Alterations in Immunity
Valentina L. Brashers
 http://evolve.elsevier.com/Rogers/pathophysiology/
http://evolve.elsevier.com/Rogers/pathophysiology/
Innate and adaptive immunity serve to fight infection, remove diseased tissue, and promote healing. The innate immune system reacts quickly to virtually any form of injury or invasion primarily through the activation of inflammation. Adaptive immunity is a slower protective response system designed to recognize and remove antigens expressed by disease causing agents and to provide long-term immunity. These defensive systems are fine-tuned networks, but they are not perfect. There are two major categories of immune dysfunction: (1) excessive or misdirected responses, and (2) inadequate responses (immunocompromise) (Algorithm 9.1).
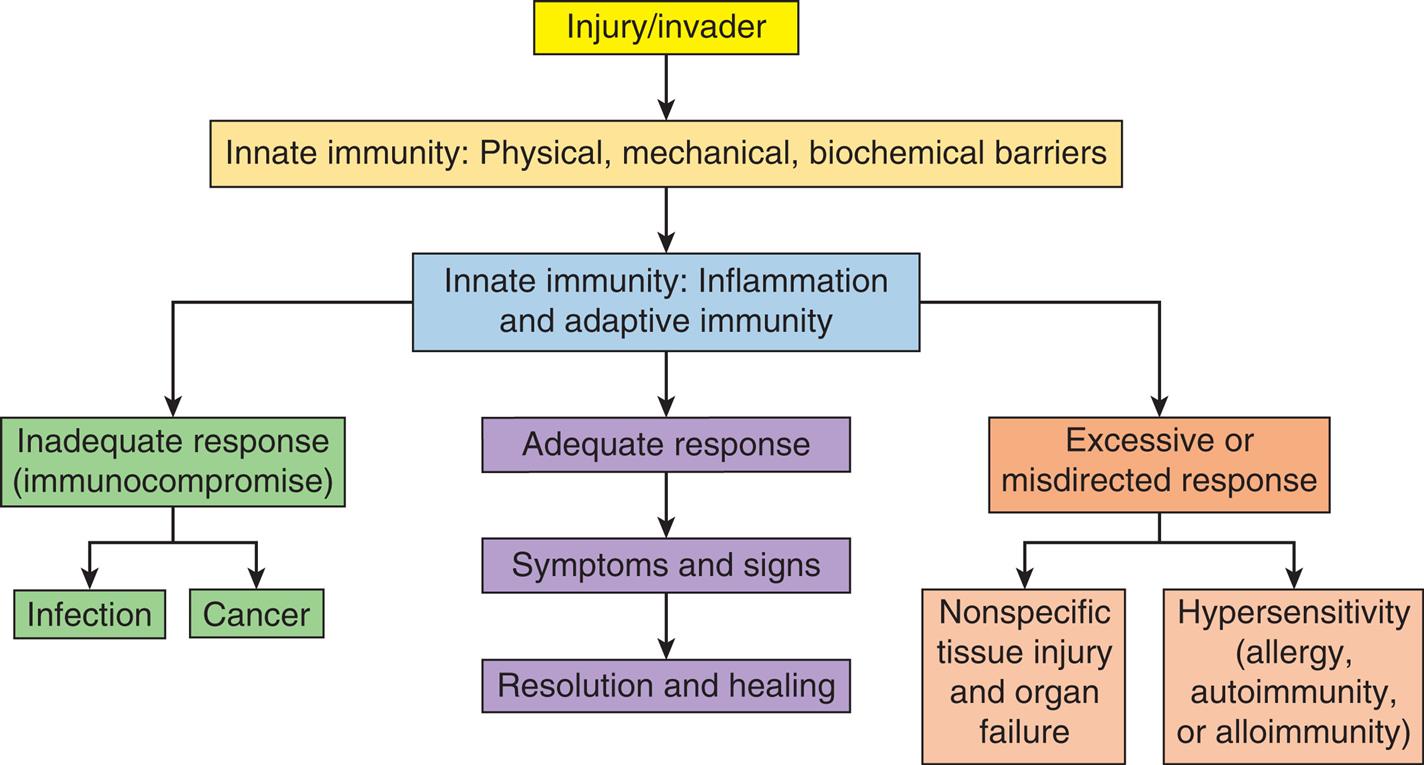
Innate and adaptive immune defense mechanisms function to fight infection, remove diseased tissue, and promote healing. An adequate immune response often causes symptoms and signs such as pain, swelling, and redness but leads to resolution of injury and healing. However, immune responses may be excessive or misdirected, resulting in tissue injury which can be nonspecific or related to hypersensitivity reactions (e.g., allergy, autoimmunity, and alloimmunity). Immune responses may also be inadequate and result in systemic immunocompromise that may lead to overwhelming infection or cancer.
A flowchart provides an overview of immune responses. The data from the flowchart are as follows. 1. Injury or invader. Leads to 2. 2. Innate immunity: physical, mechanical, biochemical barriers. Leads to 3. 3. Innate immunity: inflammation and adaptive immunity. There are three responses at level 3: • Inadequate response (immunocompromise): Infection; cancer. • Adequate response: Symptoms and signs; resolution and healing. • Excessive or misdirected response: nonspecific tissue injury and organ failure; hypersensitivity (allergy, autoimmunity, or alloimmunity).
http://evolve.elsevier.com/Rogers/pathophysiology/
Excessive or misdirected innate immunity is the cause of tissue damage in most acute and chronic diseases. For example, chronic inflammation of the blood vessels leads to atherosclerosis and heart disease. In chronic obstructive lung disease (COPD), inflammation destroys the architecture of lung tissue. An excessive innate immune response also can lead to systemic complications of acute injury or infection. In the case of severe infection, an uncontrolled systemic inflammatory response can lead to septic shock, failure of multiple organs, and death (see Chapter 48). Excessive inflammation like that seen in coronavirus disease 2019 (COVID-19) infection (see Chapter 10) results in overwhelming lung injury and respiratory failure.1
Excessive or misdirected adaptive immune responses result in what are called hypersensitivity reactions. These reactions may be (1) exaggerated against noninfectious environmental substances (allergy); (2) misdirected against the body's own cells (autoimmunity); or (3) directed against beneficial foreign issues, such as transfusions or transplants (alloimmunity). Several of these inappropriate responses can be serious or life-threatening.
Immune responses that are insufficient to protect the host against pathogens and abnormal or foreign cells (immune deficiency) may be the result of inherited or acquired immune defects. Immunocompromise often is caused by therapies aimed at treating other conditions such as cancer. This chapter will describe the mechanisms of hypersensitivity reactions with examples of allergy, autoimmunity, and alloimmunity. This will be followed by a summary of selected inherited and acquired immune deficiency diseases. Many other autoimmune and immune deficiency diseases are covered in detail in later chapters in this textbook.
Hypersensitivity Reactions
A hypersensitivity reaction is an altered immunologic response to an antigen that results in disease or damage to the individual. Hypersensitivity reactions can be classified by the immunologic mechanism that causes disease. These mechanisms have been divided into four distinct types: type I (immunoglobulin E [IgE]-mediated reactions), type II (tissue-specific reactions), type III (immune complex–mediated reactions), and type IV (cell-mediated reactions) (Table 9.1). The four mechanisms are interrelated, and in most hypersensitivity reactions, several mechanisms can be functioning simultaneously or sequentially.
Table 9.1

Immunologic Mechanisms of Hypersensitivity Reactions
Although the inflammatory responses of the innate immune system cause much of the tissue damage associated with hypersensitivity reactions, the mechanisms that initiate inflammation and tissue damage involve misdirected or excessive responses of the adaptive immune system. Hypersensitivity reactions are complicated, but an understanding of these mechanisms is essential to providing appropriate management of the many disorders that result from them.
Type I: IgE-Mediated Hypersensitivity Reactions
Type I hypersensitivity reactions are mediated by antigen-specific IgE and the products of tissue mast cells (Fig. 9.1). Most common allergic reactions are type I reactions against environmental antigens (e.g., pollen, bee venom, nuts, medications). Individuals who are genetically predisposed to this type of hypersensitivity generate an inappropriate IgE-mediated response to what would otherwise be an innocuous exposure. Most commonly, the term allergy indicates IgE-mediated reactions. However, some allergic reactions can be caused by other adaptive immune mechanisms, and IgE can contribute to some autoimmune and alloimmune diseases.
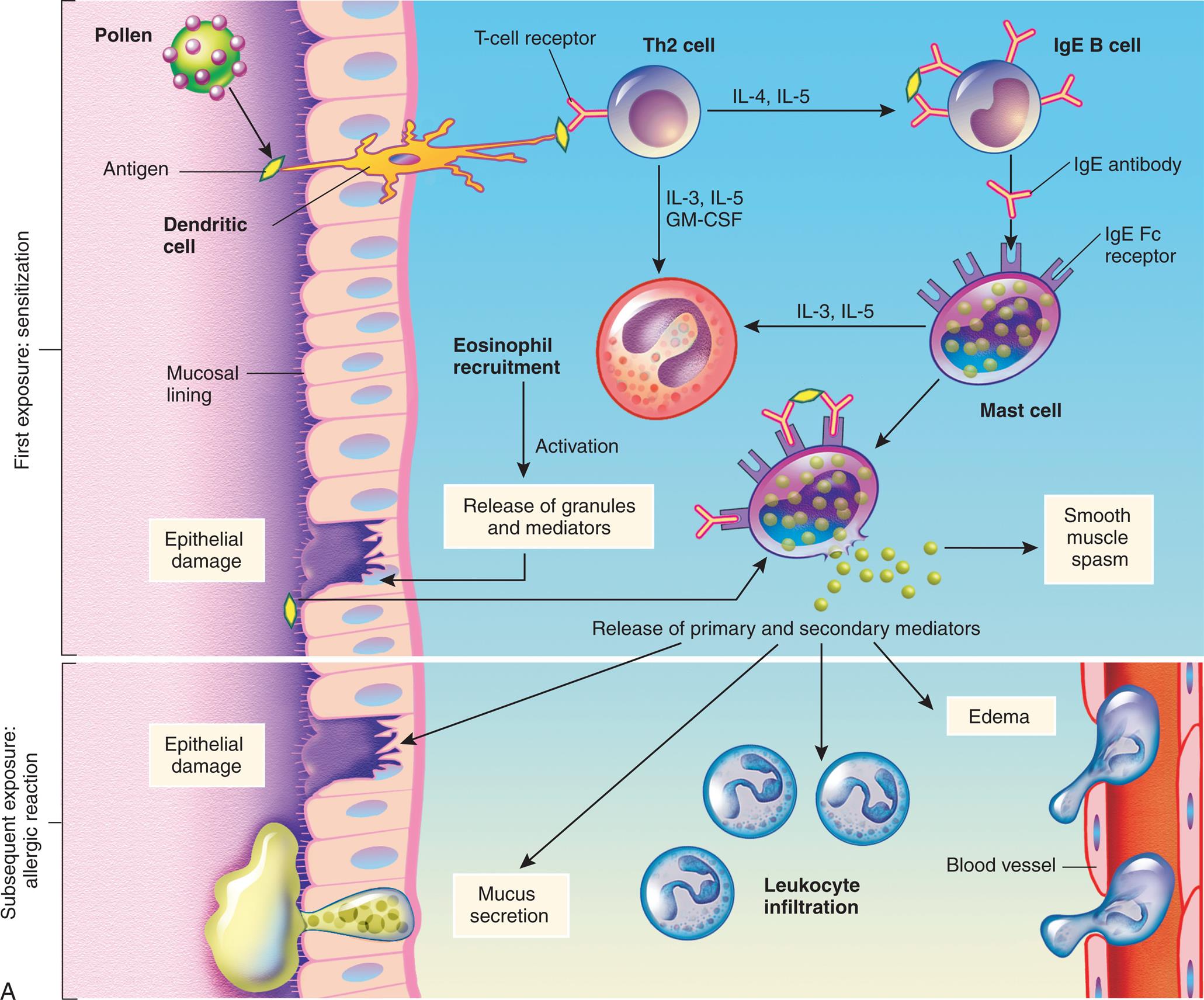
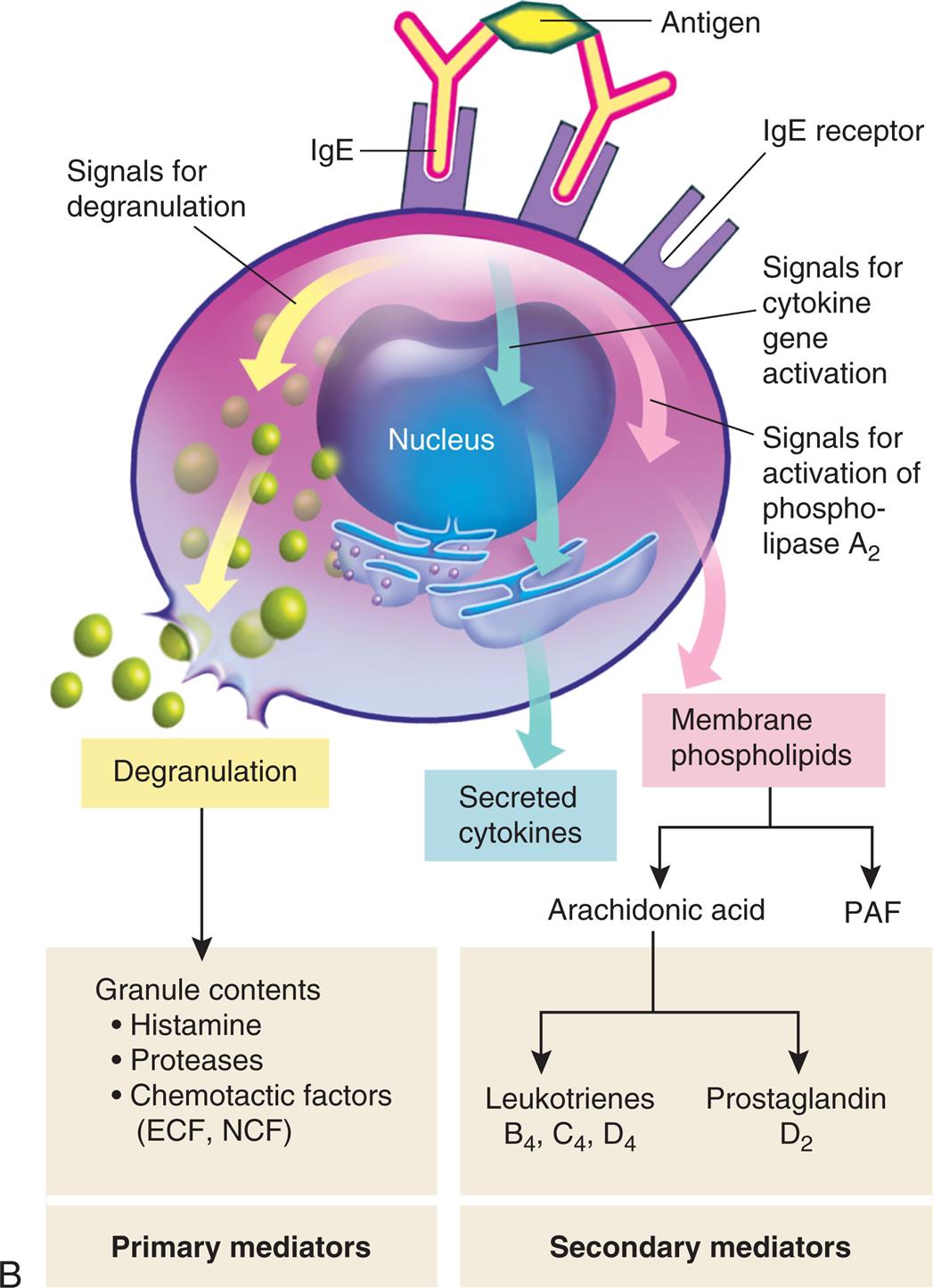
(A) Th2 cells are activated by antigen-presenting dendritic cells to produce cytokines, including IL-4 and IL-5. IL-5 attracts and promotes the survival of eosinophils. High levels of IL-4 induce B cells to class-switch to IgE-producing plasma cells. The IgE coats the surface of the mast cell by binding with IgE-specific Fc receptors on the mast cell's plasma membrane (sensitization). Further exposure to the same allergen cross-links the surface-bound IgE and activates signals from the cytoplasmic portion of the IgE Fc receptors. These signals initiate two parallel and interdependent processes: mast cell degranulation and discharge of preformed mediators (e.g., histamine, chemotactic factors) and production of newly formed mediators such as leukotrienes and prostaglandins. Type I hypersensitivity reactions have two well-defined phases. On first exposure, there is an initial phase characterized by vasodilation, vascular leakage, and, depending on the location, smooth muscle spasm. Eosinophils contribute to mucosal epithelial cell damage. Subsequent exposures are characterized by infiltration of tissues with more eosinophils and with other inflammatory leukocytes resulting in prolonged epithelial cell damage and mucus secretion. Fig. 9.1, cont’d (B) Activation of mast cells leading to degranulation of preformed mediators (primary mediators) and synthesis of newly formed (de novo) mediators (secondary mediators). ECF, Eosinophilic chemotactic factor; Fc, fragment crystallizable; Ig, immunoglobulin; IL, interleukin; NCF, neutrophil chemotactic factor; PAF, platelet-activating factor; Th, T helper.
An illustration demonstrates the mechanism of type 1, immunoglobulin E (I g E)-mediated reaction. Top panel, first exposure: sensitization. The illustration shows a tissue with mucosal lining. Pollen generates antigen that a dendritic cell transports through the epithelial layer and presents to the T-cell receptor on T h 2 cell, which leads to: • I L-4 and I L-5 present the antigen to the antibodies on the I g E B cell. • I L-3, I L-5, G M-C S F generate cytokine signals. I g E antibody from the I g E B cell binds to the I g E F c receptor in the mast cell, which leads to: • I L-3 and I L-5 recruit eosinophil and activates release of granules and mediators from the site of epithelial damage. • Smooth muscle spasm, releasing primary and secondary mediators. Eosinophil recruitment activates the release of granules and mediators at the site of epithelial damage. Antigen from this site binds to the antibodies on the I g E F c receptor of the mast cell, releasing primary and secondary mediators. Bottom panel, subsequent exposure: allergic reaction. Release of primary and secondary mediators leads to: • Epithelial damage. • Mucus secretion. • Leukocyte infiltration; enters the blood vessel. • Edema." "An illustration shows the activation of mast cells. The illustration shows 2 I g E antibodies bound to antigen through variable region and to the I g E receptor through F c region, sending the signal as follows: • Degranulation. • Cytokine gene activation leads to secretion of cytokines. • Activation of phospholipase A sub 2. Degranulation generates the following granule contents (primary mediators): • Histamine. • Proteases. • Chemotactic factors (E C F, N C F). Membrane phospholipids produce arachidonic acid (secondary mediators) or P A F. Arachidonic acid can be: • Leukotrienes: B sub 4, C sub 4, and D sub 4. • Prostaglandin: D sub 2.
Mechanisms of type I, IgE-mediated hypersensitivity reactions
Type I hypersensitivity reactions require sensitization against a particular environmental antigen (allergen) that results in a primary immune response. The response occurs when the immune system first encounters an antigen (primary exposure) and forms antigen-specific memory B cells and T cells (immunologic memory). Disease symptoms appear after secondary exposure to the offending antigen when memory cells are rapidly activated against the same antigen (see Chapter 8). These reactions occur within minutes to a few hours after exposure to antigen and are termed immediate hypersensitivity reactions.
When allergens enter the body of a genetically predisposed individual, they are detected by dendritic cells and B cells, then undergo antigen processing and presentation (Fig. 9.1A). T helper (Th) cells are activated to produce large amounts of the Th2 cytokines, especially interleukin (IL)-4, IL-5, and IL-13. High levels of IL-4 and IL-13 cause B cells to proliferate and become plasma cells that produce antigen-specific IgE. IL-5 recruits and activates eosinophils, which contain granules full of enzymes that are especially damaging to the respiratory system (see Chapter 36). Other interleukins such as IL-9 and IL-33 also play a role in exacerbating type I hypersensitivity reactions.2,3
IgE has a relatively short life span in blood because it rapidly binds to Fc receptors (antibody receptors) on mast cells. The Fc receptors on mast cells specifically bind IgE that has not previously interacted with antigen. After a large amount of IgE has bound to the mast cells, an individual is considered sensitized. When there is a secondary or reexposure of a sensitized individual to the allergen, the IgE antibodies signal the mast cells to degranulate and release mediators.
Mast cells release a variety of cytokines (Fig. 9.1B). Histamine is the most potent preformed mediator of IgE-mediated hypersensitivity. Histamine acts immediately (within 15 to 30 minutes) and affects several key target cells. The tissues most commonly affected by type I responses contain large numbers of mast cells and are sensitive to the effects of histamine released from them. These tissues are found in the gastrointestinal tract, skin, and respiratory tract. Acting through histamine 1 (H1) receptors, histamine contracts bronchial smooth muscles (bronchial constriction), increases vascular permeability (edema), and causes vasodilation (increased blood flow) (see Chapter 7).
Mast cells also synthesize secondary mediators, such as leukotrienes, prostaglandins, and platelet activating factor, which act more slowly (within hours) and have effects similar to that of histamine (see Fig. 9.1B). These newly formed mediators also attract other immune cells (e.g., eosinophils, neutrophils, basophils, monocytes), activate kinins, and initiate the complement cascade with release of the anaphylatoxins C3a, C4a, and C5a (see Chapter 7). These mediators are responsible for a late phase reaction that sets in 2 to 24 hours later even without additional exposure to antigen and may last for several days. The overall effect of these cytokines is inflammation of affected tissues leading to vasodilation, mucous secretion, bronchoconstriction, and tissue injury.
Type II: Tissue-Specific Hypersensitivity Reactions
Type II hypersensitivities are generally immune reactions against a specific cell or tissue. Cells express a variety of antigens on their surfaces, some of which are called tissue-specific antigens because they are expressed on the membranes of only certain cells. For example, platelets have groups of antigens that are found on no other cells of the body. Environmental antigens (e.g., drugs or their metabolites) may bind to the plasma membranes of specific cells and function as haptens, making them targets of type II reactions. The three general mechanisms by which type II hypersensitivity reactions can affect cells are complement mediated lysis, antibody-dependent cellular cytotoxicity, and antireceptor antibodies (Fig. 9.2). Each mechanism begins with antibody binding to tissue-specific antigens or antigens present on particular tissues. The symptoms of many type II diseases are determined by which tissue or organ expresses the particular antigen.
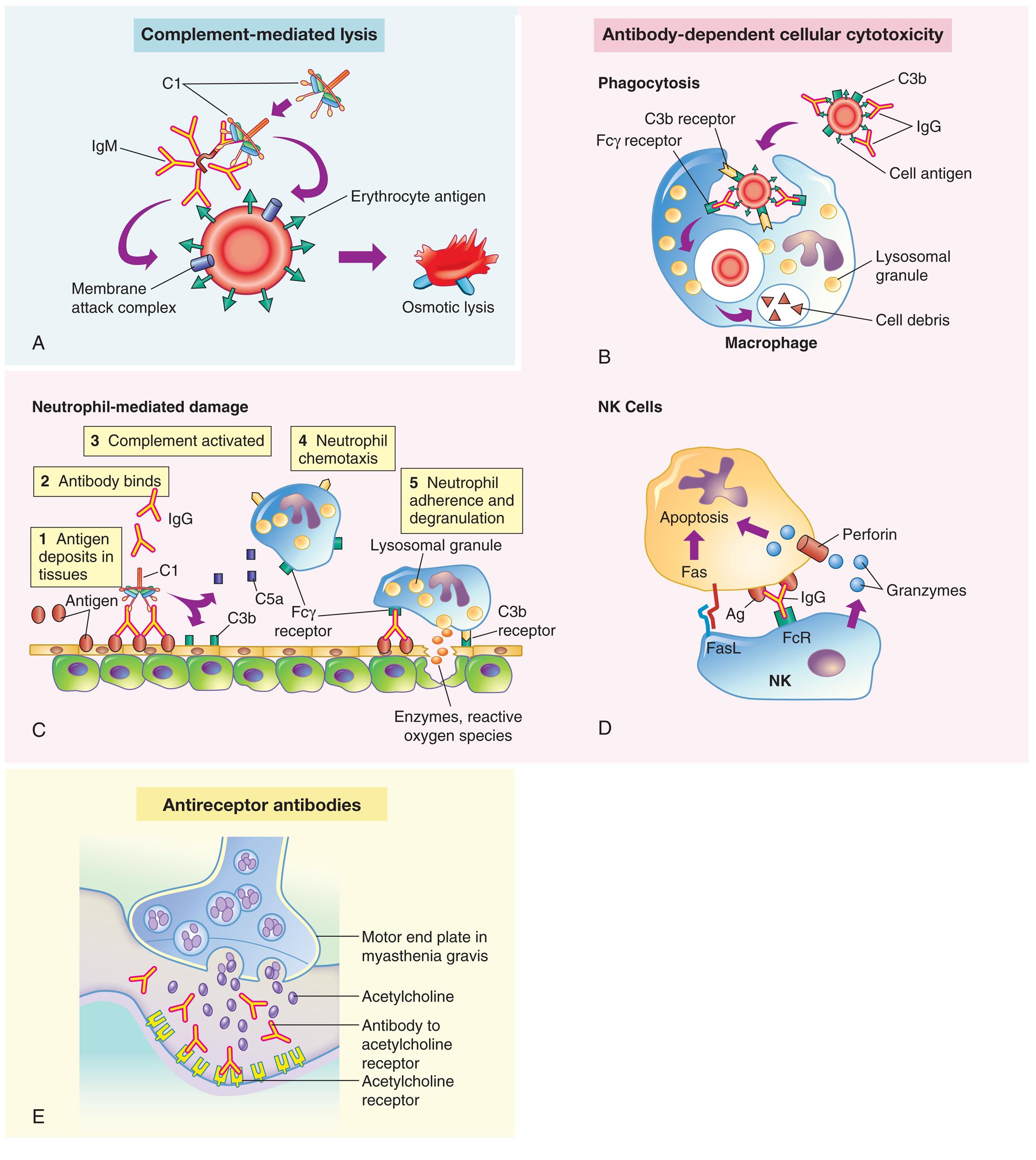
Antigens on the target cell bind with antibody and are destroyed or prevented from functioning by one of three mechanisms: Complement-mediated lysis(A) Complement binding to autoantibodies on a target cell (an erythrocyte target is illustrated here) results in destruction of the cell by the complement membrane attack complex. Antibody-dependent cellular cytotoxicity occurs through three processes: (B) Clearance (phagocytosis) by macrophages in the tissue; (C) Neutrophil-mediated immune destruction; (D) Apoptosis of target cells by natural killer (NK) cells by the release of granzymes and perforin and by the interactions of Fas ligand. Antireceptor antibodies(E) Modulation or blocking of the normal function of receptors by antireceptor antibody. This example of mechanism depicts myasthenia gravis in which acetylcholine receptor antibodies block acetylcholine from attaching to its receptors on the motor end plates of skeletal muscle, thereby impairing neuromuscular transmission and causing muscle weakness. C1, Complement component C1; C3b, complement fragment produced from C3, which acts as an opsonin; C5a, complement fragment produced from C5, which acts as a chemotactic factor for neutrophils; Fcγ receptor, cellular receptor for the Fc portion of IgG; FcR, Fc receptor.
Five illustrations, A, B, C, D, and E, demonstrate the mechanisms of type 2, tissue-specific reactions. Illustration A, complement-mediated lysis. An I g M binds C 1, leading to a membrane attack complex on a blood cell, releasing erythrocyte antigen, and resulting in osmotic lysis. Illustration B, antibody-dependent cellular cytotoxicity: phagocytosis. C 3 b is attached to the blood cell. I g G binds to the cell antigen. C 3 b receptor binds C 3 b and F c gamma receptor binds the I g G, producing lysosomal granules and cell debris in the macrophage. Illustration C, antibody-dependent cellular cytotoxicity: neutrophil-mediated damage. The illustration shows an epithelial layer and shows the following sequence of damage: • Antigen deposits in tissues. • Antibody binds. • Complement activated: C 3 b and C 5 a. • Neutrophil chemotaxis: F c gamma receptor on the macrophage. • Neutrophil adherence and degranulation: release of lysosomal granules as enzymes, reactive oxygen species. Illustration D, antibody-dependent cellular cytotoxicity: N K cells. An N K cell comprises Fas L and F c R. Granzymes from the N K cell are attracted to the perforin of the apoptosis. I g G binds F c R in N K to A g in Apoptosis. Illustration E, antireceptor antibodies. The illustration shows the motor end plate in myasthenia gravis. Acetylcholine is released. Antibody to acetylcholine receptor binds to the acetylcholine receptor.
Mechanisms of type II, tissue-specific reactions
- (1) Complement-mediated lysis occurs when antibodies (IgM or IgG) bound to tissue-specific antigens cause activation of the complement cascade through the classical pathway (Fig. 9.2A). Formation of the membrane attack complex (C5-9) damages the membrane and results in lysis of the cell. For example, erythrocytes are destroyed by complement-mediated lysis in individuals who have received mismatched transfused blood cells (see the section on Alloimmunity).
- (2) Antibody-dependent cell-mediated cytotoxicity (ADCC) (Fig. 9.2B–D) occurs when antibodies bound to tissue-specific antigens activate macrophages, neutrophils, and natural killer (NK) cells, which attack the target cells. When antibodies activate complement, there is the deposition of C3b on the target cell surface. Receptors on macrophages and neutrophils recognize and bind these opsonins (antibody plus C3b) and increase phagocytosis of the target cell (see Fig. 9.2B). Toxic products produced by neutrophils (lysozymes and toxic oxygen radicals) also cause tissue damage (Fig. 9.2C). ADCC also involves NK cells (Fig. 9.2D). Antibodies on the target cell are recognized by Fc receptors on NK cells, which release toxic substances that destroy the target cell. Examples of ADCC include autoimmune conditions in which autoantibodies are made against antigens on platelets or red blood cells causing them to be removed by phagocytes in the spleen.
- (3) Antireceptor antibodies do not destroy the target cell but rather cause the cell to malfunction (Fig. 9.2E). These autoantibodies change the function of the target receptor by blocking, overstimulating, or destroying it. For example, in Graves disease, autoantibodies called thyroid-stimulating immunoglobulins (TSIs) bind to and activate receptors for thyroid-stimulating hormone (TSH) on thyroid gland cells. TSH normally stimulates thyroid hormone secretion but is under the control of feedback mechanisms. When TSIs bind to the TSH receptors, they stimulate the thyroid cells to overproduce thyroxine, thus producing symptoms of hyperthyroidism (see Chapter 22).
Type III: Immune Complex–Mediated Hypersensitivity Reactions
Type III hypersensitivity disease reactions are caused by antigen-antibody (immune) complexes that are formed in the circulation and are deposited in vessel walls or other tissues (Fig. 9.3). The primary difference between type II and type III mechanisms is that in type II hypersensitivity, antibody binds to antigen on the cell surface, whereas in type III, antibody binds to soluble antigen that was released into blood or body fluids.
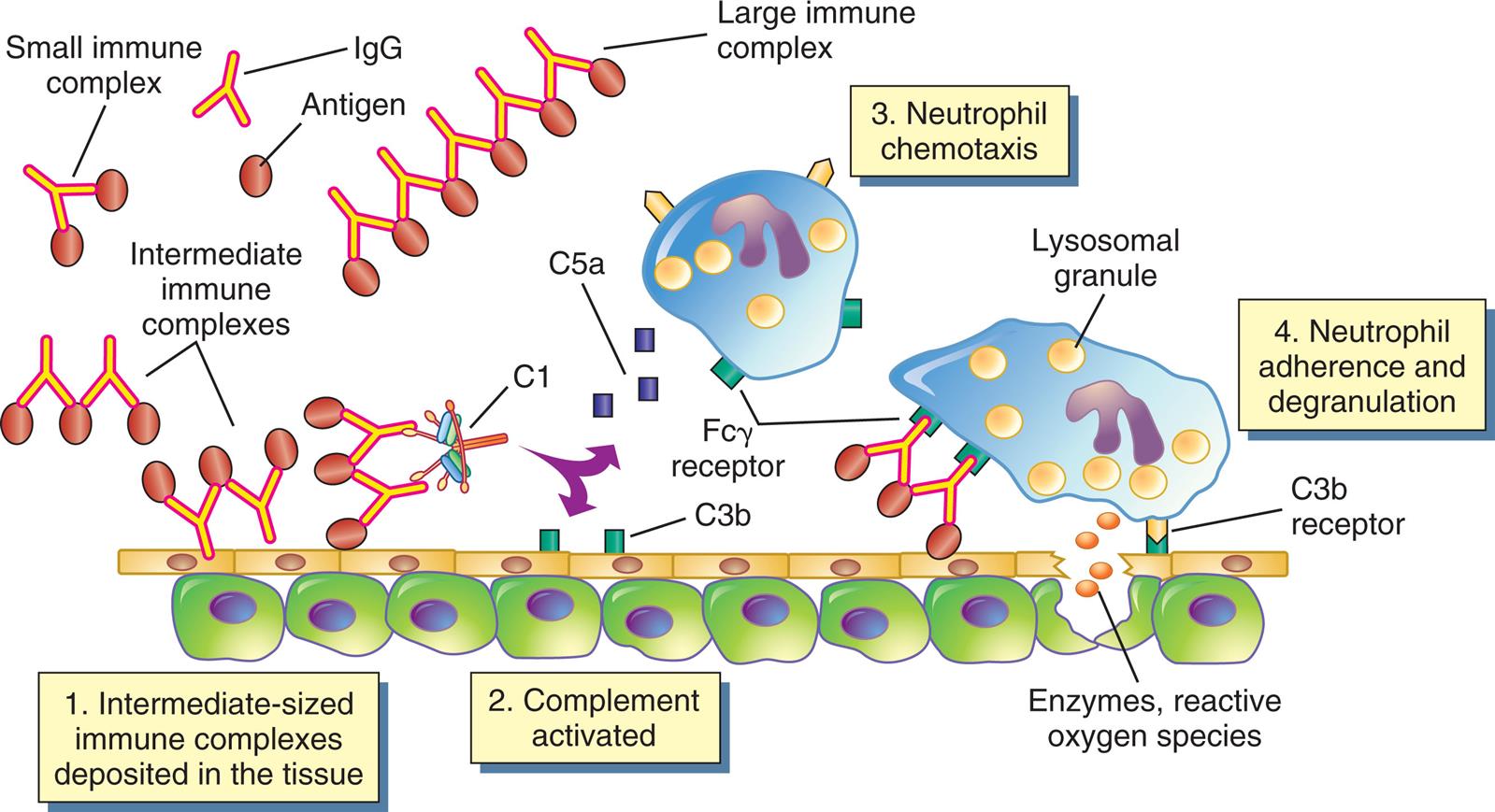
Immune complexes form in the blood from circulating antigen and antibody. Both small and large immune complexes are removed successfully from the circulation and do not cause tissue damage. Intermediate-sized complexes are deposited in certain target tissues in which the circulation is slow or filtration of blood occurs. The complexes activate the complement cascade through C1 and generate fragments, including C5a and C3b. C5a is chemotactic for neutrophils, which migrate into the inflamed area and attach to the IgG and C3b in the immune complexes. The neutrophils attempt unsuccessfully to phagocytose the tissue and, in the process, release a variety of degradative enzymes that destroy the healthy tissues. Fcγ receptor is the cellular receptor for the Fc portion of immunoglobulin G.
An illustration shows the mechanism of type 3, immune complex-mediated reactions. The illustration shows a I g G antibody and an antigen. A small immune complex is a structure with two antigens, one each on the paratopes of the I g G. Intermediate immune complexes are pairs of antibodies with antigens on the paratopes. A large immune complex is a chain of multiple antibodies with antigens on the paratopes. The mechanism of reactions are as follows. 1. Intermediate-sized immune complexes deposited in the tissue. 2. Complement (C 1, C 5 a) activated. 3. Neutrophil chemotaxis (F c gamma receptor). 4. Neutrophil adherence and degranulation (lysosomal granules, C 3 b receptor): enzymes, reactive oxygen species.
Mechanisms of type III, immune-mediated hypersensitivity reactions
When antibodies bind to circulating antigens, immune complexes are then deposited in vascular tissues. Type III reactions are not organ specific and most commonly result in a vasculitis in the skin, kidney, or lungs. The harmful effects of immune complex deposition are caused by complement activation and by neutrophils attempting to phagocytose the immune complexes. During the attempted phagocytosis, large quantities of lysosomal enzymes are released into the inflammatory site instead of into phagolysosomes. The attraction of neutrophils and the subsequent release of lysosomal enzymes cause most of the resulting tissue damage. Two prototypic models of type III hypersensitivity help to explain the variety of diseases in this category. Serum sickness is a model of systemic type III hypersensitivities, and the Arthus reaction is a model of localized or cutaneous reactions.
Serum sickness–type reactions are caused by the formation of immune complexes in the blood and their subsequent generalized deposition in target tissues. A form of serum sickness is Raynaud phenomenon, a condition caused by the temperature-dependent deposition of immune complexes in the capillary beds of the peripheral circulation. Certain immune complexes precipitate at temperatures less than normal body temperature, particularly in the tips of the fingers, toes, and nose, and are called cryoglobulins. The precipitates block the circulation and cause localized pallor and numbness, followed by cyanosis (a bluish tinge resulting from oxygen deprivation) and eventually gangrene if the circulation is not restored.
Arthus reaction is vasculitis caused by repeated local exposure to an antigen that reacts with preformed antibody and forms immune complexes in the walls of the local blood vessels. Symptoms of Arthus reaction begin within 1 hour of exposure and peak 6 to 12 hours later. The lesions are characterized by a typical inflammatory reaction, with increased vascular permeability, an accumulation of neutrophils, edema, hemorrhage, clotting, and tissue damage. For example, gluten-sensitive enteropathy (celiac disease) follows ingestion of antigen, usually gluten from wheat products (see Chapter 42). Allergic alveolitis (farmer lung disease, pigeon breeder disease) is Arthus-like acute hemorrhagic inflammation of the air sacs (alveoli) of the lungs, resulting from inhalation of fungal antigens, usually particles from moldy hay or pigeon feces (see Chapter 35).
Type IV: Cell-Mediated Hypersensitivity Reactions
Types I, II, and III hypersensitivity reactions are mediated by antibodies, whereas type IV hypersensitivity reactions are mediated by T lymphocytes and do not involve antibodies (Fig. 9.4). Type IV mechanisms occur through the presentation of antigens on major histocompatibility molecules (MHCs) to T cells. Th cells produce Th1 and Th17 cytokines. Th1 and Th17 cytokines recruit and activate macrophages and cytotoxic T lymphocytes (Tc cells) (see Chapter 8). Tc cells directly kill target cells. Macrophages release soluble factors, such as lysosomal enzymes and toxic reactive oxygen species. Together, these responses cause tissue damage. The response is delayed, occurring 24 to 72 hours after antigen reexposure, compared with an immediate type I reaction, which occurs within minutes. The response is delayed because of the time it takes for sensitized T cells to travel to the site of antigen reexposure and the time needed to produce cytokines that activate other cells including macrophages (delayed hypersensitivity).
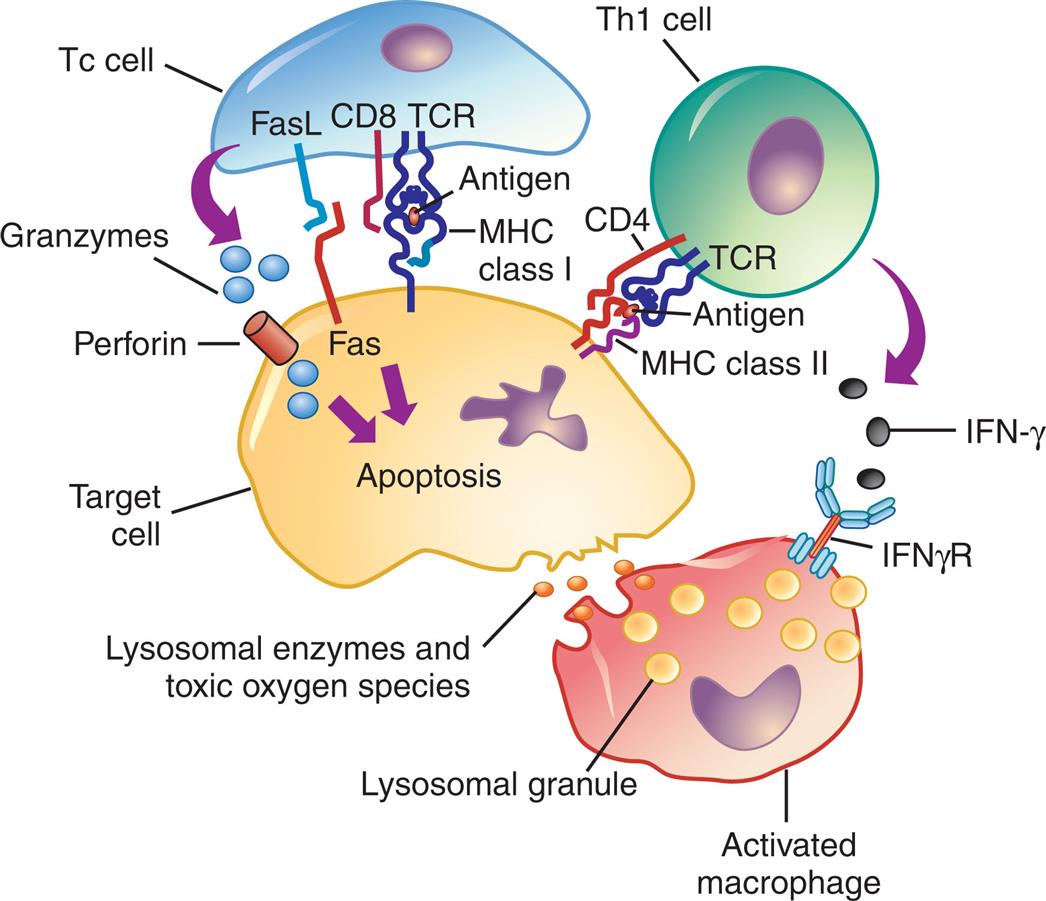
Antigens from target cells stimulate T cells to differentiate into T cytotoxic cells (Tc cells), which have direct cytotoxic activity through release of granzymes or the induction of apoptosis via Fas and Fas ligand binding. T helper cells (Th1) cells produce lymphokines (especially interferon-γ) that activate macrophages through specific receptors (e.g., IFN-γ receptor). The macrophages can attach to targets and release enzymes and reactive oxygen species that are responsible for most of the tissue destruction. FasL, Fas ligand; IFN-γ, interferon-γ; IFN-γR, interferon-γ receptor; MCH, major histocompatibility complex; TCR, T cell receptor.
An illustration shows the mechanism of type 4, cell-mediated reactions. The illustration shows a T c cell, a T h 1 cell, a target cell, and an activated macrophage. T C R on the T h 1 cell is bound to the target cell through M H C class 2 and C D 4. I F N-gamma from the T h 1 cell are attracted to the I F N gamma R. T C R on the T c cell is bound to the target cell through Fas L, Fas, C D 8, and M H C class 1. Granzymes from the T c cell are attracted to the perforin on the target cell. The target cell undergoes apoptosis, releasing lysosomal enzymes and toxic oxygen species. Lysosomal granules activate the macrophage.
In some cases, Tc cells and macrophages cannot kill or remove the offending agent. In these cases, they surround and contain the invader through the formation of a granuloma (see Chapter 7). The formation of multiple granulomas can lead to tissue damage and organ dysfunction.
Mechanisms of type IV cell-mediated hypersensitivity reactions
Clinical examples of type IV hypersensitivity reactions include graft rejection, reaction on the skin test for tuberculosis, and allergic reactions resulting from skin contact with some substances, such as poison ivy and metals. A type IV component also may be present in many autoimmune diseases. For example, T cells against type II collagen (a protein present in joint tissues) contribute to the destruction of joints in rheumatoid arthritis, and T cells against an antigen on the surface of pancreatic beta cells (the cell that normally produces insulin) are responsible for beta-cell destruction in insulin-dependent (type 1) diabetes mellitus.
In 1891, Ehrlich was the first to thoroughly describe type IV hypersensitivity reaction in the skin, leading to the development of a diagnostic skin test for tuberculosis. The reaction follows an intradermal injection of tuberculin antigen (purified protein derivative [PPD]) into an individual who has latent or active tuberculosis and is therefore sensitized (has developed adaptive immune cells against tuberculosis antigen). After 24 to 72 hours the reaction site becomes infiltrated with T lymphocytes and macrophages, resulting in a clear hard center (induration) and a reddish surrounding area (erythema). The reaction is referred to as a positive skin test result (i.e., positive PPD) for tuberculosis infection.
Allergic type IV reactions are elicited by some environmental antigens that are haptens and become immunogenic after binding to larger (carrier) proteins in the individual. In allergic contact dermatitis, the carrier protein is in the skin. The best-known example is the reaction to poison ivy (Fig. 9.5). The antigen is a plant catechol, urushiol, which reacts with normal skin proteins and evokes a cell-mediated immune response. Skin reactions to industrial chemicals, cosmetics, detergents, clothing, food, metals, and topical medicines (e.g., penicillin) are elicited by the same mechanism. Contact dermatitis consists of lesions only at the site of contact with the allergen, as in metal allergy to jewelry.
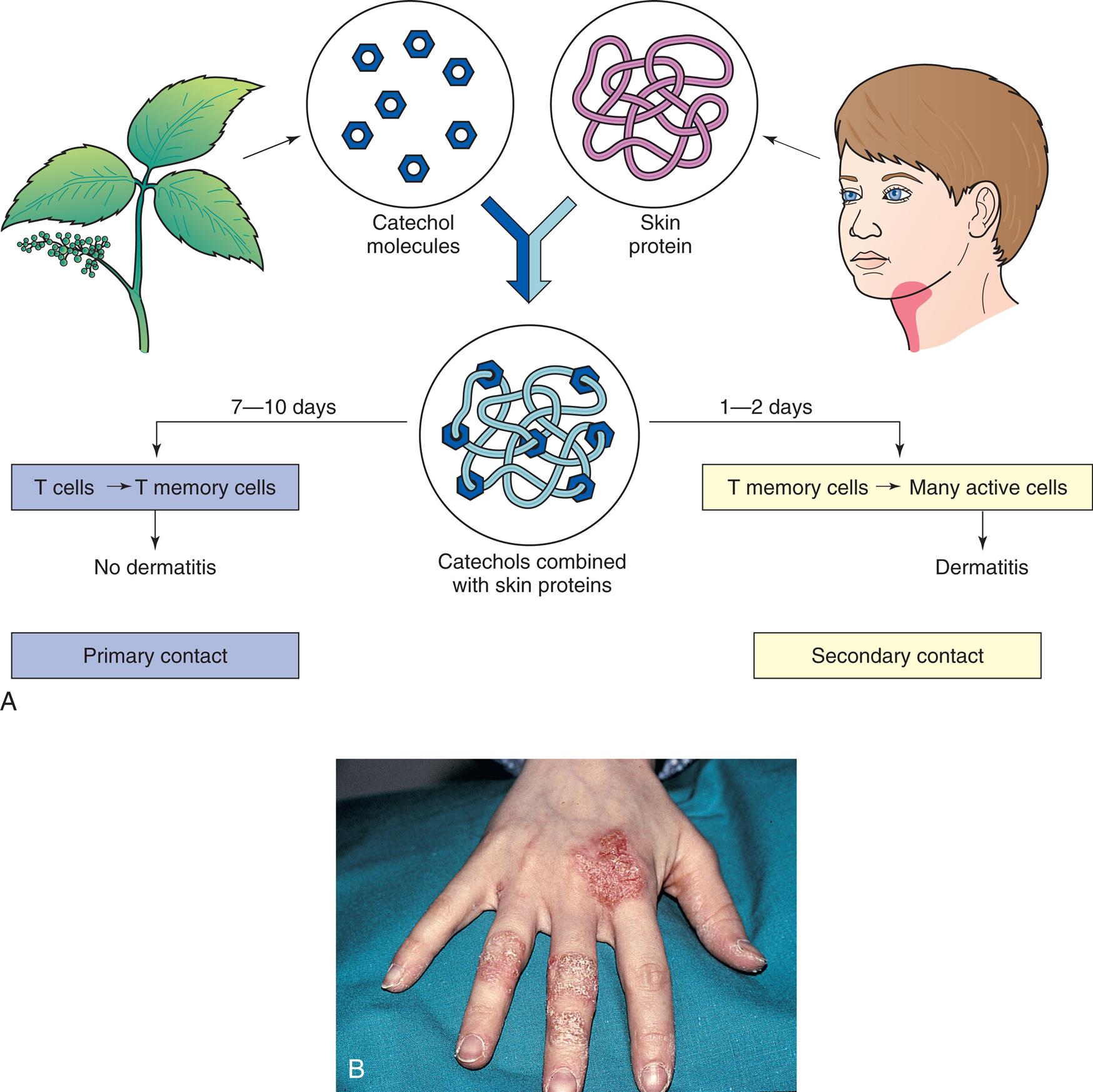
(A) The development of type IV hypersensitivity to poison ivy. The first (primary) contact with allergen sensitizes (produces reactive T cells) the individual but does not produce a rash (dermatitis). Secondary contact activates a type IV cell-mediated reaction that causes dermatitis. (B) Contact dermatitis caused by a delayed hypersensitivity reaction leading to vesicles and scaling at the sites of contact. From Damjanov I, Linder J. Anderson's pathology, 10th edition. St. Louis: Mosby; 1996.
Top panel, A, is an illustrated flowchart showing the development of type 4 hypersensitivity to poison ivy. Catechol molecules from poison ivy combines with the skin proteins on humans. • 7 to 10 days: T cells to T memory cells, no dermatitis. Primary contact. • 1 to 2 days: T memory cells to many active cells, dermatitis. Secondary contact. Bottom panel, B, is a closeup of a hand affected by dermatitis. The skin appears dry and cracked on the fingers and on the surface below the index finger.
Antigenic Targets of Hypersensitivity Reactions
The four types of hypersensitivity reactions contribute to tissue damage and clinical diseases in a variety of ways. Often, several mechanisms of hypersensitivity are active at the same time. The three major types of hypersensitivity disorders are allergy, autoimmunity, and alloimmunity. The mechanisms that initiate the onset of hypersensitivity reactions are not completely understood. It is generally accepted that genetic and environmental agents are contributing factors.
Allergy
Allergy (atopy) refers to hypersensitivity to environmental antigens. These can include medicines, natural products (e.g., pollens, bee stings), infectious agents, and any other antigen that is not naturally found in the individual. Allergies are the most common hypersensitivity reactions. The majority of allergies are type I IgE-mediated reactions. These reactions usually lead to relatively mild symptoms such as rhinitis and sneezing. However, in some individuals, these reactions can be excessive and life-threatening (anaphylaxis). Antigens that cause allergic responses are called allergens. Typical allergens include pollens (e.g., ragweed), molds and fungi (e.g., Penicillium chrysogenum), foods (e.g., milk, eggs, fish), animals (e.g., cat dander, dog dander), cigarette smoke, and components of house dust (e.g., fecal pellets of house mites). The most common forms of atopic disease are allergic rhinitis, bronchial asthma, anaphylaxis, and atopic dermatitis (eczema).
Genetic predisposition and environmental factors
Genes and environment interact in complex ways in individuals with type I hypersensitivity.4 Certain individuals are genetically predisposed to develop type I IgE-mediated hypersensitivity and are called atopic. In families in which one parent has an allergy, allergies develop in approximately 40% of the offspring. If both parents have allergies, the incidence in the offspring may be as high as 80%. Atopic individuals tend to produce higher quantities of IgE and to have more Fc receptors for IgE on their mast cells. The airways and the skin of atopic individuals are also more responsive to a wide variety of both specific and nonspecific stimuli than are the airways and skin of individuals who are not atopic. Multiple genes have been associated with the atopic state, including polymorphisms in a large variety of cytokines that regulate IgE synthesis (e.g., IL-4, IL-13) and cellular receptors.
The environment has a significant impact on allergic disease. Diet, medications, and comorbidities impact the microbiome and the health of the adaptive immune system (see Emerging Science Box: The Microbiome and Food Allergy).5 In genetically predisposed individuals, exposure to large quantities of allergens and irritants in the environment can trigger symptoms. The role of infection in allergic disease is complex and may be protective or may render organ systems such as the respiratory tract more vulnerable to allergic symptoms.
Clinical symptoms of allergy
The clinical manifestations of allergy are attributable mostly to the biologic effects of histamine. Tissues most commonly affected contain large numbers of mast cells and are sensitive to the effects of histamine released from them. These tissues are found in the gastrointestinal tract, skin, and upper and lower respiratory tracts (Fig. 9.6). The particular symptoms frequently reflect the main portal of entry for the allergen. For instance, pollens and other airborne allergens usually cause respiratory symptoms.
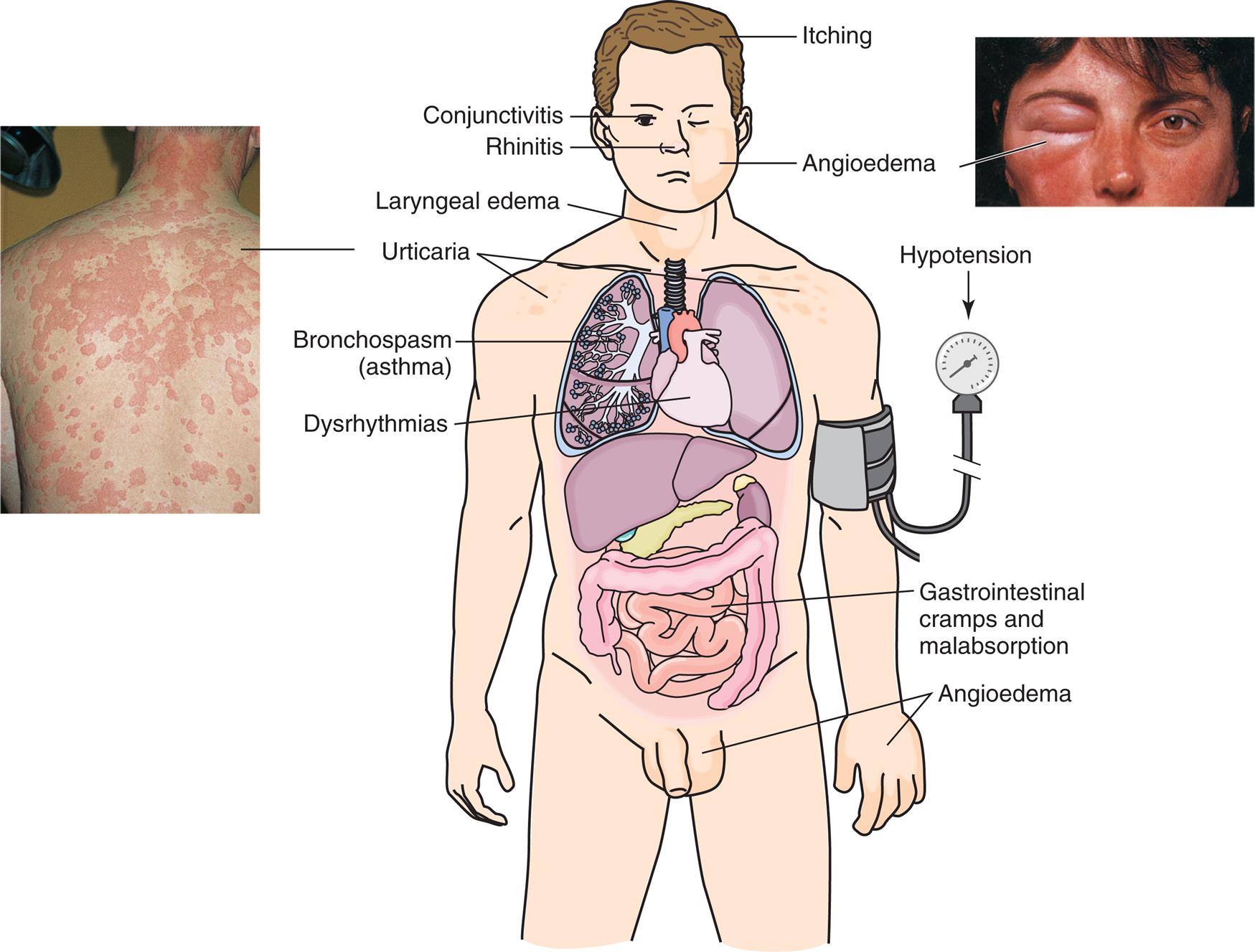
Manifestations of allergic reactions as a result of type I hypersensitivity include pruritus, angioedema (swelling caused by exudation), edema of the larynx, urticaria (hives), bronchospasm (constriction of airways in the lungs), hypotension (low blood pressure), and dysrhythmias (irregular heartbeat) because of anaphylactic shock, and gastrointestinal cramping caused by inflammation of the gastrointestinal mucosa. Photographic inserts show a diffuse allergy-like eye and skin reaction on an individual. The skin lesions have raised edges and develop within minutes or hours, with resolution occurring after about 12 hours. (Inserts from Male D, Brostoff J, Roth D, et al. Immunology, 8th edition. St. Louis: Mosby; 2013.)
An illustration shows the anterior view of a human, with the internal organs highlighted. The following conditions are highlighted on the illustration: • Head: itching. • Eye: conjunctivitis. • Nose: rhinitis. • Cheek: angioedema (an accompanying closeup shows a person’s eye swollen shut). • Neck: laryngeal edema. • Shoulders: urticaria (an accompanying closeup shows a person’s shoulders and back, covered in welts). • Lungs: bronchospasm (asthma). • Heart: dysrhythmias. • Upper arm (attached with a blood pressure gauge): hypotension. • Intestines: gastrointestinal cramps and malabsorption. • Hands and genitalia: angioedema.
Effects of allergens on the mucosa of the eyes, nose, and respiratory tract include conjunctivitis (inflammation of the membranes covering the front of the eye and the lining the eyelids), rhinitis (inflammation of the mucous membranes of the nose), and asthma (constriction and swelling of the bronchi). Symptoms of all these conditions are caused by vasodilation, hypersecretion of mucus, edema, and swelling of the mucosa. Because the mucous membranes lining the respiratory tract (accessory sinuses, nasopharynx, and upper and lower respiratory tracts) are continuous, they are all adversely affected. The degree to which each is affected determines the symptoms of the disease. One of the most common type I reactions is asthma. It is presented in detail in Chapter 35. The central problem in asthma is obstruction of the large and small airways (bronchi) of the lower respiratory tract by bronchospasm (constriction of smooth muscle in airway walls), edema, and thick secretions. This leads to ventilatory insufficiency, wheezing, and difficult or labored breathing.
Urticaria, or hives, is a dermal (skin) manifestation of type I allergic reactions (see Fig. 9.6). The underlying mechanism is the localized release of histamine and increased vascular permeability, resulting in limited areas of edema. Urticaria is characterized by white fluid-filled blisters (wheals) surrounded by areas of redness (flares). The wheal and flare reaction is usually accompanied by itching. Not all urticarial symptoms are caused by allergic (immunologic) reactions. Some, termed nonimmunologic urticaria, result from exposure to cold temperatures, emotional stress, medications, systemic diseases, hyperthyroidism, or malignancies (e.g., lymphomas).
Gastrointestinal allergies are caused primarily by allergens that enter through the mouth—usually foods or medicines. When food is the allergen, the active immunogen may be a product of food breakdown by digestive enzymes. Acute symptoms usually occur rapidly (frequently within minutes) and include vomiting, diarrhea, or abdominal pain. Prolonged or recurrent reactions may result in malabsorption or protein-losing enteropathy. Systemic symptoms may range from urticaria to life-threatening anaphylactic reactions. The most common food allergies are tree nuts, peanuts, milk, shellfish, and fish. The prevalence of food allergies is estimated at 3% to 10% of adults and 8% of children worldwide.6 The most rapid and severe allergic reaction is anaphylaxis. Anaphylaxis occurs within minutes of reexposure to the antigen and can be either systemic (generalized) or cutaneous (localized). Symptoms of systemic anaphylaxis include pruritus, erythema, vomiting, abdominal cramps, diarrhea, and breathing difficulties, and the most severe reactions may include contraction of bronchial smooth muscle, edema of the throat, and decreased blood pressure that can lead to shock and death (see Chapter 48). Examples of systemic anaphylaxis are allergic reactions to antibiotics, bee stings, or peanuts.
Diagnosis and management of allergic reactions
Several tests are available to diagnose allergic reactions, including skin tests and measurement of IgE in the blood. Reactivity to a particular allergen may be tested by controlled administration of small doses of the suspected allergen or injection of an allergen into (intradermal) or onto (epicutaneous or prick test) the skin. If the individual is allergic to a particular allergen, a local wheal and flare reaction may occur within a few minutes at the site of injection. A variety of immunoassays can be used can detect IgE antibodies in serum. These assays can be used to measure circulating levels of total IgE, or circulating levels of specific IgE antibodies against selected allergens.
If possible, avoidance of the allergen is the best method to limit allergic responses. Clinical desensitization to allergens can be achieved in some individuals. Minute quantities of the allergen are injected in increasing doses over a prolonged period. The procedure may reduce the severity of the allergic reaction in the treated individual.
Some type I allergic responses can be controlled by blocking histamine receptors with antihistamines. The effects of allergy on the respiratory system may require bronchodilators or medications that block leukotrienes (see Chapter 35). The most effective medications for allergic disease are the corticosteroids, which can be given by local administration (e.g., inhalers or topical ointments) or systemically to decrease tissue inflammation. For individuals with severe allergy, monoclonal antibodies that block IgE may be required (omalizumab). Other monoclonal antibodies that block IL-4 (dupilamab) and IL-5 (mepolizumab) are now available. In anaphylaxis, systemic administration of sympathomimetic medications such as epinephrine may be required to support the respiratory and cardiovascular systems (see Chapter 48).
Autoimmunity
Autoimmunity occurs when the immune system reacts against self-antigens to such a degree that autoantibodies or autoreactive T cells damage the individual's tissues. It is the result of a disturbance in the immunologic tolerance of self-antigens (see Chapter 8). This breakdown in tolerance is believed to occur in a genetically predisposed individual when there is some initiating event. Most often, the initiating event is unrecognized but is hypothesized to be an environmental factor such as infection or toxins, or some change in neurologic, endocrine, and/or immune status.
It is well established that autoimmune diseases can be familial. In familial cases, affected family members may not all have the same disease but rather may develop different autoimmune disorders. However, most autoimmune diseases appear as isolated events without a positive family history, and susceptibility for developing such diseases appears to be linked to a combination of multiple genes and multifactorial. Genetic associations with particular autoimmune diseases have been identified for a variety of major histocompatibility complex (MHC) genes (see Chapter 8). Some associations are strong; others are more tenuous. Autoimmunity may result from changes in the way MHC molecules present antigen. These changes may cause an inappropriate or exaggerated response to environmental antigens such as microorganisms. A large variety of non-MHC genes also have been identified as risk factors for the development of specific autoimmune diseases. Most of these genes encode for inflammatory cytokines or costimulatory molecules found on the cell surface.
Tolerance is a state of immunologic control so that the individuals do not make a detrimental immune response against their own cells and tissues. Central tolerance occurs when autoreactive lymphocytes are either eliminated or suppressed in the primary lymphoid organs during differentiation of immature T or B lymphocytes (see Algorithm 8.1 and (8.2). Peripheral tolerance is maintained in the secondary lymphoid organs through the action of T regulatory (Treg) lymphocytes (see Chapter 8). Many autoimmune diseases display defects in either the number or the function of Treg cells in peripheral blood (see Emerging Science Box: T regulatory Cells and Autoimmunity).7 In addition to Treg cells, dendritic cell processing and presentation of antigen are critical for the adaptive immune system to differentiate between self- and nonself-antigens. Defects in dendritic cell interaction with T cells have been linked to autoimmunity.8
In the vast majority of autoimmune diseases, the nature of the initiating event that results in a breakdown of tolerance is unclear.
One of the most studied of these potential initiating events is the role of foreign antigens from infectious microorganisms causing molecular mimicry. Some antigens of infectious agents so closely resemble (mimic) a particular self-antigen that antibodies or T cells produced to protect against the infection also recognize the self-antigen as foreign (cross-reactive antibody or T cell). One example is rheumatic heart disease that may occur after a group A streptococcal sore throat. In this example, the M protein and group A carbohydrate in the streptococcal bacterial capsule mimic normal antigens in the heart valves resulting in the production of antibodies and T cells that damage the valves. Most potential associations between infection and autoimmunity are much less clear. For example, many researchers believe that there is a link between viral or bacterial infection and the onset of type 1 diabetes.9 The link between infectious microorganisms and autoimmune disease continues to be investigated.
Women are far more likely to develop autoimmune disease than are men. Many theories have been proposed to explain this prevalence of autoimmunity in women. Many of the genes that code for immune function reside on the X chromosome. Although having two X chromosomes is protective against X-linked disorders (see Chapter 4), it has been proposed that hormonal changes during the life of a female individual may cause skewed activation of immune genes on X chromosomes.10,11 Another hypothesis to explain the prevalence of autoimmunity in women is that a gene called VGLL3, which is more active in female than male skin cells, is overexpressed in women with autoimmune disease. This gene influences the function of several other genes involved in immune control.12
These kinds of initiating and contributing factors result in adaptive immune responses that are misdirected toward self-antigens. Self-directed immune responses often include a combination of hypersensitivity mechanisms II, II, and IV. The innate system is also activated with inflammatory damage to tissues.
Examples of Autoimmune Diseases
Many clinical disorders are associated with autoimmunity and are collectively referred to as autoimmune diseases. Table 9.2 provides a list of a few of the many autoimmune diseases. Many of these and other autoimmune disorders will be discussed in later chapters of this textbook. In this chapter, we will discuss only two representative examples, heparin-induced thrombocytopenia (HIT) and systemic lupus erythematosus (SLE).
Table 9.2
Autoimmune disease: Heparin-induced thrombocytopenia
Heparin-induced thrombocytopenia (HIT) is a common complication associated with the use of the anticoagulant drug heparin (see Chapter 29). Heparin serves as a hapten and forms molecular complexes with the tissue-specific platelet antigen called platelet factor 4 (PF4). These hapten/antigen complexes are then attacked by autoantibodies causing type II hypersensitivity. When these autoantibodies bind to the surface of platelets, they cause two problems. The first is that they destroy platelets, leading to thrombocytopenia and bleeding. The second is that the destroyed platelets release particles that activate thrombin, forming clots that can block blood vessels (thrombosis).13
Approximately 2% of individuals treated with heparin will develop clinically significant HIT.14 To diagnose HIT, laboratory tests are done to document thrombocytopenia and/or thrombosis and to look for the presence of the autoantibodies. Management requires stopping heparin therapy and, if anticoagulation is still needed, substituting with a nonheparin anticoagulant.13
Autoimmune disease: Systemic lupus erythematosus
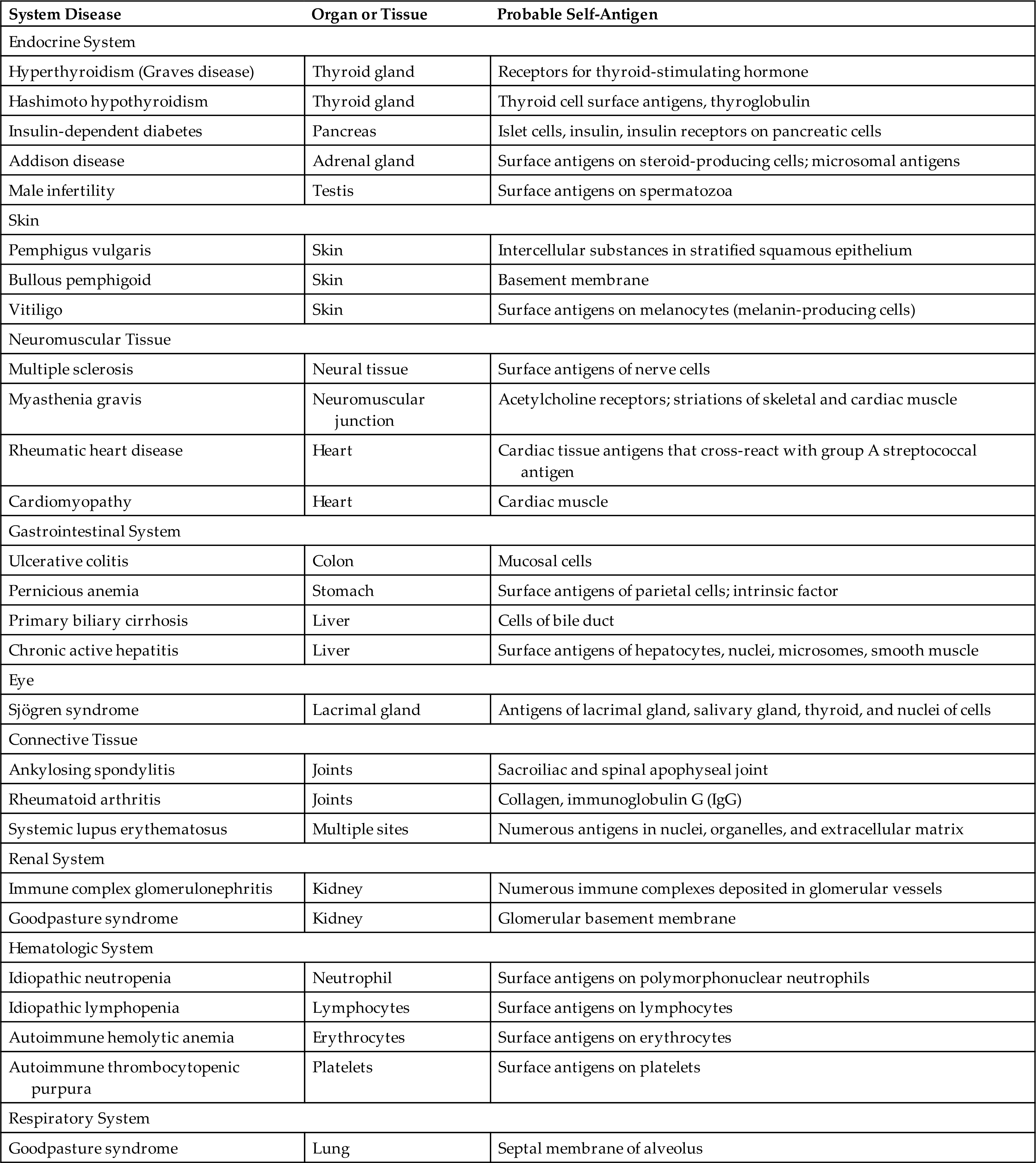
Systemic lupus erythematosus (SLE) is one of the most common, complex, and serious of the autoimmune disorders. It can affect any organ in the body. SLE is characterized by the production of a large variety of antibodies (autoantibodies) against self-antigens, including nucleic acids, erythrocytes, coagulation proteins, phospholipids, lymphocytes, platelets, and many other self-components. The most characteristic autoantibodies are against nucleic acids (e.g., single-stranded deoxyribonucleic acid [ssDNA], double-stranded DNA [dsDNS]), histones, ribonucleoproteins, and other nuclear materials. The blood normally contains many of these products of cellular turnover and breakdown. In SLE, autoantibodies react with the circulating antigen and form circulating immune complexes. The deposition of circulating DNA/anti-DNA complexes in the kidneys can cause severe kidney inflammation (Fig. 9.7). Similar reactions can occur in other systems, such as the brain, heart, spleen, lung, gastrointestinal tract, peritoneum, and skin. Thus many of the symptoms of SLE result from a type III hypersensitivity reaction and affect many systems. Other symptoms are related to type II hypersensitivity reactions and include destruction of red blood cells (anemia), lymphocytes (lymphopenia), and platelets (thrombocytopenia).
These photographs of tissue were obtained from individuals with lupus and stained with fluorescent anti-IgG. (A) Section from a kidney showing a glomerulus with deposits of IgG (arrow, indicating bright areas of staining). (B) Section of the skin showing deposition of IgG along the dermal-epidermal junction (arrow, indicating bright green staining). A courtesy Dr. Helmut Rennke, Department of Pathology, Brigham and Women's Hospital, Boston; B courtesy Dr. Richard Sontheimer, Department of Dermatology, University of Texas Southwestern Medical School, Dallas.
SLE, like most autoimmune diseases, occurs more often in women (approximately a 9:1 predominance of females), especially in the 20- to 40-year-old age group.15 Blacks are affected more often compared with Whites (approximately an eightfold increased risk). Several genes have been identified that are associated with an increased risk for SLE, including changes in MHC molecular structure.16,17 Environmental triggers (e.g., ultraviolet radiation exposure, smoking, medications, viruses such as Epstein-Barr virus [EBV], low vitamin D levels, environmental pollutants) and hormonal changes interplay with genetic predisposition in disease development and activity.18 As in many autoimmune conditions, microbiome dysbiosis is thought to negatively impact adaptive immune responses in SLE.19
As with other autoimmune diseases, clinical manifestations of SLE may wax and wane; the individual may go through periods of remission and be relatively disease free until the onset of a flare (exacerbated disease activity). Clinical manifestations of SLE depend on organ systems involvement, including skin (rashes and photosensitivity), eyes (keratoconjunctivitis, scleritis, uveitis retinopathy), mucus membranes (ulcers), joints (arthralgias), linings of the viscera (serositis, pleuritis), kidney (proteinuria), blood (anemia), gut (abdominal pain, vasculitis, hepatobiliary disease), and the neurologic system (seizures or psychosis).20 Lupus nephritis is common and carries a high risk for end-stage renal failure. Cardiovascular disease is common.21 Fever also may be present. Classification of SLE is based on the type and severity of organ system damage along with measurement of antibodies and complement levels.22
Laboratory diagnosis is usually based on a positive antinuclear antibody (ANA) screening test. This is a very sensitive test, but a substantial number of false-positive results occur in healthy individuals and those with other diseases. Detection of ANAs is usually followed by one or more specific tests (e.g., antibodies against Smith antigen [Sm], and dsDNA).20 Further diagnostic testing may be indicated such as urinalysis and serum C reactive protein and complement levels.
There is no cure for SLE or most other autoimmune diseases. Fatalities resulting from SLE are usually related to infection, organ failure, or cardiovascular disease. The goals of treatment are to control symptoms and prevent further damage by suppressing the autoimmune response. Ultraviolet light may initiate flares, and protection from sun exposure is helpful. Nonsteroidal antiinflammatory drugs (NSAIDs), such as ibuprofen, reduce inflammation and relieve pain. Hydroxychloroquine is the preferred treatment for individuals with stable disease. Corticosteroids are often prescribed for flares and more serious active disease. Immunosuppressive drugs, (e.g., methotrexate, cyclophosphamide, azathioprine, tacrolimus, or mycophenolate mofetil) are used to treat severe symptoms. Immunotherapies focused on B-cell depletion (e.g., belimumab, rituximab) are used in selected individuals.23 Many new biologic agents, such as anifrolumab (anti–type I interferon receptor antibody) and ustekinumab (antibody against IL-12/23 [p40]), are in clinical trials.24
Alloimmunity
Alloimmunity (isoimmunity) occurs when the immune system of one individual produces an immunologic reaction against tissues of another individual. Alloantigens (isoantigens) are nonself-antigens from members of the same species. No two individuals have exactly the same antigens on their tissues; therefore the introduction of one individual’s tissues and cells into another will result in a strong immune response to the foreign antigens. Alloimmunity can be observed during immunologic reactions to blood transfusions, fetal tissues, or transplanted tissue.
Alloimmune disease: Transfusion reactions
Red blood cells (erythrocytes) express several important surface antigens, which are known collectively as the blood group antigens and can be targets of alloimmune reactions. More than 80 different red blood cell antigens are grouped into several dozen blood group systems. The most important of these, because they provoke the strongest humoral alloimmune response, are the ABO and Rh systems.
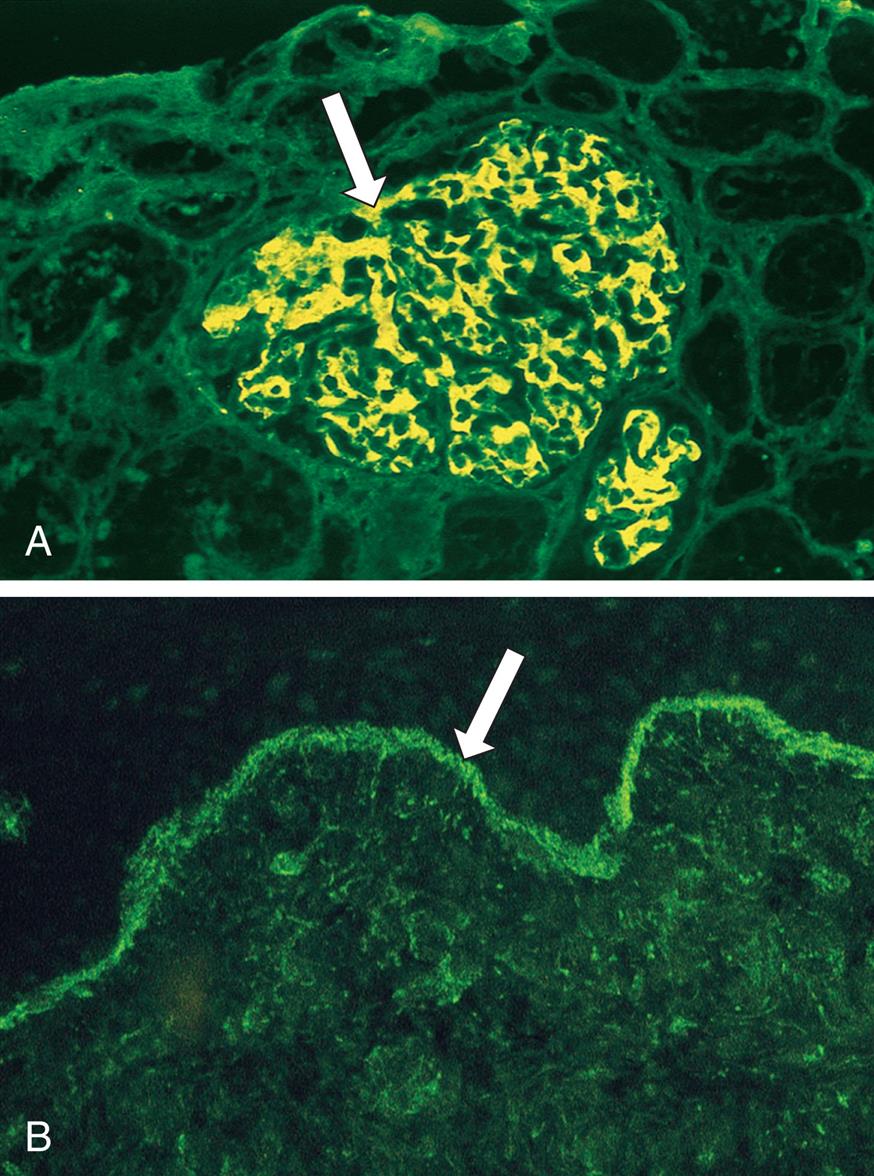
The ABO blood group consists of two major carbohydrate antigens, labeled A and B (Fig. 9.8), that are expressed on virtually all cells. These are codominant so that both A and B can be simultaneously expressed, resulting in an individual having any one of four different blood types. The erythrocytes of blood type A express the type A carbohydrate antigen, those with blood type B express the B antigen, those with blood type AB express both A and B antigens on the same cell, and those of blood type O express neither the A nor the B antigen. A person with type A blood also has circulating antibodies to the B carbohydrate antigen. If this person receives blood from a type AB or B individual, a severe transfusion reaction occurs, and the transfused erythrocytes are destroyed by agglutination or complement-mediated lysis. Similarly, a type B individual (whose blood contains anti-A antibodies) cannot receive blood from a type A or AB donor. Type O individuals, who have neither antigen but have both anti-A and anti-B antibodies, cannot accept blood from any of the other three types. These naturally occurring antibodies, called isohemagglutinins, are IgM antibodies developed early in life because of the presence of similar antigens expressed on naturally occurring bacteria in the intestinal tract.
This figure shows the relationship of antigens and antibodies associated with the ABO blood groups. The surfaces of erythrocytes of individuals with blood group O have a core carbohydrate that is present on cells of all ABO blood groups (H antigen, depicted in blue). The sera of blood group O individuals contain immunoglobulin M (IgM) antibodies against both A and B carbohydrates. In individuals of the blood group A, some of the H antigens have been modified into A antigens (depicted in green). The sera of these individuals have IgM antibodies against the B antigen. In individuals with blood group B, some of the H antigens have been modified into B antigens (depicted in yellow). These individuals have IgM antibodies against the A antigen in their sera. In individuals of the blood group AB, some of the H antigens have been modified into both the A and B antigens. These individuals do not have antibody to either A or B antigens.
A series of illustrations shows A B O blood types. The erythrocyte for O blood type comprises H antigen, and the antibody in serum comprises anti-A and anti-B. The erythrocyte for A blood type comprises A antigen and H antigen, and the antibody in serum comprises anti-B. The erythrocyte for B blood type comprises B antigen and H antigen, and the antibody in serum comprises anti-A. The erythrocyte for A B blood type comprises A antigen, B antigen, and H antigen, and the there are no antibody in serum.
Harmful transfusion reactions can be prevented by complete and careful ABO matching between donor and recipient. Because individuals with type O blood lack both types of antigens, they are considered universal donors—that is, anyone can accept their red blood cells. Similarly, type AB individuals are considered universal recipients because they lack both anti-A and anti-B antibodies and can be transfused with any ABO blood type.
Alloimmune disease: Hemolytic disease of the fetus and newborn
Hemolytic disease of the fetus and newborn (HDFN) (also known as erythroblastosis fetalis) is a condition in which maternal blood antigens do not match those of the fetus. This disorder usually results from incompatibility between maternal and fetal red blood cell (erythrocyte) Rho(D) antigens. The Rh blood group is a group of antigens expressed on red blood cells. This is the most diverse group of red blood cell antigens, consisting of at least 45 separate antigens, although only one is considered of major importance: the D antigen. Individuals who express the D antigen on their red blood cells are Rh positive, whereas individuals who do not express the D antigen are Rh negative. Approximately 85% of North Americans are Rh positive. Rh-negative individuals make IgG antibody to the D antigen (anti-D) if exposed to Rh-positive erythrocytes. In an Rh-negative mother whose fetus is Rh-positive, antibodies from the mother cross the placenta and attack fetal erythrocytes. The attack induces severe anemia in the fetus (erythroblastosis fetalis) or in the newborn (erythroblastosis neonatorum). With each successive pregnancy with an Rh-positive fetus, the mother’s immune system makes anti-D antibodies faster and in greater amounts. Diagnosis involves measurement of maternal antibodies and fetal testing. Prenatal and postnatal management may include intrauterine transfusions, exchange transfusions, intravenous immunoglobulin (IVIG), or plasmapheresis.25 The occurrence of this particular form of the disease has decreased dramatically because of the use of prophylactic anti-D immunoglobulin (i.e., RhoGAM). Administration of anti-D antibody within a few days of exposure to RhD-positive erythrocytes prevents sensitization against the D antigen (see Chapter 30).
Alloimmune disease: Transplant rejection
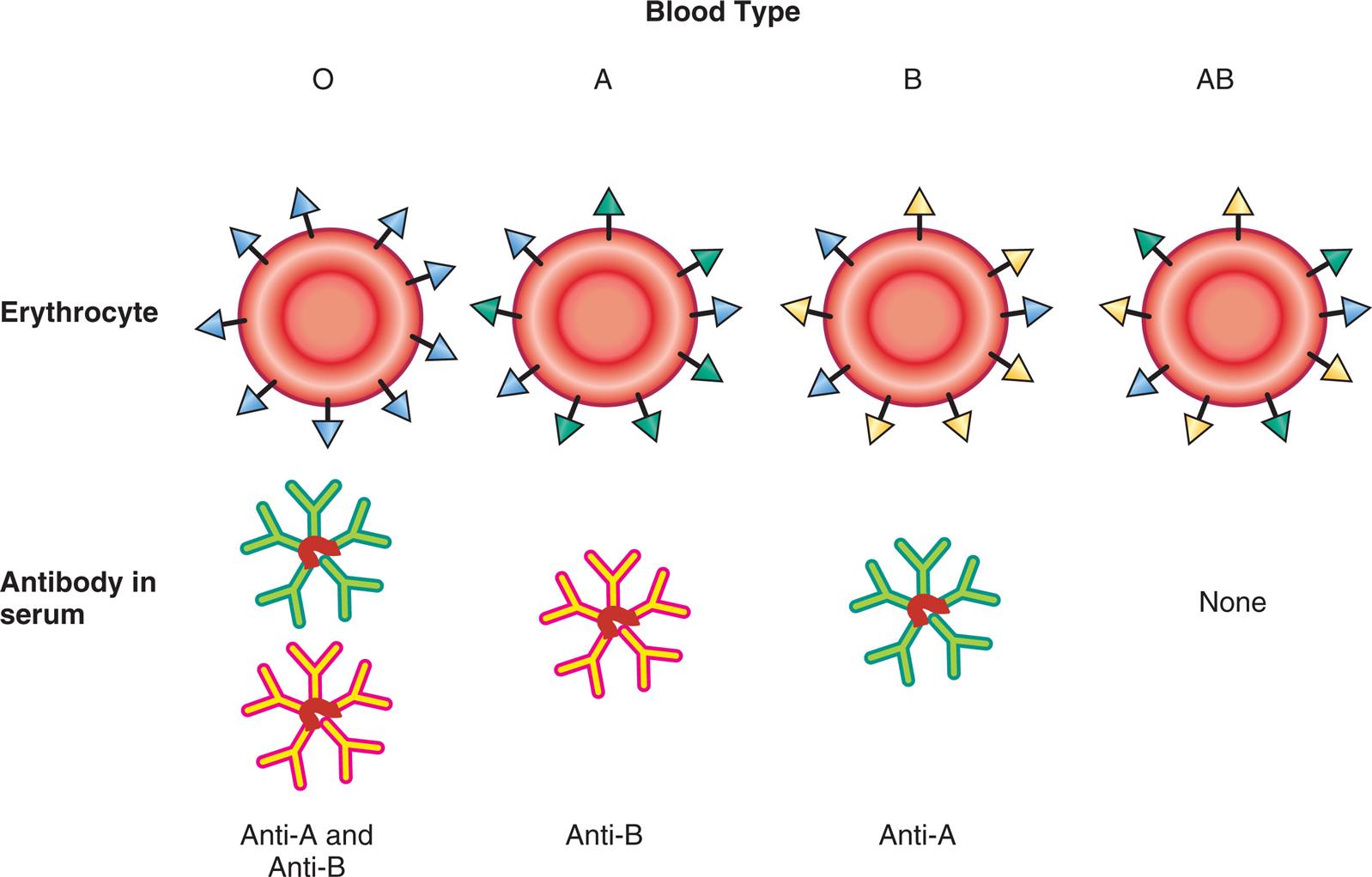
Molecules of the major histocompatibility complex (MHC) were discussed in Chapter 8 as antigen-presenting molecules (see Figs. 8.6 and 8.7). MHC molecules also are a major target of transplant rejection. The human MHC molecules are also referred to as human leukocyte antigens (HLAs), especially in the context of transplantation. The different MHC genetic loci are identified as class I: HLA-A, HLA-B, and HLA-C, and class II: HLA-DR, HLA-DQ, and HLA-DP (Fig. 9.9). Humans have two copies of each MHC locus (one inherited from each parent) that are codominant so that molecules encoded by each parent's genes are expressed on the surface of every cell, except erythrocytes. The tremendous number of possible alleles that can be expressed throughout the population makes it highly unlikely that any two unrelated individuals will have the same HLA antigens. This diversity of HLA molecules becomes clinically relevant during organ transplantation. The recipient of a transplant will mount an immune response against the foreign HLA antigens on the donor tissue, resulting in rejection. To minimize the chance of tissue rejection, the donor and recipient are tissue-typed to identify differences in HLA antigens prior to transplantation. The more similar the two individuals are in their HLA tissue type, the more likely it is that transplantation will be successful. The chance of finding a reasonably close match among siblings is much higher than the general population, and, clearly, the most successful transplants would be between identical twins because they are nearly identical genetically.
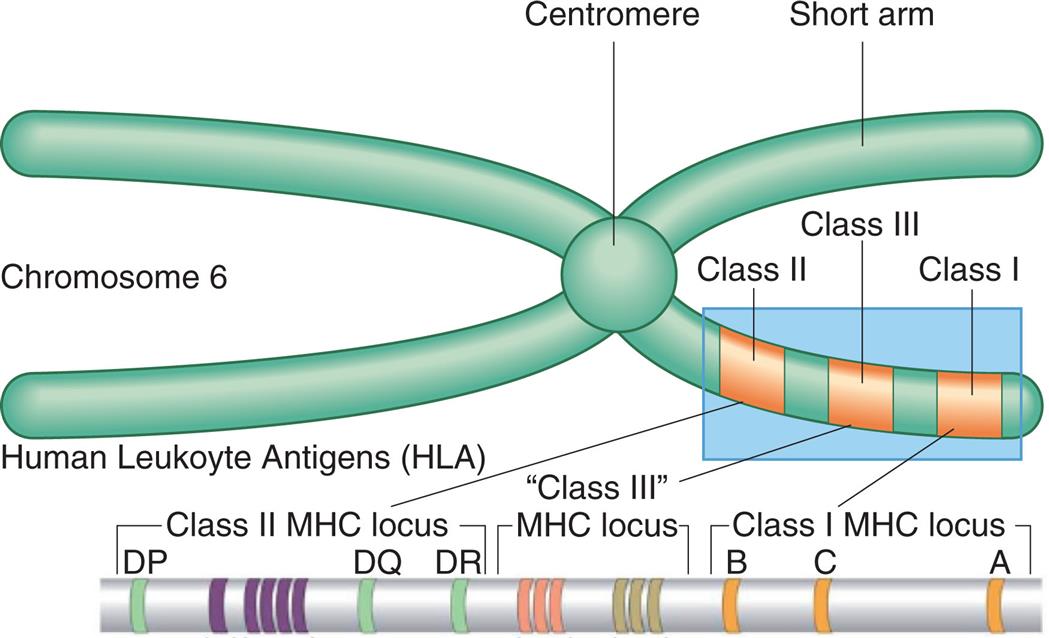
The major histocompatibility complex (MHC) is located on the short arm of chromosome 6 and contains genes (genetic loci) that code for class I antigens (found mostly on nucleated cells), class II antigens (found mostly on dendritic cells, macrophages and B lymphocytes), and class III proteins (i.e., complement proteins and cytokines). From Peakman M, Vergani D. Basic and clinical immunology, 2nd edition. London: Churchill Livingstone; 2009 and Abbas AK, Lichtman AH, Pillai S. Basic immunology, 4th edition. St. Louis: Elsevier; 2014.
An illustration of chromosome 6 is labeled with the centromere and the short arms. The following protein classes are labeled on the short arm, from the outside toward the centromere: class 1, class 3, and class 2. The classification of the human leukocyte antigens (H L A) are as follows: • Class 1 M H C locus. From the right to the left: A, C, B. • Class 3 M H C locus. • Class 2 M H C locus. From the right to the left: D R, D Q, and D P.
When donor tissue is transplanted into a recipient, the recipient’s T cells recognize alloantigens in two different ways: (1) direct recognition of foreign HLA antigens, and (2) indirect recognition of donor peptides that are presented on recipient antigen-presenting cells (see Algorithm 9.1). Both processes result in the widespread activation and proliferation of Th cells. Th cells differentiate into several subtypes that produce numerous cytokines, including IL-2 and interferon (IFN)-γ (Th1 cells), IL-4 (Th2 cells), and IL-17 (Th17 cells) (see Chapter 8). These cytokines activate T cytotoxic cells, macrophages, NK cells, and B cells such that there is an intense cell-mediated and humoral attack on the graft tissue.
Transplant rejection may be classified as hyperacute, acute, or chronic, depending on the amount of time that elapses between transplantation and rejection and the mechanisms by which rejection occurs. Hyperacute rejection is immediate and rare. Hyperacute rejection occurs because of the presence of preexisting antibodies (type II hypersensitivity) to HLA antigens on the vascular endothelial cells in the grafted tissue. These preexisting antigens are usually found in individuals who have received multiple blood transfusions or previous transplants. When the circulation is reestablished to the grafted area, the graft may immediately turn white (the so-called white graft) instead of a normal pink color. Hyperacute rejection can be avoided by testing the recipient for preexisting antibodies prior to transplantation.
Acute rejection occurs within days to months after transplantation. This type of rejection occurs when the recipient develops an immune response against unmatched HLA antigens after transplantation. Both humoral and cell-mediated immune responses play a role (Algorithm 9.2). Direct and indirect recognition of alloantigens result in release of Th1, Th2, and Th17 cytokines. Alloantibodies are formed to graft blood vessels (type II hypersensitivity) which then activate complement, resulting in necrosis of graft vessels (vasculitis). Tc cells and macrophages are activated, resulting in direct lysis of graft cells and disruption of tissue architecture; this leads to graft dysfunction and destruction (type IV hypersensitivity). NK cells and other components of innate immunity also play a role. The release of IL-2 is of particular importance in acute rejection; thus several antirejection medications target IL-2 synthesis or receptors (e.g., cyclosporin, tacrolimus, basiliximab). Other antirejection medications deplete T- and B-cell numbers, metabolism, and function (e.g., mycophenolate mofetil, rituximab, prednisone, azathioprine, belatacept), or block inflammatory cytokines such as IL-6 (e.g., tocilizumab). New methods for achieving graft tolerance include adoptive transfer of donor and recipient Treg cells and myeloid-derived suppressor cells.26,27 Studies are exploring the potential for modifying the epigenetic profiles of immune cells and the use of messenger RNA (mRNA) in the prevention of graft rejection.28 The choice of immunosuppressive medications is based on the type of organ being transplanted and the potential for recipient toxicity.
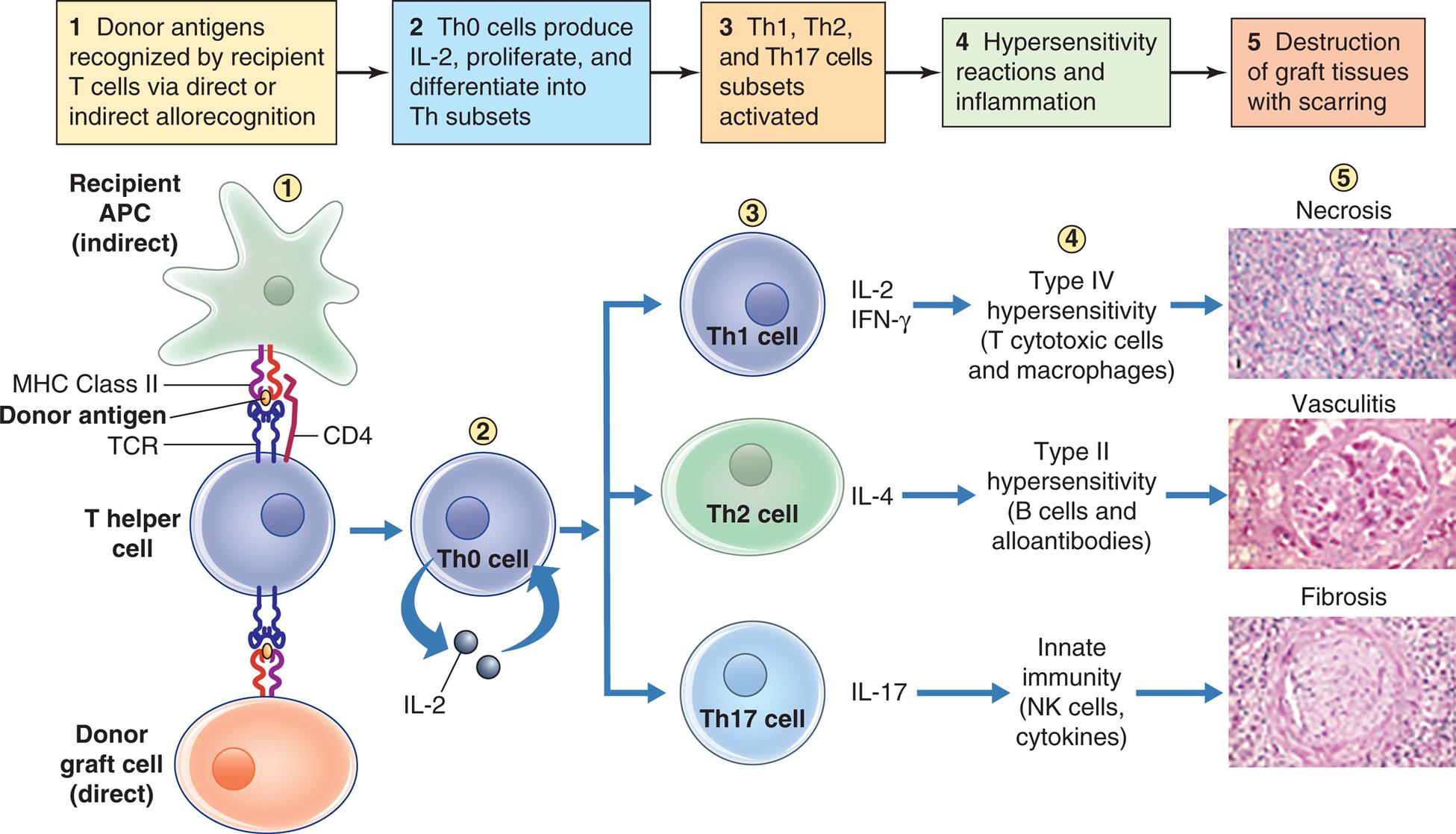
Acute graft rejection occurs days to months after transplantation. Recipient T helper (Th) cells recognize graft human leukocyte antigens (direct allorecognition) or donor antigens presented on recipient antigen-presenting cells (indirect allorecognition). These cells then secrete IL-2, proliferate, and differentiate into Th1, Th2, or Th17 cells. Th1 cells secrete IL-2 and IFN-γ which activate T cytotoxic cells and macrophages which participate in type IV hypersensitivity (cell-mediated immunity). Th2 cells secrete IL-4 which activate B cells to become alloantibody-producing plasma cells. Th17 cells secrete IL-17 which stimulates inflammation. These processes result in necrosis of graft cells, vasculitis of graft blood vessels, and fibrosis (scarring) of graft tissue. The end result is dysfunction and death of the transplanted organ. IFN-γ, Interferon-gamma; IL, interleukin; MHC, major histocompatability complex; Th, T helper; TCR, T cell receptor.
An illustration flowchart demonstrates acute graft rejection. There are five stages of rejection. 1. Donor antigens recognized by recipient T cells via direct or indirect allorecognition. Recipient A P C (indirect) and donor graft cell (direct) bind to the T helper cell through M H C class 2, donor antigen, T C R, and C D 4. 2. T h 0 cells produce I L-2, proliferate, and differentiate into T h subsets. 3. T h 1, T h2, and T h 17 cells subsets activated. 4. Hypersensitivity reactions and inflammation. 5. Destruction of graft tissue with scarring. The result of activation of the different cells are as follows: • T h 1 cell secretes I L-2, I F N-gamma. Type 4 hypersensitivity (T cytotoxic cells and macrophages) leads to necrosis. An accompanying photomicrograph shows dead cells. • T h 2 cell secretes I L-4. Type 2 hypersensitivity (B cells and alloantibodies) leads to vasculitis. An accompanying photomicrograph shows inflamed blood vessels. • T h 17 cell secretes I L-17. Innate immunity (N K cells, cytokines) leads to fibrosis. An accompanying photomicrograph shows a network of fibrous connective tissue.
Chronic rejection may occur after a period of months or years of normal function. It is characterized by slow, progressive organ failure. Chronic rejection occurs most often in recipients who were poorly matched to their donor, have comorbidities (e.g., diabetes, hypertension), received a graft that was in poor condition or was damaged during the transplantation procedure, or have required treatment for multiple acute rejection episodes.
Chronic rejection involves several mechanisms. A cell-mediated (type IV hypersensitivty) reaction against minor histocompatibility antigens on the grafted tissue contributes to persistent Tc-cell and phagocyte activation. Th17 cytokines trigger chronic inflammation. There also is binding of alloantibodies to donor graft MHC molecules, resulting in complement activation and tissue destruction (late antibody-mediated rejection).29 These processes are subacute and slowly progressive, leading to graft fibrosis (scarring), dysfunction, and tissue death. Once chronic rejection is well established, there are few effective treatments, and it may be necessary to replace the graft with a new transplanted organ.
Deficiencies in Immunity
Immune deficiency is the failure of the immune or inflammatory response to function normally, resulting in increased susceptibility to infections and cancer. Primary (congenital) immune deficiency is caused by a genetic defect, whereas secondary (acquired) immune deficiency is caused by another condition, such as cancer, infection, or normal physiologic changes, such as aging. Acquired forms of immune deficiency are far more common than the congenital forms.
Initial Clinical Presentation
The clinical hallmark of immune deficiency is a tendency to develop unusual or recurrent, severe infections. The most severe primary immune deficiencies develop in children 2 years of age and younger. Potential immune deficiencies should be considered if the individual has experienced severe, documented bouts of pneumonia, otitis media, sinusitis (sinus infection), bronchitis, septicemia (blood infection), meningitis, or infections with opportunistic microorganisms (e.g., Pneumocystis jiroveci). Infections are generally recurrent, and multiple simultaneous infections are common. Invasive fungal infections are rare in healthy individuals and strongly indicate a defective immune system. Children frequently present with failure to thrive because of diarrhea and other chronic symptoms. A familial history of immune deficiency may be found in some types of primary deficiency.
The type of recurrent infections may indicate the type of immune defect. Deficiencies in T-cell immune responses are associated with recurrent infections caused by certain viruses (e.g., varicella zoster, cytomegalovirus [CMV]), fungi, yeasts (e.g., Candida, Histoplasma), or atypical microorganisms (e.g., P. jiroveci). B-cell deficiencies and phagocyte deficiencies are suggested if the individual has documented, recurrent infections with microorganisms that require opsonization (e.g., encapsulated bacteria, such as Pneumococcus) or those with viruses against which humoral immunity is normally effective (e.g., rubella virus). Some complement deficiencies resemble defects in antibody or phagocyte function, but others are associated with disseminated infections with bacteria of the genus Neisseria (Neisseria meningitides and Neisseria gonorrhoeae).
Primary (Congenital) Immune Deficiencies
Most primary immune deficiencies are the result of single gene defects. To date, 430 gene mutations are associated with immunodeficiency disorders.30 In general, the mutations are sporadic and not inherited: a family history exists in only approximately 25% of individuals. The sporadic mutations occur before birth, but the onset of symptoms may be early or later, depending on the particular syndrome. In some instances, symptoms of immune deficiency appear within the first 2 years of life. Other immune deficiencies are slowly progressive, with the onset of symptoms appearing in the second or third decade of life.
The prevalence of primary immune deficiency diseases globally varies dramatically from country to country, and data are complicated by differences in diagnostic and reporting methods. The prevalence of diagnosed primary immune deficiency in North America is approximately 5 cases per 100,000 individuals; however, it is estimated that as many as 70% of cases are undiagnosed.31 Many are subtle with minor deficiencies, but several result in major defects and lead to recurrent life-threatening infections. Sex distribution is approximately even, although some specific diseases have a male or female predominance. Primary immune deficiencies are classified into 10 groups, based on the principal component of the immune or inflammatory systems that is defective.30 Of these 10 groups, the most common disorders are included within combined immunodeficiencies (affecting both cellular and humoral immunity). These disorders may also be associated with syndromic features. The other classification groups include predominantly antibody deficiencies, diseases of immune dysregulation, defects in phagocyte number or function, defects in innate immunity, autoinflammatory disorders, complement defects, bone marrow failure, and phenocopies of inborn errors of immunity.30 To provide a better understanding of the diversity and severity of primary immune deficiencies, a few select examples of each category will be discussed.
Combined Deficiencies
Combined deficiencies include the most life-threatening disorders and result from defects that directly affect the development of both T and B lymphocytes. The severity of each disorder depends on the degree to which B and T cells are affected.
Combined deficiencies wthout nonimmunologic abnormalities
Severe combined immunodeficiencies (SCIDs) are the most common combined deficiency without nonimmunologic abnormalities. Most often, it is inherited in an autosomal recessive pattern, or it may be X-linked.32 There are at least 20 different forms of SCID, depending on how the underlying genetic defect affects lymphocyte development and function. T-cell differentiation is defective, and, depending on the type of SCID, B cells and NK cells also are affected.32 Most individuals with SCIDs have few detectable lymphocytes or NK cells in the circulation and secondary lymphoid organs (spleen, lymph nodes). Immunoglobulin levels, especially IgM and IgA, are absent or greatly reduced. The most severe form of SCID is due to reticular dysgenesis in which a common stem cell fails to develop into mature immune cells. Most children with this form of SCID die in utero or soon after birth.
Several forms of SCID are caused by autosomal recessive enzymatic defects that result in the accumulation of toxic metabolites, and rapidly dividing cells, such as lymphocytes, are especially sensitive. For instance, deficiency of adenosine deaminase (ADA deficiency) results in the accumulation of toxic purines. Enzyme replacement therapy is available for this form of SCID.33X-linked SCID results from a common defect in important IL receptors needed for lymphocyte maturation (e.g., IL-2, IL-4, IL-7, and others). Recent reports demonstrate the remarkable effectiveness of gene therapy in which a lentivirus is used as a vector to insert a normal copy of the IL-2 receptor (IL-2R) gene into a person’s own hematopoietic stem cells.34
Even if nearly adequate numbers of B and T cells are produced, their cooperation may be defective. Bare lymphocyte syndrome is the form of SCID characterized by the inability of lymphocytes and macrophages to produce MHC class I or class II molecules. Without MHC molecules, antigen presentation and intercellular cooperation cannot occur effectively. Children with this deficiency develop serious, life-threatening infections and usually die before age 5 years.
A SCID newborn screening test is available and has been in use in the United States since 2008. After an abnormal screening, additional testing is needed to determine which type of SCID is present.35 This approach has resulted in prompt treatment and higher survival rates and is now performed for all newborns in the United States. Hematopoietic stem cell transplantation is the standard treatment for infants with SCID; unfortunately, it is not universally effective and can be associated with serious complications such as graft-versus-host disease (GVHD). New gene therapies are emerging for several forms of SCID.36
Combined deficiencies with nonimmunologic abnormalities
Wiskott-Aldrich syndrome (WAS), an X-linked disorder characterized by the clinical triad of low platelet count (thrombocytopenia), eczema, and recurrent infections, is a condition in which IgM antibody production is greatly depressed. WAS results from a mutation of the WAS gene which causes defects in the WAS protein (WASP). WASP is required for normal differentiation of B cells and several other hematopoietic cell types. For example, defective WASP affects the actin cytoskeleton, which is important for platelet function. Immunodeficiencies include reduced antibody responses against antigens that primarily elicit an IgM response, such as polysaccharide antigens from bacterial cell walls (e.g., Pseudomonas aeruginosa, Streptococcus pneumoniae, Haemophilus influenzae). Defective WASP also impacts neutrophil migration and function. Clinical manifestations include bleeding, eczematous rash, and recurrent infections (e.g., otitis media, pneumonia, herpes simplex, CMV). Autoimmune conditions (e.g., hemolytic anemia) are also common. Management includes IVIG or subcutaneous immunoglobulin (SQIG), stem cell transplantation, and gene therapy.37
Chromosome 22q11.2 deletion syndrome (DiGeorge syndrome or velocardiofacial syndrome) is another combined immunodeficiency with syndromic features. Chromosome 22q11.2 deletion is the most common microdeletion genetic syndrome in humans.38 This mutation is associated with a wide spectrum of phenotypes even within families. The deletion of a particular gene, T-box transcription factor 1 (TBX1), is thought to be responsible for many of the syndrome's characteristic signs and symptoms. The TBX1 gene provides instructions for making a protein called T-box 1. The T-box 1 protein is necessary for the development of muscles and bones of the face and neck, aorta, and the thymus and parathyroid glands.39 In most cases, hypoplasia of the thymus results in greatly decreased T-cell numbers and function. Immunocompromise with susceptibility to infection is complicated by a shift in Th-cell function to Th2 predominance resulting in allergies, as well as a reduction in Treg function resulting in autoimmune disorders. Cardiac and endocrine problems are also common. Defective development of the third and fourth pharyngeal pouches during embryonic development results in thymic defects and the absence of the parathyroid gland (causing inability to regulate calcium concentration). Low blood calcium levels cause the development of tetany or involuntary rigid muscular contraction. This syndrome frequently is associated with abnormal development of facial features that are controlled by the same embryonic pouches; these include low-set ears, fish-shaped mouth, and other altered features (Fig. 9.10). Loss of this gene may also contribute to behavioral problems such as schizophrenia and bipolar disorder. Dopaminergic neurons are also affected, leading to an increased risk for development of Parkinson disease later in life.40 Management includes monitoring and intervention for hypoparathyroidism, heart defects, facial abnormalities, infections, and neurologic complications. No specific treatment for the underlying condition is currently available.
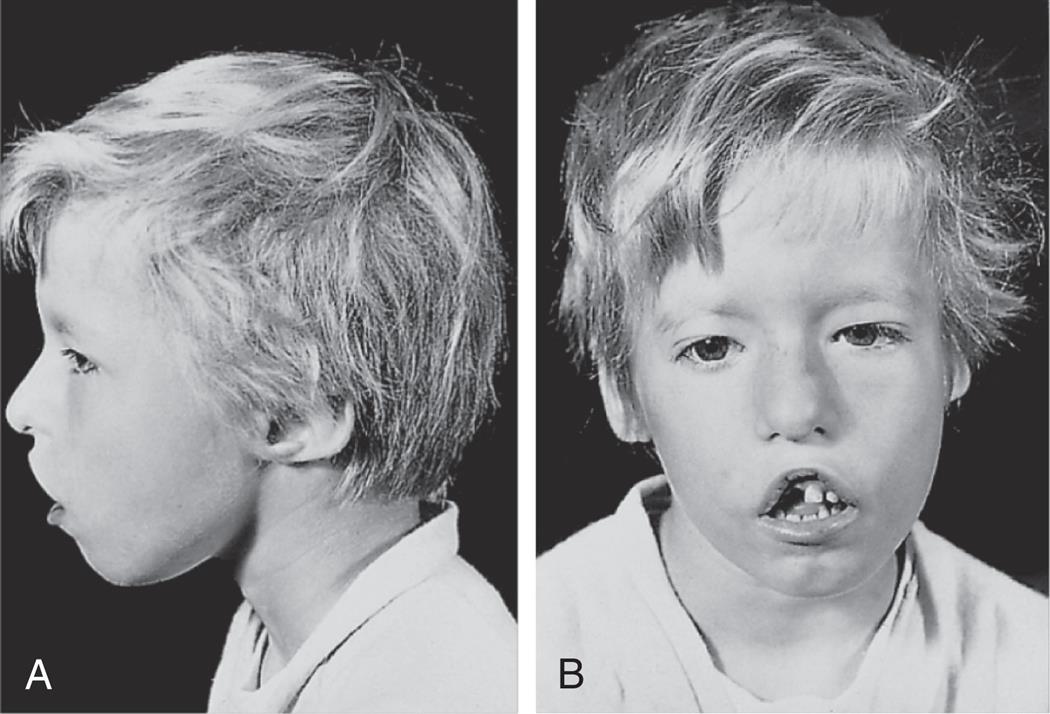
Note the wide-set eyes (B), low-set ears (A and B), shortened structure of the upper lip (B), and underdeveloped chin (A and B). From Male D, Brostoff J, Roth D, et al. Immunology, 8th edition. St. Louis: Mosby; 2013.
Predominantly Antibody Deficiencies
Predominantly antibody deficiencies result from defects in B-cell maturation or function and are the most common of immune deficiencies. T-cell immune responses are not affected in pure B-lymphocyte deficiencies. The results are lower levels of circulating immunoglobulins (hypogammaglobulinemia) or occasionally totally or nearly absent immunoglobulins (agammaglobulinemia).
Common variable immune deficiency
Common variable immunodeficiency (CVID) is the most common symptomatic primary immune deficiency, affecting up to 1 in 10,000 individuals.41 There are two peak ages of onset, one before the age of 10 and another between 30 and 40 years of age. As the name implies, the presentation is very heterogeneous. It is characterized by hypogammaglobulinemia, but the particular class of antibody that is decreased varies. Although B-cell numbers are normal, most individuals have low amounts of IgG, which may or may not be accompanied by decreased levels of IgA and IgM. Multiple genetic defects in terminal differentiation of B lymphocytes may account for this condition, although the pathogenesis remains poorly understood.42 Failure to produce sufficient immunoglobulins results in recurrent infections in 90% of individuals with CVID. Pneumonia caused by S. pneumoniae or H. influenzae, and infections with adenovirus, CMV, and varicella zoster are common.41 Secondary complications include arthritis (infectious and noninfectious), gastrointestinal symptoms (malabsorption, chronic diarrhea), autoimmune disease (anemia, thrombocytopenia, endocrine diseases), and cancer (of the lymphoid system, skin, and gastrointestinal tract).43 Interstitial lung disease is a significant cause of morbidity and mortality in CVID and is believed to be the result of dysregulated B-cell function.44 Because of the heterogeneity of clinical features, diagnosis is difficult and may be delayed for several years. The advent of new diagnostic techniques will likely improve outcomes.42
Selective IgA deficiency
Some defects may involve a particular class of antibody, such as selective IgA deficiency, in which only IgA is suppressed. It is the most prevalent of the selective antibody deficiencies and is defined as a decreased serum IgA level lower than 7 mg/dL in individuals older than 4 years with normal levels of IgM and IgG in serum and exclusion of other causes of hypogammaglobulinemia. Selective IgA deficiency occurs in 1 in 700 to 1 in 400 individuals, and familial inheritance occurs in approximately 20% of cases. There is an increased incidence of common variable immune deficiency and transient hypogammaglobulinemia of infancy among family members of those with selective IgA deficiency. It results from defects in the process of IgA class switch, production and secretion of IgA, and long-term survival of IgA-switched memory B cells and plasma cells.45 Many individuals are asymptomatic, although others have a history of recurring sinus, joint, and pulmonary infections.41 Some have gastrointestinal infection such as chronic intestinal candidiasis (infection with C. albicans). Complications of IgA deficiency include severe allergic disease and autoimmune diseases. Some individuals are at risk for life-threatening allergic reactions when they receive blood products that contain some IgA. This is thought to be due to IgG (or possibly IgE) anti-IgA antibodies, which may be found in some IgA-deficient individuals. Management includes treatment of infections and allergic reactions, but no specific treatment for this immunodeficiency is currently available.
IgG subclass deficiency
Deficiencies in certain subclasses of antibody, particularly IgG2 (IgG subclass deficiency), may result from a defect in switch to a particular subclass constant region. A reduced level of IgG2 results in an inability to adequately attack polysaccharide antigens on the surface of encapsulated bacteria. Low levels of IgG2 therefore are responsible for recurrent risk for pneumonias caused by these bacteria. Management with IVIG reduces the number of infections.46
Bruton agammaglobulinemia
Bruton agammaglobulinemia (X-linked agammaglobulinemia) is caused by blocked development of mature B cells in bone marrow. It results from mutations of genes responsible for the synthesis of Bruton tyrosine kinase (BTK). An absence of BTK causes absent precursor B-cell differentiation in the bone marrow and severe B-cell deficiency (<2%).47 There is an associated inability to generate plasma cells and antibodies of all classes (panhypogammaglobulinemia). B-lineage cells in all organs are affected resulting in reduced sizes of lymph nodes and tonsils. Encapsulated bacterial infections are common. Nearly 85% of affected individuals develop infections early in life including otitis media, skin infection, sepsis, sinusitis, acute gastroenteritis, cervical lymphadenitis, epididymitis, meningitis, osteomyelitis, urinary tract infection, and encephalitis. Treatment consists of replacement immunoglobulin and prophylactic antibiotics to prevent infections.
Immune Dysregulation
Primary immunodeficiencies categorized as immune dysregulation include disorders specific to Tc and NK cell function. This group of conditions is also characterized by autoimmune conditions as a result of alterations in tolerance.
Perforin deficiency results from a mutation in the perforin gene, the product of which is an important component of cellular killing by Tc and NK cells. Several other perforin deficiencies result from mutations in secretory vesicle function, including fusion with the cell membrane and other aspects of the process.
Chédiak-Higashi syndrome results from a defect in the movement of intracellular cytoplasmic granules and is caused by an autosomal recessive mutation in the lysosomal trafficking regulator (LYST) gene. Dysregulation of LYST function results in defects in lysosomal biogenesis.48 As a result of these defects, the granules remain in the cytoplasm and form large aggregates that are readily apparent microscopically. Leukocytes from individuals with Chédiak-Higashi syndrome have decreased chemotaxis, granular fusion, and bacterial killing. Platelet granules also may be affected, resulting in prolonged bleeding, and partial albinism can occur because of defects in melanocyte granules. Affected children develop recurrent infections of the skin, respiratory tract, and mucous membranes, especially with gram-positive bacteria.
Autoimmune conditions associated with primary immunodeficiencies caused by immune dysregulation may result from abnormalities in Treg function. For example, mutations in IL-10 or its receptor IL-10R suppress the production or response to IL-10 (an immunosuppressive cytokine from Treg cells), resulting in excessive inflammation and an increased risk for early-onset inflammatory bowel disease, or recurrent respiratory disease. Lymphoproliferative disorders (adenopathy, splenomegaly, colitis) may also occur.30
Phagocyte Defects: Number, Function or Both
Phagocyte defects include inadequate numbers of phagocytes (e.g., severe congenital neutropenia) or defects in phagocyte function. These defects result in recurrent infections caused by a wide range of microorganisms that can infect lungs, skin, bones, and other organs.
Congenital neutropenia results from the inheritance of a large range of autosomal dominant, recessive, or X-linked mutations that result in lack of neutrophil maturation and function. Without adequate neutrophils, the innate immune response is severely impacted, particularly with regard to defense against microorganisms. Congenital neutropenia results in recurrent bacterial infections commensurate with the absolute count of neutrophil granulocytes in the circulation.49 It is associated with several syndromic conditions, including oculocutaneous albinism, metabolic diseases, and bone marrow failure syndromes. Approximately 20% of people with severe congenital neutropenia develop certain cancerous conditions of the blood, particularly myelodysplastic syndrome or leukemia. Management includes in injections of granulocyte colony-stimulating factor and, in severe cases, bone marrow transplantation.
Primary autoimmune neutropenia (chronic benign neutropenia of childhood) is an idiopathic self-limiting condition affecting infants and toddlers. In most cases, mild to moderate decreases in neutrophil numbers resolve by early childhood without intervention. However, Black children have a 3.5 higher risk of having persistent neutropenia and may require treatment with granulocyte colony-stimulating factor.50
Chronic granulomatous disease (CGD) is a severe defect in the myeloperoxidase–hydrogen peroxide system (NADPH oxidase system)—a major means of bacterial destruction within the phagolysosome (see Chapter 7). Deficient production of hydrogen peroxide and other oxygen products needed for phagocytic killing results in recurrent severe pneumonias; tumor-like granulomata in the lungs, skin, and bones; and other infections with some opportunistic microorganisms, such as Staphylococcus aureus, Serratia marcescens, and Aspergillus species. These infections are characterized by recurrent episodes of cutaneous and organ abscess, pneumonia, and osteomyelitis. CGD is also associated with other phagocytic defects, including decreased Toll-like receptor (TLR) recognition of pathogens, inability to form neutrophil extracellular traps, and hyperinflammatory states (see Chapter 7).51
Defects in Innate Immunity
Some immune deficiencies are characterized by a defect in the capacity to produce an immune response against a particular antigen. Innate immune cytokines such as interferons and ILs may be affected. Depending on the specific defect, there can be increased susceptibility to infection by mycobacteria, (e.g., tuberculosis), viruses (e.g., herpes simplex), bacteria, or fungi.30 In chronic mucocutaneous candidiasis, interaction between the Th17 lymphocytes and macrophages is ineffective related to a specific infectious agent, Candida albicans. Thus the macrophage cannot be activated, and these individuals usually have mild to extremely severe recurrent Candida infections involving the mucous membranes and skin. Other defects in innate immunity include defects in TLRs and NK cells resulting in susceptibility to infections with mycobacteria, salmonella, and viruses (EBV, herpes, influenza viruses).
Autoinflammatory Disorders
Autoinflammatory disorders are characterized by abnormally high levels of inflammation secondary to mutations in genes that control inflammasome activation or in defects in cellular receptors of cytokines designed to decrease inflammation. These disorders are frequently related to diminished control of infections of epithelial surfaces. Vasculitis, inflammatory lung disorders, arthritis, inflammatory bowel disease, SLE, and other autoimmune conditions are common.30
Complement Deficiencies
Many complement deficiencies have been described. Every component of the complement cascade (see Chapter 7) may be affected.30 C3 deficiency is the most severe defect because of its central role in the complement cascade. Loss of C3b results in an inability to opsonize, causing recurrent life-threatening infections with encapsulated bacteria (e.g., H. influenzae and S. pneumoniae) at an early age. Deficiencies of any of the terminal components of the complement cascade (C5, C6, C7, C8, or C9 deficiencies) are associated with increased infections with only one group of bacteria—those of the genus Neisseria (N. meningitides or N. gonorrhoeae). Neisseria species usually cause localized infections (meningitis or gonorrhea), but terminal pathway defects result in a very high risk for systemic infections with atypical strains of these microorganisms. Management includes prompt treatment for infection. No specific treatments for complement deficiency are currently available.
Bone Marrow Failure
Some primary immune deficiencies result in failure of the bone marrow to produce hematopoietic cells, including those necessary for immune defense. One of the best described examples is Fanconi anemia. There are 22 different mutations that can cause Fanconi anemia (A-W); most are inherited in an autosomal recessive pattern.30 These mutations result in dysfunction of some of the proteins involved in DNA replication, causing bone marrow failure. Severe anemia and life-threatening infections result. It is also associated with a significant risk of cancer.52 Allogenic hematopoietic stem cell transplantation is curative for the bone marrow failure; however, those with malignant transformation prior to transplantation have a high rate of toxicity and transplant-related death.53 Stem cell transplantation with lentiviral-mediated gene corrections is emerging as a potential cure for this condition.54
Phenocopies of Inborn Errors of Immunity
This group of conditions includes a wide variety of somatic mutations affecting T- or B-cell function, or the production of autoantibodies to inflammatory cytokines.30 These abnormalities cause lymphoid changes such as splenomegaly and lymphadenopathy or may be associated with infections by mycobacteria, fungi, or staphylococci.
Evaluation of Those With Primary Immune Deficiency
The evaluation of primary immune deficiencies relies on next-generation sequencing methodology (exomes and genomes). These techniques have greatly improved genetic diagnosis with resulting improvements in care and genetic counseling.55 In addition to genetic analysis, other evaluations include a complete blood count (CBC) with a differential. The CBC provides information on the numbers of red blood cells, white blood cells, and platelets, and the differential indicates the quantities of lymphocytes, granulocytes, and monocytes in blood. Quantitative determination of immunoglobulins (IgG, IgM, IgA) is a screening test for antibody production, and an assay for total complement (total hemolytic complement, CH50) is useful if a complement defect is suspected.
Therapies for Primary Immune Deficiencies
The rapidly increasing knowledge of genetic, cellular, and molecular mechanisms that characterize primary immune deficiencies has fueled research into therapies aimed at specific immune defects.56 As has been noted previously, some primary immune deficiencies can be successfully treated by replacing the missing component of the immune system. For example, individuals with B-cell deficiencies that cause hypogammaglobulinemia or agammaglobulinemia are usually treated by administration of IVIG, antibody-rich fractions prepared from plasma pooled from large numbers of donors. Administration of IVIG replaces the individual's antibodies temporarily; these antibodies have a half-life of 3 to 4 weeks. Thus individuals must be treated repeatedly to maintain a protective level of antibodies in blood.
Bone marrow transplants containing hematopoietic stem cells have been routinely used to treat SCID, WAS, and other primary immunodeficiencies.57 However, individuals with SCID are at risk for graft-versus-host disease (GVHD). This occurs if T cells in a transplanted graft (e.g., transfused blood, bone marrow transplants) are mature and therefore capable of cell-mediated immunity against the recipient's HLA. The primary targets for GVHD are the skin (e.g., rash, loss or increase of pigment, thickening of skin), liver (e.g., damage to bile duct, hepatomegaly), mouth (e.g., dry mouth, ulcers, infections), eyes (e.g., burning, irritation, dryness), and gastrointestinal tract (e.g., severe diarrhea), and the disease may lead to death from infections. The risk of GVHD can be diminished by removing mature T cells from tissue used to treat individuals with immune deficiencies. Mesenchymal stem cells (MSCs) are present in all adult tissues and may be useful in treating GVHD. MSCs have potent immunosuppressive properties. Several clinical trials have demonstrated complete suppression of GVHD in a large number of recipients of MSCs.58
Gene therapy involves the use of viral vectors to transfer healthy genes into stem cells that are then transplanted into affected individuals. This approach provides long-term replacement of specific immune factors. Gene therapy has become safer and more effective since new viral vectors (e.g., lentiviruses) are being used. Trials have verified immune reconstitution in individuals with ADA deficiency, X-linked SCID, CGD, WAS, and Fanconi anemia, and numerous clinical trials are underway for this form of treatment.34,36,57,59,60
Secondary (Acquired) Immune Deficiencies
Secondary, or acquired, immune and inflammatory deficiencies are far more common than primary deficiencies. These deficiencies are complications of other physiologic or pathophysiologic conditions (Box 9.1). Although secondary deficiencies are common, many are not clinically relevant. In many cases, the degree of the immune deficiency is relatively minor and without any apparent increased susceptibility to infection. Alternatively, the immune system may be substantially suppressed but only for a short duration, thus minimizing the incidence of clinically relevant infections. However, some secondary immune deficiencies (e.g., acquired immunodeficiency syndrome [AIDS] or immunosuppression by cancer) are severe and may result in recurrent life-threatening infections. Management usually consists of supportive care, prompt treatment of infections, and reversal of the underlying immunocompromising condition if possible.
Psychological Stress
The relationship between emotional stress and depressed immune function is an area of intense research, and the mechanisms linking them are now beginning to be understood. Many lymphoid organs are innervated and can be affected by nerve stimulation. In addition, lymphocytes have receptors for many hormones (e.g., sex hormones, neurotransmitters, and neuropeptides) and can respond to changing levels of these chemicals with increased or decreased function. For instance, stress-induced catecholamines affect the expression of adhesion molecules and the movement of lymphocytes among lymphoid organs. (Further discussion of the effects of stress on susceptibility to disease is the subject of Chapter 11.)
Physical Trauma
Major trauma results in a systemic inflammatory response that is paralleled by a compensatory antiinflammatory response syndrome (CARS). This results in a period of critical illness in during which there is persistent inflammatory and immunosuppressive responses.61 Although the innate immune system is overactive, the adaptive immune system is suppressed. Antigen presentation by macrophages is reduced. Neutrophils and NK cells demonstrate decreased phagocytosis, cytokine production, and cytotoxic activity, as well as elevated rates of apoptosis.62 Treg activity is increased. This immune suppression is associated with poor wound healing, infection, and multiple organ dysfunction.
Dietary Insufficiencies
Nutritional status can have a profound effect on immune function, and malnutrition is the predominant cause of secondary immune deficiencies worldwide. Severe deficits in protein or calorie (protein-calorie malnutrition) intake lead to immune deficiencies. Marasmus (deficiency in calories) and kwashiorkor (deficiency in protein but adequate calories) have similar outcomes. T cell–rich areas of primary (thymus) and secondary lymphoid tissue are greatly affected, resulting in impaired T-cell function. Antibody levels are normal, but neutrophil function (chemotaxis, phagocytosis, bacterial killing), complement levels, and NK activity are impaired, resulting in infections with microorganisms that are normally destroyed by opsonization and phagocytosis. Deficiencies in specific nutritional components such as zinc and vitamins may also contribute to T- and B-cell dysfunction. In hospitalized individuals with malnutrition, modifications in the gut microbiota and decreased functioning of the gut mucosal immune system result in increased nosocomial infections and mortality.63
Chronic Diseases
Chronic diseases of the cardiovascular, gastrointestinal, endocrine, and renal systems are commonly complicated by a secondary immune suppression. For example, diabetes results in altered glucose metabolism and suppresses many aspects of the immune and inflammatory responses, including phagocytosis and chemotaxis, and lymphocyte proliferation.64 Diminished T-cell memory for pathogens, impaired B-cell function, low levels of complement, and glycation of circulating antibodies are also found in those with uncontrolled diabetes.65 Chronic heart failure is associated with poor nutrition, chronic inflammation, and adaptive immune dysfunction. Inflammatory bowel diseases, such as Crohn disease, ulcerative colitis, celiac disease, gastrointestinal infections, and cancer of the gastrointestinal tract are associated with decreased circulating levels of immunoglobulins.
Malignancies
Virtually all malignancies are complicated by immunosuppression, either through the effect of the disease itself on the body's defense mechanisms or as the result of treatment. Cancers cause both chronic inflammation and suppression of the adaptive immune system. For example, chronic inflammation releases growth factors such as vascular endothelial growth factor which causes the growth of new blood vessels (angiogenesis). At the same time, immunosuppressive macrophages and Treg cells are activated thus reducing the adaptive immune response to the tumor. Cancer cells are capable of protecting themselves by directly suppressing T lymphocytes that seek to destroy cancer cells (see Chapter 12). Cancer cells release metabolites and create a microenvironmental acidosis that hinder immune cell metabolism, proliferation, and functioning.66 Late-stage malignancies result in generalized deficiency of the immune response and greatly increased susceptibility to developing life-threatening infections. Mechanisms include malnutrition, replacement of bone marrow by cancer cells, decreased NK and T lymphocyte function, and the release of soluble immunosuppressive chemicals. In fact, many people with malignancies die as a result of infections rather than of the direct effects of the tumor. Some malignancies (e.g., lymphomas, leukemias, plasmacytomas) present with an early and specific immune depression through a direct effect on B cells.
Immunosuppressive treatments
The list of medications that affect the immune response is ever increasing and includes anesthetics, analgesics, antithyroid medications, anticonvulsants, antihistamines, antimicrobial agents, antilymphocyte antibodies, and tranquilizers. The most profound immunosuppressive treatments are those that are intentionally used to suppress the immune system to manage immune-mediated disease, to treat malignancy, or to prevent rejection of transplanted tissues.
Corticosteroids are intentionally used to control hypersensitivity diseases (especially autoimmune disease) or to prevent rejection of transplants. They predominantly inhibit T-cell function, prevent lymphocyte proliferation, inhibit production of critical cytokines, and suppress monocyte/macrophage functions. These effects greatly increase an individual's susceptibility to infection.
Many drugs and other treatments such as radiation that are used to treat cancer (e.g., chemotherapeutic agents, irradiation) are not specific for cancer cells but are designed to attack cells at susceptible stages in their cell cycles. They attack rapidly proliferating cells, thus killing immune cells as well as malignant cells. Depending on the dose of chemotherapy and/or irradiation administered, the entire immune system may be depleted.
Antirejection drugs are used to prevent immune-mediated rejection of transplanted tissue. Although more targeted treatments are becoming available, most antirejection regimens cause generalized immune suppression and increase the likelihood of infection and cancer.67 A careful therapeutic balance must be maintained between protecting the graft and preventing these complications.
Infections
Many infectious microorganisms successfully invade the human body using mechanisms for fighting off specific immune/inflammatory responses against themselves (discussed in Chapter 10). However, some infectious agents more broadly suppress the immune response. HIV is one of the few microorganisms that directly attacks the central processes involved in the development of an immune response (discussed in detail in Chapter 10). It infects and destroys CD4+ Th cells, which are necessary to provide help for the maturation of both plasma cells and Tc cells. Therefore HIV suppresses the immune response against itself and secondarily creates a generalized immune deficiency by suppressing the development of immune responses against other pathogens and opportunistic microorganisms.
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection has been shown to decrease innate immune responses, including the production of interferon, which can contribute to a lack of adequate initiation of adaptive immunity.67,68 Decreased innate immune responses to the virus have been linked to an increase in asymptomatic COVID-19, as well as an increase in severity in those with progressive disease.69 T-cell activation may be delayed, leading to decreased early symptoms, but can contribute to morbidity and mortality.70 Lymph nodes from individuals with COVID-19 were found to have a loss of germinal centers, and those remaining contained only one-third of normal T- and B-cell numbers.67
Several other viruses (e.g., measles; hepatitis B; and herpes viruses, such as EBV, CMV, and herpes simplex viruses) may suppress various components of the immune response. Measles virus can infect both B and T cells and macrophages. Infection may result in a transient lymphopenia and a suppressed T-cell response. Acute infections with herpes viruses also may transiently suppress the immune system. EBV not only infects B cells, but it also may suppress both CD4+ and CD8+ T cells and NK cells, although immunosuppression is generally not severe. Many viruses produce molecules similar in function to IL-10 that further contribute to immunosuppression. CMV infects mucosal epithelium and can infect macrophages where antigen processing and presentation may be impaired. Tumor viruses such as human papillomavirus (HPV) help to create a tumor microenvironment in which cancer cells are free to grow.71
Some fungal infections may suppress the immune response. In disseminated C. albicans infections, T-cell responses and neutrophil chemotaxis are suppressed to various degrees. Similar immunosuppression may be observed in individuals with disseminated histoplasmosis (infections with Histoplasma capsulatum). The most severe form of acute malaria (caused by the parasite Plasmodium falciparum) suppresses specific antibody responses against protein and polysaccharide antigens by dysregulation of CD4+ T-cell function and decreased IL-2 production.
Summary Review
Hypersensitivity: Allergy, Autoimmunity, and Alloimmunity
- 1. Inappropriate immune responses are exaggerated misdirected responses innocuous environmental antigens (allergy), the host's own tissues (autoimmunity), or beneficial foreign tissues (alloimmunity); or insufficient responses to protect the host (immune deficiency).
- 2. Allergy, autoimmunity, and alloimmunity are collectively known as hypersensitivity reactions.
- 3. Mechanisms of hypersensitivity are classified as type I (IgE-mediated) reactions, type II (tissue-specific) reactions, type III (immune complex–mediated) reactions, and type IV (cell-mediated) reactions.
- 4. Hypersensitivity reactions can be immediate (developing within minutes to a few hours) or delayed (developing within several hours or days).
- 5. Allergens are antigens that cause allergic responses.
- 6. Type I (IgE-mediated) hypersensitivity reactions are mediated through the binding of IgE to Fc receptors on mast cells and cross-linking of IgE by antigens that bind to the Fab portions of IgE. Cross-linking causes mast cell degranulation and the release of histamine (the most potent mediator) and other inflammatory substances.
- 7. Histamine, acting through the H1 receptor, contracts bronchial smooth muscles, causing bronchial constriction; increases vascular permeability, causing edema; and increases blood flow into the affected area, causing vasodilation. Histamine with H2 receptors results in increased gastric acid secretion and a decrease of histamine released from mast cells and basophils.
- 8. Type II (tissue-specific) hypersensitivity reactions are caused by five possible mechanisms: complement-mediated lysis, opsonization and phagocytosis, neutrophil-mediated tissue damage, antibody-dependent cell-mediated cytotoxicity, and modulation of cellular function.
- 9. Type III (immune complex–mediated) hypersensitivity reactions are caused by the formation of immune complexes that are deposited in target tissues, where they activate the complement cascade, generating chemotactic fragments that attract neutrophils into the inflammatory site. Neutrophils release lysosomal enzymes that result in tissue damage.
- 10. Immune complex disease can be a systemic reaction, such as serum sickness, or a localized response, such as the Arthus reaction.
- 11. Type IV (cell-mediated) hypersensitivity reactions are caused by cytotoxic T lymphocytes (Tc cells), lymphokine-producing Th1 cells and activated macrophages.
- 12. Typical allergens include pollen, molds and fungi, certain foods (milk, eggs, fish, peanuts), animals, certain drugs, cigarette smoke, and house dust.
- 13. Clinical manifestations of allergic reactions usually are confined to the areas of initial intake or contact with the allergen. Ingested allergens induce gastrointestinal symptoms, airborne allergens induce respiratory tract or skin manifestations, and contact allergens induce allergic responses at the site of contact.
- 14. Autoimmune diseases originate from the coincidence of an initiating event in a genetically predisposed individual leading to an autoimmune mechanism that affects specific target tissues or cells. Central tolerance develops during the embryonic period. Peripheral tolerance is maintained in secondary lymphoid organs by regulatory T lymphocytes or antigen-presenting dendritic cells.
- 15. Heparin-induced thrombocytopenia is a condition in which heparin molecules attach to proteins in the surface of platelets resulting in the formation of autoantibodies that destroy platelets (bleeding) and promote clotting (thrombosis).
- 16. Systemic lupus erythematosus (SLE) is a chronic, multisystem, inflammatory disease and is one of the most serious of the autoimmune disorders. SLE is characterized by the production of a large variety of autoantibodies.
- 17. Alloimmunity is the immune system's reaction against antigens on the tissues of other members of the same species.
- 18. Alloimmune disorders include transient neonatal disease, in which the maternal immune system becomes sensitized against antigens expressed by the fetus; transplant rejection; and transfusion reactions, in which the immune system of the recipient of an organ transplant or blood transfusion reacts against foreign antigens on the donor's cells.
- 19. Red blood cell antigens may be the targets of autoimmune or alloimmune reactions. The most important of these, because they provoke the strongest humoral immune response, are the ABO and Rh systems.
- 20. Antigens on fetal red blood cells (Rh) can cause maternal antibodies to cross the placenta and cause severe anemia in the fetus.
- 21. Hyperacute graft rejection (preexisting antibody) is immediate and rare, acute rejection is both antibody and cell mediated and occurs days to months after transplantation, and chronic rejection is caused by inflammatory damage to endothelial cells as a result of a weak cell-mediated reaction.
Deficiencies in Immunity
- 1. Disorders resulting from immune deficiency are the clinical sequelae of impaired function of components of the immune or inflammatory response, phagocytes, or complement.
- 2. Immune deficiency is the failure of mechanisms of self-defense to function in their normal capacity.
- 3. Immune deficiencies are either congenital (primary) or acquired (secondary). Primary immune deficiencies are caused by genetic defects that disrupt lymphocyte development, whereas secondary immune deficiencies are secondary to disease or other physiologic alterations.
- 4. The clinical hallmark of immune deficiency is a propensity to unusual or recurrent severe infections. The type of infection usually reflects the immune system defect.
- 5. The most common infections in individuals with defects of the cell-mediated immune response are fungal and viral, whereas infections in individuals with defects of the humoral immune response or complement function are primarily bacterial.
- 6. Severe combined immunodeficiency is a total lack of T-cell function and a severe (either partial or total) lack of B-cell function. Other combined defects may result from deficiencies in antigen-presenting molecules (bare lymphocyte syndrome) or cytoskeletal proteins (WAS).
- 7. Chromosome 22q11.2 deletion syndrome (DiGeorge syndrome) is characterized by complete or partial lack of the thymus (resulting in depressed T-cell immunity) and the parathyroid glands (resulting in hypocalcemia) and the presence of cardiac anomalies.
- 8. Defects in B-cell function are diverse, ranging from a complete lack of the human bursal equivalent function, the lymphoid organs required for B-cell maturation (as in Bruton agammaglobulinemia), to deficiencies in a single class of immunoglobulins (e.g., selective IgA deficiency).
- 9. Defects in phagocyte function, which include insufficient numbers of phagocytes or defects of chemotaxis, phagocytosis, or killing, can result in recurrent life-threatening infections such as septicemia and disseminated pyogenic lesions.
- 10. Immune dysregulation disorders are characterized by abnormally high levels of inflammation secondary to mutations in control of inflammasome activation or in defects in cellular receptors of cytokines designed to decrease inflammation. These disorders are frequently related to diminished control of infections of epithelial surfaces.
- 11. Almost any portion of the complement cascade may be defective. The most severe defect is C3 deficiency, which results in recurrent life-threatening bacterial infections. Defects in proteins of the membrane attack complex usually result in unusual, disseminated infections with bacteria of the Neisseria spp.
- 12. Bone marrow failure and somatic mutations in immune genes may also result in severe primary immunodeficiency states.
- 13. Primary immune deficiencies may sometimes be treated by replacement therapy. Deficient antibody production is treated by replacement of missing immunoglobulins with commercial gamma-globulin preparations. Lymphocyte deficiencies are treated with the replacement of host lymphocytes with bone marrow and stem cell transplants and gene therapies when available.
- 14. Acquired immunodeficiencies are caused by superimposed conditions, such as aging, malnutrition, infections, malignancies, physical or psychological trauma, environmental factors, some medical treatments, or other diseases chronic disease, or infections.