Alterations of Cardiovascular Function
Valentina L. Brashers
 http://evolve.elsevier.com/Rogers/pathophysiology/
http://evolve.elsevier.com/Rogers/pathophysiology/
Cardiovascular disease is the leading cause of death, both in the United States and worldwide. Disorders of the veins, arteries, and heart comprise the scope of cardiovascular disease. Current understanding of the pathophysiology of cardiovascular disease is focused on genetic, neurohumoral, inflammatory, and metabolic mechanisms that underlie tissue and cellular alterations.
Diseases of the Veins
Varicose Veins and Chronic Venous Insufficiency
A varicose vein is a vein in which blood has pooled, producing distended, tortuous, and palpable vessels (Fig. 32.1). Risk factors include age, female sex, family history of varicose veins, obesity, pregnancy, deep venous thrombosis (DVT), and previous leg injury. Varicose veins typically involve the saphenous veins of the leg and are caused by (1) injury or disease involving the saphenous vein valves or (2) gradual venous distention caused by the action of gravity on blood in the legs.
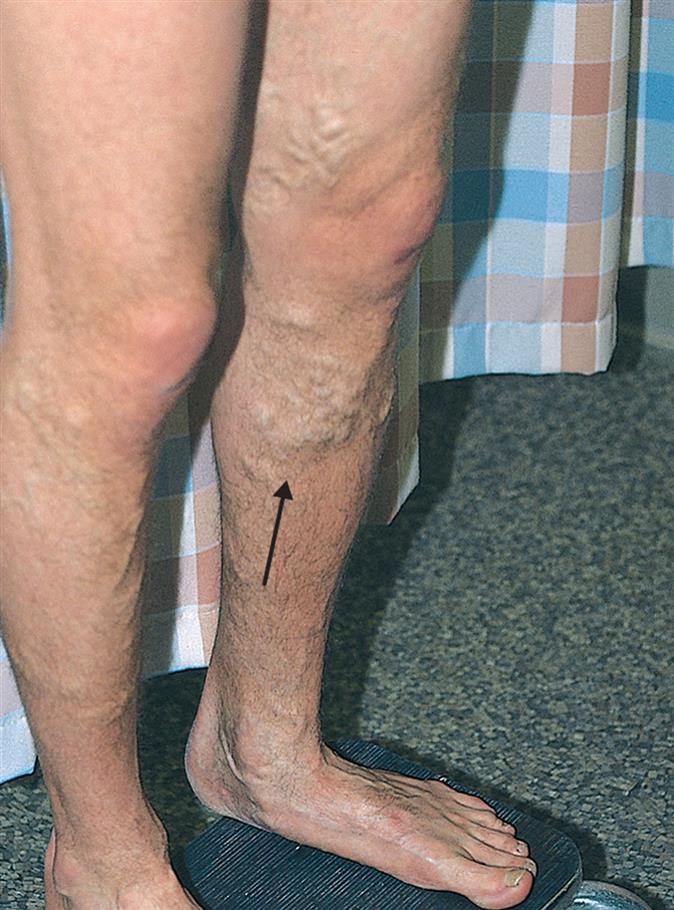
If a valve is damaged, volume and pressure increase within the vessel. The vein swells as it becomes engorged and surrounding tissue becomes edematous because increased hydrostatic pressure pushes plasma through the stretched vessel wall. Venous distention develops over time, especially in individuals who habitually stand for long periods, wear constricting garments, or cross the legs at the knees, which diminishes the action of the muscle pump (see Fig. 31.20). Genetic factors, female sex, and pregnancy are contributing factors. Eventually the pressure in the vein damages venous valves, rendering them incompetent and unable to maintain normal venous pressure.
Varicose veins and valvular incompetence can progress to chronic venous insufficiency, especially in sedentary obese individuals and those who smoke. Chronic venous insufficiency (CVI) is inadequate venous return over a long period of time. Venous hypertension, circulatory stasis, and tissue hypoxia cause an inflammatory reaction and changes in the extracellular matrix in vessels. Endothelial cells express adhesion molecules bringing leukocytes and fibroblasts to the area. Collagen synthesis increases vascular wall thickening and fibrosclerotic remodeling of the veins. The microcirculation is impaired contributing to more inflammation and tissue hypoxia. Approximately one-third of those with varicose veins develop significant skin changes.1 Symptoms include edema of the lower extremities and hyperpigmentation of the skin of the feet and ankles. Poor circulation makes tissues vulnerable to trauma and infection resulting in the formation of venous stasis ulcers (Fig. 32.2) and cellulitis.
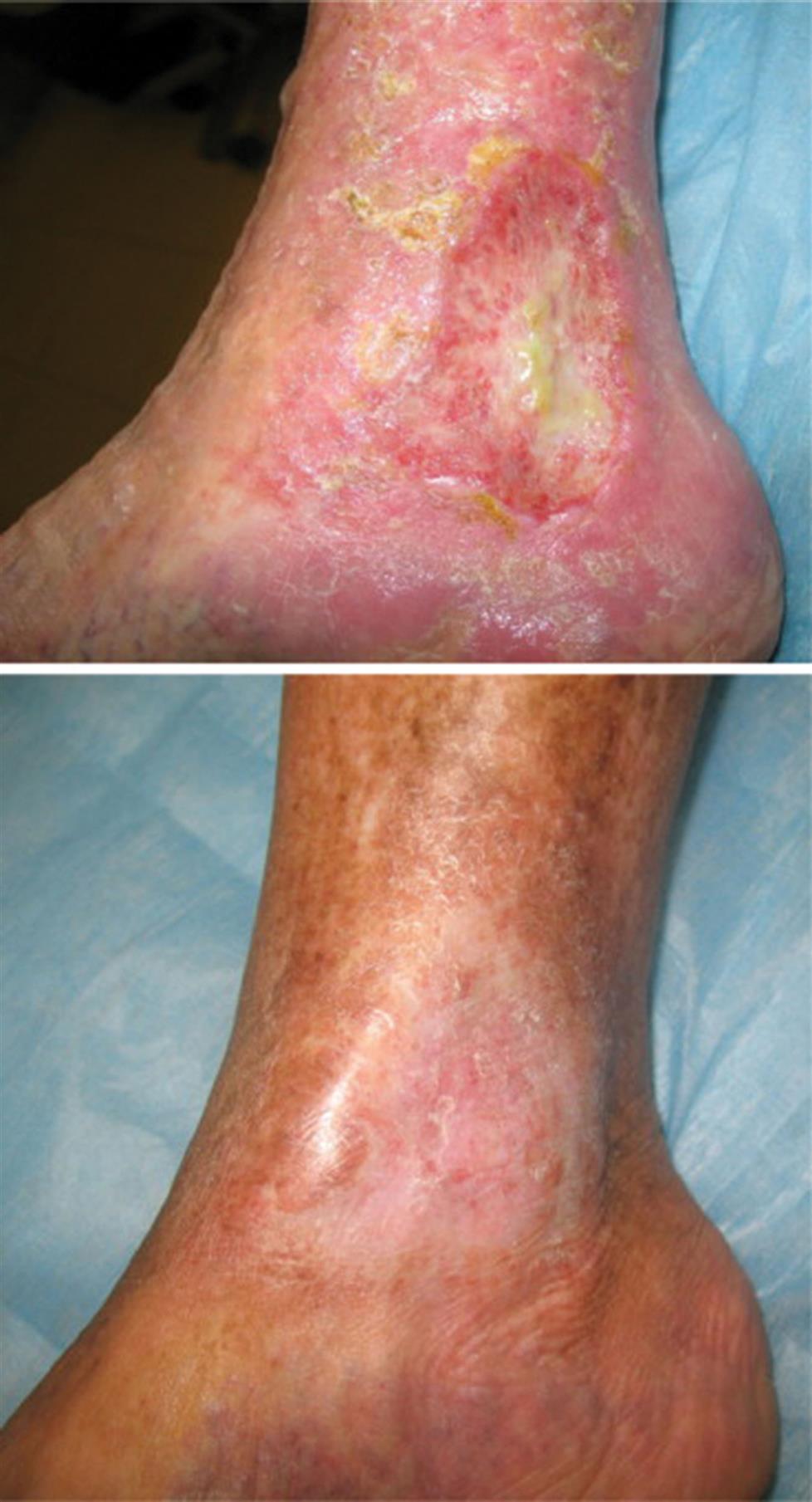
Typical venous ulcer, healed after 11 weeks of compression therapy using high pressure Unna boot bandages. (From Caprini JA, Partsch H, Simman R. Venous ulcers. Journal of the American College of Clinical Wound Specialists, 2013;4(3):54–60. doi:10.1016/j.jccw.2013.11.001.)
Evaluation of CVI includes careful physical examination of pulses and skin, followed by Doppler ultrasound and measurement of ankle brachial pressure index. Identification of the underlying cause is crucial to effective management.2 Treatment of varicose veins and CVI begins conservatively with elevating the legs, wearing compression bandages or stockings, and performing physical exercise. In those with venous ulcers, the choice of compression materials and their proper use is essential to obtaining improvement in symptoms and ulcer healing.1 Advanced wound care techniques including skin grafting, mesenchymal stem cells, and autologous platelet rich plasma may be indicated for large non-healing ulcers.2 Invasive management for venous obstruction and dilation includes endovenous ablation, sclerotherapy or surgical ligation, conservative vein resection (phlebectomy), and vein bypass.
Thrombus Formation in Veins
A thrombus is a blood clot that remains attached to a vessel wall. A detached thrombus is a thromboembolus. The Centers for Disease Control and Prevention estimates that venous thrombi and associated thromboembolism affect about 900,000 people and cause nearly 100,000 deaths in the United States per year.3 Deep venous thrombosis (DVT) occurs primarily in the lower extremity. Venous thrombi are more common than arterial thrombi because flow and pressure are lower in the veins than in the arteries. Three factors (triad of Virchow) promote venous thrombosis: (1) venous stasis, (2) venous endothelial damage, and (3) hypercoagulable states (see Chapter 29).
Venous stasis occurs in conditions that limit flow through the local or systemic venous circulation. For example, immobility (e.g., stroke, spinal cord injury, casting) prevents the muscular pump (see Fig. 31.20) from increasing blood flow from the lower extremity to the inferior vena cava and right atrium. Similarly, heart failure results in increased diastolic filling pressures causing reduced venous return. Accumulation of clotting factors and platelets leads to thrombus formation in the vein, often near a venous valve. Further platelet aggregation leads to the thrombus propagating (growing) proximally.
Damage to endothelial cells can occur due to trauma (e.g., orthopedic injury or surgery), the use of caustic intravenous medications, or the use of invasive venous procedures (e.g., peripherally inserted central catheters). The healthy vascular endothelium serves as a barrier between the blood and the prothrombotic subendothelium. It also expresses several anticoagulant factors including protein C receptors and tissue factor pathway inhibitor. In the presence of endothelial damage, endothelial cells lose their anticoagulant properties and instead express adhesion molecules that promote inflammatory cell and platelet activation.4 Polymorphonucleocytes infiltrate the vessel wall and produce neutrophil extracellular traps (NETs) (see Chapter 7) that cause additional inflammatory damage to the endothelium and promote clotting.
Many individuals develop DVT because of transient or prolonged hypercoagulability states. Pregnancy is associated with both hypercoagulability and venous stasis in the lower extremities. Active cancer is estimated to be the cause of approximately 20% of all cases of venous thromboembolism, especially hematologic malignancies and those involving the pancreas, stomach, lung, brain, ovaries, and kidneys.5 In malignancy-related DVT, a hypercoagulable state is created by the cancer cells themselves. Tissue factor produced by cancer cells activates coagulation, fibrin synthesis, and platelet activation. Many cancer cells also produce plasminogen activator inhibitor-1 which inhibits the fibrinolytic system.6 Some infections also are associated with hypercoagulability and venous thromboembolism. For example, systemic inflammation that results from infections such as bacterial sepsis or SARS-CoV-2 induced Coronavirus Disease 2019 (COVID-19) are associated with a high risk for the development of DVT.7,8 Inherited hypercoagulability states increase the risk for DVT, especially in association with other risk factors, such as immobility or pregnancy. The most common inherited hypercoagulability state is factor V Leiden mutation, which affects 3% to 8% of the population and is estimated to be the cause of 20% to 25% of venous thromboembolism cases. Factor V Leiden results from a single point mutation in the Factor V gene which causes an inadequate anticoagulant response to activated protein C.9 Individuals may develop DVT without apparent cause or after a relatively minor risk event such as airplane travel. This mutation also increases the risk of DVT recurrence after treatment. Other inherited hypercoagulability states are caused by prothrombin mutations and deficiencies of protein C, protein S, and antithrombin.
Venous thrombosis is often asymptomatic and affected individuals may be unaware that they are at risk for serious complications until they develop symptoms of thromboembolism to the lungs (pulmonary embolism, see Chapter 35). In some cases, venous inflammation causes pain and redness of the overlying tissues and skin. If the thrombus creates significant obstruction to venous blood flow, increased pressure in the vein behind the clot may lead to edema of the extremity. Most thrombi will eventually dissolve without treatment; however, untreated DVT is associated with a high risk of thromboembolization. Persistent venous obstruction may lead to CVI and postthrombotic syndrome with associated pain, edema, and ulceration of the affected limb.4
Because DVT is usually asymptomatic and difficult to detect clinically, prevention is important in at-risk individuals. Risk stratification algorithms are used to identify and manage at-risk individuals. Common approaches to prevention include early ambulation, pneumatic compression devices, and prophylactic anticoagulation. If thrombosis is suspected, a serum D dimer is measured. If it is negative, the diagnosis of DVT is unlikely. If the D-dimer is elevated, diagnosis of DVT is confirmed by a Doppler ultrasonography. Current guidelines recommend that uncomplicated DVT should be managed at home rather than in the hospital unless there is limb-threatening venous obstruction, high risk of bleeding, or severe pain.10 Direct oral anticoagulants (e.g., dabigatran, apixaban, edoxaban, rivaroxaban) have been shown to have a favorable benefit-to-risk ratio and are rapidly becoming the treatments of choice.10 Thrombolytic therapy (intravenous or catheter directed) or placement of an inferior vena cava filter may be indicated in selected individuals. Treatment is continued for 3 to 6 months at which time anticoagulation may be discontinued, or continued indefinitely for those with irreversible underlying risk factors.10
Superior Vena Cava Syndrome
Superior vena cava syndrome (SVCS) is a progressive occlusion of the superior vena cava (SVC) that leads to venous distention in the upper extremities and head. The most common cause is bronchogenic cancer followed by lymphomas and metastasis of other cancers. The incidence of device-related thromboses as a cause of SVCS is increasing.11 Other less common causes include tuberculosis, mediastinal fibrosis, and cystic fibrosis. The SVC is a relatively low-pressure vessel that lies in the closed thoracic compartment; therefore space-occupying lesions can easily compress the SVC. The SVC is surrounded by lymph nodes and abuts the right mainstem bronchus, which commonly becomes involved in thoracic cancers which may compress the SVC during tumor growth. The SVC also can be occluded by the presence of large thrombi. Invasive therapies (pacemaker wires, central venous catheters, and pulmonary artery catheters) with associated thrombosis now account for nearly half of cases of SVCS.
Clinical manifestations of SVCS are edema and venous distention in the upper extremities and face, including the ocular beds. Affected persons complain of a feeling of fullness in the head or tightness of shirt collars, necklaces, and rings. Cerebral edema may cause headache, visual disturbance, and impaired consciousness. The skin of the face and arms may become purple and taut, and capillary refill time is prolonged. Respiratory distress may be present because of bronchial compression. In infants, SVCS can lead to hydrocephalus.
Diagnosis is made by chest x-ray, Doppler studies, computed tomography (CT), magnetic resonance imaging (MRI), and ultrasound. SVCS is an oncologic emergency. Treatment for malignant disorders can include radiation therapy, surgery, chemotherapy, and the administration of diuretics, steroids, and anticoagulants, as necessary. Treatment for moderate to severe nonmalignant causes may include endovascular therapy such as balloon angioplasty or bypass surgery using various grafts. Severe symptoms require thrombolysis, balloon angioplasty, placement of intravascular stents, and/or surgery.11
Diseases of the Arteries
Hypertension
Hypertension is consistent elevation of systemic arterial blood pressure. It results from a sustained increase in peripheral vascular resistance (PVR), an increase in circulating blood volume and cardiac output, or both. Hypertension is defined as a sustained systolic blood pressure (SBP) of 130 mm Hg or a diastolic blood pressure (DBP) of 80 mm Hg or greater (Table 32.1). According to the American Heart Association, 51.7% of men and 42.8% of women over the age of 20 in the United States have hypertension, and the lifetime risk for developing hypertension is between 70% and 86%, with white females having the lowest risk and black males the highest risk.12 Most affected individuals do not have their hypertension under control. Between 2007 and 2017, hypertension-related deaths in the United States increased from 18.3 per 100,000 to 23.0 per 100,000, probably related to increasing rates of obesity and diabetes.13 The chance of developing primary hypertension increases with age, although children are being diagnosed with increasing frequency (see Chapter 33). The prevalence of hypertension is higher in Blacks and in those with diabetes. Those who fall into the category called elevated blood pressure (SBP 120 to 129 mm Hg and DBP < 80 mm Hg) are at risk for developing hypertension unless lifestyle modification is instituted. All stages of hypertension are associated with increased risk for target organ disease events, such as myocardial infarction (MI), kidney disease, and stroke.
Table 32.1
Data from Whelton PK, Carey RM, Aronow WS, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension. 2018;71(6):e13–e115.
Most cases (90% to 95%) of hypertension are diagnosed as primary hypertension (also called essential or idiopathic hypertension). Secondary hypertension is caused by an underlying disease process or medication that raises PVR or cardiac output. This form of hypertension accounts for only 5% to 10% of cases.14 Examples include renal vascular or parenchymal disease, adrenocortical tumors, adrenomedullary tumors (pheochromocytoma), and drugs (oral contraceptives, corticosteroids, antihistamines). If the cause is identified and removed before permanent structural changes occur, blood pressure returns to normal.
Primary Hypertension
A specific cause for primary hypertension has not been identified. Primary hypertension is the result of a complicated interaction of genetics and the environment mediated by a host of neurohumoral effects that influence intravascular volume and PVR (Algorithm 32.1). Genetic predisposition to hypertension is thought to be polygenic and associated with epigenetic changes influenced by diet and lifestyle. Genetic risks include defects in renal sodium excretion, insulin sensitivity, activity of the sympathetic nervous system (SNS) and the renin-angiotensin-aldosterone system (RAAS), and cell membrane sodium or calcium transport. Risk factors for primary hypertension relate to age, sex, race, and dietary factors (see Risk Factors: Primary Hypertension). Many of these factors are also risk factors for other cardiovascular disorders. In fact, obesity, hypertension, dyslipidemia, and glucose intolerance often are found together in a condition called the metabolic syndrome (see Chapter 22).
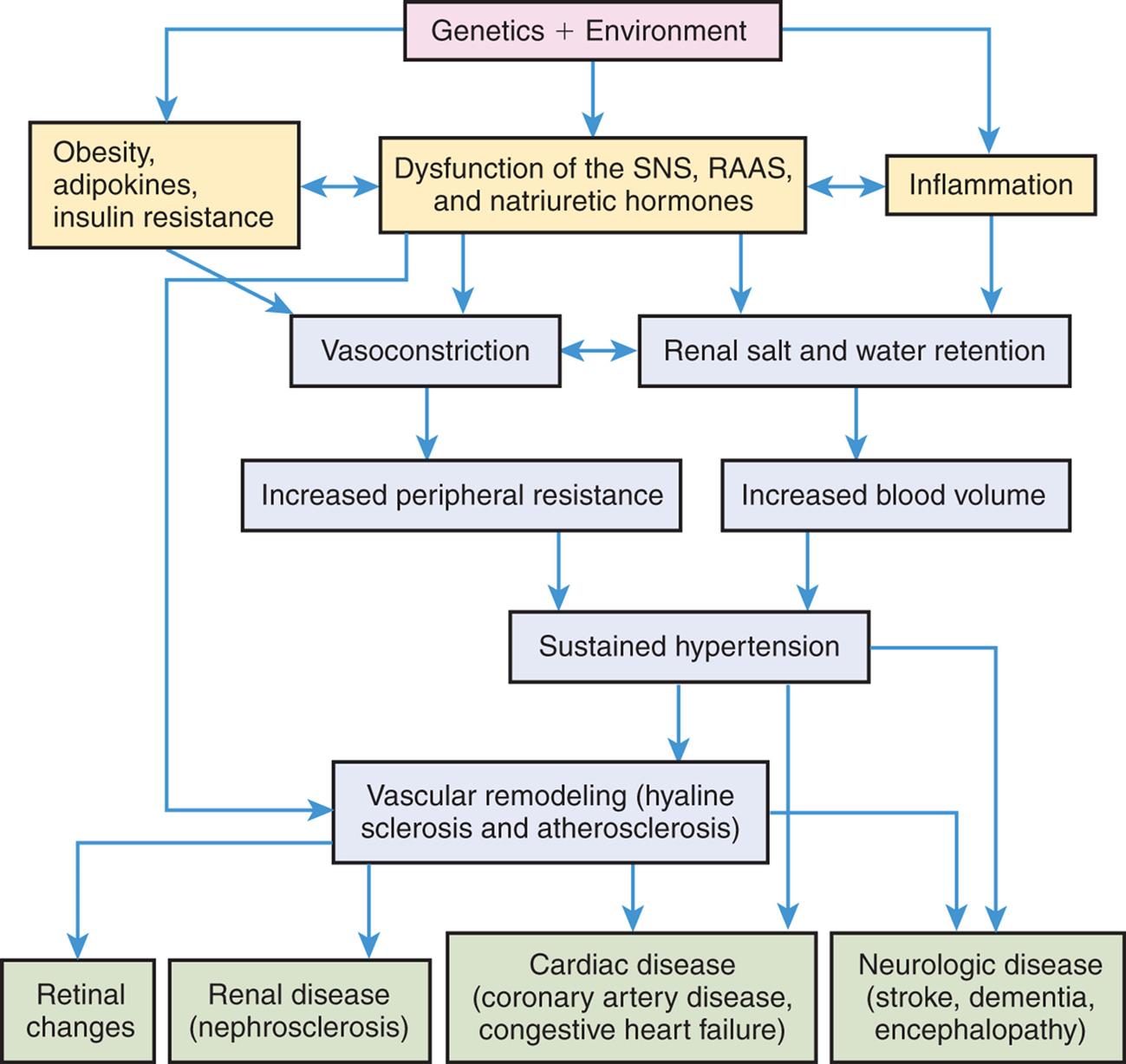
Numerous genetic vulnerabilities have been linked to hypertension and these, in combination with environmental risks, cause neurohumoral dysfunction (sympathetic nervous system [SNS], renin-angiotensin-aldosterone [RAA] system, natriuretic hormones) and promote inflammation and insulin resistance. Insulin resistance, obesity-related adipokine alterations, and neurohumoral dysfunction contribute to sustained systemic vasoconstriction and increased peripheral vascular resistance. Inflammation contributes to renal dysfunction, which, in combination with the neurohumoral alterations, results in renal salt and water retention and increased blood volume. Increased peripheral vascular resistance and increased blood volume are two primary causes of sustained hypertension. Sustained hypertension leads to blood vessel sclerosis and associated damage to the retina, kidney, heart, and brain.
A flowchart shows the pathophysiology of hypertension. 1. Genetics and environment. Leads to 2, 3, and 4. 2. Obesity, adipokines, and insulin resistance. Interrelated with 3. Leads to 5. 3. Dysfunction of the S N S, R A A S, and natriuretic hormones. Interrelated with 4. Leads to 5, 6, and 10. 4. Inflammation. Leads to 6. 5. Vasoconstriction. Interrelated with 6. Leads to 7. 6. Renal salt and water retention. Leads to 8. 7. Increased peripheral resistance. Leads to 9. 8. Increased blood volume. Leads to 9. 9. Sustained hypertension. Leads to 10, 13, and 14. 10. Vascular remodeling (hyaline sclerosis and atherosclerosis). Leads to 11, 12, 13, and 14. 11. Retinal changes. 12. Renal disease (nephrosclerosis). 13. Cardiac disease (coronary artery disease, congestive heart failure). 14. Neurologic disease (stroke dementia, encephalopathy).
Pathophysiology
Multiple mechanisms contribute to the pathophysiology of hypertension including changes in the SNS, the RAAS, and natriuretic peptides. Inflammation, endothelial dysfunction, obesity-related hormones, and insulin resistance also contribute. Increased vascular volume is related to a decrease in renal excretion of salt, often referred to as a shift in the pressure-natriuresis relationship (Fig. 32.3). This means that for a given blood pressure, individuals with hypertension tend to secrete less salt in their urine.
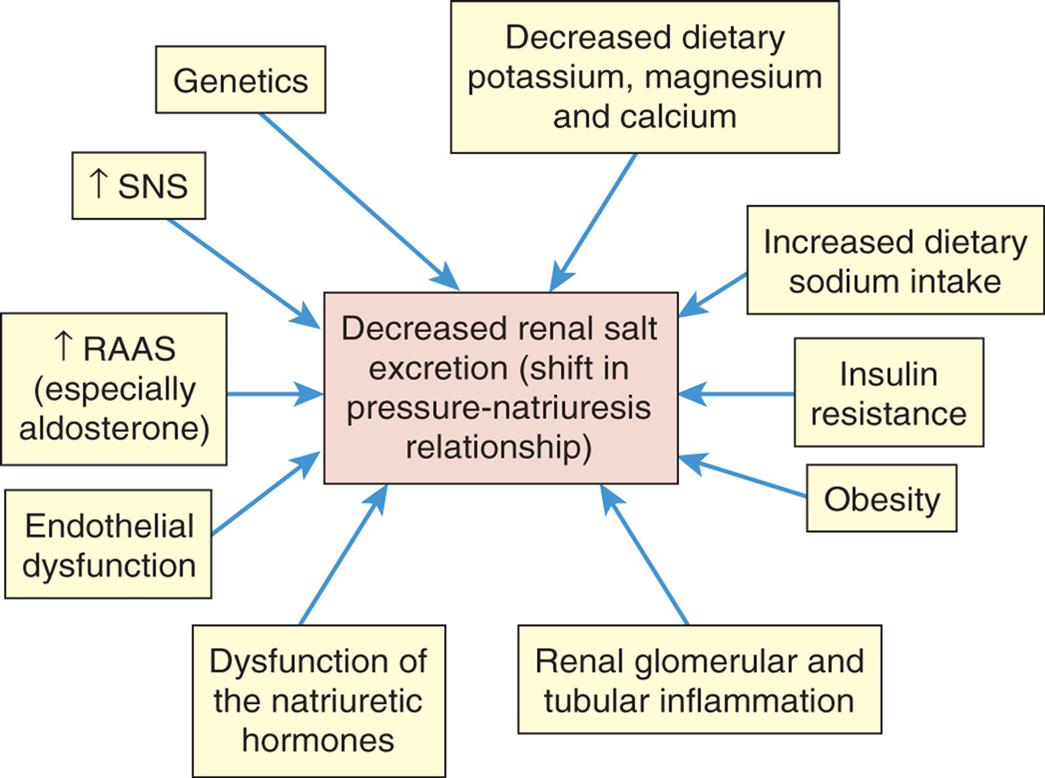
Numerous factors have been implicated in the pathogenesis of sodium retention in individuals with hypertension. These factors cause less renal excretion of salt than would normally occur with increased blood pressure. This is called a shift in the pressure-natriuresis relationship and is thought to be a central process in the pathogenesis of primary hypertension. RAAS, Renin-angiotensin-aldosterone system; SNS, sympathetic nervous system.
An illustration lists the various factors leading to decreased renal salt (shift in pressure-natriuresis relationship). • Increased dietary sodium intake. • Insulin resistance. • Obesity • Renal glomerular and tubular inflammation. • Dysfunction of the natriuretic hormones. • Endothelial dysfunction. • Increased R A A S (especially aldosterone). • Increased S N S. • Genetics. • Decreased dietary potassium, magnesium, and calcium.
As described in Algorithm 32.1, genetic and environmental risks lead to changes in neurohormones, insulin resistance, and inflammation. Obesity contributes to these changes through changes in adipokines and increased inflammation. The combination of all of these factors cause sodium and water retention and peripheral vasoconstriction leading to sustained hypertension and organ damage.
Increased SNS activity causes accelerated heart rate and systemic vasoconstriction. These changes increase both cardiac output and peripheral vascular resistance, thus raising the blood pressure. Additional mechanisms of SNS-induced hypertension include structural changes in blood vessels (vascular remodeling), renal sodium retention (shift in pressure-natriuresis curve), insulin resistance, increased renin and angiotensin levels, and procoagulant effects.
In hypertensive individuals, overactivity of the classical pathway of the RAAS directly causes salt and water retention and increased vascular resistance (Algorithm 32.2). This RAAS pathway begins when angiotensinogen is synthesized in the liver and is released into the blood. There it is cleaved to angiotensin I (Ang I) by renin which is secreted from the juxtaglomerular apparatus (JGA) in the kidney. Angiotensin-converting enzyme 1 (ACE1) in the lung and in tissues catalyzes the formation of angiotensin II (Ang II). Ang II binds with several receptors, the most important of which are AT1 and AT2. AT1 receptor binding results in vasoconstriction (increases vascular resistance) and aldosterone secretion by the adrenal cortex (increases salt and water retention) leading to hypertension and edema. AT1 receptor binding by Ang II also activates the SNS and acts as a growth factor contributing to vascular and myocardial remodeling, inflammation, insulin resistance, and platelet activation. Vascular remodeling is structural change in vessel walls that results in permanent increases in PVR and contributes to atherogenesis. Taken together, these effects contribute to atherosclerosis, ischemic heart disease, myocardial hypertrophy, and heart failure. Medications such as ACE inhibitors, angiotensin receptor blockers (ARBs), and aldosterone blockers oppose the activity of the RAAS and are effective in reducing blood pressure and protecting against target organ damage.
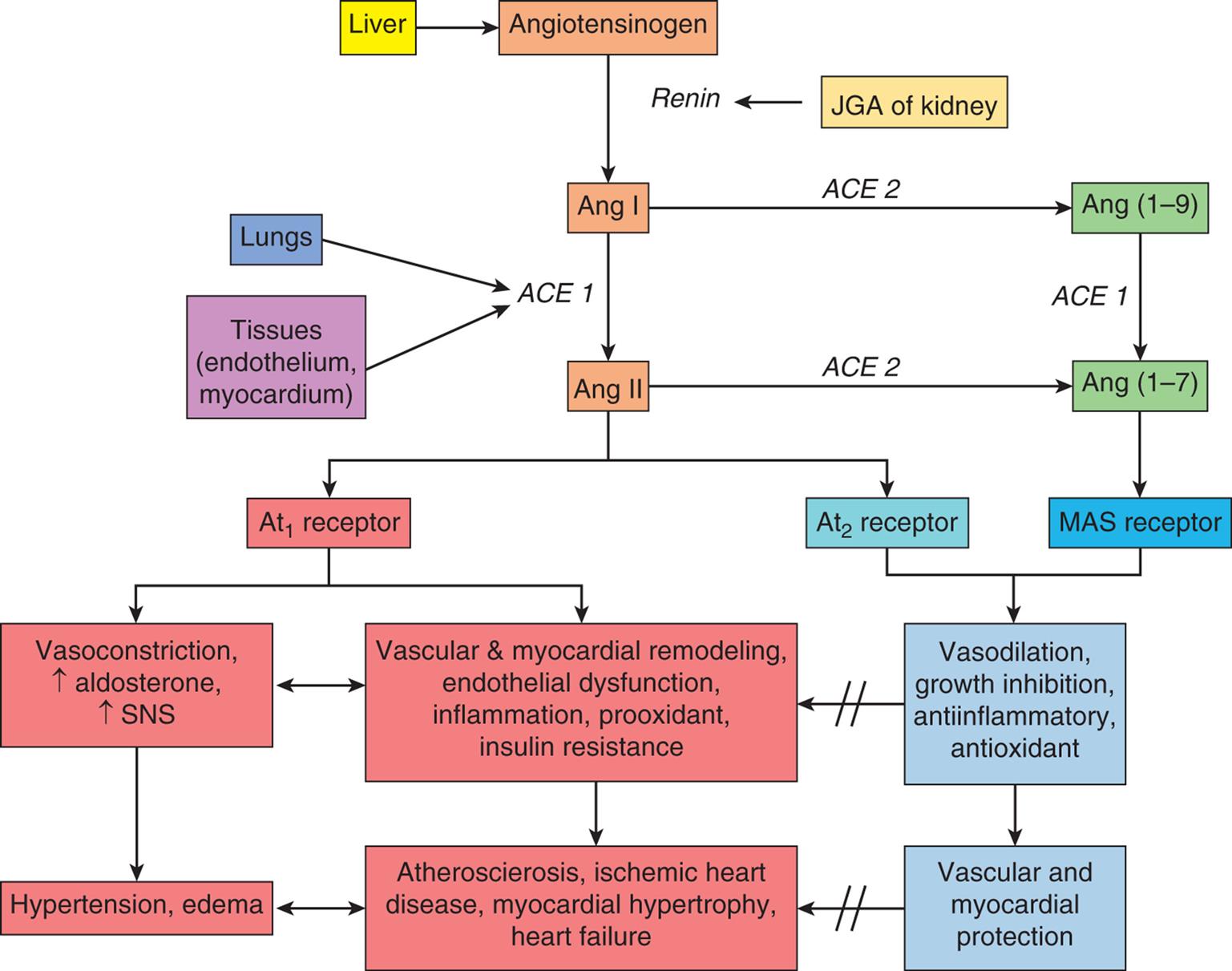
The classical pathway of the renin-angiotensin-aldosterone system (RAAS) begins when angiotensinogen is synthesized in the liver and is released into the blood. There it is cleaved to angiotensin I (Ang I) by renin which is secreted from the juxtaglomerular apparatus (JGA) in the kidney. Angiotensin-converting enzyme 1 (ACE1) in the lung and in tissues catalyzes the formation of angiotensin II (Ang II). Ang II binds with several receptors, the most important of which are AT1 and AT2. AT1 receptor binding results in vasoconstriction (increases vascular resistance) and aldosterone secretion by the adrenal cortex (increases salt and water retention) leading to hypertension and edema. AT1 receptor binding by Ang II also acts as a growth factor leading to vascular and myocardial remodeling as well as inflammation and insulin resistance. Taken together, these effects contribute to atherosclerosis, ischemic heart disease, myocardial hypertrophy and heart failure. In contrast, Ang II binding to the AT2 receptor causes vasodilation, decreased remodeling and has anti-inflammatory and antioxidant effects. Another pathway of the RAAS uses the enzyme ACE2 to form Angiotensin (1–7) from Angiotensin (1–9). Ang (1–7) binds to the MAS receptor and provides additional protection against the negative vascular and myocardial effects of Ang II. SNS, Sympathetic nervous system.
In contrast, when Ang II binds to the AT2 receptor, it functions to oppose the effects of AT1 stimulation. Ang II binding to the AT2 receptor causes vasodilation and decreased remodeling and has anti-inflammatory and antioxidant effects. An imbalance between these two receptor pathways is linked to primary hypertension (see Algorithm 32.2). A second RAAS pathway uses angiotensin converting enzyme 2 (ACE2) to create Ang (1–7) which binds to MAS receptors in vascular, cardiac, and pulmonary tissues. It serves to downregulate AT1 receptors, promotes antihypertensive vasodilation, and reduces cardiovascular remodeling.15 MAS receptor binding by Ang (1–7) also provides cerebrovascular and metabolic protective effects. In addition, the ACE2 pathway is highly expressed in lungs with the ability to mitigate cardiopulmonary diseases such as inflammatory lung disease associated with COVID-19.16 New therapies aimed at potentiating the ACE2 pathway are in development.17
Dysfunction of the natriuretic hormones plays an important role in the pathogenesis of hypertension. These hormones include atrial natriuretic peptide (ANP), B-type natriuretic peptide (BNP), C-type natriuretic peptide (CNP), and urodilatin. Together they modulate renal sodium (Na+) excretion and require adequate potassium, calcium, and magnesium intake to function properly. ANP and BNP are released when there is mechanical stretch of the myocardium. After binding to the natriuretic receptor, they stimulate salt and water loss by the kidney (natriuresis), contribute to arteriolar vasodilation, and reduce RAAS activation.18 Dysfunction of these hormones, along with alterations in the RAA system and the SNS, causes a shift in the pressure-natriuresis relationship leading to increased blood volume and blood pressure. Decreased activity of the natriuretic peptides also is linked to vascular and cardiac remodeling. With inadequate natriuretic function, there is a compensatory increase in natriuretic peptide serum levels. High levels of these peptides therefore indicate dysfunction and are linked to an increased risk for ventricular hypertrophy, atherosclerosis, and heart failure in individuals with hypertension. Salt restriction combined with adequate intake of dietary potassium, magnesium, and calcium improves natriuretic peptide function. Diuretics promote renal salt and water excretion and are a mainstay of hypertensive treatment. Drugs that block the degradation of the natriuretic peptides by blocking the enzyme neprilysin are currently used in combination with angiotensin receptor blockers (ARNi) (e.g., sacubitril/valsartan) for the treatment of heart failure, and are being evaluated for the treatment of hypertension, but may be associated with decreased cognitive function.19,20
Innate and adaptive immunity with associated inflammation play a role in the pathogenesis of hypertension. Activation of immunity results in chronic inflammation with damage to endothelial cells, decreased production of vasodilators (such as nitric oxide), vascular remodeling, and smooth muscle contraction. The contributions of diet, obesity, insulin resistance, and activation of the RAAS to the development of hypertension are likely to be mediated in part by increased systemic inflammation.21 Neuroinflammation is linked to increased SNS activity.22 Inflammation also contributes to insulin resistance, decreased natriuresis, and autonomic dysfunction.
Obesity accounts for 65% to 75% of primary hypertension.23 Obesity and increased caloric intake contribute to adipocyte dysfunction and ectopic fat deposition throughout the cardiovascular system. Adipocytes secrete adipokines, including leptin and adiponectin. The primary function of leptin is to interact with the hypothalamus to control body weight through appetite inhibition and increased metabolic rate (see Chapter 23). Adiponectin is a protein produced by adipose tissue but is reduced in obesity. With obesity, increased leptin and decreased adiponectin have been found to increase sympathetic nervous system and renin-angiotensin-aldosterone system activity, contribute to insulin resistance, decrease renal sodium excretion, promote inflammation, and stimulate myocyte hypertrophy. Other adipokines that are altered in obesity-related cardiovascular diseases include resistin, omentin, visfatin, and perivascular adipose tissue–derived relaxing factor. Obesity also is linked with endothelial dysfunction which contributes to vasoconstriction and arterial remodeling. Taken together, these obesity-related changes result in vasoconstriction, salt and water retention, and renal dysfunction that contribute to the development of hypertension. Weight loss is an essential treatment for obesity-related hypertension. In severe obesity, bariatric surgery has been shown to cause long-standing remission of hypertension in many individuals, although those with severe hypertension requiring multiple medications are less likely to benefit.24,25
Insulin resistance is common in hypertension, even in individuals without clinical diabetes. Insulin resistance is associated with decreased endothelial release of nitric oxide and other vasodilators. It also affects renal function and causes renal salt and water retention. Insulin resistance promotes overactivity of the SNS and RAAS. The interactions among obesity, hypertension, insulin resistance, and lipid disorders in metabolic syndrome result in a high risk of cardiovascular disease.26
Given the complexity of all the factors that contribute to its pathophysiology, primary hypertension is now being considered a metabolic disease that results from the interaction of genes, diet, neurohormones, adipokines, immune cytokines, and gut microbiota (see Emerging Science Box: Hypertension as a Metabolic Disease). These discoveries are leading to new approaches to hypertension
Complicated hypertension
As hypertension becomes more severe and chronic, tissue damage can occur in the blood vessels and tissues leading to target organ damage in the heart, kidney, brain, and eyes. Cardiovascular complications of sustained hypertension include left ventricular hypertrophy, angina pectoris, heart failure, coronary artery disease, myocardial infarction, and sudden death.14 Myocardial hypertrophy is mediated by the SNS and RAAS. Hypertrophy is characterized by a myocardium that is thickened, scarred, and less able to relax during diastole, leading to heart failure with preserved ejection fraction. Over time, the increased size of the heart muscle increases demand for oxygen delivery, the contractility of the heart is impaired, and the individual is at risk for myocardial infarction and heart failure with reduced ejection fraction. Vascular complications include hyaline sclerosis and accelerated atherosclerosis that can affect perfusion to any vascular bed. Hypertension also can contribute to the formation, dissection, and rupture of aneurysms (outpouchings in vessel walls). Renal manifestations of complicated hypertension include nephrosclerosis, renal arteriosclerosis, and renal insufficiency or failure. Microalbuminuria (small amounts of protein in the urine) occurs in many individuals with HTN and is now recognized as an early sign of impending renal dysfunction and increased risk for cardiovascular events. Complications specific to the retina include retinal vascular sclerosis, exudation, and hemorrhage. Cerebrovascular complications include transient ischemia, stroke, cerebral thrombosis, aneurysm, hemorrhage, and dementia. The pathologic effects of complicated hypertension are summarized in Table 32.2.
Table 32.2
RAAS, Renin-angiotensin-aldosterone system; SNS, sympathetic nervous system.
Clinical Manifestations
The early stages of hypertension have no clinical manifestations other than elevated blood pressure; for this reason, hypertension is called a silent disease. Some hypertensive individuals never develop signs, symptoms, or complications, whereas others become very ill, and hypertension can be a cause of death. If elevated blood pressure is not detected and treated, it becomes established, setting the stage for the complications of hypertension that begin to appear during the fourth, fifth, and sixth decades of life.
Most clinical manifestations of hypertensive disease are caused by complications that damage organs and tissues outside the vascular system. Besides elevated blood pressure, the signs and symptoms therefore tend to be specific for the organs or tissues affected. Evidence of heart disease, renal insufficiency, central nervous system dysfunction, impaired vision, impaired mobility, vascular occlusion, or edema can all be caused by sustained hypertension.
Evaluation and Treatment
Diagnosis of hypertension requires the measurement of blood pressure on at least two separate occasions, averaging two readings at least 2 minutes apart, with the following conditions: the person is seated, the arm is supported at heart level, the person must be at rest for at least 5 minutes, and the person should not have smoked or ingested any caffeine in the previous 30 minutes. Diagnostic tests for further evaluation of hypertension may include ambulatory 24-hour blood pressure monitoring to detect night-time blood pressure variations and the presence of white-coat or masked hypertension (blood pressures that are elevated or normal only during office blood pressure measurement) in selected individuals.27 Other evaluative studies should include measurement of electrolytes, glucose, and lipids, and an electrocardiogram (ECG). Individuals who have elevated blood pressure are assumed to have primary hypertension unless their history, physical examination, or initial diagnostic screening indicates secondary hypertension. Once the diagnosis is made, a careful evaluation for other cardiovascular risk factors and for end-organ damage should be done (e.g., echocardiography, carotid ultrasound, renal evaluation).
Treatment of primary hypertension depends on its severity. Management begins with lifestyle modification including exercise, and dietary modifications including reducing salt intake, smoking cessation, and weight loss. In 2021 the American College of Cardiology recommended that individuals with Stage 1 hypertension who are at low risk for cardiovascular disease but who do not respond to lifestyle modification should consider beginning pharmacologic therapy.28 Pharmacologic treatment is recommended for individuals with Stage 1 hypertension who have existing or are at high risk for atherosclerotic cardiovascular disease, and for those who have Stage 2 hypertension. The 2017 American College of Cardiology/American Heart Association guidelines recommend medications including diuretics, ACE inhibitors or ARBs, and calcium channel blockers.27 These guidelines were largely affirmed by the International Society of Hypertension in 2020.29 The choice of medications depends upon several factors including race and ethnicity, gender, age, and comorbidities such as heart disease, diabetes, and renal disease. Combinations of different types of antihypertensive medications may be indicated. Careful follow-up to support continued adherence, determine the response, and monitor for potential side effects of these medications is important. Other therapies for selected individuals include mineralocorticoid receptor antagonists and renal denervation.
Treatment goals include returning blood pressure to normotensive levels in most individuals, which can reduce the risk for stroke, dementia, and cardiovascular complications.27,30 Older adults with hypertension also benefit from blood pressure reduction treatments but should be managed carefully with treatment goals adjusted by age and underlying comorbidities.27
Hypertensive crisis
Hypertensive crisis (malignant hypertension) is rapidly progressive hypertension in which systolic pressure is ≥180 mm Hg and/or diastolic pressure is ≥120 mm Hg and is associated with advanced bilateral retinopathy, encephalopathy, or microangiopathy.27 It can occur in those with primary hypertension, but the reason some people develop this complication and others do not is unknown. Other causes include complications of pregnancy, cocaine or amphetamine use, reaction to certain medications, adrenal tumors, and alcohol withdrawal. High arterial pressure renders the cerebral arterioles incapable of regulating blood flow to the cerebral capillary beds. High hydrostatic pressures in the capillaries cause vascular fluid to exude into the interstitial space. Retina exhibit hemorrhages, cotton wool spots, and papilledema. If blood pressure is not reduced, cerebral edema and cerebral dysfunction (encephalopathy) increase until death occurs. Besides encephalopathy, hypertensive crisis can cause hemolysis and thrombocytopenia, angiopathy, myocardial infarction, cardiac failure with pulmonary edema, aortic dissection, uremia, and cerebrovascular accident (stroke) and is considered a medical emergency. A rapid evaluation for underlying cause and cardiovascular and neurologic complications is indicated, with rapid institution of medications such as beta- or alpha-blockers, calcium channel blockers, and nitrates.27 It is likely that in many cases, individuals who present with hypertensive crisis have poorly controlled underlying chronic hypertension, and careful follow-up and management is crucial.
Orthostatic (Postural) Hypotension
The term orthostatic (postural) hypotension (OH) refers to a decrease in SBP of at least 20 mm Hg or a decrease in DBP of at least 10 mm Hg within 3 minutes of moving to a standing position. It is a common condition in individuals accessing community and primary care clinics (prevalence 17% to 19%) and affects nearly one third of individuals in residential care or nursing homes, especially those with dementia and Parkinson disease.31 OH affects men more often than women, and usually occurs between the ages of 40 and 70 years. It is a significant risk factor for falls and associated injury and for increased mortality.
OH is often associated with disorders that affect autonomic nervous function. Normally when an individual stands, the gravitational changes on the circulation are compensated by a baroreceptor-mediated reflex that stimulates the SNS. This causes arteriolar and venous constriction and increased heart rate upon standing. Other compensatory mechanisms include mechanical factors, such as the closure of valves in the venous system, contraction of the leg muscles, and a decrease in intrathoracic pressure. These mechanisms are dysfunctional or inadequate in individuals with orthostatic hypotension; consequently, upon standing, blood pools in the lower extremities and normal arterial pressure cannot be maintained.
Orthostatic hypotension may be acute or chronic. Acute orthostatic hypotension is common in older adults and occurs when the normal regulatory mechanisms are inadequate as a result of (1) altered body chemistry, (2) drug action (e.g., antihypertensives, antidepressants), (3) prolonged immobility, (4) starvation, (5) physical exhaustion, (6) volume depletion (e.g., dehydration, diuresis, potassium or sodium depletion), or (7) any condition that results in venous pooling (e.g., pregnancy, extensive varicosities of the lower extremities).
Chronic orthostatic hypotension may be (1) secondary to a specific disease or (2) primary (idiopathic). Secondary causes include neurogenic OH which results from diseases of the central or peripheral nervous systems that affect autonomic function (e.g., spinal cord injury, Parkinson disease, multiple system atrophy, intracranial tumors, cerebral infarcts, Wernicke encephalopathy, and peripheral neuropathies). Other causes of secondary OH are adrenal insufficiency and metabolic disorders (e.g., diabetes, porphyria). Cardiovascular autonomic neuropathy is a common cause of OH in persons with diabetes affecting up to 20% of diabetic individuals and is often overlooked.32 Chronic OH also can occur in those taking antihypertensive medications. It is interesting to note that although OH is common in older individuals being treated for hypertension, those who undergo intensive treatment and achieve good control of their systolic blood pressure are less likely to develop OH than those who are treated less intensively, likely due to favorable effects of improved blood pressure on autonomic function.33 Many other medications also can cause OH including nitrates, antidepressants, phosphodiesterase inhibitors, and dopamine agonists.32
OH can be asymptomatic. In those with symptoms, it often is characterized by dizziness, blurring or loss of vision, and syncope. In neurogenic OH, symptoms often worsen during exercise and after meals. When possible, acute OH and secondary chronic OH are managed by correction of the underlying condition. Chronic primary OH and irreversible secondary OH are managed with a combination of nondrug and drug therapies.34 Nonpharmacologic interventions include avoidance of caffeine and alcohol, increased fluid and salt intake, frequent small low-glycemic meals, sleeping with the head of the bed raised, and waist-high stockings or abdominal binders. Pharmacologic management includes mineralocorticoids (e.g., fludrocortisone) and vasoconstrictors (e.g., midodrine, droxidopa). Norepinephrine reuptake inhibitors are being studied.32
Aneurysm
An aneurysm is a localized dilation or outpouching of a vessel wall or cardiac chamber. True aneurysms involve weakening in all three layers of the arterial wall (Fig. 32.4A). Most are fusiform and circumferential, whereas saccular aneurysms are basically spherical in shape. False aneurysms are an extravascular hematoma that communicates with the intravascular space. A common cause of this type of lesion is a leak between a vascular graft and a natural artery.
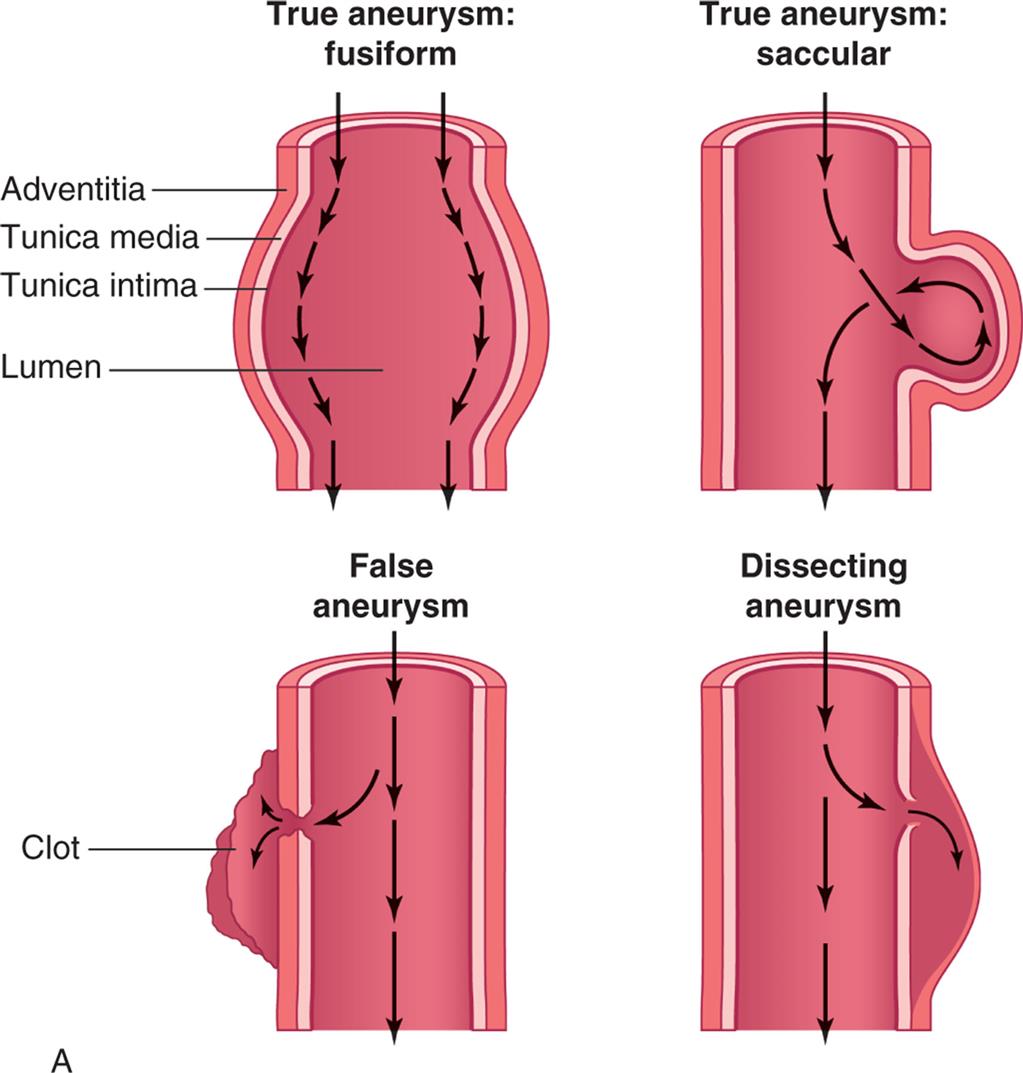
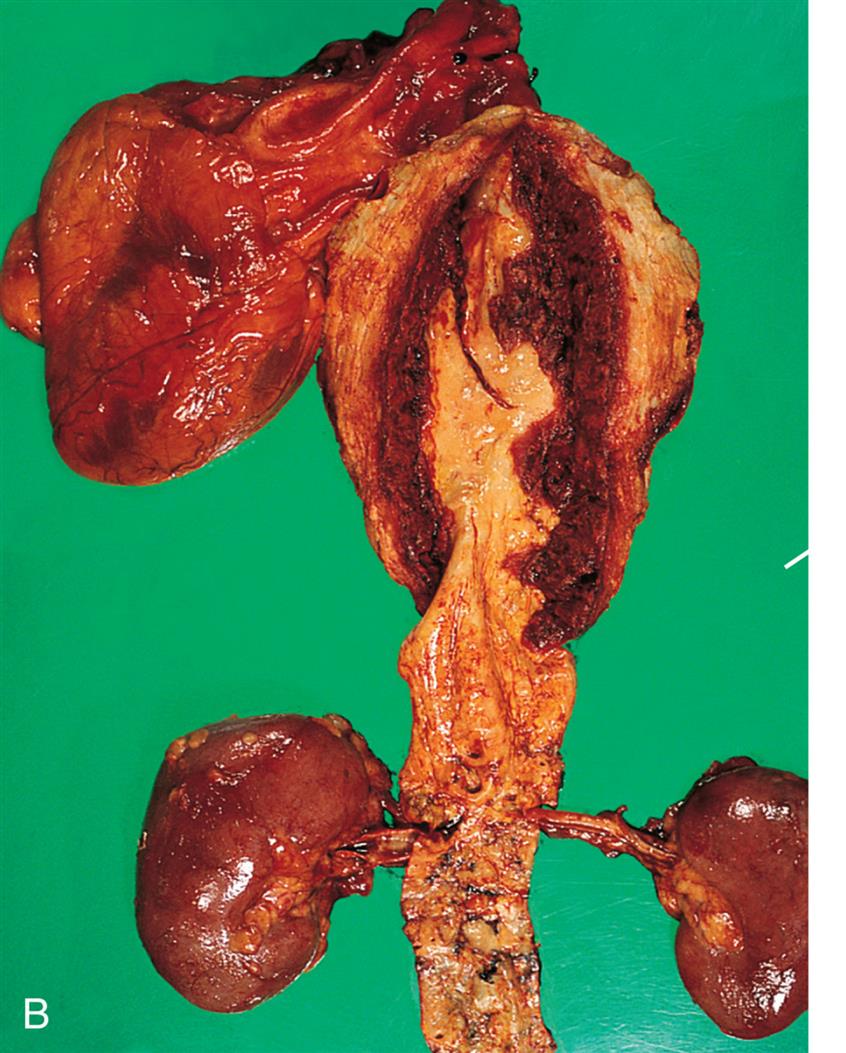
(A) True aneurysms are caused by bulging of all three layers of the vessel wall and can be circumferential (fusiform) or can form a bulge on one side of the vessel (saccular). False aneurysms are a collection of clotted blood just outside of the vessel due to a break all the way through the vessel wall, usually caused by trauma. Dissecting aneurysms result from a partial break in the vessel wall such that blood is pushed between the tunica media and the adventitia. (B) Dissecting aneurysm of thoracic aorta (arrow). (B, From Damjanov I, Linder J, eds. Anderson’s pathology, 10th edition. St. Louis: Mosby; 1996.)
Left panel, A. True aneurysm: fusiform. Illustration shows labels for adventitia, tunica media, tunica intima, and lumen. Arrows along the inner walls represent the flow. True aneurysm: saccular. Illustration shows bulging of outer three layers on one side. Arrows, representing the flow, are traced along the wall, looping through the bulge, and flowing further down. False aneurysm. Illustration shows formation of clot. Arrows along the center of the longitudinal section represent direction of flow. Part of the flow also enters the clot. Dissecting aneurysm. Illustration shows broken tunica media and tunica intima on one side. Arrows along the center of the longitudinal section represent direction of flow. Part of the flow also enters the broken section. Right panel, B. Specimen of thoracic aorta shows enlarged artery with extra growth of tissue.
Vascular aneurysms most commonly occur in the thoracic or abdominal aorta, and in the cerebrovascular system. Major risk factors for aneurysm formation include hypertension and atherosclerosis. Chronic hypertension results in mechanical and shear forces that contribute to inflammation and remodeling resulting in weakening of the vessel wall. Atherosclerotic plaque formation also causes inflammatory changes that erode the vessel wall. Additional risk factors include gene polymorphisms that are linked to the production of growth factors, myosin, and proteases.35 Infections, such as syphilis, collagen disorders (such as Marfan syndrome), and traumatic injury to the chest or abdomen, also can cause aortic aneurysms. There are rare inherited syndromes that are characterized by aneurysm formation at an early age.
The aorta is particularly susceptible to aneurysm formation because of constant stress on the vessel wall and the absence of penetrating vasa vasorum in the media layer. Chronic inflammation of the wall of the aorta is considered the primary cause of aortic aneurysms leading to weakening of the intima and medial layers. Neutrophils, macrophages, and T and B lymphocytes enter the aortic wall, and cytokines produced by these cells cause an inflammatory response. Proteases are released that destroy tissues and contribute to extracellular matrix remodeling, apoptosis and calcification of vascular smooth muscle cells, and decreased wall elasticity.36 These chronic inflammatory changes are potentiated in individuals with atherosclerosis and hypertension, which are found in more than half of all individuals with aneurysms.
Formation of a cardiac ventricular wall aneurysm most often occurs when intraventricular tension stretches noncontracting infarcted muscle. Acutely infarcted myocardium is characterized by a weak and thin layer of necrotic tissue that bulges with each contraction. With time the aneurysm becomes more fibrotic, but continues to bulge with each systole, thus acting as a “reservoir” and reducing stroke volume.
Clinical manifestations of aneurysms depend on where the aneurysm is located. Aortic aneurysms often are asymptomatic until they rupture and then cause severe pain and hypotension. Thoracic aortic aneurysms can cause dysphagia (difficulty swallowing) and dyspnea (breathlessness). An aneurysm that impairs flow to an extremity causes symptoms of ischemia. Cerebral aneurysms, which often occur in the circle of Willis, are associated with signs and symptoms of increased intracranial pressure and stroke. (Cerebral aneurysms are described in Chapter 18.) Aneurysms in the heart present with dysrhythmias, heart failure, and embolism of clots to the brain or other vital organs.
The diagnosis of an aneurysm is usually confirmed by ultrasonography, CT, MRI, or angiography. Medical treatment for small asymptomatic aortic aneurysms includes cessation of smoking, reduction of blood pressure, and initiation of cholesterol lowering medications (statins) and antiplatelet drugs. None of these treatments can fully prevent the progression of these aneurysms, and their growth must be monitored closely. Potential new medical therapies being explored include the diabetes drug metformin, blockers of specific inflammatory pathways, and stem cells.36 For aneurysms that are dilating rapidly or have become large, either open or endovascular surgical repair is indicated and usually includes replacement with a prosthetic graft.37
Aortic aneurysms can be complicated by the acute aortic syndromes, which include aortic dissection, hemorrhage into the vessel wall, or vessel rupture. Dissection of the layers of the arterial wall occurs when there is a tear in the intima and blood is pushed between the tunica media and the adventitia of the wall of the artery (see Fig. 32.4B). Dissections can involve any part of the aorta (ascending, arch, or descending) and can disrupt flow through arterial branches. Symptoms include severe pain in the neck, jaw, chest, back, or abdomen. Emergent evaluation and surgical intervention are critical. Chest CT angiography is the diagnostic technique of choice.38 Surgical intervention is often emergent.
Thrombus Formation in Arteries
As in venous thrombosis, arterial thrombi develops when intravascular conditions promote activation of coagulation or when there is stasis of blood flow. These conditions include those in which there is intimal irritation or roughening (such as in percutaneous or surgical procedures and trauma), inflammation, infection, low intravascular volume and pressures, or obstructions that cause blood stasis and pooling within the vessels. Damage to the endothelium leads to activation of the clotting cascade and platelet adherence. An anatomic change in an artery (such as an aneurysm) can contribute to thrombus formation, particularly if the change results in a pooling of arterial blood. Valvular thrombi are most commonly associated with inflammation of the endocardium (endocarditis) and rheumatic heart disease. Widespread arterial thrombus formation can occur in shock when systemic inflammation activates the intrinsic and extrinsic pathways of coagulation, resulting in microvascular thrombosis throughout the systemic arterial circulation (see Chapter 48).
Arterial thrombi pose two potential threats to the circulation. First, the thrombus may grow large enough to occlude the artery, causing ischemia in tissue supplied by the artery. Second, the thrombus may dislodge, becoming a thromboembolus that travels through the vascular system until it occludes flow into a distal systemic vascular bed.
Diagnosis of arterial thrombi is usually accomplished through the use of Doppler ultrasonography and angiography. Pharmacologic treatment involves the administration of anticoagulants or thrombolytics. A balloon-tipped catheter can be used to remove or compress an arterial thrombus.
Embolism
Embolism is the obstruction of a vessel by an embolus—a bolus of matter circulating in the bloodstream. The embolus may consist of a dislodged thrombus; an air bubble; an aggregate of amniotic fluid; an aggregate of fat, bacteria, or cancer cells; or a foreign substance. The types of emboli are summarized in Table 32.3. Most emboli arise from venous or arterial thrombi and travel in the bloodstream until they reach a vessel through which they cannot pass. Pulmonary emboli originate on the venous system (mostly from the deep veins of the legs) or in the right heart; arterial emboli most commonly originate in the left heart and are associated with thrombus formation associated with MI, valvular disease, left heart failure, endocarditis, and dysrhythmias.
Table 32.3
Embolism causes ischemia or infarction in tissues distal to the obstruction, producing organ dysfunction and pain. Infarction and subsequent necrosis of a central organ are life-threatening. For example, occlusion of a coronary artery will cause an MI whereas occlusion of a cerebral artery causes a stroke (see Chapter 18).
Peripheral Vascular Diseases
Thromboangiitis Obliterans (Buerger Disease)
Thromboangiitis obliterans (Buerger disease) is a highly inflammatory autoimmune disease of the peripheral arteries. It is strongly associated with smoking. Thromboangiitis obliterans is characterized by the formation of thrombi filled with inflammatory and immune cells. Inflammatory cytokines and toxic oxygen free radicals contribute to accompanying vasospasm. Over time, these thrombi become organized and fibrotic and result in permanent occlusion of small- and medium-sized arteries in the feet and sometimes in the hands.
The chief symptom of thromboangiitis obliterans is pain and tenderness of the affected part, usually affecting more than one extremity. Clinical manifestations include rubor (redness of the skin), which is caused by dilated capillaries under the skin, and cyanosis, which is caused by tissue ischemia. Chronic ischemia causes the skin to become thin and shiny and serious ulcers may occur. In advanced disease, profound ischemia of the extremities can cause gangrene necessitating amputation. Thromboangiitis obliterans also has been associated with cerebrovascular disease (stroke), mesenteric disease, and rheumatic symptoms (joint pain).
Diagnosis of thromboangiitis obliterans is made by identification of the following common features—age <45 years, smoking history, evidence of peripheral ischemia—and by exclusion of other causes of arterial insufficiency. The most important part of treatment is cessation of cigarette smoking. Other measures include vasodilators and exercises aimed at improving circulation to the foot or hand. Endovascular procedures can improve outcomes in selected individuals. Stem cell therapy for skin ulcers have been shown to improve healing and symptoms.39,40
Raynaud Phenomenon
Raynaud phenomenon (RP) is characterized by attacks of vasospasm in the small arteries and arterioles of the fingers and, less commonly, the toes. Approximately 90% of individuals with RP have primary Raynaud phenomenon, which is a common vasospastic disorder of unknown origin affecting up to 5% of adults.41 Genetic factors have been implicated. Secondary Raynaud phenomenon is associated with systemic diseases, particularly collagen vascular disease (progressive systemic sclerosis [scleroderma]), vasculitis, malignancy, pulmonary hypertension, chemotherapy, cocaine use, hypothyroidism, thoracic outlet syndrome, trauma, serum sickness, or long-term exposure to environmental conditions such as cold temperatures or vibrating machinery in the workplace. Blood vessels in affected individuals demonstrate dysfunctional autonomic vascular thermoregulation.41 In addition, endothelial dysfunction with an imbalance in endothelium-derived vasodilators (e.g., nitric oxide) and vasoconstrictors (e.g., endothelin-1) is seen. Platelet activation also may play a role. It tends to affect young women and is characterized by vasospastic attacks triggered by brief exposure to cold, vibration, or emotional stress. Genetic predisposition may play a role in its development.
The clinical manifestations of the vasospastic attacks of either disorder are changes in skin color and sensation caused by ischemia. Attacks tend to be bilateral, and manifestations usually begin at the tips of the digits and progress to the proximal phalanges. Vasospasm causes pallor, numbness, and the sensation of coldness in the digits. Sluggish blood flow resulting from ischemia may cause the skin to appear cyanotic. Rubor, throbbing pain, and paresthesias follow as blood flow returns. Skin color returns to normal after the attack, but frequent, prolonged attacks interfere with cellular metabolism, causing the skin of the fingertips to thicken and the nails to become brittle. In severe, chronic Raynaud phenomenon, ischemia can eventually cause ulceration and gangrene.
The diagnosis of RP is based on clinical presentation and nailfold capillaroscopy or infrared thermography. A search for underlying secondary causes should be conducted. Treatment of RP begins with avoidance of stimuli that trigger attacks (e.g., cold temperatures, emotional stress) and cessation of cigarette smoking to eliminate the vasoconstricting effects of nicotine. If attacks of vasospasm become frequent or prolonged, vasodilators (e.g., calcium channel blockers, alpha blockers, ACE inhibitors) are administered.41 Sympathectomy or botulinum injection may be indicated in severe cases.
Atherosclerosis
Arteriosclerosis is a condition characterized by thickening and hardening of the vessel wall. Atherosclerosis is a form of arteriosclerosis that is caused by the accumulation of lipid-laden macrophages within the arterial wall, which leads to the formation of a lesion called a plaque. Atherosclerosis is a pathologic process that can affect vascular systems throughout the body and is the leading cause of peripheral artery disease, CAD, and cerebrovascular disease. (Atherosclerosis of the coronary arteries is described later in this chapter, and atherosclerosis of the cerebral arteries is described in Chapter 18.)
Pathophysiology
Atherosclerosis is an inflammatory disease that begins with injury to the endothelial cells that line artery walls.42 Pathologically, the lesions progress from endothelial injury and dysfunction to fatty streak to fibrotic plaque to complicated lesion (Fig. 32.5). Possible causes of endothelial injury include the common risk factors for atherosclerosis, such as smoking, hypertension, diabetes, increased levels of low-density lipoprotein (LDL), decreased levels of high-density lipoprotein (HDL), and autoimmunity. Other “nontraditional” risk factors include increased serum markers for inflammation and thrombosis (e.g., high-sensitivity C-reactive protein [hs-CRP]), troponin I, adipokines, infection, and air pollution. These risk factors are discussed in more detail in the following section on CAD (see the section on Coronary Artery Disease, Myocardial Ischemia, and Acute Coronary Syndromes).
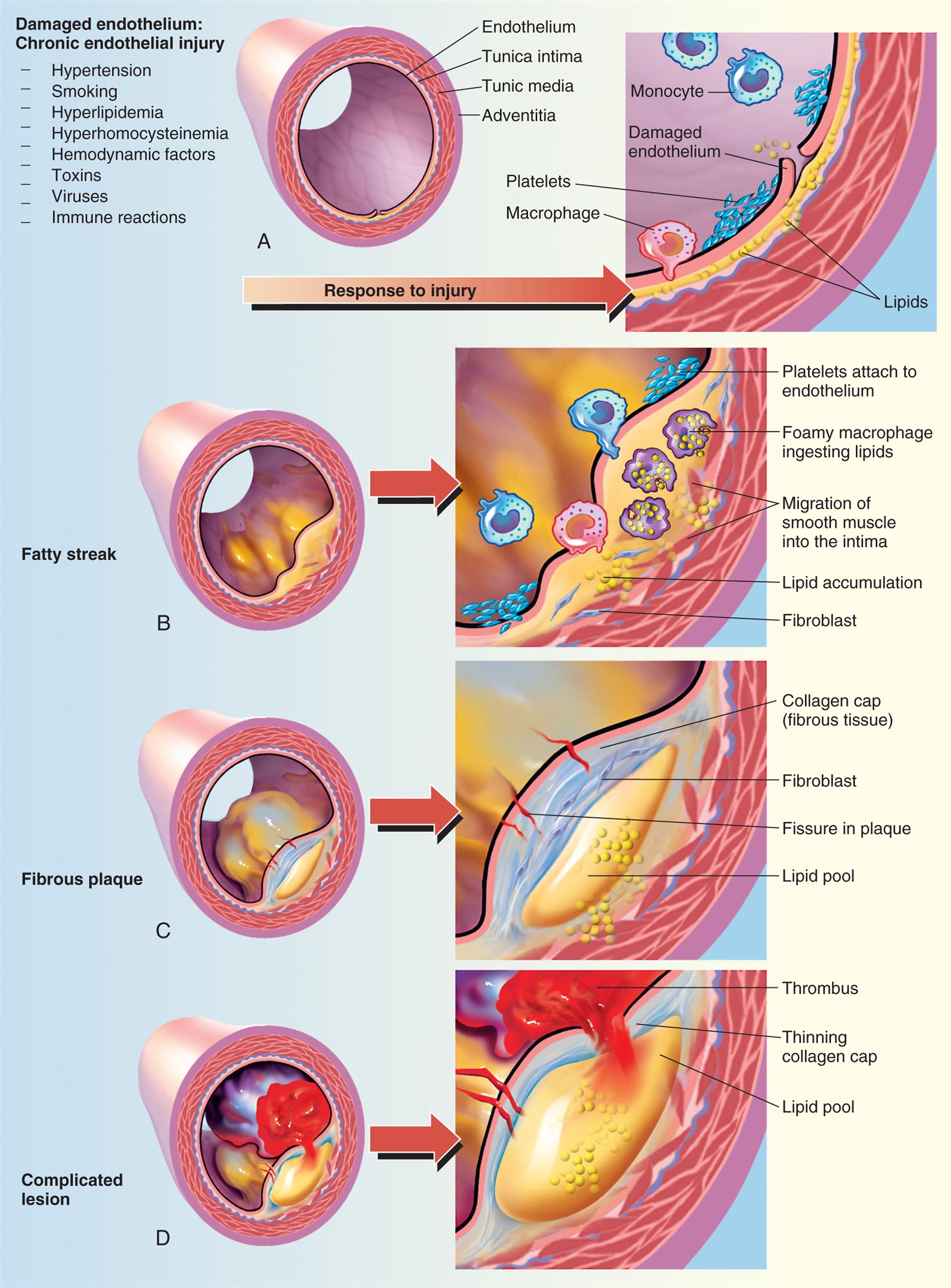
(A) Damaged endothelium. (B) Diagram of fatty streak and lipid core formation (see Fig. 32.6 for a diagram of oxidized low-density lipoprotein [LDL]). (C) Diagram of fibrous plaque. Raised plaques are visible: some are yellow; others are white. (D) Diagram of complicated lesion; thrombus is red; collagen is blue. Plaque is complicated by red thrombus deposition.
Panel a. Illustration of vessel shows labels as follows: endothelium, tunica intima, tunic media, and adventitia. The symptoms of damaged endothelium: chronic endothelial injury are hypertension, smoking, hyperlipidemia, hyperhomocysteinemia, hemodynamic factors, toxins, viruses, and immune reactions. An arrow labeled response to injury points toward a cross-section that shows labels as follows: monocyte, damaged endothelium, platelets, macrophage, and lipids. Panel b. Illustration labeled fatty streak shows buildup of plaque inside endothelium. Cross-section of fatty streak shows labels as follows: platelets attach to endothelium, foamy macrophage ingesting lipids, migration of smooth muscle into the intima, lipid accumulation, and fibroblast. Panel c. Illustration labeled fibrous plaque shows blue and yellow deposits. Cross-section of fibrous plaque shows labels as follows: collagen cap (fibrous tissue), fibroblast, fissure in plaque, and lipid pool. Panel d. Illustration labeled complicated lesion shows build-up of plaque and blood clot. Cross-section of complicated lesion shows labels as follows: thrombus, thinning collagen cap, and lipid pool.
Injured endothelial cells become inflamed. Inflamed endothelial cells cannot make normal amounts of antithrombic and vasodilating cytokines and express adhesion molecules that bind macrophages and other inflammatory and immune cells (Fig. 32.6). Macrophages release numerous inflammatory cytokines (e.g., tumor necrosis factor-alpha [TNF-α], interferons, interleukins, C-reactive protein) and enzymes that further injure the vessel wall.43 Toxic oxygen free radicals generated by the inflammatory process cause oxidation (i.e., addition of oxygen) of LDL that has accumulated in the vessel intima. Oxidized LDL causes additional adhesion molecule expression with the recruitment of monocytes that differentiate into macrophages. These macrophages penetrate into the intima, where they engulf oxidized LDL, and are then called foam cells. When they accumulate in significant amounts, they form a lesion called a fatty streak (see Figs. 32.6 and 32.7). Once formed, fatty streaks produce more toxic oxygen free radicals, and secrete additional inflammatory mediators resulting in progressive damage to the vessel wall. In addition, oxidized LDL and foam cells serve as damage-associated molecular patterns (DAMPs; see Chapter 7) and activate macrophage release of inflammatory cytokines and recruit autoreactive T cells leading to autoimmune vascular injury.44–46 Selected lipid lowering and antihypertensive medications may improve atherosclerotic disease progression through their antiinflammatory effects (see Emerging Science Box: Lipid-Lowering and Antihypertensive Medication Effects on Atherosclerosis-Associated Inflammation). Increasing understanding of the pivotal role of inflammation and autoimmunity in the pathogenesis of atherosclerosis has led to significant research into potential antiinflammatory treatments.42,47
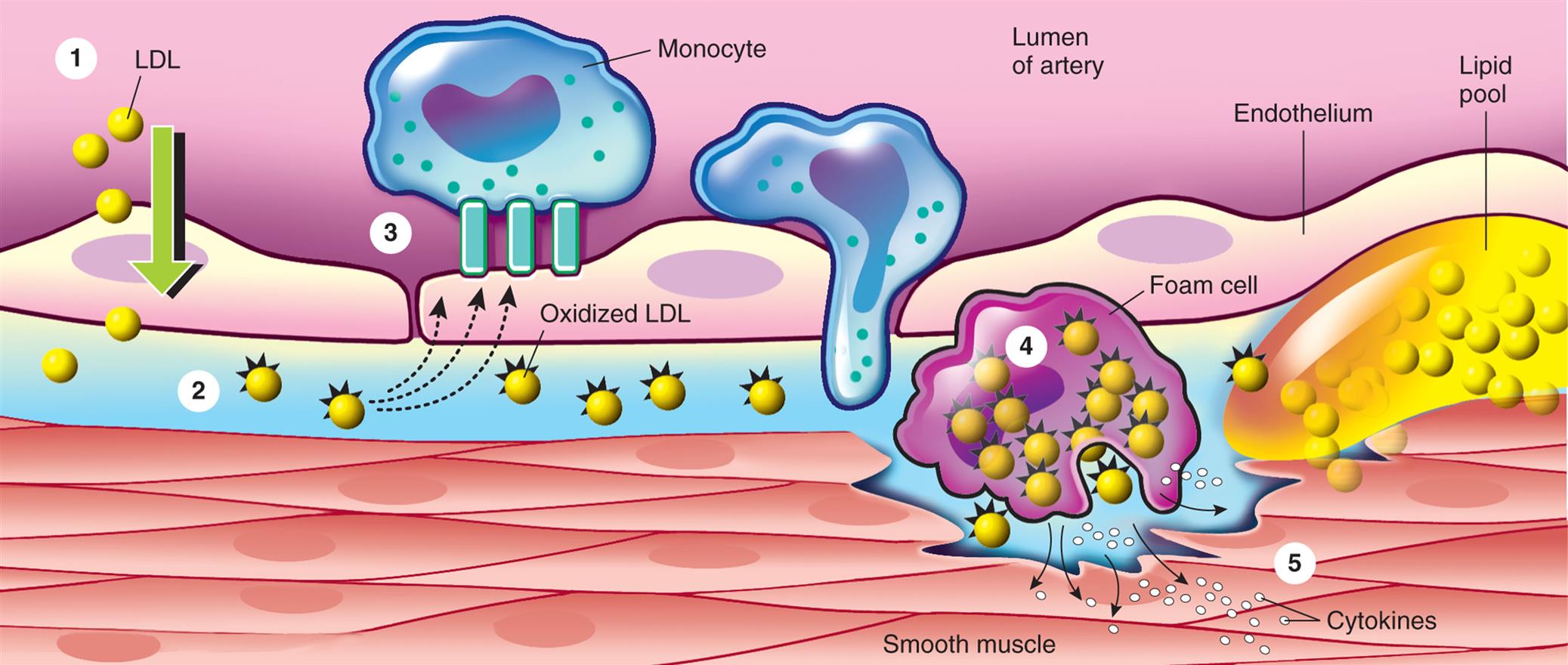
(1) Low-density lipoprotein (LDL) enters the arterial intima through an intact endothelium. (2) In hypercholesterolemia, the influx of LDL exceeds the eliminating capacity and an extracellular pool of LDL is formed. (3) Intimal LDL oxidized through the action of free oxygen radicals generates proinflammatory cytokines that induce endothelial expression of the adhesion molecules. Monocytes bind to the adhesion molecules and differentiate into macrophages, which then (4) internalize oxidized LDL and become foam cells. (5) Foam cells accumulate forming a fatty streak and release many inflammatory cytokines that damage the vessel wall. (Modified from Crawford MH, Dim Arco JP, Paulus WJ. Cardiology, 3rd edition. London: Mosby; 2010.)
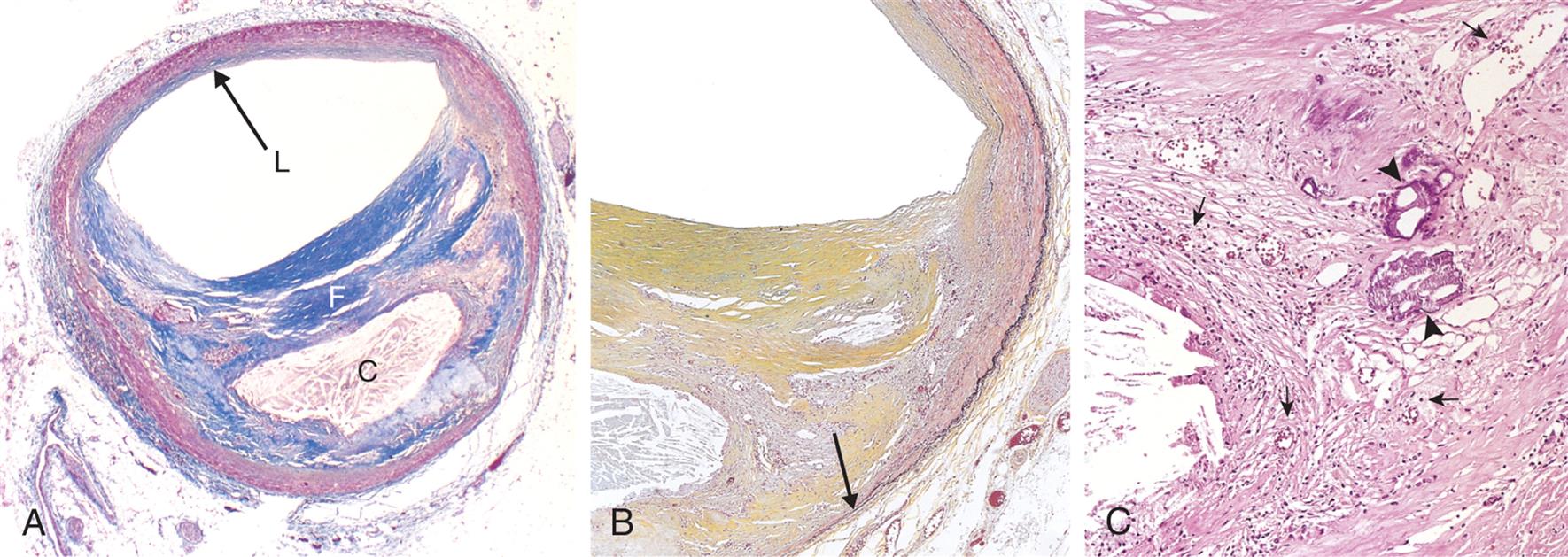
(A) Overall architecture demonstrating fibrous cap (F) and a central necrotic (largely lipid) core (C). The lumen (L) has been moderately narrowed. Note that a segment of the wall is plaque free (arrow), so that there is an eccentric lesion. In this section, collagen has been stained blue (Masson trichrome stain). (B) Higher power photograph of a section of the plaque shown in (A) stained for elastin (black), demonstrating that the internal and external elastic membranes are destroyed and the media of the artery is thinned under the most advanced plaque (arrow). (C) Higher magnification photomicrograph at the junction of the fibrous cap and core, showing scattered inflammatory cells, calcification (arrowhead), and neovascularization (small arrows). (From Kumar V, Abbas A, Aster J. Robbins basic pathology, 9th edition. St. Louis: Saunders; 2007.)
Macrophages also release growth factors that stimulate smooth muscle cell proliferation. Smooth muscle cells in the region of endothelial injury proliferate, produce collagen, and migrate over the fatty streak, forming an atherosclerotic plaque (see Fig. 32.7).48 The plaque may calcify, protrude into the vessel lumen, and obstruct blood flow to distal tissues (especially during exercise), which may cause symptoms (e.g., angina or intermittent claudication).
Many plaques are “unstable,” meaning they are prone to rupture. These plaques are clinically silent and do not affect luminal blood flow significantly until they erode and rupture (see the section on Coronary Artery Disease, Myocardial Ischemia, and Acute Coronary Syndromes). Rupture of unstable plaques occurs due to the degradative effects of inflammatory cytokines and enzymes, wall stress, and neurohumoral changes.49 Plaques that have ruptured are called complicated plaques. Once rupture occurs, exposure of underlying tissue results in platelet adhesion, initiation of the clotting cascade, and rapid thrombus formation. The thrombus may suddenly occlude the affected vessel, resulting in ischemia and infarction. Aspirin or other antithrombotic agents are used to prevent this complication of atherosclerotic disease.
Clinical Manifestations
Atherosclerosis presents with symptoms and signs that result from inadequate perfusion of tissues because of obstruction of the vessels that supply them. Partial vessel obstruction may lead to transient ischemic events, often associated with exercise or stress. As the lesion becomes complicated, increasing obstruction with superimposed thrombosis may result in tissue infarction. Obstruction of peripheral arteries can cause significant pain and disability. CAD caused by atherosclerosis is the major cause of myocardial ischemia. Atherosclerotic obstruction of the vessels supplying the brain is the major cause of stroke. Often, more than one vessel will become involved with this disease process such that an individual may present with symptoms from several ischemic tissues at the same time, and disease in one area may indicate that the individual is at risk for ischemic complications elsewhere.
Evaluation and Treatment
In evaluating individuals for the presence of atherosclerosis, obtaining a complete health history (including risk factors and symptoms of ischemia) is essential. Physical examination may reveal arterial bruits and evidence of decreased blood flow to tissues. Laboratory data that include measurements of levels of lipids, blood glucose, and hs-CRP are also indicated. Judicious use of x-ray films, electrocardiography, ultrasonography, nuclear scanning, CT, MRI, and angiography may be necessary to identify affected vessels, particularly coronary vessels.
Current management of atherosclerosis is focused on detection and treatment of preclinical lesions with drugs aimed at stabilizing and reversing plaques before they rupture. Once a lesion obstructs blood flow, the primary goal in the management of atherosclerosis is to restore adequate blood flow to the affected tissues. If an individual has presented with acute ischemia (e.g., MI, stroke), interventions are specific to the diseased area (discussed further under those topics). In situations in which the disease process does not require immediate intervention, management focuses on reduction of risk factors and prevention of plaque progression. This includes implementation of an exercise program, cessation of smoking, and control of contributing factors such as hypertension, diabetes, and dyslipidemia. Management of atherosclerotic risk factors is discussed further in the Coronary Artery Disease, Myocardial Ischemia, and Acute Coronary Syndromes section.
Peripheral Artery Disease
Peripheral artery disease (PAD) refers to atherosclerotic disease of arteries that perfuse the limbs, especially the lower extremities. PAD affects an estimated 6.5 million Americans aged > 40 years.12 The risk factors for PAD are the same as those previously described for atherosclerosis. It is especially prevalent in smokers and older adults with diabetes.
Lower extremity ischemia resulting from arterial obstruction in PAD can be gradual or acute. In most individuals, gradually increasing atherosclerotic changes in arterial walls is associated with endothelial cell dysfunction, decreases in endogenous vasodilators such as endothelin -1, and the tendency for thrombosis.50 This leads to obstruction to arterial blood flow and exercise-related ischemia. In the iliofemoral vessels, this may result in leg pain with ambulation called intermittent claudication. If a thrombus forms over the atherosclerotic lesion, complete obstruction of blood flow can occur acutely, causing severe pain, loss of pulses, and skin color changes in the affected extremity. Critical limb ischemia may lead to gangrene.
Evaluation for PAD requires a careful history and physical examination that focuses on finding evidence of atherosclerotic disease (e.g., bruits), determining a difference in blood pressure measured at the ankle versus the arm (ankle-brachial index), and measuring blood flow using duplex ultrasound, CT angiography, or magnetic resonance angiography. Treatment begins with risk factor reduction including smoking cessation, exercise, diabetes and hypertension management, and treatment for dyslipidemia.50 Symptomatic PAD should be managed with vasodilators in combination with antiplatelet medications. Newer vasodilators such as cilostazol and anticoagulants such as rivaroxaban may be indicated.51,52 If acute or refractory symptoms occur, emergent invasive catheterization followed by percutaneous or surgical revascularization may be needed.
Coronary Artery Disease, Myocardial Ischemia, and Acute Coronary Syndromes
Coronary artery disease (CAD) caused by atherosclerosis is the primary cause of heart disease in the United States. CAD can diminish the myocardial blood supply until deprivation impairs myocardial metabolism enough to cause myocardial ischemia, a local state in which the cells are temporarily deprived of blood supply. The cells remain alive but cannot function normally. Persistent ischemia or the complete occlusion of a coronary artery causes the acute coronary syndromes, including MI (heart attack).
Development of Coronary Artery Disease
An estimated 20.1 million Americans have coronary heart disease, which constitutes a prevalence of 7.2% in adults over 20 years of age and 22% to 34% in those over 80. In 2018, coronary heart disease was the leading cause of death attributable to cardiovascular disease in the United States, with mortality reaching over 365,000 deaths.12 Risk factors for CAD are the same as those for atherosclerosis and can be categorized as conventional (major) versus nontraditional (novel) and as modifiable versus nonmodifiable. Conventional or major risk factors for CAD that are nonmodifiable include (1) advanced age, (2) male sex or women after menopause, and (3) family history. Aging and menopause are associated with increased exposure to risk factors and poor endothelial healing. Family history may contribute to CAD through genetics and shared environmental exposures. Modifiable major risks include (1) dyslipidemia, (2) hypertension, (3) cigarette smoking, (4) diabetes and insulin resistance, (5) obesity, (6) sedentary lifestyle, and (7) atherogenic diet. Fortunately, modification of these factors can dramatically reduce the risk for CAD.
Genes
An individual's risk of developing CAD and MI is impacted by both genetic and lifestyle factors. Recent studies have identified 37 genes that are likely causal for CAD.53 Inheritance of genetic risks for dyslipidemia, diabetes, hypertension, and other risk factors is common. Epigenetic patterns are affected by the environment and can modulate gene expression. For example, dietary changes can alter the expression of genes related to dyslipidemia and the development of atherosclerotic lesions.54
Dyslipidemia
Using data from 2015-2018, it is estimated that over 90 million, or 38 %, of American adults have dyslipidemia.12 The link between CAD and abnormal levels of lipoproteins is well documented. The term lipoprotein refers to lipids, phospholipids, cholesterol, and triglycerides bound to carrier proteins. The cycle of lipoprotein synthesis is complex. Dietary fat is packaged into particles known as chylomicrons in the small intestine. Chylomicrons primarily contain triglycerides. Some of the triglycerides may be removed and either stored by adipose tissue or used by muscle as an energy source. The chylomicron remnants, composed mainly of cholesterol, are taken up by the liver. A series of chemical reactions in the liver results in the production of several lipoproteins that vary in density and function. These include very-low-density lipoproteins (VLDLs) composed primarily of triglycerides and protein; LDLs, composed mostly of cholesterol and protein; and HDLs, composed mainly of phospholipids and protein. Although lipoproteins are necessary for many physiologic functions, they can accumulate in abnormal amounts in the serum.
Dyslipidemia (or dyslipoproteinemia) refers to abnormal concentrations of serum lipoproteins as defined by the Third Report of the National Cholesterol Education Program.55 (Table 32.4). These abnormalities are the result of a combination of genetic and dietary factors. Primary or familial dyslipoproteinemias result from genetic defects that cause abnormalities in lipid-metabolizing enzymes and abnormal cellular lipid receptors.56 Secondary causes of dyslipidemia include the existence of several common systemic disorders, such as diabetes, hypothyroidism, pancreatitis, and renal nephrosis, as well as the use of certain medications, such as some diuretics, glucocorticoids, interferons, and antiretrovirals.
Table 32.4
| Optimal | Near-Optimal | Desirable | Low | Borderline | High | Very High | |
|---|---|---|---|---|---|---|---|
| Total cholesterol | <200 | 200–239 | ≥240 | ||||
| Low-density lipoprotein | <100 | 100–129 | 130–159 | 160-189 | ≥190 | ||
| Triglycerides | <150 | 150–199 | 200-499 | ≥500 | |||
| High-density lipoprotein | <40 | ≥60 |
aAll units are milligrams per deciliter.
Data from Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Executive summary of the third report of the National Cholesterol Education Program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III). JAMA. 2001;285:2486–2497.
LDL is responsible for the delivery of cholesterol to the tissues, and an increased serum concentration of LDL is a strong indicator of coronary risk. Serum levels of LDL are normally controlled by hepatic receptors that bind LDL and limit liver synthesis of this lipoprotein. Genetic predisposition to dyslipidemias, in combination with a high dietary intake of saturated fats, result in excess amounts of LDL in the bloodstream. Excess LDL migration into the vessel wall, oxidation, and phagocytosis by macrophages are key steps in the pathogenesis of atherosclerosis (see Fig. 32.6). LDL also plays a role in endothelial injury, inflammation, and immune responses that have been identified as being important in atherogenesis. The term LDL actually describes several types of LDL molecules. Measurement of LDL subfractions allows for a better prediction of coronary risk. For example, LDL-C and apolipoprotein B (structural protein found in both LDL and VLDL) measurements allow for the detection of the small, dense LDL particles that are the most atherogenic. Guidelines from the American Heart Association and the American College of Cardiology focus on treating dyslipidemia in the context of other risk factors.57 Diet and medication are the mainstays of treatment for elevated LDL. The most commonly used medications are the 3-hydroxy-3-methyl-glutaryl-CoA reductase medications (statins); however, side effects limit their use in some individuals. New medications, such as the proprotein convertase subtilisin/kexin 9 (PCKS9) inhibitors, also effectively lower LDL. Lipid-lowering medications may have other beneficial effects on the vasculature (see Emerging Science Box: Lipid-Lowering and Antihypertensive Medication Effects on Atherosclerosis-Associated Inflammation).
Low levels of HDL cholesterol are an indicator of increased coronary risk, whereas high levels of HDL are associated with a significant reduction in coronary risk independent of age, smoking history, LDL levels, blood pressure, or weight.58 HDL is responsible for “reverse cholesterol transport,” which returns excess cholesterol from the tissues to the liver for processing or elimination in the bile. HDL also participates in endothelial repair and decreases thrombosis. It can be fractionated into several particle densities (HDL-2 and HDL-3) that have different effects on vascular function. Exercise, weight loss, fish oil consumption, and moderate alcohol use result in modest increases in HDL level. Pharmacologic interventions to increase HDL levels have largely been ineffective in reducing CAD risk.59 Recent studies suggest that it is not only the serum levels of HDL that are key to determining CAD risk, but rather HDL functionality, which is harder to measure.60 One approach is measurement of the cholesterol efflux capacity (an indirect measure of reverse cholesterol transport) which has been found to be inversely associated with atherosclerotic risk.61
Other lipoproteins associated with increased cardiovascular risk include elevated levels of serum VLDLs (triglycerides) and increased lipoprotein(a) levels. Triglycerides are associated with an increased risk for CAD, especially in combination with other risk factors such as diabetes. Lipoprotein(a) (Lp[a]) is a genetically determined molecular complex between LDL and a serum glycoprotein called apolipoprotein A and has been shown to be an important risk factor for atherosclerosis, especially in women. Lipoprotein(a) potentially contributes to cardiovascular disease through proatherogenic, proinflammatory, and prothrombotic effects.62 Emerging therapies targeting elevated levels of Lp(a) may improve the management of individuals with CAD.63
Hypertension
Hypertension is responsible for a two- to threefold increased risk of atherosclerotic cardiovascular disease. It contributes to endothelial injury, a key step in atherogenesis. It also can cause myocardial hypertrophy, which increases myocardial demand for coronary flow (see Algorithm 32.2). Overactivity of the RAAS commonly found in hypertension also contributes to the genesis of atherosclerosis, and treatment of hypertension with medications that block the RAAS reduces CAD risk.
Cigarette smoking
Both direct and passive (environmental) smoking increase the risk of CAD. Smoking has a direct effect on endothelial cells and the generation of oxygen free radicals that contribute to atherogenesis. Nicotine stimulates the release of catecholamines (epinephrine and norepinephrine), which increase heart rate and peripheral vascular constriction. As a result, blood pressure increases, as do cardiac workload and oxygen demand. Cigarette smoking is associated with an increase in LDL levels and a decrease in HDL levels. The risk of CAD increases with heavy smoking and decreases when smoking is stopped.
Diabetes mellitus
Insulin resistance and diabetes mellitus are extremely important risk factors for CAD (see Chapter 22). Type 2 diabetes is associated with chronic vascular inflammation resulting from hyperglycemia, insulin resistance, and elevated levels of circulating insulin. Insulin resistance and diabetes have multiple effects on the cardiovascular system, including damage to the endothelium, thickening of the vessel wall, increased thrombosis, glycation of vascular proteins, and decreased production of endothelial-derived vasodilators, such as nitric oxide. Diabetes also is associated with dyslipidemia. Good diabetic control is linked to reduced risk for CAD.64
Obesity/adipokines/sedentary lifestyle
A sedentary lifestyle not only increases the risk of obesity but also has an independent effect on increasing CAD risk. Abdominal obesity has a strong link with increased CAD risk and is related to inflammation, insulin resistance, decreased HDL level, and increased blood pressure. Adipokines are a group of hormones released from adipose cells. Obesity causes increased levels of leptin and decreased levels of adiponectin that are associated with inflammation, endothelial injury, and thrombosis. Obesity-related changes in adipokines have been linked to hypertension, diabetes, and heart failure, as well as CAD. Excessive accumulation of perivascular adipose tissue leads to the paracrine release of vasoconstrictors and growth factors which have deleterious effects on vascular smooth muscle endothelial cells.65 Perivascular fat tissue also affects local adipokine levels (especially leptin), as well as releases proinflammatory signals and promotes atherosclerotic plaque formation.66,67 Weight loss, exercise, and healthy diet improve adipokine levels. Bariatric surgery procedures, such as gastric bypass, can provide sustained improvement in risk factors for cardiovascular disease, such as hypertension, dyslipidemia, and diabetes in selected individuals.68,69
Atherogenic diet
Diet plays a complex role in atherogenic risk. Diets high in salt, fats, trans-fats, and carbohydrates all have been implicated. There are many recommendations regarding diet modification to reduce coronary risk; one of the most effective is called the Mediterranean diet.
The traditional Mediterranean diet is characterized by a high intake of olive oil, fruits, nuts, vegetables, and cereals; moderate intake of fish and poultry; low intake of dairy products, red meat, processed meats, and sweets; and moderate intake of wine consumed with meals. The beneficial effects of the Mediterranean diet are hypothesized to include modulation of many different biologic components of cardiovascular health such as epigenetic control of genes associated with cardiovascular risk, modification of the gut microbiome, antiinflammatory and immune effects, and improvement in metabolic factors such as glucose tolerance and lipid metabolism. Dietary guidance to improve cardiovascular health had been published by the American Heart Association.70
Nontraditional risk factors
Nontraditional risk factors for CAD have been identified that can help with clinical decision-making about how best to manage individuals who also have established CAD or significant traditional risk factors.
Markers of inflammation and ischemia
Of the numerous markers of inflammation that have been linked to an increase in CAD risk, high-sensitivity C-reactive protein (hs-CRP) is the most important clinically. hs-CRP is a protein synthesized in the liver and is used as an indirect measure of atherosclerotic plaque–related inflammation. The primary use of hs-CRP is as an aid to decision-making about pharmacologic interventions for individuals with other risk factors for coronary disease.
Troponin I (TnI) is a serum protein whose measurement is used as a sensitive and specific diagnostic test to help identify myocardial injury during acute coronary syndromes. Highly sensitive TnI assays are used in individuals without a history of CAD to assess risk for future CHD events, mortality, and heart failure.
Chronic kidney disease
In individuals with chronic kidney disease (CKD), a decline in glomerular filtration rate is associated with an increasing risk for CAD. CKD is associated with dyslipidemia, endothelial injury, and vascular calcification, which contribute to atherogenesis.
Air pollution and ionizing radiation
Exposure to air pollution, especially roadway exposures, is strongly correlated with coronary risk. Fine particulate matter is the major cardiovascular risk component of air pollution and is considered the most important risk factor contributing to global cardiovascular mortality.71 It is postulated that toxins in pollution contribute to macrophage activation, oxidation of LDL, thrombosis, and inflammation of vessel walls. Exposure to even low levels of ionizing radiation also has been linked to increased risk for CAD. Reducing exposure to air pollutants can reduce cardiovascular risk.72
Medications
Medications may contribute to CAD through their effect on lipid metabolism (e.g., protease inhibitors, diuretics, antirejection medications), clotting (e.g., estrogens and progesterones), or other effects on vascular function and tone. Nonsteroidal antiinflammatory drugs (NSAIDs) are linked to an increase in CAD-related ischemic events that can occur within weeks of beginning their use.73 Likely mechanisms include increases in toxic oxygen radicals, vasoconstrictors, and thrombosis.
The microbiome
The microbiome is increasingly being recognized for its influence on cardiovascular disease risk. The impact of the microbiome on atherogenesis is likely related to its effects on underlying risk factors, as well as its role in modulating innate and adaptive immunity. Differences in the composition of the microbiome between those with CAD and healthy individuals have been documented. Changes in the microbiome have been found to have significant effects on the development of traditional risk factors for cardiovascular disease, such as diabetes and obesity. The identification of microbiota-controlled metabolites, such as trimethylamine N-oxide (TMAO), is shedding light on the link between diet and these risk factors.74,75
Transient Myocardial Ischemia
As described previously, CAD can diminish the myocardial blood supply causing ischemia. The process of atherosclerotic plaque progression can be gradual, which usually results in transient myocardial ischemic syndromes when demand for blood supply exceeds supply, but perfusion is restored before there is permanent damage to the heart muscle. Ischemia also can occur with coronary vasospasm even in the absence of atherosclerosis.
Pathophysiology
Myocardial ischemia develops if the flow or oxygen content of coronary blood is insufficient to meet the metabolic demands of myocardial cells (Algorithm. 32.3). Imbalances between coronary blood supply and myocardial demand can result from a number of conditions. The most common cause of decreased coronary blood flow and resultant myocardial ischemia is the formation of atherosclerotic plaques in the coronary circulation (CAD). As the plaque increases in size, it may partially occlude the vessel lumen, thus limiting coronary flow and causing ischemia especially during exercise. While these large plaques are often considered “stable,” individuals with chronic atherosclerotic changes are still at risk for adverse events. Myocardial ischemia also can result from other causes of decreased blood and oxygen delivery to the myocardium, such as coronary spasm, hypotension, dysrhythmias, and decreased oxygen-carrying capacity of the blood (e.g., anemia, hypoxemia). Common causes of increased myocardial demand for blood include tachycardia, exercise, hypertension (hypertrophy), and valvular disease.
Factors that decrease coronary artery blood flow (e.g., atherosclerosis) or increase demand for myocardial blood supply (e.g., exercise) cause myocardial ischemia. Both transient and prolonged ischemia can cause abnormal or absent myocyte responses to electrical impulses through the myocardium leading to dysrhythmias or decreased contractility. Decreased ability of the heart to pump blood results in heart failure and decreased coronary perfusion, further compromising the supply of blood to the heart. Dysrhythmias may also lead to sudden death.
A flowchart shows the cycle of myocardial ischemic events. 1. Imbalance between coronary supply and myocardial demand. Leads to 2. 2. Myocardial oxygen deficit. Leads to 3 or 4. 3. Less than 20 minutes: ischemic attack. Leads to 5. 4. Greater than 20 minutes: myocardial infraction. Leads to 5. 5. Abnormal or absent response to electrical impulses. Leads to 6 or 7. 6. Dysrhythmias. Leads to 8 or 10. 7. Failure to contract. Leads to 8. 8. Impaired cardiac pumping. Leads to 1 or 9. 9. Heart failure. 10. Sudden death.
Factors that decrease coronary artery blood flow (e.g., atherosclerosis) or increase demand for myocardial blood supply (e.g., exercise) cause myocardial ischemia. Both transient and prolonged ischemia can cause abnormal or absent myocyte responses to electrical impulses through the myocardium leading to dysrhythmias or decreased contractility. Decreased ability of the heart to pump blood results in heart failure and contributes to decreased coronary perfusion, further compromising the supply of blood to the heart. Dysrhythmias also may lead to sudden death.
Myocardial cells become ischemic within 10 seconds of coronary occlusion, thus hampering pump function and depriving the myocardium of a glucose source necessary for aerobic metabolism. Anaerobic processes take over, and lactic acid accumulates. After several minutes, the heart cells lose the ability to contract and cardiac output decreases. Cardiac cells remain viable for approximately 20 minutes under ischemic conditions. If blood flow is restored, aerobic metabolism resumes, contractility is restored, and cellular repair begins. If perfusion is not restored, myocardial infarction occurs.
Individuals with transient myocardial ischemia present clinically in several ways. Chronic coronary obstruction results in recurrent predictable chest pain called stable angina. Abnormal vasospasm of coronary vessels results in unpredictable chest pain called vasospastic (Prinzmetal) angina. Myocardial ischemia that does not cause detectable symptoms is called silent ischemia.
Stable angina pectoris
Angina is chest pain caused by myocardial ischemia. Atherosclerotic plaques partially obstruct coronary vessels, and affected vessels cannot dilate in response to increased myocardial demand associated with physical exertion or emotional stress. In stable angina pectoris, blood flow is restored with rest and the administration of nitrates, and necrosis of myocardial cells does not occur. The pain of stable angina is caused by the buildup of lactic acid or abnormal stretching of the ischemic myocardium that irritates myocardial nerve fibers. These afferent sympathetic fibers enter the spinal cord from levels C3 to T4, accounting for a variety of locations and radiation patterns of anginal pain. Discomfort may radiate to the neck, lower jaw, left arm, and left shoulder, or occasionally to the back or down the right arm.
Stable angina is typically experienced as transient substernal chest discomfort, ranging from a sensation of heaviness or pressure to moderately severe pain. Individuals often describe the sensation by clenching a fist over the left sternal border. The discomfort may be mistaken for indigestion. Pallor, diaphoresis, and dyspnea may be associated with the pain. Guidelines for the acute evaluation of chest pain recommend a careful symptom history and physical examination followed by diagnostic testing to include ECG, chest radiography, and measurement of serum biomarkers such as troponins.76
Many individuals with reversible myocardial ischemia will have a normal physical examination between events. Physical examination of those experiencing myocardial ischemia may disclose rapid pulse rate or extra heart sounds (gallops or murmurs), and pulmonary congestion indicating impaired left ventricular function. The presence of xanthelasmas (small fat deposits) around the eyelids or arcus senilis of the eyes (a yellow lipid ring around the cornea) suggests severe dyslipidemia and possible atherosclerosis. The presence of peripheral or carotid artery bruits suggests probable atherosclerotic disease and increases the likelihood that CAD is present. Electrocardiography detects distortion of electrical impulses across ischemic myocardium, and can be conducted during exercise stress testing to detect ischemic changes associated with stable angina. Transient ST segment depression and T wave inversion are characteristic signs of stable angina involving the endocardium, although ST elevation indicative of ischemia involving the full myocardial wall (transmural ischemia) may occur (Fig. 32.8). Serum troponin levels remain within normal limits. Stress radionucleotide imaging with single-photon emission computerized tomography (SPECT) is effective at identifying ischemia and estimating coronary risk. Other noninvasive tests for evaluating coronary atherosclerotic lesions include stress echocardiography and coronary CT angiography.77
(A) Normal ECG. (B) Electrocardiographic alterations associated with ischemia.
Left panel, A. Electrocardiogram labeled normal E C G deflections shows gentle peak labeled P and atrial depolarization, followed by a sharp and tall peak, marked R. The area under Q R S is labeled ventricular depolarization. It is followed by another gentle peak labeled T. Area from S to T is labeled ventricular repolarization. Right panel, B. Electrocardiogram labeled E C G alterations associated with ischemia is as follows: S T segment depression: It shows sharp and tall peak labeled R and depression at S. T wave inversion shows trough labeled T. S T segment elevation shows curve reaches at height labeled R, followed by fluctuations while sloping down.
The primary aims of therapy for myocardial ischemia and stable angina are to increase coronary blood flow, reduce myocardial oxygen consumption, and to reduce the risk for adverse cardiovascular events such as myocardial infarction. Coronary blood flow is improved by reversing vasoconstriction, reducing plaque growth and rupture, and preventing clotting. Myocardial oxygen demand is reduced by manipulation of blood pressure, heart rate, contractility, and left ventricular volume. Several classes of drugs are useful for increasing coronary flow and decreasing myocardial demand, especially nitrates, β-blockers, calcium channel blockers, and sodium ion channel inhibitors (e.g., nicorandil).77 Antiplatelet medications are used to prevent thrombus formation. Percutaneous coronary intervention (PCI) is a procedure in which stenotic (narrowed) coronary vessels are dilated with a catheter. The use of PCI for stable angina may be associated with improvements in symptoms but does not reduce the risk for future MI or death.78 Severe CAD also can be surgically treated by a coronary artery bypass graft (CABG), usually using the saphenous vein from the lower leg.77,79
Primary prevention of CAD is key to improving morbidity and mortality in those with stable angina. Primary prevention refers to reduction in CAD risk factors before the occurrence of a major ischemic event such as myocardial infarction, whereas secondary prevention refers to risk reduction after such an event. Primary prevention rests on the calculation of 10-year atherosclerotic cardiovascular disease risk, followed by the development of a comprehensive plan for lifestyle change to include exercise and dietary modifications.80 Antihypertensive and lipid lowering medications should be prescribed as indicated by current guidelines.27,28,57 In those individuals with diabetes, metformin, sodium-glucose cotransporter 2 (SGLT2) inhibitors, or a glucagon-like peptide-1 receptor agonist are recommended along with diet and exercise.80
It is estimated that half of women with stable angina do not have obstructive CAD, but rather have “microvascular angina,” which results from vasoconstriction of small coronary arterioles deep in the myocardium. This form of myocardial ischemia may present with atypical chest pain, palpitations, sense of unease, and severe fatigue rather than typical angina; thus many women with this disorder are misdiagnosed. Small intramyocardial arterioles constrict in MVA due to endothelial dysfunction, decreased endogenous vasodilators, inflammation, changes in adipokines, and platelet activation. Evaluation for microvascular angina relies upon transthoracic Doppler echocardiography and positron emission tomography. The combination of calcium channel blockers or sodium ion channel inhibitors with statins has been shown to be effective in many women.81
Vasospastic (Prinzmetal) angina
Vasospastic angina is chest pain attributable to transient ischemia of the myocardium that occurs unpredictably and often at rest. Pain is caused by vasospasm of one or more major coronary arteries and can occur in those with or without coronary artery disease. The spasm may involve the large epicardial coronary arteries, or may occur in the microvasculature. Vasospasm may result from coronary smooth muscle hypercontractility, decreased vagal activity, endothelial dysfunction, magnesium deficiency, inflammation, oxidative stress, and hyperactivity of the SNS.82 It can be triggered by hyperventilation, mental stress, smoking, alcohol, use of stimulants, or rapid eye movement sleep. Hypokalemia worsens symptoms and outcomes.83 Vasospastic angina is diagnosed by matching clinical manifestations with documented transient ischemic changes on ECG. In some individuals, confirmation with coronary angiography combined with administration of a provocative agent (e.g., ergonovine) is needed. Management includes avoidance of triggers and the use of calcium channel blockers or nitrates. Although vasospastic angina is usually a benign condition, it can cause dysrhythmias and infarction, especially in those who also have atherosclerotic coronary lesions.84
Another form of vasospastic myocardial ischemia without coronary artery disease is called Takotsubo syndrome (TTS). TTS occurs primarily in postmenopausal women during mental or physical stress and is associated with acute transient heart failure. It is believed to be the result of increased catecholamine levels coupled with increased adrenergic receptor density and sensitivity in the myocardium.85 It is usually reversible but can cause significant morbidity and mortality (see Emerging Science Box: Takotsubo Syndrome).
Silent ischemia
Silent ischemia is myocardial ischemia that does not cause detectable symptoms such as angina. Ischemia can be totally asymptomatic, or individuals may complain of fatigue, dyspnea, or a feeling of unease. It is believed to be more common than symptomatic angina.86 The primary cause of silent ischemia is abnormalities in autonomic innervation, most commonly associated with diabetes mellitus. Other causes include surgical denervation during CABG or cardiac transplantation. Also of interest is silent ischemia occurring in some individuals during mental stress. Chronic stress has been linked to an increase in inflammatory cytokines and a hypercoagulable state that may contribute to acute ischemic events (see Chapter 11). Detection and management of silent ischemia is important because it may be an indicator of future serious cardiovascular events. Treatment is based on the degree of ischemia and the extent of underlying coronary disease.86
Acute Coronary Syndromes
The process of atherosclerotic plaque progression can be gradual. However, when there is sudden coronary obstruction caused by thrombus formation over a ruptured atherosclerotic plaque, the acute coronary syndromes result (Algorithm 32.4). Unstable angina is a form of acute coronary syndrome that is a harbinger of impending infarction. Myocardial infarction (MI) results when there is prolonged ischemia causing irreversible damage to the heart muscle. MI can be further subdivided into non-ST elevation MI (non-STEMI) and ST elevation MI (STEMI). Sudden cardiac death can occur as a result of any of the acute coronary syndromes.
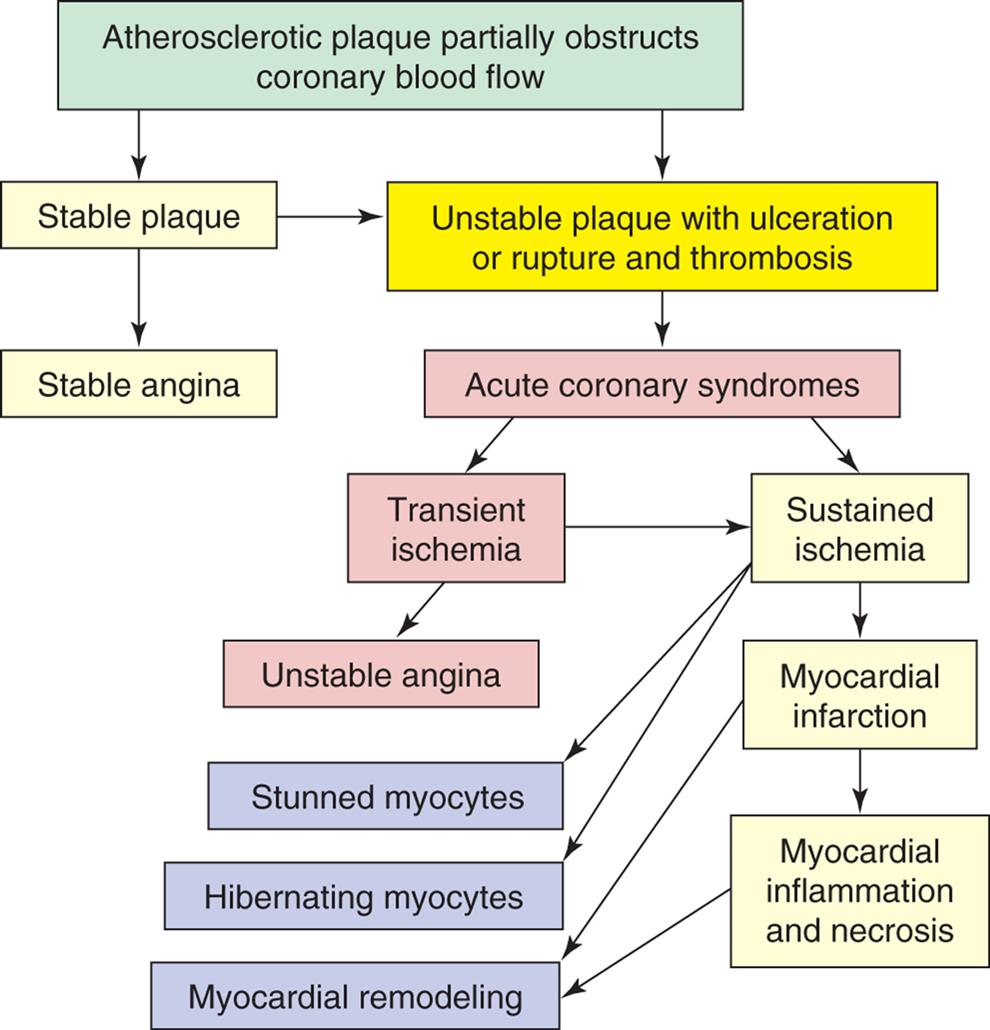
The atherosclerotic process can lead to stable plaque formation and stable angina or can result in unstable plaques that are prone to rupture and thrombus. Thrombus formation on a ruptured plaque that disperses in less than 20 minutes leads to transient ischemia and unstable angina. If the vessel obstruction is sustained, myocardial infarction (MI) with inflammation and necrosis of the myocardium results. In addition, MI is associated with other structural and functional changes, including myocyte stunning and hibernation and myocardial remodeling.
A flowchart shows the pathophysiology of acute coronary syndromes. 1. Atherosclerotic plaque partially obstructs coronary blood flow. Leads to 2 and 12. 2. Unstable plaque with ulceration or rupture and thrombosis. Leads to 3. 3. Acute coronary syndromes. Leads to 4 and 5. 4. Transient ischemia. Leads to 5 and 6. 5. Sustained ischemia. Leads to 7, 8, and 10. 6. Unstable angina. 7. Stunned myocytes. 8. Hibernating myocytes. 9. Myocardial remodeling. 10. Myocardial infarction. Leads to 9 and 11. 11. Myocardial inflammation and necrosis. 12. Stable plaque. Leads to 2 and 13. 13. Stable angina.
An unstable atherosclerotic plaque has a core that is especially rich in deposited oxidized LDL, has a thin fibrous cap, and is prone to rupture (Fig. 32.9). Calcification of these plaques further destabilizes them.87 These unstable plaques may not extend into the lumen of the vessel and may be clinically silent until they rupture. New methods are emerging for detecting unstable plaques before they rupture, thus identifying those individuals at highest risk for acute coronary syndromes.88 Plaque rupture occurs because of the effects of shear forces, inflammation with release of multiple inflammatory mediators, secretion of neutrophil- and macrophage-derived degradative enzymes, and apoptosis of cells at the edges of the lesions.44 Exposure of the plaque substrate activates the clotting cascade. The resulting thrombus can form very quickly (Fig. 32.10A). Vessel obstruction is further exacerbated by the release of vasoconstrictors, such as thromboxane A2 and endothelin. The thrombus may dissolve when only minimal myocyte damage has occurred (unstable angina), or it may persist causing prolonged ischemia with infarction of the heart muscle (MI) (see Fig. 32.10B).
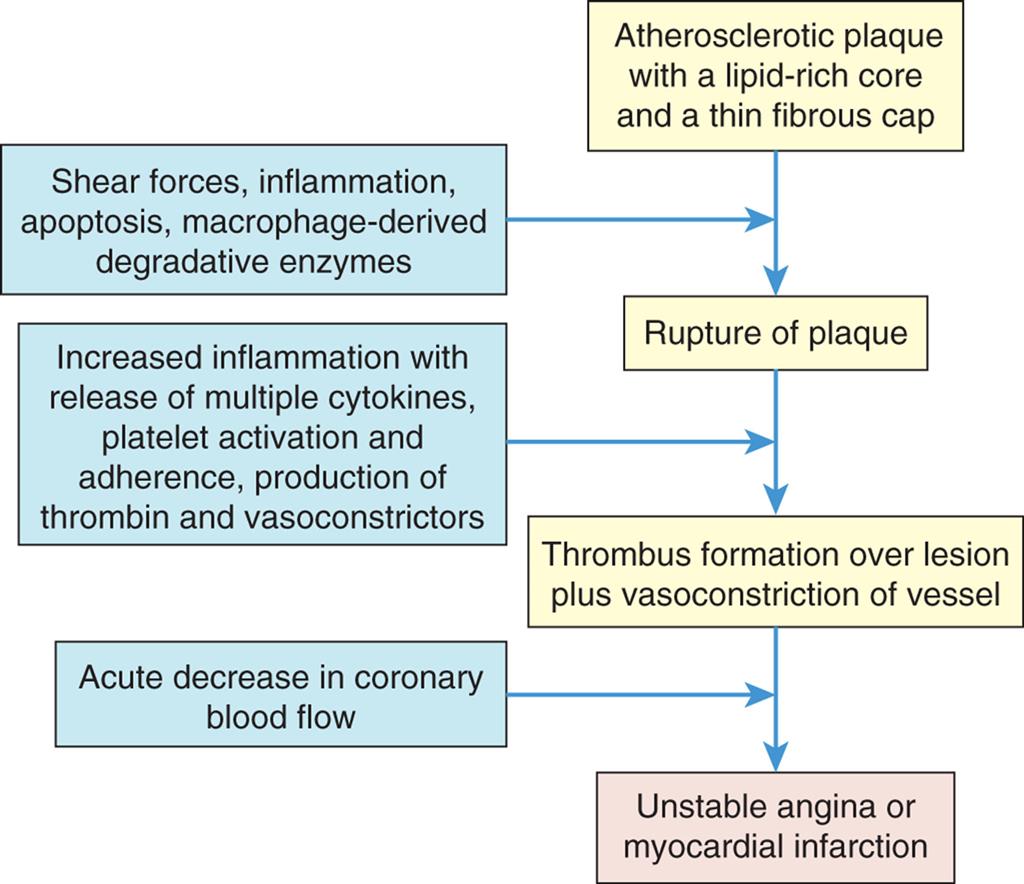
A flowchart represents the pathogenesis of unstable plaques and thrombus formation. 1. Atherosclerotic plaque with a lipid-rich core and a thin fibrous cap. Leads to 3. 2. Shear forces, inflammation, apoptosis, macrophage-derived degradative enzymes. Leads to 3. 3. Rupture of plaque. Leads to 5. 4. Increased inflammation with release of multiple cytokines, platelet activation and adherence, production of thrombin and vasoconstrictors. Leads to 5. 5. Thrombus formation over lesion plus vasoconstriction of vessel. Leads to 7. 6. Acute decrease in coronary blood. Leads to 7. 7. Unstable angina or myocardial infarction.
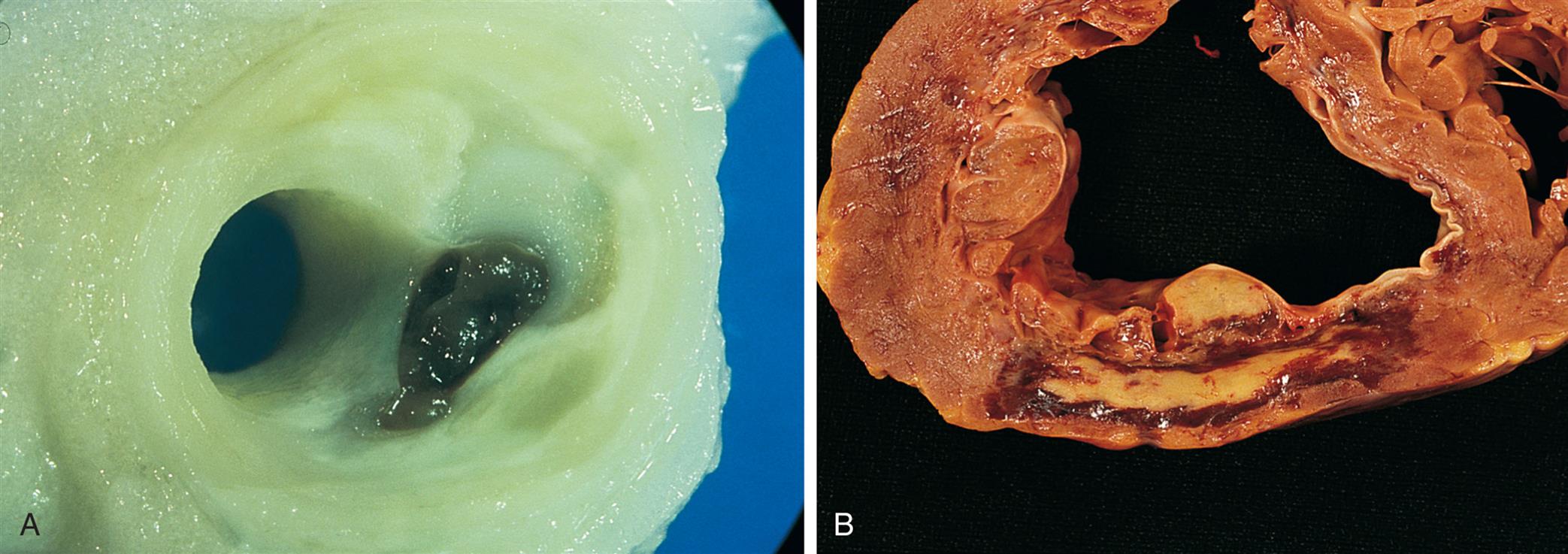
(A) Plaque disruption. The cap of the lipid-rich plaque has become torn, with the formation of a thrombus, mostly inside the plaque. (B) Myocardial infarction. This infarct is 6 days old. The center is yellow and necrotic with a hemorrhagic red rim. The responsible arterial occlusion is probably in the right coronary artery. The infarct is on the posterior wall. (From Damjanov I, Linder J, eds. Anderson’s pathology, 10th edition. St. Louis: Mosby; 1996.)
Unstable angina
Unstable angina was once considered a separate type of acute coronary syndrome characterized by reversible myocardial ischemia and the absence of myocardial cell injury. However, the use of highly sensitive measurements of myocardial damage (hs-troponin I) has resulted in the recognition that most individuals with unstable angina do have some myocardial damage. Therefore, unstable angina is the first step in a progressive spectrum of ischemia-related myocardial injury and now is grouped under the classification of non-ST elevation acute coronary syndromes. It is important to recognize this syndrome because it signals that the atherosclerotic plaque has begun to rupture and infarction may soon follow. Unstable angina occurs when a fairly small fissuring or superficial erosion of the plaque leads to transient episodes of thrombotic vessel occlusion and vasoconstriction at the site of plaque damage. This thrombus is labile and occludes the vessel for no more than 10 to 20 minutes, with return of perfusion before large amounts of myocardial necrosis occurs. Unstable angina presents as new-onset angina, angina that is occurring at rest, or angina that is increasing in severity or frequency. Individuals may experience increased dyspnea, diaphoresis, and anxiety as the angina worsens. Physical examination may reveal evidence of ischemic myocardial dysfunction such as pulmonary congestion. The ECG most commonly shows ST segment depression and T wave inversion during pain that resolve as the pain is relieved. Serum levels of hs-troponin I are often slightly increased. Management of unstable angina requires immediate hospitalization with administration of nitrates and antithrombotics (e.g., aspirin, clopidogrel, abciximab, eptifibatide). Anticoagulants, such as low-molecular-weight heparin (enoxaparin), bivalirudin, or fondaparinux, are also given.89,90 Beta-blockers, calcium channel blockers, and ACE inhibitors also may be used. Choices are based on symptoms, underlying conditions, and planned interventions. Lipid lowering medications such as statins should be administered. Rapid intervention with PCI also may be indicated if the individual's condition is refractory to medical treatment.89,90
Myocardial infarction
When coronary blood flow is interrupted for an extended period, infarction with myocyte necrosis occurs. This results in MI. Plaque progression, disruption, and subsequent clot formation are the same for MI as they are for unstable angina (see Algorithm 32.4 and Figs. 32.9 and 32.10). In this case, however, the thrombus is less labile and occludes the vessel for a prolonged period, such that myocardial ischemia progresses to myocyte necrosis and death. Pathologically, there are two major types of MI: subendocardial infarction and transmural infarction. Clinically, however, MI is categorized as non-ST segment elevation MI (non-STEMI) or ST segment elevation MI (STEMI).
If the thrombus disintegrates before complete distal tissue necrosis has occurred, the infarction will involve only the myocardium directly beneath the endocardium (subendocardial MI) (Fig. 32.11). This type of infarction usually will present with ST segment depression and T wave inversion without ST elevation; therefore, it is termed non-STEMI. It is important to recognize this form of acute coronary syndrome because recurrent clot formation on the disrupted atherosclerotic plaque is likely. If the thrombus lodges permanently in the vessel, the infarction will extend through the myocardium all the way from endocardium to epicardium (transmural MI), resulting in severe cardiac dysfunction (see Fig. 32.11). Transmural myocardial infarction usually will result in ST segment elevation on ECG, so it is called STEMI. Clinically, it is important to identify individuals with STEMI because they are at highest risk for serious complications and should receive definitive intervention without delay.
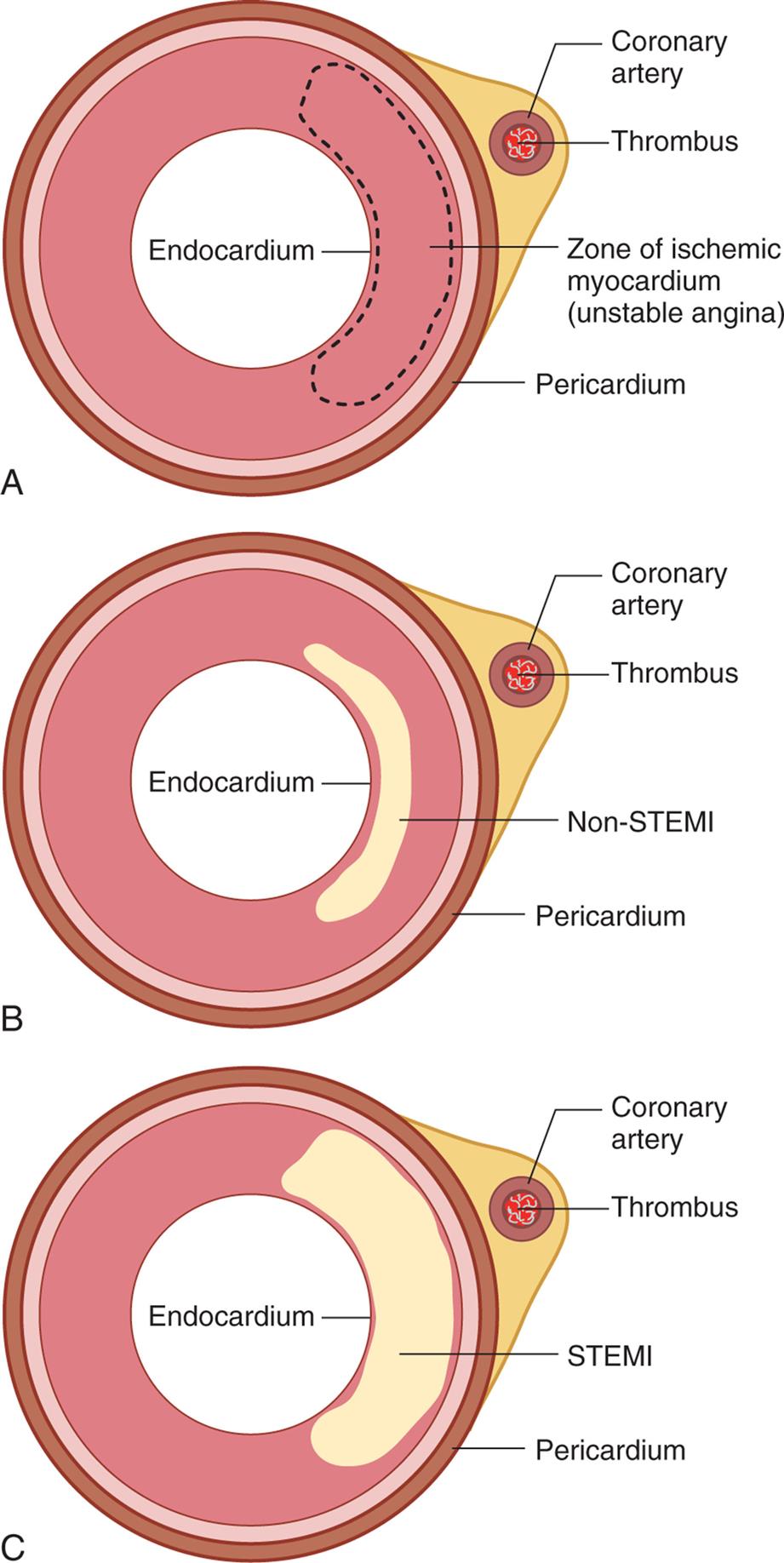
(A) Unstable angina. Coronary thrombosis leads to myocardial ischemia. (B) Non-STEMI. Persistent coronary occlusion leads to infarction of the myocardium closest to the endocardium. (C) STEMI. Continued coronary occlusion leads to transmural infarction extending from endocardium to pericardium.
Three illustrations A through B depict the cross section of coronary artery of different types of myocardial infarction. Top panel, Part A. The illustration depicts unstable angina showing the coronary thrombosis leading to myocardial infarction with parts labeled as follows. coronary artery, thrombus, zone of ischemic myocardium (unstable angina), and pericardium. Middle panel, Part B. The illustration depicts Non-S T E M I showing the coronary thrombosis leading infarction of the myocardium closest to the endocardium with labels as follows. Coronary artery, thrombus, Non-STEMI, and pericardium. Bottom panel, Part C. The illustration depicts S T E M I showing the coronary thrombosis leading infarction of the myocardium extending to pericardium with labels as follows. Coronary artery, thrombus, S T E M I, and pericardium.
Pathophysiology
After 8 to 10 seconds of decreased blood flow, myocardial oxygen reserves are used quickly. Glycogen stores decrease as anaerobic metabolism begins. Unfortunately, glycolysis can supply only 65% to 70% of the total myocardial energy requirement and produces much less adenosine triphosphate (ATP) than aerobic processes. Hydrogen ions and lactic acid accumulate, making the myocardium more vulnerable to the damaging effects of lysosomal enzymes and may suppress impulse conduction and contractile function, thereby leading to heart failure.
Oxygen deprivation also is accompanied by electrolyte disturbances, specifically the loss of potassium, calcium, and magnesium from cells. Myocardial cells deprived of necessary oxygen and nutrients lose contractility, thereby diminishing the pumping ability of the heart. Ischemia causes the myocardial cells to release catecholamines, predisposing the individual to serious imbalances of sympathetic and parasympathetic function, irregular heartbeats (dysrhythmia), and heart failure. Catecholamines cause an increase in plasma concentrations of free fatty acids and glycerol, which can have a harmful detergent effect on cell membranes. Norepinephrine elevates blood glucose levels, which also contributes to myocardial dysfunction. Ang II is released during myocardial ischemia and causes peripheral vasoconstriction, coronary spasm, and fluid retention. It also is a growth factor for vascular smooth muscle cells, myocytes, and cardiac fibroblasts, resulting in structural changes in the myocardium called remodeling. Infiltration of inflammatory cells further contributes to tissue injury.
MI results in both structural and functional changes of cardiac tissues (Fig. 32.12). Cardiac cells can withstand ischemic conditions for about 20 minutes before irreversible hypoxic injury causes cellular death (apoptosis) and myocardial infarction. Gross tissue changes at the area of infarction may not become apparent for several hours, despite almost immediate onset (within 30 to 60 seconds) of electrocardiographic changes. Myocyte injury and necrosis result in the release of intracellular enzymes, such as creatine phosphokinase-myocardial bound (CPK-MB), and myocyte proteins, such as the troponins, through the damaged cell membranes into the interstitial spaces. The lymphatics absorb the enzymes and transport them into the bloodstream, where they can be detected by serologic tests.
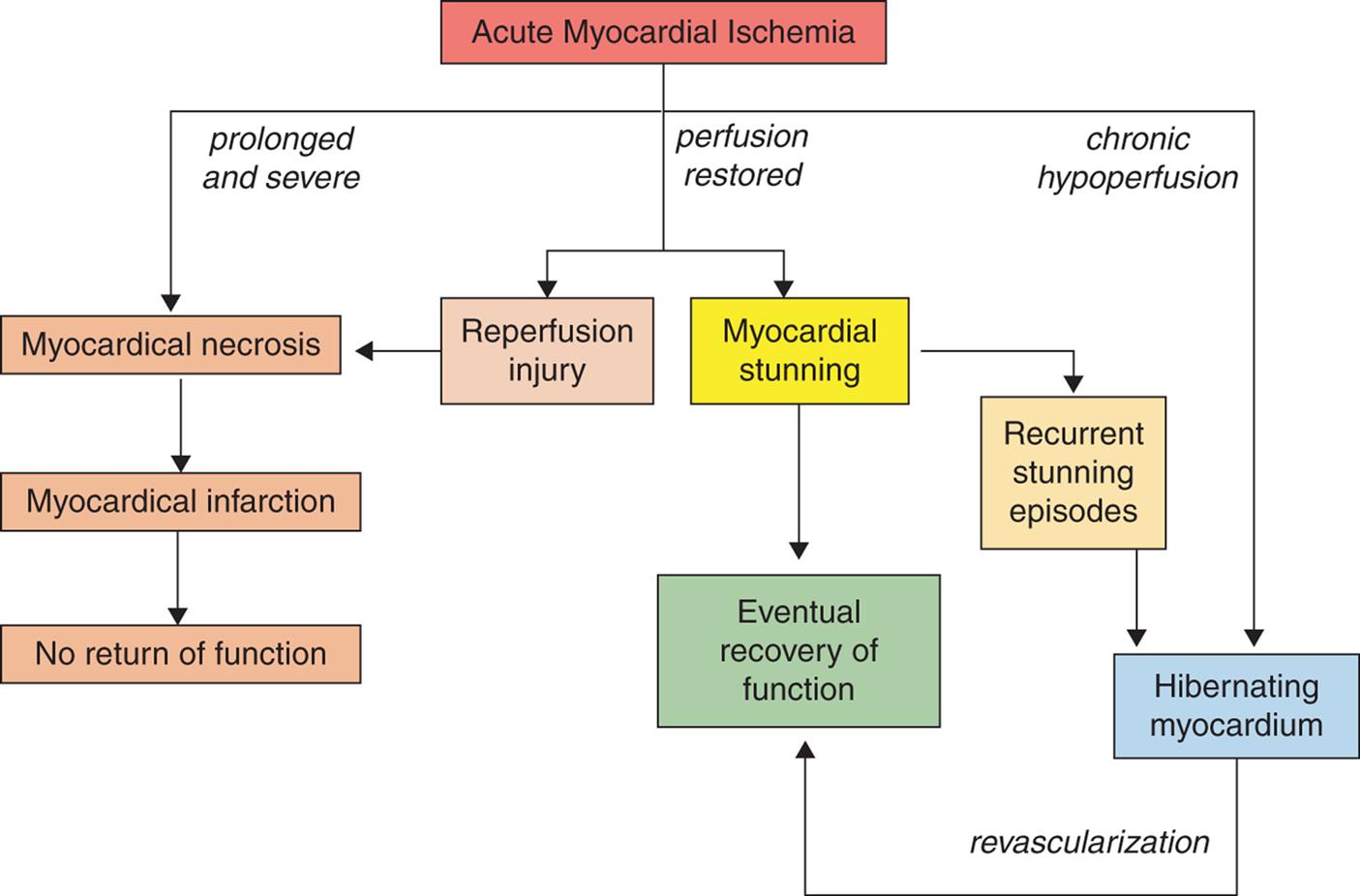
Acute myocardial ischemia can result in prolonged and severe hypoperfusion, reperfusion, or chronic hypoperfusion. As a result, an acute coronary syndrome may be characterized by infarction, reperfusion injury, stunning, or hibernating myocardium. Recovery of function is not possible once myocardial tissue is necrosed, however stunned and hibernating myocardium may regain function after days, weeks, or even months, especially after appropriate revascularization procedures. (Adapted from Kloner RA. Stunned and hibernating myocardium: Where are we nearly 4 decades later? Journal of the American Heart Association, 2020;9:e015502.)
A flowchart shows the structural and functional changes associated with acute myocardial ischemia. 1. Acute Myocardial ischemia. If prolonged and severe, leads to 2. If perfusion is restored, leads to 5 and 6. If chronic hypoperfusion, leads to 9. 2. Myocardial necrosis. Leads to 3. 3. Myocardial infraction. Leads to 4. 4. No return of function. 5. Reperfusion injury. Leads to 2. 6. Myocardial stunning. Leads to 7 and 8. 7. Recurrent stunning episodes. Leads to 9. 8. Eventual recovery of function. 9. Hibernating myocardium. If revascularization, leads to 8.
Although restoration of blood flow is crucial to reducing infarct size, reperfusion of the ischemic myocardium triggers a process called reperfusion injury which can contribute as much as 50% to the overall infarct size (see Fig. 32.12).91 Reperfusion injury involves the release of toxic oxygen free radicals, calcium flux, and pH changes that cause a sustained opening of mitochondrial permeability transition pores (mPTPs) and contribute to resultant cellular death. Ischemia and reperfusion also cause damage to the coronary circulation through endothelial injury, platelet activation, inflammation, and vasoconstriction.92 Many studies are exploring ways to reduce the extent of injury in ischemia and reperfusion injury during myocardial infarction (see Emerging Science Box: Cardioprotection Therapies for Myocardial Infarction and Reperfusion Injury).
Cardiac tissue surrounding the area of necrosed tissue in myocardial infarction also undergoes changes. Myocardial stunning is a temporary loss of contractile function that persists for hours to days after perfusion has been restored (see Fig. 32.12). This pathophysiologic state can occur both after MI and in individuals who suffer ischemia during cardiovascular procedures or during CNS trauma. Stunning is caused by the alterations in electrolyte pumps and calcium homeostasis and by the release of toxic oxygen free radicals and contributes to heart failure, shock, and dysrhythmias.93 Numerous interventions to limit the amount of stunning are being explored. Hibernating myocardium describes tissue that is persistently ischemic and undergoes metabolic adaptation to prolong myocyte survival until perfusion can be restored. Cardiomyocytes undergo downregulation of calcium-handling and mitochondrial proteins and upregulation of survival and stress proteins.93 PCI or surgery aimed at reperfusion of hibernating myocardium can restore significant cardiac function (see Fig. 32.12).
Myocardial remodeling is a process that causes myocyte hypertrophy and loss of contractile function in the areas of the heart distant from the site of infarction. It can occur over days, weeks, and months after an acute ischemic event.94 It is mediated by Ang II, aldosterone, catecholamines, adenosine, and inflammatory cytokines. It contributes to long-term myocardial dysfunction and heart failure. Remodeling can be limited through rapid restoration of coronary flow and the use of renin-angiotensin-aldosterone blockers and β-blockers after MI.
The severity of functional impairment depends on the size of the lesion and the site of infarction. Functional changes can include (1) decreased cardiac contractility with abnormal wall motion, (2) altered left ventricular compliance, (3) decreased stroke volume and ejection fraction, (4) increased left ventricular end-diastolic pressure (LVEDP), and (5) sinoatrial (SA) node malfunction. Life-threatening dysrhythmias and heart failure often follow MI.
With infarction, ventricular function is abnormal and the EF falls, resulting in increases in ventricular end-diastolic volume (VEDV). If the coronary obstruction involves the perfusion to the left ventricle, pulmonary venous congestion ensues; if the right ventricle is ischemic, increases in systemic venous pressures occur.
MI causes a severe inflammatory response that ends with wound repair (see Chapter 6).95 Damaged cells undergo degradation, fibroblasts proliferate, and scar tissue is synthesized. Within 24 hours, leukocytes infiltrate the necrotic area, and proteolytic enzymes released from scavenger neutrophils degrade the necrotic tissue. Macrophages remove dead cells and secrete growth factors which help repair myocardial injury.96 The collagen matrix that is deposited is initially weak, mushy, and vulnerable to reinjury. Unfortunately, it is at this time in the recovery period (10 to 14 days after infarction) that individuals feel more like increasing activities and may stress the newly formed scar tissue. After 6 weeks, the necrotic area is completely replaced by scar tissue, which is strong but cannot contract and relax like healthy myocardial tissue. These cellular and extracellular matrix changes contribute to sustained functional impairment of the heart often resulting in heart failure. Recent studies have shown that cardiac stem cells have regenerative capacity, and stem cell therapy may be a novel approach for cardiac muscle repair and regeneration (see Emerging Science Box: Cardioprotection Therapies for Myocardial Infarction and Reperfusion Injury).97
Clinical Manifestations
The first symptom of acute MI is usually sudden, severe chest pain. The pain is similar to that of angina pectoris but more severe and prolonged. It may be described as heavy and crushing, such as a “truck sitting on my chest.” Radiation to the neck, jaw, back, shoulder, or left arm is common. Some individuals, especially those who are older or have diabetes, experience no pain, thus having a “silent” infarction. Infarction often simulates a sensation of unrelenting indigestion. Nausea and vomiting may occur because of reflex stimulation of vomiting centers by pain fibers. Vasovagal reflexes from the area of the infarcted myocardium also may affect the gastrointestinal tract.
Various cardiovascular changes are found on physical examination:
- 1. The SNS is reflexively activated to compensate, resulting in a temporary increase in heart rate and blood pressure.
- 2. Abnormal extra heart sounds reflect left ventricular dysfunction.
- 3. Pulmonary findings of congestion, including dullness to percussion and inspiratory crackles at the lung bases, can occur if the individual develops heart failure.
- 4. Peripheral vasoconstriction may cause the skin to become cool and clammy.
The number and severity of postinfarction complications depend on the location and extent of necrosis, the individual's physiologic condition before the infarction, and the availability of swift therapeutic intervention. Table 32.5 lists the most common complications of MI. Sudden cardiac death can occur in individuals with myocardial ischemia even if infarction is absent or minimal. Risk factors for sudden death are related to an interaction among three factors: ischemia, left ventricular dysfunction, and electrical instability.
Table 32.5
Evaluation and Treatment
There are several defined categories of myocardial infarction, each with their own criteria for diagnosis.98 In general, the diagnosis of acute MI is made on the basis of history, physical examination, ECG results, and serial cardiac troponin elevations.
MI can occur in various regions of the heart wall and may be described as anterior, inferior, posterior, lateral, subendocardial, or transmural, depending on the anatomic location and extent of tissue damage from infarction. Twelve-lead ECGs help localize the affected area through identification of changes in ST segments and T waves. The infarcted myocardium is surrounded by a zone of hypoxic injury, which may progress to necrosis or return to normal, and adjacent to this zone of hypoxic injury is a zone of reversible ischemia (Fig. 32.13). A characteristic Q wave often develops on ECG some hours later in STEMI.
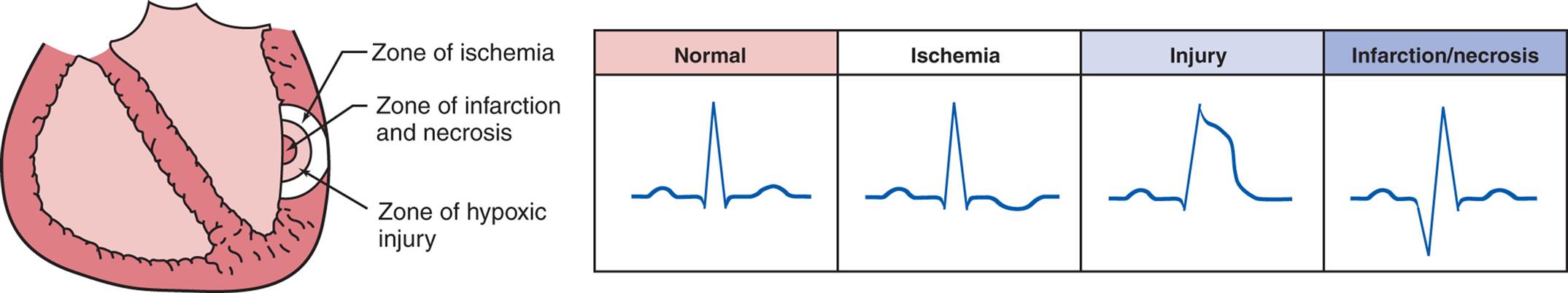
An illustration of cross-section of the heart indicating the following zones from the inside to the outside: a zone of ischemia, zone of infarction, and zone of hypoxic injury. An accompanying table comprises the electrocardiogram of normal, ischemic, injury, and infarction or necrosis.
Cardiac troponin I (cTnI) is the most specific indicator of MI, and measurement of its level should be performed on admission to the emergency department. cTnI level elevation is detectable 2 to 4 hours after onset of symptoms. Additional measurements within 6 to 9 hours and again at 12 to 24 hours are recommended if clinical suspicion is high and previous samples were negative. Troponin levels also can be used to estimate infarct size and therefore the likelihood of complications. Additional laboratory data may reveal leukocytosis and elevated C-reactive protein (CRP), both of which indicate inflammation. The individual's blood glucose level is usually elevated and the glucose tolerance level may remain abnormal for several weeks.
Acute MI requires admission to the hospital, often directly into a coronary care unit. The individual should be given an aspirin immediately (ticlopidine if allergic to aspirin) along with nitrates and morphine for pain. Continuous monitoring of cardiac rhythms and enzymatic changes is essential, because the first 24 hours after onset of symptoms is the time of highest risk for sudden death. Non-STEMI is treated in the same way as unstable angina including antithrombotics, anticoagulation or PCI, or both.89 STEMI is best managed with emergent PCI and antithrombotics.99,100 Hyperglycemia is treated with insulin, and hyperlipidemia with statins. Recent studies suggest that intravenous statins given as a bolus during acute MI management may reduce infarct size, improve systolic contraction, and reduce remodeling.101 ACE inhibitors should be prescribed as secondary prevention.79 Individuals who are in shock require aggressive fluid resuscitation, ionotropic drugs, and possible emergent invasive procedures.
Bed rest, followed by gradual return to activities of daily living, reduces the myocardial oxygen demands of the compromised heart. Individuals not receiving thrombolytic or heparin infusion must receive DVT prophylaxis as long as their activity is significantly limited. Stool softeners are given to eliminate the need for straining. Education regarding appropriate diet and caffeine intake, smoking cessation, exercise, and other aspects of risk factor reduction is crucial for secondary prevention of recurrent myocardial ischemia.
Disorders of the Heart Wall
Disorders of the Pericardium
Pericardial disease is a localized manifestation of another disorder, such as infection (bacterial, viral, fungal, rickettsial, or parasitic); trauma or surgery; neoplasm; or a metabolic, immunologic, or vascular disorder (uremia, rheumatoid arthritis, systemic lupus erythematosus, periarteritis nodosa). The pericardial response to injury from these diverse causes may consist of pericarditis, pericardial effusion, or constrictive pericarditis.
Pericarditis
Pericarditis can be categorized as acute, recurrent, or chronic. Acute pericarditis is acute inflammation of the pericardium. The etiology of acute pericarditis is most often idiopathic or caused by viral infection by coxsackie, influenza, hepatitis, measles, mumps, varicella viruses, or human immunodeficiency virus (HIV). Other causes include MI, trauma, neoplasm, bacterial infection (especially tuberculosis), connective tissue disease (especially systemic lupus erythematosus and rheumatoid arthritis), or hypothyroidism. Iatrogenic causes include radiation therapy, cardiac surgery, pacemaker insertion, radiofrequency ablation, transcatheter aortic valve implantation, and PCI.102 The pericardial membranes become inflamed with the release of inflammatory cytokines such as IL-1, and a pericardial effusion may develop that can be serous, purulent, or fibrinous (Fig. 32.14).
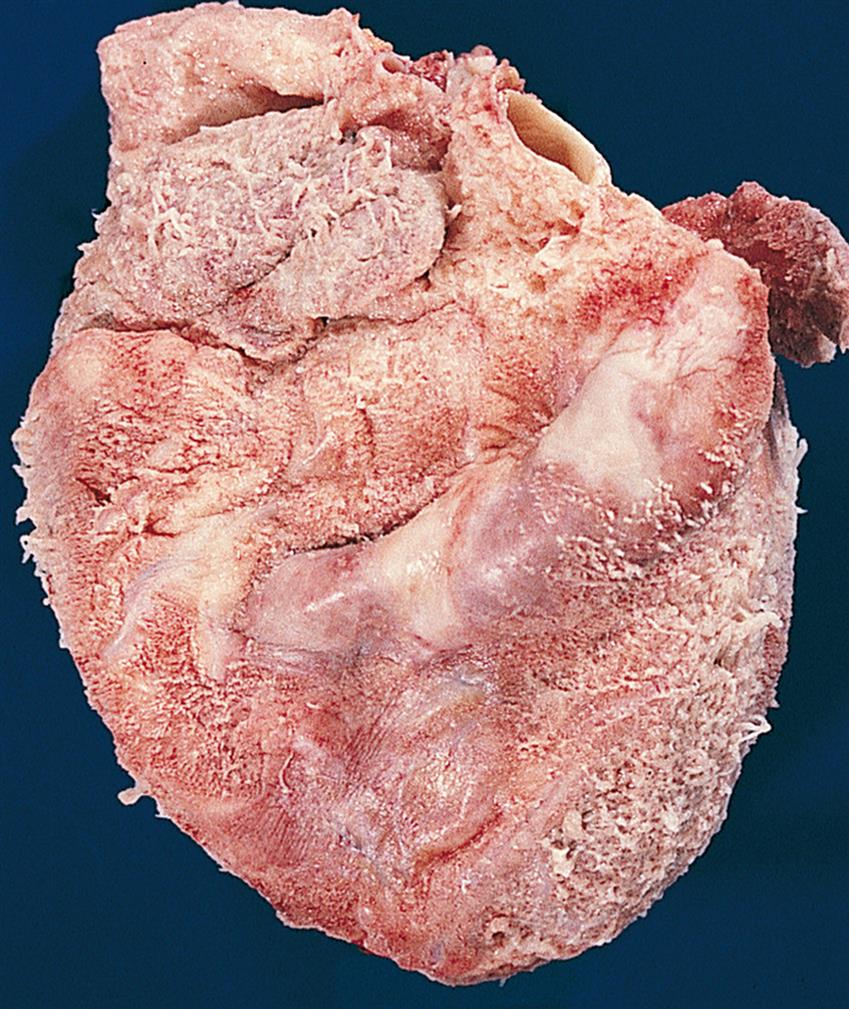
Note shaggy coat of fibers covering the surface of heart. (From Damjanov I, Linder J. Pathology: A color atlas. St. Louis: Mosby; 2000.)
Symptoms may follow several days of fever and usually begin with the sudden onset of severe retrosternal chest pain that worsens with respiratory movements and when assuming a recumbent position. The pain may radiate to the back as a result of irritation of the phrenic nerve (innervates the trapezius muscles) as it traverses the pericardium. Individuals with acute pericarditis also report dysphagia, restlessness, irritability, anxiety, weakness, and malaise.
Physical examination often reveals low-grade fever and sinus tachycardia. An intermittent friction rub—a scratchy, grating sound—may be heard at the cardiac apex and left sternal border and is highly suggestive of pericarditis. The rub is caused by the roughened pericardial membranes rubbing against each other. Evidence of underlying infection or disease may also be evident. ECG findings may reflect inflammatory processes through PR segment depression and diffuse ST segment elevation (sometimes described as “saddle shaped”) without Q waves, and they may remain abnormal for days or even weeks. Cardiac troponins may be slightly elevated in some individuals. Echocardiography is the primary imaging modality for diagnosis. Some individuals will require CT or cardiac magnetic resonance (CMR) evaluation. Acute pericarditis requires at least two of the following four criteria for diagnosis: (1) chest pain characteristics of pericarditis, (2) pericardial rub, (3) characteristic electrocardiographic changes, and (4) new or worsening pericardial effusion.103
Treatment for uncomplicated acute pericarditis consists of administration of antiinflammatory agents, such as salicylates and NSAIDs, and colchicine.102 Exploration of the underlying cause is important. Approximately one-third of cases will be complicated by the development of recurrent pericarditis (symptoms return after 4 to 6 weeks without symptoms) and may require the administration of more aggressive antiinflammatory and immunosuppressive medications such as corticosteroids.103 Other possible sequelae of pericarditis include chronic pericarditis (symptoms persist beyond 3 months), cardiac tamponade, or constrictive pericarditis.
Pericardial Effusion
Pericardial effusion is the accumulation of fluid in the pericardial cavity and can occur in all forms of pericarditis. Many are idiopathic (20%), but other causes, such as neoplasm, infection, renal failure, or hypothyroidism must be considered.104 Analysis of the fluid obtained through pericardiocentesis allows for identification of the likely source of the fluid. The fluid may be a transudate, such as the serous effusion that develops with left heart failure, overhydration, or hypoproteinemia. More often, however, the fluid is an exudate, which reflects pericardial inflammation like that seen with acute pericarditis, MI, heart surgery, some chemotherapeutic agents, infections, and autoimmune disorders such as systemic lupus erythematosus. (Types of exudate are described in Chapter 7.) If the fluid is serosanguineous, the underlying cause is likely to be tuberculosis, neoplasm, uremia, or radiation, although some remain idiopathic. Effusions of frank blood are generally related to aneurysms, trauma, or coagulation defects. If chyle leaks from the thoracic duct, it may enter the pericardium and lead to cholesterol pericarditis.
If a pericardial effusion develops gradually, the pericardium can stretch to accommodate large quantities of fluid without compressing the heart. If the fluid accumulates rapidly, however, even a small amount (50 to 100 mL) may create sufficient pressure to cause cardiac compression, a serious condition known as tamponade. In tamponade, the pressure exerted by the pericardial fluid eventually equals or even exceeds diastolic pressure within the heart chambers, which interferes with right atrial filling. This causes increased venous pressure, systemic venous congestion, and signs and symptoms of right heart failure (distention of the jugular veins, edema, hepatomegaly). Decreased atrial filling also leads to decreased ventricular filling, decreased stroke volume, and reduced cardiac output. Life-threatening circulatory collapse may occur.
An important clinical finding is pulsus paradoxus, in which arterial blood pressure during expiration exceeds arterial pressure during inspiration by more than 10 mm Hg. Pulsus paradoxus in the setting of a pericardial effusion indicates tamponade and reflects impairment of diastolic filling of the left ventricle plus reduction of blood volume within all four cardiac chambers.
Other clinical manifestations of pericardial effusion are distant or muffled heart sounds, poorly palpable apical pulse, dyspnea on exertion, and dull chest pain. A chest x-ray film may disclose a “water-bottle configuration” of the cardiac silhouette. An echocardiogram (which often may be done at the bedside) can detect an effusion as small as 20 ml and is a reliable and accurate diagnostic test, although CT scans also may be done.
Treatment of pericardial effusion or tamponade consists of pericardiocentesis (aspiration of excessive pericardial fluid) and treatment of the underlying condition.104 Persistent pain may be treated with analgesics, antiinflammatory medications, or steroids. Surgery may be required if the underlying cause of tamponade is trauma or aneurysm. A pericardial “window” may be surgically created to prevent tamponade.105,106
Constrictive Pericarditis
Constrictive pericarditis, or restrictive pericarditis (chronic pericarditis), is most commonly idiopathic or associated with viral infection, tuberculosis, radiation exposure, collagen vascular disorders, sarcoidosis, neoplasm, uremia, or cardiac surgery. In constrictive pericarditis, fibrous scarring with occasional calcification of the pericardium causes the visceral and parietal pericardial layers to adhere, obliterating the pericardial cavity. The fibrotic lesions encase the heart in a rigid shell (Fig. 32.15). Like tamponade, constrictive pericarditis compresses the heart and eventually reduces cardiac output. Unlike tamponade, however, constrictive pericarditis always develops gradually.