Alterations of Hematologic Function in Children
Lauri A. Linder and Anne Harvey
 http://evolve.elsevier.com/Rogers/pathophysiology/
http://evolve.elsevier.com/Rogers/pathophysiology/
This chapter briefly explains fetal and neonatal hematopoiesis and postnatal changes in the blood as a foundation for understanding the pathophysiology of specific blood disorders in childhood. Among the diseases that affect erythrocytes are acquired disorders, such as iron deficiency anemia (IDA), hemolytic disease of the newborn, and anemia of infectious disease; and inherited disorders, such as glucose-6-phosphate dehydrogenase (G6PD) deficiency, hereditary spherocytosis, sickle cell disease, and the thalassemias. Disorders of coagulation and platelets include inherited hemorrhagic diseases, such as the hemophilias, and antibody-mediated hemorrhagic diseases, which include immune thrombocytopenia, autoimmune neonatal thrombocytopenias, and autoimmune vascular purpuras. Finally, leukocyte disorders, such as leukemia and the lymphomas (non-Hodgkin lymphoma as well as Hodgkin lymphoma), are discussed.
Fetal and Neonatal Hematopoiesis
As the developing embryo becomes too large for oxygenation of tissues by simple diffusion, the production of erythrocytes begins within the vessels of the yolk sac. Shortly after 2 weeks of gestation, circulating erythrocytes play a significant role in delivering oxygen to the tissues. At approximately the eighth week of gestation, the site of erythrocyte production shifts from the vessels to the liver sinusoids, and the production of leukocytes and platelets begins in the liver and spleen. Erythropoiesis in the liver and, to a lesser extent, in the spleen and lymph nodes, peaks at approximately 4 months. Hepatic blood formation declines steadily thereafter but does not disappear entirely during the remainder of gestation. By the fifth month of gestation, hematopoiesis begins to occur in the bone marrow and increases rapidly until hematopoietic (red) marrow fills the entire bone marrow space. By the time of delivery, the marrow is the only significant site of hematopoiesis.
In neonates and young infants, hematopoietic marrow progressively fills the bony cavities of the entire axial skeleton (skull, vertebrae, ribs, and sternum), the long bones of the limbs, and many intramembranous bones. (These structures are described in Chapter 43). Fatty (yellow) marrow gradually replaces hematopoietic marrow in some bones. During childhood, hematopoietic tissue retreats centrally to the vertebrae, ribs, sternum, pelvis, scapulae, skull, and proximal ends of the femur and humerus.
In diseases characterized by hemolysis, erythrocyte production can increase as much as 12 times the normal level because erythropoietin causes hematopoietic marrow to increase in volume. Initially, hematopoietic marrow expands from the ends of the long bones toward the middle of the shafts, replacing fatty marrow. Next, blood cell production begins to occur outside the marrow cavities, especially in the liver and spleen. Extramedullary hematopoiesis is more likely to occur in children than in adults because the bony cavities of children already are filled with red marrow (Fig. 30.1), which is the reason hemolytic disease causes especially pronounced enlargement of the spleen and liver in children.
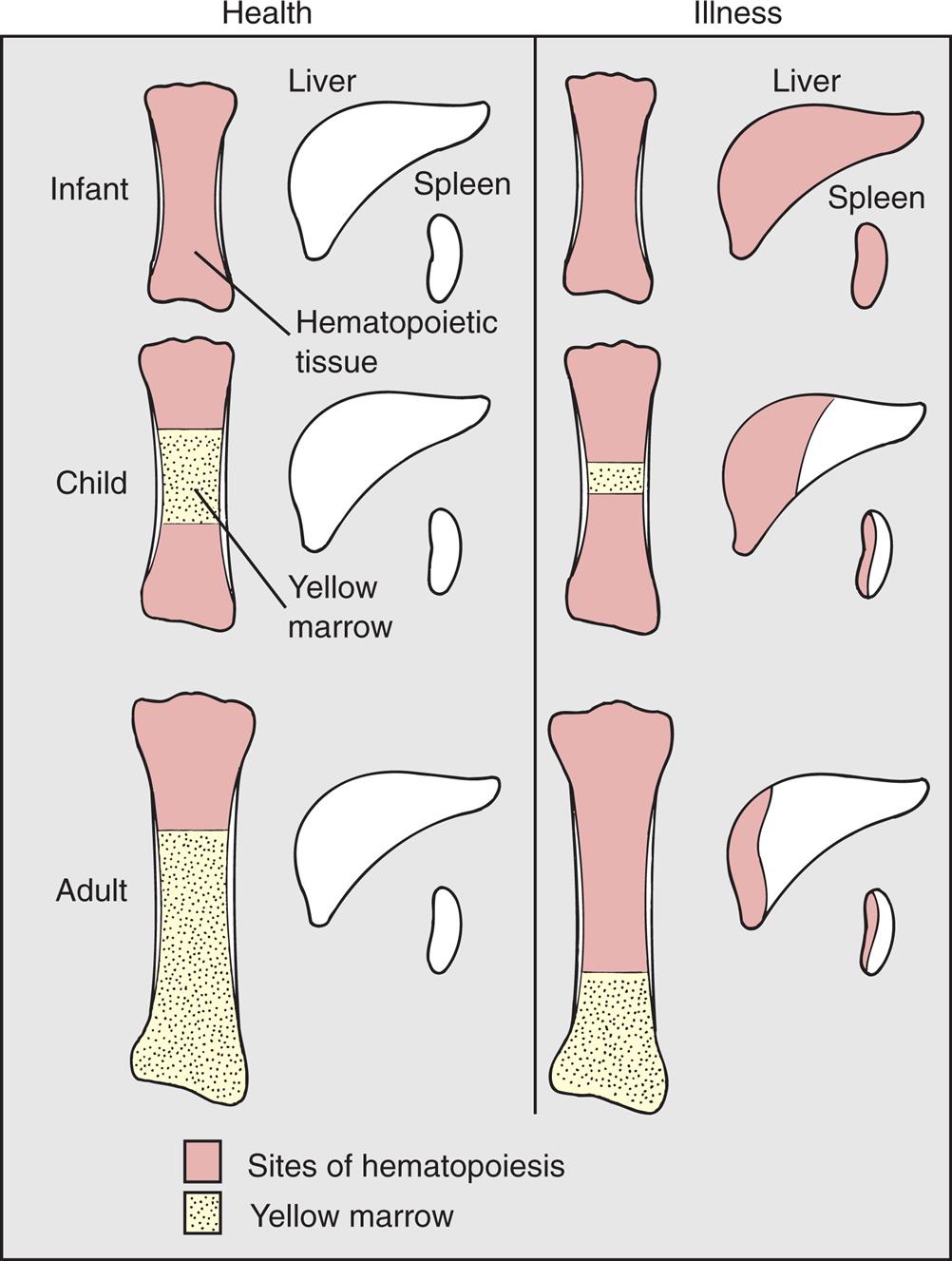
With normal maturation, red marrow is partly replaced by yellow marrow in the shafts of the long bones. In adults, red marrow is largely restricted to the proximal ends of the femur and humerus. In response to hemolysis, red marrow replaces yellow marrow in the long bones. In infants, whose long bones already are filled with red marrow, additional hematopoiesis takes place in the liver and spleen. In children and adults, red marrow can replace yellow marrow in response to hemolysis, necessitating less hematopoiesis in the liver and spleen.
A series of illustration sets compare the sites of hematopoiesis in infant, child, and adult. Each illustration set shows a long bone, the liver, and the spleen. • Healthy infant: The entire bone comprises hematopoietic tissue. The liver and spleen are normal. • Sick infant: The bone, liver, and spleen comprise hematopoietic tissue. • Healthy child: The upper and lower segments of the bone comprise hematopoietic tissue. The central part of the bone comprises yellow marrow. The liver and spleen are normal. • Sick child: The yellow marrow in the bone is narrow, compared to that in the healthy child. Half the liver and half the spleen comprise hematopoietic tissue. • Healthy adult: Three-fourths of the bone comprises hematopoietic tissue. The liver and spleen are normal. • Sick adult: One-fourths of the bone comprises hematopoietic tissue. A very small segment of the liver and about half the spleen comprises hematopoietic tissue.
The erythrocytes undergo striking changes during gestation, particularly during the first two trimesters, at which time they nearly double in numbers and in hemoglobin content. A proportionate increase in hematocrit level also occurs. By the end of gestation, the erythrocyte count has more than tripled, but the size of each erythrocyte has decreased.
A biochemically distinct type of hemoglobin is synthesized during fetal life. The three embryonic hemoglobin molecules (Gower 1, Gower 2, and Portland) have different combinations of alpha, epsilon, zeta, and gamma chains, and fetal hemoglobin (HbF) molecules are composed of two α and two γ chains of polypeptides. Adult hemoglobin HbA1 molecules are composed of two α chains and two β chains, and adult hemoglobin HbA2 molecules are composed of 2 α chains and two δ chains. (The structure of an adult hemoglobin molecule is illustrated in Fig. 28.13 and types of hemoglobin are defined in Table 28.5). A transcriptional regulation mechanism promotes γ-chain synthesis and inhibits β- and δ-chain synthesis in utero. This results in production of embryonic or fetal hemoglobin. After birth, γ-chain synthesis is inhibited, whereas β- and δ-chain synthesis is facilitated, resulting in production of adult hemoglobin types.
Fetal hemoglobin has greater affinity for oxygen than does adult hemoglobin because it interacts less readily with an enzyme (2,3-diphosphoglycerate [2,3-DPG]) that inhibits hemoglobin-oxygen binding. The decreased inhibitory effects of 2,3-DPG enable fetal blood to transport oxygen despite the relative lack of oxygen in the intrauterine environment. The increased affinity for oxygen enables HbF to bind with maternal oxygen in the placental circulation.
During the first trimester, nearly all of the hemoglobin in the fetus is embryonic, but some HbA can be detected. Therefore, some disorders of adult hemoglobin, such as sickle cell anemia and thalassemia major, can be identified as early as 16 to 20 weeks of gestation. In the 6-month fetus, HbF constitutes 90% of the total hemoglobin. This percentage then declines. At birth, neonatal hemoglobin consists of 70% HbF, 29% HbA, and 1% HbA2. Between 6 and 12 months of age, normal adult hemoglobin percentages are established (see Chapter 28).
Postnatal Changes in the Blood
Blood cell counts tend to rise to higher than adult levels at birth and then decline gradually throughout childhood. Table 30.1 lists normal ranges during infancy and childhood. The immediate rise in values is the result of the accelerated hematopoiesis during fetal life, the increased numbers of cells that result from the trauma of birth, and the cutting of the umbilical cord. These events surrounding the birth also are accompanied by the presence of large numbers of immature erythrocytes and leukocytes (particularly granulocytes) in peripheral blood (see Chapter 28). As the infant develops over the first 2 to 3 months of life, the numbers of these immature blood cells decrease.
Table 30.1
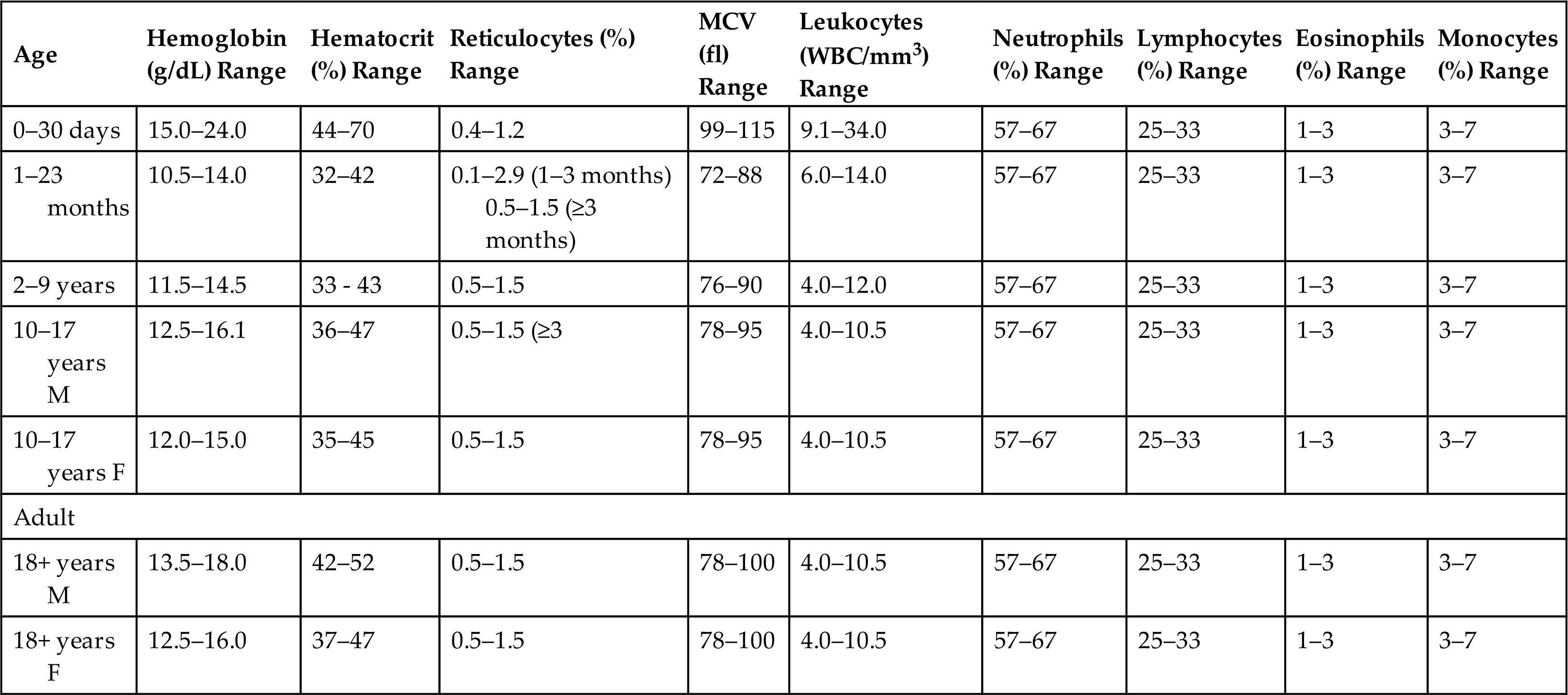
fl, Femtoliters; MCV, mean corpuscular volume; WBC, white blood cells.
From Kliegman RM, et al., eds. Nelson textbook of pediatrics, 21st edition. Philadelphia: Saunders; 2020.
Average blood volume in the full-term neonate is 85 mL/kg of body weight. The premature infant has a proportionately larger blood volume of 90 to 100 mL/kg of body weight. In both full-term and premature infants, blood volume relative to body weight decreases during the first few months. By 3 years of age, a child's average blood volume is 75 to 77 mL/kg, which is similar to that of older children and adults.
Erythrocytes
The hypoxic intrauterine environment stimulates erythropoietin production in the fetus. This accelerates fetal erythropoiesis, producing polycythemia (excessive proliferation of erythrocyte precursors) of the newborn. After birth, the oxygen from the lungs saturates arterial blood, and the amount of oxygen delivered to the tissues increases. In response to the change from a placental to a pulmonary oxygen supply during the first few days of life, levels of erythropoietin and the rate of blood cell formation decrease. The very active rate of fetal erythropoiesis is reflected by the large numbers of immature erythrocytes (reticulocytes) in the peripheral blood of full-term neonates. The number of reticulocytes decreases abruptly during the first few days after birth, which is associated with decreased erythropoietin production.1 Finding an elevated reticulocyte count after the first week of life is rare. A decrease in extramedullary hematopoiesis also occurs at this time. In the peripheral blood, the erythrocyte count drops for 6 to 8 weeks after birth. During this period of rapid growth, the rate of erythrocyte destruction is greater than that in later childhood and adulthood. In full-term infants, the normal erythrocyte life span is 60 to 80 days; in premature infants it may be as short as 20 to 30 days; and in children and adolescents, it is the same as that in adults—120 days (see Mechanisms of hemolysis are described in Chapter 28).
In the premature infant, the postnatal decrease in hemoglobin and hematocrit values is more marked than in the full-term infant. In the preschool and school-age child, hemoglobin, hematocrit, and red blood cell (RBC) count values rise gradually. In males and females, these values first begin to diverge in adolescence. In the female, the gradual hemoglobin level increase continues into early puberty, at which time it stabilizes. In the male, the hemoglobin level increase keeps pace with growth and maturation and eventually surpasses that of the female. This higher value of hemoglobin level in the mature male is related to androgen secretion.
Metabolic processes within the erythrocytes of neonates differ significantly from those of erythrocytes in the normal adult. The relatively young population of erythrocytes in the newborn consumes more glucose than do erythrocytes in adults. Several enzymes that regulate glucose consumption are increased in the erythrocytes of neonates, with a subsequent increase in the rate of glycolysis.
Leukocytes and Platelets
The lymphocytes of children tend to have more cytoplasm and less compact nuclear chromatin than do the lymphocytes of adults. The significance of these differences is unknown. One possible explanation is that children tend to have more frequent viral infections, which are associated with atypical lymphocytes. Even minor infections, in which the child fails to exhibit clinical manifestations of illness, or administration of immunizations may result in lymphocyte changes.2
The lymphocyte count is high at birth and continues to rise in some healthy infants during the first year of life. A steady decline occurs throughout childhood and adolescence until lower adult values are reached. Whether these developmental variations are physiologic or are a pathologic response to frequent viral infections and immunizations in children is not known.
In healthy neonates, the neutrophil count peaks 6 to 12 hours after birth and then declines over the next few days of life.3 Neutrophil counts also are slightly higher in female neonates compared with males. After 2 weeks of life, neutrophil counts fall to within or below normal adult ranges. By approximately 4 years of age, the neutrophil count is similar to that of an adult. White children have slightly higher counts than black children.4
The eosinophil count is elevated in the first year of life relative to children, teenagers, or adults.5,6 Monocyte counts are elevated through the preschool years and then decrease to adult levels. No relationship between age and basophil count has been found. Platelet counts in full-term neonates are comparable to platelet counts in adults and remain so throughout infancy and childhood.5
Disorders of Erythrocytes
Anemia is the most common blood disorder in children. Although anemia is not a disease state in and of itself, its presence may be associated with an underlying pathophysiologic process. Like anemia in adults, anemias occurring in children result from inadequate erythropoiesis or early destruction of erythrocytes. Iron deficiency is the most common cause of inadequate erythropoiesis. Iron deficiency can result from insufficient dietary intake or chronic loss of iron caused by bleeding. The hemolytic anemias of childhood are either inherited, acquired, or both. They may be divided into disorders that result from destruction caused by (1) intrinsic abnormalities of the erythrocytes and (2) damaging factors external to the erythrocytes.
The most dramatic form of acquired congenital hemolytic anemia is hemolytic disease of the fetus and newborn (HDFN), also termed erythroblastosis fetalis. HDFN is an alloimmune disorder in which the maternal blood and fetal blood are antigenically incompatible, causing the mother's immune system to produce antibodies against fetal erythrocytes. Fetal erythrocytes that have been bound to maternal antibodies are recognized as foreign or defective by the fetal mononuclear phagocyte system and are removed from the circulation by phagocytosis, usually in the fetal spleen. (For a complete discussion of HDFN, see Hemolytic Disease of the Fetus and Newborn.) Other acquired hemolytic anemias—some of which begin in utero—include those caused by infections or the presence of toxins.
The inherited forms of hemolytic anemia result from intrinsic defects of the child's erythrocytes, any of which can lead to erythrocyte destruction by the mononuclear phagocyte system. Structural defects include abnormal RBC size and abnormalities of plasma membrane structure (spherocytosis). Intracellular defects include enzyme deficiencies, the most common of which is glucose-6-phosphate dehydrogenase (G6PD) deficiency; and defects of hemoglobin synthesis, which manifest as sickle cell disease or thalassemia, depending on which component of hemoglobin is defective. These and other causes of childhood anemia, some more common than others, are listed in Table 30.2.
Table 30.2
| Cause | Examples of Anemic Condition |
|---|---|
| Blood Loss | |
| Iron deficiency anemia | |
| Decreased Red Cell Production or Hemoglobin Synthesis | |
| Decreased stem cell population in marrow (congenital or acquired pure red cell aplasia) | Normocytic-normochromic anemia |
| Decreased erythropoiesis despite normal stem cell population in marrow (infection, inflammation, cancer, chronic renal disease, congenital dyserythropoiesis) | Normocytic-normochromic anemia |
| Deficiency of a Factor or Nutrient Needed for Erythropoiesis | |
| Cobalamin (vitamin B12), folate | Megaloblastic anemia |
| Iron | Microcytic-hypochromic anemia |
| Increased or Premature Hemolysis | |
| Alloimmune disease (maternal-fetal Rh, ABO, or minor blood group incompatibility) | Autoimmune hemolytic anemia |
| Autoimmune disease (idiopathic autoimmune hemolytic anemia, symptomatic systemic lupus erythematosus, lymphoma, drug-induced autoimmune processes) | Autoimmune hemolytic anemia |
| Inherited defects of plasma membrane structure (spherocytosis, elliptocytosis, stomatocytosis) or cellular size or both (pyknocytosis) | Hemolytic anemia |
| Infection (bacterial sepsis, congenital syphilis, malaria, cytomegalovirus infection, rubella, toxoplasmosis, disseminated herpes) | Hemolytic anemia |
| Intrinsic and inherited enzymatic defects (deficiencies) of G6PD, pyruvate kinase, 5′-nucleotidase, glucose phosphate isomerase | Hemolytic anemia |
| Inherited Defects of Hemoglobin Synthesis | |
| Structurally abnormal globins | Sickle cell anemia |
| Deficient globin synthesis | Thalassemia |
| Other Anemias | |
| Disseminated intravascular coagulation (see Chapter 29) | Hemolytic anemia |
| Galactosemia | Hemolytic anemia |
| Prolonged or recurrent respiratory or metabolic acidosis | Hemolytic anemia |
| Blood vessel disorders (cavernous hemangiomas, large vessel thrombus, renal artery stenosis, severe coarctation of aorta) | Hemolytic anemia |
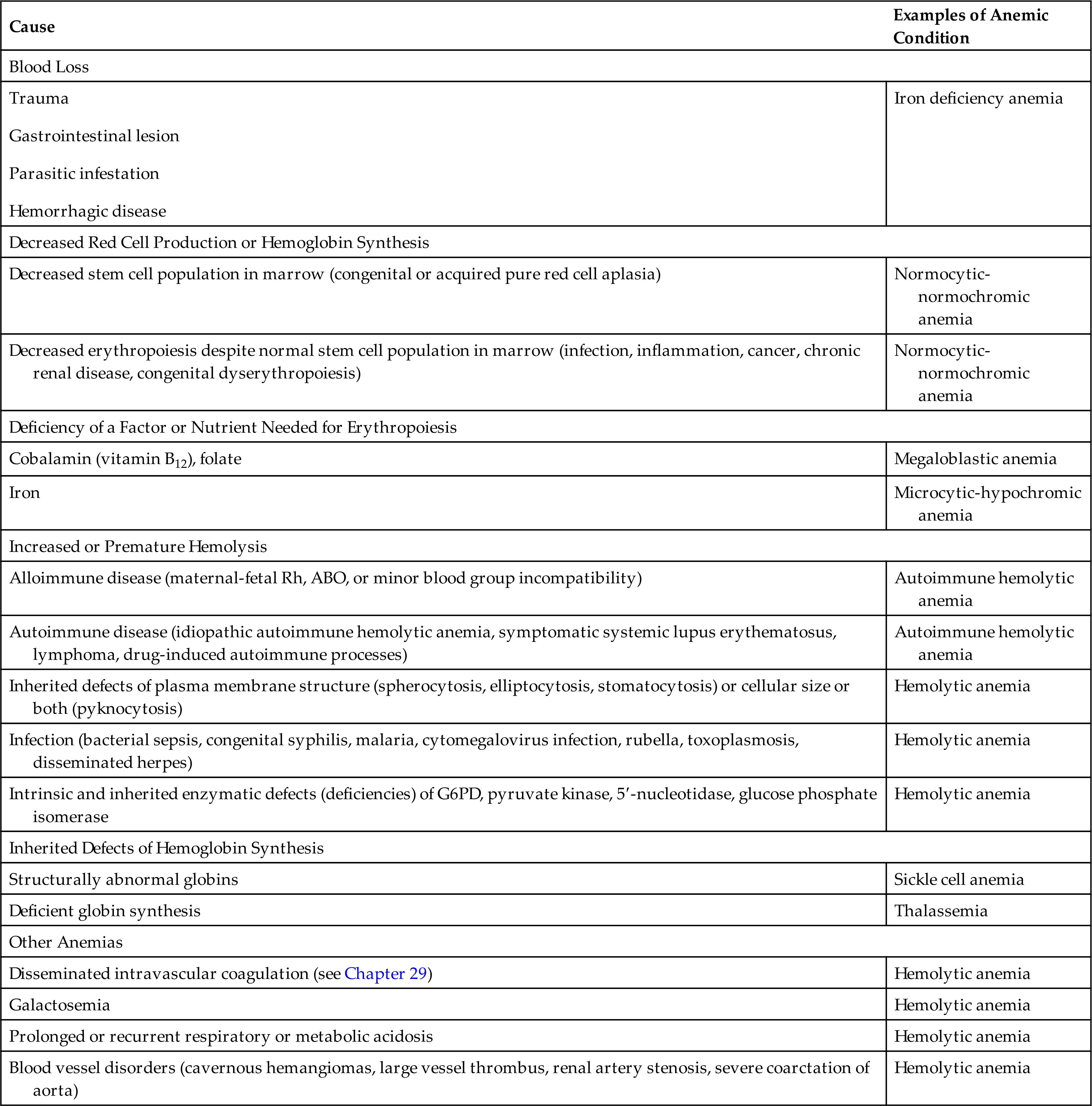
ABO; Type A, type B, type O blood; G6PD; glucose-6-phosphate dehydrogenase.
Acquired Disorders
Iron Deficiency Anemia
IDA is the most common nutritional disorder worldwide, with the highest incidence occurring between 6 months and 2 years of age. Its prevalence in the United States is greatest among toddlers, adolescent girls, and women of childbearing age. Iron is critical to the developing child, especially for normal brain development. Without it, the damage from the periods of IDA is irreversible. The clinical manifestations of IDA are mostly related to inadequate hemoglobin synthesis. The incidence of IDA is not related to gender or race; however, socioeconomic factors are important because they affect nutrition.
IDA can result from (1) dietary lack of iron, (2) problems with iron absorption, (3) blood loss, and (4) increased requirement for iron. During the first few years of life, IDA most often results from inadequate iron intake. During childhood and adolescence, blood loss is the most common cause of IDA. Chronic IDA from occult (hidden) blood loss may be caused by a gastrointestinal lesion, parasitic infestation, or hemorrhagic disease. A reasonable hypothesis for infants and young children who develop IDA is that they have chronic intestinal blood loss induced by exposure to a heat-labile protein in cow's milk. Such exposure causes an inflammatory gastrointestinal reaction that damages the mucosa and results in diffuse microhemorrhage. Growing evidence indicates that cellular components of both innate and adaptive immunity play significant roles during the pathogenesis of cow's milk allergy.7,8
Dietary lack of iron is less common in developed countries, where iron is readily absorbed from heme found in meat. In developing countries, food may be less available. Although iron is found in plants, it is a more poorly absorbed form.9 In the United States, rates of IDA have remained relatively unchanged over the past 10 years.10 Infants are at increased risk for IDA because milk has only very small amounts of iron. The bioavailability of iron from breast milk, however, is higher than that from cow's milk. In developed countries, including the United States, excessive consumption of cow’s milk remains an important predisposing factor for severe IDA.11 Presently, only low-quality evidence for use of micronutrient powders in pregnant women and in the general population is available.12,13 Impaired absorption is found in the context of chronic diarrhea, fat malabsorption, and celiac disease. Evidence also is emerging from international studies regarding genetic polymorphisms that may contribute to altered iron absorption in cases of refractory IDA with a familial component.14,15
Children in developing countries are often affected by chronic parasite infestations that result in blood and iron loss greater than dietary intake. However, a recent systematic review addressing outcomes of anti-helminthic treatments in adolescent girls and women failed to show an effect on anemia and iron deficiency.16 Newer evidence is emerging related to a potential protective benefit of iron deficiency against malaria in parts of the world where malaria is endemic.17 Additional research is needed to establish causality and to determine interventions to minimize adverse health consequences of IDA while maintaining the protective benefit. The association of IDA with lead (Pb) poisoning remains controversial. The role of Helicobacter pylori infection in relation to IDA is an area of ongoing investigation. While gastric colonization with H. pylori containing the sabA gene is associated with IDA, an understanding of its underlying pathophysiology is limited.18
Pathophysiology
No matter the cause, a deficiency of iron produces a hypochromic-microcytic anemia. In the early stages, however, the body may respond by increasing RBC activity in the bone marrow, which may temporarily prevent the development of anemia. As the body's iron stores are depleted, anemia develops (also see Chapter 29). Alterations in hepcidin have an essential role in diseases involving disturbances of iron metabolism (see Chapters 28 and 29). Low serum levels of ferritin and transferrin saturation lead to lowered hemoglobin and hematocrit levels (see Mechanisms of iron depletion are described in Chapter 29).
Clinical Manifestations
The symptoms of mild anemia—listlessness and fatigue—may go unnoticed in infants and young children, who are unable to describe these symptoms. Clinical indicators of anemia also are nonspecific, such as general irritability, decreased activity tolerance, weakness, and lack of interest in play, and may be attributed to other causes. As a result, parents may not note persistent changes in the child's behavior until moderate anemia has developed. In mild to moderate IDA (hemoglobin level of 6 to 10 g/dL), compensatory mechanisms of tissue oxygenation, such as increased amounts of 2,3-DPG within erythrocytes and a shift of the oxyhemoglobin dissociation curve, may be so effective that few clinical manifestations are apparent. Other clinical manifestations, such as pallor, anorexia, tachycardia, and systolic murmurs, are often not present until hemoglobin levels fall below 5 g/dl.
Other symptoms and signs of chronic IDA include splenomegaly, widened skull sutures, decreased physical growth, developmental delays, pica (a behavior in which nonfood substances, such as clay or ice, are eaten). Weight is not necessarily an indicator of IDA because children may be obese, underweight, or normal weight. Consequences of IDA are significant and may include altered neurologic and intellectual function, especially involving attention span, alertness, and learning ability.
Evaluation and Treatment
The evaluation and treatment of IDA are similar in children and adults (see Chapter 29). The diagnosis of IDA is confirmed by laboratory tests. These tests include hemoglobin, hematocrit, serum iron, ferritin levels and the total iron binding capacity. Obtaining a thorough history of the child's present illness and dietary history and performing a complete physical examination also are essential to the evaluation and subsequent clinical management of IDA. Oral administration of a simple ferrous salt is usually sufficient. Taking iron supplements with a vitamin C source helps promote absorption.19 If liquid iron supplements are used, they should be given with a straw or a dropper placed back on the tongue to prevent staining the teeth. Iron therapy is continued for at least 2 months after erythrocyte indexes have returned to normal to replenish iron stores.20
Dietary modification is required to prevent recurrences of IDA.
Intake of iron-rich foods is increased, and the intake of cow's milk may be restricted, with the exact amount restricted to 16 to 32 ounces per day depending on the child's age. Limiting milk intake makes the child hungrier for other iron-rich foods and prevents gastrointestinal blood loss in children whose anemia is aggravated or caused by inflammatory reactions to proteins in cow's milk.
Hemolytic Disease of the Fetus and Newborn
The most common cause of hemolytic anemia in newborns is alloimmune disease. Hemolytic disease of the fetus and newborn (HDFN) (erythroblastosis fetalis) can occur only if antigens on fetal erythrocytes differ from antigens on maternal erythrocytes. The antigens present on erythrocytes are determined genetically. They may be type A, B, or O, and they may or may not include Rh antigen D. Erythrocytes that express Rh antigen D are called Rh-positive; those that do not are called Rh-negative. The frequency of Rh negativity is higher in whites (15%) than in blacks (5%), and it is rare in Asians. Maternal-fetal incompatibility exists if mother and fetus differ in ABO blood type or if the fetus is Rh-positive and the mother is Rh-negative. (The antigenic properties of erythrocytes are described in Chapter 9).
Most cases of HDFN are caused by ABO incompatibility, which occurs if the mother and fetus have different ABO blood types. Although ABO incompatibility occurs in about 20% to 25% of all pregnancies, only 1 in 10 cases of ABO incompatibility results in HDFN. About 1 in 3 cases of HDFN is caused by Rh incompatibility, which occurs when the fetus is Rh-positive and the mother is Rh-negative. Rh incompatibility occurs in less than 10% of pregnancies and rarely causes HDFN in the first incompatible fetus. Typically, erythrocytes from the first incompatible fetus cause the mother's immune system to produce antibodies. These antibodies then can affect the fetuses of subsequent incompatible pregnancies. Even after five or more pregnancies, however, only 5% of women have babies with hemolytic disease. Some minor blood antigens also may be involved.
Pathophysiology
Three conditions must be present for HDFN to occur:
- 1. the mother's blood contains preformed antibodies against fetal erythrocytes or produces them when exposed to fetal erythrocytes,
- 2. sufficient amounts of antibody (usually immunoglobulin G [IgG] class) cross the placenta and enter fetal blood, and
- 3. IgG binds with sufficient numbers of fetal erythrocytes to cause widespread antibody-mediated hemolysis or splenic removal (antibody-mediated cellular destruction is described in Chapter 9).
In most cases of HDFN, the mother has blood type O, and the fetus has blood type A or B. Maternal antibodies may be formed against type B fetal erythrocytes if the mother has blood type A or against type A fetal erythrocytes if the mother has blood type B.
ABO incompatibility can cause HDFN even if fetal erythrocytes do not escape into the maternal circulation during pregnancy because the blood of most adults already contains anti-A or anti-B antibodies. These antibodies are produced on exposure to certain foods or infection by gram-negative bacteria. As a result, IgG against type A or B erythrocytes is usually already present in maternal blood and can enter the fetal circulation during the first incompatible pregnancy. Anti-O antibodies do not exist because type O erythrocytes are not antigenic.
Anti-Rh antibodies, on the other hand, form only in response to the presence of Rh-positive erythrocytes from the fetus in the blood of an Rh-negative mother. This exposure typically occurs when fetal blood is mixed with the mother's blood at the time of delivery. Exposure may also occur through transfused blood, and, rarely, previous sensitization of the mother by her own mother's incompatible blood.
The first Rh-incompatible pregnancy generally presents no difficulties for the fetus because few fetal erythrocytes cross the placental barrier during the pregnancy. When the placenta detaches at birth, many fetal erythrocytes often enter the mother's bloodstream. If the mother is Rh-negative and the fetus is Rh-positive, the mother produces anti-Rh antibodies. The capacity of the mother's immune system to produce anti-Rh antibodies depends on many factors, including her genetic capacity to make antibodies against the Rh antigen D, the amount of fetal-to-maternal bleeding, and the occurrence of any bleeding earlier in the pregnancy. These anti-Rh antibodies persist in the mother's bloodstream for a long time. If the next offspring is Rh-positive, the mother's anti-Rh antibodies can enter the bloodstream of the fetus and destroy the erythrocytes. Antibodies against Rh antigen D are of the IgG class and easily cross the placenta.
Antibody-coated fetal erythrocytes are usually destroyed through extravascular hemolysis, primarily by phagocytic cells in the spleen. As hemolysis proceeds, the fetus becomes anemic. Erythropoiesis accelerates, particularly in the liver and spleen. Immature nucleated cells (erythroblasts) are released into the bloodstream, hence the name erythroblastosis fetalis. The degree of anemia depends on several factors: (1) the length of time the antibody has been in the fetal circulation, (2) the concentration of the antibody, and (3) the ability of the fetus to compensate for increased hemolysis. During the pregnancy, unconjugated (indirect) bilirubin, which forms during the breakdown of hemoglobin, is transported across the placental barrier into the maternal circulation and is excreted by the mother. Hyperbilirubinemia, an increase in bilirubin concentration in the blood occurs in the neonate shortly after birth because lipid-soluble unconjugated bilirubin is no longer excreted through the placenta.
HDFN is typically more severe in Rh incompatibility than in ABO incompatibility. ABO incompatibility may resolve after birth without life-threatening complications. Maternal-fetal incompatibility in which a mother with type O blood has a child with type A or B blood usually is so mild that it does not require treatment.
Rh incompatibility is more likely to result in severe or even life-threatening anemia, death in utero, or damage to the central nervous system (CNS). Severe anemia alone can cause death because of cardiovascular complications. Extensive hemolysis can result in increased levels of unconjugated bilirubin in the neonate's circulation. If bilirubin levels exceed the liver's ability to conjugate and excrete bilirubin, it can be deposited in the brain, a condition known as kernicterus, causing cellular damage and, eventually, death if the neonate does not receive exchange transfusions.
Fetuses that do not survive anemia in utero are usually stillborn with gross edema in the entire body, a condition called hydrops fetalis. Death can occur as early as 17 weeks’ gestation and results in spontaneous abortion.
Clinical Manifestations
Neonates with mild HDFN may appear healthy or slightly pale, with slight enlargement of the liver or spleen. Pronounced pallor, splenomegaly, and hepatomegaly indicate severe anemia, which predisposes the neonate to cardiovascular failure and shock. Life-threatening symptoms as a consequence of Rh incompatibility, however, are rare, largely because of the routine use of Rh immunoglobulin.
Because the maternal antibodies remain in the neonate's circulatory system after birth, erythrocyte destruction can continue causing hyperbilirubinemia and icterus neonatorum (neonatal jaundice) that occurs shortly after birth. Without replacement transfusions, in which the child receives Rh-negative erythrocytes, the bilirubin can be deposited in the brain, causing a condition termed kernicterus. If kernicterus develops, it can cause cerebral damage, including intellectual disabilities, cerebral palsy, high-frequency deafness, and even death (icterus gravis neonatorum).21
Evaluation and Treatment
Fetuses and neonates with ABO incompatibility typically do not require additional monitoring or treatment. Fetuses and infants at risk for HDFN because of Rh incompatibility may require additional monitoring and treatment. Routine evaluation of fetuses at risk for HDFN includes the Coombs test. The indirect Coombs test measures antibody in the mother's circulation and indicates whether the fetus is at risk for HDFN. The direct Coombs test measures antibody already bound to the surfaces of fetal erythrocytes. It is used primarily to confirm the diagnosis of antibody-mediated HDFN. If a prior history of fetal hemolytic disease is present, additional diagnostic tests are done to determine risk with the current pregnancy. These include maternal antibody titers, fetal blood sampling, amniotic fluid spectrophotometry, and ultrasound fetal assessment.22
Prevention is the key to managing HDFN that results from Rh incompatibility. Immunoprophylaxis through the use of Rh immune globulin (RhoGAM), a preparation of antibody against Rh antigen D (anti-D Ig), prevents an Rh-negative woman from producing antibodies. If an Rh-negative woman is given Rh immune globulin within 72 hours of exposure to Rh-positive erythrocytes, she will not produce antibody against the D antigen. As a result, the next Rh-positive baby she conceives will be protected. Updated United States and United Kingdom guidelines also state that if anti-D Ig is not given within 72 hours, every effort should be made to administer it within 10 days.23,24
The injected (anti-D Ig) antibodies remain in the mother's bloodstream long enough to prevent her immune system from producing its own anti-Rh antibodies but not long enough to affect subsequent offspring. The mother must be given Rh immune globulin injections after the birth of each Rh-positive baby and after a miscarriage. The mother also must be especially careful not to receive a transfusion containing Rh-positive blood because this would stimulate production of anti-Rh antibodies.
If antigenic incompatibility of the mother's erythrocytes is not discovered in time to administer Rh immune globulin and a child is born with HDFN, treatment consists of exchange transfusions in which the neonate's blood is replaced with new Rh-positive blood that is not contaminated with anti-Rh antibodies. This treatment is instituted during the first 24 hours of extrauterine life to prevent kernicterus. Phototherapy also is used to reduce the toxic effects of unconjugated bilirubin.
Jaundice and indirect hyperbilirubinemia are reduced when the infant is exposed to high-intensity light in the visible spectrum from 460 to 490 nm.25 Bilirubin in the skin absorbs light energy, which converts the toxic unconjugated bilirubin into its conjugated water-soluble form that is excreted in the bile. Phototherapy also causes autosensitization that results in oxidation reactions. Breakdown products from the oxidation reactions are excreted by the liver and kidney without need for conjugation. The therapeutic effect of phototherapy depends on the light energy emitted in the effective wavelengths, the distance between the infant and the light source, and the amount of skin exposed. The rate of hemolysis and the infant's ability to excrete bilirubin also are factors in determining the effectiveness of phototherapy in lowering serum bilirubin levels.
Anemia of Infectious Disease
Infections of the newborn, often initially acquired by the mother and transmitted to the fetus, may result in a hemolytic anemia with clinical manifestations similar to those of HDFN. Congenital syphilis, toxoplasmosis, cytomegalic inclusion disease, rubella, coxsackievirus B infection, herpesvirus infection, and bacterial sepsis can cause hemolytic anemia in the neonate.
The exact mechanism of anemia caused by congenital infections is unclear. In some instances, it is related to direct injury of erythrocyte membranes or erythrocyte precursors by the infectious microorganism. In other instances, it results from traumatic destruction of erythrocytes during their passage through inflamed capillaries.
Anemia in Critically Ill Children
Anemia is a common occurrence in critically ill children (see Chapter 49). The causes are numerous and include decreased erythropoietin activity, poor iron use by the body, and blood loss from diverse conditions and consequences of treatment. A topic of ongoing discussion is whether transfusion of blood products, particularly packed RBCs, improves outcomes in critically ill children because of problems related to blood storage. New research is ongoing and needed to understand these problems, the development of new blood transfusion strategies, and blood substitutes.26
Inherited Disorders
A number of inherited and intrinsic erythrocyte conditions are known to cause hemolytic disease or increased hemolysis (see Table 30.2). These conditions may result from enzymatic abnormalities that disrupt metabolic processes and prevent normal biochemical balance within the cell, alterations of hemoglobin structure or synthesis, or plasma membrane defects accompanied by changes in erythrocyte size or shape.
Glucose-6-Phosphate Dehydrogenase Deficiency
Glucose-6-phosphate dehydrogenase (G6PD) deficiency is an inherited disorder caused by a genetic defect in the RBC enzyme G6PD, which is involved in the normal processing of carbohydrates. It is the most common disorder of RBCs, estimated to affect 200 to 400 million people worldwide. The deficiency occurs most often in tropical and subtropical regions of the Eastern Hemisphere including the Mediterranean region, Africa, the Middle East, and Asia. G6PD deficiency is an X-linked recessive disorder, most fully expressed in homozygous males, although partial expression is possible in heterozygous females because of mosaicism resulting from X-inactivation. (X-linked inheritance is discussed in Chapter 4). More than 200 mutations have been identified in the G6PD gene, almost all of which result in changes in single amino acids. The consequences of these mutations are decreased quantities of the G6PD enzyme or an enzyme with abnormal structure and function. The type of mutation that is present will determine the degree of deficiency and the associated clinical manifestations.27
G6PD is responsible for the first step in a pathway that converts glucose to ribose-5-phosphate. These initial chemical reactions usually produce another enzyme, nicotinamide adenine dinucleotide phosphate (NADPH), which helps protect cells from oxidative stress from reactive oxygen species. The deficiency of G6PD leads to damaged RBCs that can hemolyze, or rupture and break down prematurely.
Pathophysiology
G6PD enables erythrocytes to maintain normal metabolic processes despite injury from oxidative stressors. As a result, G6PD deficiency is usually asymptomatic unless exposure to a source of oxidative stress occurs. Deficient or abnormal enzyme function can cause abnormalities in the hexose monophosphate shunt or glutathione metabolism that impairs the ability of RBCs to protect themselves against oxidative stress injuries. Such injury can cause both an intravascular and an extravascular hemolysis. Sources of oxidative stress include exposure to certain classes of drugs (sulfonamides, nitrofurantoins, antimalarial agents, salicylates, or naphthaquinolones), ingestion of fava beans (a dietary staple in some Mediterranean areas and parts of Africa), hypoxemia, fever, acidosis, or infection. Infections associated with initiation of hemolysis include viral hepatitis, pneumonia, and typhoid fever. Erythrocyte damage in affected children begins after intense or prolonged exposure to one or more sources of oxidative stress and ceases when the stressors are removed. In black males, the G6PD defect becomes more pronounced as the erythrocyte ages. In other population groups the defect is profound even in young erythrocytes. A fetus with G6PD deficiency may experience an episode of hemolysis if the mother ingests a substance with oxidant properties, such as a salicylate (aspirin).
In the absence of G6PD, oxidative stressors damage hemoglobin and the plasma membranes of erythrocytes and possibly interfere with the activities of other enzymes within the cell. Hemoglobin is oxidized progressively to methemoglobin, sulfmethemoglobin, and denatured globin-glutathione complexes. Exposure to oxidizing substances results in the precipitation of insoluble hemoglobin inclusions, called Heinz bodies, within the cell. Plasma membrane damage and the presence of Heinz bodies cause hemolysis, primarily in the spleen.
Clinical Manifestations
In infants, G6PD deficiency may present as icterus neonatorum. The most common clinical manifestation of G6PD deficiency is acute hemolytic anemia, usually after infections or the ingestion of certain oxidative drugs or foods. Ingestion of fava beans can produce a severe hemolytic reaction in children with G6PD deficiency.
Hemolytic episodes are characterized by pallor, icterus, dark urine, back pain, and, in severe cases, shock, cardiovascular collapse, and death. When the child is not exposed to sources of oxidative stress, the child does not have anemia, and erythrocyte survival is normal.
Evaluation and Treatment
Reduced G6PD activity in erythrocytes is required for diagnosis. Immediately after a hemolytic episode, reticulocytes and young erythrocytes are evident. Because young erythrocytes have significantly higher enzyme activity than do older cells, laboratory evaluation should not be performed after a crisis. Doing so can result in a low to normal range, a false-negative result. Testing for suspected G6PD deficiency should be done when hemoglobin and reticulocytes are normal. G6PD deficiency also can be detected by electrophoretic analysis.
Prevention of hemolysis is the most important therapeutic measure. Prevention includes avoiding medications and dietary substances associated with hemolysis. Because of the high frequency of G6PD deficiency in areas of the world that are endemic for malaria, the World Health Organization (WHO) currently recommends testing for G6PD deficiency before administration of antimalarial medications in these regions.28 When hemolysis occurs, supportive treatment may include blood transfusions and oral iron therapy. Spontaneous recovery generally follows treatment.
Hereditary Spherocytosis
Hereditary spherocytosis (HS) is an inherited disorder caused by defects in the membrane skeleton of RBCs. The changes cause RBCs to become spherical, less deformable, and vulnerable to destruction. Mutations in at least five genes that produce proteins necessary for the RBC membrane (ANK1, EPB42, SLC4A1, SPTA1, and SPTB) result in the HS phenotype. Proteins produced by these genes act as transporters for molecules in and out of the cells, attach to other proteins, and maintain the RBC’s biconcave disc shape. Thus, these proteins help the cells to be flexible for RBC mobility from large blood vessels to capillaries. Altered RBC cell membranes result in a more rigid and spherical cell shape. These misshapen cells, or spherocytes, are vulnerable to destruction or hemolysis particularly in the splenic vessels. Ultimately, these damaged cells are removed from circulation in the spleen.
Pathophysiology
HS is transmitted as an autosomal dominant trait in about 75% of cases. The defect results from properties of its specialized membrane skeleton, which lies close to the internal surface of the plasma membrane. The affected proteins include spectrins and ankyrin, and their intrinsic defects in the membrane cause less deformability and increased vulnerability to splenic sequestration and destruction.
The spleen is intimately involved in the hemolytic process. The spherocyte is relatively rigid and passes with difficulty through the small openings between the splenic cords and sinuses initiating macrophage response. Circulation of blood to the spleen creates repeated circulation through a metabolic environment that results in sequestration and destruction of spherocytes.
Clinical Manifestations
The presenting signs of HS are anemia, jaundice, and splenomegaly, which can be mild to severe depending on the individual's physiologic compensation. In these cases, the reticulocyte count will be elevated. In infancy, hemolytic anemia and hyperbilirubinemia may be severe.29 Infants and children may have life-threatening anemia with clinical symptoms ranging from difficulty feeding, circumoral pallor, tachycardia, nasal flaring, and diaphoresis to lethargy. They also are at increased risk for gallstones because of the presence of extra bile pigment from hemolysis. Infection (specifically parvovirus),30 fever, and stress stimulate the spleen to destroy more RBCs than usual, leading to a worsening anemia in a child with baseline anemia. The parvovirus also infects erythroid progenitor cells in the bone marrow resulting in an aplastic crisis.31
Evaluation and Treatment
Ascertaining a family history of spherocytosis is important. Laboratory findings include spherocytes in the peripheral blood smear (spherocytosis), elevated reticulocyte count (with or without anemia), indirect hyperbilirubinemia, and a positive osmotic fragility test. An osmotic fragility test is performed by placing RBCs in a saline solution for 24 hours. Spherocytes do not tolerate saline solutions; as a result, they burst more readily than normal RBCs. In addition, flow cytometry can detect reductions in RBC transmembrane proteins. Band 3 Protein Reduction testing can confirm the diagnosis of HS. Treatment of HS is based on disease severity and some children or adolescents may require RBC transfusions; however, this is somewhat rare. Management includes regular monitoring for anemia and splenomegaly. Daily folic acid supplementation supports the increased production of healthy RBCs. In the past, splenectomy was the first line of treatment. Currently, however, splenectomy is only recommended for those children more than 5 years of age with severe anemia or splenomegaly. Some children who develop gallstones may undergo splenectomy and cholecystectomy. Partial splenectomy, in which only a portion of the spleen is removed, is being performed on children with HS to decrease the risk of postsplenectomy complications.31
Sickle Cell Disease
Sickle cell disease is a group of autosomal recessive disorders characterized by the production of an atypical type of hemoglobin, hemoglobin S (HbS; sickle hemoglobin) within the erythrocytes. The hemoglobin molecule consists of four protein subunits called α-globin and two subunits called β-globin. The hemoglobin B (HbB) gene provides instructions for making protein β-globin. HbS is formed as a result of a genetic point mutation (missense mutation) in β-globin in which one amino acid (valine) replaces another (glutamic acid) (Fig. 30.2). Abnormal versions of β-globin can cause erythrocytes to distort into a sickle shape. Other mutations in the HbB gene lead to other versions of β-globin, such as hemoglobin C (HbC) and hemoglobin E (HbE). HbB gene mutations also can affect the quantity of β-globin, such as low levels of β-globin found in β-thalassemia.
![Top panel, A. The illustration shows the structure of a normal hemoglobin beta-chain and a sickle cell anemia hemoglobin beta-chain. The normal hemoglobin beta-chain shows the following sequence: valine, histidine, leucine, threonine, proline, glutamic acid, and glutamic acid. The sickle cell anemia hemoglobin beta-chain shows the following sequence: valine, histidine, leucine, threonine, proline, valine, and glutamic acid. Bottom panel, B. Two adjacent photomicrographs show a cluster of normal red blood cells on the left and a sickle red blood cell on the right. An accompanying illustration, C, shows and labels the following structures: macrophage-sheathed capillaries and splenic sinusoid. The sequence of mechanisms is as follows. 1. Retrograde obstruction by irreversibly sickled cells is a consequence of reduction in blood flow that aggravates the obstruction because oxygen tension decreases. 2. Preferential adhesion of sickled cells to endothelial cell surfaces increases with peripheral resistance and causes narrowing of the vascular lumen. 3. Dense trapping of sickled cells in splenic sinusoids. 4. Hemolysis caused by precipitation of Hb and dissociation of the red blood cell plasma membrane from the subjacent cytoskeleton. 5. Macrophages phagocytose remnants of hemolytic sickled cells. Blood flows from the left to the right. Text below reads as follows. Sickle cell anemia is determined by the substitution of normal hemoglobin (H b A) by hemoglobin S (H b S) caused by a point mutation (replacement of the nucleotide triplet C T C coding glutamic acid at the m R N A level [G A G] by the C A C triplet [G U G] coding for valine) that modifies the physicochemical properties of the beta¬-globin chain of hemoglobin. All hemoglobin is abnormal in homozygous individuals for the mutant gene, and red blood cells show a sickling deformity and hemolytic anemia in the presence or absence of normal oxygen tension. Heterozygous individuals contain a mixture of H b A and H b S, and sickling and anemia are observed when the tension of oxygen decreases. Irreversibly sickled red blood cells are trapped within the splenic sinusoids and are destroyed by adjacent macrophages. Hemolysis may also occur in the macrophage-sheathed capillaries of the red pulp.](../images/F000306f30-02-9780323789875.jpg)
Top panel, A. The illustration shows the structure of a normal hemoglobin beta-chain and a sickle cell anemia hemoglobin beta-chain. The normal hemoglobin beta-chain shows the following sequence: valine, histidine, leucine, threonine, proline, glutamic acid, and glutamic acid. The sickle cell anemia hemoglobin beta-chain shows the following sequence: valine, histidine, leucine, threonine, proline, valine, and glutamic acid. Bottom panel, B. Two adjacent photomicrographs show a cluster of normal red blood cells on the left and a sickle red blood cell on the right. An accompanying illustration, C, shows and labels the following structures: macrophage-sheathed capillaries and splenic sinusoid. The sequence of mechanisms is as follows. 1. Retrograde obstruction by irreversibly sickled cells is a consequence of reduction in blood flow that aggravates the obstruction because oxygen tension decreases. 2. Preferential adhesion of sickled cells to endothelial cell surfaces increases with peripheral resistance and causes narrowing of the vascular lumen. 3. Dense trapping of sickled cells in splenic sinusoids. 4. Hemolysis caused by precipitation of Hb and dissociation of the red blood cell plasma membrane from the subjacent cytoskeleton. 5. Macrophages phagocytose remnants of hemolytic sickled cells. Blood flows from the left to the right. Text below reads as follows. Sickle cell anemia is determined by the substitution of normal hemoglobin (H b A) by hemoglobin S (H b S) caused by a point mutation (replacement of the nucleotide triplet C T C coding glutamic acid at the m R N A level [G A G] by the C A C triplet [G U G] coding for valine) that modifies the physicochemical properties of the beta¬-globin chain of hemoglobin. All hemoglobin is abnormal in homozygous individuals for the mutant gene, and red blood cells show a sickling deformity and hemolytic anemia in the presence or absence of normal oxygen tension. Heterozygous individuals contain a mixture of H b A and H b S, and sickling and anemia are observed when the tension of oxygen decreases. Irreversibly sickled red blood cells are trapped within the splenic sinusoids and are destroyed by adjacent macrophages. Hemolysis may also occur in the macrophage-sheathed capillaries of the red pulp.
The most prevalent types of sickle cell disease are sickle cell anemia, sickle cell–thalassemia disease, and sickle cell–HbC disease (Table 30.3). (See Chapter 4 for a discussion of genetic inheritance of disease.) SCD is inherited in an autosomal recessive pattern in which each parent carries one copy of the mutated gene. Sickle cell anemia (SCA; HbSS), a homozygous form, is the most severe.32 The most prevalent SCD genotypes include homozygous hemoglobin SS (HbSS, or sickle cell anemia) and the compound heterozygous conditions hemoglobin Sβ0-thalassemia (Hbβ0-thalassemia), hemoglobin Sβ-thalassemia (HbSβ+-thalassemia), and hemoglobin SC disease (HbSC).33 HbSS and HbSβ0-thalassemia are clinically similar and are, therefore, commonly referred to as sickle cell anemia (SCA); these genotypes are associated with the most severe clinical manifestations. Sickle cell trait (HbAS), in which the child inherits HbS from one parent and normal hemoglobin (HbA) from the other. This heterozygous carrier state rarely has clinical manifestations and is not regarded as a form of SCD. All forms of SCD are lifelong conditions.34
Table 30.3
Sickle cell disease affects millions of people worldwide and is most common among persons with ancestry from sub-Saharan Africa. Although less common, it also is present among individuals with ancestry from Mediterranean countries, the Arabian Peninsula, parts of India, and Spanish-speaking areas of South America. Approximately 100,000 individuals living in the United States have SCD,33 and most infants with SCD born in the United States are now identified by routine neonatal screening. In the United States, sickle cell anemia is most common in black people, with a reported incidence of around 1 in 365 live births.33 In the general population, the risk of two black parents having a child with sickle cell anemia is 0.7%. Sickle cell–HbC disease occurs in 1 in 800 births, and sickle cell–thalassemia is even less common (1 in 1700 births).
Between 1 and 3 million Americans and more than 100 million individuals worldwide are estimated to be heterozygous carriers for the sickle cell trait (HbAS).34 Sickle cell trait is present in 7% to 13% of African Americans. Its prevalence in African countries, such as Nigeria and the Democratic Republic of Congo, may be as high as 30%.35 The sickle cell trait may provide protection against lethal forms of malaria. This results in a genetic advantage for carriers who reside in regions of the world that are endemic for malaria, such as sub-Saharan Africa and some Mediterranean countries.
Under conditions of decreased oxygen tension and dehydration, HbS stretches and elongates. Repeated cycles of deoxygenation and oxygenation cause the HbS molecule to polymerize and stiffen. These polymers can damage the RBC structure, leading to sickle-shaped RBCs. This change causes several pathologic consequences: the sickle-shaped RBCs die prematurely leading to hemolytic anemia, microvascular obstruction, and ischemic tissue damage (Fig. 30.3).
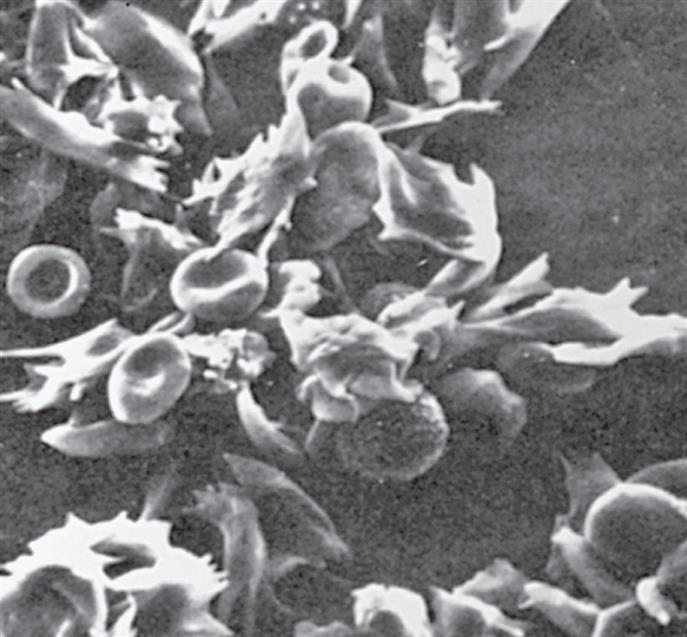
The irregularly shaped cells are the sickle cells; the circular cells are the normal blood cells. (From Raven PH, Johnson GB. Biology, 3rd edition. St. Louis: Mosby; 1992.)
Two effective disease therapies for SCD are hydroxyurea and chronic transfusion.32 The role of curative treatments including hematopoietic stem cell transplantation (HSCT) and gene therapy (GT) for SCD are increasing. See Emerging Science Box: Gene Therapy for Sickle Cell. Although data are encouraging, additional studies are needed to assess efficacy and long-term outcomes.36
Pathophysiology
The sickling process is an occasional, intermittent phenomenon that can be triggered or sustained by one or more of the following stressors: decreased oxygen tension (PO2) of the blood (e.g., hypoxemia), acidosis (decreased pH), increased plasma osmolality, decreased plasma volume, and low temperature (Fig. 30.4). Low temperatures can precipitate sickle crisis, presumably because of vasoconstriction.37
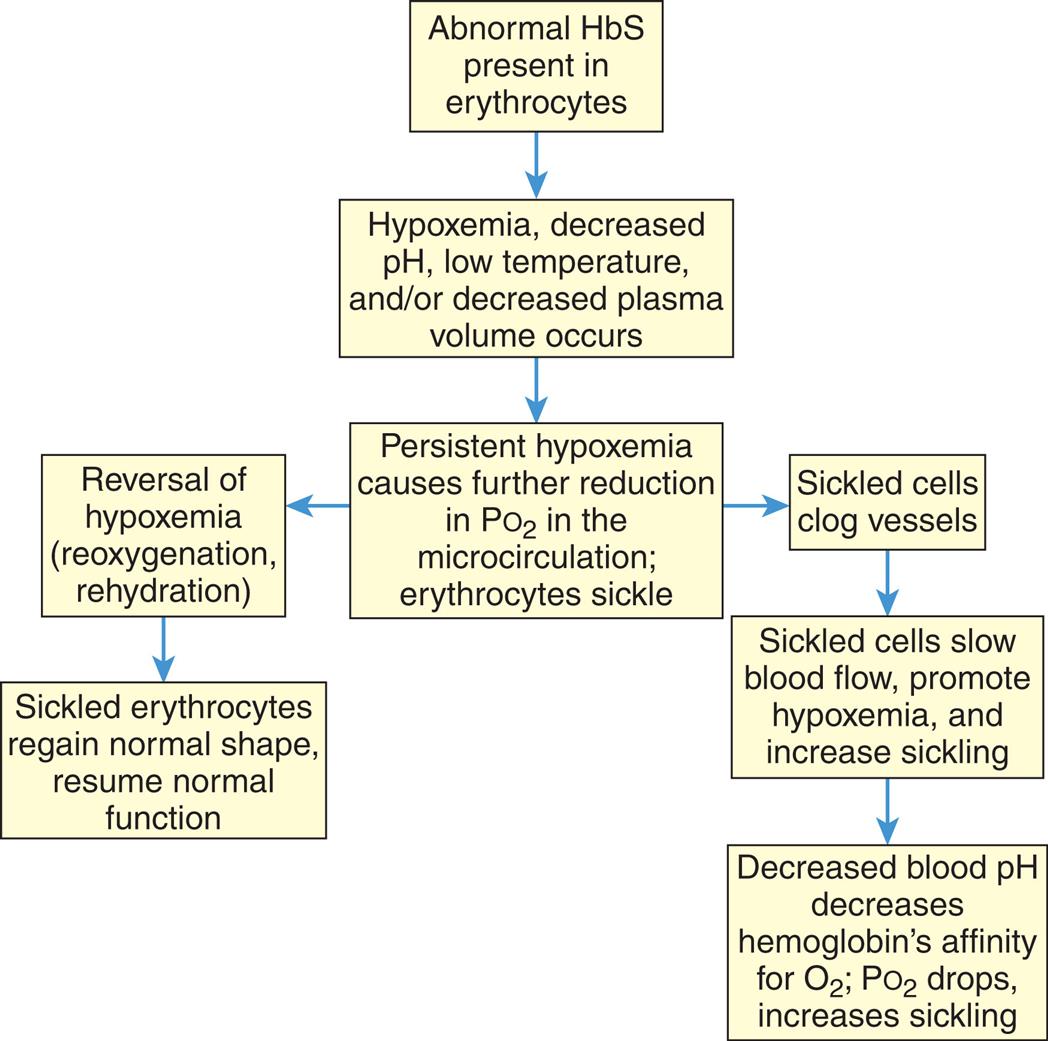
A flowchart shows the sequence of sickling of erythrocytes. 1. Abnormal H b S present in erythrocytes. Leads to 2. 2. Hypoxemia, decreased p H, low temperature, and or decreased plasma volume occurs. Leads to 3. 3. Persistent hypoxemia causes further reduction in partial pressure of oxygen in the microcirculation; erythrocytes sickle. Leads to 4 and 6. 4. Reversal of hypoxemia (reoxygenation, rehydration). Leads to 5. 5. Sickled erythrocytes regain normal shape, resume normal function. 6. Sickled cells clog vessels. Leads to 7. 7. Sickled cells slow blood flow, promote hypoxemia, and increase sickling. Leads to 8. 8. Decreased blood p H decreases hemoglobin’s affinity for oxygen; partial pressure of oxygen drops, increases sickling.
The pathophysiology of the sickling process includes erythrocyte derangement, chronic hemolysis (hemolytic anemia), microvascular occlusions, and tissue damage. HbS is soluble and usually causes no problem when it is properly oxygenated; therefore, deoxygenation is probably the most important variable in determining the occurrence of sickling. Other significant variables that affect sickling include interaction of HbS with other types of hemoglobin in the cell, mean cell hemoglobin concentration (MCHC), intracellular pH, and transit times of erythrocytes through the microcirculation. Among individuals who are heterozygous for sickle cell trait, the presence of other types of Hb prevents sickling except under conditions of severe hypoxia.
As the sickling process begins, intracellular dehydration increases the MCHC, which increases sickling. A decrease in pH reduces the oxygen affinity of hemoglobin, resulting in a further increase in the quantity of deoxygenated HbS regardless of the oxygen tension. These changes, in turn, increase the sickling process. Inflammation in the microcirculation will slow erythrocyte transit times because blood flow is sluggish with adhesion of leukocytes to activated endothelial cells. Increased osmolality of the plasma draws water out of the erythrocytes. The result is sickling by raising the relative HbS content in erythrocytes. Investigators are studying the optimal intravenous fluid to increase erythrocyte deformability and biomechanical properties.38
Sickling causes damage to erythrocytes through several mechanisms, including (1) membrane derangements occur because as HbS units (polymers) grow they protrude through the membrane skeleton by only the lipid layer, causing changes in membrane structure: (2) membrane derangement leads to changes in ionic flow with an influx in Ca++ ions, which induces cross-linking of membrane proteins and activation of an ion channel that induces the efflux of K+ and H2O; and (3) with time the damaged cells are converted to end-stage, nondeformable or stiff and irreversibly sickled cells (Fig. 30.5). Recent studies have shown that elevated red cell levels of the enzyme sphingosine kinase 1 (SPH1) underlie sickling and disease progression by increasing sphingosine 1-phosphate (S1P) production in the blood.39,40S1P level, a bioactive lipid enriched in red cells, is elevated in red cells and plasma of mice and humans with SCD.40 S1P also is a signaling molecule that regulates diverse biologic functions including inflammation.41 Additionally, investigators demonstrated that the compound 5 C can inhibit SPHK1 and, thus, has antisickling properties.42 These data are important for identifying the structure of the sickling process to assess potential new therapeutics.
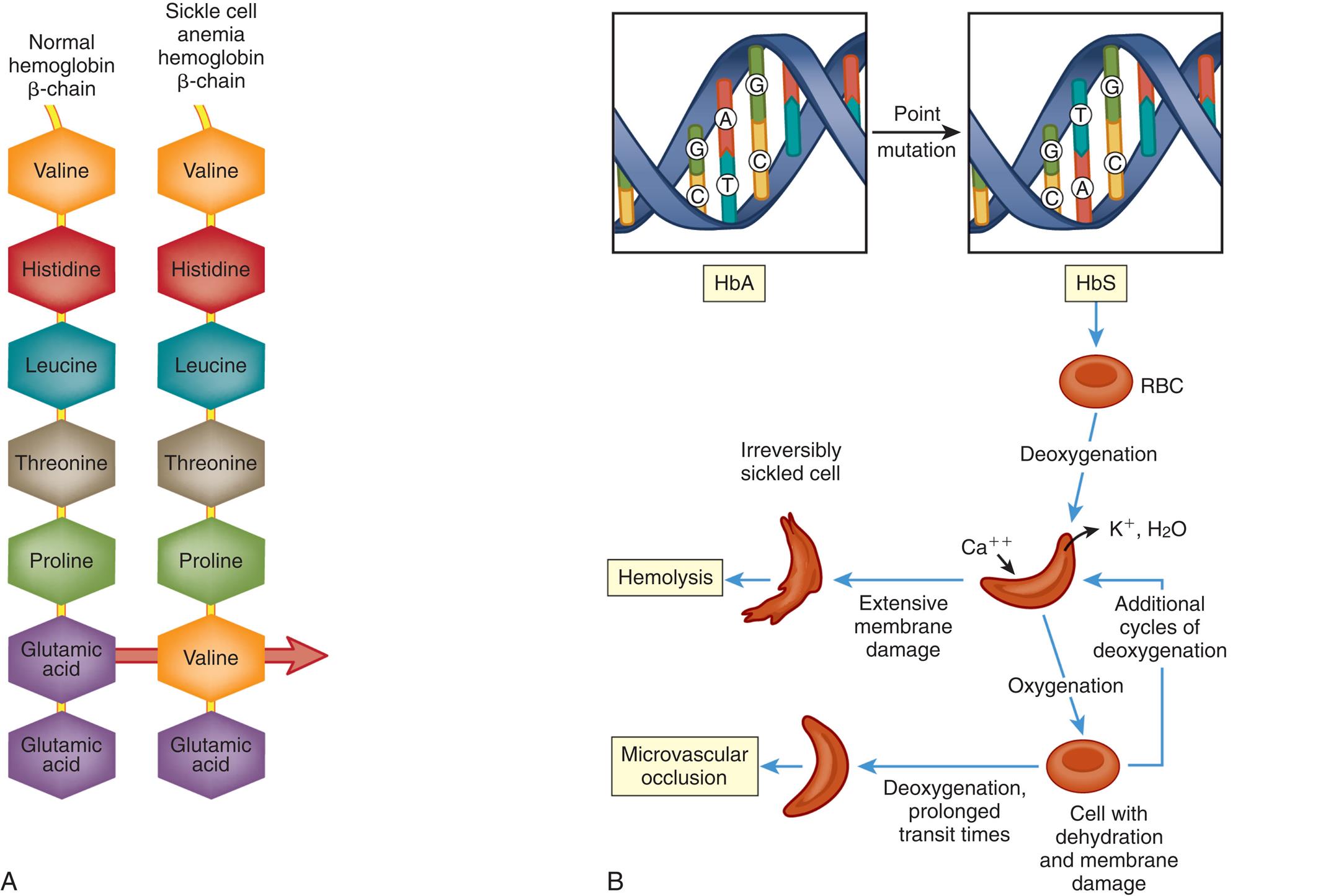
See text for discussion. (A and B, Adapted from Kumar V, Abbas AK, Aster JC. Robbins and Cotran pathologic basis of disease, 9th edition. Philadelphia: Elsevier Saunders; 2015.)
Left panel, A. The illustration shows a normal hemoglobin beta-chain and sickle cell anemia hemoglobin beta-chain. The normal hemoglobin beta-chain shows the following sequence: valine, histidine, leucine, threonine, proline, glutamic acid, and glutamic acid. The sickle cell anemia hemoglobin beta-chain shows the following sequence: valine, histidine, leucine, threonine, proline, valine, and glutamic acid. Right panel, B. The illustration shows the point mutation of H b A to H b S in a D N A helical structure with G C, T A, and G C. The sequence of pathogenesis is as follows. • R B C undergoes deoxygenation. • Cell absorbs calcium ions and releases potassium ions and water. • Hemolysis: Deoxygenated cell undergoes extensive membrane damage and becomes an irreversibly sickled cell. • Microvascular occlusion: Deoxygenated cell undergoes oxygenation and becomes cell with dehydration and membrane damage. Deoxygenation and prolonged transit times of dehydrated cell results in microvascular occlusion. • Cyclic mechanism: Deoxygenated cell undergoes oxygenation resulting in a dehydrated cell. Dehydrated cell undergoes additional cycles of deoxygenation.
Some cells remain sickled even with full oxygenation and are vulnerable to hemolysis. Polymerization of sickled hemoglobin is central to the disorder. Polymerization stiffens the sickled erythrocyte, changing it from a flexible, beneficial cell to an inflexible one where HbS molecules stack into polymers that starve and damage tissues. The pathogenesis of SCD derives from the tendency of HbS molecules to stack into polymers even when deoxygenated and assemble into needle-like fibers within cells, producing the distorted crescent-like sickle or holly-leaf shape (see Figs. 30.2 and 30.3). Sickled cells undergo hemolysis in the spleen or become sequestered there, causing blood pooling and infarction of splenic vessels. The anemia that follows triggers erythropoiesis in the marrow and, in extreme cases, in the liver.43,44
The pathogenesis of microvascular occlusions, a main feature of SCD is not fully understood. This process is responsible for the most serious and urgent manifestations of SCD. Microvascular occlusions are not related to the quantity of irreversibly sickled cells in the blood. Rather, they are dependent on understated RBC membrane damages and other local factors, such as inflammation or vasoconstriction, that tend to decrease blood flow or arrest red cells through the microcirculation.45 Sickled RBCs express higher than normal amounts of adhesion molecules and are sticky. During inflammatory reactions, leukocyte release of meditators increases the expression of adhesion molecules on endothelial cells. These reactions further promote sickled erythrocytes to become arrested during movement through the microvasculature.45 The sluggish and stagnant red cells within the inflamed vascular vessels result in extended exposure to low oxygen tension, sickling, and vascular obstruction. Lysed sickle erythrocytes release hemoglobin. This free hemoglobin can bind and inactivate nitric oxide (NO), which is a powerful vasodilator and inhibitor of platelet aggregation. A decrease in blood pH reduces hemoglobin's affinity for oxygen leading to an increasing fraction of deoxygenated HbS at any oxygen tension and predisposition to sickling. As less oxygen is taken up by hemoglobin in the lungs, the PO2 drops, promoting additional sickling.
Sickling usually is not permanent. Most sickled erythrocytes regain a normal shape after reoxygenation, a return of the PO2 to normal, and rehydration. Irreversible sickling is caused by permanent plasma membrane damage, which in turn is caused by sickling. In persons with sickle cell anemia, in which the erythrocytes contain a high percentage of HbS (75% to 95%), up to 30% of the erythrocytes can become irreversibly sickled.
The extent, severity, and clinical manifestations of sickling depend to a great extent on the percentage of hemoglobin that is HbS. That is why homozygous inheritance of HbS produces the severest form of SCD—sickle cell anemia. The presence of sickle cell trait rarely results in sickling because these individuals also produce normal HbF and HbA which do not contribute to sickling at all. Anemia persists because HbF does not live 120 days.
Clinical Manifestations
The clinical manifestations of sickle cell disease can vary. Some individuals have mild symptoms; others suffer from repeated vasoocclusive crises. The clinical manifestations of sickle cell disease usually do not appear until the infant is at least 6 months old. At this time, postnatal concentrations of HbF decrease, causing concentrations of HbS to rise (Fig. 30.6). Two key attributes of SCD contribute to its presentation: the first is its nature to be a chronic disease with acute exacerbations; the second is that it is a condition affecting RBCs that supply oxygen to all cells of the body. As a consequence, SCD can affect any part of the body. Sites of specific dysfunction are shown in Fig. 30.7.
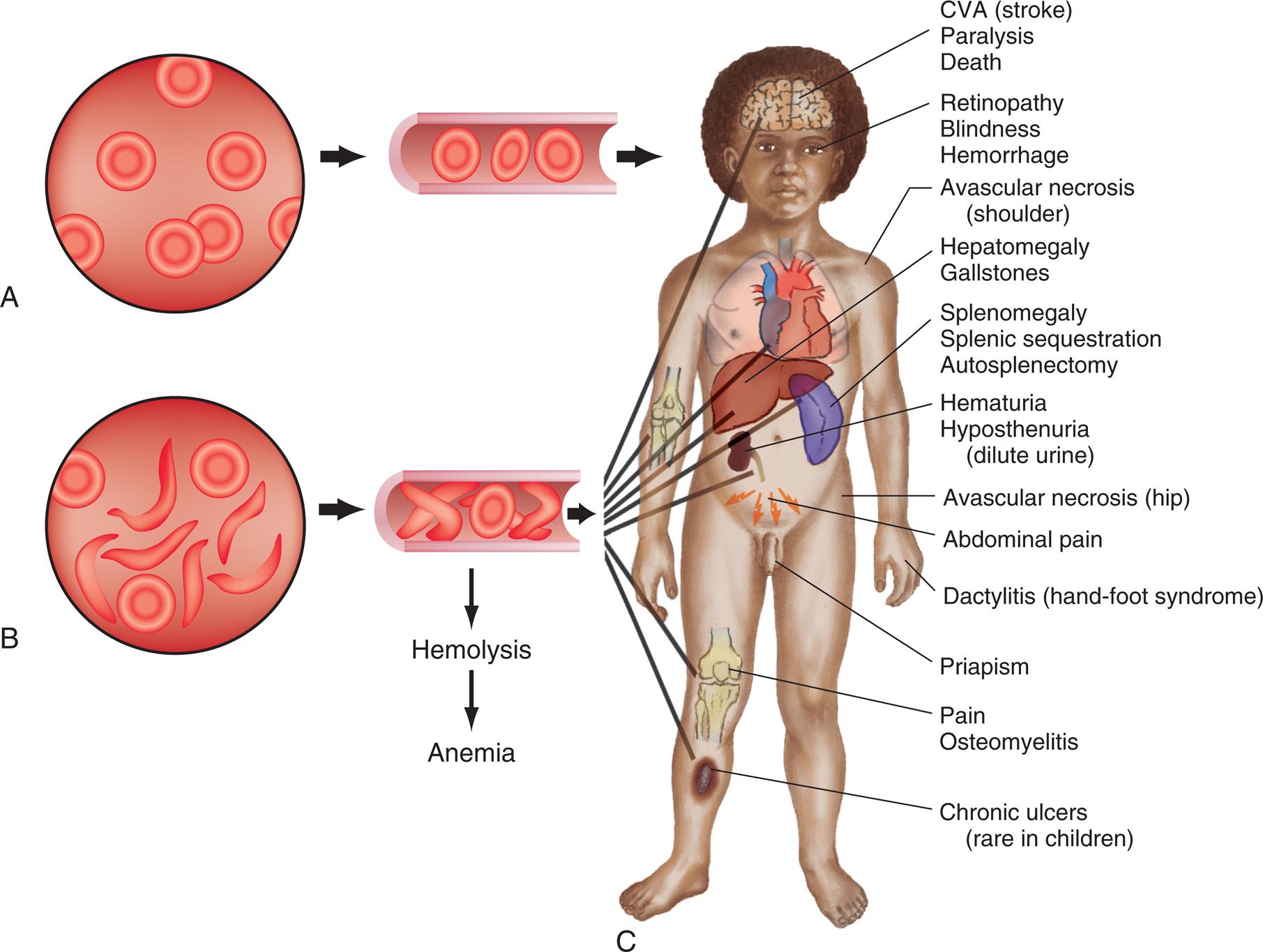
Top-left panel, A. The illustration shows normal red blood cells. Top-right panel, B. The illustration shows sickle and normal red blood cells. Right panel, C. An illustration of the anterior view of the human body identifies the following tissue effects from the top to the bottom. • C V A (stroke), paralysis, and death. • Retinopathy, blindness, and hemorrhage. • Avascular necrosis (shoulder). • Hepatomegaly and gallstones. • Splenomegaly, splenic sequestration, and autosplenectomy. • Hematuria and hyposthenuria (dilute urine). • Avascular necrosis (hip). • Abdominal pain. • Dactylitis (hand-foot syndrome). • Priapism. • Pain and osteomyelitis. • Chronic ulcers (rare in children).
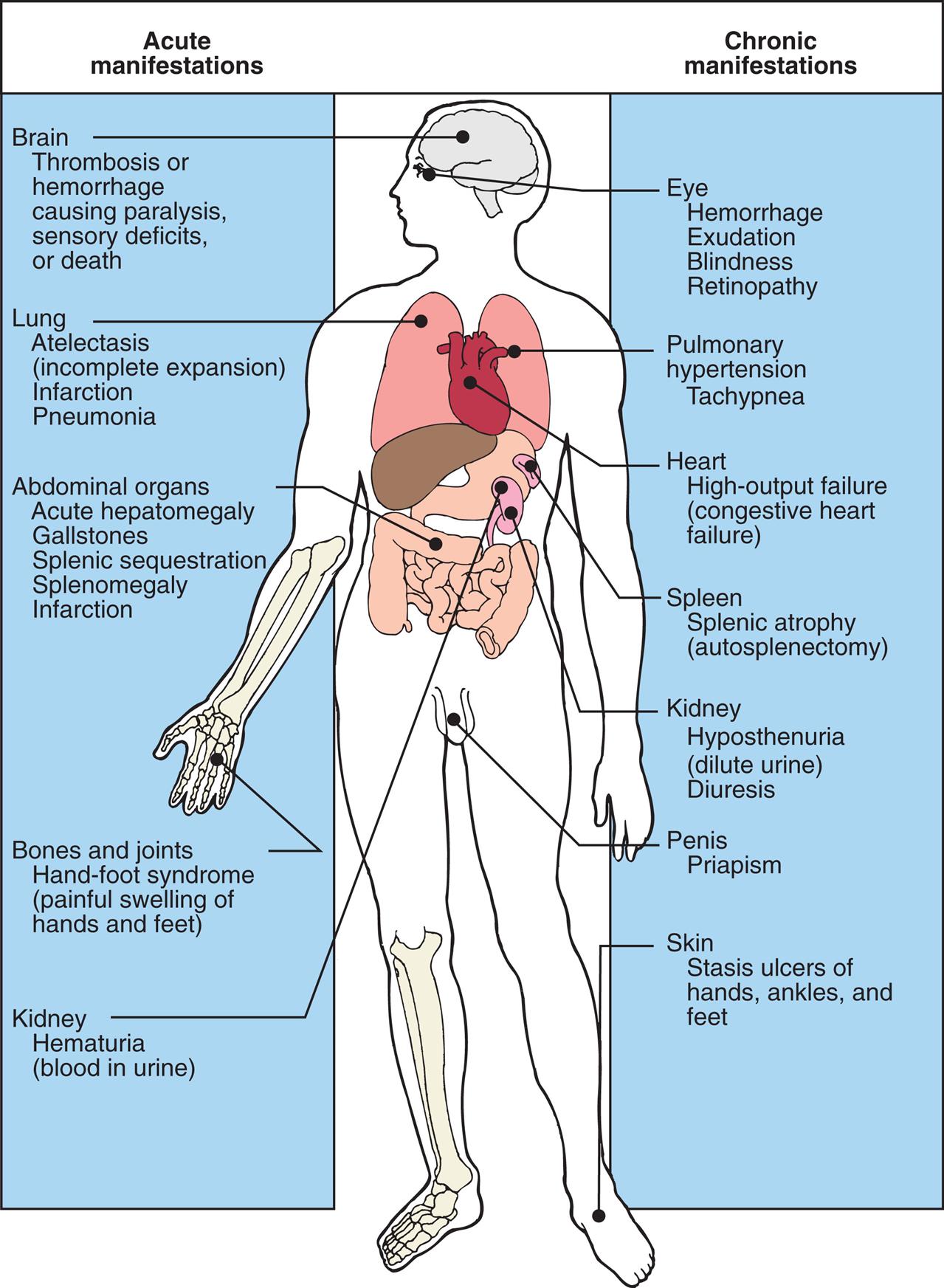
An illustration of the anterior view of the human body shows acute and chronic manifestations by organ. Acute manifestations are as follows. • Brain: thrombosis or hemorrhage causing paralysis; sensory deficits or death. • Lung: atelectasis (incomplete expansion); infarction; and pneumonia. • Abdominal organs: acute hepatomegaly; gallstones; splenic sequestration; splenomegaly; and infarction. • Bones and joints: hand-foot- syndrome (painful swelling of hands and feet). • Kidney: hematuria (blood in urine). Chronic manifestations are as follows. • Eye: hemorrhage; exudation; blindness; and retinopathy. • Pulmonary: hypertension and tachypnea. • Heart: high-output failure (congestive heart failure). • Spleen: splenic atrophy (autosplenectomy). • Kidney: hyposthenuria (dilute urine) and diuresis. • Penis: priapism. • Skin: stasis ulcers of hands, ankles, and feet.
The general manifestations of hemolytic anemia from the sickling process include pallor, fatigue, jaundice, and irritability. Extensive sickling can precipitate four types of acute manifestations, often referred to as crises: (1) vaso-occlusive crisis, (2) aplastic crisis, (3) sequestration crisis, or rarely (4) hyperhemolytic crisis.
Vasoocclusive crisis (pain crisis). This type of crisis involves hypoxic injury and infarction that can cause severe pain in the microcirculation. The frequency of this type of crisis is variable and unpredictable because it may develop spontaneously or be precipitated by infection, exposure to cold, dehydration, low PO2, acidosis (low pH), or localized hypoxemia. Vaso-occlusive crises are extremely painful, and the specific cause of this sensory pain is not well characterized.46,47
As blood flow is obstructed by sickled cells, vasospasm occurs, and a logjam effect blocks all blood flow through the vessel. Unless the process is reversed, thrombosis and infarction of local tissue occur. Vasoocclusive crisis may last for days or even weeks, with an average duration of 4 to 6 days. The most common sites include bones, lungs, spleen, liver, brain, and penis. Painful bone crises are common in young children and are difficult to distinguish from acute osteomyelitis. These bone alterations can manifest as painful swelling of the hands and feet (hand-foot syndrome or dactylitis).
A high-risk type of vaso-occlusive crisis involving the lungs is known as acute chest syndrome. It typically presents with fever, cough, chest pain, and accumulations of lung infiltrates. The complications in the lungs create a worsening cycle of hypoxemia, sickling, and vaso-occlusion. Acute chest syndrome remains a leading cause of death among people with SCD.48,49Priapism, or prolonged erection of the penis, can lead to hypoxic damage and erectile dysfunction. Vaso-occlusion in vessels to the brain can result in stroke. Chronic vaso-occlusion in vessels to the kidneys results in end-stage renal disease.
Aplastic crisis. This type of crisis involves profound anemia caused by a transient cessation in RBC production despite a need for new RBCs. In sickle cell anemia, erythrocyte survival is only 10 to 20 days; however, the bone marrow is typically able to compensate to replace RBCs that are lost through hemolysis. Aplastic crisis is often precipitated by a viral infection, such as parvovirus B1. The virus causes temporary shutdown of RBC production with an accompanying sudden drop in hemoglobin level with an extremely low reticulocyte count. This type of crisis typically lasts 7 to 10 days.
Sequestration crisis. This type of crisis is typically seen only in children less than 5 years of age. It occurs when large amounts of sickled RBCs become acutely pooled in the liver and spleen. Because the spleen can hold as much as one-fifth of the body's blood supply, hypovolemia and, sometimes shock, can occur (see Chapter 49). The risk of mortality is high if the condition is not recognized and managed appropriately. Clinical management may include exchange transfusions.32 Approximately half of children who experience sequestration crises will have recurrent episodes.
Hyperhemolytic crisis. This type of crisis, associated with an accelerated rate of RBC destruction, is unusual, but it may occur in association with certain drugs or infections. It is characterized by anemia, jaundice, and reticulocytosis. The concomitant presence of G6PD deficiency (see Glucose-6-Phosphate Dehydrogenase Deficiency) contributes to hyperhemolytic episodes, especially when combined with infections. It has also been reported as an acute or chronic reaction following a blood transfusion.
Infection is the most common cause of death related to sickle cell disease. Infection is also an important cause of disease-related morbidity, particularly for children with impaired splenic function. Sepsis and meningitis develop in as many as 10% of children with sickle cell anemia during the first 5 years of life. Splenic congestion and poor blood flow compromise splenic function and lead to splenic infarction. As a consequence, the risk of infection from Pneumococcus pneumoniae and Haemophilus influenzae is increased.
Glomerular disease, characterized by damage to the glomeruli allowing protein and often RBCs to leak into the urine, is caused by sickling of RBCs in the kidneys. Extensive damage to the glomeruli results in nephropathy that may progress to renal failure. The earliest manifestation of SCD in the kidney is hyposthenuria, or the inability of the tubules of the kidneys to concentrate urine. As a result, the specific gravity of the urine tends to be very low. In young children, hyposthenuria can result in bed-wetting. Proteinuria also is an early manifestation of nephropathy associated with sickle cell disease.
Cholecystitis, inflammation of the gallbladder, occurs when a gallstone blocks the cystic duct. It can also be caused by hemolysis resulting in an increase of bilirubin concentration, which in turn causes the formation of gallstones in the gallbladder. The presence of gallstones can cause right upper quadrant pain, nausea, vomiting, and an elevated white blood cell count and alkaline phosphatase level. Cholecystectomy may be required.
Sickle cell–hemoglobin C (HbC) disease is usually milder than sickle cell anemia. HbC results when lysine is substituted for glutamic acid in the amino acid chain. HbC is less soluble than HbA; however, it does not polymerize under conditions of decreased oxygen tension as does HbS. The main clinical problems are related to vasoocclusive crises, which are thought to result from higher hematocrit values and viscosity. In older children, sickle cell retinopathy, renal necrosis, and aseptic necrosis of the femoral heads can occur along with obstructive crises.
Sickle cell–thalassemia has the mildest clinical manifestations of all the sickle cell diseases. Individuals with sickle cell–thalassemia have mutations in each allele coding for hemoglobin. One mutation results in HbS formation, and the other is associated with β-thalassemia, which results in decreased production of hemoglobin. Even though most of the child's hemoglobin is HbS (60% to 90%), normal hemoglobins (HbA and HbF) also are present. The normal hemoglobins, particularly HbF, inhibit sickling. The erythrocytes tend to be small (microcytic) and to contain relatively little hemoglobin (hypochromic). As a result, these cells are less likely to occlude the microcirculation, even when in a sickled state.
Evaluation and Treatment
The parents’ hematologic history and clinical manifestations may suggest that a child has sickle cell disease, but hematologic tests are necessary to confirm the diagnosis. If the sickle solubility test confirms the presence of HbS in peripheral blood, hemoglobin electrophoresis will be performed to provide information about the amount of HbS in erythrocytes. Prenatal diagnosis can be made by chorionic villus sampling (CVS) as early as 8 to 10 weeks’ gestation or by amniotic fluid analysis at 15 weeks’ gestation (Fig. 30.8). Hemoglobinopathies, including sickle cell disease, are now included as part of routine newborn screening in all 50 states and the District of Columbia.
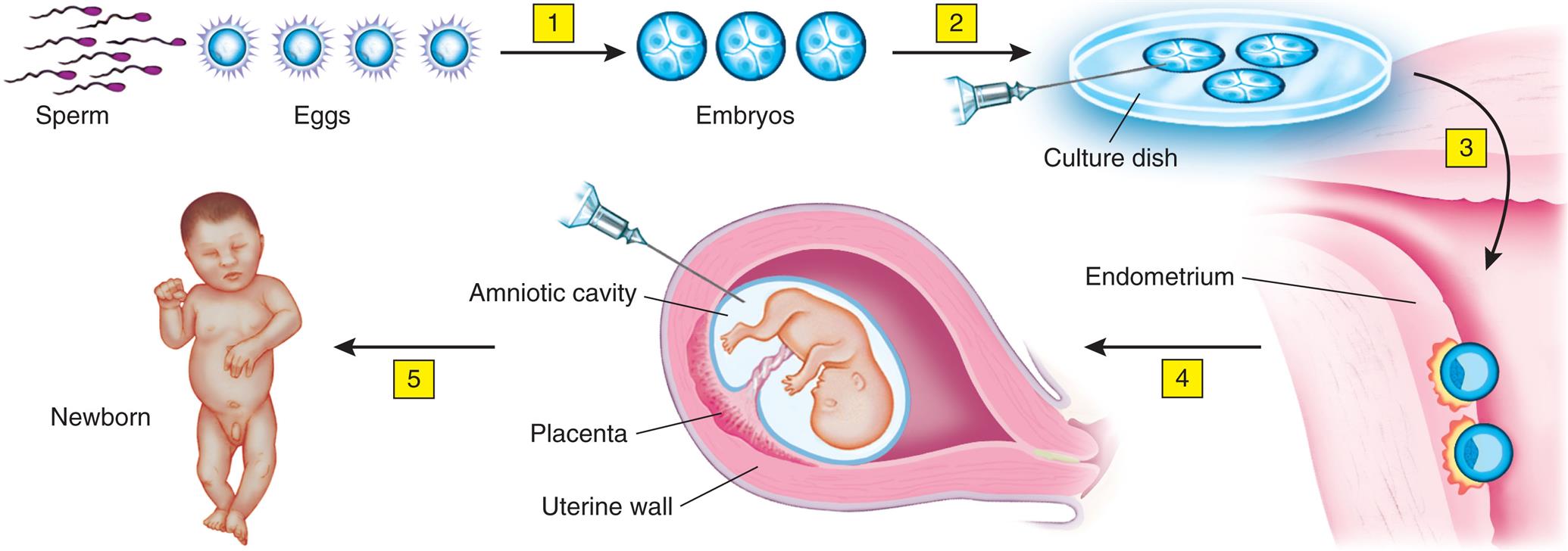
This technique has potential for detection of other inherited diseases. (1) Fertilization produces several embryos. (2) The embryos are tested for the presence of the gene. (3) The embryos without the gene are implanted. (4) Amniocentesis confirms whether the fetus (or fetuses) has the sickle cell gene. (5) Woman has a normal child.
An illustrated flowchart shows the prepregnancy sickle cell test. The illustration shows sperm and eggs. Fertilization of sperm and eggs leads to embryos (1). The illustration shows embryonic cells in a culture dish (2). The embryos are implanted in the endometrium (3). The embryo develops into a fetus (4). A syringe collects sample from the amniotic cavity. Birth of a newborn (5). The placenta and the uterine wall are identified. The illustration shows a newborn.
Advances in identification of SCD and supportive care have led to decreased morbidity and improved survival of children with SCD. Supportive care emphasizes preventing consequences of anemia and avoiding crises, including adequate hydration, infection prevention, and pain management. Genetic counseling and psychologic support are important for the child and family.
Health maintenance activities are key to reduce the risk of crises and other aspects of disease-related morbidity. In addition to regular childhood vaccines, children with SCD should receive vaccination against pneumococcus and other encapsulated microorganisms. Children under 5 years of age may also be prescribed prophylactic penicillin to further reduce the risk of infection.32 Because of the high infection-related mortality, individuals with SCD and their families should be instructed to seek immediate medical attention in the event of fever. Evidence-based guidance for screening for nephropathy, hypertension, retinopathy, pulmonary disease, and the risk of stroke have been published by the National Heart, Lung, and Blood Institute.32
Sickle cell trait typically does not affect life expectancy or interfere with daily activities. On rare occasions, however, severe hypoxia caused by shock, vigorous exercising at high altitudes, flying at high altitudes in unpressurized aircraft, or undergoing anesthesia may precipitate vasoocclusive episodes in persons with sickle cell trait. Persons with sickle cell trait are at risk for hyphema with traumatic injury to the eye. Untreated hyphema may result in glaucoma and potential vision loss. In 2010, the National Collegiate Athletic Association implemented testing for sickle cell trait for all incoming student athletes. While having sickle cell trait does not disqualify the individual from participation in athletics, the goal of this testing is to ensure the safety of student athletes.50
Treatment advances since the late 1980s have significantly decreased morbidity and mortality in children with SCD. Aggressive management of fever, early diagnosis of acute chest syndrome (hypoxia, anemia, progressive multilobar pneumonia, fat emboli), and proper pain management can improve quality of life and prognosis for these children. Treatment of SCD consists of supportive care aimed at preventing consequences of anemia and avoiding crises. Crises can be prevented by avoiding fever, infection, acidosis, dehydration, constricting clothes, and exposure to cold. Immediate correction of acidosis and dehydration with appropriate intravenous fluids is imperative. Infections require aggressive antibiotic therapy, and infections can be reduced by vaccination. Oxygen is not needed unless the child becomes hypoxic. Pain associated with SCD is very complex, requiring accurate assessment and multimodal management.51,52
A common treatment for sickle cell disease is hydroxyurea. Hydroxyurea inhibits deoxyribonucleic acid (DNA) synthesis, which causes an increase in HbF concentration. HbF will not sickle. It also provides an anti-inflammatory effect by decreasing leukocyte production. These outcomes are thought to decrease mechanisms that lead to crises.
Two new medications were approved by the US Food and Drug Administration for use in individuals with sickle cell disease in 2019.32 Voxelotor is an oral medication approved for individuals 12 years of age and older. It is proposed to inhibit HbS polymerization and the sickling process by modifying the affinity between hemoglobin and oxygen.53 Crizanlizumab is a monoclonal antibody against the adhesion molecule P-selectin. P-selectin is involved in the underlying process by which blood cells adhere to blood vessel walls, thereby contributing to vaso-occlusion.54 Crizanlizumab is administered as an IV infusion.
Transfusion therapy can decrease morbidity and mortality associated with sickle cell disease, particularly in those at increased risk for stroke.32 Despite these benefits, it can result in iron overload in the liver, heart, and endocrine glands causing disruption of normal function, including delayed physical and sexual development and heart disease. Chelation therapy to remove excess iron is often required for individuals with SCD who require chronic transfusion of RBCs
HSCT offers the only cure for sickle cell disease; however, it is not without important risks. Observational studies to date have demonstrated improved survival and prevention of long-term complications. Additional studies are needed to evaluate benefits and risks by comparing individuals based on disease severity.55 Current research is seeking to reduce the toxicities associated with transplantation while optimizing long-term outcomes. Clinical trials evaluating the feasibility of GT using an RNA viral vector are presently underway. One type of trial seeks to inhibit the BCL11a gene with the goal of increasing hemoglobin F production, thereby creating a sickle cell trait phenotype.56
Thalassemias
The alpha- and beta-thalassemias are autosomal recessive disorders that result in impaired synthesis of one of the two chains—α or β—of adult hemoglobin (HbA). The disorder was named thalassemia, which is derived from the Greek word for sea, because it was initially described in people with origins near the Mediterranean Sea. Beta-thalassemia, in which synthesis of the β-globin chain is slowed or defective, is most prevalent among Greek, Italian, some Arab, and Sephardic Jewish people. Alpha-thalassemia, in which the α chain is affected, is most common among Chinese, Vietnamese, Cambodian, and Laotian people. Both alpha- and beta-thalassemias are common among black people.
Beta-thalassemias are more common than alpha-thalassemias. The classification is based on the genes that control α- or β-chain synthesis. It also is based on the combination of mutations within these genes and whether they are homozygous or heterozygous. The anemia associated with both alpha- and beta-thalassemia is a microcytic-hypochromic hemolytic anemia.
Pathophysiology
The β-thalassemias are caused by mutations in the HBB gene that decrease the synthesis of β-globin chains and lead to anemia, tissue hypoxia, and RBC hemolysis. The β-thalassemias are classified into three types depending on the severity of symptoms: thalassemia major, intermedia, or minor. Both beta-thalassemia major (Cooley anemia) and beta-thalassemia intermedia are inherited in an autosomal recessive pattern in which both copies of the HBB gene in each cell have mutations; however, beta thalassemia major is more severe. β-globin chain production is moderately depressed in the heterozygous form, beta-thalassemia minor, and is known as beta-thalassemia trait. In a small percentage of families, the HBB gene mutation, associated with beta-thalassemia major, is inherited in an autosomal dominant manner.57
The mutations associated with beta-thalassemia are classified as (1) β0 mutations or absent β-globin synthesis and (2) β+ mutations with reduced amounts of β-globin synthesis. More than 200 different causative mutations have been identified, and most are point (missense) mutations. The clinical classification of β-thalassemias is based on the severity of the anemia, which is dependent on the type of mutation (β0 or β+ allele) and whether the individual is homozygous or heterozygous for mutations.57 Individuals who have β-thalassemia major, a β0/β0 genotype, will lack HbA and have severe anemia. Those with β+/β+ or β0/β+ genotypes, β-thalassemia intermedia, may produce small amounts of HbA. β-Chain production is depressed in the heterozygous form, thalassemia minor (β0 or β+ allele); and anemia, if present, is mild. The predominant type of hemoglobin is HbF and typically comprises more than 90% of the total hemoglobin among individuals who are homozygous for mutations. HbA2 levels are variable among individuals who are homozygous but may be increased among individuals with beta thalassemia minor.58
The depression of β-chain synthesis that occurs in beta-thalassemia results in erythrocytes having a reduced amount of hemoglobin and accumulations of free α chains (Fig. 30.9). Free α-chains are unstable and easily precipitate in the cell. Most erythroblasts that contain precipitates are destroyed by mononuclear phagocytes in the marrow. Destruction results in ineffective erythropoiesis and anemia. Some of the precipitate-carrying cells mature and enter the bloodstream. These cells are destroyed prematurely in the spleen, resulting in mild hemolytic anemia.
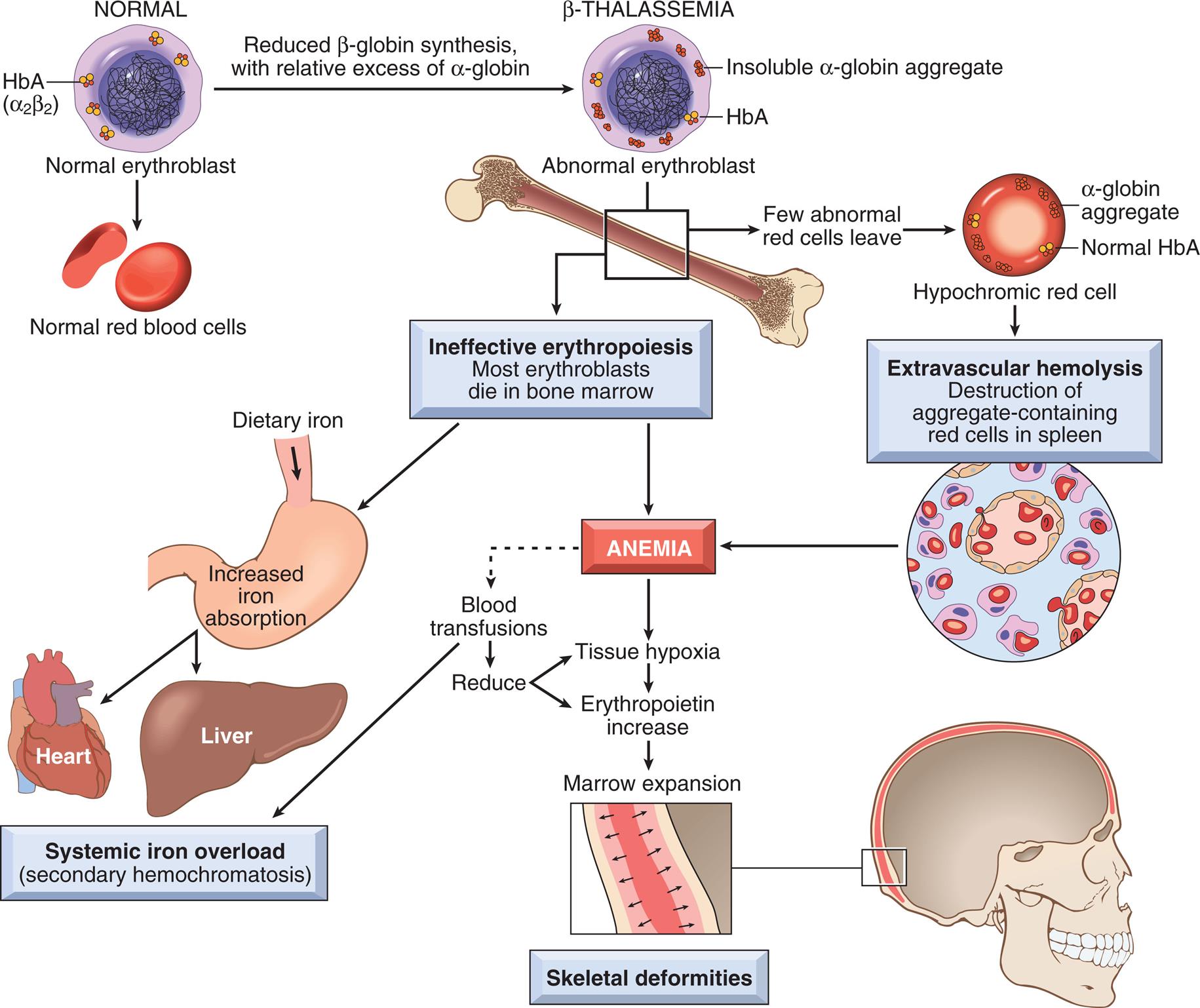
The aggregates of unpaired α-globin chains are a hallmark of the disease. Blood transfusions can diminish the anemia, but they add to the systemic iron overload. (From Kumar V, Abbas AK, Aster JC. Robbins and Cotran pathologic basis of disease, 9th edition. Philadelphia: Elsevier Saunders; 2015.)
An illustrated flowchart shows the pathogenesis of beta-thalassemia major. • The illustration shows a normal cell with H b A (alpha sub 2 beta sub 2). • Normal erythroblast results in normal red blood cells. • Normal cell undergoes reduced beta-globin synthesis, with relative excess of alpha-globin, leading to beta-thalassemia. • The illustration of the abnormal erythroblast shows a cell with insoluble alpha-globin aggregate and H b A. • The illustration shows a bone. Few abnormal red cell leave the bone, forming a hypochromic red cell with alpha-globin aggregate and normal H b A. • Hypochromic red cell leads to extravascular hemolysis (destruction of aggregate-containing red cells in spleen), leading to anemia. • Ineffective erythropoiesis in bone marrow leads to most erythroblasts dying in bone marrow, resulting in anemia. • Ineffective erythropoiesis leads to increased iron absorption (from dietary iron), which leads to systemic iron overload (secondary hemochromatosis) in heart and liver. • Anemia leads to tissue hypoxia, which leads to erythropoietin increase, which leads to marrow expansion and skeletal deformities. • Blood transfusions reduce tissue hypoxia and erythropoietin. • Blood transfusions leads to systemic iron overload.
The alpha (α)thalassemias result from deletions involving the HBA1 and HBA2 genes. These genes provide instructions for making a protein called α-globin, a subunit of hemoglobin. The different types of α-thalassemia result from mutations in one or more of the four alleles involved in producing α-globin. These alleles include the two copies of the HBA1 gene and the two copies of the HBA2 gene that are present in each cell. The characteristic features of α-thalassemia are anemia, weakness, fatigue, and other more severe complications. The four types of α-thalassemia include
- 1. Hb Bart syndrome is the most severe form of α-thalassemia and is caused by loss-of-function mutations in all four α-globin alleles. In an attempt to compensate, Hb Barts, which contains γ-globin chains, is produced. These γ-globin chains have a high affinity for oxygen; however, they do not deliver oxygen to the tissues of the developing fetus. As a result, hydrops fetalis develops, and signs of fetal distress may become evident in the third trimester of pregnancy. Although most cases are fatal, intrauterine transfusions may save the baby.59
- 2. HbH disease results from a loss-of-function in three of the four α-globin alleles. These individuals have an excess of beta-globin chains within the RBCs that form beta-4 tetramers, known as hemoglobin H (HbH). HbH also does not carry oxygen effectively, which leads to anemia and other related consequences.59,60
- 3. Alpha thalassemia minor or alpha thalassemia trait occurs when mutations are present in two alleles. Although the amount of α-globin will be reduced, some normal hemoglobin will still be produced and therefore cause fewer health problems. Affected individuals may have microcytic RBCs and slight anemia.59
- 4. Silent α-thalassemia carrier results from a mutation in one of the four α-globin alleles. These individuals typically have no related clinical manifestations but can pass the mutated allele on to their children.59
Clinical Manifestations
Beta-thalassemia minor causes mild to moderate microcytic-hypochromic anemia. The degree of reticulocytosis depends on the severity of the anemia. Hemolysis of immature, and therefore fragile, erythrocytes may cause a slight elevation in serum iron and indirect bilirubin levels. Persons with beta-thalassemia minor may be asymptomatic; however, they may experience mild splenomegaly, bronze coloring of the skin, and hyperplasia of the bone marrow. These individuals are less likely to experience life-threatening complications.
If untreated, persons with beta-thalassemia major may become quite ill and have impaired physical growth and development. Skeletal changes begin in infancy and include spinal impairment that impedes linear growth and causes subsequent upper and lower limb-length discrepancy. Deformity of the facial bones in response to hyperplastic marrow results in a characteristic chipmunk-like facial appearance, as the nasal bridge, mandible, and maxilla widen. Osteopenia, osteochondrosis, or both also may develop. The severe anemia resulting from this condition can cause a significant cardiovascular burden with high-output congestive heart failure. For persons with thalassemia that require regular transfusions, progressive hemosiderosis will occur. If not managed carefully from infancy into adulthood, death may occur. Today, careful management with regular blood transfusions and compliance with chelation therapy can increase the life span significantly.
Persons who inherit the mildest form of α-thalassemia (silent α-thalassemia trait) usually are symptom free or have mild microcytosis. Alpha-thalassemia minor has clinical manifestations similar to those of beta-thalassemia minor: mild microcytic-hypochromic anemia, reticulocytosis, bone marrow hyperplasia, increased serum iron concentrations, and mild to moderate splenomegaly.
The clinical manifestations of HbH disease are variable based on the specific characteristics of the mutations that are present in the three affected alleles.
Alpha-thalassemia major (Hb Barts) causes hydrops fetalis, whereby the developing fetus suffers from severe tissue anoxia and may develop fulminant intrauterine congestive heart failure. Signs of fetal distress became evident by the third trimester of pregnancy. In the past, severe tissue anoxia led to death in utero; now many such infants are saved by intrauterine transfusions.
Evaluation and Treatment
Evaluation of thalassemia is based on the familial disease history, clinical manifestations, and blood tests. Diagnostic tests include peripheral blood smears that show a microcytic, hypochromic anemia and target cells. Hemoglobin electrophoresis typically indicates decreased amounts of HbA and increased amounts of HbF after age 12 months and may be useful in diagnosing beta thalassemia. However, to diagnose α-thalassemia, targeted deletion analysis is recommended.
Prenatal diagnosis may be performed, and families with an affected fetus will be referred for genetic counseling. Identification of thalassemia is now included as part of routine newborn screening for hemoglobinopathies in all 50 states and the District of Columbia. Molecular genetic testing of at-risk siblings should be offered to allow for early diagnosis and appropriate treatment. Women with thalassemia intermedia who have never received a blood transfusion or who received a minimal quantity of blood are at risk for severe alloimmune anemia if blood transfusions are required during pregnancy.61 Treatment for thalassemia is largely supportive. Typical treatment involves a regular transfusion program and chelation therapy to reduce transfusion iron overload. Individuals with milder forms of thalassemia rarely require transfusion. These individuals are at risk for iron overload secondary to increased intestinal absorption of iron from ineffective erythropoiesis.61 Optimal clinical management may decrease the need for splenectomy. Treatment of thalassemia intermedia is based on the individual’s symptoms, and splenectomy may be performed for those who are more symptomatic. These individuals may also require sporadic red cell transfusions, folic acid supplementation, and iron chelation.61 The only available definitive cure for thalassemia major is allogeneic HSCT from a matched family or unrelated donor or cord blood transplantation from a related donor.61 In addition, on-going clinical trials are investigating the use of GT strategies for thalassemia.62,63
Disorders of Coagulation and Platelets
Inherited Hemorrhagic Disease
Table 30.4 provides a summary of the coagulation factors and deficiencies associated with clinical bleeding. Not all the disorders are discussed in this chapter because some are extremely rare (congenital dysfibrinogenemias), and others have no clinical significance. For example, Hageman factor deficiency is a condition with profound laboratory deficiency of factor XII yet is associated with no clinical defects in humans.
Table 30.4
Hemophilias
Awareness of a serious bleeding disorder in males was documented nearly 2000 years ago in the Babylonian Talmud, which exempted those boys having male relatives prone to excessive bleeding from the rite of circumcision. In 1803, the first description of this disorder appeared in the medical literature, where it was noted to be X-linked and associated with joint bleeding and crippling.
The hemophilias are a group of inherited bleeding disorders resulting from mutations in the genes responsible for coagulation factors. Until 1952 the term hemophilia was reserved for deficiency of factor VIII (antihemophilic factor). Since then, two additional coagulation proteins, factor IX (plasma thromboplastin component) and factor XI (plasma thromboplastin antecedent), have been identified. Deficiencies in these two clotting factors are associated with similar clinical manifestations and are regarded as hemophilia B and hemophilia C, respectively. Collectively, inherited deficiencies of these three plasma clotting factors—VIII, IX, XI—account for 90% to 95% of the hemorrhagic bleeding disorders.
Types of hemophilia
Hemophilia has a wide range of clinical severity. The most prevalent types of hemophilia are hemophilia A (classic hemophilia or factor VIII deficiency) and hemophilia B (Christmas disease or factor IX deficiency). Hemophilia A results from mutations in the F8 gene, which codes for factor VIII, an essential cofactor for factor IX in the coagulation cascade. It is the most common hereditary disease associated with life-threatening bleeding. Hemophilia B results from a mutation in the F9 gene, which codes for factor IX. Because both factors VIII and IX function together to activate factor X, hemophilias A and B are clinically indistinguishable. The alterations or deficiencies of these coagulation factors decrease the ability to form blood clots in response to injury. The decreased or ineffective blood clotting leads to continuous bleeding.
Hemophilias A and B occur with varying degrees of clinical severity, depending on concentrations of clotting factor VIII or IX in the blood. Severe hemophilia (concentration of clotting factors less than 1% of normal) is associated with spontaneous bleeding. This manifestation of hemophilia is associated with mutations that eliminate the activity of factors VIII or XI. In moderate hemophilia (1% to 5% of normal), bleeding may occur with injury, trauma, or surgery; in the mild form (6% to 50% of normal), bleeding typically occurs only after severe trauma or surgery. Moderate and mild hemophilia are associated with mutations that reduce but do not eliminate the activity of these coagulation factors.63
Hemophilia is equally prevalent across racial groups. The incidence of hemophilia A is approximately 1 in 5000 male births. Hemophilia B is five times less common, with an incidence of approximately 1 in 20,000 male births.64 Hemophilia C, which results from a deficiency of Factor XI is inherited as an autosomal recessive condition and is extremely rare. It affects approximately 1 in 1,000,000 individuals.65 The worldwide prevalence of severe hemophilia is estimated to be around 400,000 people. A recent meta-analysis of worldwide registry data, however, suggests that approximately 1,125,000 males are living with hemophilia, with manifestations ranging from mild to severe disease.66
Pathophysiology
As X-linked recessive conditions, hemophilias A and B are most frequently inherited from a mother who is heterozygous, that is, a carrier, for a mutation in either the F8 or F9 gene. Approximately 30% of cases, however, result from a new mutation. This new mutation can occur in either a carrier female or in an affected male. Multiple types of genetic mutations are associated with hemophilias A and B. More than 2000 unique mutations have been identified for hemophilia A,67 and more than 1000 mutations have been described for hemophilia B.68 The type of mutation also affects the phenotype, or severity, of the disease. Mutations tend to be identical among affected members of a given family; however, mutations often differ across families.69
The F8 and F9 genes are located on the long arm of the X chromosome. A mutation in either of these genes typically results in either deficient or abnormal function of the corresponding clotting factor. Because males have only one copy of the X chromosome, the mutation results in the clinical manifestations of hemophilia. Females who are heterozygous carriers typically do not experience excessive bleeding. However, 50% of female carriers have lower than normal clotting factor levels. Those females without a sufficient quantity of normal functioning clotting factor may have bleeding similar to a male with mild hemophilia. Because X-inactivation, or lyonization (see Chapter 4), is a random process, phenotypes of females who are heterozygous carriers can vary. Although very uncommon, it is plausible for a female to be homozygous for mutations in the F8 or F9 gene and therefore have hemophilia.
Inversions in introns 1 and 22 of the factor VIII gene are the most frequently observed mutations and account for the most severe cases of hemophilia A.68 Point mutations, in which a single base in the DNA is inserted in the place of another base, are another type of mutation that causes hemophilia. This type of mutation comprises the majority of unique mutations associated with hemophilia.67 When a point mutation gives rise to a de novo stop codon (nonsense mutation), translation of the protein ceases, and a shortened version of the protein is synthesized. Usually, the protein is destroyed intracellularly and never reaches the plasma. A point mutation defect also is associated with severe hemophilia, that is, with coagulant activity levels below 1%. Point mutations resulting in the substitution of one amino acid for another can cause phenotypes of varying severity. The altered amino acid chain can destroy protein function, activation, or folding; inhibit intracellular processing; or cause protein clearance.69
Clinical Manifestations
The clinical manifestations and severity of hemophilia depend largely on the level of factor VIII and IX activity. Children with severe hemophilia also manifest prolonged bleeding times at different ages. Many boys with hemophilia are circumcised without excessive bleeding. Normal hemostasis is achieved in these infants because clotting is activated through the extrinsic coagulation cascade, which does not involve factors VIII, IX, or XI.
During the first year of life, spontaneous bleeding often is minimal, but hematoma formation may result from immunizations and from firm holding (e.g., under the arms), and joint bleeding may occur. Most children with severe hemophilia initiate prophylactic factor administration around the time they become mobile (e.g., crawling, pulling to stand).
Joint bleeding, or hemarthrosis, is the most characteristic type of bleeding in hemophilia. The joints most often affected are the knees, ankles, and elbows.70 Hemarthrosis causes pain, limits joint mobility, and predisposes the child to degenerative joint changes. Bleeding into muscles, usually from trauma, also can occur. Oral bleeding is common in the setting of traumatic injury to the mouth or dental surgery. Spontaneous painless hematuria is relatively common in hemophilia; it does not result in significant blood loss but requires evaluation. Hematuria accompanied by pain requires prompt evaluation and treatment. Spontaneous epistaxis may also occur. Although troublesome, it tends to be a minor complication.
Recurrent bleeding—spontaneous and after minor trauma—is a lifelong problem. Intracranial bleeding, bleeding of internal organs, and bleeding into the tissues of the neck, chest, or abdomen are all life-threatening. Delayed or suboptimal treatment of these bleeding episodes may lead to permanent brain injury, loss of organ function, or death.
Evaluation and Treatment
Because hemophilia most often manifests as an inherited disease, a positive family history may expedite a diagnosis of hemophilia. When a mother who is a known or suspected carrier is pregnant, prenatal genetic testing through CVS or amniocentesis may reveal a diagnosis of hemophilia. In the absence of a positive family history, a personal bleed history, laboratory testing, family history, and physical assessment contribute to a thorough evaluation and accurate diagnosis. In general, those with hemophilia A or B will have a prolonged partial thromboplastin time (PTT) and the prothrombin time (PT) will be normal. Measuring factor VIII and factor IX levels also is necessary for diagnosis.
Most children with hemophilia A can be treated with recombinant factor VIII, and most children with hemophilia B can be treated with recombinant factor IX. Recombinant factor is reconstituted in a small volume of diluent, administered by slow intravenous push, and raises the factor level almost immediately.
Primary prophylaxis consists of regular infusions of factor VIII or IX with the goal of preventing joint bleeding. It is usually given to children with severe hemophilia. A 5-year, multicenter trial, which has now become the standard of care,71,72 found prophylaxis initiated in children between 6 and 30 months of age to be effective in the prevention of joint bleeding, structural joint damage, and frequency of bleeding in boys with factor VIII deficiency. Recent multisite trials have demonstrated the safety and efficacy of pegylated, recombinant factors VIII and IX. These pegylated products have extended half-lives and require less frequent dosing.73,74 Newer generation products are fully produced in human cell lines and help reduce the risk of the development of inhibitors that limit the efficacy of the infused product.75 Recently approved substitution therapies using monoclonal antibodies have shown great promise in the management of bleeding in hemophilia. Ongoing investigation of rebalancing therapies and GT may transform disease severity and management.
von Willebrand Disease
von Willebrand disease is an inherited condition with variable clinical manifestations and hematologic findings resulting from a deficiency or dysfunction in von Willebrand factor. von Willebrand factor binds factor VIII and platelets to the blood vessel wall as part of the clotting process. As a result, factor VIII activity may be decreased in individuals with von Willebrand disease. Over 80% of cases of von Willebrand disease demonstrate an autosomal dominant pattern of inheritance; however, some cases demonstrate an autosomal recessive or compound heterozygous pattern. The pattern of inheritance depends on the type of mutation that is present.67 The clinical management of von Willebrand disease will depend on the underlying subtype that is present.
Congenital Hypercoagulability and Thrombosis
Hereditary bleeding disorders, such as hemophilia, have been recognized and treated for centuries; however, the counterpart of these disorders, thrombophilia, has not been recognized until the past 50 years. The inherited thrombophilic conditions generally are caused by defects in the clotting factors that inhibit clot formation; thus, the balance between bleeding and clotting is directed toward the clotting aspects of hemostasis. In general, thromboses in children in the absence of some type of triggering event such as a vascular access catheter, infection, or heart disease are rare and comprise less than 5% of thrombotic events in children. For these spontaneous events, approximately 60% have some type of hereditary basis.76 A recent study investigating genetic mutations in 115 adolescents and young adults presenting with thrombotic events identified single-gene mutations in two-thirds of this sample, and some individuals had more than one mutation.77 Ongoing studies are needed to further investigate the specific roles of these mutations as well as the clinical management of children and adolescents who present with a thrombotic event. Current guidance supports testing for inherited thrombophilia if (1) identification of a thrombophilic mutation or defect might explain the basis for the event, (2) a positive test would have implications for the child or adolescent’s clinical management, or (3) testing might help identify asymptomatic family members and provide guidance in avoiding risk factors for future thrombotic events.78
Defects in specific proteins (C and S) and antithrombin (AT) and resistance to activated protein C (APC) and hyperhomocysteinemia are the main recognized causes of inherited thrombophilia. Both proteins C and S inhibit coagulation and depend on vitamin K for synthesis in the hepatocytes of the liver. Decreased levels of either of these proteins interfere with the normal homeostatic balance of procoagulant and anticoagulant activity at the endothelial level. Protein C and S deficiency states predispose affected individuals to thrombosis, especially venous thrombosis of the lower extremities.
Protein C deficiency is inherited in an autosomal dominant manner and results from mutations in the PROC gene. Its prevalence is approximately 0.2% to 0.4% in the general population. Genetic mutations may result in quantitative (type I) or qualitative (type II) deficiencies of protein C. Type I deficiencies involve a reduction in both biologic and immunologic activity of protein C. Type I is caused by deletion of the entire gene. Type II deficiencies are less common and result in decreased functional levels of protein C activity despite the presence of a normal level of protein C.79
Individuals who are heterozygous for protein C mutations have levels that are 50% to 60% of normal. They may develop superficial thrombophlebitis, deep venous thrombosis, or pulmonary embolism in their late teens and early 20 s. Individuals with homozygous mutations have less than 1% of normal levels of protein C and tend to develop thrombosis of the cutaneous vessels with large areas of skin necrosis. Arterial thrombosis is rare among individuals with protein C deficiency.
Neonatal purpura fulminans is a fatal syndrome found in neonates who are homozygous or compound heterozygous for types I and II protein deficiency. Ecchymosis becomes apparent on the first day of life and develops around the head, trunk, and extremities and often is accompanied by cerebral thrombosis and infarction. The ecchymotic areas often coalesce, and ulceration and necrosis develop. Treatment is largely supportive and includes administration of fresh frozen plasma (FFP) and heparinization; however, the condition is often fatal.
Treatment for protein C deficiency depends on the clinical manifestations. Individuals who are asymptomatic may not require therapy unless a strong family history of thrombosis is present. Heparin is the treatment for acute episodes of thrombosis caused by protein C deficiency. The need for long-term therapy is varied and will be individualized to the person with attention to prior family history. Supplemental protein C concentrates (human) also are available and have been approved for use in children.80
Protein S deficiency results from mutations in the PROS1 gene and is similar to protein C deficiency. Its autosomal dominant pattern of inheritance is also similar. The severity of the clinical manifestations varies. Approximately 60% to 80% of affected individuals will develop a venous thrombosis at some point during their lives.81
Protein S deficiency exists in three forms: type I, type II, and type III.82Type I deficiency is identified as a quantitative deficiency and manifests as low levels of total protein S antigen and free protein S antigen. Type II deficiency is identified as a qualitative deficiency with normal levels of free and total protein S antigen but reduced protein S activity. Type III deficiency results in low free protein S levels; however, the total plasma concentration of protein S is normal. Type III deficiency has not been established as a risk factor for venous or arterial thromboembolism.
The clinical manifestations of Protein S deficiency vary across individuals. Approximately half of individuals who are heterozygous for mutations will experience a thrombotic event before age 55. Other manifestations include superficial thrombophlebitis and pulmonary emboli. Although clots may occur spontaneously in individuals with protein S deficiency, risk factors, such as increasing age and immobility, can raise the risk of thrombus formation.
Homozygotes demonstrate severe manifestations of the condition and may develop a form of purpura fulminans in the neonatal period. The homozygous state also may lead to intrauterine death. Treatment with heparin, warfarin (Coumadin), and protein C concentrate is similar to that for protein C deficiency.
Antithrombin III (AT III) deficiency is inherited as an autosomal dominant condition. Most individuals are heterozygous for a mutation. AT III deficiency results from mutations in the SERPINC1 gene and exists in forms type I and type II. Type I is a quantitative deficiency of the AT III antigen resulting from decreased production. Type II is characterized by decreased functional activity. Normal levels of AT III are present; however, their activity is reduced.83
Individuals with AT III deficiency are at risk for early development of venous thrombosis and pulmonary embolism. These events often occur in the middle to late teens, but they can occur as early as 10 years of age. The deep veins of the lower extremities are usually involved, most commonly the iliofemoral vein. Other sites include the mesenteric veins, vena cava, renal veins, and retinal veins. Cerebral thromboses also have been described, and arterial thrombotic events are rare. In some cases, thrombosis is precipitated by surgery, trauma, pregnancy, oral contraceptives, and infection.
The treatment of choice for AT III deficiency is heparin. Antiplatelet agents (e.g., aspirin, dipyridamole) may be used, as well as AT III concentrates.
Antibody-Mediated Hemorrhagic Disease
Antibody-mediated hemorrhagic diseases are caused by the immune response. Antibody-mediated destruction of platelets or antibody-mediated inflammatory reactions to allergens damage blood vessels and cause seepage into tissues. The thrombocytopenic purpuras may be intrinsic or idiopathic. They also may be transient phenomena transmitted from mother to fetus. The inflammatory, or “allergic,” purpuras, although rare, occur in response to allergens in the blood. All of these disorders first appear during infancy or childhood.
Primary Immune Thrombocytopenia
Primary immune thrombocytopenia (ITP) (previously referred to as idiopathic thrombocytopenic purpura) is the most common of the thrombocytopenic purpuras of childhood. It is a disorder of platelet consumption in which autoantibodies bind to the plasma membranes of platelets. Antibody binding causes platelet sequestration and destruction by mononuclear phagocytes in the spleen and other lymphoid tissues at a rate that exceeds the ability of the bone marrow to produce them. The destruction of platelets is triggered by drugs, infections, lymphomas, or an unknown cause.
ITP is defined further based on the phases of the illness and established criteria for evaluating the response to therapy.84 Specific phases include newly diagnosed ITP, within 3 months of diagnosis; persistent ITP, describing individuals with 3 to 12 months of diagnosis who have not achieved remission or complete response off therapy; chronic ITP, symptoms lasting longer than 12 months; and severe ITP, the presence of bleeding that requires treatment either at diagnosis or following initiation of treatment.
Pathophysiology
The autoantibodies that produce the destruction are often of the IgG class and are usually against the platelet membrane glycoproteins (IIb-IIIa or Ib-IX). Approximately 70% of cases of ITP are preceded by a viral illness (e.g., cytomegalovirus [CMV], Epstein-Barr virus [EBV], parvovirus, or respiratory tract infection), suggesting that viral sensitization has occurred. The typical interval between infection and onset of purpura is 1 to 3 weeks.
Clinical Manifestations
Bruising and a generalized petechial rash often develop acutely about 1 to 3 weeks after a viral illness. Petechiae can develop into ecchymoses. Asymmetric bruising is typical and is found most often on the legs and trunk. Hemorrhagic bullae of the gums, lips, and other mucous membranes may be prominent. Epistaxis (nose bleeding) may be severe and difficult to control. Except for signs of bleeding, the child appears well. The principal changes are found in the spleen, bone marrow, and blood. The acute phase of the disease associated with spontaneous hemorrhages lasts 1 to 2 weeks, but thrombocytopenia often persists. Intracranial hemorrhage is the most serious complication of ITP; however, the incidence is less than 1%. In some cases, the onset of ITP is more gradual, and clinical manifestations consist of moderate bruising and a few petechiae.
Evaluation and Treatment
Laboratory examination reveals an isolated low platelet count. The few platelets observed on a peripheral smear are large, reflecting increased bone marrow production. The Ivy bleeding time is prolonged. Bone marrow aspiration is not recommended for children with typical features of ITP. The primary management for children newly diagnosed with ITP, with no or mild bleeding (skin manifestations), is only observation regardless of the platelet count. When non-life-threatening mucosal bleeding or diminished quality of life is present, a short burst of corticosteroids is recommended.84
Even without treatment, the prognosis for children with ITP is excellent: 75% recover completely within 3 months. After the initial acute phase, spontaneous clinical manifestations subside. By 6 months after onset, 80% to 90% of affected children have regained normal platelet counts. ITP that persists longer than 12 months in children is considered chronic. Thrombopoietin receptor agonists (TPO-RA) are recommended for the management of children with ITP who are unresponsive to first-line therapies.84
Autoimmune Neonatal Thrombocytopenias
Antibody-mediated thrombocytopenic purpura occurs in neonates in either autoimmune or alloimmune form. Both forms are characterized by the immunologic destruction of platelets by autoantibodies (IgG) against tissue-specific antigens expressed by the platelets (i.e., platelet-specific antigens).
Autoimmune neonatal thrombocytopenia was first noted in the early 1950s when it was observed that mothers with ITP often delivered infants who were transiently thrombocytopenic. Neonatal thrombocytopenia was observed in approximately 50% of infants at risk and lasted an average of 1 month. As platelet counts returned to normal, a concomitant drop in the level of maternal antiplatelet antibody on the child's platelets occurred. The antibody is directed against antigens common to maternal and neonatal platelets. The prognosis generally is favorable, and the frequency of intracranial hemorrhage is rare (1% to 3% of cases). Medical management of affected infants is to prevent the severe thrombocytopenia that can cause significant morbidity by administering intravenous immunoglobulins.
Neonatal alloimmune thrombocytopenia purpura (NAIT) is less common and is estimated to occur in 1 to 2 per 1000 live births. NAIT is caused by maternal immunization against fetal paternally derived platelet-specific antigens (similar to rhesus [Rh] disease). The mother has a normal platelet count, whereas the fetus can be severely thrombocytopenic.
The diagnosis of NAIT is confirmed by detection in the maternal serum of antibody that reacts with platelets from the infant and father but not with platelets from the mother. In approximately 75% to 85% of cases, NAIT recurs in subsequent pregnancies. Purpura usually develops in the affected infant shortly after delivery, and intracranial, renal, and gastrointestinal hemorrhages are possible. The mortality rate from intracranial hemorrhage has been estimated at 10% to 15%. Management of the newborn with NAIT includes an immediate cranial ultrasound because of the significant risk of intracranial hemorrhage. Severely thrombocytopenic newborns (<10,000/μl) or newborns with intracranial or visceral hemorrhages should receive a matched platelet transfusion (maternal or homozygous human platelet antigen 1b [HPA-1b] donor) as soon as possible. If maternal platelets are used, they must be processed to remove platelet alloantibodies. Newborn thrombocytopenia is difficult to predict because newborn platelet counts do not correlate with maternal platelet counts or antiplatelet antibody titers.85
Most of the life-threatening clinical manifestations of transient neonatal thrombocytopenia and NAIT can be avoided through cesarean delivery. If the mother has antiplatelet disease, however, surgery can result in hemorrhage and serious maternal morbidity. Maternal morbidity resulting from NAIT during pregnancy is low (less than 5%): the principal maternal risk is bleeding from surgical incisions during cesarean delivery. The incidence of transient thrombocytopenia in infants born to mothers with NAIT is about 50%. If all deliveries were cesarean, half the mothers would undergo cesarean delivery unnecessarily. Conversely, if all deliveries were vaginal, half the infants—those with thrombocytopenia—would be at risk for intracranial bleeding. Therefore, in the absence of any clear benefit to the neonate (given the low rate of intracranial hemorrhage in infants born to mothers with ITP), cesarean delivery should be reserved for the usual obstetric indications.
Autoimmune Vascular Purpura
Autoimmune vascular purpura (allergic purpura) is caused by antibody-mediated injury of blood vessel walls, typically arterioles and capillaries. The exact cause of the inflammatory reaction is unknown but is believed to result from exposure to foreign proteins or chemicals in the blood such as microorganisms, drugs, or other chemicals.
Autoimmune vascular purpura usually is seen in young children. Its incidence decreases among adolescents and adults and occurs only rarely in older adults. The average age at onset is 5 years, with a slightly higher proportion of males affected. Purpura occurs as vessel integrity is disrupted by inflammatory processes, causing effusion of serosanguineous exudate to perivascular tissues.
Clinical manifestations include headache, anorexia, fever, abdominal pain, constipation, arthralgias and urticaria, and erythema that are located symmetrically on the proximal portions of the extremities, particularly on the legs and buttocks, and may be accompanied by itching or paresthesias. Abdominal pain results from hemorrhage into the bowel, which may lead to colic, nausea, and vomiting. These symptoms may precede the appearance of skin lesions. The pain usually is midabdominal but may radiate to other parts of the abdomen. Joint pain and tenderness may be present, but hemarthrosis does not occur. Periarticular swelling and edema of the hands and feet are common and may precede the onset of abdominal pain and purpura. Subacute glomerulonephritis occurs in some cases but usually is reversible.
The characteristic skin lesions (purpura and cutaneous manifestations of allergy), accompanied by a history of joint and abdominal pain, are suspicious for diagnosis. Laboratory test results often reveal no major abnormalities. Attacks may last several weeks and may recur at odd intervals and with changing manifestations with each episode. Treatment, if necessary, consists of symptom management, often with analgesics and non-steroidal anti-inflammatory medications.
Neoplastic Disorders
Leukemia
Leukemia is cancer of the blood-forming tissues, such as the bone marrow, that most often produces abnormal white blood cells called leukemic cells. Once in the blood, leukemic cells can spread to other organs, such as the lymph nodes, spleen, and brain. Leukemia is the most common malignancy in children and adolescents. Of the types of acute childhood leukemia, 75% to 80% of leukemias in children are derived from the lymphoid line and are classified as acute lymphoblastic leukemia (ALL). The remaining 20% to 25% are derived from the myeloid line and are classified as acute myeloid leukemia (AML) and related neoplasms. The related neoplasms include acute promyelocytic, myelomonocytic, and myelomonoblastic leukemias, as well as the very rare erythroid leukemia.86 A very small percentage of acute leukemias have characteristics of both the lymphoid and myeloid line and are classified as acute leukemia of ambiguous lineage. Chronic leukemias are rare in children and account for fewer than 5% of cases. Leukemia accounts for 25% of cases of cancer in black children and 34% of cases of cancer in white children.
In the United States, over 50% of all cases of ALL are diagnosed in individuals less than 20 years of age with new cases most prevalent among children between 2 and 6 years of age.86 The incidence of ALL is greatest among children aged 2 to 3 years (>90 cases per 1 million per year) with rates decreasing to fewer than 30 cases per million by age 8. Since 1975, the incidence of ALL has gradually increased.87 New cases of leukemia have been rising on average 0.3% a year in the last 10 years. ALL affects more white and Hispanic than black children and more males than females. The incidence of ALL is also higher in Western and industrialized nations.88 ALL is more common in boys than girls and among Hispanic and white children than among black and Asian American children. AML is slightly more common during the first 2 years of life and during the teenage years. It occurs about equally among boys and girls of all races; however, recent studies suggest increasing rates among Asian children and those of Hispanic ethnicity.89,90
Types of Leukemia
A number of different classifications are used for the leukemias. First, acute leukemia is differentiated from chronic leukemia. Second, the cell line determines whether lymphoid cells or myeloid cells are involved. In acute leukemia, this difference separates ALL from AML and vice versa. Categories of ALL are further identified based on their presumed origin from bursa-equivalent cells (B cells) of cases of ALL and thymic cells (T cells) of normal lymphocytes. B-cell ALL comprises 80% to 85% of cases of ALL, and approximately 15% to 20% of cases are T-cell ALL. (See Chapter 29 for a discussion of leukemias in adults.)
Cytogenic studies of leukemic cells are performed routinely at most major treatment centers during the diagnostic process. Abnormal morphologic characteristics, as well as abnormalities in the number of copies of chromosomes, are often found in leukemic cells. Hyperdiploidy (the presence of greater than the diploid [46] number of chromosomes) is associated with a good prognosis. Common translocations associated with ALL are TEL-AML1, MLL rearrangements, and BCR-ABL. TEL-AML1 is the most common abnormality (in 20% to 30% of cases) and occurs when the TEL gene on chromosome 12p13 fuses with the AML1 gene on chromosome 21q22. TEL-AML1 is associated with a favorable outcome. MLL rearrangements are translocations between the MLL gene on chromosome 11q23 and other partner chromosomes. One of the most noted MLL rearrangements involves chromosome 4 as the partner chromosome t(4;11). This specific MLL rearrangement occurs most frequently among leukemia in infants less than 1 year of age and is associated with a poor prognosis despite intensive therapy.91 A translocation involving the long arms of chromosomes 9 and 22, t(9;22)(q34;q11), also described as the Philadelphia chromosome, occurs in more than 95% of cases of chronic myeloid leukemia (CML) yet is also present in about 2% to 3% of cases of ALL.86 The specific breakpoint, however, varies in CML and ALL. The translocation results in the generation of the BCR-ABL fusion gene. The bcr-abl protein that is produced by this fusion gene results in unregulated tyrosine kinase activity.
Classification of childhood leukemia has become a complex but essential process to determine treatment. The WHO has developed a classification scheme based on a comprehensive system that uses morphology, immunophenotyping, cytogenetic, and clinical features.86 Immunophenotyping using flow cytometry is used to distinguish between lymphoblastic and nonlymphoblastic leukemia. Molecular cytogenetic techniques, such as fluorescence in situ hybridization (FISH), allow for further detection of specific genetic abnormalities associated with leukemia subtypes. Next-generation sequencing (NGS) techniques, including whole genome and whole exome sequencing, are supporting further investigation of the molecular basis of childhood leukemia as well as planning treatment.92 (See Emerging Science Box: Molecular Characterization of Childhood Leukemia and Treatment Considerations.)
Etiology
The exact cause of most childhood cancer, including leukemia, is unknown. The occurrence of leukemia in monozygotic twins is estimated as being as high as 25%. About 5% of all childhood cancers are caused by inherited mutations. Genetic mutations that predispose the child to cancer development can occur during fetal development. Genetic conditions associated with leukemia include Down syndrome, neurofibromatosis, Li-Fraumeni syndrome, Shwachman-Diamond syndrome, Bloom syndrome, Fanconi anemia, and ataxia-telangiectasia.93 AML in children is sometimes associated with loss or deletion of chromosome 7 in the leukemia cells. AML can develop from preexisting myeloproliferative disorders that also are preleukemia syndromes, such as myelodysplastic syndrome. Epigenetic modifications, including DNA methylation, have been proposed as mediating events between environmental exposures and subsequent disease development.94,95
Multiple types of exposures have been investigated in relation to the development of leukemia. Many studies have shown that exposure to ionizing radiation (prenatal exposure to x-rays and postnatal exposure to high doses) can lead to the development of childhood leukemia and possibly other cancers.96 There is recent concern for performing computed tomography (CT) scans in children. The increased use of these scans combined with wide variability in radiation doses has resulted in many children receiving a high dose of radiation.97 Studies of other possible environmental risk factors, including parental exposure to cancer-causing chemicals, prenatal exposure to pesticides, childhood exposure to common infectious agents, and living near a nuclear power plant, have so far produced inconsistent results. Higher risks of cancer have not been seen in children of individuals treated for sporadic cancer (cancer not caused by an inherited mutation).98,99
Exposure to prenatal and postnatal smoking has also been investigated in relation to the development of leukemia and other childhood cancers. A recent meta-analysis identified an association between the development of ALL and paternal smoking in both the preconception and prenatal periods.100 Paternal smoking before, during, and after a pregnancy was identified as contributing to an increased risk of both ALL and AML in another recent meta-analysis; however, maternal smoking was not.101
Prenatal exposure to pesticides is hypothesized to increase the risk of childhood leukemia by resulting in oxidative stress leading to the generation of free radicals that induce breaks in the DNA of early hematopoietic stem cells. Results of recent meta-analyses support associations between parental exposure to pesticides and other potential environmental toxins before or during pregnancy and subsequent development of childhood leukemia.102,103
Chemicals such as benzene have been associated with the development of AML in adults. Recent meta-analyses provide evidence of a possible association between benzene exposure, occurring in the context of traffic-related air pollution and childhood leukemia.104,105 Previous treatment with chemotherapy for other cancer types is also a risk factor for developing ALL and AML.91
Studies investigating other types of exposures have had inconclusive results. Leukemic “clusters” that represent a greater number of leukemia cases occurring in a particular geographic location have raised speculation about environmental factors or infectious patterns of transmission. Follow-up studies, however, have not provided evidence of increased risk in the context of these exposures.106,107
Pathophysiology
ALL involves immature B (pre-B) or T (pre-T) cells called lymphoblasts. As leukemia develops, the bone marrow becomes dense with lymphoblasts that replace the normal marrow and disrupt normal function. Many of the chromosomal abnormalities documented in ALL cause dysregulation of the expression and function of transcription factors required for normal B-cell and T-cell development.108,109 The mutations can include both gain of function and loss of function that are required for normal development.
AML is caused by acquired oncogenic mutations that impair differentiation, resulting in the accumulation of immature myeloid blasts in the marrow and other organs. Epigenetic alterations are frequent in AML and have a central role. The bone marrow crowding by blast cells produces marrow failure and complications, including anemia, thrombocytopenia, and neutropenia. AML is very heterogeneous because myeloid cell differentiation is very complex. Leukemia, whether ALL or AML, is typically distinguished from lymphoma by the presence of greater than 20% leukemic blasts in the bone marrow.
Clinical Manifestations
The onset of leukemia may be abrupt or insidious. Children with leukemia may present with symptoms only a few weeks before diagnosis. Regardless of how leukemia develops, the most common symptoms reflect consequences of bone marrow failure. These include decreased levels of RBCs and platelets, as well as changes in white blood cells. Pallor, fatigue, petechiae, purpura, bleeding, and fever generally are present. Approximately 45% of children present with a hemoglobin level below 7 g/dL. Three-quarters of children with ALL have platelet counts less than 100,000/mm3 at diagnosis, and 28% have platelet counts less than 20,000/mm3. Epistaxis, excessive bruising, and hematuria may occur in children with severe thrombocytopenia. Half of all children newly diagnosed with AML have platelet counts less than 50,000/mm3. Disseminated intravascular coagulation occurs more commonly with AML, particularly with promyelocytic leukemia. The granules in the leukemic promyelocytes likely possess thromboplastin activity.
Fever can be present as a result of (1) infection associated with the decrease in functional neutrophils and (2) hypermetabolism associated with the ongoing rapid growth and destruction of leukemic cells. In most children with ALL, the total white blood count at diagnosis is less than 10,000/mm3, and with AML, most present with white cell counts less than 50,000/mm3. In a few children, however, the peripheral white blood count at presentation may be greater than 100,000/mm3. White blood cell counts greater than 200,000/mm3 can cause leukostasis, an intravascular clumping of cells resulting in infarction and hemorrhage, usually in the brain and lung. The three most important favorable prognostic factors are age at diagnosis (2 to 10 years), initial leukocyte count (<50,000/mm3), and initial response to treatment.
Renal failure as a result of hyperuricemia (high uric acid levels) can be associated with ALL, particularly at diagnosis or during the initial phase of treatment. Uric acid levels rise as an end product of purine metabolism from cellular destruction. Because the major excretory pathway is through the kidney, urates can precipitate in renal tubules or ureters and can lead to oliguria and acute renal failure. Renal failure is typically preventable if uric acid levels are monitored, and initial management is aimed at optimal hydration and blockage of further uric acid formation by administration of the drug allopurinol. Rasburicase, a medication that directly inhibits existing uric acid, may be administered to children with elevated uric acid levels and deemed at increased risk for renal failure.110
Extramedullary invasion with leukemic cells can occur in nearly all body tissue. Most children with ALL have some extramedullary involvement at diagnosis. Leukemic invasion of tissue other than bone marrow is believed to represent metastatic infiltration. Hepatosplenomegaly and lymphadenopathy, resulting from extramedullary hematopoiesis, occur in nearly half of children with ALL, but they are less common in children with AML.
The CNS is a common site of infiltration of extramedullary leukemia. Less than 10% of children with ALL, however, will have CNS involvement at diagnosis. The most common symptoms of CNS involvement relate to increased intracranial pressure, causing early morning headaches, nausea, vomiting, irritability, and lethargy. Gonadal involvement with testicular infiltration also may occur.
Leukemic infiltration into bones and joints is common. Reports of bone or joint pain actually lead to the diagnosis of leukemia in some children. In most children, bone pain is characterized as migratory, vague, and without areas of swelling or inflammation. In some cases, however, joint pain is the primary symptom, and some swelling is associated with the pain. Occasionally, these children are initially misdiagnosed as having rheumatoid arthritis. Other organs reported to be sites of leukemic invasion include the kidneys, heart, lungs, thymus, eyes, skin, and gastrointestinal tract. Skin involvement is more common in AML than in ALL.
Evaluation and Treatment
Leukemia is diagnosed through blood tests and examination of peripheral blood smears. A bone marrow aspiration is usually performed to further characterize the leukemia. The blast cell is the hallmark of acute leukemia (Fig. 30.10A). The blast cell is a relatively undifferentiated cell characterized by diffusely distributed nuclear chromatin, with one or more nucleoli and basophilic cytoplasm (Fig. 30.10B).
(A) Bone marrow smear with numerous blast cells and (B) blast cells (shown in purple) with basophilic cytoplasm. (From Naeim F, Nagesh Rao P, Song SX, et al. Acute myeloid leukemia, not otherwise specified. Atlas of Hematopathology, 2018; 345–374. https://doi.org/10.1016/b978-0-12–809843-1.00022-x.)
Two photomicrographs of bone marrow smears A and B show the morphology of acute myeloid leukemia. Photomicrograph A shows large blast cells with a low nucleus. Photomicrograph B shows numerous blasts cell with round nuclei.
Healthy children have less than 5% blast cells in the bone marrow and none in the peripheral blood. In ALL, the bone marrow often is replaced by 80% to 100% blast cells. The bone marrow, at the time of diagnosis, is typically considered hypercellular and is composed of a homogeneous population of cells. Counts of normally developing RBCs, granulocytes, and platelets are typically reduced. Occasionally, the marrow appears hypocellular, making the diagnosis difficult to differentiate from aplastic anemia. When this occurs, bone marrow biopsy or biopsy of extramedullary sites is necessary to confirm the diagnosis.
Approximately 85% of children with ALL will become 5-year survivors of their illness. Chemotherapy, using a combination of medications, is the treatment of choice for acute leukemia. Radiation of the CNS is used only in selected cases. Identification of various risk groups among children with ALL has led to the development of different intensities of drug protocols. As a result, treatment can be targeted specifically for a particular risk group. For example, enhanced technologies to detect minimal residual disease have improved the ability to identify children who are at increased risk of relapse. These children are then able to receive intensified treatment with emerging therapeutic approaches, including bi-specific antibodies earlier in their course of treatment.111 Children who experience relapses of ALL may receive hematopoietic stem cell transplants. Treatment with chimeric antigen receptor T cells (CAR-T cells) is also showing promise.93
AML is more difficult to treat than ALL. Combination chemotherapy is the most common approach to treatment. Those children with unfavorable cytogenetic markers and those who experience a relapse of their disease will often undergo HSCT.112
CML accounts for less than 5% of childhood leukemias. Biologically targeted therapies, specifically tyrosine kinase inhibitors (TKIs), are becoming the mainstay of treatment, specifically for individuals whose disease has the BCR/ABL translocation.112 TKIs are administered orally, and several are now approved for use in children. Treatment requires continued adherence to the medication regimen, and the health impact of long-term TKI therapy is not yet known.
Lymphomas
Lymphoma (Hodgkin lymphoma [HL] and non-Hodgkin lymphoma [NHL]) develops from the proliferation of malignant lymphocytes in the lymphoid system (see Chapters 14 and 29). Lymphomas arise from discrete tissue masses and involve some recognizable stage of lymphocyte B- or T-cell differentiation. The WHO provides a classification scheme for lymphoma that was updated in 2016 (also see Chapter 29).113
Lymphomas are the third most common type of childhood cancer, behind leukemia and CNS tumors, and represent about 11% of all cases of childhood cancer. Around 1800 children younger than 20 years of age are diagnosed with lymphoma in the United States each year.114 NHL (including Burkitt lymphoma) occurs more often than Hodgkin lymphoma. Either group of diseases is rare before the age of 5 years, and the relative incidence increases throughout childhood. Boys are more likely to be diagnosed with lymphoma than are girls. Children with inherited or acquired immunodeficiency syndromes, such as Wiskott-Aldrich syndrome, ataxia-telangiectasia, and Bloom syndrome, are at particular risk for developing NHL.
Non-Hodgkin Lymphoma
Non-Hodgkin lymphomas (NHLs) are cancers of immune cells. NHLs are a large and diverse group of tumors. NHL typically arises at extranodal sites and spreads in a noncontiguous or unpredictable manner. Some tumors develop more slowly, whereas others develop more quickly and aggressively. Some mature B-cell lymphomas present more like leukemia. Childhood NHL typically becomes evident as a diffuse disease and can be further subdivided into four major types: (1) mature B-cell non-Hodgkin lymphoma (Burkitt and Burkitt-like lymphoma and Burkitt leukemia); (2) lymphoblastic lymphoma; and (3) anaplastic large cell lymphoma.115 The common types of NHL in children are different from those in adults. The most common types of NHL in children are Burkitt lymphoma (40%), lymphoblastic lymphoma (25% to 30%), and large cell lymphoma (10%).
Pathophysiology
Burkitt lymphoma will be discussed as an example of the pathogenesis of NHL in children. Burkitt lymphoma includes three clinical variants: sporadic, endemic, and immunodeficiency associated. Each of these variants is associated with translocations of the MYC gene on chromosome 8 that lead to increased MYC protein levels.115 MYC is a transcription factor, meaning that it influences the rate of transcription of DNA into messenger RNA. Overexpression of MYC further increases the expression of genes required for aerobic glycolysis, called the Warburg effect (see Chapter 12). Many Burkitt lymphomas are associated with latent infection of the EBV.116 EBV is present in nearly all cases of endemic Burkitt lymphoma, which occur mostly among individuals living in areas of the world where malaria is highly prevalent. Malaria is believed to reduce resistance to EBV, thereby increasing susceptibility to lymphoma. EBV is also present in 15% to 20% of sporadic cases of Burkitt lymphoma and about 25% of tumors associated with human immunodeficiency virus (HIV) infection.116,117 Other immunodeficiency conditions that increase the risk for Burkitt and other NHLs include receipt of a solid organ transplant, Wiskott-Aldrich syndrome, ataxia-telangiectasia, and Bloom syndrome.
Clinical Manifestations
NHL can arise from any lymphoid tissue. Signs and symptoms therefore are specific for the involved site. Some children have such widespread involvement that no original site can be determined. Because some childhood NHL is rapidly progressive, symptoms are often present only a few weeks before diagnosis is made. Rapidly enlarging lymphoid tissue and painless lymphadenopathy are common in about one-third of children with abdominal sites of involvement, usually representing a gastrointestinal origin for the disease. Symptoms often include abdominal pain and vomiting, but a palpable mass is not always present. The other common site of childhood NHL is the chest region. Associated signs of NHL include swelling of the lymph nodes in the neck, underarm, stomach, or groin; trouble swallowing; a painless lump or swelling in a testicle; weight loss for unknown reason; night sweats; and possibly trouble breathing. Involvement of facial bones, particularly the jaw, is common in endemic Burkitt lymphoma.
Evaluation and Treatment
Diagnosis is made by physical exam and health history, followed by a needle biopsy of disease sites, usually the involved lymph nodes, tonsils, spleen, liver, bowel, or skin. Burkitt lymphoma is very aggressive and responds well to treatment. With intensive chemotherapy, most children and young adults can be cured.
Hodgkin Lymphoma
Hodgkin lymphoma (HL) is a group of lymphoid neoplasms that, unlike NHL, arises in a single chain of lymph nodes and spreads first in a contiguous way to lymphoid tissue. HL is characterized by the presence of Hodgkin and Reed-Sternberg cells (HRS), which are large cells that come from the germinal center of B cells (Fig. 30.11). HL is a common type of cancer in young adults and adolescents but rare in childhood. The WHO has identified five types of HL: (1) nodular sclerosis, (2) mixed cellularity, (3) lymphocyte rich, (4) lymphocyte depletion, and (5) lymphocyte predominance (also see Chapter 29). Similar expression of HRS cells occurs in the first four types; therefore, they are considered the classic types. In the lymphocyte predominance type, the HRS cell is distinctive but different than the others. Risk factors for HL include having EBV, being infected with HIV or other immunologic disorders (immunodeficiencies, autoimmune lymphoproliferative syndrome), having a personal history of mononucleosis, and having a parent or sibling with a personal history of HL.118 Relatively few cases of HL are diagnosed in children before age 5 years, and HL is much more prevalent among males than females (M:F = 5.3) in this age group. Among adolescents 15 to 19 years of age, however, the incidence of HL is slightly greater in females (M:F = 0.8). The incidence of HL gradually increases through age 11 years, with a marked increase through adolescence that continues into the 30 s.
A large multinucleated or multilobated cell with inclusion body–like nucleoli (arrow) surrounded by a halo of clear nucleoplasm. (From Damjanov I, Linder J. Pathology: A color atlas. St. Louis: Mosby; 2000.)
Pathophysiology
The HRS cells fail to express most of the normal B-cell markers, as well as those of T-cells. The causes of the genetic rearrangements or reprogramming are not fully known but are thought to be the result of widespread epigenetic changes.
The abnormal pattern of gene expression in HRS cells suggests that the activity of many transcription factors is also altered.119 NGS studies have identified mutations in signaling pathways, including transcription factor nuclear factor-kappa B (NF-κB) and other epigenetic regulators. Abnormalities in the activation of NF-κB may be influenced by EBV infection. NF-κB is involved in many biologic processes, including inflammation, immunity, cell growth, differentiation, and apoptosis. EBV-infected B cells, resembling Reed-Sternberg cells, are found in lymph nodes in individuals with infectious mononucleosis, suggesting that the EBV proteins may have a role in changes of the B cells into HRS cells.120 Altered expression of major histocompatibility class antigens may allow HRS cells to avoid the normal host immune response.121
Clinical Manifestations
Painless lymphadenopathy in the lower cervical chain, with or without fever, is the most common symptom in children. These nodes are firm and rubbery and may be sensitive to palpation if they have grown rapidly. Other lymph nodes and organs also may be involved (Fig. 30.12). At least two-thirds of individuals have mediastinal involvement that may cause symptoms ranging from a nonproductive cough to tracheal or bronchial compression leading to airway obstruction. Extranodal primary sites in Hodgkin lymphoma are rare. Systemic symptoms at the time of diagnosis may include fatigue, anorexia, weight loss, malaise, drenching night sweats, and pruritus. Intermittent fever is present in 30% of children, and weight loss may be present.
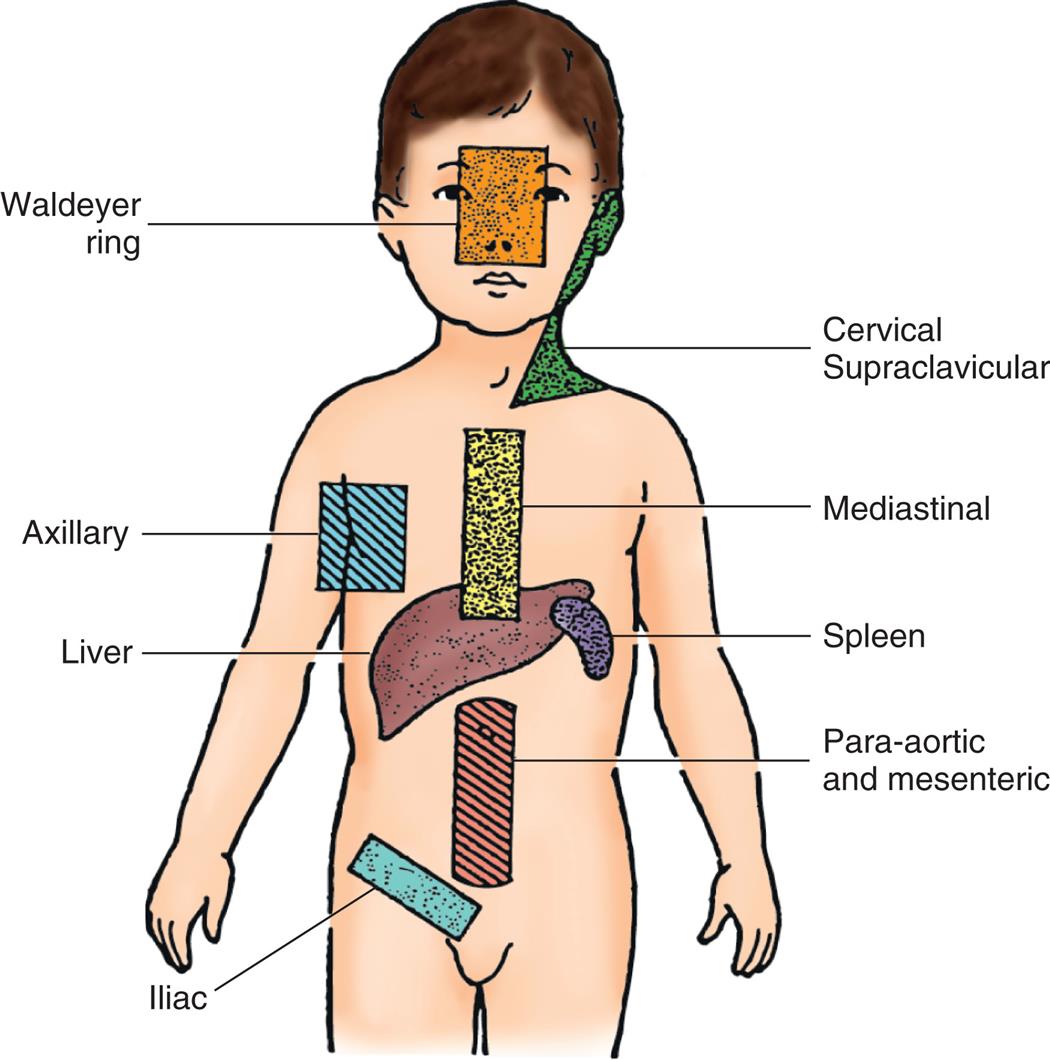
An illustration of the anterior view of the human body shows the main areas of lymphadenopathy and organ involvement in Hodgkin lymphoma: Waldeyer ring, cervical supraclavicular, mediastinal, axillary, liver, spleen, para-aortic and mesenteric, and iliac.
Evaluation and Treatment
The Ann Arbor staging system considers extent and location of disease, as well as substage classifications that consider systemic symptoms (presence of fever of 38°C [100.4°F] for 3 consecutive days, drenching night sweats, or unexplained loss of 10% or more of body weight in the 6 months preceding diagnosis), extranodal manifestations, or bulky disease. Combination chemotherapy used with or without low-dose radiation has been an effective treatment, with long-term cure rates reported from 90% to 95%.118
Historically, survivors had a much greater risk of developing a secondary cancer, such as lung cancer, melanoma, and breast cancer. Treatment protocols have been modified to minimize the use of radiotherapy and use less toxic chemotherapy. Targeted therapies, including monoclonal antibodies such as brentuximab vedotin and immune checkpoint inhibitors, may have a greater role in treating Hodgkin lymphoma.
Summary Review
Disorders of Erythrocytes
- 1. Anemia is the most common blood disorder in children. Like the anemias of adulthood, the anemias of childhood are caused by ineffective erythropoiesis or premature destruction of erythrocytes.
- 2. Iron deficiency anemia (IDA) is the most common nutritional disorder worldwide. Its incidence is greatest among children between 6 months and 2 years of age. Iron is critical for the developing child and without it, damage from the periods of IDA is irreversible.
- 3. Regardless of its cause, IDA produces a hypochromic-microcytic anemia. Symptoms of mild anemia are often nonspecific, so parents may not notice changes until moderate anemia has developed.
- 4. Hemolytic disease of the fetus and newborn (HDFN) results from incompatibility between the maternal and the fetal Rh factors or blood type (ABO). Maternal antibodies (anti-Rh antibodies) form in response to the presence of fetal incompatible (Rh-positive) erythrocytes in the blood of an Rh-negative mother. The maternal antibodies then enter the fetal circulation and cause hemolysis of fetal erythrocytes. ABO incompatibility can cause HDFN even if fetal erythrocytes do not escape into the maternal circulation during pregnancy.
- 5. The key to treatment of HDFN resulting from Rh incompatibilities lies in prevention or immunoprophylaxis.
- 6. Sickle cell disease is a group of disorders characterized by the production of abnormal hemoglobin S (HbS) within the erythrocytes. It is most common among people with ancestry from sub-Saharan Africa.
- 7. Sickle cell disease is an inherited, autosomal recessive disorder expressed as sickle cell anemia, sickle cell–thalassemia disease, or sickle cell–HbC disease, depending on mode of inheritance. Sickle cell anemia, in which the individual is homozygous for HbS, is the most severe. Sickle cell–thalassemia and sickle cell–HbC disease are compound heterozygous forms in which the child inherits HbS from one parent or another type of abnormal hemoglobin from the other parent. All forms of sickle cell disease are lifelong conditions.
- 8. Sickle cell trait, in which the child inherits HbS from one parent and normal hemoglobin (HbA) from the other, is a heterozygous carrier state that rarely has clinical manifestations.
- 9. Sickle cell disease causes a change in the shape of red blood cells into the sickle shape. Sickling is triggered by decreased oxygen or dehydration. Most sickled erythrocytes regain a normal shape after reoxygenation and rehydration.
- 10. The alpha- and beta-thalassemias are inherited autosomal recessive disorders. These conditions result in an impaired rate of synthesis of one of the two chains—α or β—of adult hemoglobin (HbA).
Disorders of Coagulation and Platelets
- 1. Hemorrhagic diseases can be either inherited (hemophilias) or antibody-mediated (primary immune thrombocytopenia [ITP]).
- 2. The hemophilias are a group of inherited bleeding disorders resulting from mutations in the genes responsible for coagulation factors.
- 3. Hemophilia A (factor VIII deficiency) and hemophilia B (factor IX deficiency) are caused by mutations in the genes coding for factors VIII and IX, factors essential in the coagulation cascade. Because factors VIII and IX function together, hemophilia A and B are clinically indistinguishable. Hemophilia A is the most common hereditary disease associated with life-threatening bleeding.
- 4. Hemophilias A and B are inherited as X-linked recessive conditions. Approximately one-third of cases, however, are the result of a spontaneous mutation in the involved gene.
- 5. The antibody-mediated hemorrhagic diseases are a group of disorders caused by the immune response. Antibody-mediated destruction of platelets or antibody-mediated inflammatory reactions to allergens damage blood vessels and cause seepage into tissues.
- 6. ITP is the most common disorder of platelet consumption in which antiplatelet antibodies bind to the plasma membranes of platelets. ITP results in platelet sequestration and destruction by mononuclear phagocytes at a rate that exceeds the ability of the bone marrow to produce them.
Neoplastic Disorders
- 1. Leukemia is cancer of the blood-forming tissues, such as the bone marrow, that most often produces abnormal white blood cells called leukemic cells.
- 2. About 75% of childhood leukemias are acute lymphoblastic leukemia (ALL). The remaining cases are classified as acute myeloid leukemia (AML) and related neoplasms. Chronic leukemias are rare in children.
- 3. The cause of most childhood cancer, including leukemia, is unknown. About 5% of all childhood cancers are caused by inherited mutations. Genetic mutations that predispose the child to cancer development can occur during fetal development.
- 4. Exposure to ionizing radiation can lead to the development of childhood leukemia and possibly other cancers.
- 5. ALL causes dysregulation of the expression and function of transcription factors required for normal B- and T-cell development.
- 6. Epigenetic alterations are frequent in AML and have a central role in its development.
- 7. The onset of leukemia may be abrupt or insidious. The most common symptoms reflect consequences of bone marrow failure and can include decreased levels of red blood cells and platelets, as well as changes in white blood cells.
- 8. The lymphomas of childhood are Hodgkin lymphoma (HL) and non-Hodgkin lymphoma (NHL).
- 9. Lymphomas are proliferations of malignant lymphocytes that arise from discrete tissue masses. Lymphoid neoplasms involve some recognizable stage of lymphocyte B- or T-cell differentiation.
- 10. Some lymphomas occasionally have leukemic presentations, and evolution to leukemia is not unusual during the progression of incurable lymphoma.
- 11. The lymphomas of childhood are Hodgkin lymphoma (HL) and non-Hodgkin lymphoma (NHL).
- 12. NHLs are cancers of immune cells. Children with inherited or acquired immunodeficiency syndromes have an increased risk of developing NHL.
- 13. The most common types of NHL in children are Burkitt lymphoma, lymphoblastic lymphoma, and large cell lymphoma. Most Burkitt lymphomas are latently infected with the Epstein-Barr virus (EBV).
- 14. HL is a group of lymphoid cancers. HL arises in a single chain of lymph nodes and spreads first in a contiguous way to lymphoid tissue.
- 15. HL is characterized by the presence of Reed-Sternberg cells, which are large cells derived from the germinal center of B cells.