Alterations of Hormonal Regulation
Jodi A. Allen
 http://evolve.elsevier.com/Rogers/pathophysiology/
http://evolve.elsevier.com/Rogers/pathophysiology/
Functions of the endocrine system involve complex interactions between hormones and most body systems that maintain dynamic steady states and influence tissue growth and reproductive capabilities. Endocrine system dysfunction is usually caused by hypersecretion or hyposecretion of the various hormones, leading to abnormal hormone concentrations in the blood. Dysfunction also may result from abnormal cell receptor function or from altered intracellular response to the hormone–receptor complex.
Mechanisms of Hormonal Alterations
Significantly elevated or significantly depressed hormone levels may result from two primary mechanisms: (1) inappropriate amounts of hormone delivered to the target cell, or (2) inappropriate responses by the target cell (Table 22.1). Inappropriate amounts of hormone can result from disorders of endocrine glands, causing them to synthesize too little or too much hormone, failure of feedback systems designed to control hormone release, dysfunctional or ectopically produced hormones (hormones produced from nonendocrine sources), or defects in delivery of the hormone in the bloodstream.
Table 22.1
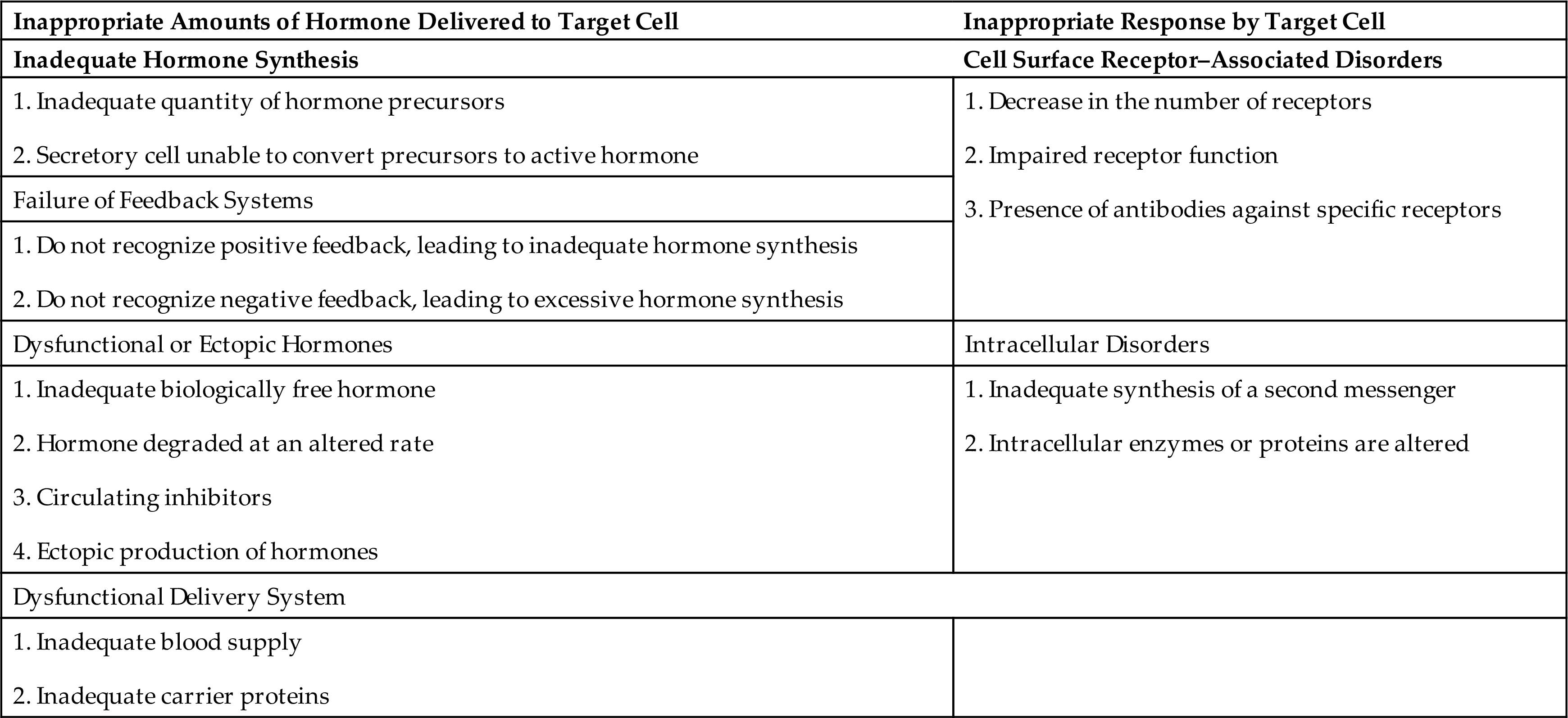
Target cells may not respond appropriately to hormonal stimulation for a number of reasons. Target cells may have too many or too few cell surface receptors. Those receptors may be abnormal and insensitive, or they may be blocked or stimulated by antibodies. There also may be intracellular disorders that cause a failure of the target cell to respond to receptor stimulation. For example, there may be inadequate synthesis of second messengers, such as cyclic adenosine monophosphate (cAMP), or the cell may respond abnormally to the second messenger if levels of intracellular enzymes or proteins are altered (second messengers for various hormones are listed in Table 21.2).
Alterations of the Hypothalamic–Pituitary System
The most common cause of apparent hypothalamic dysfunction is interruption of the pituitary stalk. Such interruptions prevent hypothalamic hormones from reaching the pituitary gland. Damage to the pituitary stalk can be caused by destructive lesions, rupture after head injury, surgical transection, or tumor. Without hypothalamic hormones, the pituitary releases inadequate amounts of follicle-stimulating hormone (FSH), luteinizing hormone (LH), adrenocorticotropic hormone (ACTH), thyroid-stimulating hormone (TSH), and growth hormone (GH) (Fig. 22.1). The control of prolactin is predominantly inhibitory by hypothalamic dopamine. It does not have endocrine target tissue and therefore lacks a regulatory feedback pathway. The effects of these changes in pituitary hormones are discussed later in this chapter.
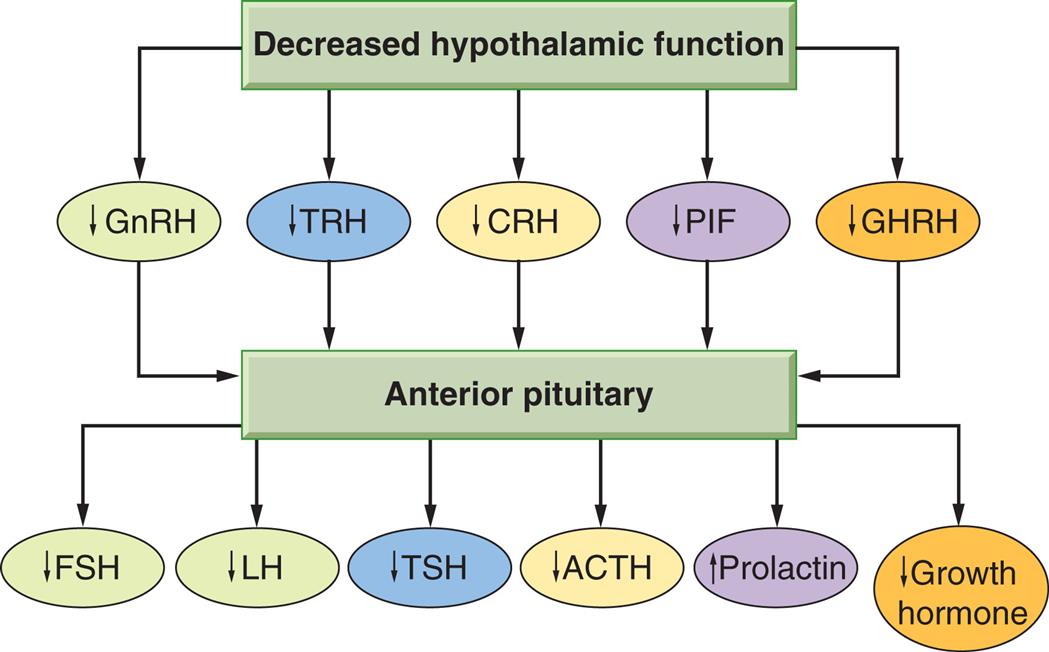
ACTH, Adrenocorticotropic hormone; CRH, corticotropin-releasing hormone; FSH, follicle-stimulating hormone; GHRH, growth hormone–releasing hormone; GnRH, gonadotropin-releasing hormone; LH, luteinizing hormone; PIF, prolactin inhibitory factor (probably dopamine); TRH, thyrotropin-releasing hormone; TSH, thyroid-stimulating hormone.
A flowchart represents the loss of hypothalamic hormones. Decreased hypothalamic functions include decreased G n R H, T R H, C R H, P I F, and G H R H, which trigger the anterior pituitary in releasing the following: • Decreased F S H. • Decreased L H. • Decreased T S H. • Decreased A C T H. • Increased Prolactin. • Decreased Growth Hormone.
Diseases of the Posterior Pituitary
Diseases of the posterior pituitary cause either increased or decreased secretion of antidiuretic hormone (ADH, arginine vasopressin [AVP]). An excess amount of this hormone results in water retention, whereas deficiencies in the amount or response to ADH result in water loss. These complex pathophysiologic states not only have significant clinical effects on the modulation of body fluids and electrolytes, but also affect cognitive and emotional responses to stress.
Syndrome of Inappropriate Antidiuretic Hormone Secretion
The syndrome of inappropriate ADH secretion (SIADH) results from ADH release in the absence of normal physiologic stimuli from the pituitary gland, nonpituitary sources of ADH, or the continued action of ADH on vasopressin receptors.1,2 SIADH is characterized by hypotonic and euvolemic hyponatremia along with urinary hyperosmolarity. SIADH can be idiopathic or due to multiple causes including neurological disease, pulmonary disease, malignant diseases, medications, or acute conditions such as stress, pain, and general anesthesia. Non-osmotic stimuli also mediate release of ADH via the afferent parasympathetic nervous system from high- and low-pressure receptors in the atria and carotid and artery during hemodynamic stress. Central nervous system disorders that may cause SIADH include encephalitis, meningitis, intracranial hemorrhage, tumors, and trauma. Pulmonary disorders associated with SIADH include pneumonia (e.g., tuberculosis), asthma, cystic fibrosis, and respiratory failure requiring mechanical ventilation. A common cause of SIADH is the ectopic production of ADH by tumors, such as cancers of the lung, stomach, pancreas, bladder, prostate, and endometrium; lymphomas; and sarcomas.
Iatrogenic causes of SIADH include surgery and medications. Any surgery can result in increased ADH secretion for as long as 5 to 7 days after surgery. The precise mechanism is uncertain, but likely related to fluid and volume changes after surgery, the amount and type of intravenous fluids given, and the use of narcotic analgesics. Medications are an important cause of SIADH, especially in the elderly. These include narcotics, general anesthetics, antidepressants, antipsychotics, chemotherapeutic agents, and nonsteroidal anti-inflammatory drugs.2
Pathophysiology
The cardinal features of SIADH are the result of enhanced renal free water retention. ADH induces the insertion of a water channel protein, called aquaporin-2, into the tubular luminal membrane, which increases water reabsorption by the kidneys (renal function is discussed in Chapter 37). This results in an expansion of extracellular fluid volume that leads to dilutional hyponatremia (low serum sodium concentration), high urinary osmolarity, high urinary sodium concentration, absence of edema, or clinical signs of volume depletion.3 Urine is inappropriately concentrated with respect to serum osmolarity because water is reabsorbed that normally would be excreted.
Clinical Manifestations
The symptoms of SIADH result from hyponatremia (see Chapter 3) and are determined by its severity and rapidity of onset. Thirst, impaired taste, anorexia, dyspnea on exertion, fatigue, and dulled sensorium occur when the serum sodium level decreases rapidly. Gastrointestinal (GI) symptoms, including vomiting and abdominal cramps, occur with a drop in sodium concentration from 130 to 120 mEq/L. There is weight gain from water retention, but peripheral edema is absent. Serum sodium levels below 115 mEq/L cause confusion, lethargy, muscle twitching, and convulsions; severe and sometimes irreversible neurologic damage may occur. Symptoms usually resolve with correction of hyponatremia.
Evaluation and Treatment
A diagnosis of SIADH is based on these findings: (1) serum hypoosmolality and hyponatremia, (2) urine hyperosmolarity (i.e., urine osmolality is greater than expected for the concomitant serum osmolality), and (3) the absence of conditions that can alter volume status (e.g., adrenal or thyroid dysfunction, congestive heart failure, or renal insufficiency) (Table 22.2). It is important to remember that the diagnosis of SIADH requires normal renal, cardiac, hepatic, adrenal, and thyroid function. In particular, hypothyroidism and adrenal insufficiency must be excluded.4
Table 22.2
DI, Diabetes insipidus; SIADH, syndrome of inappropriate antidiuretic hormone.
The treatment of SIADH involves the correction of any underlying causal problems, as well as fluid restriction with careful monitoring of serum sodium and neurologic symptoms. In severe SIADH, emergency correction of severe hyponatremia by careful administration of hypertonic saline and administration of vasopressin receptor antagonists (vaptans) may be required. Resolution usually occurs within 3 days. If hyponatremia is corrected too rapidly, a severe neurologic syndrome called central pontine myelinolysis can ensue.
Diabetes Insipidus
Diabetes Insipidus (DI) is characterized by insufficient ADH activity, leading to polyuria (frequent urination) and polydipsia (frequent drinking). The three major forms of DI are:
- 1. Neurogenic or central DI. Caused by the insufficient secretion of ADH, it occurs when any organic lesion of the hypothalamus, pituitary stalk, or posterior pituitary interferes with ADH synthesis, transport, or release. Causative lesions include primary brain tumors, traumatic brain injury, hypophysectomy, aneurysms, thrombosis, infections, and immunologic disorders. It can also be caused by hereditary disorders that affect ADH genes or result in structural changes in the pituitary gland.
- 2. Nephrogenic DI. Caused by inadequate response of the renal tubules to ADH. Acquired nephrogenic DI is caused by disorders and drugs that damage the renal tubules. These disorders include pyelonephritis, amyloidosis, destructive uropathies, and polycystic kidney disease. Drugs that may induce a reversible form of nephrogenic DI include lithium carbonate, colchicine, amphotericin B, loop diuretics, general anesthetics, and demeclocycline. There are several genetic causes of nephrogenic DI, including a mutation in the gene that codes for aquaporin-2, which is one of the water transport channels in the renal tubule.5
- 3. Primary polydipsia or Excessive thirst appreciation. A rare form of DI, called gestational DI, is associated with pregnancy in which the level of the vasopressin-degrading enzyme vasopressinase is increased, resulting in ADH deficiency. Clinical manifestations are usually mild and do not require treatment.
Pathophysiology
Individuals with DI have a partial to total inability to concentrate urine. Insufficient ADH activity causes excretion of large volumes of dilute urine, leading to increased plasma osmolality. In conscious individuals, the thirst mechanism is stimulated and induces polydipsia. Dehydration develops rapidly without ongoing fluid replacement. If the individual with DI cannot conserve as much water as is lost in the urine, serum hypernatremia and hyperosmolality occur. Concentrations of other serum electrolytes generally are not affected. Neurogenic (central) DI is the clinical manifestation of destruction of the neurons of the hypothalamus/posterior pituitary axis, with consequent loss of AVP secretion. The syndrome is caused by a wide variety of acquired or congenital lesions.6
Clinical Manifestations
The clinical manifestations of DI include polyuria, nocturia, continuous thirst, and polydipsia. The urine output is varied but can increase from the normal output of 1 to 2 L/day to as much as 8 to 12 L/day. Individuals with long-standing DI develop a large bladder capacity and hydronephrosis (see Chapter 38). Neurogenic DI usually has an abrupt onset, whereas nephrogenic DI usually has a more gradual onset. Table 22.2 compares the signs and symptoms of DI and SIADH. A history of lithium therapy may raise the possibility of NDI, whereas previous brain injury or neurosurgical intervention may indicate neurogenic (central) DI.6
Evaluation and Treatment
The criteria for the diagnosis of DI include polyuria, polydipsia, low urine specific gravity (<1.010), low urine osmolality (<200 mOsm/kg), hypernatremia, high serum osmolality (300 mOsm or more depending on water intake), and continued diuresis despite a serum sodium level of 145 mEq/L or greater. The diagnosis of DI is generally confirmed through a two-step water deprivation testing in which urine output is maintained despite dehydration. Other measurements that are useful in diagnosis are serum and urine osmolality, serum electrolytes, ADH levels, and radioimmunoassay for AVP. DI must be distinguished from other polyuric states, including diabetes mellitus, osmotically induced diuresis, and psychogenic polydipsia. Therefore, an evaluation for underlying reversible central or renal causes should be undertaken.
Dipsogenic or primary polydipsia may be confused with DI, but it is particularly important not to misdiagnose primary polydipsia as neurogenic (central) DI, because inappropriate therapy with desmopressin can lead to dangerous hyponatremia in individuals who continue to drink excessively.6 Primary polydipsia is usually the result of a psychiatric condition that causes the chronic ingestion of extremely large quantities of fluid, which washes out the renal concentration gradient, resulting in partial resistance to ADH.
Treatment of neurogenic (central) DI is based on the extent of the ADH deficiency. Fluid replacement using oral or intravenous routes is usually adequate. Some individuals require ADH replacement with the synthetic vasopressin analog desmopressin (DDAVP). Once a diagnosis of neurogenic (central) DI is made, magnetic resonance imaging (MRI) of the hypothalamic-pituitary region is required to establish whether there is a structural lesion that is responsible for AVP deficiency. Management of nephrogenic DI requires treatment of any reversible underlying disorders, discontinuation of etiologic medications, and correction of associated electrolyte disorders.6 Drugs that potentiate the action of otherwise insufficient amounts of endogenous ADH, such as thiazide diuretics, chlorpropamide, carbamazepine, and clofibrate, may be used in individuals with incomplete ADH deficiency.
Diseases of the Anterior Pituitary
Hypopituitarism
Hypopituitarism is characterized by the absence of one or more anterior pituitary hormones or the complete failure of all anterior pituitary hormone functions.7 The most common causes of hypopituitarism are pituitary infarction or space-occupying lesions. Pituitary infarction may occur when there is significant blood loss or hypovolemic shock. This also may occur in women during the postpartum period (Sheehan syndrome). Space-occupying lesions include pituitary adenomas or aneurysms, which can enlarge and compress the pituitary gland. Other causes of hypopituitarism include traumatic brain injury, removal or destruction of the gland, infections (e.g., meningitis, syphilis, tuberculosis), autoimmune hypophysitis, and certain drugs (e.g., bexarotene, carbamazepine, ipilimumab). A rare congenital form of hypopituitarism results from an early mutation of the prophet of pituitary transcription factor (PROP-1) gene, which affects embryonic pituitary development.
Pathophysiology
The pituitary gland is highly vascular and relies heavily on portal blood flow from the hypothalamus. It is therefore vulnerable to ischemia and infarction. Infarction results in tissue necrosis and edema with swelling of the gland. Over time, fibrosis of pituitary tissue occurs, and the symptoms of hypopituitarism develop. Adenomas and aneurysms may compress otherwise normal secreting pituitary cells and lead to compromised hormonal output. These lesions further impede blood supply because of enlargement of the pituitary within the fixed compartment of the sella turcica.
Clinical Manifestations
The signs and symptoms of hypofunction of the anterior pituitary are variable and depend on which hormones are affected. In panhypopituitarism, all hormones are deficient, and the individual suffers from multiple complications.
ACTH deficiency with associated loss of cortisol is a potentially life-threatening disorder. Symptoms of cortisol insufficiency include nausea, vomiting, anorexia, fatigue, and weakness. Hypoglycemia results from increased insulin sensitivity, decreased glycogen reserves, and decreased gluconeogenesis associated with hypocortisolism. ACTH deficiency also is associated with changes in aldosterone secretion, with resulting decreases in the glomerular filtration rate and urine output.
TSH deficiency causes cold intolerance, skin dryness, lethargy, and decreased metabolic rate. The symptoms usually are less severe than those of primary hypothyroidism.
The onset of FSH and LH deficiencies in women of reproductive age is associated with amenorrhea and atrophy of the vagina, uterus, and breasts. In postpubertal males, the testicles atrophy and facial hair growth is diminished. Both men and women experience decreased body hair and diminished libido.
GH deficiency in children is manifested by growth failure and a condition known as hypopituitary dwarfism (Fig. 22.2). Symptoms of chronic adult GH deficiency syndrome include increased body fat, decreased strength and lean body mass, osteoporosis, reduced sweating, dry skin, and psychological problems, including depression, social withdrawal, fatigue, loss of motivation, and a diminished feeling of well-being. Dyslipidemias and atherosclerotic cardiovascular disease may occur.

A pituitary giant and dwarf contrasted with normal-size men. Excessive secretion of growth hormone by the anterior lobe of the pituitary gland during the early years of life produces giants of this type, whereas deficient secretion of this substance produces well-formed dwarfs. (From Enderle A. Dwarfism and gigantism in historical picture postcards. Journal of the Royal Society of Medicine, 1998;91(5):273–278. https://doi.org/10.1177/014107689809100511)
A black-and-white historical photo shows the brothers, Hugo and Adrien, both 230 centimeters tall, standing together. While the brothers have pituitary giantism. Adrien, 69 centimeters, standing between them, has hypopituitary dwarfism and stands just as tall as the brothers’ knees.
Evaluation and Treatment
The diagnostic evaluation of suspected pituitary disease includes simultaneous measurements of the levels of tropic hormones from the pituitary and target endocrine glands. Imaging of the pituitary (MRI or computed tomography [CT] scans) is critical to assess for anatomic lesions, such as tumors.
Management of hypopituitarism requires correction of the underlying disorder as quickly as possible. Replacement of target gland hormones that are deficient is essential (such as cortisol, thyroid hormone (TH), GH, and sex-specific steroid hormones). In cases of acute circulatory collapse, immediate therapy with glucocorticoids and intravenous fluids is critical.
Hyperpituitarism: Primary Pituitary Neuroendocrine Tumors
Pituitary neuroendocrine tumors (previously known as pituitary adenomas) usually are benign, slow-growing tumors that arise from cells of the anterior pituitary.8 The cause of pituitary neuroendocrine tumors is not known, and most occur sporadically. Altered gene expression is commonly detected, and familial pituitary neuroendocrine tumors occur as part of syndromes affecting other organs, such as multiple endocrine neoplasia type 1. About 50% are microscopic (microadenomas) found incidentally, are hormonally silent, and do not pose significant hazards to the individual. Larger tumors (macroadenomas) are associated with morbidity and mortality attributable to alterations in hormone secretion or to invasion or impingement of surrounding structures.9,10
Pathophysiology
Local expansion of the neuroendocrine tumor may impinge on the optic chiasma and cause various visual disturbances. If the tumor is locally aggressive, invasion of the cavernous sinuses may occur, resulting in compromise of cranial nerve function. Extension to the hypothalamus disturbs control of wakefulness, thirst, appetite, and temperature.
Hormonal effects of a neuroendocrine tumor include hypersecretion from the tumor itself and hyposecretion from surrounding pituitary cells. The adenomatous tissue secretes the hormone of the cell type from which it arose, without regard to regulatory feedback mechanisms (autonomous function). Because of the pressure exerted by the tumor in the unexpandable bony sella turcica, hyposecretion from those cells that are most sensitive to pressure is common (FSH-and LH-secreting cells).
Clinical Manifestations
The clinical manifestations of pituitary neuroendocrine tumors are related to tumor growth and hormone hypersecretion or hyposecretion. Increased tumor size causes headache, fatigue, neck pain or stiffness, and seizures. Visual changes include visual field impairments (often beginning in one eye and progressing to the other) and temporary blindness. If the tumor infiltrates other cranial nerves, neuromuscular function is affected.
Pituitary neuroendocrine tumors are most often associated with increased secretion of GH and prolactin. Gonadotropic hyposecretion results in menstrual irregularity in women, decreased libido, and receding secondary sex characteristics in both men and women. If the tumor exerts sufficient pressure, thyroid and adrenal hypofunction may occur because of a lack of TSH and ACTH, resulting in the symptoms of hypothyroidism and hypocortisolism, respectively.
Evaluation and Treatment
Diagnosis of pituitary neuroendocrine tumor involves physical and laboratory evaluations, including pertinent hormone assays and radiographic examination of the skull (MRI [preferred] or contrast-enhanced CT). The goal of treatment is to protect the individual from the effects of tumor growth and to control hormone hypersecretion while minimizing damage to appropriately secreting portions of the pituitary. Depending on tumor size and type, individuals may be treated by administration of specific medications to suppress tumor growth, transsphenoidal tumor resection, or radiation therapy, including stereotactic treatments.
Hypersecretion of Growth Hormone: Acromegaly
Acromegaly results from increased release of GH and insulin-like growth factor 1 (IGF-1). It almost always is caused by a GH-secreting pituitary neuroendocrine tumor.11 Acromegaly is usually diagnosed in adults in the 40- to 59-year-old age group, although it is often present for years before diagnosis. It is a slow progressive disease and, if untreated, is associated with a decreased life expectancy. Deaths from acromegaly are caused by heart disease secondary to hypertension and coronary artery disease, stroke, diabetes mellitus, or malignancy (colon or lung cancers).
Pathophysiology
With a GH-secreting neuroendocrine tumor, only slight elevations of GH and IGF-1 can stimulate growth. In children and adolescents whose epiphyseal plates have not yet closed, the effect of increased GH levels is termed giantism (see Fig. 22.2). Skeletal growth is excessive, with some individuals becoming 8 or 9 feet tall. In the adult, epiphyseal closure has occurred, and increased amounts of GH and IGF-1 cause connective tissue proliferation and increased cytoplasmic matrix, as well as bony proliferation that results in the characteristic appearance of acromegaly (Fig. 22.3).
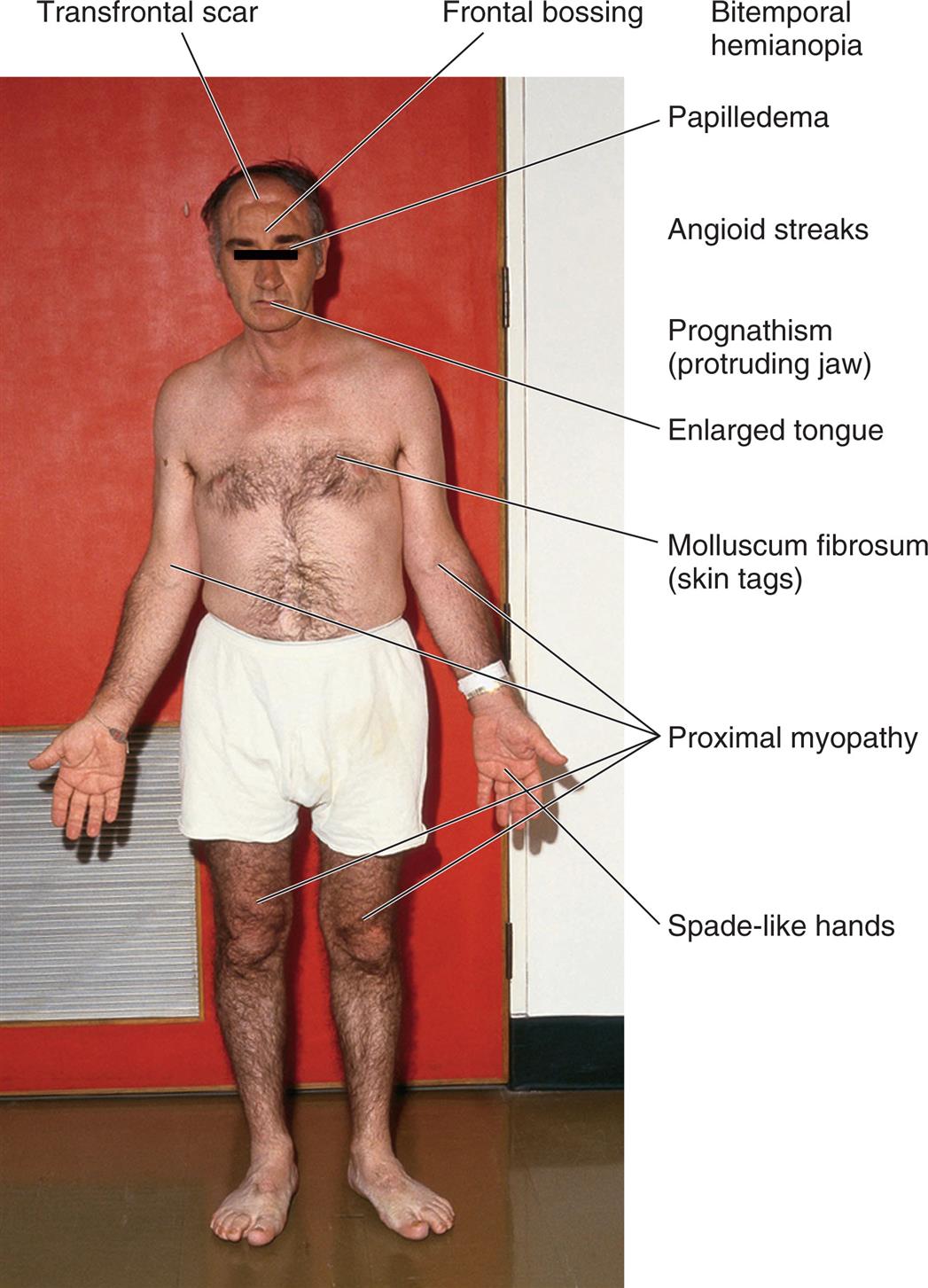
A photo of the anterior profile of a person with acromegaly shows and labels the different conditions on the body, from the top to the bottom: transfrontal scar, frontal bossing, bitemporal hemianopia, papilledema, angioid streaks, prognathism (protruding jaw), enlarged tongue, molluscum fibrosum (skin tags), proximal myopathy, and spade-like hands.
GH also has significant effects on glucose, lipid, and protein metabolism. Hyperglycemia results from adipocyte inflammation and GH inhibition of peripheral glucose uptake and increased hepatic glucose production, followed by compensatory hyperinsulinism and, finally, insulin resistance. Diabetes mellitus occurs when the pancreas cannot secrete enough insulin to offset the effects of GH. Excessive levels of GH and IGF-1 also affect the cardiovascular system with calcium influx in cardiomyocytes, enhanced cardiac contractility, and cardiomyocyte growth. Hypertension, cardiomegaly, and left ventricular heart failure are seen in one-third to one-half of individuals with acromegaly. GH also acts on the renal tubules to increase phosphate reabsorption, leading to mild hyperphosphatemia. Approximately 20% of GH-secreting tumors also secrete prolactin. Because the adenoma becomes increasingly a space-occupying lesion, hypopituitarism may occur because of compression of surrounding hormone-secreting cells.
Clinical Manifestations
With connective tissue proliferation, individuals with acromegaly have an enlarged tongue, interstitial edema, enlarged and overactive sebaceous and sweat glands (leading to increased body odor), and coarse skin and body hair. Bony proliferation involves periosteal vertebral growth and enlargement of the bones of the face, hands, and feet (see Fig. 22.3). The lower jaw and forehead also protrude.
Increased IGF-1 levels cause ribs to elongate at the bone–cartilage junction, leading to a barrel-chested appearance, and increased proliferation of cartilage in joints, which causes backache and arthralgias. With bony and soft tissue overgrowth, nerve entrapment occurs, leading to peripheral nerve damage manifested by weakness, muscular atrophy, foot drop, and sensory changes in the hands.
Symptoms of diabetes mellitus, such as polyuria and polydipsia, may occur. Acromegaly-associated hypertension is usually asymptomatic until heart failure symptoms develop. Increased tumor size results in central nervous system symptoms of headache, seizure activity, visual disturbances, and papilledema. If compression hypopituitarism occurs, gonadotropin secretion may be affected, causing amenorrhea in women and sexual dysfunction in men. Increased prolactin results in hypogonadism.
Evaluation and Treatment
Diagnosis is confirmed by clinical features of the disease, MRI scans, and elevated levels of GH and IGF-1. The goals of treatment are to normalize or reduce GH secretion and relieve or prevent complications related to tumor expansion. The treatment of choice is transsphenoidal surgical removal of the GH-secreting neuroendocrine tumor. Radiation therapy may be effective when rapid control of GH levels is not essential, when the individual is not a good surgical candidate, or when hyperfunction persists after subtotal resection. Somatostatin analogs (e.g., octreotide or lanreotide) normalize IGF-1 and GH levels. Dopaminergic agonists may be helpful, especially if the tumor also secretes prolactin. Cardiovascular, metabolic, and symptoms of tumor compression often improve with treatment. Skeletal abnormalities are irreversible.
Prolactinoma
Pituitary tumors that secrete prolactin, prolactinomas, are the most common hormonally active pituitary tumors. Other conditions, such as renal failure, polycystic ovarian disease, primary hypothyroidism, breast stimulation, or even the stress of venipuncture, can increase prolactin levels. Prolactin is under tonic inhibitory hypothalamic control through the secretion of dopamine. Thus, medications that block the effects of dopamine can increase prolactin levels. These include some antipsychotics, metoclopramide, tricyclic antidepressants, and methyldopa. Estrogens can increase prolactin concentration by stimulating hyperplasia of prolactin-secreting cells. Any process that interferes with the delivery of dopamine from the hypothalamus to the lactotrophs (pituitary stalk tumor, pituitary stalk transection, or compressive pituitary tumor) also results in hyperprolactinemia. Because thyrotropin-releasing hormone (TRH) stimulates prolactin secretion, prolactin may be elevated in individuals with primary hypothyroidism.
Pathophysiology
The hallmark of a prolactinoma is sustained increases in the levels of serum prolactin, also known as hyperprolactinemia, which has multiple effects on female and male reproductive organs.12 Pathologic elevations of prolactin suppress LH and FSH, resulting in estrogen and progesterone deficiency in women and low testosterone levels in men.
Hypopituitarism may occur because of the compression of surrounding hormone-secreting cells. Central nervous system symptoms may develop because of growth and pressure of the tumor within the sella turcica. These complications are especially common with what are called macro (>1 cm in diameter) or giant (>4 cm in diameter) prolactinomas.
Clinical Manifestations
Women with hyperprolactinemia generally present with galactorrhea (nonpuerperal milk production) and menstrual disturbances, including amenorrhea. Estrogen deficiency also may cause hirsutism, and fractures may occur because of osteopenia or osteoporosis. Hyperprolactinemia in men causes gynecomastia, hypogonadism, and erectile dysfunction, although they often are not diagnosed until they develop symptoms related to the increasing size of the tumor (i.e., headache or visual impairment).
Evaluation and Treatment
The diagnostic evaluation of hyperprolactinemia includes a careful history to exclude medications that may cause elevations in prolactin concentration. Screening for hypothyroidism is mandatory. MRI scanning of the pituitary is indicated to determine the size and location of an adenoma.
Dopaminergic agonists (bromocriptine and cabergoline) are the treatment of choice for prolactinomas. Decreases in tumor size and restoration of fertility in previously anovulatory women are common. In individuals resistant or intolerant to these medications, transsphenoidal surgery and radiotherapy are options. New chemotherapeutic and targeted molecular therapies are being explored in selected cases.
Alterations of Thyroid Function
Disorders of thyroid function develop as a result of primary dysfunction or disease of the thyroid gland or, secondarily, as a result of pituitary or hypothalamic alterations. Primary thyroid disorders result in either increased or decreased TH levels. These disorders also cause secondary feedback effects on pituitary TSH. For example, when there are primary elevations in the TH level, the TSH level will secondarily decrease because of negative feedback. When the TH level is decreased because of a condition affecting the thyroid gland, the TSH level will be elevated. Central (secondary) thyroid disorders are related to disorders of the anterior pituitary gland that affect thyroid function. When there is excessive TSH production, the thyroid follicular cells are stimulated and secrete excessive THs. The TH levels become elevated secondary to the primary elevation of the TSH concentration. The reverse is true with inadequate TSH production.
The majority of primary thyroid diseases are idiopathic and caused by autoimmune mechanisms that affect the gland. Although the exact genetic and environmental influences are not known, some individuals experience a predominantly cellular autoimmune response (some antithyroid autoantibodies also are involved) with resultant destruction of the thyroid gland, leading to hypothyroidism. Others experience a predominantly antibody-mediated autoimmune response that stimulates the gland, leading to hyperthyroidism. The most common autoimmune hypothyroid condition is called chronic lymphocytic thyroiditis (also called Hashimoto thyroiditis) with thyroid infiltration of T lymphocytes and follicular destruction.13 The most common autoimmune hyperthyroid condition is called Graves disease (Fig. 22.4).14,15
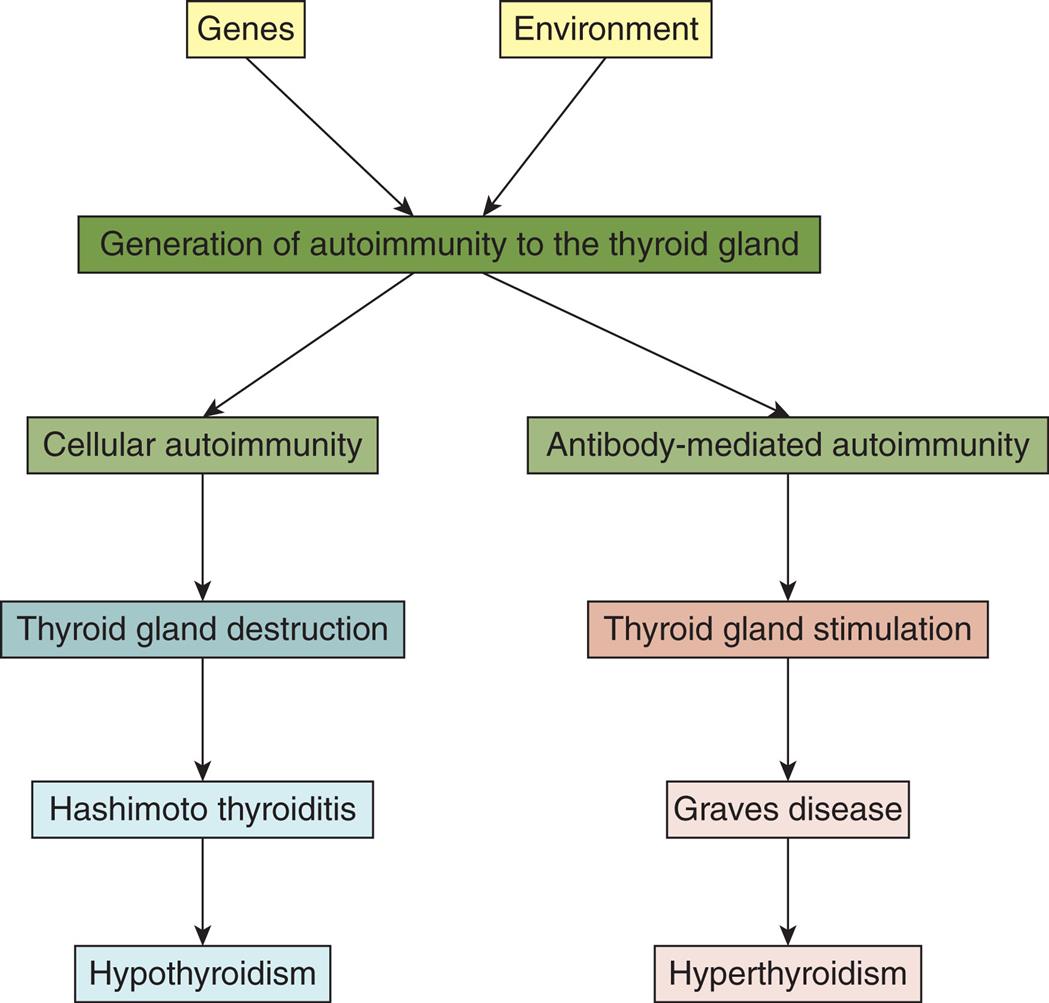
A flowchart represents the autoimmune mechanisms in primary thyroid disease. Genes and environment impact the generation of autoimmunity to the thyroid gland. Two pathways are charted. The pathway leading to hypothyroidism is as follows. • Cellular autoimmunity. • Thyroid gland destruction. • Hashimoto thyroiditis. • Hypothyroidism. The pathway leading to hyperthyroidism is as follows. • Antibody-mediated autoimmunity. • Thyroid gland stimulation. • Graves disease. • Hyperthyroidism.
Thyrotoxicosis/Hyperthyroidism
Pathophysiology
Thyrotoxicosis is a condition that results from any cause of increased TH levels and can result from dysfunction of the pituitary, the thyroid gland, ectopic thyroid tissue, or the ingestion of excessive amounts of TH medication. Hyperthyroidism is a form of thyrotoxicosis in which excess amounts of TH are secreted from the thyroid gland (Fig. 22.5).16Primary hyperthyroidism results from thyroid gland dysfunction and is most commonly caused by Graves disease, toxic multinodular goiter, and solitary toxic adenoma. Central (secondary) hyperthyroidism is less common and is caused by TSH-secreting pituitary adenomas. Each condition is associated with a specific pathophysiology and manifestations; however, all forms of thyrotoxicosis share some common characteristics.
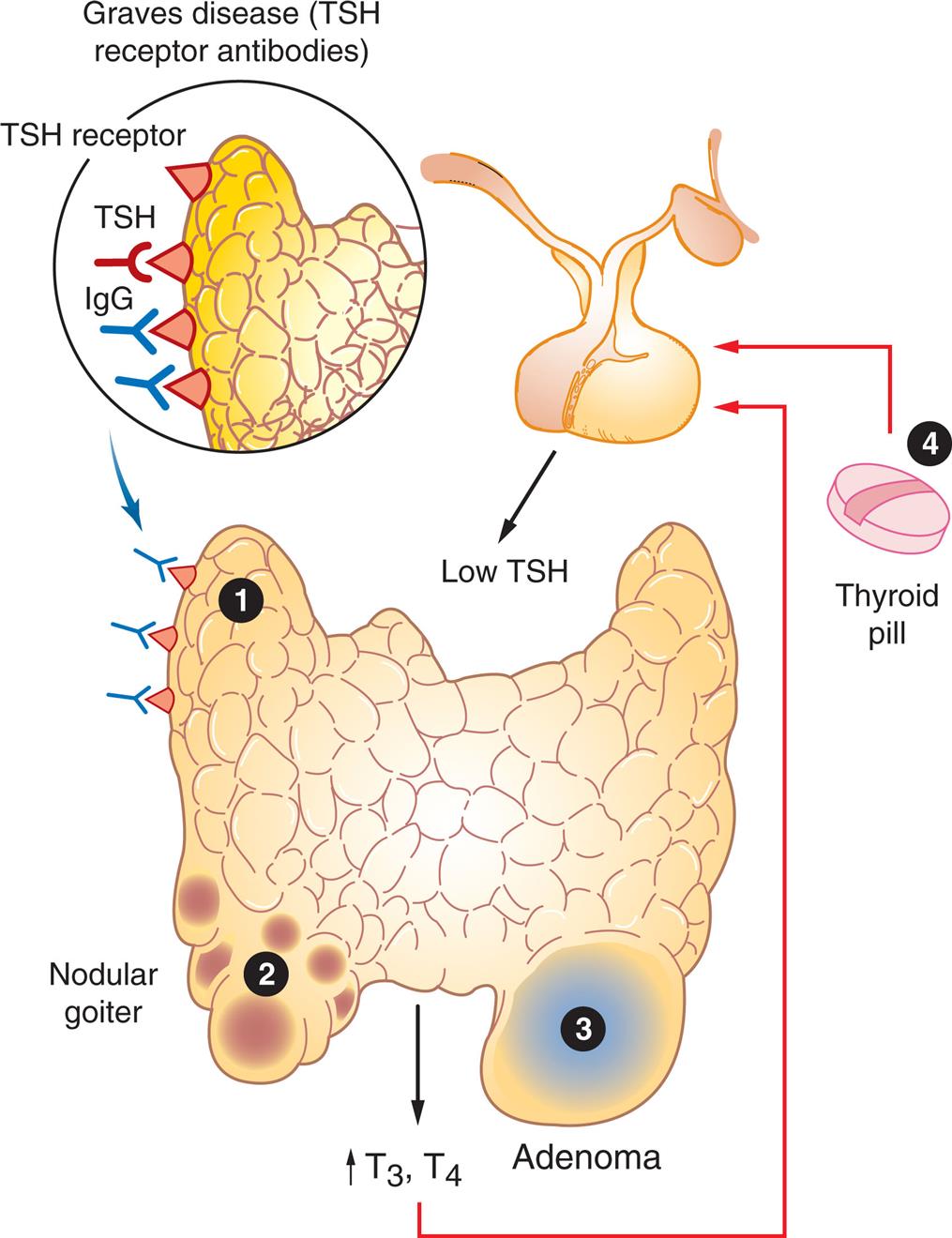
Hyperthyroidism may have several causes, among them: 1, Graves disease; 2, toxic multinodular goiter; 3, follicular adenoma; 4, thyroid medication. TSH, thyroid stimulating hormone. (Adapted from Damjanov I. Pathology for the health professions, 4th edition. St Louis: Saunders; 2012.)
An illustration of the thyroid highlights the following common causes of hyperthyroidism. 1. Graves disease (T S H receptor antibodies). T S H receptors attract T S H and I g G. 2. Nodular goiter. The illustration shows multiple fluid-filled lumps on the thyroid. 3. Adenoma. The illustration shows a large tumor on the thyroid. 4. Thyroid pill. The illustration shows that the ingestion of the pill results in a low T S H, leading to increased T 3 and T 4, which provides a feedback loop.
Clinical Manifestations
The clinical features of thyrotoxicosis are attributable to the metabolic effects of increased circulating levels of THs. This results in an increased metabolic rate, with heat intolerance and increased tissue sensitivity to stimulation by the sympathetic nervous system. The major manifestations are summarized in Fig. 22.6 and Table 22.3.
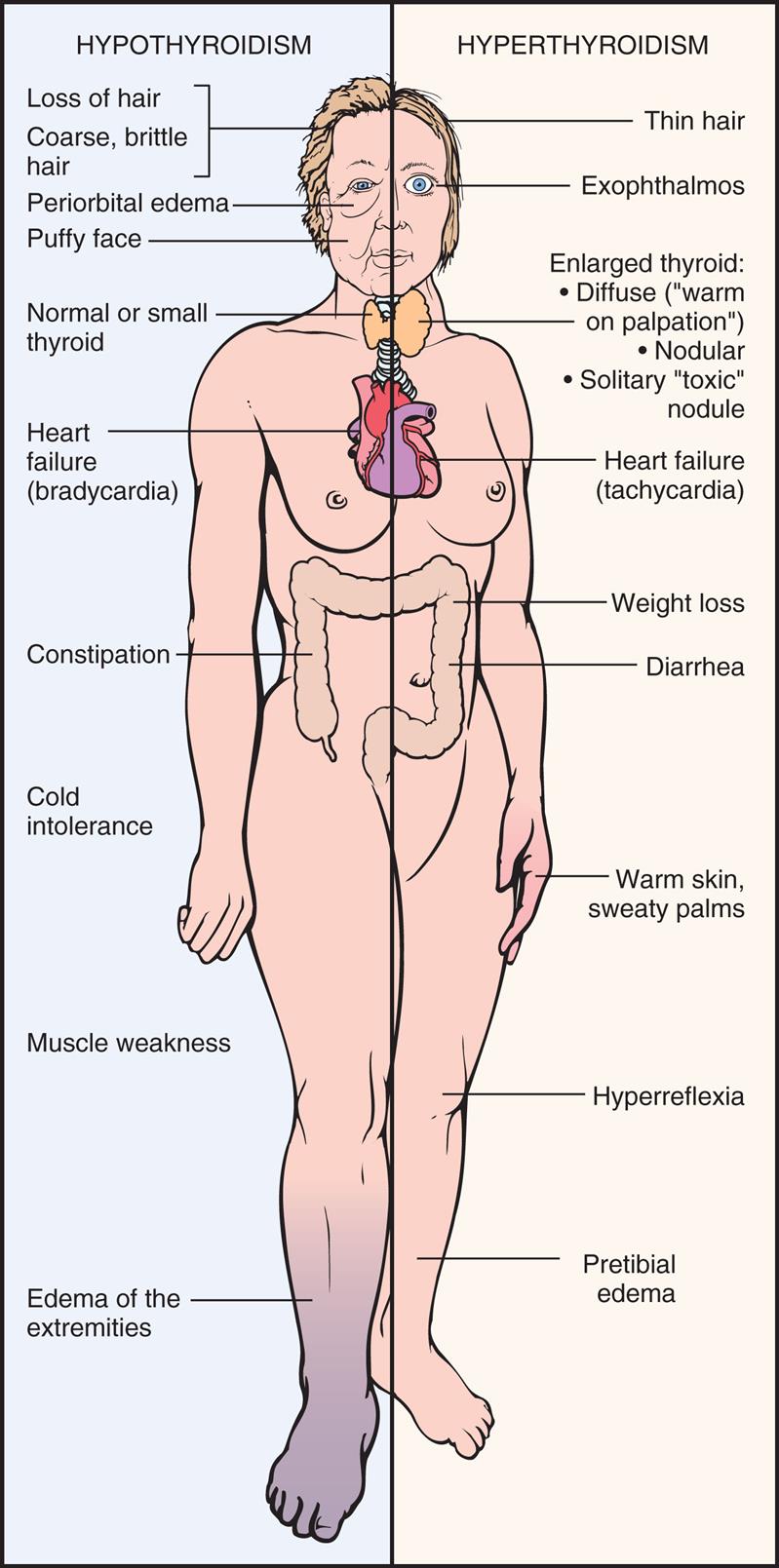
An illustration of the anterior view of a person highlights the manifestations for hypothyroidism and hyperthyroidism. The manifestations for hypothyroidism, identified from the top to the bottom, are as follows. • Loss of hair or coarse, brittle hair. • Periorbital edema. • Puffy face. • Normal or small thyroid. • Heart failure (bradycardia). • Constipation. • Cold intolerance. • Muscle weakness. • Edema of the extremities. The manifestations for hyperthyroidism, identified from the top to the bottom, are as follows. • Thin hair. • Exophthalmos. • Enlarged thyroid: diffuse (warm on palpation), nodular, solitary toxic nodule. • Heart failure (tachycardia). • Weight loss. • Diarrhea. • Warm, skin, sweaty palms. • Hyperreflexia. • Pretibial edema.
Table 22.3
Evaluation and Treatment. Elevated serum thyroxine (T4) and triiodothyronine (T3) levels and suppressed serum TSH levels are diagnostic for primary hyperthyroidism. By contrast, central (secondary) hyperthyroidism caused by TSH-secreting pituitary tumors is characterized by normal to increased TSH levels despite elevated TH concentrations. Treatment is directed at controlling excessive TH production, secretion, or action and involves antithyroid drug therapy, radioactive iodine therapy (absorbed only by thyroid tissue, causing death of cells), or surgical removal of nodules or part of the thyroid gland. A major complication of all forms of treatment for hyperthyroidism is excessive ablation of the gland, leading to hypothyroidism.
Hyperthyroid Conditions
Graves disease
Graves disease is the underlying cause of 60% to 80% of cases of hyperthyroidism with a prevalence of approximately 3% of women and 0.5% of men in the United States.17 Although the exact cause of Graves disease is not known, genetic factors interacting with environmental triggers play an important role in the pathogenesis. Graves disease is classified as an autoimmune disease and results from a form of type II hypersensitivity (see Chapter 9) in which there is stimulation of the thyroid by autoantibodies directed against the TSH receptor. These autoantibodies, called thyroid-stimulating immunoglobulins (TSIs; also called thyroid-stimulating antibodies [TSAbs] or thyroid receptor antibodies [TRAbs]), override the normal regulatory mechanisms. TSI stimulation of TSH receptors in the gland results in hyperplasia of the gland (goiter) and increased synthesis of TH, especially of T3. Increased levels of TH result in the classic signs and symptoms of hyperthyroidism illustrated in Fig. 22.6. TSH production by the pituitary is inhibited through the usual negative feedback loop.
Autoimmunity also contributes to the two major distinguishing clinical manifestations of Graves disease (ophthalmopathy and dermopathy [pretibial myxedema]) (Fig. 22.7). Two categories of ophthalmopathy associated with Graves disease are (1) functional abnormalities resulting from hyperactivity of the sympathetic division of the autonomic nervous system (lag of the globe on upward gaze and of the upper lid on downward gaze) and (2) infiltrative changes involving the orbital contents with enlargement of the ocular muscles. These changes affect more than half of individuals with Graves disease. Orbital connective tissue accumulation, inflammation, and edema of the orbital contents result in exophthalmos (protrusion of the eyeball), periorbital edema, and extraocular muscle weakness leading to diplopia (double vision). The individual may experience irritation, pain, lacrimation, photophobia, blurred vision, decreased visual acuity, papilledema, visual field impairment, exposure keratosis, and corneal ulceration.
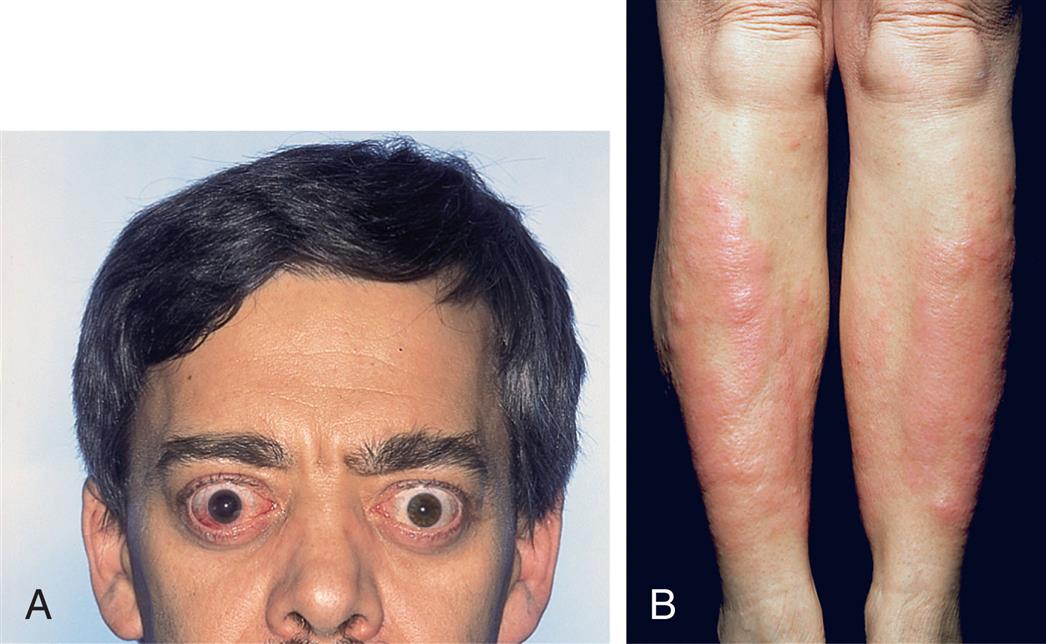
(A) Exophthalmos (large and protruding eyeballs, often in association with a large goiter). (B) Pretibial myxedema associated with Graves disease; note lumpy and swollen appearance from accumulation of connective tissue and pinkish purple discoloration. (A, From Belchetz P, Hammond P. Mosby’s color atlas and text of diabetes and endocrinology. Edinburgh: Mosby; 2003; B, From Habif T. Clinical dermatology, 5th edition. St Louis: Mosby; 2009.)
A small number of individuals with Graves disease who have very high levels of TSI experience pretibial myxedema (Graves dermopathy), characterized by subcutaneous swelling on the anterior portions of the legs and by indurated and erythematous skin. Graves dermopathy is associated with TSI stimulation of fibroblasts and T lymphocytes, causing excessive amounts of hyaluronic acid production in the dermis and subcutaneous tissue. These manifestations occasionally appear on the hands, giving the appearance of clubbing of the fingers (thyroid acropachy). Fig. 22.8 provides an overview of the pathophysiology of Graves disease.
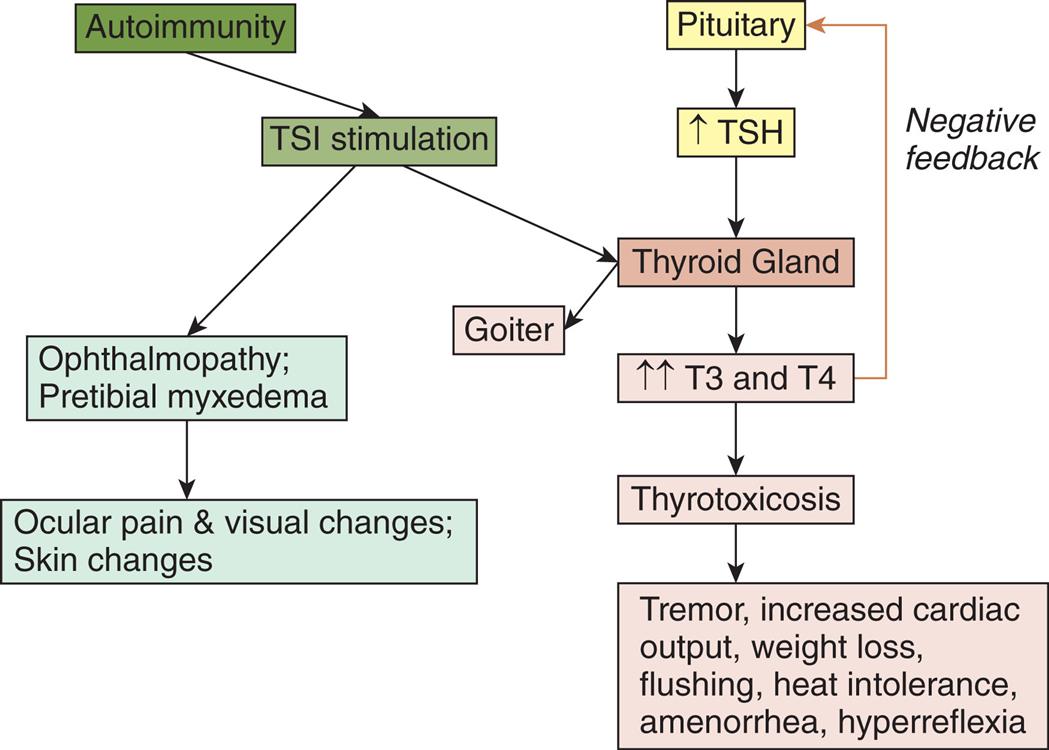
TSI, Thyroid-stimulating immunoglobulins (antibodies); TSH, thyroid-stimulating hormone; T3, triiodothyronine; T4, tetraiodothyronine.
A flowchart shows the pathophysiology of Graves disease. There are three pathways. The first pathway is as follows. • Autoimmunity. • T S I stimulation. • Ophthalmopathy; pretibial myxedema. • Ocular pain and visual changes; skin changes. The second pathway is as follows. • Autoimmunity. • T S I stimulation. • Thyroid gland. • Goiter. The third pathway is as follows. • Pituitary. • Increased T S H. • Thyroid gland. • Increased T 3 and T 4. (negative feedback to pituitary) • Thyrotoxicosis. • Tremor, increased cardiac output, weight loss, flushing, heat intolerance, amenorrhea, and hyperreflexia.
Hyperthyroidism resulting from nodular thyroid disease
The thyroid gland normally enlarges in response to the increased demand for TH that occurs in puberty, pregnancy, and iodine-deficient states, as well as in individuals with immunologic, viral, or genetic disorders. When the condition resulting in increased TH resolves, TSH secretion normally subsides, and the thyroid gland returns to its original size.
Irreversible changes can occur in some follicular cells so that these cells form nodules that function autonomously and produce excessive amounts of TH. Toxic multinodular goiter occurs when there are several hyperfunctioning nodules leading to hyperthyroidism. Unlike Graves disease, there is absence of an autoimmune stimulus. If only one nodule is hyperfunctioning, it is termed toxic adenoma.
The classic clinical manifestations of hyperthyroidism (see Fig. 22.6) usually develop slowly, and exophthalmos and pretibial myxedema do not occur. Nodules may be palpable on physical examination, and there is increased uptake of radioactive iodine.
There is an increased incidence of malignancy in toxic nodular goiter, so most individuals should undergo a fine-needle aspiration biopsy of suspicious nodules before treatment. Treatment consists of a combination of radioactive iodine (may increase risk of solid cancer-related death, including breast cancer death),18 surgery, and antithyroid medications.
Thyrotoxic crisis
Thyrotoxic crisis (thyroid storm) is a rare but dangerous worsening of the thyrotoxic state in which TH levels rise dramatically and death can occur within 48 hours without treatment. The condition may develop spontaneously but usually occurs in individuals who have undiagnosed or partially treated Graves disease and who are subjected to physiologic stress, such as infection, pulmonary or cardiovascular disorders, trauma, seizures, surgery (especially thyroid surgery), obstetric complications, or dialysis.
The systemic manifestations of thyrotoxic crisis include hyperthermia; tachycardia, especially atrial tachydysrhythmias; heart failure; agitation or delirium; and nausea, vomiting, or diarrhea contributing to fluid volume depletion. Treatment includes drugs that block TH synthesis (i.e., propylthiouracil or methimazole), β blockers, glucocorticoids, iodine, and supportive care.
Hypothyroidism
Hypothyroidism results from deficient production of TH by the thyroid gland. Primary hypothyroidism is the most common disorder of thyroid function, affecting 4% to 7% of the U.S. population, and occurs more commonly in women.19 It may be primary, central, or subclinical. Primary hypothyroidism accounts for the majority of all cases. Central (secondary) hypothyroidism is much less common and is related to either pituitary or hypothalamic failure. Subclinical hypothyroidism is a mild thyroid failure estimated to occur in 13 million U.S. adults, most commonly in women and older adults.20 It is characterized by elevated serum TSH levels with normal levels of circulating TH (T3, Free T4, Total T4). Progression to blatant hypothyroidism is common over time.19
Pathophysiology
In primary hypothyroidism, loss of thyroid function leads to decreased production of TH and increased secretion of TSH and TRH. The most common causes of primary hypothyroidism in adults include autoimmune thyroiditis (chronic lymphocytic thyroiditis), iatrogenic loss of thyroid tissue after surgical or radioactive treatment for hyperthyroidism or after head and neck radiation therapy, medications (e.g., lithium and amiodarone), and endemic iodine deficiency. Iodine, required for the production of TH, is not produced naturally by the body and is obtained through dietary intake. Iodine deficiency is relatively rare in the United States because of the use of iodized salt and fortified foods.19 Infants and children may present with hypothyroidism because of congenital defects. Central (secondary) hypothyroidism is caused by the pituitary's failure to synthesize adequate amounts of TSH or a lack of TRH. Pituitary tumors that compress surrounding pituitary cells or the consequences of their treatment are the most common causes of central hypothyroidism. Other causes include traumatic brain injury, subarachnoid hemorrhage, pituitary infarction, or metabolic disorders including insulin resistance or hyperglycemia, dyslipidemia, obesity, and endothelial dysfunction.21 Hypothalamic dysfunction results in low levels of TH, TSH, and TRH.
Clinical Manifestations
Hypothyroidism generally affects all body systems and occurs insidiously over months or years. Decreased TH levels lower energy metabolism and heat production. The individual develops a low basal metabolic rate, cold intolerance, lethargy, and slightly lowered basal body temperature (see Fig. 22.6). The decrease in the level of TH leads to excessive TSH production, which stimulates thyroid tissue and causes a goiter (Table 22.4).
Table 22.4
| System | Clinical Manifestations | Mechanisms Underlying Clinical Manifestations |
|---|---|---|
| Neurologic | Confusion, syncope, slowed speech and thinking, memory loss; lethargy, headaches, hearing loss, night blindness; slow, clumsy movements; cerebellar ataxia; slow α-wave activity and loss of amplitude in EEG; reduced cAMP response to epinephrine, glucagons, and PTH stimulation; decreased appetite | Decreased cerebral blood flow leading to cerebral hypoxia; reduced intracellular processes caused by decreased β-adrenergic activity that may be related to a decrease in the number of β-adrenergic receptor sites |
| Endocrine | Increased TSH production in primary hypothyroidism; enlarged pituitary thyrotropes, increase in serum prolactin levels with galactorrhea; decreased rate of cortisol turnover but with normal serum cortisol levels | Impaired TH synthesis or defects in iodide trapping leading to compensatory TSH production; chronic overstimulation of thyrotropes of TRH and by TSH synthesis; stimulation of lactotropes by TRH related to increased prolactin levels; decreased deactivation of cortisol |
| Reproductive | Decreased androgen secretion in men, increased estriol formation in women; low total hormone values but with increased amounts of unbound hormone; anovulation, decreased libido, menorrhagia, and a high incidence of spontaneous abortion in women; erectile dysfunction, decreased libido, and oligospermia in men | Altered metabolism of estrogens and androgens; decreased levels of sex hormone–binding globulin |
| Hematologic | Decrease in red cell mass leading to normocytic, normochromic anemia; macrocytic anemia associated with vitamin B12 deficiency and inadequate folate or iron absorption in the GI tract | Decreased basal metabolic rate and reduced oxygen requirements; decreased production of erythropoietin; possible relationship between TH and optimal hematologic response to vitamin B12 |
| Cardiovascular | Reduction in stroke volume and heart rate causing lowered cardiac output; increased peripheral vascular resistance to maintain systolic blood pressure can cause hypertension; normal response to exercise but with alterations in circulatory system at rest (prolonged circulation time and decreased blood flow to tissues); cool skin and cold tolerance; enlarged heart; decreased intensity of heart sounds and variety of ECG changes (sinus bradycardia, prolonged PR interval, depressed P waves, flattened or inverted T waves, and low-amplitude QRS complexes); cardiac tamponade (although rare) (see Chapter 32) | Decreased metabolic demands and loss of regulatory and rate-setting effects of TH; protein-mucopolysaccharide–rich fluid in the pericardial sac associated with enlarged heart; pericardial effusions associated with heart sounds and ECG changesIncreases in peripheral vascular resistance and increased blood volume can cause hypertension |
| Pulmonary | Dyspnea; myxedematous changes in respiratory muscles leading to hypoventilation and carbon dioxide retention, which contribute to myxedema coma | Pleural effusions associated with dyspnea, although effusions may be asymptomatic |
| Renal | Reduced renal blood flow and glomerular filtration rate leading to decreased renal excretion of water; increase in total body water and dilutional hyponatremia; reduced production of erythropoietin | Hemodynamic alterations associated with reduced blood flow and filtration; increased total body water related to decreased excretion and mucinous deposits in tissue |
| GI | Constipation, weight gain, and fluid retention; decreased absorption of most nutrients; decreased protein metabolism leading to retarded skeletal and soft tissue growth and slightly positive nitrogen balance; edema; decreased glucose absorption and delayed glucose uptake; elevated serum lipid values | Reduced intake and reduced peristaltic activity that may progress to fecal impaction; water absorption related to prolonged transit time; fluid retention associated with myxedematous changes; edema associated with high concentrations of exchangeable albumin in the extravascular space caused by increased capillary permeability to proteins; depressed insulin degradation; depressed lipid synthesis and degradation |
| Musculoskeletal | Muscle aching and stiffness; slow movement and slow tendon jerk reflexes; decreased bone formation and resorption, increased bone density; aching and stiffness in joints | Decreased rate of muscle contraction and relaxation contributing to slow movement and reflexes |
| Integumentary | Dry, flaky skin; dry, brittle head and body hair; reduced growth of nails and hair; slow wound healing | Reduced sweat and sebaceous gland secretion |
| Myxedema | Accumulation of hyaluronic acid, which binds water and causes a puffy appearance | |
| Cool skin | Decreased circulation to skin |
cAMP, Cyclic adenosine monophosphate; ECG, electrocardiogram; EEG, electroencephalogram; GI, gastrointestinal; PTH, parathyroid hormone; TH, thyroid hormone; TRH, thyrotropin-releasing hormone; TSH, thyroid-stimulating hormone.
The characteristic sign of severe or long-standing hypothyroidism is myxedema, which results from the altered composition of the dermis and other tissues. The connective tissue fibers are separated by large amounts of protein and mucopolysaccharide. This complex binds water, producing nonpitting, boggy edema, especially around the eyes, hands, and feet and in the supraclavicular fossae (Fig. 22.9). The tongue and laryngeal and pharyngeal mucous membranes thicken, producing thick, slurred speech and hoarseness. Myxedema coma, a medical emergency, is a diminished level of consciousness associated with severe hypothyroidism. Signs and symptoms include hypothermia without shivering, hypoventilation, hypotension, hypoglycemia, and lactic acidosis. Older individuals with comorbid conditions, such as pulmonary or urinary infections, congestive heart failure, or cerebrovascular accident, and with moderate or untreated hypothyroidism are particularly at risk for developing myxedema coma. It also may occur after overuse of narcotics or sedatives or after an acute illness in hypothyroid individuals. Symptoms of hypothyroidism in older adults should not be attributed to expected age-related changes.
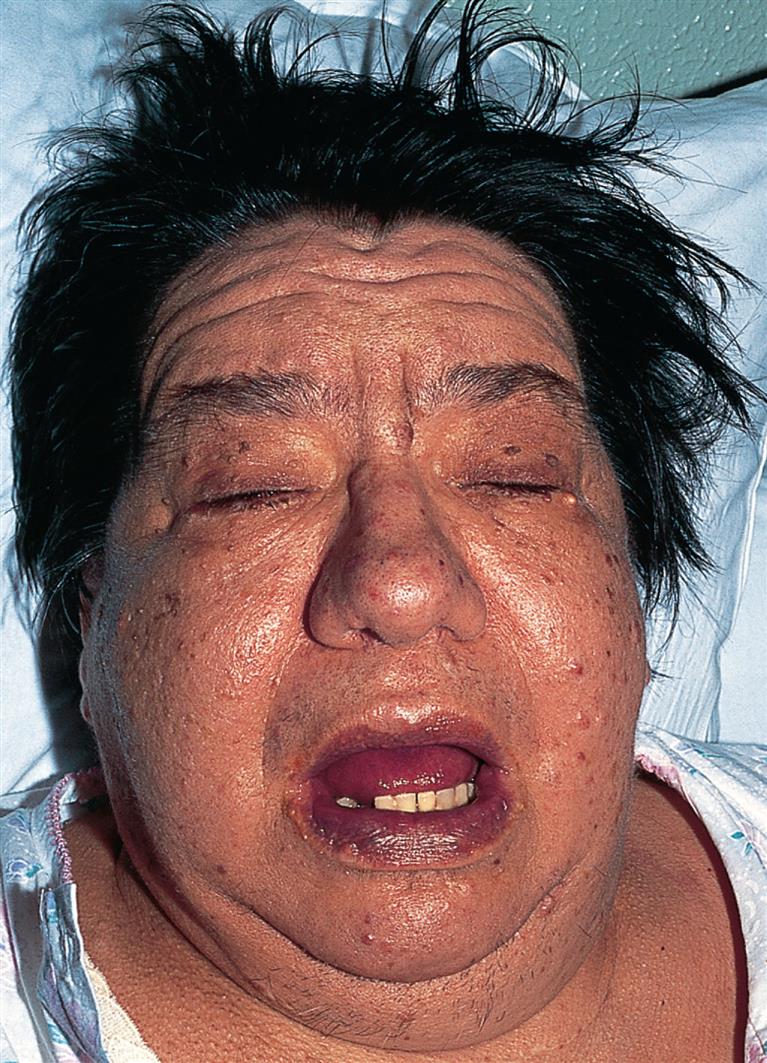
The hair is dry. (From Bolognia JL, et al. Dermatology, 3rd edition. St. Louis: Mosby; 2012.)
Evaluation and Treatment
The diagnosis of primary hypothyroidism is made by documentation of the clinical symptoms of hypothyroidism and measurement of increased serum levels of TSH and decreased serum levels of TH (total T3 and both total and free T4). Central hypothyroidism is diagnosed by finding low TH and low serum TSH levels. Alluded to earlier, subclinical hypothyroidism is diagnosed by an elevation in TSH level with normal levels of circulating TH. Hormone replacement therapy with the hormone levothyroxine is the treatment of choice for both primary and central thyroid disorders (Emerging Science Box: Combination Therapy for the Treatment of Hypothyroidism).
Hypothyroid Conditions
Chronic lymphocytic thyroiditis (Hashimoto disease)
The most common cause of primary hypothyroidism in the United States is chronic lymphocytic thyroiditis (Hashimoto disease), which results in gradual, inflammatory destruction of thyroid tissue. This disorder is linked with several genetic risk factors and is often associated with other autoimmune conditions. Infiltration of the thyroid with autoreactive T lymphocytes, antithyroid antibodies (antithyroid peroxidase and antithyroglobulin antibodies), and natural killer cells induces inflammation, glandular apoptosis, and tissue destruction (Fig. 22.10).
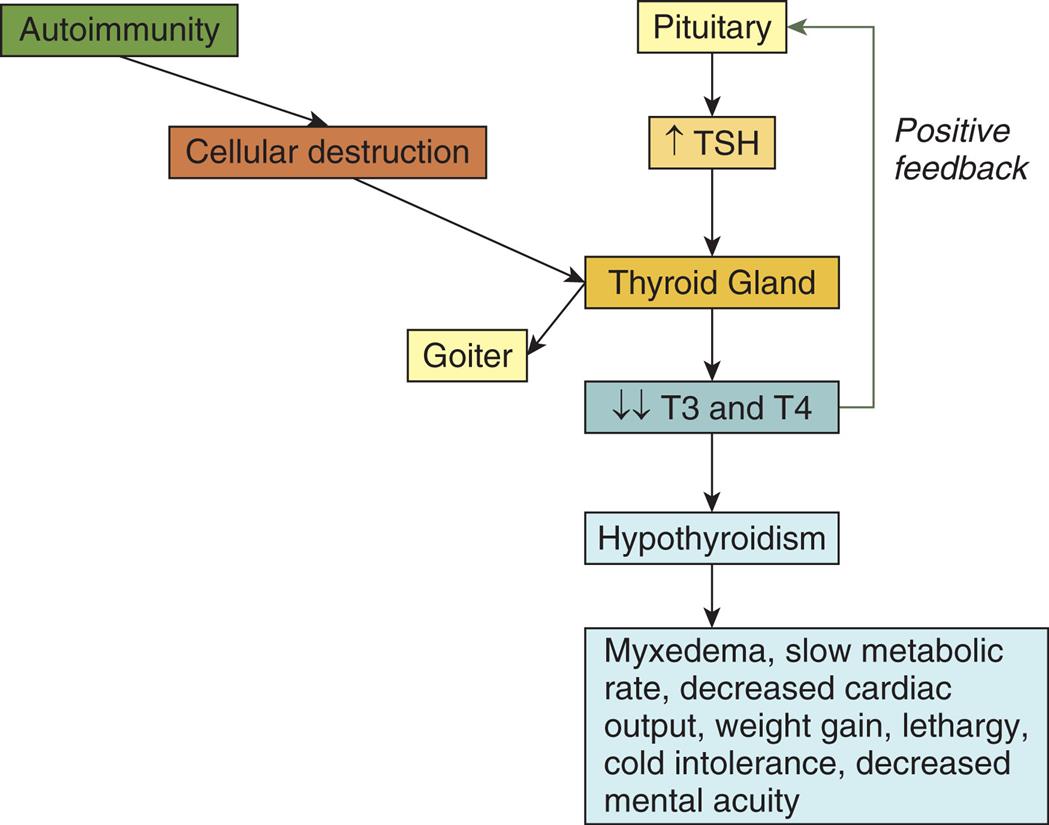
TSH, Thyroid-stimulating hormone; T3, triiodothyronine; T4, tetraiodothyronine.
A flowchart shows two pathways representing the pathophysiology of Hashimoto thyroiditis. One of the pathways is as follows. • Autoimmunity. • Cellular destruction. • Thyroid gland. • Goiter. The other pathway is as follows. • Pituitary. • Increased T S H. • Thyroid gland. • Decreased T 3 and T 4. (positive feedback to pituitary) • Hypothyroidism. • Myxedema, slow metabolic rate, decreased cardiac output, weight gain, lethargy, cold intolerance, and decreased mental acuity.
Uncommon causes of hypothyroidism
Other, less common causes of hypothyroidism are subacute thyroiditis and postpartum thyroiditis. Subacute thyroiditis (de Quervain thyroiditis) is a rare nonbacterial inflammation of the thyroid gland often preceded by a viral infection. It is accompanied by fever, tenderness, an increase in erythrocyte sedimentation rate, enlargement of the thyroid gland, and transient hypothyroidism before the gland recovers normal activity. Symptoms may last for 2 to 4 months, and nonsteroidal anti-inflammatory drugs or corticosteroids usually resolve symptoms. Postpartum thyroiditis (PPT) is an autoimmune disease associated with antibodies to thyroid peroxidase. PPT has a prevalence of about 5% and generally occurs up to 6 months after birthing with a course similar to that seen in subacute thyroiditis. Individuals who develop PPT need to be monitored for permanent hypothyroidism. Iatrogenic hypothyroidism results from ablation of the thyroid gland during treatment for hyperthyroid conditions.
Congenital hypothyroidism
Hypothyroidism in infants occurs when thyroid tissue is absent (thyroid dysgenesis) or with hereditary defects in TH synthesis. Thyroid dysgenesis occurs more often in female infants, with permanent abnormalities in about 1 of every 4000 live births. The affected fetus is dependent on maternal thyroxine for the first 20 weeks of gestation, then becomes deficient in TH. TH is essential for fetal growth and for the development of brain tissue, so the infant will suffer developmental and cognitive disabilities if left untreated. Hypothyroidism may not be evident at birth. Symptoms may include high birth weight, hypothermia, delay in passing meconium, and neonatal jaundice. Cord blood can be examined in the first days of life for measurement of T4 and TSH levels. The probability of normal growth and intellectual function is high if treatment with levothyroxine is started before the child is 3 or 4 months old. The earlier TH replacement is initiated, the better the child's outcome.
Without early screening, hypothyroidism may not be evident until after 4 months of age. Symptoms include difficulty eating, hoarse cry, and protruding tongue caused by myxedema of oral tissues and vocal cords; hypotonic muscles of the abdomen with constipation, abdominal protrusion, and umbilical hernia; subnormal temperature; lethargy; excessive sleeping; slow pulse rate; and cold, mottled skin. Skeletal growth is stunted because of impaired protein synthesis, poor absorption of nutrients, and lack of bone mineralization. The child will be dwarfed with short limbs, if not treated. Dentition is often delayed. Cognitive disability varies with the severity of hypothyroidism and the length of delay before treatment is initiated.
Thyroid carcinoma
Thyroid carcinoma is the most common endocrine malignancy and is the seventh most common cancer in the United States in women.22 Exposure to ionizing radiation, especially during childhood, is the most consistent causal factor. Papillary and follicular thyroid carcinomas are the most frequent, and medullary and anaplastic thyroid carcinomas are less common.
The cancer is typically discovered as a small thyroid nodule or metastatic tumor in the lungs, brain, or bone. Changes in voice and swallowing and difficulty breathing are related to tumor growth impinging on the trachea or esophagus. The diagnosis of thyroid cancer is generally made by ultrasonography and then by fine-needle aspiration of a thyroid nodule. Most individuals with thyroid carcinoma have normal T3 and T4 levels and are therefore euthyroid.
Treatment may include partial or total thyroidectomy, TSH suppression therapy (levothyroxine), radioactive iodine therapy (in iodine-concentrating tumors), postoperative radiation therapy, and chemotherapy (especially in anaplastic carcinoma).23 New insights into the molecular pathogenesis of thyroid carcinoma are leading to new therapies.
Alterations of Parathyroid Function
Hyperparathyroidism
Hyperparathyroidism is characterized by greater than normal secretion of parathyroid hormone (PTH) with associated hypercalcemia. Hyperparathyroidism is classified as primary, secondary, or tertiary.
Pathophysiology
Primary hyperparathyroidism is characterized by inappropriate excess secretion of PTH by one or more of the parathyroid glands.24 It is one of the most common endocrine disorders. Approximately 80% to 85% of cases are caused by parathyroid adenomas, another 10% to 15% result from parathyroid hyperplasia, and approximately 1% of cases are caused by parathyroid carcinoma. In addition, primary hyperparathyroidism may be caused by a variety of genetic causes, especially the genes that cause multiple endocrine neoplasia.
In primary hyperparathyroidism, PTH secretion is increased and is not under the usual feedback control mechanisms. The calcium level in the blood rises because of increased bone resorption and GI absorption of calcium but fails to inhibit PTH secretion by the parathyroid gland. Some individuals with primary hyperparathyroidism maintain normal levels of calcium despite elevated levels of PTH and are diagnosed only when they develop osteoporosis.
Secondary hyperparathyroidism is a compensatory response of the parathyroid glands to chronic hypocalcemia, which is commonly associated with decreased activation of vitamin D in individuals with renal failure (see Chapter 38). Secretion of PTH is elevated, but not enough to achieve normal calcium levels because of insufficient levels of activated vitamin D. Other causes of secondary hyperparathyroidism include a dietary deficiency of vitamin D or calcium; decreased intestinal absorption of vitamin D or calcium; and ingestion of drugs, such as phenytoin, phenobarbital, and laxatives, which either accelerate the metabolism of vitamin D or decrease intestinal absorption of calcium.
Tertiary hyperparathyroidism can develop after any long-standing period of hypocalcemia, such as is seen with chronic dialysis, renal transplantation, or GI malabsorption. Parathyroid chief cell hyperplasia leads to excessive secretion of PTH and may cause hypercalcemia (cellular adaptation is discussed in Chapter 2).
Clinical Manifestations
Hypercalcemia and hypophosphatemia are the hallmarks of primary hyperparathyroidism. Hypercalcemia and hypophosphatemia may be asymptomatic or affected individuals may present with symptoms related to the muscular, nervous, and GI systems, including fatigue, headache, depression, anorexia, and nausea and vomiting. Excessive osteoclastic and osteocytic activity causes bone resorption, resulting in osteoporosis, pathologic fractures, kyphosis of the dorsal spine, and compression fractures of the vertebral bodies (bone resorption is discussed in Chapter 44).
Hypercalcemia means that the renal tubules must filter large amounts of calcium, leading to hypercalciuria and production of an abnormally alkaline urine. PTH hypersecretion enhances renal phosphate excretion and results in hypophosphatemia and hyperphosphaturia (see Chapter 3). The combination of these three variables—hypercalciuria, alkaline urine, and hyperphosphaturia—predisposes the individual to the formation of calcium stones, particularly in the renal pelvis or renal collecting ducts. These stones may be associated with infections and impaired renal function. Chronic hypercalcemia also is associated with mild insulin resistance, necessitating increased insulin secretion to maintain normal glucose levels (Table 22.5).
Table 22.5
Data from Flint PW, et al. Cummings otolaryngology: Head & neck surgery, 5th edition. St. Louis: Mosby.
Secondary hyperparathyroidism, caused by renal disease, presents clinically not only with the complications of bone resorption but also with the symptoms of hypocalcemia and hyperphosphatemia, such as muscle spasms and cardiovascular complications (see Chapter 3).
Evaluation and Treatment
The diagnosis of primary hyperparathyroidism is suggested by the concurrent findings of elevated PTH levels and an increased ionized calcium concentration. Imaging procedures are used to localize adenomas before surgery. Observation of asymptomatic individuals with mild hypercalcemia is recommended; these individuals are advised to avoid dehydration and limit dietary calcium intake. Definitive treatment of more severe primary hyperparathyroidism involves surgical removal of the solitary adenoma or, in the case of hyperplasia, complete removal of three and partial removal of the fourth hyperplastic parathyroid glands.
If the serum calcium concentration is low despite elevated levels of PTH, secondary hyperparathyroidism is likely. Evaluation for renal function may indicate chronic renal disease. Treatment for secondary hyperparathyroidism in chronic renal disease requires calcium replacement, dietary phosphate restriction and phosphate binders, and vitamin D replacement. Treatment also may include calcimimetics, which work to increase parathyroid calcium receptor sensitivity, thus lowering PTH levels.
Hypoparathyroidism
Hypoparathyroidism (abnormally low PTH levels) is most commonly caused by damage to the parathyroid glands during thyroid surgery. This occurs because of the anatomic proximity of the parathyroid glands to the thyroid (Fig. 21.11). Hypomagnesemia is another cause of low PTH levels. Hypoparathyroidism also is associated with genetic syndromes, including familial hypoparathyroidism and DiGeorge syndrome (see Chapter 9). There is an inherited condition called pseudohypoparathyroidism that causes a defect in tissue responsiveness to PTH. Pseudohypoparathyroidism is associated with hypocalcemia despite normal to elevated levels of PTH.
Pathophysiology
No matter the cause, the absence of PTH impairs resorption of calcium from bone and the renal tubules, leading to hypocalcemia. Deficient PTH also stimulates increased renal reabsorption of phosphate, leading to hyperphosphatemia. Hyperphosphatemia further lowers the calcium concentration by inhibiting the activation of vitamin D, thereby lowering the GI absorption of calcium.
Hypomagnesemia inhibits PTH secretion. Hypomagnesemia may be related to chronic alcoholism, malnutrition, malabsorption, increased renal clearance of magnesium caused by the use of aminoglycoside antibiotics or certain chemotherapeutic agents, or prolonged magnesium-deficient parenteral nutritional therapy. When serum magnesium levels return to normal, however, PTH secretion returns to normal.
Clinical Manifestations
Symptoms associated with hypopara thyroidism are primarily those of hypocalcemia (see Chapter 3). Hypocalcemia causes muscle spasms, which can progress to tetany, dry skin, and loss of body and scalp hair. Irreversible complications include hypoplasia of developing teeth, horizontal ridges on the nails, cataracts, basal ganglia calcifications (which may be associated with a parkinsonian syndrome), and bone deformities, including brachydactyly and bowing of the long bones.
Evaluation and Treatment
A low PTH level, along with a low serum calcium concentration and a high phosphorus level in the absence of renal failure, intestinal disorders, or nutritional deficiencies, suggests hypoparathyroidism. Measurement of the serum magnesium level and urinary calcium excretion also can help in diagnosis. Treatment is directed toward alleviation of the hypocalcemia. In acute states, this involves parenteral administration of calcium, which corrects the serum calcium concentration within minutes. Chronic maintenance of the serum calcium level is achieved with pharmacologic doses of cholecalciferol (vitamin D3) and oral calcium. PTH hormone replacement with recombinant human parathyroid hormone (rhPTH) is safe and effective.
Dysfunction of the Endocrine Pancreas: Diabetes Mellitus
Diabetes mellitus is a group of metabolic diseases characterized by hyperglycemia resulting from defects in insulin secretion, insulin action, or both. In 2018, an estimated 34.2 million individuals (10.5%) in the United States had diabetes, and another 7.3 million (21.4%) were estimated to be undiagnosed.25 The American Diabetes Association (ADA)26 classifies four categories of diabetes mellitus:
- 1. Type 1 diabetes (caused by autoimmune β-cell destruction, usually leading to absolute insulin deficiency)
- 2. Type 2 diabetes (caused by progressive loss of β-cell insulin secretion, frequently with a background of insulin resistance)27
- 3. Gestational diabetes mellitus (GDM) (diabetes diagnosed in the second or third trimester of pregnancy that was not clearly overt diabetes prior to gestation)
- 4. Specific types of diabetes mellitus due to other causes
Specific types of diabetes include monogenic diabetes syndromes (e.g., neonatal diabetes and maturity-onset diabetes of the young [MODY]), disease of the exocrine pancreas (e.g., cystic fibrosis and pancreatitis), and drug- or chemical-induced diabetes (e.g., glucocorticoid use in the treatment of human immunodeficiency virus [HIV] infection and/or acquired immunodeficiency syndrome [AIDS] or after organ transplantation) (Table 22.6).
Table 22.6
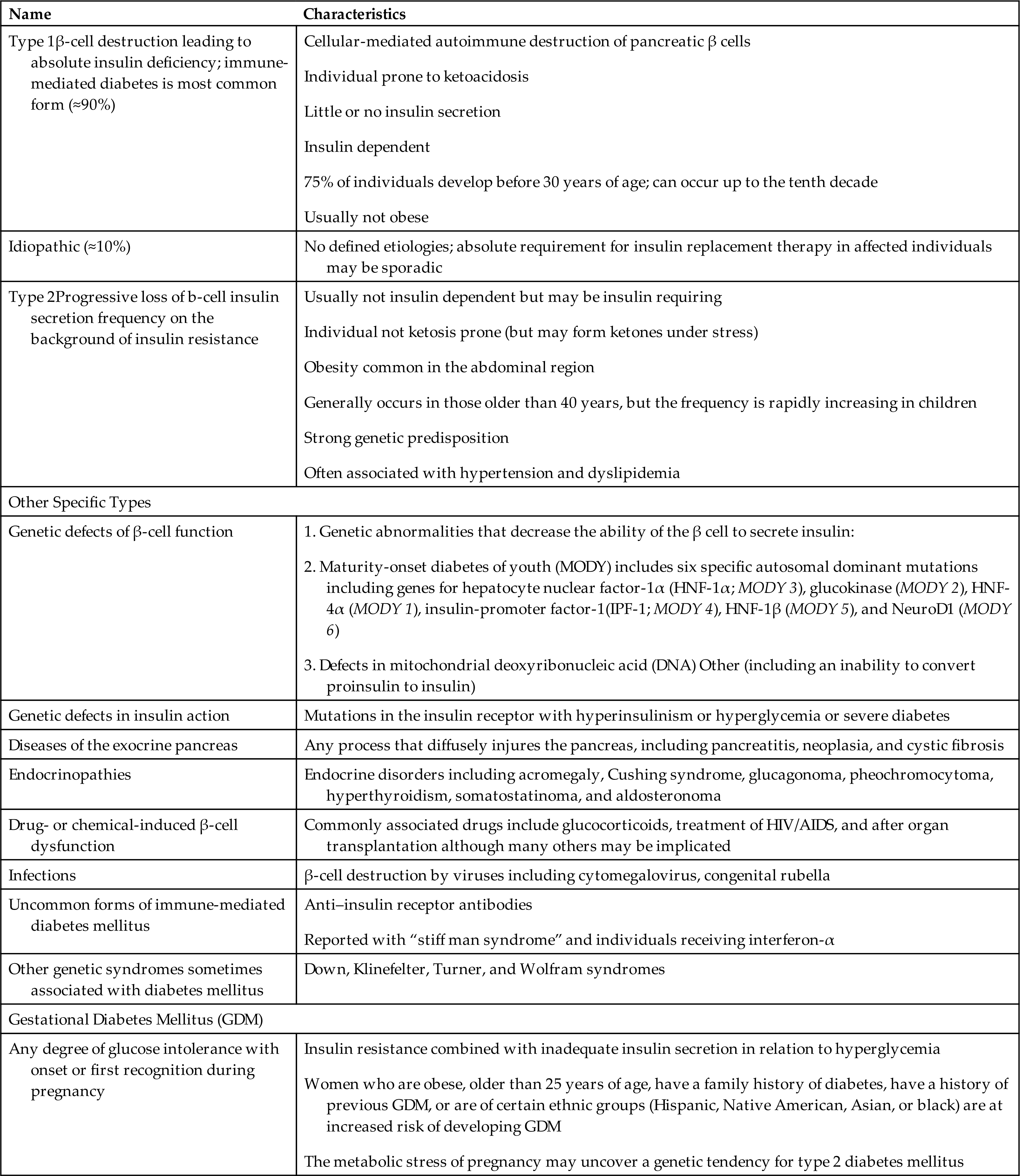
GDM, Gestational diabetes mellitus; MODY, maturity-onset diabetes of youth.
Data from American Diabetes Association. Diabetes Care. 2017;40(Suppl. 1):S11–S24.
The diagnosis of diabetes mellitus is based on glycated hemoglobin (HbA1C) levels; fasting plasma glucose (FPG) levels; oral glucose tolerance testing (OGTT); or random glucose levels in an individual with symptoms (Box 22.1).26 Glycated hemoglobin refers to the permanent attachment of glucose to hemoglobin molecules and reflects the average plasma glucose exposure over the life of a red blood cell (RBC) (approximately 120 days). It provides a more accurate measure for monitoring long-term control of blood glucose levels.
The ADA classification “categories at increased risk for diabetes” (or prediabetes) describes nondiabetic elevations of the HbA1C, FPG, or 2-hour plasma glucose value during an OGTT (see Box 22.1).26 The Centers for Disease Control and Prevention (CDC) estimates that 88 million U.S. adults (34.5%) aged 18 years or older have prediabetes.24
This classification includes impaired glucose tolerance (IGT), which results from diminished insulin secretion; and impaired fasting glucose (IFG), which is caused by enhanced hepatic glucose output. Individuals with IGT and IFG are at increased risk of cardiovascular disease and premature death and carry up to a 50% 5-year risk of developing diabetes, particularly type 2 diabetes. Thus, prevention of diabetes with lifestyle interventions is essential.
Types of Diabetes Mellitus
Diabetes Mellitus Type 1
Diabetes mellitus type 1 accounts for 5% to 10% of diabetes cases and is the most common pediatric chronic disease. It currently affects approximately 187,000 U.S. children younger than 20, and the incidence is increasing.25 Between 10% and 13% of individuals with newly diagnosed type 1 diabetes have a first-degree relative (parent or sibling) with type 1 diabetes, and there is a 50% concordance rate in twins. Diagnosis is rare during the first 9 months of life and peaks at 12 years of age (Table 22.7).
Table 22.7
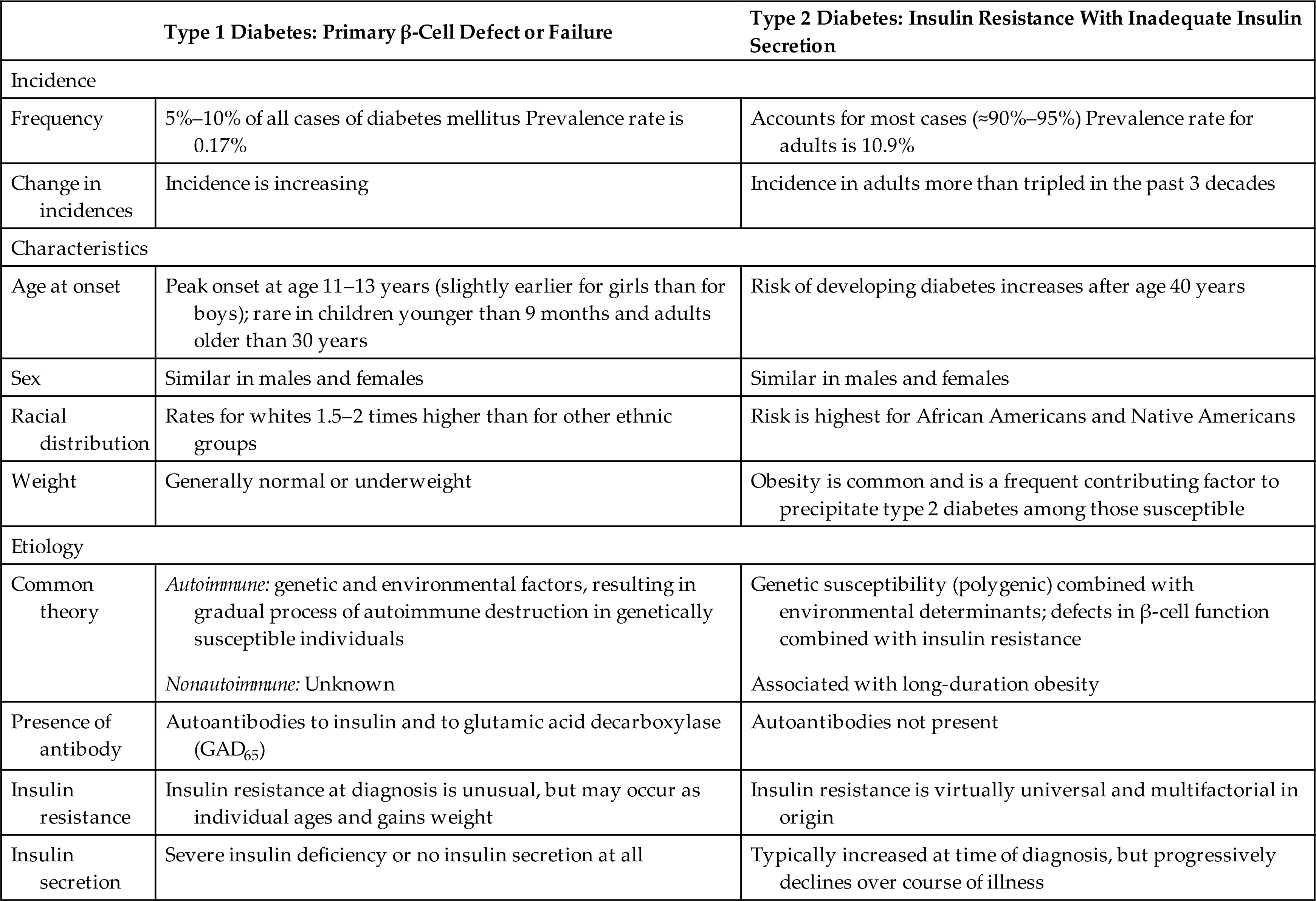
Data from American Diabetes Association. Standards of medical care in diabetes—2020. Diabetes Care. 2020;43(Suppl. 1):S1–S212. https://care.diabetesjournals.org/content/diacare/suppl/2019/12/20/43.Supplement_1.DC1/Standards_of_Care_2020.pdf.; Centers for Disease Control and Prevention (CDC). National diabetes statistics report. https://www.cdc.gov/diabetes/data/statistics/statistics-report.html.
Pathophysiology
Two distinct types of type 1 diabetes have been identified: idiopathic and autoimmune. Idiopathic diabetes mellitus type 1 is far less common than autoimmune diabetes, has a strong genetic component, and occurs mostly in people of Asian or African descent. Affected individuals have no evidence of β-cell autoimmunity and have varying degrees of insulin deficiency.
Autoimmune diabetes mellitus type 1 is a slowly progressive disease that destroys β cells of the pancreas. There are strong genetic associations with histocompatibility leukocyte antigen (HLA) class II alleles HLA-DQ and HLA-DR. Environmental factors that have been implicated include exposure to certain drugs, foods, and viruses. These gene-environment interactions result in the formation of autoantigens that are expressed on the surface of pancreatic β cells and circulate in the bloodstream and lymphatics (see Fig. 22.11). Cellular immunity (T-cytotoxic cells and macrophages) and humoral immunity (autoantibodies against islet cells, insulin, glutamic acid decarboxylase [GAD], and other cytoplasmic proteins) are stimulated, resulting in β-cell destruction and apoptosis. Over time, 80% to 90% of the insulin-secreting β cells of the islet of Langerhans are destroyed, insulin synthesis declines, and hyperglycemia develops.
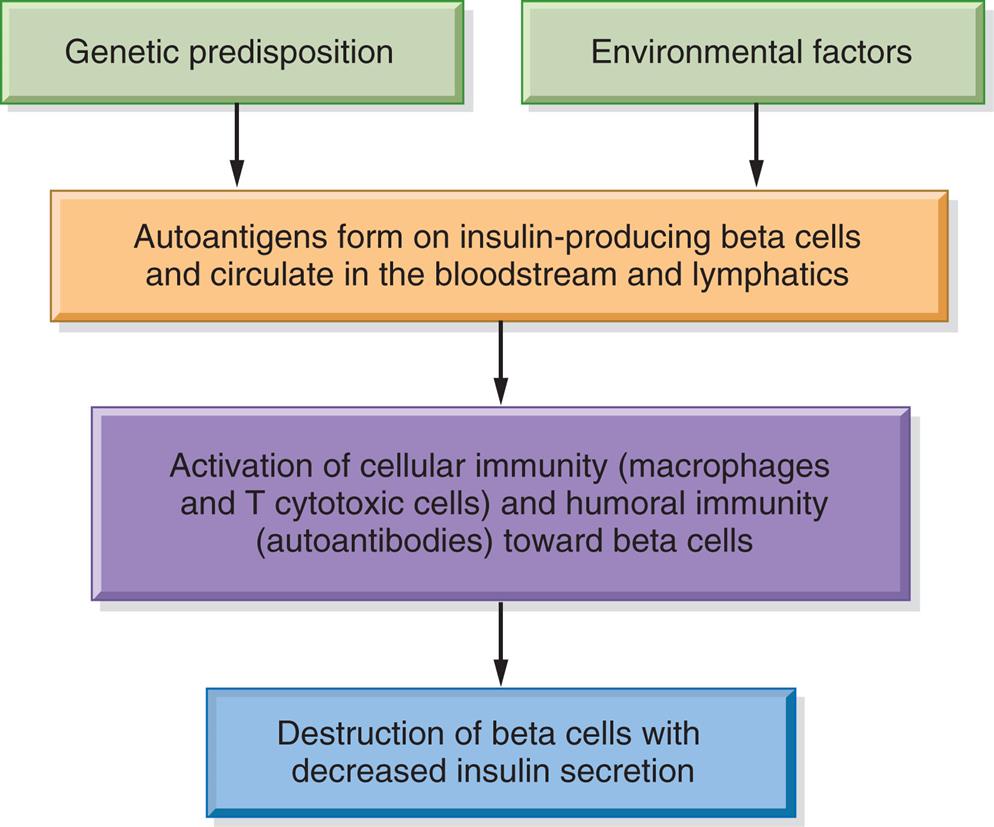
A flowchart represents the pathophysiology of type 1 diabetes mellitus. Genetic predisposition and environmental factors impact the autoantigens form on insulin-producing beta cells and circulate in the bloodstream and lymphatics, which leads to the following sequence. • Activation of cellular immunity (macrophages and T cytotoxic cells) and humoral immunity (autoantibodies) toward beta cells. • Destruction of beta cells with decreased insulin secretion.
Insulin normally suppresses secretion of glucagon, and thus hypoinsulinemia leads to a marked increase in glucagon secretion. In addition to the decline in insulin secretion, there is decreased secretion of amylin (another β-cell hormone), which also leads to an increase in glucagon. Glucagon, a hormone produced by the α cells of the islets, acts in the liver to increase the blood glucose level by stimulating glycogenolysis and gluconeogenesis. Thus, both a lack of insulin and a relative excess of glucagon contribute to hyperglycemia in type 1 diabetes.
The natural history of type 1 diabetes involves a long preclinical period before insulin deficiency and hyperglycemia develop. Glucose accumulates in the blood and appears in the urine as the renal threshold for glucose is exceeded, producing an osmotic diuresis and symptoms of polyuria and thirst (Table 22.8). Wide fluctuations in blood glucose levels occur. Insulin deficiency also causes protein and fat breakdown, resulting in weight loss. Excessive metabolism of fats and proteins leads to high levels of circulating ketones, causing a condition known as diabetic ketoacidosis (DKA) (see the Acute Complications of Diabetes section).
Table 22.8
Although most individuals with type 1 diabetes are of normal or decreased weight, there are increasing numbers of individuals who have both type 1 diabetes and the clinical manifestations of metabolic syndrome, including obesity, dyslipidemia, and hypertension (Box 22.2). These individuals are at high risk for chronic complications of diabetes, including heart disease and stroke.
Clinical Manifestations
The common clinical manifestations of type 1 diabetes result from both insulin deficiency and hyperglycemia. These manifestations are described in Table 22.8. Acute complications also may include hypoglycemia and DKA, which are described later in this chapter. Chronic complications include renal, nervous system, cardiac, peripheral vascular, retinal, and bony tissue dysfunction.
Evaluation and Treatment
The criteria for diagnosis of type 1 diabetes are the same as those for type 2 diabetes (see Box 22.1). To estimate the severity of β-cell destruction, C-peptide, a component of proinsulin released during insulin production, can be measured in the serum as a surrogate for insulin levels. An individual with residual pancreatic β cell function will have detectable levels of C-peptide, but a negligible amount of C-peptide can point to the diagnosis of type 1 diabetes. Individuals with hyperglycemia and at risk for type 1 diabetes can be tested for a variety of autoantibodies, such as zinc transporter 8 (ZnT8Ab) or tyrosine phosphatases. If two or more of the autoantibodies are positive in conjunction with diagnostic hyperglycemia, then the diagnosis of type 1 diabetes is confirmed. It is important to note that antibody testing itself is not a diagnostic requirement for type 1 diabetes. Other important aspects of evaluation include looking for evidence of acute and chronic complications of type 1 diabetes.
There are no approved treatments for preventing destruction of the β cells. Currently, treatment regimens are designed to achieve optimal glucose level control (as measured by the HbA1C value) without causing episodes of significant hypoglycemia. A comprehensive, person-centered collaborative management plan is essential.26 Management requires individual planning according to type of disease, age, and activity level, but all individuals require some combination of insulin therapy, meal planning, an exercise regimen, and glucose monitoring. There are several different types of insulin preparations available, and there are new technologies for more physiologic insulin delivery systems. Many different kinds of therapies are being tested to prevent the autoimmune destruction of β cells, including immunosuppression with antirejection drugs and stem cell transplantation (Emerging Science Box: Pancreatic β Cell Regeneration as a Plausible Diabetic Therapy).
Diabetes Mellitus Type 2
Diabetes mellitus type 2 (non–insulin-dependent diabetes mellitus) is a metabolic disorder that has reached pandemic dimensions all over the world, affecting 400 million people worldwide, and 10.9% of adults in the United States.26,28 Prevalence is highest among American Indians and Alaska Natives (14.7%) and lowest among non-Hispanic whites (7.5%). There also is an increased prevalence of type 2 diabetes in children, especially in obese children (see Table 22.7).
Genetic abnormalities combined with environmental influences result in the basic pathophysiologic mechanisms of type 2 diabetes, which are insulin resistance and decreased insulin secretion by β cells (Fig. 22.12). The most well-recognized risk factors are family history, age, obesity, hypertension, poor diet, and physical inactivity. More than 60 genes have been identified that are associated with type 2 diabetes, including those that code for β-cell mass, β-cell function (ability to sense blood glucose levels, insulin synthesis, and insulin secretion), proinsulin and insulin molecular structures, insulin receptors, hepatic synthesis of glucose, glucagon synthesis, and cellular responsiveness to insulin stimulation.
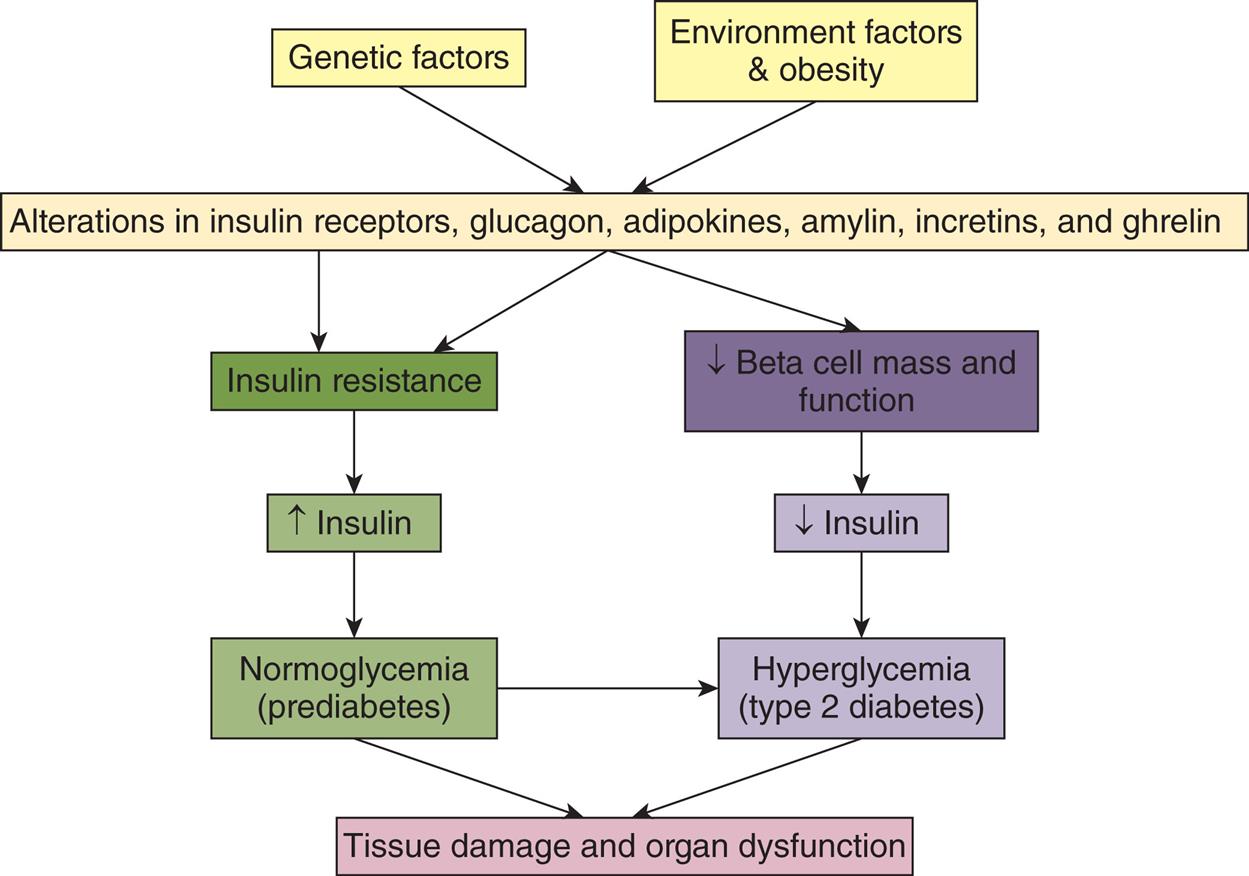
A flowchart represents the pathophysiology of type 2 diabetes mellitus. Genetic factors and environmental factors and obesity impact the alterations in insulin receptors, glucagon, adipokines, amylin, incretins, and ghrelin. Two pathways are charted; both of which lead to tissue damage and organ dysfunction. One pathway is as follows. • Insulin resistance. • Increased insulin. • Normoglycemia (prediabetes) (also to hyperglycemia). The other pathway is as follows. • Decreased beta cell mass and function. • Decreased insulin. • Hyperglycemia (type 2 diabetes).
There is increasing evidence that diet, including diet during pregnancy (both undernutrition and overnutrition), influences the long-term risk of type 2 diabetes in children and adults. Diets high in fruits, vegetables, fiber, and nuts reduce risk; diets high in simple carbohydrates, saturated fats, and red meat are associated with an increased risk. Weight gain and a lack of exercise also contribute to the risk for diabetes mellitus type 2.
Pathophysiology
Diabetes mellitus type 2 is a chronic and metabolic disease characterized by defects in pancreatic insulin secretion and (or) insulin effect on target tissues, generating a persistent state of hyperglycemia, inducing metabolic alteration, cell death, and inflammation.28 Insulin resistance is defined as a suboptimal response of insulin-sensitive tissues (especially liver, muscle, and adipose tissue) to insulin. Several mechanisms are involved in abnormalities of the insulin signaling pathway and contribute to insulin resistance. These include an abnormality of the insulin molecule, high amounts of insulin antagonists, down-regulation of the insulin receptor, and alteration of glucose transporter (GLUT) proteins.
Obesity is one of the most important contributors to insulin resistance and diabetes and acts through several important mechanisms:
- 1. Adipokines (cytokines produced by adipose tissue): Increased serum levels of leptin (leptin resistance) and decreased levels of adiponectin result in inflammation and decreased insulin sensitivity.
- 2. Free fatty acids (FFAs): Increased FFAs, along with intracellular deposits of triglycerides and cholesterol, lead to decreased tissue responses to insulin.
- 3. Inflammation: Adipocyte-associated proinflammatory macrophages and inflammatory cytokines released from adipocytes induce insulin resistance and are cytotoxic to β cells.
- 4. Mitochondrial dysfunction: Decreased insulin-induced mitochondrial activity leads to insulin resistance.
- 5. Hyperinsulinemia: Obesity is correlated with hyperinsulinemia and decreased insulin receptor density.
Compensatory hyperinsulinemia prevents the clinical appearance of diabetes for many years. Eventually, however, a decrease in β-cell mass and a reduction in normal β-cell function develops and leads to a relative deficiency of insulin activity.
The glucagon concentration is increased in type 2 diabetes because pancreatic α cells become less responsive to glucose inhibition, resulting in an increase in glucagon secretion. These abnormally high levels of glucagon increase the blood glucose level by stimulating glycogenolysis and gluconeogenesis. As was discussed under type 1 diabetes, type 2 diabetes also is associated with a deficiency in amylin, further increasing glucagon levels. Pramlintide, a synthetic analog of amylin, is used for treatment in type 2 diabetes.
Hormones released from the GI tract play a role in insulin resistance, β-cell function, and diabetes. Ghrelin is a peptide produced in the stomach and pancreatic islets that regulates food intake, energy balance, and hormonal secretion. Decreased levels of circulating ghrelin have been associated with insulin resistance and increased fasting insulin levels. The incretins are a class of peptides that are released from the GI tract in response to food intake and function to increase the secretion of insulin and have many other positive effects on metabolism. The most studied incretin is called glucagon-like peptide 1 (GLP-1), and studies have demonstrated that β-cell responsiveness to GLP-1 is reduced both in prediabetes and in type 2 diabetes. Incretin therapies include GLP-1 receptor agonists (GLP-1 RAs) and dipeptidyl peptidase IV (DPP-IV) inhibitors, which can help control postprandial glucose levels by promoting glucose-dependent insulin secretion.
The kidneys also influence the pathophysiology of type 2 diabetes. Renal reabsorption of glucose through the sodium-glucose cotransporter 2 (SGLT2) is an important controller of serum glucose levels, and medications aimed at blocking it have resulted in decreased measurements for blood glucose level, weight, and blood pressure.
As you have learned, many organs contribute to insulin resistance, chronic hyperglycemia, and the consequences of type 2 diabetes. These causes and consequences are summarized in Fig. 22.13, and the complications of type 2 diabetes will be discussed later in this chapter.
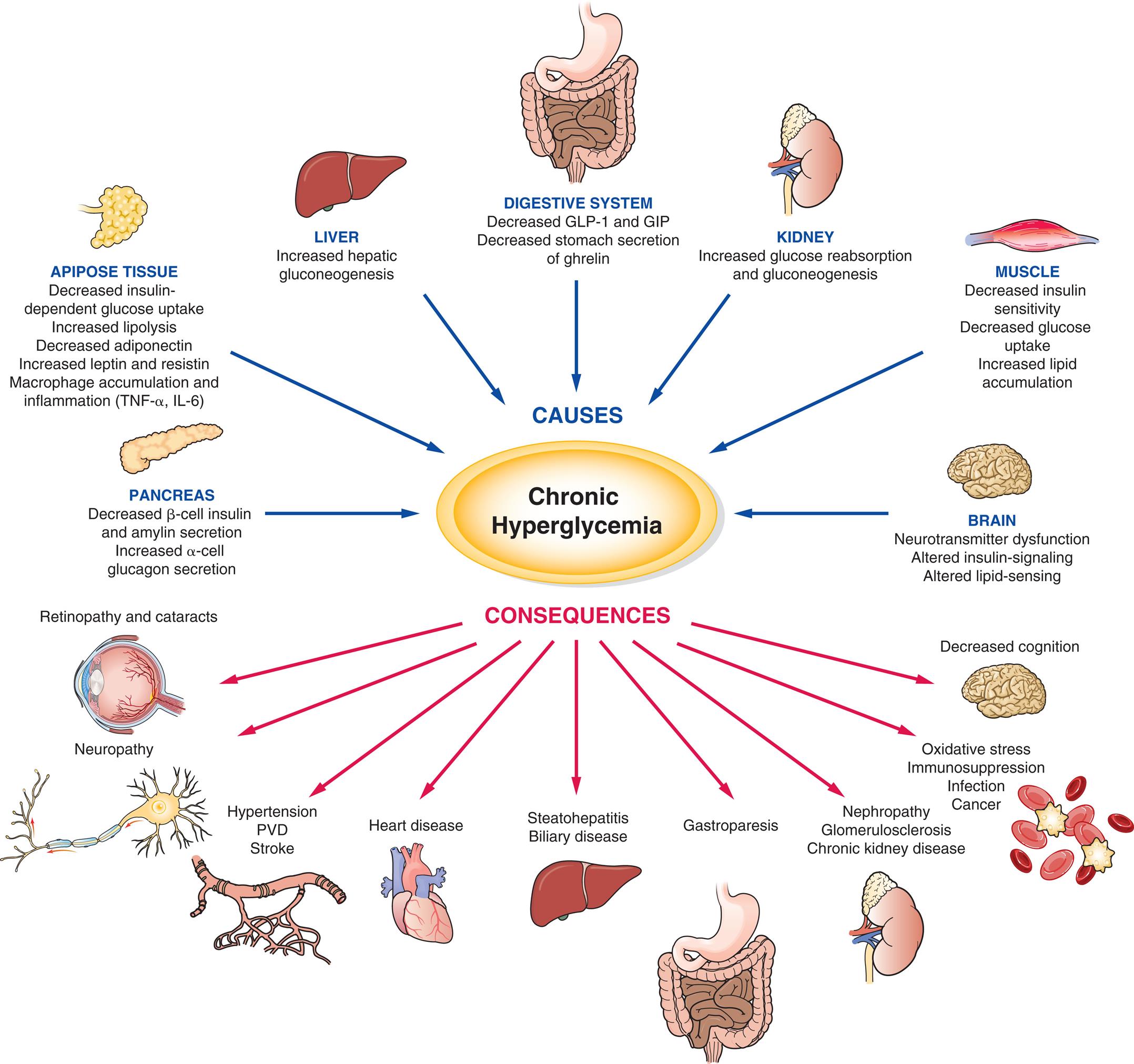
GLP-1, Glucagon-like peptide 1; GIP, gastric inhibitory polypeptide; IL, interleukin; PVD, peripheral vascular disease; TNF, tumor necrosis factor.
An illustration lists the causes and consequences of chronic hyperglycemia. The causes of chronic hyperglycemia are as follows. • Pancreas. Decreased beta-cell insulin and amylin secretion. Increased alpha-cell glucagon secretion. • Adipose tissue. Decreased insulin-dependent glucose uptake. Increased lipolysis. Decreased adiponectin. Increased leptin and resistin. Macrophage accumulation and inflammation (T N F-alpha, I L-6). • Liver. Increased hepatic gluconeogenesis. • Digestive system. Decreased G L P-1 and G I P. Decreased stomach secretion of ghrelin. • Kidney. Increased glucose reabsorption and gluconeogenesis. • Muscle. Decreased insulin sensitivity. Decreased glucose uptake. Increased lipid accumulation. • Brain. Neurotransmitter dysfunction. Altered insulin-signaling. Altered lipid-sensing. The consequences of chronic hyperglycemia are as follows. • Decreased cognition. • Oxidative stress. Immunosuppression. Infection. Cancer. • Nephropathy. Glomerulosclerosis. Chronic kidney disease. • Gastroparesis. • Steatohepatitis. Biliary disease. • Heart disease. • Hypertension. P V D. Stroke. • Neuropathy. • Retinopathy and cataracts.
Clinical Manifestations
The clinical manifestations of type 2 diabetes are nonspecific. The individual with type 2 diabetes may show some classic symptoms of diabetes, such as polyuria and polydipsia, but more often will have nonspecific symptoms, such as fatigue, pruritus, recurrent infections, visual changes, or symptoms of neuropathy (paresthesia or weakness). The affected individual is often overweight, dyslipidemic, and hypertensive. The increased morbidity and mortality rates related to diabetes mellitus type 2 are often associated with vascular complications, such as cardiovascular diseases, nephropathy, retinopathy, endothelial dysfunction, dyslipidemia, and an increase of oxidative stress, which is also due to inflammation (Emerging Science Box: Inflammation and Its Importance to the Pathophysiology of Insulin Resistance in Prediabetes and Diabetes Mellitus Type 2).29,30
Evaluation and Treatment
The diagnostic criteria for type 2 diabetes are the same as those for type 1 (see Box 22.1). Metabolic syndrome is a constellation of disorders (central obesity, dyslipidemia, prehypertension, and an elevated FPG) that confer a high risk of developing type 2 diabetes and associated cardiovascular complications (see Box 22.2).
As with type 1 diabetes, the goal of treatment for individuals with type 2 diabetes (and metabolic syndrome) is the restoration of near-euglycemia (normal blood glucose) levels and correction of related metabolic disorders. Prevention and treatment of type 2 diabetes, especially in those individuals with prediabetes, focuses on lifestyle modifications as the cornerstone modality.31 Diet should match activity levels and include more complex carbohydrates (rather than simple sugars), foods low in fats, adequate protein, and fiber. Obesity management results in improved glucose tolerance. Bariatric surgery improves glycemic control, decreases the risk of cardiovascular disease, and promotes weight loss in those morbidly obese.26
For individuals who require further intervention, oral and/or non-insulin antidiabetic agents are indicated (Table 22.9).26 Currently, metformin is considered the primary pharmacologic choice for the treatment of type 2 diabetes. If the HbA1C target is not maintained over 3 months, a GLP-1 receptor agonist (incretin) may be added.26,31
Table 22.9
CNS, Central nervous system; DPP-IV, dipeptidyl peptidase-IV; GLP-1, glucagon-like peptide-1; SGLT2, sodium-glucose cotransporter 2.
Data from Brietzke SA. Oral antihyperglycemic treatment options for type 2 diabetes mellitus. Medical Clinics of North America, 2015;99(1):87–106; Tran L, Zielinski A, Roach AH, et al. Pharmacologic treatment of type 2 diabetes: Oral medications. Annals of Pharmacotherapy, 2015;49(5):540–556.
If treatment goals are still not met, a combination of drugs, including SGLT2 inhibitors, may be required. Insulin therapy may be needed in the later stage of type 2 diabetes because of loss of β-cell function, which is progressive over time (Emerging Science Box: Smart Pens for Improved Safety and Efficacy of Insulin Therapy).
Gestational Diabetes Mellitus and Specific Types of Diabetes Mellitus From Other Causes
As listed in Table 22.7 the ADA classification of diabetes mellitus not only includes the most common forms of diabetes (type 1 and type 2) but also encompasses GDM and specific types of diabetes mellitus from other causes.
Gestational diabetes mellitus (GDM) complicates approximately 7% of pregnancies and is defined as diabetes diagnosed in the second or third trimester of pregnancy that was not clearly overt diabetes prior to gestation.26 In most cases, this hyperglycemia is the result of IGT due to pancreatic β-cell dysfunction on a background of chronic insulin resistance. Risk factors for GDM include overweight and obesity, advanced maternal age, and a family history of any form of diabetes.32 An OGTT is used to confirm the diagnosis. However, this definition means that many women with previously undiagnosed type 1 or type 2 diabetes are diagnosed with GDM, and many of them have progressive disease after delivery. Therefore, the ADA recommends that women with risk factors for diabetes be screened at their initial prenatal visit and that women diagnosed during pregnancy should continue to be monitored postpartum and at least every 3 years for persistent or recurrent diabetes.26 Careful glucose control prenatally, during pregnancy, and after delivery is essential to the short- and long-term health of both mother and baby.
Specific types of diabetes from other causes include monogenic diabetes syndromes (e.g., MODY), disease of the exocrine pancreas (e.g., cystic fibrosis), and drug- or chemical-induced diabetes (e.g., with glucocorticoid use, in the treatment of HIV/AIDS, or after organ transplantation). Maturity-onset diabetes of youth (MODY) usually presents before 25 years of age and includes at least 13 genetic mutations that affect critical enzymes involved in β-cell function but with little impact on insulin action.26 The diagnosis of MODY should be considered in individuals who have atypical clinical presentations, who are diagnosed within the first 6 months of life (neonatal diabetes), and who have a strong family history of diabetes. Diagnosis includes genetic testing for the three most common forms of MODY. Management is similar to that used for type 2 diabetes. Diabetes also can result from pancreatic damage incurred through the effects of cystic fibrosis, medications, and immunosuppression after organ transplantation.
Acute Complications of Diabetes Mellitus
The major acute complications of diabetes mellitus are hypoglycemia, DKA, and hyperosmolar hyperglycemic nonketotic syndrome (HHNKS).33 A comparison of these complications is summarized in Table 22.10.
Table 22.10
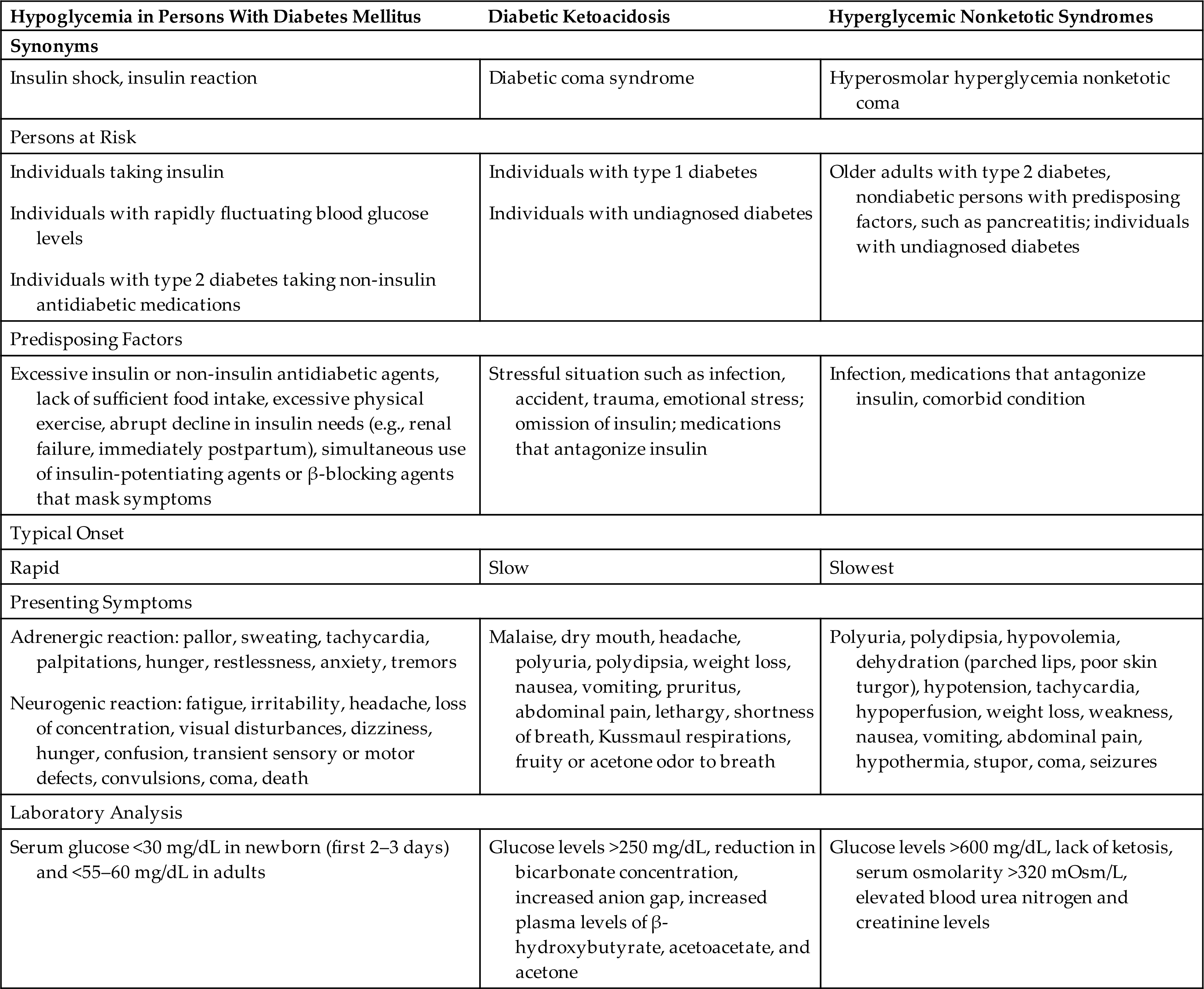
Hypoglycemia in diabetes is sometimes called insulin shock or insulin reaction. Individuals with type 1 diabetes require insulin to manage their diabetes and are at more risk for hypoglycemia than those with type 2 diabetes because they have more severe deficits in their ability to control serum glucose levels. Significant drops in blood sugar most often occur when there is an unexpected change in caloric intake or exercise without appropriate modification of insulin dosing. Hypoglycemia sometimes occurs in type 2 diabetes during treatment with insulin or non-insulin antidiabetic medications. Symptoms include pallor, tremor, anxiety, tachycardia, palpitations, diaphoresis, headache, dizziness, irritability, fatigue, poor judgment, confusion, visual disturbances, hunger, seizures, and coma. Treatment requires immediate replacement of glucose either orally or intravenously. Glucagon for home use can be prescribed for individuals who are at high risk. Prevention is achieved with individualized management of medications and diet, monitoring of blood glucose levels, and education.
Diabetic ketoacidosis (DKA) is a serious complication related to a deficiency of insulin and an increase in the levels of insulin counterregulatory hormones (catecholamines, cortisol, glucagon, GH) (Fig. 22.14). DKA is much more common in type 1 diabetes because insulin is more deficient (see Table 22.10). It is characterized by hyperglycemia, acidosis, and ketonuria. Insulin normally stimulates lipogenesis and inhibits lipolysis, thus preventing fat catabolism. With insulin deficiency, lipolysis is enhanced and there is an increase in the amount of nonesterified fatty acids delivered to the liver. The consequence is increased glyconeogenesis, contributing to hyperglycemia and the production of ketone bodies (acetoacetate, hydroxybutyrate, and acetone) by the mitochondria of the liver at a rate that exceeds peripheral use. Accumulation of ketone bodies causes a drop in pH, resulting in metabolic acidosis and transient hyperkalemia. Symptoms of DKA include Kussmaul respirations (hyperventilation in an attempt to compensate for the acidosis), postural dizziness, central nervous system depression, ketonuria, anorexia, nausea, abdominal pain, thirst, and polyuria. DKA is managed with a combination of fluids, insulin, and electrolyte replacement.
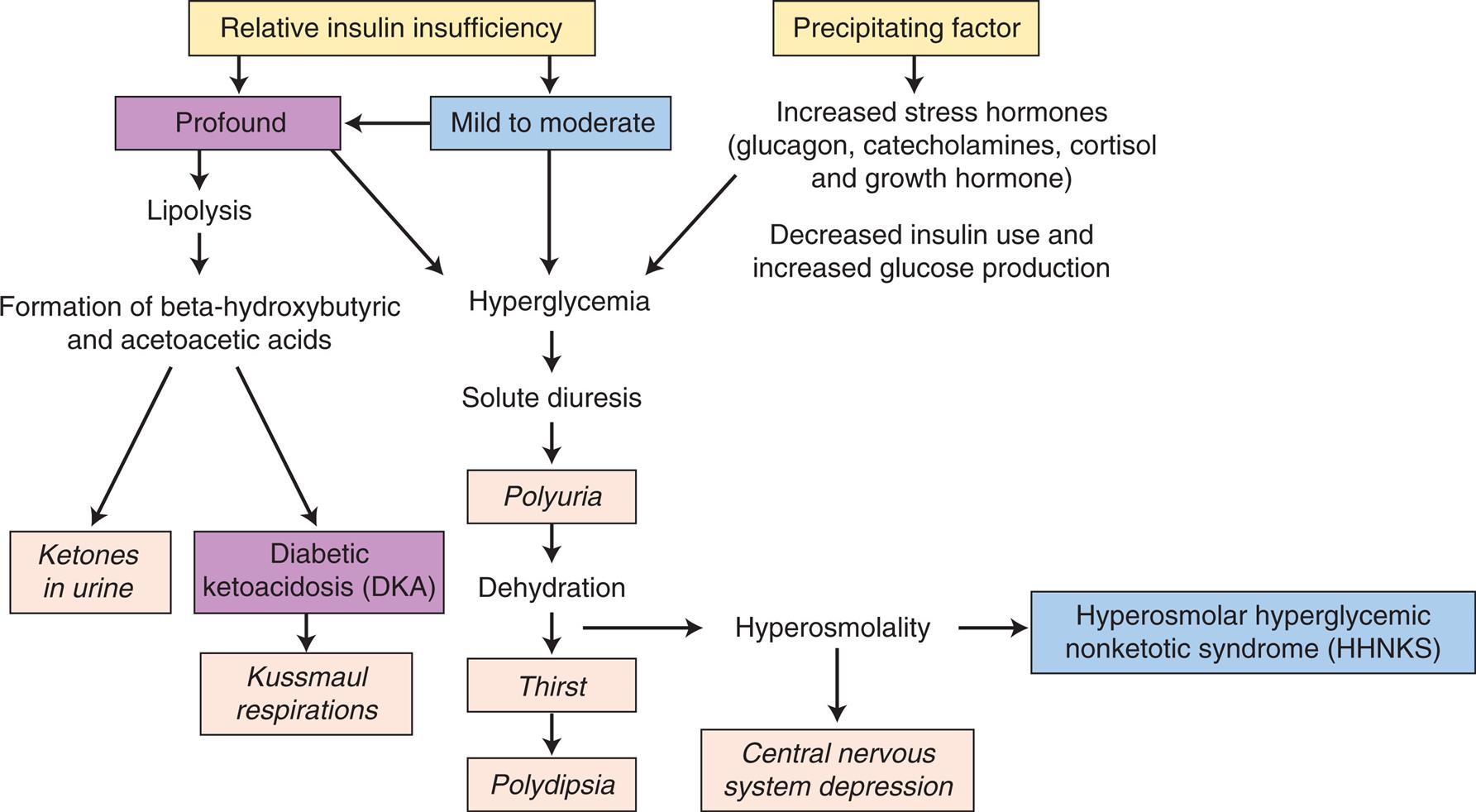
A flowchart represents the pathophysiology of diabetic ketoacidosis and hyperosmolar hyperglycemia nonketotic syndrome in diabetes mellitus. The sequence and relationships are as follows. 1. Relative insulin insufficiency. Could be of type 2 and 3. 2. Profound. Leads to 4 and 9. 3. Mid to moderate. Could aggravate to 2 or lead to 10. 4. Lipolysis. Leads to 5. 5. Formation of beta-hydroxybutyric and acetoacetic acids. Leads 6 and 7. 6. Ketones in urine. 7. Diabetic ketoacidosis (D K A). Leads to 8. 8. Kussmaul respirations. 9. Hyperglycemia. Leads to 10. 10. Solute diuresis. Leads to 11. 11. Polyuria. Leads to 12. 12. Dehydration. Leads 13 and 15. 13. Thirst. Leads to 14. 14. Polydipsia. 15. Hyperosmolality. Leads to 16 and 17. 16. Hyperosmolar hyperglycemic nonketotic syndrome (H H N K S). 17. Central nervous system depression. 18. Precipitating factor. Leads to 19 and 20. 19. Increased stress hormones (glucagon, catecholamines, cortisol and growth hormone). 20. Decreased insulin use and increased glucose production.
Hyperosmolar hyperglycemic nonketotic syndrome (HHNKS) is an uncommon but significant complication of diabetes mellitus type 2 with a high overall mortality. It occurs more often in elderly individuals who have other comorbidities, including infections or cardiovascular or renal disease. HHNKS differs from DKA because type 2 diabetes is characterized by a lesser degree of insulin deficiency, which therefore prevents lipolysis and the production of ketones (see Fig. 22.14). However, hyperglycemia is usually more profound in HHNKS leading to more polyuria and fluid deficiency. Therefore, the clinical features of HHNKS include a very high serum glucose concentration and osmolarity without metabolic acidosis. Clinical manifestations include severe dehydration; loss of electrolytes (especially potassium); and neurologic changes, such as stupor. Management includes fluid, insulin, and electrolyte replacement.
Chronic Complications of Diabetes Mellitus
A number of serious complications are associated with any type of poorly controlled diabetes mellitus. Most complications are associated with insulin resistance or deficit and chronic hyperglycemia. Insulin resistance or deficit renders cells unable to process glucose normally, so they resort to a number of alternative pathways for glucose metabolism, particularly through activation of the polyol pathway. This pathway results in the accumulation of sorbitol and glutathione, both of which contribute to chronic tissue damage. Hyperglycemia leads to a number of deleterious effects, including abnormalities in intracellular communication (e.g., protein kinase C pathways) and the attachment of glucose to proteins, lipids, and nucleic acids (glycation). Therefore, glycation creates what are called advanced glycation end products (AGEs), which interfere with many crucial cellular processes. The cumulative effect of these abnormal processes leads to the microvascular (damage to capillaries; retinopathies, nephropathies, and neuropathies) and macrovascular (damage to larger vessels; coronary artery, peripheral vascular, and cerebral vascular disease) complications of chronic diabetes mellitus (Table 22.11).
Table 22.11
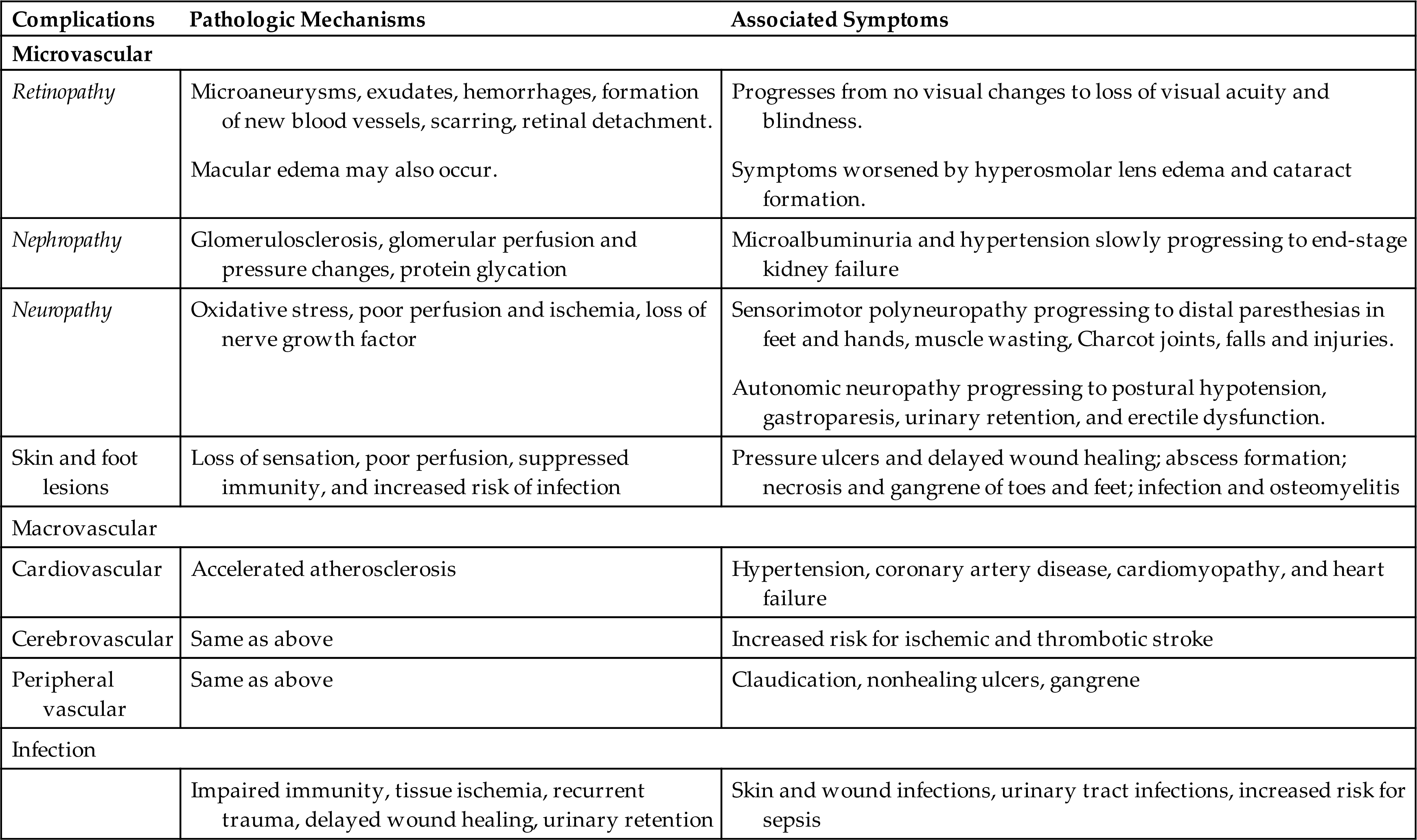
Microvascular Disease
Diabetic microvascular complications are characterized by occlusion of capillaries, with associated ischemia of tissues.34 It is a leading cause of blindness, diabetic nephropathy (including end-stage kidney failure), and various neuropathies. Approximately half of the individuals diagnosed with diabetes mellitus type 2 develop peripheral neuropathy. This progressive disorder is chronic, affecting the peripheral nervous system and characterized by allodynia, pain, and paresthesia symptoms. Neuropathies are caused by axonal degeneration and segmental demyelination. The frequency and severity of complications appear to be proportional to the duration of the disease (more or less than 10 years) and the status of glycemic control. Hypoxia and ischemia accompany microvascular disease, especially in the eye, kidney, and nerves. Many individuals with type 2 diabetes will present with microvascular complications because of the long duration of asymptomatic hyperglycemia that generally precedes diagnosis. This underscores the need to screen for diabetes.
Diabetic retinopathy
Diabetic retinopathy is a leading cause of blindness worldwide and is a common complication of type 2 diabetes because of the likelihood of long-standing hyperglycemia before diagnosis. Most individuals with diabetes will eventually develop retinopathy and also are more likely to develop cataracts and glaucoma (see Chapter 16).
Diabetic retinopathy results from relative hypoxemia, damage to retinal blood vessels, RBC aggregation, and hypertension (Fig. 22.15).35 The three stages of retinopathy that lead to loss of vision are nonproliferative (stage I), characterized by an increase in retinal capillary permeability, vein dilation, microaneurysm formation, and superficial (flame-shaped) and deep (blot) hemorrhages; preproliferative (stage II), a progression of retinal ischemia, with areas of poor perfusion that culminate in infarcts; and proliferative (stage III), the result of neovascularization (angiogenesis) and fibrous tissue formation within the retina or optic disc. Traction of the new vessels on the vitreous humor may cause retinal detachment or hemorrhage into the vitreous humor, with severe blurring or loss of vision. Macular edema is the leading cause of blurred vision among persons with diabetes. Blurring of vision also can be a consequence of hyperglycemia and sorbitol accumulation in the lens. Dehydration of the lens, aqueous humor, and vitreous humor also reduces visual acuity.
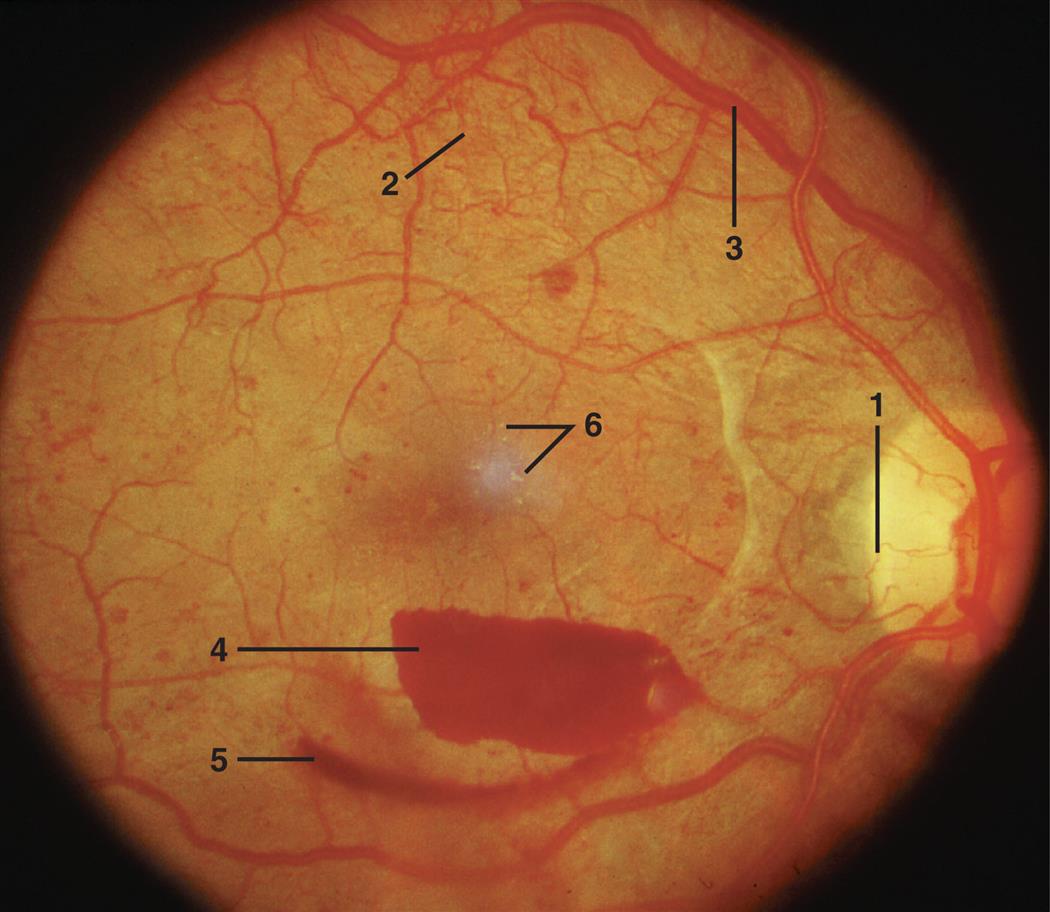
Neovascularization is present at the optic nerve (1) and along vascular pathways (2). Retinal veins are engorged (3) and a preretinal boat-shaped hemorrhage (4) is present below the fovea. A more diffuse mild vitreous hemorrhage (5) is present below the preretinal hemorrhage. A few small, hard exudates are visible in the fovea (6). (From Palay DA, Krachmer JH. Primary care ophthalmology, 2nd edition. St. Louis: Mosby; 2006.)
Diabetic nephropathy
Diabetes is the most common cause of chronic kidney disease and end-stage renal disease in the world. Nearly half of individuals with diabetes mellitus develop diabetic nephropathy.36 Renal glomerular changes occur early in diabetes mellitus, occasionally preceding the overt manifestation of the disease. The glomeruli are injured by hyperglycemia with protein glycation (bonding of a glucose molecule to a protein), high renal blood flow (hyperfiltration), and intraglomerular hypertension exacerbated by systemic hypertension (Fig. 22.16). There is progressive glomerulosclerosis and decreased glomerular blood flow and glomerular filtration that can progress to renal failure. Tubulointerstitial fibrosis also develops in advanced stages of disease. Loss of autoregulation of renal blood flow and anemia contribute to hypoxia. Hyperglycemia and glomerular hyperfiltration increase tubular reabsorption of glucose promoting inflammation and activation of growth factors and fibroblasts. The consequence is tubulointerstitial fibrosis which contributes to renal failure.37
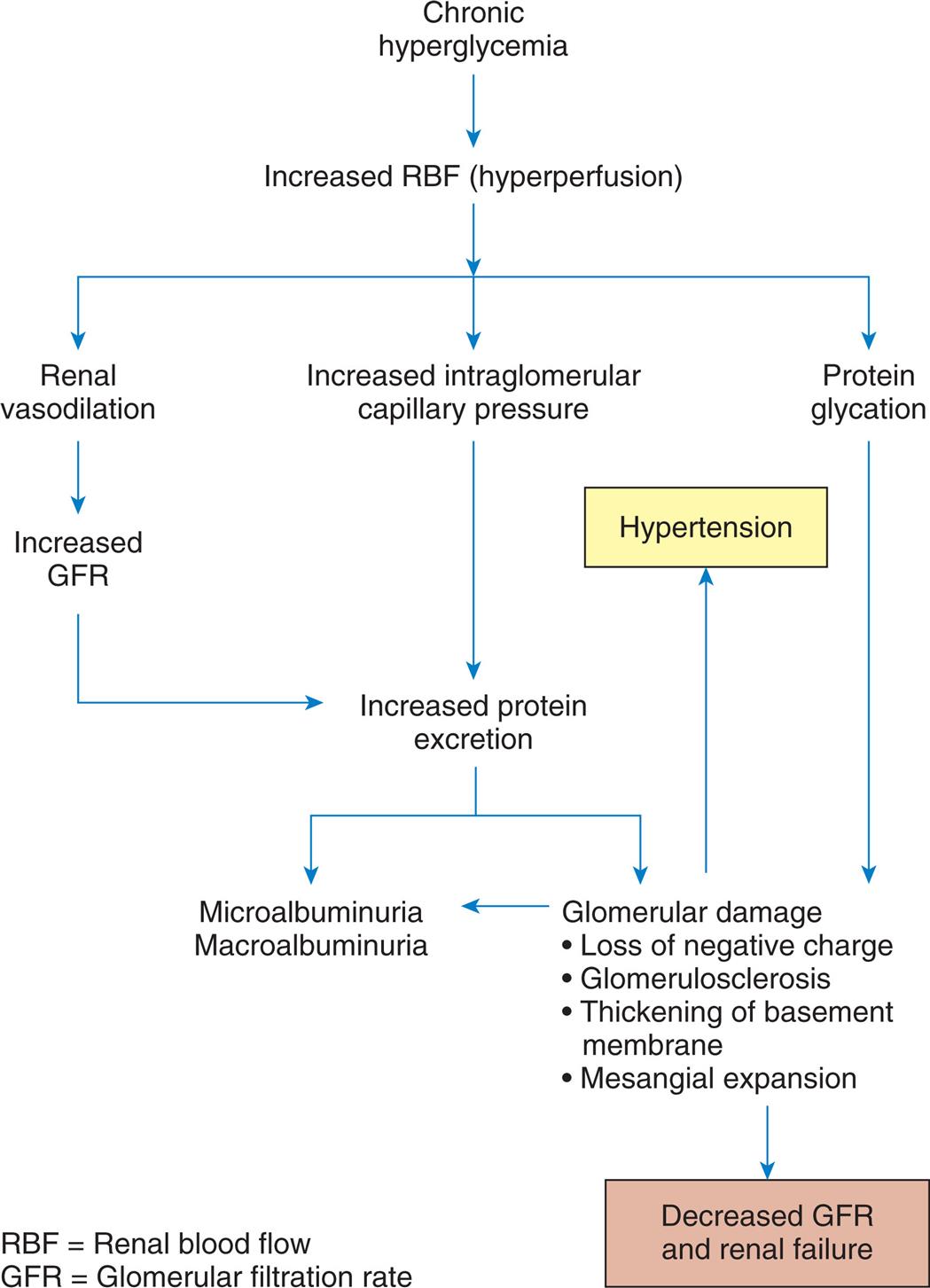
A flowchart represents diabetic nephropathy. 1. Chronic hyperglycemia. Leads to 2. 2. Increased R B F (hyperperfusion). Leads to 3, 5, and 7. 3. Renal vasodilation. Leads to 4. 4. Increased G F R. Leads to 8. 5. Increased intraglomerular capillary pressure. Leads to 6. 6. Increased protein excretion. Leads to 8 and 9. 7. Protein glycation. Leads to 9. 8. Microalbuminuria, macroalbuminuria. 9. Glomerular damage: loss of negative charge; glomerulosclerosis; thickening of basement membrane; mesangial expansion. Leads to 8, 10, and 11. 10. Decreased G F R and renal failure. 11. Hypertension.
Microalbuminuria is the first manifestation of diabetic kidney dysfunction and signals the onset of systemic diabetic complications. Before proteinuria, no clinical signs or symptoms of progressive glomerulosclerosis are likely to be evident. Later, hypoproteinemia, reduction in plasma oncotic pressure, fluid overload, anasarca (generalized body edema), and hypertension may occur. As renal function continues to deteriorate, individuals with type 1 diabetes may experience hypoglycemia (because of loss of renal insulin metabolism), which necessitates a decrease in insulin therapy. As the glomerular filtration rate drops below 10 mL/min, uremic signs, such as nausea, lethargy, acidosis, anemia, and uncontrolled hypertension, occur (see Chapter 38 for a discussion of renal failure). Early diagnosis and control of hypertension and hyperglycemia decreases the severity of nephropathy and delays the onset of end-stage kidney disease.
Diabetic neuropathies
Diabetic neuropathy is the most common complication of diabetes. The underlying pathologic mechanism includes metabolic and vascular factors related to chronic hyperglycemia with ischemia and demyelination contributing to neural changes and delayed conduction. Both somatic and peripheral nerve cells show diffuse or focal damage, resulting in polyneuropathy.38,39 Loss of pain, temperature, and vibration sensation is more common than motor involvement and often involves the extremities first in the hands and feet. Motor neuropathies can affect muscle groups, particularly of the legs and feet, contributing to deformity and unstable balance. Peripheral neuropathy can cause Charcot arthropathy, a progressive deterioration of weight-bearing joints, typically in the foot and ankle. Distal neuropathies combined with vascular complications, infection, or injury can lead to amputation (Fig. 22.17).
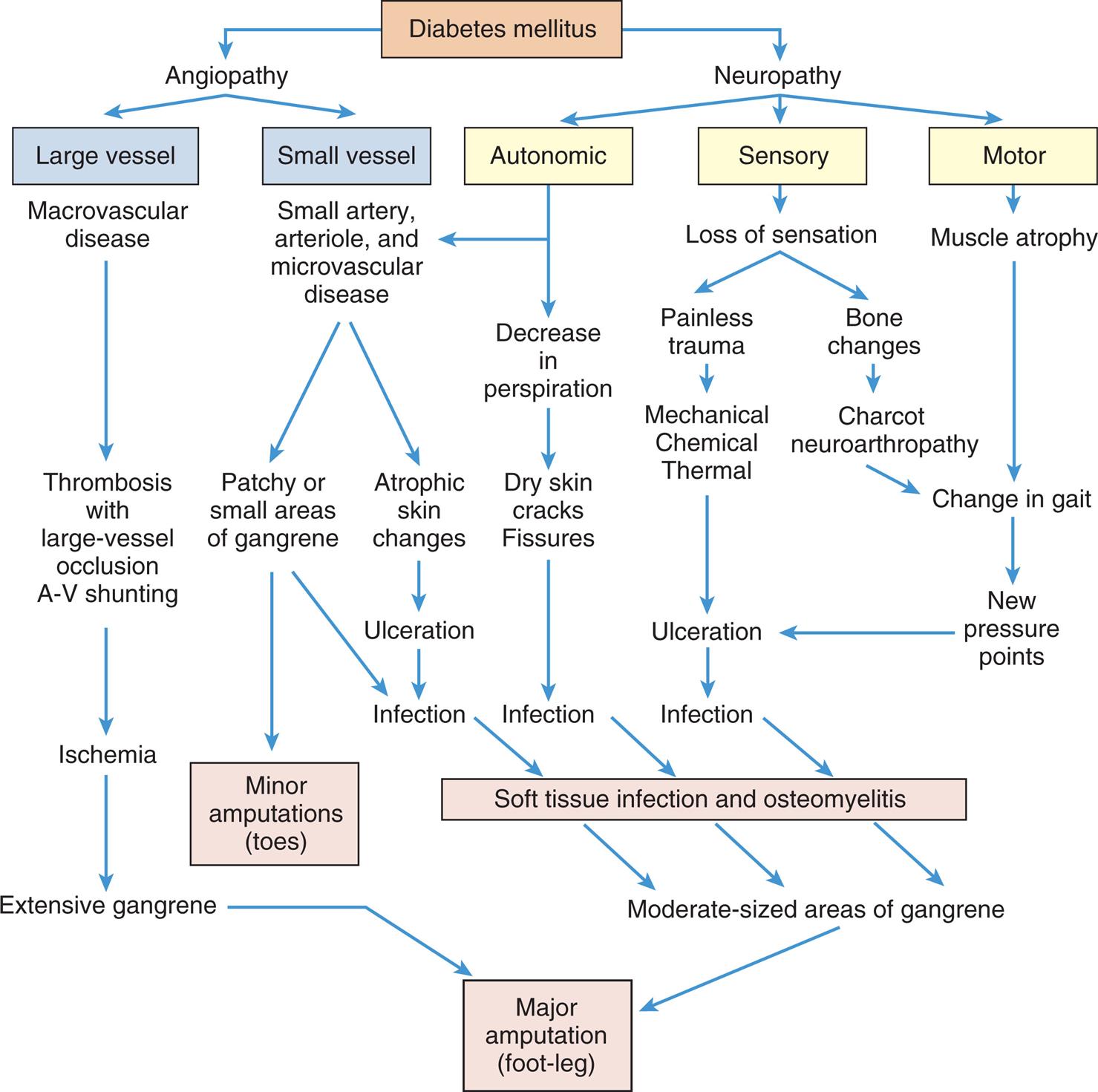
How Foot Lesions of Diabetes Lead to Amputation. (From Levin ME, et al. The diabetic foot, 5th edition. St. Louis: Mosby; 1993.)
A flowchart shows how foot lesions of diabetes lead to amputation. Diabetes can lead to angiopathy or neuropathy. The pathways that lead to amputation (foot-leg) are as follows. 1. Large vessel (from angiopathy); macrovascular disease. Leads to 2. 2. Thrombosis with large-vessel occlusion A-V shunting. Leads to 3. 3. Ischemia. Leads to 4. 4. Extensive gangrene. Leads major amputation (foot-leg). 5. Small vessel (small artery, arteriole, and microvascular disease). Leads to 6 and 7. 6. Patchy or small areas of gangrene. Leads to minor amputations (toes) and 9. 7. Atrophic skin changes. Leads to 8. 8. Ulceration. Leads to 9. 9. Infection. Leads to 10. 10. Soft tissue infection and osteomyelitis. Leads to 11. 11. Moderate-sized areas of gangrene. Leads to major amputation (foot-leg). 12. Autonomic (from neuropathy). Leads to 5 and 13. 13. Decrease in perspiration. Leads to 14. 14. Dry skin cracks; fissures. Leads to 9. 15. Sensory (from neuropathy). Leads to 16. 16. Loss of sensation. Leads to 17 and 18. 17. Painless trauma. Leads to 19. 18. Bone changes. Leads to 21. 19. Mechanical, chemical, thermal. Leads to 20. 20. Ulceration. Leads to 9. 21. Charcot neuroarthropathy. Leads to 24. 22. Motor (from neuropathy). Leads to 23. 23. Muscle atrophy. Leads to 24. 24. Change in gait. Leads to 25. 25. New pressure points. Leads to 20.
Autonomic neuropathies include delayed gastric emptying, diabetic diarrhea, altered bladder function (e.g., decreased sensation of bladder fullness, urge or overflow incontinence), impotence, orthostatic hypotension, and heart rate variability, with both tachycardia and bradycardia. Neuropathy may occur during periods of “good” glucose control and may be the initial clinical manifestation of diabetes mellitus type 2. Chronic hyperglycemia also can cause cognitive dysfunction with alterations in learning and memory.
Macrovascular Disease
Macrovascular disease (lesions in large- and medium-sized arteries) increases morbidity and mortality and increases the risk for hypertension, accelerated atherosclerosis, cardiovascular disease, stroke, and peripheral vascular disease (PVD), particularly among individuals with diabetes mellitus type 2 (atherosclerosis is discussed in Chapter 32). Children with poorly controlled diabetes have higher risk for macrovascular complications within 1 to 2 decades. The process tends to be more severe and accelerated in the presence of other risk factors, including obesity, hyperlipidemia, and smoking.
Cardiovascular disease
Cardiovascular disease is the primary cause of death of people with diabetes, with higher risk for women. Hypertension often coexists with diabetes mellitus. Diabetes also is associated with deleterious changes in serum lipids, including high levels of low-density lipoproteins (LDLs), and low levels of high-density lipoproteins (HDLs). The combination of diabetes, hypertension dyslipidemia, and obesity (metabolic syndrome) greatly increases the risk for atherosclerotic cardiovascular disease, particularly CAD and stroke. CAD results from vessel injury related to insulin resistance and hyperglycemia, leading to accelerated atherosclerosis (see Chapter 32). In general, the prevalence of CAD increases with the duration but not the severity of diabetes, and the onset can be silent.
The incidence of congestive heart failure is higher in individuals with diabetes, even without myocardial infarction. This may be related to cardiomyopathy and the presence of increased amounts of collagen in the ventricular wall and ventricular hypertrophy. There is reduced mechanical compliance of the heart during filling with diastolic and, eventually, systolic failure (heart disease is described in Chapter 32).
Stroke
Stroke is twice as common in those with diabetes (particularly type 2 diabetes) as in the nondiabetic population. As in CAD, accelerated atherosclerosis of the cerebral vessels results from insulin resistance and hyperglycemia, especially when associated with hypertension and hyperlipidemia (see Chapter 18).
Peripheral vascular disease
Diabetes mellitus increases the incidence of PVD. Age, duration of diabetes, genetics, and additional risk factors (smoking, hyperlipidemia, hypertension) influence the development and management of PVD. PVD in those with diabetes is more diffuse and often involves arteries below the knee. Occlusions of the small arteries and arterioles can lead to claudication (pain from reduced blood flow during exercise), ulcers, gangrene, and amputation. The lesions begin as ulcers and progress to osteomyelitis or gangrene, requiring amputation (see Fig. 22.17). Peripheral neuropathies and an increased risk for infection advance the disease.
Infection
Diabetic individuals are more prone to develop common infections, like cystitis, enteric infections, skin and soft tissue infections, external otitis, pneumonia, appendicitis, and peritonitis as well as rare and severe infections, such as emphysematous pyelonephritis.40
The individual with diabetes is at an increased risk for infection throughout the body for several reasons:
- 1. The senses. Impaired vision caused by retinal changes and impaired touch caused by neuropathy lead to loss of protection, with injury and repeated trauma, open wounds, and soft tissue or osseous infection, particularly in the legs and feet.
- 2. Hypoxia. Once skin integrity is compromised, susceptibility to infection increases as a result of hypoxia. In addition, the glycated hemoglobin in the RBCs impedes the release of oxygen to tissues and contributes to delayed wound healing.
- 3. Pathogens. Some pathogens proliferate rapidly because of increased glucose in body fluids, which provides an excellent source of energy.
- 4. Blood supply. Decreased blood supply results from vascular changes and reduces the supply of white blood cells to the affected area.
- 5. Suppressed immune response. Chronic hyperglycemia impairs both innate and adaptive immune responses, including abnormal chemotaxis and vasoactive responses, and defective phagocytosis.
Alterations of Adrenal Function
Disorders of the Adrenal Cortex
Disorders of the adrenal cortex are related either to hyperfunction or to hypofunction. Hyperfunction that causes increased secretion of cortisol (hypercortisolism) leads to Cushing disease or Cushing syndrome. Hyperfunction that causes increased secretion of adrenal androgens or estrogens leads to virilization or feminization. Hyperfunction that causes increased levels of aldosterone leads to hyperaldosteronism, which may be primary or secondary. These syndromes often have overlapping features. Hypofunction of the adrenal cortex leads to Addison disease.
Hypercortical Function (Cushing Syndrome, Cushing Disease)
Cushing syndrome (chronic hypercortisolism) refers to the clinical manifestations resulting from chronic exposure to excess cortisol regardless of cause. Cushing disease refers to excess endogenous secretion of ACTH.41,42ACTH-dependent hypercortisolism (about 80% of cases) results from overproduction of pituitary ACTH by a primary pituitary corticotroph adenoma (most common and can occur at any age) or by an ectopic secreting nonpituitary tumor, such as a small cell carcinoma of the lung (more common in older adults). ACTH-independent hypercortisolism is caused by cortisol secretion from a rare benign or malignant tumor of one or both adrenal glands (more common in children). A Cushing-like syndrome may develop as a side effect of long-term pharmacologic administration of glucocorticoids.
Pathophysiology
With ACTH-dependent hypercortisolism, the excess ACTH stimulates excess production of cortisol and there is loss of feedback control of ACTH secretion. In individuals with ACTH-dependent hypercortisolism, secretion of both cortisol and adrenal androgens is increased, and cortisol-releasing hormone is inhibited. In contrast, ACTH-independent secreting tumors of the adrenal cortex generally secrete only cortisol. Elevated cortisol levels suppress CRH and ACTH levels. When the secretion of cortisol by the tumor exceeds normal cortisol levels, symptoms of hypercortisolism develop.
Clinical Manifestations
Weight gain is the most common feature and results from the accumulation of adipose tissue in the trunk, facial, and cervical areas. These characteristic patterns of fat deposition have been respectively described as “truncal obesity,” “moon face,” and “buffalo hump” (Figs. 22.18 and 22.19).
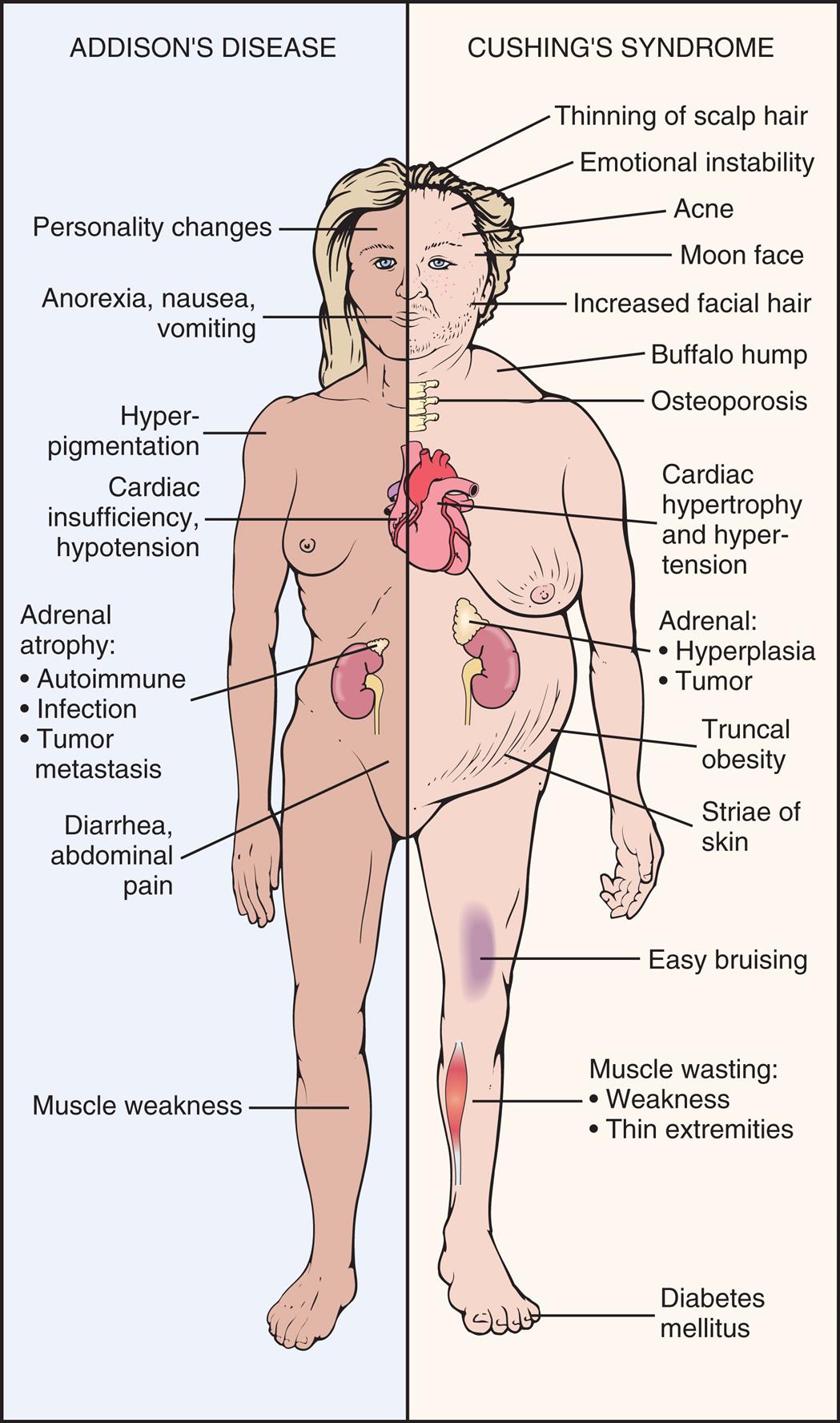
Symptoms of Addison and Cushing Diseases. (From Goodman CC, Kelly Snyder TE. Differential diagnosis for physical therapists, 5th edition. Philadelphia: Saunders; 2013.)
An illustration of the anterior view of a person identifies and labels the symptoms for Addison’s disease and Cushing’s syndrome. The symptoms of Addison’s disease are as follows. • Personality changes. • Anorexia, nausea, vomiting. • Hyperpigmentation. • Cardiac insufficiency, hypotension. • Adrenal atrophy: autoimmune, infection, and tumor metastasis. • Diarrhea, abdominal pain. • Muscle weakness. The symptoms of Cushing’s syndrome are as follows. • Thinning of scalp hair. • Emotional instability. • Acne. • Moon face. • Increased facial hair. • Buffalo hump. • Osteoporosis. • Cardiac hypertrophy and hypertension. • Adrenal: hyperplasia and tumor. • Truncal obesity. • Striae of skin. • Easy bruising. • Muscle wasting: weakness and thin extremities. • Diabetes mellitus.
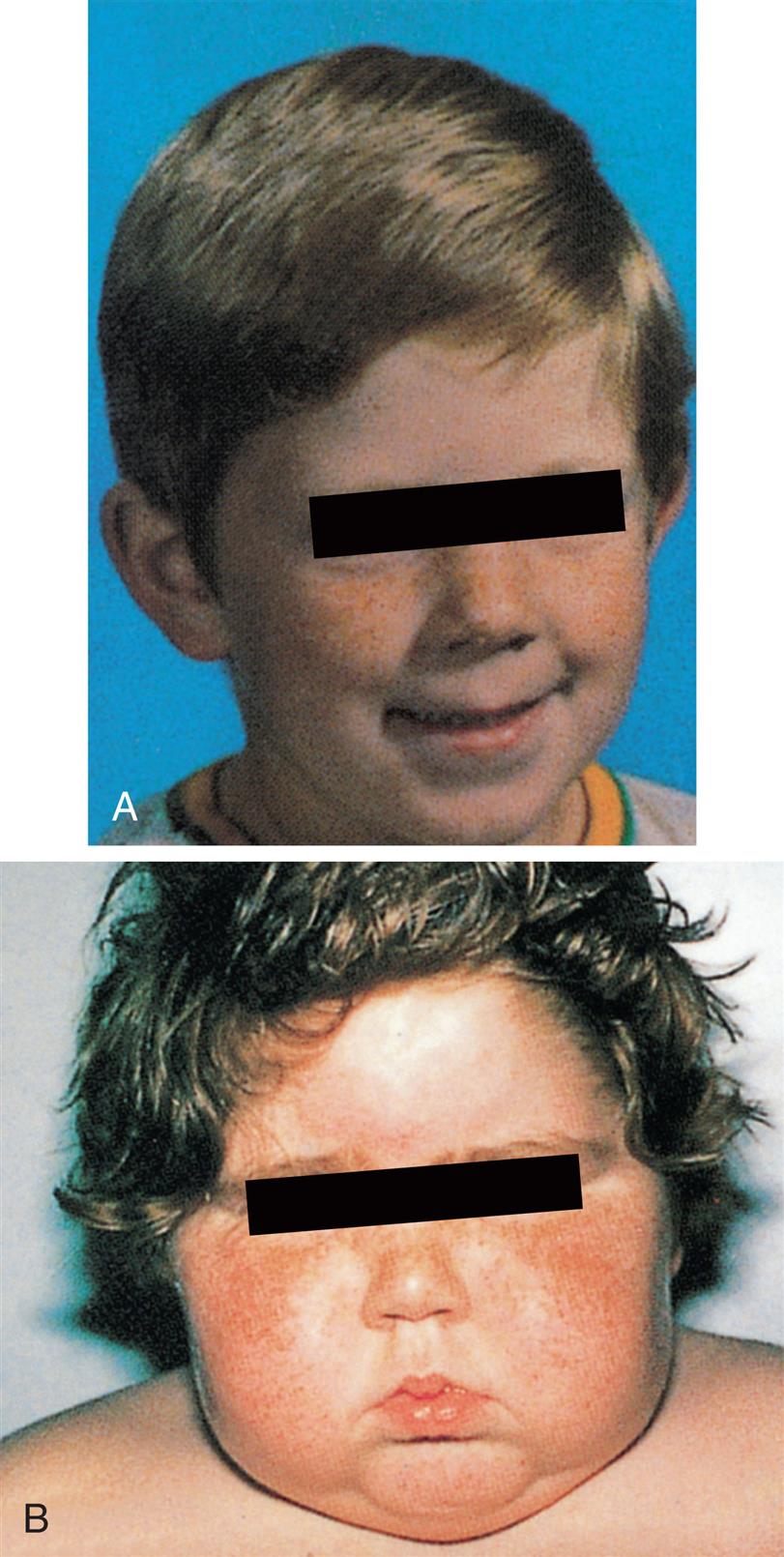
Glucose intolerance occurs because of cortisol-induced insulin resistance and increased gluconeogenesis and glycogen storage by the liver. Overt diabetes mellitus develops in approximately 20% of individuals with hypercortisolism. Polyuria is a manifestation of hyperglycemia and resultant glycosuria.
Protein wasting is caused by the catabolic effects of cortisol on peripheral tissues. Muscle wasting leads to muscle weakness. In bone, loss of the protein matrix leads to osteoporosis, with pathologic fractures, vertebral compression fractures, bone and back pain, kyphosis, and reduced height. Cortisol interferes with the action of GH in long bones; thus, children who present with short stature may be experiencing growth retardation related to Cushing syndrome rather than GH deficiency. Bone disease may contribute to hypercalciuria and resulting renal stones.
In the skin, loss of collagen leads to thin, weakened integumentary tissues through which capillaries are more visible and are easily stretched by adipose deposits. Together, these changes account for the characteristic purple striae seen in the trunk area. Loss of collagenous support around small vessels makes them susceptible to rupture, leading to easy bruising, even with minor trauma. Thin, atrophied skin is also easily damaged, leading to skin breaks and ulcerations. Bronze or brownish hyperpigmentation of the skin, mucous membranes, and hair occurs when there are very high levels of ACTH.
With elevated cortisol levels, vascular sensitivity to catecholamines increases significantly, leading to vasoconstriction and hypertension. Mineralocorticoid effects promote hypokalemia and sodium and water retention with transient weight gain. Suppression of the immune system and increased susceptibility to infections also occur. Approximately 50% of individuals with Cushing syndrome experience irritability and depression, disturbed sleep, difficulty concentrating, memory loss, and, rarely, schizophrenia-like psychosis. Females with ACTH-dependent hypercortisolism may experience symptoms of increased adrenal androgen levels (virilism), with increased hair growth (especially facial hair), acne, and oligomenorrhea. Rarely, unless an adrenal carcinoma is involved, do androgen levels become high enough to cause changes of the voice, recession of the hairline, and hypertrophy of the clitoris.
Evaluation and Treatment
Routine laboratory examinations may reveal hyperglycemia, glycosuria, hypokalemia, and metabolic alkalosis. A variety of laboratory tests are used to confirm the diagnosis of hypercortisolism and to determine the underlying disorder. These include serum and urinary cortisol levels, serum ACTH levels, and dexamethasone suppression testing. Late-evening salivary cortisol levels are used as a screening. Tumors are diagnosed using imaging procedures.
Treatment is specific for the cause of hypercorticoadrenalism and includes surgery, medication, and radiation. Differentiation between pituitary, ectopic, and adrenal causes is essential for effective treatment. Without treatment, individuals with Cushing syndrome have a high risk for developing overwhelming infection and significant complications from generalized arteriosclerosis and hypertensive disease.
Congenital Adrenal Hyperplasia
Congenital adrenal hyperplasia results from an inherited deficiency of an enzyme that is critical in cortisol biosynthesis in the adrenal gland. Because cortisol is not produced efficiently, the concentration of ACTH increases and causes adrenal hyperplasia, which results in the overproduction of mineralocorticoids or androgens, or both. The most common form is a 21-hydroxylase deficiency, which involves both mineralocorticoid and cortisol synthesis. Affected female children are virilized, and infants of both sexes exhibit salt wasting. Prenatal diagnosis is available, and treatment guidelines have been developed. Disease management requires treatment with glucocorticoids and mineralocorticoids and management of sex steroid excess.
Hyperaldosteronism
Hyperaldosteronism is characterized by excessive adrenal secretion of aldosterone. Both primary and secondary forms of hyperaldosteronism can occur.
Primary hyperaldosteronism (Conn syndrome, primary aldosteronism) is caused by excessive secretion of aldosterone from an abnormality of the adrenal cortex, usually a single benign aldosterone-producing adrenal adenoma.43,44 Bilateral adrenal nodular hyperplasia and adrenal carcinomas account for the remainder of cases.
Secondary hyperaldosteronism results from an extra-adrenal stimulus of aldosterone secretion, most often through the secretion of excess angiotensin II in response to decreased circulating blood volume (e.g., in dehydration, shock, or hypoalbuminemia) and decreased delivery of blood to the kidneys (e.g., renal artery stenosis, heart failure, or hepatic cirrhosis). Here, the activation of the renin-angiotensin system and subsequent aldosterone secretion may be seen as compensatory, although in some instances (e.g., congestive heart failure) the increased circulating volume further worsens the condition. Other causes of secondary hyperaldosteronism are Bartter syndrome, a renal tubular defect causing hypokalemia, and renin-secreting tumors of the kidney.
Pathophysiology
In primary hyperaldosteronism, pathophysiologic alterations are caused by excessive aldosterone secretion and the fluid and electrolyte imbalances that ensue. Hyperaldosteronism promotes (1) increased renal sodium and water reabsorption with corresponding hypervolemia (see Chapter 3) and hypertension and (2) renal excretion of hydrogen and potassium (see Chapter 3). The extracellular fluid volume overload, hypertension, and suppression of renin secretion are characteristic of primary disorders. Hypokalemic alkalosis, changes in myocardial conduction, and skeletal muscle weakness may be seen, particularly with severe potassium depletion.
In secondary hyperaldosteronism, renin secretion is stimulated by pressure-initiated cellular changes at the juxtaglomerular apparatus (see Chapter 37). This leads to an increase in angiotensin II and aldosterone with sodium and water retention and increased circulating blood volume.
Clinical Manifestations
Hypertension, hypokalemia, and hypervolemia are the hallmarks of primary hyperaldosteronism, and renin levels are suppressed. Hypertension is resistant to treatment and can lead to the development of left ventricular dilation and hypertrophy, vascular disease, and kidney disease. Edema is often absent.
Evaluation and Treatment
Various clinical and laboratory evaluations are useful in assessing hyperaldosteronism:
- 1. Measurement of blood pressure: hypertension is usually present.
- 2. Serum and urinary electrolyte levels: hypernatremia and metabolic alkalosis may be present.
- 3. Plasma aldosterone-to-renin ratio and aldosterone suppression testing: an increased aldosterone-to-renin ratio is present in primary hyperaldosteronism.
- 4. Imaging techniques may be used to localize an aldosterone-secreting adenoma.
Treatment includes management of hypertension, hypervolemia, and hypokalemia, most often with aldosterone receptor antagonists, such as spironolactone, eplerenone, or angiotensin-converting enzyme (ACE) inhibitors. If an aldosterone-secreting adenoma is present, it must be surgically removed.
Hypersecretion of Adrenal Androgens and Estrogens
Hypersecretion of adrenal androgens and estrogens may be caused by adrenal tumors (adenomas or carcinomas), Cushing syndrome, or defects in steroid synthesis. The clinical syndrome that is manifested depends on the hormone secreted, the sex of the individual, and the age at which the hypersecretion is initiated. Hypersecretion of estrogens causes feminization, the development of female secondary sex characteristics. Hypersecretion of androgens causes virilization, the development of male secondary sex characteristics (Fig. 22.20).
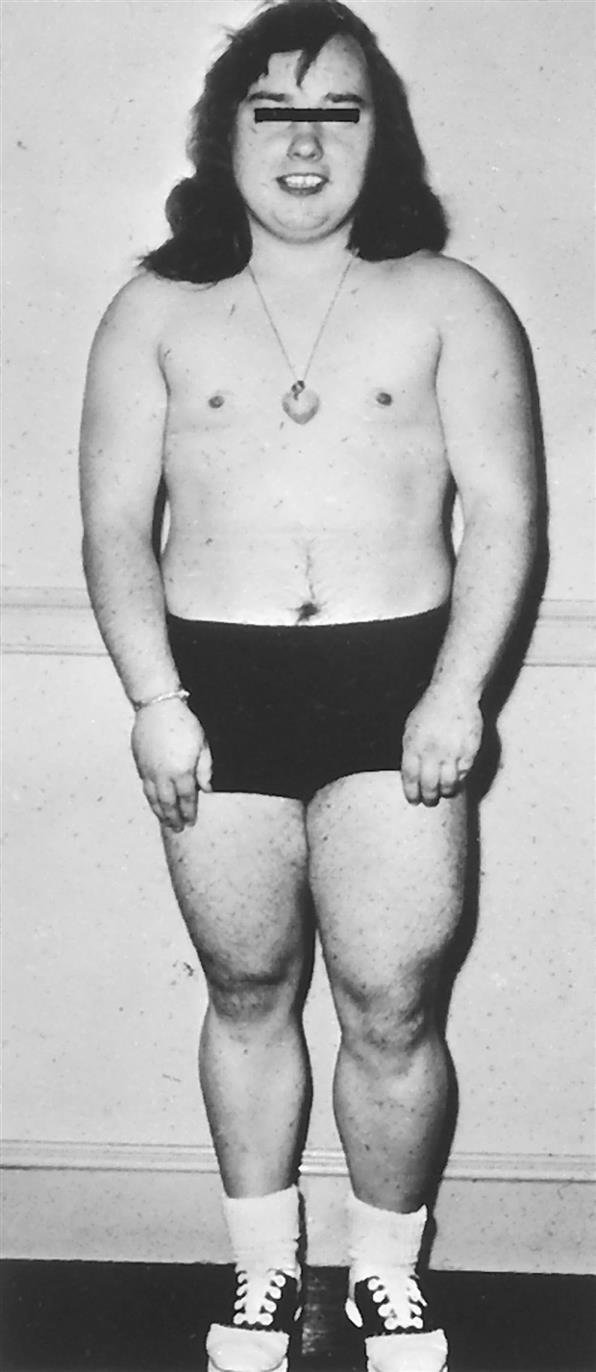
Virilization of a young girl by an androgen-secreting tumor of the adrenal cortex. Masculine features include lack of breast development, increased muscle bulk, and hirsutism (excessive hair). (From Thibodeau GA, Patton KT. The human body in health & disease, 4th edition. St. Louis: Mosby; 2010.)
The effects of an estrogen-secreting tumor are most evident in males and result in gynecomastia, testicular atrophy, and decreased libido. In female children, such tumors may lead to early development of secondary sex characteristics. The changes caused by an androgen-secreting tumor are more easily observed in females and include excessive face and body hair (hirsutism), clitoral enlargement, deepening of the voice, amenorrhea, acne, and breast atrophy. In children, virilizing tumors promote precocious sexual development and bone aging. Treatment of adrenal tumors usually involves surgical excision.
Adrenocortical Hypofunction
Hypocortisolism (low levels of cortisol secretion) develops either because of inadequate stimulation of the adrenal glands by ACTH or because of a primary inability of the adrenals to produce and secrete the adrenocortical hormones. Sometimes there is partial dysfunction of the adrenal cortex, so only synthesis of cortisol, aldosterone, or the adrenal androgens is affected.
Addison disease
Primary adrenal insufficiency is termed Addison disease. It is relatively rare, occurring most often in adults aged 30 to 60 years, although it may appear at any time. In the United States, Addison disease is most commonly caused by autoimmune mechanisms that destroy adrenal cortical cells. Chronic infections that affect the adrenal gland, such as tuberculosis, account for the majority of cases of primary adrenal insufficiency in underdeveloped countries.
Pathophysiology
Addison disease is characterized by inadequate corticosteroid and mineralocorticoid synthesis and elevated levels of serum ACTH (loss of negative feedback). Idiopathic Addison disease (organ-specific autoimmune adrenalitis) causes adrenal atrophy and hypofunction and is an organ-specific autoimmune disease resulting from autoantibodies and autoreactive T cells that attach the adrenal cortical cells. It may occur in childhood (type 1) or adulthood (type 2) and is associated with other autoimmune diseases, especially Hashimoto thyroiditis, pernicious anemia, and idiopathic hypoparathyroidism. In some cases, Addison disease may be inherited as an autosomal recessive trait (Mechanisms of inheritance are described in Chapter 4). The adrenal glands in idiopathic Addison disease are smaller than normal and may be misshapen.
Clinical Manifestations
The symptoms of Addison disease are the result of hypocortisolism and hypoaldosteronism and are often nonspecific. With mild to moderate hypocortisolism, symptoms begin with weakness and easy fatigability. Skin changes, including hyperpigmentation and vitiligo, may occur. As the condition progresses, anorexia, nausea, vomiting, and diarrhea may develop. Of greatest concern is the development of hypotension that can progress to complete vascular collapse and shock. This is known as adrenal crisis, or Addisonian crisis, and most often develops in individuals who have undiagnosed disease and experience a physiologic stress.
Evaluation and Treatment
Serum and urine levels of cortisol are depressed with primary hypocortisolism, and ACTH levels are increased. Because of dehydration, blood urea nitrogen levels may increase. The serum glucose level is low. Eosinophil and lymphocyte counts often are elevated. Hyperkalemia is seen in Addison disease and may cause mild alkalosis (see Chapter 3). The ACTH stimulation test may be used to evaluate serum cortisol levels.
The treatment of Addison disease involves lifetime glucocorticoid and mineralocorticoid replacement therapy, together with dietary modifications and correction of any underlying disorders. The individual's diet should include at least 150 mEq of sodium per day; sodium intake should be increased if the individual experiences excessive sweating or diarrhea. With acute stressors (e.g., infection, surgery, or trauma), additional cortisol must be administered.
Secondary hypocortisolism
Secondary hypocortisolism commonly results from prolonged administration of exogenous glucocorticoids; they suppress ACTH secretion and cause adrenal atrophy, resulting in inadequate corticosteroidogenesis once the exogenous glucocorticoids are withdrawn. Decreased ACTH secretion also can result from pituitary infarction, pituitary tumors that compress ACTH-secreting cells, or hypophysectomy. In all instances of low ACTH levels, adrenal atrophy occurs, and endogenous adrenal steroidogenesis is depressed. Clinical manifestations of secondary hypocortisolism are similar to those of Addison disease, although hyperpigmentation usually does not occur. The renin-angiotensin system usually is normal, so aldosterone and potassium levels also tend to be normal.
Tumors of the Adrenal Medulla
Hyperfunction of the adrenal medulla is caused by pheochromocytomas (chromaffin cell tumors) or sympathetic paragangliomas of the adrenal medulla. These are rare tumors, but 10% to 20% are malignant and may metastasize. Occurrence is usually sporadic, although nearly half are associated with genetic markers that may be inherited.
Pathophysiology
Pheochromocytomas and sympathetic paragangliomas cause excessive production of norepinephrine, although large tumors secrete both epinephrine and norepinephrine. Approximately 5% of people with these tumors have no symptoms, apparently because the tumor is nonfunctioning. Such tumors can, however, release catecholamines, especially in response to a stressor, such as surgery.
Clinical Manifestations
The clinical manifestations of a pheochromocytoma and sympathetic paragangliomas are related to the chronic effects of catecholamine secretion and include persistent hypertension, headache, pallor, diaphoresis, tachycardia, and palpitations. Hypertension results from increased peripheral vascular resistance and may be sustained or paroxysmal. An acute episode of hypertension related to hypersecretion of catecholamines may follow specific events, such as exercise, excessive ingestion of tyrosine-containing foods (aged cheese, red wine, beer, yogurt), ingestion of caffeine-containing foods, external pressure on the tumor, and induction of anesthesia. Hypertension unresponsive to drug therapy is often the first indication of a pheochromocytoma. Headaches appear because of sudden changes in catecholamine levels in the blood, affecting cerebral blood flow. Hypermetabolism and sweating are related to chronic activation of sympathetic receptors in adipocytes, hepatocytes, and other tissues. Glucose intolerance may occur because of catecholamine-induced inhibition of insulin release by the pancreas. These tumors tend to be extremely vascular and can rupture, causing massive and potentially fatal hemorrhage.
Evaluation and Treatment
Symptoms of pheochromocytoma can be insidious or intermittent and difficult to diagnose. A diagnosis is made when increased catecholamine production is found in the blood or urine. The site of the tumor is then determined using abdominal imaging techniques. Because of the possibility of metastasis, whole-body scanning may be done. Genetic testing can guide therapy.
Management of catecholamine excess is essential to prevent hypertensive emergencies and requires the use of α- and β-adrenergic blockers. The usual treatment of pheochromocytoma is surgical excision of the tumor. Medical therapy is continued to stabilize blood pressure before, during, or after surgery. Malignant pheochromocytoma is rarely curable and is usually managed by a combination of surgical debulking of the tumor combined with chemotherapy.
Summary Review
Mechanisms of Hormonal Alterations
- 1. Abnormalities in endocrine function may be caused by elevated or depressed hormone levels that result from (1) disorders within glands that cause them to synthesize abnormal amounts of hormone, (2) faulty feedback systems, (3) dysfunctional or ectopically produced hormones, or (4) defects in carrying the hormone in the bloodstream.
- 2. Target cells may fail to respond to hormones because of (1) cell surface receptor–associated disorders, or (2) intracellular disorders.
Alterations of the Hypothalamic–Pituitary System
- 1. Dysfunction in the action of hypothalamic hormones is most commonly related to interruption of the connection between the hypothalamus and pituitary—the pituitary stalk.
- 2. Disorders of the posterior pituitary include syndrome of inappropriate antidiuretic hormone (ADH) secretion (SIADH) and diabetes insipidus. SIADH is characterized by abnormally high ADH secretion; diabetes insipidus is characterized by abnormally low ADH secretion.
- 3. In SIADH, high ADH levels cause retention of excess free water, leading to hyponatremia and hypoosmolality. SIADH is caused by ectopic production of ADH by tumors, surgical procedures, pulmonary disorders, and central nervous system disorders.
- 4. Diabetes insipidus may be neurogenic, also known as central (caused by insufficient amounts of ADH), or nephrogenic (caused by an inadequate response to ADH). Low ADH results in excess free water loss leading to hypernatremia and hyperosmolality. Its principal clinical features are polyuria and polydipsia.
- 5. Diseases of the anterior pituitary include hypopituitarism, hyperpituitarism, acromegaly, and prolactinoma.
- 6. Hypopituitarism is characterized by the absence of anterior pituitary hormones or the complete failure of all anterior pituitary hormone functions. The most common causes of hypopituitarism are pituitary infarction or space-occupying lesions.
- 7. Hypopituitarism can affect any or all of the pituitary hormones and symptoms may range from mild to life-threatening, depending on the hormone affected. Adrenocorticotropic hormone (ACTH) deficiency is a potentially life-threatening disorder. Growth hormone (GH) deficiency causes increased body fat, decreased muscle mass, and psychologic problems in adults, and hypopituitary dwarfism in children.
- 8. Hyperpituitarism is caused by pituitary neuroendocrine tumors. These are usually benign, slow-growing tumors that arise from cells of the anterior pituitary.
- 9. Expansion of a pituitary neuroendocrine tumor causes both neurologic and secretory effects. Pressure from the expanding tumor causes hyposecretion of surrounding cells, dysfunction of the optic chiasma (leading to visual disturbances), dysfunction of the hypothalamus, and some cranial nerves.
- 10. Hypersecretion of GH in adults causes acromegaly and is most commonly the result of a pituitary neuroendocrine tumor. Prolonged, abnormally high levels of GH lead to proliferation of body and connective tissue and slowly developing renal, thyroid, and reproductive dysfunction.
- 11. Excessive GH secretion in children with open epiphyseal plates causes giantism.
- 12. Prolactinomas, pituitary tumors that secrete prolactin, result in galactorrhea, hirsutism, amenorrhea, hypogonadism, and osteopenia.
Alterations of Thyroid Function
- 1. Alterations in thyroid function can be primary or secondary. Primary thyroid diseases are caused by dysfunction or disease of the thyroid gland, usually the result of autoimmunity, leading to either increased or decreased thyroid hormone (TH) levels. Secondary thyroid disorders are related to disorders of pituitary gland thyroid-stimulating hormone (TSH) function.
- 2. Thyrotoxicosis is a general condition resulting from elevated TH levels. Hyperthyroidism is defined as excessive secretion of TH from the thyroid gland. Common clinical manifestations include increased metabolic rate, heat intolerance, and weight loss.
- 3. Graves disease, the most common form of hyperthyroidism, is caused by an autoimmune mechanism that stimulates the TSH receptors on the thyroid gland. It is characterized by thyrotoxicosis and circulating thyroid-stimulating immunoglobulins. The two distinguishing clinical manifestations are ophthalmopathy and dermopathy (pretibial myxedema).
- 4. Toxic nodular goiter and toxic multinodular goiter occur when there are independently functioning follicular cells that form adenomas, which produce excessive TH.
- 5. Thyrotoxic crisis is a severe form of hyperthyroidism that is often associated with physiologic stress.
- 6. Hypothyroidism is caused by deficient production of TH by the thyroid gland. Primary hypothyroidism has increased levels of TSH and is caused by autoimmune thyroiditis, loss of thyroid tissue, medications, and iodine deficiency. Secondary hypothyroidism has decreased levels of TSH and is caused by hypothalamic or pituitary dysfunction.
- 7. Symptoms of hypothyroidism depend on the degree of TH deficiency. Common manifestations include decreased energy metabolism, cold intolerance, decreased heat production, lethargy, and myxedema.
- 8. Myxedema is a sign of hypothyroidism caused by alterations in connective tissue with water-binding proteins that lead to edema and thickened mucous membranes. Myxedema coma is a severe form of hypothyroidism that may be life-threatening without emergency medical treatment.
- 9. Autoimmune thyroiditis (Hashimoto disease) is the most common cause of primary hypothyroidism in the United States and involves autoimmune destruction of the thyroid gland and gradual loss of thyroid function.
- 10. Subacute thyroiditis is a nonbacterial inflammation of the thyroid gland that is often preceded by a viral infection.
- 11. Postpartum thyroiditis generally occurs up to 6 months after giving birth. Iatrogenic hypothyroidism results from ablation of the thyroid gland during treatment for hyperthyroid conditions.
- 12. Congenital hypothyroidism is the absence of thyroid tissue during fetal development or defects in hormone synthesis and, if untreated, can lead to severe physical and cognitive disorders.
- 13. Thyroid carcinoma is associated with exposure to ionizing radiation, especially in childhood.
Alterations of Parathyroid Function
- 1. Hyperparathyroidism, which may be primary, secondary, or tertiary, is characterized by greater than normal secretion of parathyroid hormone (PTH). Hyperparathyroidism leads to neuromuscular symptoms, bone damage, and renal stones.
- 2. Primary hyperparathyroidism is caused by an interruption of the normal mechanisms that regulate calcium and PTH levels. Hallmark manifestations include hypercalcemia and hypophosphatemia.
- 3. Secondary hyperparathyroidism is a compensatory response to hypocalcemia and often occurs with chronic renal failure and vitamin D deficiency.
- 4. Tertiary hyperparathyroidism develops after long-standing hypocalcemia, especially in individuals undergoing renal dialysis or transplantation.
- 5. Hypoparathyroidism, defined by abnormally low PTH levels, is caused by thyroid surgery, autoimmunity, or genetic mechanisms.
- 6. The lack of circulating PTH in hypoparathyroidism causes hypocalcemia, hyperphosphatemia, and decreased bone resorption.
Dysfunction of the Endocrine Pancreas: Diabetes Mellitus
- 1. Diabetes mellitus is a group of metabolic diseases characterized by hyperglycemia resulting from defects in insulin secretion, insulin action, or both.
- 2. A diagnosis of diabetes mellitus is based on elevated plasma glucose concentrations and measurement of glycated hemoglobin. Classic signs and symptoms are often present as well.
- 3. Diabetes mellitus is classified as type 1, type 2, or gestational.
- 4. Diabetes mellitus type 1 is caused by autoimmune β-cell destruction, usually leading to absolute insulin deficiency. It is characterized by loss of β cells, presence of islet cell antibodies, lack of insulin, excess of glucagon, and altered metabolism of fat, protein, and carbohydrates.
- 5. In diabetes mellitus type 1, hyperglycemia causes polyuria and polydipsia resulting from osmotic diuresis. Hypoglycemia and diabetic ketoacidosis may be seen.
- 6. Diabetes mellitus type 2 is caused by progressive loss of β-cell insulin secretion frequently on the background of insulin resistance. Genetic susceptibility is triggered by environmental factors. The most compelling environmental risk factor for type 2 diabetes is obesity.
- 7. In the obese, many factors, including metabolic syndrome, altered adipokines, increased fatty acids, inflammation, and hyperinsulinemia, contribute to the development of insulin resistance and hyperglycemia.
- 8. Early in the course of diabetes mellitus type 2, hyperinsulinemia occurs in order to overcome underlying insulin resistance. Over time, however, the weight and number of β cells decrease, and insulin levels decline. There also are decreased levels of amylin, ghrelin, and incretins and glucagon concentration is increased. All contribute to chronic hyperglycemia.
- 9. Gestational diabetes is glucose intolerance diagnosed during the second or third trimester of pregnancy.
- 10. The category other specific types of diabetes include monogenetic forms of diabetes called maturity-onset diabetes of youth (MODY).
- 11. Acute complications of diabetes mellitus include hypoglycemia, diabetic ketoacidosis, and hyperosmolar hyperglycemic nonketotic syndrome.
- 12. Hypoglycemia in diabetes is a complication related to insulin treatment.
- 13. Diabetic ketoacidosis (DKA) develops when there is an absolute or relative deficiency of insulin and an increase in the insulin counterregulatory hormones of catecholamines—cortisol, glucagon, and growth hormone. DKA presents with hyperglycemia, acidosis, and ketonuria.
- 14. Hyperosmolar hyperglycemic nonketotic syndrome is pathophysiologically similar to DKA, although a lack of ketosis resulting in acidosis indicates some level of insulin action. Severe dehydration and electrolyte imbalance are present.
- 15. Chronic complications of diabetes mellitus include microvascular disease (e.g., neuropathy, retinopathy, nephropathy), macrovascular disease (e.g., coronary artery disease, stroke, peripheral vascular disease), and infection.
- 16. Microvascular disease is characterized by obstruction of capillaries and decreased tissue perfusion.
- 17. Macrovascular disease associated with diabetes mellitus is most often related to the proliferation of atherosclerotic plaques in the arterial wall.
- 18. The incidence of coronary heart disease, peripheral vascular disease, and stroke is greater in those with diabetes than in nondiabetic individuals.
- 19. Individuals with diabetes are at risk for a variety of infections. Infection may be related to sensory impairment and resulting injury, hypoxia, increased proliferation of pathogens in elevated concentrations of glucose, decreased blood supply associated with vascular damage, and impaired immune protection.
Alterations of Adrenal Function
- 1. Disorders of the adrenal cortex are related to hyperfunction (secreting too much hormone) or hypofunction (secreting too little hormone).
- 2. Increased secretion of cortisol (hypercortisolism) is usually caused by Cushing disease (pituitary-dependent) and very rarely can be caused by ectopic production of ACTH. Complications include obesity, diabetes, protein wasting, immune suppression, and mental status changes.
- 3. Congenital adrenal hyperplasia is a genetic disorder with overproduction of mineralocorticoids or androgens, or both.
- 4. Excessive aldosterone secretion causes hyperaldosteronism, which may be primary or secondary. Primary hyperaldosteronism is caused by an abnormality of the adrenal cortex. Secondary hyperaldosteronism involves renin and angiotensin secretion in response to renal underperfusion.
- 5. Hyperaldosteronism promotes increased sodium reabsorption (with corresponding hypervolemia), increased extracellular volume (which is variable), hypokalemia related to renal reabsorption of sodium, and excretion of potassium.
- 6. Hypersecretion of adrenal androgens and estrogens can be a result of adrenal tumors, either adenomas or carcinomas; Cushing syndrome; or defects in steroid synthesis. Hypersecretion of estrogens causes feminization, the development of female secondary sexual characteristics. Hypersecretion of androgens causes virilization, the development of male secondary sexual characteristics.
- 7. Hypocortisolism, or low levels of cortisol, is caused secondarily by inadequate adrenal stimulation by ACTH or because of primary adrenal insufficiency, termed Addison disease.
- 8. Addison disease is characterized by elevated ACTH levels with inadequate corticosteroid and mineralocorticoid synthesis.
- 9. Manifestations of Addison disease are related to hypocortisolism and hypoaldosteronism. Symptoms include weakness, fatigability, hyperpigmentation, vitiligo, anorexia, vomiting, and diarrhea. The development of hypotension can progress to complete vascular collapse and shock, known as adrenal crisis, or Addisonian crisis.
- 10. Secondary hypocortisolism commonly results from prolonged administration of exogenous glucocorticoids that suppress ACTH secretion. Decreased ACTH secretion also can result from pituitary infarction, pituitary tumors, or hypophysectomy. In all instances of low ACTH levels, adrenal atrophy occurs, and endogenous adrenal steroidogenesis is depressed.
- 11. Hyperfunction of the adrenal medulla is usually caused by a pheochromocytoma, a catecholamine-producing tumor. Symptoms of catecholamine excess include hypertension, palpitations, tachycardia, glucose intolerance, excessive sweating, and constipation.