Alterations of Neurologic Function in Children
John Ruge
 http://evolve.elsevier.com/Rogers/pathophysiology/
http://evolve.elsevier.com/Rogers/pathophysiology/
Neurologic disorders in children can occur from infancy through adolescence and include congenital malformations, genetic defects in metabolism, brain injuries, infection, tumors, and other disorders that affect neurologic function.
Development of the Nervous System in Children
The nervous system develops from the embryonic ectoderm through a complex, sequential process. A neural tube starts to form 2 and one-half weeks from conception and “zippers shut” by 4 weeks. The spinal cord, spine, brain, and skull form from this neural tube. By approximately day 175, the brain has developed into all of its parts (Fig. 20.1A–C). The brain continues the formation of network connections and synapses from birth to many years postnatally. Many different events happen simultaneously, and critical periods must pass uninterrupted if the vulnerable fetus is to develop normally. Genetic and environmental factors (e.g., nutrition, hormones, oxygen levels, toxins, alcohol, drugs, maternal infections, maternal disease) can have a significant effect on neural development.1,2
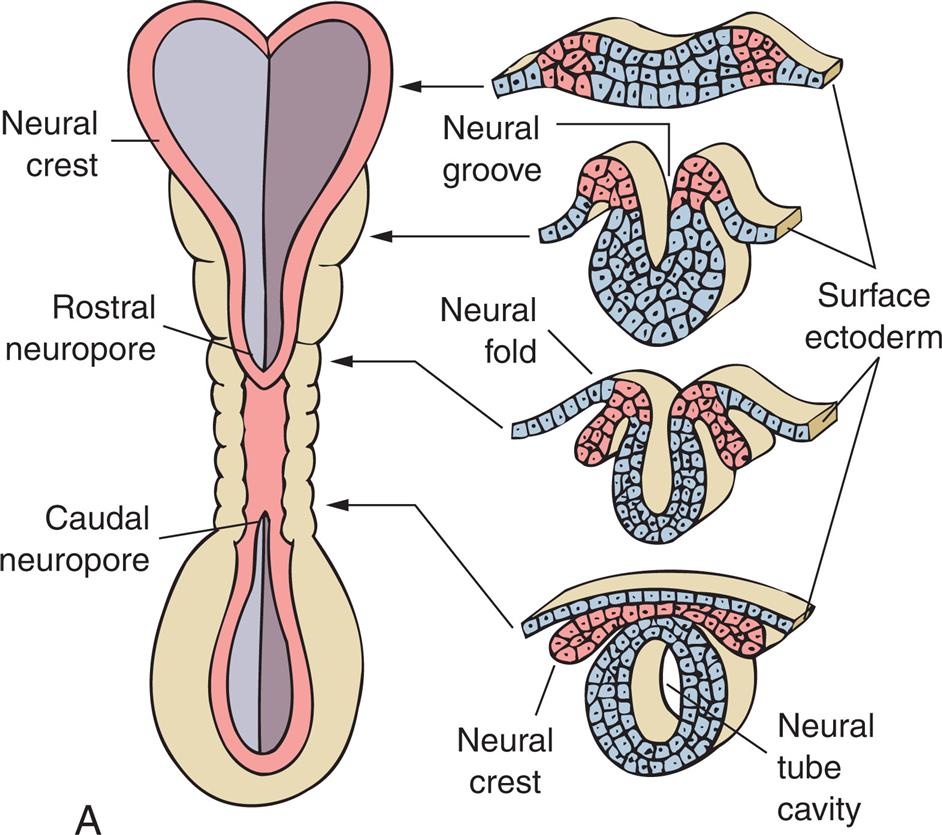
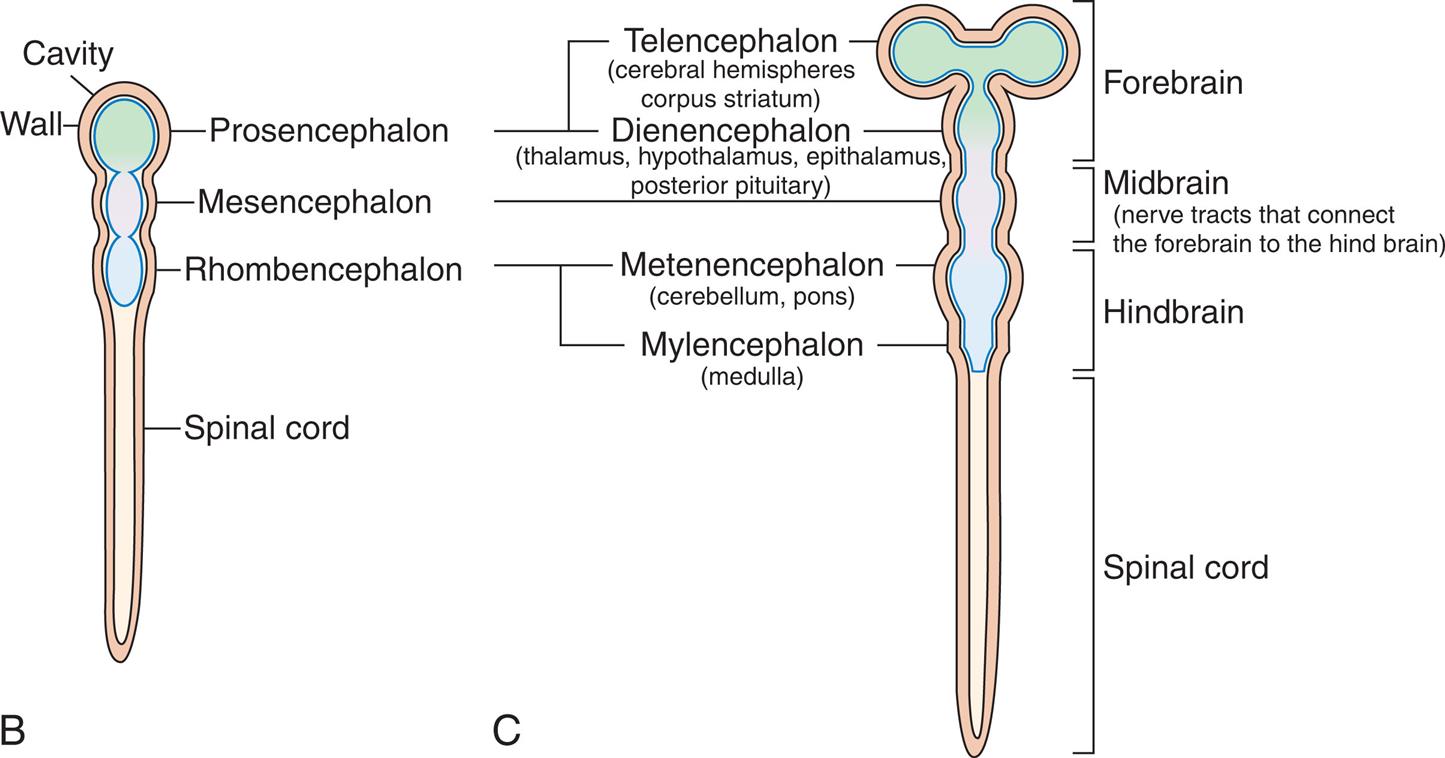
(A) Neural tube at 3 weeks’ gestation. Neural folds have begun to fuse at the cervical level of the future spinal cord. Right, Cross sections of the neural tube at four different levels; at any given level the embryonic central nervous system goes through a series of stages resembling these four cross sections. Total length of neural tube at this time is approximately 2.5 mm. (B) The anterior part of the neural tube enlarges to form the 3 primary vesicles of the brain. The narrow posterior part forms the spinal cord. (C) Primary and secondary vesicles develop into various parts of the brain.
Illustration A shows the lateral and cross-sectional views of the neural crest, the rostral neuropore, and the caudal neuropore. Accompanying illustrations show the stages of the folding of the neural cells and the surface ectoderm. • The illustration shows wavy layers of tissues made of alternating neural and ectoderm cells. • The illustration shows the neural cells forming a groove and the ectoderm cells clustered. • The illustration shows the neural fold with the ectoderm cells curving inward. • The illustration shows a neural tube cavity with a neural crest and a surface ectoderm. Illustration B shows and labels the following structures in a neural tube from the top to the bottom: cavity, wall, prosencephalon, mesencephalon, rhombencephalon, and spinal cord. Illustration C shows a neural tube develop into the primary and secondary vesicles. The following structures are labeled from the top to the bottom: forebrain, midbrain (nerve tracts that connect the forebrain to the hind brain), hindbrain, and spinal cord. The forebrain comprises the telencephalon (cerebral hemispheres corpus striatum) and the diencephalon (thalamus, hypothalamus, epithalamus, posterior pituitary). The hindbrain comprises metencephalon (cerebellum, pons) and myelencephalon (medulla).
The growth and development of the brain occur rapidly from the third month of gestation through the first year of life, reflecting the proliferation of neurons and glial cells. Although basically all of the neurons that an individual will ever have are present at birth, development of skills, such as walking, talking, and thinking, depends on these cells making correct connections with other cells and on myelination of the axons making those connections. The head is the fastest-growing body part during infancy. One-half of postnatal brain growth is achieved by the first year and is 90% complete by age 6 years. The cortex thickens with maturation, and the sulci deepen as a result of rapid expansion of the surface area of the brain. Cerebral blood flow and oxygen consumption during these years are approximately twice those of the adult brain.
The bones of the infant's skull are separated at the suture lines, forming two fontanelles, or “soft spots”: one diamond-shaped anterior fontanelle and one triangular-shaped posterior fontanelle. The sutures allow for expansion of the rapidly growing brain. The sutures remain open, allowing for brain and skull growth into early and mid-adulthood. The posterior fontanelle may be open until 2 to 3 months of age; the anterior fontanelle normally does not fully close until 18 months of age (Fig. 20.2). Head growth almost always reflects brain growth. Monitoring the fontanelles and careful measurement and plotting of the head circumference on standardized growth charts are essential elements of the pediatric examination.
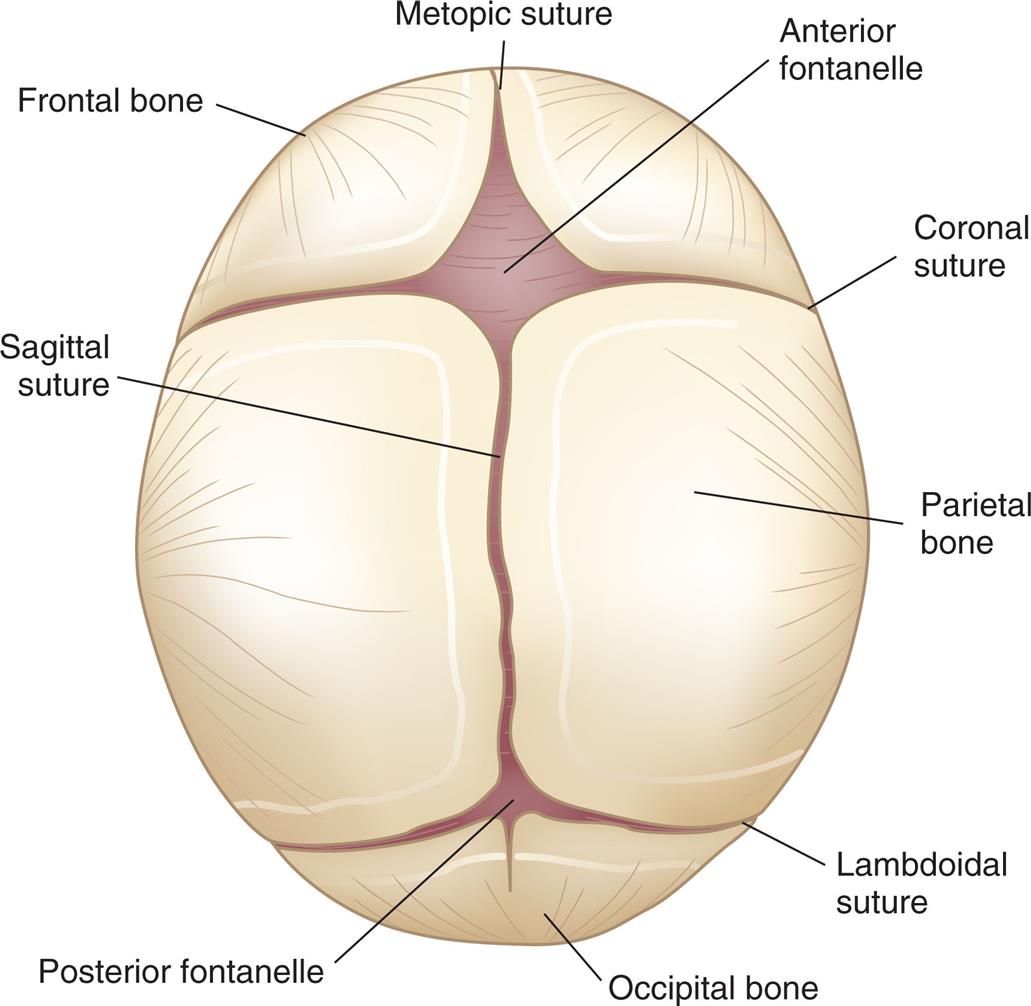
Fibrous union of the suture lines and interlocking of the serrated edges occurs by 6 months; solid union requires approximately 12 years. (Head growth charts are available from the Centers for Disease Control and Prevention at www.cdc.gov/nchs/data/series/sr_11/sr11_246.pdf).
A coronal view of the skull shows and labels the following parts, clockwise from the top: metopic suture, anterior fontanelle, coronal suture, parietal bone, lambdoidal suture, occipital bone, posterior fontanelle, sagittal suture, and frontal lobe.
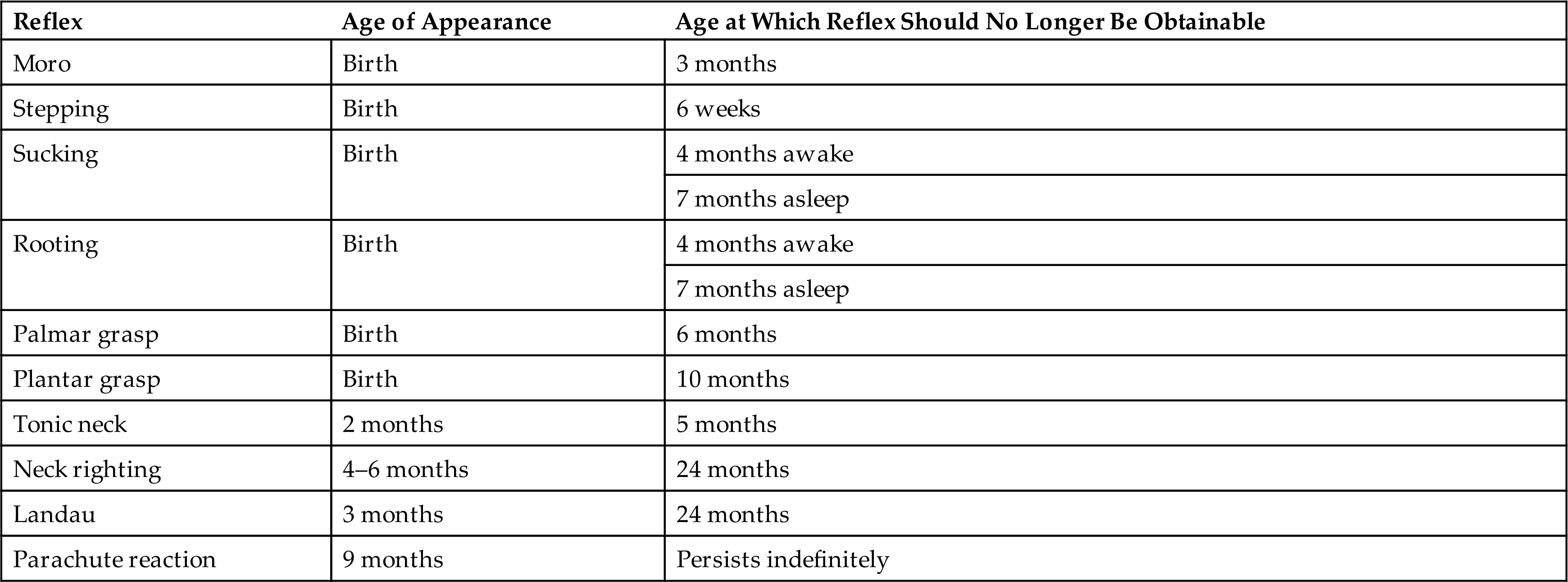
Because of the immaturity of much of the human forebrain at birth, neurologic examination of the infant detects mostly reflex responses that require an intact spinal cord and brainstem. A maturing brain tends to inhibit most reflexes, so as the brain matures, reflexes of infancy are normally lost. Persistent infantile reflexes can indicate developmental delay or a central nervous system (CNS) lesion. Absence of expected reflex responses at the appropriate age indicates general depression of central or peripheral motor functions. Asymmetric responses may indicate lesions in the motor cortex or peripheral nerves or may occur with fractures of bones after traumatic delivery or postnatal injury (Table 20.1).
Table 20.1
Structural Malformations
Malformations of the CNS affect approximately 5 to 10 children per 1000 births. CNS malformations are responsible for 75% of fetal deaths and 40% of deaths during the first year of life. CNS malformations account for 33% of all apparent congenital malformations, and 90% of CNS malformations are defects of neural tube closure.
Defects of Neural Tube Closure
Neural tube defects (NTDs) are an arrest of the normal development of the brain and spinal cord during the first month of embryonic development. They occur in approximately 3000 pregnancies in the United States each year, although there are significant regional prevalence variations.3 Fetal death often occurs in the more severe forms, thereby reducing the actual prevalence of neural defects at birth.
The cause of NTDs is believed to be multifactorial (a combination of genes and environment). No single gene has been found to cause NTDs. Folic acid deficiency during preconception and early stages of pregnancy increases the risk for NTDs, and supplementation (400 mcg of folic acid per day) ensures adequate folate status. In 1996 the United States mandated folate fortification in many foods, and since that time NTDs have decreased by 20% to 30%. Guidelines are available for folic acid supplementation for women at high risk for NTDs.4 Other risk factors include a previous NTD pregnancy, maternal diabetes or obesity, use of anticonvulsant drugs (particularly valproic acid), and maternal hyperthermia.5 Defects of neural tube closure are divided into two categories: (1) anterior midline defects (ventral induction) and (2) posterior defects (dorsal induction). Anterior midline defects may cause brain and face abnormalities, with the most extreme form being cyclopia, in which the child has a single midline orbit and eye with a protruding nose-like proboscis above the orbit. Anencephaly (an, “without”; enkephalos, “brain”) and encephalocele are also anterior midline defects. Spina bifida (split spine) is the most common NTD. Vertebrae fail to close in spina bifida, and the defect includes meningocele, myelomeningocele, and spina bifida occulta. Disorders of embryonic neural development are summarized in Fig. 20.3.
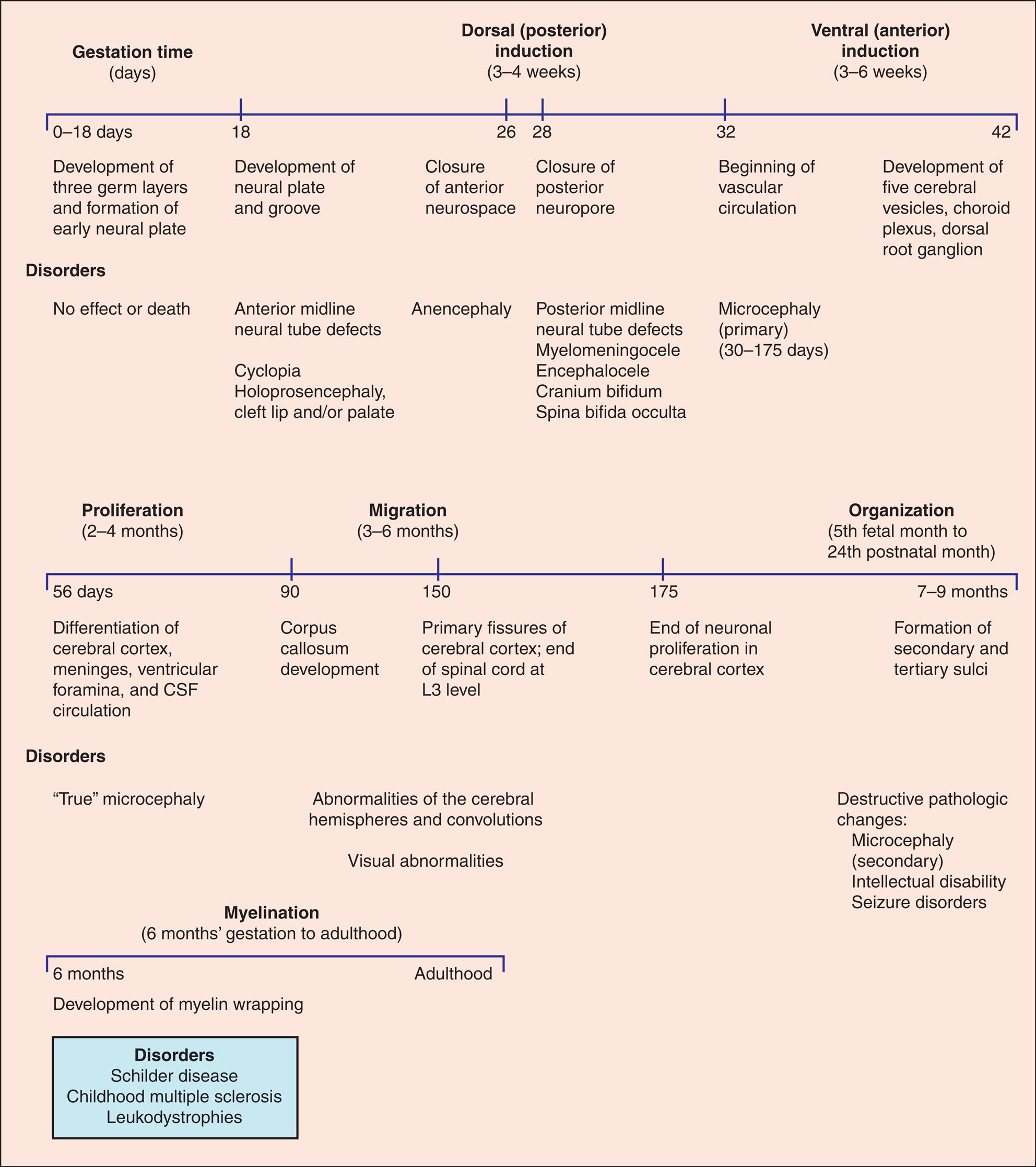
CSF, Cerebrospinal fluid.
Three timelines show disorders associated with specific stages of embryonic development. Top panel. The timeline shows three sections: gestation time (days), dorsal (posterior) induction (3 to 4 weeks), and ventral (anterior) induction (3 to 6 weeks). The data from the timeline are as follows. • 0 to 18 days. Development of three germ layers and formation of early neural plate. Disorders have no effect or death. • 18 days. Development of neural plate and groove. Disorders include anterior midline neural tube defects, cyclopia holoprosencephaly, and cleft lip and or palate. • 26 days. Closure of anterior neurospace. Disorder includes anencephaly. • 28 days. Closure of posterior neuropore. Disorders include posterior midline neural tube defects, myelomeningocele, encephalocele, cranium bifidum, and spina bifida occulta. • 32 days. Beginning of vascular circulation. Disorder includes microcephaly (primary; 30 to 175 days). • 42 days. Development of five cerebral vesicles, choroid plexus, and dorsal root ganglion. Middle panel. The timeline shows three sections: proliferation (2 to 4 months), migration (3 to 6 months), and organization (fifth fetal month to twenty-fourth postnatal month). The data from the timeline are as follows. • 56 days. Differentiation of cerebral cortex, meninges, ventricular foramina, and C S F circulation. Disorder includes true microcephaly. • 90 days. Corpus callosum development. • 150 days. Primary fissures of cerebral cortex; end of spinal cord at L 3 level. Disorders include abnormalities of the cerebral hemispheres and convolutions, and visual abnormalities. • 175 days. End of neuronal proliferation in cerebral cortex. • 7 to 9 months. Formation of secondary and tertiary sulci. The disorder includes destructive pathologic changes: microcephaly (secondary), intellectual disability, and seizure disorders. Bottom panel. The timeline shows two segments between myelination (6 months’ gestation to adulthood). The data from the timeline are as follows. • 6 months. Development of myelin wrapping. Disorders include Schilder disease, childhood multiple sclerosis, and leukodystrophies. • Adulthood.
Anencephaly is an anomaly in which the soft, bony component of the skull and part of the brain are missing. This disorder occurs in approximately 1 in 4600 births in the United States each year.6 These infants are stillborn or die within a few days after birth. The pathologic mechanism is unknown. Diagnosis is often made prenatally by using ultrasound or evaluating the maternal serum alpha fetoprotein (AFP).
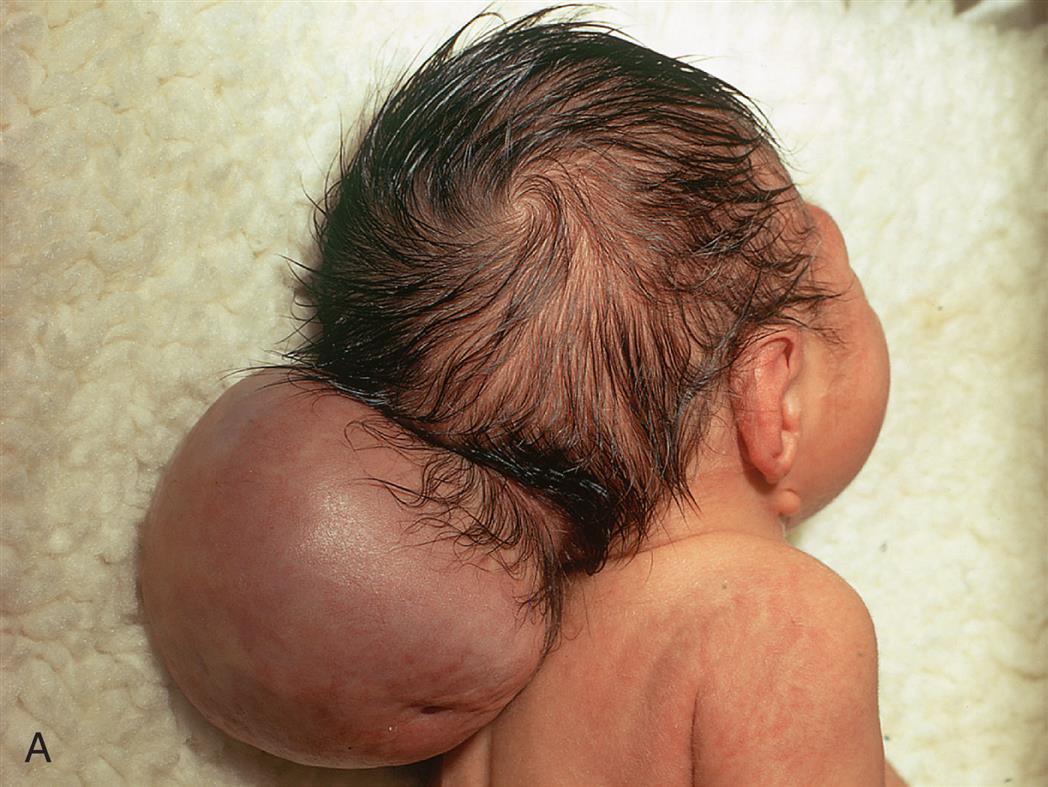
Encephalocele refers to a herniation or protrusion of the brain and meninges through a defect in the skull, resulting in a sac-like structure. The incidence is approximately 1 in 10,500 live births in the United States each year.7 Encephalocele treatment involves surgical closure of the skull opening. The complexity of the surgery depends on the location and content of the defect. Often several surgeries may be needed with variable long-term neurologic sequelae.
Meningocele is a sac-like cyst of meninges filled with spinal fluid and is a mild form of spina bifida (Fig. 20.4). It develops during the first 4 weeks of pregnancy when the neural tube fails to close completely and is the rarest type of spina bifida. The cystic dilation of meninges protrudes through the vertebral defect but does not involve the spinal cord or nerve roots and may produce no neurologic deficit or symptoms. Meningoceles occur most commonly in the lumbar spine.
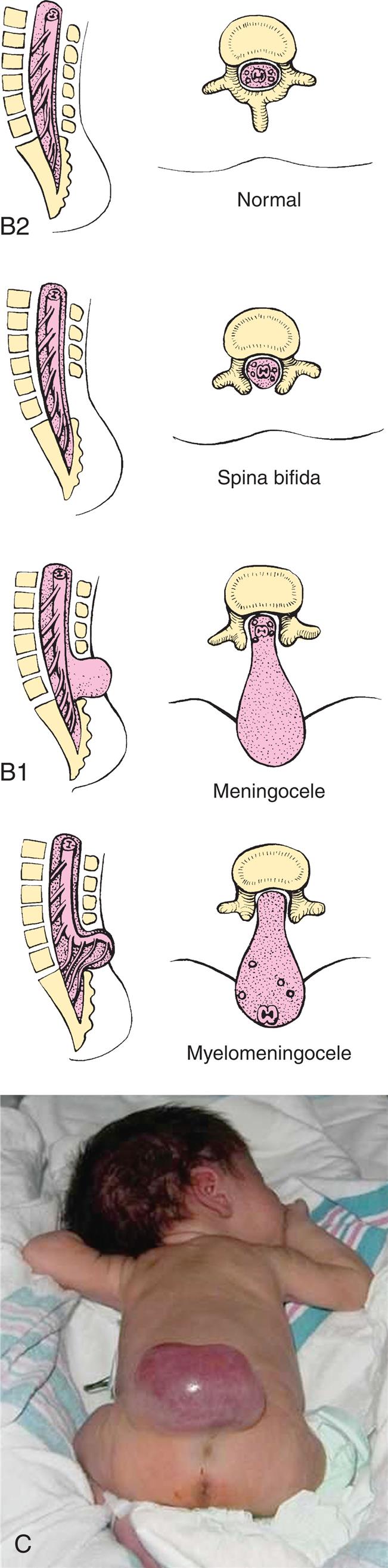
Closeup A shows an infant on the side with a large herniation, the size of the infant’s head, at the base of the head. Two pairs of accompanying illustrations show lateral and cross-sectional views of the spine. One pair shows meningocele and the other pair shows myelomeningocele. Closeup B shows an infant lying on its stomach with the legs curled toward to the abdomen. There is a large herniation on the lower back. Two pairs of accompanying illustrations show lateral and cross-sectional views of a normal spine and a spine affected by spina bifidia. Some of the vertebrae are missing in the spine with spina bifida.
Myelomeningocele (meningomyelocele; spina bifida cystica) is a hernial protrusion of a sac-like cyst (containing meninges, spinal fluid, and a portion of the spinal cord with its nerves) through a defect in the posterior arch of a vertebra. It is most commonly located as a sac on the lumbar and lumbosacral regions, the last regions of the neural tube to close. Myelomeningocele is one of the most common and severe developmental anomalies of the nervous system, with an incidence of approximately 1 per 4000 live births.8 It accounts for 75% of all spina bifida cases.
Meningocele and myelomeningoceles are evident at birth as a pronounced skin defect on the infant's back (see Fig. 20.4). The bony prominences of the unfused neural arches can be palpated at the lateral border of the defect. The defect usually is covered by a transparent membrane that may have neural tissue attached to its inner surface. This membrane may be intact at birth or may leak cerebrospinal fluid (CSF), thereby increasing the risks of infection and neuronal damage. The spinal cord and nerve roots are malformed below the level of the lesion, resulting in loss of motor, sensory, reflex, and autonomic functions. A brief neurologic examination concentrating on motor function in the legs, reflexes, and sphincter tone is usually sufficient to determine the level above which spinal cord and nerve root function is preserved (Table 20.2). This is useful to predict if the child will ambulate, require bladder catheterization, or be at high risk for developing scoliosis (see Chapter 45).
Table 20.2
Modified from Sandler AD. Children with spina bifida: Key clinical issues. Pediatric Clinics of North America, 2010;57(4):879–892.
Hydrocephalus occurs in 85% of infants with myelomeningocele.9 Seizures also occur in 30% of those with myelomeningoceles. Visual and perceptual problems, including ocular palsies, astigmatism, and visuoperceptual deficits, are common. Motor and sensory functions below the level of the lesions are altered. These problems should not worsen as the child grows. If a neurologic decline is identified in a child with spina bifida, the cause needs to be investigated.
Myelomeningoceles are almost always associated with the Chiari II malformation.9 This is a complex malformation of the brainstem and cerebellum in which the cerebellar tonsils are displaced downward through the foramen magnum and into the cervical spinal canal. The upper medulla and lower pons are elongated and thin, and the medulla is also displaced downward and sometimes has a “kink” (Fig. 20.5). The Chiari II malformation is associated with hydrocephalus from pressure that blocks the flow of CSF. The Chiari II malformation is also associated with syringomyelia, an abnormality resulting from CSF migrating into the center of the spinal cord forming a pocket or cyst (syrinx) that can expand and cause pressure on the spinal cord, resulting in bowel, bladder, sensory, and motor dysfunction. The incidence of Chiari II malformation is reduced in those children operated on in utero for myelomeningocele.
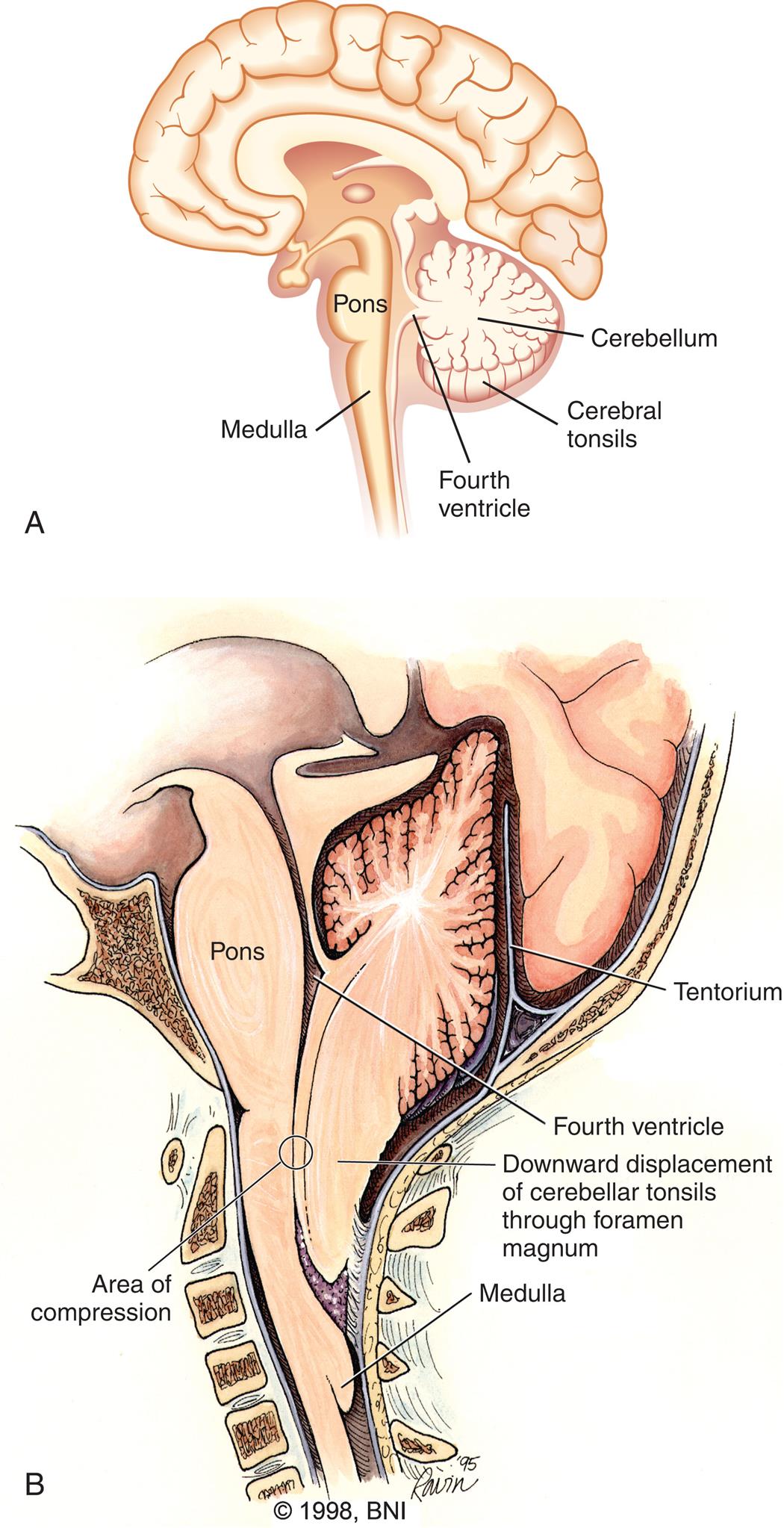
(A) Diagram of a normal brain. (B) Diagram of an Arnold-Chiari II malformation, showing downward displacement of the cerebellar tonsils and medulla through the foramen magnum causing, compression and obstruction of cerebrospinal fluid flow. (B, Modified from Barrow Neurological Institute of St Joseph's Hospital and Medical Center. Reprinted with permission.)
Illustration A is a left lateral view of the brain with the following parts labeled: cerebellum, cerebral tonsils, fourth ventricle, pons, and medulla. Illustration B is a left lateral view of the base of the brain with the following parts labeled: tentorium, fourth ventricle, pons, downward displacement of cerebellar tonsils through foramen magnum, area of compression, and medulla.
Other types of Chiari malformations are not associated with spina bifida. In type I Chiari malformation the cerebellar tonsils protrude into the foramen magnum and the brainstem is not involved. Type I Chiari is the most common of all Chiari malformations and may be asymptomatic or symptomatic (e.g., neck pain, dizziness, difficulty with gait and swallowing), sometimes requiring neurosurgical intervention. In type III, the brainstem or cerebellum extends into a high cervical myelomeningocele and can cause serious neurologic defects. Type IV is characterized by lack of cerebellar development (cerebellar hypoplasia).
Most cases of meningocele and myelomeningocele are diagnosed prenatally by a combination of maternal serologic testing (AFP) and prenatal ultrasound. In these cases, the fetus is usually delivered by elective cesarean section to minimize trauma during labor. Surgical repair is critical and can be performed by in utero fetal surgery or during the first 72 hours of life.10 It is possible for a defect to occur without any visible exposure of meninges or neural tissue, and the term spina bifida occulta is then used. The defect is common and occurs to some degree in 10% to 25% of infants. Spina bifida occulta usually causes no neurologic dysfunction because the spinal cord and spinal nerves are normal. A sacral dimple on examination of a newborn infant can be associated with spina bifida occulta and should be further evaluated. Any midline skin abnormality such as a hairy tuft, hemangioma type lesion, or unusual dimple may indicate underlying spinal pathology and should be evaluated. Tethered cord syndrome may develop after surgical correction for myelomeningocele. The cord becomes abnormally attached or tethered in the lumbar cistern as a result of scar tissue as the cord ascends in the vertebral canal with growth.11 Although radiographically almost all persons affected by myelomeningocele will appear to be tethered, only those with symptoms or new neurologic decline warrant further investigation, and prompt neurosurgical release of the tethering is appropriate. Special attention should be given to children whose myelomeningocele was corrected in utero, because their risks of spinal cord retethering is increased (see Emerging Science Box: In Utero Myelomeningocele Surgery).
Craniosynostosis
Skull malformations range from minor, insignificant defects to major defects that are incompatible with life. Craniosynostosis (craniostenosis) is the premature closure of one or more of the cranial sutures (sagittal, coronal, lambdoid, metopic) during the first 18 to 20 months of the infant's life. The incidence of craniosynostosis is approximately 1 per 2500 live births.12 Males are affected twice as often as females. Fusion of a cranial suture prevents growth of the skull perpendicular to the suture line, resulting in an asymmetric shape of the skull. The general term plagiocephaly, meaning “misshapen skull,” is used to describe deformities that result from craniosynostosis or from asymmetric head posture (positional). When a single coronal suture fuses prematurely, the head is flattened on that side in front. When the sagittal suture fuses prematurely, the head is elongated in the anteroposterior direction (known as scaphocephaly). Single suture craniosynostosis requires early surgical intervention because it is not just a cosmetic issue. When multiple sutures fuse prematurely, surgical intervention is necessary. Brain growth may be restricted, and surgical repair may prevent neurologic dysfunction (Fig. 20.6). Syndromic craniosynostosis involves deformities in other systems (i.e., the heart, limbs, and CNS).
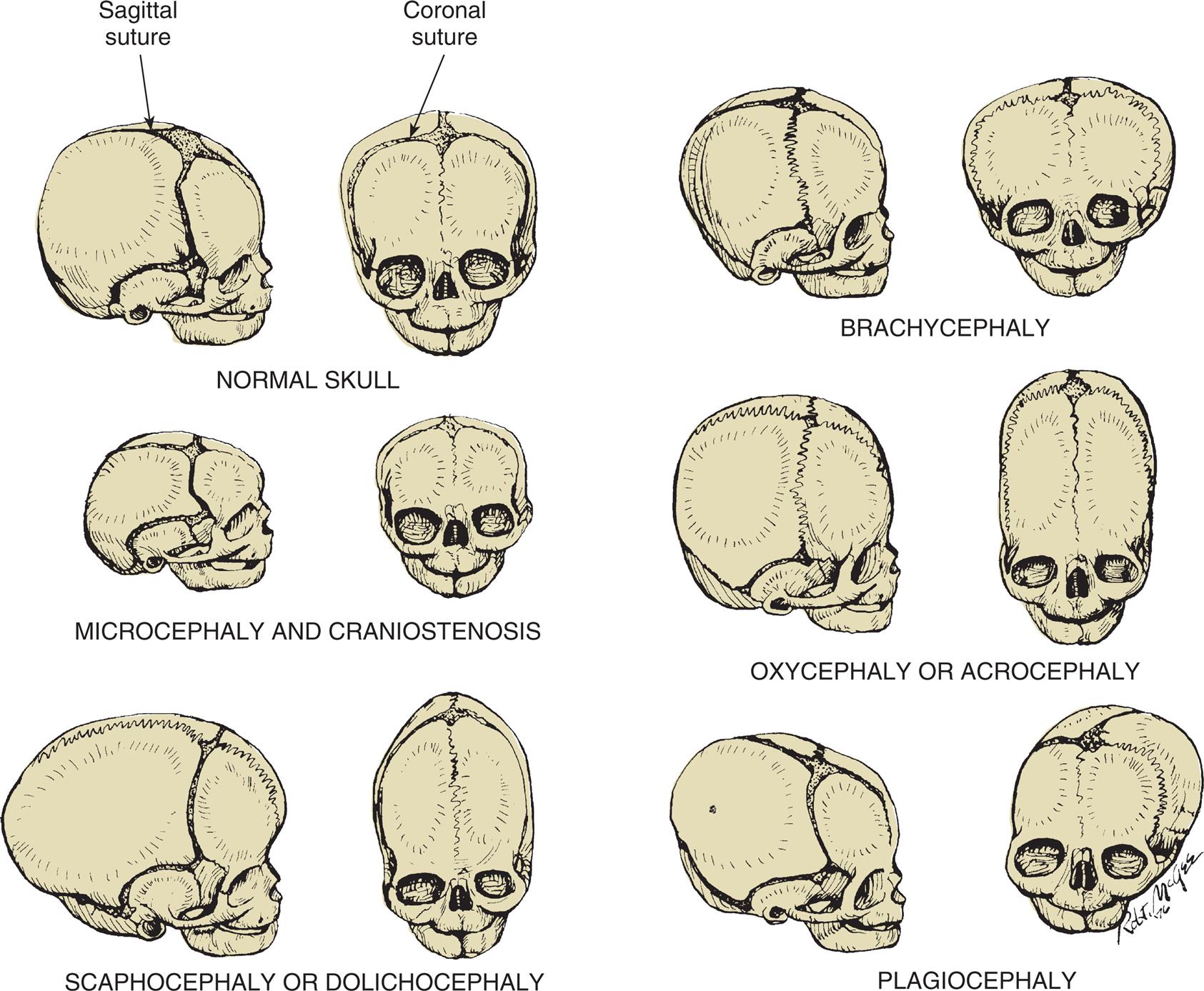
Normal skull: The bones are separated by membranous seams until the sutures gradually close. Microcephaly and craniostenosis: In microcephaly, the head circumference is more than 2 standard deviations below the mean for age, gender, race, and gestation, reflecting a small brain. Craniosynostosis is premature closure of the sutures. Scaphocephaly or dolichocephaly (frequency 56%): Premature closure of the sagittal suture, resulting in restricted lateral growth. Brachycephaly: Premature closure of the coronal suture, resulting in excessive lateral growth. Oxycephaly or acrocephaly (frequency 5.8% to 12%): Premature closure of all coronal and sagittal sutures, resulting in accelerated upward growth and a small head circumference. Plagiocephaly (frequency 13%): Unilateral premature closure of the coronal suture, resulting in asymmetric growth. (From Hockenberry MJ, Wilson D. Wong’s nursing care of infants and children, 10th edition. St. Louis: Mosby; 2015.)
Six pairs of illustrations of skulls show normal and abnormal head configurations. Each pair shows the right lateral view and the anterior view of the skull. Top left, normal skull. The sagittal suture is identified in the lateral view. The coronal suture is identified in the anterior view. Top right, brachycephaly. The sagittal suture in the lateral view is shrunken. The coronal suture in the anterior view is almost non-existent. The head is unusually large. Middle left, microcephaly and craniostenosis. The skulls are smaller than normal. The sagittal suture in the lateral view is shrunken. The coronal suture in the anterior view is minimal. Middle right, oxycephaly or acrocephaly. The sagittal suture in the lateral view and the coronal suture are almost non-existent. The head is almost twice as long as normal. Bottom left, scaphocephaly or dolichocephaly. The skulls are larger and narrower. The sagittal suture and the coronal suture are minimal. Bottom right, plagiocephaly. The skulls are misshapen. The left and the right side are disproportional. The sagittal suture is present. The coronal suture is only partially present.
Malformations of Brain Development
Reduced proliferation or accelerated apoptosis (programmed cell death) of brain cells causes congenital microcephaly (microencephaly—small brain). Increased proliferation of brain cells causes megalencephaly (macrencephaly—abnormally large brain). Both disorders are rare. Diagnosis is made by clinical history, family history, and brain imaging.13,14
Microcephaly (small head) is associated with a defect in brain growth as a whole (see Fig. 20.6). Cranial size is significantly below average for the infant's age, gender, race, and gestation. The small size of the skull reflects a small brain (microencephaly), which is caused by reduced proliferation or accelerated apoptosis. True (primary) microcephaly is usually caused by an autosomal recessive genetic or chromosomal defect. Secondary (acquired) microcephaly is associated with various causes, such as intrauterine infection (including emergence of the Zika virus)15; trauma; metabolic disorders; maternal anorexia experienced during the third trimester of pregnancy; in utero exposure to alcohol, toxins, or certain medications; and the presence of other genetic syndromes. Children with microcephaly are usually developmentally delayed. Microcephaly can be familial and, if so, can be associated with normal development.
Cortical dysplasias (malformations) are defects in brain development and related to failure of embryonic neurons to migrate to the right places in the brain. These disorders may range from a small area of abnormal tissue to an entire brain that is smooth and without the normal configuration of gyri and sulci, known as lissencephaly (Fig. 20.7), or a brain that has too many small gyri (folds), known as polymicrogyria. There is a specific genetic defect for some of these disorders; others are multifactorial or acquired (e.g., intrauterine trauma or infection). Cortical dysplasias increase the risk for seizures that are difficult to control and cause developmental delay and motor dysfunction. Genetic testing assesses risk in other family members and guides therapy.16,17 Congenital hydrocephalus is present at birth and characterized by increased CSF pressure and enlargement of the ventricles. The overall incidence of hydrocephalus is approximately 1 to 2 per 1000 live births.18 Hydrocephalus may be caused by blockage within the ventricular system where the CSF flows, an imbalance in the production of CSF, or a reduced reabsorption of CSF. The increased pressure within the ventricular system dilates the ventricles and pushes and compresses the brain tissue against the skull cavity (Fig. 20.8A and B). When hydrocephalus develops before fusion of the cranial sutures, the skull can expand to accommodate this additional space-occupying volume and preserve neuronal function (Types of hydrocephalus are discussed in Chapter 17).
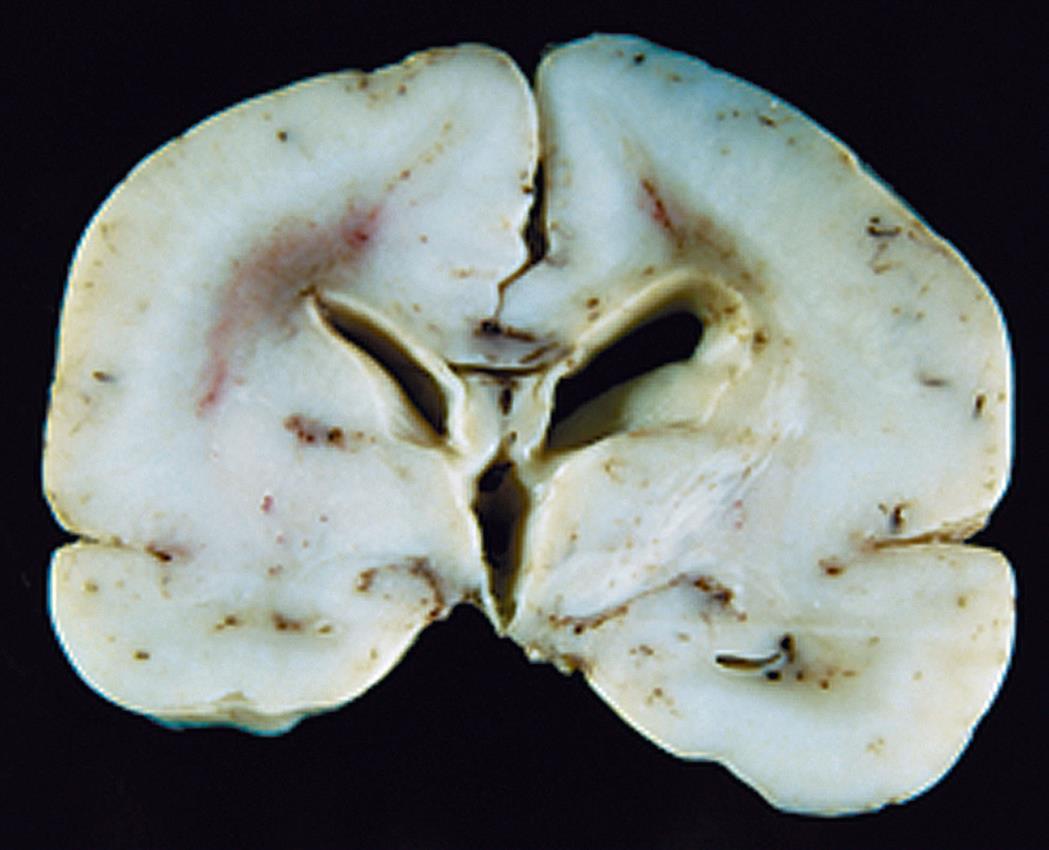
The absence of cortical gyri defines this abnormality, seen here in the brain from a full-term infant. (From Kumar V, et al. Robbins & Cotran pathologic basis of disease, 8th edition. Philadelphia: Saunders; 2010.)
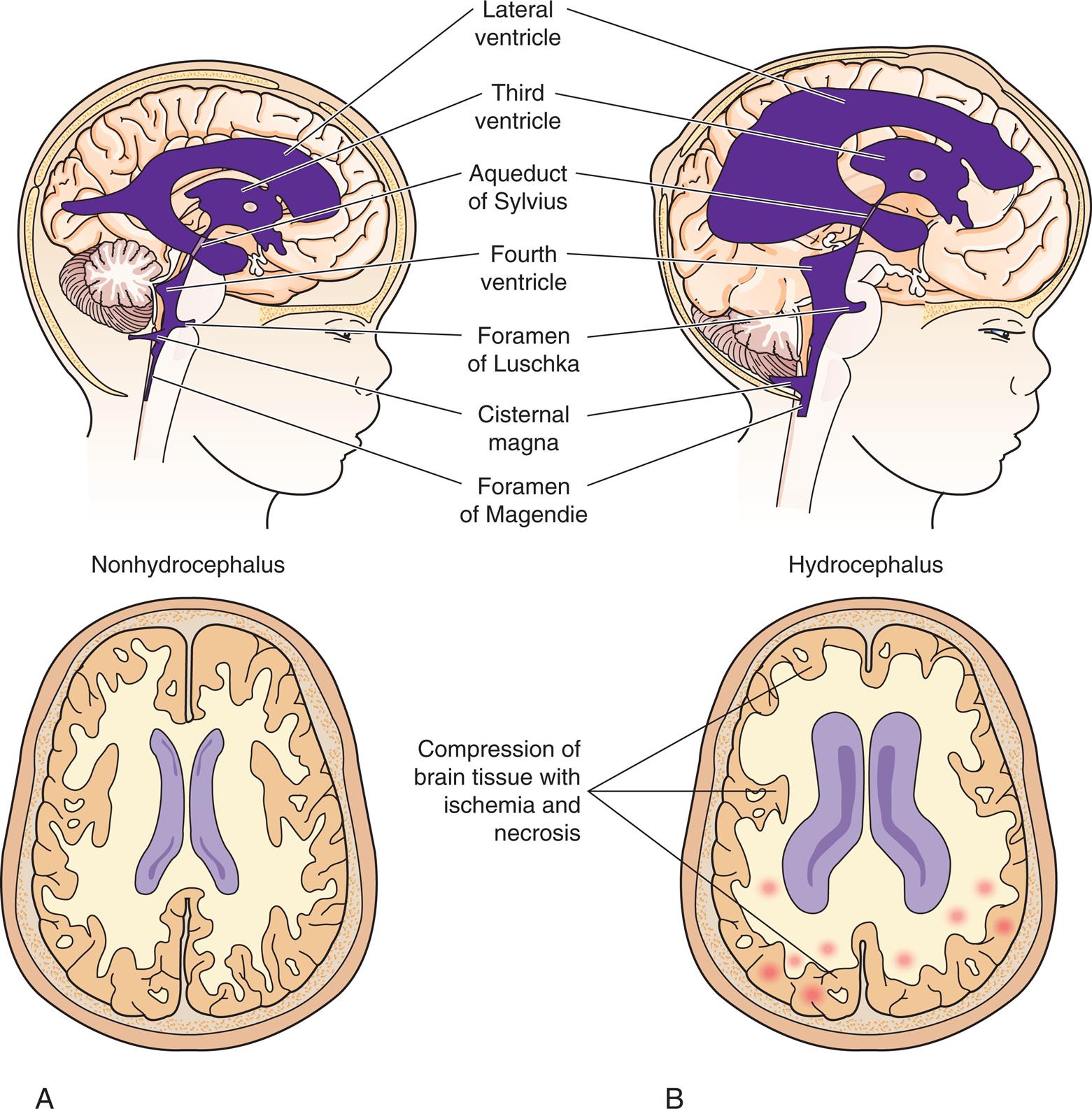

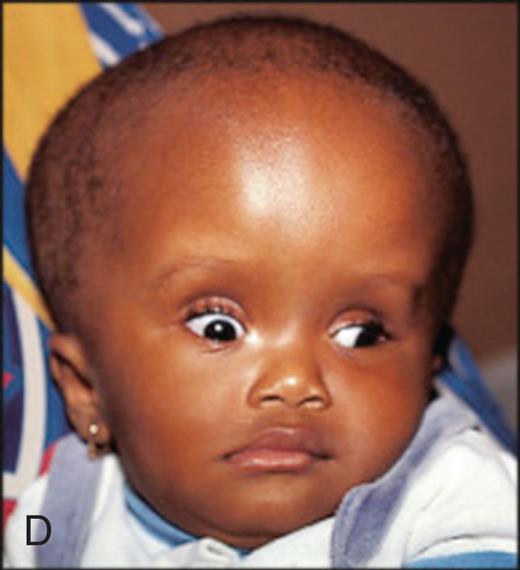
Fluid accumulation in the brain because of blockage in the flow of cerebrospinal fluid (CSF). (A) Patent cerebrospinal fluid circulation. (B) Enlarged lateral and third ventricles caused by obstruction of circulation of CSF—stenosis of aqueduct of Sylvius. (C) Child with enlarged head caused by hydrocephalus. (D) Congenital hydrocephalus with sunsetting of the eyes. (C, From McLaurin DC. Pediatric neurosurgery, 2nd edition. Philadelphia: Saunders; 1989. D, From Lissauer T, ed. Illustrated textbook of paediatrics, 5th edition. Elsevier; 2018.)
Illustration A shows the right lateral view of a child’s head and a superior view of the brain, both with nonhydrocephalus. Illustration B shows the right lateral view of a child’s head and a superior view of the brain, both with hydrocephalus. The structures in the hydrocephalus are different from those in the nonhydrocephalus in the following ways. • Lateral ventricle covers most of the brain. • Third ventricle is almost twice as large. • Aqueduct of Sylvius is thinner. • Fourth ventricle is about thrice as large. • Foramen of Luschka is bigger. • Cisternal magna is bigger. • Foramen of Magendie is shorter. • Compression of brain tissue with ischemia and necrosis. Closeup C shows the right lateral profile of an infant with hydrocephalus. The infant’s head is abnormally large. Closeup D shows the front profile of an infant with hydrocephalus. The infant’s head is abnormally large.
Congenital hydrocephalus may cause fetal death in utero, or the increased head circumference may require cesarean delivery of the infant. Symptoms depend directly on the cause and rate of hydrocephalus development. When there is separation of the cranial sutures, a resonant note sounds when the skull is tapped, a manifestation called the Macewen sign, or “cracked pot” sign. The eyes may assume a staring expression, unable to look upwards, with sclera visible above the cornea, called sunsetting (see Fig. 20.8D). Treatment outcomes are variable. Cognitive impairment in children with hydrocephalus is often related to associated brain malformations or episodes of shunt failure or infection. Approximately 40% to 65% of children with uncomplicated congenital hydrocephalus complete schooling and are employed when treated successfully with shunting or endoscopic third ventriculostomy with or without choroid plexus cauterization.19–21
The Dandy-Walker malformation (DWM) is a congenital defect of the cerebellum characterized by a large posterior fossacyst that communicates with the fourth ventricle, cerebellar hypoplasia, and macrocephaly. DWM is commonly associated with hydrocephalus. Other causes of obstructions within the ventricular system that can result in hydrocephalus include brain tumors, cysts, trauma, arteriovenous malformations, blood clots, infections, and the Chiari malformations (Emerging Science Box: Gene Therapy for Pediatric Neurologic Conditions).
Alterations In Function: Encephalopathies
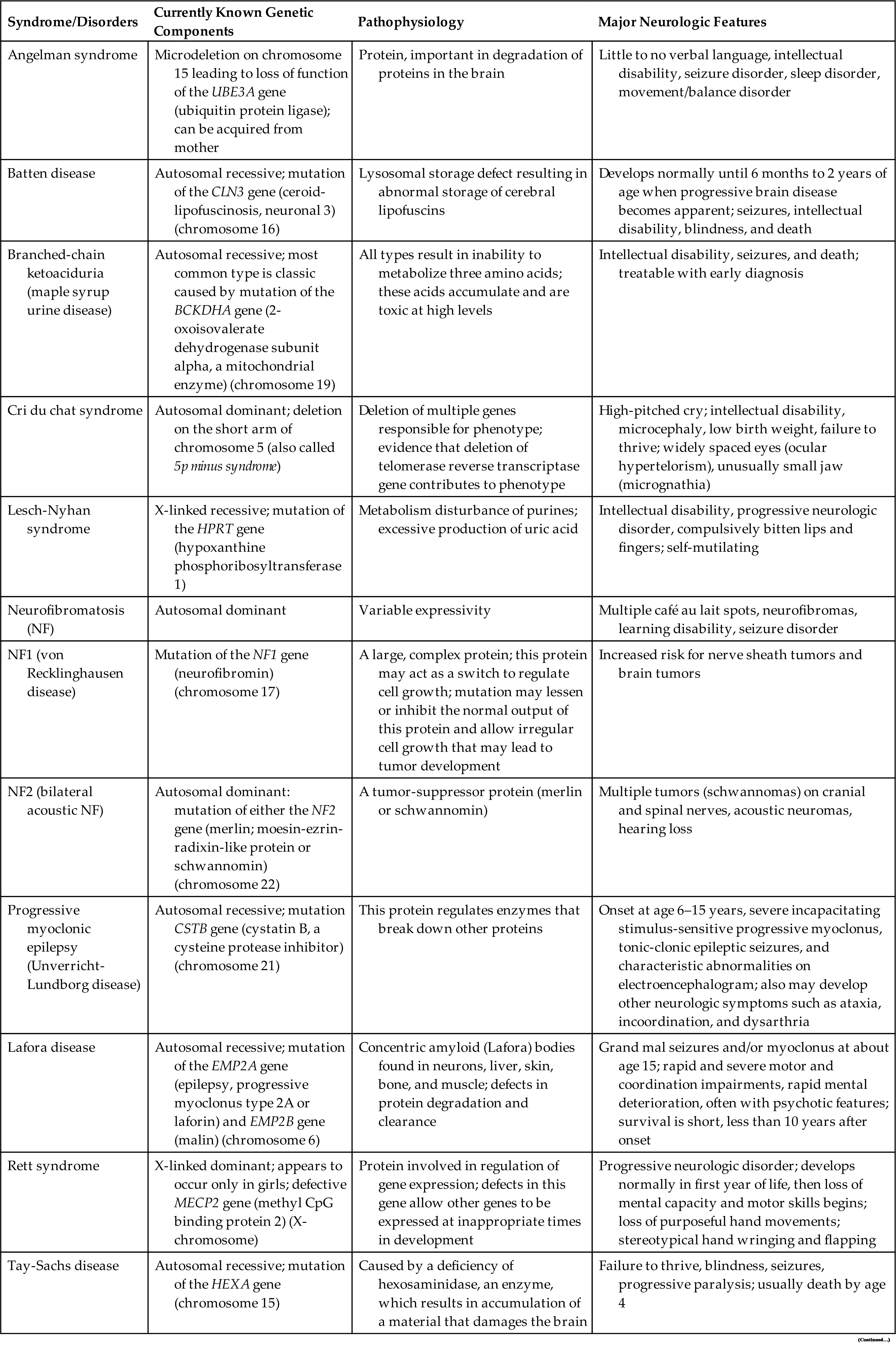
Encephalopathy, meaning brain pathology, is a general category that includes a number of syndromes and diseases (see Chapter 18). These disorders may be acute or chronic, as well as static or progressive. Some common neurologic disorders with a genetic basis are summarized in Table 20.3.
Table 20.3

Static (Nonprogressive) Encephalopathies
Static or nonprogressive encephalopathy describes a neurologic condition caused by a fixed lesion without active and ongoing disease. Causes include brain malformations or brain injury that may occur during gestation or birth or at any time during childhood. The degree of neurologic impairment is directly related to the extent of the injury or malformation and the stage of development at the time of injury. Anoxia, trauma, and infections are the most common causes of injury to the nervous system in the perinatal period. Infections, metabolic disturbances (acquired or genetic), trauma, toxins, and vascular disease may injure the nervous system in the postnatal period.22
Cerebral palsy is a disorder of movement, muscle tone, or posture that is caused by injury or abnormal development in the immature brain, before, during, or after birth up to 1 year of age. Cerebral palsy is one of the most common crippling disorders of childhood, affecting nearly 764,000 children in the United States alone. Although the exact incidence is unknown, studies suggest that the prevalence is approximately 1 in 345 children in the United States.23
Risk factors include prenatal or perinatal cerebral hypoxia, hemorrhage, infection, genetic abnormalities, or low birth weight (Table 20.4). Cerebral palsy can be classified on the basis of neurologic signs and motor symptoms, with the major types being spasticity, dystonia, ataxia, or a combination of these symptoms (mixed). Diplegia, hemiplegia, or tetraplegia may be present.
Table 20.4
Pyramidal/spastic cerebral palsy is the most common. It results from damage to corticospinal pathways (upper motor neurons) and is associated with increased muscle tone, persistent primitive reflexes, hyperactive deep tendon reflexes, clonus, rigidity of the extremities, scoliosis, and contractures. Extrapyramidal/nonspastic cerebral palsy is less common and is caused by damage to cells in the basal ganglia or cerebellum and includes two subtypes: dystonic and ataxic. Dystonic (dyskinetic) cerebral palsy is associated with injury to the basal ganglia with extreme difficulty in fine motor coordination and purposeful movements. Movements are stiff, uncontrolled, and abrupt. Ataxic cerebral palsy is caused by damage to the cerebellum, with alterations in coordination and movement. There is a broad-based gait in an attempt to maintain balance, and tremor is common with intentional movements. A child may have symptoms of each of these cerebral palsy types, which leads to a mixed disorder.24
Children with cerebral palsy often have associated neurologic disorders, such as seizures and intellectual impairment ranging from mild to severe. Other complications include visual impairment, communication disorders, respiratory problems, bowel and bladder problems, and orthopedic disabilities, such as hip pain or dislocation, balance problems, or hand dysfunction.25
Inherited Metabolic Disorders of the Central Nervous System
A large number of inherited metabolic disorders have been identified, typically leading to diffuse brain dysfunction. Early diagnosis and treatment are vital if these infants are to survive without severe neurologic problems. Newborn metabolic screening for 28 metabolic conditions (in most states) has led to most of these children being identified before symptoms develop. Inborn errors of metabolism are present at birth, and most cause disturbances of the nervous system, although they may not manifest until childhood or even adulthood. Defects in amino acid and lipid metabolism are among the most common and are presented here.
Defects in Amino Acid Metabolism
Defects in amino acid metabolism are caused by genetic defects resulting in lack of a normal protein and absence of enzymatic activity. The defects include (1) those in which the transport of an amino acid is impaired; (2) those involving an enzyme or cofactor deficiency; and (3) those encompassing certain chemical components, such as branched-chain or sulfur-containing amino acids.
Phenylketonuria
Phenylketonuria (PKU) is an example of an inborn error of metabolism characterized by phenylalanine hydroxylase deficiency and the inability of the body to convert the essential amino acid phenylalanine to tyrosine (Fig. 20.9). PKU is an autosomal recessive inborn error of metabolism characterized by mutations of the phenylalanine hydroxylase (PAH) gene. PKU has an incidence of 1 per 10,000 to 15,000 live births in the United States.19
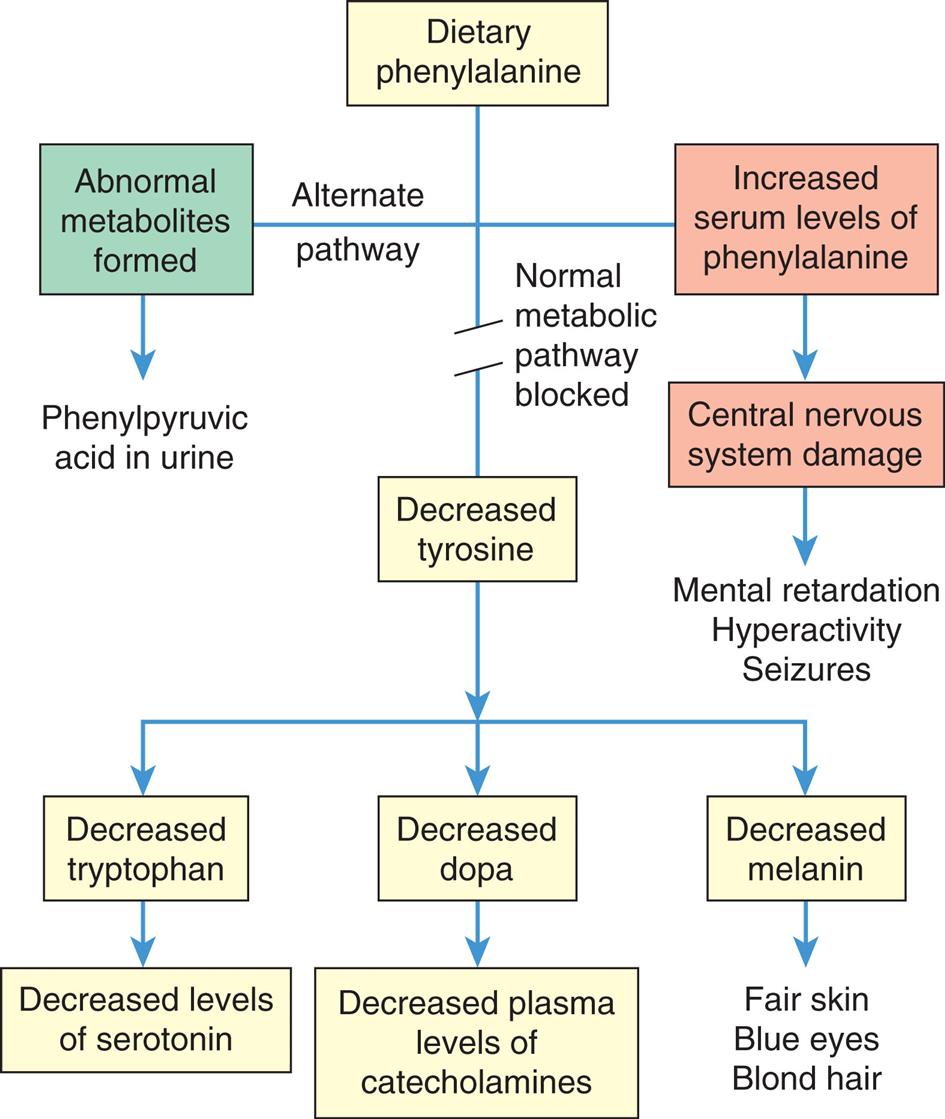
A flowchart shows metabolic error and consequences in phenylketonuria. There are three pathways from dietary phenylalanine. • Alternate pathway: abnormal metabolites formed, leading to phenyl pyruvic acid in urine. • Normal metabolic pathway blocked. • Increased serum levels of phenylalanine lead to central nervous system damage and subsequently to mental retardation, hyperactivity, and seizures. Decreased tyrosine leads to the following conditions. • Decreased tryptophan, leading to decreased levels of serotonin. • Decreased dopa, leading to decreased plasma levels of catecholamines. • Decreased melanin, leading to fair skin, blue eyes, and blond hair.
Most natural food proteins contain approximately 15% phenylalanine, an essential amino acid. Phenylalanine hydroxylase controls the conversion of this essential amino acid to tyrosine in the liver. The body uses tyrosine in the biosynthesis of proteins, melanin, thyroxine, and the catecholamines in the brain and adrenal medulla. Phenylalanine hydroxylase deficiency causes an accumulation of phenylalanine in the serum. Elevated phenylalanine levels result in developmental abnormalities of the cerebral cortical layers, defective myelination, and cystic degeneration of the gray and white matter. Unfortunately, brain damage occurs before the metabolites can be detected in the urine, and damage continues as long as phenylalanine levels remain high. Nonselective newborn screening is used to detect PKU in the United States and in more than 30 other countries. Treatment, consisting of reduction of dietary phenylalanine (PKU diet), is effective and allows for normal development. Mutations in the PAH gene are by far the most common cause of PKU, although there are other types of PKU. In one such variation, there is impaired synthesis of cofactors (e.g., tetrahydrobiopterin [BH4]), which contributes to elevated levels of phenylalanine. Individuals with impaired synthesis of BH4 have a positive response when sapropterin, a synthetic form of BH4, is included in their treatment.20
Defects in Lipid Metabolism
Disorders of lipid metabolism are termed lysosomal storage diseases because each disorder in this group can be traced to a missing lysosomal enzyme needed for the breakdown and recycling of many cellular products. Lysosomal storage disorders include more than 50 known genetic disorders. The incidence of lysosomal storage disorders is approximately 1 in 5000 live births.21 These disorders cause an excessive accumulation of a particular cell product, occurring in the brain, liver, spleen, bone, and lung and thus involving several organ systems; these disorders often are highly debilitating and cause progressive physical and neurologic disability. In general, these disorders are not included in newborn screening. Some of these disorders may be treated with enzyme replacement therapy.26 Perhaps the best known of the lysosomal storage disorders is GM2 gangliosidosis (Tay-Sachs disease), an autosomal recessive disorder caused by deficiency of the lysosomal enzyme hexosaminidase A (HexA), an enzyme that degrades GM2 gangliosides (fatty acids) within nerve cell lysosomes. Eighty percent of individuals diagnosed are of Jewish ancestry, although sporadic cases appear in the non-Jewish population. Onset of this disease usually occurs when the infant is 4 to 6 months old. Symptoms of Tay-Sachs include an exaggerated startle response to loud noise, seizures, developmental regression, dementia, and blindness. Death from this disease is almost universal and occurs by 5 years of age. Screening for carriers of the gene defect concomitant with counseling to prevent disease transmission is possible.27
Intoxications of the Central Nervous System
Drug-induced encephalopathies must always be considered a possibility in the child with unexplained neurologic changes. Such encephalopathies may result from accidental ingestion, therapeutic overdose, intentional overdose, or ingestion of environmental toxins. There were 37.7/1000 children younger than 6 years of age with poison exposures in 2018. Approximately 44.2% of all poison exposures occurred in children younger than 6 years of age, with the highest incidence being 1- and 2-year-old children. The most commonly ingested poisons in children younger than 6 years are cosmetics and personal care products, cleaning substances, and over-the-counter analgesics and other medications (Table 20.5).28
Table 20.5
Lead poisoning results in high blood levels of lead. If lead poisoning is untreated, lead encephalopathy results and is responsible for serious and irreversible neurologic damage. Those at greatest risk are children ages 2 to 3 years and children prone to the practice of pica—the habitual, purposeful, and compulsive ingestion of nonfood substances, such as clay, soil, and paint chips or paint dust. Lead intoxication also may occur from chronic exposure to lead in cosmetics, inhalation of gasoline vapors, and ingestion of airborne lead.29 Fetal neurotoxicity occurs with maternal lead exposure, particularly during the first trimester.30 Details related to lead intoxication are described in Chapter 2. Refer to Fig. 20.10 for a summary of toxic lead effects.
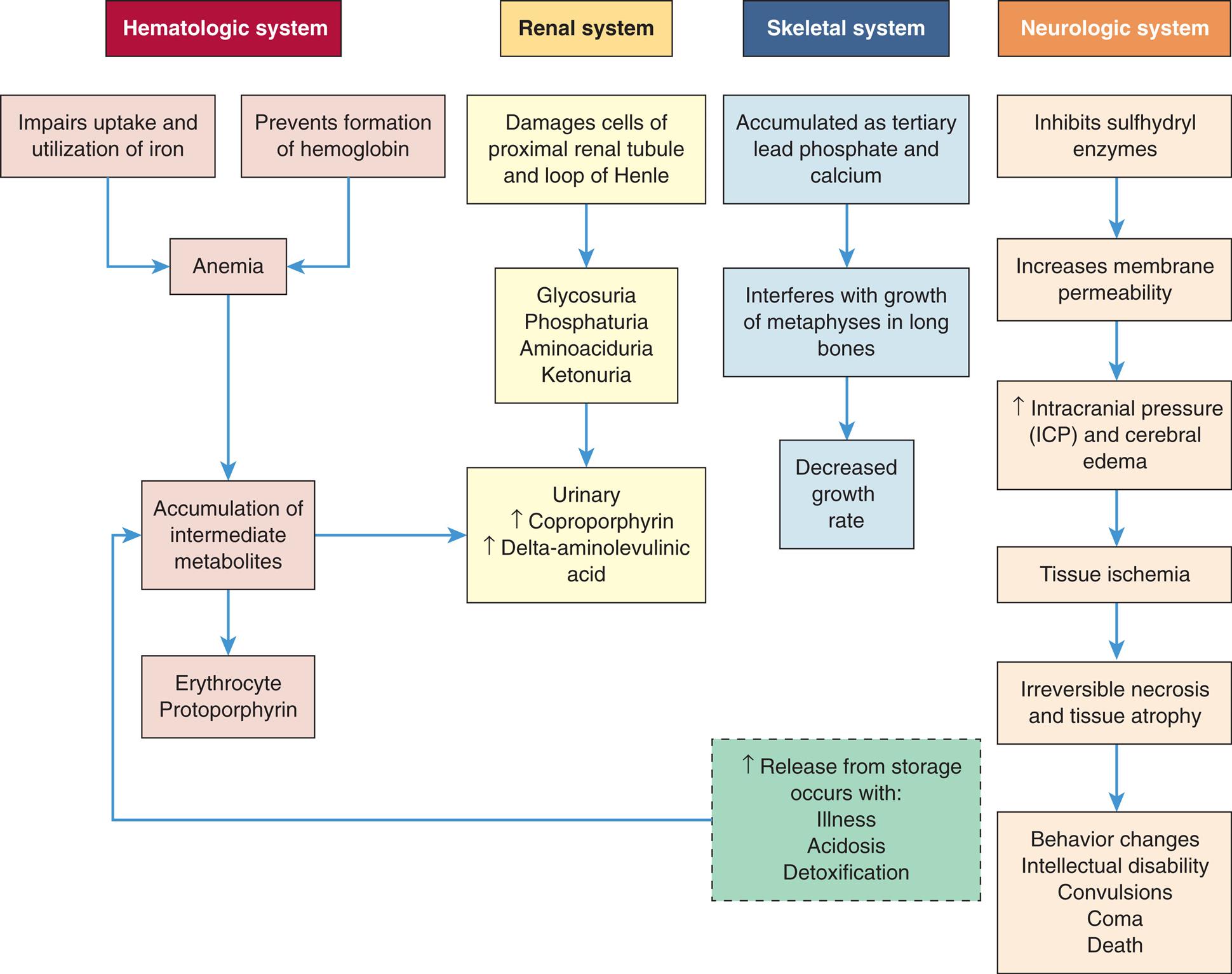
A flowchart tracks a systemic effects of increased lead absorption in children. The flowchart shows four systems. The hematologic system is as follows. Impaired update and utilization of iron and prevention of formation of hemoglobin leads to anemia. Anemia leads to accumulation of intermediate metabolites, which in turn results in either of the following: • Erythrocyte, protoporphyrin. • Urinary, increased coproporphyrin, increased delta-aminolaevulinic acid. The renal system undergoes the following changes. • Damages cells of proximal rental tubule and loop of Henle. • Glycosuria, phosphaturia, aminoaciduria, and ketonuria. • Urinary, increased coproporphyrin, increased delta-aminolaevulinic acid. The skeletal system undergoes the following changes. • Accumulated as tertiary lead phosphate and calcium. • Interferes with growth of metaphysis in long bones. • Decreased growth rate. The neurologic system undergoes the following changes. • Inhibits sulfhydryl enzymes. • Increases membrane permeability. • Increased intracranial pressure (I C P) and cerebral edema. • Tissue ischemia. • Irreversible necrosis and tissue atrophy. • Behavior changes, intellectual disability, convulsions, coma, and death. Increased release from storage occurs with illness, acidosis, and detoxification leads to accumulation of intermediate metabolites.
Infections of the Central Nervous System
Meningitis is an infection of the meninges and subarachnoid space of the brain and spinal cord. Encephalitis is inflammation within the brain. In many infections of the meninges, encephalitis also is present and the term meningoencephalitis is used. The origin of such inflammation and acute encephalopathy can be caused by bacteria, viruses, or other microorganisms. Aseptic meningitis has no evidence of bacterial infection but may be associated with viral infection, systemic disease, or drugs.
Bacterial meningitis
Acute bacterial meningitis is one of the most serious infections to which infants and children are susceptible. The introduction of conjugate vaccines against Haemophilus influenzae type B, Streptococcus pneumoniae, and Neisseria meningitidis (meningococcus) has decreased the incidence of bacterial meningitis.31 Vaccines for serogroup B N. meningitidis are now available.32
Group B Streptococcus causes lethal meningitis and sepsis in neonates; it is transmitted to the child from the mother's birth canal and is the most common cause of meningitis in newborns. S. pneumoniae is the most common microorganism in children 1 to 23 months of age. Staphylococcal or streptococcal meningitis can occur in children of any age but shows a predilection for children who have had neurosurgery, skull fracture, or a complication of systemic bacterial infection. Infections that originate in the middle ear, sinuses, or mastoid cells also may lead to S. pneumoniae infection in children. Children with sickle cell disease or who have had a splenectomy are particularly at high risk for infection.
Escherichia coli and group B beta-hemolytic streptococci are the most common causes of meningitis in the newborn period. The second most common microorganism causing bacterial meningitis, particularly in children younger than 4 years, is N. meningitidis (meningococcus) and it has the potential to occur in epidemics. Approximately 2% to 5% of healthy children are carriers of N. meningitidis. Because the incidence of N. meningitidis infection increases in adolescence and with crowded environments, such as in dormitories and among military personnel, it is recommended that all individuals 11 to 12 years of age receive two immunizations against this pathogen. Teens and young adults (16 through 23 year olds) also may be vaccinated with a serogroup B meningococcal vaccine.33
Pathophysiology
Pathogens enter the nervous system by direct extension from a contiguous source (e.g., paranasal sinuses or mastoid cells) or, more commonly, by hematogenous spread (e.g., infective endocarditis, pneumonia, neurosurgical procedures, severe burns). Pathogens then cross the blood-brain barrier, enter the CSF, and multiply. Bacterial toxins increase cerebrovascular permeability, causing alterations in blood flow and edema. Increased intracranial pressure (ICP) may be increased further by obstruction to the CSF circulation. Herniation of the brainstem causes death.
Clinical manifestations
Acute bacterial meningitis often is preceded by an upper respiratory tract or a gastrointestinal infection. Inflammation leads to the general symptoms of fever, headache, vomiting, and irritability and the CNS symptoms of photophobia, nuchal and spinal rigidity, decreased level of consciousness, and seizures. Irritation of the meninges and spinal roots causes pain and resistance to neck flexion (nuchal rigidity), a positive Kernig sign (resistance to knee extension in the supine position with the hips and knees flexed against the body), and a positive Brudzinski sign (flexion of the knees and hips when the neck is flexed forward rapidly). With severe meningeal irritation the child may demonstrate opisthotonic posturing (rigid arching of the back with the head extended). Infants may have bulging fontanelles. Meningococcal meningitis can produce a characteristic petechial rash.
Viral meningitis may result from a direct infection caused by a virus, or it may be secondary to disease, such as measles, mumps, herpes, or leukemia. The hallmark of viral meningitis, or aseptic meningitis, is a mononuclear response in the CSF and the presence of normal glucose levels. The clinical manifestations are similar to those in bacterial meningitis, although usually milder.
Evaluation and treatment
A definitive diagnosis is made by examination of CSF obtained from a lumbar puncture and by CSF and blood cultures. The principles of treatment are similar to those followed for adults (see Chapter 18) and are based on the culture results in which the causative microorganism is identified. Empirical antibiotic therapy should be initiated and continued until CSF cultures are negative or changed if the empirical therapy is not correct for the bacteria cultured from the CSF. Bacterial lysis induced by antibiotics can cause subarachnoid inflammation; the severity may be reduced with corticosteroid treatment. The factors that influence outcomes are the age of the child (mortality is highest in infants younger than 1 year), the infective microorganisms (the lowest mortality is in meningococcal meningitis and the highest in meningitis caused by gram-negative enteric microorganisms), and the duration and extent of inflammation before treatment. Approximately 8% of children with H. influenzae meningitis die; 35% of the survivors have serious and permanent sensory or motor dysfunction caused by pressure on the peripheral nerves during the early phases of the illness. Approximately 5% of the children who survive meningitis have hearing deficits; 15% to 30% have cerebral damage, hydrocephalus, motor deficits, or sensory impairments.
Viral encephalitis in children is similar to viral encephalitis in adults (see Chapter 18, and Fig. 18.28) and can be difficult to distinguish from viral meningitis. Viruses are obligate intracellular pathogens which can directly invade the brain, causing inflammation. Postinfectious encephalitis can develop as a result of an autoimmune response. Inflammatory and autoimmune causes of encephalitis may be treatable if identified early in an individual’s course of illness.34 (Encephalopathy resulting from infection with the human immunodeficiency virus [HIV] is discussed in Chapters 9 and 18.)
Cerebrovascular Disease in Children
Perinatal Stroke
Perinatal stroke is defined as stroke occurring from 28 weeks’ gestation to 28 days postnatal.35 Perinatal arterial ischemic stroke is estimated at 1 in 4000 live births and is a leading cause of perinatal brain injury, cerebral palsy, and lifelong disability. Although a cause for perinatal stroke is usually not found, clotting abnormalities may make the child prone to further vascular events. Up to 80% are ischemic with the remainder secondary to hemorrhage or cerebral sinovenous thrombosis (CSVT).
Childhood Stroke
Childhood stroke occurs in 1.3 to 1.6 per 100,000 children per year and may be divided into two categories: ischemic and hemorrhagic.35,36 According to the Center for Disease Control and Prevention, cerebrovascular disease (stroke) is one of the 10 leading causes of death in children and adolescents from 1 to 24 years of age. Ischemic (occlusive) stroke in children may result from embolism, Cerebral sinovenous thrombosis (CSVT), or congenital or iatrogenic narrowing of vessels, leading to decreased flow of blood and oxygen to areas of the brain. Although rare, it is more common than hemorrhagic stroke. Children with arterial ischemic stroke do not have the typical adult risk factors of atherosclerosis and hypertension. Many children with acute ischemic stroke have no identifiable risk factors.35 Arteriopathies, cardiac anomalies, sickle cell disease, infections, head and neck disorders, systemic conditions, and prothrombic states are the common disorders associated with arterial ischemic stroke.35
Hemorrhagic stroke is most commonly caused by bleeding from congenital cerebral arteriovenous malformations and is rare in children younger than 19 years. Intraventricular hemorrhage associated with premature birth is related to immature blood vessels and unstable blood pressure. There is a high risk of developing posthemorrhagic hydrocephalus in very low birth weight (<1000 g) preterm infants with increased risk of developmental disabilities.37
The clinical presentation of stroke varies according to the vessels involved and the age of the individual. Symptoms include hemiplegia, weakness, seizures, headaches, high fever, nuchal rigidity, hemianopia, sensory changes, facial palsy, and temporary aphasia. Obtaining a thorough history of evolving symptoms and risk factors is important for diagnosis. Laboratory studies may be indicated. Neuroimaging studies assist in determining the extent of the disease. Surgery is an option for treatment, and anticoagulants and antithrombotics may be used in selected cases.
Moyamoya disease is a rare, chronic, progressive vascular stenosis of the circle of Willis of unknown etiology. There is obstruction of arterial flow to the brain and the development of small basal arterial collateral vessels that vascularize the hypoperfused brain distal to the occluded vessels.38 Moyamoya means a puff of smoke in Japanese and represents the appearance of these small vessels on cerebral angiography. The disease is most common in East Asia and is associated with genetic factors, particularly those associated with vascular growth factors. These children often present with transient ischemic attacks (TIAs) because the new vessels fail to provide adequate perfusions.39 Diagnosis is made by cerebral angiography, and the most effective treatment is surgical revascularization.
Epilepsy and Seizure Disorders in Children
The incidence of epilepsy varies greatly with age, geographic location, and study design. Approximately 470,000 children in the United States had epilepsy in 2015.40 Seizures are the abnormal discharge of electrical activity within the brain. When a sufficient number of neurons become overexcited, they discharge abnormally, which sometimes results in clinical manifestations (seizures) with alterations in motor function, sensation, autonomic function, behavior, and consciousness. The manifestations depend on the site and spread of abnormal electrical activity within the brain. If a child has more than two unprovoked seizures more than 24 hours apart, that child is said to have epilepsy, although there are a few exceptions—one example being febrile seizures (see Chapter 16). Seizures may result from diseases that are primarily neurologic (CNS) or are systemic and affect CNS function secondarily (e.g., diabetes). Seizures can be caused by structural abnormalities of the brain, hypoxia, intracranial hemorrhage, CNS infection, traumatic injury, electrolyte imbalance, or inborn metabolic disturbances (see Chapter 18). Febrile Seizures are benign and the most common type of childhood seizure. Seizures are sometimes clearly familial. Often the cause of epilepsy is unknown and presumed to have a genetic basis. (Table 18.10 summarizes the classification of seizures.)
Antiepileptic drugs (AEDs) stabilize ion channels and neurotransmitters and are effective in treating seizures in 70% of persons. The 30% of individuals with medication-refractory epilepsy are increasingly effectively treated with other modalities. A ketogenic diet with high-fat and low-carbohydrate consumption replaces energy metabolism with ketone bodies instead of glucose and appears to increase the seizure threshold.41 Neuromodulation (e.g., vagus nerve stimulation) can be effective to suppress epileptogenic activity.42 Surgery to remove epileptic brain areas or disconnect eliptogenic pathways can reduce seizure activity.43
Epileptic spasms (also known as infantile spasms) are rare, severe types of seizures associated with electroencephalogram (EEG) abnormalities and varying clinical manifestations. The cause can be related to underlying brain abnormalities such as intrauterine stroke, cortical dysplasias, or tuberous sclerosis (a benign tumor), whereas others may be caused by mutations in one of several genes. Clinical manifestations of the resulting spasms may range from subtle head nods to violent body contractions, commonly referred to as jackknife seizures. Onset of infantile spasms usually is during infancy or early childhood. An EEG will display a classic hypsarrhythmic pattern on a slow, disorganized chaotic background. After infantile spasms begin, there is usually a typical clinical course. The “spasms” usually occur in clusters and transpire 5 to 150 times per day. They are commonly worse when the infant is waking up or falling asleep. Once begun, the seizure activity increases in intensity and severity over time. Invariably, a loss of developmental milestones and disability is associated with this syndrome. Treatment is the administration of adrenocorticotrophic hormone, vigabatrin (particularly for children with tuberous sclerosis), and corticosteroids. Surgery is an option when pharmacologic treatment has failed.44
Childhood Tumors
Brain Tumors
Brain tumors are the most common solid tumor, are the second most common primary neoplasm in children (after leukemia), and can be benign or malignant.45,53 They are the leading cause of cancer-related death in children, and the 5-year survival is approximately 74%, varying significantly by tumor type. Primary brain tumors arise from brain tissue and do not metastasize outside the brain. The cause of brain tumors is unknown, although genetic, environmental, and immune factors have been investigated. Exposure to radiation therapy has been the only environmental factor consistently related to the development of brain tumors.46,53
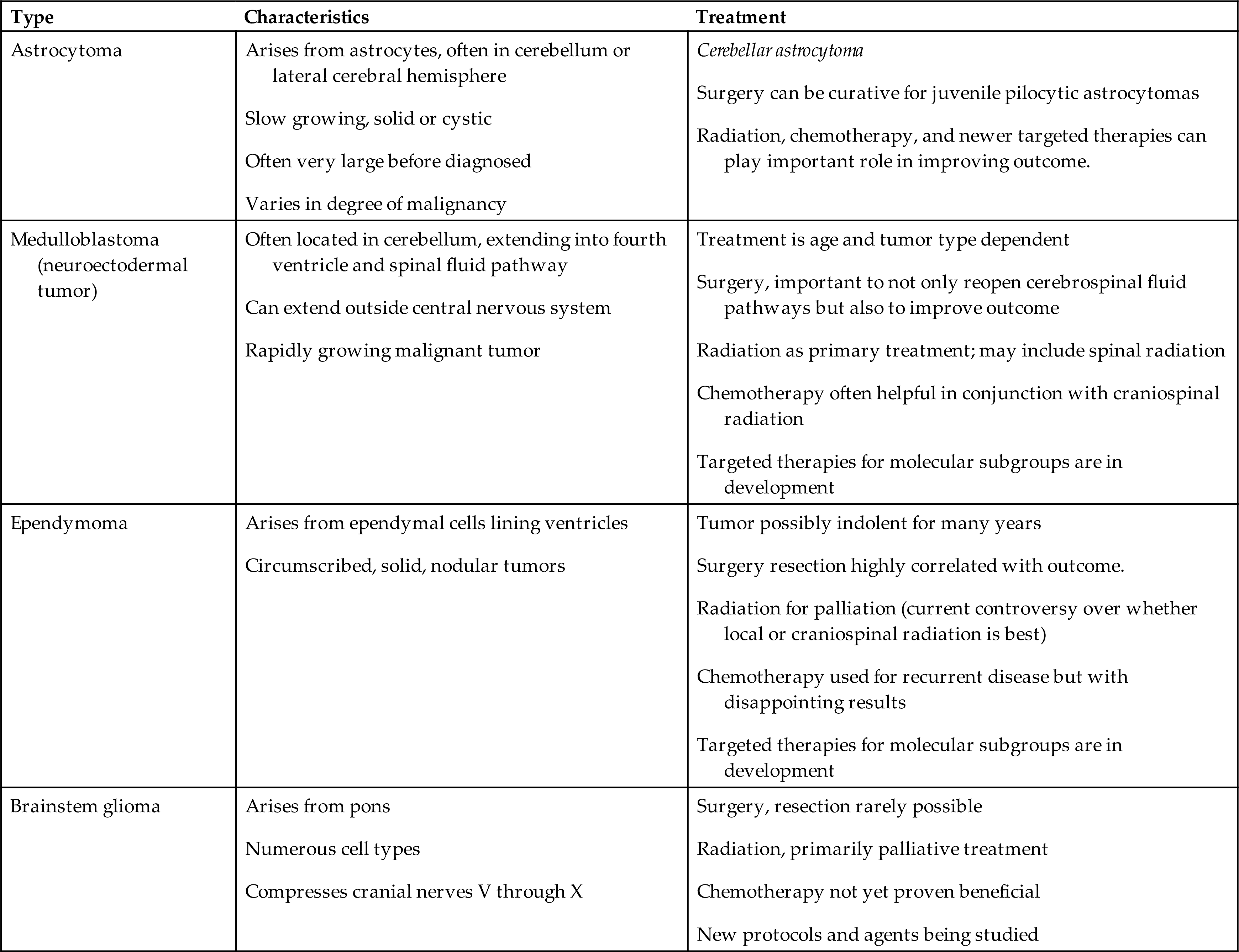
Brain tumors can arise from any CNS cell, and tumors are classified by cell type and molecular markers. The most common tumors are embryonal tumors, including medulloblastoma, atypical medulloblastoma, atypical teratoid rhabdoid tumors, CNS primitive neuroectodermal tumors, and high-grade gliomas, including glioblastomas, diffuse pontine gliomas, and other malignant astrocytomas. Other tumor types are less common, including low-grade astrocytomas (especially pilocytic astrocytomas), neuronal and mixed neuronal-glial tumors, and ependymal tumors (ependymomas). Brain tumors in children are often located in the posterior fossa (Fig. 20.11). The types and characteristics of the more common childhood brain tumors are summarized in Table 20.6.
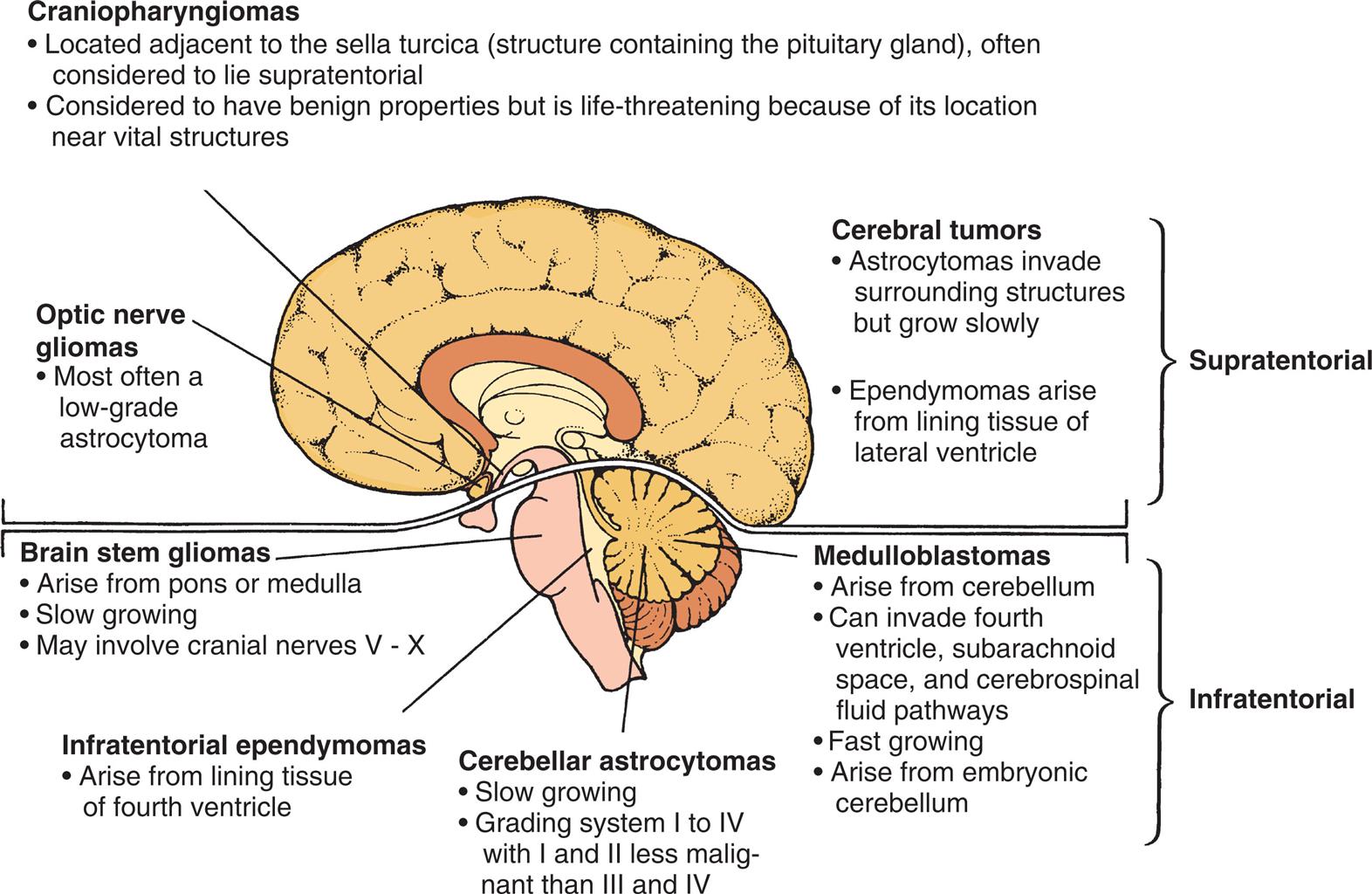
An illustration of the left lateral view of a child’s brain identifies the location of brain tumors. The data from the illustration are as follows. Craniopharyngiomas. • Located adjacent to the Sella turcica (structure containing the pituitary gland), often considered to lie supratentorial. • Considered to have benign properties but is life threatening because of its location near vital structures. Optic nerve gliomas. • Most often a low-grade astrocytoma. Cerebral tumors (supratentorial). • Astrocytomas invade surrounding structures but grow slowly. • Ependymomas arise from lining tissue of lateral ventricle. Brain stem gliomas. • Arise from pons or medulla. • Slow growing. • May involve cranial nerves V to X. Medulloblastomas (infratentorial). • Arise from cerebellum. • Can invade fourth ventricle, subarachnoid space, and cerebrospinal fluid pathways. • Fast growing. • Arise from embryonic cerebellum. Infratentorial ependymomas. • Arise from lining tissue of fourth ventricle. Cerebellar Astrocytomas. • Slow growing. • Grading system 1 to 4 with 1 and 2 less malignant than 3 and 4.
Table 20.6
Signs and symptoms of brain tumors in children vary from generalized and vague to localized and related specifically to an anatomic area. Signs of elevated ICP may occur, including headache, vomiting, lethargy, and irritability. If a young child complains of repeated and worsening headache, a thorough investigation should take place because headache is an uncommon complaint in young children. Headache caused by elevated ICP usually is worse in the morning and gradually improves during the day, when the child is upright and venous drainage is enhanced. The frequency of headache and other symptoms increases as the tumor grows. Irritability or possible apathy and increased somnolence also may result. Like headache, vomiting occurs more commonly in the morning. Often it is not preceded by nausea and may become projectile, differing from a gastrointestinal disturbance in that the child may be ready to eat immediately after vomiting. Persistent headaches with vomiting in a child should be evaluated promptly. Other signs and symptoms include increased head circumference with bulging fontanelles in the child younger than 2 years, cranial nerve palsies, and papilledema (Box 20.1).
Localized findings relate to the degree of disturbance in physiologic functioning in the area where the tumor is located. The majority of pediatric brain tumors involve the cerebellum, brain stem, and fourth ventricle affecting all pediatric age groups. Children with infratentorial tumors (posterior fossa) exhibit localized signs of impaired coordination and balance, including ataxia, gait difficulties, truncal ataxia, and loss of balance. Supratentorial tumors of the cerebral hemispheres are more common in children younger than 3 years of age.46 Most pediatric brain tumors require surgical resection. Aggressive radiation and chemotherapy are required for high-grade tumors, although radiation therapy is rarely offered to children younger than 4 years of age, due to significant long-term side effects. Advances are being made in areas of immunotherapy and targeted molecular therapy (see Emerging Science Box: The New Evolving Classification System for Pediatric Brain Tumors).47
Embryonal Tumors
Neuroblastoma
Neuroblastoma is an embryonal tumor originating from neural crest tissues of the sympathetic nervous system outside the CNS. It is the most common cancer in infants younger than 1 year of age. Although it accounts for only approximately 6% of pediatric malignancies (approximately 800 new cases per year), neuroblastoma causes approximately 15% of cancer deaths in children.48 More than with any other cancer, neuroblastoma has been associated with spontaneous remission, commonly in infants. Prognosis is worse for children older than 2 years of age with disseminated disease. Although familial tendency has been noted in individual cases, a nonfamilial or sporadic pattern is found in most children with neuroblastoma. Familial cases of neuroblastoma are considered to have an autosomal dominant pattern of inheritance (mechanisms of inheritance are discussed in Chapter 4).
The most common location of neuroblastoma is in the retroperitoneal region, most often in the adrenal medulla. The tumor is evident as an abdominal mass and may cause anorexia, bowel and bladder alteration, and sometimes spinal cord compression. The second most common location of neuroblastoma is the mediastinum (15% of cases), where the tumor may cause dyspnea or infection related to airway obstruction. Rarely, neuroblastoma may arise from the cervical sympathetic ganglion.
A number of systemic signs and symptoms are characteristic of neuroblastoma, including weight loss, irritability, fatigue, and fever. Intractable diarrhea occurs in some children and is caused by tumor secretion of a hormone called vasoactive intestinal polypeptide (VIP). Most children with neuroblastoma have increased amounts of catecholamines and associated metabolites in their urine (e.g., urinary vanillylmandelic acid [VMA] and homovanillic acid [HVA]). High levels of urinary catecholamines and serum ferritin are associated with a poor prognosis. Immunotherapeutic treatments are undergoing continuing investigation and hold promise for good outcomes without toxicity.49
Retinoblastoma
Retinoblastoma (RB) is the most common intraocular congenital eye tumor of young children that originates in the retina of one or both eyes (Fig. 20.12A and B). Two forms of RB are exhibited: inherited and acquired. Both forms of the disease are a consequence of loss of function of the RB gene (RB1). The inherited form of the disease generally is diagnosed during the first year of life. The acquired disease is diagnosed before the age of 5 in 90% of the cases.50 Approximately 40% of RBs are inherited as an autosomal dominant trait with incomplete penetrance (see Fig. 4.24). The remaining 60% are acquired somatic mutations. The “two-hit” hypothesis explains the occurrence of both hereditary and acquired forms of the disease.51 This hypothesis predicts that two separate transforming events or “hits” must occur in a normal retinoblast cell to cause the cancer. Furthermore, it proposes that in the inherited form, the first hit or mutation occurs in the germ cell (inherited from either parent), and the mutation is contained in every cell of the child's body. Only a second, random mutation in a retinoblast cell is needed to transform that cell into cancer. Multiple tumors are observed in the inherited form because these second mutations are likely to occur in several of the approximately 1 to 2 million retinoblast cells. In contrast, the acquired form of RB requires two independent hits or mutations to occur in the same somatic cell (after the egg is fertilized) for the transformation to cancer. This is much less likely to happen. Fig. 20.13 illustrates the “two-hit” model for these two patterns of mutation. Epigenetic mechanisms (i.e., inactivating the gene without changing its DNA sequence) also play a role in dysregulation of the tumor suppressor and oncogenic pathways of RB, and they may become markers for difficult to diagnose tumors.52 The primary sign of RB is leukocoria, a white pupillary reflex (white reflex) also called cat's eye reflex, which is caused by the mass behind the lens (see Fig. 20.12B). This easy to identify sign can be missed. Other signs and symptoms include strabismus; a red, painful eye; and limited vision.
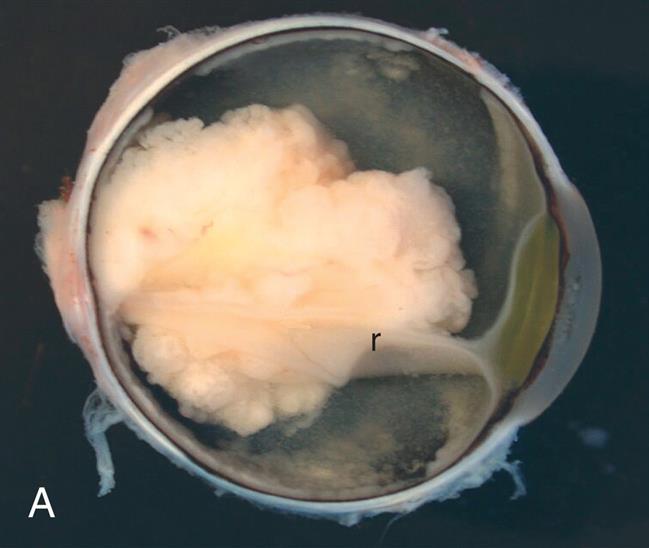
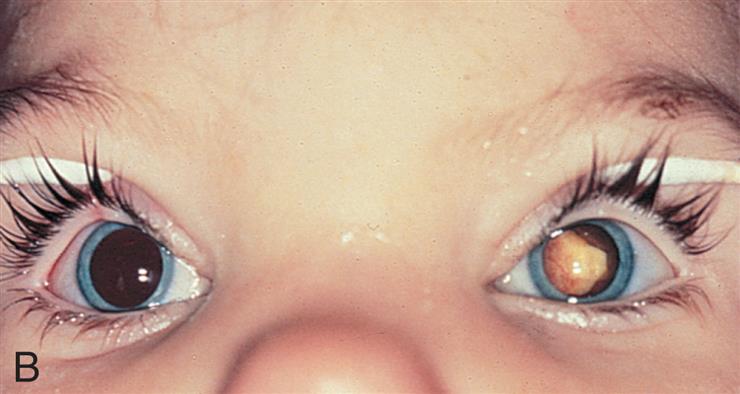
(A) Presence of white mass surrounding the detached retina (r). (B) The tumor occupies a large portion of the inside of the eye globe. (A, From Roberts F. Macroscopic techniques for ophthalmic tumor specimens. Seminars in Diagnostic Pathology, 2016;33(3):114–121. B, From Damjanov I. Pathology for the health professions, 3rd edition. St. Louis: Saunders; 2006. Courtesy Dr. Walter Richardson and Dr. Jamsheed Khan, Kansas City, KS.)
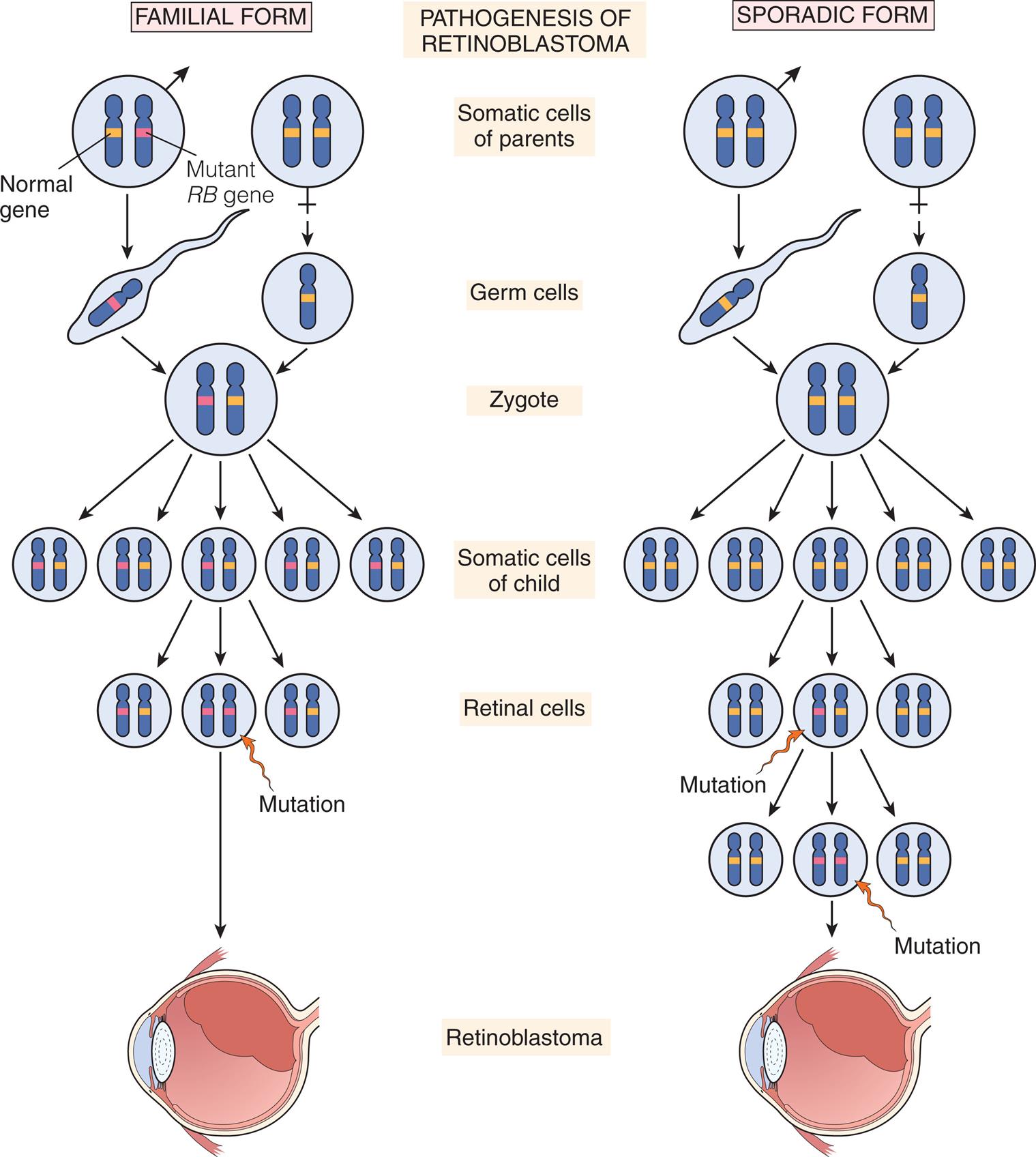
In inherited retinoblastoma, the first mutation is transmitted through the germline of an affected parent. The second mutation occurs somatically in a retinal cell, leading to development of the tumor. In sporadic retinoblastoma, development of a tumor requires two somatic mutations.
Two sets of illustrations show mutations leading to retinoblastoma development. Left panel, familial form. Somatic cells of parents show a pair of chromosomes each. Male cell contains 1 chromosome containing normal gene and 1 containing mutant gene. Mutant gene is passed into the zygote, multiplying into pairs, where each pair has one normal gene and one mutated gene. Mutation of the somatic cells of the child result in a pair of two mutated genes, leading to retinoblastoma. Right panel, sporadic form. Somatic cells of parents are both normal, that form a normal zygote, that multiply into normal somatic cells of the child. Mutation in one pair of cells leads to one mutated gene and one normal gene. Additional mutation leads to a cell with two mutated genes, leading to retinoblastoma.
Because RB is a treatable tumor, dual priorities are saving the child's life and restoring useful vision. The prognosis for most children with RB is excellent, with a greater than 90% long-term survival.
Summary Review
Development of the Nervous System in Children
- 1. Growth and development of the brain occur most rapidly during fetal development and during the first year of life.
- 2. The bones of the skull are joined by sutures, and the wide, membranous junctions of the sutures (known as fontanelles) allow for brain growth and close by 18 months of age typically.
- 3. At birth, the forebrain is immature, so neurologic examination is primarily of reflex responses that require an intact spinal cord and brainstem.
Structural Malformations
- 1. Neural tube defects (NTDs) are an arrest of the normal development of the brain and spinal cord during the first month of embryonic development. Risk factors include folic acid deficiency, previous NTD pregnancy, maternal diabetes or obesity, use of anticonvulsants, and maternal hyperthermia.
- 2. Spina bifida (failure of vertebral closure) is the most common NTD and includes meningocele (a sac-like meningeal cyst of spinal fluid that protrudes through a vertebral defect), myelomeningocele (like a meningocele that also contains a portion of the spinal cord with its nerves), and spina bifida occulta.
- 3. Premature closure of the cranial sutures causes craniosynostosis, resulting in an asymmetric skull shape. If multiple sutures fuse prematurely, brain growth may be restricted.
- 4. Microencephaly is a defect in brain cell proliferation that leads to a small brain, small skull, and usually developmental delays.
- 5. Cortical dysplasias are defects in brain development caused by defects in neuronal cell migration and subsequent abnormalities in connection between cells.
- 6. Congenital hydrocephalus, characterized by increased cerebrospinal fluid (CSF) pressure, results from impaired absorption or blockage of circulation of CSF and rarely overproduction of CSF. Dandy-Walker deformity, which is commonly associated with hydrocephalus, is associated with cystic dilation of the fourth ventricle and aqueductal compression.
Alterations in Function: Encephalopathies
- 1. Static (nonprogressive) encephalopathies (e.g., cerebral palsy and epilepsy) are disorders of the brain caused by a fixed lesion without active and ongoing disease. They can occur during gestation, birth, or childhood and can be caused by endogenous or exogenous factors.
- 2. Cerebral palsy (CP) can be caused by prenatal or perinatal cerebral hypoxia or perinatal trauma, with symptoms of motor dysfunction (including increased muscle tone, increased reflexes, and loss of fine motor coordination), mental retardation, seizure disorders, or developmental disabilities. CP can be extrapyramidal/nonspastic or pyramidal/spastic.
- 3. Inherited metabolic disorders that damage the nervous system include defects in amino acid metabolism (phenylketonuria) and lysosomal storage defects (GM2 gangliosidosis/Tay-Sachs disease) and result in abnormal behavior, seizures, and deficient psychomotor development.
- 4. Seizure disorders are abnormal discharges of electrical activity within the brain. They are associated with numerous nervous system disorders and more often are a generalized rather than a partial type of seizure.
- 5. Accidental ingestion, therapeutic overdose, intentional overdose, or ingestion of environmental toxins, such as in lead poisoning, can cause serious neurologic damage.
- 6. Bacterial meningitis is commonly caused by Neisseria meningitidis or Streptococcus pneumoniae and may result from respiratory tract or gastrointestinal infections; symptoms include fever, headaches, photophobia, seizures, rigidity, and stupor.
- 7. Viral meningitis may result from direct infection or be secondary to a systemic viral infection (e.g., measles, mumps, herpes, or leukemia).
Cerebrovascular Disease in Children
- 1. Perinatal arterial ischemic stroke is a leading cause of perinatal brain injury, cerebral palsy, and lifelong disability.
- 2. Ischemic (occlusive) stroke is rare in children but can occur from embolism, sickle cell disease, cerebral arteriopathies, and cardiac anomalies.
- 3. Hemorrhagic stroke can occur in association with immature blood vessel associated with prematurity or cerebral arteriovenous malformations.
- 4. Moyamoya is a rare, progressive vascular stenosis of the circle of Willis that obstructs arterial blood flow to the brain and is associated with transient ischemic attacks.
Epilepsy and Seizure Disorders in Children
- 1. Seizures are abnormal discharges of electrical activity within the brain. Epilepsy is the occurrence of more than two unprovoked seizures separated by over 24 hours.
- 2. Seizures can be caused by structural abnormalities of the brain, hypoxia, intracranial hemorrhage, CNS infection, traumatic injury, electrolyte imbalance, or inborn metabolic disturbances.
- 3. Epileptic spasms (infantile spasms) are a rare severe type of seizure associated with brain injury that occur in clusters of 10 to 150 spasms affecting the whole body or just the arms and legs.
Childhood Brain Tumors
- 1. Brain tumors are the second most common type of childhood cancer and are the leading cause of cancer related death in children.
- 2. Symptoms of brain tumors may be generalized or localized. The most common general symptoms are the result of increased intracranial pressure and include headache, irritability, vomiting, somnolence, and bulging of fontanelles.
- 3. Localized findings relate to the degree of disturbance in physiologic functioning in the area where the tumor is located. Localized signs of infratentorial tumors in the cerebellum include impaired coordination and balance.
- 4. Neuroblastoma is an embryonal tumor of the sympathetic nervous system and can be located anywhere there is sympathetic nervous tissue. Symptoms include weight loss, irritability, fatigue, fever, and intractable diarrhea. Neuroblastoma has been associated with spontaneous remission in infants.
- 5. Retinoblastoma is a congenital eye tumor that has two forms: inherited and acquired; both are related to loss of function of the RB gene.
Emerging Science Box In Utero Myelomeningocele Surgery
Operating on a fetus in utero with spina bifida reduces the risk of developing hydrocephalus and Chiarimalformation and may improve motor function. This can have a very positive impact for the child in terms quality of life. Prenatal repair of myelomeningocele continues to evolve since first reported by Bruner and Tulipan in 1997. Typically after myelomeningocele has been confirmed and the mother is 18 years of age or older and between 19 and 25.9 weeks’ gestation, the surgery is performed either endoscopically or by small open hysterotomy. Candidates for this surgery are typically screened very carefully, and the spinal defect must be below thoracic level 1. Maternal and fetal risks are increasingly being reduced with further experience. Preterm delivery remains a significant problem such that this operation is best performed by an experienced team with delivery by cesarean section. The child may have increased risk of retethering and intraspinal dermoid cysts. Long-term evaluation of neurologic function and quality of life are ongoing.

Data from: Bruner JP, et al. Endoscopic coverage of fetalopen myelomeingocelein utero. Am J ObstetGynecol.1997;176:256–257:; Eggink AJ, et al. Neural tube anomalies: An update on the pathophysiology and prevention. In: Fetal therapy: Scientific basis and critical appraisal of clinical benefits, Cambridge: Cambridge University Press. January 2, 2020:449–455; Dewan MC, et al. Fetal surgery for spina bifida. Journal of Neurosurgery. Pediatrics, August 1, 2019;24(2):105–114.
Emerging Science Box Gene Therapy for Pediatric Neurologic Conditions
Much has been learned from studies and trials from the 1980’s and 1990’s. Due to the high risk of germ line gene therapy, where changes made to correct the genetic variants of the reproductive cells would be passed on to offspring, only somatic gene therapies are under trial. Somatic gene therapy involves either in vivo or ex vivo introduction of genetic material into cells in the body or those harvested, altered and then placed back into the body. Three pediatric FDA approved therapies exist at this time for spinal muscular atrophy, B-cell acute lymphoblastic leukemia and a specific retinal dystrophy. Many other trials are underway and being developed due to the great promise this therapy has for certain genetic neurologic conditions. A recent search of the National Cancer Institute website for gene therapy or gene transfer yields 10 trials for CNS childhood tumors, 13 trials for pediatric neurologic disorders including Duchenne muscular dystrophy, Hunter Syndrome, gangliosidosis, Danon disease and others. Currently the Clinical Trials.gov web site lists 45 studies involving children receiving or being evaluated for the outcomes of gene therapy for specific diseases or conditions (Accesses April 20, 2021, Available at https://clinicaltrials.gov/ct2/results?cond=Pediatric+ALL&term=+gene+therapy&cntry=&state=&city=&dist=&Search=Search&recrs=a&recrs=b&age=0).
In 2012 a revolutionary new gene editing technique called CRISPR (clusters of regularly interspaced short palindromic [reads the same in both directions] repeats [sequences of nucleotides]) was first introduced. This technique allows for precise gene editing by inserting a cut or break in the DNA and allowing a cell’s natural DNA repair mechanisms to introduce the desired changes. Gene editing is being investigated for neurologic disorders such as genetic blindness, muscular dystrophy, Huntington disease. cancer, blood disorders, cystic fibrosis and other medical conditions that may likely benefit from CRISPR.

Data from U.S. Food and Drug Administration News Release: FDA approves innovative gene therapy to treat pediatric patients with spinal muscular atrophy, a rare disease and leading genetic cause of infant mortality, May 24, 2019 Available at https://www.fda.gov/news-events/press-announcements/fda-approves-innovative-gene-therapy-treat-pediatric-patients-spinal-muscular-atrophy-rare-disease;Safarzadeh Kozani P, et al. Novel antigens of CAR T cell therapy: New roads; old destination. Transl Oncol. 2021 Apr 13;14(7):101079. Ameri H. Prospect of retinal gene therapy following commercialization of voretigene neparvovec-rzyl for retinal dystrophy mediated by RPE65 mutation. J Curr Ophthalmol. 2018 Feb 16;30(1):1–2. Sharma G, et al. CRISPR-Cas9: A Preclinical and Clinical Perspective for the Treatment of Human Diseases. Mol Ther. 2021 Feb 3;29(2):571–586.