Structure and Function of the Hematologic System
Karen C. Turner
 http://evolve.elsevier.com/Rogers/pathophysiology/
http://evolve.elsevier.com/Rogers/pathophysiology/
All the body’s tissues and organs require oxygen and nutrients to survive. These essential needs are provided by the blood that circulates through miles of vessels found throughout the human body. The red blood cells provide the oxygen and the fluid portion of the blood carries the nutrients. The blood cleans discarded waste from the tissues; transports hormones; moves white blood cells (WBCs), platelets, and other components necessary to protect the body from injury and infection and initiate healing; and provides thermal regulation to maintain organs and tissues within an acceptable range of temperatures.
Components of the Hematologic System
Composition of Blood
Blood consists of various cells that circulate suspended in a solution of protein and inorganic materials (plasma), which is approximately 91% water and 9% dissolved substances (solutes) (Fig. 28.1). The blood volume amounts to about 6 quarts (5.5 L) in adults. The continuous movement of blood guarantees that critical components are available to all parts of the body to carry out their chief functions: (1) delivery of substances needed for cellular metabolism in the tissues, (2) removal of the wastes of cellular metabolism, (3) defense against invading microorganisms and injury, and (4) maintenance of acid–base balance.
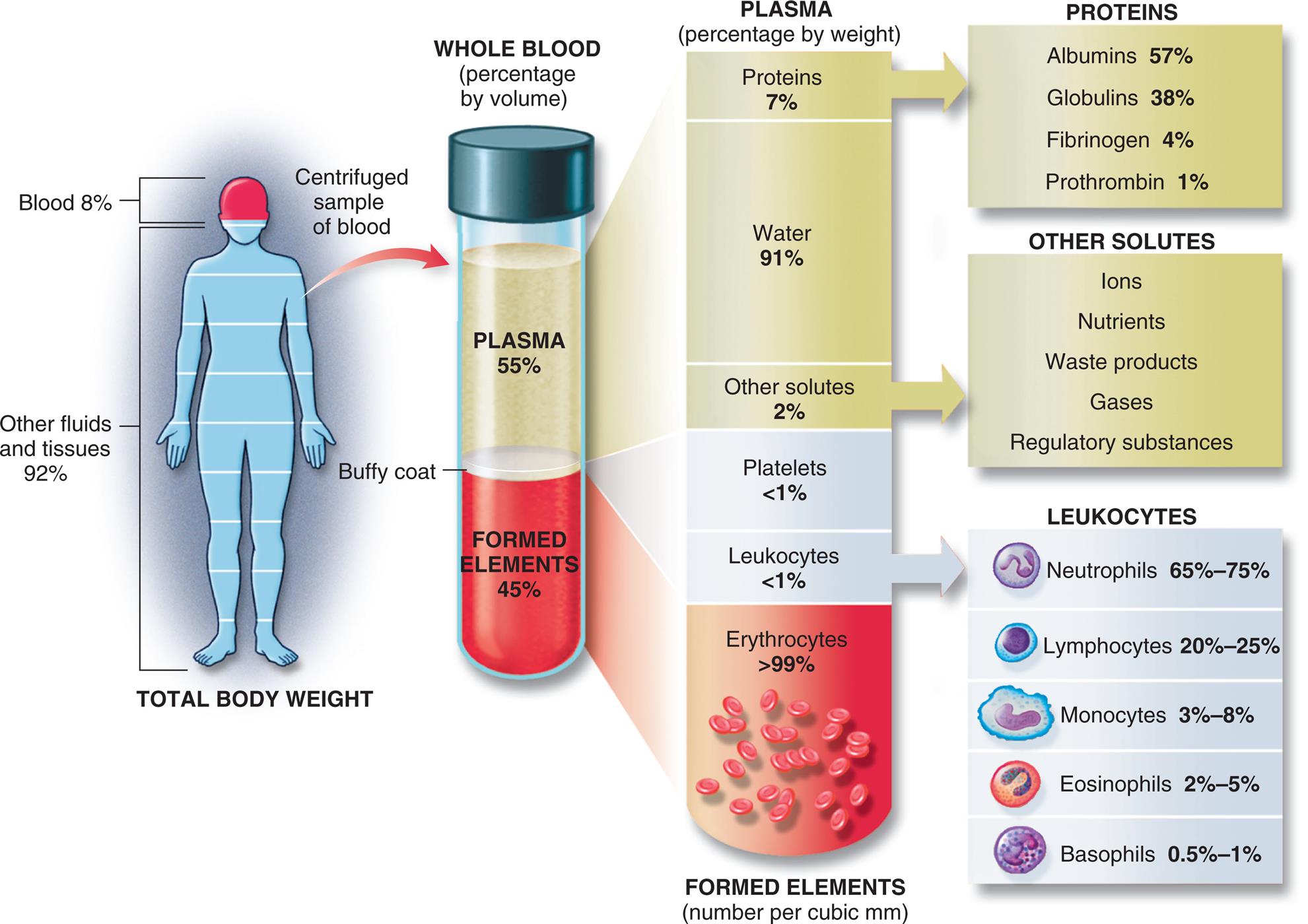
Approximate values for the components of blood in a normal adult. (From Patton KT, Thibodeau GA. The human body in health & disease, 7th edition. St. Louis: Mosby; 2018.)
An illustration shows the composition of the whole blood. The total body weight has the following composition. • Blood, 8 percent. • Other fluids and tissues, 92 percent. The whole blood (percentage by volume) has the following composition. • Plasma, 55 percent. • Buffy coat. • Formed elements, 45 percent. The plasma (percentage by weight) has the following composition: • Proteins (plasma), 7 percent. • Water (plasma), 91 percent. • Other solutes (plasma), 2 percent. • Platelets (buffy coat), less than 1 percent. • Leukocytes, (buffy coat), less than 1 percent. • Erythrocytes (formed elements), greater than 99 percent, in number per cubic millimeters. The composition of proteins in plasma is as follows. • Albumins, 57 percent. • Globulins, 38 percent. • Fibrinogen, 4 percent. • Prothrombin, 1 percent. The composition of other solutes in plasma is as follows. • Ions. • Nutrients. • Waste products. • Gases. • Regulatory substances. The composition of leukocytes in buffy coat is as follows. • Neutrophils, 65 to 75 percent. • Lymphocytes, 20 to 25 percent. • Monocytes, 3 to 8 percent. • Eosinophils, 2 to 5 percent. • Basophils, 0.5 to 1 percent.
Plasma and Plasma Proteins
In adults, plasma accounts for 50% to 55% of blood volume (see Fig. 28.1). Plasma is a complex aqueous liquid containing a variety of organic and inorganic elements (Table 28.1). The concentration of these elements varies, depending on diet, metabolic demand, hormones, and vitamins. Plasma differs from serum in that serum is free of clotting proteins (e.g., fibrinogen). The clotting proteins may interfere with some diagnostic tests.
Table 28.1
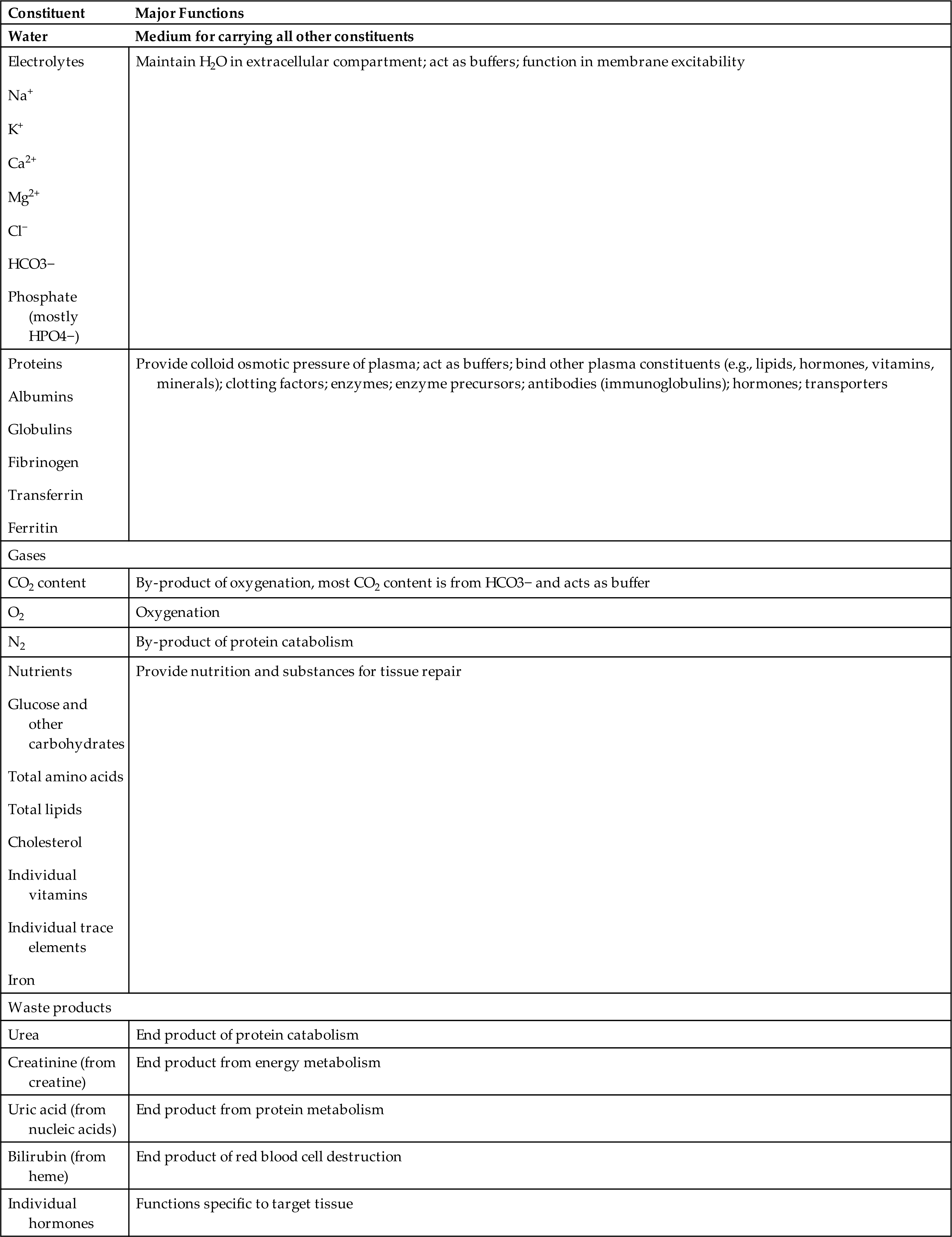
BUN, Blood urea nitrogen; Ca2+, calcium; Cl−, chloride; CO2, carbon dioxide; H2O, water;HCO3−, bicarbonate; HPO4−, phosphorus; K+, potassium; Mg2+, magnesium; N2, nitrogen; Na+, sodium.
Data from Vander AJ, et al. Human physiology: The mechanisms of body function. New York: McGraw-Hill; 2001.
Plasma contains a large number of proteins (plasma proteins) that make up about 7% of the total plasma weight. These vary in structure and function and can be classified primarily into two major groups, albumin and globulins, plus a small amount of clotting proteins. Most plasma proteins are produced by the liver. The major exception is antibody, which is produced by plasma cells (B cells) in the lymph nodes and other lymphoid tissues (see Chapter 8).
Albumin (about 57% of total plasma protein) serves as a carrier molecule for the normal components of blood and for drugs that have low solubility in water (e.g., free fatty acids, lipid-soluble hormones, thyroid hormones, bile salts). Its most essential role is regulation of the passage of water and solutes through the capillaries. Albumin molecules are large and do not diffuse freely through the vascular endothelium, and thus they maintain the critical colloidal osmotic pressure (or oncotic pressure) that regulates the passage of fluids and electrolytes into the surrounding tissues (see Chapters 1 and 3). Water and solute particles tend to diffuse out of the arterial portions of the capillaries because blood pressure (hydrostatic pressure) is greater in arterial than in venous blood vessels. Water and solutes move from tissues into the venous portions of the capillaries, where the pressures are reversed, oncotic pressure being greater than intravascular pressure or hydrostatic pressure (see Fig. 3.1). In the case of decreased production of albumin (e.g., cirrhosis, other diffuse liver diseases, protein malnutrition) or excessive loss of albumin (e.g., certain kidney diseases, extensive burns), the reduced plasma oncotic pressure leads to excessive movement of water and solutes into the tissue and decreased blood volume.
Most of the remaining plasma proteins are globulins (about 38% of total plasma protein), which are often classified by their properties in an electric field (serum electrophoresis). Under the normal conditions used to perform serum electrophoresis, albumin is the most rapidly moving protein. The globulins are classified into alpha, beta, and gamma groups by their movement relative to albumin. Alpha (α) globulins (those moving most closely to albumin) include high-density lipoproteins [HDLs], prothrombin, and proteins that transport hormones. Beta (β) globulins include low-density lipoproteins (LDLs). Gamma (γ) globulins (those with the least movement) consist primarily of immunoglobulin G (IgG) (see Chapter 8).
Plasma proteins also can be classified by function: clotting, defense, transport, or regulation. The clotting factors or proteins promote coagulation and stop bleeding from damaged blood vessels. Fibrinogen is the most plentiful of the clotting factors and is the precursor of the fibrin clot (see Fig. 28.19). Proteins involved in defense, or protection, against infection include antibodies (immunoglobulins) and complement proteins (see Chapters 8 and 9). Transport proteins specifically bind and carry a variety of inorganic and organic molecules, including iron (transferrin), copper (ceruloplasmin), lipids and steroid hormones (lipoproteins) (see Chapter 1), and vitamins (e.g., retinol-binding protein for vitamin A transport). The plasma lipids, triglycerides, phospholipids, cholesterol, and fatty acids are transported through the blood as complexes with plasma proteins; they are known as lipoproteins (see Chapters 1 and 32). Regulatory proteins include a variety of enzymatic inhibitors (e.g., α1-antitrypsin) that protect the tissues from damage; precursor molecules (e.g., kininogen) that are converted into active biologic molecules when needed; and protein hormones (e.g., cytokines) that communicate between cells.
Plasma also contains several charged inorganic ions that regulate cell function, osmotic pressure, and blood pH. These include the electrolytes sodium, potassium, calcium, chloride, and phosphate (Electrolytes are described in Chapters 1 and 3).
Cellular Components of the Blood
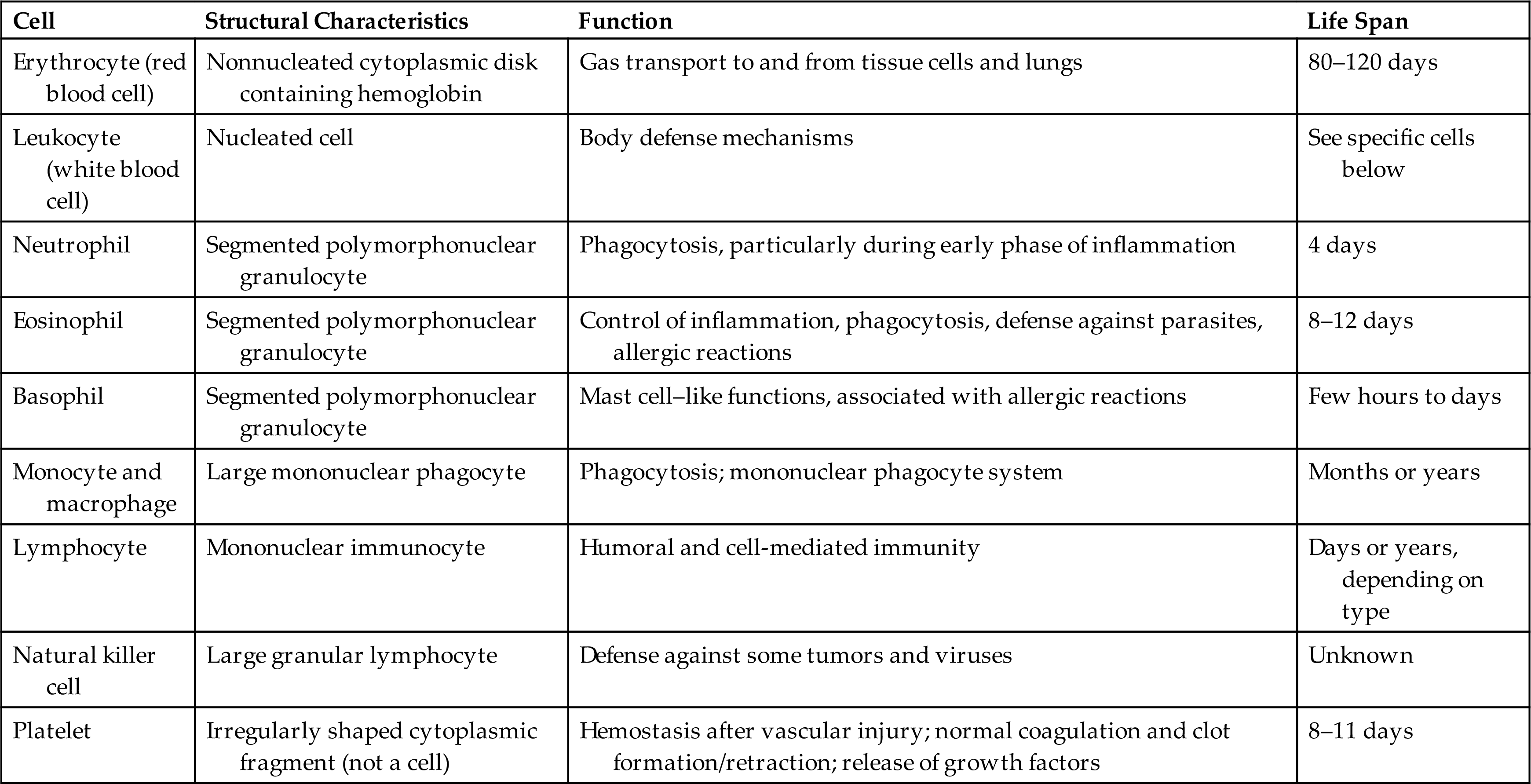
The cellular elements of the blood are broadly classified as red blood cells (erythrocytes), WBCs (leukocytes), and platelets (thrombocytes) (Fig. 28.2). The components of the blood are listed in Table 28.2.
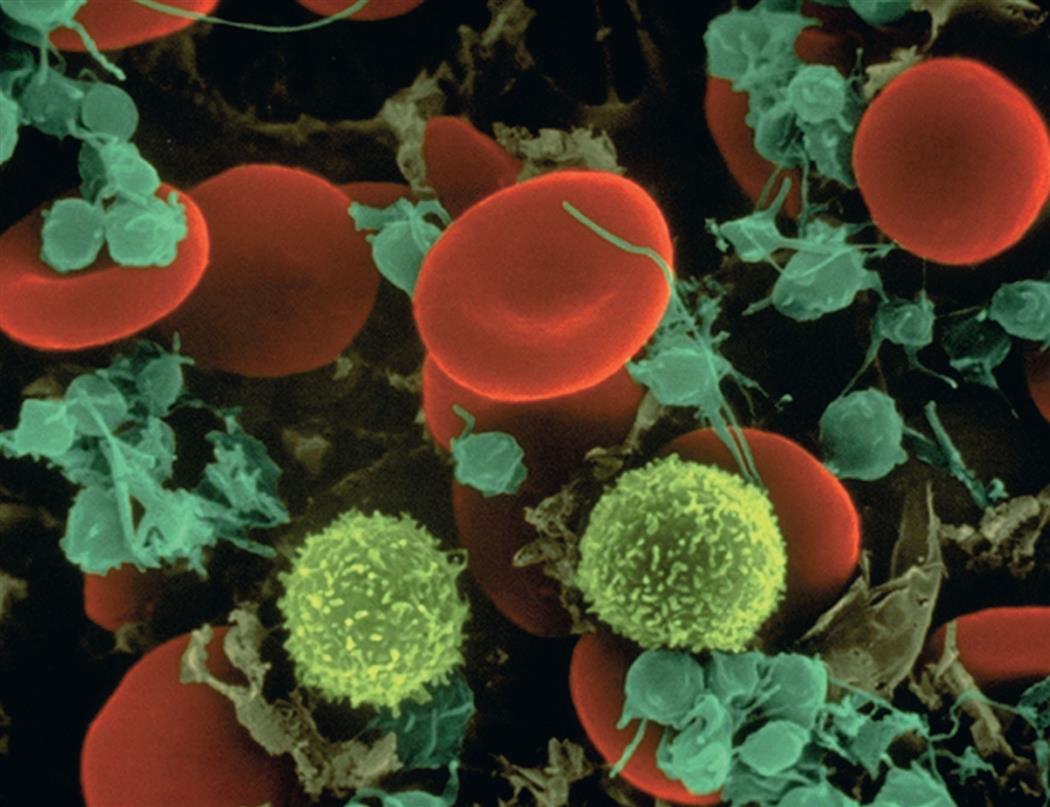
Leukocytes are spherical and have irregular surfaces with numerous extending pili. Leukocytes are the cotton candy–like cells (yellow). Erythrocytes are flattened spheres with a depressed center (red). Platelets are the smaller (green) cell fragments. (Copyright Dennis Kunkel Microscopy, Inc.)
Table 28.2
Erythrocytes
Erythrocytes (red blood cells [RBCs]) are the most abundant cells of the blood, occupying approximately 48% of the blood volume in men and about 42% in women. Erythrocytes are formed in the bone marrow, and there are 4.2 to 6.2 million erythrocytes/mm3 circulating in normal blood. Erythrocytes are primarily responsible for tissue oxygenation. The erythrocyte contains hemoglobin (Hb), which carries the gases, and electrolytes, which regulate gas diffusion through the cell’s plasma membrane. The mature erythrocyte lacks a nucleus and cytoplasmic organelles (e.g., mitochondria), so it cannot synthesize protein or carry out oxidative reactions. Because it cannot undergo mitotic division, the erythrocyte has a limited life span (approximately 100 to 120 days), ages, and is removed from the circulation, primarily in the spleen, to be replaced by new erythrocytes.
The erythrocyte’s size and shape is ideally suited to its function as a gas carrier. It is a small disk with two unique properties: (1) a biconcave shape and (2) the capacity to be reversibly deformed. The flattened, biconcave shape (see Fig. 28.2) provides a surface area/volume ratio that is optimal for gas diffusion into and out of the cell and for deformity. During its life span, the erythrocyte, which is 6 to 8 μm in diameter, repeatedly circulates through splenic sinusoids and capillaries that are only 2 μm in diameter. Reversible deformity enables the erythrocyte to assume a more compact torpedo-like shape, squeeze through the microcirculation (diapedesis), and return to normal (Fig. 28.3).
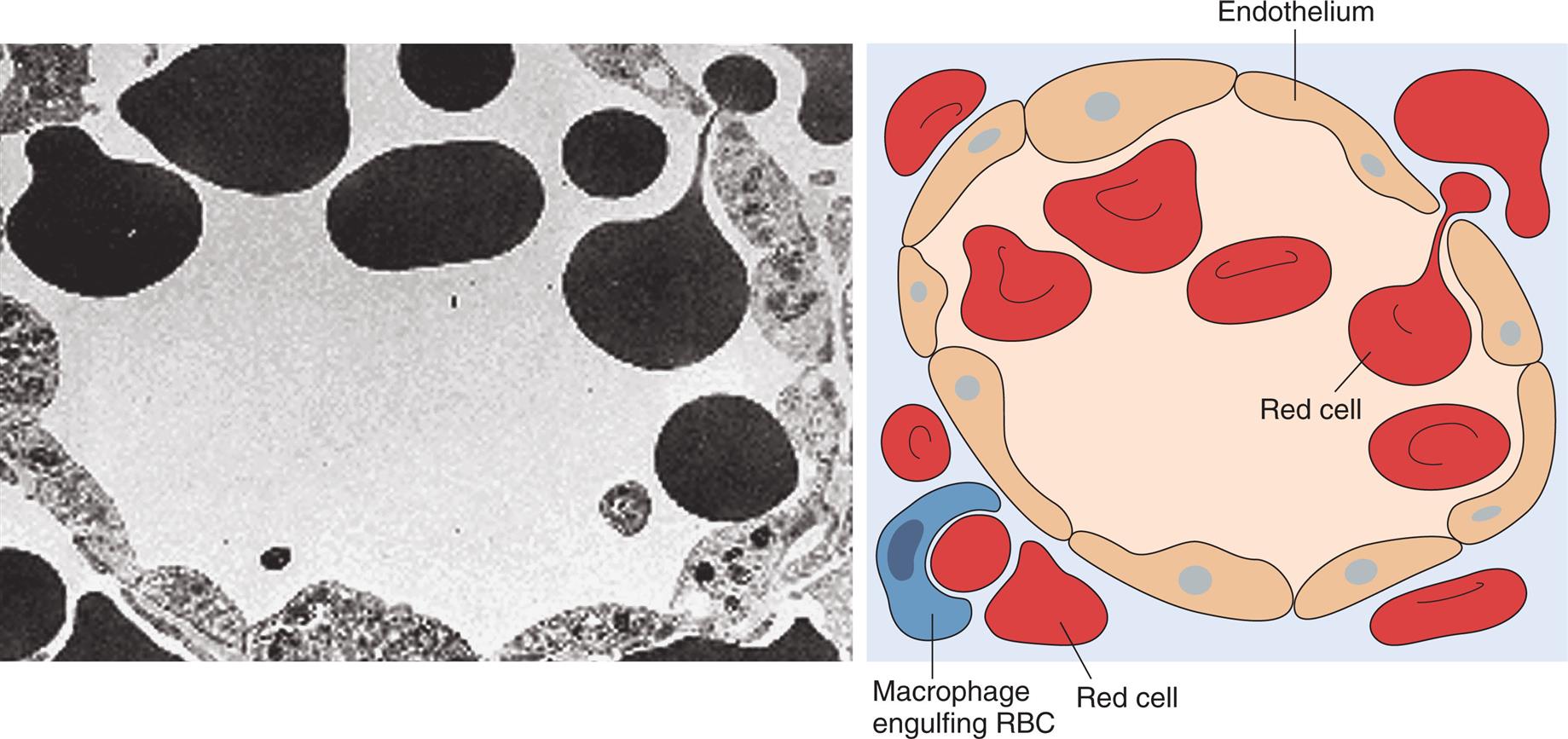
Transmission electron micrograph and schematic drawing of erythrocytes in the process of moving by diapedesis from the red pulp cords into the sinus lumen. They resume their normal shape after squeezing through. Note the degree of deformability required for red cells to pass through the wall of the sinus. RBC, Red blood cell. (From Damjanov I, Linder J, eds. Anderson’s pathology, 10th edition. St. Louis: Mosby; 1996. Schematic from Kumar V, Fausto N, Abbas A. Robbins & Cotran pathologic basis of disease, 7th edition. St. Louis: Saunders; 2005.)
A photomicrograph shows a ring of endothelium cells, surrounded by red cells. An accompanying illustration shows a similar ring of endothelium cells, surrounded by red cells. A macrophage engulfing R B C is also identified in the illustration.
Erythrocytes are also part of the innate immune system. Erythrocytes catch bacteria in the blood stream by electric charge attraction and kill them by oxycytosis, the release of oxygen from oxyhemoglobin (oxygen-dependent killing). The decomposed bacteria are then cleared in the liver and spleen. Oxycytosis is complimentary to leukocytosis (phagocytosis) as leukocytes cannot recognize and attach bacteria in the blood because of the velocity of blood flow.1
Leukocytes
Leukocytes (WBCs) (see Fig. 28.2) defend the body against organisms that cause infection and also remove debris, including dead or injured host cells of all kinds. The leukocytes act primarily in the tissues but are transported in the circulation. The average adult has approximately 5000 to 10,000 leukocytes/mm3 of blood.
Leukocytes are classified according to structure as either granulocytes or agranulocytes and according to function as either phagocytes or immunocytes (cells that create immunity; see Chapter 8). The granulocytes, which include neutrophils, basophils, eosinophils, and mast cells, are all phagocytes. (Phagocytic action is described in Chapter 7.) Of the agranulocytes, the monocytes and macrophages are phagocytes, whereas the lymphocytes are immunocytes. Fig. 28.4 shows a microscopic image of the various types of leukocytes.
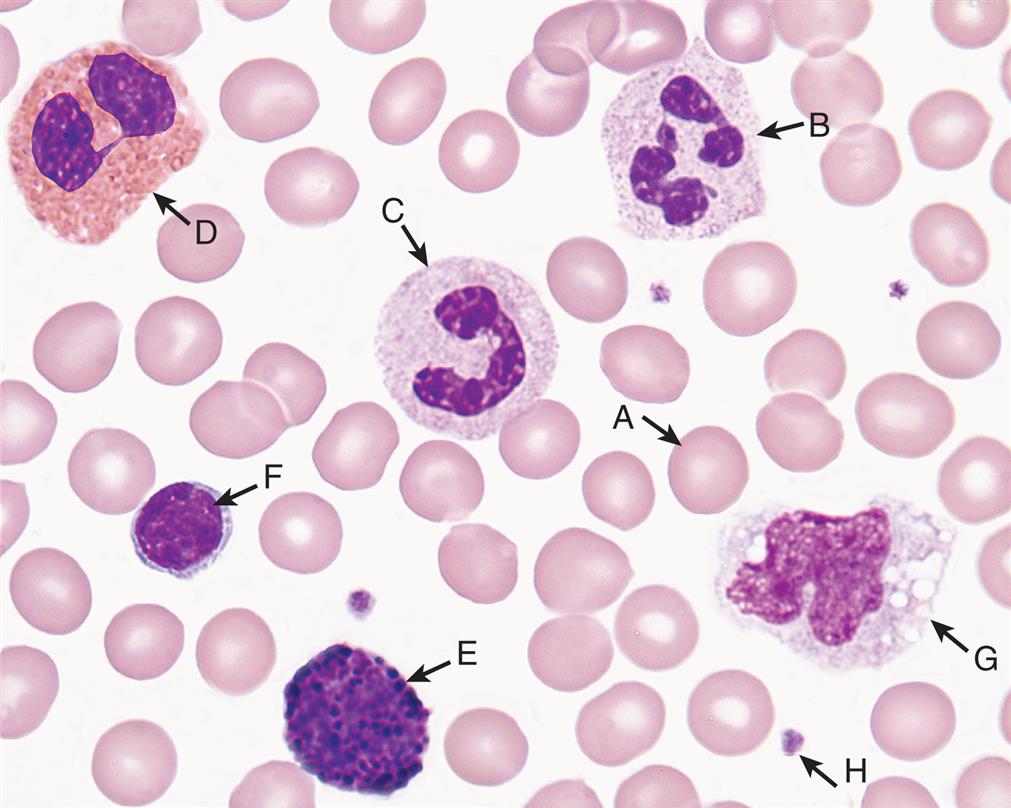
Normal cells in peripheral blood. A, Erythrocyte (red blood cell); B, neutrophil (segmented); C, neutrophil (banded); D, eosinophil; E, basophil; F, lymphocyte; G, monocyte; H, platelet. (From Keohane E, Smith L, Walenga J. Rodak’s hemotology, 5th edition. St. Louis: Saunders; 2016.)
Granulocytes
The granulocytes are formed in the red bone marrow and have a nucleus with several lobes (polymorphonuclear) and many membrane-bound granules in their cytoplasm. These granules contain enzymes capable of killing microorganisms and catabolizing debris ingested during phagocytosis. The granules also contain powerful biochemical mediators with inflammatory and immune functions. These mediators, along with the digestive enzymes, are released from granulocytes in response to specific stimuli and affect other cells in the circulation. The biochemical mediators have vascular and intercellular effects, and the enzymes participate in the breakdown of debris from sites of infection or injury. Granulocytes are capable of amoeboid movement, by which they migrate through vessel walls (diapedesis) and then to sites where their action is needed. They have a life span of only a few hours or days.
The neutrophil (polymorphonuclear neutrophil [PMN]) is the most numerous and best understood of the granulocytes. Neutrophils constitute about 65% to 75% of the total leukocyte count in adults. The results vary by laboratory and populations studied.
The cytoplasm of neutrophils contains small lysosomal granules and a central nucleus with two to five distinct lobes. Immature neutrophils are called bands or stabs. Mature neutrophils are called segmented neutrophils because of the characteristic appearance of their nucleus (see Fig. 28.4). Neutrophils reach a fully mature state in the bone marrow, where these mature neutrophils are called the marrow neutrophil reserve. Normally it takes about 14 days for neutrophils to develop from early precursors, but this process is accelerated by infection and treatment with colony-stimulating factors.
Neutrophils are the chief phagocytes of early inflammation. Soon after bacterial invasion or tissue injury, neutrophils migrate out of the capillaries and into the damaged tissue, where they phagocytize and destroy contaminating microorganisms and debris. Neutrophils also release nuclear DNA onto extracellular pathogens to control their growth and proliferation and aid the killing of bacteria. These extracellular DNA deposits are termed neutrophil extracellular traps (NETs), and are a phagocytosis-independent anti-microbial pathway. In addition to their crucial role in fighting infection, neutrophils play a complex role in tumors. (see Emerging Science Box: Tumor Associated Neutrophils). Neutrophils are sensitive to the environment in damaged tissue (e.g., low pH, enzymes released from damaged cells) and die in 1 or 2 days. The breakdown of dead neutrophils releases digestive enzymes from their cytoplasmic granules. These enzymes dissolve cellular debris and prepare the site for healing. (This final function, called débridement, is described in Chapter 7.)
Eosinophils, which have large, coarse granules, constitute only about 2% to 5% of the normal leukocyte count in adults. Using a spectrum of pattern-recognition receptors, eosinophils are capable of amoeboid movement and phagocytosis. Unlike neutrophils, eosinophils ingest antigen-antibody complexes and are induced by immunoglobulin E (IgE)–mediated hypersensitivity reactions to attack parasites by release of toxic molecules. Eosinophils also mediate immunity against bacterial, viral, and fungal infections by presenting bacterial, viral, and parasitic antigen to cytotoxic T cells. IL-5 promotes the maturation and migration of eosinophils during the inflammatory response. Eosinophilic inflammation mediated by IL-5, particularly from T helper cells, and other cytokines, contributes to the type 2 hypersensitivity response associated with asthma and other chronic eosinophilic inflammatory diseases (see Chapter 9). During inflammation, eosinophil death and cytolysis is known as extracellular trap cell death (ETosis), which differs from the processes of accidental necrosis or apoptosis. Eosinophil extracellular trap products consist of damage-associated molecular patterns (DAMPs) that are able to activate both the innate and adaptive immune systems (see Chapters 7 and 8).2 Eosinophils also release IL-4 important for tissue repair and regeneration in muscle and liver.3
Eosinophil secondary granules contain toxic chemicals (e.g., major basic protein, eosinophil cationic protein, eosinophil peroxidase, eosinophil-derived neurotoxin) that are highly destructive to parasites and viruses. Eosinophil granules also contain a variety of enzymes (e.g., histaminase) that help control inflammatory processes. Eosinophils also release proinflammatory molecules, including leukotrienes, prostaglandins, platelet-activating factor (PAF), and a variety of cytokines (e.g., IL-1, IL-4, IL-6, tumor necrosis factor-alpha [TNF-α], and granulocyte-macrophage colony-stimulating factor [GM-CSF]). Type I and type II hypersensitivity allergic reactions and asthma are characterized by high circulating eosinophil counts (see Chapters 9 and 36). The eosinophils may be involved in a dual role: regulation of inflammation and contribution to the destructive inflammatory processes observed in allergic reactions (e.g., the lungs of persons with asthma and eosinophilic esophagitis).
Basophils, which make up less than 1% of leukocytes, are structurally similar to the mast cells (see Fig. 28.4). Basophils contain cytoplasmic granules that have an abundant mixture of biochemical mediators, including histamine, chemotactic factors, proteolytic enzymes (e.g., elastase, lysophospholipase), and an anticoagulant (heparin). Stimulation of basophils also induces synthesis of vasoactive lipid molecules (e.g., leukotrienes) and cytokines, including IL-6, which affects differentiation of type 1 T-helper cells (Th1 cells) (focus on intracellular parasites) and type 2 T-helper cell (Th2 cells) (focus on extracellular parasites). Basophils are a particularly rich source of the cytokine IL-4, which preferentially guides B-cell differentiation toward plasma cells that secrete IgE (see Chapter 8).
The number of basophils is often increased at sites of allergic inflammatory reactions and parasitic infection, particularly ectoparasites (e.g., ticks). IgE receptors on the basophil induce degranulation at sites of IgE-mediated hypersensitivity reactions and contribute to the local inflammatory response.
Mast cells are highly similar to basophils but are generated from a different set of precursor cells in the bone marrow, from which they migrate in an immature form into tissues. Mast cells reside in vascularized connective tissues just beneath body epithelial surfaces, including the submucosal tissues of the respiratory and gastrointestinal tracts, as well as in the dermal layer that lies just below the surface of the skin. Mast cells play a central role in inflammation. Activation and degranulation of the mast cells affect a great number of other cells, including those involved in inflammation (e.g., vascular endothelial cells, smooth muscle cells, circulating platelets and leukocytes, nerves) and healing (e.g., fibroblasts), as well as glandular cells and cells of the immune system. Their activation contributes greatly to increased permeability of blood vessels and synthesis of mediators producing smooth muscle contraction (see Fig 7.4).
Agranulocytes
The agranulocytes—monocytes, macrophages, and lymphocytes—contain relatively fewer granules than granulocytes. Monocytes and macrophages make up the mononuclear phagocyte system (MPS) (see Chapter 7). Both monocytes and macrophages participate in the immune and inflammatory response, being powerful phagocytes. They also ingest dead or defective host cells, particularly blood cells.
The mononuclear phagocyte system (MPS), also known as the reticuloendothelial system or macrophage system, consists of specialized endothelial cells: promonocytes and their precursors in the bone marrow; monocytes in the peripheral blood; and a portion of macrophages that reside in the tissues, remaining there for months or perhaps years. Cells of the MPS ingest and destroy (by phagocytosis) unwanted materials, such as foreign protein particles, circulating immune complexes, microorganisms, debris from dead or injured cells, defective or injured erythrocytes, and dead neutrophils.
Monocytes are immature macrophages. They are the largest normal blood cell and have a horseshoe-shaped nucleus (see Fig. 28.4). Monocytes are formed and released by the bone marrow into the bloodstream. As they mature, monocytes migrate into a variety of tissues (e.g., liver [Kupffer cells], spleen, lymph nodes, peritoneum, gastrointestinal tract, brain [microglia], and bone) and differentiate into tissue macrophages with tissue-specific functions (Table 28.3). Monocytes also differentiate into myeloid linage dendritic cells and disseminate to lymphoid organs and body surfaces such as the skin and mucous membranes. Both macrophages and dendritic cells are part of the MPS. Other monocytes may migrate out of blood vessels and differentiate into macrophages in response to infection or inflammation. Macrophages are generally larger and are more active as phagocytes than monocytes. Dendritic cells frequently extend projections (dendrites) into the tissue and assume a “neuron-like” appearance. The origin and turnover of many of the tissue macrophages are not precisely known. It seems clear that once monocytes leave the circulation, they do not return. They can survive many months or even years.
Table 28.3
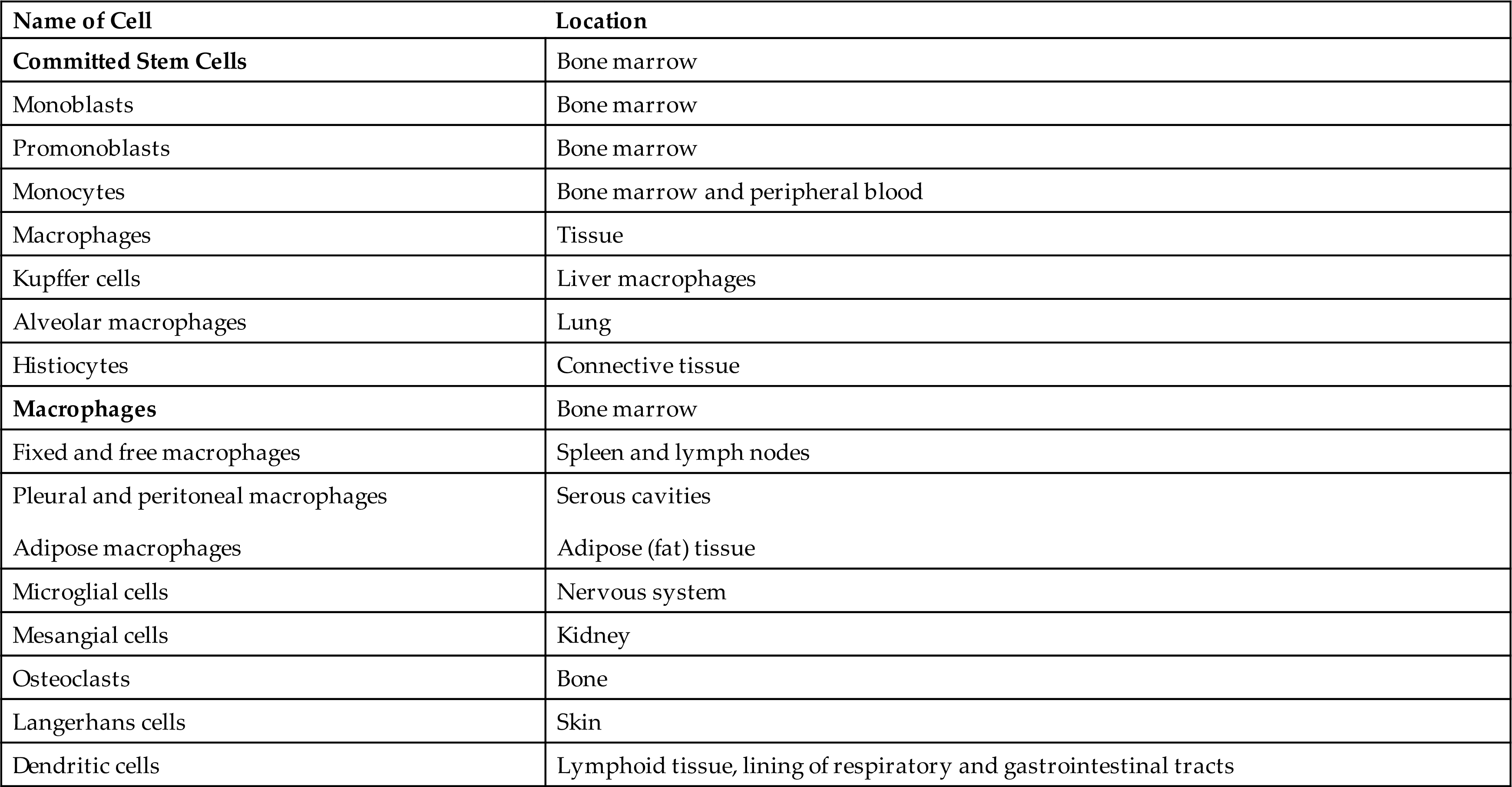
Modified from Kumar V, et al. Robbins & Cotran pathologic basis of disease, 9th edition. Philadelphia: Saunders; 2015.
The normal role of macrophages is to remove old and damaged cells and large molecular substances from the blood. Cellular targets of macrophage phagocytosis include circulating senescent or damaged erythrocytes and platelets (removed primarily in spleen), dead neutrophils (in the circulation and at sites of inflammation), and cells undergoing apoptosis. Noncellular targets include antigen-antibody complexes, cellular debris, products of coagulation, and macromolecules (such as lipids and carbohydrates synthesized by the body as the result of faulty metabolism, as in storage diseases). Macrophages remove and kill contaminating microorganisms in the blood (mostly in the liver and spleen) and at sites of infection. Macrophages and, particularly, dendritic cells are the major “antigen-processing” and “antigen-presenting” cells that initiate immune responses (see Chapter 8). Macrophages initiate wound healing and tissue remodeling and, if activated by cytokines from T cells, secrete a large array of biologically active chemicals that if uncontrolled result in chronic inflammation and tissue injury (see Chapter 7).
Lymphocytes constitute about 20% to 25% of the total leukocyte count and are the primary cells of the immune response (see Fig. 28.4 and Chapter 8). Lymphocytes arise from lymphoid stem cells in the bone marrow and subsequently develop and reproduce in the lymphatic tissues as mature T cells, B cells, or plasma cells. The lymphocytes do not contain any enzyme-filled digestive vacuoles. The life span of the lymphocyte can be days, months, or years, depending on its type and subtype.
Natural killer (NK) cells, which resemble large granular lymphocytes, recognize cells that do not express “self” proteins (self-antigens) on their plasma membrane or that contain foreign or abnormal antigens such as cancer cells or some virus-infected cells without prior exposure to these antigens (see Chapters 7 and 8). Hence, they are named natural killer cells as they do not require exposure to antigen for activation to differentiate them from T-cytotoxic cells which are induced by antigen. NK cells also have the capacity to activate T cells and phagocytes and produce a variety of cytokines that can regulate immune responses. The predominant form of NK cells develops in the bone marrow and circulates in the blood, and is found mainly in the peripheral blood and spleen where it accounts for 5% to 10% of the circulating lymphoid pool. NK cells develop independent of the thymus, although some NK precursors are found in the thymus and may develop into NK T cells, which will then have markers of both NK cells and T cells.4
Platelets
Platelets (thrombocytes) are not true cells, but rather irregularly discoid-shaped cytoplasmic fragments that lack a nucleus and deoxyribonucleic acid (DNA) and are incapable of mitotic division. Platelets are essential for blood coagulation and control of bleeding. They are formed in the bone marrow by fragmentation of very large cells (40 to 100 μm in diameter) known as megakaryocytes. They contain cytoplasmic granules (e.g., dense granules, alpha granules, and lysosomes) and can release adhesive proteins and coagulation and growth factors when stimulated by injury to a blood vessel. Platelets can assume different shapes and form adhesive pseudopodia (long surface extensions) that increase their surface area and promote interconnectedness and adherence to collagen fibers in damaged vascular walls, plugging vascular openings to control bleeding (Fig. 28.5).
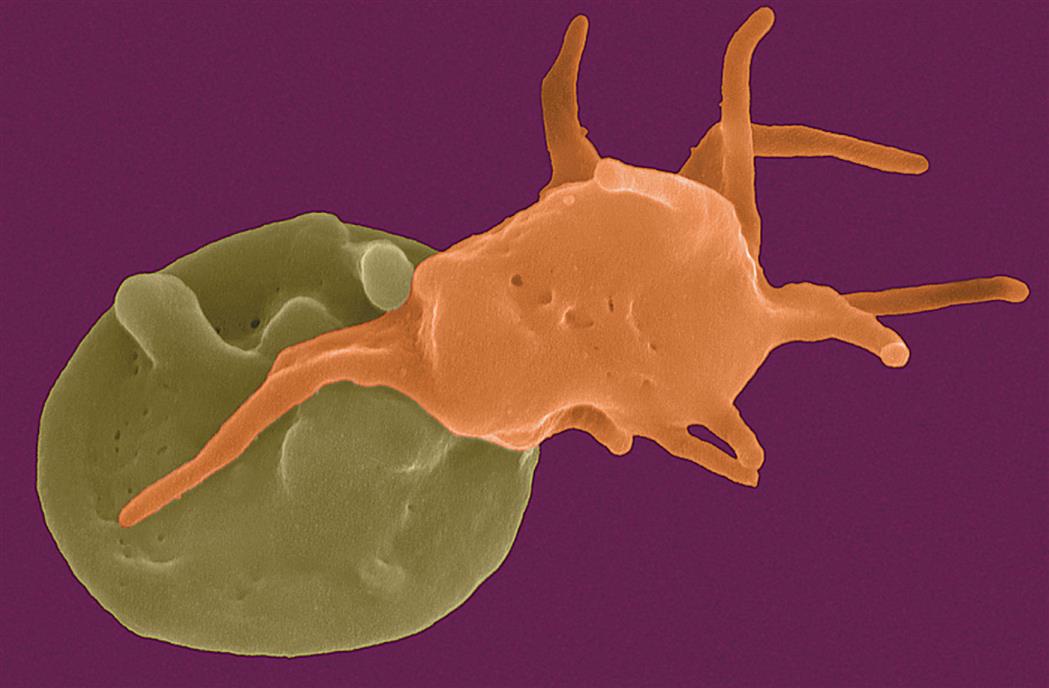
The platelet on the left is moderately activated, with a generally round shape and the beginning of formation of pseudopodia (foot-like extensions from the membrane). The platelet on the right is fully activated, with extensive pseudopodia. (Copyright Dennis Kunkel Microscopy, Inc.)
The normal platelet concentration is about 150,000 to 400,000 platelets/mm3 of circulating blood, although the normal ranges may vary slightly from laboratory to laboratory. An additional one-third of the body’s available platelets are in a reserve pool in the spleen. A platelet circulates for approximately 8 to 11 days, and is then removed by macrophages, mostly in the spleen.
Lymphoid Organs
The lymphoid system is closely integrated with the circulatory system. The lymphoid organs, some of which are merely aggregations of lymphoid tissue, are classified as primary or secondary. The primary lymphoid organs are the thymus and the bone marrow. The secondary lymphoid organs consist of the spleen, lymph nodes, tonsils, and Peyer patches in the ileum of the small intestine. All of the lymphoid organs link the hematologic and immune systems in that they are sites of residence, proliferation, differentiation, or function of lymphocytes and mononuclear phagocytes (monocytes and macrophages) (see Chapter 8). The liver, which is primarily a digestive organ and is described in Chapter 40, synthesizes both coagulant and anticoagulant proteins.
Spleen
The spleen is the largest of the lymphoid organs. It serves as a site of fetal hematopoiesis, filters blood-borne antigens, cleanses the blood through the action of mononuclear phagocytes, initiates an immune response to blood-borne microorganisms, destroys aged erythrocytes, and serves as a reservoir for blood.
The spleen is a concave, encapsulated organ that weighs about 150 g and is about the size of a fist. It is located in the left upper abdominal cavity, curved around a portion of the stomach. Strands of connective tissue (trabeculae) extend throughout the spleen from the splenic capsule, dividing it into compartments that contain masses of lymphoid tissue called splenic pulp (white and red) (Fig. 28.6). The spleen is interlaced with many blood vessels, some of which can distend to store blood. Blood that circulates through the spleen comes from the splenic artery, which branches from the descending aorta and reenters the circulatory system through the splenic vein and into the portal vein.
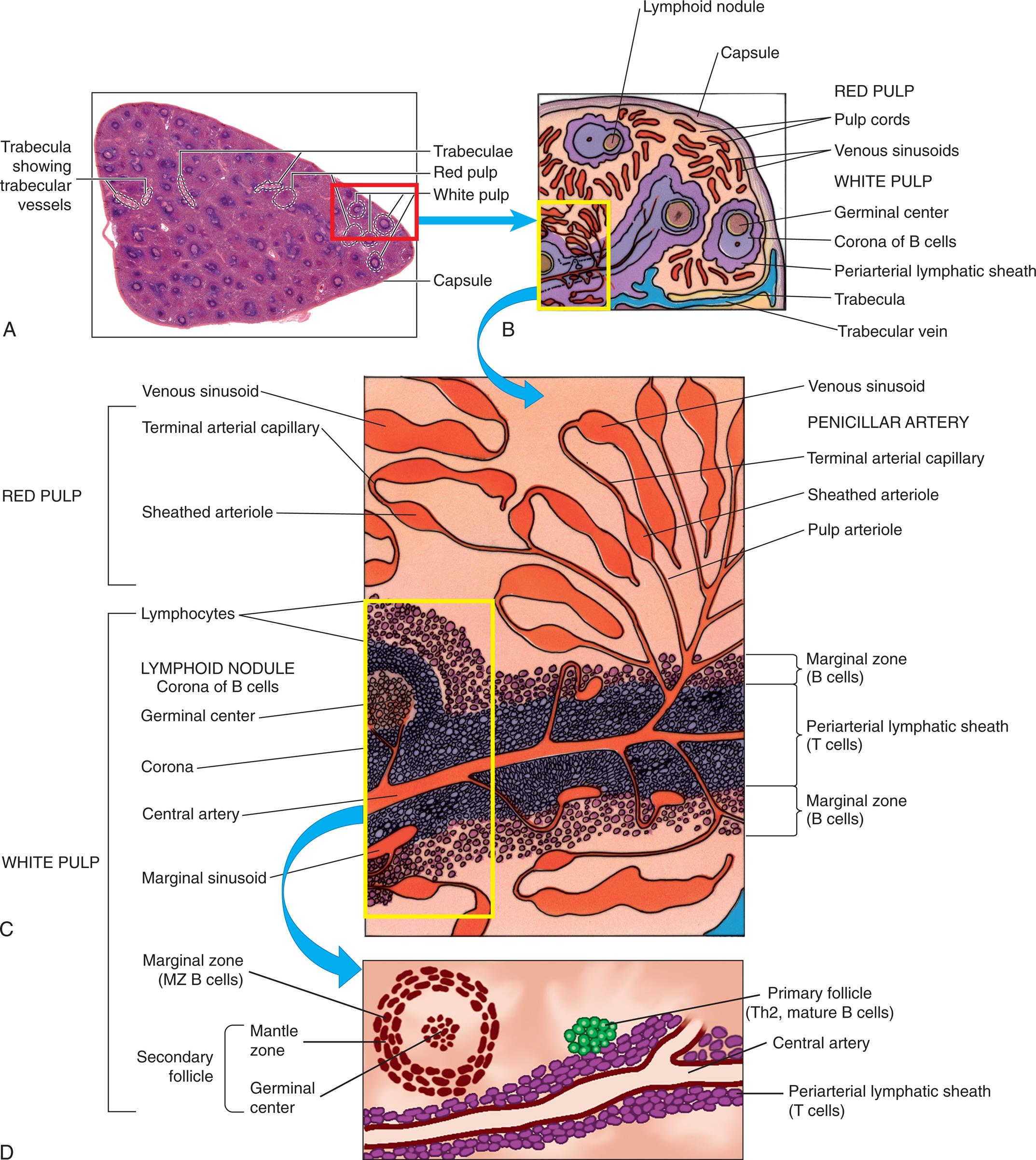
(A) Cross-section of the spleen. The spleen is enclosed in a capsule with the interior pulp divided into compartments by strands of connective tissue. The splenic pulp contains regions that are rich in lymphocytes (white pulp) and those containing erythrocytes (red pulp). (B) Subcapsular splenic structures. (C) Magnified view of red and white pulp structures. (D) The splenic artery branches into trabecular arteries and then to central arteries frequently surrounded by a periarterial lymphoid sheath, primarily containing T cells. The secondary lymphoid follicles contain B cells that are proliferating in response to antigen. Th2, T-helper 2 cells. (A, From Telser AG, Young JK, Baldwin KM. Elsevier’s integrated histology. St. Louis: Mosby; 2007; B and C, From Gartner LP, Hiatt JL. Color textbook of histology, 3rd edition. Philadelphia: Elsevier; 2007; D, From Hoffman R, et al. Hematology: Basic principles and practice, 6th edition. Philadelphia: Churchill Livingstone; 2013.)
Three illustrations, A, B, C, and D, shows the splenic architecture. Top-left panel, A. The illustration shows a cross-section of the spleen and highlights the following structures: trabecula showing trabecular vessels, trabeculae, red pulp, white pulp, and capsule. Top-right panel, B. The illustration shows and labels the following subcapsular splenic structures, clockwise from the top: lymphoid nodule, capsule, red pulp (pulp cords, venous sinusoids), and white pulp (germinal center, corona of B cells, periarterial lymphatic sheath, trabecula, and trabecular vein. Middle panel, C. The illustration shows magnified view of the red pulp and the white pulp. The red pulp comprises venous sinusoid, terminal arterial capillary, sheathed arteriole, venous sinusoid, penciller artery, terminal artery capillary, sheathed arteriole, and pulp arteriole. The white pulp comprises lymphocytes, lymphoid nodule corona of B cells, germinal center, corona, central artery, marginal sinusoid, marginal zone (B cells), periarterial lymphatic sheath (T cells), and marginal zone (B cells). Bottom panel, D. The illustration shows the splenic arteries and the trabecular arteries. The following structures on the illustration are labeled, marginal zone (M Z B cells), primary follicle (T h 2, mature B cells), secondary follicle (mantle zone and germinal center), and periarterial lymphatic sheath (T cells).
Arterial blood that enters the spleen first encounters the white splenic pulp, which consists of masses of lymphoid tissue containing macrophages and lymphocytes, primarily T lymphocytes in proximity to the arterioles (the periarterial lymphoid sheath) (see Fig. 28.6). Cellular clumps (lymphoid follicles) are formed in the white pulp around the splenic arterioles. The lymphoid follicles consist primarily of B lymphocytes and are the chief sites of immune function within the spleen. Here blood-borne antigens encounter lymphocytes, initiating the immune response and the conversion of lymphoid follicles into germinal centers in the middle of the follicle where B cells proliferate and differentiate during the humoral immune response (see Chapter 8).
Some of the blood that enters the terminal capillaries continues through the microcirculation and enters highly distensible storage areas, called venous sinuses, in the red pulp of the spleen. The venous sinuses (and the red pulp) can store more than 300 mL of blood. Passive dilation of the venous sinuses enables the spleen to increase its storage capacity as needed by the body. Sudden reductions in blood pressure cause the sympathetic nervous system to stimulate constriction of the sinuses and expel as much as 200 mL of blood into the venous circulation, helping restore blood volume or pressure in the circulation and increasing the hematocrit by as much as 4%.
The endothelial lining of the venous sinuses is discontinuous (having gaps between endothelial cells) and therefore extremely permeable, allowing blood cells to exit the circulation. The red pulp contains a system of loosely interconnected resident macrophages that provide the principal site of splenic filtration. Because of the slow circulation in the sinuses, the macrophages easily phagocytose old, damaged, or dead blood cells of all kinds (but chiefly erythrocytes), microorganisms, macromolecules, and particles of debris. Hemoglobin from phagocytosed erythrocytes is catabolized, and heme (iron) is stored in the cytoplasm of the macrophages or released back into the blood (see Fig. 28.15). The macrophages also can remove particulate inclusions containing denatured hemoglobin (Heinz bodies) from erythrocytes without harming the cells themselves. Blood that filters through the red pulp then moves through the venous sinuses and into the portal circulation (blood flowing to the liver).
The spleen is not absolutely necessary for life or for adequate hematologic function. However, splenic absence as a result of any cause (atrophy, traumatic injury, or removal because of disease) has several secondary effects on the body. For example, leukocytosis (high levels of circulating leukocytes) often occurs after splenectomy, suggesting that the spleen exerts some control over the rate of proliferation of leukocyte stem cells in the bone marrow or their release into the bloodstream. Circulating levels of iron also may decrease, reflecting the spleen’s role in the iron cycle (see the Iron Cycle section). The immune response to encapsulated bacteria (e.g., Streptococcus pneumoniae [pneumococcus], Neisseria meningitidis [meningococcus], and Haemophilus influenzae), which is primarily an IgM response, may be severely diminished, resulting in increased susceptibility to disseminated infections and sepsis. An increase in morphologically defective blood cells within the circulation after the loss of the spleen helps confirm the spleen’s role in removing old or damaged cells. Thrombocytosis and thrombosis are complications of splenectomy and requires anticoagulation therapy.5
Lymph Nodes
Structurally lymph nodes are part of the lymphatic system. Lymphatic vessels collect interstitial fluid from the tissues and transport it, as lymph, through vessels of increasing size to the thoracic duct, which drains into the superior vena cava and returns the lymph to the circulation. Lymph nodes are distributed throughout the body and provide filtration of the lymph during its journey through the lymphatics. Each lymph node is enclosed in a fibrous capsule, branches of which (trabeculae) extend inward to partition the node into several compartments. Reticular fibers of connective tissue divide the compartments into a meshwork throughout the lymph node. The node consists of outer (cortex) and inner (paracortex) cortical areas and an inner medulla. Lymph enters through multiple small afferent lymphatic vessels into the subcapsular sinus, just beneath the capsule, and drains into the cortical sinuses to the medullary sinuses, from which the lymph is collected and leaves the node by way of the efferent lymphatic vessel. Blood flows into the lymph nodes through the lymphatic artery, which ends in groups of postcapillary venules distributed throughout the outer cortex. The blood is drained through the lymphatic vein.
Functionally, lymph nodes are part of the hematologic and immune systems and are the primary site for the first encounter between antigen and lymphocytes. Lymphocytes enter the lymph node from the blood through specialized postcapillary venules called high endothelial venules (HEVs) by means of diapedesis across the endothelial lining (Fig. 28.7). Within the secondary lymph nodes, naïve T and B lymphocytes are segregated into different regions. B lymphocytes tend to migrate preferentially to nodes in the cortex and medulla, whereas T lymphocytes predominantly migrate to the paracortex. Macrophages reside in the lymph node; help filter the lymph of debris, foreign substances, and microorganisms; and provide antigen-processing functions. The dendritic cells encounter and process antigens (see Chapter 8) and microorganisms in other tissues, then enter the lymph node through the afferent lymph vessels, and migrate throughout the nodes. The reticular network provides adhesive surfaces for trapping large numbers of phagocytes and lymphocytes and facilitates their organization into follicles or primary nodules. The presence of antigen, either removed from the lymph by macrophages or presented on the surface of dendritic cells, results in the production of secondary nodules containing germinal centers. In the germinal centers lymphocytes, particularly B cells, respond to antigenic stimulation by undergoing proliferation and further differentiation into memory cells and plasma cells (see Chapter 8). Plasma cells migrate to the medullary cords. The B-lymphocyte proliferation in response to a great deal of antigen (e.g., during infection) may result in lymph node enlargement and tenderness (reactive lymph node), which can be palpable during a physical examination.
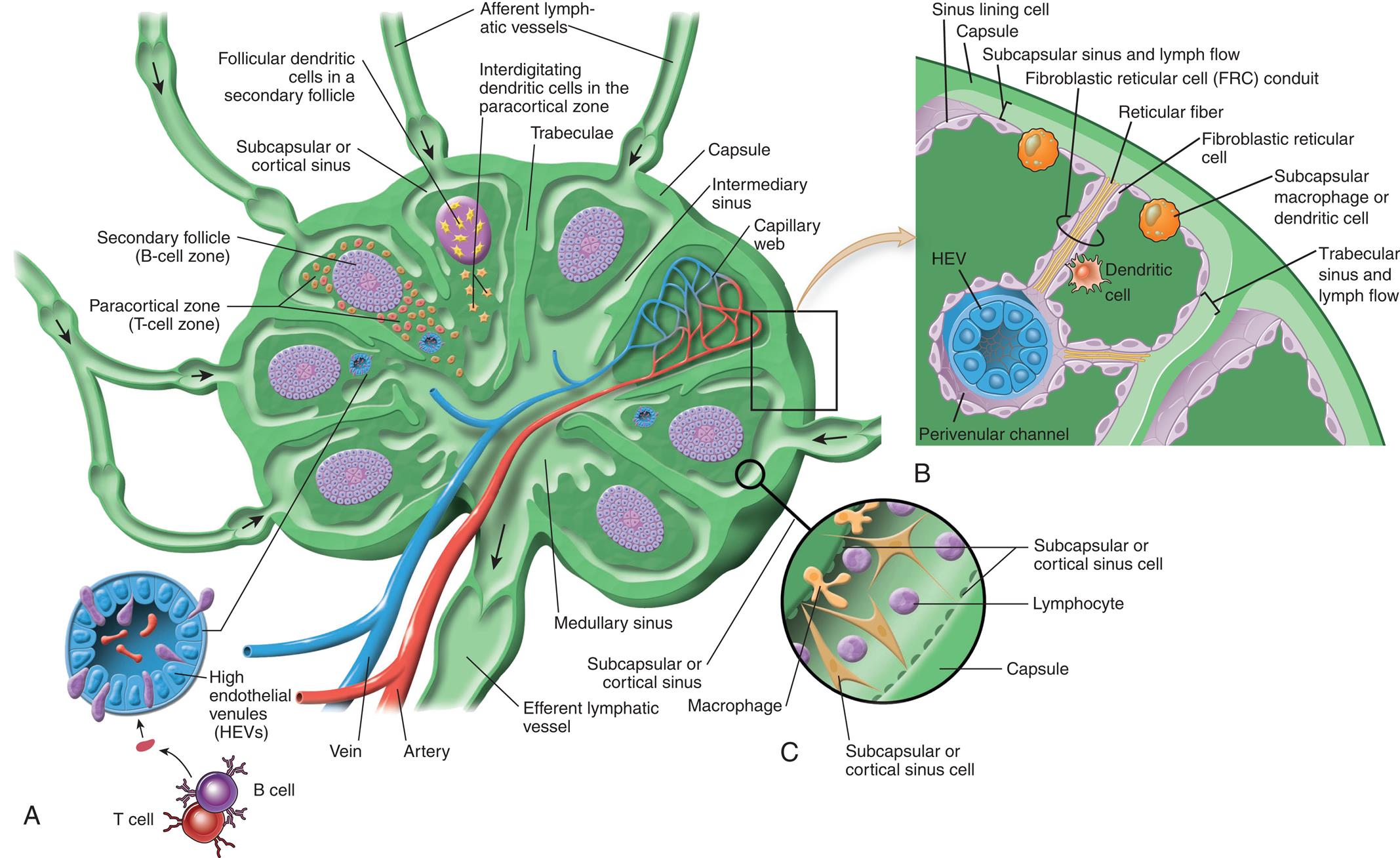
(A) Cross section showing ingoing and outgoing lymph vessels (afferent and efferent), blood supply, compartmentalization of the B-cell region (secondary follicle), T-cell region (paracortical zone) with precapillary or high endothelium venules, follicular interdigitating dendritic cells, medullary sinus, intermediate sinus, and subcapsular or cortical sinus. Naive B and T lymphocytes enter through the high endothelial venule (HEV), shown in cross section, and are drawn to different areas of the node by chemokines that are produced in these areas and bind selectively to either cell type. (B) Cellular composition of the lymph node cortex depicting the route of lymph drainage from the subcapsular sinus, through fibroreticular cell conduits, to the perivenular channel around the high endothelial venule (HEV). Dendritic cells pick up antigen from peripheral sites of antigen entry and enter the lymph node through the afferent lymph vessels and migrate to the T-cell region of the node. (C) Cellular composition of subcapsular sinus. (A and C, From Paulsen F. Sobottta atlas of human anatomy. Vol. 1, 15th edition. Philadelphia: Elsevier; 2013; B, From Abbas AK, Pillai S. Cellular and molecular immunology, 9th edition. Philadelphia: Saunders; 2015.)
Three illustrations, A, B, and C, show the architecture of the lymph node. Left panel, A. The illustration shows a cross-section of the lymph vessel. The following structures on the illustration are labeled, clockwise from the top: afferent lymphatic vessels, interdigitating dendritic cells in the paracortical zone, trabeculae, capsule, intermediary sinus, capillary web, medullary sinus, efferent lymphatic vessel, artery, vein, high endothelial venules (H E Vs), paracortical zone (T-cell zone), secondary follicle (B-cell zone), follicular dendritic cells in a secondary follicle, and subcapsular cortical sinus. Top-right panel, B. The illustration shows the cellular composition of the lymph node. The following structures on the illustration, clockwise from the top: sinus lining cell, capsule, subcapsular sinus and lymph flow, fibroblastic reticular cell (F R C) conduit, reticular fiber, fibroblastic reticular cell, subcapsular macrophage or dendritic cell, trabecular sinus and lymph flow, dendritic cell, H E V, and perivenular channel. Bottom-right panel, C. The illustration shows the subcapsular sinus and labels the following parts: subcapsular or cortical sinus cell, lymphocyte, capsule, subcapsular or cortical sinus cell, macrophage, and subcapsular or cortical sinus.
Development of Blood Cells
Bone Marrow
Bone marrow is confined to the cavities of bone and is the primary site of residence of hematopoietic stem cells (HSCs). The bone marrow environment provides HSC self-renewal, proliferation, differentiation, and apoptosis, and supports the developing progenitor cells.
Adults have two kinds of bone marrow: red marrow (active or hematopoietic marrow; also called myeloid tissue) and yellow marrow (inactive marrow). Bone cavities contain only red marrow at birth. The large quantity of fat in adult marrow is responsible for its yellow appearance. Not all bones contain active marrow. In adults, active marrow is found primarily in the flat bones of the pelvis (34%), vertebrae (28%), cranium and mandible (13%), sternum and ribs (10%), and in the extreme proximal portions of the humerus and femur (4% to 8%). Stem cells for transplant are commonly harvested from the flat bones of the pelvis. Inactive marrow predominates in cavities of other bones. (Bones are discussed further in Chapter 43.)
The hematologic compartment of the bone marrow consists of a variety of cellular and specialized molecular microenvironments, called bone marrow niches (Fig. 28.8). The niches provide essential autocrine, endocrine, and paracrine signals as well as direct cell-to-cell interactions necessary for the ability of HSCs to self-renew and to differentiate into all blood-cell lineages.
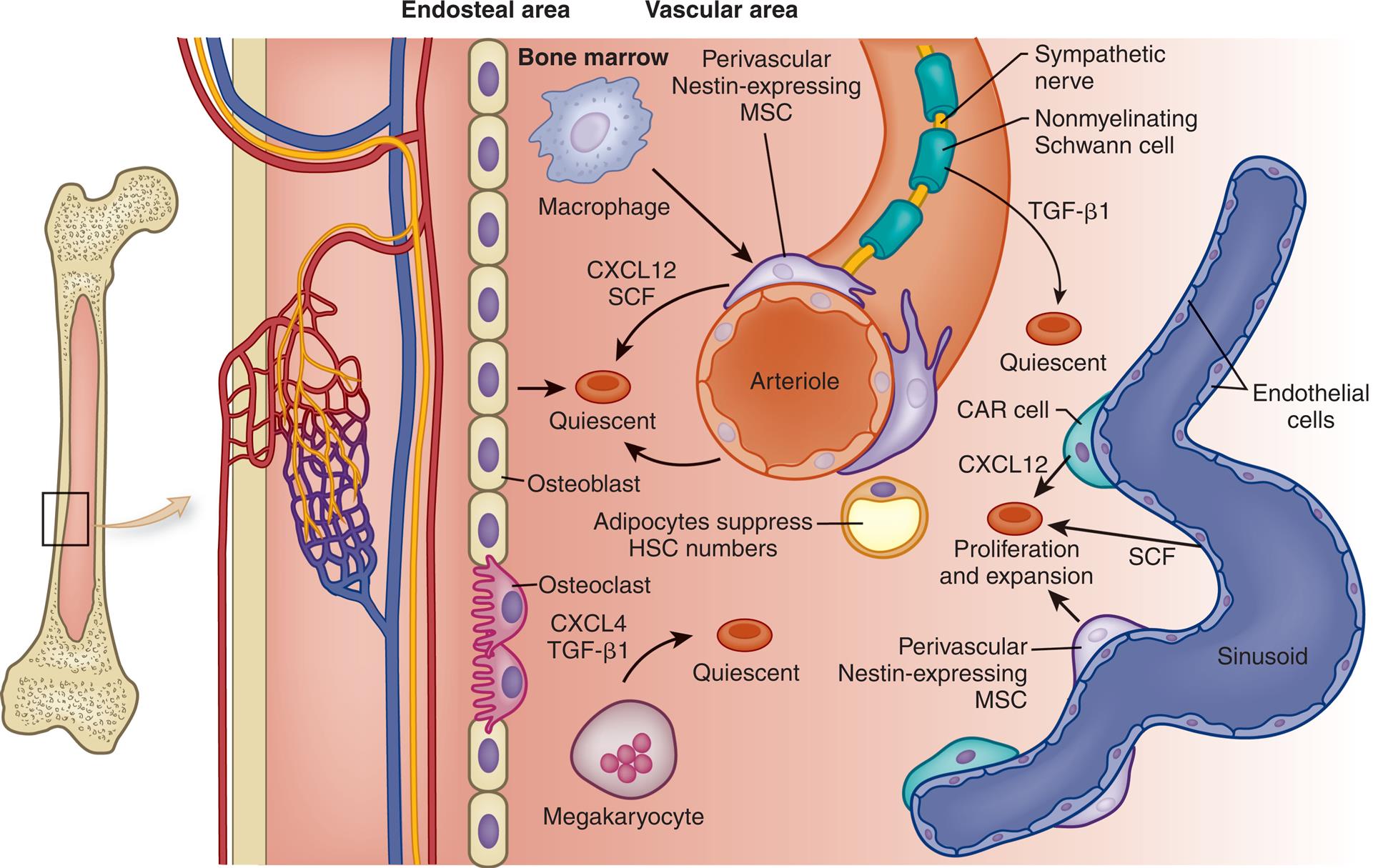
Stem cell niches are microenvironments where stem cells undergo hematopoiesis into all forms of blood cells. Stem cell niches retain and maintain adult quiescent hematopoietic stem cells (HSCs) and are activated to promote cell self-renewal, proliferation, and differentiation to form new cells. The fate of individual HSCs is determined by interactions with specialized cells within the niches. CAR, CXCL12-abundant reticular cells; CXCL4, chemokine ligand 4 (also known as platelet factor 4); CXLC12, chemokine ligand 12; MSC, mesenchymal stem cells; SCF, stem cell factor; TGF-β1, transforming growth factor-beta 1. (Adapted from Boulais PE, Frenette PS. Making sense of hematopoietic stem cell niches. Blood, 2015;125(17):2621–2629; Schepers K, Campbell TB, Passegué E. Normal and leukemic stem cell niches: insights and therapeutic opportunities. Cell Stem Cell, 2015;16(3):254–267.)
An illustration shows a magnified view of a section of a bone. From the left to the right, the structure comprises an endosteal area and a vascular area. The endosteal area comprises a layer of osteoblast cells embedded with osteoclast cells. The vascular area comprises an arteriole. The following structures on the arteriole are identified: sympathetic nerve, nonmyelinating Schwann cell, T G F-beta 1 (quiescent), perivascular nestin-expressing M S C, bone marrow, macrophage, and C X C L 12, S C F. The adipocytes suppress H S C numbers. The megakaryocyte release C X C L 4 and T G F-beta 1 to the quiescent. The following structures on the sinusoid are identified: endothelial cells, C A R cell, C A C L 12, S C F, and perivascular nestin-expressing M S C, which contribute to proliferation and expansion of the quiescent.
At least two populations of stem cells are found in bone marrow niches. Hematopoietic stem cells (HSCs) are progenitors of all the cell types of myeloid and lymphoid lineages (see Fig. 28.10). Mesenchymal stem cells (MSCs) are stromal cells (connective tissue cells) that can differentiate into a variety of cells, including osteoblasts (produce bone), adipocytes (store fat), chondrocytes (produce cartilage), sinusoidal endothelial cells, fibroblasts, and other stromal cells (connective tissue cells). The stromal cells secrete substances that form an extracellular matrix critical for cell growth and for anchoring developing blood cell progenitors in the bone marrow. Both populations of stem cells undergo continuous proliferation and self-renewal in the niches of the microenvironment of the bone marrow so that additional HSCs and MSCs are produced to replace those undergoing differentiation.
Hematopoietic niches are specialized microenvironments within the bone marrow that promote the maintenance of stem cells and regulate their function. The two main niches are the endosteal niche located close to endosteal bone, and the perivascular niche located more centrally near blood vessels and venous sinusoids. Different cell types reside in each niche. The primary cells of the endosteal niche are osteocytes, osteoblasts, osteoclasts, megakaryocytes, and macrophages. Osteoblasts are derived from MSCs and are responsible for construction of bone and support myelopoiesis through the release of soluble factors. Osteocytes are terminally differentiated osteoblasts and are the mature cell of the bone matrix. They are indirectly involved in the maintenance of the HSC pool through their interaction with osteoblasts. Osteoclasts are cells which originate from mononuclear cells of the HSC lineage and remodel bone by resorption. Additionally, osteoblasts can promote differentiation of B cells and osteoclasts can present antigen to T cells and are phagocytic, providing a link to immune function.6
Megakaryocytes and macrophages promote HSC migration and proliferation in the perivascular niches of the bone marrow. Megakaryocytes lie close to vascular sinuses where they produce platelets releasing them into the blood stream. Macrophages are also important for erythroid proliferation, differentiation, and maturation and supplying iron. Bone marrow adipocytes are a subpopulation of stromal adipocytes located throughout the bone marrow and support HSC maintenance.
Perivascular niches contain two specialized MSCs: chemokine ligand 12 (CXCL12)-abundant sinusoidal reticular (CAR) cells and periarteriolar and sinusoidal nestin-expressing cells. These cells closely interact with HSCs and provide intercellular signaling and cross-talk through several HSC regulatory molecules important for retention, expansion, maintenance, and quiescence or dormancy (prevents cell exhaustion and maintain their numbers throughout life) of HSCs. These regulatory molecules include CXCL12 (promotes retention and expansion of HSCs), stem cell factor (SCF, stimulates hematopoiesis), vascular cell adhesion molecule 1 (VCAM-1, promotes HSC retention), secreted protein angiopoietin 1 (ANG1, promotes quiescence or HSC stemness or ability to self-replicate), and transforming growth factor-beta (TGF-β, regulates quiescence and self-renewal).7 The sympathetic nervous system acts with SCF to promote HSC maintenance and retention and regulates HSC mobilization.8 The perivascular niche is also essential for gas exchange and delivery of nutrients and waste removal in the bone marrow.9
Hematopoietic marrow is vascularized by the primary arteries of the bones. Arterial vessels enter the bone, branch near the endosteum into smaller arterioles and capillaries, and drain into venous sinusoids. These sinuses coalesce into a large central sinus in the central cavity. Hematopoietic marrow and yellow marrow fill the spaces surrounding the network of venous sinuses. Newly produced blood cells traverse narrow openings between endothelial cells (diapedesis) in the venous sinus walls and thus enter the circulation. The low blood flow of the arteriole/sinusoid network limits gas exchange, creates a relatively hypoxic microenvironment, and maintains HSC dormancy.
Cellular Differentiation
All humans originate from a single cell (the fertilized egg) that has the capacity to proliferate and eventually differentiate into the huge diversity of cells of the human body. After fertilization, the egg divides over a 5-day period to form a hollow ball (blastocyst) that implants on the uterus. Until about 3 days after fertilization, each cell (blastomere) is undifferentiated and retains the capacity to differentiate into any cell type. During this period, the outer layer of cells of the blastocyte has undergone differentiation and commitment to become the placenta. Cells of the inner cell mass, however, continue to have unlimited differentiation potential (currently referred to as being embryonic pluripotent stem cells) and can grow into different kinds of tissue—blood, nerves, heart, bone, and so forth. After implantation, cells of the inner cell mass begin differentiation into other cell types. Differentiation is a multistep process and results in intermediate groups of stem cells with more limited, but still impressive, abilities to differentiate into many different types of cells. These are known as multipotent stem cells and include the HSCs (Fig. 28.9).
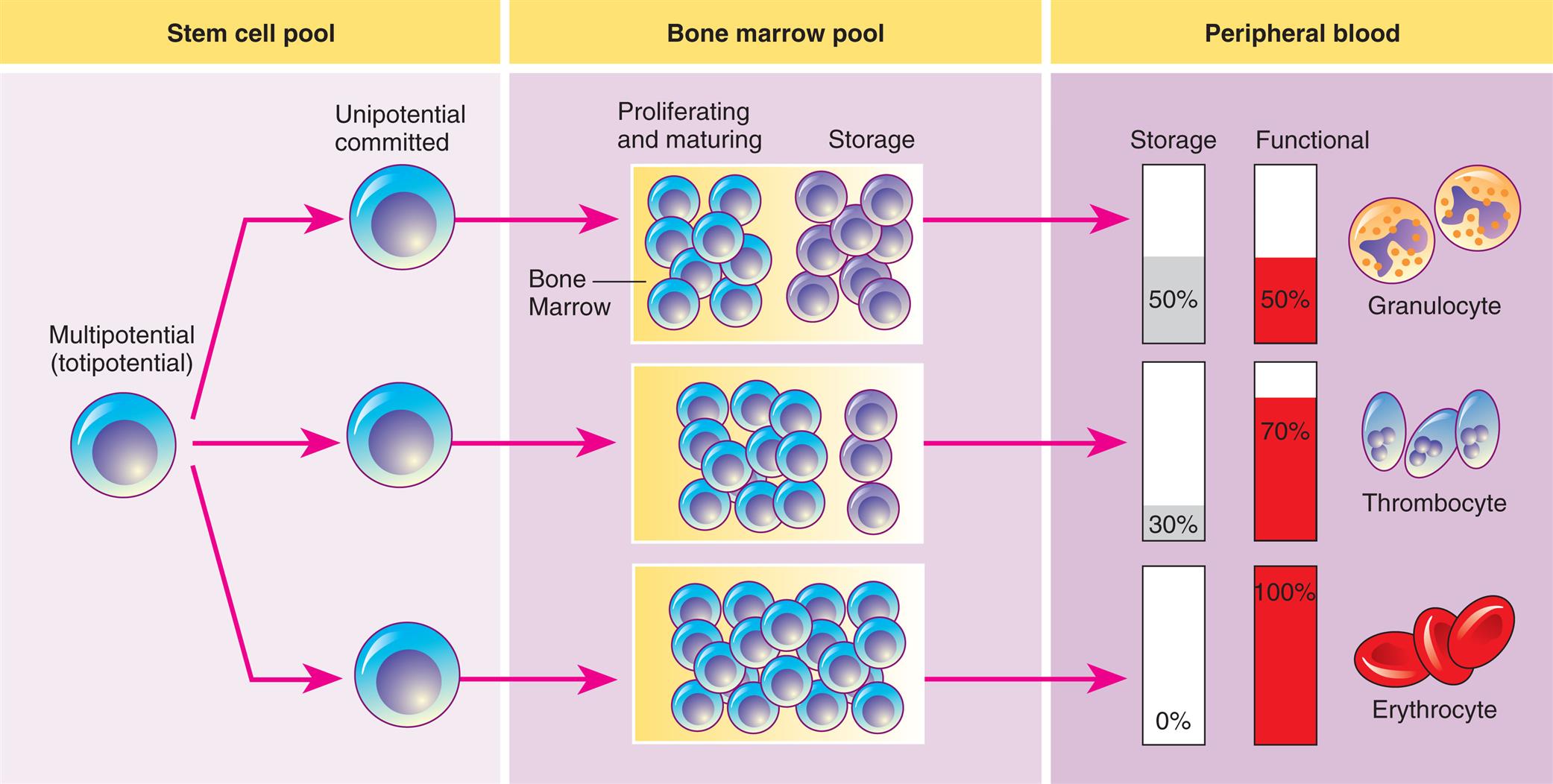
Hematopoiesis from the stem cell pool; activity mainly in the bone marrow and in the peripheral blood.
A three-panel illustration shows the process of hematopoiesis. Left panel, stem cell pool. A multipotential (totipotential) cell differentiates to unipotential committed cells. Middle panel, bone marrow pool. Half the pool proliferates and matures, while the other half is stored; most of the pool proliferates and matures, while the rest is stored; or the entire pool proliferates and matures. Right panel, peripheral blood. There are three types of cells. • Granulocyte: 50 percent stored and 50 percent functional. • Thrombocyte: 30 percent stored and 70 percent functional. • Erythrocyte: 0 percent stored and 100 percent functional.
Within the bone marrow niches, each type of blood cell originates from HSCs that proliferate and differentiate under control of a variety of cytokines and growth factors (Table 28.4). As with all stem cells, the HSCs are self-renewing (they have the ability to proliferate without further differentiation) so that a relatively constant population of stem cells is available. Some HSCs will continue differentiation into hematopoietic progenitor cells. Progenitor cells retain proliferative capacity but are committed to possible further differentiation into particular (unipotent) lineages of hematologic cells. The cell lineages include lymphoid lineages (T and B lymphocytes, NK cells) and myeloid lineages (monocytes, macrophages, neutrophils, basophils, eosinophils, megakaryocyte/platelets, and erythrocytes) (Fig. 28.10).
Table 28.4
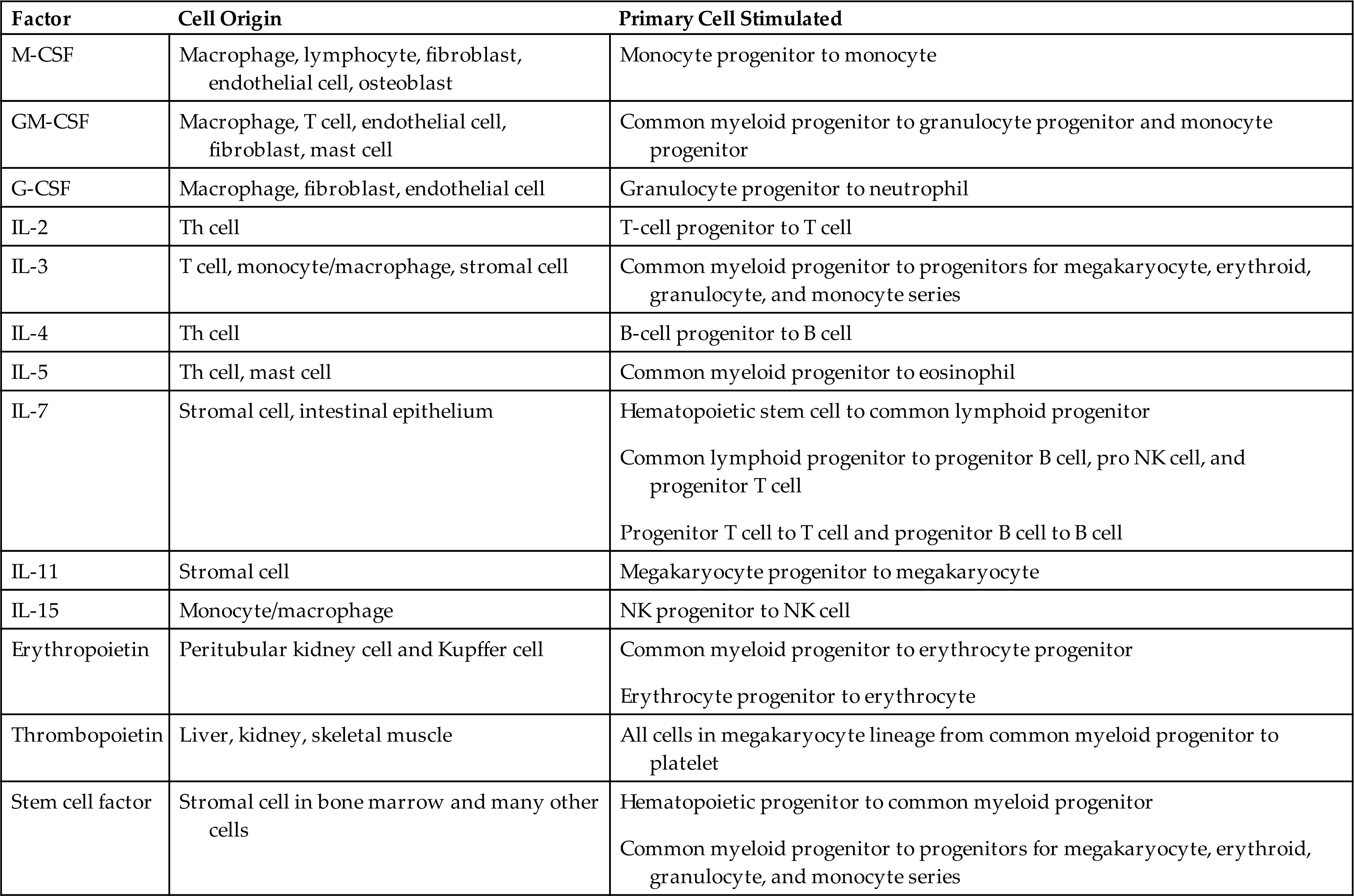
G-CSF, Granulocyte colony-stimulating factor; GM-CSF, granulocyte-macrophage colony–stimulating factor; IL, interleukin; M-CSF, macrophage colony-stimulating factor; NK, natural killer; pro, progenitor; Th, T helper.
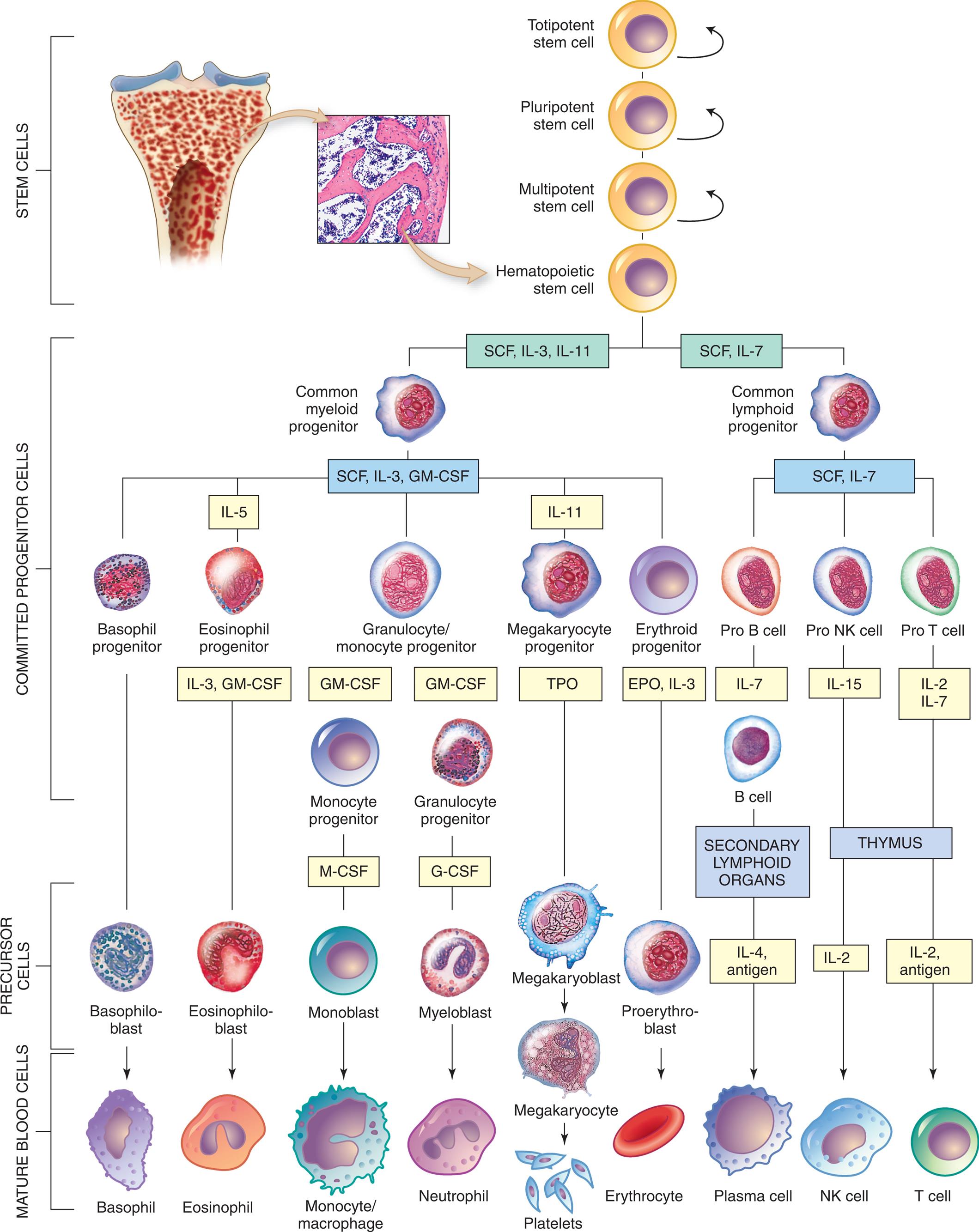
Curved arrows indicate proliferation and expansion of prehematopoietic stem cell populations. Yellow boxes are the hematopoietic growth factors needed for proliferation and differentiation (see Table 28.4) EPO, Erythropoietin; G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte-macrophage colony–stimulating factor; IL, interleukin; M-CSF, macrophage colony-stimulating factor; NK, natural killer; SCF, stem cell factor; TPO, thrombopoietin. (Modified from Patton K. Anatomy and Physiology. 10th ed. Elsevier; 2019.)
An illustrated flowchart tracks the differentiation of hematopoietic cells from stem cells through committed progenitor cells and precursor cells to mature blood cells. Stem cells: The cells can be totipotent, pluripotent, multipotent, or hematopoietic. The differentiation of the committed progenitor cells is as follows. 1. Hematopoietic cells. Leads to 2 and 3. 2. S C F, I L-3, I L-11. Leads to 4. 3. S C F, I L-7. Leads to 5. 4. Common myeloid progenitor. Leads to 6, 7, 8, 9, and 10. 5. Common lymphoid progenitor. Leads to 11, 12, and 13. 6. Basophil progenitor. 7. I L-5: Eosinophil progenitor (I L-3, G M-C S F). 8. Granulocyte or monocyte progenitor (G M-C S F). 9. I L-11: Megakaryocyte progenitor (T P O). 10. Erythroid progenitor (E P O, I L-3). 11. Pro B cell (I L-7). 12. Pro N K cell (I L-15). 13. Pro T cell (I L-2, I L-7). The differentiation of the precursor cells and mature blood cells is as follows. • Basophiloblast from basophil progenitor; to basophil. • Eosinophiloblast from I L-3 and G M-C S F; to eosinophil. • Monoblast from M-C S F of monocyte progenitor (G M-C S F); to monocyte or macrophage. • Myeloblast from G-C S F of granulocyte progenitor (G M-C S F); to neutrophil. • Megakaryoblast from T P O; to megakaryocyte and subsequently to platelets. • Proerythroblast from E P O and I L-3; to erythrocyte. • I L-4 and antigen from secondary lymphoid of B cell (I L-7); to plasma cell. • I L 2 from thymus (I L -15); to N K cell. • I L-2 and antigen from thymus (I L-2 and I L-7); to T cell.
Several cytokines participate in hematopoiesis, particularly colony-stimulating factors (CSFs or hematopoietic growth factors), which stimulate the proliferation of progenitor cells and their progeny and initiate the maturation events necessary to produce fully mature cells (see Table 28.4 and Fig. 28.10). Multiple cell types in hematopoietic organs, including endothelial cells, fibroblasts, granulocytes, monocytes, and lymphocytes, produce the necessary CSFs
Hematopoiesis in the bone marrow occurs in two separate pools: the stem cell pool and the bone marrow pool (see Fig. 28.9). The stem cell pool is the product of self-renewal that maintains the number of pluripotent stem cells and partially committed progenitor cells. The bone marrow pool, the largest pool, contains cells that are proliferating and maturing in preparation for release into the circulation and mature cells that are stored for later release into the peripheral blood. The peripheral blood also contains two pools of cells: those in the circulation and those stored around the walls of the blood vessels (often called the marginating storage pool). The marginating storage pool primarily consists of neutrophils that adhere to the endothelium in vessels where the blood flow is relatively slow. These cells can rapidly move into tissues and mucous membranes when needed (e.g., during an inflammatory response).
Under certain conditions, the levels of circulating hematologic cells need to be rapidly replenished. Medullary hematopoiesis can be accelerated by any or all of three mechanisms: (1) conversion of yellow bone marrow, which does not produce blood cells, to red marrow, by the actions of erythropoietin (a hormone produced primarily by the kidney that stimulates erythrocyte production); (2) faster proliferation of stem cells into progenitor cells; and (3) faster differentiation of progenitor cells.
Hematopoiesis
The typical human requires about 100 billion new blood cells per day. Hematopoiesis is the production of blood cells. Blood cell production is constantly ongoing, occurring in the liver and spleen of the fetus, but only within the bone marrow (medullary hematopoiesis) after birth. This process involves the biochemical stimulation of populations of relatively undifferentiated cells to undergo mitotic division (i.e., proliferation) and maturation (i.e., differentiation) into mature hematologic cells (Fig. 28.10). Although proliferation and differentiation are usually sequential, certain blood cells proliferate and differentiate simultaneously. Erythrocytes and neutrophils generally differentiate fully before entering the blood, but monocytes and lymphocytes continue to mature in the blood and in secondary lymphatic organs.
Hematopoiesis continues throughout life, increasing in response to a need to replenish aged or destroyed circulating cells or in response to infection. In general, long-term stimuli, such as chronic diseases, cause a greater increase in hematopoiesis than do acute conditions, such as hemorrhage. Various abnormalities in medullary hematopoiesis have been identified and are discussed in Chapter 29.
Extramedullary hematopoiesis is blood cell production in tissues other than bone marrow, primarily the liver and spleen, but less frequently in the lymph nodes, adrenal glands, cartilage, adipose tissue, intrathoracic areas, and kidneys. Extramedullary hematopoiesis is usually a sign of disease and can occur in pernicious anemia, sickle cell anemia, thalassemia, hemolytic disease of the newborn (erythroblastosis fetalis), hereditary spherocytosis (abnormal sphere-shaped RBCs), and certain leukemias.
Development of Erythrocytes
Erythropoiesis is the development of RBCs. In the confines of the bone marrow, progenitor cells proliferate and differentiate into large, nucleated proerythroblasts, which are committed to producing erythroid cells (Fig. 28.11). The proerythroblast, which has ribosomes and can produce protein, differentiates through several intermediate forms of erythroblast (sometimes called normoblast) while progressively eliminating most intracellular structures (including the nucleus), synthesizing hemoglobin, and becoming more compact, eventually assuming the shape and characteristics of an erythrocyte.
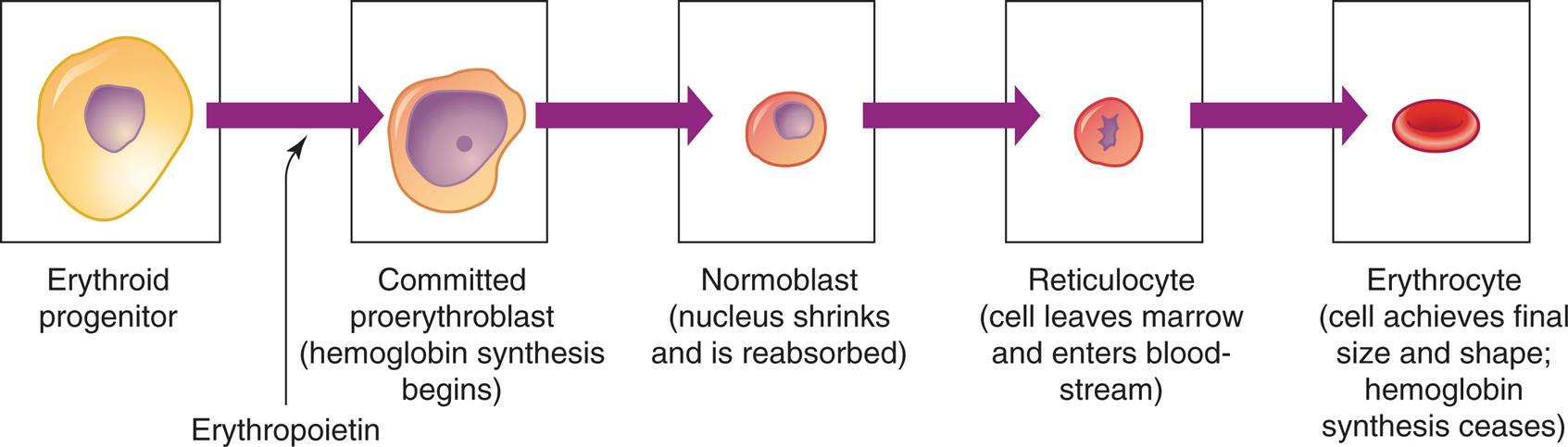
Erythrocyte differentiation from large, nucleated progenitor cells to small nonnucleated erythrocytes.
A series of illustration shows the differentiation of erythrocyte. The sequence of differentiation is as follows. • Erythroid progenitor. • Erythroid progenitor in the presence of erythropoietin develops into committed proerythroblast. • Committed proerythroblast (hemoglobin synthesis begins). • Normoblast (nucleus shrinks and is reabsorbed). • Reticulocyte (cell leaves marrow and enters blood-stream). • Erythrocyte (cell achieves final size and shape; hemoglobin synthesis ceases).
The last immature form is the reticulocyte, which is anucleate and contains a mesh-like (reticular) network of ribosomal ribonucleic acid (rRNA). The reticulocyte contains polyribosomes (for globin synthesis) and mitochondria (for oxidative metabolism and heme synthesis). The reticulocyte matures into an erythrocyte within 24 to 48 hours. During this period, mitochondria and ribosomes disappear and the cell becomes smaller and more disc-like. With these final changes, the erythrocyte loses its capacity for hemoglobin synthesis and oxidative metabolism. Reticulocytes remain in the marrow approximately 1 day and are released into the venous sinuses. They continue to mature in the bloodstream and may travel to the spleen for several days of additional maturation. The normal reticulocyte count is 1% of the total RBC count. Approximately 1% of the body’s circulating erythrocyte mass normally is generated every 24 hours. Therefore, the reticulocyte count is a useful clinical index of erythropoietic activity and indicates whether new red cells are being produced.
Regulation of erythropoiesis
In healthy humans, the total volume of circulating erythrocytes remains surprisingly constant. Most steps of erythropoiesis are primarily under the control of a feedback loop involving erythropoietin (EPO) and other cytokines (see Table 28.4). In conditions of tissue hypoxia, EPO is secreted primarily by the peritubular cells of the kidney (Fig. 28.12). Rising circulating levels of EPO cause a compensatory increase in erythrocyte production. The normal steady-state rate of production of 2.5 million erythrocytes per second can increase to 17 million per second under anemic or low-oxygen states, such as high-altitude environments or pulmonary disease. Thus the body responds to reduced oxygenation of blood in two ways: (1) by increasing the intake of oxygen through increased respiration and (2) by increasing the oxygen-carrying capacity of the blood through increased erythropoiesis.
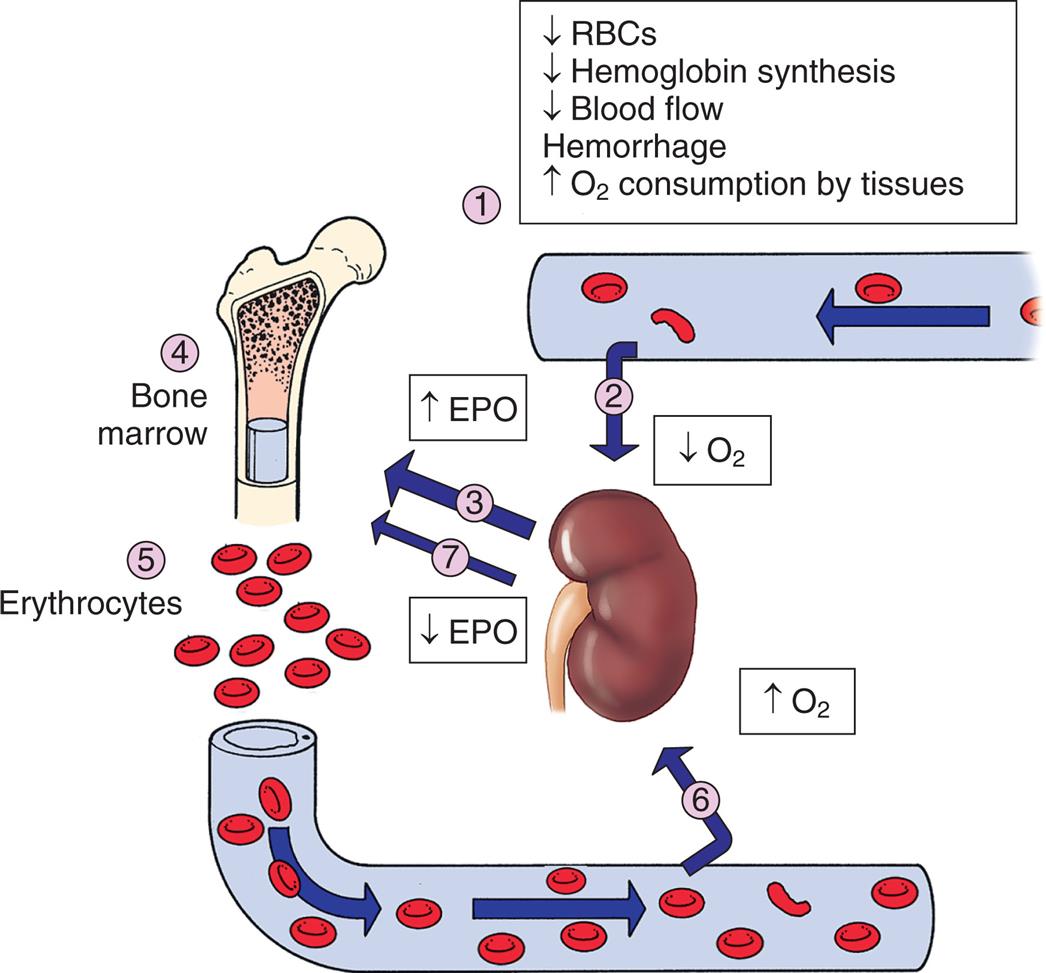
(1) Decreased arterial oxygen levels result in decreased tissue oxygen (hypoxia) that (2) stimulates the kidney to increase production (3) of erythropoietin. Erythropoietin is carried to the bone marrow (4) and binds to erythropoietin receptors on proerythroblasts, resulting in increased red cell production (5). The increased release of red cells into the circulation frequently corrects the hypoxia in the tissues. Perception of normal oxygen levels by the kidney (6) causes diminished production (7) of erythropoietin (negative feedback) and a return to normal levels of erythrocyte production. EPO, Erythropoietin; O2, oxygen in the blood and tissue; RBCs, red blood cells.
An illustration shows the role of erythropoietin in regulation of erythropoiesis. 1. Decreased R B Cs, decreased hemoglobin synthesis, decreased blood flow, hemorrhage, and increased oxygen consumption by tissues stimulate the kidney. 2. Oxygen level decreases. 3. E P O increases. 4. Triggers the bone marrow. 5. Erythrocytes increase. 6. Oxygen level increases. 7. E P O decreases.
Recombinant human erythropoietin (r-HuEPO) is used in individuals with anemia secondary to decreased EPO from chronic renal failure. An immediate effect of EPO administration is an increase in the blood reticulocyte count, followed by increasing levels of erythrocytes. The most significant side effect is increased blood pressure.
Hemoglobin synthesis
Erythrocytes form hemoglobin in the bone marrow. Hemoglobin (Hb), the oxygen-carrying protein of the erythrocyte, constitutes approximately 90% of the cell’s dry weight. Hb-packed blood cells take up oxygen in the lungs and exchange it for carbon dioxide in the tissues. A single erythrocyte can contain as many as 300 hemoglobin molecules. Hb increases the oxygen-carrying capacity of blood by 100-fold. Each Hb molecule is composed of two pairs of polypeptide chains (the globins) and four colorful complexes of iron plus protoporphyrin (the hemes), which are responsible for the blood’s ruby-red color and oxygen-carrying capacity (Fig. 28.13).
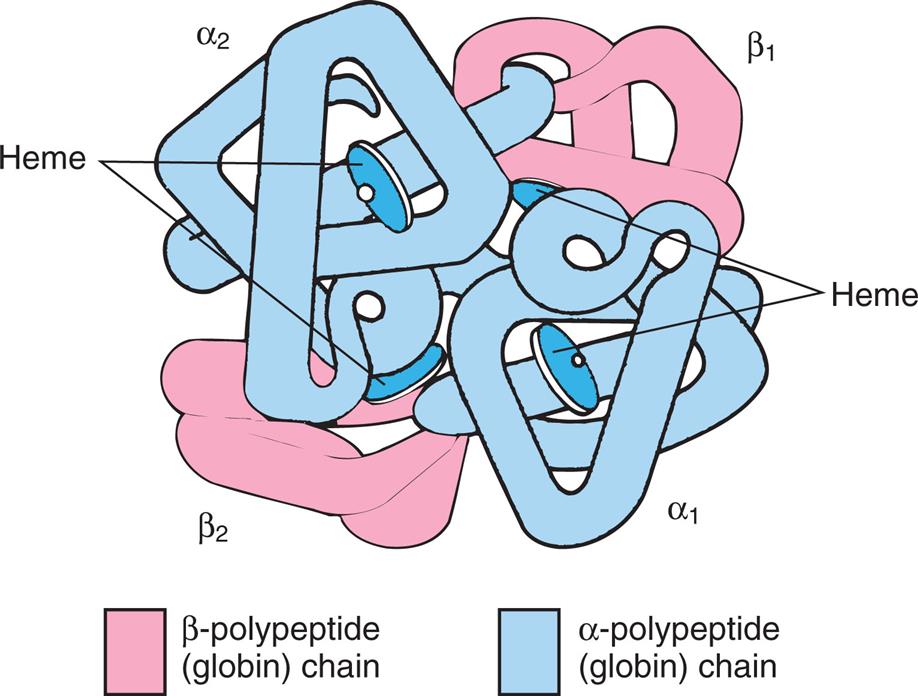
Molecule is spherical and contains a pair of α-polypeptide chains, a pair of β-polypeptide chains, and four heme groups.
Each polypeptide chain contains approximately 150 amino acids and is arranged in a knotted-sausage configuration. The chains assemble to form a tetrahedron containing two pairs of identical chains. Several variants of Hb exist (Table 28.5), and they differ only slightly in primary structure based on the use of different polypeptide chains: alpha, beta, gamma, delta, epsilon, or zeta (α, β, γ, δ, ε, or ζ, respectively). Hb A, the most common type in adults, is composed of two α- and two β-polypeptide chains (α2β2) (see Fig. 28.13). A normal variant, fetal Hb (Hb F), is a complex of two α- and two γ-polypeptide chains (α2γ2) that binds oxygen with a much greater affinity than adult Hb. Other variants are genetic mutations associated with various diseases (see Chapter 29).10
Table 28.5
| Type of Hemoglobin (Hb) | Identity of Polypeptide Chain | Significance |
|---|---|---|
| HbA | α2β2 | 92% of adult Hb |
| HbA1c | α2 (β-NH-glucose) | 5% of adult Hb; increased in diabetes (see Chapter 22) |
| HbA2 | α2δ2 | 2% of adult Hb; increased in β-thalassemia (see Chapter 29) |
| HbF | α2γ2 | Major fetal Hb from the third through ninth month of gestation; promotes oxygen transfer across platelets; increased in β-thalassemia |
| Hb Gower I | ε4 or ζ2β2 | Present in early embryo; function unknown |
| Hb Gower II | α2ε2 | Present in early embryo; function unknown |
| Hb Portland | ζ2γ2 | Present in early embryo; function unknown |
Heme is a large, flat, iron-protoporphyrin disc that is synthesized in the mitochondria and can carry one molecule of oxygen (O2). Thus, an individual Hb molecule with its four hemes can carry four oxygen molecules. If all four oxygen-binding sites are occupied by oxygen, the molecule is said to be saturated. Through a series of biochemical reactions, protoporphyrin, a complex four-ringed molecule, is produced and bound with ferrous iron. It is crucial that the iron be correctly charged; reduced ferrous iron (Fe2+) can bind oxygen in the lungs and release it in the tissues, whereas ferric iron (Fe3+) cannot. Binding of O2 to ferrous iron temporarily oxidizes Fe2+ to Fe3+ (oxyhemoglobin), but after the release of O2 the body reduces the iron to Fe2+ and reactivates the Hb (deoxyhemoglobin [reduced hemoglobin]). Without reactivation, the Fe3+-containing Hb (methemoglobin) cannot bind O2. An excess of ferric iron occurs with certain drugs and chemicals, such as nitrates and sulfonamides, and reduces oxygen-carrying capacity.
Hb undergoes a conformational change when binding O2. When one of the iron molecules binds O2, the porphyrin ring changes shape, increasing the exposure of the three remaining iron atoms to O2. This greatly increases the affinity for the oxygen-carrying capacity of Hb, as occurs in the lungs. When oxygen is unloaded from Hb, the oxygen-carrying capacity of hemoglobin is low, facilitating the transport of carbon dioxide back to the lungs.
Several other molecules can competitively bind to deoxyhemoglobin. Carbon monoxide (CO) directly competes with O2 for binding to Fe2+ with an affinity that is about 200-fold greater than that of O2. Thus even a small amount of CO can dramatically decrease the ability of Hb to bind and transport O2. Hb also binds carbon dioxide (CO2), but at a binding site separate from where O2 binds. In the lungs, CO2 is released, allowing hemoglobin to bind O2.
Erythrocytes may also play a role in the maintenance of vascular relaxation and blood flow. Nitric oxide (NO) produced by blood vessels is a major mediator of relaxation and dilation of the vessel walls. In the lungs, Hb can concurrently bind O2 to the Fe2+ and NO to cysteine residues in the globins. As Hb transfers its O2 to tissue, it may also shed small amounts of NO, contributing to dilation of the blood vessels and helping transfer of the O2 into tissues.
Nutritional requirements for erythropoiesis
Normal development of erythrocytes and synthesis of hemoglobin depend on an optimal biochemical state and adequate supplies of the necessary building blocks, including protein, vitamins, and minerals (Table 28.6). If these components are lacking for a prolonged time, erythrocyte production slows and anemia (insufficient numbers of functional erythrocytes) may result (see Chapter 29).
Table 28.6
| Nutrient | Role in Erythropoiesis | Consequence of Deficiencya |
|---|---|---|
| Protein (amino acids) | Structural component of plasma membrane | Decreased strength, elasticity, and flexibility of membrane; hemolytic anemia |
| Synthesis of hemoglobin | Decreased erythropoiesis and life span of erythrocytes | |
| Cobalamin (vitamin B12) | Synthesis of DNA, maturation of erythrocytes, facilitator of folate metabolism | Macrocytic (megaloblastic) anemia |
| Folate (folic acid) | Synthesis of DNA and RNA, maturation of erythrocytes | Macrocytic (megaloblastic) anemia |
| Vitamin B6 (pyridoxine) | Heme synthesis | Microcytic-hypochromic anemia |
| Vitamin B2 (riboflavin) | Oxidative reactions | Normocytic-normochromic anemia |
| Vitamin C (ascorbic acid) | Iron metabolism, acts as reducing agent to maintain iron in its ferrous (Fe2+) form | Normocytic-normochromic anemia |
| Pantothenic acid | Heme synthesis | Unknown in humansb |
| Niacin | None, but needed for respiration in mature erythrocytes | Unknown in humans |
| Vitamin E | Heme synthesis (?); protection against oxidative damage in mature erythrocytes | Hemolytic anemia with increased cell membrane fragility; shortens life span of erythrocytes in individual with cystic fibrosis |
| Iron | Hemoglobin synthesis | Iron deficiency anemia |
| Copper | Optimal mobilization of iron from tissues to plasma | Microcytic-hypochromic anemia |
DNA, Deoxyribonucleic acid; RNA, ribonucleic acid.
aSee Chapter 29.
bAlthough pantothenic acid is important for optimal synthesis of heme, experimentally induced deficiency failed to produce anemia or other hematopoietic disturbances.
Data from Lee GR, et al. Wintrobe’s Clinical Hematology, 9th edition. Philadelphia: Lea & Febiger; 1993; Harmening DM. Clinical hematology and fundamentals of hemostasis, 3rd edition. Philadelphia: FA Davis; 1997.
Erythropoiesis cannot proceed in the absence of vitamins, especially B12, folate (folic acid), B6, riboflavin, pantothenic acid, niacin, ascorbic acid, and vitamin E. Dietary vitamin B12 is a large molecule that requires a protein secreted by parietal cells into the stomach (intrinsic factor [IF]) for transport across the ileum. Once absorbed, vitamin B12 is stored in the liver and used as needed in erythropoiesis. Defects in IF production lead to decreased B12 absorption, which may lead to pernicious (megaloblastic) anemia.
Folate is the second most important vitamin for erythrocyte production and maturation. Folate is necessary for DNA and ribonucleic acid (RNA) synthesis in precursor RBCs. Lack of folate results in an increase in abnormally large, nucleated precursor RBCs (megaloblasts) with a reduced capacity to carry O2. Folate is absorbed principally in the upper small intestine and is stored in the liver. Folate deficiency is more common than vitamin B12 deficiency and occurs more rapidly. Folate stores can be depleted within a few months, whereas vitamin B12 depletion can take years. Folate supplements are prescribed for pregnant women because pregnancy increases the demand for folate. Supplements may prevent anemia and can protect against neural tube defects because folate is also required for embryonic neural tube closure (see Chapter 20).
Normal destruction of senescent erythrocytes
After about 120 days in the circulation, old erythrocytes are removed by tissue macrophages, primarily in the spleen.11 Although mature erythrocytes lack nuclei, mitochondria, and the endoplasmic reticulum, they do have cytoplasmic enzymes capable of glycolysis (anaerobic glucose metabolism) and production of small quantities of adenosine triphosphate (ATP). ATP provides the energy needed to maintain cell function and keep its plasma membrane pliable. Metabolic processes diminish as the erythrocyte ages, so less ATP is available to maintain plasma membrane function. The aged or senescent red cell becomes increasingly fragile and loses its reversible deformability due to disruption of the anchorage between the cytoskeleton and the plasma membrane, thus becoming susceptible to rupture while passing through narrowed regions of the microcirculation.
Additionally, the plasma membrane of senescent red cells undergoes phospholipid rearrangement, signaling macrophages (primarily in the spleen) to selectively remove and sequester the senescent red cells. If the spleen is dysfunctional or absent, macrophages in the liver (Kupffer cells) assume control of this process. The erythrocytes are digested by proteolytic and lipolytic enzymes in the phagolysosomes (digestive vacuoles) of the macrophage. The heme and globin dissociate easily, and the globin is broken down into its component amino acids. The iron in hemoglobin is oxidized, forming Fe3+ (methemoglobin), and recycled.
During digestion of Hb in the macrophage, porphyrin reduces to unconjugated bilirubin, which is conjugated (made water soluble) and excreted from the liver into the intestine or into bile (Fig. 28.14). Bacteria in the intestinal lumen transform conjugated bilirubin into urobilinogen. Although a small portion is reabsorbed and excreted by the kidneys, most urobilinogen is excreted in feces. Conditions causing accelerated erythrocyte destruction increase the load of serum unconjugated bilirubin, increased liver conjugation of bilirubin, and increased urinary excretion of urobilinogen. Gallstones (cholelithiasis) can result from a chronically elevated rate of bilirubin excretion.
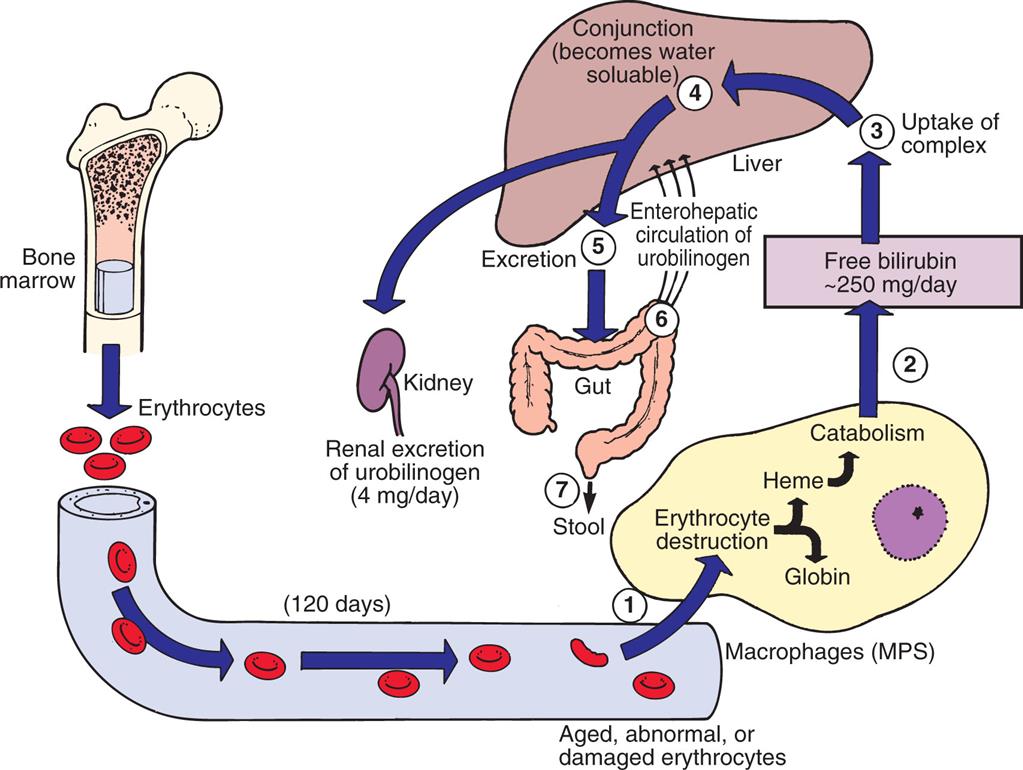
(1) Senescent red blood cells (RBCs) are consumed by macrophages. During digestion, (2) the porphyrin in the RBC reduces to bilirubin. (3) Bilirubin is taken up by the liver, where it is (4) conjugated by glucuronyl transferase and then is (5) excreted as bile. (6) Bacteria in the intestine transform conjugated bilirubin into urobilinogen, (7) most of which is excreted in feces. MPS, Mononuclear phagocyte system.
An illustration shows the metabolism of bilirubin released by heme. Erythrocytes from the bone marrow, at 120 days, become aged, abnormal, or damaged. 1. Erythrocytes are attracted to the macrophages (M P S), where they are destructed as heme or globin. 2. Heme undergoes catabolism releasing free bilirubin by 250 milligrams per day. 3. Uptake of complex. 4. Conjunction (becomes water soluble). 5. Excretion. 6. Enterohepatic circulation of urobilinogen. 7. Urobilinogen is excreted as stool. Conjunction also triggers the kidney, supporting the renal excretion of urobilinogen of 4 milligrams per day.
Iron cycle
Approximately 67% of total body iron is bound to heme in erythrocytes (Hb), and about 5% to 10% is bound to heme-containing myoglobin in muscle cells. Approximately 30% is stored in mononuclear phagocytes (i.e., macrophages) and hepatic parenchymal cells as either ferritin (iron bound to protein) or hemosiderin (intracellular bound iron). The remaining 3% (less than 1 mg) is lost daily in urine, sweat, bile, sloughing of epithelial cells from the skin and intestinal mucosa, and minor bleeding, as occurs with menstruation in women. There is no excretory mechanism for iron. Unbound, or free, iron is toxic to human cells. Approximately 25 mg of iron is required daily for erythropoiesis; only 1 to 2 mg of iron is dietary, and the remainder is obtained from continual recycling of iron from erythrocytes through a process known as the iron cycle.12 Pregnant women require more iron to meet the needs of the placenta and growing fetus.
The methemoglobin released from the breakdown of senescent or damaged erythrocytes is dissociated by the enzyme heme oxygenase, and the iron is released into the bloodstream, where it binds again to transferrin or is stored in the macrophage’s cytoplasm as ferritin or hemosiderin (Fig. 28.15). A minute amount of iron is stored in muscle cells by the heme-containing protein myoglobin. Unavailable stores of iron are present in cytochromes, catalases, and peroxidase enzymes.
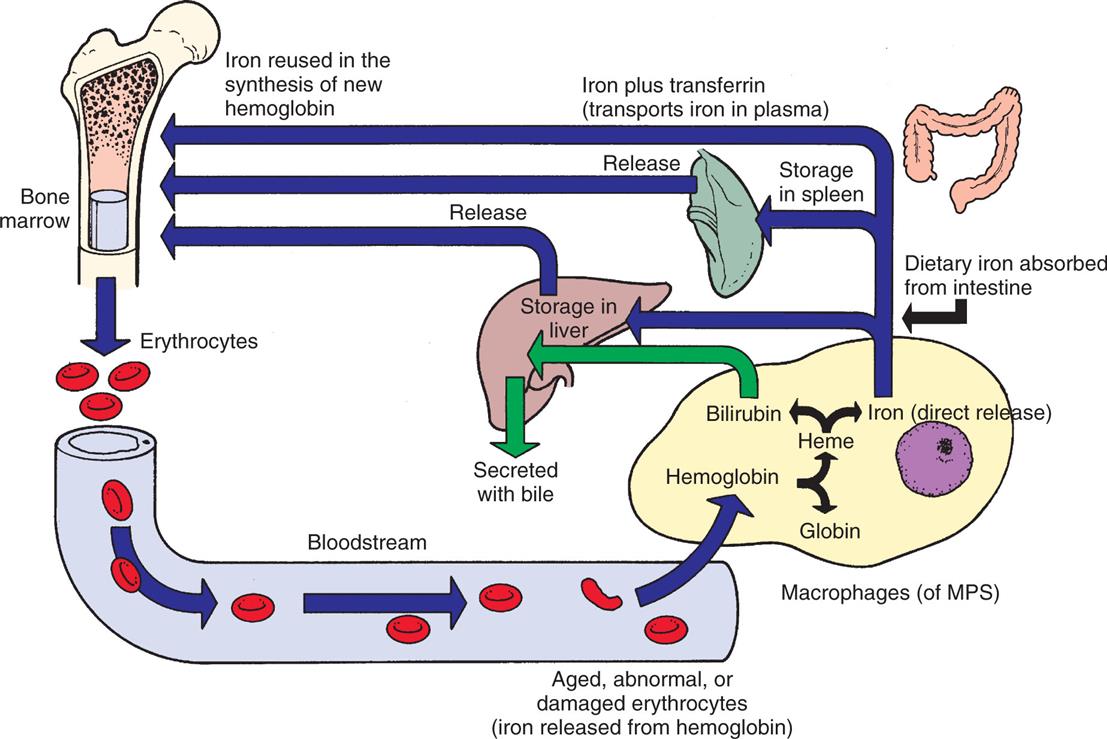
Iron released from gastrointestinal epithelial cells circulates in the bloodstream associated with its plasma carrier, transferrin. It is delivered to erythroblasts in bone marrow, where most of it is incorporated into hemoglobin. Mature erythrocytes circulate for approximately 120 days, after which they become senescent and are removed by the mononuclear phagocyte system (MPS). Macrophages of the MPS (mostly in the spleen) break down ingested erythrocytes and return iron to the bloodstream directly or after storing it as ferritin or hemosiderin.
An illustration shows the iron cycle. Erythrocytes from the bone marrow enter the blood stream and tend to become aged, abnormal, or damaged (iron released from hemoglobin). These erythrocytes are consumed by macrophages (M P S), where the hemoglobin is destructed as heme or globin. The heme destructs as bilirubin, which is stored in the liver or secreted with bile, or as iron, which is released directly. Dietary iron absorbed from the intestine is: • Stored in the liver or released into the bone marrow. • Stored in the spleen or released into the bone marrow. • Iron plus transferrin (transports iron in plasma). • Iron reused in the synthesis of new hemoglobin in the bone marrow.
Ferritin is the major intracellular iron storage protein. Apoferritin is ferritin without attached iron and can store thousands of atoms of iron. Apoferritin binds to free Fe2+ and stores it as Fe3+, becoming ferritin. Excess accumulations of iron produce large intracellular iron storage complexes, known as hemosiderin. Hemosiderin is visible as an iron-based pigment under a light microscope as cell inclusions. The iron within deposits of hemosiderin is poorly available because it is not water-soluble, but it is available when iron requirements increase. The most common cause of hemosiderin deposition is simple bruising. Hemosiderin in small amounts within iron-rich tissues (i.e., spleen, liver, bone marrow) is considered normal. Large aggregates or its presence in tissue, such as the lungs or subcutaneous tissue, suggests a pathologic condition known as hemosiderosis.
Iron from either dietary sources, release of iron stores, or erythrocyte catabolism is transported in the blood bound to apotransferrin, thus becoming transferrin. Under normal conditions, only one-third of the iron-binding sites on transferrin molecules are occupied. Apotransferrin is a glycoprotein synthesized primarily by hepatocytes in the liver but also produced in small quantities by tissue macrophages, submaxillary and mammary glands, and ovaries or testes. Iron for hemoglobin production is carried by transferrin to the bone marrow, where it binds to transferrin receptors on erythroblasts. Transferrin receptors are on the plasma membrane of all nucleated cells, but at particularly high levels on erythroid precursors and rapidly proliferating cells (e.g., lymphocytes), and are thought to be the only route of cellular entry for transferrin-attached iron. Transferrin is recycled (transferrin cycle) by intracellular dissociation of iron from transferrin and secretion of the resultant apotransferrin to the bloodstream. The iron is transported to the erythroblast’s mitochondria (the site of Hb production), where the enzyme heme synthetase inserts Fe2+ into protoporphyrin to form heme. Heme then is bound to globin to form Hb. Iron not used in erythropoiesis is stored temporarily as ferritin or hemosiderin and later excreted or recycled.
Splenic red pulp macrophages are specialized for iron recycling with increased expression of proteins for the uptake of hemoglobin, the breakdown of heme, and the export of iron.13 The body’s iron homeostasis is primarily controlled by the hormone hepcidin. Hepcidin is the regulator of the systemic transportation of iron.14 It is a protein synthesized in the liver and released into the plasma, where it is bound with high affinity to α2-macroglobulin and with less affinity to albumin. Hepatocellular hepcidin production is regulated physiologically by the levels of iron in the body, rate of erythropoiesis, and percentage of oxygen saturation. Hepatocytes (liver cells) sense levels of circulating iron by means of receptors for transferrin. Excess iron is stored in hepatocytes and macrophages, and hepatocytes sense these levels by means of receptors for bone morphogenetic protein (BMP), most likely BMP-6, which is a growth factor produced to a large extent by bone marrow sinusoid endothelial cells. Hepcidin production also can be induced by inflammation through IL-6 and other inflammatory cytokines, produced in response to infection or chronic conditions such as cancer.
Hepcidin regulates iron levels through its binding capacity to ferroportin, which is a transmembrane iron exporter found in the plasma membrane of cells that transport or store iron, including macrophages, hepatocytes, enterocytes (intestinal cells), and erythrocytes. The body’s total iron balance is maintained through controlled absorption rather than excretion. Dietary iron (primarily as Fe2+) is transported directly across the membranes of enterocytes in the duodenum and proximal jejunum and is carried in the plasma by transferrin. (Transport mechanisms are described in Chapter 1.) Hepcidin induces internalization and degradation of ferroportin, thus leading to increased intracellular iron stores, decreased dietary iron absorption, and decreased levels of circulating iron. Decreased production of hepcidin leads to release of stored iron and increased dietary absorption. Thus, if the body’s iron stores are low or the demand for erythropoiesis increases, dietary iron is transported rapidly through the epithelial cell and into the plasma. If body stores are high and erythropoiesis is not increased, iron transport is stopped. Hepcidin production also can be induced by inflammation. During inflammation, increased levels of hepcidin lead to sequestering of iron as a result of down-regulation of ferroportin expression on macrophages and enterocytes and thus can cause anemia.
Development of Leukocytes
Leukocytes consist of lymphocytes, granulocytes, and monocytes (their pathways of differentiation are shown in Fig. 28.10). Most of the leukocytes arise from stem cells in the bone marrow. HSCs differentiate into two populations of progenitor cells: common lymphoid progenitors and common myeloid progenitors. Lymphopoiesis (lymphocytopoiesis) is the generation of lymphocytes from lymphoid progenitor cells released into the bloodstream to undergo further maturation in the primary and secondary lymphoid organs (see Chapter 8).
Myelopoiesis is the development of granulocytes (neutrophils, eosinophils, and basophils) and monocytes from the differentiation of myeloid progenitor cells in the bone marrow. The common myeloid progenitors differentiate into progenitors for basophils, mast cells, eosinophils, and megakaryocytes, and into granulocyte/monocyte progenitors. The granulocyte/monocyte progenitors further differentiate into monocyte progenitors and granulocyte progenitors, which develop into monocytes/macrophages and neutrophils, respectively. Development from HSCs to common granulocyte-monocyte progenitors primarily is under the control of SCF, IL-3, and GM-CSF, whereas further differentiation into granulocytic and monocytic progenitors is controlled by granulocyte colony stimulating factor (G-CSF) and macrophage stimulating factor (M-CSF), respectively (see Table 28.4 and Fig. 28.10).
Monocytic progenitors undergo development into monocytes within 24 hours and are released into the circulation. Monocytes mature into various forms of macrophages, a process that is usually completed within 1 or 2 days after their release.
Progenitor cells for granulocytes normally fully mature in the bone marrow into neutrophils, eosinophils, and basophils. The ultimate phenotype is determined by relative local bone marrow concentrations of early- and late-acting cytokines, including GM-CSF, G-CSF, IL-3, IL-5, SCF, and others (see Table 28.4). Granulocytes are released into the blood within 10 to 14 days of development. The bone marrow selectively retains immature granulocytes as a reserve pool that can be rapidly mobilized in response to the body’s needs.
Most leukocytes exist in the body from days to years, depending on type (e.g., mature neutrophils live for hours and some macrophages live for years). Maintenance of optimal levels of granulocytes and monocytes in the blood depends on the availability of pluripotent stem cells in the marrow, induction of these into committed stem cells, timely release of new cells from the marrow, and mobilization of the granulocyte reserve pool. Leukocyte production increases in response to infection, to the presence of steroids, and to reduction or depletion of reserves in the marrow. It is also associated with strenuous exercise, convulsive seizures, heat, intense radiation, paroxysmal tachycardias, pain, nausea and vomiting, and anxiety.
Development of Platelets
Thrombopoiesis is the development of platelets. Platelets (thrombocytes) are derived from stem cells and progenitor cells that differentiate into megakaryocytes (see Fig. 28.10). During thrombopoiesis, the megakaryocyte nucleus enlarges and becomes extremely polyploidy (up to 100-fold or more of the normal amount of DNA) without cellular division. Concurrently, the numbers of cytoplasmic organelles (e.g., internal membranes, granules) increase and the cell develops cellular surface elongations and branches that progressively fragment into platelets. A single large (up to 100 μm) megakaryocyte may produce thousands of smaller platelets (2 to 3 μm). Like erythrocytes, platelets released from the bone marrow lack nuclei but contain granules that promote their stickiness. About two-thirds of platelets enter the circulation, and the remainder resides in the splenic pool.
Thrombopoietin (TPO), a hormone growth factor, stimulates the production and differentiation of megakaryocytes and is the main regulator of the circulating platelet numbers.15 TPO is primarily produced by the liver and induces platelet production in the bone marrow. Platelets express receptors for TPO and, when circulating platelet levels are normal, TPO is adsorbed onto the platelet surface and prevented from accessing the bone marrow and initiating further platelet production. When platelet levels are low, however, the amount of TPO exceeds the number of available platelet TPO receptors, and free TPO can enter the bone marrow. During inflammation IL-6 induces increased production of TPO, which increases production of newly formed platelets, which are more thrombogenic.
Platelets circulate in the bloodstream for about 8 to 10 days before beginning to lose their ability to perform thrombogenic activity and biochemical reactions. Senescent platelets are sequestered and destroyed in the spleen by macrophage phagocytosis.
Mechanisms of Hemostasis
Hemostasis is the arrest of bleeding by formation of blood clots at sites of vascular injury (Fig. 28.16). As a result of hemostasis, damaged blood vessels may maintain a relatively steady state of blood volume, pressure, and flow. Three equally important interactive components of hemostasis are the vasculature (endothelial cells and subendothelial matrix), platelets, and clotting factors. The general sequence of events in hemostasis is as follows:
- 1. Vascular injury leads to a transient arteriolar vasoconstriction to limit blood flow to the affected site.
- 2. Damage to the endothelial cell lining of the vessel exposes prothrombogenic subendothelial connective tissue matrix, leading to platelet adherence and activation and formation of a hemostatic plug to prevent further bleeding (primary hemostasis).
- 3. Tissue factor, produced by the endothelium, collaborates with secreted platelet factors and activated platelets to activate the clotting (coagulation) system to form fibrin clots and further prevent bleeding (secondary hemostasis).
- 4. The fibrin/platelet clot contracts to form a more permanent plug, and regulatory pathways are activated (fibrinolysis) to limit the size of the plug and begin the healing process.
After endothelial denudation, platelets and leukocytes (blue arrows) adhere to the subendothelium in a monolayer fashion. (From Zipes DP, et al. Braunwald’s heart disease: A textbook of cardiovascular medicine, 8th edition. Philadelphia: Saunders; 2019, as reproduced from Faggiotto A, Ross R. Studies of hypercholesterolemia in the nonhuman primate. II. Fatty streak conversion to fibrous plaque. Arteriosclerosis. 1984;4:341–356.)
The relative importance of the hemostatic mechanisms clearly varies with vessel size. Damage to large vessels cannot easily be controlled by hemostasis but requires vascular contraction and dramatically decreased blood flow into the damaged vessels (Table 28.7).
Table 28.7

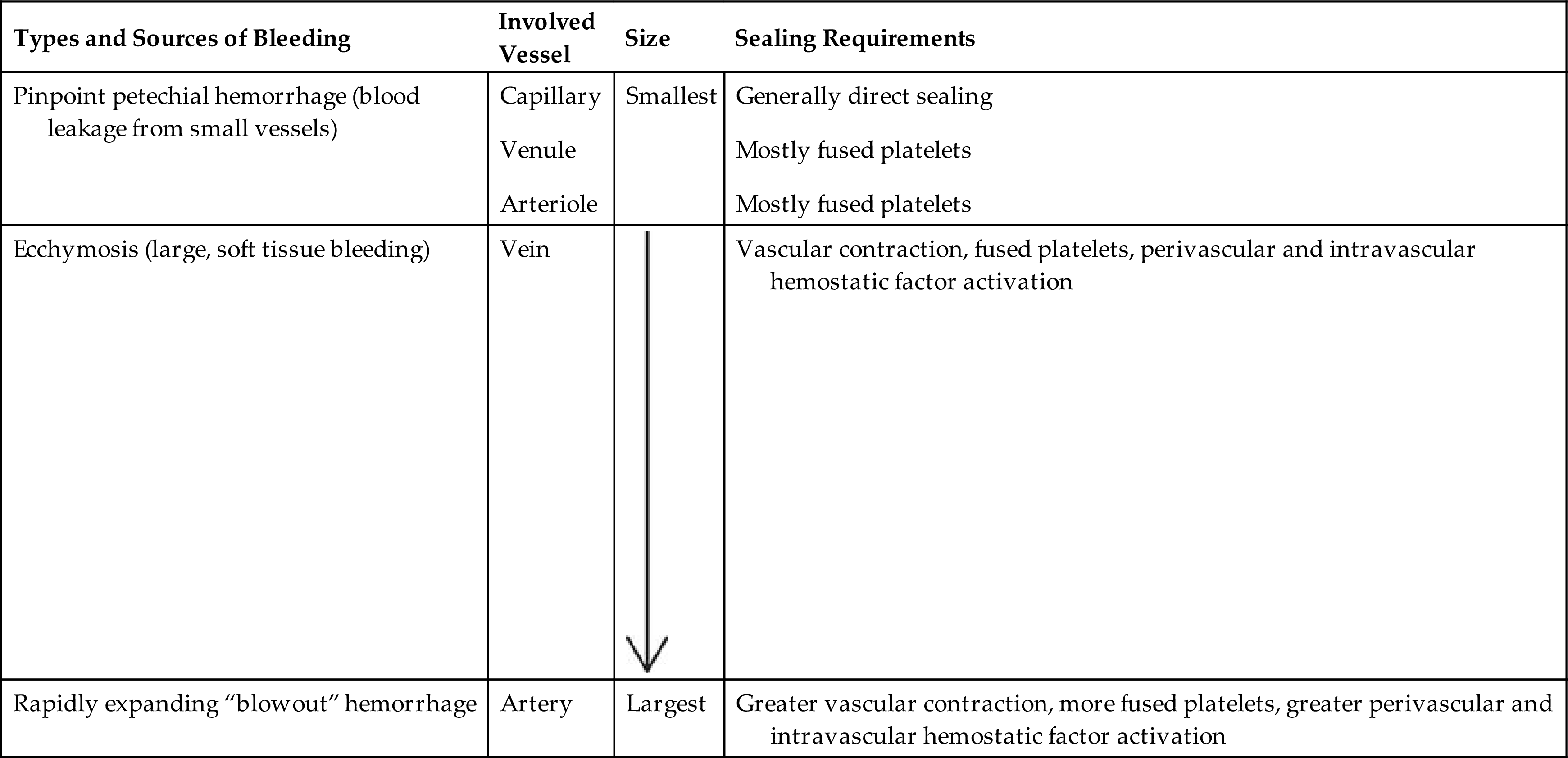
Modified from Harmening DM, ed. Clinical hematology and fundamentals of hemostasis, 3rd edition. Philadelphia: FA Davis; 1997.
Function of Blood Vessel Endothelium
The vessel walls consist of a layer of endothelial cells that adhere to an underlying subendothelial matrix of connective tissue. The matrix contains a variety of proteins, including collagen, fibronectin, and laminins. Endothelial cells adhere to the matrix and to each other through receptors that are expressed only on the intercellular and basal surfaces.
Under normal conditions the endothelium actively regulates blood flow and prevents spontaneous activation of platelets and the clotting system. Endothelial cells produce nitric oxide (NO), also known as endothelium-derived vasorelaxant factor (EDRF),16 and synthesize prostacyclin (prostaglandin I2 [PGI2]), both of which are vasodilators that modulate blood flow and pressure and maintain platelets in an inactive state. Synergism between PGI2 and NO is significant. PGI2 production varies a great deal in response to stimuli, whereas NO is released continually to regulate vascular tone. Endothelium also produces adenosine diphosphatase, which degrades adenosine diphosphate (ADP), a potent activator of platelets.
The endothelial cell surface contains antithrombotic molecules, such as glycosaminoglycans (e.g., heparan sulfate), thrombomodulin, and plasminogen activators. These limit platelet activation and fibrin deposition. Although thrombomodulin and plasminogen activators help control hemostasis in normal vessels, their effects are magnified during vascular damage and clot formation.
As a result of damage to the vessels, the endothelial cell barrier is frequently compromised, the remaining endothelial cells are activated by products of tissue damage, and the underlying subendothelial matrix is exposed. Endothelial cells contain intracellular structures that contain von Willebrand factor (vWF, clotting factor VIII), which is released during vascular injury and activates platelets for hemostasis.
Function of Platelets
Platelets normally circulate freely, suspended in plasma, in an unactivated state. The roles of platelets are to (1) contribute to regulation of blood flow into a damaged site by induction of vasoconstriction (vasospasm); (2) initiate platelet–platelet interactions (adhesion and aggregation) and attachment to injured endothelium, resulting in formation of a platelet plug to stop further bleeding; (3) release granules that activate the coagulation (or clotting) cascade to stabilize the platelet plug; and (4) initiate repair processes, including clot retraction and clot dissolution (fibrinolysis) and release of growth factors.17
The normal platelet count ranges from 150,000 to 400,000/mm3. Thrombocytopenia (abnormally low numbers of platelets) develops if the platelet count drops below 100,000/mm3, and an individual may experience longer than normal clotting times. Spontaneous major bleeding episodes do not generally occur unless the platelet count falls below 20,000/mm3. If platelet numbers are elevated (thrombocytosis), the risk for spontaneous blood clots (thrombosis), stroke, or heart attack is increased.
Damage to the vessel initiates a process of platelet activation: (1) increased platelet adhesion to the damaged vascular wall; (2) activation leading to platelet degranulation, which stimulates changes in platelet shape and biochemistry; (3) aggregation as platelet–vascular wall and platelet-platelet adherence increases; and (4) activation of the clotting system and development of an immobilizing meshwork of platelets and fibrin.
Adhesion
Normally platelets are generally observed “rolling” along the margins of vessels. At sites of vessel injury, however, platelets become adherent to the site of endothelial damage, where the subendothelial matrix is exposed and endothelial cells have released vWF and decreased their antithrombotic activities (Fig. 28.17). Platelet adhesion is mostly mediated by the binding of platelet surface receptor glycoprotein Ib (GPIb) to vWF. The vWF protein is found in the subendothelial matrix and is released by endothelial cells and platelets. Platelet adhesion narrows the diameter of the blood vessel, resulting in increasing shear forces that could strip platelets off the vessel surface. However, those same forces induce conformational changes in the vWF molecule that result in increased affinity with the adhesive GPIb, thus stabilizing the adherent platelet.
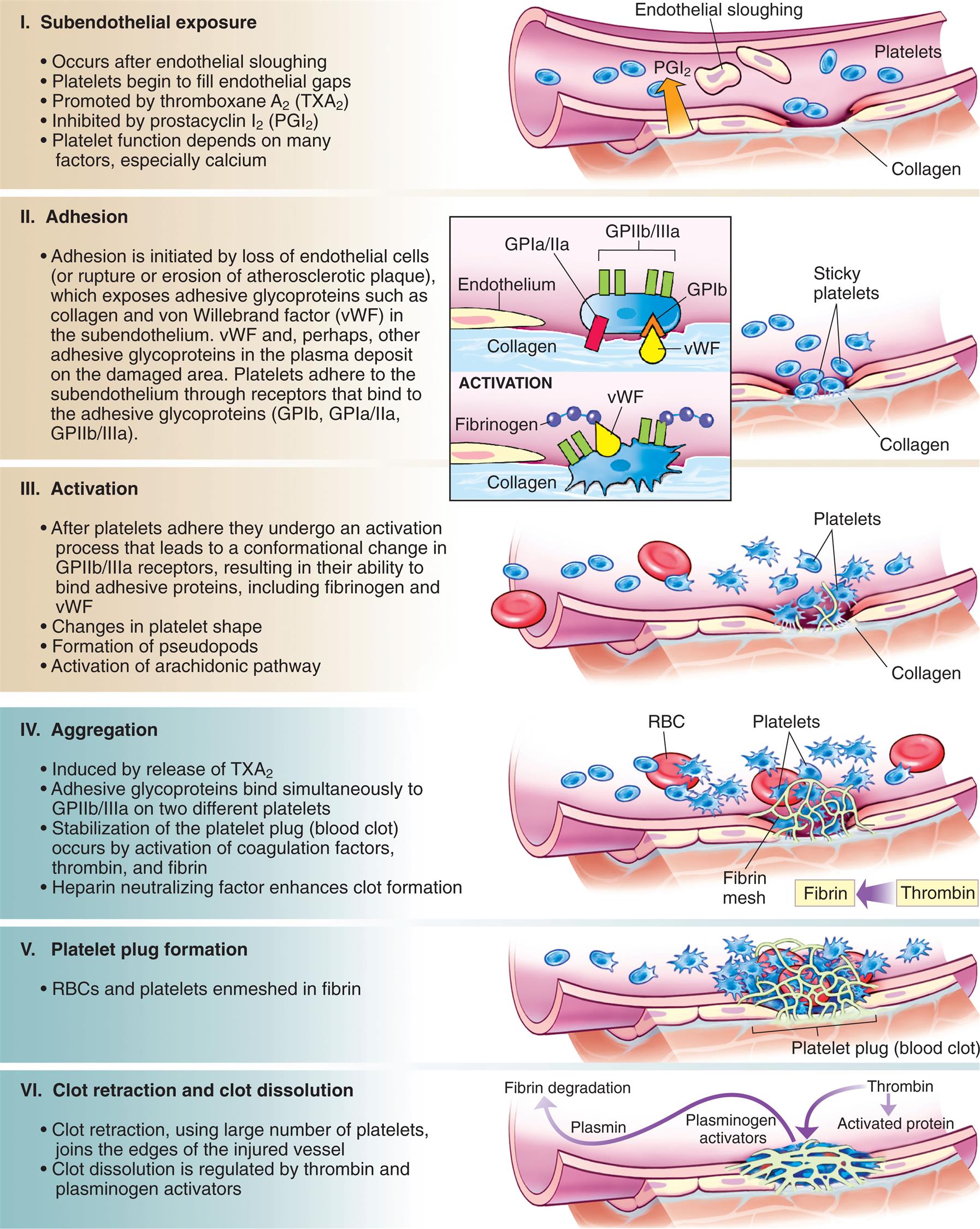
A series of illustrations show blood vessel damage, platelet activation, blood clot, and clot dissolution. Subendothelial exposure. The illustration shows collagen, P G I sub 2, platelets, and endothelial sloughing. • Occurs after endothelial sloughing. • Platelets begin to fill endothelial gaps. • Promoted by thromboxane A sub 2 (T X A sub 2). • Inhibited by prostacyclin I sub 2 (P G I sub 2). • Platelet function depends on many factors, especially calcium. Adhesion. The illustration shows collagen and sticky platelets. An accompanying magnified view identifies a G P 1 b with G P 1 a or 2 a and G P 2 b or 3 a on the endothelium and collagen with v W F attached to fibrinogen. Adhesion is initiated by loss of endothelial cells (or rupture or erosion of atherosclerotic plaque), which exposes adhesive glycoproteins such as collagen and von Willebrand factor (v W F) in the subendothelium. v W F and, perhaps, other adhesive glycoproteins in the plasma deposit on the damaged area. Platelets adhere to the subendothelium through receptors that bind to the adhesive glycoproteins (G P 1 b, G P 1 a or 2 a, G P 2 b or 3 a). Activation. The illustration shows platelets concentrated at the collagen. • After platelets adhere they undergo an activation process that leads to a conformational change in G P 2 b or 3 a receptors, resulting in their ability to bind adhesive proteins, including fibrinogen and v W F. • Changes in platelet shape. • Formation of pseudopods. • Activation of arachidonic pathway. Aggregation. The illustration shows platelets attached to the R B C. Thrombin converted to fibrin, which forms a mesh on the platelets. • Induced by release of T X A sub 2. • Adhesive glycoproteins bind simultaneously to G P 2 b or 3 a on two different platelets. • Stabilization of the platelet plug (blood clot) occurs by activation of coagulation factors, thrombin, and fibrin. • Heparin neutralizing factor enhances clot formation. Platelet plug formation. The illustration shows a fibrin mesh covering a cluster of platelets attached to R B Cs. RBCs and platelets enmeshed in fibrin. Clot retraction and clot dissolution. The illustration shows a thick fibrin mesh. Thrombin activates protein and triggers the mesh generating plasminogen activators that produce plasmin for fibrin degradation. • Clot retraction, using large number of platelets, joins the edges of the injured vessel. • Clot dissolution is regulated by thrombin and plasminogen activators.
Platelet adhesion is also facilitated by other interactions between platelet receptors and exposed molecules of the subendothelial matrix. For instance, adhesion is increased through binding of the platelet collagen receptors GPVI and integrin α2β1 to exposed collagen in the matrix.
Activation
As a result of interactions with the endothelium or the subendothelial matrix, as well as exposure to inflammatory mediators produced by the endothelium and other cells, the platelets are activated. Activation results in reorganization of the platelet cytoskeleton, leading to dynamic changes in platelet shape from smooth spheres to those with spiny projections (increases surface area) (see Fig. 28.5) and degranulation (also called the platelet-release reaction) and resulting in the release of various potent biochemicals.
Platelets contain three types of granules: dense bodies, alpha granules, and lysosomes. The contents of the dense bodies and alpha granules are particularly important in hemostasis. The dense bodies contain ADP, serotonin, and calcium. ADP recruits and activates other platelets through specific receptors. During activation the platelet plasma membrane experiences several important changes, including becoming ruffled and sticky; undergoing cellular spreading to make tight contacts between neighboring platelets, causing the platelet plug to seal the injured endothelium; and externalizing the phospholipid phosphatidylserine, which provides a matrix for activation of clotting factors. Serotonin is a vasoactive amine that functions like histamine and increases vasodilation and vascular permeability. Calcium is necessary for many of the adhesive interactions as well as for intracellular signaling mechanisms that control platelet activation.
Alpha granules contain a mixture of clotting factors (e.g., fibrinogen), growth and angiogenic factors (e.g., platelet-derived growth factor [PDGF], vascular endothelial growth factor [VEGF], basic fibroblast growth factor), and angiogenesis inhibitors (e.g., platelet factor 4, thrombospondin, inhibitors of metalloproteinases). Platelet factor 4 also is a heparin-binding protein and enhances clot formation at the site of injury. Depending on the particular stimulus, platelets may selectively release promoters or inhibitors of angiogenesis. Many of these mediators either promote or inhibit platelet activity and the eventual process of clot formation. PDGF stimulates smooth muscle cells and promotes tissue repair.
Platelet lysosomes have a role in the digestion of phagocytic and cytosolic components, similar to that in nucleated cells. Lysosomal contents also may have important extracellular functions, such as supporting receptor cleavage, fibrinolysis and degradation of extracellular matrix components, and remodeling of the vasculature.18
Platelets also initiate production of the prostaglandin derivative thromboxane A2 (TXA2), which counters the effects of PGI2 produced by endothelial cells. TXA2 causes vasoconstriction and promotes the degranulation of platelets, increases expression of platelet fibrinogen receptors, and stimulates platelet aggregation, whereas PGI2 promotes vasodilation and inhibits platelet degranulation. Arachidonic acid metabolism by cyclooxygenase (COX) is a major pathway for blood platelet activation. An isoform of cyclooxygenase (COX-1) converts arachidonic acid to TXA2 in platelets. Aspirin at low doses specifically and irreversibly inactivates COX-1, decreasing production of TXA2 and decreasing platelet activation and aggregation.19 Daily intake of low doses of aspirin leads to more than 95% inhibition of TXA2 in just a few days.
Other stimuli of platelet activation include epinephrine, thrombin, and collagen. Thrombin and collagen are particularly strong stimuli. Thrombin cleaves the extracellular domain of G-protein–coupled protease-activated receptors (PARs), thereby initiating transmembrane signaling.
Aggregation
Platelet aggregation is stimulated primarily by TXA2 and ADP, which induce functional fibrinogen receptors on the platelet. The glycoprotein IIb/IIIa (GPIIb/IIIa) complex (also called integrin αIIbβ3) undergoes a conformational change during activation to become a calcium-dependent receptor for fibrinogen, allowing it to bind other matrix proteins (e.g., fibronectin, fibrinogen, thrombospondin). Although the GPIIb/IIIa complex is the most abundant fibrinogen receptor on the platelet, receptors for vWF and collagen also contribute to the process. Interplatelet aggregation and clot retraction, which forms the primary hemostatic plug, are facilitated by fibrinogen bridges between receptors on the platelets. The GPIIb/IIIa–fibrinogen pathway is essential for the formation of a thrombus and as such is an important therapeutic target for blockage by antiplatelet drugs. In addition, fibrin strands within the clot shorten and become denser and stronger, helping the clot to approximate the edges of the injured vessel wall and sealing the site of injury. Contraction of myosin and actin filaments in the platelet cytoskeleton mediates platelet contraction and fusion of the platelet mass into a secondary hemostatic plug. Contraction expels serum from the fibrin meshwork, resulting in greater packing and increased strength.
If blood vessel injury is minor, hemostasis is achieved temporarily by formation of the platelet plug within 3 to 5 minutes of injury. Platelet plugs adhere to damaged vessel walls and seal the many minute ruptures that occur daily in the microcirculation, particularly in capillaries. With too few platelets, numerous small hemorrhagic areas called purpuras develop under the skin and throughout the tissues (see Chapter 29). If primary hemostasis is inadequate to prevent further bleeding, the process proceeds through secondary hemostasis to create a larger complex of more tightly interactive platelets within a matrix created by activation of the clotting system.
Function of Clotting Factors
A blood clot is a meshwork of protein strands that stabilizes the platelet plug and traps other cells, such as erythrocytes, phagocytes, and microorganisms (see Fig. 28.16). The strands are made of fibrin, which is produced by the clotting (coagulation) system. The clotting system was described in Chapter 7 and consists of a family of proteins that circulate in the blood in inactive forms (proenzymes). The proteins of the clotting system are all produced in liver hepatocytes. Initiation of the system results in sequential activation (cascade) of proteolytic enzymes which activate downstream enzymes in the system until a fibrin clot is created (Table 28.8).
Table 28.8
The clotting system is commonly presented as two pathways of initiation: the extrinsic pathway (also known as the tissue factor pathway) and the intrinsic pathway (also known as the contact activation pathway). These two pathways join to form a common pathway, with activation of factor X, which proceeds to thrombin, fibrin, and clot formation. These pathways are controlled by anticoagulant proteins. The extrinsic pathway is activated when there is vascular injury and blood escapes into the tissue, with activation of tissue factor. The process is a normal mechanism of hemostasis. Tissue factor (TF) (also called tissue thromboplastin) is abundant in vascular subendothelium, in the surrounding larger blood vessels, and body organs, particularly the skin, brain, lungs, kidney, and placenta. There also are receptors for TF on activated monocytes. With vessel trauma and bleeding, TF binds with high affinity to circulating activated factor VII (TF/VIIa), initiating the clotting pathway. TF mediates a number of changes in disease-related activities, including inflammation, tumor angiogenesis metastasis, cell migration, and clotting associated with atherosclerosis (Fig. 28.18). The extrinsic pathway is evaluated in vitro by adding TF to a sample of plasma and measuring the prothrombin time (time to form a fibrin clot).
Factor VIIa signaling through TF expressed on the surface of cells mediates changes in disease-related cellular activities. EC, Endothelial cell; MC, monocyte/macrophage; SMC, smooth muscle cell. (Modified from Rao LV, Pendurthi UR. Tissue factor-factor VIIa signaling. Arteriosclerosis, Thrombosis, and Vascular Biologyl, 2005;25(1):47–56.)
A flowchart illustrates tissue factor (T F) and disease. 1. T F or factor 7 a. Leads to 2, 3, and 4. 2. E C. Leads to 5. 3. Tumor cell. Leads to 8. 4. S M C. Leads to 9. 5. Activated E C. Leads to 6. 6. Angiogenesis. Leads to 7. 7. Metastasis. 8. Tumor growth or survival. Leads to 7. 9. Proliferation and migration. Leads to 10. 10. Atherosclerosis and vascular remodeling. 11. M C. Leads to 12. 12. Activated M C. Leads to 7, 13, and 14. 13. Leukocyte recruitment. Leads to 15. 14. Inflammation and septic shock. 15. Lung injury. Leads to 16. 16. Fibrosis. 17. Fibroblast. Leads to 16 and 19. 18. T F on keratinocyte. Leads to 19. 19. Wound healing.
Factor XIIa (Hageman factor), prekallikrein (PK), and high-molecular-weight kininogen (HK; activates factor XII) initiate the intrinsic coagulation pathway through activation of the downstream substrate factor XI. The terms intrinsic or contact mean that the pathway is activated within the blood vessel by contact with anionic (negatively charged) surfaces (e.g., exposure of subendothelial collagen from vessel damage) or other substances without the action of TF. The intrinsic pathway is not a significant pathway for normal hemostasis but is a pathophysiologic surface defense mechanism against foreign proteins, microbial pathogens, and artificial materials. Individuals with deficiencies in intrinsic pathway components (e.g., factor XI, factor XII) do not have disruption of hemostasis or prolonged bleeding with the exception of hemophilia C (deficiency of factor XI that usually causes mild bleeding).
In the activation of the intrinsic pathway, factor XIIa recruits HK that is complexed with PK. Factor XIIa activates PK to kallikrein, which in turn generates more factor XIIa and amplifies the pathway. Factor XIIa also activates HK to release bradykinin (a proinflammatory mediator). Other exogenous artificial surfaces also can activate factor XII (e.g., use of tubing for blood flow during hemodialysis, cardiopulmonary bypass, or extracorporeal membrane oxygenation) and require anticoagulation therapy. More recently it has been discovered that naturally occurring polyphosphates (e.g., RNA and DNA from injury in arterioles), activated platelets, neutrophil extracellular traps, and cell membranes from bacteria can activate the intrinsic pathway and promote thrombin formation and thrombosis. The intrinsic pathway also may be essential for stabilization of thrombi and prevention of embolism.20
It is important to recognize that factor XIa and factor IXa can be generated by routes independent of factor XIIa (e.g., thrombin also can activate factor XI, as well as factors V and VIII). Thrombin activation of XI and IX is a feedback mechanism that amplifies the coagulation pathway and maintains the clot over time.
Activated platelets are important participants in clotting as previously described. The phosphatidylserine-rich surface produced during platelet activation provides a matrix on which several important complexes of clotting factors are formed. These include the intrinsic pathway’s tenase complex (factor X and activated factors VIII and IX) that activates factor X and the prothrombinase complex (prothrombin and activated factors X and V) that activates prothrombin into thrombin. Thrombin then converts fibrinogen into fibrin, which polymerizes into a fibrin clot (see Fig. 28.16). Thrombin has broad activity in the inflammatory response. In addition to producing fibrin, thrombin is an activator of other coagulation proteins (e.g., factors V, VIII, XI, XIII), platelets (e.g., aggregation, degranulation), endothelial cells (e.g., up-regulation of adhesion molecules for leukocytes, increased NO, PGI2, PDGF), and monocytes (e.g., cytokine secretion and increased receptors for endothelial cells). In addition to these pathways, RBCs can also promote clotting, particularly when there are diseases that affect the vasculature or there is an increase in the concentration of RBCs (see Emerging Science Box: Red Bloods Cells and Hemostasis).
Control of Hemostatic Mechanisms
Blood normally remains fluid despite the continual presence of clotting factors and platelets in the circulation. This is because under normal conditions, spontaneous activation of hemostasis is prevented by factors residing on the endothelial cell surface. These include thrombin inhibitors (e.g., antithrombin III), tissue factor inhibitors (e.g., tissue factor pathway inhibitor), and mechanisms for degrading activated clotting factors (e.g., protein C). When the endothelium is damaged, it transitions to the hemostatic action (vasoconstriction, platelet and clotting factor activation) described previously.
Antithrombin III (AT-III) is a circulating inhibitor of plasma serine proteases. AT-III is produced by the liver and binds to heparin sulfate found naturally on the surface of endothelial cells, or with heparin administered clinically to prevent thrombosis. Heparin induces a change in AT-III that greatly enhances its capacity to inhibit thrombin and other activated clotting factors. Under normal conditions the presence of endothelial cell heparan sulfate and the available AT-III in the circulation cooperate to protect the vessels from the effects of spontaneously activated thrombin. Acquired AT-III deficiencies can result from infection with bacteria that produce AT-III inhibitors, sepsis, liver disease, and nephrotic syndrome and lead to venous thrombosis and pulmonary embolism (PE).
Tissue factor pathway inhibitor (TFPI) is produced by endothelial cells and platelets and is the primary inhibitor of the initiation of blood clotting. TFPI forms complexes with and reversibly inhibits factor Xa. The resultant TFPI/Xa complex inhibits the extrinsic coagulation initiation complex TF/VIIa and prothrombinase (converts prothrombin to thrombin) and thus inhibits clotting.21
Thrombomodulin is a transmembrane thrombin-binding protein on the surface of endothelial cells and a receptor for thrombin. When thrombin binds to thrombomodulin, it no longer serves as a procoagulant and cannot activate platelets, convert fibrinogen to fibrin, or amplify fibrin generation. Protein C in the circulation binds to thrombomodulin-thrombin complex and is rapidly converted to activated protein C. Activated protein C, in association with a cofactor (protein S), degrades factors Va and VIIIa, inhibits fibrin formation, and inhibits coagulation. Deficiencies of AT-III, protein C, or protein S are important inherited causes of hypercoagulation (increased clotting). Expression of thrombomodulin and the endothelial cell protein C receptor is down-regulated by cytokines and other products of inflammation (e.g., IL-1α, TNF-α, endotoxin), thereby enhancing clot formation. Activated protein C inhibits the adhesion of neutrophils to the endothelium, but during inflammation neutrophil elastase enzymatically removes thrombomodulin from the endothelial cell surface and regulates clot formation.22
Retraction and Lysis of Blood Clots
After a clot is formed, it retracts, or “solidifies.” Fibrin strands shorten, becoming denser and stronger, which approximates the edges of the injured vessel wall and seals the site of injury. Retraction is facilitated by the large numbers of platelets trapped within the fibrin meshwork. The platelets contract and “pull” the fibrin threads closer together while releasing a factor that stabilizes the fibrin. Contraction expels serum from the fibrin meshwork. This process usually begins within a few minutes after a clot has formed, and most of the serum is expelled within 20 to 60 minutes.23
Lysis (breakdown) of blood clots is carried out by the fibrinolytic system (Fig. 28.19). Another plasma protein, plasminogen, is converted to plasmin by several products of coagulation and inflammation, especially by the enzymatic action of tissue plasminogen activator (t-PA). Endothelial cells express t-PA, which is activated maximally after binding to fibrin. Another activator of plasminogen is urokinase-like plasminogen activator (u-PA). The u-PA binds to a specific cellular u-PA receptor (u-PAR), causing activation of plasminogen. This urokinase is the major activator of fibrinolysis in the extravascular, or tissue, compartment, whereas t-PA is largely involved in intravascular fibrinolysis. Several cancers appear to use membrane-bound u-PA to digest intercellular matrix and greatly facilitate tumor invasion and metastasis. Both t-PA and u-PA have been used clinically to treat diseases associated with a blood clot (e.g., PE, myocardial infarction, stroke).
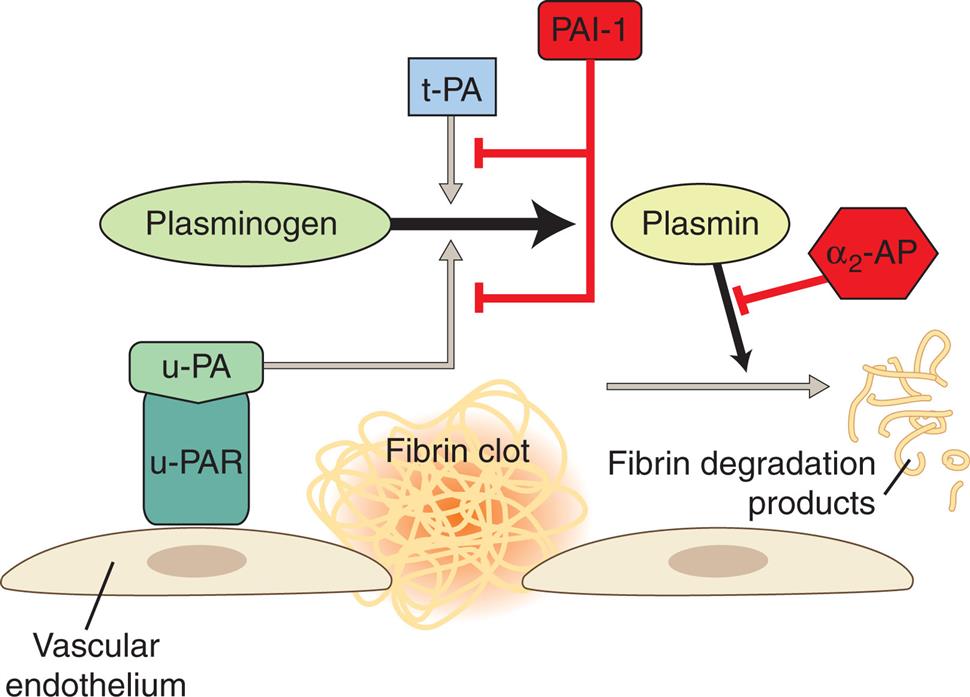
Fibrinolysis is initiated by the binding of plasminogen to fibrin. Although tissue plasminogen activator (t-PA) initiates intravascular fibrinolysis, urokinase plasminogen activator (u-PA) is the major activator of fibrinolysis in tissue (extravascular). Plasmin digests the fibrin into smaller soluble pieces (fibrin degradation products). u-PAR, Urokinase-like plasminogen activator receptor.
An illustration, representing the fibrinolytic system, shows a layer of vascular endothelium with a fibrin clot between two cells. u-P A R attracts u-P A. P A I-1 and alpha 2-A P are inhibitors, former inhibits plasminogen and later inhibits plasmin.
As with most components of inflammation, plasmin interacts greatly with other factors. In addition to activation by t-PA and u-PA, plasminogen is activated to plasmin by thrombin, fibrin, factor XIIa, factor XIa, and kallikrein. Plasmin is proteolytic to several substrates, and cleaves fibronectin, fibrin, thrombospondin, laminin, and vWF. Cross-linked fibrin is deposited in tissues around wounds, inflammatory sites, and tumors. Fibrin removal is an important biologic process, for intravascular and extravascular spaces, with various controlling mechanisms that can lead to abnormalities of fibrin accumulation and thrombotic events and can be a structural barrier to tumor invasion.
Plasmin is an enzyme that dissolves clots (fibrinolysis) by degrading fibrin and fibrinogen into fibrin degradation products (FDPs). A major FDP is D-dimer. Measurement of levels of circulating d-dimer (a small protein fragment of fibrin degradation) has been used for diagnosis because D-dimer can be elevated in cases of deep venous thrombosis (DVT) or PE.
Clinical Evaluation of the Hematologic System
Tests of Bone Marrow Function
Several abnormal conditions in the numbers or morphology of circulating blood cells or suspected infection of the marrow may justify further investigation of the bone marrow. Usually bone marrow is aspirated from the sternum or pelvis using a needle. In children a bone marrow aspirate can be obtained from the vertebrae or the femur. The aspirate is examined microscopically and may be cultured if infection (e.g., fungi, mycobacteria, brucellosis, typhoid fever) is suspected. Microscopic evaluation may also include flow cytometry, chromosome analysis, or polymerase chain reaction (PCR) related to the presence of atypical cells, the presence of atypical numbers of normal cells, and the absence of particular cell types. A normal bone marrow aspirate contains stromal cells (fibroblasts, macrophages, osteoblasts, adipocytes), stem cells (hematopoietic, mesenchymal, and endothelial), and immature and mature forms of blood cells (erythrocytes, leukocytes, platelets) (Fig. 28.20A). The differential cell count of a bone marrow aspirate involves examining approximately 400 nucleated cells under oil-immersion magnification and counting populations of cells differentiated on the basis of morphology. The relative number of each type of stem cell is expressed as a fraction of 400 (Table 28.9).
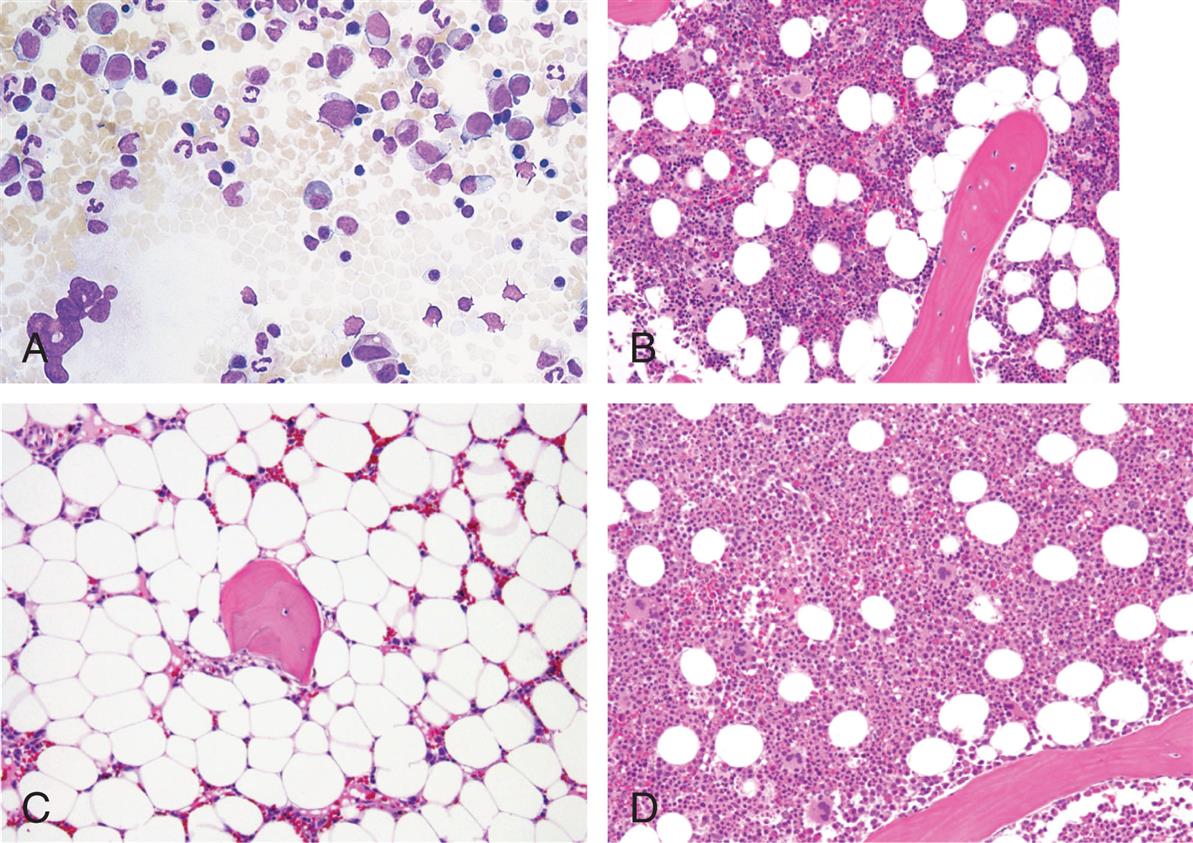
(A) Normal bone marrow aspirate stained by the May-Grünwald/Giemsa technique. (B–D) Bone marrow biopsies representing conditions of normal cellularity (B), hypocellularity (C) as would be seen in conditions of bone marrow depletion such as aplastic anemia, and hypercellularity (D) as seen in malignant conditions. (A and C, From Hoffbrand V, et al. Color atlas of clinical hematology, 4th edition. Philadelphia: Mosby; 2013; B–D, From Jaffe E. Hematology. Philadelphia: Saunders; 2010.)
Four photomicrographs, A, B, C, and D, show bone marrow samples. A. Normal bone marrow. There is a normal amount of hematopoietic cells. B. Hypocellularity. There is a decreased amount of hematopoietic cells. C. Aplastic Anemia or hypercellularity. There is an increased amount of large hematopoietic cells. D. Malignant. There are very less hematopoietic cells.
Table 28.9
NOTE: Values are percentages of cell types counted during examination of a marrow specimen containing approximately 400 nucleated cells.
Bone marrow iron stores, primarily in macrophages, can be examined using special stains (e.g., Prussian blue) for iron-containing granules. A direct measure of iron stores also can be obtained only from liver biopsy specimens, although bone marrow is preferred because it is a safer procedure and because bone marrow is the immediate source of iron destined for erythrocyte production.
Bone marrow aspiration is an important diagnostic test for severe central defects in hematopoiesis (e.g., aplastic anemia, metabolic anemias arising from insufficient iron or inadequate EPO, thrombocytopenia, and neutropenia). Examination of the bone marrow is also useful to diagnose B-lymphocyte immune deficiencies, nonmalignant myeloproliferative disorders (e.g., polycythemia vera), and lymphoid/monocytic malignancies (e.g., leukemias, myelomas, lymphomas). This test can also be used to monitor the effects of chemotherapy on malignancies that have invaded the bone marrow. A marrow aspirate that is richly cellular implies normal or increased hematopoiesis but does not indicate whether marrow activity is effective.
Results from bone marrow aspiration are sometimes limited because this technique disturbs the architecture of the marrow and only provides an analysis of the general cellularity (numbers of constituent cells) of the marrow. On occasion, analysis of an aspirate may only suggest the presence of a malignancy or a central defect in hematopoiesis without being clearly confirmatory, or the sample may be inadequate for diagnosis of bone marrow fibrosis. In these cases the need for a bone marrow biopsy may be indicated.
During a biopsy, a special needle is used to obtain a “core” or cylindrical sample of bone and marrow in which the three-dimensional structure of the marrow is preserved. The biopsy specimens provide the most reliable and complete information about marrow cellularity (see Fig. 28.20B–D). Obtaining a bone marrow biopsy is, however, usually more painful and expensive than aspiration. Therefore biopsy is not performed unless insufficient information is obtained from aspiration.
Blood Tests
Blood tests provide information about the absolute and relative numbers of blood cells in a specimen of blood, as well as various structural and functional characteristics of the cells, and usually provide the initial justification for performing a bone marrow aspiration. Deviations from the normal differential distribution and the presence of abnormal or immature cells can reflect disease, physiologic states (e.g., pregnancy, infancy, old age), injury, or dysfunction in almost any part of the body. Blood tests that reflect chiefly hematologic disorders are listed in Table 28.10.
Table 28.10
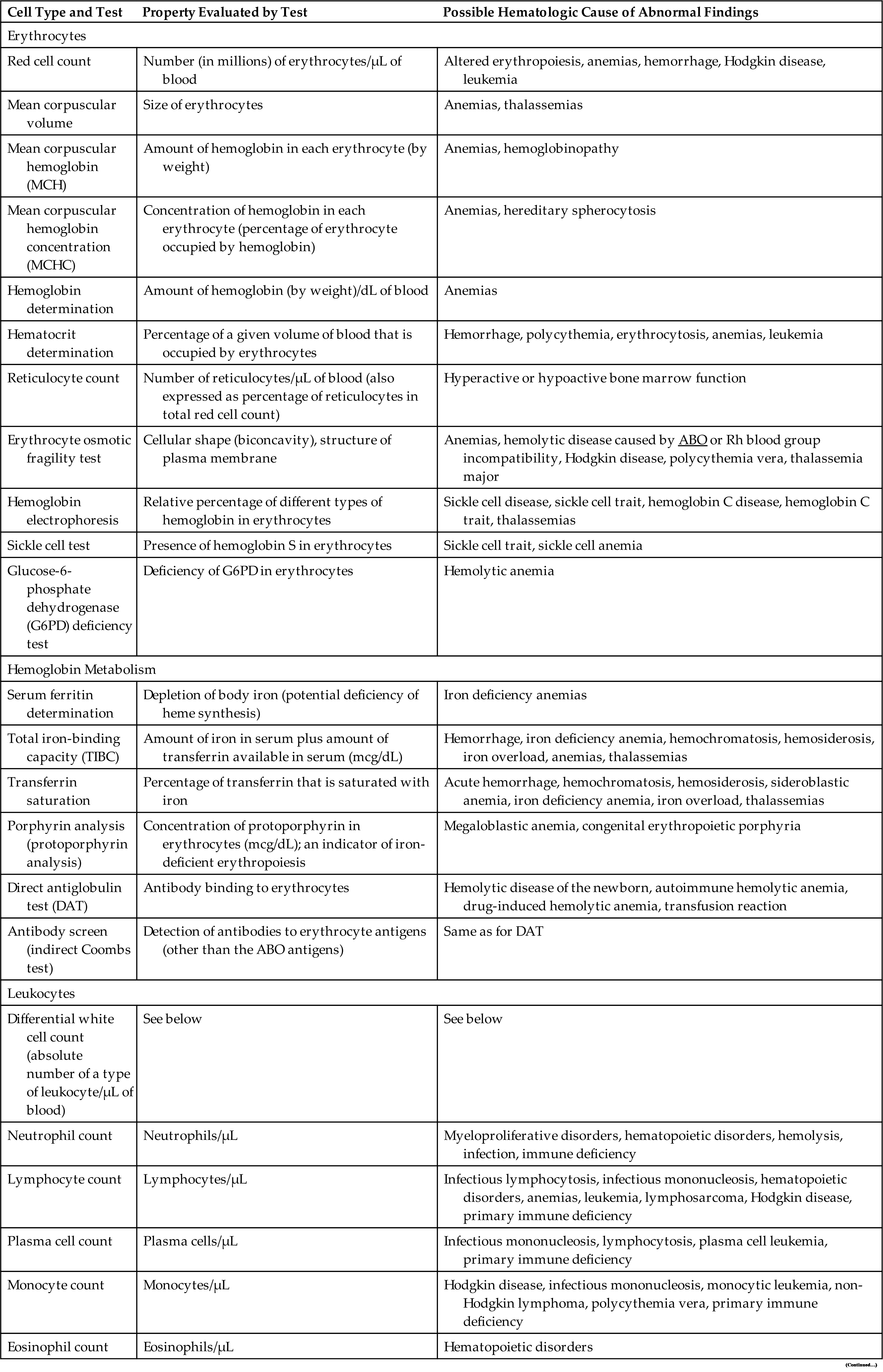
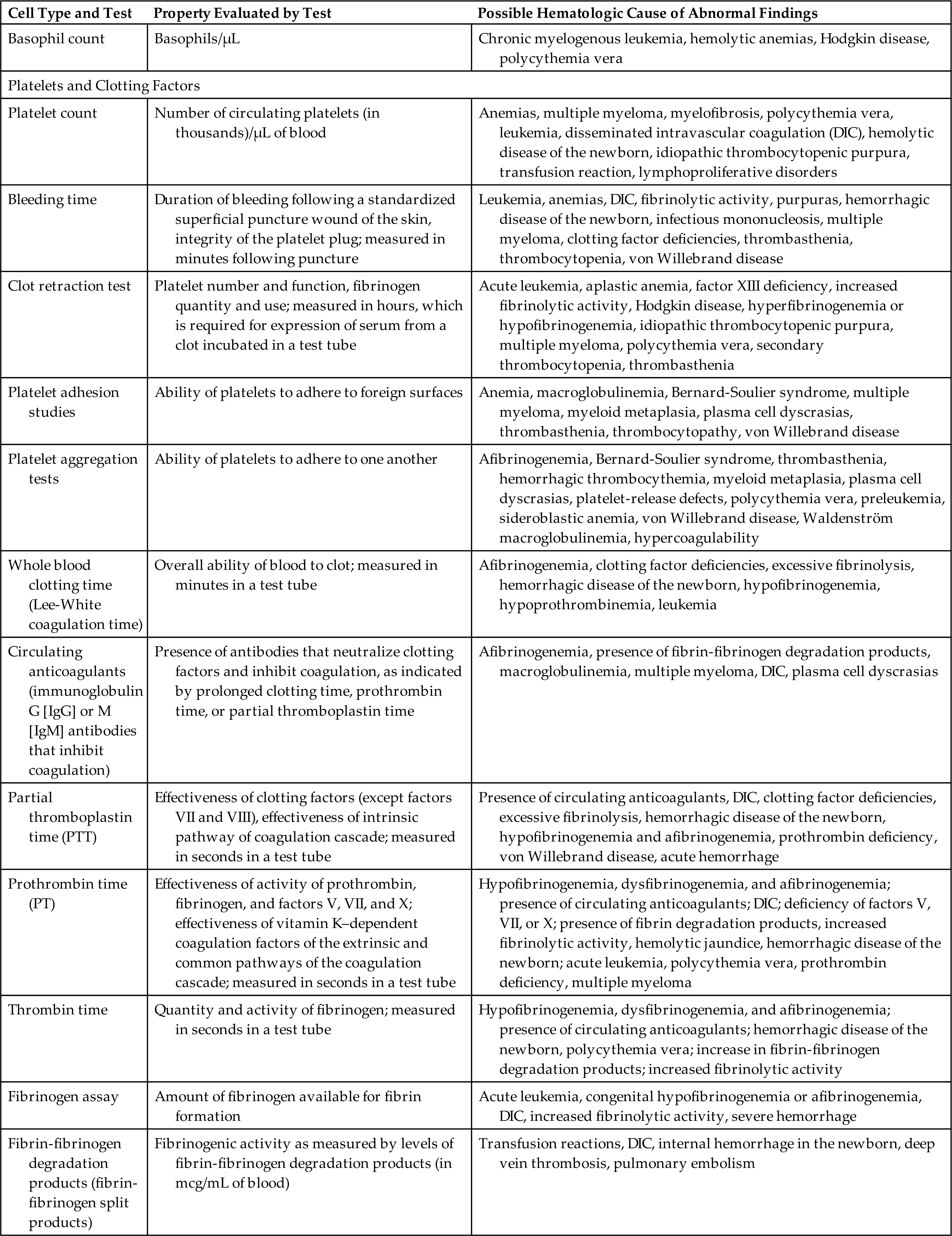
DAT, Direct antiglobulin test; DIC, disseminated intravascular coagulation.
Data from Garrels M, Oatis CS. Laboratory and diagnostic testing in ambulatory care: A guide for health care professionals, 3rd edition. St. Louis: Saunders; 2015; Hudnall D. Hematology: A pathophysiologic approach. St. Louis: Mosby; 2012.
Pediatrics and the Hematologic System
Blood cell counts tend to rise above adult levels at birth and then decline gradually throughout childhood. Table 28.11 lists normal ranges during infancy and childhood. The immediate rise in values is the result of accelerated hematopoiesis during fetal life and the increased numbers of cells that result from the trauma of birth and cutting of the umbilical cord.
Table 28.11
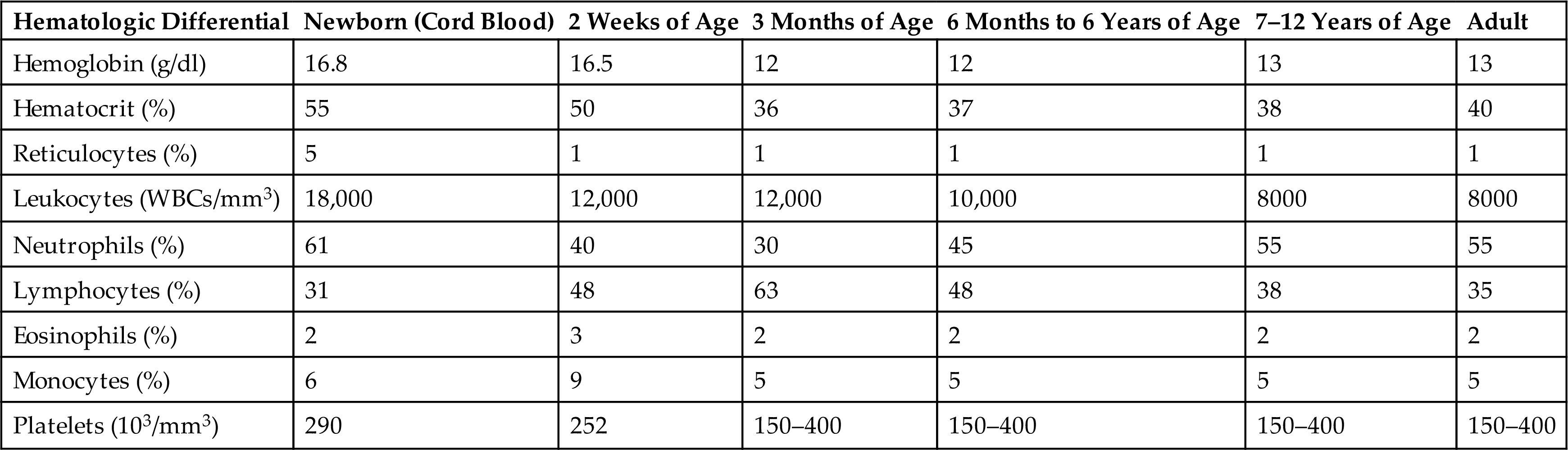
Average blood volume in the full-term neonate is about 85 mL/kg of body weight. The premature infant has a slightly larger blood volume of 90 mL/kg of body weight. In both full-term and premature infants, blood volume decreases during the first few months. Thereafter the average blood volume is about 75 to 80 mL/kg, which is similar to that of older children and adults.
The hypoxic intrauterine environment stimulates EPO production in the fetus and accelerates fetal erythropoiesis, producing polycythemia (excessive proliferation of erythrocyte precursors) in the newborn. After birth, the oxygen from the lungs saturates arterial blood, and more oxygen is delivered to the tissues. In response to the change from a placental to a pulmonary oxygen supply during the first few days of life, levels of EPO and the rate of blood cell formation decrease. The active rate of fetal erythropoiesis is reflected by the large numbers of immature erythrocytes (reticulocytes) in the peripheral blood of full-term neonates. After birth, the number of reticulocytes decreases by 50% every 12 hours, so it is rare to find an elevated reticulocyte count after the first week of life. During this period of rapid growth, the rate of erythrocyte destruction is greater than that in later childhood and adulthood. In full-term infants, the normal erythrocyte life span is 60 to 80 days; in premature infants, it may be as short as 20 to 30 days; and in children and adolescents, it is the same as that in adults—100 to 120 days.
The postnatal fall in hemoglobin and hematocrit values is more marked in premature infants than it is in full-term infants. Fetal hemoglobin has higher affinity for oxygen compared to adult globins, which presumably allows for efficient maternal–embryonic gas exchange in the placental environment. In preschool and school-aged children, hemoglobin, hematocrit, and RBC counts gradually rise. Metabolic processes within the erythrocytes of neonates differ significantly from those found in erythrocytes of normal adults. The relatively young population of erythrocytes in newborns consumes greater quantities of glucose than do erythrocytes in adults. Platelet counts in full-term neonates are comparable with those in adults and remain so throughout infancy and childhood.
Most coagulation factors (both procoagulants and anticoagulants) in children reach adult levels by 6 months of age, and some not until adolescence. Children have quantitative and qualitative differences in clotting factors that result in decreased risk for thrombosis without increased risk for bleeding (developmental hemostasis) because the reduction of procoagulant and anticoagulant proteins is balanced. Laboratory values for hemostasis (e.g., prothrombin time and activated partial thromboplastin time) are referenced by age.24
WBCs develop and mature during fetal life, including neutrophils, monocyte-macrophages, eosinophils, and lymphocytes, but the function of the cells at birth is weak because the cells are immature. At birth the lymphocyte count is high, and it continues to rise during the first year of life. Then it steadily declines until the lower value seen in adults is reached. The lymphocytes of children also tend to have more cytoplasm and less compact nuclear chromatin than do the lymphocytes of adults. A possible explanation is that children tend to have more frequent viral infections, some of which are subclinical, and are receiving vaccinations, both of which are associated with atypical lymphocytes.
The neutrophil count, like the lymphocyte count, is high at birth and rises during the first days of life. After 2 weeks, the neutrophil count falls to within or below the normal adult range. Although the exact age can vary, by approximately 7 years of age, the neutrophil count is the same as that of an adult. The eosinophil count is higher in the first year of life and higher in children than in teenagers or adults. Monocyte counts also are high in the first year of life but then decrease to adult levels.
Newborn infants are at risk for impaired phagocytosis, bacterial infections, and delayed wound healing. Immature lymphocytes also increase the risk for viral infection, and there is a weak response to the production of antibodies and a poor response to foreign antigens. Protection against infectious disease is provided by passive IgG antibody transfer from the mother transplacentally and in breast milk. By 2 months of age the WBCs have matured enough for the infant to begin receiving protection from infectious disease by vaccination. As the child ages, immunity is further enhanced by intercurrent infection and vaccinations, with the formation of antibodies and memory T cells that provide lifelong protection.
Aging and the Hematologic System
Blood composition changes little with age, although some components may be altered by iron deficiency. Values of total serum iron level, total iron-binding capacity, and intestinal iron absorption are all decreased somewhat in elderly persons. The erythrocyte life span is normal, although the erythrocytes are replenished more slowly after bleeding and their deformability decreases with advanced age.25 Hemoglobin levels may be low, and the plasma membranes of erythrocytes become increasingly fragile, with portions being lost, presumably because of physical trauma inflicted during circulation. Chronic inflammation and the presence of chronic disease, including diabetes mellitus, chronic heart disease, chronic renal insufficiency, and Parkinson disease, are associated with suppressed erythropoiesis and anemia occurring in older adults.26 Lymphocyte function decreases with age, causing changes in cellular immunity and some decline in T-cell function. The humoral immune system is less able to respond to antigenic challenge and to vaccination.
Platelet count remains relatively stable during middle age (25 to 60 years old) but falls in older people (greater than 60 years),27 yet platelet adhesiveness increases, which may be related to oxidative stress. Fibrinogen levels and levels of factors V, VII, and IX and also vWF tend to be increased. In addition, thrombin generation and platelet activation are enhanced in older adults. Consequently, there is an increased incidence of venous thromboembolism.28
Summary Review
Components of the Hematologic System
- 1. Blood consists of a variety of components—about 91% water and 9% solutes. In adults, the total blood volume is approximately 5.5 L.
- 2. Plasma, a complex aqueous liquid, contains two major groups of plasma proteins: albumins and globulins.
- 3. The cellular elements of blood are the red blood cells (erythrocytes), white blood cells (leukocytes), and platelets.
- 4. Erythrocytes are the most abundant cells of the blood, occupying approximately 48% of the blood volume in men and approximately 42% in women. Erythrocytes are responsible for tissue oxygenation.
- 5. Leukocytes are fewer in number than erythrocytes and constitute approximately 5000 to 10,000 cells/mm3 of blood. Leukocytes defend the body against infection and remove dead or injured host cells.
- 6. Leukocytes are classified as either granulocytes (neutrophils, eosinophils, basophils) or agranulocytes (monocytes/macrophages, lymphocytes, natural killer cells).
- 7. The mononuclear phagocyte system (MPS) is composed of monocytes in bone marrow and peripheral blood and macrophages in tissue. Cells of the MPS ingest and destroy unwanted materials, such as foreign protein particles, circulating immune complexes, microorganisms, debris from dead or injured cells, defective or injured erythrocytes, and dead neutrophils.
- 8. Platelets are irregularly shaped anuclear cytoplasmic fragments. Platelets are essential for blood coagulation and control of bleeding.
- 9. The lymphoid organs are sites of residence, proliferation, differentiation, or function of lymphocytes and mononuclear phagocytes.
- 10. 10 The spleen is the largest lymphoid organ and functions as the site of fetal hematopoiesis, filters and cleanses the blood, and acts as a reservoir for lymphocytes and other blood cells.
- 11. The lymph nodes are the site of development or activity of large numbers of lymphocytes, monocytes, and macrophages.
Development of Blood Cells
- 1. Bone marrow consists of red (active or hematopoietic) marrow and yellow (inactive) marrow.
- 2. The hematopoietic marrow consists of a variety of cellular and molecular microenvironments called niches. Niches support the hematopoietic stem cells by direct cell-to-cell signaling and production of growth factors and cytokines important for retention, expansion, maintenance, and quiescence.
- 3. The bone marrow contains multiple populations of stem cells. Hematopoietic stem cells develop into blood cells. Mesenchymal stem cells develop into osteoblasts, adipocytes, chondrocytes, sinusoidal endothelial cells, fibroblasts, and other stromal cells.
- 4. Osteoblasts (responsible for construction of bone) and osteoclasts (remodel bone by resorption) produce cytokines that affect proliferation and maintenance of hematopoietic cells.
- 5. Specific hematopoietic growth factors (e.g., colony-stimulating factors) are necessary for the adequate production of myeloid, erythroid, lymphoid, and megakaryocytic lineages.
- 6. Hematopoiesis, or blood cell production, occurs in the liver and spleen of the fetus and in the bone marrow after birth.
- 7. Hematopoiesis involves two stages: mitotic division (i.e., proliferation) and maturation (i.e., differentiation). Each type of blood cell has parent cells called stem cells.
- 8. Hematopoiesis continues throughout life to replace blood cells that grow old and die, are killed by disease, or are lost through bleeding.
- 9. Regulation of erythropoiesis (development of red blood cells) is mediated by erythropoietin, which is secreted by the kidneys in response to tissue hypoxia. Erythropoietin causes a compensatory increase in erythrocyte production if the oxygen content of the blood decreases because of anemia, high altitude, or pulmonary disease.
- 10. Hemoglobin, the oxygen-carrying protein of the erythrocyte, enables the blood to transport 100 times more oxygen than could be transported dissolved in plasma alone.
- 11. Erythropoiesis depends on the presence of vitamins (especially vitamin B12, folate, vitamin B6, riboflavin, pantothenic acid, niacin, ascorbic acid, and vitamin E).
- 12. The iron cycle reutilizes iron released from old or damaged erythrocytes. Iron binds to transferrin in the blood, is transported to macrophages, and is stored in the cytoplasm as ferritin.
- 13. Iron homeostasis is controlled by hepcidin, a small hormone produced by hepatocytes, which regulates ferroportin, the principal transporter of iron from stores in hepatocytes and macrophages and from intestinal cells that absorb dietary iron.
- 14. Myelopoiesis is the development of granulocytes (neutrophils, eosinophils, and basophils) and monocytes from the differentiation of myeloid progenitor cells in the bone marrow. Granulocytes are released into the blood and are either functioning cells that circulate in the blood or stored cells that are stored around the walls of blood vessels (marginating storage pool). Monocytes are released into the blood and travel to various tissues to become tissue macrophages and dendritic cells.
- 15. Lymphocytes, which are generated from lymphoid progenitor cells in a process called lymphopoiesis, are released into the bloodstream to undergo further maturation in the primary and secondary lymphoid organs.
- 16. Platelets develop from megakaryocytes by a process called thrombopoiesis, which is controlled by thrombopoietin. During thrombopoiesis, megakaryocytes undergo mitosis but not cell division and the cytoplasm and plasma membrane fragment into platelets.
Mechanisms of Hemostasis
- 1. Hemostasis, or arrest of bleeding, involves (1) vasoconstriction (vasospasm), (2) formation of a platelet plug, (3) activation of the clotting cascade, (4) formation of a blood clot, and (5) clot retraction and clot dissolution.
- 2. The normal vascular endothelium prevents spontaneous clotting by producing factors such as nitric oxide (NO) and prostacyclin I2 (PGI2) that relax the vessels and prevent platelet activation.
- 3. Platelet activation involves three linked processes: (1) adhesion, (2) activation, and (3) aggregation.
- 4. A blood clot is a meshwork of protein strands that stabilizes the platelet plug. The strands are made of fibrin. Fibrin is the end product of the coagulation cascade.
- 5. Lysis of blood clots is the function of the fibrinolytic system. Plasmin, a proteolytic enzyme, splits fibrin and fibrinogen into fibrin degradation products that dissolve the clot.
Clinical Evaluation of the Hematologic System
Pediatrics and the Hematologic System
- 1. Blood cell counts tend to rise above adult levels at birth and then decline gradually throughout childhood. Platelet counts in full-term neonates are comparable with those in adults and remain so throughout infancy and childhood.
- 2. White blood cells develop during fetal life but the function of the cells at birth is weak as the cells are immature.
- 3. Newborn infants are at risk for impaired phagocytosis, bacterial infections, and delayed wound healing. Immature lymphocytes also increase risk for viral infection and there is a weak response to the production of antibodies and a poor response to foreign antigens.
Aging and the Hematologic System
- 1. Blood composition changes little with age. Erythrocyte replenishment may be delayed after bleeding, presumably because of iron deficiency.
- 2. Lymphocyte function appears to decrease with age. Particularly affected is a decrease in cellular immunity.
- 3. Platelet numbers decrease with age, but adhesiveness increases.