The Cellular Environment: Fluids and Electrolytes, Acids and Bases
Linda Felver
 http://evolve.elsevier.com/Rogers/pathophysiology/
http://evolve.elsevier.com/Rogers/pathophysiology/
The cells of the body live in a fluid environment that requires electrolyte and acid-base concentrations to be maintained within a very narrow range. A balance is maintained by an integration of renal, hormonal, neural, and respiratory functions. Changes in electrolyte concentration affect the electrical activity of nerve and muscle cells and cause shifts of fluid from one compartment to another. Fluid fluctuations also affect blood volume and therefore blood pressure. Alterations in acid-base balance disrupt the cellular function of enzyme systems and can cause cell injury. Disturbances in fluid and electrolyte or acid-base balance are common and can be life-threatening. Understanding how alterations occur and the body's ability to compensate or correct the disturbance is important for comprehending many pathophysiologic conditions.
Distribution of Body Fluids
Body fluids are distributed among functional compartments, or spaces, and provide a transport medium for cellular and tissue function. Intracellular fluid (ICF) is all the fluid within cells and comprises approximately two-thirds of total body water (TBW). Extracellular fluid (ECF) is all the fluid outside the cells and comprises approximately one-third of TBW. ECF includes the interstitial fluid, the intravascular fluid, and the various transcellular fluids. The interstitial fluid is the fluid surrounding the cells and found in the spaces between cells but not within the blood vessels. The intravascular fluid is the fluid found within blood vessels; it is more commonly known as the blood plasma. The transcellular fluids, the smallest component of ECFs, are the fluids contained within epithelial-lined cavities of the body. The major transcellular fluids are summarized in Table 3.1. Other transcellular fluids include pleural, synovial, peritoneal, pericardial, and intraocular fluids.
Table 3.1
| Fluid | Na+ (mEq/L) | K+ (mEq/L) | Cl− (mEq/L) |  HCO−3 (mEq/L) HCO−3 (mEq/L) |
|---|---|---|---|---|
| Saliva | 33 | 20 | 34 | 40 |
| Gastric juicea | 60 | 9 | 84 | 0 |
| Bile | 149 | 5 | 101 | 45 |
| Pancreatic juice | 141 | 5 | 77 | 92 |
| Ileal fluid | 129 | 11 | 116 | 29 |
| Cecal fluid | 80 | 21 | 48 | 22 |
| Cerebrospinal fluid | 141 | 3 | 127 | 23 |
| Sweat | 45 | 5 | 58 | 0 |
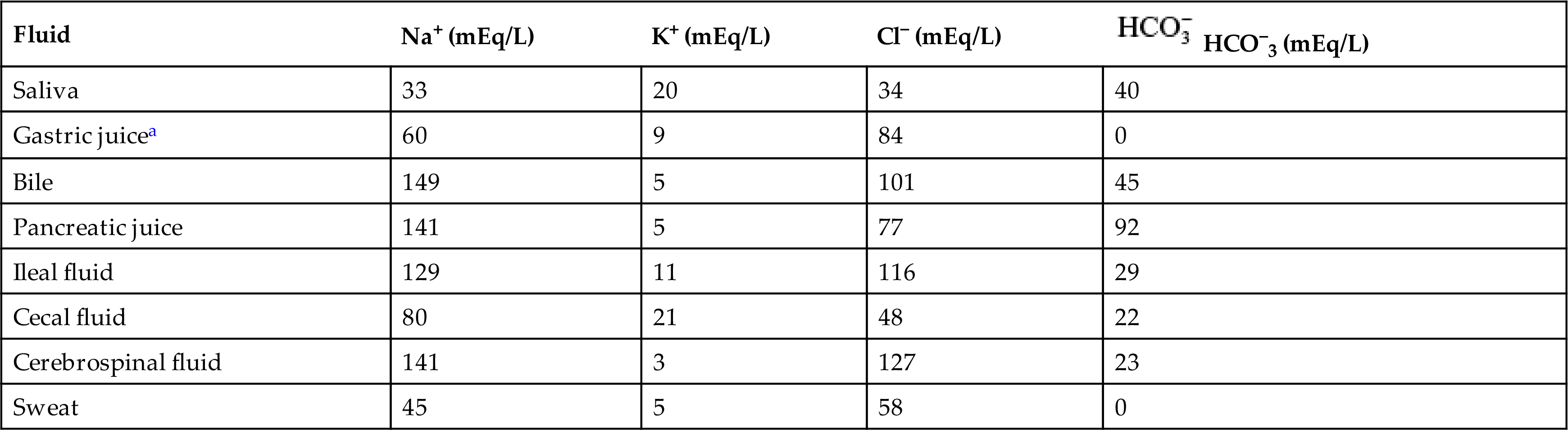
aThe Cl− concentration exceeds the Na+, K+ concentration by 15 mEq/L in gastric juice. This largely represents the secretions of hydrochloric acid by parietal cells.
The sum of fluids within all compartments constitutes the total body water (TBW) (Table 3.2). The volume of TBW is usually expressed as a percentage of body weight in kilograms. Average TBW volumes are summarized in Table 3.3.
Table 3.2
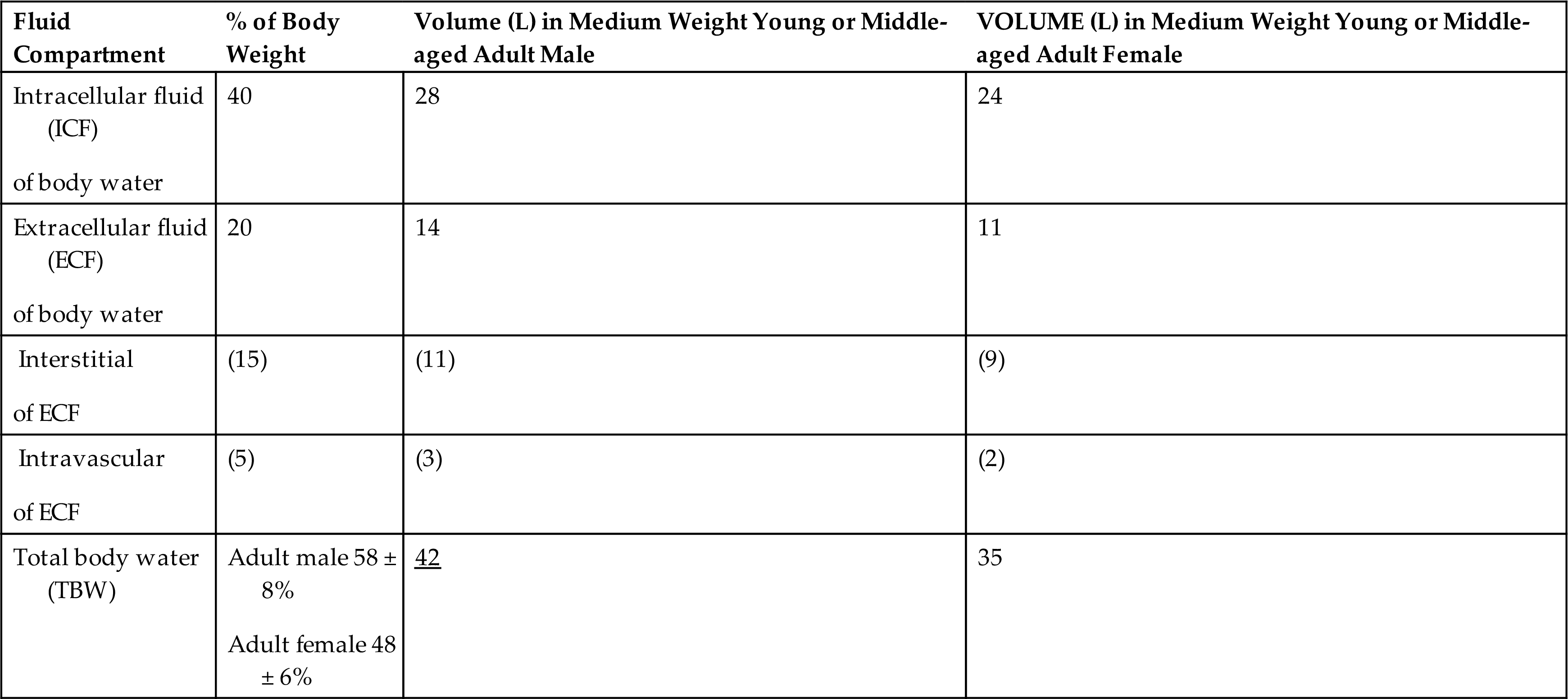
The values listed are based on the average total water in the body.
Table 3.3

NOTE: The values listed are based on the average total water in the body as a percentage of body weight. Note: Formulas for calculating estimates of total body water considering sex, age, height, and weight are available at: MD App, Total Body Water (TBW) Calculator; https://www.mdapp.co/total-body-water-tbw-calculator-448/. Data from Buchkremer F, Segerer S. Body surface area, creatinine excretion rate, and total body water: Reference data for adults in the United States. Kidney Medicine. 2021;3(2):312–313; Watson PE, Watson ID, Batt RD. Total body water volumes for adult males and females estimated from simple anthropometric measurements. The American Journal of Clinical Nutrition, 1980;33(1):27–39; Hume R, Weyers E. Relationship between total body water and surface area in normal and obese subjects. Journal of Clinical Pathology. 1971;24:234–238; Mendley SR, Majkowski NL, Schoeller DA. Validation of estimates of total body water in pediatric dialysis patients by deuterium dilution. Kidney International. 2005;67(5):2056-2062.
Although daily fluid intake may fluctuate widely, the body regulates water volume within a relatively narrow range. The primary sources of body water are drinking of fluids, ingestion of water in food, and derivation of water from oxidative metabolism. Most water is lost through renal excretion. Lesser amounts are eliminated through the stool and through vaporization from the skin and lungs. Skin losses of water include visible sweat and insensible (invisible) water loss (Table 3.4). Fluid loss from the body must be replaced daily to maintain normal fluid balance.
Table 3.4
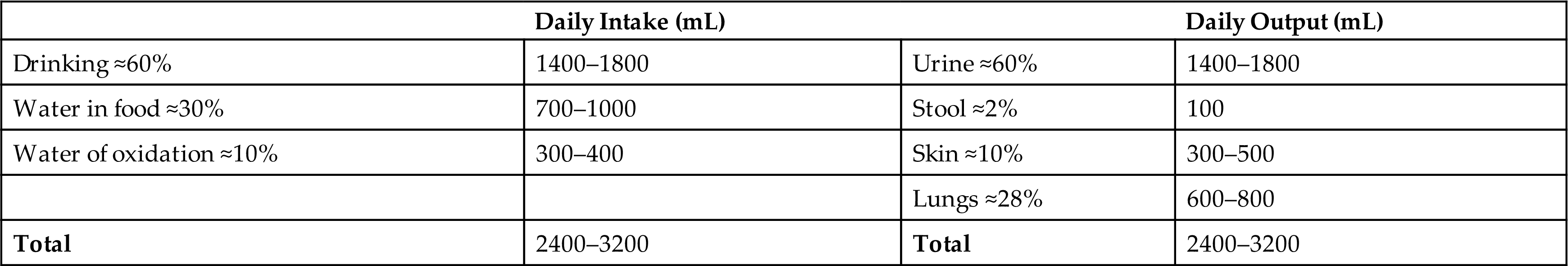
The amount of fluid within the various compartments is relatively constant with solutes (e.g., salts) and water being exchanged between compartments to maintain their unique compositions. The percentage of TBW varies with the amount of body fat and age. Fat is hydrophobic (water repelling), and very little water is contained in adipose (fat) cells. Individuals with more body fat have proportionately less TBW and tend to be more susceptible to dehydration.
Aging and Distribution of Body Fluids
The distribution and amount of TBW change with age (see Table 3.3). In newborn infants, TBW is approximately 70% to 80% of body weight because infants store less fat. In the immediate postnatal period, a physiologic loss of body water occurs, equivalent to approximately 5% of body weight as the infant adjusts to a new environment. Infants are particularly susceptible to significant changes in TBW because of their high metabolic rate and potential for evaporative fluid loss attributable to their greater body surface area in proportion to total body size. Loss of fluids from diarrhea can represent a significant proportion of body weight in infants. Renal mechanisms that regulate fluid and electrolyte conservation may not be mature enough to counter the losses, so dehydration can develop rapidly.
During childhood, TBW slowly decreases to 60% to 65% of body weight. At adolescence the percentage of TBW approaches adult proportions, and sex differences begin to appear. Males eventually have a greater percentage of body water as a function of increasing muscle mass. Females have more body fat and less muscle as a function of estrogens and therefore have less body water.
With increasing age, the percentage of TBW declines further still. The decrease is caused in part by an increased amount of fat and a decreased amount of muscle and by a reduced ability to regulate sodium and water balance. With older age, the kidneys become less efficient at conserving sodium and therefore have difficulty concentrating the urine. Insensible water loss through the skin may increase and thirst perception may be impaired. The normal reduction of TBW in older adults becomes clinically important when the body is under physiologic stress, such as development of fever or dehydration; loss of body fluids at such times can be severe and life-threatening.
Water Movement Between ICF and ECF
Water moves between ICF and ECF compartments primarily as a function of osmotic forces. (See Chapter 1.) Water moves freely by diffusion through the lipid bilayer cell membrane and through aquaporins, a family of water channel proteins that provide permeability to water.1 Sodium is responsible for the osmotic balance of the ECF space and potassium helps maintain the ICF osmotic balance. The osmotic force of ICF proteins and other nondiffusible substances is balanced by the active transport of ions out of the cell. Water crosses cell membranes freely; thus the osmolality of TBW normally is at equilibrium. Under normal conditions, the ICF is not subject to rapid changes in osmolality; however, when the ECF osmolality changes, water moves from one compartment to another until osmotic equilibrium is reestablished.
Water Movement Between Plasma and Interstitial Fluid
The distribution of water and the movement of nutrients and waste products between the plasma in the tissue capillaries and interstitial spaces occurs as a result of changes in hydrostatic pressure and osmotic/oncotic pressure at the arterial and venous ends of the capillary (see Fig. 1.25). Hydrostatic pressure pushes water out of the capillaries; osmotic/oncotic pressure pulls water into the capillaries. Water, sodium, and glucose readily move across the capillary membrane. Under normal conditions, plasma proteins (particularly albumin) do not cross the capillary membrane. They maintain physiologic osmolality by generating oncotic pressure within the plasma.
Filtration refers to fluid movement out of the capillary and into the interstitial space. Reabsorption refers to fluid movement into the capillary from the interstitial space. As plasma flows from the arterial to the venous end of the capillary, four forces, sometimes called Starling forces, determine whether the net effect is filtration or reabsorption.
- 1. Capillary hydrostatic pressure (blood pressure) facilitates the outward movement of water from the capillary to the interstitial space.
- 2. Capillary (plasma) oncotic pressure osmotically attracts water from the interstitial space back into the capillary.
- 3. Interstitial hydrostatic pressure facilitates the inward movement of water from the interstitial space into the capillary.
- 4. Interstitial oncotic pressure osmotically attracts water from the capillary into the interstitial space.
The forces controlling the net movement of fluid across the capillary wall are summarized by these equations:
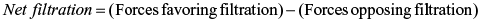
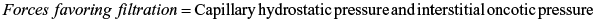
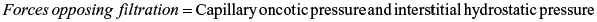
At the arterial end of the capillary, hydrostatic pressure exceeds capillary oncotic pressure and water moves into the interstitial space (filtration). At the venous end of the capillary, oncotic pressure within the capillary exceeds capillary hydrostatic pressure; thus fluids move into the capillary to enter into the circulation (reabsorption). Interstitial hydrostatic pressure promotes the movement of approximately 10% of the interstitial fluid, along with small amounts of protein, into the lymphatic vessels, which is then returned to the systemic circulation. These two systems, the lymphatic and circulatory systems, connect at a location near the left internal jugular vein where the lymphatic thoracic duct joins the left subclavian vein. Because significant amounts of albumin do not normally cross the capillary membrane, interstitial oncotic pressure normally is minimal. Fig. 3.1 illustrates net filtration.
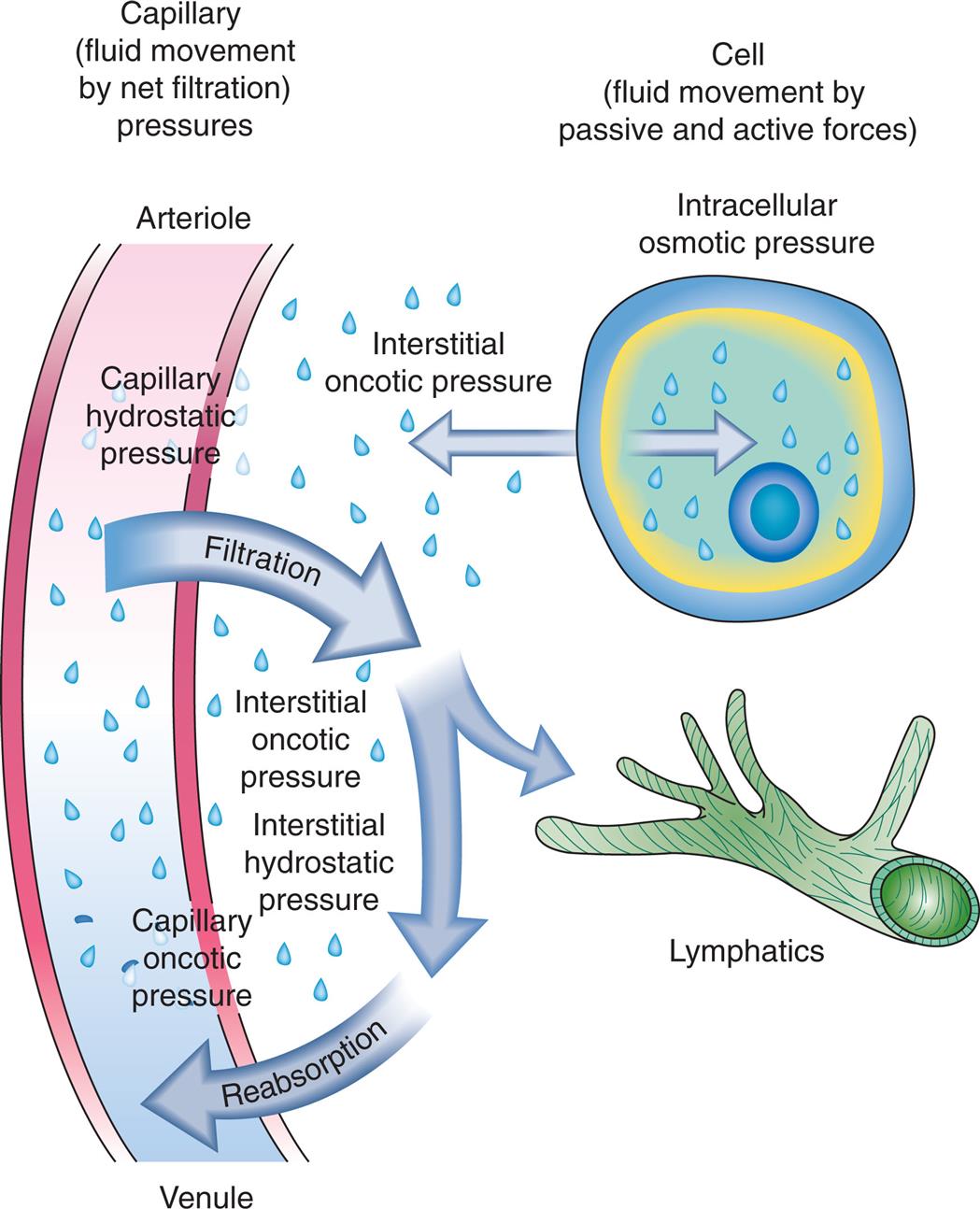
Water, electrolytes, and small molecules exchange freely between the vascular compartment and the interstitial space at the site of capillaries and small venules. The rate and amount of exchange are driven by the physical forces of hydrostatic and oncotic pressures and the permeability and surface area of the capillary membranes. The two opposing hydrostatic pressures are capillary hydrostatic pressure and interstitial hydrostatic pressure. The two opposing oncotic pressures are capillary oncotic pressure and interstitial oncotic pressure. The forces that favor filtration from the capillary are capillary hydrostatic pressure and interstitial oncotic pressure, and the forces that oppose filtration are capillary oncotic pressure and interstitial hydrostatic pressure. The sum of these forces is known as net filtration pressure. In the example of normal exchange illustrated here, a small amount of fluid moves to the lymph vessels, which accounts for the net filtration difference between the arterial and venous ends of the capillary.
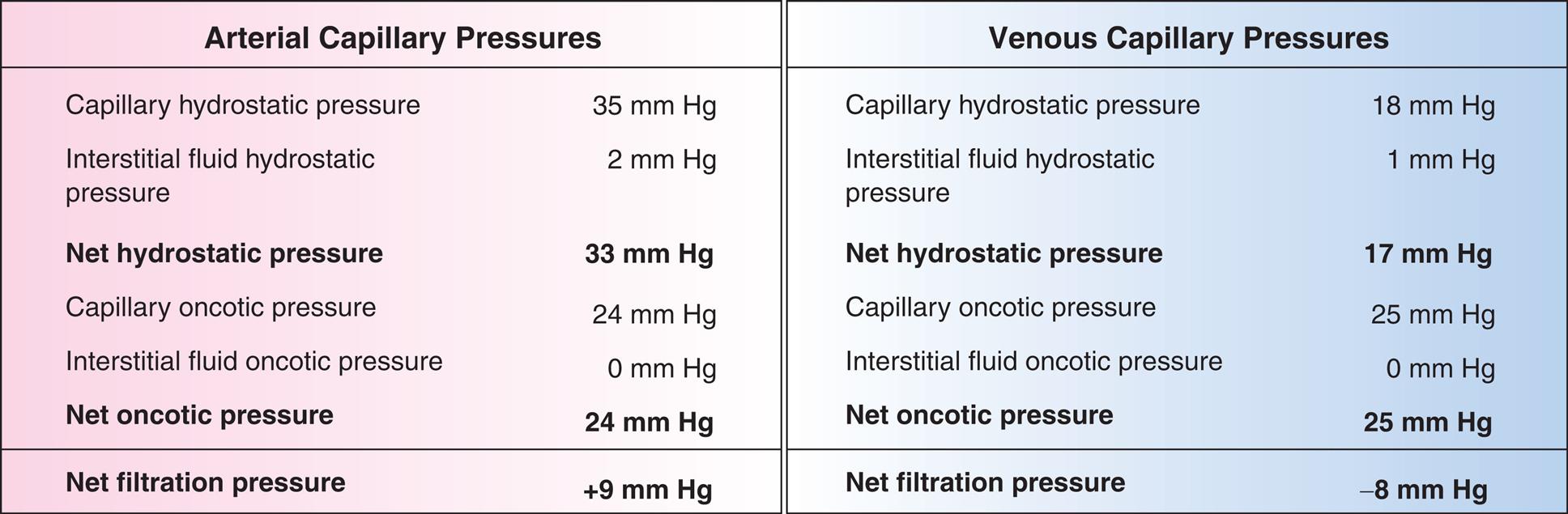
A three-part illustration and a two-part table demonstrate capillary filtration process. The first part of the illustration shows capillary fluid movement by net filtration pressures. The arteriole is identified at the top and the venule is identified at the bottom. Capillary hydrostatic pressure at the arteriole undergoes filtration, interstitial oncotic pressure, interstitial hydrostatic pressure, and capillary oncotic pressure, before reabsorption at the venule. The second part of the illustration shows a call with fluid movement by passive and active forces: intracellular osmotic pressure and interstitial oncotic pressure. The third part of the illustration shows lymphatics. The first part of the table shows arterial capillary pressures, and the second part of the table shows venous capillary pressures. The data from the table are as follows. Capillary hydrostatic pressure: 35 millimeters of mercury in arteriole and 18 millimeters of mercury in venule. Interstitial fluid hydrostatic pressure: 2 millimeters of mercury in arteriole and 1 millimeters of mercury in venule. Net hydrostatic pressure: 22 millimeters of mercury in arteriole and 17 millimeters of mercury in venule. Capillary oncotic pressure: 24 millimeters of mercury in arteriole and 25 millimeters of mercury in venule. Interstitial fluid oncotic pressure: 0 millimeters of mercury in arteriole and 0 millimeters of mercury in venule. Net oncotic pressure: 24 millimeters of mercury in arteriole and 25 millimeters of mercury in venule. Net filtration pressure: positive 9 millimeter of mercury in arteriole and negative 8 millimeters of mercury in venule.
An important factor in capillary filtration of fluid is the integrity of the capillary membrane. Increases in membrane permeability may permit the escape of plasma proteins into the interstitial space. The normal net filtration is altered, with the osmotic movement of water into the interstitial space causing tissue edema.
Alterations in Water Movement
Edema
Edema is the excessive accumulation of fluid within the interstitial spaces. It results from a shift of fluid from the capillaries (intravascular fluid) or lymphatic vessels into the tissues (Fig. 3.2). The four most common mechanisms are:
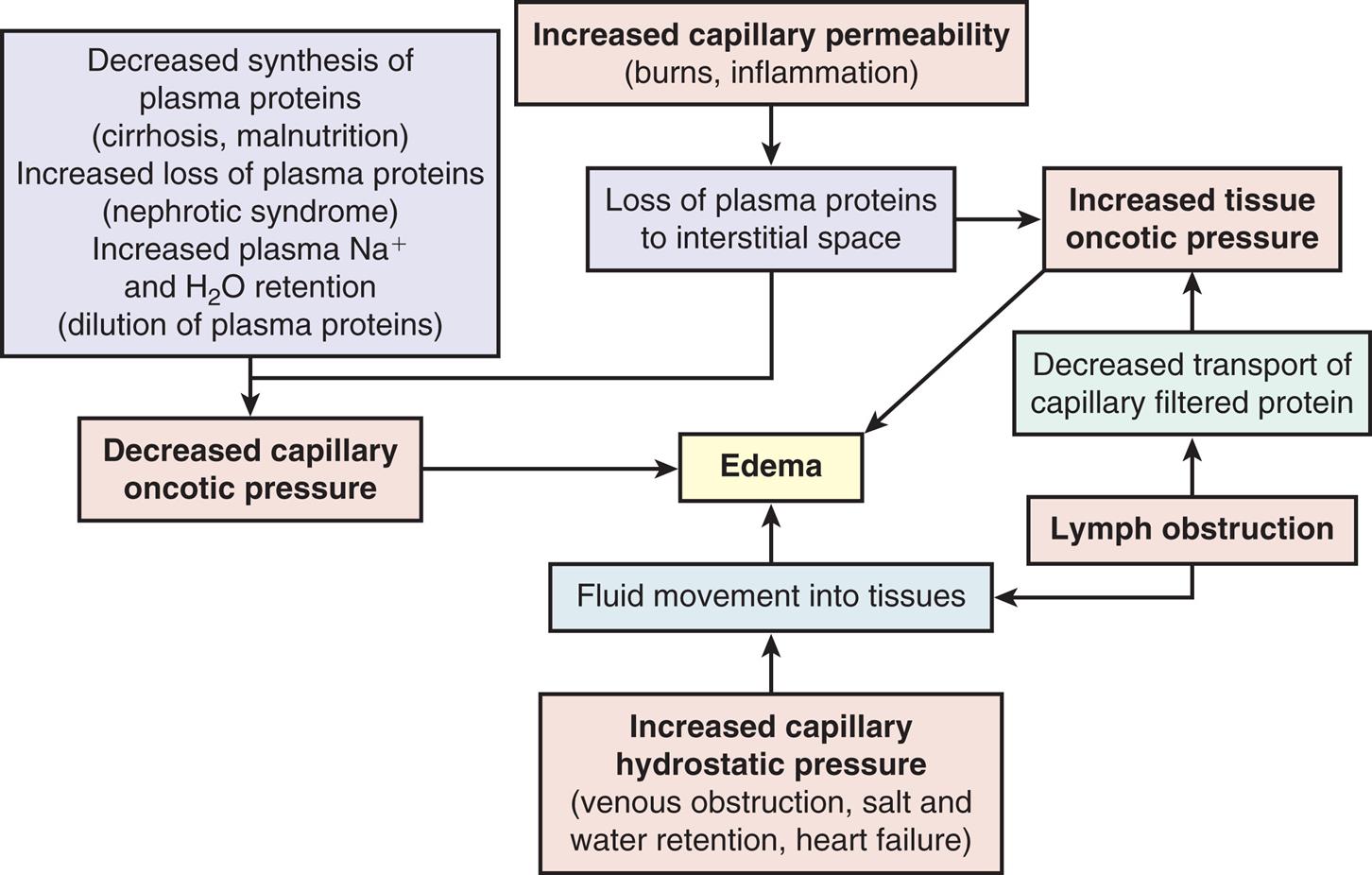
A process diagram illustrates the mechanisms of edema formation. 1. Increased capillary permeability, including burns and inflammation. 1 leads to 2. 2. Loss of plasma proteins to interstitial space. 2 leads to 3 and 5. 3. Increased tissue oncotic pressure. 4. Decreased synthesis of plasma proteins, including cirrhosis, malnutrition. Increased loss of plasma proteins, including nephrotic syndrome. Increased plasma sodium ions and water retention, including dilution of plasma proteins. 4 also leads to 5. 5. Decreased capillary oncotic pressure. 6. Increased capillary hydrostatic pressure, including venous obstruction, salt and water retention, and heart failure. 6 leads to 8. 7. Lymph obstruction. 7 leads to 8 and 9. 8. Fluid movement into tissues. 3, 5, and 8 lead to 10. 9. Decreased transport of capillary filtered protein. 10. Edema.
1.Increased capillary hydrostatic pressure
2.Decreased capillary oncotic pressure
3.Increased capillary membrane permeability
4.Lymph channel obstruction
Pathophysiology
Increased capillary hydrostatic pressure results from either venous obstruction or sodium and water retention. Venous obstruction results in an increased hydrostatic pressure behind the obstruction, pushing fluid from the capillaries into the interstitial spaces. Common causes of venous obstruction include thrombophlebitis, blood clots, hepatic venous outflow obstruction, right heart failure, tight clothing around the extremities, and prolonged standing. Excessive sodium and water retention result in edema secondary to plasma volume overload. The overload produces an increased capillary hydrostatic pressure. Common predisposing causes include heart failure, oliguric kidney disease, and cirrhosis of the liver. In these circumstances, the volume of interstitial fluid exceeds the capacity of the lymphatics to return fluid to the vascular system.
Decreased capillary oncotic pressure occurs when plasma proteins, especially albumin, are lost or production is diminished. Decreased synthesis of plasma proteins occurs with severe liver disease or protein malnutrition. Losses of plasma proteins occur with glomerular diseases of the kidney (nephrotic syndrome), hemorrhage, serous drainage from open wounds or burns, and occasionally from protein-losing enteropathies.2 Decreased oncotic attraction of fluid within the capillary causes more fluid to filter into the interstitial space than is reabsorbed into the systemic circulation through the capillaries. As fluid accumulates within the tissues, edema worsens.
Increased capillary membrane permeability resulting in edema occurs with inflammation and immune responses. (Immunity is discussed in Chapters 7, 8, and 9; inflammation is discussed in Chapters 7 and 9.) The increased vessel permeability permits significant amounts of proteins to escape from within blood vessels (the vascular space) into the interstitial space, producing edema. This edema results primarily from interstitial fluid protein accumulation, which increases the oncotic pressure in the interstitial space; localized decreased capillary oncotic pressure may also contribute. These responses are often the result of trauma such as burns or crushing injuries, neoplastic disease, allergic reactions, and infection.
Lymphatic channel obstruction causing edema occurs when the lymphatic channels are blocked. The lymphatic system normally absorbs interstitial fluid, along with small amounts of protein. These substances travel through a one-way system of progressively larger lymphatic vessels until they are returned to the systemic circulation. When lymphatic channels are blocked or surgically removed, proteins and fluids are not reabsorbed and accumulate in the interstitial space, causing lymphedema. Lymphedema of the arm or leg can occur after surgical removal of axillary or femoral lymph nodes, associated with the surgical treatment of cancer.3
Clinical Manifestations
Edema may be localized or generalized. Localized edema is usually limited to the site of tissue injury, as in a sprained joint. Local edema can also occur within particular organs, causing for example, vasogenic cerebral edema in the brain, pulmonary edema in the lungs, or laryngeal edema. Edema of specific organs, such as the brain, lung, or larynx, can be life-threatening. Generalized edema is manifested by a more uniform distribution of fluid in interstitial spaces throughout the body. Anasarca refers to a severe generalized edema. Dependent edema, in which fluid accumulates in gravity-dependent areas of the body, might appear in the feet and legs when standing and in the sacral area and buttocks when supine. Dependent edema can be identified by using the fingers to press into edematous fluid in tissues overlying bony prominences. A pit will be left in the skin, hence the term pitting edema (Fig. 3.3).
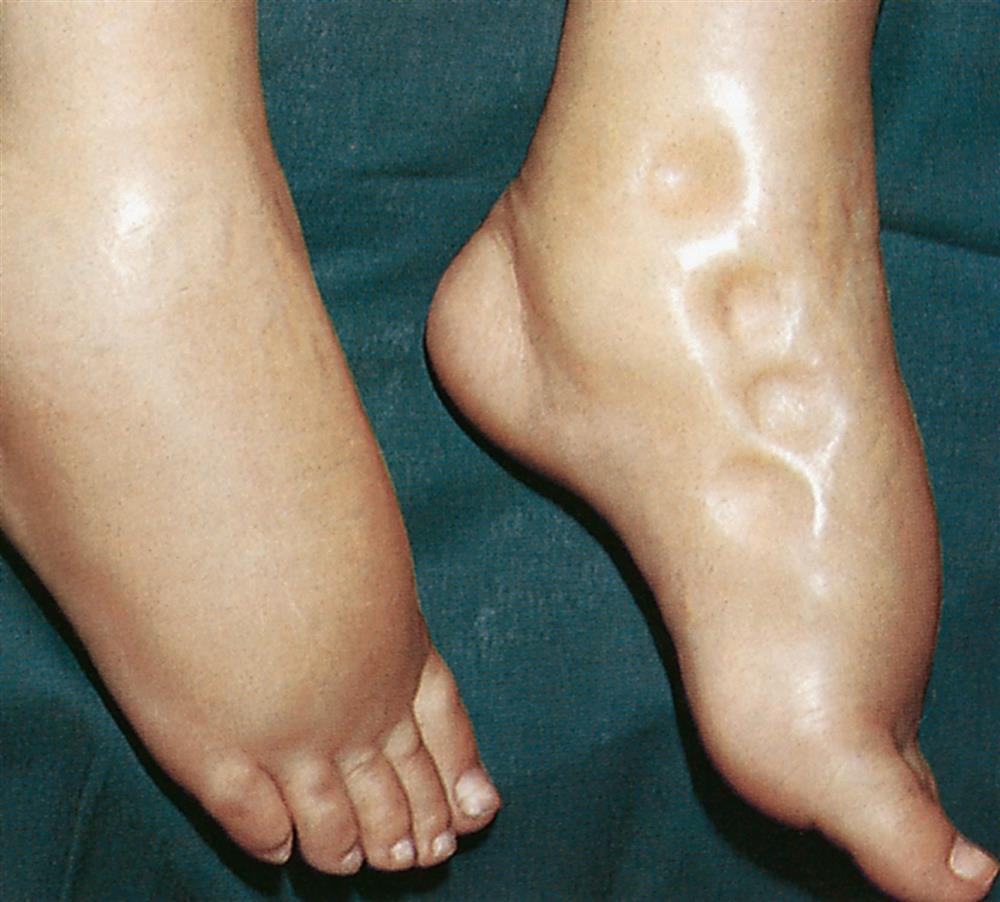
Edema is associated with sudden weight gain, swelling and puffiness, tight-fitting clothes and shoes, and limited movement of affected joints. It usually presents along with symptoms associated with the underlying pathologic condition. Fluid accumulation increases the distance required for nutrients, oxygen, and wastes to move between capillaries and cells in the tissues. Increased tissue pressure also may diminish capillary blood flow, leading to ischemia. Therefore wounds heal more slowly and risk of infection or pressure injury increases.
Fluid accumulation within a body cavity or space is referred to as an effusion. Examples include a pleural effusion (fluid accumulation in the pleural space) and a pericardial effusion (fluid accumulation within the membrane surrounding the heart). Fluid accumulation in the peritoneal cavity is called ascites. The region where edema fluid or an effusion accumulates sometimes is referred to as a third space; the term third spacing is used to refer to the process of this fluid accumulation. Fluid trapped in such spaces is unavailable for metabolic processes or perfusion. Common third spacing situations include ascites, large local interstitial edema, pleural effusion, pericardial effusion, and rapid movement of fluid into the intestinal lumen with an acute intestinal obstruction. Although a person with generalized edema appears swollen because of excess fluid, hypovolemia (decreased intravascular volume) can develop as a result of the fluid sequestering. With severe burns, large amounts of vascular fluid are lost into the interstitial spaces and shock often occurs secondary to the resulting reduced blood volume (see Chapter 48).
Clinically, lymphedema presents differently from edema secondary to fluid accumulation from the capillaries. With edema from capillary sources, the edematous tissue is easily compressed (i.e., pitting edema). By contrast, lymphedema is firm and noncompressible. In severe cases, lymphedema can be profound and causes gross enlargement and distortion of the body parts affected.
Evaluation and Treatment
Medical treatment for edema varies, depending on the underlying conditions that cause fluid retention. Generalized edema often is treated symptomatically, commonly with diuretics. Diuretics may be used until the underlying disorder has been corrected or continued chronically if the underlying disorder persists. In addition to diuretics, symptomatic treatment includes elevating edematous limbs, applying compression stockings, avoiding prolonged standing, and restricting salt intake.
Sodium, Chloride, and Water Balance
The combined influences of the kidneys and hormones have a central role in maintaining sodium and water balance. Because water follows the osmotic gradients established by changes in salt concentration, sodium concentration and water balance are integrally related. The amount of sodium in the ECF is regulated by the renal effects of aldosterone from the adrenal cortex and natriuretic peptides from the heart. Water balance is primarily regulated by the renal response to antidiuretic hormone (ADH; also known as arginine-vasopressin) from the posterior pituitary and is discussed in the Water Balance section.
Sodium accounts for 90% of the ECF cations (positively charged ions) (Table 3.5). The amount of sodium in the ECF is regulated by renal tubular reabsorption of sodium and water within the kidney in response to neural and hormonal influences. That process is a major regulator of ECF volume. The concentration of sodium in the ECF reflects water balance, which is regulated by ADH and is discussed later. Sodium in concert with chloride and bicarbonate, the two major anions (negatively charged ions), influences water distribution by contributing to extracellular osmotic forces. Sodium has an important role in other physiologic functions, including nerve impulse conduction, regulation of acid-base balance, cellular chemical reactions, and transport of substances across the cellular membrane (see Chapter 1).
Table 3.5
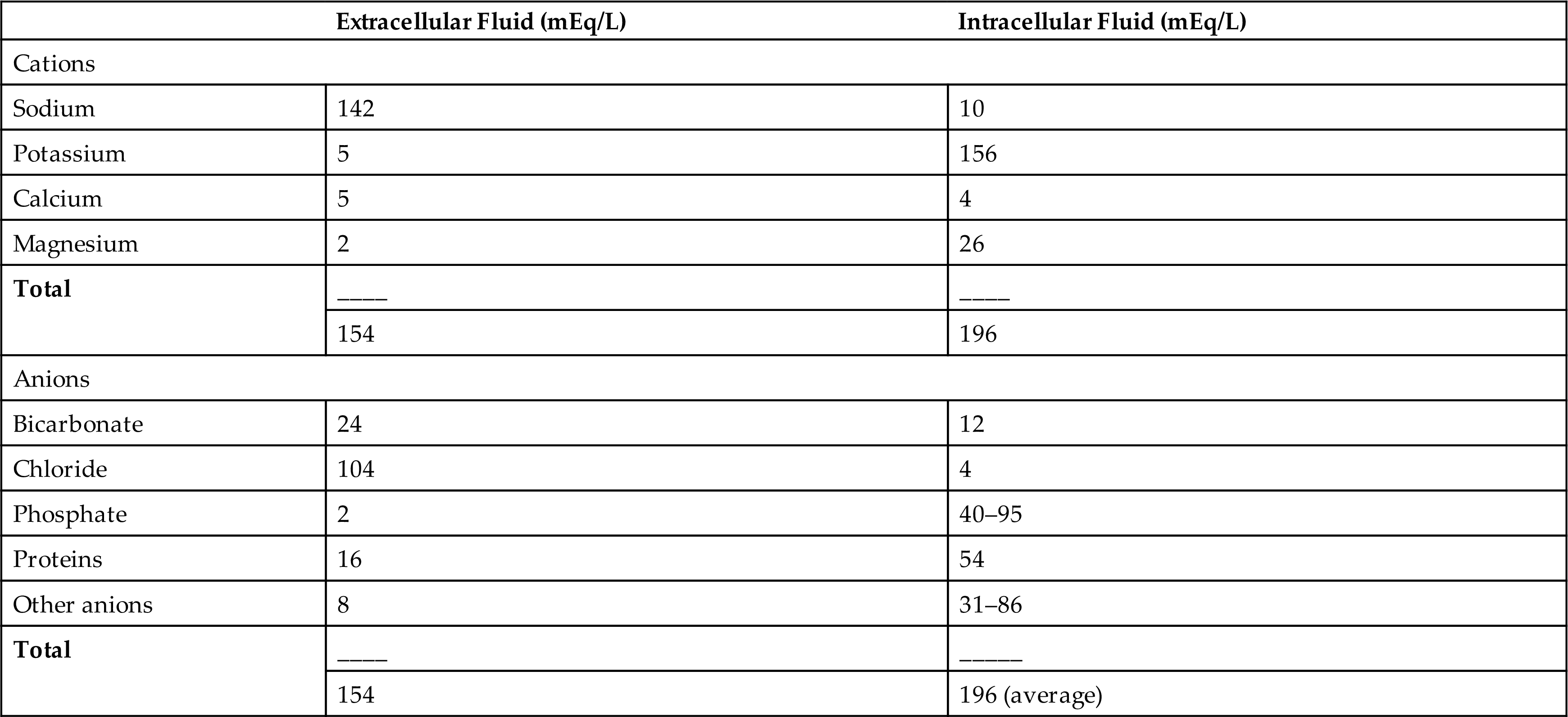
In general, sodium intake matches sodium excretion. The average dietary intake of sodium ranges from 5 to 6 g/day; the minimal daily requirement of sodium is 500 mg. Sweating depletes sodium and water volume and increases the body's sodium requirement.
Hormonal regulation of sodium (and potassium) balance is mediated by aldosterone, a mineralocorticoid (steroid) synthesized and secreted from the adrenal cortex. Aldosterone is a component of the renin-angiotensin-aldosterone system (Fig. 3.4). Aldosterone secretion is influenced by a number of factors, including circulating blood volume, blood pressure, and plasma concentrations of sodium and potassium. When the circulating blood volume or blood pressure is reduced, increased amounts of renin, an enzyme secreted by the juxtaglomerular cells of the kidney, are released. More renin also is released when sodium levels in the renal tubules are depressed or there is increased firing of the renal sympathetic nerves. Once released, renin stimulates the formation of angiotensin I, an inactive polypeptide from angiotensinogen, a substance secreted by the liver. Angiotensin-converting enzyme (ACE), found primarily in pulmonary vessels and to a lesser extent in endothelial and renal epithelial cells, converts angiotensin I to angiotensin II, a potent vasoconstrictor. Vasoconstriction elevates blood pressure and restores renal perfusion. Restoring renal perfusion inhibits further release of renin. Angiotensin II also stimulates both the secretion of aldosterone from the adrenal cortex and ADH from the posterior pituitary. Aldosterone promotes sodium and water reabsorption, in addition to the excretion of potassium within the renal tubules. The net effect is to increase blood volume and blood pressure (see Fig. 3.4).
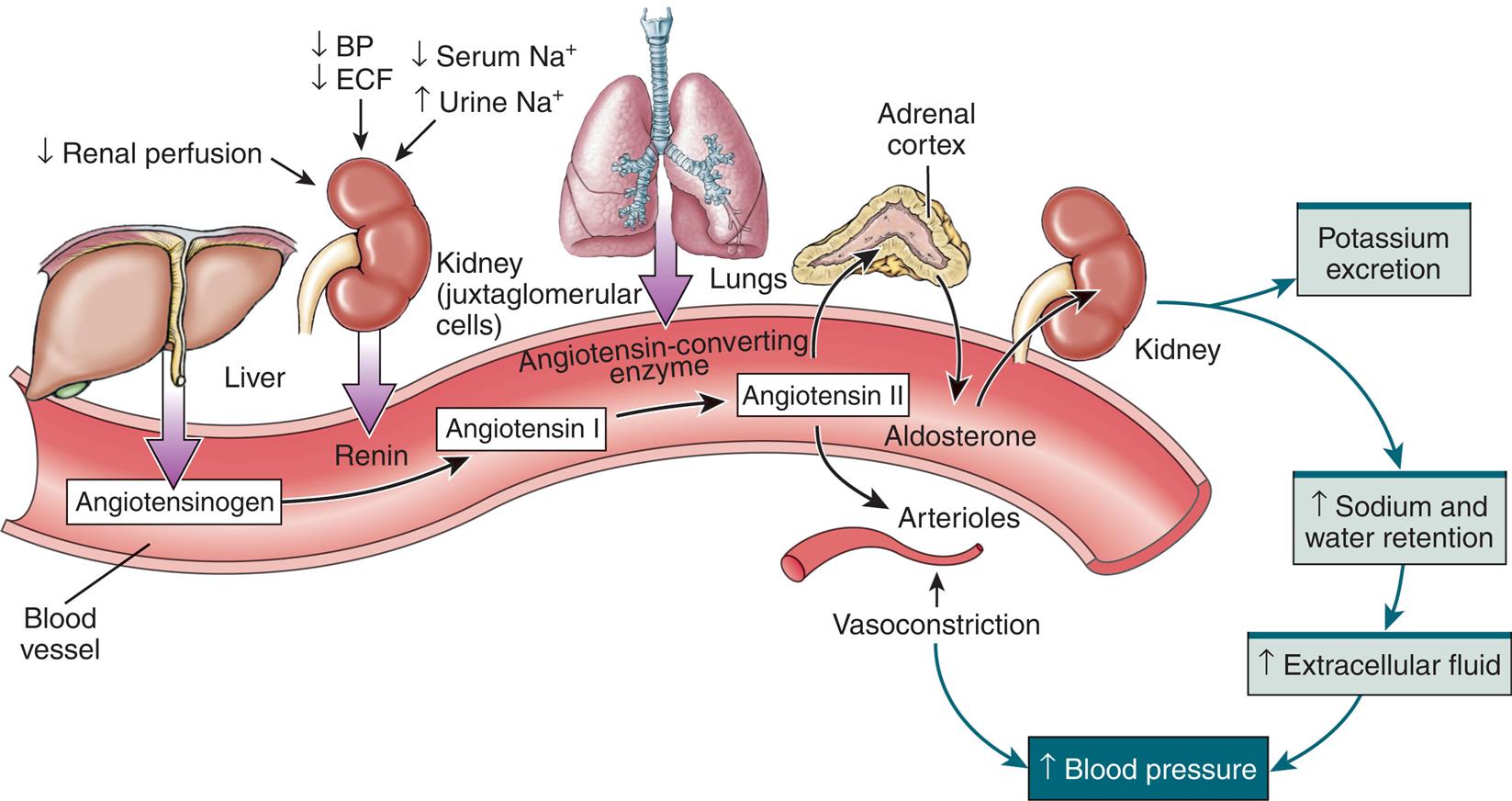
BP, Blood pressure; ECF, extracellular fluid; Na+, sodium ion. (Modified from Herlihy B, Maebius N. The human body in health and disease, 4th edition. Philadelphia: Saunders; 2011. Borrowed from Lewis, et al. Medical-surgical nursing: Assessment and management of clinical problems, 9th edition. St. Louis: Mosby; 2014.)
An illustrated flow diagram represents the renin-angiotensin-aldosterone system. The illustration shows a blood vessel. Liver secretes angiotensinogen in the blood vessel. Decreased renal perfusion, decreased blood pressure, decreased extracellular fluid, decreased serum sodium ions, and increased urine sodium ions in the juxtaglomerular cells of the kidney secretes renin in the blood vessel, converts angiotensinogen to angiotensin 1. Angiotensin-converting enzyme from the lung contribute to angiotensin 2. Angiotensin 2 affects adrenal cortex and arterioles. Adrenal cortex secretes to aldosterone, which affects the kidney. Kidney excretes potassium and leads to increased sodium and water retention, which results in increased extracellular fluid, contributing to increased blood pressure. Angiotensin 2 acts on arterioles, causing vasoconstriction, and results in increased blood pressure.
Drugs used for the treatment of hypertension include ACE inhibitors and angiotensin receptor blockers (ARBs). Both of these classes of drugs inhibit the renin-angiotensin-aldosterone system (RAAS) and lower blood pressure.
Natriuretic peptides are hormones primarily produced by the myocardium. Atrial natriuretic peptide (ANP) is produced by the atria. B-type natriuretic peptide (BNP) is produced by the ventricles. Urodilatin (an ANP analogue) is synthesized within the kidney. Natriuretic peptides are released from the heart when the transmural atrial pressure increases (increased volume) or the tension in the left ventricular wall increases, which commonly occurs with heart failure, or when the mean arterial pressure increases (Fig. 3.5). Measurement of BNP is used as a means to assist with the initial diagnosis and management of people with heart failure. Urodilatin is released from distal tubular kidney cells when there is increased arterial pressure and increased renal blood flow. These hormones are natural antagonists to the RAAS; although they are less powerful than aldosterone, they can moderate its physiologic effect on extracellular volume. Natriuretic peptides cause vasodilation and increase renal sodium and water excretion, decreasing blood pressure. Natriuretic peptides sometimes are called a “third factor” in sodium regulation. An increased glomerular filtration rate is considered to be the first factor, and aldosterone is considered to be the second factor.
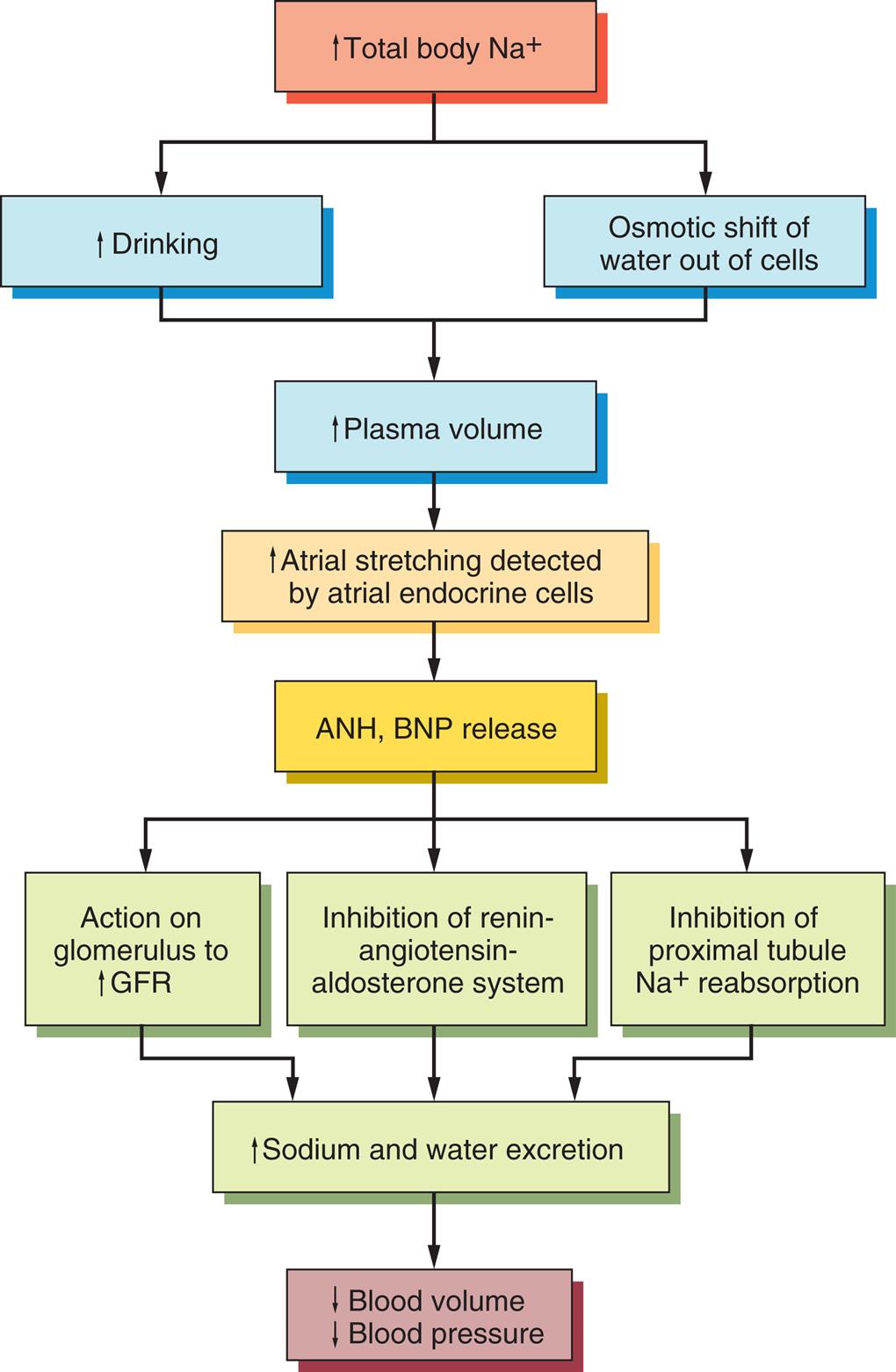
ANP, Atrial natriuretic peptide; BNP, brain natriuretic peptide; GFR, glomerular filtration rate.
A flowchart represents the natriuretic peptide system as: • Increase total body sodium ions. • Increased drinking and osmotic shift of water out of cells. These give out 3 results (leading to each other) as: • Increased plasma volume. • Increased atrial stretching detected by atrial endocrine cells. • Atrial natriuretic peptide, brain natriuretic peptide release. This leads to: • Action on glomerulus to increase G F R; inhibition of renin-angiotensin-aldosterone system; inhibition of proximal tubule sodium ions reabsorption. These give out 2 results (leading to each other) as: • Increased sodium and water excretion. • Decreased blood volume and decreased blood pressure.
Chloride (Cl−) is the major anion in the ECF and provides electroneutrality, particularly in relation to sodium. Chloride transport is generally passive, except in the renal tubules, and follows the active transport of sodium so that increases or decreases in chloride concentration are proportional to changes in sodium concentration. Chloride concentration tends to vary inversely with changes in the concentration of bicarbonate ( ), the other major ECF anion.
), the other major ECF anion.
Water balance is regulated by the secretion of ADH, also known as arginine-vasopressin. ADH is synthesized by neurons with cell bodies in the hypothalamus that release ADH from their axon terminals in the posterior pituitary. It is secreted in larger amounts when plasma osmolality increases (the major stimulus) or if circulating blood volume decreases enough to cause a drop in blood pressure (Fig. 3.6). Increased plasma osmolality occurs when there is a decrease in water or an excess amount of sodium in relation to TBW. The increased osmolality stimulates hypothalamic osmoreceptors, which cause increased release of ADH. This hormone increases water reabsorption from the renal distal tubules and collecting ducts into the plasma (see Chapter 37). Stimulation of the hypothalamic osmoreceptors also causes thirst, which stimulates the individual to consume liquids. The net effect of both increased water intake and increased renal reabsorption of water is to decrease plasma osmolality, returning it to a normal status. ADH increases urine concentration and decreases urine volume as less water is excreted into the urine. The restoration of plasma osmolality, blood volume, and blood pressure then reduces ADH secretion back to its baseline level.
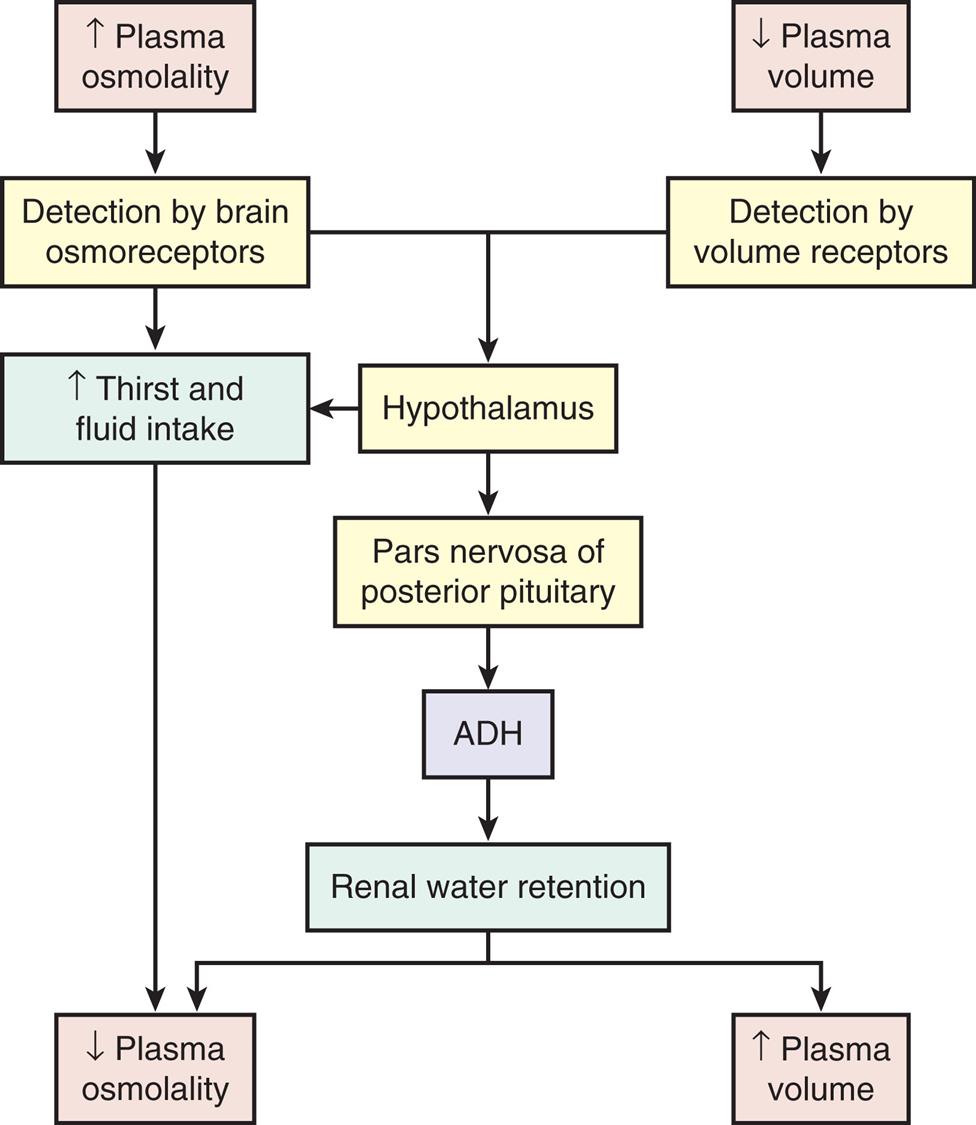
A flowchart represents the sequence of processes within the antidiuretic hormone or A D H system. The data from the flowchart are summarizes as follows. Increased plasma osmolality is detected by brain osmoreceptors, which spikes thirst and fluid intake, leading to decreased plasma osmolality. Increased plasma osmolality, detected by brain osmoreceptors, and decreased plasma volume, detected by volume receptors, affects the hypothalamus either by spiking thirst and fluid intake or by causing pars nervosa of posterior pituitary. Increased thirst and fluid intake reduces plasma osmolality. Pars nervosa of posterior pituitary causes A D H to retain renal water. Renal water retention leads to decreased plasma osmolality or increased plasma volume.
With fluid loss secondary to vomiting, diarrhea, or excessive sweating, a decrease in systemic blood volume and blood pressure often follows. Volume-sensitive receptors and baroreceptors, both of which are nerve endings sensitive to changes in blood volume and pressure, also stimulate thirst and the release of ADH, which prompts fluid consumption. The volume receptors are located in the right and left atria and in the thoracic vessels; baroreceptors are found in the aorta, pulmonary arteries, and carotid sinus. When arterial and atrial pressure drops, baroreceptors signal the hypothalamus to release ADH. The reabsorption of water mediated by the renal response to ADH promotes the restoration of plasma volume and blood pressure (see Fig. 3.6). Higher concentrations of ADH stimulate peripheral arterial vasoconstriction, thus increasing arterial blood pressure.
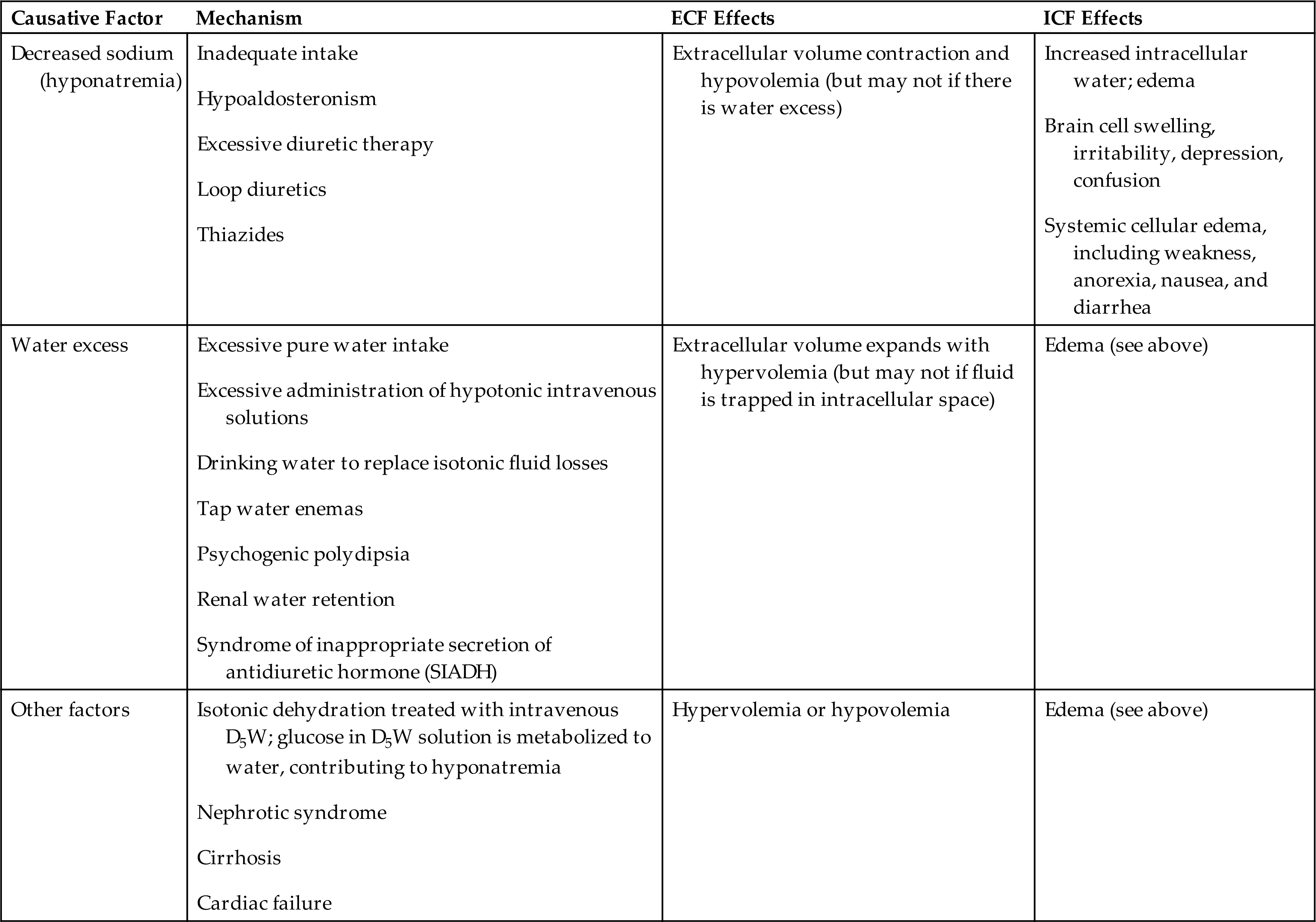
Alterations in Sodium, Water, and Chloride Balance
Alterations in sodium and water balance are closely related. Serum sodium imbalances occur with gains or losses of body water. Extracellular volume imbalances develop with gains or losses of salt. These alterations can be classified as changes in tonicity (i.e., the change in the concentration of solutes in relation to the amount of water present).4 Normal plasma osmolality is 275 to 295 milliosmoles (mOsm)/kg. Solutions are classified as isotonic, hypertonic, or hypotonic as a function of the solute concentration compared with that of normal body cells (Table 3.6 and Fig. 3.7). Isotonic solutions have solute concentrations that are equal to that of normal cells; hypertonic or hypotonic solutions have higher or lower solute concentration, respectively. Changes in tonicity (solute concentration of solutes that cannot cross semipermeable membranes) affect the volume of water within the extracellular compartment, resulting in isovolemia, hypervolemia, or hypovolemia (i.e., normal volume, excess volume, or less than normal volume in the blood, respectively).
Table 3.6

BUN, Blood serum urea nitrogen (mg/dL); [Glu], serum glucose concentration (mg/dL); [Na], serum sodium concentration (mEq/dL).
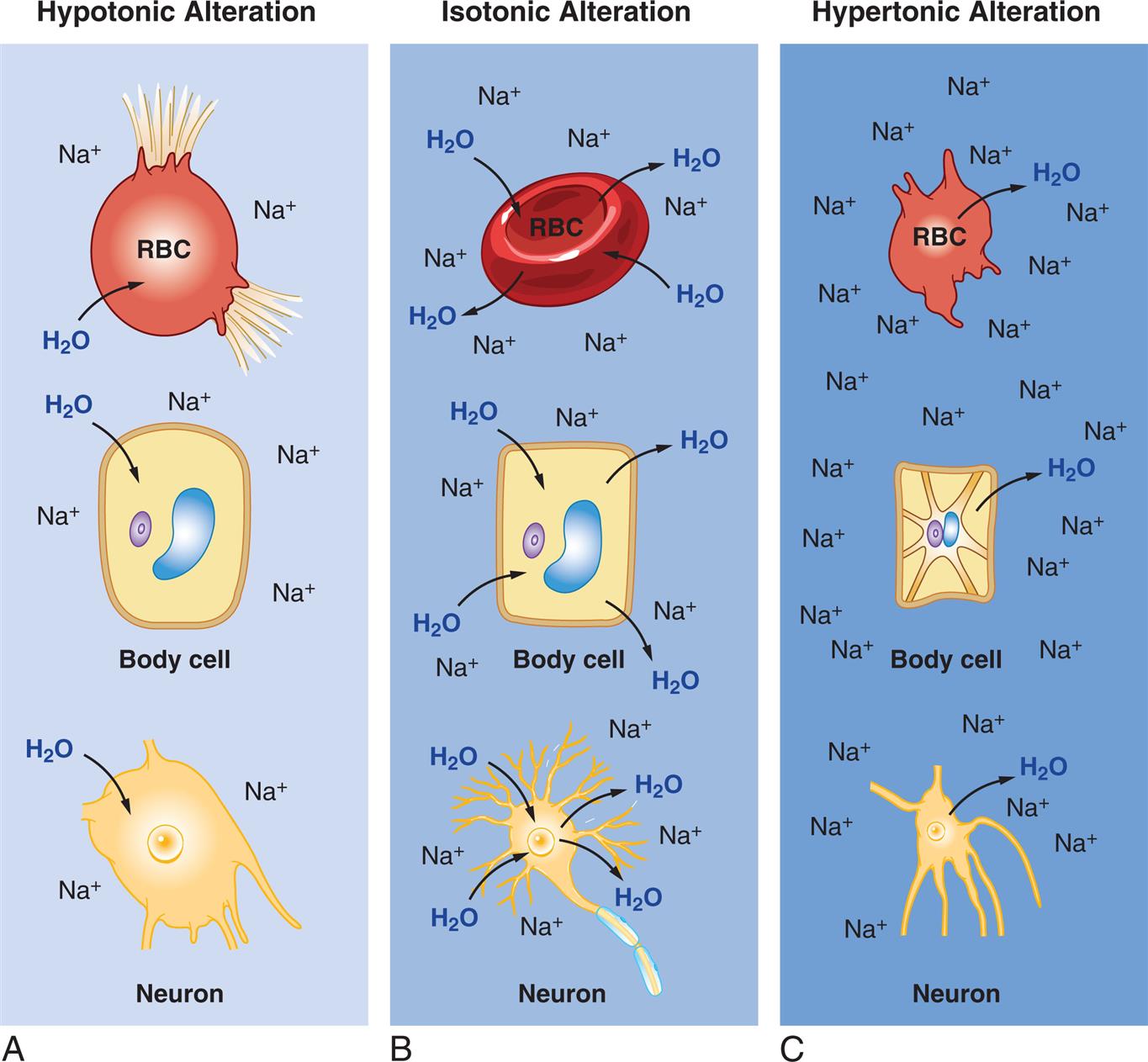
(A)Hypotonic Alteration: Decrease in ECF sodium ion (Na+) concentration (hyponatremia) results in ICF osmotic attraction of water with swelling and potential busting of cells. (B)Isotonic Alteration: Normal concentration of sodium in the ECF and no change in shifts of fluid in or out of cells. (C)Hypertonic Alteration: An increase in ECF sodium concentration (hypernatremia) results in osmotic attraction of water out of cells with cell shrinkage. ECF, Extracellular fluid; ICF, intracellular fluid; RBC, red blood cell.
Three illustrations, A, B, and C, demonstrate the effects of hypotonic, isotonic, and hypertonic alterations, respectively, in R B C, body cell, and neuron. Illustration A, hypotonic alteration. When the concentration of sodium ions is less, the R B C, the body cell, and the neuron attract water, leading to swelling and or busting of the cells. Illustration B, isotonic alteration. When the concentration of sodium ions is normal, the osmolality of water in R B C, body cell, and neuron, is even. Illustration C, hypertonic alteration. When the concentration of sodium ions is high, the R B C, the body cell, and the neuron expel water, leading to shrinking of the cells.
Isotonic Alterations
Isotonic alterations are the most common and occur when changes in TBW are accompanied by proportional changes in the concentration of electrolytes. Isotonic fluid loss causes hypovolemia. For example, if an individual loses pure plasma or ECF, fluid volume is depleted, but the number and type of electrolytes (e.g., sodium) and the osmolality remain within a normal range (275 to 295 mOsm/kg). Causes include hemorrhage, severe wound drainage, and excessive diaphoresis (sweating). There is loss of extracellular volume with weight loss, dryness of skin and mucous membranes, decreased urine output, increased hematocrit value, and symptoms of hypovolemia. Indicators of hypovolemia include a rapid heart rate and flattened neck veins and can present with a normal or decreased blood pressure. In severe states, hypovolemic shock (severe hypotension) can occur. Isotonic fluids containing electrolytes and glucose are given orally, intravenously (IV) (i.e., 0.9% saline solution or Ringer’s lactate [RL] solution), and in some cases, subcutaneously (hypodermoclysis). A variety of isotonic oral fluid replacement solutions are available as beverages and pediatric oral replacement solutions without a prescription.
Isotonic fluid excesses cause hypervolemia. Causes include excessive administration of sodium-containing IV fluids, hypersecretion of aldosterone, the effects of drugs such as prednisone and other corticosteroids (cause renal reabsorption of sodium and water), or oliguric kidney disease. Increased dietary sodium intake causes mild hypervolemia in many individuals , until the resulting decreased aldosterone secretion allows renal excretion of the excess sodium and water. As plasma volume expands, hypervolemia with sudden weight gain develops, because a liter of fluid weighs 1 kg. The diluting effect of excess plasma volume leads to a decreased hematocrit and decreased plasma protein concentration. The neck veins may distend, and the blood pressure increases. Increased capillary hydrostatic pressure leads to edema formation. If the plasma volume is great enough, pulmonary edema and heart failure develop. These presentations may be acute or chronic, depending on the underlying pathophysiology causing the fluid overload. Diuretics are commonly used for treatment.
Hypertonic Alterations
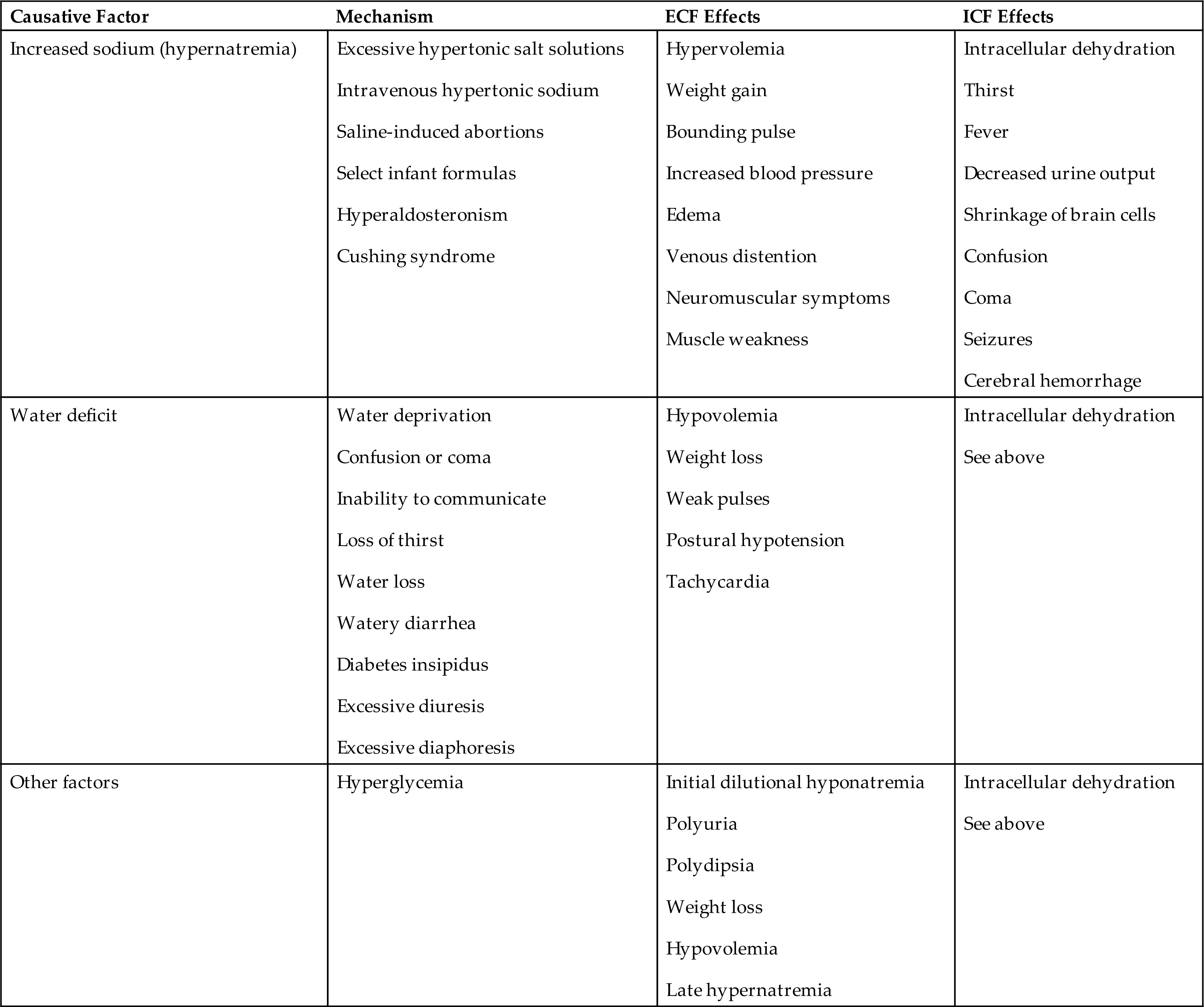
Hypertonic fluid alterations develop when the osmolality of the ECF is elevated above normal (greater than 295 mOsm/kg). The most common causes are hypernatremia (increased concentration of ECF sodium), a deficit of ECF water, or both. In either case the ECF hypertonicity attracts water from the intracellular space, causing ICF dehydration. A primary increase in the amount of ECF sodium causes an osmotic attraction of water and symptoms of hypervolemia. In contrast, a hypertonic state caused primarily by loss of electrolyte-free water leads to hypovolemia (Table 3.7).
Table 3.7
Hypernatremia
Pathophysiology
Hypernatremia occurs when serum sodium levels exceed 145 mEq/L. Increased levels of serum sodium cause hypertonicity. Hypernatremia can be hypovolemic, euvolemic, or hypervolemic, depending on the accompanying ECF water volume. Risk factors include advanced age, impaired mental state, fever, diarrhea, and vomiting.
Hypovolemic hypernatremia occurs when a loss of sodium is accompanied by a relatively greater loss of body water. Causes include use of loop diuretics (diuretics that inhibit sodium and chloride reabsorption in the kidney loop of Henle) or diuretic stages of renal disease in which the kidneys fail to concentrate urine and excrete a large volume of urine, and osmotic diuresis from diabetes mellitus induced hyperglycemia or use of mannitol (an osmotic diuretic).
Isovolemic hypernatremia is the most common and occurs when there is a loss of electrolyte-free water with a near normal body sodium concentration. Causes include inadequate water intake; excessive sweating (sweat is hypotonic); fever with hyperventilation and increased water loss from lungs; burns; vomiting; diarrhea; and central or nephrogenic diabetes insipidus (lack of ADH or inadequate renal response to ADH). Infants with severe diarrhea are vulnerable and at increased risk because they cannot communicate thirst. Insufficient water intake occurs particularly in individuals who are comatose, confused, immobilized, or receiving gastric feedings. The term dehydration can refer to water deficit but also is commonly used to indicate both sodium and water loss (isotonic or isoosmolar dehydration) or even hypovolemic hypernatremia.
Hypervolemic hypernatremia is rare. It occurs when there is an increase in TBW accompanied by a greater increase in total body sodium, resulting in hypervolemia with elevated serum sodium concentration. One cause is infusion of hypertonic saline solutions in an effort to replace sodium in cases of salt depletion that can occur with renal impairment or heart failure. Other causes include oversecretion of adrenocorticotropic hormone (ACTH) or aldosterone as can occur with Cushing syndrome or adrenal hyperplasia and deliberate ingestion of large amounts of soy sauce or concentrated salt water. High amounts of dietary sodium rarely cause hypervolemic hypernatremia in a healthy individual because the aldosterone and ADH responses allow the kidneys to correct the osmolality. Symptoms of hypervolemic hypernatremia include weight gain, bounding pulse, increased blood pressure, and may include lethargy and other cerebral manifestations of hypernatremia.
Because chloride follows sodium, hyperchloremia (elevation of serum chloride concentration greater than 105 mEq/L) often accompanies hypernatremia. Plasma bicarbonate deficits, as occur in hyperchloremic metabolic acidosis, are discussed later in the chapter. There are no specific symptoms for chloride excess, and treatment is related to management of the underlying disorder.
Clinical Manifestations
Central nervous system signs are the most serious signs of hypernatremia and are related to shrinking of brain cells, brain dehydration, and alterations in membrane potentials. Signs include weakness, lethargy, muscle twitching, and hyperreflexia (hyperactive reflexes). Confusion, coma, and seizures can occur. Hypernatremia with marked water deficit is manifested by signs and symptoms of both intracellular and extracellular dehydration with volume depletion (Box 3.1).
Evaluation and Treatment
Serum sodium levels are greater than 145 mEq/L and urine specific gravity is greater than 1.030, unless the cause is diabetes insipidus. Hematocrit and plasma protein levels will be elevated if there is an accompanying water loss. The history, physical examination, and laboratory values provide information about underlying disorders and events. The treatment for isovolemic and hypovolemic hypernatremia is to give oral water or an isotonic salt-free fluid (5% dextrose in water) until the serum sodium concentration returns to normal. Fluid replacement must be given slowly to prevent cerebral edema. Treatment for hypervolemic hypernatremia is to administer loop diuretics. Serum sodium levels need to be closely monitored and the underlying clinical condition managed.
Hypotonic Alterations
Hypotonic fluid imbalances occur when the osmolality of the ECF is less than 275 mOsm/kg. The most common causes are decreased sodium concentration (hyponatremia) or excess of electrolyte-free water (water intoxication). Either problem leads to an intracellular overhydration (cell swelling). When there is a decreased sodium concentration, the osmotic pressure of the ECF decreases; water moves into the cell, where the higher osmotic pressure pulls it in (see Fig. 3.7A). As water leaves the ECF, plasma volume decreases, resulting in symptoms of hypovolemia. When there is electrolyte-free water excess (water intoxication), the ECF volume is elevated, causing symptoms of hypervolemia (Table 3.8). Cerebral and pulmonary edema occurs in conjunction with extreme water intoxication.
Table 3.8
D5W, Dextrose 5% in water; ECF, extracellular fluid; ICF, intracellular fluid.
Hyponatremia
Pathophysiology
Hyponatremia develops when the serum sodium concentration decreases to less than 135 mEq/L. Hyponatremia results from a loss of sodium, inadequate intake of sodium, or dilution of sodium by water excess. Sodium depletion usually causes hypoosmolality with an associated movement of water into cells (see Fig. 3.7A). Cell dysfunction or even membrane rupture may then occur.
Hypovolemic hyponatremia occurs with a loss of total body fluid that involves a greater loss of body sodium (hypotonic hyponatremia) than body water. The extracellular volume is decreased. Causes include prolonged vomiting, severe diarrhea, inadequate secretion of aldosterone (e.g., adrenal insufficiency), and renal losses from diuretics. ADH will be released to facilitate repletion of blood volume.
Isovolemic hyponatremia occurs when there is loss of sodium without a significant loss of water (pure sodium deficit). Causes can include water retention secondary to syndrome of inappropriate antidiuretic hormone (SIADH; see Chapter 22, which enhances water retention), hypothyroidism, pneumonia, and glucocorticoid deficiency. Inadequate intake of dietary sodium is rare but possible in individuals consuming low-sodium diets, particularly when diuretics are also being used.
Dilutional hypotonic hyponatremia (water intoxication) occurs when there is intake of large amounts of electrolyte-free water or replacement of fluid loss with IV 5% dextrose in water, which dilutes sodium. The glucose is metabolized to carbon dioxide and water, leaving a hypotonic solution with a diluting effect. Excessive sweating stimulates thirst and intake of large amounts of electrolyte-free water, as can occur in endurance athletes. Some individuals with psychogenic disorders develop water intoxication from compulsive water drinking. Other causes can include tap water enemas, aspiration of fresh water from nonfatal drowning, use of selective-serotonin reuptake inhibitors (SSRIs), and SIADH. When the body is functioning normally, it is quite difficult to produce an excess of TBW by water intoxication because water balance is regulated by ADH action on the kidneys.
Hypervolemic hyponatremia occurs when the total body sodium amount is increased and there is excess water. The increased sodium leads to an increase in ECF volume, and the serum sodium concentration is decreased. Causes include heart failure, cirrhosis of the liver, and nephrotic syndrome. Edema typically is present in these cases.
Hypertonic hyponatremia develops with the shift of water from the ICF to the ECF, as occurs with hyperglycemia, hyperlipidemia, and hyperproteinemia. The osmotic fluid shift to the ECF in turn dilutes the concentration of sodium (pseudohyponatremia) and other electrolytes.
Clinical Manifestations
Serum sodium concentration is less than 135 mEq/L, and with severe sodium deficits, the concentration is less than 120 mEq/L. The high concentration of intracellular solutes, compared with the low concentration of extracellular solutes (hyponatremia), causes an osmotic shift of water into the cells, and cell swelling occurs. The decrease in the ECF sodium concentration changes the cell’s ability to depolarize and repolarize normally, altering the action potential in neurons and muscle (see Chapter 1). Clinical manifestations are related to impaired nerve conduction and neurologic changes (see Table 3.8). Hyponatremia is a major cause of morbidity and mortality among individuals hospitalized in intensive care units and in older adults.
Nausea and vomiting are more common with less severe hyponatremia (i.e., decreases between 125 and 130 mEq/L). Neurologic symptoms occur with severe hyponatremia (i.e., decreases less than 125 mEq/L or less severe very rapid decreases) and include lethargy, headache, confusion, apprehension, seizures, and coma.5 Hypovolemic hyponatremia with pure sodium loss is accompanied by loss of ECF with symptoms of hypotension, tachycardia, and decreased urine output. Hypervolemic hyponatremia is accompanied by weight gain, edema, ascites, and jugular vein distention. Cerebral edema can be a life-threatening complication of hyponatremia caused by increased shifts of fluid to the intracellular space and increased intracranial pressure.
Evaluation and Treatment
Evaluation of hyponatremia includes the history, physical examination, and laboratory tests for serum and urine sodium. In hyponatremic states, serum sodium concentration decreases to less than 135 mEq/L. With pure sodium deficits, the hematocrit and plasma protein levels may be elevated. Urine specific gravity is less than 1.010 when renal function is normal because sodium is maximally conserved. Evaluation of urine sodium concentration and urine osmolality assists with differential diagnosis. High urine sodium level (normal is 40 to 220 mEq/L in 24 hours) and high urine osmolality are associated with cerebral salt wasting syndrome and adrenal insufficiency. Low urine sodium level (30 mEq/L in 24 hours) and high urine osmolality are associated with extrarenal losses, such as vomiting and diarrhea or severe burns, heart failure, or cirrhosis. Serum osmolality is usually decreased, but secondary conditions of hyperlipidemia, hyperglycemia, or hyperproteinemia can increase serum osmolality.
Treatment of hyponatremia depends on the contributing disorder and severity and acuity of sodium loss. Losses of sodium and water volume are calculated from the clinical evaluation, and appropriate solutions then are selected for replacement. Restriction of water intake is required in most cases of dilutional hyponatremia because body sodium levels may be normal or increased even though serum concentrations are low. Hypertonic saline solutions are used cautiously with severe hyponatremia or the presence of symptoms such as seizures. Rapid correction of chronic hyponatremia can lead to osmotic demyelination syndrome with axonal damage in the brain resulting in neurologic disability or death.6 Arginine-vasopressin (ADH) receptor antagonists (vaptans) are a class of drugs used for the treatment of hypervolemic and isovolemic hyponatremia, particularly with SIADH.7 Serum sodium concentration must be monitored to prevent overcorrection.
Hypochloremia
Hypochloremia, a low level of serum chloride (less than 97 mEq/L), is rare and usually occurs with hyponatremia or an elevated bicarbonate concentration, as in metabolic alkalosis. Sodium deficit associated with restricted intake, use of diuretics, and vomiting is accompanied by chloride deficiency. Cystic fibrosis is a genetic disease characterized by hypochloremia (see Chapters 36 and 42). In all cases, treatment of the underlying cause is required.
Alterations in Potassium, Calcium, Phosphate, and Magnesium Balance
Potassium
Potassium (K+) is the major intracellular electrolyte and is found in most body fluids (see Table 3.5). Total body potassium content is approximately 4000 mEq, with most of it located in the cells. The ICF concentration of K+ is approximately 150 to 160 mEq/L; the ECF concentration is approximately 3.5 to 5.0 mEq/L. This concentration gradient is maintained by an active transport system, the sodium-potassium adenosine triphosphatase pump (Na+-K+ ATPase pump) (see Chapter 1). The ratio of ICF K+ concentration to ECF K+ concentration is the major determinant of the resting potential of cells, which makes K+ balance crucial for the transmission and conduction of nerve impulses, maintenance of normal cardiac rhythms, and contraction of skeletal and smooth muscles (Figs. 3.8A and 1.27). Daily dietary intake of potassium is 40 to 150 mEq/day, with an average of 1.5 mEq/kg body weight. Approximately 90% of dietary potassium is absorbed in the gastrointestinal tract and is transported rapidly into the cells after ingestion. The distribution of potassium between the intracellular and ECFs is influenced by several factors. Insulin, epinephrine, and alkalosis all facilitate the shift of potassium into cells. Insulin deficiency, aldosterone deficiency, some types of acidosis, cell lysis, and strenuous exercise promote a shift of potassium out of cells. Glucagon blocks entry of potassium into cells; glucocorticoids promote potassium excretion. Aldosterone promotes renal excretion of K+ by the distal tubules, accounting for 90% to 95% of K+ excretion.8
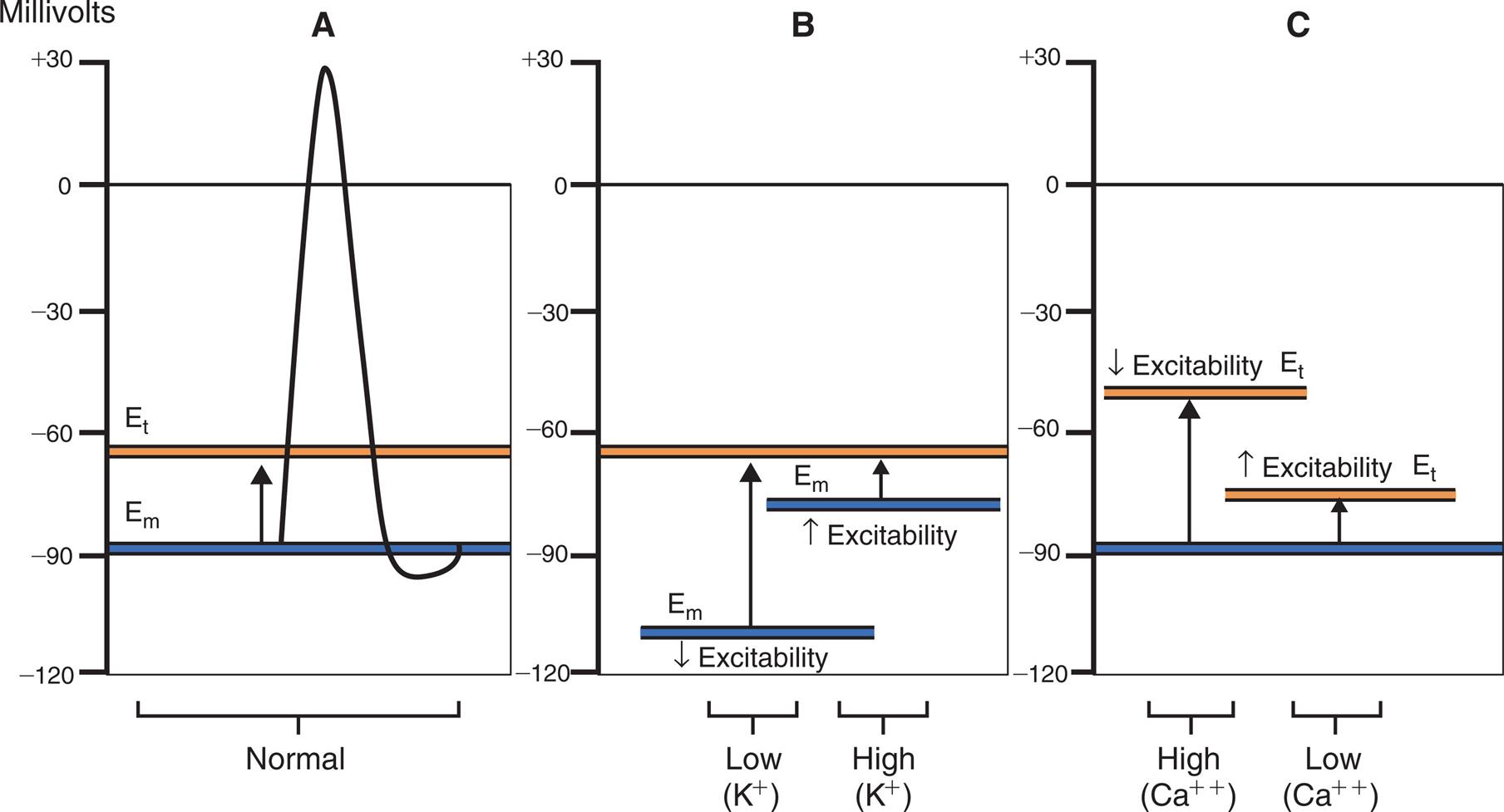
(A) Normal membrane excitability: Potassium affects the resting membrane potential (Em), and calcium affects the threshold potential (Et). (B) Effects of potassium ion (K+) changes on membrane potential. (C) Effect of calcium ion (Ca++) on threshold potential. NOTE: Hyperpolarization can be caused by either hypokalemia (Em more negative) or hypercalcemia (Et less negative)—the distance between Em and Et is increased (decreased excitability); and hypopolarization can be caused by either mild hyperkalemia (Em less negative) or hypocalcemia (Et more negative)—the distance between Em and Et is decreased (increased excitability).
Three graphs, A, B, and C, plot the effects of potassium and calcium ions on membrane excitability. The vertical axes represent millivolts and range from negative 120 to positive 30, in increments of 30. The horizontal axes represent membrane excitability. Graph A. The horizontal axis represents normal membrane excitability. The resting membrane potential, E sub m, is a solid horizontal line at about negative 90 millivolts. The threshold potential, E sub t, is a solid horizontal line at about negative 55 millivolts. An upward arrow from E sub m reaches E sub t. A narrow, sharp peak shoots from E sub m to positive 30 millivolts at the center of the graph. It then falls sharply to a little below E sub m before returning to E sub m. Graph B. The horizontal axis represents the concentrations of the potassium ions. E sub t is a solid horizontal line at about negative 55 millivolts. At low concentrations, E sub m is a solid horizontal line at about negative 110 millivolts, with decreased excitability. An upward arrow from the line reaches E sub m. At high concentrations, E sub m is a solid horizontal line at about negative 75 millivolts, with increased excitability. An upward arrow from the line reaches E sub t. Graph C. The horizontal axis represents the concentrations of the calcium ions. E sub m is a solid horizontal line at about negative 90 millivolts. At high concentrations, E sub t is a solid horizontal line at about negative 40 millivolts, with decreased excitability. An upward arrow from E sub m reaches E sub t. At low concentrations, E sub t is a solid horizontal line at about negative 75 millivolts, with increased excitability. An upward arrow from E sub m reaches E sub t.
As the predominant ICF ion, potassium exerts a major influence on the regulation of ICF osmolality and fluid balance as well as on intracellular electrical neutrality in relation to hydrogen (H+) and Na+ levels. Potassium can move out of cells, along with water, with an increased ECF osmolarity.
Insulin contributes to the regulation of plasma potassium levels by stimulating the Na+-K+ ATPase pump, thereby promoting the movement of K+ into liver and muscle cells. Clinically, insulin (usually with dextrose to prevent hypoglycemia) is used to treat severe hyperkalemia. In cases of hyperglycemia, dangerously low levels of plasma potassium can result when insulin is administered to treat elevated glucose levels. Particularly in emergency settings, it is crucial to check potassium levels before administering insulin for hyperglycemia. Monitoring potassium status is a key factor in any diabetes treatment regimen involving the administration of insulin.
In addition to conserving sodium, aldosterone regulates the potassium concentration. Aldosterone is secreted from the adrenal cortex in response to increases in plasma potassium concentration or stimulation by angiotensin II, part of the RAAS; it stimulates the secretion of potassium into the urine by the distal renal tubules. Aldosterone also increases the secretion of potassium from the colon and sweat glands.
Although potassium is found in most body fluids, the kidney is the most significant regulator of potassium balance. Potassium is freely filtered by the renal glomerulus; 90% is reabsorbed by the proximal tubule and loop of Henle. In the distal tubules, principal cells secrete potassium under the influence of aldosterone, and intercalated cells reabsorb potassium. The net effect of secretion and reabsorption determines the amount of potassium excreted from the body. Evidence suggests that the gut also may sense the amount of K+ ingested and stimulate renal K+ excretion, independent of aldosterone.9
The potassium concentration in the distal tubular cells is determined primarily by the plasma concentration of potassium in the peritubular capillaries. When plasma potassium concentration increases, potassium is secreted into the urine by the distal tubules, which is facilitated by the action of aldosterone. Decreased levels of plasma potassium result in decreased distal tubular secretion, although approximately 5 to 15 mEq/day will continue to be lost. Changes in the rate of filtrate (urine) flow through the distal tubule also influence the concentration gradient for potassium secretion. When the urine flow rate is high (e.g., with diuretic use), the potassium concentration in the distal tubular urine is lower; more potassium subsequently is secreted into the urine.10
Changes in pH (hydrogen ion concentration) also affect the potassium balance. During acute acidosis, hydrogen ions accumulate in the ICF. Unless the anion portion of the acid (e.g., acetoacetate or lactate) has entered the ICF, the accumulated H+ would create a charge imbalance. To maintain ionic balance, potassium shifts out of the cell and into the ECF. This shift occurs, in part, because of a decrease in the Na+-K+ ATPase pump activity and can contribute to hyperkalemia. Decreased ICF potassium also results in decreased potassium secretion by the distal tubular cells, increasing the potential for hyperkalemia. In acute alkalosis, hydrogen ions within the ICF move to the ECF, resulting in a shift of potassium into the cell to maintain ionic balance. This contributes to development of hypokalemia; in addition, potassium secretion increases within the distal tubular cells, which increases the potential for hypokalemia. The management of alterations associated with acid-base imbalances requires concurrent treatment of changes in K+ concentration.
In summary, renal regulation of potassium includes:
Potassium loss occurs through normal body functions, but without causing hypokalemia, unless the individual has very low dietary potassium intake. Average daily losses of K+ are as follows:
| Location | Daily Loss (mEq/L) |
|---|---|
| Stool | 5–10 |
| Sweat | 0–20 |
| Urine | 40–120 |
Potassium adaptation is the ability of the body to adapt to increased levels of K+ intake over time. A sudden increase in K+ level may be fatal. If the intake is slow, the kidney can increase the urinary excretion of K+ and maintain K+ balance.
Hypokalemia
Pathophysiology
Potassium deficiency, or hypokalemia, develops when the serum K+ concentration decreases to less than 3.5 mEq/L. Because intracellular and total body stores of K+ are difficult to measure, changes in K+ balance are reflected by the plasma concentration, although not always accurately. In general, lowered serum K+ level indicates a loss of total body K+. When K+ is lost from the ECF, the change in the concentration gradient favors the movement of K+ from the cell into the ECF. The ICF/ECF concentration ratio is maintained, but the amount of total body K+ is depleted.
Factors contributing to the development of hypokalemia include a reduced intake of potassium, an increased entry of K+ into cells, and increased losses of K+. A dietary deficiency of K+ may occur in people who have inadequate intake of potassium-rich fruits and vegetables (influenced by fad diets, food insecurity, or lack of transportation to purchase fresh produce), and in individuals with alcoholism or eating disorders. A reduced K+ intake generally becomes a problem when combined with other causes of K+ depletion.
ECF hypokalemia can develop without losses of total body K+. As discussed previously, with some types of acidosis, potassium shifts from the ECF to the ICF in exchange for hydrogen ions to maintain the plasma acid-base balance. In alkalosis, ECF hydrogen moves out of the cell to correct the alkalosis, and K+ moves into the cell to maintain an ionic balance. Insulin promotes cellular uptake of K+, and insulin administration can cause an ECF potassium deficit, particularly with the intake of high carbohydrate loads. For this reason, it is crucial to evaluate potassium status in emergency settings when treating a person with diabetes who presents with severe hyperglycemia and/or diabetic ketoacidosis (DKA). Failure to do so before administering IV insulin can result in life-threatening hypokalemia. Treating DKA typically requires administration of supplemental potassium simultaneously with IV insulin and rehydration therapy. With DKA, the overall insulin deficit results in potassium shifts from the ICF to the ECF, due to lack of insulin action on the Na+-K+ ATPase pump. A normal serum potassium level usually is maintained; however, potassium excretion through the kidney continues, resulting in a deficit of total body potassium. The deficit becomes clinically evident when insulin treatment and rehydration therapy are initiated. Accordingly, in the treatment of hyperglycemia with or without DKA, the standard of care is potassium supplementation with close monitoring.
Treatment of pernicious anemia with vitamin B12 or folate also may precipitate hypokalemia if the formation of new red blood cells causes enough K+ uptake to effect an extracellular decrease in K+ concentration. Familial hypokalemic periodic paralysis is a rare genetically transmitted disease that causes K+ to shift into the intracellular space with episodes of extreme muscle weakness.
Losses of K+ from body stores are most commonly caused by gastrointestinal and renal disorders. Diarrhea, intestinal drainage tubes, fistulae, excessive ingestion of black licorice, and laxative overuse can all result in hypokalemia. Normally, only 5 to 10 mEq of potassium and approximately 100 mL of water are excreted in the stool each day. With diarrhea, fluid and electrolyte losses can be voluminous, with several liters of fluid and 100 to 200 mEq of K+ lost per day. Vomiting or continuous nasogastric suction frequently is associated with K+ depletion. The loss occurs in part because of the K+ lost from the gastric fluid. However, the loss principally is caused by renal compensation for volume depletion and metabolic alkalosis, which occurs secondary to losses of sodium, chloride, and hydrogen ion. The loss of fluid and sodium stimulates the secretion of aldosterone, which in turn results in renal loss of K+.
Renal losses of K+ are related to increased secretion of K+ by the distal tubule. Predisposing factors include the use of diuretics, excessive aldosterone secretion, an increased distal tubular flow rate, and a low plasma magnesium concentration. Many diuretics inhibit the reabsorption of sodium chloride, resulting in increased urine production. With enhanced fluid excretion, the increased flow through the distal tubule also promotes potassium excretion. If sodium loss is severe, the compensating aldosterone secretion (which causes secondary hyperaldosteronism) may further deplete K+ stores. Primary hyperaldosteronism with excessive secretion of aldosterone from an adrenal adenoma also causes K+ wasting. Many kidney diseases impair the kidney’s ability to conserve sodium. The decreased sodium reabsorption produces a diuretic effect. As a result, the increased flow through the distal tubule promotes the secretion of K+. Magnesium deficiency increases loop of Henle and distal potassium secretion, causing secondary hypokalemia.11 Several medications, including amphotericin B, gentamicin, and nafcillin, cause hypokalemia by increasing the rate of potassium excretion. Rare hereditary defects in renal potassium transport (Bartter and Gitelman syndromes) also can result in hypokalemia (Table 3.9).
Table 3.9
ECF, Extracellular fluid; ICF, intracellular fluid; K+, potassium.
Clinical Manifestations
Mild losses of K+ are usually asymptomatic. With severe hypokalemia (<2.5 mEq/L), neuromuscular excitability decreases, causing skeletal muscle weakness, smooth muscle atony, cardiac dysrhythmias, glucose intolerance, and impaired urinary concentrating ability.
Symptoms occur in proportion to the rate of potassium depletion. The body can accommodate slow losses of potassium. Decreases in the ECF potassium concentration may facilitate a shift in potassium away from the intracellular space and into the ICF. This dynamic promotes the return of the potassium concentration gradient toward a more normal status, reducing neuromuscular symptoms. With acute and severe potassium loss, the changes in neuromuscular excitability are more profound. Skeletal muscle weakness initially occurs in the larger muscles of the legs and arms and ultimately affects the diaphragm, compromising ventilation. With severe losses, paralysis and respiratory arrest may occur. Loss of smooth muscle tone may result in a variety of gastrointestinal manifestations, such as constipation, intestinal distention, anorexia, nausea, vomiting, and paralytic ileus (paralysis of the intestinal muscles). Table 3.10 contains a summary of K+ alterations.
Table 3.10
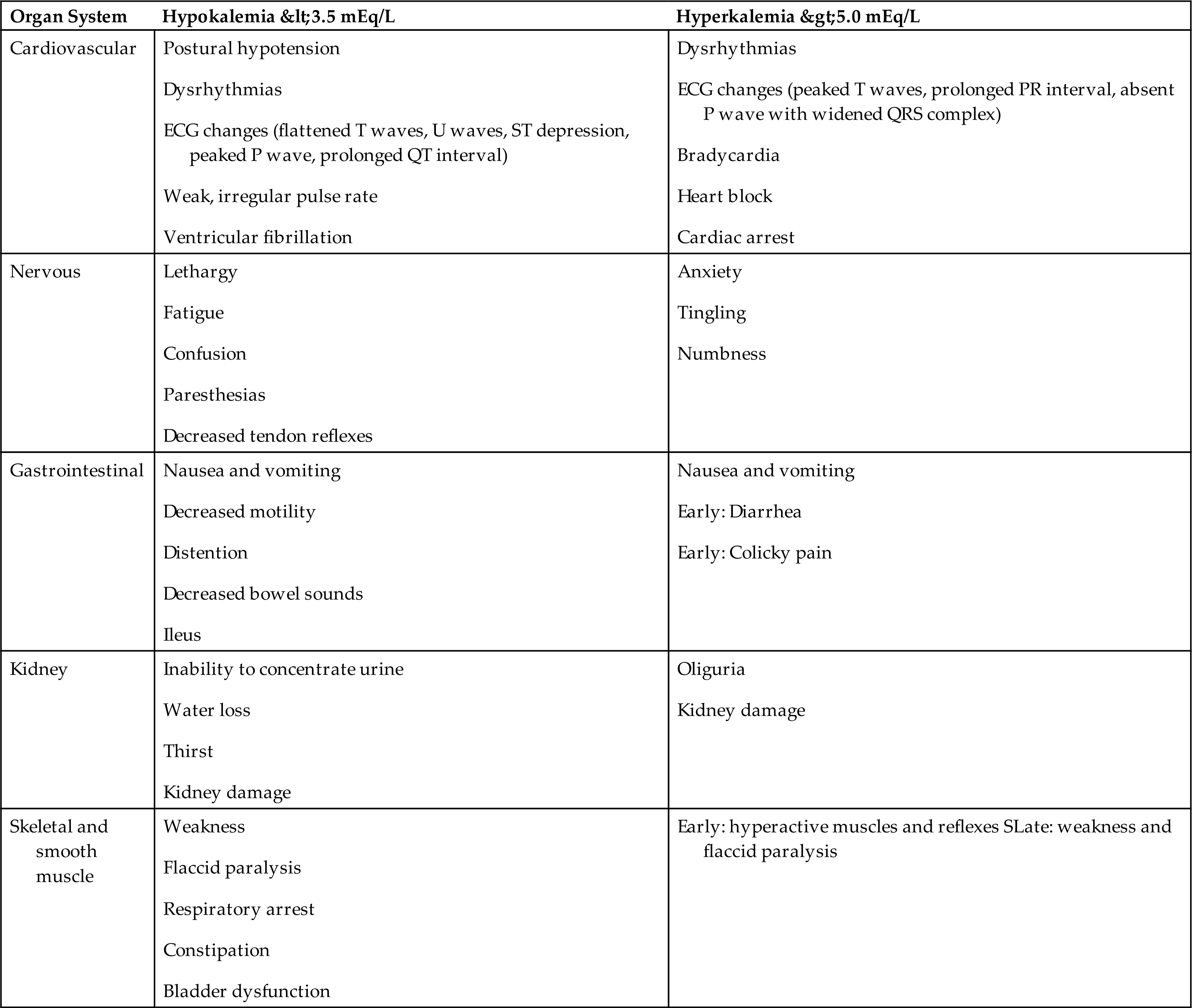
The cardiac effects of hypokalemia are related to changes in membrane excitability. As the ECF potassium concentration decreases, the resting membrane potential becomes more negative (i.e., hyperpolarized; e.g., from −90 to −100 millivolts). A hyperpolarized membrane requires a greater stimulus to trigger an action potential (Fig. 3.8B). Potassium also contributes to the repolarization phase of the action potential; hypokalemia delays ventricular repolarization. Consequently, hypokalemia may result in various dysrhythmias, including sinus bradycardia, atrioventricular block, and paroxysmal atrial tachycardia. The characteristic changes in the electrocardiogram (ECG) reflect delayed ventricular repolarization with slowed conduction and pacemaker activity. The amplitude of the T wave decreases, the amplitude of the U wave increases, and the ST segment is depressed (Fig. 3.9). In severe states of hypokalemia, P waves peak, the QT interval is prolonged, and T-wave inversions may be seen. Hypokalemia enhances the therapeutic effect of digitalis by slowing the Na+-K+ pump and excessively increasing intracellular calcium and sodium concentrations. The risk of digitalis toxicity is increased.
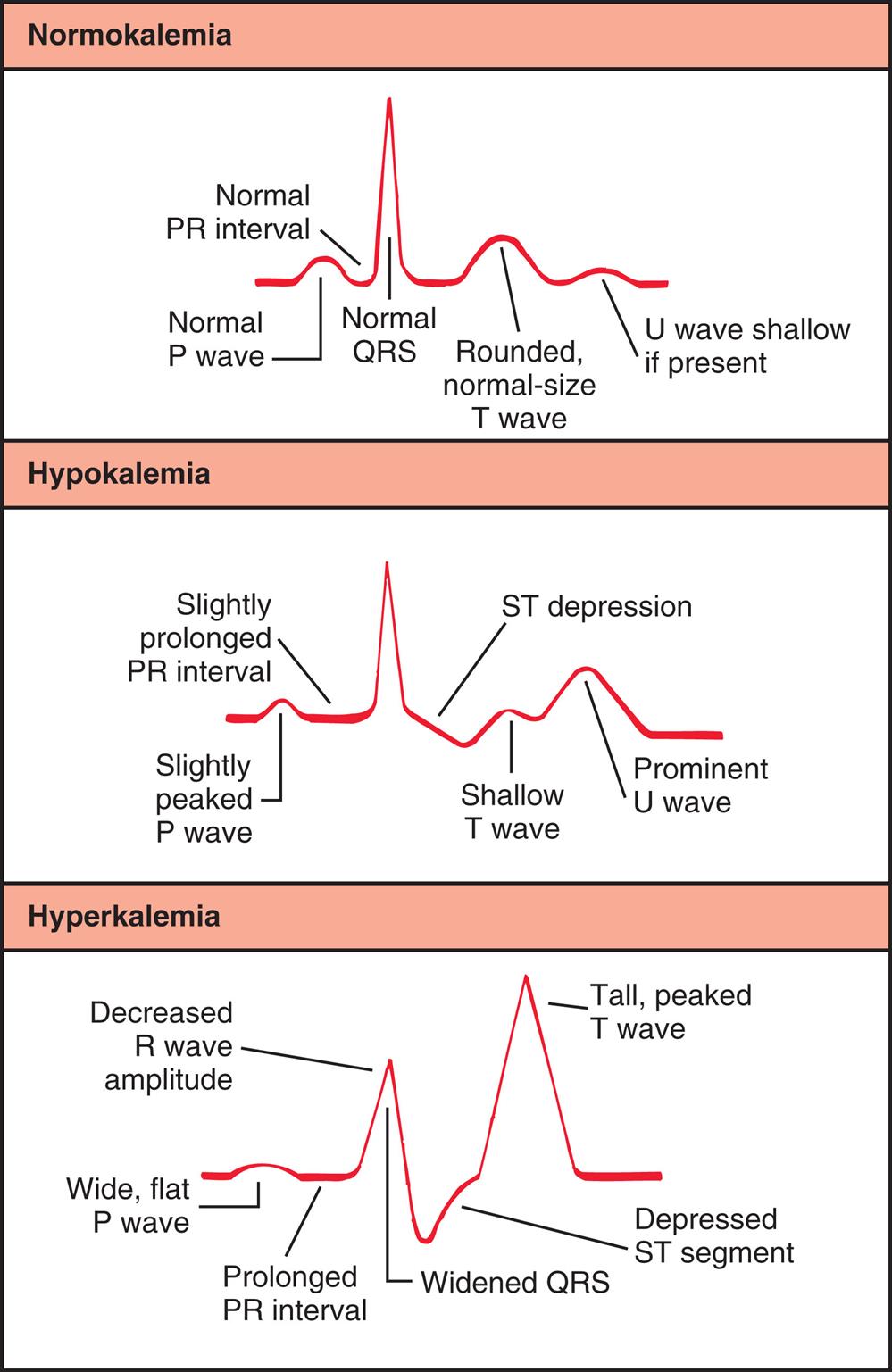
A three-panel illustration graphs tracing the electrocardiogram changes with potassium imbalance. Each graph traces waves, intervals, and peaks. Top panel, normokalaemia. The order of tracing, from the left to the right, is as follows: Normal P wave, normal P R interval, normal Q R S, rounded, normal-size T wave, and U wave shallow if present. Middle panel, hypokalemia. The order of tracing, from the left to the right, is as follows: slightly peaked P wave, slightly prolonged P R interval, S T depression, shallow T wave, and prominent U wave. Bottom panel, hyperkalemia. The order of tracing, from the left to the right, is as follows: wide, flat P wave, prolonged P R interval, decreased R wave amplitude, widened Q R S, depressed S T segment, and tall, peaked T wave.
Concurrent alterations in plasma calcium concentration also contribute to changes in neuromuscular excitability associated with hypokalemia. Increases in ECF calcium concentration tend to make the threshold potential (Et) less negative. The result is decreased membrane excitability and potentiation of hyperpolarization, amplifying the neuromuscular effects of hypokalemia (see Fig. 3.8C).
A wide range of metabolic dysfunctions may result from potassium deficiency. Carbohydrate metabolism is affected. Hypokalemia depresses insulin secretion and alters hepatic and skeletal muscle glycogen synthesis. Renal function is impaired by a decreased responsiveness to ADH, resulting in a decreased ability to concentrate urine, causing polyuria (increased urine). Polydipsia (increased thirst) may occur. Chronic potassium deficits lasting more than 1 month may damage renal tissue, with resulting interstitial fibrosis and tubular atrophy.
Evaluation and Treatment
The diagnosis of hypokalemia is based on serum K+ levels. Hypokalemia can result from disorders associated with potassium loss or from shifts of extracellular potassium into the intracellular space. Treatment involves replacing lost potassium to restore normal levels and correcting the associated fluid and acid-base imbalances. Once these have been corrected, further potassium loss should be prevented by correcting the underlying mechanism. In particular, individuals should be encouraged to eat potassium-rich foods. With normal renal function, the maximal rate of oral replacement is 40 to 80 mEq/day. A maximal safe rate of IV replacement is 20 mEq/h. Potassium is irritating to blood vessels and can result in considerable pain for the individual. Accordingly, IV infusions containing potassium should not exceed 40 mEq/L. Replacement therapy requires close monitoring of the plasma potassium concentration. Hypokalemia concurrent with hypomagnesemia is refractory to treatment until magnesium levels are corrected.
Hyperkalemia
Pathophysiology
Hyperkalemia is defined as an ECF potassium concentration greater than 5.5 mEq/L.11a Increases in the total body potassium level are relatively rare, largely because of efficient renal excretion. Acute increases in the serum potassium level are handled quickly through increased renal excretion of potassium, with some uptake also into cells.
Hyperkalemia may be caused by excessive intake, a shift of potassium from the ICF to the ECF, or decreased renal excretion.12 If renal function is normal, slow and long-term increases in K+ intake are usually well tolerated through K+ adaptation. Dietary excesses of potassium are uncommon, but accidental ingestion of potassium salt substitutes can cause toxicity. Acute K+ loading can exceed renal excretion rates. Use of stored whole blood, administration of IV boluses of penicillin G, or excessive replacement of K+ can precipitate hyperkalemia, particularly if renal function is impaired. Drugs that decrease renal potassium excretion (e.g., ACE inhibitors, ARBs, potassium-sparing diuretics, and aldosterone antagonists) also may contribute to hyperkalemia.
Potassium shifts from the ICF to the ECF occur when there is a change in cell membrane permeability caused by cell hypoxia, some types of acidosis, or insulin deficiency. Hypoxia can lead to hyperkalemia by diminishing the efficiency of cell membrane active transport, resulting in the escape of K+ to the ECF. Burns, massive crush injuries, and extensive surgeries can cause cell trauma and release of ICF potassium to the ECF. If renal function is sustained, K+ will be excreted. As cell repair begins, hypokalemia may develop if the excreted K+ is not replaced.
In states of acidosis, hydrogen ions shift into the cells in exchange for ICF potassium, unless the anion portion of the acid also enters cells; therefore hyperkalemia and acidosis often occur together. Insulin promotes cellular entry of K+; consequently, insulin deficits, which occur with conditions such as DKA, are often accompanied by hyperkalemia. Hyperkalemia may result secondary to digitalis toxicity. High levels of digitalis inhibit the Na+-K+ ATPase transport pump, allowing potassium to remain outside the cell.
Decreased renal function is commonly associated with hyperkalemia. Oliguria (urine output <30 mL/h) secondary to acute kidney injury or end-stage renal disease typically presents with elevations of serum K+ concentration. The severity of hyperkalemia is a function of the amount of K+ intake, the degree of acidosis, and the rate of renal cell damage. Hypoaldosteronism can cause decreases in the urinary excretion of K+. For example, Addison disease, characterized by adrenal cortical insufficiency, often presents with hyperkalemia secondary to decreased aldosterone secretion.
Clinical Manifestations
Symptoms vary with the severity of hyperkalemia. With a mild presentation, increased neuromuscular irritability may manifest as restlessness, intestinal cramping, and diarrhea. Severe hyperkalemia decreases the resting membrane potential from −90 to −70 millivolts, resulting in muscle weakness, loss of muscle tone, and paralysis. In mild states of hyperkalemia, myocardial cell repolarization is more rapid and reflected in the ECG as narrow and taller T waves with a shortened QT interval. Severe hyperkalemia (serum levels ≥6 mEq/L) causes delayed cardiac conduction, preventing repolarization of heart muscle. There is a decrease in conduction velocity, depressed ST segment, prolonged PR interval, and widening of the QRS complex (loss of atrial activity) (see Fig. 3.9). Brady dysrhythmias and delayed conduction are common in hyperkalemia; severe hyperkalemia can cause ventricular fibrillation or cardiac arrest.
Changes in the ratio of intracellular to extracellular K+ concentration contribute to the clinical presentation of hyperkalemia (see Table 3.9). If extracellular K+ concentration increases without a significant change in intracellular K+ concentration, the resting membrane potential becomes more positive (e.g., changes from −90 to −80 millivolts) and the cell membrane is hypopolarized (the inside of the cell becomes less negative or partially depolarized [increased excitability]) (see Fig. 3.8B). With mild elevations in extracellular K+ concentration, the cell more rapidly repolarizes and becomes more irritable (peaked T waves). An action potential then is initiated more rapidly because the distance between the resting membrane potential and the threshold potential has been decreased. With more severe hyperkalemia, the resting membrane potential approaches or exceeds the threshold potential (wide QRS merging with T wave). In this case the cell is not able to repolarize and therefore does not respond to excitation stimuli. The most serious consequence is cardiac arrest.
Like the effects of hypokalemia, the neuromuscular effects of hyperkalemia are related to the rate of increase in the ECF potassium concentration and the presence of other contributing factors, such as acidosis and calcium balance. Long-term increases in ECF potassium concentration result in shifts of K+ into the cell because the tendency is to maintain a normal ratio of intracellular/extracellular potassium concentrations. Acute elevations of extracellular K+ concentration affect neuromuscular irritability because this ratio is disrupted.
Because calcium influences the threshold potential, changes in ECF calcium concentration can augment or override the effects of hyperkalemia. With hypocalcemia the threshold potential becomes more negative, enhancing the neuromuscular effects of hyperkalemia. Hypercalcemia causes the threshold potential to become less negative, counteracting the effects of hyperkalemia on resting membrane potential (see Fig. 3.8C).
Evaluation and Treatment
Hyperkalemia is a common finding in many clinical settings (e.g., renal disease, massive trauma, insulin deficiency, Addison disease, use of potassium salt substitutes, or some types of metabolic acidosis). How rapidly symptoms evolve often is a function of the underlying cause. An ECG will identify conduction abnormalities or dysrhythmias.
Management of hyperkalemia includes both treating the contributing causes and correcting excessive potassium concentration. Normalizing the extracellular potassium concentration can be achieved with a variety of methods; the treatment chosen is related to the cause and severity of the problem. Calcium gluconate can be administered to restore membrane excitability when serum potassium levels are dangerously high. Administration of glucose, which readily stimulates insulin secretion, or administration of glucose and insulin for those with diabetes, facilitates cellular entry of potassium. Renin-angiotensin-aldosterone system inhibitor therapy and use of the newer oral potassium binders optimize therapy. Buffered solutions correct metabolic acidosis and lower serum potassium level. Dialysis effectively removes potassium in cases of renal dysfunction.13
Calcium and Phosphate
The total body content of calcium is approximately 1200 g. Most calcium (99%) is located in bone as hydroxyapatite (an inorganic compound that contributes to bone rigidity), and the remainder is in the plasma and body cells. The total fraction of calcium circulating in the blood is small (9.0 to 10.5 mg/dL), and approximately 50% is bound to plasma proteins, primarily albumin. Approximately 40% is in the free or ionized form (5.5 to 5.6 mg/dL). Ionized calcium has the most important physiologic functions. Approximately 20% of ingested calcium is absorbed in the small intestine, primarily in the duodenum.
Calcium (Ca2+) is a necessary ion for many fundamental metabolic and cellular processes. In bound form, it is the major cation associated with the structure of bones and teeth. The ionized form serves as an enzymatic cofactor for blood clotting and is required for hormone secretion and the function of cell receptors. Plasma membrane stability, permeability, and repair are directly related to calcium ions, as is the transmission of nerve impulses and the contraction of muscles. Calcium metabolism is linked to phosphate and magnesium metabolism.
Phosphate ( ) is found primarily in bone (85%), with smaller amounts found within the intracellular and extracellular spaces. In the plasma, phosphate exists in phospholipids and phosphate esters and as inorganic phosphate, which is the ionized form. The normal serum levels of inorganic phosphate range from 2.5 to 4.5 mg/dL and may be as high as 6.0 to 7.0 mg/dL in infants and young children. Intracellular phosphate has many metabolic forms, including the high-energy structures creatine phosphate and adenosine triphosphate (ATP). Phosphate acts as an intracellular and extracellular anion buffer in the regulation of acid-base balance; in the form of ATP, it provides energy for muscle contraction.
) is found primarily in bone (85%), with smaller amounts found within the intracellular and extracellular spaces. In the plasma, phosphate exists in phospholipids and phosphate esters and as inorganic phosphate, which is the ionized form. The normal serum levels of inorganic phosphate range from 2.5 to 4.5 mg/dL and may be as high as 6.0 to 7.0 mg/dL in infants and young children. Intracellular phosphate has many metabolic forms, including the high-energy structures creatine phosphate and adenosine triphosphate (ATP). Phosphate acts as an intracellular and extracellular anion buffer in the regulation of acid-base balance; in the form of ATP, it provides energy for muscle contraction.
Calcium and phosphate concentrations are rigidly controlled. They are related by the product of calcium and phosphate ( ) concentrations, which is a constant (K) [Ca2+) ×
) concentrations, which is a constant (K) [Ca2+) ×  = K]. Thus, if the concentration of one ion increases or decreases, that of the other normally increases or decreases.
= K]. Thus, if the concentration of one ion increases or decreases, that of the other normally increases or decreases.
Calcium and phosphate balance is regulated by three hormones: parathyroid hormone (PTH), vitamin D, and calcitonin. Acting together, these substances determine the amount of dietary calcium and phosphate absorbed from the intestine, the deposition and absorption of calcium and phosphate from bone, and the renal reabsorption and excretion of calcium and phosphate by the kidney.
The parathyroid glands secrete PTH in response to low levels of serum calcium. (The specific actions of PTH in relation to calcium and phosphate are described in Chapter 21.) Parathyroid hormone (PTH) controls levels of ionized calcium and phosphate in the blood and other ECFs. Renal regulation of calcium and phosphate balance requires PTH. PTH stimulates reabsorption of calcium along the distal tubule of the nephron and inhibits phosphate reabsorption by the proximal tubule of the nephron. The net result is an increase in serum calcium concentration and increased urinary excretion of phosphate. Fig. 3.10 summarizes hormonal regulation of calcium.
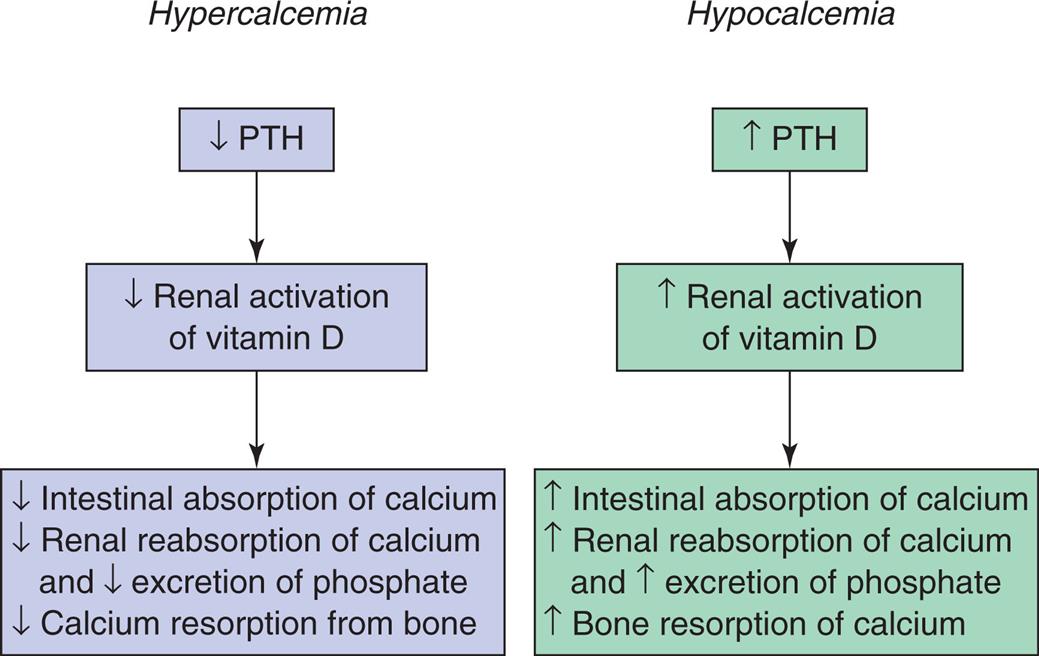
PTH, Parathyroid hormone.
Two flowcharts represent hormonal regulation of calcium balance. Flowchart on the left, hypercalcemia. • Decreased P T H. • Decreased renal activation of vitamin D. • Decreased intestinal absorption of calcium. Decreased reabsorption of calcium and decreased excretion of phosphate. Decreased calcium resorption from bone. Flowchart on the right, hypocalcemia. • Increased P T H. • Increased renal activation of vitamin D. • Increased intestinal absorption of calcium. Increased renal reabsorption of calcium and increased excretion of phosphate. Increased bone resorption of calcium.
Another compound important to calcium and phosphate regulation is vitamin D. Vitamin D (cholecalciferol) is a fat-soluble steroid ingested in food or synthesized in the skin in the presence of ultraviolet light. Several steps of activation are required before vitamin D can act on target tissues. The first step occurs in the liver; final activation is in the kidney. The renal activation of vitamin D begins when the serum calcium level decreases and stimulates secretion of PTH. PTH then acts to increase calcium reabsorption and enhance renal excretion of phosphate, producing decreased phosphate levels. The combination of low calcium level and increased PTH secretion causes the renal activation of vitamin D. The activated vitamin D (vitamin D3—calcitriol) then circulates as a hormone in the plasma and acts to increase absorption of calcium and phosphate in the small intestine, enhance bone calcification, increase renal tubular reabsorption of calcium, and increase excretion of phosphate. When end-stage renal disease occurs, vitamin D is not activated; serum calcium levels decrease; and phosphate levels increase.
As calcium levels increase, an opposite adaptation occurs, leading to suppression of PTH secretion, decreased renal vitamin D activation, decreased intestinal calcium absorption, and increased renal phosphate reabsorption. Calcitonin (produced by C cells of the thyroid gland) decreases calcium levels by inhibiting osteoclastic activity in bone and increasing renal calcium and phosphate excretion.
The fractions of serum calcium that are freely ionized or bound to plasma proteins are influenced by pH. In states of acidosis, levels of ionized calcium increase. When alkalosis develops, with an increase in pH, the amount of protein-bound calcium increases and the physiologically active, ionized calcium level decreases. The decreased concentration of ionized calcium may be great enough to cause symptoms of hypocalcemia, such as tetany.
Hypocalcemia
Pathophysiology
Hypocalcemia occurs when serum total calcium concentrations are less than 9.0 mg/dL and ionized levels are less than 5.5 mg/dL. In general, deficits in calcium are related to inadequate intestinal absorption, decreases in levels of PTH and vitamin D, or deposition of ionized calcium into bone or soft tissue.14
Nutritional deficiencies of calcium can occur in the instance of inadequate sources of dairy products or green, leafy vegetables, eating disorders, and malabsorption syndromes (celiac disease or short bowel syndrome). Excessive amounts of dietary phosphorus also bind with calcium in the gastrointestinal tract, so neither mineral is absorbed when such an excess occurs. Removal of the parathyroid glands (e.g., during total thyroidectomy) with the resulting loss of PTH also causes hypocalcemia. Severe hypomagnesemia suppresses PTH secretion, also causing hypocalcemia. Vitamin D deficiency, which can result from inadequate intake or lack of exposure to sunlight, causes decreased intestinal absorption of calcium. Malabsorption of fat, including fat-soluble vitamin D, also may contribute to calcium deficiency. Neoplastic bone metastases may inhibit bone resorption and increase calcium deposition into bone, thereby decreasing serum calcium levels.
Blood transfusions are also a common cause of hypocalcemia because the citrate solution used in storing whole blood binds with calcium and makes it unavailable to the tissues. Pancreatitis causes release of lipases into soft tissue spaces, so the free fatty acids that are formed bind calcium, causing a decrease in the concentration of ionized calcium. Metabolic or respiratory alkalosis causes symptoms of hypocalcemia because the change in pH enhances protein binding of ionized calcium. Hypoalbuminemia lowers total serum calcium levels by decreasing the amount of bound calcium in the plasma.
Clinical Manifestations
Many individuals with chronic hypocalcemia are asymptomatic. The clinical manifestations are a function of severity and rapidity of onset. Severe manifestations are caused by an increase in neuromuscular excitability with partial depolarization of nerves and muscles. As the threshold potential becomes more negative and approaches the resting membrane potential (hypopolarization) (see Fig. 3.8C), a smaller stimulus is required for initiating an action potential. The symptoms include paresthesias around the mouth and in the digits, carpopedal spasm (muscle spasms in the hands and feet), hyperreflexia, seizures, laryngospasm, and anxiety.
Two clinical signs of increased neuromuscular excitability are Chvostek sign and Trousseau sign. Chvostek sign is elicited by tapping on the facial nerve over the zygomatic arch. A positive sign is a strong twitch of the nose or lip. Trousseau sign is contraction of the hand and fingers when the arterial blood flow in the arm is occluded for 3 to 5 minutes with the use of a blood pressure cuff.
The characteristic ECG change is a prolonged QT interval, indicating prolonged ventricular depolarization and decreased cardiac contractility. Intestinal cramping and hyperactive bowel sounds also may be present because hypocalcemia affects the smooth muscles of the gastrointestinal tract. Table 3.11 contains a summary of the manifestations of calcium level alterations.
Table 3.11
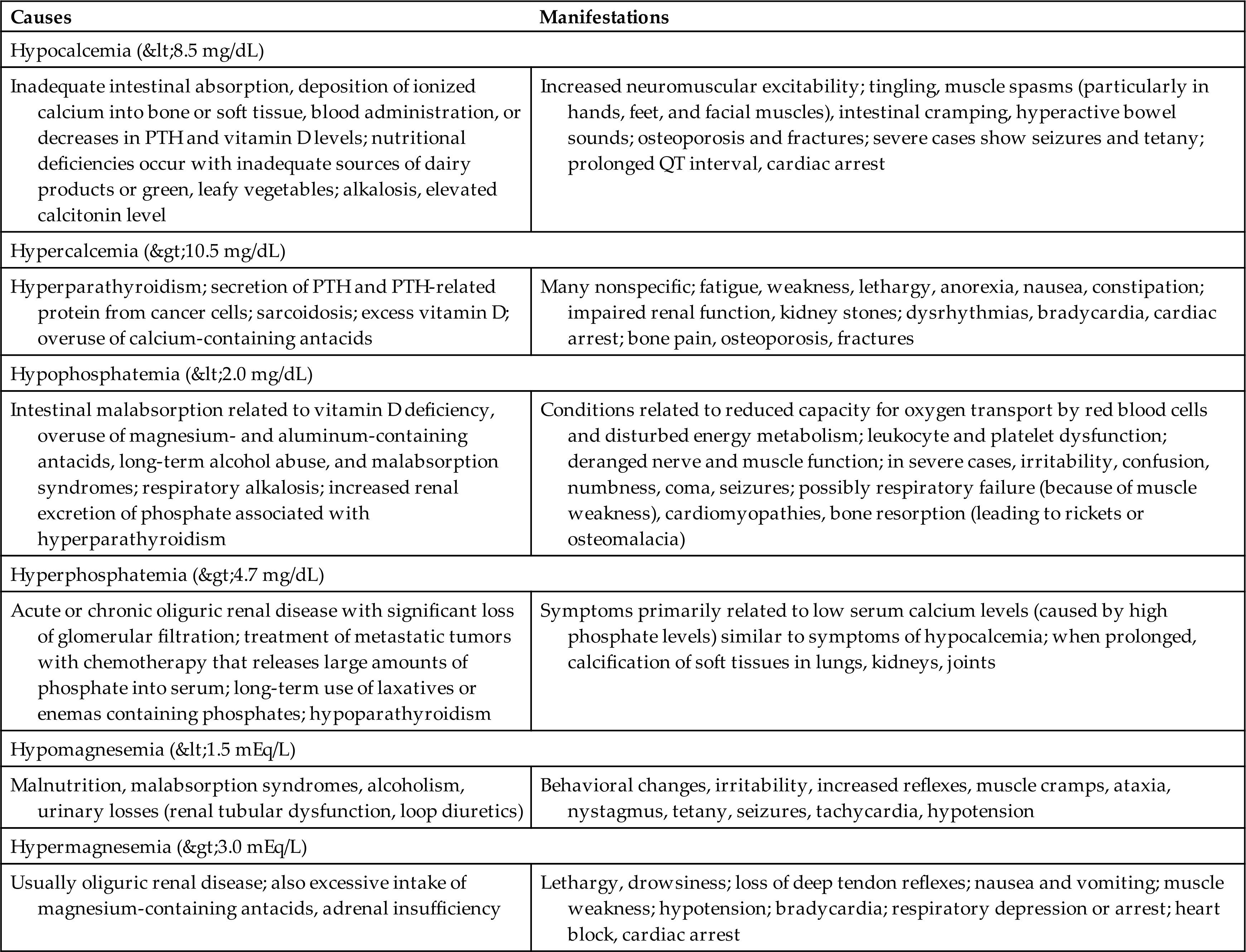
Evaluation and Treatment
Serum and ionized calcium, albumin, phosphate, and magnesium levels are evaluated. Further evaluation includes renal function and measurement of PTH and vitamin D. Severe symptoms of hypocalcemia require emergency treatment with IV 10% calcium gluconate, volume repletion, and ECG monitoring. The underlying cause must be identified. Oral calcium replacement should be initiated, and serum calcium levels should be monitored. Decreasing phosphate intake facilitates long-term management of hypocalcemia.
Hypercalcemia
Pathophysiology
Hypercalcemia with total serum calcium concentrations exceeding 10.5 mg/dL (5.2 mEq/L) can be caused by a number of diseases. The most common among these are hyperparathyroidism (which can be associated with thyrotoxicosis); many different types of cancer; sarcoidosis; and vitamin D toxicity. Many malignant tumors produce PTH or PTH-related protein, which causes bone resorption, thus elevating the serum calcium levels. Mild hypomagnesemia also stimulates PTH secretion and increases serum calcium. Sarcoidosis appears to increase vitamin D levels. Prolonged immobilization can also lead to hypercalcemia from enhanced bone resorption and decreased calcium deposition into bone. Acidosis decreases serum binding of calcium to albumin, increasing ionized calcium levels.
Clinical Manifestations
Many symptoms of hypercalcemia are nonspecific and related to severity and rapidity of onset. Because serum calcium levels are increased, a greater amount of calcium is also contained inside the cells. The threshold potential becomes more positive (hyperpolarized) (e.g., moves from −60 to −50 millivolts) and the cell membrane becomes refractory to depolarization (decreased excitability) and results in a greater difference between threshold potential and resting membrane potential (see Fig. 3.8C). Thus, many of the symptoms are related to loss of cell membrane excitability. Fatigue, weakness, lethargy, anorexia, nausea, and constipation are common.
Mental status changes and confusion may occur. Impaired renal function frequently develops with polyuria or formation of kidney stones from precipitates of calcium salts. A shortened QT segment and depressed widened T waves also may be observed on the ECG, with bradycardia and varying degrees of heart block. Table 3.11 contains a summary of the manifestations of alterations in calcium levels.
Evaluation and Treatment
With elevated serum calcium levels, often a reciprocal decrease in serum phosphate values occurs. Specific diagnostic procedures to identify the contributing pathologic condition are required.
Treatment is related to the severity of symptoms and the underlying disease. When renal function is normal, oral phosphate administration is effective. When acute illness and high calcium levels are present, treatment options include IV administration of large amounts of normal saline to enhance renal excretion of calcium, administration of bisphosphonates in the absence of renal dysfunction, and administration of calcitonin. Bisphosphonates and denosumab are used for malignancy-associated hypercalcemia, and cinacalcet is approved for the reduction of hypercalcemia associated with parathyroid carcinoma.15 Ultimately, the underlying pathologic condition must be treated.
Hypophosphatemia
Pathophysiology
Hypophosphatemia is a serum phosphate level less than 2.0 mg/dL and is usually an indication of phosphate deficiency. In some conditions, total body phosphate concentration is normal, but serum concentrations are low. The most common causes are intestinal malabsorption and increased renal excretion of phosphate. Inadequate absorption is associated with vitamin D deficiency, use of magnesium- and aluminum-containing antacids (which bind with phosphorus), long-term alcohol abuse, malabsorption syndromes, and refeeding syndromes after starvation. Respiratory alkalosis can cause severe hypophosphatemia because of cellular use of phosphorus for accelerated glycolysis (ATP) formation. Increased renal excretion of phosphorus is associated with hyperparathyroidism. Risk of hypophosphatemia has also been associated with IV iron therapy for treatment of pernicious anemia.16 Genetic mutations in fibroblast growth factor 23 (FGF23) are associated with decreased renal reabsorption of phosphorus.
Clinical Manifestations
The consequences of phosphate deficiency are not clinically evident until hypophosphatemia is severe, and multiple organ systems can be affected. There is reduced capacity for oxygen transport by red blood cells and disturbed energy metabolism. When phosphate is depleted, erythrocyte 2,3-DPG (diphosphoglycerate) and ATP levels become low and diminish the release of oxygen to the tissues, leading to hypoxia with bradycardia and varying degrees of heart block.
Leukocyte and platelet dysfunctions also are associated with hypophosphatemia. There is a greater risk of infection and blood-clotting impairment, with potential for hemorrhage. Nerve and muscle function can be affected because of derangement in energy metabolism. Muscle weakness may become serious enough to cause respiratory failure, and cardiomyopathies also can develop. Irritability, confusion, numbness, coma, and seizures develop with severe phosphate losses. In response to low phosphate levels, bone resorption occurs and may lead to rickets or osteomalacia. (Table 3.11 contains a summary of the manifestations of phosphate level alterations).
Evaluation and Treatment
To correct the condition, the underlying cause must be identified and treated. The rate and amount of replacement are determined by the cause and presenting symptoms.17
Hyperphosphatemia
Pathophysiology
Hyperphosphatemia, or an elevated serum phosphate level of more than 4.7 mg/dL, develops with exogenous or endogenous addition of phosphorus to the ECF or with significant loss of glomerular filtration and chronic kidney disease. Because most phosphate is located in cells, rapid cell destruction associated with treatment of metastatic tumors with chemotherapy can release large amounts of phosphate into the ECF. Long-term use of phosphate-containing enemas or laxatives also may lead to hyperphosphatemia. Hypoparathyroidism can cause elevated phosphate levels by increasing renal tubular reabsorption of phosphate.
High levels of serum phosphate also lower serum calcium levels, and increased amounts of phosphate and calcium are deposited in bone and soft tissues. Serum calcium levels may become low enough to cause symptoms of hypocalcemia, including tetany.
Clinical Manifestations
Symptoms of hyperphosphatemia are related primarily to low serum calcium levels and thus are comparable with symptoms of hypocalcemia. With prolonged hyperphosphatemia, calcification of soft tissues occurs in the lungs, kidneys, and joints. (See Table 3.11 contains a summary of the manifestations of alterations in phosphate concentration.)
Evaluation and Treatment
To correct hyperphosphatemia, the underlying pathologic condition must be identified and treated. Aluminum hydroxide may be administered because it binds phosphate in the gastrointestinal tract and is then eliminated; however, aluminum can be toxic, causing encephalopathy and osteomalacia. Nonaluminum, non–calcium phosphate binders (e.g., lanthanum carbonate or sevelamer) are used but can have serious side effects and are costly.18 Newer iron-based binders are being evaluated.19 Dialysis is required for management of end-stage chronic kidney disease.
Magnesium
Magnesium (Mg++) is a major intracellular cation, second to potassium. Approximately 40% to 60% is stored in muscle and bone, with 30% in the cells. A small amount (1%) is in the plasma. Plasma concentration is 1.5 to 3.0 mg/dL with approximately one-third bound to plasma proteins and the rest in ionized form. Regulation of magnesium metabolism is balanced by dietary intake, small intestinal absorption, and renal reabsorption and excretion. Low serum levels cause renal conservation of magnesium. Magnesium is a cofactor in intracellular enzymatic reactions, protein synthesis, and nucleic acid stability. Extracellular magnesium is necessary for normal neuromuscular excitability and nerve conduction. Magnesium improves myocardial metabolism and cell function; influences vascular smooth muscle tone (reduces peripheral vascular resistance and afterload); reduces cardiac dysrhythmias; and improves lipid and glucose metabolism. Magnesium also reduces vulnerability to oxygen-derived free radicals and systemic inflammation, improves human endothelial function, and inhibits platelet function, including platelet aggregation and adhesion.
Magnesium has been shown to counteract vascular calcification by inhibiting hyperphosphatemic-induced crystalline calciprotein particle formation in vascular smooth muscle.20 Calcium and magnesium often interact in reactions at the intracellular level, with magnesium being an antagonist of calcium. Mg++ reduces PTH secretion mainly when a moderate-low calcium concentration is present.
Hypomagnesemia occurs when serum magnesium concentration is less than 1.5 mEq/L and clinical symptoms are present.11 Malnutrition, malabsorption syndromes, alcoholism, renal tubular dysfunction, metabolic acidosis, use of loop and thiazide diuretics, and prolonged use of proton pump inhibitors can cause magnesium losses. Hypomagnesemia is associated with insulin resistance, diabetes mellitus, left ventricular hypertrophy, systemic inflammation, hypoalbuminemia, and osteoporosis. Because extracellular magnesium inhibits potassium channels, loss of magnesium results in movement of potassium out of the cell, with renal excretion resulting in hypokalemia. Signs and symptoms of hypomagnesemia are often absent and develop with severe losses. Clinical manifestations are similar to those of hypocalcemia. Neuromuscular irritability, positive Chvostek and Trousseau signs, increased reflexes, muscle weakness, ataxia, nystagmus, tetany, seizures, and tachydysrhythmias may be observed. Treatment is intramuscular or IV administration of magnesium sulfate. Magnesium supplementation may be beneficial in the treatment of preeclampsia, migraine, depression, cardiovascular disease, metabolic syndrome, and asthma.21–23
Hypermagnesemia, in which magnesium concentration is greater than 3.0 mEq/L, is rare and usually is caused by oliguric renal disease. Magnesium-containing antacids or cathartics can potentiate excess magnesium levels. Excess magnesium concentration inhibits calcium-mediated nerve conduction and depresses skeletal muscle contraction and nerve function. Signs and symptoms include nausea and vomiting, decreased deep tendon reflexes, muscle weakness, hypotension, bradycardia, and respiratory depression. Treatment is avoidance of magnesium-containing substances and removal of magnesium by dialysis.24 (See Table 3.11 contains a summary of the manifestations of magnesium level alterations.)
ACID-Base Balance
Acid-base balance and hydrogen ion concentration must be regulated within a narrow range for normal body function. Slight changes in amounts of hydrogen ion and pH changes can significantly alter biologic processes in cells and tissues. Hydrogen ions are required to maintain membrane integrity and the speed of enzymatic reactions.25 Many pathologic conditions disturb acid-base balance, and the degree of severity may be more harmful than the disease process itself.
Hydrogen Ion and pH
The concentration of hydrogen ions in body fluids is very small—approximately 0.0000001 mg/L. This number is expressed as the negative logarithm 10−7 mg/L and is indicated on the pH scale as pH 7.0, a neutral status. Values less than 7.0 are more acid. Values more than 7.0 are more alkaline. Accordingly, the pH number indicates the acidity or alkalinity of a solution. The pH scale is logarithmic, not linear, meaning the difference between numbers on the scale is not constant. Each number on the scale is 10 times more acid or more alkaline than the preceding number. Accordingly, a pH of 5 is 10 times more acid than a pH of 6; a pH of 4 is 10 times more acid than a pH of 5 and 100 times more acid than a pH of 6. As the pH changes by one unit from a pH 7 to a pH 6, the H+ increases 10-fold, making the solution much more acid than a neutral solution with a pH of 7.0. Solutions with an excess of hydrogen ions are acidic in nature; solutions with an excess of hydroxide ions (OH−) are basic, or alkaline, in nature. In arterial blood, the normal pH range is 7.35 to 7.45, with 7.4 being the midpoint of that normal range. Accordingly, a blood pH less than 7.35 is defined as acidic, and a pH greater than 7.45 is defined as alkaline, or basic (Table 3.12).
Table 3.12
The average person generates 50 to 100 mEq/day of acid from the metabolism of proteins, carbohydrates, and fats, as well as from loss of alkaline fluids in the stool. To maintain a normal pH, acids must be balanced by base substances within the body. A base is a substance that accepts hydrogen ions; an acid is a substance that donates hydrogen ions. Body acids exist in two forms: volatile acids (substances that can be eliminated as carbon dioxide [CO2] gas) and nonvolatile acids (substances that can be eliminated only by the kidney). The sole volatile acid formed in the body is carbonic acid (H2CO3), a weak acid, which means that in body fluids, some of the carbonic acid molecules release their hydrogen ions, but other carbonic molecules do not. In the presence of the enzyme carbonic anhydrase, it readily dissociates into CO2 and water (H2O). The CO2 is eliminated through pulmonary ventilation. The rest of the body acids are nonvolatile acids, such as lactic acid, phosphoric acid, sulfuric acid, acetoacetic acid, and beta-hydroxybutyric acid. A few nonvolatile acids are strong acids, meaning they readily release their hydrogen ions; however, most of the nonvolatile acids are weak acids. Nonvolatile acids are secreted into the urine by the renal tubules in amounts of approximately 50 to 100 mEq of hydrogen per day, or approximately 1 mEq/kg of body weight.
Mechanisms to Maintain Normal pH
The body has three mechanisms, or lines of defense, to maintain the acid-base balance: (1) physiologic (chemical) buffer systems (plasma bicarbonate, phosphate, hemoglobin [Hb], and protein), the first line of defense; (2) respiratory acid-base control, the second line of defense; and (3) renal acid-base control, the third line of defense. The physiologic buffers function instantaneously to correct alterations in the acid-base balance. The lungs and the kidneys work in concert to maintain a normal pH. The lungs respond relatively quickly (within seconds to minutes), but the kidneys require more time (hours to days) to bring the system into balance. Although the lungs respond more quickly, mechanisms involving the kidney produce more long-term acid-base balance.
Buffer Systems
Buffer systems resist changes in pH and maintain pH within the normal range. Metabolic processes primarily generate H+ (acid) ions. Unchecked, these ions would alter the pH of the body. Buffers can absorb excessive H+ ions (acid) or hydroxide ions (OH−) (base) to minimize fluctuations in pH. The buffer systems are located in both the ICF and the ECF compartments, and function at different rates (Table 3.13). The most important are the plasma buffer systems, which have two components: carbonic acid–bicarbonate and the protein Hb. The important intracellular buffers are phosphate and protein. Ammonia and phosphate can attach hydrogen ions and are important renal buffers.
Table 3.13





 H+ + Pr−
H+ + Pr−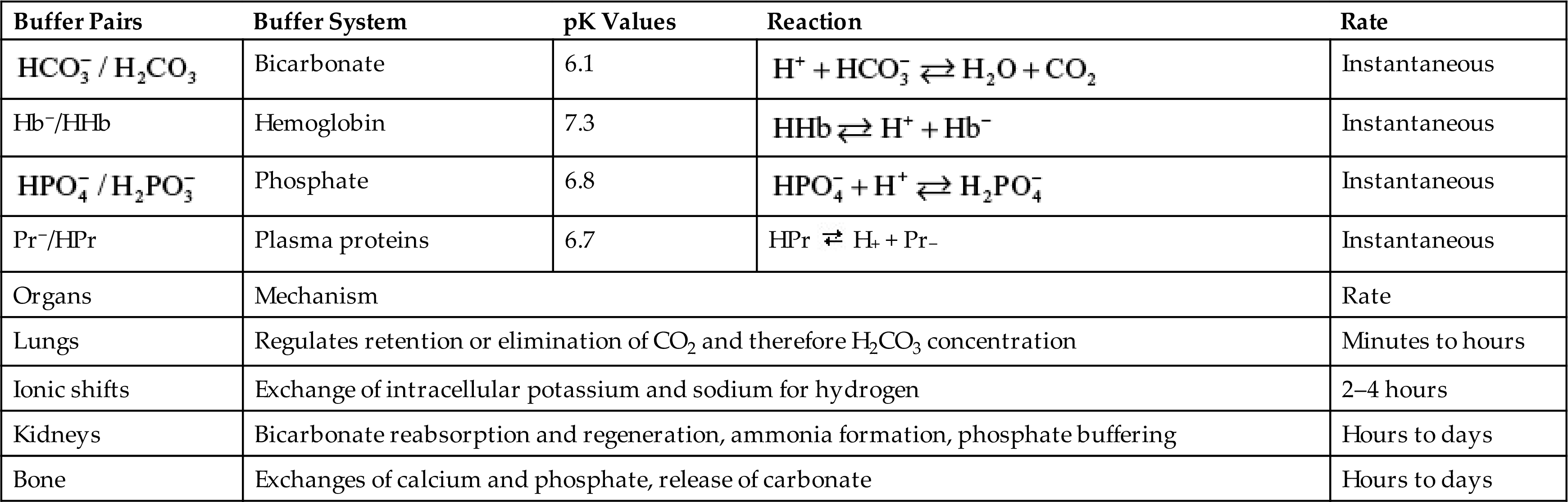
H+, Hydrogen ion;  , bicarbonate; H2CO3, carbonic acid; Hb−, hemoglobin; HHb, hydrogenated hemoglobin;
, bicarbonate; H2CO3, carbonic acid; Hb−, hemoglobin; HHb, hydrogenated hemoglobin;  , monobasic phosphate;
, monobasic phosphate;  , dibasic phosphate; HPr, hydrogenated protein; Pr−, protein.
, dibasic phosphate; HPr, hydrogenated protein; Pr−, protein.
Carbonic Acid–Bicarbonate Buffering
Cellular respiration results in the production of CO2, which combines with water to form carbonic acid (H2CO3). H2CO3 dissociates, forming one hydrogen (H+) ion and one bicarbonate ion ( ) in the blood. The bicarbonate ion is a base component of the buffer system, and the hydrogen ion is the acid component. These reactions are readily reversible, depending on whether an acid or a base environment exists. The reactions can move in one or the other direction to maintain a neutral pH. Accordingly, these reactions can correct for imbalances in pH by releasing hydrogen ions or absorbing hydrogen ions as the need arises. If excess H+ ions are present, the buffer absorbs these ions. If excess bicarbonate ions are present, the buffer releases H+ ions. The relationship between bicarbonate (
) in the blood. The bicarbonate ion is a base component of the buffer system, and the hydrogen ion is the acid component. These reactions are readily reversible, depending on whether an acid or a base environment exists. The reactions can move in one or the other direction to maintain a neutral pH. Accordingly, these reactions can correct for imbalances in pH by releasing hydrogen ions or absorbing hydrogen ions as the need arises. If excess H+ ions are present, the buffer absorbs these ions. If excess bicarbonate ions are present, the buffer releases H+ ions. The relationship between bicarbonate ( ) and carbonic acid (H2CO3) usually is expressed as a ratio. Under normal conditions, the bicarbonate level is approximately 24 mEq/L and the carbonic acid level is approximately 1.2 mEq/L when the arterial CO2 (PaCO2) is 40 mm Hg. Therefore the ratio of bicarbonate to carbonic acid is 20:1 (24/1.2). This ratio maintains a normal pH of 7.40 (Fig. 3.11).
) and carbonic acid (H2CO3) usually is expressed as a ratio. Under normal conditions, the bicarbonate level is approximately 24 mEq/L and the carbonic acid level is approximately 1.2 mEq/L when the arterial CO2 (PaCO2) is 40 mm Hg. Therefore the ratio of bicarbonate to carbonic acid is 20:1 (24/1.2). This ratio maintains a normal pH of 7.40 (Fig. 3.11).
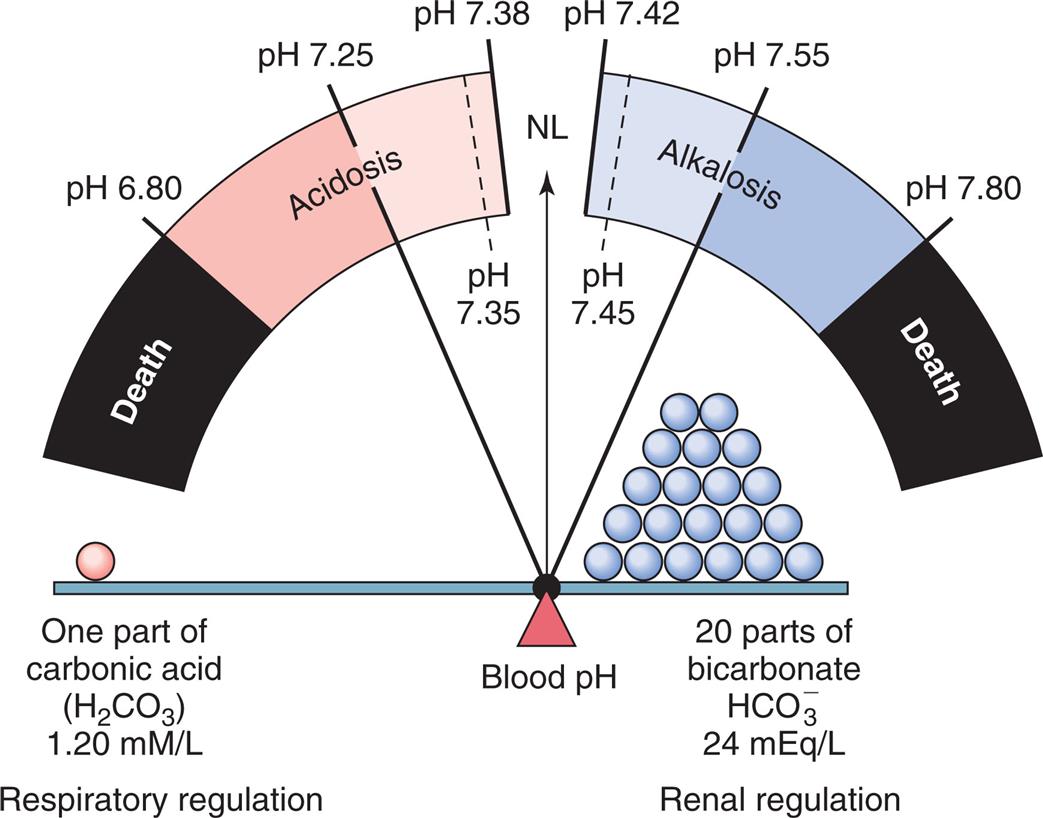
An increase in H2CO3 or decrease in
 concentration causes acidosis. A decrease in H2CO3 or increase in
concentration causes acidosis. A decrease in H2CO3 or increase in  concentration causes alkalosis. NL, Normal. (From Monahan, FD, et al. Medical-surgical nursing: Health and illness perspectives, 8th edition. St. Louis: Mosby; 2007.)
concentration causes alkalosis. NL, Normal. (From Monahan, FD, et al. Medical-surgical nursing: Health and illness perspectives, 8th edition. St. Louis: Mosby; 2007.)A fulcrum balance shows one part of carbonic acid, H 2 C O 3, on the left pan, and 20 parts of bicarbonate, H C O 3 ion, on the right pan. The fulcrum, blood p H, is closer to the right pan. The left pan represents respiratory regulation and measures 1.20 millimoles per liter. The right pan represents renal regulation and measures 24 milliequivalents per liter. A gauge traced from the fulcrum represents the p H limits. From the left to the right, the gauge shows the following segments: • Up to p H 6.80: death. • p H 6.80 to p H 7.25: high acidosis. • p H 7.25 to p H 7.38: low acidosis. • p H 7.35 to p H 7.45: N L, normal. • p H 7.42 to p H 7.55 low alkalosis. • p H 7.55 to p H 7.80: high alkalosis. • beyond p H 7.80: death.
Both the lungs and the kidneys augment the action of the bicarbonate–carbonic acid buffer system. The lungs eliminate CO2 and water and can increase the amount of CO2 eliminated by increasing the rate and depth of ventilation. Although the lungs do not respond as rapidly as the physiologic buffering system, they have twice the ability to correct for pH imbalances compared with all the chemical buffering systems combined. Similarly, the kidneys augment the carbonic acid–bicarbonate buffer system by reabsorbing or regenerating bicarbonate from CO2 and water in the renal tubules or excreting hydrogen ions into the urine. The renal response takes considerably longer than does the respiratory response. However, the kidneys’ ability to regulate and maintain a normal pH is necessary because the kidneys are the only organs that can excrete nonvolatile acids.
The pH ratio equation of 20/1 can be symbolically expressed as follows:
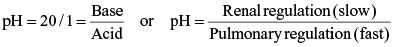
Changes in either the numerator or the denominator will change the pH. For example, if the amount of bicarbonate is decreased, the pH also decreases, causing a state of acidosis (Fig. 3.12). The pH can be returned to a normal range if the value of the denominator or the amount of carbonic acid also decreases. When a disease process causes an alteration in the bicarbonate/carbonic acid ratio, the kidneys or lungs (i.e., the organ not responsible for causing the alteration) respond to restore the ratio and maintain a normal pH.
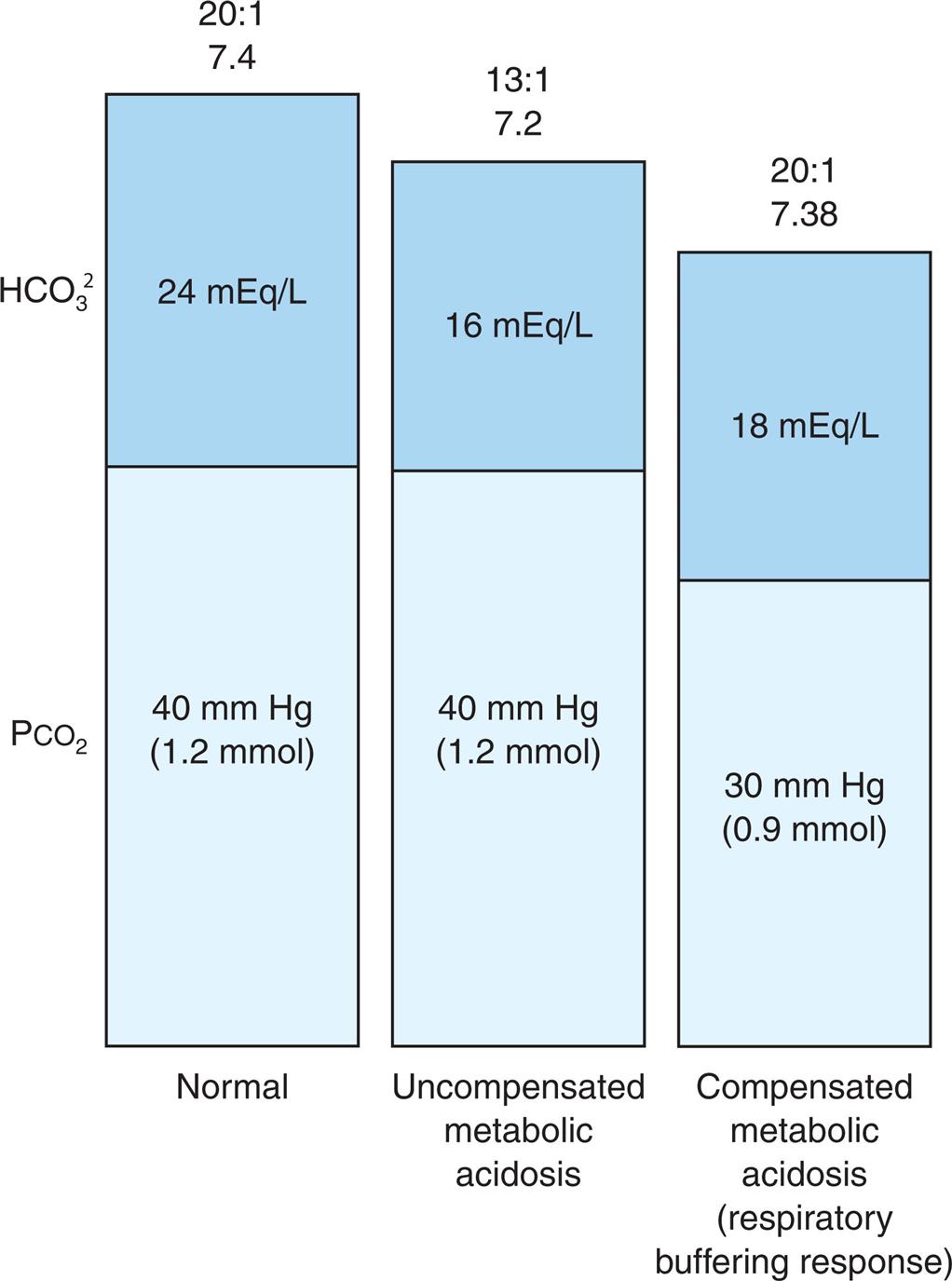
 ]/PCO2 (H2CO3) Ratio in Metabolic Acidosis.
]/PCO2 (H2CO3) Ratio in Metabolic Acidosis.Three stacked bars represent ratios of partial pressure of carbon dioxide and bicarbonate in metabolic acidosis. Left bar, normal in the ratio 20 to 1 of 7.4. P C O sub 2, 40 millimeters of mercury or 1.2 millimoles. H C O sub 3 sup 2, 24 milliequivalents per liter. Middle bar, uncompensated metabolic acidosis in the ratio 13 to 1 of 7.2. P C O sub 2, 40 millimeters of mercury of 1.2 millimoles. H C O sub 3 sup 2, 16 milliequivalents per liter. Right bar, compensated metabolic acidosis or respiratory buffering response in the ration 20 to 1 of 7.38. P C O sub 2, 30 millimeters of mercury or 0.9 millimoles. H C O sub 3 sup 2, 18 milliequivalents per liter.
Protein Buffering
All proteins can attach or release a hydrogen ion. Most proteins are inside cells; hence protein-based buffering is primarily an intracellular buffer system. Hb in erythrocytes is an excellent blood protein buffer. As the pH increases, Hb loses hydrogen ions, and the reverse happens when the pH decreases. Hb also affects the pH through a different mechanism when it binds CO2 to form carbaminohemoglobin (HHbCO2). The bound CO2 is transported to the lungs, where it is released from the body through ventilation. This dynamic is important because CO2 is a potential acid. Unbound CO2 can react in water to form H2CO3, a weak acid. Carbonic acid releases hydrogen ion, lowering the pH. By binding CO2, the Hb is preventing CO2 from becoming carbonic acid; it thereby prevents the release of excess H+ ions into the environment. The pH control system is illustrated in Fig. 3.13.
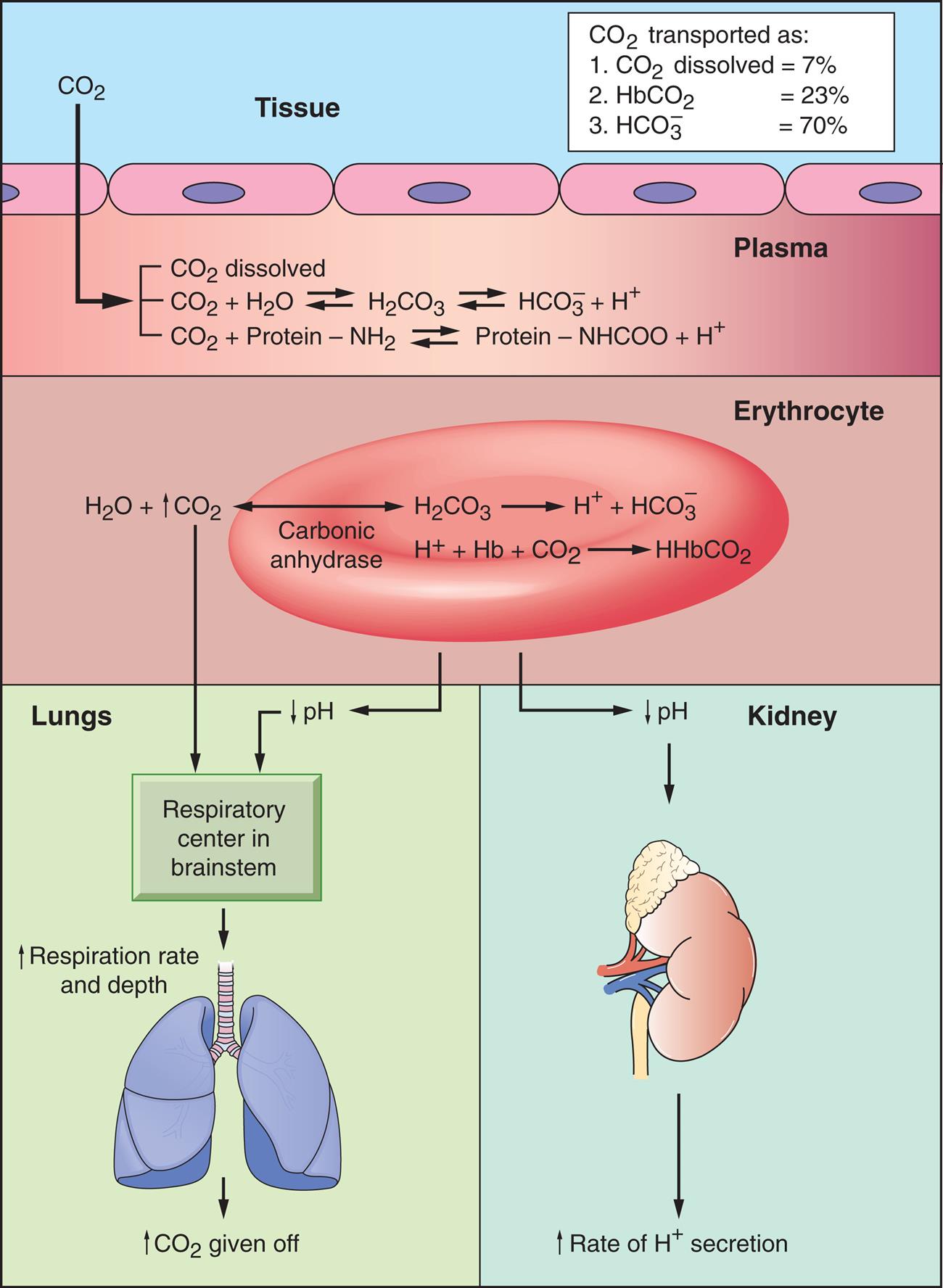
CO2 is produced in tissue cells and diffuses to plasma, where it is transported as dissolved CO2, or it combines with water to form carbonic acid (H2CO3), or it combines with protein from which hydrogen has been released. Most of the CO2 diffuses into the red blood cells and combines with water to form H2CO3. The H2CO3 dissociates to form hydrogen ion (H+) and bicarbonate (
 ). Hydrogen ion combines with hemoglobin that has released its oxygen to form hydrogenated hemoglobin (HHb), which buffers the hydrogen ion and makes venous blood slightly more acidic than arterial blood. The increase in H+ concentration coupled with elevated CO2 levels results in HHbCO2 and an increase in the respiratory rate (eliminates CO2) and secretion of H+ by the kidneys.
). Hydrogen ion combines with hemoglobin that has released its oxygen to form hydrogenated hemoglobin (HHb), which buffers the hydrogen ion and makes venous blood slightly more acidic than arterial blood. The increase in H+ concentration coupled with elevated CO2 levels results in HHbCO2 and an increase in the respiratory rate (eliminates CO2) and secretion of H+ by the kidneys.An illustration demonstrates the integration of p H control mechanisms in tissue, plasma, erythrocyte, lungs, and kidney. Tissue. C O 2 is transported as: 1. C O 2 dissolved equals 7 percent. 2. H b C O 2 equals 23 percent. 3. H C O 3 ion equals 70 percent. Plasma. C O 2 from tissue. • C O 2 dissolved. • C O 2 plus H 2 O reversible with H 2 C O 3 reversible with H C O 3 ion and H ion. • C O 2 plus protein minus N H 2 reversible with protein minus N H C O O and H ion. Erythrocyte. • H 2 O and expelled C O 2 reversible under carbonic anhydrase with H 2 C O 3, which produces H ion and H C O 3 ion. • H ion with H b and C O 2 produces H H b C O 2. Lungs. Expelled C O 2 and decreased p H from erythrocyte reach the respiratory center in brainstem. This increases respiration rate and depth, giving off C O 2. Kidney. Decreased p H from erythrocyte reaches kidney, increasing rate of H ion secretion.
Respiratory and Renal Buffering
The respiratory system regulates acid-base balance by controlling the rate of ventilation when there is metabolic acidosis or alkalosis. Central chemoreceptors sense increases or decreases in pH and PaCO2 When acidemia exists, the respiratory rate increases (eliminating CO2 and thus reducing carbonic acid concentration) (see Fig. 3.13). When alkalemia occurs, the respiratory rate decreases (retaining CO2 and increasing carbonic acid concentration).
The renal buffering of H+ ions requires the use of carbonic anhydrase in renal tubular cells to combine CO2 and H2O to form H2CO3. The H+ ion is secreted from the tubular cell and buffered in the lumen by phosphate and ammonia. The bicarbonate is reabsorbed. The end effect is the addition of new bicarbonate to the blood, which contributes to the alkalinity of the plasma, because the hydrogen ion is excreted from the body (Fig. 3.14-1).
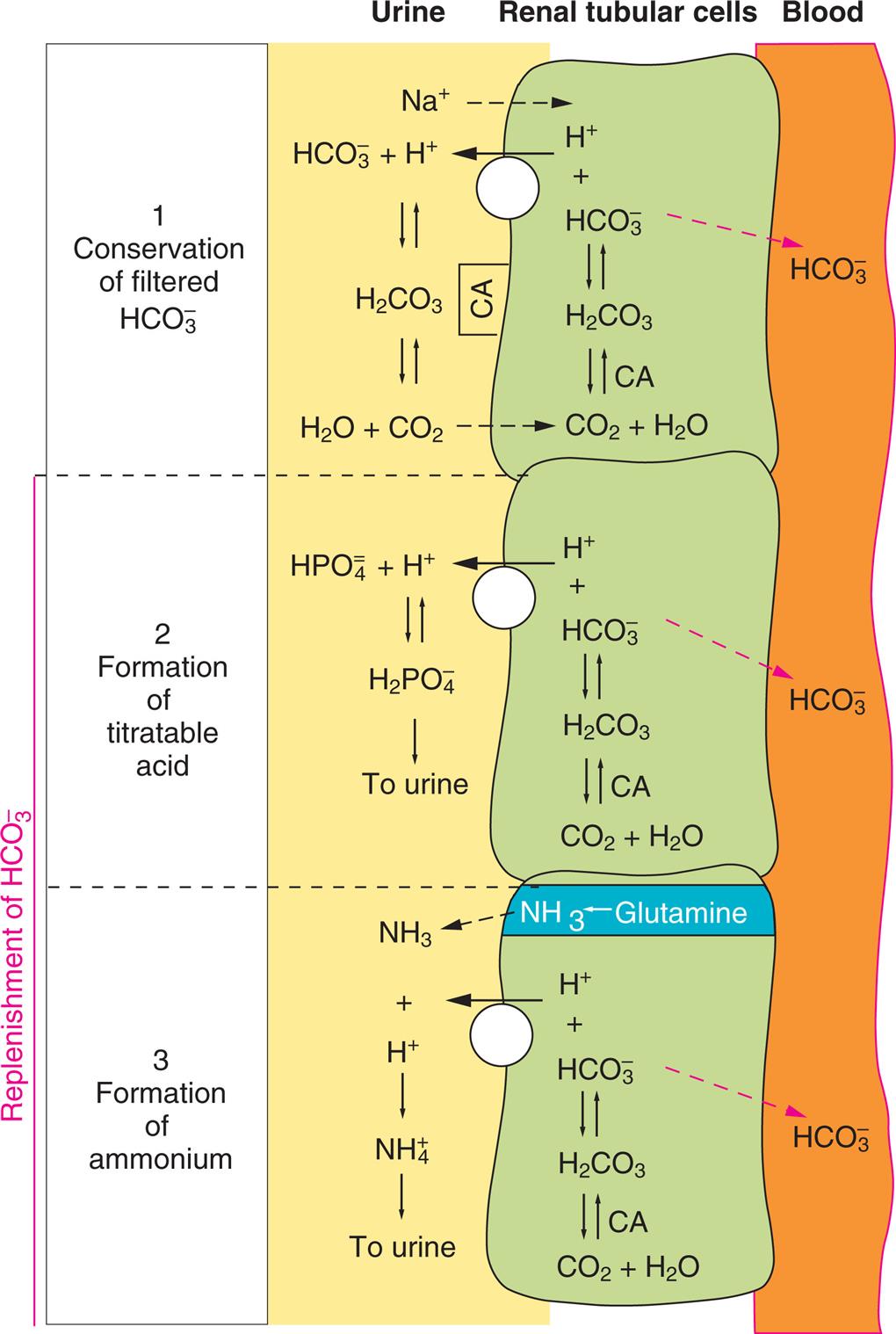
1, Conservation of Filtered Bicarbonate. Filtered bicarbonate (
 ) combines with secreted hydrogen ion (H+) to form carbonic acid (H2CO3), which then is converted to water (H2O) and carbon dioxide (CO2) by carbonic anhydrase (CA); both diffuse into the renal tubular cell. The CO2 and H2O combine to form H2CO3 in the presence of CA, and the resulting bicarbonate ion (
) combines with secreted hydrogen ion (H+) to form carbonic acid (H2CO3), which then is converted to water (H2O) and carbon dioxide (CO2) by carbonic anhydrase (CA); both diffuse into the renal tubular cell. The CO2 and H2O combine to form H2CO3 in the presence of CA, and the resulting bicarbonate ion ( ) is reabsorbed into the blood. 2, Formation of Titratable Acid. Hydrogen ion (H+) is secreted and combines with dibasic phosphate (
) is reabsorbed into the blood. 2, Formation of Titratable Acid. Hydrogen ion (H+) is secreted and combines with dibasic phosphate ( ) to form monobasic phosphate (
) to form monobasic phosphate ( ). The secreted hydrogen ion (H+) is formed from the dissociation of H2CO3, and the remaining
). The secreted hydrogen ion (H+) is formed from the dissociation of H2CO3, and the remaining  is reabsorbed into the blood. 3, Formation of Ammonium Ions. Ammonia (NH3) is produced from glutamine in the renal tubular cell and diffuses to the urine where it combines with H+ to form ammonium ion (
is reabsorbed into the blood. 3, Formation of Ammonium Ions. Ammonia (NH3) is produced from glutamine in the renal tubular cell and diffuses to the urine where it combines with H+ to form ammonium ion ( ). Once
). Once  has been formed it cannot return to the renal tubular cell (diffusional trapping) and the bicarbonate remaining in the renal tubular cell is reabsorbed into the blood. NOTE: The white circles with the arrows on top represent a renal tubular cell active transport pump.
has been formed it cannot return to the renal tubular cell (diffusional trapping) and the bicarbonate remaining in the renal tubular cell is reabsorbed into the blood. NOTE: The white circles with the arrows on top represent a renal tubular cell active transport pump.An illustration of renal tubular cells between urine and blood demonstrates the renal excretion of acid in three stages.1. Conservation of filtered H C O 3 ion. • Urine. N a ion from urine to renal tubular cell and H ion from renal tubular cell to urine. H C O 3 ion and H ion reversible with H 2 C O 3 reversible with H 2 O and C O 2. • Renal tubular cell. H ion and H C O 3 ion reversible with H 2 C O 3 reversible under C A with C O 2 and H 2 O. • Blood. H C O 3 ion from renal tubular cell to blood.2. Formation of titratable acid. • Urine. H ion from renal tubular cell to urine. H P O 4 ion and H ion reversible with H 2 P O 4 ion to urine. • Renal tubular cell. H ion and H C O 3 ion reversible with H 2 C O 3 reversible under C A with C O 2 and H 2 O. • Blood. H C O 3 ion from renal tubular cell to blood.3. Formation of ammonium. • Urine. N H 3 and H ion from renal tubular cell reversible with N H 4 to urine. • Renal tubular cell. Glutamine to N H 3. H ion and H C O 3 ion reversible with H 2 C O 3 reversible under C A with C O 2 and H 2 O. • Blood. H C O 3 ion from renal tubular cell to blood.
Two additional buffer systems are active in the renal tubules, the phosphate buffer and the ammonia buffer. Once H+ has reacted with all available, any additional H+ ions react with either the phosphate or the ammonia buffer systems. The phosphate buffer functions both in the renal tubules and in the ICF. The two components of this system, monobasic phosphate ( ) and dibasic phosphate (
) and dibasic phosphate ( ), usually function in association with sodium to form a sodium salt. As with any buffer, these components bind or release H+ to maintain physiologic neutrality (pH 7.40) (Fig. 3.14-2). The ammonia buffer functions within the renal tubules. The components for this system are ammonia (NH3) and ammonium ion (
), usually function in association with sodium to form a sodium salt. As with any buffer, these components bind or release H+ to maintain physiologic neutrality (pH 7.40) (Fig. 3.14-2). The ammonia buffer functions within the renal tubules. The components for this system are ammonia (NH3) and ammonium ion ( ) (Fig. 3.14-3). Both monobasic phosphate and ammonium ion (
) (Fig. 3.14-3). Both monobasic phosphate and ammonium ion ( ) are lipid insoluble, preventing them from diffusing back across the tubular cells and into the blood; thus, they are excreted in the urine.
) are lipid insoluble, preventing them from diffusing back across the tubular cells and into the blood; thus, they are excreted in the urine.
Other Buffers
A cellular ion exchange mechanism is also an important buffering system. The best example is the shift of potassium in exchange for hydrogen during states of acidosis or alkalosis. During acidosis, potassium tends to leave the intracellular space in exchange for hydrogen. The reverse occurs during alkalosis. Although the ionic shifts facilitate buffering, the changes in intracellular or extracellular potassium concentrations may have serious consequences (e.g., hyperkalemia or hypokalemia).
Acid-Base Imbalances
Pathophysiologic changes in the concentration of H+ ion or base in the blood lead to acid-base imbalances. Acidemia is a state in which the pH of arterial blood is less than 7.35. A systemic increase in hydrogen ion concentration or a loss of base is termed acidosis. Alkalemia is a state in which the pH of arterial blood is greater than 7.45. A systemic decrease in hydrogen ion concentration or an excess of base is termed alkalosis. These changes may be caused by either respiratory or metabolic processes, or both. If the altered pH occurs secondary to biochemical processes within the body or to renal dysfunction, it is metabolic. If the alteration in pH is secondary to an alteration with ventilation, it is respiratory. Accordingly, four clinical alterations in the acid-base status can develop: respiratory acidosis, metabolic acidosis, respiratory alkalosis, and metabolic alkalosis.25
Arterial blood gases are measured to determine which of the four alterations is present. This measurement typically includes four parameters: the blood pH, PaO2 (oxygen), PaCO2 (carbon dioxide), and (bicarbonate).26 The PaO2 provides information about the person’s oxygenation status, but it does not directly provide information about the acid-base status. The blood pH, PaCO2, and bicarbonate level, when evaluated together, provide information about whether an individual is experiencing acidosis or alkalosis, and whether the cause is respiratory or metabolic. Fig. 3.15 summarizes the relationships among pH, PaCO2, and bicarbonate during different acid-base alterations. Fig. 3.11 summarizes the relationship among pH, the partial pressure of carbon dioxide (respiratory regulation), and the concentration of bicarbonate (renal regulation) during alkalosis and acidosis. An individual can experience both metabolic and respiratory disorders at the same time. In such cases, the person may have a mixed acid-base alteration.
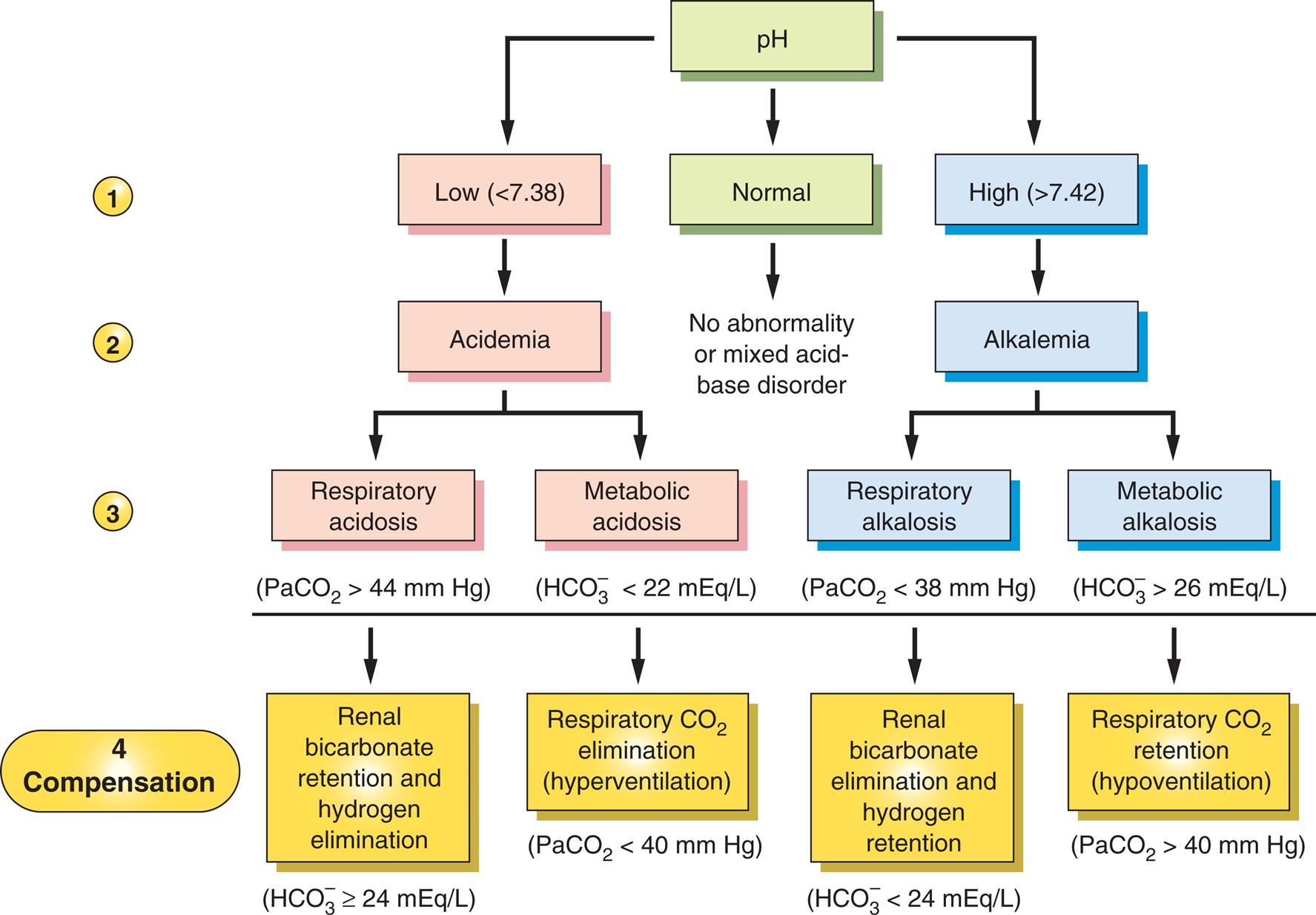
A systematic approach can be used to interpret the cause of an acid-base imbalance. 1, Is the pH low or high? 2, If the pH is low, there is acidemia; if the pH is high, there is alkalemia. 3, If the pH is low (acidemia), is the cause respiratory (high PaCO2) or metabolic (low
 )? If the pH is high (alkalemia), is the cause respiratory (low PaCO2) or metabolic (high
)? If the pH is high (alkalemia), is the cause respiratory (low PaCO2) or metabolic (high  )? 4, Is there compensation for the primary acid-base disorder? (a)
)? 4, Is there compensation for the primary acid-base disorder? (a) will be ≥24 mEq/L if there is renal compensation for a primary respiratory acidosis; (b) PaCO2 will be <40 mm Hg if there is respiratory compensation of a primary metabolic acidosis; (c)
will be ≥24 mEq/L if there is renal compensation for a primary respiratory acidosis; (b) PaCO2 will be <40 mm Hg if there is respiratory compensation of a primary metabolic acidosis; (c) will ≤24 mEq/L if there is renal compensation for primary respiratory alkalosis; (d) PaCO2 will be >40 mm Hg if there is respiratory compensation for primary metabolic alkalosis. NOTE: Examine the pH first. Then examine the changes in
will ≤24 mEq/L if there is renal compensation for primary respiratory alkalosis; (d) PaCO2 will be >40 mm Hg if there is respiratory compensation for primary metabolic alkalosis. NOTE: Examine the pH first. Then examine the changes in  and PaCO2.
and PaCO2.  concentration will be elevated when there is primary metabolic alkalosis or renal compensation for primary respiratory acidosis.
concentration will be elevated when there is primary metabolic alkalosis or renal compensation for primary respiratory acidosis.  concentration will be decreased when there is primary metabolic acidosis or renal compensation for primary respiratory alkalosis. PaCO2 will be elevated when there is primary respiratory acidosis or respiratory compensation for primary metabolic alkalosis. PaCO2 will be decreased when there is primary respiratory alkalosis or respiratory compensation for metabolic acidosis.
concentration will be decreased when there is primary metabolic acidosis or renal compensation for primary respiratory alkalosis. PaCO2 will be elevated when there is primary respiratory acidosis or respiratory compensation for primary metabolic alkalosis. PaCO2 will be decreased when there is primary respiratory alkalosis or respiratory compensation for metabolic acidosis.A hierarchy diagram, comprising four levels, represents the primary and compensatory acid-base changes of p H. Level 1. p H can be low (below 7.38), normal, or high (above 7.42). Level 2. Low p H causes acidemia. Normal p H does not cause any abnormality of mixed acid-base disorder. High p H causes alkalemia. Level 3. Acidemia leads to respiratory acidosis or metabolic acidosis. Alkalemia leads to respiratory alkalosis or metabolic alkalosis. Level 4. • Under respiratory acidosis, when P a C O 2 greater than 44 millimeters of mercury, renal bicarbonate retention and hydrogen elimination, where H C O 3 ion is greater than or equal to 23 milliequivalents per liter. • Under metabolic acidosis, when H C O 3 ion less than 22 milliequivalents per liter, respiratory C O 2 elimination or hyperventilation, where P a C O 2 less than 40 millimeters of mercury. • Under respiratory alkalosis, when P a C O 2 less than 38 millimeters of mercury, renal bicarbonate elimination and hydrogen retention, where H C O 3 ion less than 24 milliequivalents per liter. • Under metabolic alkalosis, when H C O 3 ion greater than 26 milliequivalents per liter, respiratory C O 2 retention or hypoventilation, where P a C O 2 is greater than 40 millimeters of mercury.
Renal and respiratory adjustments to primary changes in pH are known as compensation. With compensation, a 20:1 ratio may be achieved, but the actual values for  and H2CO3 concentrations are not normal (see Fig. 3.12). The respiratory system compensates for changes in pH by increasing or decreasing ventilation, a rapid response occurring within minutes to hours. The renal system compensates by producing more acidic or more alkaline urine, which may take hours to days. Correction occurs when the values for both components of the buffer pair ratio (bicarbonate and carbonic acid) return to normal.
and H2CO3 concentrations are not normal (see Fig. 3.12). The respiratory system compensates for changes in pH by increasing or decreasing ventilation, a rapid response occurring within minutes to hours. The renal system compensates by producing more acidic or more alkaline urine, which may take hours to days. Correction occurs when the values for both components of the buffer pair ratio (bicarbonate and carbonic acid) return to normal.
Carbon dioxide is referred to as a potential acid because it is readily converted to carbonic acid. Breathing more shallowly and slowly (hypoventilation) causes a retention of CO2 (hypercapnia), whereas breathing more rapidly and deeply (hyperventilation) blows off CO2 (hypocapnia). In this manner, the lungs compensate for excess acidity or alkalinity of the blood. The PaCO2 and bicarbonate levels provide information on the source of the altered pH and also indicate whether any compensation is ongoing. A low blood pH signals acidosis. If the PaCO2 is high, the source of the problem is respiratory (e.g., retained CO2). Such a scenario might occur with an opioid overdose, in which respirations are depressed and CO2 is retained. This individual is described as having a primary respiratory acidosis. If the pH is low and the PaCO2 is normal or low, the acidosis is not caused by respiratory factors. Instead, it is caused by metabolic processes within the body. In this case, the individual is described as having a primary metabolic acidosis. An example of a primary metabolic acidosis is an individual experiencing DKA from a lack of insulin secretion or administration. The diabetic individual, lacking insulin, cannot metabolize glucose. The body metabolizes fat stores, and as a result, acid by-products are released into the blood, causing metabolic acidosis.
Metabolic Acidosis
Pathophysiology
In metabolic acidosis, the concentration of nonvolatile (non–carbonic) acids increase. Less commonly this condition can result if bicarbonate (base) is lost from the ECF or cannot be regenerated by the kidney (Table 3.14). Metabolic acidosis can occur quickly, as in lactic acidosis secondary to poor perfusion or hypoxemia. It can also occur more slowly, as in oliguric renal disease (failure to excrete acid), ingestion of acid precursors that are metabolized to acids, starvation states (excess production of ketoacids from excess metabolism of fats and proteins), or DKA (excess production of ketoacids from lack of insulin) (see Chapter 22).
Table 3.14
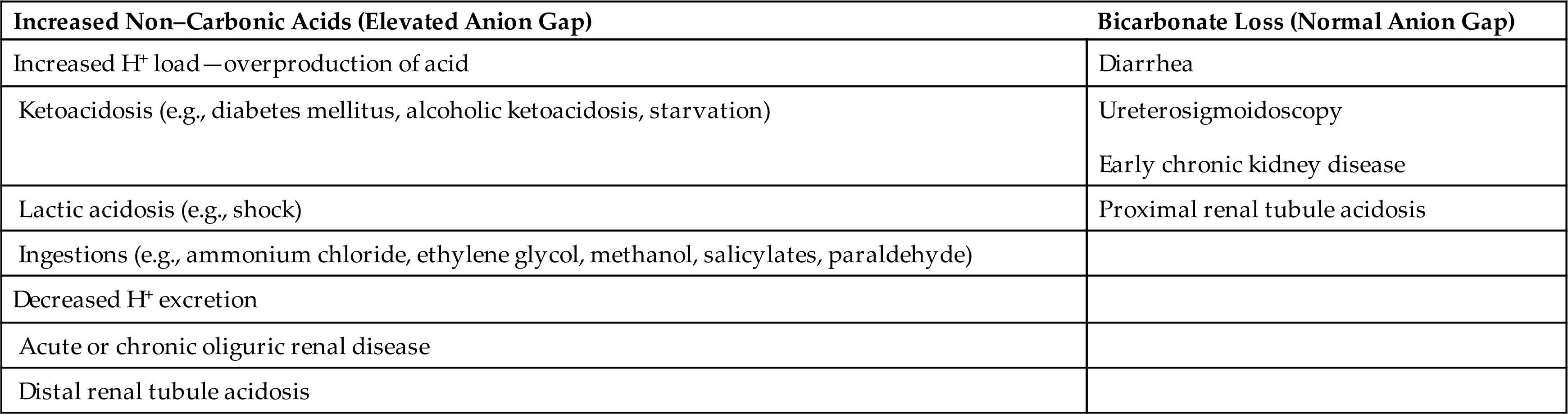
The buffer systems manage the excess acid and attempt to maintain the arterial pH within a normal range. Hydrogen ions will move to the intracellular space, and potassium will move to the extracellular space to maintain an ionic balance. Buffering by bicarbonate lowers the serum value of hydrogen ions and increases the previously decreased pH. The respiratory system compensates for a metabolic acidosis as the reduced pH stimulates hyperventilation, lowering the PaCO2 and the amount of H2CO3 circulating in the blood. The kidneys excrete the excess acid as  and titratable acid (
and titratable acid ( ). When acidosis is severe, buffers become depleted and cannot compensate for the increasing H+ load and the pH continues to decrease. The ratio of bicarbonate to carbonic acid decreases to less than 20:1 (Fig. 3.16). In states of metabolic acidosis, potassium may be redistributed from the intracellular to the extracellular space and is reabsorbed at the apical membrane of the renal collecting tubule. There is also an increase in the levels of ionized calcium because acidosis decreases the amount of calcium bound to albumin.
). When acidosis is severe, buffers become depleted and cannot compensate for the increasing H+ load and the pH continues to decrease. The ratio of bicarbonate to carbonic acid decreases to less than 20:1 (Fig. 3.16). In states of metabolic acidosis, potassium may be redistributed from the intracellular to the extracellular space and is reabsorbed at the apical membrane of the renal collecting tubule. There is also an increase in the levels of ionized calcium because acidosis decreases the amount of calcium bound to albumin.
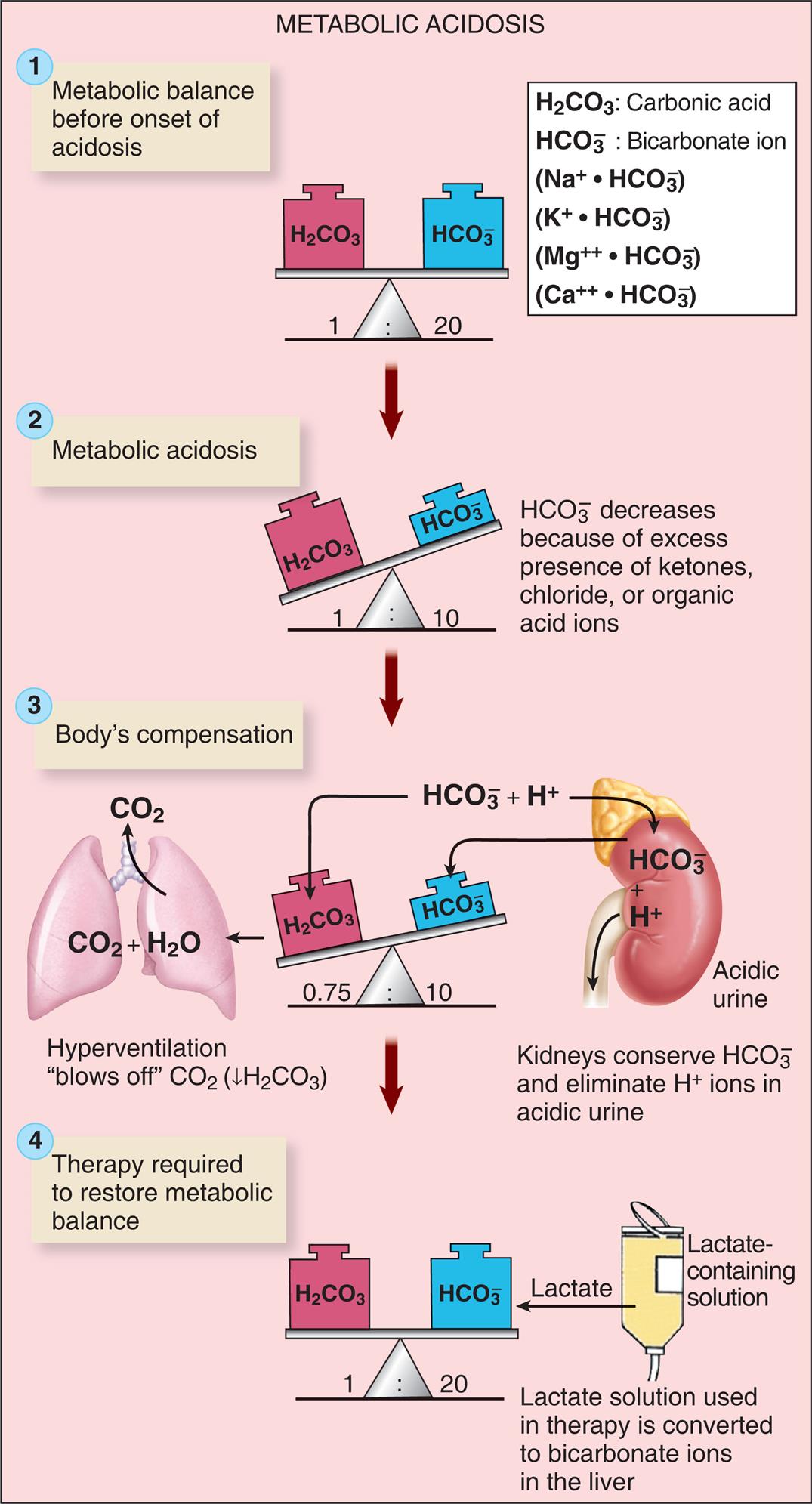
See text for abbreviations. (From Patton KT, Thibodeau, GA. Anatomy & physiology, 9th edition. St. Louis: Mosby; 2016.)
An illustrated flowchart shows the metabolic acidosis process with compensation and correction. There are four stages in the process. 1. Metabolic balance before onset of acidosis. Carbonic acid and bicarbonate ion are balanced in the ratio 1 to 20. 2. Metabolic acidosis. Carbonic acid and bicarbonate ion are unbalanced in the ratio 1 to 10. Carbonic acid decreases because of excess presence of ketones, chloride, or organic acid ions. 3. Body’s compensation. Carbonic acid and bicarbonate ion are unbalanced in the ratio 0.75 to 10. Hyperventilation blows off C O 2, decreasing H 2 C O 3. Kidneys conserve H C O 3 and eliminate H ions in acidic urine. 4. Therapy required to restore metabolic balance. Carbonic acid and bicarbonate ion are balanced in the ratio 1 to 20. Lactate-containing solution used in therapy is converted to bicarbonate ions in the liver.
The evaluation of the anion gap can be helpful when used cautiously to distinguish different factors contributing to metabolic acidosis.27 Normally, the concentrations of cations and anions in the plasma are equivalent. However, some anions, such as protein, sulfates, phosphates, and organic acids, are not measured in the common laboratory evaluations of the blood. Therefore the normal anion gap represents these unmeasured negative ions (sulfate, phosphate, lactate, ketoacids, albumin). A convenient measure of the anion gap is the difference between the sum of Na+ and K+ concentrations and the sum of  and Cl− concentrations, or approximately 10 to 12 mEq/L:
and Cl− concentrations, or approximately 10 to 12 mEq/L:

In metabolic acidosis a normal anion gap is characteristic of conditions related to bicarbonate loss with retention of chloride to maintain an ionic balance. This is called hyperchloremic metabolic acidosis, and it occurs with chronic kidney disease or prolonged diarrhea with bicarbonate loss. An elevated anion gap is characteristic of acidosis associated with accumulation of anions other than chloride (see Table 3.14). With severe pH disturbances, changes in the concentration of serum albumin, which behaves as an anion, can have a significant impact on the anion gap and a correction for this measure can be used in these cases.28
Clinical Manifestations
Metabolic acidosis is manifested by changes in the neurologic, respiratory, gastrointestinal, and cardiovascular systems. Early symptoms include headache and lethargy, which progress to confusion and coma in severe acidosis. The respiratory system’s efforts to compensate for the increase in metabolic acids result in what are termed Kussmaul respirations, a form of hyperventilation in which ventilations are deep and rapid. This represents the body’s attempt to increase the pH by expelling carbon dioxide (respiratory compensation), which in turn decreases the carbonic acid concentration. Other symptoms include anorexia, nausea, vomiting, diarrhea, and abdominal discomfort. Death can result in the most severe and prolonged cases, preceded by dysrhythmias and hypotension.
Evaluation and Treatment
The diagnosis of metabolic acidosis is established from the health history, clinical symptoms, and laboratory findings. Arterial blood pH is less than 7.35, and bicarbonate concentration is less than 22 mEq/L. The anion gap can facilitate identifying the specific cause. The oxyhemoglobin curve is shifted to the right (see Fig. 34.16), reducing Hb affinity for oxygen and impairing tissue oxygenation.
The underlying condition must be determined to establish effective treatment with a buffering solution. During severe acidosis (pH ≤ 7.1), base administration is required to elevate the pH to a safe level, particularly if there is oliguric renal disease. Accompanying sodium and water deficits must also be corrected.28 Use of bicarbonate for alkali therapy for treatment of chronic metabolic acidosis can be controversial, and a new drug is currently under investigation as an alternative treatment option (Emerging Science Box: Veverimer for Treatment of Chronic Metabolic Acidosis).
Metabolic Alkalosis
Pathophysiology
Metabolic alkalosis occurs when bicarbonate concentration is increased, usually caused by excessive loss of metabolic acids or, less commonly, by large intake of bicarbonate or substances that are metabolized to bicarbonate. With alkalemia, hydrogen ions are redistributed from the intracellular to the extracellular space and potassium moves to the intracellular space to preserve electroneutrality.29 When acid loss is caused by vomiting or gastric suctioning, renal compensation is not very effective because loss of chloride (an anion) in hydrochloric acid (HCl) stimulates renal retention of bicarbonate (an anion). The result is known as hypochloremic metabolic alkalosis (Fig. 3.17). Renal compensation is not very effective because the volume depletion and loss of electrolytes stimulate a paradoxical response by the kidneys. The kidneys increase bicarbonate reabsorption to maintain an anionic balance because the ECF chloride concentration is decreased. The resulting excretion of H+ and reabsorption of bicarbonate prevent correction of the alkalosis.
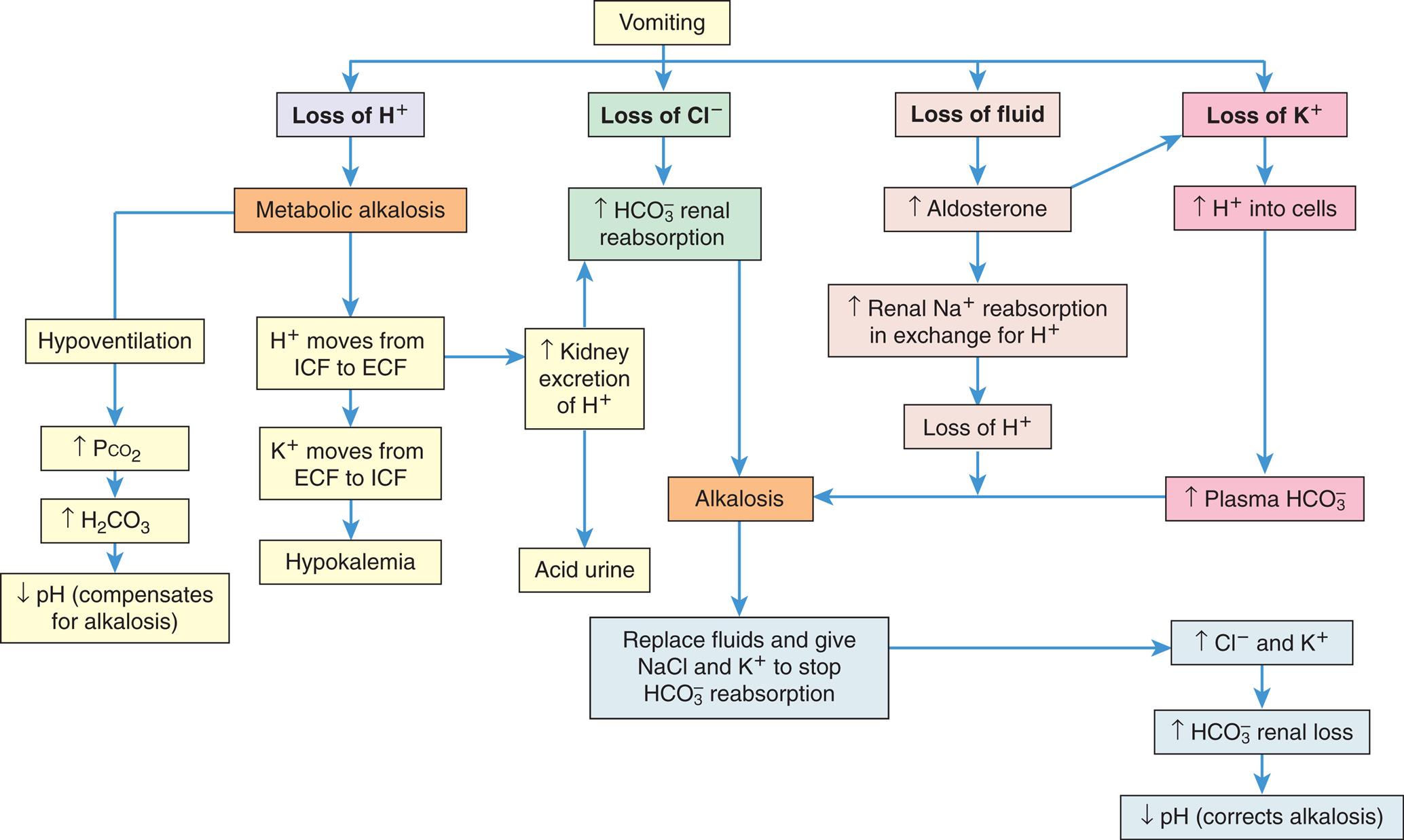
See text for abbreviations. (From Patton KT, Thibodeau, GA. Anatomy & physiology, 9th edition. St. Louis: Mosby; 2016.)
A flowchart shows the metabolic alkalosis with compensation and correction. Vomiting can cause loss of H ions, chlorine ions, fluid, or potassium ion. Loss of H ions lead to metabolic alkalosis, which can cause: • Hypoventilation. Increased P C O 2. Increased H 2 C O 3. Decreased p H (compensates for alkalosis). • H ion moves from I C F to E C F. K ions moves from E C F to I CF. Hypokalemia. • H ion moves from I C F to E C F. Increased kidney excretion of H ion. Acidic urine or increased H C O 3 ion renal reabsorption. Loss of C l ions leads to increased H C O 3 ion renal reabsorption, which can cause alkalosis and the following changes: • Replace fluids and give N a C l and K ion to stop H C O 3 ion reabsorption. • Increased C l and K ions. • Increased H C O 3 ion renal loss. • Decreased p H (corrects alkalosis). Loss of fluid leads to: • Increased aldosterone (can also lead to loss of potassium ions). • Increased renal N a ion reabsorption in exchange for H ion. • Loss of H ions. • Alkalosis and subsequent changes. Loss of potassium ions leads to: • Increased H ions into cells. • Increased plasma H C O 3 ions. • Alkalosis and subsequent changes.
Hyperaldosteronism also can lead to alkalosis as a result of sodium bicarbonate retention and loss of hydrogen and potassium. Diuretics may produce a mild alkalosis because they promote greater excretion of sodium, potassium, and chloride than of bicarbonate. Overuse of baking soda (sodium bicarbonate) as an antacid also can cause metabolic alkalosis, as can massive blood transfusions, which contain citrate that the liver metabolizes to bicarbonate.
Respiratory compensation for metabolic alkalosis occurs when the elevated pH inhibits the respiratory center. The rate and depth of ventilation are decreased, causing retention of carbon dioxide. The ratio of  concentration to H2CO3 concentration is reduced toward normal. Respiratory compensation is not very efficient because the need for oxygen limits this compensation. Chronic or severe metabolic alkalosis requires therapeutic intervention (Fig. 3.18).
concentration to H2CO3 concentration is reduced toward normal. Respiratory compensation is not very efficient because the need for oxygen limits this compensation. Chronic or severe metabolic alkalosis requires therapeutic intervention (Fig. 3.18).
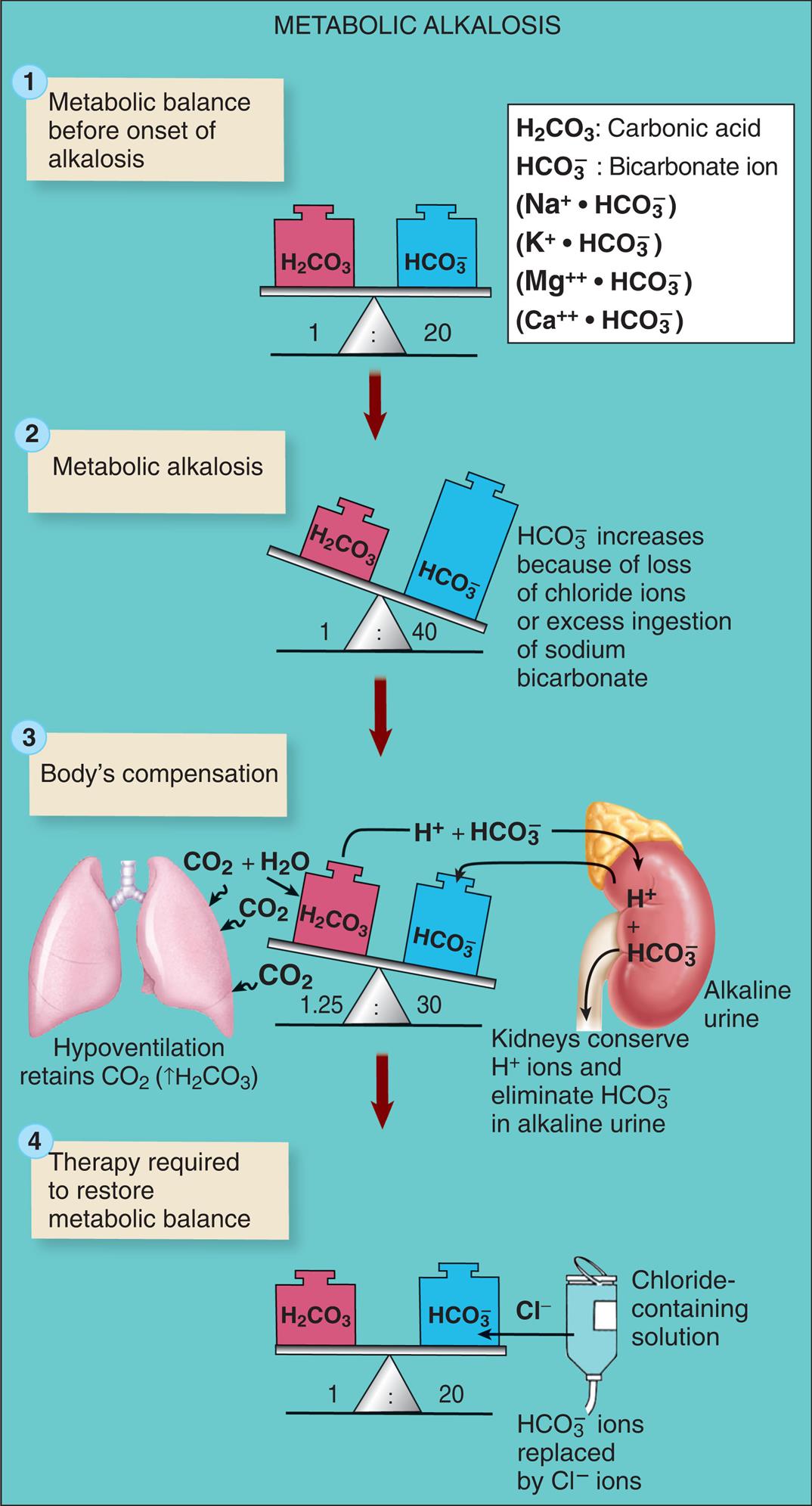
See text for abbreviations.
An illustrated flowchart shows hypochloremic metabolic alkalosis. There are four stages in the process. 1. Metabolic balance before onset of alkalosis. Carbonic acid and bicarbonate ion are balanced in the ratio 1 to 20. 2. Metabolic alkalosis. Carbonic acid and bicarbonate ion are unbalanced in the ratio 1 to 40. Carbonic acid increases because of loss of chloride ions or excess ingestion of sodium bicarbonate. 3. Body’s compensation. Carbonic acid and bicarbonate ion are unbalanced in the ratio 1.25 to 30. Kidneys conserve H ions and eliminate H C O 3 in alkaline urine. Hypoventilation retains C O 2 (increased H 2 C O 3). 4. Therapy required to restore metabolic balance. Carbonic acid and bicarbonate ion are balanced in the ratio 1 to 20. Chloride-containing solution used to replace H C O 3 ions with chlorine ions.
Clinical Manifestations
Because of the many causes of metabolic alkalosis, the symptoms vary. Some common symptoms, such as weakness, muscle cramps, and hyperactive reflexes, are related to volume depletion and electrolyte losses. Because alkalosis increases binding of Ca++ to plasma proteins (albumin), ionized calcium concentration decreases, causing excitable cells to become hypopolarized, which initiates an action potential more easily. Paresthesia (especially numbness/tingling of the fingertips and perioral area), tetany, and seizures may develop (see section on Hypocalcemia).
Respirations are slow and shallow to increase carbon dioxide retention. The oxyhemoglobin curve is shifted to the left, decreasing the dissociation of oxyhemoglobin, promoting hypoxemia, and increasing the risk of dysrhythmias (see Fig. 34.16). Atrial tachycardia is a potential problem.
Evaluation and Treatment
The health history provides significant clues to the diagnosis of metabolic alkalosis. The arterial pH is greater than 7.45, and bicarbonate levels exceed 26 mEq/L. With respiratory compensation, the PaCO2 increases to greater than 40 mm Hg. With hypochloremic metabolic alkalosis, serum chloride values are less than normal. Serum potassium levels are usually depleted because H+ is released from the cells in exchange for potassium to help regulate the pH level. The potassium is then secreted from renal distal tubule cells into the urine.
With hypochloremic alkalosis or contraction alkalosis with volume depletion, a sodium chloride solution is required for correction. The renal stimulus to increase ECF volume by retaining Na+ is diminished, and  can be excreted as NaHCO3 in the urine. The administration of potassium corrects alkalosis caused by hyperaldosteronism or hypokalemia. The potassium causes H+ to move back into the ECF and decreases loss of H+ from the distal tubule.
can be excreted as NaHCO3 in the urine. The administration of potassium corrects alkalosis caused by hyperaldosteronism or hypokalemia. The potassium causes H+ to move back into the ECF and decreases loss of H+ from the distal tubule.
Respiratory Acidosis
Pathophysiology
Respiratory acidosis occurs when there is alveolar hypoventilation resulting in hypercapnia (an excess of carbon dioxide in the blood). The arterial carbon dioxide pressure (PaCO2) is greater than 45 mm Hg and the pH is less than 7.35 (Fig. 3.19). Decreased alveolar ventilation, in relation to the metabolic production of carbon dioxide, produces an increased concentration of carbonic acid, which results in respiratory acidosis. The common causes include depression of the respiratory center (brainstem trauma, oversedation), paralysis of the respiratory muscles, disorders of the chest wall (kyphoscoliosis, obesity hypoventilation syndrome, flail chest), and disorders of the lung parenchyma (e.g., pneumonitis, pulmonary edema, and chronic obstructive lung disease).
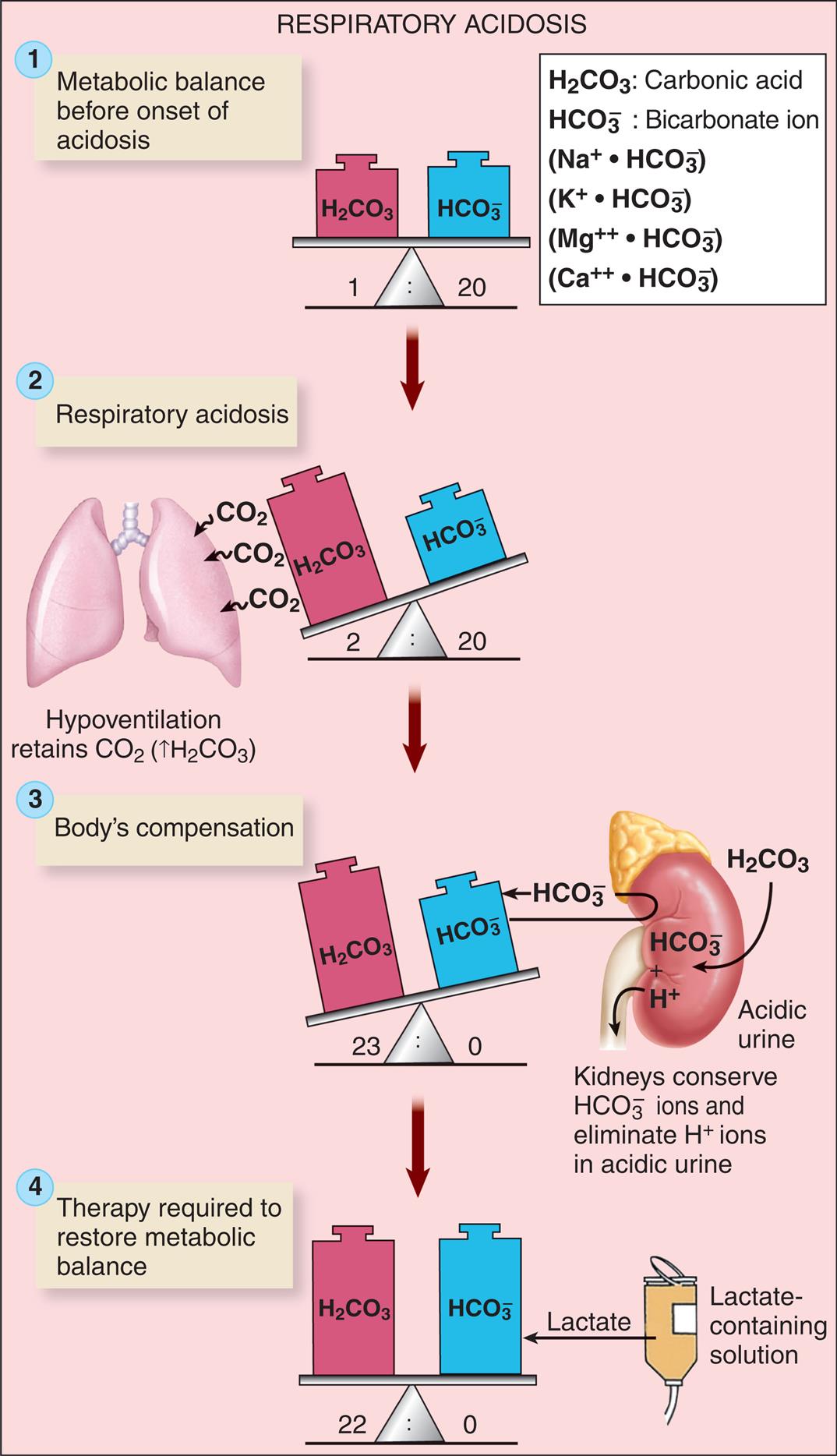
See text for abbreviations. (From Patton KT, Thibodeau, GA. Anatomy & physiology, 9th edition. St. Louis: Mosby; 2016.)
An illustrated flowchart shows the respiratory acidosis process with compensation and correction. There are four stages in the process. 1. Metabolic balance before onset of acidosis. Carbonic acid and bicarbonate ion are balanced in the ratio 1 to 20. 2. Respiratory acidosis. Carbonic acid and bicarbonate ion are unbalanced in the ratio 2 to 20. Hypoventilation retains C O 2 (increased H 2 C O 3). 3. Body’s compensation. Carbonic acid and bicarbonate ion are unbalanced in the ratio 23 to 0. Kidneys conserve H C O 3 ions and eliminate H ions in acidic urine. 4. Therapy required to restore metabolic balance. Carbonic acid and bicarbonate ion are balanced in the ratio 22 to 0. Lactate-containing solution is used in therapy.
Respiratory acidosis may be acute or chronic. Airway obstruction is the most common cause of acute respiratory acidosis (e.g., acute airway edema and bronchoconstriction from an acute asthma episode, or upper airway angioedema). Acute compensation for respiratory acidosis is not effective because the renal buffer mechanism takes time to function. Furthermore, the protein buffers provide marginal assistance, and  is not a good buffer for its conjugate acid (carbonic acid). Acute uncompensated respiratory acidosis is characterized by decreased arterial pH, elevated PaCO2, and normal or slightly increased bicarbonate concentration.
is not a good buffer for its conjugate acid (carbonic acid). Acute uncompensated respiratory acidosis is characterized by decreased arterial pH, elevated PaCO2, and normal or slightly increased bicarbonate concentration.
Chronic respiratory acidosis is commonly associated with chronic obstructive pulmonary disease and deformities of the chest wall or neuromuscular disorders. Renal compensation is effective and is established over several days. The acidosis produced from CO2 retention stimulates the kidney to secrete H+ ions and regenerate bicarbonate. Serum bicarbonate and PaCO2levels are both elevated, and pH is restored toward normal (see Fig. 3.19).
Clinical Manifestations
The symptoms of respiratory acidosis are related to acuity of onset and severity of PaCO2 retention. Initial symptoms include headache, restlessness, blurred vision, and apprehension. Lethargy, muscle twitching, tremors, seizures, and coma may follow. Chronic acidosis causes myocardial depression, dysrhythmias, and hypotension. Neurologic symptoms are caused by a decrease in the pH of cerebrospinal fluid and cerebral vasodilation. CO2 readily moves into vascular smooth muscle, causing both cerebral and peripheral vasodilation. The respiratory rate may be rapid at first and gradually becomes depressed, because over time, the respiratory center adapts to increasing levels of CO2. Cyanosis does not occur unless there is an accompanying hypoxemia, and the skin may instead be pink from vasodilation caused by the elevated CO2 level.
Evaluation and Treatment
The primary diagnostic indicators are an arterial pH less than 7.35 and hypercapnia. Acute respiratory acidosis must be distinguished from chronic respiratory acidosis and any accompanying metabolic acidosis. The health history and clinical laboratory data are helpful in making a determination whether the respiratory acidosis is acute, chronic, or acute on chronic.
In many cases, restoration of adequate alveolar ventilation removes excess CO2. If alveolar ventilation cannot be maintained spontaneously because of drug overdose or neuromuscular disorders, mechanical ventilation is required. When the hypercapnia is caused by alterations in gas diffusion at the alveolar-capillary membrane, ventilation may not be effective. The values of arterial pH, PCO2, PO2, and  must be carefully monitored. The reduction of PaCO2 must not be done too rapidly because of the associated risks, including respiratory alkalosis with seizures and death.30
must be carefully monitored. The reduction of PaCO2 must not be done too rapidly because of the associated risks, including respiratory alkalosis with seizures and death.30
The underlying diseases are treated to achieve maximal ventilation. In the presence of hypoxemia and hypercapnia, oxygen can function as a respiratory depressant when the respiratory center is no longer stimulated by the lower pH and elevated PaCO2 value. Therefore, when oxygen is administered in this situation, the individual should be monitored for respiratory depression.
Respiratory Alkalosis
Pathophysiology
Respiratory alkalosis occurs when there is alveolar hyperventilation and decreased concentration of plasma carbon dioxide (termed hypocapnia), thus increasing the ratio of  to PCO2 (H2CO3). Hyperventilation can result from a variety of causes, including hypoxemia (e.g., high altitudes); hypermetabolic states such as fever, severe anemia, and thyrotoxicosis; early salicylate intoxication; and anxiety or panic disorder. Improper settings on mechanical ventilators can cause iatrogenic respiratory alkalosis. Secondary respiratory alkalosis may develop from hyperventilation stimulated by metabolic acidosis, causing a mixed acid-base disorder.
to PCO2 (H2CO3). Hyperventilation can result from a variety of causes, including hypoxemia (e.g., high altitudes); hypermetabolic states such as fever, severe anemia, and thyrotoxicosis; early salicylate intoxication; and anxiety or panic disorder. Improper settings on mechanical ventilators can cause iatrogenic respiratory alkalosis. Secondary respiratory alkalosis may develop from hyperventilation stimulated by metabolic acidosis, causing a mixed acid-base disorder.
The onset of acute respiratory alkalosis occurs within minutes of hyperventilation. Cellular buffers provide immediate response (i.e., protein and shifts of H+ from ICF to ECF). The H+ shifts are not very effective if the PaCO2 level is significantly decreased. Renal compensation occurs with chronic respiratory alkalosis and is characterized by decreased hydrogen excretion and decreased bicarbonate reabsorption (Fig. 3.20).
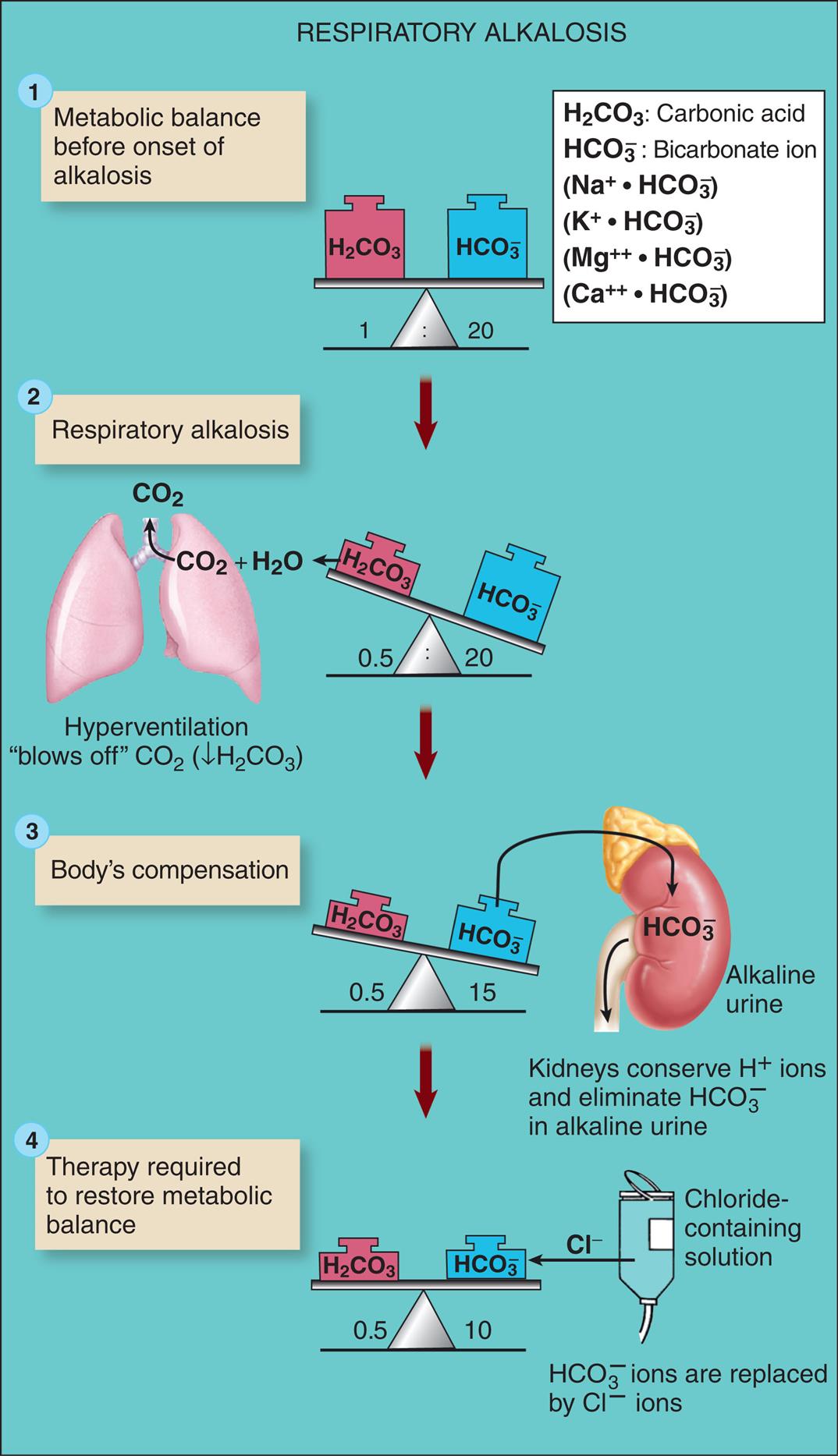
See text for abbreviations. (From Patton KT, Thibodeau, GA. Anatomy & physiology, 9th edition. St. Louis: Mosby; 2016.)
An illustrated flowchart shows the respiratory alkalosis process with compensation and correction. There are four stages in the process. 1. Metabolic balance before onset of alkalosis. Carbonic acid and bicarbonate ion are balanced in the ratio 1 to 20. 2. Respiratory alkalosis. Carbonic acid and bicarbonate ion are unbalanced in the ratio 0.5 to 20. Hyperventilation blows off C O 2 (decreased H 2 C O 3). 3. Body’s compensation. Carbonic acid and bicarbonate ion are unbalanced in the ratio 0.5 to 15. Kidneys conserve H ions and eliminate H C O 3 in alkaline urine. 4. Therapy required to restore metabolic balance. Carbonic acid and bicarbonate ion are balanced in the ratio 0.5 to 10. Chloride-containing solution used to replace H C O 3 ions with C l ions.
Clinical Manifestations
Respiratory alkalosis, like metabolic alkalosis, is irritating to the central and peripheral nervous systems. Symptoms include dizziness, confusion, tingling of extremities (paresthesias), seizures, and coma. Cerebral vasoconstriction reduces the cerebral blood flow. Carpopedal spasm and other symptoms of hypocalcemia are similar to those of metabolic alkalosis. Deep and rapid respirations (tachypnea) are primary symptoms of the disorders that cause respiratory alkalosis.
Evaluation and Treatment
The underlying disturbance must be identified. The arterial pH is greater than 7.45, and the PaCO2 is less than 38 mm Hg. In acute states, bicarbonate levels are normal. With chronic respiratory alkalosis, a compensatory decrease in the bicarbonate level occurs and the pH is closer to normal.
Treating the underlying disturbance is the most effective treatment. Hypoxemia must be corrected and hypermetabolic states reversed. Symptoms from hyperventilation caused by anxiety or acute pain can be corrected by managing the acute pain, anxiety-reducing conversation, assisting the individual to breathe more slowly while counting, or occasionally by rebreathing from a paper bag, which increases the concentration of inspired CO2 and reverses the respiratory alkalosis. However, the use of paper bag rebreathing is associated with hypoxemia and can harm individuals with underlying cardiac and respiratory disorders. It may even worsen the situation if the hyperventilation is a compensatory response to a metabolic acidosis.
Mixed Acid-Base Disorders
Mixed acid-base disorders are two or more primary acid-base disorders occurring at the same time. They are more common in hospitalized individuals, often those in critical care with comorbid conditions (i.e., combined metabolic and respiratory disorders or combinations of acute and chronic disorders). For these individuals, the clinical history and analysis of electrolytes, medications, the anion gap, and the osmolality of plasma and urine are informative. The primary disorder is assessed, and then the degree of compensation is evaluated to determine if it is adequate, greater, or lesser than expected. Renal and respiratory compensation rarely returns the pH to normal. Therefore mixed acid-base disorders can have alterations in PaCO2 and/or bicarbonate and a normal pH. Examples of conditions with mixed acid-base disorders include individuals with chronic obstructive pulmonary disease who develop heart failure and are treated with high doses of diuretics, or individuals who have acute kidney injury and vomiting or severe hypoxia with extracellular volume depletion.31,32
Summary Review
Distribution of Body Fluids
- 1. Body fluids are distributed among functional compartments and are classified as ICF or ECF.
- 2. The sum of all fluids is the TBW, which varies with age and amount of body fat and is higher in infants because they have less body fat. With older age, the percent of TBW decreases as there is loss of muscle and an increase in body fat.
- 3. Water moves between the ICF and ECF compartments principally by osmosis.
- 4. Water moves between the plasma and interstitial fluid by osmosis and hydrostatic pressure, which occur across the capillary membrane.
- 5. Movement across the capillary wall is called net filtration and is described according to Starling forces (forces favoring filtration minus forces opposing filtration).
Alterations in Water Movement
- 1. Edema is the excessive accumulation of fluid within the interstitial spaces.
- 2. Edema is caused by venous or lymphatic obstruction or increased vascular volume (increases hydrostatic pressure), plasma protein losses, or increased capillary membrane permeability (decreases plasma oncotic pressure).
- 3. The pathophysiologic process that leads to edema is related to an increase in forces favoring fluid filtration from the capillaries into the tissues.
- 4. Edema may be localized or generalized and usually is associated with swelling and puffiness, tighter-fitting clothes and shoes, limited movement of the affected area, and, in severe cases, weight gain.
- 5. An effusion is fluid accumulation within a body cavity or space sometimes called a third space.
Sodium, Chloride, and Water Balance
- 1. Sodium accounts for 90% of the ECF cations.
- 2. Sodium balance and water balance are intimately related; chloride levels are generally proportional to changes in sodium levels.
- 3. Sodium balance is regulated by aldosterone, which increases reabsorption of sodium by the distal tubule of the kidney.
- 4. Renin and angiotensin are enzymes that promote or inhibit secretion of aldosterone and thus regulate sodium and water balance.
- 5. ANH is also involved in decreasing renal tubular resorption and promoting urinary excretion of sodium.
- 6. Water balance is regulated by the sensation of thirst and by the level of ADH, which is initiated by an increase in plasma osmolality or a decrease in circulating blood volume.
Alterations in Sodium, Water, and Chloride Balance
- 1. Alterations in water balance may be classified as isotonic, hypertonic, or hypotonic.
- 2. Isotonic alterations occur when changes in TBW are accompanied by proportional changes in concentrations of electrolytes.
- 3. Hypertonic alterations develop when the osmolality of the ECF is increased to greater than normal, usually because of an increased concentration of ECF sodium or a deficit of ECF water.
- 4. Hypernatremia occurs when ECF sodium levels exceed 145 mEq/L.
- 5. Hypernatremia can be hypovolemic, euvolemic, or hypervolemic, depending on the accompanying ECF water volume.
- 6. Hyperchloremia occurs when ECF chloride exceeds 105 mEq/L and often accompanies hypernatremia.
- 7. Hypotonic alterations occur when the osmolality of the ECF is less than normal.
- 8. Hyponatremia occurs when the serum sodium concentration decreases to less than 135 mEq/L and may be caused by inadequate intake of sodium or dilution of the body's sodium level.
- 9. Water excess is rare but can be caused by compulsive water drinking, decreased urine formation, or the syndrome of inappropriate secretion of ADH.
- 10. Hyponatremia usually causes movement of water into cells.
- 11. Hypochloremia is usually the result of hyponatremia or elevated bicarbonate concentrations.
Alterations in Potassium, Calcium, Phosphate, and Magnesium Balance
- 1. Potassium is the predominant ICF ion; it functions to regulate ICF osmolality, maintain the resting membrane potential, and deposit glycogen in liver and skeletal muscle cells.
- 2. Potassium balance is regulated by the kidney, by aldosterone and insulin secretion, and by changes in pH.
- 3. A mechanism known as potassium adaptation allows the body to accommodate slowly to increased levels of potassium intake.
- 4. Hypokalemia (serum potassium concentration <3.5 mEq/L) indicates loss of total body potassium, although ECF hypokalemia can develop without losses of total body potassium, and plasma K+ levels may be normal or elevated when the amount of total body potassium is depleted.
- 5. Hypokalemia may be caused by reduced potassium intake, increased ICF to ECF potassium concentration, loss of potassium from body stores, increased aldosterone secretion (e.g., caused by hypernatremia), and increased renal excretion.
- 6. Hyperkalemia (potassium levels >5.0 mEq/L) may be caused by increased potassium intake, a shift from ICF to ECF potassium, or decreased renal excretion.
- 7. Calcium is a necessary ion in the structure of bones and teeth, in blood clotting, in hormone secretion and the function of cell receptors, and in membrane stability.
- 8. Phosphate acts as a buffer in acid-base regulation and provides energy for muscle contraction.
- 9. Calcium and phosphate concentrations are rigidly controlled by PTH, vitamin D, and calcitonin.
- 10. Hypocalcemia (total serum calcium concentration <9.0 mg/dL) is related to inadequate intestinal absorption, deposition of ionized calcium into bone or soft tissue, blood administration, or decreased PTH and vitamin D levels.
- 11. Hypercalcemia (serum calcium concentration >10.5 mg/dL) can be caused by a number of diseases, including hyperparathyroidism, bone metastases, sarcoidosis, and excess vitamin D.
- 12. Hypophosphatemia is a serum phosphate level less than 2.0 mg/dL and is usually caused by intestinal malabsorption and increased renal excretion of phosphate.
- 13. Hyperphosphatemia is a serum phosphate level more than 4.7 mg/dL and develops with acute or chronic renal failure with significant loss of glomerular filtration.
- 14. Magnesium is a major intracellular cation.
- 15. Magnesium functions in enzymatic reactions and often interacts with calcium at the cellular level.
- 16. Hypomagnesemia (serum magnesium concentrations <1.5 mEq/L) may be caused by malabsorption syndromes.
- 17. Hypermagnesemia (serum magnesium concentrations >3.0 mEq/L) is rare and is usually caused by renal failure.
Acid-Base Balance
- 1. Hydrogen ions, which maintain membrane integrity and the speed of enzymatic reactions, must be concentrated within a narrow range if the body is to function normally.
- 2. Hydrogen ion concentration is expressed as pH, which represents the negative logarithm of hydrogen ions in solution (e.g., a negative logarithm 10−7 mg/L is indicated on the pH scale as pH 7.0, a neutral status). In arterial blood, the normal pH range is 7.35 to 7.45
- 3. Different body fluids have different pH values.
- 4. The renal and respiratory systems, together with the body's buffer systems, are the principal regulators of acid-base balance.
- 5. Buffers are substances that can absorb excessive acid or base to minimize fluctuations in pH.
- 6. The principal plasma buffers are bicarbonate, protein (Hb), and phosphate.
- 7. The lungs and kidneys act to compensate for changes in pH by increasing or decreasing ventilation (increasing or decreasing carbon dioxide in the form of carbonic acid) and by producing more acidic or more alkaline urine.
- 8. Correction is a process different from compensation; correction occurs when the values for both components of the buffer pair are returned to normal.
- 9. Acid-base imbalances are caused by changes in the concentration of H+ in the blood; an increase causes acidosis, and a decrease causes alkalosis.
- 10. An abnormal increase or decrease in bicarbonate concentration causes metabolic acidosis or metabolic alkalosis; changes in the rate of alveolar ventilation produce respiratory acidosis or respiratory alkalosis.
- 11. Metabolic acidosis is caused by an increase in the concentrations of non–carbonic acids or by loss of bicarbonate from the ECF.
- 12. Metabolic alkalosis occurs with an increase in bicarbonate concentration usually caused by loss of metabolic acids from conditions such as vomiting, gastrointestinal suctioning, excessive bicarbonate intake, hyperaldosteronism, and diuretic therapy.
- 13. Respiratory acidosis occurs with a decrease of alveolar ventilation and an increase in levels of carbon dioxide, or hypercapnia.
- 14. Respiratory alkalosis occurs with alveolar hyperventilation and excessive reduction of carbon dioxide concentration, or hypocapnia.