Obesity, Starvation, and Anorexia of Aging
Jodi A. Allen and Julia L. Rogers
 http://evolve.elsevier.com/Rogers/pathophysiology/
http://evolve.elsevier.com/Rogers/pathophysiology/
Obesity is one of the most common and costly chronic diseases in the world. The causes are complex, multifactorial, and associated with an increased risk of many comorbid diseases. Alternatively, starvation and anorexia of aging also are common conditions. This chapter presents an overview of the function of adipose tissue and the pathophysiology of obesity, starvation, and anorexia of aging.
Adipose Tissue
Adipose tissue provides insulation and mechanical support, secretes hormone like signaling molecules known as adipokines, and contributes to immune cell function. It is the body's major energy reserve to fuel other tissues. Adipocytes are fat-storing cells that store calories in the form of triglycerides (triglycerol), synthesize triglycerides from glucose, and mobilize energy in the form of free fatty acids (FFAs) and glycerol. Adipose tissue is classified according to color as white adipose tissue (WAT), brown adipose tissue (BAT), and beige adipose tissue (bAT). These tissue types are found in different locations within the body and have different functions.1 Most adipose tissue in the body is WAT.
White adipocytes, which are derived from connective tissue, are located in visceral (central) and subcutaneous (peripheral) stores of WAT. WAT also is found in muscle, providing mechanical protection and sliding of muscle bundles, as well as in bone marrow. WAT contains various cells, including macrophages, mast cells, neutrophils, fibroblasts, endothelial cells, blood vessels, nerves, and precursor adipocytes. White adipocytes contain a single triglyceride fat droplet or vacuole. A low nutritional state, stimulation of the beta adrenergic sympathetic nervous system and release of catecholamines (epinephrine and norepinephrine), activates lipolysis in WAT to release FFAs and glycerol into the circulation. FFAs and glycerol can then be used for energy metabolism.
Subcutaneous or peripheral adipose tissue expands by increased adipocyte size (hypertrophy) and number (hyperplasia).2 The alterations in precursor cell commitment and subcutaneous adipose tissue adipogenesis are associated with metabolic complications of obesity. Adipogenesis is the formation of new fat cells from preadipocytes. These adipocytes are smaller and have a greater fat storage capacity. Subcutaneous fat has higher leptin production, lower adiponectin production, lower production of inflammatory cytokines, and lower association with insulin resistance. Expansion of subcutaneous fat is a healthier expansion of fat tissue and is associated with fewer complications of obesity.
Visceral adipose tissue is located in the abdomen and surrounding intra-abdominal organs. Visceral WAT is more likely to store fat by adipocyte hypertrophy. Visceral adipocytes store fat as triglycerides, primarily in the form of very-low-density lipoprotein (VLDL), derived from hepatic and dietary sources. Visceral fat is more hormonally active than subcutaneous fat and releases leptin and inflammatory mediators. Excess visceral fat is associated with impaired lipid and glucose metabolism, insulin resistance, metabolic syndrome, and an increased risk of cardiovascular disease and cancer.1 Thus, the complications of obesity are related to where fat is stored, not just the accumulation of fat stores.
Estrogen and estrogen receptors have a role in fat metabolism. They enhance the deposition of WAT in the subcutaneous tissue and inhibit it in visceral tissue. This may explain the higher incidence of peripheral obesity among premenopausal women and the increase in central obesity with menopause and in men.3
Bone marrow adipose tissue (MAT) is found in all bones, and it increases with obesity and age in long bones. With obesity, MAT releases adipokines that affect osteoblast and osteoclast function and alters hematopoiesis. In general, excessive MAT is associated with osteoporosis and fractures.4
Brown adipocytes form BAT and are derived from muscle tissue and have multiple lipid droplets. They are rich in mitochondria that contain iron, which gives them a brown color. Exposure to cold, activation of the sympathetic nervous system, catecholamines, and activation of triiodothyronine (T3) stimulate BAT to generate heat rapidly through the oxidation of FFAs, acids, and glucose. This is known as nonshivering thermogenesis; it occurs at a rate 50-fold greater than in WAT and protects against obesity and metabolic syndrome.5 Estrogen-related receptors also participate in nonshivering thermogenesis in BAT.6 Neonates generate body heat from BAT primarily located in the interscapular and perirenal regions. It traditionally was thought that BAT did not persist into adult life. However, positron emission tomography (PET) scanning has shown that adults also have BAT. It is most common in lean individuals, usually located in the neck, supraclavicular, axillary, paravertebral, and mediastinal regions.7
There is an inverse relationship between the amount of BAT and both body mass index (BMI) and age. Interindividual differences in BAT-mediated thermogenesis may explain some of the variability in obesity susceptibility and the increased prevalence of obesity with aging. Variation in BAT and bAT also may be factors in the natural regulation of weight reduction.
Located within WAT, particularly in subcutaneous fat stores, are beige—or “brite” (from “brown in white”)—adipocytes that form bAT. Beige adipocytes are a subpopulation of white adipocytes that also contain multiple mitochondria but not in the numbers associated with BAT. Beige adipocytes emerge within WAT with chronic exposure to cold and exercise, produce heat, and increase energy expenditure. This is known as the beiging or browning of WAT. The bAT disappear with elevated ambient temperatures and with warm adaptation revert to WAT.8 Leptin and insulin together promote bAT, increasing energy expenditure and weight loss. bAT is diminished in obesity. Because the beiging (browning) of adipose tissue protects against obesity and metabolic syndrome, efforts are in progress to discover whether there is a therapeutic way to stimulate the synthesis and activity of BAT and bAT as an approach to preventing or treating obesity and diabetes mellitus.9
Adipose Tissue as an Endocrine Organ
Adipose tissue is an endocrine organ, and adipocytes secrete adipokines. Adipokines are cell-signaling proteins that function like hormones, having autocrine, paracrine, and endocrine actions. Adipokines include all the biologically active substances synthesized by WAT. They are necessary for numerous functions in body tissues.10 These substances function in the regulation of appetite, food intake and energy expenditure, lipid storage, insulin secretion and sensitivity, immune and inflammatory responses, coagulation, fibrinolysis, angiogenesis, fertility, vascular homeostasis, blood pressure regulation, and bone metabolism. Excess WAT causes dysregulation of the secretion and function of adipokines, contributing to the many complications of obesity.
Adipokines are important to understand because they are targets of both experimental and currently available drugs and weight loss programs used to treat obesity and its complications. Examples of adipokines are provided as a reference in Box 23.1 (a summary of adipokines and their role in caloric intake, energy metabolism, and obesity is presented later in this chapter). The regulation of food intake and energy balance is summarized next.
Regulation of Food Intake and Energy Balance
Regulation of food intake and energy balance is a complex process controlled by central and peripheral physiological signals.11 Centrally, the arcuate nucleus (ARC) in the hypothalamus regulates food intake and energy metabolism by balancing the opposing effects of two sets of neurons. One set of neurons promotes appetite, stimulates eating, and decreases metabolism (anabolic). These are known as orexigenic neurons, which are stimulated by molecules called orexins. Another set of neurons suppresses appetite, inhibits eating, and increases metabolism. These are known as anorexigenic neurons, which are stimulated by molecules called anorexins. The hypothalamic orexin and anorexin signaling pathways are transmitted through the autonomic nervous and endocrine systems to regulate and balance appetite, food intake, energy metabolism, and body temperature. The hypothalamus also communicates with higher brain centers related to reward, pleasure, memory, and addictive behavior. These higher centers can override hypothalamic control of food intake and satiety, which increases consumption of highly palatable foods and results in increased fat stores.12 Peripherally, the gastrointestinal tract secretes a number of hormones (Box 23.2) that also control hunger and satiety. In addition, adipokines can function as orexins or anorexins and provide peripheral signals for the control of food intake and energy expenditure. They are described in the Adipokines and Obesity section later in the chapter.
Obesity
Obesity is an increase in body adipose tissue and an endocrine and metabolic disorder that has become epidemic worldwide. Obesity is defined differently in adults and children. In adults, it is a BMI that exceeds 30 kg/m2. In children, it is a BMI greater than or equal to the age- and sex-specific 95th percentile of the growth charts published in 2000 by the Centers for Disease Control and Prevention (CDC).13
Obesity develops when caloric intake exceeds caloric expenditure in genetically susceptible individuals. Between 2017 and 2018, the prevalence of obesity among U.S. adults was 42.4% and 19.3% among children and adolescents between ages 2 and 19 years.14,15 Children tend to become obese adults. Ethnic differences also are seen in the rates of obesity: Hispanics (44.8%), non-Hispanic blacks (49.6%), non-Hispanic whites (42.2%), and non-Hispanic Asians (17.4%).15
Obesity is the fifth leading cause of death globally and accounts for high health care costs worldwide.16 Three leading causes of death in the United States are associated with obesity: cardiovascular disease, type 2 diabetes mellitus, and cancer (liver, advanced prostate, ovarian, gallbladder, kidney, colorectal, esophageal, postmenopausal breast, pancreatic, endometrial, and stomach).17 Obesity is also a risk factor for hypertension, stroke, hyperlipidemia, gallstones, nonalcoholic steatohepatitis (NASH), gastroesophageal reflux, hiatal hernia, osteoarthritis, infectious disease, asthma, obstructive sleep apnea, and chronic kidney disease.18 However, some studies have shown that mild obesity in older individuals is associated with lower mortality (the obesity paradox), particularly in individuals with hypertension and coronary heart disease, but the mechanisms are not clear.19 The causes and consequences of obesity are multiple and complex, and rapidly advancing research is underway on the causal mechanisms, complications, and treatment.
Genotype and gene–environment interactions are important predisposing factors. Single-gene defects (monogenic defects) are rare, and obesity is usually polygenic and associated with other phenotypes, such as endocrine disorders (e.g., diabetes mellitus and hypothyroidism) and intellectual disability (e.g., Down and Prader-Willi syndromes).20 Metabolic abnormalities that contribute to obesity include Cushing syndrome, Cushing disease, polycystic ovary syndrome, growth hormone (GH) deficiency, hypothyroidism, and hypothalamic injury. Contributing environmental factors include food intake (low nutrient, energy-dense foods), physical inactivity, obesogens, and socioeconomic status (both high and low income). Obesity also is associated with adverse social and psychological consequences, including depression and mood disorders.21
.
Pathophysiology
The pathophysiology of obesity is complex and involves the interaction of peripheral and central neuroendocrine pathways, numerous adipokines, hormones, and neurotransmitters (Fig. 23.1).22,23 The adipocyte is the cellular basis of obesity. Excess fat is stored in mature white adipocytes when energy balance is positive (excess caloric intake in relation to energy expenditure). These adipocytes undergo hypertrophy and adipogenesis (hyperplasia), store triglycerol, and secrete adipokines. Adipokines circulate in the blood at concentrations that increase or decrease in relation to body fat mass and provide signals to the central nervous system for regulation of hunger, satiety, and energy balance, as described previously. WAT accumulation causes dysfunction in the regulation and interaction of this signaling system and contributes to the complications and consequences of obesity.24
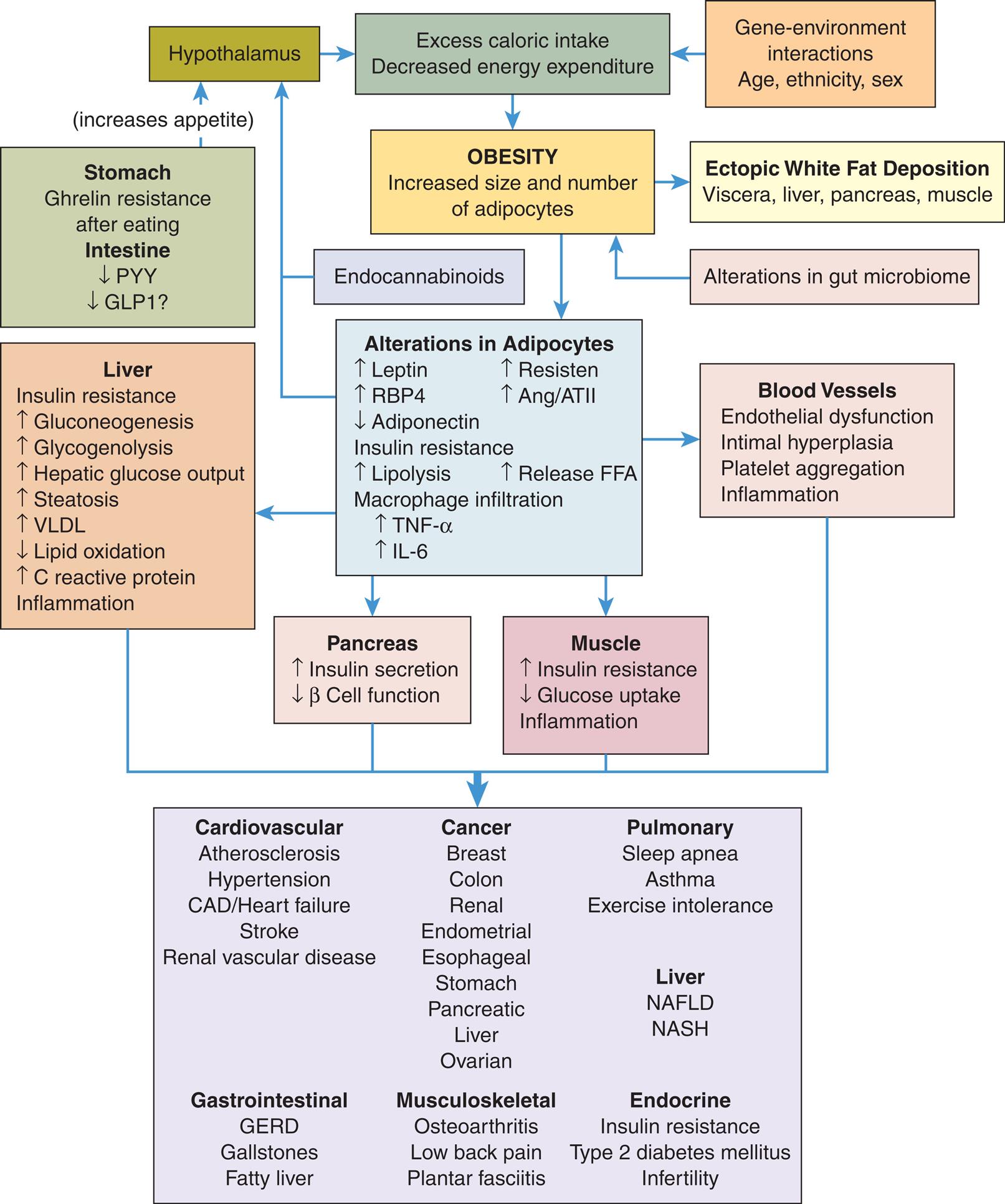
See text for details. Ang/ATII, Angiotensinogen/angiotensin 2; CAD, coronary artery disease; FFA, free fatty acids; GERD, gastroesophageal reflux disease; GLP1, glucagon-like peptide 1; IL-6, interleukin-6; PYY, intestinal peptide YY; RBP4, retinol-binding protein 4; TNF-α, tumor necrosis factor-alpha; VLDL, very-low-density lipoprotein. NAFLD, nonalcoholic fatty liver disease and NASH, nonalcoholic steatohepatitis.
“A flowchart represents the pathophysiology and common complications of obesity. 1. Hypothalamus. Leads to 3. 2. Gene-environment interactions. Age, ethnicity, sex. Leads to 3. 3. Excess caloric intake. Decreased energy expenditure. Leads to 4. 4. Obesity: increased size and number of adipocytes. Leads to 5 and 7. 5. Ectopic white fat deposition. Viscera, liver, pancreas, muscle. 6. Alterations in gut microbiome. Leads to 4. 7. Alterations in adipocytes. Increased leptin, increased R B P 4, decreased adiponectin, increased resisten, and increased Ang or A T I I. Insulin resistance: increase lipolysis and increased release F F A. Macrophage infiltration: increased T N F-alpha and I L-6. Leads to 8, 9, 10, and 11. 8. Pancreas. Increased insulin secretion; decreased beta cell function. Leads 12. 9. Muscle. Increased insulin resistance; decreased glucose uptake; inflammation. Leads 12. 10. Blood vessels. Endothelial dysfunction; intimal hyperplasia; platelet aggregation; inflammation. Leads 12. 11. Liver. Insulin resistance: increased gluconeogenesis, increased glycogenolysis, increased hepatic glucose output, increased steatosis, increased V L D L, decreased lipid oxidation, increased C reactive protein; inflammation. Leads 12. 12. Cardiovascular (atherosclerosis, hypertension, C A D or heart failure, stroke, and renal vascular disease); cancer (breast, colon, renal, endometrial, esophageal, stomach, pancreatic, liver, and ovarian); pulmonary (sleep apnea, asthma, exercise intolerance); liver (N A F L D, N A S H); gastrointestinal (G E R D, gallstones, fatty liver); musculoskeletal (osteoarthritis, low back pain, and plantar fasciitis); endocrine (insulin resistance, type 2 diabetes mellitus, infertility). 13. Stomach. Ghrelin resistance after eating. Intestine: decreased P Y Y, decreased G L P 1. Leads to 14. 14. Increases appetite. Leads to 1. 15. Endocannabinoids. Leads to 1.”
Adipokines and Obesity
Leptin is a product of the obesity gene (Ob gene) and is expressed primarily by adipocytes. Leptin levels increase after eating and act on the hypothalamus to inhibit orexigenic neurons and stimulate anorexigenic neurons to suppress appetite and increase energy expenditure. At low leptin levels (i.e., during fasting), leptin stimulates food intake and reduces energy expenditure. This balance regulates body weight and energy expenditure within a fairly narrow range. Leptin levels increase as the number of adipocytes increases. However, high leptin levels are ineffective at decreasing appetite and energy expenditure, a condition associated with obesity and known as central leptin resistance (Fig. 23.2).
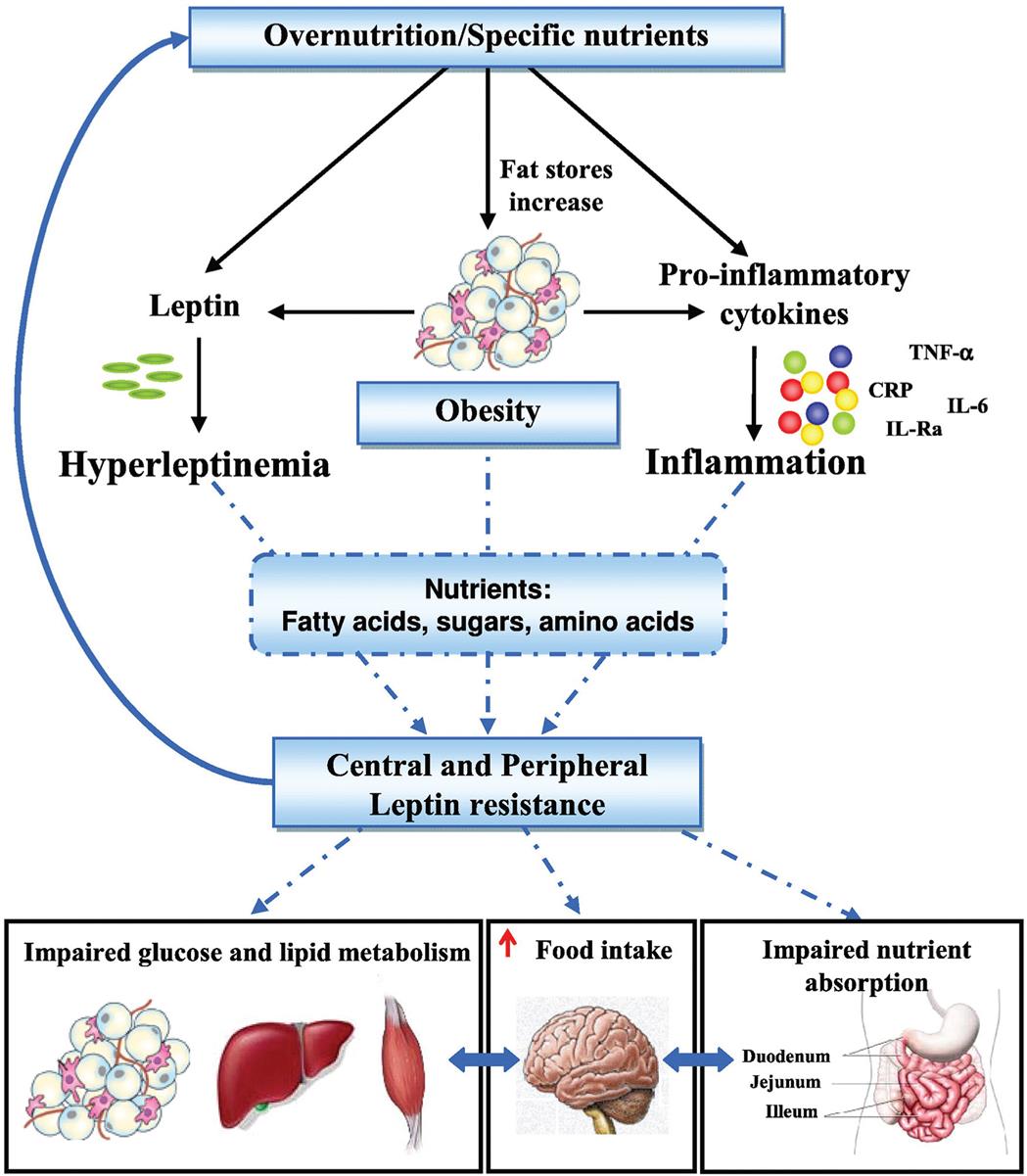
IL, Interleukin; TNF-α, tumor necrosis factor-alpha; CRP, C-reactive protein. (From Sáinz N, Barrenetxe J, Moreno-Aliaga MJ, et al. Leptin resistance and diet-induced obesity: Central and peripheral actions of leptin. Metabolism, 2015;64(1):35–46. https://doi.org/10.1016/j.metabol.2014.10.015)
“A flowchart represents pathways to leptin resistance and obesity. 1. Overnutrition or specific nutrients. Leads 2, 3, and 4. 2. Leptin. Leads to 5. 3. Fat stores increase. Leads to 6. 4. Pro-inflammatory cytokines (T N F-alpha, C R P, I L-6, I L-R a). Leads to 7. 5. Hyperleptinemia. Leads to 8. 6. Obesity. Leads to 8. 7. Inflammation. Leads to 8. 8. Nutrients: fatty acids, sugars, amino acids. 9. Central and peripheral; leptin resistance. Leads to 1, 10, 11, and 12. 10. Impaired glucose and lipid metabolism. Interrelated to 11. 11. Increased food intake. Interrelated to 12. 12. Impaired nutrient absorption (duodenum, jejunum, and ileum).”
Leptin resistance fails to inhibit orexigenic hypothalamic satiety signaling and promotes overeating and excessive weight gain. Leptin also regulates hepatic gluconeogenesis, insulin sensitivity, and glucose and lipid metabolism in liver, muscle, and adipose tissue. Peripheral leptin resistance (i.e., in muscle and adipose tissue) results in hyperglycemia, hyperinsulinemia, and hyperlipidemia and also stimulates macrophages and endothelial cells to produce proinflammatory mediators. The cause of leptin resistance is unknown. It may be related to a defect in leptin transport, an inability of leptin to cross the blood–brain barrier, an alteration in the permissive effect of leptin, or a defect in or suppression of the leptin receptor. The low-grade inflammation that accompanies obesity also is thought to contribute to leptin resistance. Chronic hyperleptinemia also stimulates the sympathetic nervous system, oxidative stress, chronic low-grade inflammation, and ventricular hypertrophy and contributes to the pathogenesis of hypertension, atherosclerosis, cardiovascular disease, type 2 diabetes, and cancer associated with obesity.25,26
Adiponectin, which is produced primarily by visceral adipose tissue but also by cardiomyocytes and skeletal muscle, increases energy expenditure. It also has insulin-sensitizing and anti-inflammatory properties. Plasma levels of adiponectin decrease with visceral obesity, and resistance to adiponectin action develops. Decreased adiponectin levels are associated with increased hepatic gluconeogenesis, insulin resistance, decreased skeletal muscle glucose uptake, and increased levels of inflammatory mediators, such as interleukin-6 (IL-6) and tumor necrosis factor-alpha (TNF-α). Adiponectin serves as an anti-inflammatory and antiatherogenic plasma protein; it also has an important role in vascular remodeling, and it is cardioprotective. Decreased levels of adiponectin are associated with type 2 diabetes mellitus and an increased risk for coronary artery disease resulting from hyperlipidemia, hypertension, and factors that promote thrombosis and inflammation.27
Decreased beta cell function and insulin resistance are associated with obesity. The mechanisms are not clear, but an association exists between hyperlipidemia and increased fat storage, macrophages and inflammation, and alterations in adipokines. Leptin resistance and decreased adiponectin also contribute to insulin resistance. Insulin resistance results in hyperinsulinemia, hyperglycemia, and a predisposition to type 2 diabetes mellitus. Retinol-binding protein 4 (binds vitamin A) is an adipokine produced both in the liver and by adipocytes. It is increased in visceral adiposity and contributes to inflammation and insulin resistance in the liver and muscles; it also is associated with cardiovascular disease and may contribute to hepatic steatosis (fatty liver).28
Endocannabinoids (i.e., anandamide) are arachidonic acid derivatives (unsaturated, essential fatty acids) expressed in both the brain and peripheral nerve tissues. They have effects on endocannabinoid (CB) receptors in orexigenic pathways. They increase appetite, enhance nutrient absorption, stimulate lipogenesis, and increase WAT accumulation by acting at both central (CB1 receptor) and peripheral sites (CB2 receptor). They also inhibit energy expenditure and thermogenesis. An increase in endocannabinoids is proposed to be associated with visceral obesity.29 Angiotensinogen (AGT) is produced in the liver and by adipocytes and is increased in obesity. AGT is the precursor to angiotensin 1 (AGTI), which is then converted to angiotensin 2 (AGTII). The effects of AGTII include vasoconstriction, renal retention of sodium and water, and release of aldosterone. Increased AGTII from adipose tissue also promotes lipogenesis, oxidative stress, inflammation, and insulin resistance. All of these effects contribute to the complications associated with obesity.30 Gastrointestinal hormones also play a role in the complex pathophysiology of obesity (see Box 23.2). The most significant ones are reviewed next.
Ghrelin is produced by the stomach gastric mucosa. Ghrelin stimulates food intake and fat storage and prevents life-threatening hypoglycemia by inhibiting insulin secretion and stimulating glucagon secretion. It increases in response to fasting and chronic caloric restriction and decreases after food intake. Ghrelin is thought to have antilipolytic effects and stimulates lipogenesis in visceral WAT, leading to an increase in body weight and body fat mass. Ghrelin also stimulates the release of GH from anterior pituitary cells, the release of gastric acid, gastrointestinal motility, and pancreatic secretion of insulin. It has satiety, vasodilatory, and cardioprotective effects. An elevation in FFAs and GH after eating normally decreases the release of ghrelin. However, obesity is associated with a decreased plasma level of ghrelin, and plasma ghrelin levels do not fall after eating. This is known as ghrelin resistance. The mechanisms for this response are not clear, and the role of ghrelin in obesity has yet to be clearly defined.31
Glucagon-like peptide 1 (GLP-1) is an anorexigenic hormone secreted by intestinal endocrine cells when nutrients enter the small intestine. GLP-1 stimulates pancreatic glucose-dependent insulin secretion, decreases blood glucose levels, delays gastric emptying, suppresses appetite, increases satiety, and increases energy expenditure. GLP-1 levels may be decreased in obese individuals, and a GLP-1 receptor analogue has been approved to treat both obesity and type 2 diabetes mellitus.32
Peptide YY (PYY) is released from intestinal endocrine cells in response to nutrients entering the intestine. PPY inhibits gastric motility and mucosal secretion and increases satiety and decreases appetite by acting on the medullary brain stem. PYY decreases with obesity.33
Cholecystokinin (CCK) is secreted by proximal small intestinal cells after food intake. Its actions include gallbladder contraction, release of pancreatic enzymes and insulin, satiation, and reduced food intake. Work is in progress to develop type 1 CCK receptor (CCK1R) agonists for the treatment of obesity.34
Lipotoxicity
Chronic positive energy balance and obesity can overwhelm fat storage by adipocytes, resulting in altered lipid metabolism and insulin resistance. Normally, insulin can inhibit lipolysis by activation of insulin receptors in adipocytes. With obesity, adipocytes are resistant to insulin inhibition of lipolysis. With a chronic increased energy intake, the increase in adipocyte size and number in visceral WAT exceeds the supporting vascular supply, resulting in hypoxia and inflamed and fibrotic adipose tissues. Excess lipolysis and FFAs are distributed to nonadipose cells (e.g., kidney, liver, heart, skeletal muscle). When their utilization capacity is exceeded, cellular dysfunction or death occurs, and this is known as lipotoxicity.35
Obesity and Inflammation
Obesity produces a state of chronic, low-grade inflammation in WAT. The enlarged adipose cell size results in apoptosis, local hypoxia, cell and mechanical stress, and promotes inflammation. Proinflammatory macrophages, lymphocytes, neutrophils, and mast cells infiltrate enlarged adipocytes and release inflammatory cytokines (e.g., TNF-α, and IL-6) (see Box 23.1).36 The inflammatory state is supported by increased leptin, decreased adiponectin, and increased resistin (a hormone released by macrophages that promotes insulin resistance and inflammation). The inflammatory state, immune dysregulation, accelerated lipolysis, and lipotoxicity contribute to the development of insulin resistance, metabolic syndrome (see Chapter 22 and Box 22.2), and the complications of obesity, including type 2 diabetes mellitus, cardiovascular disease, kidney disease, NASH, and cancer.2
Obesity and the Gut Microbiome
Changes in the intestinal microbiome also are associated with and are a contributing cause of obesity, although the mechanisms are not clear. Microbes (mostly bacteria) are found in high concentration in the lower gastrointestinal tract, and the bacterial composition is affected by genetics, diet, geographical location, use of antibiotics and other medications, and energy balance. These bacteria have considerable variability among individuals and participate in the breakdown of complex carbohydrates, nutrient absorption, vitamin synthesis, inflammatory responses, gut permeability, and bile acid metabolism. Gut microbial fermentation of dietary fiber produces short chain fatty acids (acetate, butyrate, and propionate), which function as energy sources and signaling molecules that affect the host's energy metabolism and inflammation.
More studies are needed to determine how changes in microbiota contribute to body weight regulation, metabolism, low-grade inflammation, and increased adiposity and how manipulation of the gut microbiota can assist in preventing or treating obesity.37
.
Clinical Manifestations
Obesity usually presents with different forms or phenotypes of adipose tissue distribution and/or extent of metabolic abnormalities.38 Visceral obesity (also known as intra-abdominal, central, or masculine obesity) occurs when the distribution of body fat is localized around the abdomen and upper body, resulting in an apple shape. Visceral obesity is associated with accelerated lipolysis and has an increased risk for chronic systemic inflammation, metabolic syndrome, obstructive sleep apnea syndrome, type 2 diabetes mellitus, cardiovascular complications, osteoarthritis, and cancer. Visceral venous blood drains into the portal vein, contributing to higher liver synthesis of plasma lipids and increasing the risk of nonalcoholic fatty liver disease (NAFLD) and NASH.39
Peripheral obesity (also known as subcutaneous, gluteal-femoral, or feminine obesity) occurs when the distribution of body fat is extraperitoneal and distributed around the thighs and buttocks and through the muscle, resulting in a pear shape. It is more common in premenopausal women. Peripheral and subcutaneous fat is less metabolically active and lipolytic and releases fewer adipokines (particularly adiponectin) than visceral fat. Risk factors are still present for the complications of obesity, but they are less severe than for visceral obesity.40
Obesity has been described in relation to the extent of metabolic abnormalities that are present. However, there is no universally accepted definition or guidelines to describe metabolic health for the phenotypes or the potentially transient nature of a metabolically healthy status. Depending on the phenotype, there are associated differences in adipose tissue distribution, metabolism, lipid profiles, and gut microbiota. There are modifiable lifestyle factors (diet, exercise, smoking, alcohol use, psychological stress, geographical location), and unmodifiable factors (age, sex, ethnicity, and genetics) that also play a role, all of which add to the complexity of a uniform definition. Continued development of clarity and consensus regarding criteria and parameter guidelines will facilitate research designs and personalized treatment decisions. Box 23.3 presents a summary of the proposed phenotype characteristics.
Evaluation and Treatment
All children and adults should be screened for obesity. Several methods are available for estimating or measuring the amount of adipose tissue: anthropometric measurements, including weight, height, and circumferences or various body diameters (i.e., waist-to-hip ratios and waist circumference); skinfold thickness (measured with skinfold calipers); ultrasound to measure peripheral body fat; and bioelectric impedance and underwater hydrostatic weighing to calculate total body fat. The only method for directly measuring total body fat is by dual-energy x-ray absorptiometry (DXA) scanning, an enhanced type of x-ray imaging.
In clinical practice, anthropometric and body diameter measures are most commonly used to calculate the BMI because they are the easiest to measure and most cost-effective. Body mass indices have been established based on height, weight, age, sex, and ethnicity. Overweight is defined as a BMI greater than 25 kg/m2, and obesity is a BMI greater than 30 kg/m2. BMI charts are available for children ages 2 to 20 years; these can be used for comparison during adulthood, because obese children generally become obese adults.
However, the BMI does not measure the amount and location of body fat. The waist circumference (more than 40 inches [102 cm] for men and more than 35 inches [88 cm] for women) adds information to assist with disease risk assessment in general practice. Obesity risk assessment is available from the American Association of Clinical Endocrinologists and the American College of Endocrinology.41 No specific diagnostic criteria for obesity have been established.
Obesity is a chronic disease for which various treatment approaches have been used, including correction of metabolic abnormalities and individually tailored lifestyle interventions, such as weight reduction diets and exercise programs. Additional treatments include psychotherapy, behavioral modification, self-motivation, and support systems.42,43 Several drugs have been approved for the pharmacologic management of obesity.44
Currently, bariatric surgical procedures (i.e., the Roux-en-Y gastric bypass, adjustable gastric banding, and sleeve gastrectomy) offer the most significant reduction in weight, reduction in comorbidities, and decrease in insulin resistance for the treatment of obesity.45 Efforts are continuing to identify the molecular and neuroendocrine causes of obesity. This will lead to more specific and personalized prevention and treatment strategies (see Emerging Science Box: SGLT-2 Use for the Treatment of Obesity).
Starvation
Malnutrition is lack of nourishment from inadequate amounts of calories, protein, vitamins, or minerals and is caused by an improper diet, alterations in digestion or absorption, chronic disease, or a combination of these factors. Starvation is a reduction in energy intake related to inadequate food sources that leads to weight loss. Short-term starvation and long-term starvation have different effects. Therapeutic short-term starvation is part of many weight reduction programs because it causes an initial rapid weight loss that reinforces the individual's motivation to diet (see Emerging Science Box: Weight Loss for more information). Therapeutic long-term starvation is used in medically controlled environments to facilitate rapid weight loss in morbidly obese individuals. Pathologic long-term starvation can be caused by poverty; food shortages; chronic diseases of the cardiovascular, pulmonary, hepatic, and digestive systems; malabsorption syndromes; human immunodeficiency virus (HIV) infection; cancer; and anorexia nervosa.46
Short-term starvation, or extended fasting, consists of several days of total dietary abstinence or deprivation. The body responds with mechanisms to protect protein mass. For 4 to 6 hours after the last meal, the body is in a well-fed state and its energy requirements are supplied by glucose from recently ingested carbohydrates. Once all available energy has been absorbed from the intestine, glycogen in the liver is converted to glucose through glycogenolysis—the splitting of glycogen into glucose. This process peaks within 4 to 8 hours, and gluconeogenesis begins. Gluconeogenesis is the formation of glucose from noncarbohydrate molecules: lactate, pyruvate, amino acids, and the glycerol portion of fats from lipolysis. Like glycogenolysis, gluconeogenesis takes place within the liver. Both of these processes deplete stored nutrients and, thus, cannot meet the body's energy needs indefinitely. Lipolysis and proteins continue to be catabolized to a minimal degree, providing carbon for the synthesis of glucose.47
Long-term starvation begins after several days of dietary abstinence and eventually causes death from proteolysis. Absolute deprivation of food (e.g., that associated with famine) causes marasmus, or protein-energy malnutrition (loss of muscle mass, body fat depletion, and absence of edema). Protein deprivation in the presence of carbohydrate intake is called kwashiorkor (loss of muscle mass with sustained body fat and peripheral edema). Marasmic kwashiorkor (edematous, severe childhood malnutrition) is a combination of chronic energy deficiency and chronic or acute protein deficiency and inadequate micronutrients.48,49 These conditions are described in Chapter 42.
Anorexia is loss of appetite. Anorexia nervosa is a psychological cause of long-term starvation (Box 23.4). Cachexia (also known as cytokine-induced malnutrition) is skeletal muscle wasting, causing muscle atrophy with involuntary loss of weight leading to, fatigue, and weakness. Muscle atrophy is a result of negative protein and energy balance secondary to reduced food intake and abnormal metabolism.50 Systemic inflammation caused by inflammatory mediators (e.g., TNF-α, interferon-γ, IL-1, IL-6, and IL-8), an increased catabolic response, and metabolic abnormalities are associated with the cachexia.51 Cancer, congestive heart failure, chronic obstructive pulmonary disease, acquired immunodeficiency syndrome (AIDS), rheumatoid arthritis, tuberculosis, and other major chronic progressive proinflammatory diseases are contributable pathophysiological processes. While individuals may experience one or the other, anorexia and cachexia often occur together.51 (Fig. 23.3).
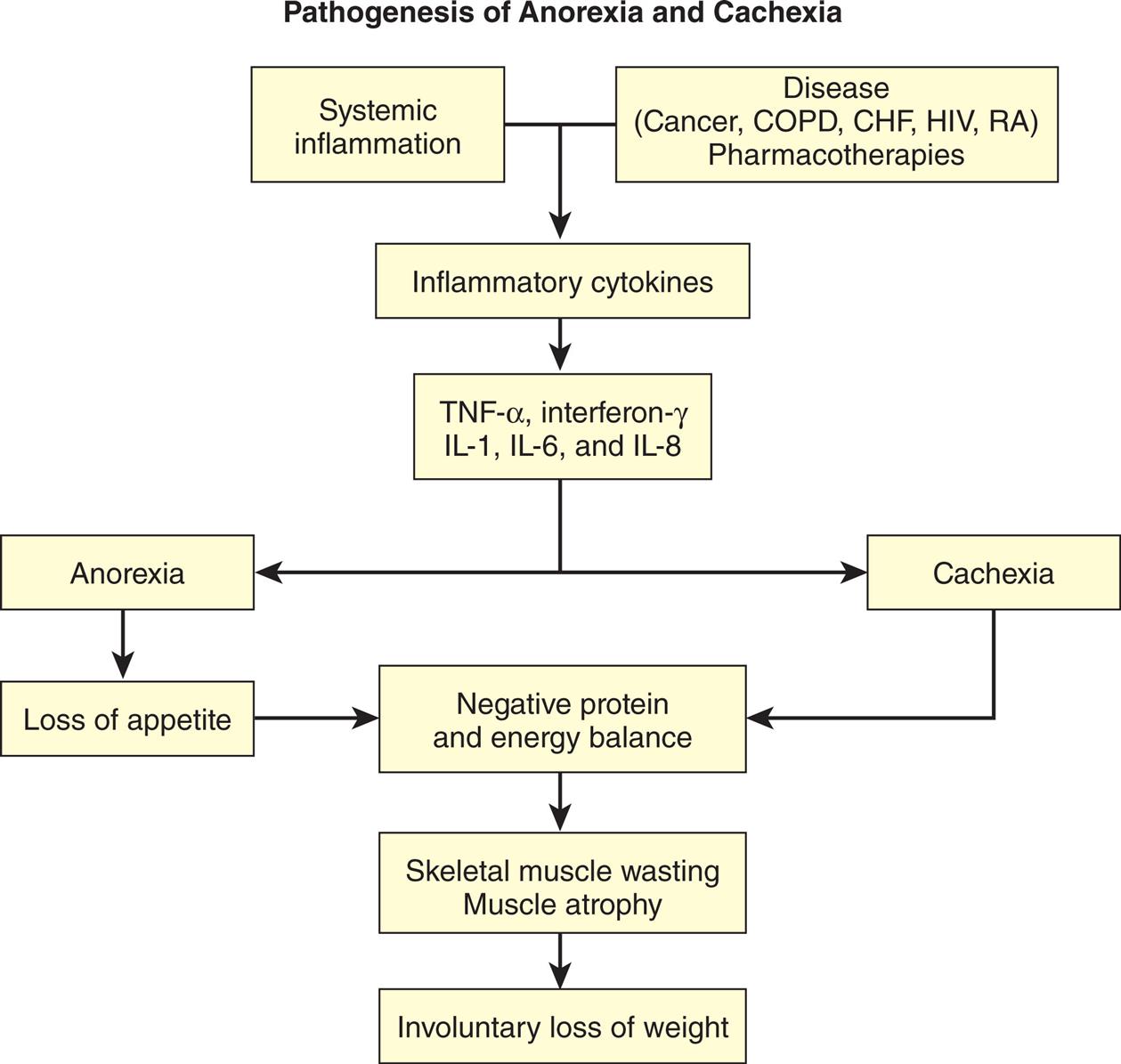
“A flow chart represents the pathogenesis of anorexia and cachexia. 1. Systemic inflammation. Leads to 3. 2. Disease (cancer, C O P D, C H F, H I V, R A); pharmacotherapies. Leads to 3. 3. Inflammatory cytokines. Leads to 4. 4. T N Y-alpha, interferon-gamma, I L-1, I L-6, and I L-8. Leads to 5 and 6. 5. Anorexia. Leads to 7. 6. Cachexia. Leads to 8. 7. Loss of appetite. Leads to 8. 8. Negative protein and energy balance. Leads to 9. 9. Skeletal muscle wasting; muscle atrophy. Leads to 10. 10. Involuntary loss of weight.”
The major metabolic characteristic of long-term starvation is a decreased dependence on gluconeogenesis and an increased use of ketone bodies (products of lipid and pyruvate metabolism) as a cellular energy source. During long-term starvation, depressed insulin levels and increased levels of glucagon, cortisone, epinephrine, and GHs promote lipolysis in adipose tissue. Lipolysis liberates fatty acids—which supply energy to cardiac and skeletal muscle cells—and ketone bodies—which sustain brain tissue. Fatty acid, or ketone body, oxidation meets most of the energy needs of the cells. (Some glucose is still needed as fuel for brain tissue and red blood cells.) Once the supply of adipose tissue is depleted, proteolysis begins. The breakdown of muscle and visceral protein is the last process the body engages in to supply energy for life. Death results from severe alterations in electrolyte balance and loss of renal, pulmonary, and cardiac function.52
Adequate ingestion of appropriate nutrients is the obvious treatment for starvation. In medically induced starvation, the body is maintained in a ketotic state until the desired amount of adipose tissue has been lysed. Starvation imposed by chronic disease, long-term illness, malabsorption syndromes, and chronic eating disorders is treated with enteral or parenteral nutrition (Box 23.5). Perioperative or critical care management of nutrition is necessary to prevent unnecessary starvation.53 Care must be taken to prevent refeeding syndrome (Box 23.6) during the treatment of long-term starvation.
Complications of Anorexia Nervosa and Bulimia
| Anorexia Nervosa Complications | |
|---|---|
| Cardiac | |
| Bradycardia | |
| Dysrhythmia | |
| Left ventricular atrophy | |
| Mitral valve prolapse | |
| Pericardial effusion | |
| Sudden cardiac death | |
| Dermatological | |
| Acrocyanosis | |
| Hair thinning | |
| Xerosis | |
| Endocrine | |
| Amenorrhea | |
| Estrogen decreased in females and testosterone decreased in males | |
| Euthyroid sick syndrome | |
| Hypoglycemia | |
| Hypogonadal related to reduced pulsed hypothalamic gonadotropin releasing hormone secretion | |
| Hypothyroidism: cold intolerance | |
| Low libido | |
| Preterm birth | |
| Thermoregulation impaired | |
| Gastrointestinal | |
| Abdominal bloating | |
| Abdominal pain | |
| Constipation related to slowed peristalsis | |
| Dysphagia related to weakened oropharyngeal muscles | |
| Early Satiety | |
| Gastroparesis | |
| Liver function test abnormalities related to malnutrition; elevated transaminases | |
| Pelvic floor dysfunction | |
| Postprandial fullness | |
| Hematological | |
| Anemia | |
| Leukopenia | |
| Thrombocytopenia | |
| Metabolic | |
| Electrolyte Imbalances (hypokalemia; hypophosphatemia; hypomagnesemia; hyponatremia) | |
| Musculoskeletal | |
| Osteoporosis: decreased bone mineral density | |
| Stress fracture risk from decreased bone formation and increased bone resorption | |
| Neurological | |
| Brain atrophy | |
| Cognitive function impairment | |
| Taste and smell abnormalities | |
| Bulimia Complications | |
| Self-Induced Vomiting | |
| Dyspepsia | |
| Dysphagia | |
| Electrolyte and acid base disorders (hypokalemia; metabolic alkalosis) | |
| Gastric reflux | |
| Hoarse voice | |
| Parotid gland enlargement | |
| Tooth erosion | |
| Vocal cord inflammation | |
| Laxative Abuse | |
| Diarrhea | |
| Electrolyte and acid base disorders (hypokalemia; hyperphosphatemia; metabolic alkalosis) | |
| Hemorrhoids | |
| Rectal Prolapse | |


Data from Donaldson AA, Gordon CM. Skeletal complications of eating disorders. Metabolism, 2015;64(9):943–951; Sangvai D. Eating disorders in the primary care setting. Primary Care, 2016;43(2):301–312; Sato Y, Fukudo S. Gastrointestinal symptoms and disorders in patients with eating disorders. Clinical Journal of Gastroenterology, 2015;8(5):255–263; Westmoreland P, Krantz MJ, Mehler PS. Medical complications of anorexia nervosa and bulimia. American Journal of Medicine, 2016;129(1):30–37; Cass K, McGuire C, Bjork I, et al. Medical complications of anorexia nervosa. Psychosomatics, 2020;61(6):625–631; Gibson D, Watters A, Cost J, et al. Extreme anorexia nervosa: Medical findings, outcomes, and inferences from a retrospective cohort. Journal of Eating Disorders, 2020;8(1):1–10.
Anorexia of Aging
Anorexia of aging describes a decrease in appetite or food intake in older adults. It can occur in illness-free individuals and with an adequate food supply. The resulting undernutrition leads to adverse outcomes and may affect up to 30% of independently living elders with higher incidence occurring in hospital and extended care nursing faciltities.54 The anorexia of aging results from multiple age-related changes, including reduced energy needs, waning hunger, diminished senses of smell and taste, decreased production of saliva, altered gastrointestinal satiety control mechanisms, and the presence of comorbidities (Fig. 23.4). The neuroendocrine system physiologically regulates appetite with multiple hormones implicated as mediators of hunger and satiety.54
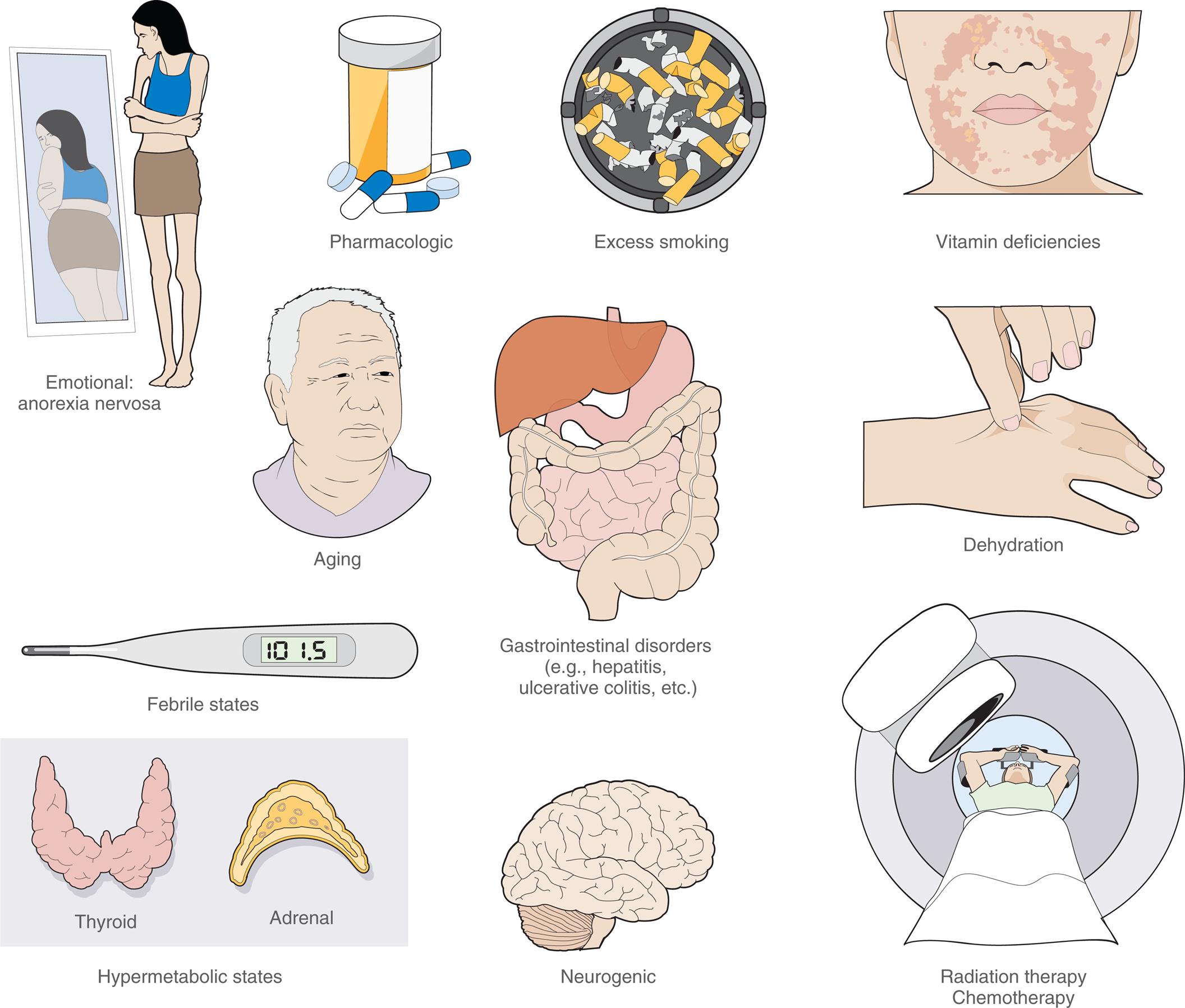
“A series of illustration shows the causes of anorexia. • Emotional: anorexia nervosa. The illustration shows a thin woman looking at a fat reflection of herself in the mirror. • Pharmacologic. The illustration shows a bottle of pills. • Excess smoking. The illustration shows a lot of cigarette buts in an ash tray. • Vitamin deficiencies. The illustration shows rashes on the jaws of a person. • Aging. The illustration shows the bust of an elderly person. • Febrile states. The illustration shows a thermometer registering a temperature of 101.5. • Gastrointestinal disorders (example, hepatitis, ulcerative colitis, et cetera) The illustration shows the digestive system. • Dehydration. The illustration shows a hand pinching the back of another hand. • Hypermetabolic states. The illustration shows the thyroid and the adrenal glands. • Neurogenic. The illustration shows a lateral view of the brain. • Radiation therapy, chemotherapy. The illustration shows a person underdoing a radiation treatment.”
Centrally, aging is associated with decreased orexigenic signals (e.g., levels of ghrelin or ghrelin resistance and reduced hypothalamic receptors, or both) and increased anorexigenic signals (e.g., decreased levels of leptin, insulin, PYY, and CCK), which lead to loss of appetite and diminished food intake (see Box 23.2). Chronic low-grade inflammation with elevated cytokines also can contribute to delayed gastric emptying and decreased motility of the small intestine. Risk factors for the anorexia of aging include functional impairments and deficiencies (e.g., loss of vision, poor dentition, dysphagia, inability to prepare foods); medical and psychiatric conditions (e.g., malabsorption syndromes, chronic disease, cancer, and depression); loneliness and grief; medications, including polypharmacy; social isolation; and abuse or neglect.55 Progression of the underlying causes of anorexia (e.g., chronic obstructive pulmonary disease, congestive heart failure) increases the workload of breathing, which decreases energy reserves required to eat and digest food. The consequences of anorexia of aging include malnutrition, physical frailty, mitochondrial dysfunction, reduced regenerative capacity, increased oxidative stress, and imbalanced hormones. There are several tools available to screen for anorexia of aging in elderly individual. Early intervention improves the overall well-being of older individuals experiencing anorexia. Currently, no specific treatments exist for the anorexia of aging, although exercise, nutrition, and other supportive strategies are used. Exercise is a key strategy in treating elderly individuals with anorexia. Exercise improves appetite and oral intake, but has other beneficial effects, such as elevating mood and building muscle and strength. Nutrition also plays a vital role. It is recommended that older adults aged 65 years and above maintain a dietary protein intake of 0.9 to 1.2 g/kg/day and individuals with acute or chronic diseases should intake 1.2 to 1.5 g/kg/day.56 Other supportive interventions to help mitigate the effects of anorexia of the aging include improved food access and appearance, dental and eye care, and social stimulation. Pharmaceutical agents (e.g., megestrol acetate and dronabinol) are not recommended in elderly persons because the risk does not outweigh the benefit. Anorexia of aging is common; however, it should not be accepted as a normal consequence of aging. Mortality rates have been shown to be higher in those with anorexia of aging and unintentional weight loss.57
Summary Review
Adipose Tissue
- 1. Adipose tissue provides insulation and tissue support and is the body's major energy reserve, storing and releasing triglycerides and glycerol.
- 2. Adipose tissue is classified according to color: WAT, BAT, and bAT.
- 3. WAT contains macrophages, mast cells, neutrophils, fibroblasts, endothelial cells, blood vessels, nerves, and precursor adipocytes.
- 4. WAT is the largest fat depot and is located in visceral (central) and subcutaneous (peripheral) sites. It also is located in muscle and bone marrow.
- 5. White adipocytes store fat as a single lipid droplet or vacuole.
- 6. With a positive energy balance, WAT storage increases by adipocyte hypertrophy (more common in visceral fat) and adipogenesis (hyperplasia, more common in subcutaneous fat).
- 7. Estrogen enhances the deposition of subcutaneous fat compared to visceral fat.
- 8. BAT has multiple lipid droplets that are rich in mitochondria containing iron, which gives them a brown color. Exposure to cold, sympathetic activation and release of catecholamines, and activation of T3 generates heat through free fatty acid oxidation (nonshivering thermogenesis).
- 9. Both neonates and adults have BAT, but not in the amounts of WAT.
- 10. bAT emerges within WAT with exposure to cold and exercise. This is known as the beiging of WAT.
- 11. BAT and bAT both protect against obesity and metabolic syndrome.
- 12. MAT is found in all bones. With obesity, MAT releases adipokines that affect osteoblast and osteoclast function. Excessive MAT is associated with osteoporosis and fractures.
- 13. Adipose tissue is an endocrine organ that secretes hormones, called adipokines, with autocrine, paracrine, and endocrine actions necessary for metabolic function and immune responses.
- 14. Regulation of food intake and energy balance are accomplished by balancing opposing sets of neurons in the arcuate nucleus: orexigenic neurons (promote appetite, stimulate eating, and decrease metabolism) and anorexigenic neurons (suppress appetite, inhibit eating, and increase metabolism). Peripherally, gastrointestinal hormones and adipokines also control food intake and energy expenditure.
- 15. Brain centers related to reward, pleasure, memory, and addictive behavior can override hypothalamic control of food intake and satiety, causing increased fat stores by increasing consumption of highly palatable foods.
Obesity
- 1. Obesity is an increase in body adipose tissue and an endocrine and metabolic disorder that develops when caloric intake exceeds energy expenditure. Obesity is defined as a BMI greater than 30 kg/m2 in adults and a BMI greater than or equal to the age- and sex-specific 95th percentile of the 2000 CDC growth charts in children.
- 2. Obesity is an epidemic that has occurred worldwide in both adults and children. It is the fifth leading cause of death globally. Three leading causes of death in the United States are associated with obesity: cardiovascular disease, type 2 diabetes mellitus, and certain cancers. Obesity also increases the risk for numerous other systemic disorders.
- 3. Single-gene (rare) and polygenic disorders and metabolic disorders are associated with obesity, as are gene–environment interactions.
- 4. Adipokines and gastrointestinal hormones are altered with obesity and contribute to associated complications.
- 5. Leptin resistance occurs when leptin levels increase with obesity and promote overeating and excessive weight gain, hyperglycemia, hyperinsulinemia, and hyperlipidemia. It also stimulates macrophages and endothelial cells to produce proinflammatory mediators.
- 6. Adiponectin levels decrease with obesity, contributing to insulin resistance, inflammation, and hyperlipidemia.
- 7. Retinol-binding protein 4 levels are increased in visceral adiposity and promote insulin resistance and hepatic steatosis.
- 8. Endocannabinoids are increased in obesity and promote appetite, enhance nutrient absorption, stimulate lipogenesis, and inhibit energy expenditure.
- 9. Angiotensinogen and angiotensin 2 increase in obesity, promoting vasoconstriction, inflammation, lipogenesis, oxidative stress, and insulin resistance.
- 10. Gastrointestinal hormones (ghrelin, GLP-1, PYY, and CCK) also provide signals that control food intake and energy expenditure and are involved in the pathophysiology of obesity.
- 11. Ghrelin increases with obesity and stimulates food intake, promotes the release of growth hormone, and stimulates lipogenesis. It also has satiety, vasodilatory, cardioprotective, and antiproliferative effects; its role in obesity is not clear.
- 12. GLP-1 promotes insulin secretion, delays gastric emptying, suppresses appetite, increases satiety, increases energy expenditure, and is decreased with obesity.
- 13. PYY inhibits gastric motility, decreases appetite, and is decreased with obesity.
- 14. CCK is released after food intake and stimulates insulin secretion and satiation, decreases food intake, and is reduced in obesity.
- 15. Lipotoxicity occurs with excess adipocyte fat storage and lipolysis with distribution of free fatty acids to peripheral organs, resulting in cellular dysfunction and death.
- 16. Obesity is a state of chronic low-grade inflammation caused by expansion of adipocyte macrophages, neutrophils, lymphocytes, and mast cells, which release inflammatory mediators.
- 17. The chronic inflammation, alterations in adipokine action, and accelerated lipolysis related to excessive fat contribute to the complications of obesity, particularly insulin resistance, type 2 diabetes mellitus, cardiovascular disease, and cancer.
- 18. Changes in the intestinal microbiome also are associated with and are a contributing cause of obesity, although the mechanisms are not clear.
- 19. Obesity has two major phenotypes: visceral obesity (also known as intra-abdominal, central, or masculine obesity) and peripheral obesity (also known as subcutaneous, gluteal-femoral, or feminine obesity). Visceral obesity has the greatest risk for accelerated lipolysis, chronic inflammation, insulin resistance, and associated complications.
- 20. Normal-weight obesity describes individuals with a normal body weight and BMI but with greater than 30% body fat. These individuals are at risk for metabolic dysregulation, increases in inflammatory cytokines, insulin resistance, increased risk for cardiovascular disease, and higher mortality.
- 21. Treatment of obesity may include correction of metabolic abnormalities and individually tailored lifestyle interventions (diets, exercise, behavioral modifications, self-motivation) and psychotherapy. The current most effective treatment for extreme obesity is bariatric surgery.
- 22. New drugs are being developed that target specific molecules and will provide a personalized approach to treatment.
Starvation
- 1. The body responds to short-term starvation (several days of total dietary abstinence or deprivation) with mechanisms to protect protein mass, using the processes of glycogenolysis and gluconeogenesis. Neither of these processes can meet the body's energy needs indefinitely because they deplete stored nutrients.
- 2. Long-term starvation (begins after several days of dietary abstinence) results in an initial decreased dependence on gluconeogenesis and an increased use of ketone bodies as a cellular energy source, followed by lipolysis in adipose tissue. In the absence of adequate nutrition, long-term starvation results in proteolysis, with death resulting from severe alterations in electrolyte balance and loss of renal, pulmonary, and cardiac function.
- 3. Anorexia is a loss of appetite, whereas anorexia nervosa is a psychological cause of long-term starvation.
- 4. Cachexia is involuntary weight loss with associated muscle wasting and atrophy. The atrophy is due to negative protein and energy balance. Cachexia can be caused by chronic progressive proinflammatory disease processes.
- 5. The major metabolic characteristic of long-term starvation is a decreased dependence on gluconeogenesis and an increased use of ketone bodies (products of lipid and pyruvate metabolism) as a cellular energy source.
- 6. Treatment is management of nutrition through parenteral nutrition; however, it is important to prevent refeeding syndrome.
Anorexia of Aging
- 1. Anorexia of aging is a decrease in appetite or food intake in older adults that leads to undernutrition, resulting in a decline in function and an increased risk for morbidity and mortality.
- 2. Contributing factors related to aging include diminished sensory functions, poor dentition, decreased gastric emptying, decreased hunger and satiety, effects of medications, and social isolation and neglect.
- 3. Consequences of anorexia of aging include malnutrition, physical frailty, mitochondrial dysfunction, reduced regenerative capacity, increased oxidative stress, and imbalanced hormones.
- 4. No specific treatments exist for the anorexia of aging, although exercise, nutrition, and other supportive strategies are used.