Innate Immunity Inflammation and Wound Healing
Valentina L. Brashers
 http://evolve.elsevier.com/Rogers/pathophysiology/
http://evolve.elsevier.com/Rogers/pathophysiology/
The human body is continually exposed to a large variety of toxic substances, physical trauma, and infectious agents (viruses, bacteria, fungi, and parasites) that can cause damage to cells and tissues. Damage can also come from within, such as with cancers. In response, the human body has developed a highly sophisticated system of interactive immune defense mechanisms. Innate immunity refers to defense mechanisms that are present at birth and provide the initial nonspecific response to invasion and injury. Adaptive immunity refers to immunity that develops over the lifetime of the individual and provides long-term protection against specific invaders.
Innate Immunity
The human body has several ways to protect itself from injury and infection. Innate immunity includes natural barriers and inflammation. Innate barriers form the first line of defense at the body’s surfaces. They serve to prevent damage by the environment and thwart infection by pathogenic microorganisms. If these barriers are breached, the second line of defense, the inflammatory response, is activated to protect the body from further injury, fight infection, and promote healing. The third line of defense, adaptive immunity (also known as acquired or specific immunity), is induced through a slower and more specific process and targets particular invaders and diseased tissues for the purpose of eradicating them. Adaptive immunity also involves “memory,” which results in a more rapid response during future exposure to the same invader. Comparisons among defense mechanisms are described in Table 7.1.
Table 7.1
While these defense mechanisms are essential for maintaining health and preventing disease, uncontrolled or chronic immune activation and inflammation play a role in virtually all diseases, including heart, lung, and kidney diseases; cancer; neurodegenerative disorders; and rheumatologic disease. The information presented in this chapter introduces the components and processes of innate immunity and sets the stage for Chapter 8, which presents an overview of adaptive immunity; Chapter 9, which discusses alterations in immunity; and Chapter 10, which reviews infection. Innate immunity in the newborn and changes associated with aging are reviewed in the Pediatric Considerations and Geriatric Considerations boxes at the end of the chapter.
First Line Of Defense: Physical and Biochemical Barriers and the Human Microbiome
Innate barriers form the first line of defense at the body’s surfaces. These barriers can be physical, mechanical, and biochemical. Surface barriers also house a group of beneficial microorganisms known as the normal microbiome that protect us from pathogenic microorganisms.
Physical and Mechanical Barriers
Physical barriers offer considerable protection from tissue damage and infection. These barriers are comprised of tightly associated epithelial cells of the skin and of the linings of the gastrointestinal (GI), genitourinary, and respiratory tracts (Fig. 7.1). When pathogens attempt to penetrate such barriers, they may be removed by mechanical means. For example, microorganisms may be sloughed off with dead skin cells (which are then replaced). Epithelial cells of the upper respiratory tract can trap microorganisms through the production of mucus and remove them through the action of hair-like cilia, which move the mucus upward where it is expelled through coughing or sneezing. Invading microorganisms can be removed from the GI tract through vomiting or defecation and from the urinary tract through urination. Other protective mechanisms of innate barriers include the relatively low temperature present on the skin and the low pH found in the stomach and the vagina, both of which inhibit the growth of pathogenic microorganisms.
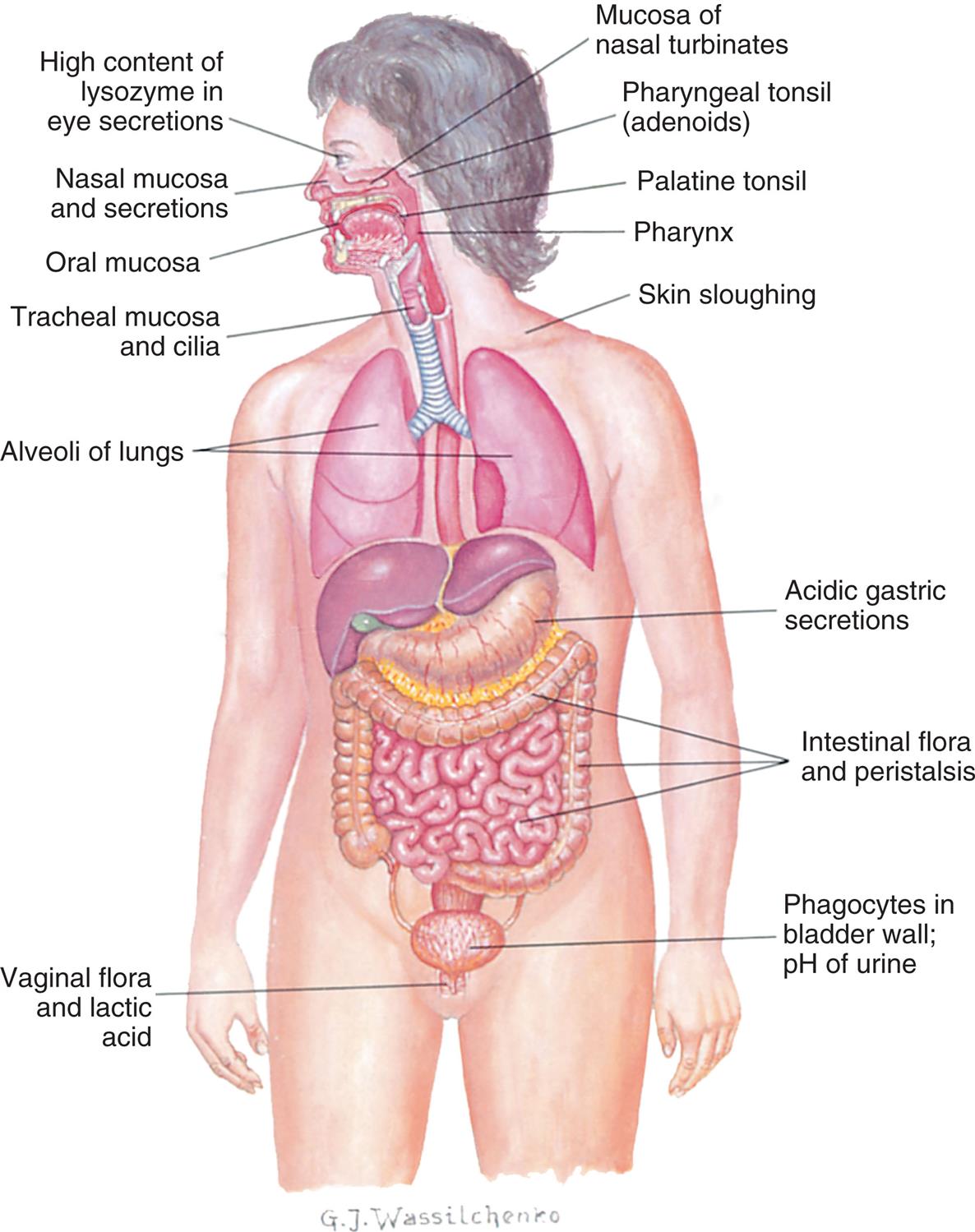
An illustration shows the anterior view of the internal organs in the human body. The following structures are labeled on the illustration, from the top to the bottom: high content lysozyme in eye secretions, nasal mucosa and secretions, mucosa of nasal turbinate, pharyngeal tonsil (adenoids), oral mucosa, palatine tonsil, pharynx, tracheal mucosa and cilia, skin sloughing, alveoli of lungs, acidic gastric secretions, intestinal flora and peristalsis, phagocytes in bladder wall or p H of urine, and vaginal flora and lactic acid.
Biochemical Barriers
Epithelial surfaces also provide biochemical barriers by synthesizing and secreting substances meant to trap or destroy microorganisms (epithelial-derived chemicals). Mucus, perspiration, saliva, tears, and earwax are all examples of biochemical secretions that can trap and kill potential disease-causing microorganisms. Sebaceous glands in the skin secrete fatty acids and lactic acid, substances which kill bacteria and fungi. These secretions also create an acidic environment on the skin surface (pH 3 to pH 5) which is inhospitable to most bacteria.
Epithelial cell secretions contain antimicrobial peptides, substances that kill or inhibit the growth of disease-causing bacteria, fungi, and viruses. Defensins are antimicrobial peptides produced by neutrophils and epithelial cells that defend against bacterial infection by disrupting bacterial membranes. Collectins are soluble glycoproteins that facilitate the ability of macrophages to recognize and kill pathogenic microorganisms and can activate the complement system (see the Plasma Protein Systems and Inflammation section). They are produced by various organs, including the lungs (e.g., surfactant is a type of collectin).
The Normal Microbiome
The body’s surfaces are colonized with an array of microorganisms, called the normal microbiome (previously referred to as normal flora). Molecular profiling of these microorganisms has shown that each individual has a unique suite of microbial strains that is acquired early in life, beginning in utero and continuing with exposure to the vaginal microbiome during birth.1 With subsequent environmental exposures such as diet, antimicrobials, toxins, and animals, the composition of the microbiome diversifies. Changes in the microbial make-up of the infant microbiome can have lifelong impacts on health and disease.2
Body surfaces that have their own specialized microbiome include the skin; mucous membranes of the eye, nose, and mouth; upper and lower GI tracts; upper respiratory tract; urethra; and vagina. These surfaces are colonized by a combination of bacteria and fungi unique to the anatomic location and the particular individual (Fig. 7.2).
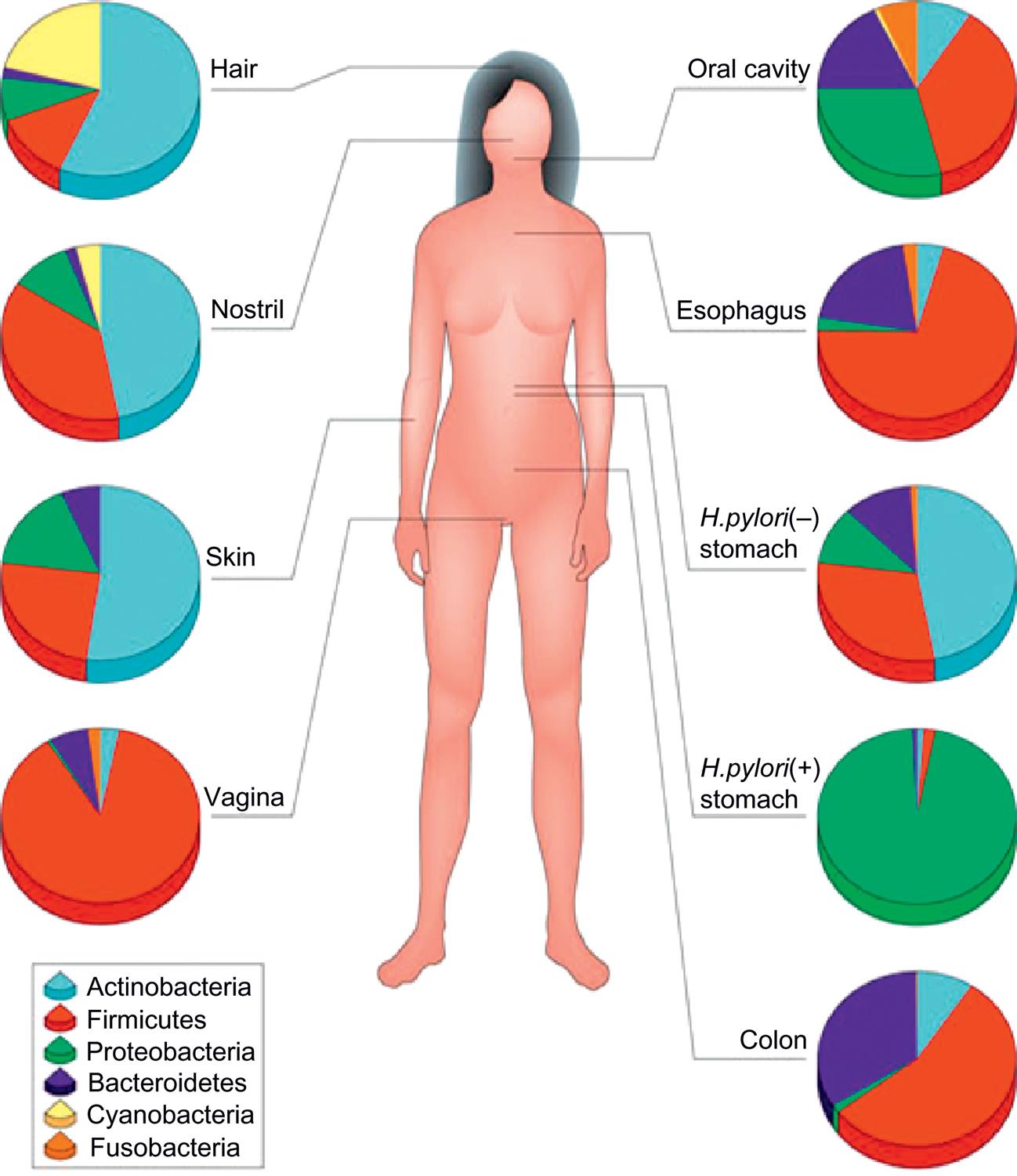
Typical phylum-level composition of the human microbiota that vary at each site (From Cho I, Blaser MJ. The human microbiome: At the interface of health and disease. Nature Reviews. Genetics, 2012; 13:260–270.)
An illustration shows the anterior view of the human body and shows pie charts against its different parts for identifying the distribution of the human microbiome. The data from the illustration, in decreasing distribution of microbiome, for each part are as follows. • Hair: Actinobacteria, cyanobacteria, firmicutes, proteobacteria, and Bacteroidetes. • Nostril: Actinobacteria, cyanobacteria, firmicutes, proteobacteria, and Bacteroidetes. • Oral cavity: Firmicutes, proteobacteria, Bacteroidetes, actinobacteria, fusobacteria. • Esophagus: Firmicutes, Bacteroidetes, actinobacteria, fusobacteria, and proteobacteria. • Skin: Actinobacteria, cyanobacteria, firmicutes, and proteobacteria. • H.pylori (negative), stomach: Actinobacteria, firmicutes, proteobacteria, Bacteroidetes, and fusobacteria. • H.pylori (positive), stomach: Proteobacteria, firmicutes, actinobacteria, and Bacteroidetes. • Colon: Firmicutes, Bacteroidetes, actinobacteria, and proteobacteria. • Vagina: Firmicutes, Bacteroidetes, actinobacteria, fusobacteria, and proteobacteria.
The microorganisms in the microbiome do not normally cause disease. The normal microbiome benefits the human body in a number of ways, and the human body provides an ideal environment for the microbiome to grow.3 The interaction between host cells and the microbiome plays an essential role in fostering healthy innate and immune defense systems.1,4,5
Mechanisms by which the microbiome contributes to health and immunity include:
- 1. Produces enzymes which facilitate digestion of fatty acids and large polysaccharides
- 2. Synthesizes essential metabolites (e.g., vitamins K and B)
- 3. Releases antibacterial substances that are toxic to pathogenic microorganisms (e.g., ammonia, phenols, and indoles)
- 4. Competes with pathogens for nutrients and blocks attachment of the pathogens to the epithelium, an obligatory first step in the infectious process
- 5. Fosters healthy innate and adaptive immunity by inducing protective immune responses to pathogens, while at the same time maintaining regulatory pathways that support immune tolerance of innocuous microorganisms.
The health of the microbiome can be positively or negatively influenced by a wide range of environmental exposures (see Emerging Science Box: The Microbiome and Disease). For example, prolonged treatment with broad-spectrum antibiotics can alter the normal microbiome, decreasing its protective activity and leading to an overgrowth of pathogenic microorganisms. In the intestine, for example, overgrowth of the yeast Candida albicans or the bacterium Clostridioides difficile (the cause of pseudomembranous colitis) may occur with antibiotic use. Some members of the normal bacterial microbiome are opportunistic pathogens—that is, they are harmless under normal conditions but can cause disease in immunocompromised individuals who lack the usual defense mechanisms. For example, Pseudomonas aeruginosa is a member of the normal microbiome of the skin, where it produces a toxin that protects the skin from infections caused by pathologic bacteria. With severe burns or critical illnesses in which the integrity of the skin and mucous membranes is compromised, Pseudomonas may enter the bloodstream and cause life-threatening systemic infections.
Epidemiologic studies have been used to associate negative changes in the microbiome (dysbiosis) with disease states (see Emerging Science Box: The Microbiome and Disease). Included in the long list of dysbiosis-associated diseases are mental health disorders, obesity, hypertension, heart failure, asthma, emphysema, rheumatologic conditions, diabetes, bowel disease, and cancer. Studies aimed at altering the microbiome for health promotion, disease prevention, and improved responses to treatment are ongoing.1
Second Line of Defense: Inflammation
Inflammation is a protective response that supports recovery from injury and disease. The inflammatory process is activated by virtually any injury to vascularized tissues. Triggers include infection and tissue damage (e.g., ischemia, trauma, chemical injury, foreign bodies, and immune reactions). When pathogen invasion or tissue damage occurs, inflammation is initiated by guardian cells (mast cells, macrophages, and dendritic cells) that are located near epithelial surfaces, in lymph nodes, and near blood vessels. Inflammatory mediators are released, causing both a vascular and a cellular response. The ultimate result of these responses is the migration of leukocytes, platelets, plasma proteins, and other biochemical mediators from the circulation into the nearby damaged tissue, where they can work together to destroy invaders, limit tissue injury, and promote healing. Algorithm 7.1 summarizes the process.
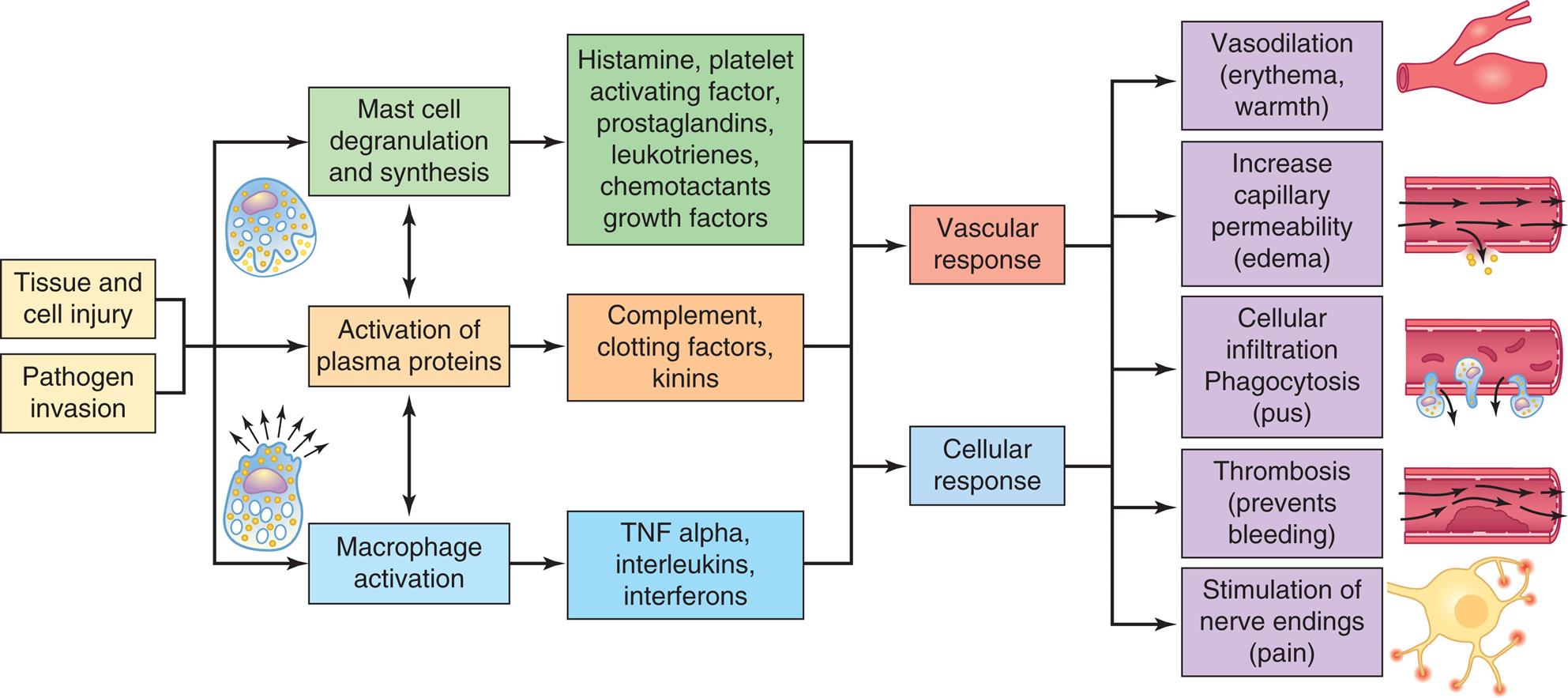
Inflammation is usually initiated by cellular injury and may be complicated by infection. The inflammatory response involves mast cell degranulation, the activation of three plasma protein systems, and the activation of macrophages that release numerous cytokines. These systems are interdependent so that induction of one (e.g., mast cell degranulation) can result in the induction of the other two. The result is the development of the characteristic microscopic and clinical hallmarks of inflammation. TNF alpha, Tumor necrosis factor-alpha.
An illustrated flowchart tracks the process of acute inflammatory response. The data from the flowchart is as follows. Tissue and cell injury and pathogen invasion leads to the following three processes: 1. Mast cell degranulation and synthesis. 2. Activation of plasma proteins; also leads to the other two processes. 3. Macrophage activation. Conditions from process 1: histamine, platelet activating factor, prostaglandins, leukotrienes, and chemotactants growth factors. Conditions from process 2: complement, clotting factors, and kinins. Conditions from process 3: T N F alpha, interleukins, and interferons. All responses lead the following vascular and cellular responses: • Vasodilation (erythema, warmth). • Increase capillary permeability (edema). • Cellular infiltration phagocytosis (pus). • Thrombosis (prevents bleeding). • Stimulation of nerve endings (pain).
The inflammatory response is characterized by the following:
- 1. The process occurs in vascularized tissues (i.e., tissues with a blood supply).
- 2. Activation is rapid (within seconds) after damage occurs.
- 3. The response includes chemical, vascular, and cellular components.
- 4. The response is nonspecific—that is, it takes place in approximately the same way regardless of the type of injury (stimulus) and whether or not the same stimulus has occurred in the past.
Inflammation, although producing pain and functional limitations in the affected individual, also results in numerous physiologic benefits. Protective functions of inflammation include the following:
- 1. Prevention of infection and further damage caused by invading microorganisms: Inflammatory exudates dilute the toxins produced by both bacteria and dying cells. The activation of plasma protein systems (e.g., complement and clotting systems) serves to contain and destroy invading microorganisms. The influx of phagocytes (neutrophils and macrophages) removes toxic cellular debris and pathogenic microorganisms, preventing them from further harming the body.
- 2. Preparation of injury for healing and repair: This involves removal of bacterial microorganisms, dead cells, and other physiologic debris through the epithelial channels and the lymphatic vessels that drain the region. Mechanisms of healing and repair are initiated.
- 3. Facilitation of the development of adaptive immunity: As fluid and cellular debris drain through the lymphatic vessels, lymphoid tissue and lymph nodes, microbial antigens encounter macrophages and lymphocytes concentrated in these areas. Adaptive immunity is initiated and helps fight off invaders and protect the body from future exposures to particular pathogens (see Chapter 8).
Plasma Protein Systems and Inflammation
Plasma protein systems are essential to an effective vascular and cellular inflammatory response (see Algorithm 7.1). There are three plasma protein systems that are especially important to innate immunity: the complement system, the clotting system, and the kinin system (Fig. 7.3). Although each system has a unique role in inflammation, all three systems have many similarities as well. Each system consists of multiple proteins and enzymes usually present in blood as inactive forms. The proteins are activated early in inflammation by enzymes, which circulate as proenzymes until some form of tissue damage triggers the system initiating the sequence. Activation of the first components results in sequential activation of other components of the system, leading to a protective biologic function. This sequential activation is referred to as a cascade, hence the common terminology that references the complement cascade, the clotting cascade, or the kinin cascade.
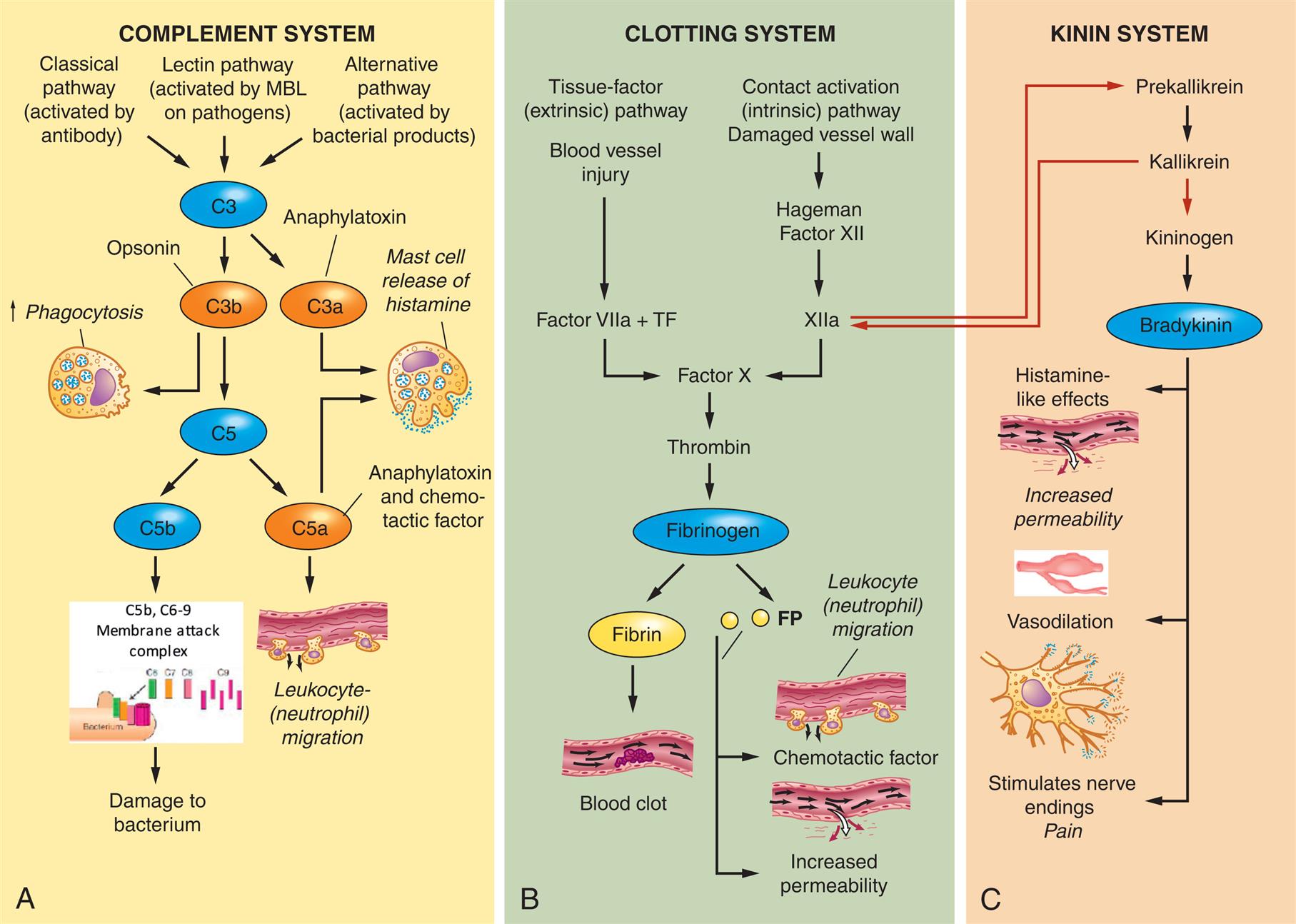
Each plasma protein system consists of a family of proteins that are activated in sequence to create potent biologic effects. (A) The complement system can be activated by three mechanisms, each of which results in proteolytic activation of C3. The fragments of C3 activation, C3a and C3b, are major components of inflammation. C3a is a potent anaphylatoxin, which induces degranulation of mast cells. C3b can bind to the surface or cells, such as bacteria, and either serve as an opsonin for phagocytosis or proteolytically activate the next component of the complement cascade, C5. The smaller fragment of C5 activation is C5a, a powerful anaphylatoxin, and is also chemotactic for neutrophils, attracting them to the site of inflammation. The larger fragment, C5b, activates the components of the membrane attack complex (C5b–C9), which damage the bacterial membrane and kill the bacteria. (B) The clotting system can be activated by the tissue factor (extrinsic) pathway and the contact activation (intrinsic) pathway. All routes of clotting initiation lead to activation of factor X and thrombin. Thrombin is an enzyme that activates fibrinogen to form fibrin and small FPs. Fibrin comes together to form a clot, and the FPs are highly active as chemotactic factors and cause increased vascular permeability. (C) Factor XIIa produced by the clotting system can also be activated by kallikrein of the kinin system (red arrow). Prekallikrein is enzymatically converted to kininogen, which activates bradykinin. Bradykinin functions similar to histamine and increases vascular permeability. Bradykinin can also stimulate nerve endings to cause pain. FP, Fibrinopeptide; MBL, mannose binding lectin; TF, tissue factor.
Three illustrated panels, A, B, and C, demonstrate plasma protein systems in inflammation in complement system, clotting system, and kinin system, respectively. Panel A, complement system. Three mechanisms activate C 3: 1. Classical pathway (activated by antibody) 2. Lectin pathway (activated by M B L on pathogens) 3. Alternative pathway (activated by bacterial products) Activated C 3 results in two fragments: 1. C 3 a or anaphylatoxin. 2. C 3 b or opsonin. C 3 b can lead to increased phagocytosis or activate the component C 5, which produces two fragments: 1. C 5a or anaphylatoxin and chemotactic factor. 2. C 5 b. C 3 a and C 5 a can cause mast cell release of histamine. C 5 a can cause leukocyte (neutrophil migration). C 5 b and C 6 to C 9 can activate the membrane attack complex, causing damage to bacterium. Panel B, clotting system. There are two pathways to the activation of Factor X. 1. Tissue-factor (extrinsic) pathway. Blood vessel injury. Factor villa and T F. 2. Contact activation (intrinsic) pathway. Damaged vessel wall. Hageman factor 12. 12 a. Factor X activates thrombin and subsequently fibrinogen: 1. Fibrin. Blood clot. 2. F P. Chemotactic factor or leukocyte (neutrophil migration) and increased permeability. Panel C, kinin system. 1. Prekallikrein; also activated by 12 a. 2. Kallikrein; also activates 12 a. 3. Kininogen. 4. Bradykinin. 5. Histamine like effects. 6. Increased permeability. 7. Vasodilation. 8. Stimulates nerve endings, pain.
Complement System
The complement system intensifies or complements the capacity of cells of the innate and adaptive immune systems to clear pathogens and damaged cells and to activate inflammation.6 Complement consists of a large number of proteins (sometimes called complement factors), which, together, constitute about 10% of the total circulating serum protein. Activation of the complement system initiates a cascade of proteolytic steps that result in the formation of several substances that can destroy pathogens directly or can eradicate pathogens through enhancing the activity of other components of the immune response.
The activation of C3 and C5, two important components of the complement cascade, results in the creation of several potent molecules critical to the immune response:
- 1. C3b—along with antibodies, serves as opsonins which coat the surface of bacteria increasing their susceptibility to phagocytosis by inflammatory cells (i.e., phagocytes [neutrophils and macrophages]).
- 2. C5a—functions as a chemotactic factor which serves as a form of chemical signaling that causes leukocytes to move to the area of inflammation.
- 3. C3a and C5a—sometimes called anaphylatoxins, induce rapid degranulation of mast cells to release histamine, a substance which induces vasodilation and increased capillary permeability.
- 4. Membrane attack complex (MAC)—composed of elements C6 through C9 and leads to bacterial destruction and tissue injury by creating pores in the outer membranes of cells or bacteria. These pores facilitate the infusion of water into the cells culminating in cellular death.
Three major pathways or cascades control the activation of complement (see Fig. 7.3A): the classical pathway, the alternative pathway, and the lectin pathway. The classical pathway is activated by antibodies, which are components of the adaptive immune system. Antibodies bind to antigens, which are typically proteins or carbohydrates produced by infectious microorganisms. Such antibodies activate the first complement component, C1, which, in turn, leads to the sequential activation of other components of the complement cascade, specifically C3 and C5, triggering inflammation.
The alternative pathway is activated directly by substances found on the surface of infectious microorganisms. These substances would include lipopolysaccharides (endotoxins) found on bacterial membranes and carbohydrates (zymosan) found on yeast cell walls. This pathway uses unique proteins (factor B, factor D, and properdin) to form a complex that sequentially activates the complement proteins C3 and C5. At this point, the process converges with the classical pathway. The alternative pathway provides a mechanism whereby the complement system can be activated in the absence of antibodies.
The lectin pathway, like the alternative pathway, is independent of antibodies and is activated by several plasma proteins, particularly mannose-binding lectin (MBL). MBL binds to bacterial polysaccharides that contain the carbohydrate mannose and activates the complement cascade. Infectious agents that do not activate the alternative pathway may still be susceptible to the complement system through the lectin pathway.
In summary, the complement pathway or cascade is a sequential series of cellular events that serve as defense mechanisms for the body. It can be activated by at least three different pathways, resulting in one or more of four protective functions: opsonization (C3b), anaphylatoxic activity from mast cell degranulation (C3a, C5a), leukocyte chemotaxis (C5a), and cell lysis (C5b–C9; membrane attack complex). Because of this potent defense system, successful pathogens must develop mechanisms that resist complement activity in order to cause disease.7
While highly beneficial to host defense, the complement system can contribute to tissue damage in a wide range of acute and chronic inflammatory diseases (see Chapter 9). New therapies aimed at blocking complement are proving to be beneficial for a number of these conditions.8
Clotting
The clotting system (also known as the coagulation system) is a group of plasma proteins, which, when activated sequentially, form a blood clot which is a meshwork of fibrin strands and platelets. The clotting system can be activated by a variety of substances released during tissue injury or infection, such as collagen, enzymes, and bacterial toxins. Clots serve to plug damaged vessels and stop bleeding (hemostasis), trap microorganisms to prevent their spread to adjacent tissues, and provide a framework for future repair and healing. When the wall of a blood vessel is injured, two converging pathways lead to clot formation (see Fig. 7.3B):
- 1. The tissue factor (extrinsic) pathway is activated by tissue factor (TF), also called tissue thromboplastin, a substance released by damaged endothelial cells of the blood vessels. It reacts with activated factor VII (VIIa).
- 2. The contact activation (intrinsic) pathway is activated when vessel wall damage causes negatively charged subendothelial substances to come into contact with Hageman factor (factor XII) found in plasma.
Both pathways converge at factor X and thrombin. Thrombin activates fibrinogen to form fibrin. Fibrin, in turn, comes together to form a fibrin clot. The purposes of the fibrin clot are to:
- 1. Stop bleeding.
- 2. Help prevent the spread of inflammation and infection.
- 3. Keep the invader near the site of injury, thus maximizing the access of inflammatory cells and proteins.
- 4. Initiate the formation of two fibrinopeptides (A and B) that are released during the formation of fibrin and are chemotactic for neutrophils and increase vascular permeability.
- 5. Provide a framework for repair and healing.9
Platelets also play a crucial role in inflammation, including the secretion of immunoregulatory cytokines and the ability to bind infectious pathogens.10 Additional details concerning the clotting system are discussed in Chapter 28.
Kinin System
The third plasma protein system, the kinin system (see Fig. 7.3C), interacts closely with the clotting system. Both the clotting and kinin systems can be initiated through activation of Hageman factor (factor XII), which, in turn, results in the formation of factor XIIa, also known as prekallikrein activator. Factor XIIa activates prekallikrein, the first component of the kinin system. The next product in the cascade is kallikrein, followed by kininogen, a precursor molecule, and then bradykinin, the final product of the kinin system. Bradykinin causes dilation of blood vessels. Bradykinin also acts in concert with prostaglandins to induce pain, trigger smooth muscle cell contraction (i.e., bronchoconstriction), and increase vascular permeability.
Control and Interaction of Plasma Protein Systems
The three plasma protein systems are highly interactive. The activation of one system results in the production of a large number of biologically active substances that activate other systems. Tight regulation of these processes is essential for two reasons. First, the inflammatory process is critical for an individual’s survival; therefore, efficient activation must occur regardless of the cause of tissue injury. The interaction among the three systems means that the entire inflammatory response may be activated regardless of which system initially triggers the sequence. Second, the biochemical mediators released during the inflammatory response are potent and potentially harmful to the individual. Their actions must be controlled and confined to injured or infected tissues.
Multiple mechanisms are available to regulate the plasma protein systems, activating or inactivating the reactions to maintain balance, and contain the inflammatory response. Plasma entering the tissues during inflammation contains enzymes that destroy the mediators of inflammation and downregulate the inflammatory response. These enzymes include protease inhibitors (e.g., C1-inhibitor) and carboxypeptidase, which inhibit the complement system; and kininase and histaminase, which inhibit the kinin system. The formation of clots activates a fibrinolytic system. This system serves to limit the size of the clot and degrade the clot after bleeding has ceased. In this system thrombin activates plasminogen, forming the enzyme plasmin, which degrades the fibrin polymers in clots and has been implicated in a wide range of inflammatory diseases.11 Defects in these mechanisms can be genetic or acquired. For example, a genetic defect in the protease C1 inhibitor (C1-inh) results in hereditary angioedema, a self-limiting edema of cutaneous and mucosal layers. Acquired defects in plasminogen activation and control, which is seen in severe infection, can result in widespread clotting.12
Initial Cellular Responders in Inflammation
Mast cells and macrophages can be thought of as guardian cells and are the primary cellular initiators of the inflammatory response (see Algorithm 7.1). Also present are dendritic cells which are a specialized type of macrophage that connects the innate and adaptive immune responses. These three types of cells reside near epithelial tissues, blood vessels, and lymphatics. They are equipped with receptors that allow them to detect pathogens and the cellular products of tissue damage (Algorithm 7.2).
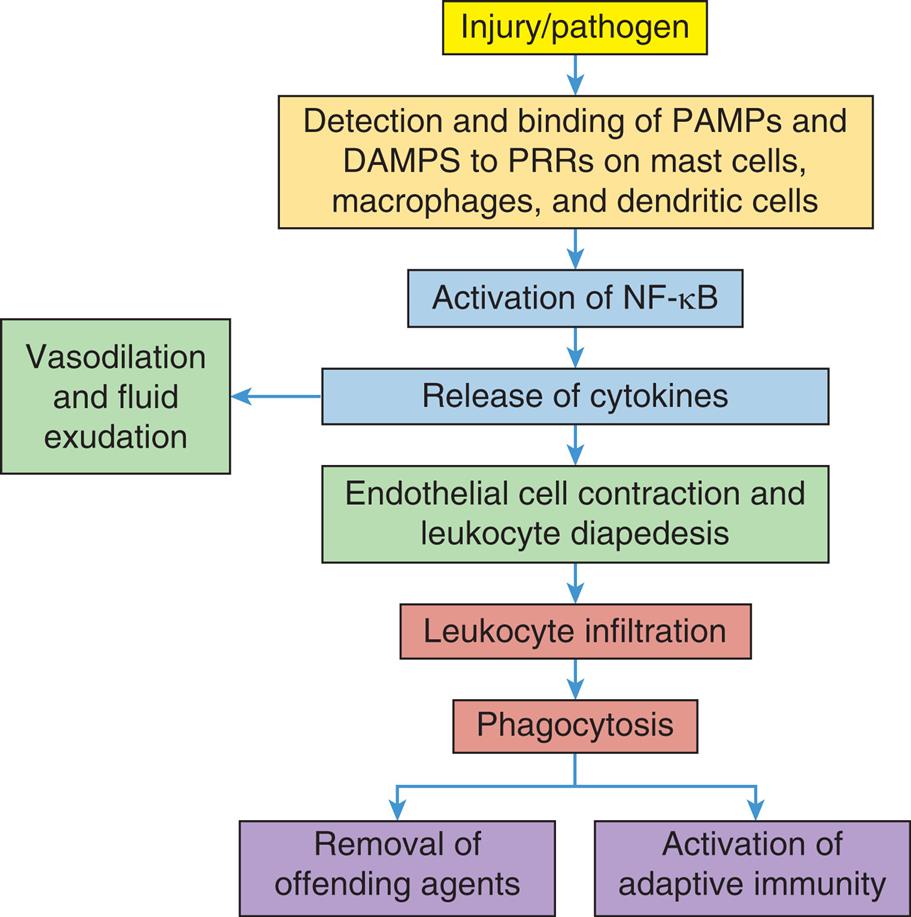
As a result of tissue injury or pathogen invasion, pattern recognition receptors (PRRs) on mast cells, macrophages, and dendritic cells are activated by binding to pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs). This leads to activation of NF-κB and the release of multiple cytokines that initiate vascular and cellular responses that work to bring immune cells to the area of injury/invasion, remove offending agents, activate adaptive immunity, and prepare the tissues for healing.
A flowchart provides an overview of the sequence of events in the acute inflammatory response. The sequence of events is as follows. 1. Injury or pathogen. 2. Detection and binding of P A M Ps and D A M Ps to P R Rs on mast cells, macrophages, and dendritic cells. 3. Activation of N F sub negative k B. 4. Release of cytokines; Vasodilation and fluid exudation. 5. Endothelial cell contraction and leukocyte diapedesis. 6. Leukocyte infiltration. 7. Phagocytosis: Removal of offending agents and activation of adaptive immunity.
Cellular Receptors
Mast cells, macrophages, and dendritic cells have evolved a set of receptors referred to as pattern recognition receptors (PRRs) (Table 7.2). PRRs are generally expressed on cells in tissues near the body’s surfaces (skin, respiratory tract, GI tract, genitourinary tract), where they monitor the environment for products of cellular damage and infectious microorganisms. PRRs can also be found on mucosal epithelial cells, neutrophils, and some lymphocytes. PRRs recognize two types of molecular patterns:
- 1. Pathogen-associated molecular patterns (PAMPs), which are molecules expressed by infectious agents either found on their surface or released as soluble molecules.
- 2. Damage-associated molecular patterns (DAMPs), which are products of cellular damage. Accordingly, cells of the innate immune system can respond to both sterile tissue damage (DAMPs) and septic tissue damage (PAMPs and DAMPs). It is estimated that at least 100 different PRRs are found on innate immune cells, rendering them capable of recognizing more than 1000 different molecules.
Table 7.2
| Receptors | Number of Receptors | What They Recognize | Where They Are Found |
|---|---|---|---|
| Toll-like receptors (TLRs) | 10 | PAMPs and DAMPs | Outer membrane of innate immune cells especially macrophages and dendritic cells |
| NOD-like receptors and NLR receptors | ~22 | PAMPs and DAMPs | Cytoplasm of innate immune cells |
| Scavenger receptors (including C-type lectin receptors (CLRs) | 12 | PAMPs, DAMPS, Cell membrane phospholipids | Outer membrane of phagocytes |
| Complement and Cytokine receptorsa | Multiple | C3a, C3b, C5a, TNF-α, interleukins, interferons, chemotactic factors, prostaglandins, platelet activating factor, IL-10, TGF-β | Outer membrane of innate immune cells, platelets, epithelial cells, vascular smooth muscle |
DAMP, Damage-associated molecular patterns; IL-10, interleukin-10; NOD, nucleotide oligomerization domain-like receptors; NRL, nucleotide-binding-like receptors; PAMP, pathogen-associated molecular patterns; TGF-β, transforming growth factor-beta; TNF-α, tumor necrosis factor-alpha).
aComplement and cytokine receptors are not true pattern recognition receptors but are essential for innate immune cell function
An important group of PRRs are Toll-like receptors (TLRs), which recognize a large variety of PAMPs located on the surface of microorganisms (e.g., bacterial lipopolysaccharide, peptidoglycans, lipoproteins, yeast zymosan, flagellin, bacterial or viral nucleic acid, and viral coat proteins). TLRs are expressed on the surface of many cells that have direct and early contact with potential pathogens. Such cells include mucosal epithelial cells, mast cells, macrophages, dendritic cells, neutrophils, and lymphocytes. TLRs also are activated by oxidative stress, tissue injury, and damaged cellular contents (DAMPs). Activation of TLRs initiates a cascade of intracellular signaling pathways leading to the activation of nuclear factor-[kappa]B (NF-κB) in the cell nucleus.13 NF-κB activation results in increased transcription and subsequent release of numerous inflammatory cytokines. Among these are tumor necrosis factor (TNF), interleukins, and interferons (IFNs) which play key roles in the inflammatory process, activation of the adaptive immune response, and the destruction of many pathogenic microorganisms (these cytokines are discussed in detail later in this chapter). Pathophysiologic changes in TLR stimulation and NF-κB activation have been implicated in numerous conditions such as neuroinflammation, heart disease, and cancer13 (see Emerging Science Box: The Role of Toll-like Receptors [TLRs] and Nuclear Factor-kappaB [NF-κB] in Disease).13
Nucleotide-binding-like receptors (NLRs) (including nucleotide oligomerization domain–like [NOD-like] receptors) are cytoplasmic (intracellular) receptors in lymphocytes, macrophages, and dendritic cells. At least 23 NLRs have been identified in humans. They recognize intracellular microorganisms and damaged cells and initiate the production of proinflammatory mediators. NLRs also detect cells that are forming inflammasomes, which are large cytoplasmic complexes that activate cytokines. Inflammasomes are implicated in a wide variety of diseases including Coronavirus Disease 2019 (COVID-19) inflammatory lung damage.14
Scavenger receptors are membrane receptors primarily expressed on macrophages and are categorized into 12 classes.15 They have multiple functions including the recognition and subsequent phagocytosis of bacterial pathogens and damaged cells. They also recognize soluble lipoproteins, such as high-density lipoprotein (HDL), low-density lipoprotein (LDL), and oxidized LDL, and have been implicated in atherosclerotic vascular diseases.16 C-type lectin receptors (CLRs) are a type of scavenger receptor that binds to both PAMPs and DAMPs. There are many types of CLRs, such as macrophage mannose, Dectin 1 and 2, and mannose-binding lectin receptors. They are particularly important in recognizing fungal antigens and subsequent activation of innate immune cells.
Complement and cytokine receptors are found on many cells involved in the immune response, as well as on some epithelial cells. These are not true PRRs but are essential to innate immune cell function. Complement receptors recognize several fragments produced through activation of the complement system, particularly C3a, C5a, and C3b. This results in chemotaxis and activation of innate immune cells. Cytokine receptors recognize both proinflammatory and anti-inflammatory cytokines (these cytokines are discussed later in this chapter).
Mast Cells
Mast cells are significant and potent activators of the inflammatory response. They have abundant granules containing biochemical mediators, which are released in instances of pathogen invasion and tissue injury (Fig. 7.4). Mast cells are activated by PAMPs and DAMPs binding to PRRs, as well as by direct physical, chemical, and thermal injury. They also can be activated by components of the complement cascade and by immunoglobulin E (IgE) antibodies produced in allergies (see Chapter 9). Located in connective tissue and close to vessels, they can be found near the body's surfaces (skin, GI, and respiratory tract linings). A variety of stimuli associated with tissue injury can induce inflammation by triggering the release of potent soluble substances from mast cells. These substances are released in two ways:
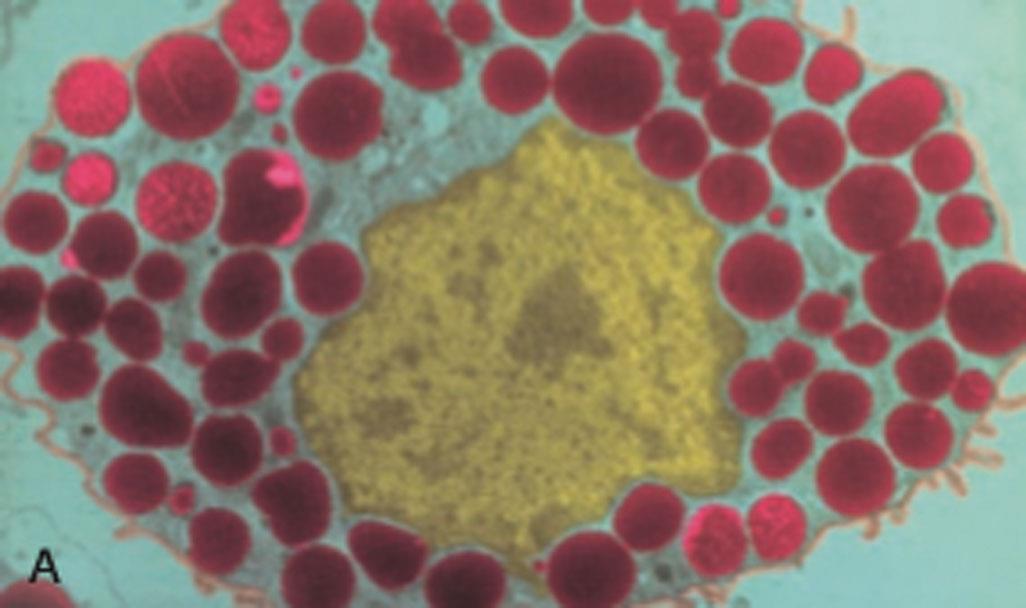
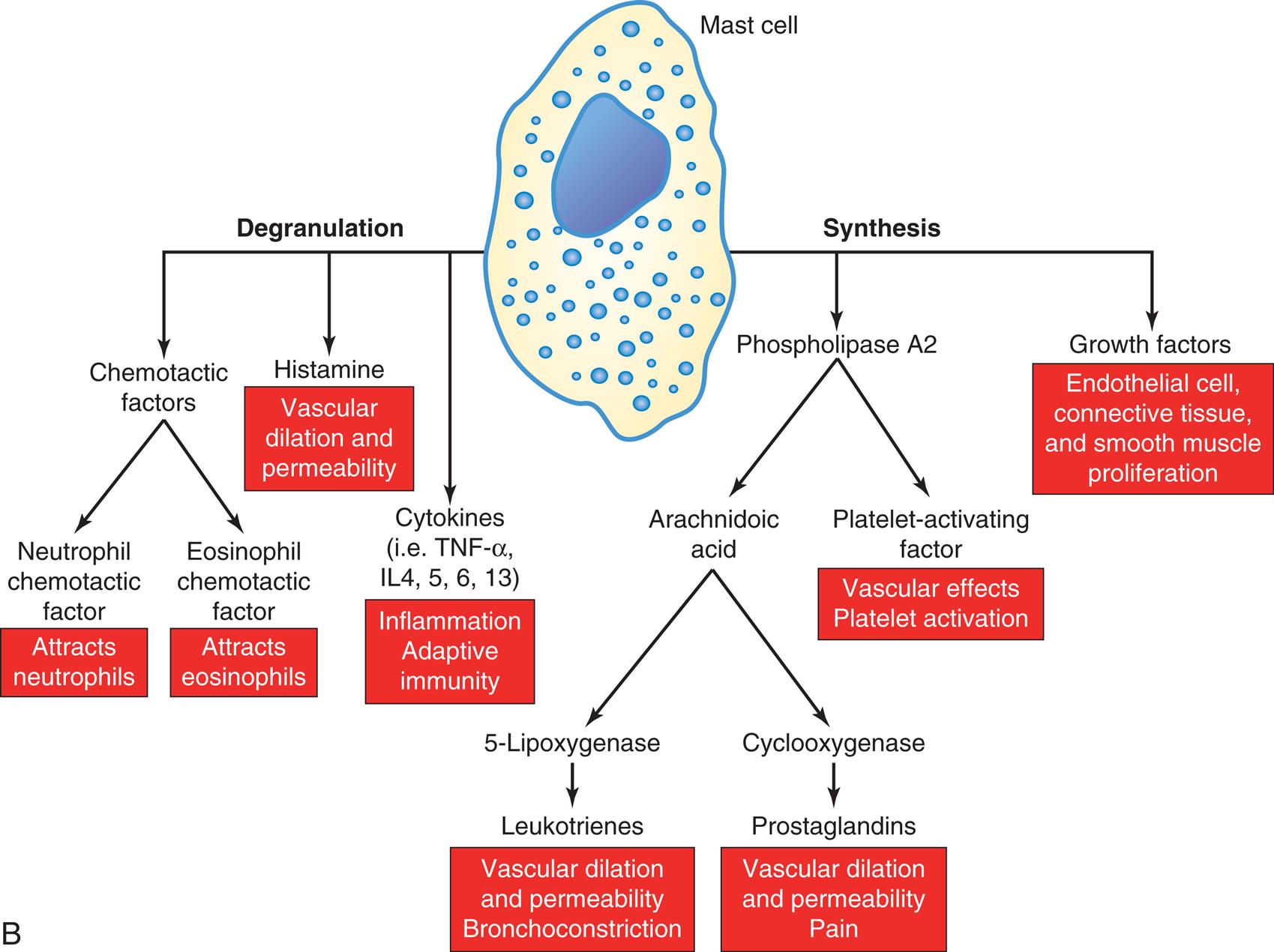
(A) Colorized photomicrograph of mast cell; dense granules contain biologically active substances. Among these are histamine, which is a major initiator of vascular changes, and a variety of chemotactic factors. (B) Mast cell degranulation (left) and synthesis (right). Histamine and other biologically active substances are released immediately after stimulation of mast cells. Long-term responses are mediated by the synthesis and release of products of phospholipase A metabolism (leukotrienes, prostaglandins, and platelet-activating factor) and growth factors that promote tissue proliferation. IL, Interleukin; TNF-α, tumor necrosis factor-alpha. (A, From Roitt IM, Brostoff J, Male DK. Immunology, 3rd edition. St. Louis: Mosby, 1993.)
Panel A shows a colorized photomicrograph of a mast cell at the center surrounded densely by intracellular granules. Panel B shows flowchart tracking the degranulation and synthesis of a mast cell. Degranulation releases the following substances: 1. Chemotactic factors include neutrophil chemotactic factor that attracts neutrophils, and eosinophil chemotactic factor that attracts eosinophils. 2. Histamine causes vascular dilation and permeability. 3. Cytokines, for example, T N F alpha, I L 4, 5, 6, and 13, include inflammation and adaptive immunity. Synthesis releases the following substances: 1. Phospholipase A 2 includes arachidonic acid, and platelet-activating factor that causes vascular effects and platelet activation. Arachidonic acid, in turn, releases the following substances: 1. 5-lipoxygenase include leukotrienes that cause vascular dilation and permeability and bronchoconstriction, and cyclooxygenase include prostaglandins that cause vascular dilation and permeability, and pain. 2. Growth factors include endothelial cell, connective tissue, and smooth muscle proliferation.
Degranulation
Mast cells release biochemical mediators from their granules into the surrounding tissues within seconds of a stimulus. Substances within the granules include histamine, chemotactic factors, and cytokines. Their effects occur immediately.
Histamine is a small-molecular-weight molecule with potent effects on many other cells, particularly those that control the circulation. Histamine binds to histamine receptors (H1, H2, H3, and H4 receptors) on various cell surfaces. The binding of histamine to the H1 receptor is proinflammatory (Fig. 7.5). This causes a rapid, temporary constriction of smooth muscle, along with dilation of the postcapillary venules. The net result of these two effects is increased blood flow within the microvasculature. H1 receptor binding also causes increased vascular permeability secondary to the retraction of endothelial cells lining the capillaries (opening the tight junctions between them), and increased leukocyte adherence to the endothelial walls. The H1 receptor also is present on smooth muscle cells within the bronchi. H1 stimulation results in bronchial smooth muscle contraction and bronchoconstriction often seen in asthma. Antihistamines are drugs that block the binding of histamine to the H1 receptor, resulting in decreased vascular effects. H4 receptors are highly expressed on mast cells where their stimulation is proinflammatory by promoting histamine and cytokine generation, and H4 receptor blockers are being developed in an effort to better treat many allergic and inflammatory disorders.17 The role of histamine receptors and hypersensitivity is discussed in Chapter 9.
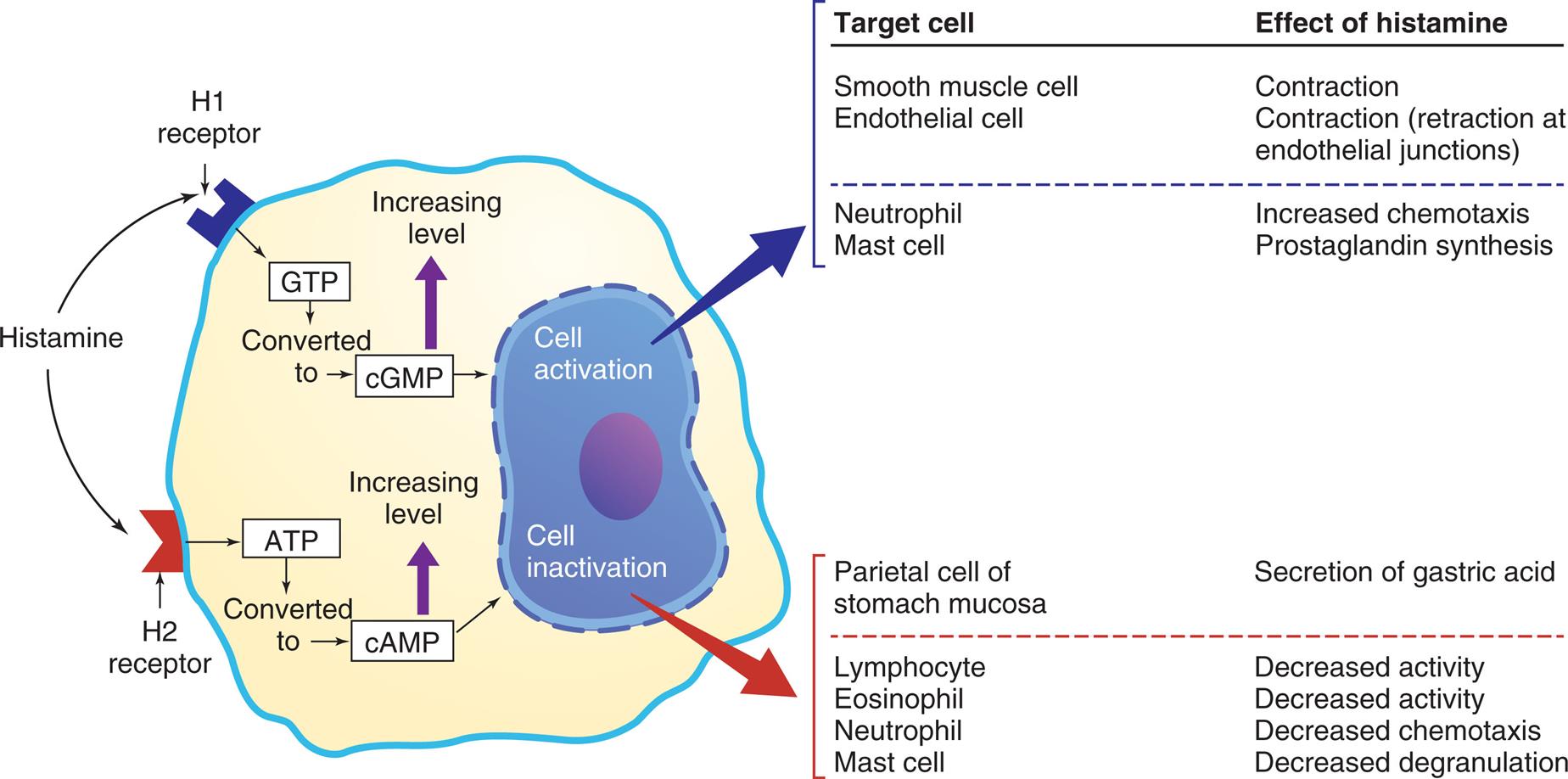
The effects depend on (1) the density and affinity of H1 or H2 receptors on the target cell and (2) the identity of the target cell. ATP, Adenosine triphosphate; cAMP, cyclic adenosine monophosphate; cGMP, cyclic guanosine monophosphate; GTP, guanosine triphosphate.
An illustrated table summarizes the effects of histamine through H 1 and H 2 receptors. The illustration of a cell shows histamine being attracted to H 1 receptor and H 2 receptor. When bound to H 1 receptor, G T P is converted to c G M P, increasing its level and leading to cell activation. When bound to H 2 receptor, A T P is converted to c A M P, increasing its level and leading to cell inactivation. The table at the top identifies the target cell and lists the effect of histamine on the target cell. The data from the table for cell activation are as follows. • Smooth muscle cell: Contraction. • Endothelial cell: Contraction (retraction at endothelial junctions) • Neutrophil: Increased chemotaxis. • Mast cell: Prostaglandin synthesis. The data from the table for cell inactivation are as follows. • Parietal cell of stomach mucosa: secretion of gastric acid. • Lymphocyte: Decreased activity. • Eosinophil: Decreased activity. • Neutrophil: Decreased chemotaxis. • Mast cell: Decreased degranulation.
In contrast, histamine binding to the H2 receptor is generally anti-inflammatory because it results in the suppression of leukocyte function. Both H1 and H2 receptors are often present on the same immune cells where they may act in an antagonistic fashion. For example, stimulation of H1 receptors on neutrophils results in an augmentation of neutrophil chemotaxis, whereas stimulation of the H2 receptor results in its inhibition. The H2 receptor also is abundant on parietal cells of the stomach mucosa, where stimulation induces the secretion of gastric acid as part of the normal physiology of the stomach. H3 receptors inhibit the release of histamine and other neurotransmitters on neurons in the central nervous system. They are involved in blood–brain barrier function, and non-inflammatory processes including cognition, sleep, and regulation of food intake.18 Research into the role of H3 receptors in health and disease are revealing potential uses treating neurodegeneration (Alzheimer and Parkinson disease), migraine, narcolepsy, and control of eating in obesity.19
Mast cell granules also contain chemotactic factors (chemokines). Chemotaxis is directional movement of cells along a chemical gradient formed by a chemotactic factor. Two important factors are neutrophil chemotactic factor (NCF) and eosinophil chemotactic factor of anaphylaxis (ECF-A). Neutrophils are the predominant cells that destroy bacteria in inflammation. Eosinophils help regulate the inflammatory response. Both cells are discussed in more detail later in this chapter.
Synthesis of mediators
Activated mast cells initiate synthesis of other mediators of inflammation. These mediators include leukotrienes, prostaglandins, platelet-activating factor and growth factors (see Fig. 7.4B).
- 1. Leukotrienes (slow-reacting substances of anaphylaxis [SRS-A]) are products of arachidonic acid. Arachidonic acid is released from mast cell membranes by intracellular phospholipase A2 which acts on membrane phospholipids. Leukotrienes are produced from arachidonic acid by the action of lipoxygenase and consist of several different subtypes (Fig. 7.6) Leukotrienes induce smooth muscle contraction (especially bronchoconstriction) and increased vascular permeability. Leukotrienes appear to be important in the later stages of the inflammatory response because they stimulate slower and more prolonged inflammatory responses than does the rapid-acting histamine.
- 2. Prostaglandins also are products of arachidonic acid. They cause increased vascular permeability, neutrophil chemotaxis, and pain. Pain results from the direct effects on nerves. Prostaglandins are long-chain, unsaturated fatty acids produced by the action of cyclooxygenase (COX). COX exists in two different forms: COX-1, which produces prostaglandins that activate platelets and protect the stomach lining, and COX-2, which activates prostaglandins associated with inflammation (see Fig. 7.6). Aspirin, acetaminophen, and nonsteroidal anti-inflammatory drugs (NSAIDs) inhibit COX-1 and/or COX-2. They suppress inflammation and improve symptoms but have associated side effects. NSAIDs can cause GI tract bleeding, hypertension, renal dysfunction, and cardiovascular disease especially in older individuals who take them regularly for underlying conditions such as osteoarthritis.20
- 3. Platelet-activating factor (PAF) molecules are produced by neutrophils, monocytes, endothelial cells, mast cells, and platelets. The biologic activity of PAF is similar to that of leukotrienes. It causes increased vascular permeability, leukocyte adhesion to endothelial cells, and platelet activation (see Fig. 7.6).
- 4. Growth factors synthesized and released by mast cells include vascular endothelial growth factor (VEGF) which promotes endothelial cell proliferation, and platelet-derived growth factor (PDGF) which promotes connective tissue and smooth muscle proliferation (see Fig. 7.4B). These growth factors contribute to wound healing.
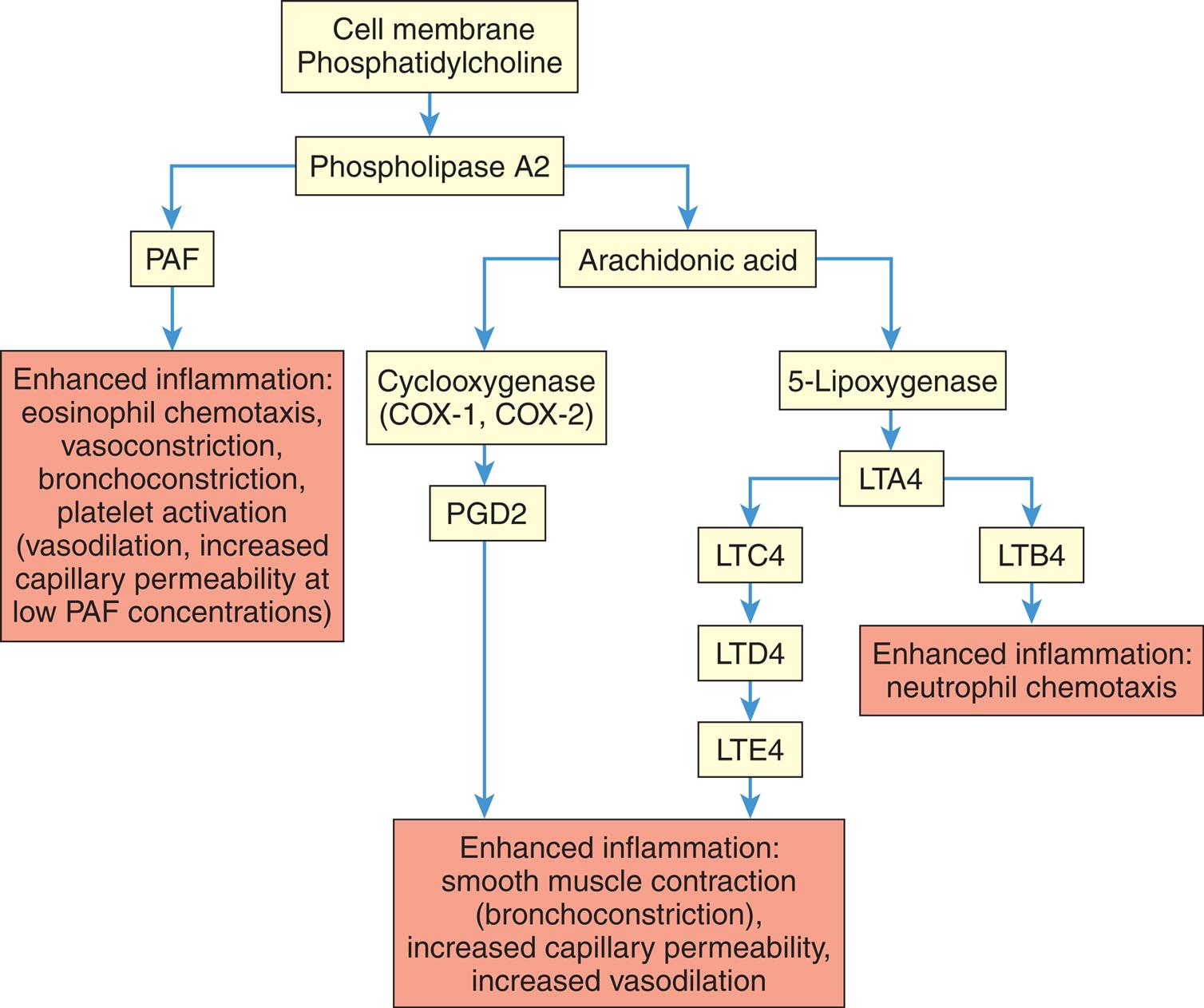
LTA4, LTC4, LTD4, LTE4, LTB4, Various leukotriene molecules; PAF, platelet-activating factor; PGD2, prostaglandin D2.
A flowchart tracks the production of lipid vasoactive substances by mast cells. The data from the flow chart is as follows. • Cell membrane phosphatidylcholine: phospholipase A 2. • Phospholipase A 2: P A F and arachidonic acid. • P A F: Enhanced inflammation. Eosinophil chemotaxis, vasoconstriction, bronchoconstriction, platelet activation (vasodilation, increased capillary permeability at low PAF concentrations). • Arachidonic acid: Cyclooxygenase and 5-Lipoxygenase. • Cyclooxygenase (C O X-1, C O X 2): P G D 2. Enhanced inflammation. Smooth muscle contraction (bronchoconstriction), increased capillary permeability, increased vasodilation. • 5-lipoxygenase: L T A 4. • L T A 4: L T C 4 and L T B 4. • L T C 4: L T D 4. L T E 4. Enhanced inflammation. Smooth muscle contraction (bronchoconstriction), increased capillary permeability, increased vasodilation. • L T B 4: Enhanced inflammation. Neutrophil chemotaxis.
Macrophages
Macrophages are derived from circulating monocytes. As with other blood cell types, monocytes are produced in bone marrow and enter into the circulation. They migrate to tissues throughout the body where they transform to macrophages. These tissue macrophages have many different names depending on where they are located, including Kupffer cells (liver), alveolar macrophages (lungs), and microglia (brain) (see Table 28.3). Tissue macrophages are important initial mediators of the inflammatory response.
Macrophages and dendritic cells have an abundance of PRRs on their surface that can recognize a wide range of pathogens and molecules released from damaged tissues. PRR receptor binding to PAMPs and DAMPs results in intracellular communication leading to activation of NF-κB and the subsequent release of proinflammatory chemokines and cytokines. Chemokines (a type of cytokine) are members of a family of low-molecular-weight peptides that function primarily to induce leukocyte chemotaxis. They facilitate the movement of phagocytes from the blood stream through the vessel wall into the area of injury. Cytokines constitute a family of intercellular signaling molecules that are secreted, bind to specific membrane receptors, and regulate innate and adaptive immunity. Macrophages release both proinflammatory and antiinflammatory cytokines (Fig. 7.7). The most important proinflammatory cytokines secreted by macrophages are tumor necrosis factor-alpha (TNF-α), interleukins, and IFNs.
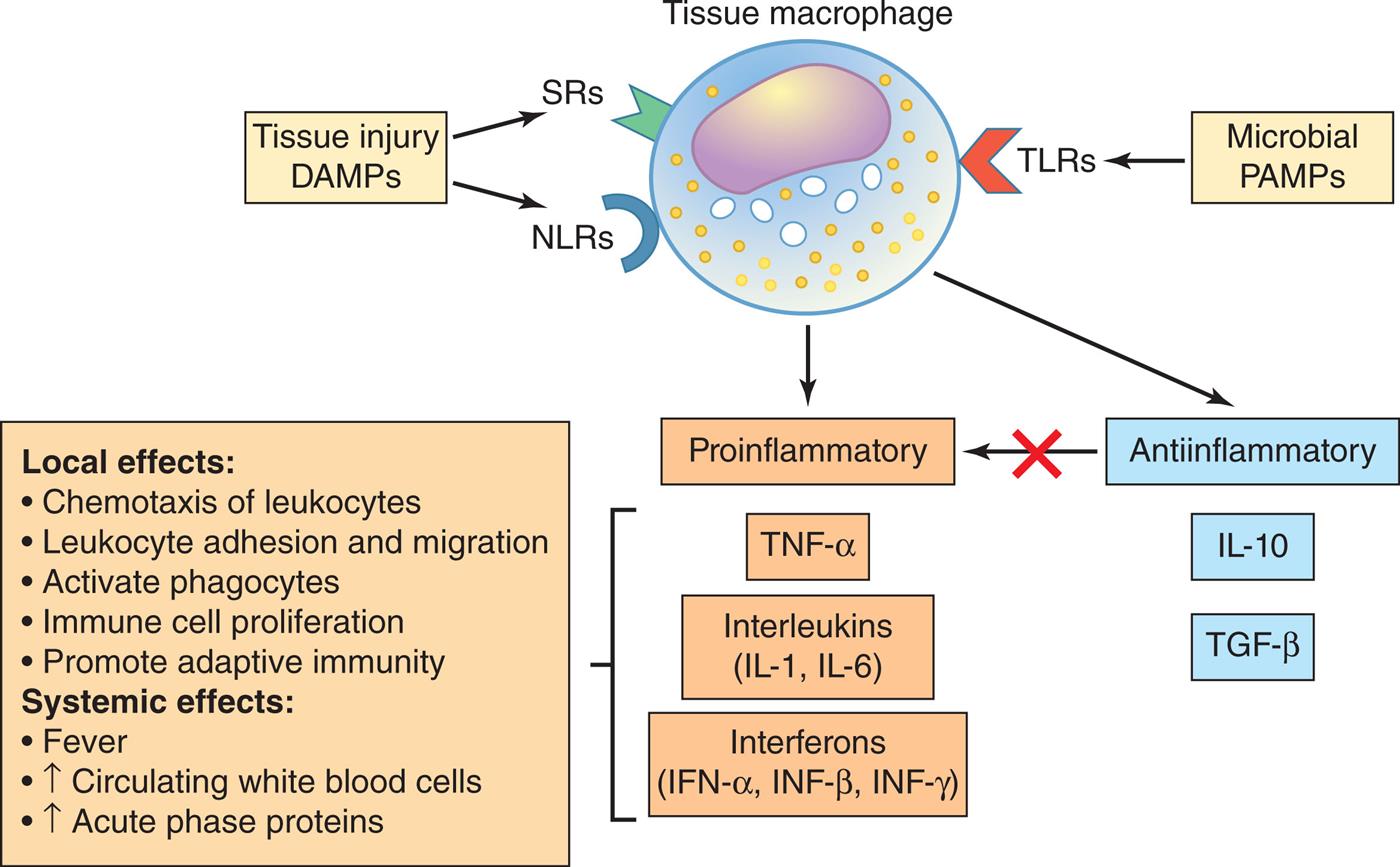
When stimulated by pathogen invasion or tissue injury, macrophages release proinflammatory cytokines that have a wide range of local and systemic effects. Antiinflammatory cytokines are also released which limit the effects on inflammation. DAMPs, Damage-associated molecular patterns; IFN, interferon; IL, interleukin; NLRs, nucleotide-binding-like receptors; PAMPs, pathogen-associated molecular patterns; SRs, scavenger receptors; TLRs, Toll-like receptors; TNF-α, tumor necrosis factor alpha.
An illustrated flowchart summarizes the tissue macrophage-derived cytokines. Tissue macrophage is a result of S Rs and N L Rs from tissues injury D A M Ps and T L Rs from microbial P A M Ps, and it causes: • Proinflammatory: T N F-alpha, interleukins (I L-1, I L-6), interferons (I F N-alpha, I N F-beta, I N F-gamma). • Anti-inflammatory: Interleukins (I L-10), Interferons (T G F-beta). Local effects from proinflammation: • Chemotaxis of leukocytes. • Leukocyte adhesion and migration. • Activate phagocytes. • Immune cell proliferation. • Promote adaptive immunity. Systemic effects from proinflammation: • Fever. • Increase in circulating white blood cells. • Increase in acute phase proteins.
Tumor necrosis factor-alpha
Tumor necrosis factor-alpha (TNF-α) is secreted primarily by activated macrophages, but is also released by mast cells, neutrophils, and lymphocytes. When PRRs on the surface of macrophages bind to PAMPs or DAMPs, NF-κB is activated and TNF-α is one of the most important proinflammatory cytokines that is then produced. It plays a role in promoting the innate response to virtually any injury or infection including chemotaxis and adherence of neutrophils, phagocytosis, and inflammatory and adaptive immune cell proliferation.21 TNF-α has systemic effects, including the induction of fever, increased liver synthesis of inflammation-related serum proteins (acute phase proteins), muscle wasting (cachexia), and intravascular thrombosis. These effects can be deleterious in cases of severe or chronic infection and cancer. TNF-α contributes to the damaging effects of severe acute inflammatory conditions such as sepsis and to many chronic inflammatory diseases.
Interleukins
Interleukins (ILs) are produced predominantly by macrophages and lymphocytes in response to stimulation of PRRs or by cytokines secreted by other immune cells. More than 30 interleukins have been identified. Their effects include the following:
- 1. Regulation of cell adhesion molecules (CAMs), which are proteins that facilitate leukocyte binding with other cells or with the extracellular matrix
- 2. Attraction of leukocytes to a site of inflammation (chemotaxis)
- 3. Induction, proliferation, and maturation of leukocytes in bone marrow
- 4. General enhancement or suppression of inflammation and the adaptive immune response
Two major proinflammatory interleukins are interleukin-1 (IL-1) and interleukin-6 (IL-6). IL-1 is produced mainly by macrophages and activates monocytes, other macrophages, and lymphocytes, thereby enhancing both innate and adaptive immunity. It also acts as a growth factor for many cells. IL-1 is an endogenous pyrogen (fever-causing cytokine) which reacts with receptors on cells of the hypothalamus resulting in increased body temperature. IL-6 is produced by macrophages, lymphocytes, fibroblasts, and other cells. IL-6 directly induces hepatocytes in the liver to produce many of the proteins needed for inflammation (acute phase proteins). IL-6 also stimulates growth and differentiation of blood cells in the bone marrow and the growth of fibroblasts required for wound healing. IL-1 and IL-6 play important roles in numerous inflammatory diseases and infections, and monoclonal antibodies that block these cytokines are used to treat many of them. For example, in severe COVID-19 infection, blockade of IL-1 and IL-6 resulted in decreased mortality.22
Interferons
Interferons (IFNs) are members of a family of cytokines that protect against viral infections and modulate the immune response. They are considered essential components of both innate and adaptive immunity. Type I IFNs (primarily IFN-α, IFN-β) are produced and released by virally infected cells. These IFNs do not kill viruses; rather they induce antiviral proteins in neighboring healthy cells. Type II IFN (IFN-γ) is produced primarily by lymphocytes. It activates macrophages and increases their capacity to detect and process invaders and abnormal cells so that they can be removed by the adaptive immune system.
Antiinflammatory cytokines
Some cytokines are antiinflammatory and diminish and control the inflammatory response. Two of the most important are IL-10 and TGF-β. Interleukin-10 (IL-10) is primarily produced by lymphocytes. It suppresses the activation and proliferation of other lymphocytes and limits the production of proinflammatory cytokines by macrophages. The result is a downregulation of both the inflammatory and the adaptive immune responses. Recent evidence suggests that IL-10 also may have proinflammatory effects in certain severe conditions such as Crohn disease and COVID-19.23 Transforming growth factor-beta (TGF-β) is produced by many cells in response to inflammation. Its primary role in immunity is to suppress the activity of lymphocytes and downregulate the production of proinflammatory cytokines by macrophages.
Dendritic Cells
Dendritic cells can be considered a specialized type of macrophage derived from a common precursor stem cell in the bone marrow. Some, like macrophages, differentiate from circulating monocytes. They provide one of the major links between the innate and adaptive immune responses. They are located in tissues that have contact with the outside environment, e.g. the gut, respiratory tract, and skin. Like tissue macrophages, they recognize invaders with PRRs and phagocytose them. They are different from tissue macrophages in that they migrate from the tissues through lymphatic vessels to lymph nodes, where they present antigens from the phagocytosed invaders to T lymphocytes resulting in an adaptive immune response (see Chapter 8). Dendritic cells produce many of the same cytokines as macrophages.
The Vascular Response in Inflammation
After initiation by the plasma protein systems, mast cells, tissue macrophages, and dendritic cells, the inflammatory response has an almost immediate effect on blood vessels (see Algorithm 7.1). Inflammatory changes in the microcirculation (arterioles, capillaries, and venules) surrounding the site of an injury happen within seconds. They include the following processes:
- 1. Hemostasis (coagulation): Injury to blood vessels initiates the clotting cascade and activates platelets. Clotting slows blood flow, walls off injury, and provides a meshwork for healing.
- 2. Vasodilation: Arteriole and venule dilation increase the diameter of blood vessels, the volume of blood delivered to the injured site, and slows the velocity of blood flow. This action allows more time for the movement of fluids, chemicals, and cells into surrounding tissues, resulting in erythema (redness) and warmth in the area of injury.
- 3. Increased vascular permeability: Blood vessels become porous, secondary to retraction of endothelial cells, thus opening vascular tight junctions and enlarging the spaces between these cells. This results in exudation (the leaking of fluid and cells from vessels) and edema (tissue swelling from fluid leakage) of the area surrounding the injury.
- 4. Leukocyte cell adhesion: Leukocytes adhere to the inner walls of vessels, where they migrate through the enlarged spaces between endothelial cells and into the surrounding tissue (diapedesis). Accordingly, there is an influx of phagocytes (neutrophils and macrophages) to the injured tissue, where they target invaders and damaged cells.
Together, these vascular changes deliver leukocytes (particularly neutrophils and macrophages), plasma proteins, and other biochemical mediators to the site of injury. Chemical mediators activate pain fibers. Tissue injury, pain, and swelling contribute to loss of function.
Lymphatic vessels, which drain the extravascular fluid to lymph nodes, may become secondarily inflamed. The resulting lymphangitis (inflammation of lymph vessels) or lymphadenitis (inflammation of nodes) can present as enlarged and painful lymph nodes. For example, in an individual with a sore throat, the infected and inflamed pharynx can result in enlarged and painful lymph nodes, which are readily palpable in the neck region.
The Cellular Response in Inflammation
Once the inflammatory response has been initiated by plasma proteins, mast cells, tissue macrophages, and dendritic cells, other leukocytes move out of the blood vessels and travel quickly to the area of invasion or injury. Neutrophils are the most important phagocyte in the cellular response in inflammation and arrive early at the scene in large numbers where they begin the process of removing microorganisms and cellular debris. Monocyte-derived macrophages play an essential role in removing debris and initiating the healing process. Both of these cell types function primarily through the process of phagocytosis.
Neutrophils
The neutrophil, or polymorphonuclear neutrophil (PMN), is a granulocyte named for its characteristic staining pattern as well as its multilobed nucleus. Neutrophils circulate in the blood in large numbers and are attracted to the area of injury by chemotactic factors, such as mast cell cytokines, chemokines, and complement. Neutrophils are the predominant phagocytes in early inflammation, arriving at the inflammatory site within 6 to 12 hours after the initial injury. The primary role of the neutrophil is to phagocytize pathogenic microbes and remove cellular debris and dead cells from lesions. The neutrophil is a mature cell incapable of division and is sensitive to the acidic environment found at inflammatory sites. Accordingly, it is short-lived and rapidly becomes a component of the purulent exudate (pus) removed from the body through the epithelium or drained from the infected site through the lymphatic system. They are present in sterile lesions, which drain “sterile pus,” as well as in septic (nonsterile) lesions found in bacterial infections.
In addition to phagocytosis, neutrophils can form neutrophil extracellular traps (NETs) which are crucial in attacking pathogens. NETs are formed of material from cell nucleus breakdown (chromatin and histones) and neutrophil granules which contribute numerous enzymes that can degrade microorganisms. These enzymes include neutrophil elastase, myeloperoxidase, matrix metalloproteinases, and others.24 NET formation usually leads to the destruction of the neutrophil. NETs may also contribute to tissue damage and autoimmune diseases.25
Monocyte-Derived Macrophages
Monocyte-derived macrophages are also essential phagocytes in inflammation. They begin appearing at the inflammatory site as soon as 24 hours after the initial neutrophil infiltration but do not arrive in large numbers until 3 to 7 days later. They are attracted by chemotactic factors released by the neutrophils that are already present at the site. Macrophage activation results in two subpopulations, M1 and M2 macrophages, each with specialized functions. M1 macrophages are proinflammatory and have bactericidal activity, promote adaptive immunity, and can attack cancer cells.26M2 macrophages are primarily involved in tissue healing and repair. Macrophages can survive and divide in the acidic environment of the inflammatory site and so are capable of conducting prolonged phagocytosis of microorganisms and damaged cells. They play an essential role in removing debris and promoting the formation of new blood vessels (angiogenesis) and secretion of growth factors that prepare the tissue for healing.27
Phagocytosis
Phagocytosis is the ingestion of microbes, foreign particles, or cell fragments. The two most important cells for phagocytizing pathogens or damaged cells are neutrophils and macrophages. Inflammatory cytokines and chemokines serve to activate these cells and through chemotactic factors cause them to move toward the area of injury. These cytokines also open the tight junctions between endothelial cells, creating spaces between them. Additionally, inflammation induces endothelial nitric oxide synthase (iNOS), increasing the amount of nitric oxide (NO) production. Effects of NO in inflammation include vasodilation and changes in inflammatory cell function.28
The first step in the migration of phagocytes to the area of invasion or injury starts when endothelial cells, monocytes, and neutrophils begin expressing new CAMs on their surfaces. The most important CAMs related to vascular inflammation are selectins, integrins, and intracellular adhesion molecules (ICAMs). These CAMs increase the adhesion, or stickiness, between phagocytes and endothelial cells in the walls of the capillaries. At first, the phagocytes roll down the endothelial surface. This is followed by tight binding to the endothelium in a process called margination, or pavementing (Fig. 7.8A). Other adhesion molecules such as platelet endothelial cell adhesion molecules (PECAMs) then are expressed leading to diapedesis or emigration of the cells through the openings between endothelial cells. The leukocytes digest the basement membrane and migrate into the surrounding tissues.29 Monoclonal antibodies that block adhesion molecules are used to reduce inflammation in a variety of conditions including cancer (see Chapter 9).30
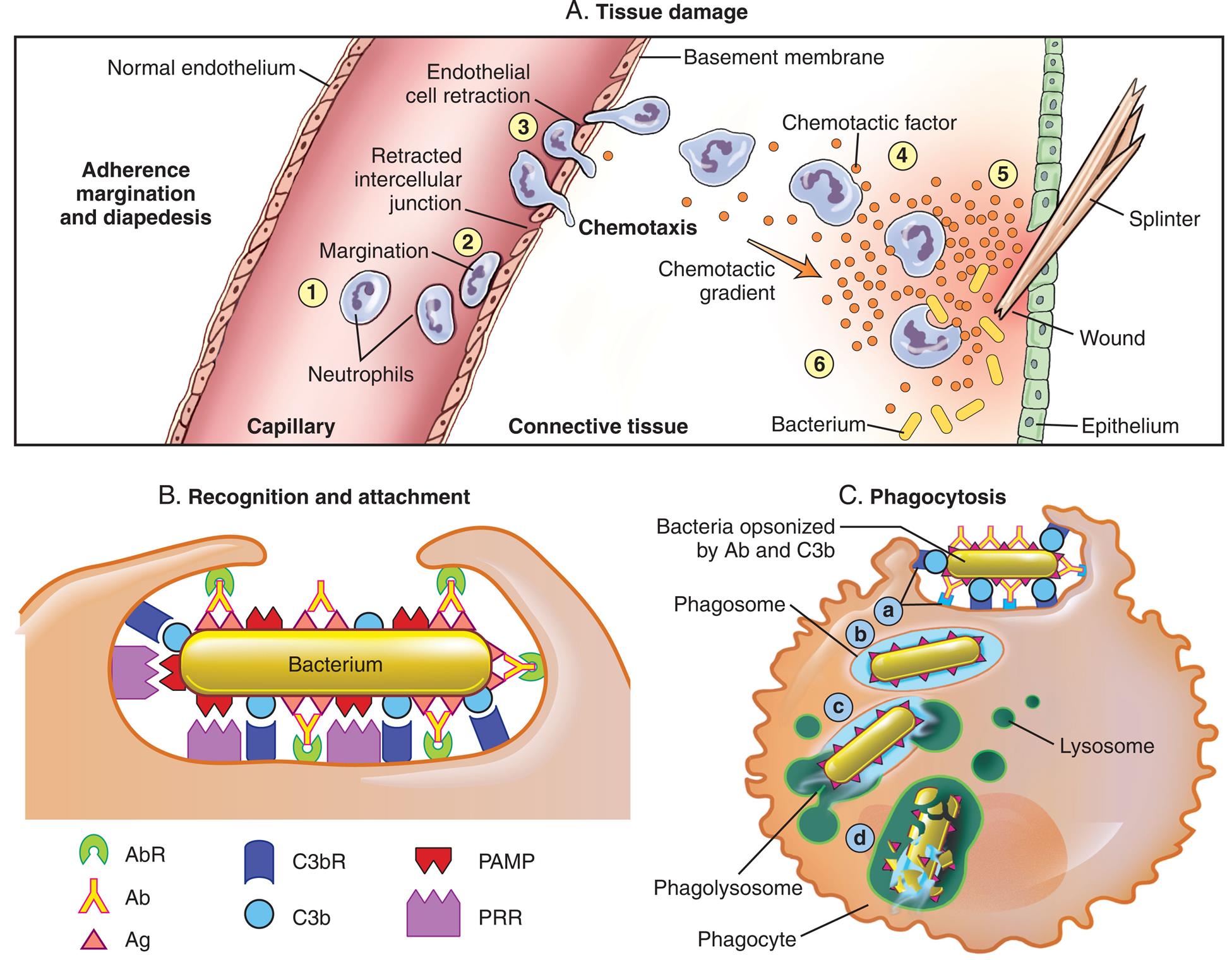
The process that results in phagocytosis is characterized by three interrelated steps: adherence and diapedesis, tissue invasion by chemotaxis, and phagocytosis. (A) Tissue damage. Adherence, margination, and diapedesis: The primary phagocyte in the blood is the neutrophil, which usually moves freely within the vessel (1). At sites of inflammation, neutrophils and endothelial cells respond to inflammatory cytokines by expressing adhesion molecules (selectins, integrins, and ICAMs). This causes the neutrophils to progressively develop increased adherence to the endothelium, leading to accumulation along the vessel wall (margination or pavementing) (2). At sites of endothelial cell retraction, other adhesion molecules such as PECAMs allow the neutrophil to move through the vessel wall by means of diapedesis (3). Chemotaxis: In the tissues, the neutrophil detects chemotactic factor gradients through surface receptors (4) and migrates toward higher concentrations of the factors (5). The high concentration of chemotactic factors at the site of inflammation immobilizes the neutrophil (6). (B) Recognition and attachment. Specific receptors are expressed on the surface of phagocytes that facilitate recognition and attachment to ligands found on the surface of target invaders such as bacteria. (C) Phagocytosis. (a) Antibodies and complement serve to opsonize the surface of microorganisms which are then more easily recognized and bound to the surface of a phagocyte. (b) The microorganism is engulfed (ingested) into a phagocytic vacuole, or phagosome. (c) Lysosomes fuse with the phagosome, resulting in the formation of a phagolysosome. (d) The microorganism is killed and digested. Ab, Antibody; AbR, antibody receptor; Ag, antigen; C3b, complement component C3b; C3bR, complement C3b receptor; PAMP, pathogen-associated molecular pattern; PRR, pattern recognition receptor.
Three panels, A, B, and C, illustrate tissue damage, recognition and attachment, and phagocytosis, respectively. Top panel, tissue damage. An illustration shows a splinter punctured through the epithelium, causing a wound. A capillary with normal endothelium and a basement membrane contains white blood cells. Adherence margination and diapedesis of the blood cells is illustrated inside the capillary. 1. Neutrophils in the capillary. 2. Margination: cell moves closer to the endothelium. 3. Retracted intercellular junction: an open space between two endothelium cells. 4. Endothelial cell retraction: cell passe through the junction, chemotaxis. Bacterium from the site of the wound enters the connective tissue, which is combated by the white blood cells as a result of chemotactic gradient. Bottom left panel, recognition and attachment. An illustration shows a bacterium. Ligands and receptors binding to the ligands around the bacterium are as follows: • A g: A b R through A b. • C 3 b: C 3 b R. • P A M P: P R R. Bottom right panel, phagocytosis. An illustration shows bacterium being engulfed into the cell. The following structures in the cell are identified. • Bacteria opsonized by A b and C 3 b. • Phagosome. • Phagolysosome. • Phagocyte. • Lysosome.
Once inside the connective tissue in the perivascular space, phagocytes migrate to the inflammatory site by means of chemotaxis. They detect chemotactic factors (chemokines) in the environment through chemoreceptors on their plasma membranes and migrate in the direction of highest concentration (see Fig. 7.8A). The most important chemotactic factors include bacterial products, complement fragments C3a and C5a, and chemokines. Neutrophils also respond to chemotactic factors released from mast cells (NCF). Monocyte-derived macrophages are attracted to monocyte chemotactic factor (MCF) that has already been released by neutrophils at the site of injury.
Phagocytosis at the inflammatory site involves four steps (see Fig. 7.8C):
Most phagocytes can trap and engulf pathogens using their PRRs which recognize PAMPs on the microorganisms (see Fig. 7.8B). However, some pathogens such as encapsulated bacteria (see Chapter 10) are more difficult to engulf. Opsonization greatly enhances adherence of the phagocyte to the target microorganism or cell. Opsonins that coat the target bacteria or cell act as a “glue” tightening the affinity between the phagocyte and the target, making phagocytosis more effective. The most efficient opsonins are C3b of the complement system and antibodies produced during the adaptive immune response.
Engulfment (endocytosis) is a process whereby the microorganism is drawn into the interior of a phagocytic cell. Engulfment is carried out by small pseudopods that extend from the plasma membrane of the phagocyte to surround the target, forming an intracellular phagocytic vacuole, or phagosome. Upon formation of a phagosome, the lysosomes (containing destructive enzymes) converge, fuse with the phagosome, and discharge their contents to create a phagolysosome. Destruction of the target takes place within the phagolysosome. Destruction is accomplished by both oxygen-dependent and oxygen-independent mechanisms.
Oxygen-dependent killing mechanisms result from the production of toxic oxygen species (e.g., hydrogen peroxide, singlet oxygen, or hydroxyl radicals). Phagocytosis is accompanied by a burst of oxygen uptake by the phagocyte. Chemical mediators and other reactive oxygen species are highly damaging to bacteria.
Oxygen-independent mechanisms of microbial killing include (1) the acidic pH (3.5 to 4.0) of the phagolysosome, (2) cationic proteins that bind to and damage target cell membranes, (3) enzymatic attack of the microorganism's cell wall by lysozyme and other enzymes, and (4) inhibition of bacterial growth by lactoferrin binding of iron (iron is essential for bacterial growth). Throughout the process, these toxic substances are isolated within membrane-bound vesicles. This isolation protects the phagocyte from harm secondary to the phagocyte's own enzymes.
Several bacteria are resistant to destruction by phagocytes and can survive inside macrophages. Microorganisms, such as Mycobacterium tuberculosis (tuberculosis), Mycobacterium leprae (leprosy), Salmonella typhi (typhoid fever), Brucella abortus (brucellosis), and Listeria monocytogenes (listeriosis), can remain dormant or multiply inside the phagolysosomes of macrophages.31
In addition to removing pathogens and damaged cells, phagocytes (especially macrophages) are responsible for removing cells that have completed their normal life cycle. For example, white blood cells and erythrocytes are programmed to live for only weeks or months. These dead cells number in the billions per day and must be recognized and removed from the body via phagocytosis.32 Phagocytosis of a red blood cell is illustrated in Fig. 7.9.
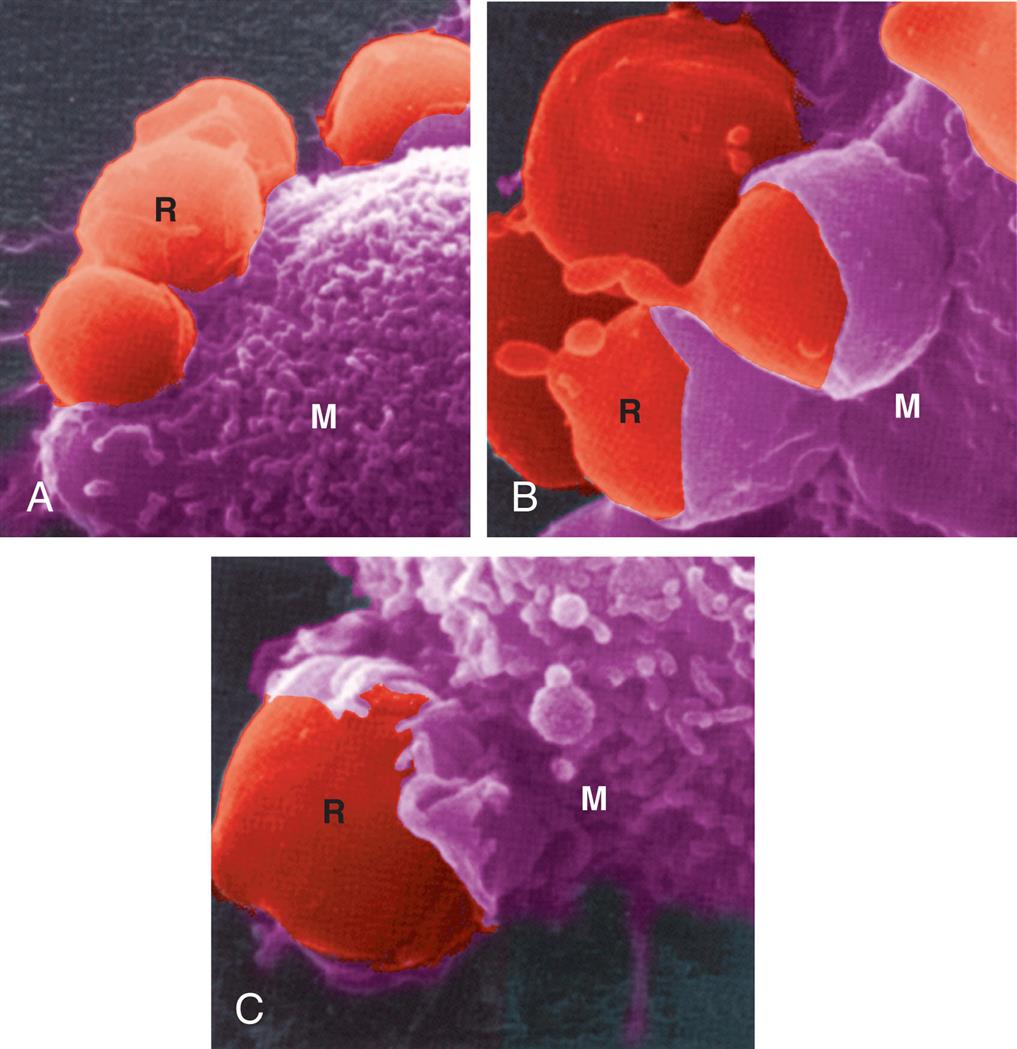
This scanning electron micrograph shows the progressive steps in phagocytosis. (A) Red blood cells (R) attach to the surface of a macrophage (M). (B) Part of the macrophage (M) membrane starts to enclose the red cell (R). (C) The red blood cells are almost totally engulfed by the macrophage. (From King DW, Fenoglio CM, Lefwitch JH. General pathology: Principles and dynamics. Philadelphia: Lea & Febiger; 1983.)
Dying phagocytes can cause tissue damage at the site of inflammation. When a phagocyte dies at an inflammatory site, it frequently lyses (breaks open), releasing its enzymatic contents into the tissue. These enzymes can cause inflammation-associated tissue destruction. The destructive effects of enzymes released by dying phagocytes are minimized by natural inhibitors found in blood. Examples of such inhibitors would include catalase, which breaks down hydrogen peroxide, and alpha1-antitrypsin (α1-antitrypsin), a protease inhibitor produced by the liver. An inherited deficiency of α1-antitrypsin, a disease known as alpha-1 antitrypsin deficiency, results in pulmonary emphysema secondary to chronic lung inflammation, even in nonsmokers (see Chapter 35).33
Other Cells of Inflammation
Another type of innate immune cell is the eosinophil. Although eosinophils are only mildly phagocytic, they serve as the body's primary defense against parasites and regulate vascular mediators released from mast cells. Lysosomal granules within the eosinophil contain enzymes that degrade vasoactive substances from mast cells, thus limiting and controlling the vascular effects of inflammation. Eosinophils also are important components of allergic conditions and can contribute to tissue damage; these effects are discussed in Chapter 9.
The basophil is the least prevalent granulocyte in the blood. It is similar to mast cells with respect to the contents of its granules. Basophilic granules also contain heparin, a naturally occurring anticoagulation product. Basophils release histamine which, as discussed previously, has potent vasoactive properties. Basophils are an important source of cytokines involved in the adaptive immune response, particularly responses associated with allergies and asthma.
Lymphocytes, another type of leukocyte, initiate specific, protective immune responses against pathogens and cancer. B lymphocytes produce antibodies, and T lymphocytes regulate other immune cells and kill viruses and cancer cells (see Chapter 8). Natural killer (NK) cells, a type of lymphocyte, eliminate virally infected and cancerous cells. NK cells have inhibitory and activating receptors that allow differentiation between infected or tumor cells and normal cells. If the NK cell binds to a target cell through activating receptors, it produces several cytokines and toxic molecules, which, in turn, kill the target cell. Innate lymphoid cells (ILCs) also are derived from lymphocyte precursors that reside near mucosal surfaces. There are three major groups of ILCs (ILC1s, ILC2s, and ILC3s) which contribute in different ways to preventing pathogen invasion and activating both the innate and adaptive immune responses (see Chapter 8).34
Acute and Chronic Inflammation
Inflammation can be divided into two phases: acute inflammation and chronic inflammation (Algorithm 7.3). The acute inflammatory response is self-limiting; it continues only until the threat to the body is eliminated. The process usually takes 8 to 10 days from onset of injury to healing. If the acute inflammatory response proves inadequate or does not resolve appropriately, chronic inflammation may develop and persist for weeks or months. The characteristics of the acute inflammatory response differ from those of a chronic response. Each phase involves different biochemical mediators and cells, all of which function in a coordinated manner. Depending on the successful containment of tissue damage and infection, both the acute and chronic phases may eventually lead to healing.
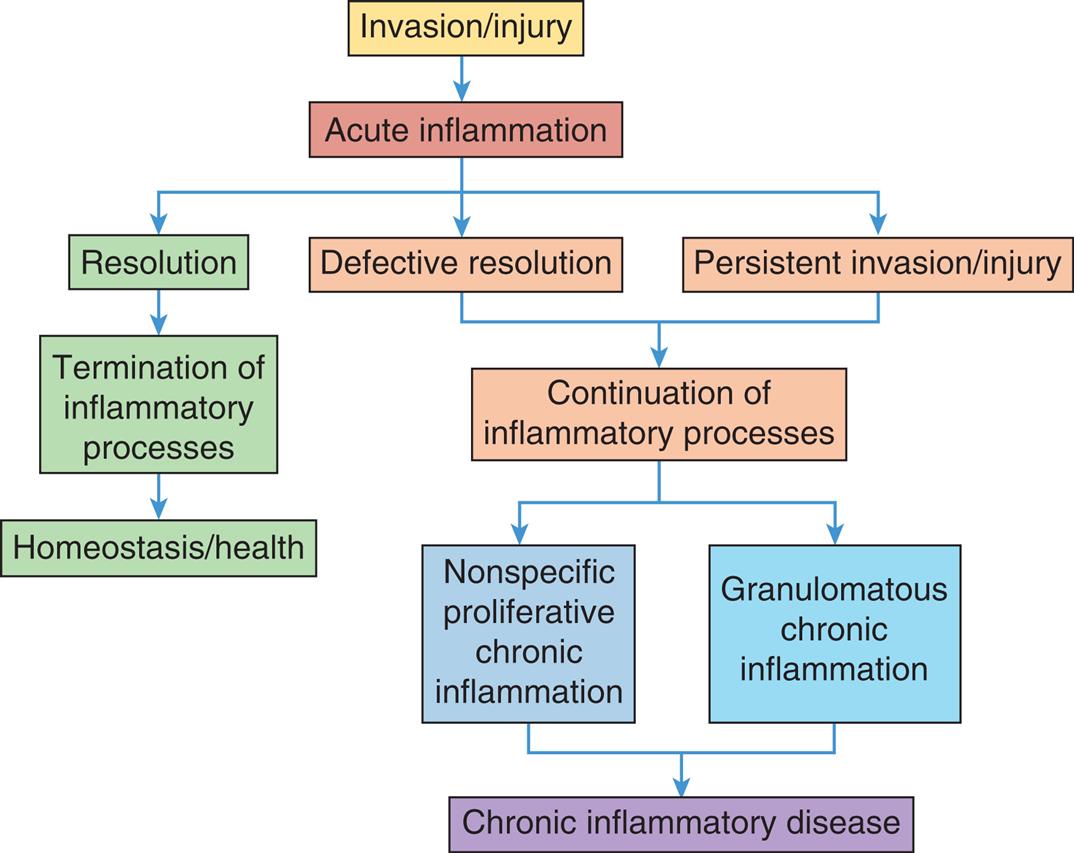
The acute inflammatory response to invasion or injury usually leads to resolution of the inciting event, reduction of inflammatory cytokines and cells, and restoration of tissue homeostasis and health. However, defects in the resolution process or persistent invasion and injury lead to chronic inflammatory damage to tissues and resultant disease.
A flowchart represents the chronic inflammatory response. The data from the flowchart are as follows from top to bottom. • Invasion or injury leads to acute inflammation. • Acute inflammation leads to resolution, defective resolution, and persistent invasion or injury. • Resolution leads to termination of inflammatory processes, which, in turn, leads to homeostasis or health. • Defective resolution and persistent invasion or injury leads to continuation of inflammatory processes. • Continuation of inflammatory processes leads to nonspecific proliferative chronic inflammation and granulomatous chronic inflammation, which, in turn finally leads to chronic inflammatory disease.
Manifestations of Local and Systemic Acute Inflammation
The cells and plasma protein systems involved in the inflammatory response interact to produce the characteristic changes of inflammation, whether local or systemic. They also determine the duration of inflammation, either acute or chronic.
The classic or cardinal signs of the acute local inflammatory response were described in the first century by a Roman named Celsus. They include the following:
Inflammatory exudates result from increased vascular permeability and the leakage of fluid into tissues. Exudates vary in composition, depending on the stage of inflammation, and, to a lesser extent, the triggering event. In early or mild inflammation, the exudate may be a serous exudate (watery) with very few plasma proteins or leukocytes (e.g., fluid in a blister). In more severe or advanced inflammation, the exudate may be a fibrinous exudate (thick and clotted) (e.g., the fluid exudates in the lungs of an individual with pericarditis). A purulent exudate (pus) is the accumulation of a large number of leukocytes, as occurs in bacterial infections (e.g., sputum in persons with pneumonia). A purulent exudate can also occur within walled-off lesions, known as cysts or abscesses. When bleeding occurs, the exudate is filled with erythrocytes and is described as a hemorrhagic exudate.
Systemic manifestations of acute inflammation include three primary systemic changes: fever, leukocytosis (a transient increase in the levels of circulating leukocytes), and increased levels of circulating plasma proteins. It is important to realize that individuals who are immunocompromised (e.g., those taking corticosteroids, chemotherapy agents, and antirejection medications) may not demonstrate these clinical manifestations, making it difficult to recognize acute infection or tissue injury.
Fever
Fever is partially induced by specific cytokines (e.g., TNF-α and IL-1) released from neutrophils and macrophages. These cytokines are known as endogenous pyrogens to differentiate them from pathogen-produced exogenous pyrogens (e.g., endotoxins produced by some bacteria). Pyrogens act directly on the hypothalamus, the portion of the brain which controls the body's thermostat. A fever can be beneficial, as many microorganisms are sensitive to small increases in body temperature. For example, the microorganisms causing syphilis or gonococcal urethritis are highly sensitive to small increases in body temperature. However, fever may have harmful side effects when it enhances the body's susceptibility to endotoxins associated with severe bacterial infections.
Leukocytosis
Leukocytosis is an increase in the number of circulating white blood cells beyond the upper limit of normal (11,000/mL3 in adults) (see Chapter 29). Inflammation stimulates the proliferation and release of granulocyte and monocyte precursors in bone marrow. The cells migrate into the systemic circulation resulting in both an increase in the total number of circulating leukocytes and an increased ratio of immature to mature cell forms. In particular, increased numbers of immature neutrophil forms such as band cells, metamyelocytes, and (rarely) myelocytes are released from the marrow into blood causing an abnormal complete blood count (CBC), a common laboratory analysis of blood cells. The differential count is a measure of the ratio or proportion of each of the various blood cell types circulating in the blood. An increase in the total number of white blood cells in combination with an abnormal differential count is a clue that there may be an ongoing infection.
Plasma Protein Synthesis
The hepatic synthesis of many plasma proteins is increased during inflammation. These proteins, which can be either pro-inflammatory or anti-inflammatory in nature, are referred to as acute-phase reactants (proteins). Acute-phase reactants reach maximal circulating levels within 10 to 40 hours after the onset of inflammation.
Laboratory tests that measure levels of acute-phase reactants are available. For example, an increase in blood levels of the acute-phase reactants fibrinogen is associated with an increased adhesiveness of erythrocytes. Erythrocytes adhere to one another under these circumstances, forming large clumps that are buoyant and slow to settle to the bottom of a test tube of blood. This results in an increased value for the laboratory test referred to as erythrocyte sedimentation rate (ESR) or, as it is more commonly known, the “sed rate.” Although this increased ESR is a nonspecific reaction, persons found to have an elevated ESR will likely have an inflammatory process going on somewhere within the body. Another common laboratory measure of inflammation is the C-reactive protein (CRP), a laboratory measurement which also is increased during an inflammatory response. CRP is used to look for subclinical inflammation in individuals at risk for heart disease and to estimate the severity of certain infectious and autoimmune diseases.35
Chronic Inflammation
In its simplest terms, the difference between acute and chronic inflammation is duration. Chronic inflammation is slow, long-term inflammation lasting for prolonged periods of several months to years. It is characterized by low-grade persistent inflammatory changes that result in collateral damage to tissues.36 Virtually all chronic diseases are characterized by a component of chronic inflammation. Although the cause of defective resolution and a persistent inflammatory response is often unknown, some common conditions associated with chronic inflammation include increasing age, cardiac and neurologic disorders, malignancy, autoimmune disorders, and chronic infection.
Chronic inflammation is most often thought to be preceded by an unsuccessful acute inflammatory response, although that initial insult may not have been recognized (see Algorithm 7.3). For example, if bacterial contamination or foreign objects (dirt, wood splinter, silica, glass, etc.) persist in a wound, the inflammatory response will be prolonged. Some microorganisms are resistant to killing by the acute inflammatory response or may produce toxins which damage tissue and cause persistent inflammation, even after the original microorganism is killed. Chronic inflammation also can occur as a result of defective resolution of acute inflammation, due to an inability to appropriately dampen the innate immune response.37 Trained immunity in which innate immune cells remain hyperreactive to inflammatory stimuli may also play a role (see Emerging Science Box: Understanding the Causes of Chronic Inflammatory Disease).38
There are two major types of chronic inflammation (Fig. 7.10). Nonspecific proliferative chronic inflammation is characterized by the infiltration of lymphocytes and macrophages followed by a proliferation of fibroblasts, connective tissue, and epithelial cells. This often leads to scar or polyp formation. Granulomatous chronic inflammation occurs when macrophages and eosinophils attempt to wall off and isolate a foreign body or an infected area by forming a granuloma (Fig. 7.11). Infections caused by certain bacteria (tuberculosis, listeriosis, brucellosis), fungi (histoplasmosis, coccidioidomycosis), and parasites (leishmaniasis, schistosomiasis, toxoplasmosis) commonly result in granuloma formation. Many autoimmune conditions also are associated with granulomatous tissue changes.39
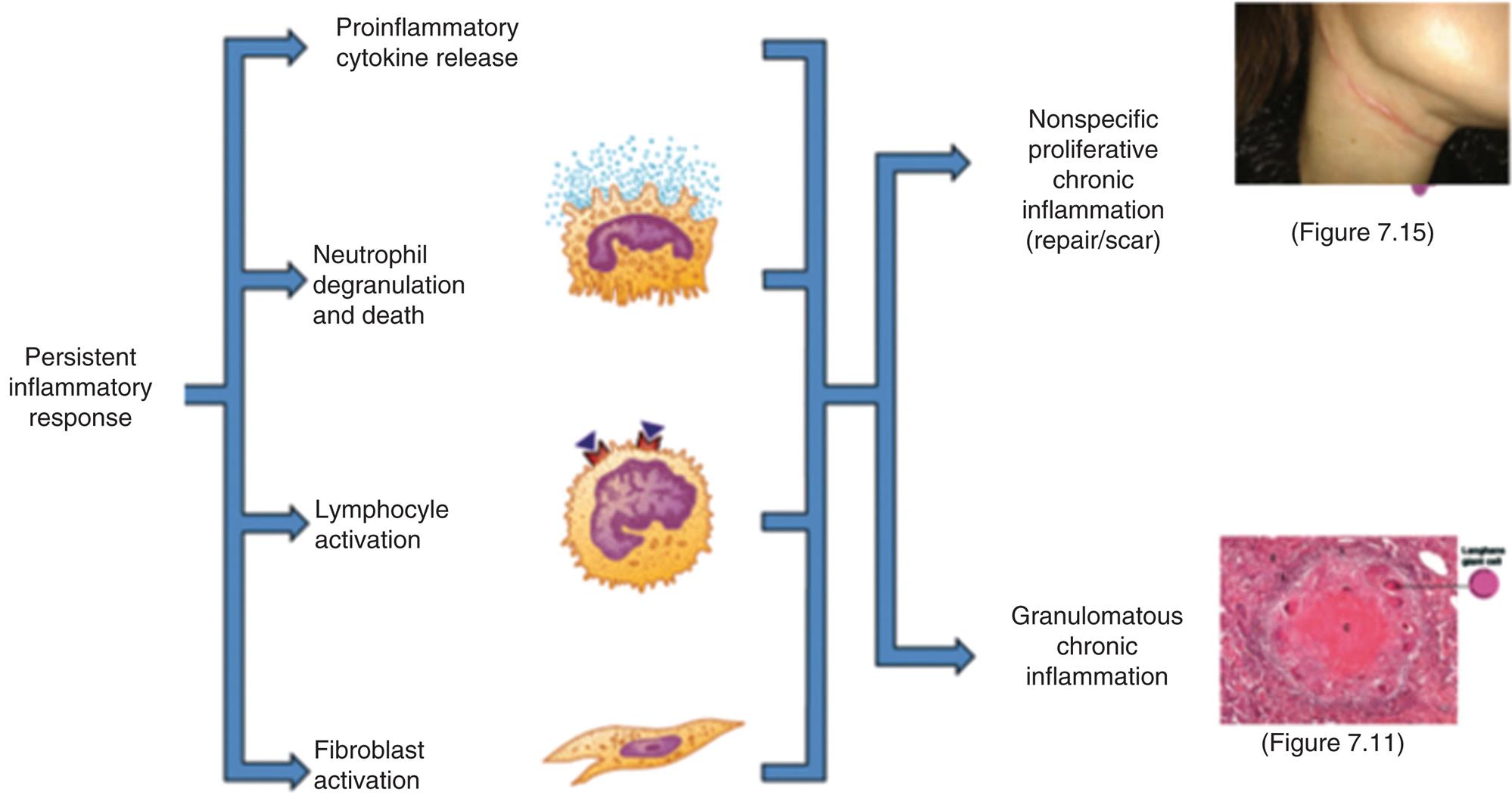
Chronic inflammation is characterized by the persistence of many of the processes of acute inflammation. In addition, large amounts of neutrophil degranulation and death, the activation of lymphocytes, and the concurrent activation of fibroblasts result in the release of mediators that induce the infiltration of more lymphocytes and monocytes/macrophages leading to two types of chronic inflammatory outcomes.
An illustration identifies the types of chronic inflammation. Persistent inflammatory responses: • Proinflammatory cytokine release. • Neutrophil degranulation and death. • Lymphocyte activation. • Fibroblast activation. All responses lead to nonspecific proliferative chronic inflammation (repair or scar) and granulomatous chronic inflammation. A closeup of a hypertrophic scar shows a thin line of raised fibrous tissue running around the upper throat of a person. A photomicrograph of cells affected by tuberculous granuloma shows a smooth central area (amorphous caseous necrosis) and some pits indicating langhans giant cells, surrounded by epithelioid macrophage cells and lymphocytes.
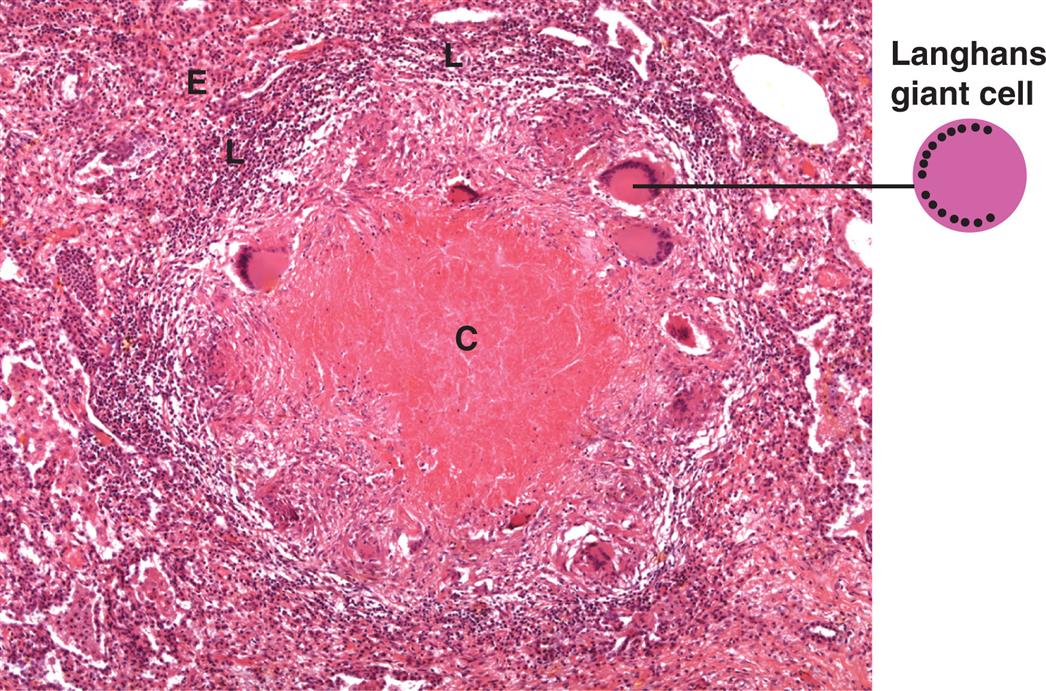
A central area of amorphous caseous necrosis (C) is surrounded by a zone of lymphocytes (L) and enlarged epithelioid macrophage cells (E). Activated macrophages frequently fuse to form multinucleated cells (Langerhans giant cells). In tuberculoid granulomas the nuclei of the giant cells move to the cellular margins in a horseshoe-like formation. (From Kumar V, Abbas AK, Aster JC. Robbins basic pathology, 9th edition. Philadelphia: Saunders; 2013.)
A photomicrograph of cells affected by tuberculous granuloma shows a smooth central area (amorphous caseous necrosis) and some pits indicating langhans giant cells, surrounded by epithelioid macrophage cells and lymphocytes.
Granuloma formation is mediated by TNF-α and other proinflammatory cytokines. Macrophages are called to the area of persistent inflammation, where some differentiate into large epithelioid cells, which specialize in taking up debris and other small particles. Other macrophages fuse into multinucleated giant cells, which are active phagocytes, which can engulf particles larger than those that can be engulfed by a single
macrophage. These two types of specialized cells form the center of the granuloma and are surrounded by lymphocytes. The granuloma itself is often encapsulated by fibrous deposits of collagen. It may become cartilaginous or even calcified by deposits of calcium carbonate and calcium phosphate.
The classic granuloma associated with tuberculosis is characterized by a wall of lymphocytes and epithelioid cells surrounding the mycobacteria. Death of cells within the granuloma results in the release of acids and enzymatic lysosomal contents from dead phagocytes forming a cheese-like proteinaceous center which consists of decaying tissue (caseous necrosis). In this inhospitable environment, the cellular debris is broken down into its basic constituents, forming a clear fluid within the granuloma (liquefaction necrosis). Eventually, this fluid diffuses out, leaving a hollow, thick-walled structure that has replaced normal tissue, resulting in the reduced lung function seen with tuberculosis infections.
Wound Healing
The most favorable outcome of inflammation is healing with a return to normal structure and function. Some destroyed tissues are capable of regeneration, a process where damaged tissue is replaced with healthy tissue of the original type. Regeneration occurs within the epithelia of the skin and intestines as well as in some organs, notably the liver. This restoration is an example of effective resolution of the acute inflammatory response.
Regeneration does not always follow inflammation and may not be possible when extensive damage has occurred and at sites where the original tissue is not capable of complete restoration of normal tissue structure. Where resolution is not possible, tissue repair occurs. Repair refers to the replacement of destroyed tissue with scar tissue. Scar tissue is composed primarily of collagen, a substance which fills in the lesion and restores tissue integrity and strength but cannot carry out the physiologic functions of the tissue it has replaced, resulting in loss of function.
A clean incision, such as a paper cut or a sutured surgical wound, heals primarily through the process of primary intention. Because this type of wound has minimal tissue loss and close apposition of the wound edges, healing can occur quickly (Fig. 7.12).
Phases of wound healing (coagulation, inflammation, proliferation/tissue formation, remodeling, and maturation) and steps in wound healing by primary intention (left) and secondary intention (right). Note the large amounts of granulation tissue and wound contraction in healing by secondary intention. (From Roberts JR, Custalow CB. Roberts and Hedges’ clinical procedures in emergency medicine, 6th edition. Philadelphia: Elsevier/Saunders; 2013.)
Three pairs of illustrations show wound healing by primary and secondary intention by stage. Top pair of illustrations, 24 hours. There are two stages of healing. 1, hemostasis. Platelets surround the fibrin clot. 2, Inflammation. Neutrophils bind to the site of injury in healing by primary intention and the necrotic tissue in healing by secondary intention. Middle pair of illustrations, 3 days to 14 days. There is one stage of healing, proliferation. It involves epithelial mitosis, granulation tissue, macrophage, lymphocyte, fibroblast, and new capillary. Bottom pair of illustrations, weeks to months. There is one stage of healing, remodeling maturation. It involves reepithelialization and fibrous union (scar). In healing by secondary intention, the wound contracts.
Other wounds do not heal as readily. Healing of an open wound, such as a severe burn, requires a great deal of tissue replacement. In these cases, healing occurs through secondary intention (see Fig. 7.12). Healing by either primary or secondary intention may occur at different rates for different types of tissue injury.
Epidermal wounds that heal by secondary intention are not completely restored by healing. At best, repaired tissue regains 80% of its original tensile strength. In fibrous connective tissue, such as joints and ligaments, normal healing results in replacement of the original tissue with new tissue which does not exactly replicate the structure or function of the original. Some tissues heal without replacement of any functional cells as those cells are not capable of replication. For example, damage resulting from myocardial infarction heals with fibrous scar tissue rather than with cardiac muscle. Accordingly, cardiac function is not restored completely and the individual surviving the infarction will have varying amounts of impaired myocardial contractility and cardiac function depending on the size of the scar.
Wound healing occurs in four overlapping phases or stages: (1) hemostasis (coagulation), (2) inflammation, (3) proliferation with new tissue formation, and (4) remodeling and maturation (Fig. 7.13).
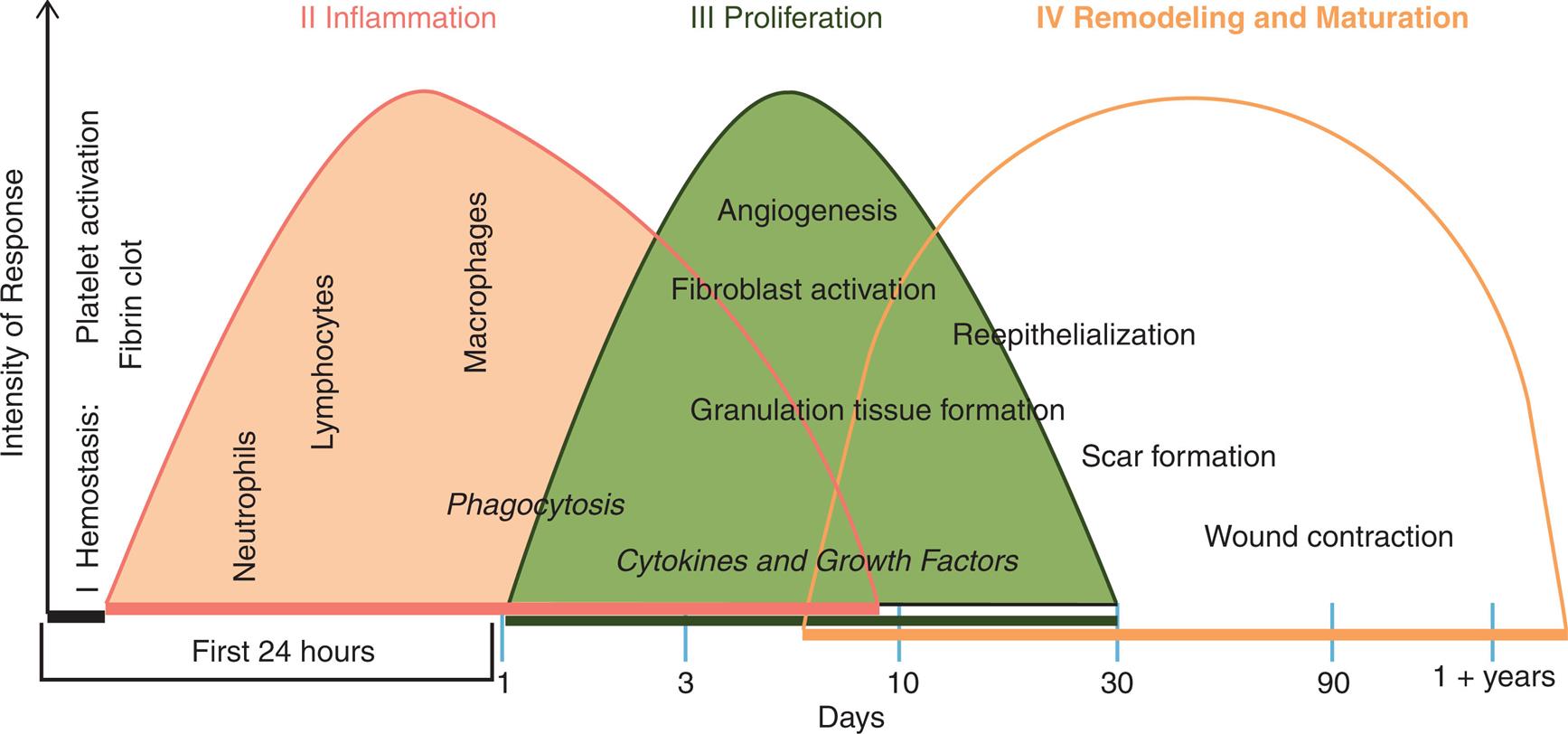
A graph plotting intensity of response over time period shows three curves representing the three phases of wound healing. The horizontal axis represents days: 1, 3, 10, 30, 90, and 1 plus years. There are four phases of wound healing. Phase 1, hemostasis. Occurs immediately and includes platelet activation and fibrin clot. Phase 2, inflammation or phagocytosis. The curve is very gently skewed to the left and spans about 9 days. It peaks through neutrophils, lymphocytes, and macrophages. Phase 3, proliferation or cytokines and growth factors. The curve is normal and spans between 1 day and 30 days. It involves angiogenesis, fibroblast activation, and granulation tissue formation. Phase 4, remodeling and maturation. The curve is wide with a broad peak and spans between 6 days and 1 plus years. It involves reepithelialization, scar formation, and wound contraction.
Phase I: Hemostasis (Coagulation)
Tissue injury damages capillaries and blood vessels and causes bleeding into the wound. Damage to blood vessels causes immediate vasoconstriction followed by vasodilation and initiates the coagulation cascade and activation of platelets. The fibrin mesh of the blood clot acts as a scaffold for cells that participate in healing. Platelets contribute to clot formation. As the platelets degranulate, they release factors which cause increased capillary permeability and promote growth factors which initiate proliferation in undamaged cells. In addition to activation of clotting, there is activation of complement and kinins.
Phase II: Inflammation
The inflammatory phase begins within minutes. As has been described in detail previously, macrophages and mast cells release numerous vasoactive cytokines that increase blood flow to the wound and bring needed cells and proteins to the area of injury. DAMPs and chemokines released by injured cells recruit and activate inflammatory cells, especially neutrophils.40 Neutrophils infiltrate the area of injury and initiate the process of clearing the wound of debris and bacteria. Lymphocytes initiate an adaptive immune response.
Phase III: Proliferation and New Tissue Formation
The proliferative phase begins 3 to 4 days after the injury and continues for as long as 2 weeks. The wound is sealed, and the fibrin clot is replaced by normal tissue or scar tissue during this phase. The proliferative phase is characterized by an invasion of macrophages. During this phase, both monocyte-derived and tissue macrophages are stimulated by a variety of cytokines to become M2 macrophages.41 They clear debris, release numerous growth factors, recruit fibroblasts, and promote collagen synthesis and angiogenesis. Angiogenesis describes the process of the formation of new blood vessels and is one of the most important steps in wound healing. When blood vessels are injured, growth factors such as VGEF and PDGF secreted by M2 macrophages cause endothelial cells to sprout new capillaries. This brings nutrients and oxygen to the healing wound.40
The cleanup of the lesion, which also involves dissolution of fibrin clots (or scabs), is called debridement. After debridement, the remaining debris is drained away by blood and lymphatic vessels and the vascular dilation and permeability associated with early inflammation are reversed, thus preparing the lesion for either regeneration or repair. An important role for macrophages during this phase is the engulfment of neutrophils (efferocytosis) to clear them and promote resolution of inflammation.40
As wound healing progresses, granulation tissue grows into the wound from surrounding healthy connective tissue. It consists of invasive cells, new lymphatic vessels, and new capillaries, all derived from the analogous structures found in the surrounding healthy tissue. These components lend a red and granular appearance to the newly formed granulation tissue. Capillary buds sprout from vascular endothelial cells around the wound and extend into the débrided areas. Loops form when the young capillaries join (anastomose). The loops are more fragile and permeable than mature vessels, resulting in leakage of erythrocytes and neutrophils. The erythrocytes are phagocytosed by macrophages, and the neutrophils assist in further debridement of the inflammatory lesion. Many of the new capillaries differentiate into larger vessels as repair continues, promoting influx of nutrients and removal of metabolic wastes. New lymphatic vessels also grow into the granulation tissue by a similar process.
Epithelialization is the process by which epithelial stem cells proliferate and grow into the wound from surrounding healthy tissue. Epithelial cells migrate under the clot or scab using matrix metalloproteinases to unravel the collagen. The migrating epithelial cells connect with similar cells from all sides of the wound and seal it. The epithelial cells remain active, undergoing differentiation to give rise to the various epidermal layers. Epithelialization of a skin wound can be hastened if the wound is kept moist, preventing the fibrin clot from becoming a scab.
Fibroblasts are important cells during healing because they secrete collagen and other connective tissue proteins. Fibroblasts deposit connective tissue proteins in debrided areas, about 6 days after they have entered a lesion. Collagen is the most abundant protein in the body. It contains high concentrations of the amino acids glycine, proline, and lysine, many of which are enzymatically modified during the healing process. Additionally, wound healing requires iron, ascorbic acid (vitamin C), and molecular oxygen (O2). The absence of any of these components results in impaired wound healing. As healing progresses, collagen molecules form collagen fibrils which further modify to form collagen fibers. The complete process takes several months.
In granulation tissue, some fibroblasts transition into myofibroblasts, specialized cells responsible for wound contraction. Myofibroblasts have features of both smooth muscle cells and fibroblasts. Wound contraction occurs as extensions from the plasma membrane of myofibroblasts establish connections between neighboring cells. They anchor themselves to the wound bed then contract their fibers so as to exert tension on the neighboring cells. Wound contraction is necessary for closure of all wounds, especially those that heal by secondary intention. Contraction is noticeable 6 to 12 days after injury.
Phase IV: Remodeling and Maturation
Tissue remodeling and maturation are a process that begins several weeks after injury. Tissue regeneration and wound contraction continue in this phase during which there is a continuation of cellular differentiation, scar formation, and scar remodeling. The fibroblast is the major cell involved in tissue remodeling, where it functions to deposit collagen into an organized matrix. For wounds that heal by scarring, scar tissue is remodeled, and capillaries disappear, resulting in an avascular scar. Within 2 to 3 weeks after maturation has begun, the scar tissue has gained about two-thirds of its eventual maximal strength, which may be less than the strength prior to injury.
Dysfunctional Wound Healing
Dysfunctional wound healing and impaired epithelialization may occur during any phase of the healing process. The causes of dysfunctional wound healing include ischemia; excessive bleeding; excessive fibrin deposition; a predisposing disorder, such as diabetes mellitus; obesity; wound infection; inadequate nutrition; use of certain drugs; and tobacco smoking as well as history of keloid formation.
Ischemic (oxygen-deprived) tissue is susceptible to cellular death and infection, which prolongs inflammation and delays healing. Ischemia (both acute following injury as well as chronic) reduces energy production and impairs collagen synthesis as well as the tensile strength of regenerating connective tissue.
Excessive bleeding delays healing. Large clots increase the amount of space that granulation tissue must fill. Clots also serve as mechanical barriers to oxygen diffusion further amplified by tissue ischemia. Accumulated blood is an excellent growth medium for bacteria. Bacteria predispose the wound to infection and prolong inflammation through increased exudation and pus formation. Under normal conditions, vessels dilate, delivering inflammatory cells, nutrients, and oxygen to the site of injury. Decreased blood volume, associated with bleeding, inhibits inflammation secondary to vessel constriction.
Excessive fibrin deposition is detrimental to healing. Fibrin released in response to injury must eventually be reabsorbed to prevent the formation of fibrous adhesions. Adhesions that form in the pleural, pericardial, or abdominal cavities can bind together, forming fibrous bands. Such bands distort, impinge on, or strangulate the affected organs. An adhesion, so formed, may bind an organ to an adjacent organ resulting in both pain and/or impaired organ function (Fig. 7.14).
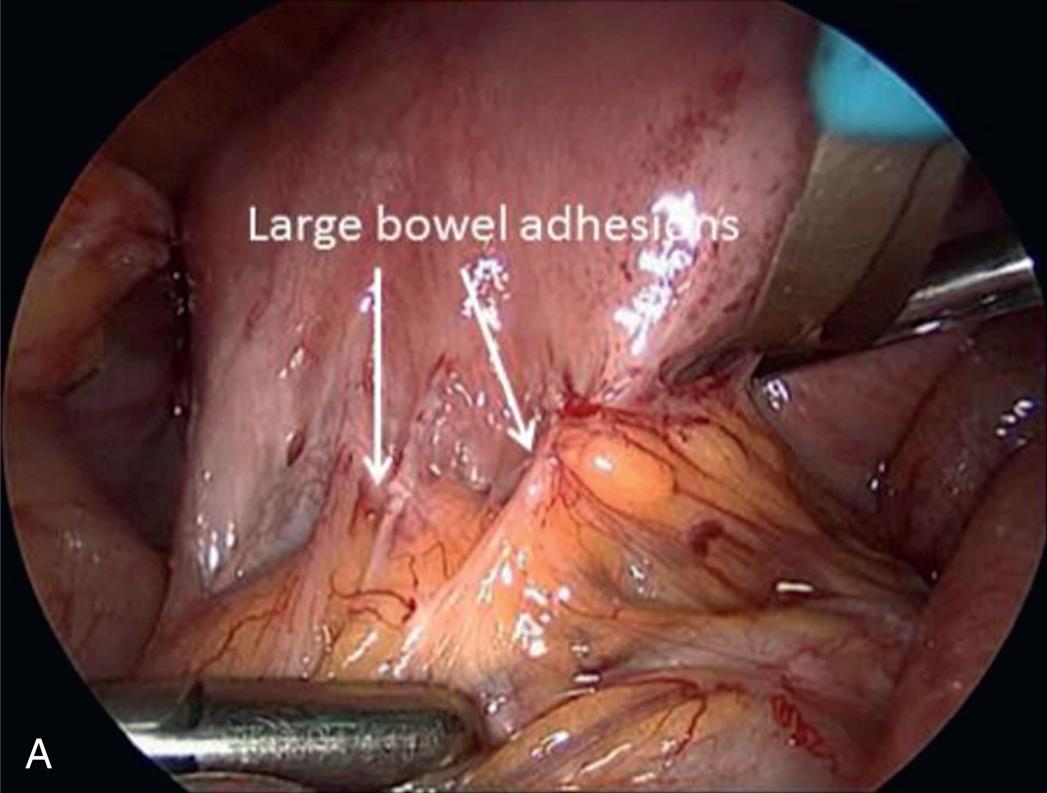
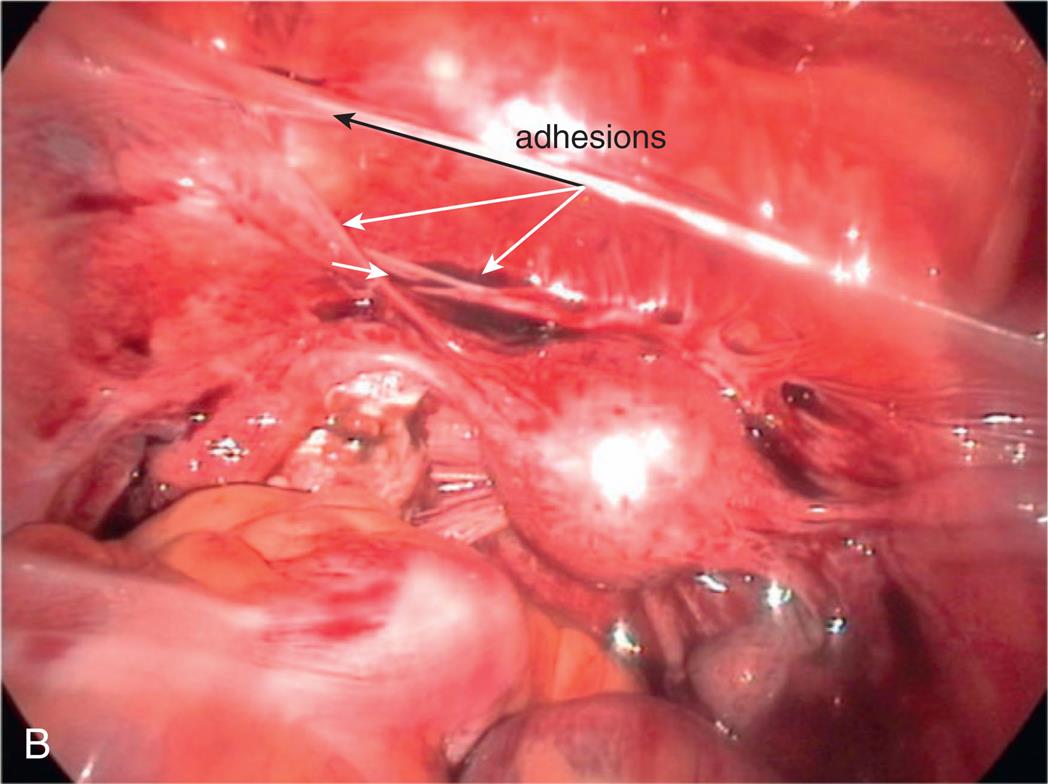
Closeup A shows the fused cells in large bowel adhesions. The fusion between the intestines and the abdominal wall causes the surrounding fibers and cells to stretch. The sites of large bowel adhesions are indicated by arrows. Closeup B shows the fused cells in uterine adhesion. Fibrous bands stretch between the uterine wall, the liver, and the abdominal wall. The sites of adhesions in the abdominal wall are indicated by arrows.
Obesity delays wound healing through impairing leukocyte function, which, in turn, predisposes the wound to infection, decreased growth factor production, and increases in proinflammatory cytokines. Additionally, there is a dysregulation of collagen synthesis and a decrease in angiogenesis.
Persons with diabetes are at risk for prolonged wound healing and wound infection, especially if they have persistent hyperglycemia. The causes of poor wound healing in diabetes are multifactorial and complex. Ischemia, hyperglycemia, infection, and decreased immune function all play a role 42 (see Emerging Science Box: Mechanisms of Impaired Wound Healing in Diabetes).
Wound infection is caused by the infiltration of pathogens into the injured or compromised tissues. Infection is promoted by the necrotic debris, low oxygen tension, and a decreased host immune response. Biofilms may form within the wound and prevent healing.43 Biofilms are accumulations of microbes that adhere to the wound surface and secrete a matrix of polysaccharides that protect the pathogens from the immune response and from antibiotics (see Chapter 10). Such pathogens damage cells, stimulate the continued release of inflammatory mediators, consume nutrients, and delay wound healing.43
Optimal nutrition is important during all phases of healing because of the increased metabolic needs of the affected tissues. Hypoproteinemia with decreased levels of amino acids, such as arginine and glutamine, impairs fibroblast proliferation and collagen synthesis.44 Prolonged lack of vitamins A and C, which are cofactors required for collagen synthesis, results in poorly formed connective tissue and significantly impaired healing. Other cofactors for collagen synthesis include iron, zinc, manganese, and copper. Accordingly, malnutrition results in an increased risk for wound infection, delayed healing, and reduced wound tensile strength.
Medications associated with delayed or dysfunctional wound healing include antineoplastic (anticancer) agents, NSAIDs, and steroids. Antineoplastic agents slow cell division and inhibit angiogenesis. NSAIDs suppress acute inflammation and inhibit prostaglandin production, resulting in delayed wound healing, especially healing involving bone tissue.45 Corticosteroids (e.g., prednisone) disrupt wound healing by preventing macrophages from migrating to the site of injury and through inhibiting the release of both collagenase and plasminogen activator. Steroids also inhibit fibroblast migration into the wound during the proliferative phase, thus delaying epithelialization.
Toxic agents in tobacco smoke (carbon monoxide and hydrogen cyanide) delay wound healing. Nicotine is a potent vasoconstrictor that compromises wound healing by predisposing the tissue to ischemia and infection.
Dysfunctional collagen synthesis may involve excessive production of collagen, leading to a hypertrophic scar or keloid. Myofibroblasts release growth factors and inflammatory cytokines and contribute to excessive tissue contraction and extracellular matrix deposition.46 A hypertrophic scar is raised but remains within the original boundaries of the wound. It tends to regress over time (Fig. 7.15A). A keloid is a raised scar that extends beyond the original boundaries of the wound, invading surrounding tissue. Keloids are fibrous lesions and are often characterized by pruritis, pain, and psychological discomfort.40 They are likely to recur after surgical removal and treatment is often ineffective (see Fig. 7.15B). A familial tendency to keloid formation has been observed, with a greater incidence in persons with darker skin pigmentation, especially African Americans.
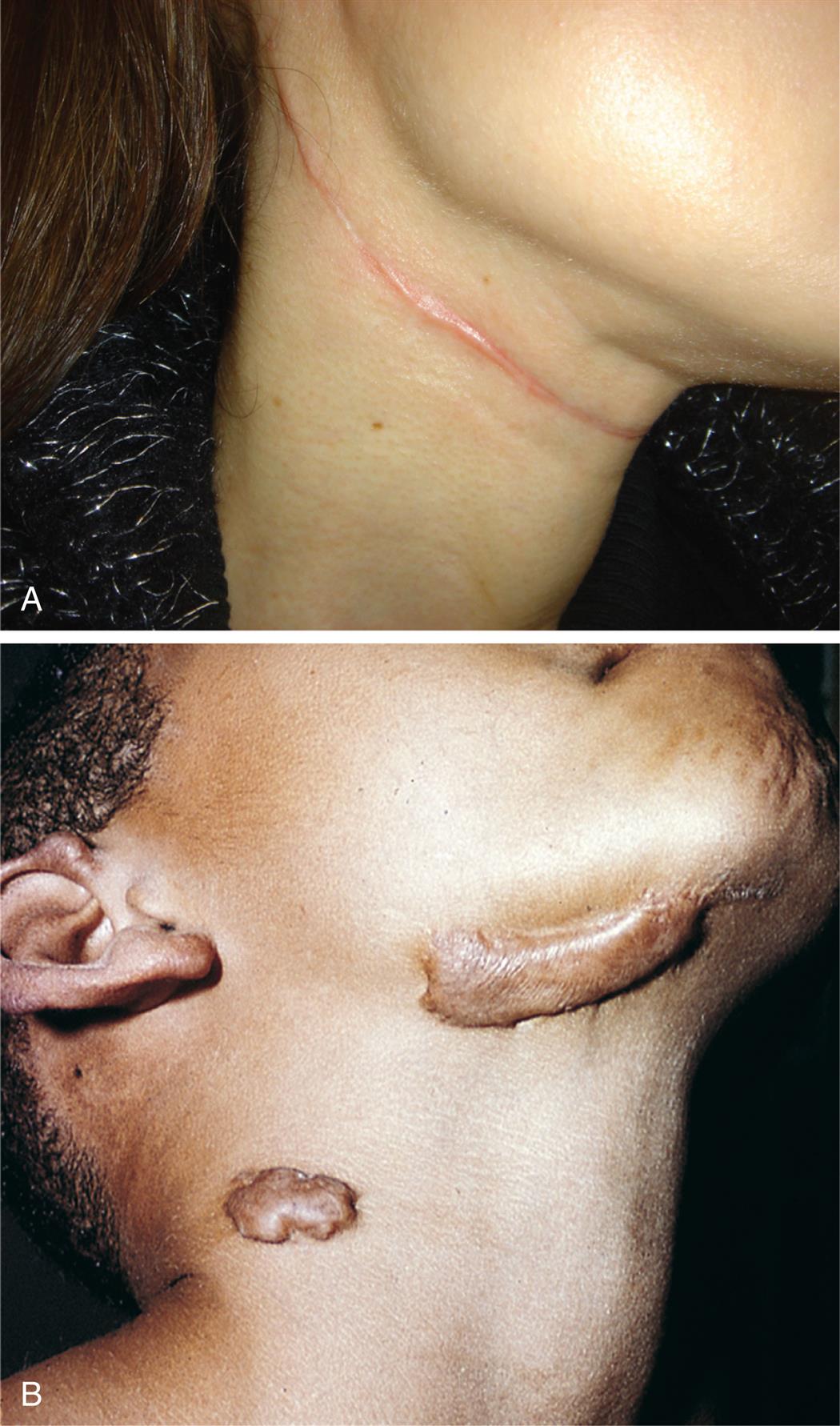
Hypertrophic scar (A) and keloid scar (B) caused by excessive synthesis of collagen at suture sites. (A, From Flint PW, Haughey B, Lund V, et al. Cummings otolaryngology: Head and neck surgery, 6th edition. Philadelphia: Mosby; 2015; B, From Damjanov I, Linder J. Anderson's pathology, 10th edition. St. Louis: Mosby; 1996.)
Closeup A is a hypertrophic scar that shows a thin line of raised fibrous tissue running around the upper throat of a person. Closeup B is a pair of keloid scars: one under a person’s chin and the other at the base of the person’s throat. Both scars are large, raised, discolored bumps on the skin surface.
Wound Disruption
Even after the healing process has occurred, wounds can be disrupted in a variety of ways that may reverse the healing process or negatively impact function. These complications are more likely if wound healing was dysfunctional.
Dehiscence
A potential complication of wounds that are closed with sutures is dehiscence, a scenario where the wound pulls apart at the suture line. Dehiscence generally occurs 5 to 12 days after suturing, at a time when collagen synthesis is at its peak. Approximately half of dehiscence occurrences are associated with wound infection. Other instances result from suture rupture caused by excessive strain. Adipose tissue is difficult to suture, increasing the risk of dehiscence in individuals who are obese. Wound dehiscence is usually heralded by increased serous drainage from the wound; there often is a perception by the individual that “something gave way.” Prompt surgical attention is required.
Contracture
Wound contraction, necessary for healing, may become excessive where it results in an anatomic deformity or contracture of scar tissue. Burns of the skin are especially prone to contracture, particularly at joints with a resultant loss of joint function. Internal contractures include duodenal strictures, caused by dysfunctional healing of a peptic ulcer; esophageal strictures, caused by chemical burns, such as lye ingestion; or abdominal adhesions, caused by surgery, infection, or radiation. Contracture may occur in cirrhosis of the liver, where constricted vascular flow contributes to the development of portal hypertension and esophageal varices. Proper positioning, range-of-motion exercises, compression, and surgery are among the strategies used to prevent or treat skin contractures. Internal contractures, such as those caused by fibrous adhesions between loops of bowel, cause pain or result in a loss of function and are released with surgery.
Summary Review
Human Defense Mechanisms
- 1. Innate defenses are the first line of defense, are present at birth, and include the surface barriers skin and mucous membranes.
- 2. Inflammation is the second line of defense and is activated with injury or infectious disease.
- 3. Adaptive (acquired) immunity is the third line of defense, is specific to particular antigens, and has memory.
Innate Immunity
- 1. There are three layers of human defense: physiologic barriers, the inflammatory response, and adaptive (acquired) immunity.
- 2. Physical barriers are the first lines of defense functioning to prevent damage to the individual and thwart the entrance of pathogens. These barriers include the skin and mucous membranes.
- 3. Antibacterial peptides are found in mucous secretions, perspiration, saliva, tears, and other secretions. They provide a biochemical barrier against pathogenic microorganisms.
- 4. The skin, mucous membranes, and the lining of the gastrointestinal (GI) tract are colonized by commensal or mutualistic microorganisms called the microbiome. These microorganisms provide protection by releasing biochemical compounds which facilitate immune responses and prevent colonization by pathogens. Within the gut, they also facilitate digestion in the GI tract.
- 5. The second line of defense is the inflammatory response, a rapid and nonspecific protective response to cellular injury resulting from any cause. It can occur only in vascularized tissues.
- 6. Inflammation is mediated by three key plasma protein systems: the complement system, the clotting system, and the kinin system. The components of all three systems are a series of inactive proteins which are activated sequentially in the presence of tissue injury.
- 7. The complement system can be activated by antigen–antibody reactions (through the classical pathway) or by other products, especially bacterial polysaccharides (through the lectin pathway or the alternative pathway). The lectin and alternative pathways do not require antibody activation to recruit phagocytes, activate mast cells, and destroy pathogens.
- 8. The most biologically potent products of the complement system are C3b (opsonin), C3a (anaphylatoxin), and C5a (anaphylatoxin, chemotactic factor).
- 9. The clotting system stops bleeding, localizes microorganisms, and provides a meshwork for repair and healing.
- 10. Bradykinin is the most important product of the kinin system and causes vascular permeability, smooth muscle contraction, and pain.
- 11. Control of inflammation regulates inflammatory cells and enzymes and localizes the inflammatory response to the area of injury or infection.
- 12. Carboxypeptidase, histaminase, kinase, and C1 inhibitor are inactivating enzymes. The fibrinolytic system and plasmin facilitate clot degradation after bleeding is stopped.
- 13. Mast cells and macrophages are the most important cells for initiating the inflammatory response.
- 14. These cells express plasma membrane pattern recognition receptors (PRRs) which recognize molecules produced by infectious microorganisms. These molecules include pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs). Toll-like receptors (TLRs) are transmembrane receptors and nucleotide-binding-like receptors (NLR-like), and nucleotide oligomerization domain-like (NOD-like) receptors are cytoplasmic receptors. They are expressed by many inflammatory cells and recognize both PAMPs and DAMPs. Upon recognition, they promote the release of cytokines and inflammatory mediators, which, in turn, eliminate damaged cells and protect against invasion by microbes.
- 15. Mast cells are near epithelial surfaces and capillaries. Mast cells initiate inflammation by releasing biochemical mediators (histamine and chemotactic factors) from cytoplasmic granules. They also synthesize other mediators (prostaglandins, leukotrienes, and platelet-activating factor [PAF]) in response to stimuli. Basophils, found in blood, function in a manner that is similar to mast cells.
- 16. Histamine is the major vasoactive amine released from mast cells. It increases vascular permeability through dilation of capillaries and retraction of endothelial cells lining the capillaries.
- 17. Tissue macrophages use PRRs to identify microorganisms and molecules from damaged tissue, and then secrete many biochemical mediators (cytokines), which are responsible for activating other cells and regulating the inflammatory response. These cytokines include TNF-α, interleukins, interferons, and other molecules.
- 18. TNF-α is produced when PRR binding sends intracellular messengers to the nucleus of the macrophage which activates NF-κB. TNF-α has multiple pro-inflammatory effects including vascular effects, chemotaxis, cellular proliferation, and systemic inflammatory changes.
- 19. Interleukins are produced primarily by lymphocytes and macrophages. They activate the growth and differentiation of leukocytes and contribute to systemic inflammatory changes such as fever.
- 20. The most important proinflammatory interleukins are interleukin-1 (IL-1) and interleukin-6.
- 21. Interferons are produced by cells that are infected by viruses. Once released from infected cells, interferons can stimulate neighboring healthy cells to produce substances that prevent viral infection.
- 22. There are also anti-inflammatory cytokines such as TGF-β and IL-10 which downregulate the inflammatory response.
- 23. The vascular responses to inflammation are vasodilation, increased capillary permeability, and an accumulation of fluid and cells at the inflammatory site.
- 24. The cellular response to inflammation includes neutrophils, monocyte-derived macrophages, and other inflammatory cells.
- 25. The endothelial cells lining the circulatory system (vascular endothelium) regulate circulating components of the inflammatory system, maintaining normal blood flow. During inflammation, the endothelium expresses receptors that stimulate leukocytes to exit the vessel. The endothelial cell body also retracts to allow fluid to pass into the tissues.
- 26. The polymorphonuclear neutrophil (PMN), the predominant phagocytic cell in the early inflammation, exits the circulation, through retracted endothelial junctions, by diapedesis. On exiting, it moves to the inflammatory site by chemotaxis.
- 27. The monocyte-derived macrophage, the predominant cell in the late inflammatory response, is highly phagocytic. Additionally, it is responsive to cytokines, which promote wound healing.
- 28. Phagocytosis is a multistep cellular process, which usually results in the destruction of pathogens and foreign debris. The steps include recognition and attachment, engulfment, formation of a phagosome, formation of a phagolysosome, and eventual destruction of the pathogen or foreign debris. Phagocytic cells engulf microorganisms, enclosing them within phagocytic vacuoles (phagolysosomes). The vacuoles contain toxins (especially metabolites of oxygen) and/or enzymes that kill and digest the microorganisms.
- 29. Opsonins are molecules which enhance phagocytosis by coating the antigen. This activity results in a stronger attraction between the microorganism and the phagocyte (“marking” the organism). It also enhances the affinity with which the phagocyte binds to the microorganism. Examples include antibodies and the complement component C3b.
- 30. Eosinophils release products that control the inflammatory response, and they are the principal cells that destroy parasitic organisms.
- 31. Other cells of innate immunity include dendritic cells which function as messengers between the innate and acquired (adaptive) immune systems, NK cells which detect certain invaders and cancer cells, and innate lymphoid cells which modulate many aspects of innate immunity.
Acute and Chronic Inflammation
- 1. Acute inflammation is self-limiting and usually resolves within 8 to 10 days.
- 2. Local manifestations of inflammation include the classic signs of redness, heat, swelling, pain, and loss of function. They are the result of vascular changes associated with the inflammatory process, including vasodilation and increased capillary permeability.
- 3. The principal systemic effects of inflammation are fever, leukocytosis (increased levels of circulating leukocytes), and an increase in plasma proteins, primarily the acute-phase reactants, IL-1, and IL-6.
- 4. Chronic inflammation is the persistence of the inflammatory response often contributing to tissue damage.
- 5. Chronic inflammation is characterized by a dense infiltration of lymphocytes and macrophages. It can take two forms, nonspecific proliferative chronic inflammation and granulomatous chronic inflammation.
- 6. Nonspecific proliferative chronic inflammation occurs when the acute inflammatory response fails to eliminate the invader/injury or there is dysfunctional resolution of the acute inflammatory response.
- 7. Granuloma formation is a process wherein the body walls off and isolates certain infectious microorganisms or foreign bodies that could not be removed by acute inflammation. It serves to protect the body from further tissue damage.
- 8. Both forms of chronic inflammation can contribute to tissue dysfunction and organ damage and are the major causes of chronic disease.
Wound Healing
- 1. Resolution (regeneration) is the return of tissue to nearly normal structure and function. Repair is healing by scar tissue formation.
- 2. Resolution occurs when little tissue has been lost or where the injured tissue is capable of regeneration. This type of healing is called healing by primary intention.
- 3. Tissues that have sustained extensive damage or tissue types that are incapable of regeneration heal by repair, a process which results in the formation of a scar. This process is called healing by secondary intention.
- 4. Wound healing occurs in four overlapping phases; hemostasis, inflammation, proliferation with new tissue formation, and remodeling or maturation. Each of these phases is characterized by the complex interaction of multiple cells including platelets, neutrophils, macrophages fibroblasts, endothelial cells, and epithelial cells.
- 5. Dysfunctional wound healing can be secondary to ischemia, excessive bleeding, excessive fibrin deposition, predisposing disorders (e.g., diabetes mellitus), wound infection, inadequate nutrients, use of NSAIDs and steroids, or altered collagen synthesis.
- 6. Dehiscence is a disruption where the wound pulls apart at the suture line.
- 7. A contracture is a structural deformity caused by the excessive shortening of collagen in scar tissue.
Pediatric and Geriatric Considerations
- 1. Neonates often have transiently depressed inflammatory function, particularly decreased phagocyte chemotaxis and killing of microorganisms and limited complement activity.
- 2. Aging impairs the immune system due to a process called immunosenescence which causes impairment of cellular function in both immunity and wound healing.
- 3. Impaired wound healing in the aging population is multifactorial and includes changes in innate immunity, cellular metabolism, and tissue integrity.
- 4. The elderly are also at risk for excessive and disordered innate immune responses called inflammaging that contribute to many chronic diseases.