (p. 59)
Atrophy is a reduction in the size of an organ or tissue due to a decrease in cell size and/or number. Physiologic atrophy is common during normal development; some embryonic structures, such as the notochord and thyroglossal duct, undergo atrophy during fetal development. Pathologic atrophy has several causes:
- • Decreased workload (disuse atrophy). When a limb is immobilized in a plaster cast, skeletal muscle atrophy rapidly ensues. The initial decrease in cell size is reversible once activity is resumed. With more prolonged disuse, skeletal muscle fibers can decrease in number, too (due to apoptosis).
- • Loss of innervation (denervation atrophy). The structure and function of skeletal muscle depends on its nerve supply; damage to the nerves leads to atrophy of the muscle fibers (Chapter 27).
- • Diminished blood supply. A gradual decrease in blood supply (chronic ischemia) to a tissue results in atrophy.
- • Inadequate nutrition. Profound protein-calorie malnutrition (marasmus) leads to catabolism of skeletal muscle proteins to provide a source of energy. This results in marked muscle wasting (cachexia; Chapter 9). Cachexia can also occur with chronic inflammatory diseases and cancer due to overproduction of the inflammatory cytokine tumor necrosis factor.
- • Loss of endocrine stimulation. Hormone-responsive tissues, e.g., breast and reproductive organs, are dependent on endocrine stimulation for normal structure and function. Loss of estrogen stimulation (e.g., due to menopause) results in atrophy of the endometrium, vaginal epithelium, and breast.
- • Pressure. Tissue compression for any length of time can cause atrophy, probably as a result of ischemic changes caused by vascular compromise.
The fundamental cellular changes associated with atrophy are similar in all of these settings. The initial response is a decrease in cell size and organelles, reducing the metabolic needs of the cell sufficiently to permit its survival. By bringing into balance the cell’s metabolic demands and the lower levels of blood supply, nutrition, or trophic stimulation, a new equilibrium is achieved.
Initially, atrophic cells and tissues have diminished function, but cell death is minimal. However, atrophy caused by gradually reduced blood supply or atrophy of endocrine organs after hormone withdrawal may progress to cell death by apoptosis.
Mechanisms of Atrophy (p. 60)
Atrophy results from decreased protein synthesis and/or increased protein degradation in cells. Protein degradation occurs mainly via the ubiquitin-proteasome pathway; nutrient deficiency or disuse may activate ubiquitin ligases to target certain proteins for proteasome degradation. This is also likely responsible for cancer cachexia. Atrophy can also occur through increased autophagy. Some of the cell debris within the autophagic vacuoles may resist digestion and persist as membrane-bound residual bodies, e.g., lipofuscin granules, discussed later.
Metaplasia (p. 61)
Metaplasia is a reversible change in which one differentiated cell type is replaced by another cell type. It can be an adaptive response when a cell type that is sensitive to a particular stress is replaced by another cell type that is better able to withstand the adverse environment.
An example is a columnar-to-squamous metaplasia occurring in the respiratory tract in response to chronic irritation (e.g., cigarette smoke) (Fig. 2.17). Presumably, the more rugged stratified squamous epithelium is able to survive where the more fragile specialized columnar epithelium might succumb. However, metaplasia can ultimately be maladaptive; thus although the epithelial lining in the respiratory tract becomes more durable, mucus secretion and the
ciliary action of the columnar epithelium are lost. Metaplasia from squamous-to-columnar type may also occur, as in Barrett esophagus where the esophageal squamous epithelium is replaced by intestinal-like columnar cells under the influence of refluxed gastric acid. Importantly, the influences that predispose to metaplasia, if persistent, can initiate malignant transformation in metaplastic epithelium.
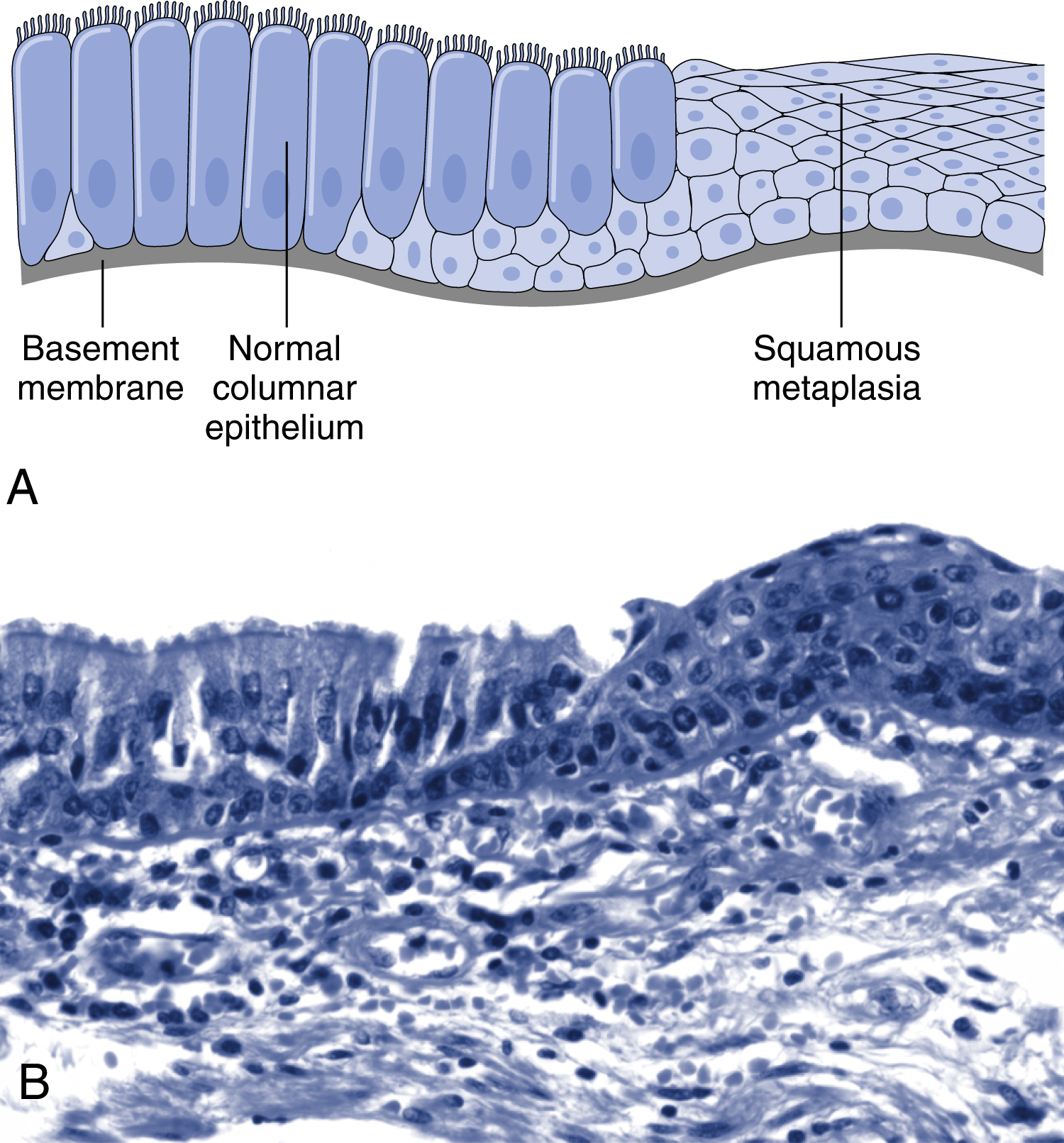
Connective tissue metaplasia occurs when cartilage, bone, or fat (mesenchymal tissues) arise in tissues that normally do not contain these elements. Thus bone formation in muscle, designated myositis ossificans, occasionally occurs after intramuscular hemorrhage. Unlike epithelial metaplasia, this is not associated with increased cancer risk.
Mechanisms of Metaplasia (p. 61)
Metaplasia results from either reprogramming of local tissue stem cells or by colonization from nearby differentiated cell populations. It is stimulated by cytokines, growth factors, and extracellular matrix components that promote the expression of genes that drive cellular differentiation pathways. A direct link between transcription factor dysregulation and metaplasia is seen with the squamous metaplasia (in the respiratory epithelium and in the cornea) that occurs with vitamin A (retinoic acid) deficiency. Retinoic acid regulates gene transcription directly through nuclear retinoid receptors (Chapter 9), which can influence the differentiation of progenitors derived from tissue stem cells.
Intracellular Accumulations (p. 62) (Fig. 2.18)
Cells may accumulate abnormal amounts of various substances.
- • Inadequate removal of a normal endogenous substance due to packaging or transport defects (e.g., fat accumulation in liver cells).
- • Accumulation of an endogenous substance due to genetic or acquired defects in folding, packaging, transport, or secretion (e.g., α1-antitrypsin disease).
- • Failure to degrade a normal substance due to genetic or acquired defects in its metabolism (e.g., lysosomal storage diseases; Chapter 5).
- • Abnormal exogenous substances may accumulate because cells lack the machinery to degrade them (e.g., macrophages laden with environmental carbon).
Lipids (p. 62)
Triglycerides (the most common), cholesterol and cholesterol esters, and phospholipids can accumulate in cells.
Steatosis (Fatty Change) (p. 62)
Steatosis is an abnormal accumulation of triglycerides within parenchymal cells either due to excessive entry or defective metabolism and export. It can occur in heart, muscle, or kidney, but is most common in the liver. Hepatic causes include alcohol abuse (most common in the United States), protein malnutrition, diabetes mellitus, obesity, toxins, and anoxia.
Cholesterol and Cholesterol Esters (p. 62)
Cholesterol is normally required for cell membrane or lipid-soluble hormone synthesis; production is tightly regulated but accumulation (seen as intracellular cytoplasmic vacuoles) can be present in a variety of pathologic states:

- • Atherosclerosis: Cholesterol and cholesterol esters accumulate in arterial wall smooth muscle cells and macrophages (Chapter 11). Extracellular accumulations appear microscopically as cleft-like cavities formed when cholesterol crystals are dissolved during normal histologic processing.
- • Xanthomas: In acquired and hereditary hyperlipidemias, cholesterol accumulates in clusters of “foamy” macrophages and mesenchymal cells.
- • Cholesterolosis: Focal accumulations of cholesterol-laden macrophages within the lamina propria of gallbladders.
- • Niemann-Pick disease, type C: a lysosomal storage disease due to mutations of an enzyme involved in cholesterol catabolism (Chapter 5).
Proteins (p. 63)
Intracellular protein accumulation occurs with excessive synthesis, absorption, or defects in cellular transport. Morphologically visible accumulations appear as rounded, eosinophilic cytoplasmic droplets. In some disorders (e.g., amyloidosis; Chapter 6), abnormal proteins deposit primarily in the extracellular space.
- • Reabsorption droplets of proteins accumulate in proximal renal tubules in the setting of chronic proteinuria.
- • Normally secreted proteins can accumulate if produced in excessive amounts (e.g., immunoglobulin within plasma cells).
- • Defective intracellular transport and secretion leads to pathology not only from the unfolded protein response and apoptosis (see preceding discussion), but also loss of protein function.
- • Accumulated cytoskeletal proteins. Excess intermediate filaments (e.g., keratin or certain neurofilaments) are hallmarks of cell injury; thus keratin intermediate filaments coalesce into cytoplasmic eosinophilic inclusions called alcoholic hyaline (Chapter 18), and the neurofibrillary tangle in Alzheimer disease contains neurofilaments (Chapter 28).
- • Aggregates of abnormal proteins. Aggregation of abnormally folded proteins (e.g., due to genetic mutations, aging) can cause pathologic change.
Hyaline Change (p. 64)
Hyaline change refers to any deposit that imparts a homogeneous, glassy pink appearance in H&E-stained histologic sections. Examples of intracellular hyaline change include proximal tubule epithelial protein droplets, viral inclusions, and alcoholic hyaline. For example, extracellular hyaline change occurs in damaged arterioles (e.g., due to chronic hypertension), presumably due to extravasated proteins.
Glycogen (p. 64)
Glycogen is commonly stored within cells as a ready energy source. Excessive intracellular deposits (seen as clear vacuoles) are seen with abnormalities of glycogen storage (so-called glycogenosis; Chapter 5) and glucose metabolism (diabetes mellitus).
Pigments (p. 64)
Pigments are colored substances that can be exogenous (e.g., coal dust) or endogenous, such as melanin or hemosiderin.
- • Exogenous pigments include carbon or coal dust (most common); when visibly accumulated within pulmonary macrophages and lymph nodes these deposits are called anthracosis. Pigments from tattooing are taken up by macrophages and persist for the life of the cell.
- • Endogenous pigments include:
-
- • Lipofuscin, so-called “wear-and-tear” pigment and usually associated with cellular and tissue atrophy (brown atrophy). The pigment is composed of complex lipids, phospholipids, and proteins derived from cell membrane peroxidation.
- • Melanin, a normal endogenous brown-black pigment formed by enzymatic oxidation of tyrosine to dihydroxyphenylalanine in melanocytes.
- • Homogentisic acid is a black pigment formed in patients with alkaptonuria (lacking homogentisic oxidase) that deposits in skin and connective tissue; the pigmentation is called ochronosis.
- • Hemosiderin is a hemoglobin-derived, golden yellow-brown, granular intracellular pigment composed of aggregated ferritin. Accumulation can be localized (e.g., macrophage-mediated breakdown of blood in a bruise) or systemic (i.e., due to increased dietary iron absorption, primary hemochromatosis), impaired utilization (e.g., thalassemia), hemolysis, or chronic transfusions (Chapter 18)
Pathologic Calcification (p. 65)
Pathologic calcification—the abnormal tissue deposition of calcium salts—occurs in two forms: dystrophic calcification arises in nonviable tissues in the presence of normal calcium serum levels, whereas metastatic calcification happens in viable tissues in the setting of hypercalcemia.
Dystrophic Calcification (p. 65)
While frequently only a marker of prior injury, it can also be a source of significant pathology. Dystrophic calcification occurs in atherosclerosis, in damaged heart valves, and in areas of necrosis. Calcium can be intracellular and extracellular. Deposition ultimately involves precipitation of a crystalline calcium phosphate similar to bone hydroxyapatite:
- • Initiation (nucleation) occurs extracellularly or intracellularly. Extracellular initiation occurs on membrane-bound vesicles from dead or dying cells that concentrate calcium due to their content of charged phospholipids; membrane-bound phosphatases then generate phosphates that form calcium-phosphate complexes; the cycle of calcium and phosphate binding is repeated, eventually producing a deposit. Initiation of intracellular calcification occurs in mitochondria of dead or dying cells.
- • Propagation of crystal formation depends on the concentration of calcium and phosphates, the presence of inhibitors, and structural components of the extracellular matrix.
Metastatic Calcification (p. 66)
These calcium deposits occur as amorphous basophilic densities that can be present widely throughout the body. They usually have no clinical sequelae, although massive deposition can cause renal and lung deficits. Metastatic calcification results from hypercalcemia (four principal causes):
- • Elevated parathyroid hormone (PTH) (e.g., hyperparathyroidism due to parathyroid tumors or ectopic PTH-related protein secreted by other neoplasms)
- • Bone destruction, as in primary marrow malignancies (e.g., multiple myeloma) or by diffuse skeletal metastasis (e.g., breast cancer), by accelerated bone turnover (Paget disease), or by immobilization
- • Vitamin D-related disorders, including vitamin D intoxication and systemic sarcoidosis (macrophages activate a vitamin D precursor)
- • Renal failure, causing secondary hyperparathyroidism due to phosphate retention and the resulting hypocalcemia
Cellular Aging (p. 66)
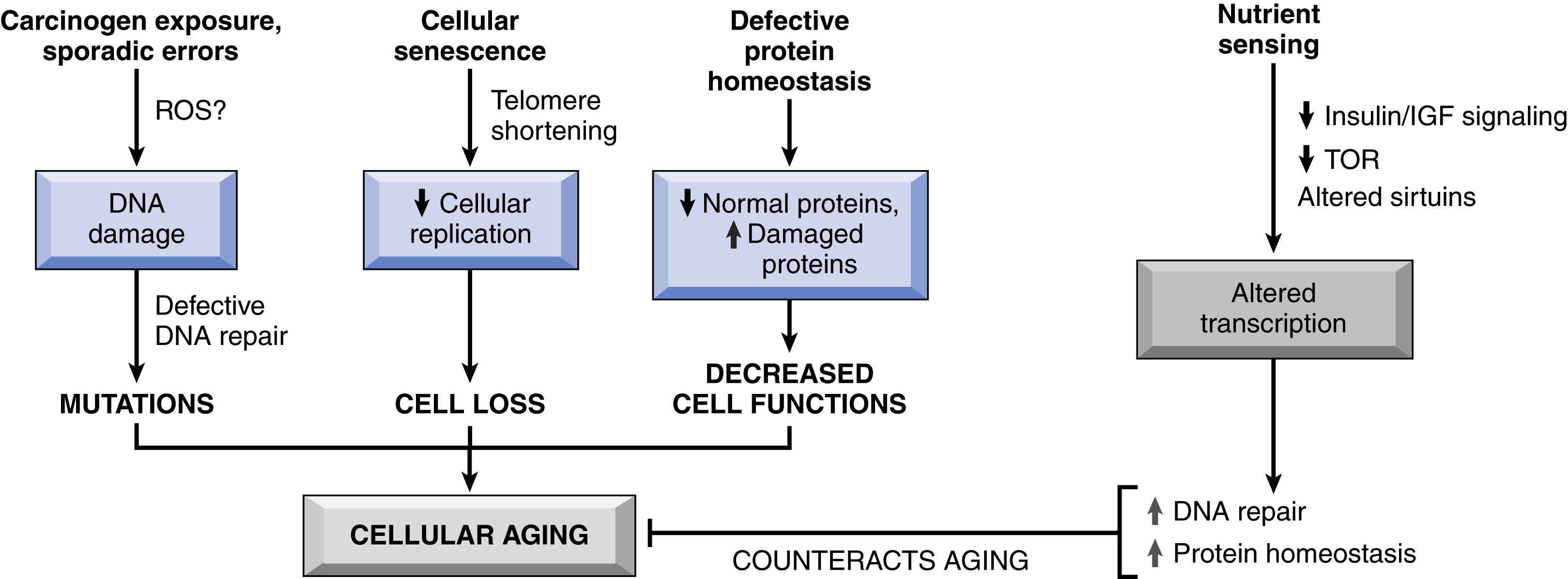
With increasing age, degenerative changes impact the structure and physiologic function of all organ systems. The tempo and severity of such changes in any given individual are influenced by genetic factors, diet, social conditions, and the impact of other comorbidities, such as atherosclerosis, diabetes, and osteoarthritis. Cellular aging—reflecting the progressive accumulation of sublethal cellular and molecular damage due to both genetic and exogenous influences—leads to cell death and diminished capacity to respond to injury; it is a critical component of the aging of the entire organism (Fig. 2.19).
Aging—at least in model systems—appears to be a regulated process influenced by a limited number of genes; this, in turn, implies that aging can potentially be parsed into definable mechanistic alterations.
- • DNA damage (p. 67). Imperfect DNA repair is an important element of aging. Nuclear and mitochondrial DNA are under constant attack by both exogenous (physical, chemical, and biologic agents) and endogenous (e.g., ROS) agents. Although most damage is successfully repaired, any residual defects become fixed in the primary sequence and will accumulate as cells age.
Premature aging is a feature of disorders associated with abnormal DNA repair (e.g., due to mutations in DNA helicase [Werner syndrome]) or defects in the repair of double-strand breaks (Bloom syndrome and ataxia-telangiectasia).
- • Cellular senescence (p. 67). Cells have a limited capacity for replication; after a fixed number of divisions, cells arrest in a terminally nondividing state. This phenomenon is reflected in the observation that cells from children exhibit more rounds of replication than cells from geriatrics. Cellular senescence is driven by:
-
- • Telomere attrition (p. 67). Telomeres are short repeated sequences of DNA that comprise the termini of chromosomes; they ensure complete replication of genes at the ends of chromosomes and also protect the chromosome tips from fusion and degradation. A small segment of telomere is lost with each cell division. Consequently, as somatic cells repeatedly divide, their telomeres progressively shorten until they no longer adequately protect the chromosome tips; this signals a growth checkpoint where cells become senescent. Accelerated telomere shortening has been associated with diseases such as pulmonary fibrosis and aplastic anemia.
Germ cells, and to a lesser extent stem cells, maintain sufficient telomere length to ensure unlimited replication through the activity of telomerase, an RNA-protein enzyme complex that uses its own RNA as a template to add nucleotides to the ends of chromosomes. Telomerase is typically undetectable in somatic cells but in cancer cells can become reactivated, allowing telomere stabilization and indefinite proliferation (Chapter 7).
- • Activation of tumor suppressor genes (p. 68). Replicative senescence is also regulated by certain tumor suppressor genes, particularly those at the INK4a/ARF locus that regulate G1-to-S phase transition in cell cycling.
- • Defective protein homeostasis (p. 68). The correct folding of proteins is maintained by chaperones; if that mechanism is not adequate to the task, misfolded proteins are degraded through the autophagy-lysosome and/or ubiquitin-proteasome systems. Defects in these systems contribute to aging through effects on replication, cell function, or apoptosis.
- • Dysregulated nutrient sensing (p. 68). Caloric restriction increases lifespan, suggesting that aging is also intimately associated with nutritional status and metabolism. The longevity effects of caloric restriction are attributed to the inhibition of the insulin-like growth factor 1 (IGF-1) signaling pathway and by increasing sirtuins.
-
- • Insulin and IGF-1 signaling pathway (p. 68). Both mediators signal glucose availability, promoting an anabolic state, as well as cell growth and replication. Among multiple downstream targets, IGF-1 induces mammalian target of rapamycin (mTOR) and Akt (also known as protein kinase B) kinase activities. Notably, some of the beneficial effects of caloric restriction can be mimicked by inhibiting mTOR (e.g., with rapamycin).
- • Sirtuins (p. 68). These are members of a family of NAD-dependent protein deacetylases that allow cellular adaptation to exogenous stressors, including food deprivation and DNA damage. Sirtuins induce the expression of a variety of genes that cumulatively promote longevity (e.g., by reducing apoptosis, stimulating protein folding, and inhibiting the effects of ROS); they also increase insulin sensitivity and inhibit some metabolic activities.