Alterations of Digestive Function
Scarlet R. Spain
 http://evolve.elsevier.com/Rogers/pathophysiology/
http://evolve.elsevier.com/Rogers/pathophysiology/
Disorders of the gastrointestinal (GI) tract disrupt one or more of its structures and functions. The GI tract is a continuous, hollow organ that extends from the mouth to the anus. It includes the esophagus, stomach, small intestine, large intestine, and rectum. The accessory organs of digestion include the salivary glands, liver, gallbladder, and pancreas. Structural and neural abnormalities can slow, obstruct, or accelerate the movement of intestinal contents at any level of the GI tract. Inflammatory and ulcerative conditions of the GI wall disrupt secretion, motility, and absorption. Inflammation or obstruction of the liver, pancreas, or gallbladder can alter metabolism and result in local and systemic symptoms. Many clinical manifestations of GI tract disorders are nonspecific and can be caused by a variety of impairments.
Disorders of the Gastrointestinal Tract
Clinical Manifestations of Gastrointestinal Dysfunction
Anorexia
Anorexia is the lack of desire to eat despite physiologic stimuli that would normally produce hunger (see Chapter 23). This nonspecific symptom is often associated with nausea, abdominal pain, diarrhea, psychological stress, and weight loss. Side effects of drugs and pathologic disease processes such as cancer, heart disease, renal disease, and liver disease are often accompanied by anorexia. Anorexia can lead to weight loss, protein energy malnutrition, sarcopenia, functional decline, and is often associated with cachexia.1 The aging population exhibits a high prevalence of anorexia and is an independent predictor of morbidity and mortality in the community and in clinical care settings.2,3
Vomiting
Vomiting (emesis) is an involuntary, forceful emptying of stomach and intestinal contents (chyme) through the mouth. The vomiting center, called the area postrema, lies in the medulla oblongata. The vomiting center contains receptors that may be stimulated to cause vomiting. Stimulation of the vomiting center occurs by either irritants or indirect stimuli. Some causes of indirect stimulation involve the cerebral cortex and thalamus (e.g., anxiety and pain); the vestibular system through the eighth cranial nerve (e.g., motion sickness); and several types of intestinal, vagal, as well as differing types of sympathetic input. Examples of vomiting caused by intestinal, vagal, or sympathetic input include presence of ipecac in the duodenum after ingestion; side effects of certain drugs; distention of the stomach or duodenum; and torsion or trauma affecting the ovaries, testes, uterus, bladder, or kidney. Serotonin (5-hydroxytryptamine [5-HT]) may also stimulate the vomiting center and appears to be released from enterochromaffin cells that lie in the intestinal wall. These cells activate vagal afferents leading to the chemoreceptor trigger zone (CTZ), which leads to vomiting by triggering receptors for dopamine (D2), opioids, acetylcholine, substance P, serotonin (5-HT type 3), and neurokinin-1.
Nausea and retching (dry heaves) are distinct events that usually precede vomiting. Nausea is a subjective experience associated with various conditions, including abnormal pain, used of opioids, and labyrinthine stimulation (i.e., motion). Specific neural pathways that cause nausea have not been identified, but hypersalivation and tachycardia are common associated symptoms. Retching is the muscular event of vomiting without the expulsion of vomitus.
Vomiting begins with deep inspiration. The glottis closes, the intrathoracic pressure decreases, and the esophagus becomes distended. Simultaneously, the abdominal muscles contract, creating a pressure gradient from abdomen to thorax. The lower esophageal sphincter (LES) and body of the stomach relax, but the duodenum and antrum of the stomach spasm. The reverse peristalsis and pressure gradient force chyme from the stomach and duodenum up into the esophagus. Because the upper esophageal sphincter is closed, chyme does not enter the mouth. When the stomach is full of gastric contents, the diaphragm is forced high into the thoracic cavity by strong contractions of the abdominal muscles. The higher intrathoracic pressure forces the upper esophageal sphincter to open, and chyme is expelled from the mouth. Then the stomach relaxes and the upper part of the esophagus contracts, forcing the remaining chyme back into the stomach. The LES then closes. The cycle is repeated if there is a volume of chyme remaining in the stomach. A diffuse sympathetic discharge causes the tachycardia, tachypnea, and diaphoresis that accompany retching and vomiting. The parasympathetic system mediates copious salivation, increased gastric motility, and relaxation of the upper and LESs.
Spontaneous vomiting not preceded by nausea or retching is called projectile vomiting. It is caused by direct stimulation of the vomiting center by neurologic lesions (e.g., increased intracranial pressure, tumors, or aneurysms) involving the brainstem (see Chapter 17), or it can be a symptom of GI obstruction (pyloric stenosis). The metabolic consequences of vomiting are fluid, electrolyte, and acid-base disturbances, including hyponatremia, hypokalemia, hypochloremia, and metabolic alkalosis (see Chapter 3). The management of nausea and vomiting includes fluid and electrolyte maintenance, use of medications, and complimentary nonpharmacologic therapies.4
Constipation
Constipation is difficult or infrequent defecation. It is a common problem, afflicting 24% of the United States population. Although it affects all age groups, prevalence increases with age.5
Constipation means a decrease in the number of bowel movements per week, hard stools, straining, abdominal pain, and difficult evacuation. The definition of constipation must be individually determined because normal bowel habits range from one to three evacuations per day to one per week.
Pathophysiology
Constipation can occur as a primary or secondary condition. Chronic idiopathic or primary constipation is generally classified into three categories: functional defecation disorder, slow transit constipation (STC), and constipation-predominant irritable bowel syndrome (IBS-C). Overlap may exist between these three classifications, and the classifications are not mutually exclusive.6Functional constipation is similar between children and adults, but differences exist regarding the symptomology and pathophysiology of disease, as well as differences in required diagnostic work-up and treatment modalities.7 Functional constipation involves a normal rate of stool passage but difficulty with stool evacuation. STC involves impaired colonic motor activity with symptoms of infrequent bowel movements, straining to defecate, mild abdominal distention, and palpable stool in the sigmoid colon. IBS-C is associated with chronic constipation and abdominal pain. The exact cause of IBS-C is poorly understood, but various factors that may contribute to the pathology of the disease include diet, genetics, colonic motility, absorption, socioeconomic status, daily behaviors, and medication use.8 Lack of access to toilet facilities, consistent suppression of the urge to empty the bowel, pelvic floor dyssynergia, and dehydration may be other causes of primary constipation.
Secondary constipation can be caused by diet, medications, or neurogenic disorders (e.g., stroke, Parkinson disease, spinal cord lesions, multiple sclerosis, and Hirschsprung disease) in which neural pathways or neurotransmitters are diseased or degenerated, resulting in altered or delayed colon transit time. Rectal fissures, strictures, or hemorrhoids also may cause constipation. Antacids containing calcium carbonate or aluminum hydroxide, anticholinergics, iron, and bismuth tend to inhibit bowel motility causing constipation. Opioid-induced constipation is caused by drugs that activate μ-opioid receptors in the gut and slow transit time. Endocrine or metabolic disorders, which may also be associated with constipation, include hypothyroidism, diabetes mellitus, hypokalemia, and hypercalcemia. Pelvic hiatal hernia (herniation of the bowel through the floor of the pelvis), diverticula, irritable bowel syndrome (IBS) (constipation predominant), and pregnancy are associated with constipation. Aging may result in decreased mobility, changes in neuromuscular function, use of medications, and comorbid medical conditions causing constipation. Pain or weakness of the abdominal muscles may interfere with the generation of adequate intra-abdominal pressure needed to evacuate stool from the rectum. Depression may also impair bowel evacuation due to a sedentary lifestyle and diet changes. It is important to remember that constipation or a notable change in bowel habits can be an indication of colorectal cancer (CRC).
Clinical Manifestations
Indicators of primary or functional constipation (not including IBS-C) are guided by the Rome IV criteria. The Rome IV criteria define chronic constipation as including at least two of the following symptoms: straining with defecation at least 25% of the time; lumpy or hard stools at least 25% of the time; sensation of incomplete emptying at least 25% of the time; feeling of anorectal obstruction/blockage at least 25% of the time; manual maneuvers to facilitate stool evacuation for at least 25% of defecations; and fewer than three bowel movements per week (see Table 41.1 for Rome IV criteria).6,8 Changes in bowel evacuation patterns, such as less frequent defecation, smaller stool volume, hard stools, difficulty passing stools (e.g., straining), a feeling of bowel fullness and discomfort, or blood in the stools, require further assessment. Straining to evacuate stool may cause engorgement of the hemorrhoidal veins and hemorrhoidal disease or thrombosis with rectal pain, bleeding, and itching. Passage of hard stools can cause painful anal fissures. Fecal impaction (hard, dry stool retained in the rectum) is associated with rectal bleeding, abdominal or cramping type pain, nausea and vomiting, weight loss, and episodes of diarrhea. If left untreated, fecal impaction may cause increased pressure on the intralumen of the colon that may lead to ischemia with possible perforation of the colon and even death.9
Table 41.1

Evaluation and Treatment
The history, current use of medications, physical examination, and stool diaries provide precise clues regarding the nature of constipation. The individual’s description of the duration of symptoms, frequency of bowel movements, stool consistency, difficult rectal evacuation, sense of incomplete evacuation, presence of blood with stools, and if evacuation was stimulated by enemas or laxatives is important. It is also important to note any abdominal pain or bloating and if any type of digital evacuation to remove stool has been performed. The clinician should also discuss comorbidities, previous GI disorders or surgeries, diet, fluid intake, and physical activity. As mentioned, sudden-onset constipation may signify a new or developing mass and requires careful evaluation for CRC. Abdominal palpation may disclose colonic distention, masses, and tenderness. Digital examination of the rectum and anorectal manometry are performed to assess sphincter tone and detect anal lesions. Colonic transit time and imaging techniques can assist in identifying the cause of constipation. Colonoscopy is used to visualize the bowel lumen directly and can help with identification of polyps, inflammatory bowel diseases (IBDs), or other suspicious lesions including tumors.
The treatment for constipation is to manage the underlying cause or disease. Lifestyle modifications can often help immensely. Management of constipation usually consists of bowel retraining, in which the individual establishes a satisfactory bowel evacuation routine without becoming preoccupied with bowel movements. The individual also may need to engage in moderate exercise and increase fluid and fiber intake. Fiber supplements and stool softeners are useful for some individuals. Many different types of laxatives are available; however, studies are lacking comparing the efficacy and safety of different categories of laxatives. Choice of laxative for an individual should be guided by a healthcare professional and should take into account individual preferences and cost.5 Enemas can be used to establish a bowel routine, but they should not be used routinely. Biofeedback may be beneficial in some instances for forming new bowel evacuation habits. Colectomy with ileorectal anastomosis is rarely performed but may be done with individuals with severe symptoms that have not responded to other treatments.5
Diarrhea
Diarrhea is the presence of loose, watery stools and may be acute, persistent, or chronic. Diarrhea is defined as the passage of three or more loose or liquid stools per day, or more frequent passing of stools than what has routinely been “normal” for a specific individual.10 Acute diarrhea is more than three loose stools developing within 24 hours and lasting less than 14 days. Persistent diarrhea is diarrhea in an individual that lasts longer than 14 to 30 days and chronic diarrhea is diarrhea that lasts longer than 4 weeks. Diarrhea can have high rates of morbidity and mortality in children younger than 5 years of age, particularly in developing countries (see Chapter 42), and in the elderly. Diarrheal disease is the second leading cause of death in children younger than age 5.10
Many factors determine stool volume, including water content of the colon, diet, presence of nonabsorbed food or material, and intestinal secretions. Stool volume in the normal adult averages less than 200 g/day. Stool volume in children depends on age and size. An infant may pass up to 100 g/day. The adult intestine processes approximately 9 L of luminal contents per day: 2 L are ingested, and the remaining 7 L consist of intestinal secretions. Of this volume, most of the fluid is absorbed: 90% (7 to 8 L) in the small intestine and a smaller amount 9% (1 to 2 L) in the colon. Normally, approximately 150 mL of water is excreted daily in the stool.
Pathophysiology
The intestinal mucosa is made up of a complex epithelium where absorption and secretion occur. The majority of water and electrolyte absorption occurs in the small intestine.11 Diarrhea in which the volume of feces is increased is called large-volume diarrhea. It generally is caused by excessive amounts of water or secretions in the intestines. Small-volume diarrhea, in which the volume of feces is not increased, usually results from excessive intestinal motility and may be caused by an inflammatory disorder of the intestine, such as ulcerative colitis (UC), Crohn disease (CD), or microscopic colitis, but also can result from colon cancer or fecal impaction.
The three major mechanisms of diarrhea are osmotic, secretory, and motile.
- 1. Osmotic diarrhea. A nonabsorbable substance in the intestine draws excess water into the lumen of the intestine by osmosis and increases stool weight and volume, producing large-volume diarrhea. Large oral doses of poorly absorbed ions, such as magnesium, sulfate, and phosphate, can increase intraluminal osmotic pressure. Excessive ingestion of synthetic, nonabsorbable sugars (e.g., sorbitol); introduction of full-strength tube feeding formulas; and dumping syndrome associated with gastric resection draw water into the intestinal lumen. Once ingestion of the osmotic substance stops, the osmotic diarrhea will stop. Malabsorption related to lactase deficiency, pancreatic enzyme, or bile salt deficiency, small intestine bacterial overgrowth, or celiac disease also causes osmotic diarrhea.
- 2. Secretory diarrhea. A form of large-volume diarrhea caused by excessive mucosal secretions of chloride or bicarbonate-rich fluid or overall inhibition of net sodium absorption. Infectious causes include viruses (e.g., rotavirus), bacterial enterotoxins (e.g., Escherichia coli, Vibrio cholera, Shiga toxin), exotoxins (e.g., overgrowth of Clostridioides difficile following antibiotic therapy), or small bowel bacterial overgrowth. These infections cause secretion of transmitters from enteroendocrine cells (e.g., cells found in the wall of the bowel that have numerous processes in the body), activation of afferent neurons that stimulate submucosal secretomotor neurons, and altered sodium and chloride transport that results in decreased water absorption. Certain neoplasms (e.g., gastrinoma and thyroid carcinoma) also produce hormones that stimulate intestinal secretion, causing diarrhea.
Small-volume diarrhea is usually caused by an inflammatory bowel disorder, such as UC or CD. Inflammation of the colon causes smooth muscle contraction, cramping type pain, bowel urgency, and frequency. Small-volume diarrhea also can be caused by fecal impaction. The diarrhea caused from a fecal impaction consists of secretions of mucus of fluid produced by the colon to lubricate the impacted feces and move it toward the anal canal. These secretions flow around the impaction and cause low-volume, secretory diarrhea. - 3. Motility diarrhea. Excessive motility decreases transit time and the opportunity for fluid absorption, resulting in diarrhea. This type of diarrhea is caused by resection of the small intestine (short bowel syndrome), surgical bypass of an area of the intestine, fistula formation between loops of intestine, IBS–diarrhea predominant, diabetic neuropathy, hyperthyroidism, and laxative abuse.
Clinical Manifestations
Diarrhea can be acute or chronic, depending on the cause. Systemic effects of prolonged diarrhea are dehydration, electrolyte imbalance (hyponatremia, hypokalemia), metabolic acidosis, and weight loss. Manifestations of acute bacterial or viral infection include fever, with or without vomiting or cramping pain. Most diarrhea caused by infectious organisms lasts less than 2 weeks, although some causes of bacterial gastroenteritis may last longer, such as C. difficile, Aeromonas, or Yersinia enterocolitica. Fever, cramping pain, and bloody stools accompany chronic diarrhea caused by IBD or dysentery. Steatorrhea (fat in the stools), bloating, and diarrhea are common signs of malabsorption syndromes. Diarrhea may also cause anal and perineal skin irritation.
Evaluation and Treatment
A thorough history is taken to document the onset, frequency, volume of stools, duration of diarrhea, and presence of blood in the stools. Documentation of recent travel is important to obtain in the history. Iatrogenic diarrhea is suggested if the individual has undergone abdominal radiation therapy, intestinal resection, or treatment with selected drugs (e.g., antibiotics, diuretics, antihypertensives, laxatives, anticoagulants, or chemotherapy). A thorough physical examination should be completed and can help identify underlying systemic disease. Stool studies, abdominal imaging, endoscopy, and intestinal biopsies provide more specific data, particularly for persistent diarrhea.
Treatment for diarrhea includes restoration of fluid and electrolyte balance, administration of antimotility (e.g., loperamide) and/or water-absorbent (e.g., attapulgite and polycarbophil) medications, and treatment of causal factors. Natural bran and commercial preparations of psyllium are inexpensive and effective treatments for mild diarrhea. Probiotics can be useful for preventing and treating C. difficile–associated diarrhea as an approach to restoring normal microflora in addition to antibiotic therapy. Fecal transplantation can be used for cases that are resistant to conventional therapies, particularly C. difficile–associated diarrhea. Nutritional deficiencies need to be corrected in cases of chronic diarrhea or malabsorption.12
Abdominal Pain
Abdominal pain is the presenting symptom of several GI diseases and can be acute or chronic. The causal mechanisms of abdominal pain are mechanical, inflammatory, or ischemic. Abdominal organs are sensitive to stretching and distention. This stretching activates nerve endings in both hollow and solid structures, causing pain. Pain accompanies rapid distention rather than gradual distention. Traction on the peritoneum caused by adhesions, distention of the common bile duct, or forceful peristalsis resulting from intestinal obstruction causes pain because of increased tension. Capsules that surround solid organs, such as the liver and gallbladder, contain pain fibers that are stimulated by stretching if these organs swell. Abdominal pain may be generalized to the abdomen or localized to a particular abdominal quadrant. The nature of the pain is often described as sharp, dull, or colicky.
Abdominal pain is usually associated with tissue injury and inflammation. Biochemical mediators of the inflammatory response, such as histamine, bradykinin, and serotonin, stimulate organic nerve endings and produce abdominal pain. The edema and vascular congestion that accompany chemical, bacterial, or viral inflammation also cause painful stretching. Hindrance of blood flow from the distention of bowel obstruction or mesenteric vessel thrombosis produces the pain of ischemia and increased concentrations of tissue metabolites stimulate pain receptors.
Abdominal pain can be parietal (somatic), visceral, or referred. Parietal pain, originating from the parietal peritoneum, is more localized and intense than visceral pain, which arises from the organs themselves. Nerve fibers from the parietal peritoneum are predominantly A-delta fibers and travel with somatic peripheral nerves to the spinal cord. Parietal pain is caused by an irritation of fibers of the peritoneal peritoneum or lining. The sensation of pain is localized to the dermatome superficial to the area of painful stimuli. Visceral pain arises from a stimulus (distention, inflammation, ischemia) causing stretching, damage, or disruption of the organ or organ tissue involved and is transmitted via sympathetic fibers. Inflammatory mediators associated with chronic low-grade inflammation can cause pain hypersensitivity, and they include neurokinins, histamine, serotonin, and proteases.13 These mediators can activate voltage-gated sodium ion channels.14 Pain is usually near the midline in the epigastrium, midabdomen, or lower abdomen because sensory afferents enter the spinal cord bilaterally and lack specificity. The pain is usually poorly localized, diffuse, or vague with a radiating pattern because nerve endings in abdominal organs are sparse and multisegmented. Pain arising from the stomach, for example, is experienced as a sensation of fullness, cramping, or gnawing in the midepigastric area. Referred pain is visceral pain felt at some distance from a diseased or affected organ. It is usually well localized and is felt in the skin dermatomes or deeper tissues that share a central afferent pathway with the affected organ. For example, acute cholecystitis may have pain referred to the right shoulder or scapula.
Gastrointestinal Bleeding
Upper GI bleeding is bleeding in the esophagus, stomach, or duodenum and is characterized by frank, bright red bleeding or dark, grainy digested blood (“coffee grounds”) in the stool (Table 41.2). Upper GI bleeding is commonly caused by bleeding esophageal or gastric varices, peptic ulcers, arteriovenous malformations, or a Mallory-Weiss tear at the esophageal-gastric junction caused by severe retching. Upper GI bleeding may also be associated with use of nonsteroidal antiinflammatory drugs (NSAIDs), selective serotonin reuptake inhibitors, and antiplatelet and anticoagulant drugs.15,16
Table 41.2
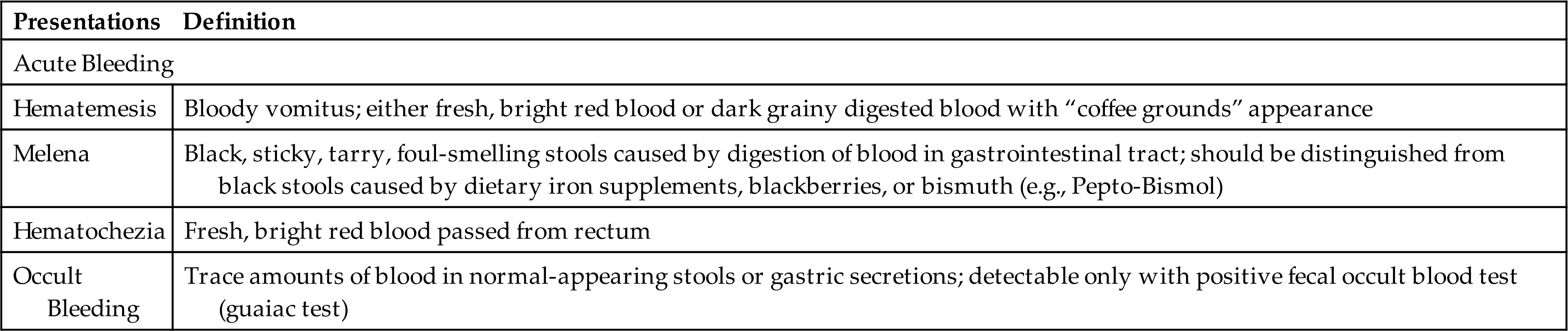
Lower GI bleeding, or bleeding from the jejunum, ileum, colon, or rectum, can be caused by polyps, diverticulitis, inflammatory disease, cancer, or hemorrhoids. Occult bleeding is usually caused by slow, chronic blood loss that is not obvious and results in iron deficiency anemia as iron stores in the bone marrow are slowly depleted.
Physiologic response to GI bleeding depends on the amount and rate of the loss. Acute, severe GI bleeding can be life threatening, depending on the volume and rate of blood loss, associated diseases, the age of the individual, and the effectiveness of treatment. Changes in blood pressure and heart rate are the best indicators of massive blood loss in the GI tract. During the early stages of blood volume depletion, the peripheral arteries and arterioles constrict to shunt blood to vital organs, including the brain. Signs of large-volume blood loss are postural hypotension (a drop in blood pressure that occurs with a change from the recumbent position to a sitting or upright position), lightheadedness, and loss of vision. Tachycardia develops as a compensatory response to maintain cardiac output and tissue perfusion. If blood loss continues, hypovolemic shock develops (see Chapters 32 and 48). Diminished blood flow to the kidneys causes decreased urine output and may lead to oliguria (low urine output), tubular necrosis, and renal failure. Ultimately, insufficient cerebral and coronary blood flow causes irreversible anoxia and death (Fig. 41.1).
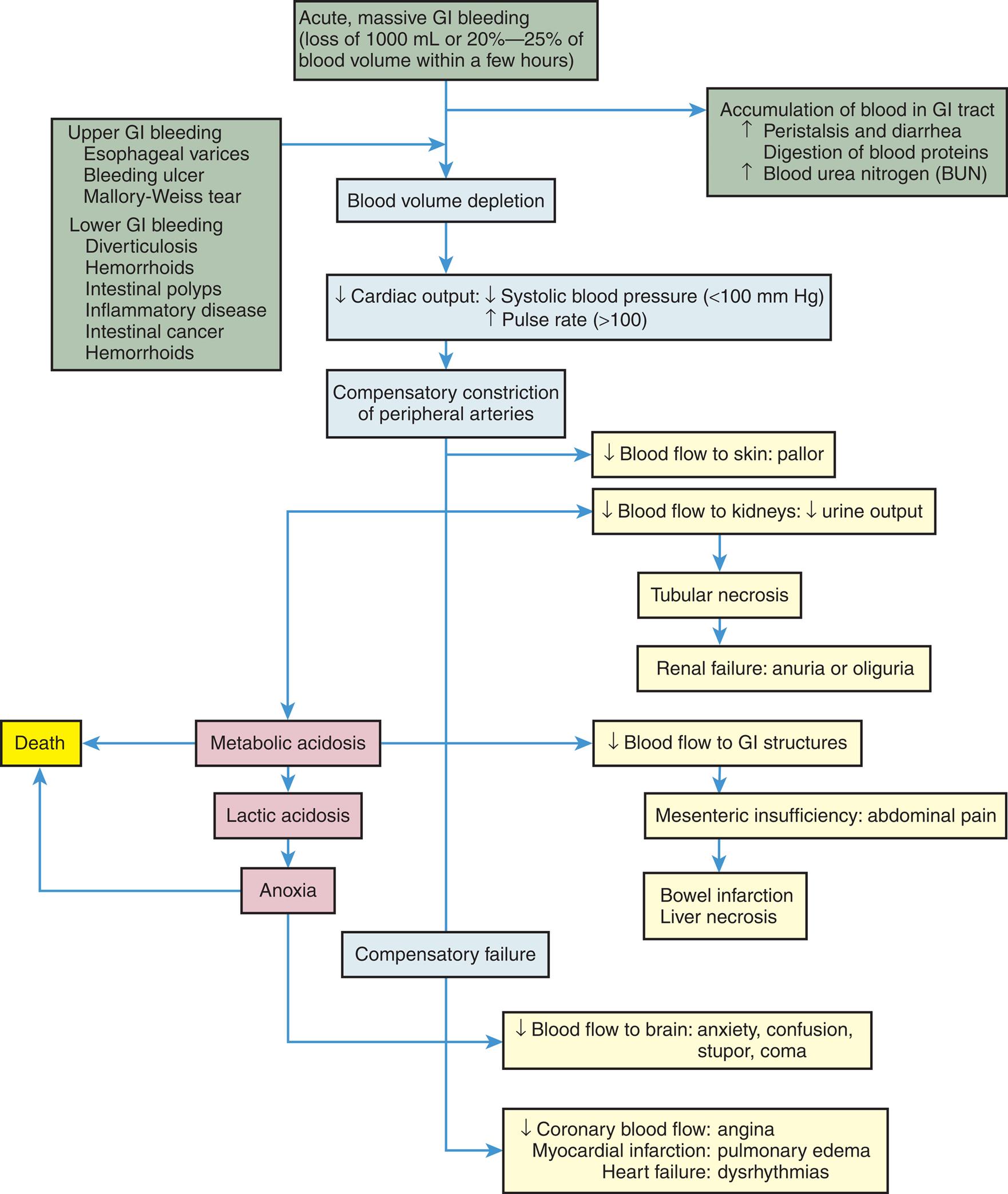
A flow diagram summarizes the pathophysiology of gastrointestinal bleeding, which beings as follows. • Upper G I bleeding consisting esophageal varices, bleeding ulcer, Mallory-Weiss tear; lower G I bleeding consisting of intestinal polyps, inflammatory disease, intestinal cancer, hemorrhoids; acute or massive G I bleeding consisting of loss of 1000 milliliters or 20 percent to 25 percent of blood volume within a few hours leads to accumulation of blood in G I tract which increases peristalsis and diarrhea, digestion of blood proteins and blood urea nitrogen which further leads to blood volume depletion. • Blood volume depletion leads to a decrease in cardiac output, which consists of a decrease in systolic blood pressure of less than 100 millimeters of mercury and an increase in pulse rate of greater than 100. • Decrease cardiac output leads to compensatory constriction of peripheral arteries, which leads to compensatory failure due to decreased coronary blood flow: angina; myocardial infarction: pulmonary edema; heart failure: dysrhythmias. • Compensatory constriction of peripheral arteries includes a decrease in blood flow to skin: pallor; decreased blood flow to kidneys with decreased urine output leads to tubular necrosis and metabolic acidosis. • The tubular necrosis leads to renal failure: anuria or oliguria. • Metabolic acidosis leads to death due to a decrease in blood flow to G I structure, leads to mesenteric insufficiency or abdominal pain. • Mesenteric insufficiency or abdominal pain leads to bowel infarction or liver necrosis. • Metabolic acidosis leads to lactic acidosis. • Lactic acidosis leads to anoxia due to a decrease in blood flow to the brain: anxiety, confusion, stupor, coma, and anoxia lead to death.
The presentations of GI bleeding are summarized in Table 41.2. The accumulation of blood in the GI tract is irritating and increases peristalsis, causing vomiting or diarrhea, or both. Hematemesis may be present. If bleeding is from the lower GI tract, hematochezia (bloody stools) may be present. Bleeding from the upper GI tract may also be rapid enough to produce hematochezia, but generally some digestion of the blood components will have occurred, producing melena. The digestion of blood proteins originating from massive upper GI bleeding is reflected by an increase in blood urea nitrogen (BUN) levels.
The hematocrit and hemoglobin values are not the best indicators of acute GI bleeding because plasma volume and red cell volume are lost proportionately. As the plasma volume is replaced, the hematocrit and hemoglobin values begin to reflect the extent of blood loss. The interpretation of these values is modified to account for exogenous replacement of fluids and the hydration status of the tissues. Anemia associated with chronic GI bleeding is caused by iron depletion. Evaluation and treatment involve identifying and treating the source of the bleeding and replacing iron losses. Administration of blood products may be used for massive hemorrhage. Guidelines are available for the diagnosis and management of GI bleeding that may include endoscopic management with upper GI bleeds.16–18
Disorders of Motility
Dysphagia
Pathophysiology
Dysphagia is difficulty swallowing or the perception of obstruction while swallowing. It can result from mechanical obstruction of the esophagus or from a functional disorder that impairs esophageal motility. Intrinsic obstructions originate in the wall of the esophageal lumen (e.g., esophageal dysphagia) and include tumors, strictures, and diverticular herniations (e.g., outpouchings). Extrinsic mechanical obstructions originate outside the esophageal lumen and narrow the esophagus by pressing inward on the esophageal wall. The most common cause of extrinsic mechanical obstruction is tumor.
Functional dysphagia is caused by neural or muscular disorders that interfere with voluntary swallowing or peristalsis. Disorders that affect the striated muscles of the hypopharyngeal area and upper esophagus interfere with the oropharyngeal (voluntary) phase of swallowing (oropharyngeal dysphagia). Typical causes are dermatomyositis (a muscle disease) and neurologic impairments caused by cerebrovascular accidents, Parkinson disease, multiple sclerosis, muscular dystrophy, or achalasia.
Achalasia is a rare form of dysphagia related to loss of inhibitory neurons in the myenteric plexus with smooth muscle atrophy in the middle and lower portions of the esophagus. A proposed mechanism is that myenteric neurons are attacked by a cell-mediated and antibody-mediated immune response against an unknown antigen (e.g., a virus). This leads to altered esophageal peristalsis and failure of the LES to relax, causing functional obstruction of the lower esophagus with varying severity. Food accumulates above the obstruction, distends the esophagus, and causes dysphagia. Cough and aspiration can occur. As hydrostatic pressure increases, food is slowly forced past the obstruction into the stomach. Chronic inflammation from esophageal food retention can increase risk for esophageal cancer.
Clinical Manifestations
Clinical manifestations of dysphagia vary according to the location of the obstruction. Distention and spasm of the esophageal muscles during eating or drinking may cause a mild or severe stabbing pain at the level of obstruction. Discomfort occurring 2 to 4 seconds after swallowing is associated with upper esophageal obstruction. Discomfort occurring 10 to 15 seconds after swallowing is more common in obstructions of the lower esophagus. If obstruction results from a growing tumor, dysphagia begins with difficulty swallowing solids and advances to difficulty swallowing semisolids and liquids. If motor function is impaired, both solids and liquids are difficult to swallow. Regurgitation of undigested food, an unpleasant taste sensation, vomiting, aspiration, and weight loss are common manifestations of all types of dysphagia. Aspiration of esophageal contents can lead to cough and pneumonia.
Evaluation and Treatment
Knowledge of the individual’s history and clinical manifestations contributes significantly to a diagnosis of dysphagia. Imaging is used to visualize the contours of the esophagus and identify potential structural defects. Esophageal motility testing documents abnormal pressure changes associated with obstruction or loss of neural regulation. Esophageal endoscopy is performed to examine the esophageal mucosa and obtain biopsy specimens.
The individual is taught to manage symptoms by eating small meals slowly, taking fluid with meals, and sleeping with the head elevated to prevent regurgitation and aspiration. Food and medications may need to be formulated with a thickening agent so they can be swallowed. Tube feedings may be required for some individuals, particularly following stroke. Mechanical dilation of the esophageal sphincter and surgical separation of the lower esophageal muscles with a longitudinal incision (myotomy) may be an effective treatment for achalasia.19
Gastroesophageal Reflux Disease
Gastroesophageal reflux disease (GERD) is the reflux of acid and pepsin or bile salts from the stomach into the esophagus, causing esophagitis. The prevalence of GERD is estimated at 18% to 27% in North America.20 Risk factors for GERD include increasing age, obesity, hiatal hernia, and drugs or chemicals that relax the LES (anticholinergics, nitrates, calcium channel blockers, nicotine). GERD may be a trigger for asthma or chronic cough. Gastroesophageal reflux that does not cause symptoms is known as physiologic reflux. In nonerosive reflux disease (NERD), individuals have symptoms of reflux disease but no visible or minimal esophageal mucosal injury (functional heartburn).
Pathophysiology
Abnormalities in LES function, esophageal motility, and gastric motility or emptying can cause GERD. The resting tone of the LES has an average pressure of approximately 20 mm Hg that prevents gastric content from refluxing into the esophagus. Spontaneous relaxation of the LES may be triggered by gastric distention after meals and trigger acid reflux. Acid reflux may be triggered by diet and lifestyle factors such as food intake that causes delayed gastric emptying, acidic foods, and obesity. Sliding hiatal hernia facilitates reflux.21 Vomiting, coughing, lifting, bending, and pregnancy also increase abdominal pressure, contributing to the development of reflux esophagitis.
The severity of the esophagitis depends on the composition of the gastric contents and the esophageal mucosa exposure time. If the gastric contents are highly acidic or contain bile salts and pancreatic or intestinal enzymes, reflux esophagitis can be severe. In individuals with weak esophageal peristalsis, refluxed chyme remains in the esophagus longer than usual. The refluxate causes mucosal injury and inflammation, with hyperemia, increased capillary permeability, edema, tissue fragility, and erosion (Fig. 41.2). Fibrosis and thickening may develop. Precancerous lesions (Barrett esophagus [BE]; see the Esophageal Cancer section) can be a long-term consequence. Precancerous lesions can progress to adenocarcinoma.
Clinical Manifestations
The clinical manifestations of erosive reflux esophagitis are related to mucosal injury from acid regurgitation. Manifestations are heartburn (e.g., pyrosis) and acid regurgitation. Dysphagia, chest pain, chronic cough, asthma attacks (see Chapter 35), laryngitis, hoarseness, and upper abdominal pain that occurs within 1 hour of eating are less common. The symptoms worsen if the individual lies down or if intra-abdominal pressure increases (e.g., as a result of coughing, vomiting, or straining at stool). Edema, strictures, esophageal spasm, or decreased esophageal motility may result in dysphagia with weight loss. Alcohol or acid-containing foods, such as citrus fruits, can cause discomfort during swallowing.
Evaluation and Treatment
The diagnosis of GERD is based on the history and clinical manifestations. Esophageal endoscopy shows hyperemia, edema, erosion, and strictures. Dysplastic changes, such as occurs with BE (see section on esophageal carcinoma), can be identified by tissue biopsy. Impedance/pH monitoring measures the movement of stomach contents upward into the esophagus and the acidity of the refluxate. Heartburn may be experienced as chest pain, so cardiac ischemia must be ruled out.
Treatment includes once-daily proton pump inhibitors (PPIs) for 4 weeks, and continuing therapy if esophagitis or BE is present.21 Weight reduction, smoking cessation, elevation of the head of the bed 6 inches, and avoiding tight clothing may also help to alleviate symptoms. The most common surgical treatment is laparoscopic fundoplication. Emerging surgical treatments include magnetic sphincter augmentation (a device placed around the distal esophagus and comprises titanium beads with magnets in the center that augment lower esophageal tone and thus prevent reflux), radiofrequency ablation, and transoral incisionless fundoplication.
Eosinophilic esophagitis (EoE) is an idiopathic chronic allergic/immune disease of the esophagus characterized by infiltration of eosinophils in the esophagus. EoE is most associated with atopic disease, including asthma, allergic rhinitis, eczema, and food allergies that occur in both children and adults, but the symptoms may vary by age. EoE causes many white blood cells to be found in the inner lining of the esophagus. Typically, eosinophils are not found in the esophagus, although other conditions (e.g., acid reflux disease) may contribute to the presence of eosinophils in the esophagus. Manifestations of the disease are caused by esophageal inflammation (See Chapter 42).22 Dysphagia, decreased appetite, recurring abdominal pain, vomiting, and weight loss are common symptoms. Diagnosis is made by endoscopy with biopsy that identifies the eosinophilic infiltration and differentiates this condition from GERD. Treatment is symptomatic and includes acid inhibitors, elimination diets, and corticosteroids.22 Other conditions associated with EoE, such as food allergies, asthma, or eczema, must also be treated appropriately (see Chapter 42).
Hiatal Hernia
Pathophysiology
Hiatal hernia is a common disorder characterized by a protrusion or bulging of an abdominal structure into the thoracic cavity. Causation is from a weakening of the diaphragm muscle.23 (Fig. 41.3) The most common type is a sliding hiatal hernia (type 1) (see Fig. 41.3 A). In this type of hernia, the proximal portion of the stomach moves into the thoracic cavity through the esophageal hiatus. The esophageal hiatus is an opening in the diaphragm for the esophagus and vagus nerves. A congenitally short esophagus, fibrosis, excessive vagal nerve stimulation, or weakening of the diaphragmatic muscles at the gastroesophageal junction contributes to this type of hernia. Laying in the supine position causes the lower esophagus and stomach to be pulled into the thorax. As an individual stands, the organs slide back into the abdomen. Coughing, bending, tight clothing, ascites, obesity, and pregnancy accentuate the hernia in association with the resting pressure of the LES.
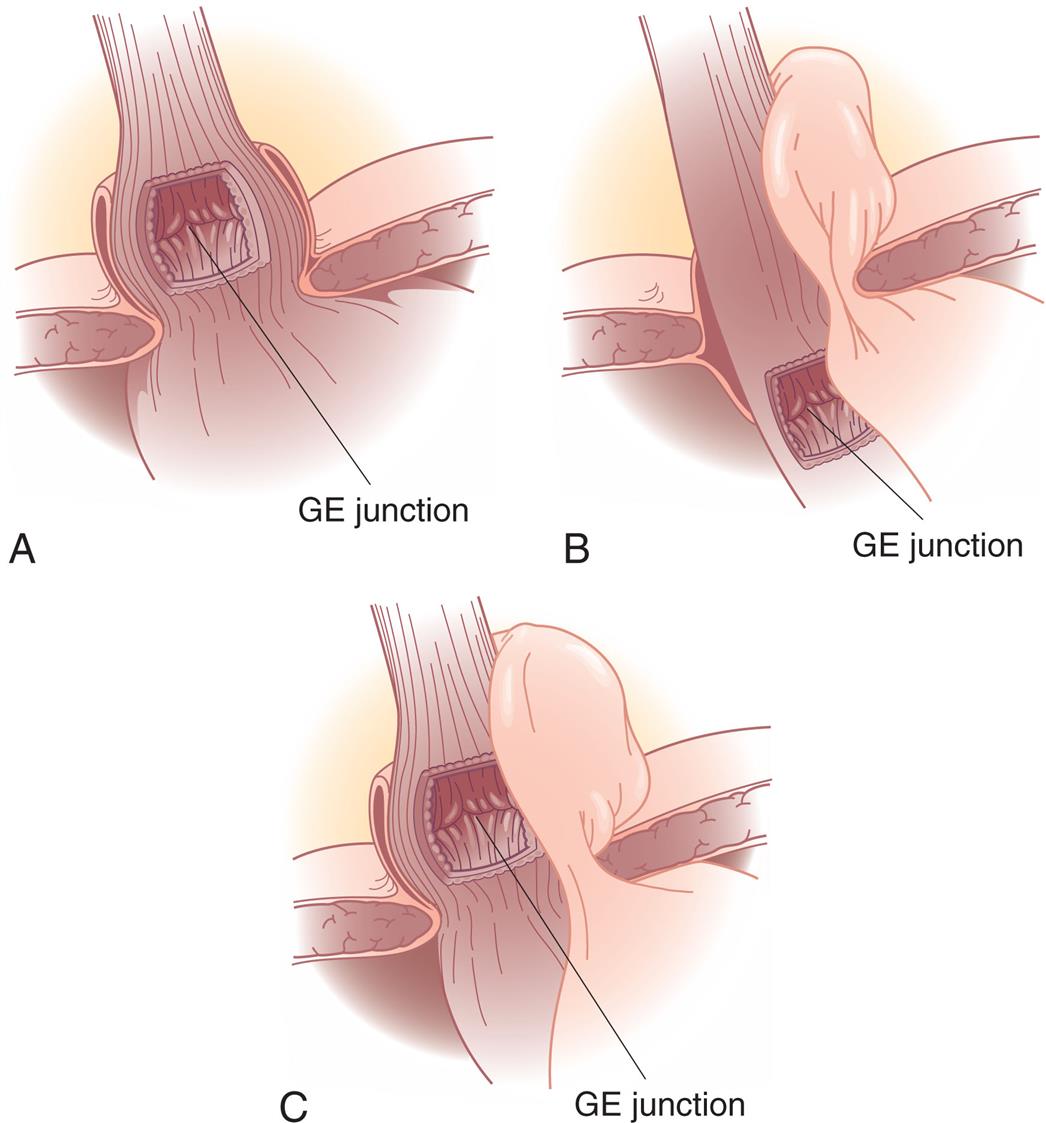
(A) Type I—sliding hernia. The visceral peritoneum remains intact and restrains the size of the hernia in sliding hiatal hernia. (B) Type II—paraesophageal or rolling hernia. The membrane becomes thinner or defective in a paraesophageal hernia, allowing a true peritoneal sac to protrude into the posterior mediastinum, where negative intrathoracic pressure causes it to enlarge. (C) Type III—mixed hernia. GE, Gastroesophageal. Note: Type IV—complex paraesophageal hernia is not shown. (From Townsend CM, et al. Sabiston textbook of surgery: The biologic basis surgical practice, 21st edition. St. Louis: Elsevier; 2022)
Three illustrations depict the three types of hiatal hernia. Illustration A depicts the sliding hernia, which shows the G E junction above the level of diaphragmatic hiatus. Illustration B depicts the rolling hernias, which shows a normal position to the G E junction, but a portion of the fundus above the hiatus. Illustration C depicts the mixed hernia, which shows displacement of both the G E junction and fundus above the hiatus.
Paraoesophageal hiatal hernia (type 2) is a herniation of the greater curvature of the stomach through a secondary opening in the diaphragm alongside the esophagus that moves into the thorax above the diaphragm (see Fig. 41.3 B). This abnormal positioning of a portion of the stomach causes congestion of mucosal blood flow, leading to gastritis and ulcer formation. Reflux is uncommon with this type of hernia. Strangulation of the hernia is a major complication that results with occlusion of blood vessels and causes vascular engorgement with resulting edema, ischemia, and hemorrhage. Manifestations or symptoms of this type of hernia include vomiting and epigastric/retrosternal epigastric pain and is a surgical emergency.
Mixed hiatal hernia (type 3), less common, is a combination of sliding and paraoesophageal hiatal hernias (see Fig. 41.3 C). It tends to occur in conjunction with several other diseases, including reflux esophagitis, peptic ulcer, cholecystitis, cholelithiasis, chronic pancreatitis, and diverticulosis. A mixed hiatal hernia may progress to a type 4 hernia. This type of hernia involves the presence of a structure other than the stomach (e.g., omentum, colon, or small bowel) within the hernia sac.23
Clinical Manifestations
Hiatal hernias are often asymptomatic. In general, a wide variety of symptoms develop later in life; symptoms are associated with other GI disorders, including GERD, and include heartburn, regurgitation, dysphagia, and epigastric pain. Ischemia from hernia strangulation causes acute pain that may include severe chest or epigastric pain and other associated symptoms of nausea, vomiting, and GI bleeding.
Evaluation and Treatment
Many diagnostic procedures may be indicated in diagnosing a hiatal hernia. The mainstays of evaluation are upper endoscopy and barium swallows.23 Plain chest radiographs, contrast studies, esophagogastroduodenoscopy (EGD), manometry, pH testing, and nuclear medicine studies may also be ordered. Computed tomography (CT) scan may be indicated in an urgent situation for an individual with suspected complications.23
Treatment for a sliding hiatal hernia is usually conservative. The individual can diminish reflux by eating small, frequent meals and avoiding the recumbent position after eating. Abdominal supports and tight clothing should be avoided, and weight control is recommended for obese individuals. Antacids may help to alleviate reflux esophagitis. Individuals who are uncomfortable at night may benefit from sleeping with the head of the bed elevated 6 inches. PPIs alleviate reflux esophagitis. Histamine 2 (H2) receptor antagonists and antacids are typically less effective treatments. Drugs that relax the LES, such as anticholinergic type drugs, nitrates, and calcium channel blockers, are contraindicated due to delaying gastric emptying. If medical management fails to provide symptom control or a paraesophageal hiatal hernia is present, a laparoscopic fundoplication may be indicated, and permanent mesh maybe used to prevent recurrence.24
Gastroparesis is delayed gastric emptying in the absence of a mechanical gastric outlet obstruction. It is most associated with diabetes mellitus, surgical vagotomy, or fundoplication but may be idiopathic. The pathophysiology is not well understood but involves abnormalities of the autonomic nervous system, smooth muscle cells, enteric neurons, and GI hormones. Diabetic gastroparesis represents a form of neuropathy involving the vagus nerve. Symptoms include nausea, vomiting, abdominal pain, and postprandial fullness or bloating. Treatment options for gastroparesis are challenging due to the availability of therapies demonstrating poor evidence of efficacy or long-term safety concerns.25 Current treatments include dietary management, prokinetic drugs, endoscopic techniques, and, in some cases, gastric electrical stimulation or surgical venting gastrostomy.25–27
Pyloric Obstruction
Pathophysiology
Pyloric obstruction (gastric outlet obstruction) is the consequence of diseases causing narrowing or blocking of the opening between the stomach and the duodenum. This condition can be congenital (e.g., infantile hypertrophic pyloric stenosis; see Chapter 42) or acquired. Acquired obstruction is caused by peptic ulcer disease or carcinoma near the pylorus. Duodenal ulcers are more likely than gastric ulcers to obstruct the pylorus. Ulceration causes obstruction resulting from inflammation, edema, spasm, fibrosis, or scarring. Tumors cause obstruction by growing into the pylorus.
Clinical Manifestations
Early in the course of pyloric obstruction, the individual experiences vague epigastric fullness, which becomes more distressing after eating and at the end of the day. Nausea and epigastric pain may occur as the muscles of the stomach contract in an attempt to force chyme (pulpy acidic gastric secretions) past the obstruction. These symptoms disappear when the chyme finally moves into the duodenum. As the obstruction progresses, anorexia and accompanying weight loss may occur. Severe obstruction causes gastric distention and atony (lack of muscle tone and gastric motility). Gastric distention stimulates gastric secretion, which increases the feeling of fullness. Rolling or jarring of the abdomen produces a sloshing sound called the succussion splash. At this stage, vomiting is a cardinal sign of obstruction. It is usually copious and occurs several hours after eating. The vomitus contains undigested food but no bile. Prolonged vomiting leads to dehydration, which is accompanied by a hypokalemic and hypochloremic metabolic alkalosis caused by loss of gastric potassium and acid. Food is not able to enter the intestine, making stools infrequent and small. Prolonged pyloric obstruction causes severe malnutrition, dehydration, and extreme debilitation.
Evaluation and Treatment
Diagnosis is based on clinical manifestations, a history of ulcer disease, and examination of residual gastric contents. Endoscopy is performed if gastric carcinoma is the suggested cause of pyloric obstruction.
Obstructions resulting from ulceration often resolve with conservative management. A nasogastric tube is used to aspirate stomach contents and relieve distention. Nasogastric suction is typically placed to decompress the stomach and to help restore normal motility. Gastric secretions that contribute to inflammation and edema can be suppressed with PPIs or H2-receptor antagonists. Fluids and electrolytes (saline and potassium) are given intravenously to promote rehydration and correct hypokalemia and alkalosis (see Chapter 3). Severely malnourished individuals may require parenteral hyperalimentation (artificial nutrients, usually intravenous nutrition). Surgery or the placement of pyloric stents may be required to treat gastric carcinoma or persistent obstruction caused by fibrosis and scarring.28
Intestinal Obstruction and Paralytic Ileus
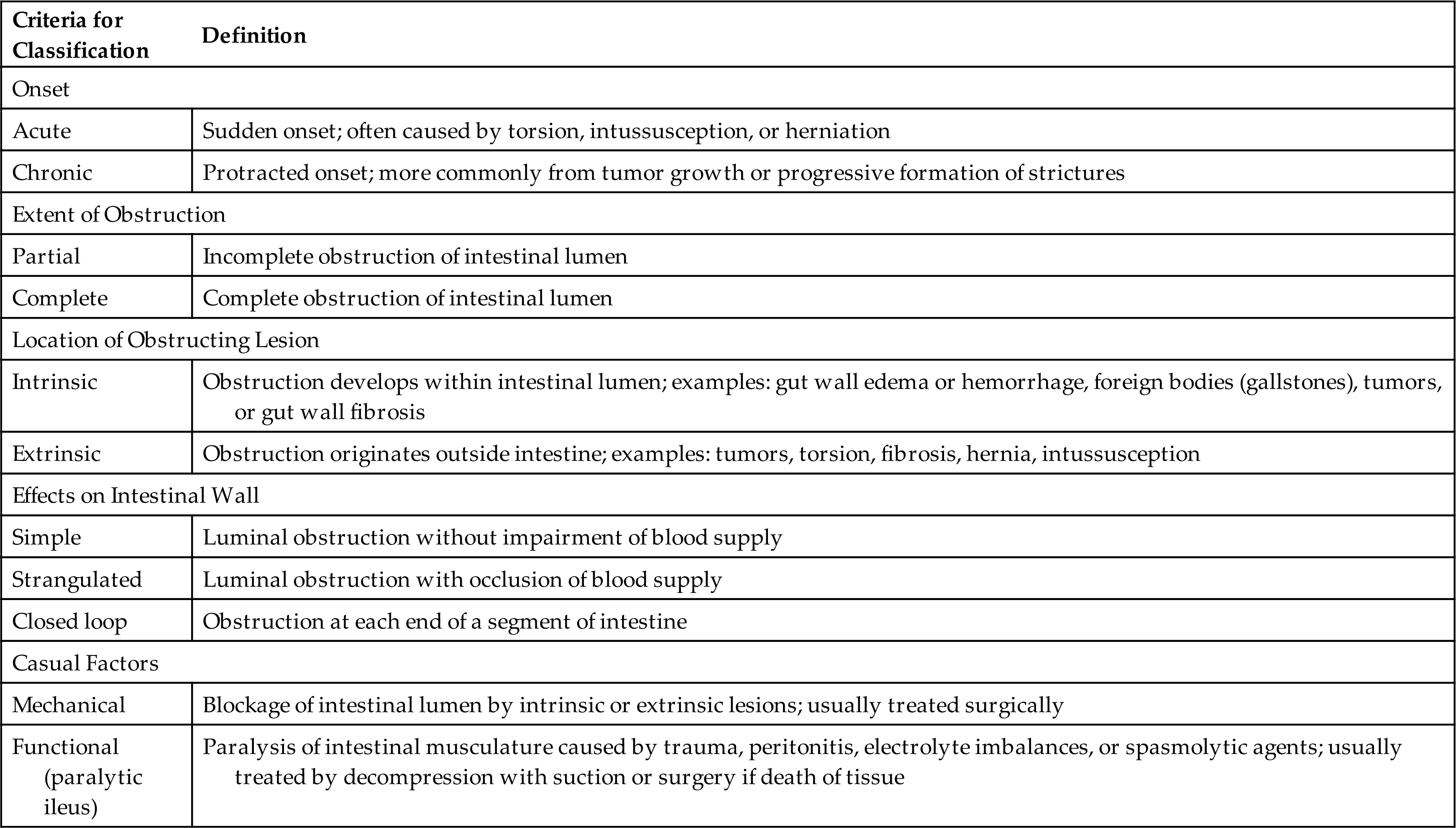
Intestinal obstruction can be caused by any condition that prevents the normal flow of chyme through the intestinal lumen (Table 41.3). Obstructions can occur in either the small or the large intestine (Table 41.4). The small intestine is more commonly obstructed because of its narrower lumen. Classifications of intestinal obstruction are summarized in Table 41.5. Intestinal obstruction is classified by cause as simple or functional. Simple obstruction caused by fibrous adhesions of the small intestine is the most common type of intestinal obstruction. Paralytic ileus, or functional or pseudo-obstruction, is a failure of normal intestinal motility often occurring after intestinal or abdominal surgery, acute pancreatitis, intestinal infection, cardiac dysfunction, or hypokalemia. Acute obstructions usually have mechanical causes, such as adhesions or hernias (Fig. 41.4). In a strangulated obstruction, blood flow is compromised, leading to intestinal ischemia and possible necrosis and perforation if left untreated. Chronic pseudo-obstruction is often idiopathic and partial obstructions are often associated with tumors or inflammatory disorders, particularly of the large intestine.29–31
Table 41.3
| Cause | Pathophysiology |
|---|---|
| Hernia | Protrusion of intestine through weakness in abdominal muscles or through inguinal ring |
| Intussusception | Telescoping of one part of intestine into another; this usually causes strangulation of the blood supply; more common in infants 10–15 months of age than in adults (see Fig. 41.4D) |
| Torsion (volvulus) | Twisting of the intestine on its mesenteric pedicle, with occlusion of the blood supply; often associated with fibrous adhesions; occurs most often in middle-aged and elderly men |
| Diverticulosis | Inflamed saccular herniations (diverticula) of mucosa and submucosa through tunica muscularis of the colon; diverticula are interspersed between thick, circular, fibrous bands; most common in obese individuals older than 60 years (see Fig. 41.14) |
| Tumor | Tumor growth into intestinal lumen; adenocarcinoma of the colon and the rectum is the most common tumoral obstruction; most common in individuals older than 60 years |
| Paralytic (adynamic) ileus | Loss of peristaltic motor activity in intestine; associated with abdominal surgery, peritonitis, hypokalemia, ischemic bowel, spinal trauma, or pneumonia |
| Fibrous adhesions | Peritoneal irritation from surgery, trauma, or Crohn disease leads to the formation of fibrin and adhesions that attach to intestine, omentum, or peritoneum and can cause obstruction; most common in small intestine |
Table 41.4
| Type of Obstruction | Cause |
|---|---|
| Small bowel obstruction | |
| Large bowel obstruction |
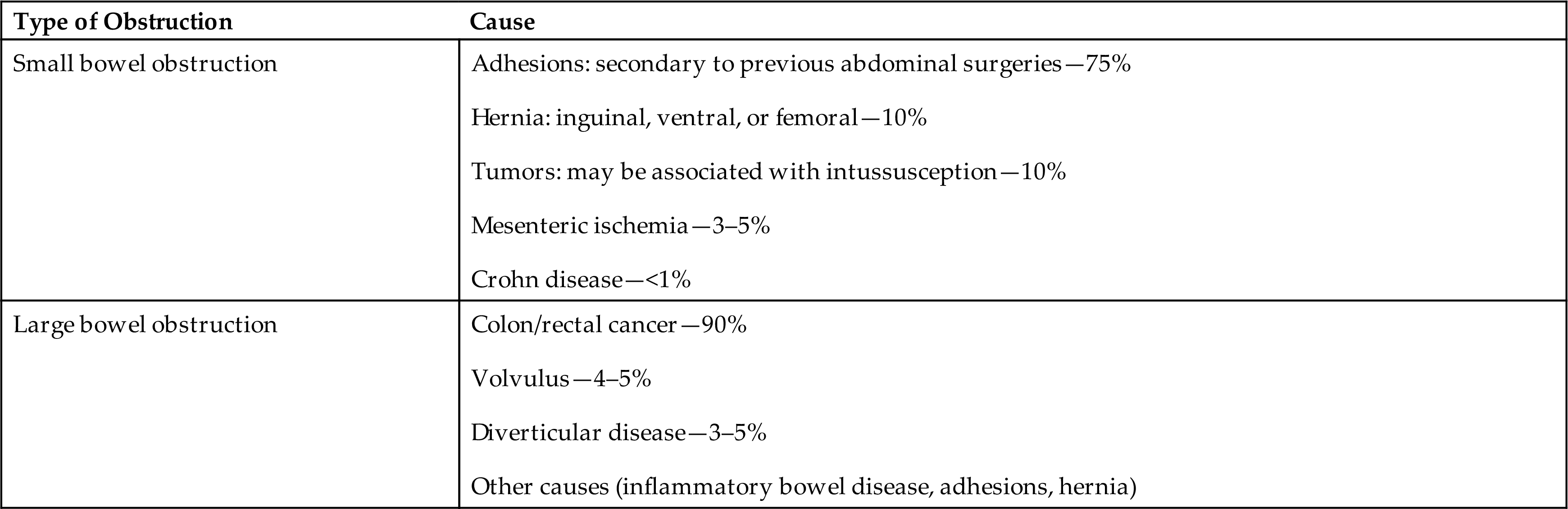
Data from Mizell JS, Turnage RH. Intestinal obstruction. In: Feldman M, et al, eds. Sleisenger & Fordtran’s gastrointestinal and liver disease, 10th edition. Philadelphia: Saunders; 2016: pp 2154–2170.
Table 41.5
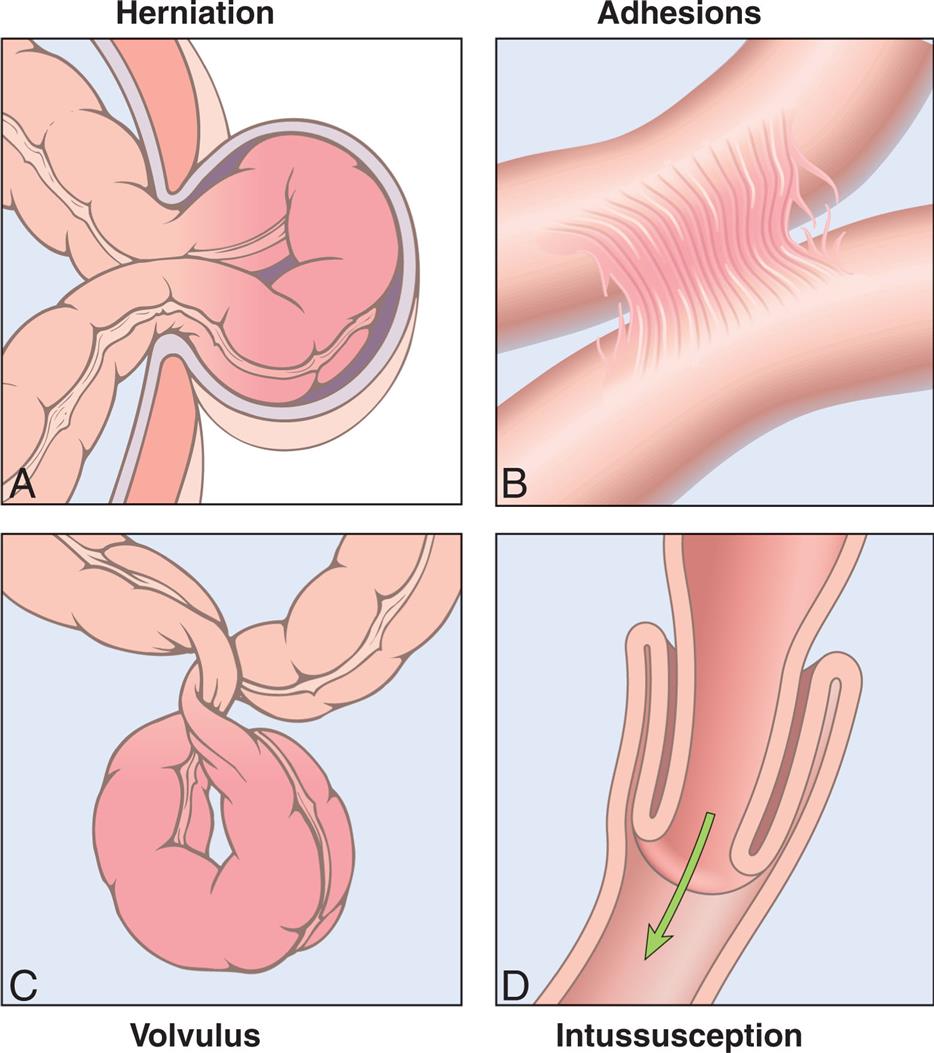
(A) Hernia. (B) Constrictions from adhesions. (C) Volvulus. (D) Intussusception. (From Kumar V, et al. Robbins basic pathology, 10th edition. Philadelphia: Elsevier; 2018.)
Four illustrations, A through D, depicts the intestinal obstruction. Illustration A depicts the herniation, which shows the small intestine pushing through the abdominal wall. Illustration B depicts the adhesion, which shows fibrous bands formed between the tissues and organs. Illustration C depicts the volvulus, which shows the twisting of the bowel upon itself. Illustration D depicts the intussusception, which shows the small intestine in the large intestine.
Pathophysiology
The consequences of intestinal obstruction are related to the onset and location of the obstruction, as well as the presence and severity of associated ischemia. The major pathophysiologic alterations are presented in Fig. 41.5. The exact cause of postoperative paralytic ileus remains unknown, but it is thought to be a multifactorial and complex interaction between the autonomic and central nervous system that alters the equilibrium of the intestine, resulting in disorganized electrical activity and paralysis.32
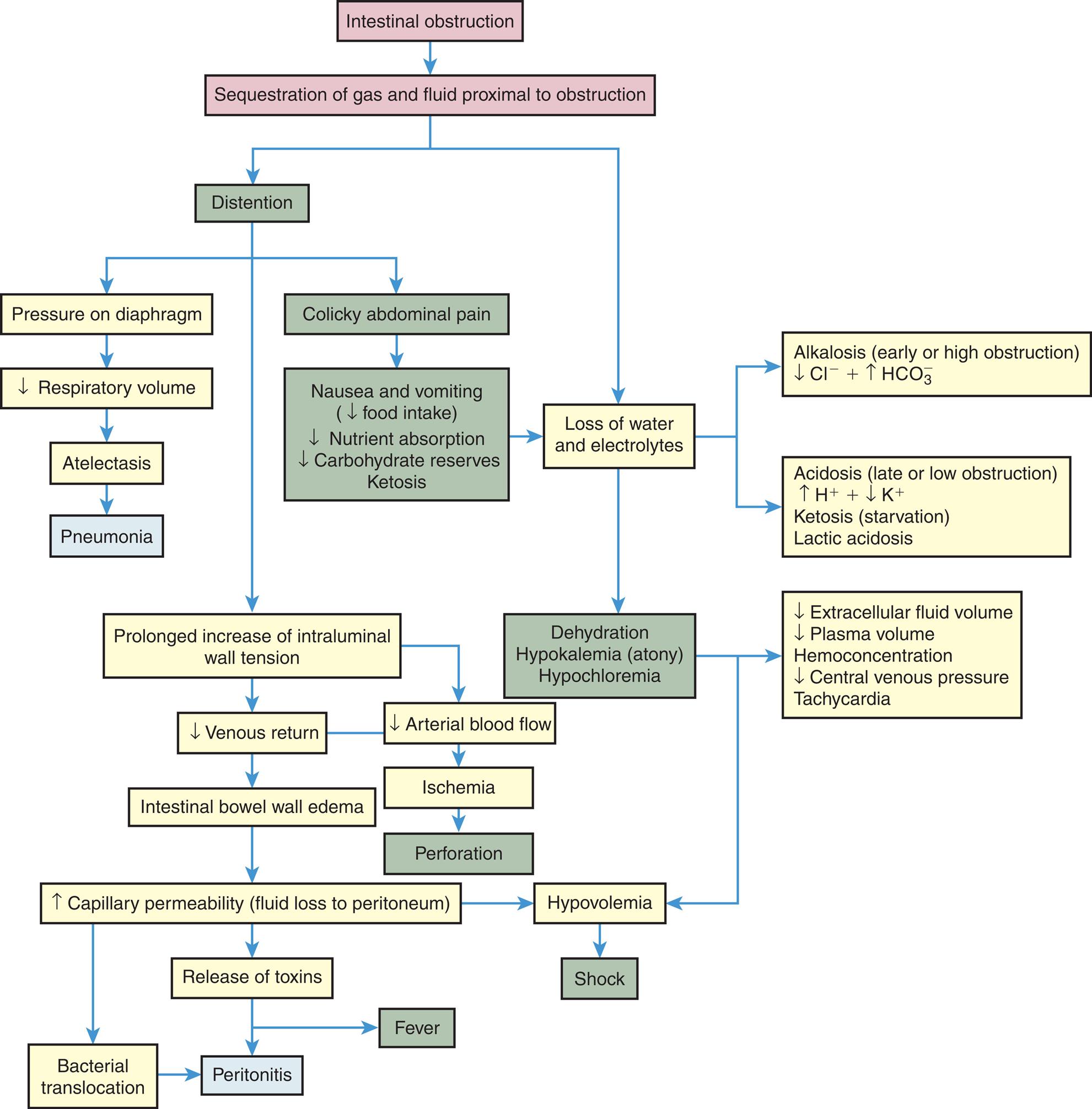
A flow diagram illustrates the pathophysiology of intestinal obstruction. The diagram begins as follows. • Intestinal obstruction due to sequestration of gas and fluid proximal to obstruction leads to distention and loss of water and electrolytes. • Distention leads to a prolonged increase of intraluminal wall tension, pressure on the diaphragm, and colicky abdominal pain. •Pressure on the diaphragm leads to decreased respiratory volume, leading to atelectasis and causing pneumonia. • Colicky abdominal pain leads to nausea and vomiting with decreased food intake, nutrient absorption, carbohydrates reserves ketosis leads to loss of water and electrolytes, causing dehydration, hypokalemia, and hypochloremia. • Prolonged increase of intraluminal wall tension leads to decrease venous return, arterial BF causing intestinal bowel wall edema and ischemia. • Ischemia leads to perforation. • Intestinal bowel wall edema leads to increase capillary permeability or fluid loss to the peritoneum, causing hypovolemia which leads to shock. • An increase in capillary permeability also leads to the release of toxins and bacterial translocation, causing peritonitis and fever. • The loss of water and electrolytes leads to alkalosis, acidosis, increased ketosis, and lactic acidosis. • Dehydration, hypokalemia, and hypochloremia lead to a decrease in extracellular fluid volume, plasma volume, hemoconcentration, central venous pressure tachycardia, causing hypovolemia which leads to shock.
Small bowel obstruction (SBO) is often caused by postoperative adhesions, tumors, CD, and hernias. SBO leads to distention caused by impaired absorption and increased secretion with the accumulation of fluid and gas inside the lumen proximal to the obstruction.33 Distention decreases the intestine's ability to absorb water and electrolytes and increases the net secretion of these substances into the lumen. Copious vomiting or sequestration of fluids in the intestinal lumen prevents their reabsorption and produces severe fluid and electrolyte disturbances. Extracellular fluid volume and plasma volume decrease, causing dehydration, increased hematocrit level, hypotension, and tachycardia. Severe dehydration leads to hypovolemic shock. Metabolic alkalosis initially develops because of excessive loss of hydrogen ions that would normally be reabsorbed from the gastric juice and vomiting. Prolonged obstruction or obstruction lower in the intestine may contribute to metabolic acidosis because bicarbonate from pancreatic secretions and bile cannot be reabsorbed. Hypokalemia from vomiting and decreased potassium absorption can be extreme, promoting acidosis and atony of the intestinal wall. Metabolic acidosis also may be accentuated by ketosis, which is the result of declining carbohydrate stores caused by starvation. In addition, lack of circulation permits the buildup of significant amounts of lactic acid, which worsens the metabolic acidosis. If pressure from the distention is severe enough, it occludes arterial circulation and causes ischemia, necrosis, perforation, and peritonitis. Fever and leukocytosis are often associated with overgrowth of bacteria, ischemia, and bowel necrosis. Bacterial proliferation and translocation across the mucosa to the systemic circulation cause peritonitis or sepsis. The release of inflammatory mediators into the circulation causes remote organ failure.
Large bowel obstruction is less common and often related to cancer. Diverticulitis, IBD, and other causes of obstruction are less common. Acute colonic pseudo-obstruction (Ogilvie syndrome) is a pathologic massive dilation of the colon without underlying mechanical obstruction or other identified organic causes. This occurs mostly in individuals with serious comorbidities. The pathologic basis remains unclear but may be caused from a functional disturbance in the enteric nervous system.34
Clinical Manifestations
Signs and symptoms of small intestine obstruction include distention and colicky type pain, followed by nausea and vomiting. Pain usually intensifies for seconds or minutes as a peristaltic wave of muscle contraction meets the obstruction. Pain may be continuous with severe distention and then diminish in intensity. If ischemia occurs, the pain loses its colicky character and becomes more constant and severe. Sweating and tachycardia occur as a sympathetic nervous system response to hypotension. Fever, severe leukocytosis, abdominal distention, and rebound tenderness develop as ischemia progresses to necrosis, perforation, and peritonitis.
Obstruction at the pylorus causes early, profuse vomiting. Obstruction in the proximal small intestine causes mild distention and vomiting of bile-stained fluid. Lower obstruction in the small intestine causes more pronounced distention because a greater length of intestine is proximal to the obstruction. In this case, vomiting may occur later and contain fecal material. Partial obstruction can cause diarrhea or constipation, whereas complete obstruction usually causes constipation only. Complete obstruction increases the number of bowel sounds, which may be accompanied by peristaltic rushes and crampy type abdominal pain. Signs of hypovolemia and metabolic acidosis may be observed as early as 24 hours after the occurrence of complete obstruction. Distention may be severe enough to push against the diaphragm and decrease lung volume. This can lead to atelectasis and pneumonia, particularly in debilitated individuals.
Large bowel obstruction usually presents with hypogastric type pain and abdominal distention. Pain can vary from vague to excruciating, depending on the degree of ischemia and the development of peritonitis. Vomiting occurs late in the obstructive process. Small and large intestinal perforation presents with the same acute, persistent type abdominal pain, nausea, vomiting, and fever. Acute colonic pseudo-obstruction has the absence of mechanical obstruction and is characterized by abdominal distention, abdominal pain, nausea, and vomiting. Bowel sounds are usually present.
Evaluation and Treatment
Evaluation is based on clinical manifestations and imaging studies. Successful management requires early identification of the location and type of obstruction. Replacement of fluid and electrolytes and decompression of the lumen with gastric or intestinal suction are essential forms of therapy. Laparoscopic procedures can release adhesions. Immediate surgical intervention is required for strangulation, complete obstruction, or perforation. Colonic stents may be placed for malignant obstruction. If conservative methods are not successful, neostigmine, a parasympathomimetic, may be used for colonic pseudo-obstruction. Neostigmine increases the activation of muscarinic receptors by inhibition of the breakdown of acetylcholine. This stimulates colonic motor activity and increases intestinal transit time. Pseudo-obstruction is often managed symptomatically.31,35
Gastritis
Gastritis is a nonspecific inflammatory disorder of the gastric mucosa. Gastritis can present as an acute manifestation or may be chronic and often will progress to chronic gastritis if not treated in the acute phase.36 The most common causes of gastritis are use of NSAIDs, Helicobacter pylori infection, and physiologic stress–related mucosal changes. Alcohol, digitalis, and metabolic disorders, such as uremia, also are contributing factors.
Acute gastritis is caused by injury of the protective mucosal barrier. NSAIDs (e.g., ibuprofen, naproxen, indomethacin, and aspirin) cause gastritis by inhibition of prostaglandin synthesis, which normally stimulates the secretion of mucus. Alcohol, histamine, digitalis, and metabolic disorders, such as uremia, are contributing factors. H. pylori–associated acute gastritis causes inflammation, increased gastric secretion in antral gastritis, decreased gastric secretion in fundal gastritis, pain, nausea, and vomiting (Box 41.1 and Fig. 41.6). The clinical manifestations of acute gastritis can include vague abdominal discomfort, epigastric tenderness, and bleeding. Healing usually occurs spontaneously within a few days. Discontinuing injurious drugs, using antacids, or decreasing acid secretion with H2 receptor antagonist or PPIs facilitates healing.

Acute erosive gastritis is shown in the opened stomach. The mucosa appears hyperemic, and the foci of superficial ulceration are manifested as scattered, small, red areas termed erosions. (From Kumar V, et al. Pathologic basis of disease, 7th edition. Philadelphia: Saunders; 2006.)
Chronic gastritis causes chronic inflammation of the gastric mucosa which progresses to atrophic gastritis, characterized by the loss of normal mucosal glands.36 Chronic gastritis is classified as type A immune (fundal) or type B nonimmune (antral), depending on the pathogenesis and location of the lesions. When both types of chronic gastritis occur, it is known as type AB, or pangastritis, and the antrum is more severely involved. Type C gastritis is associated with reflux of bile and pancreatic secretions into the stomach, causing chemical injury.
Chronic immune (fundal) gastritis (autoimmune gastritis) is the rarest form of gastritis and is a recessive, multigenetic disease. It is associated with the loss of T-cell tolerance and the development of autoantibodies to acid-secreting parietal cells. H. pylori infection may trigger the immune response through molecular mimicry (a mechanism of autoimmune disease with similarities between foreign and self-antigens sufficient to result in the cross-activation of autoreactive T or B cells).37 The gastric mucosa degenerates extensively in the fundus (body) of the stomach, leading to gastric atrophy. Loss of parietal cells diminishes acid and intrinsic factor secretion. Pernicious anemia can develop from decreased vitamin B12 absorption (see Chapter 29). The feedback mechanism that normally inhibits gastrin secretion (i.e., loss of acid secretion) is also impaired, causing elevated plasma levels of gastrin, thus stimulating gastric secretion. Chronic fundal gastritis occurs in association with other autoimmune diseases (e.g., rheumatoid arthritis, autoimmune thyroid disease, or type 1 diabetes mellitus) and is a risk factor for gastric carcinoma, particularly in individuals who develop pernicious anemia.
Chronic nonimmune (antral) gastritis generally involves the antrum only and is more common than fundal gastritis.38 Chronic use of alcohol, tobacco, and NSAIDs and H. pylori infection are contributing factors. There are high levels of hydrochloric acid secretion with an increased risk of duodenal ulcers. H. pylori infection also can progress to autoimmune atrophic gastritis and involve the fundus, thus becoming pangastritis.39 There is greater risk for the development of gastric cancer in these cases.
Clinical Manifestations.Signs and symptoms of chronic gastritis often include vague symptoms, such as anorexia, fullness, nausea, vomiting, and epigastric pain. Gastric bleeding may be the only clinical manifestation of gastritis. Gastroscopic examination and biopsy may show a long-standing inflammatory process and gastric atrophy in an individual with no history of abdominal distress. Gastric secretion analysis confirms achlorhydria (the absence of hydrochloric acid) and loss of intrinsic factor. Pernicious anemia can develop because intrinsic factor is less available to facilitate vitamin B12 absorption. Iron deficiency may also be present. The presence of antiparietal cell antibody and elevated plasma ghrelin level are specified for atrophic gastritis. H. pylori infection is evidence for H. pylori chronic gastritis with infiltration of neutrophils and lymphocytes. Eradication of H. pylori is recommended for the prevention of gastric carcinoma.40–42
Evaluation and Treatment.Symptoms can usually be managed by eating smaller meals in conjunction with a soft, bland diet and by avoiding alcohol and NSAIDs. H. pylori infection is treated with antibiotics, and vitamin B12 is administered to correct pernicious anemia.
Peptic Ulcer Disease
A peptic ulcer is a break or ulceration in the protective mucosal lining, usually located in the stomach or proximal duodenum; however, they can be found in the esophagus (see Figs. 41.7 and 41.8). Ulcers develop when mucosal protective factors are overcome by erosive factors such as gastric acid secretion or pepsin. This causes an imbalance between the gastric mucosal protective factors and the destructive factors. Peptic ulcer disease has various causes; however, most cases are caused by H. pylori and NSAIDs. Additional risk factors for peptic ulcer disease include the use of the following medications: corticosteroids, bisphosphonates, potassium chloride, and fluorouracil. Smoking, alcohol consumption, and certain disease processes that can make the gastric lining a hypersecretory environment, such as Zollinger-Ellison syndrome, systemic mastocytosis, cystic fibrosis, hyperparathyroidism, and antral G-cell hyperplasia, are also risk factors.43
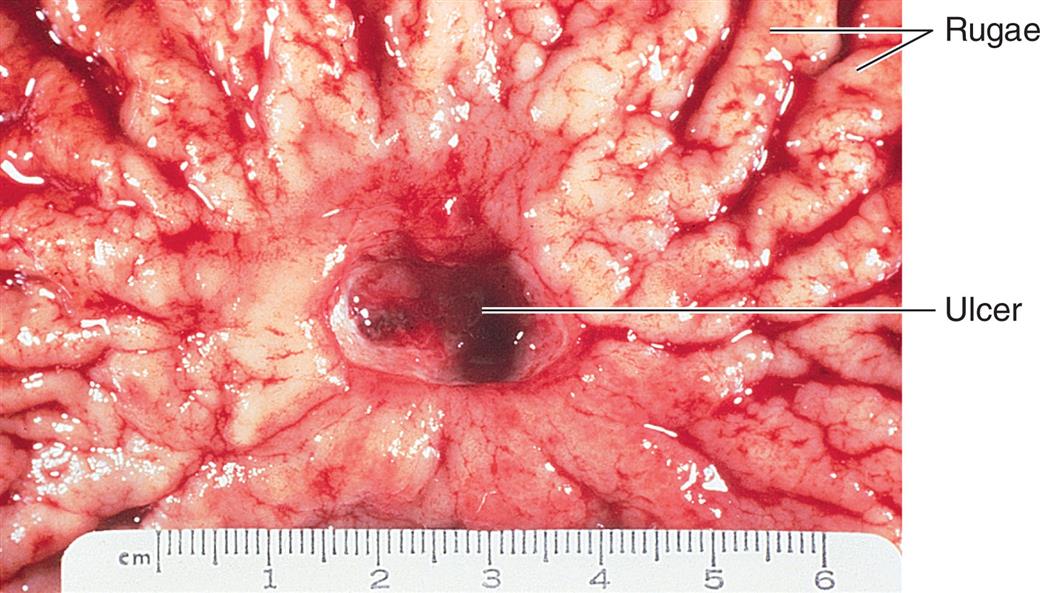
Gross photograph of a chronic peptic ulcer located in the lesser curvature, straddling the antrum and corpus of the stomach. (From Damjanov I, Linder J, eds. Anderson’s pathology, 10th edition. St. Louis: Mosby; 1996.)
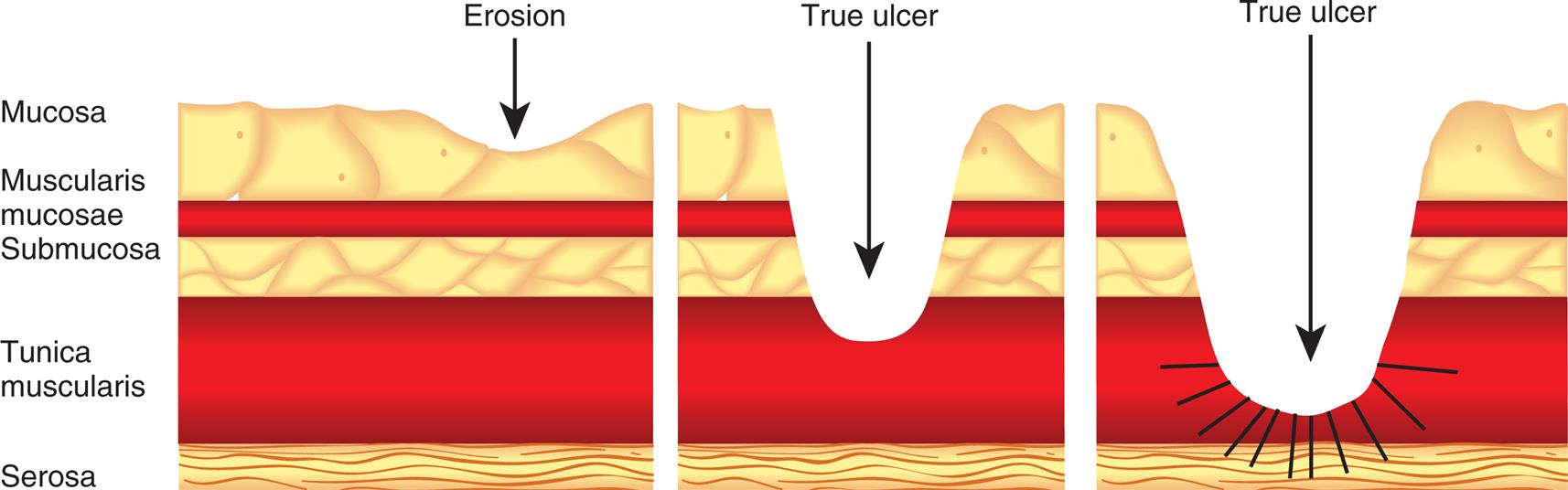
Three illustrations depict lesions by peptic ulcer disease, which show the layers of the stomach labeled mucosa, muscularis mucosae, submucosa, tunica muscularis, and serosa. The first illustration depicts erosion where the lesion occurs in the mucosa. The second illustration depicts the true ulcer where the lesion extends to tunica muscularis. The third illustration depicts the true ulcer where the lesion extends to the serosa.
Peptic ulcers can be single or multiple, acute, or chronic, and superficial or deep. Superficial ulcerations are called erosions because they erode the mucosa but do not penetrate the muscularis mucosa. True ulcers extend through the muscularis mucosae and damage blood vessels, causing hemorrhage and possible perforation of the GI wall. Successful antibiotic treatment of H. pylori infection and the use of mucosal protecting agents during NSAID and H. pylori treatment have significantly reduced the incidence of peptic ulcer disease.
Zollinger-Ellison syndrome is a rare syndrome that also is associated with peptic ulcers caused by a gastrin-secreting neuroendocrine tumor or multiple tumors of the pancreas or duodenum that release large amounts of acid. The body normally releases small amounts of gastrin after eating; gastrin then triggers the stomach to make gastric acid. Gastrin stimulates a proliferation of gastric parietal cells and chronic secretion of gastric acid. The resulting excess acid causes gastric and duodenal ulcers, gastroesophageal reflux with abdominal pain, diarrhea, bloating, burping, weight loss, and poor appetite. Diagnosis includes secretin or calcium- stimulated measures of gastrin levels, gastric pH levels less than 2, and symptomatic evidence of peptic ulcer disease. PPIs reduce gastric acid secretion, and surgical removal of tumors limits metastasis.44
Duodenal Ulcers
Duodenal ulcers occur with greater frequency than other types of peptic ulcers and are generally caused by H. pylori infection and NSAID use. Idiopathic duodenal ulcers are rare and can be associated with altered mucosal defenses, rapid gastric emptying, elevated serum gastrin levels, or acid production stimulated by smoking.45
Pathophysiology
Causative factors, independently or in combination, cause acid and pepsin concentrations in the duodenum to increase and penetrate the mucosal barrier, causing ulceration (Fig. 41.9). The host response to chronic stomach antral H. pylori infection is increased levels of gastrin resulting in increased stomach acid secretion and an increased acid load in the duodenum. The increased duodenal acid promotes gastric metaplasia in the duodenum and favors H. pylori colonization. Both H. pylori and the increased acid result in decreased duodenal bicarbonate production. In addition, H. pylori infection activates immune cells (T and B lymphocytes with the infiltration of neutrophils) and the release of inflammatory cytokines which damage the mucosa. H. pylori also produces a toxin that causes loss of protective mucosal cells, resulting in ulceration. H. pylori mucosal infection can promote gastric cancer, but the incidence is lower for duodenal ulcer than for gastric ulcer, and the mechanism is unknown.46
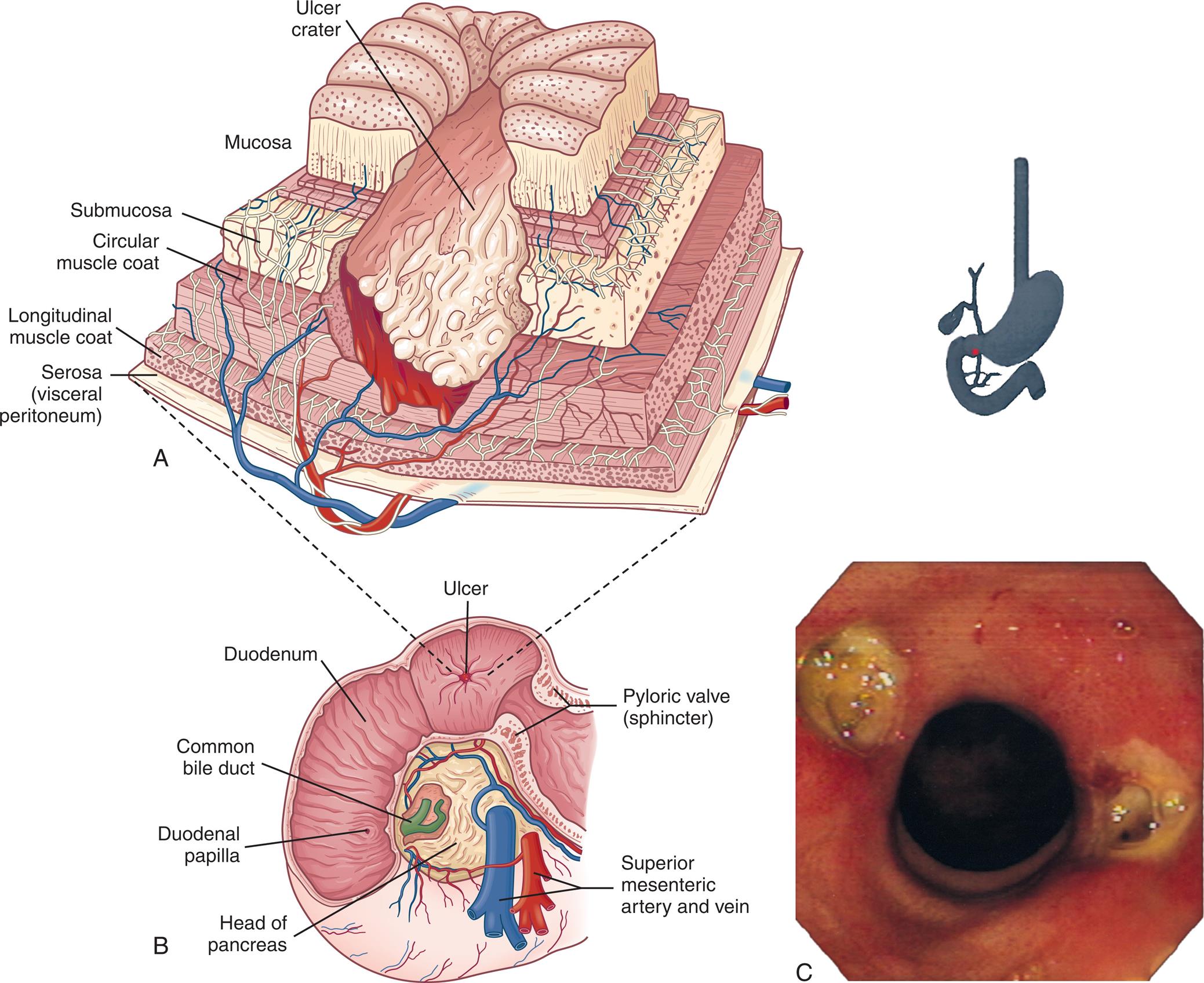
(A) A deep ulceration in the duodenal wall extending as a crater through the entire mucosa and into the muscle layers. (B) Sequence of ulcerations from normal mucosa to duodenal ulcer. (C) Bilateral (kissing) duodenal ulcers in a person using nonsteroidal antiinflammatory drugs. (C, Courtesy David Bjorkman, MD, University of Utah School of Medicine, Department of Gastroenterology, Salt Lake City, UT.)
Illustration A depicts the cutaway section of the ulcerated duodenal wall with labels indicating ulcer crater, mucosa, submucosa, circular muscle coat, longitudinal muscle coat, and serosa or visceral peritoneum. Illustration B depicts the cross-section of the stomach with labels indicating ulcer, duodenum, common bile duct, duodenal papilla, head of the pancreas, superior mesenteric artery and vein, and pyloric valve or sphincter. Image C represents the endoscopic view of bilateral duodenal ulcers.
Clinical Manifestations
The characteristic manifestation of a duodenal ulcer is chronic, intermittent pain in the epigastric area. The pain begins 2 or 3 hours after eating, when the stomach is empty. It is not unusual for pain to occur in the middle of the night and disappear by morning. Pain is relieved rapidly by ingestion of food or antacids, creating a typical pain-food-relief pattern. Some individuals with a duodenal ulcer may have no symptoms; the first manifestation may be hemorrhage or perforation, particularly with a history of NSAID or anticoagulant use. Complications of a duodenal ulcer include bleeding, perforation, and obstruction of the duodenum or outlet of the stomach. Bleeding is the most common cause of mortality, particularly among the elderly. Bleeding from duodenal ulcers causes hematemesis or melena. Perforation occurs with destruction of all layers of the duodenal wall and causes sudden, severe epigastric pain. Obstruction may be the result of edema from inflammation or scarring from chronic injury. Duodenal ulcers often heal spontaneously. However, repeat imaging is indicated if the ulcer was large, associated complications were present, or the individual has continued pain.47
Evaluation and Treatment
Several diagnostic approaches are used to differentiate duodenal ulcers from gastric ulcers or gastric carcinoma. Endoscopic evaluation allows visualization of lesions and biopsy. Radioimmune assays of gastrin levels are evaluated to identify ulcers associated with gastric carcinomas. H. pylori is detected using the urea breath test, H. pylori–specific serum immunoglobulin G (IgG) and IgA antibodies, and the measurement of H. pylori stool antigen levels. Findings from the gastric biopsy detect H. pylori infection and can also confirm eradication after treatment. Polymerase chain reaction testing provides additional virulence and antibiotic sensitivity profiling.48
The management of duodenal ulcers is aimed at relieving the causes of the ulceration. The effects associated with the hyperacidity and pepsin present in the gut should be managed with diet and pharmacotherapy. Antacids neutralize gastric contents and relieve pain. Acid secretion can be suppressed with drugs that block H2 receptors and inhibit the secretion of acid. PPIs inhibit acid production. H. pylori is treated with a combination of antibiotics and PPIs, but antibiotic resistance is an increasing problem.49 Surgical resection may be required for bleeding or perforating ulcers, obstruction, or peritonitis.
Gastric Ulcers
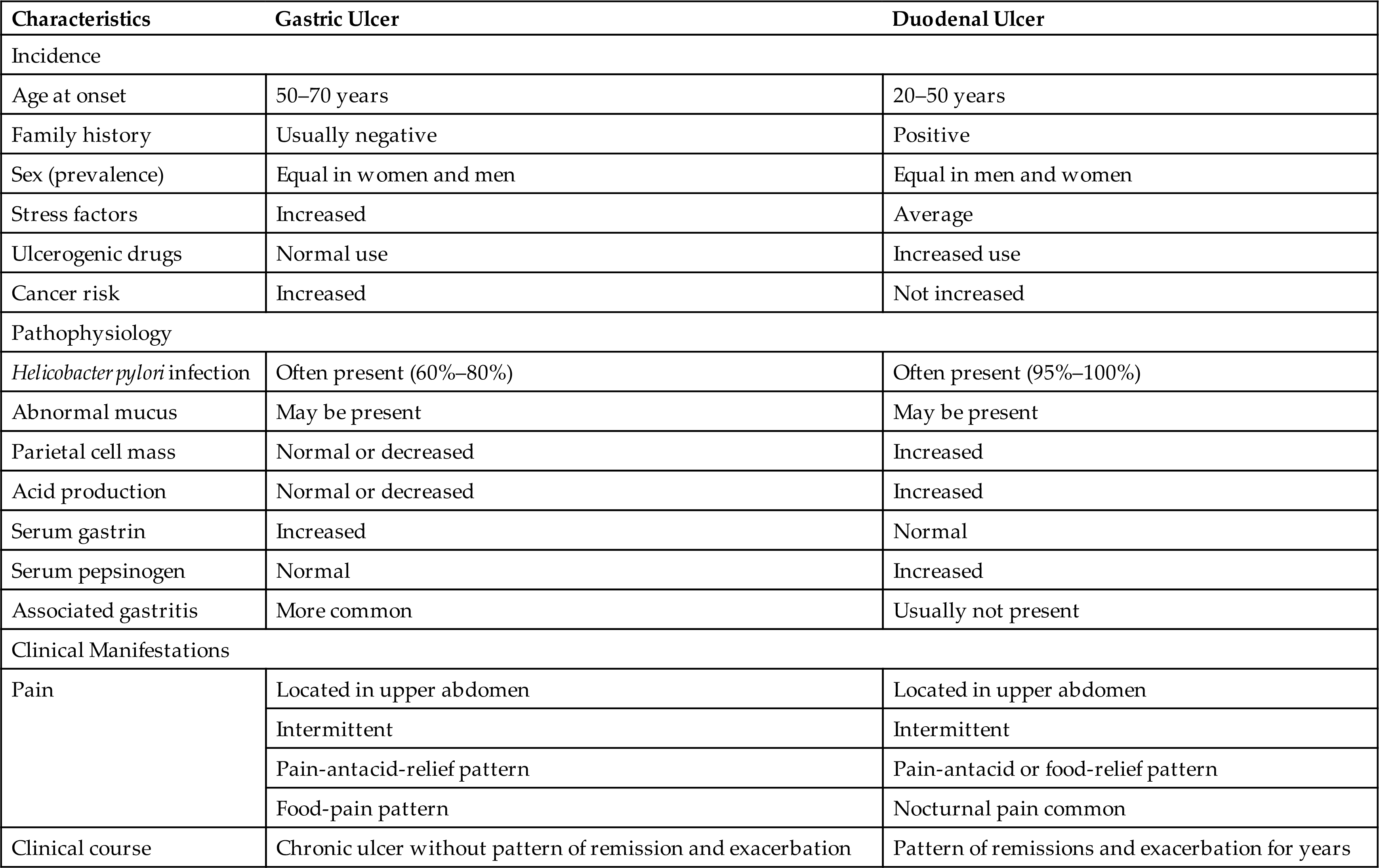
Gastric ulcers are ulcers of the stomach. They occur about equally in males and females, usually between the ages of 55 and 65 years. They are less common than duodenal ulcers (Table 41.6).
Table 41.6
Pathophysiology
In general, gastric ulcers develop in the antral region, adjacent to the acid-secreting mucosa of the body. The primary defect is an abnormality that increases the mucosal barrier's permeability to hydrogen ions. Gastric secretion may be normal or less than normal, and there may be a decreased mass of parietal cells. Chronic gastritis is often associated with the development of gastric ulcers and may precipitate ulcer formation by limiting the mucosa's ability to secrete a protective layer of mucus (Fig. 41.10). Other factors include:
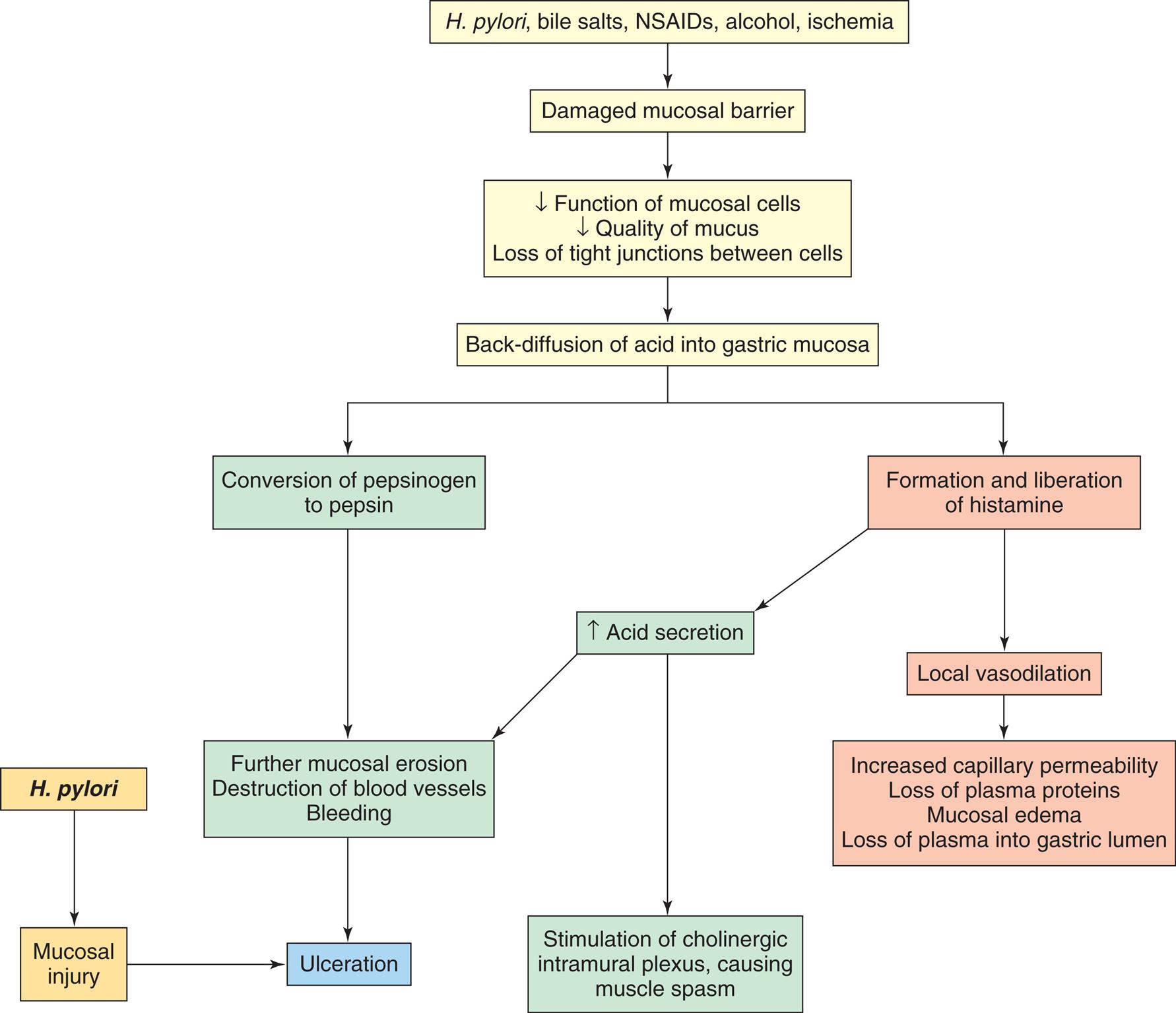
NSAIDs, Nonsteroidal antiinflammatory drugs.
A flow diagram illustrates the pathophysiology of gastric ulcer formation. The diagram begins as follows. • H. pylori, bile salts, N S A I Ds, alcohol, ischemia leads to damaged mucosal barrier. • Damaged mucosal barrier leads to decrease function of mucosal cells, quality of mucus, loss of tight junctions between cells which further leads to back-diffusion of acid into gastric mucosa. • The diffusion of acid into gastric mucosa leads to the conversion of pepsinogen to pepsin and the formation and liberation of histamine. •Conversion of pepsinogen leads to further mucosal erosion, destruction of blood vessels, and bleeding, causing ulceration. • H. pylori leads to mucosal injury and also causes ulceration. • Formation and liberation of histamine increase acid secretion, leads to stimulation of intramural cholinergic plexus causes muscle spasms and further mucosal erosion, destruction of blood vessels, and bleeding. • Formation and liberation of histamine lead to local vasodilation and causes increased capillary permeability, loss of plasma proteins, mucosal edema, and loss of plasma into the gastric lumen.
A break in the mucosal barrier permits hydrogen ions to diffuse into the mucosa, where they disrupt permeability and cellular structure. A vicious cycle can be established as the damaged mucosa liberates histamine, which stimulates the increase of acid and pepsinogen production, blood flow, and capillary permeability. The disrupted mucosa becomes edematous and loses plasma proteins. Destruction of small vessels causes bleeding.
Clinical Manifestations
The clinical manifestations of gastric ulcers are similar to those of duodenal ulcers (see Table 41.6). The pattern of pain is common, but the pain of gastric ulcers also occurs immediately after eating. Gastric ulcers also tend to be chronic rather than alternating between periods of remission and exacerbation, and they cause more anorexia and vomiting than duodenal ulcers. The pain associated with eating tends to suppress food intake, resulting in weight loss. The evaluation and treatment of gastric ulcers are similar to those for duodenal ulcers. However, long-term use of PPIs is a reported risk factor for gastric cancer after H. pylori eradication and is related to hypergastrinemia and hyperplasia of enterochromaffin-like cells that promote the secretion of gastric acid.50
Stress-Related Mucosal Disease
A stress-related mucosal disease (stress ulcer) is an acute form of peptic ulcer that tends to accompany the physiologic stress of severe illness or major trauma. Usually, multiple sites of ulceration are distributed within the stomach or duodenum. Stress ulcers may be classified as ischemic ulcers or Cushing ulcers.
Ischemic ulcers develop within hours of an event such as hemorrhage, multisystem trauma, severe burns, heart failure, or sepsis. Shock, anoxia, inflammation, and sympathetic responses cause ischemia of the stomach and duodenal mucosa, disrupting the mucosal barrier. Stress ulcers that develop as a result of burn injury are often called Curling ulcers. Cushing ulcer is a stress ulcer associated with severe brain trauma or brain surgery. Decreased mucosal blood flow and hypersecretion of acid caused by overstimulation of the vagal nuclei damage the mucosal barrier, causing erosions and ulceration.
The primary clinical manifestation of stress-related mucosal disease is bleeding, which is uncommon, but occurs more readily with the presence of coagulopathy and in the presence of more than 48 hours of mechanical ventilation. Prophylactic treatment regimens are used to prevent this disease. Stress ulcers seldom become chronic.51
Surgical Treatment of Ulcer
Advances in the medical treatment of peptic ulcer disease with acid suppression and eradication of H. pylori have reduced the number of cases requiring surgery. The most common indications for ulcer surgery are recurrent or uncontrolled bleeding and perforation of the stomach or duodenum. The primary objectives of surgical treatment are to reduce stimuli for acid secretion, decrease the number of acid-secreting cells in the stomach, and correct complications of ulcer disease.
Acute complications of gastrectomy or anastomosis are relatively uncommon except in debilitated persons. However, chronic complications are likely to develop if a large portion of the stomach has been removed. These complications and their pathophysiologic mechanisms are described in the next section.
Postgastrectomy Syndromes
Postgastrectomy syndromes are a group of signs and symptoms that occur after gastric resection for the treatment of peptic ulcer, gastric carcinoma, or bariatric surgery for extreme obesity. They are caused by anatomic and functional changes in the stomach and upper small intestine52 and include the following conditions.
Dumping syndrome is the rapid emptying of hypertonic chyme from the surgically created residual stomach (i.e., the smaller stomach component remaining after surgical resection following gastric or bariatric surgery) into the small intestine 10 to 20 minutes after eating. It occurs with varying severity and is promoted by loss of gastric capacity, loss of emptying control when pylorus is removed, and loss of feedback control by the duodenum once removed. Rapid gastric emptying and a creation of a nonphysiologic, high osmotic gradient within the small intestine cause a sudden shift of fluid from the vascular compartment to the intestinal lumen. Plasma volume decreases and rapid distention of the intestine occurs, producing symptoms such as cramping type pain, nausea, vomiting, osmotic diarrhea, hypotension, weakness, and pallor.
Late dumping syndrome occurs 1 to 3 hours after eating a high carbohydrate meal and is related to hyperinsulinemia with hypoglycemia. The symptoms of late dumping syndrome include weakness, diaphoresis, and confusion. Most cases of dumping syndrome respond to dietary management. Individuals should eat frequent small meals high in protein and low in carbohydrates.
Alkaline (bile) reflux gastritis occurs when there is a disruption of the mucosal barrier in the remnant stomach. Reflux of bile and alkaline pancreatic secretions containing proteolytic enzyme disrupts the mucosal barrier in the remnant stomach causing inflammation. Symptoms include nausea, bilious vomiting, and sustained epigastric pain that worsen after eating and is not relieved by antacids. It responds somewhat to avoidance of aspirin and alcohol, but surgical correction may be required.
Afferent loop obstruction is a rare complication of Billroth gastrojejunostomy. Symptoms include intermittent severe pain and epigastric fullness after eating because of volvulus, hernia, adhesion, or stenosis of the duodenal stump on the proximal side of the gastrojejunostomy. Vomiting typically relieves symptoms. Management includes low-fat diet, but decompression or surgery revision is required for complete obstruction.
Diarrhea is related to rapid gastric emptying and osmotic attraction of water into the gut, especially after a large intake of high-carbohydrate liquids. Small, dry meals and anticholinergic drugs are effective control measures.
Weight loss is commonly caused by inadequate caloric intake because the individual cannot tolerate carbohydrates or a normal-sized meal. The stomach also is less able to mix, churn, and break down food. In the case of bariatric surgery for extreme obesity, weight loss is the intended outcome, but nutrients, including vitamins and minerals, must be monitored and supplemented to prevent deficiencies.53
Anemia may occur if iron malabsorption results from decreased acid secretion or lack of duodenum after a Billroth II procedure (gastrojejunostomy). Deficiencies of iron, vitamin B12, or folate also may result.
Bone and mineral disorders are related to altered calcium absorption and metabolism. This causes an increased risk of fractures and deformity, and malabsorption of vitamins and nutrients, such as vitamin D.
Malabsorption Syndromes
Malabsorption syndromes interfere with nutrient absorption in the small intestine. Historically they have been classified as maldigestion or malabsorption. Maldigestion is failure of the chemical processes of digestion that take place in the intestinal lumen or at the brush border of the intestinal mucosa. Malabsorption is failure of the intestinal mucosa to absorb the digested nutrients. Often these two syndromes are interrelated or occur together, making classification difficult. In general, maldigestion is caused by deficiencies of the enzymes needed for digestion or inadequate secretion of bile salts and inadequate reabsorption of bile in the ileum. Malabsorption is the result of mucosal disruption caused by gastric or intestinal resection, vascular disorders, or intestinal disease (also see Chapter 42).
Pancreatic Exocrine Insufficiency
The pancreatic enzymes (lipase, amylase, trypsin, chymotrypsin) are required for the digestion of proteins, carbohydrates, and fats. Pancreatic insufficiency is the deficient production of these enzymes, particularly lipase, by the pancreas. Causes include chronic pancreatitis, pancreatic carcinoma, pancreatic resection, and cystic fibrosis. Significant damage to or loss of pancreatic tissue must occur before enzyme levels decrease sufficiently to cause maldigestion. Although pancreatic insufficiency causes poor digestion of all nutrients, fat maldigestion is the chief problem. Absence of pancreatic bicarbonate in the duodenum and jejunum causes an acidic pH that worsens maldigestion by precipitating bile salts and preventing activation of the pancreatic enzymes that are present. A large amount of fat in the stool (steatorrhea) is the most common sign of pancreatic insufficiency. There is also a deficit of fat-soluble vitamins (A, D, E, and K) and weight loss. Several diagnostic tests are available to diagnose pancreatic exocrine insufficiency; however, there remains confusion regarding which testing procedure demonstrates the best approach. Treatment consists of dietary management and lifestyle changes and pancreatic enzyme replacement therapy.54
Lactase Deficiency (Lactose Intolerance)
A deficiency of disaccharidase at the brush border of the small intestine is caused by a genetic defect in which a single enzyme, usually lactase, is lacking. Lactase deficiency inhibits the breakdown of lactose (milk sugar) into monosaccharides and therefore prevents lactose digestion and absorption across the intestinal wall. Lactase deficiency is most common in Blacks, Latinos, and Native Americans and usually does not develop until adulthood. Secondary (acquired) lactase deficiency can be caused by several diseases of the intestine, including gluten-sensitive enteropathy, enteritis, and bacterial overgrowth.
The undigested lactose remains in the intestine, where bacterial fermentation causes formation of gases. Undigested lactose also increases the osmotic gradient in the intestine, causing irritation and osmotic diarrhea. Clinical manifestations of lactose consumption with lactase deficiency are bloating, cramping type pain, diarrhea, and flatulence. The disorder is diagnosed by a lactose-tolerance test. Avoiding milk products and adhering to a lactose-free diet relieves symptoms.55
Bile Salt Deficiency and Malabsorption
Pathophysiology.Conjugated bile acids (BAs) (bile salts) are necessary for the digestion and absorption of fats. Bile salts are conjugated in the bile that is secreted from the liver. When bile enters the duodenum, the bile salts aggregate with fatty acids and monoglycerides to form micelles. Micelle formation makes fat molecules more soluble and allows them to pass through the unstirred layer at the brush border of the small intestinal villi (see Chapter 40). A minimum concentration of bile salts, termed the critical micelle concentration, is required to allow formation of micelles. Therefore conditions that decrease the production or secretion of bile result in bile salt deficiency and decreased micelle formation and fat malabsorption. These conditions include advanced liver disease, which decreases the production of bile salts; obstruction of the common bile duct, which decreases flow of bile into the duodenum (cholestasis); intestinal stasis (lack of motility), which permits overgrowth of intestinal bacteria that deconjugate bile salts; and diseases of the ileum, which prevent the reabsorption and recycling of bile salts (enterohepatic circulation).56
Clinical Manifestations.Clinical manifestations of bile salt deficiency are related to poor intestinal absorption of fat and fat-soluble vitamins (A, D, E, and K). Increased fat in the stools (steatorrhea) leads to diarrhea and decreased levels of plasma proteins. The losses of fat-soluble vitamins and their effects include:
- • Vitamin A deficiency results in night blindness.
- • Vitamin D deficiency results in decreased calcium absorption with bone demineralization (osteoporosis), bone pain, and fractures.
- • Vitamin K deficiency prolongs prothrombin time, leading to spontaneous development of purpura (bruising) and petechiae.
- • Vitamin E deficiency has uncertain effects but may cause testicular atrophy and neurologic defects in children.
Evaluation and Treatment.The most effective treatment for fat-soluble vitamin deficiency is to increase consumption of medium-chain triglycerides in the diet, for example, by using coconut oil for cooking. Vitamins A, D, and K may be given parenterally. Oral bile salts are also an effective therapy.
Bile malabsorption is associated with a number is disorders, including ileal resection or inflammation, small intestinal bacterial overgrowth, and celiac disease. The accumulation of bile in the colon results in watery diarrhea known as BA diarrhea. Cholestyramine and colesevelam, which bind BA in the colon, are used for treatment. In the future, farsenoid X receptor agonists may also be effective because they regulate BA synthesis, conjugation, and transport.57
Inflammatory Bowel Disease
UC and CD are major types of chronic relapsing IBDs, and the cause is unknown. The prevalence of IBD is approximately 1.4 million people in the United States, with approximately 30,000 new cases annually.58
IBD causes an inflammation of the intestinal mucosa, which causes episodic abdominal pain, diarrhea, bloody stools, and weight loss. Inflammation and ulceration are caused by an influx of neutrophils and macrophages that produce proinflammatory cytokines, proteolytic enzymes, and free radicals (Table 41.7).59
Table 41.7
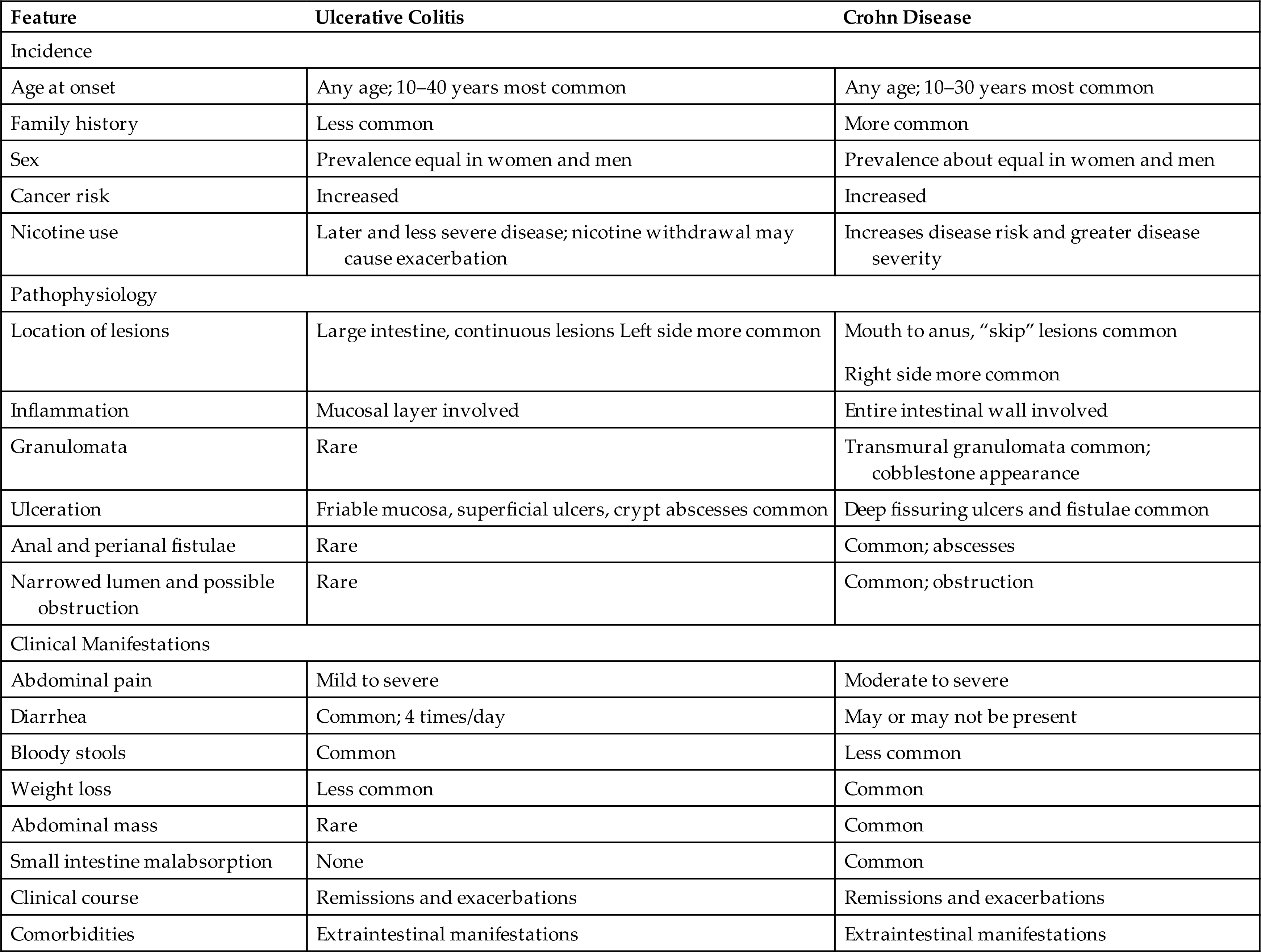
Environmental factors or infections are thought to alter the barrier function of the mucosal epithelium, leading to loss of immune tolerance to normal intestinal antigens. Environmental triggers may include diet, psychological stress, viruses, and smoking.58 There is possible loss of discrimination of potentially harmful pathogens from commensal microorganisms in the intestinal mucosa. The loss of tolerance activates immune cells. Production of proinflammatory mediators also damages the intestinal epithelium.
The risk of colon cancer has been significantly reduced with current approaches to treatment.60 Future research is directed at an integration of these factors to refine our understanding of disease cause and trajectory, particularly interactions between genetics, the microflora, mucosa, and immune responses. The clinical manifestations of UC and CD are similar, but there are different pathologic features and extent of inflammatory involvement.
Ulcerative Colitis
Ulcerative colitis (UC) is a chronic inflammatory disease that causes ulceration of the colonic mucosa, most commonly in the rectum and sigmoid colon (Fig. 41.11). The lesions appear in susceptible individuals between 20 and 40 years of age. UC is less common in people who smoke or have had an appendectomy, and the mechanisms are not clearly known.61
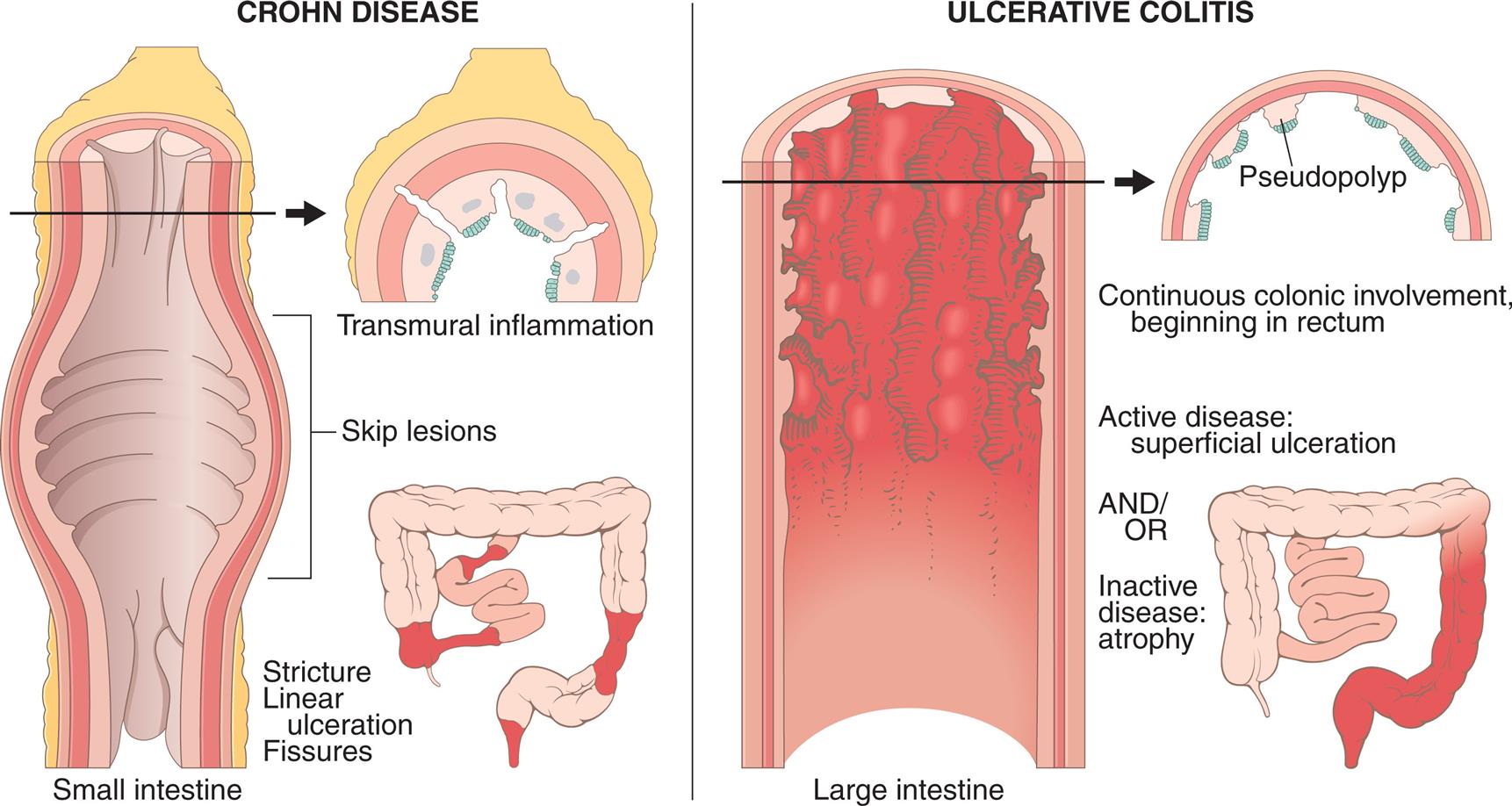
Comparison of distribution patterns of Crohn disease and ulcerative colitis as well as different conformations of ulcers and wall thickenings. (From Kumar V, et al. Robbins basic pathology, 8th edition. St. Louis: Mosby; 2008.)
The illustration on the left depicts the distribution pattern of Crohn's disease, which shows the sectional view of the small intestine with labels indicating the skip lesion, stricture, linear ulceration, and fissures accompanied by the illustrations of a horizontal sectional view small intestine indicating the transmural inflammation. The illustration on the right depicts the distribution pattern of ulcerative colitis with labels indicating continuous colonic involvement, beginning in rectum; active disease: superficial ulceration; and or inactive disease: atrophy accompanied by the horizontal section of the large intestine indicating the pseudopolyp.
Pathophysiology
The primary lesion of UC begins with inflammation at the base of the crypt of Lieberkühn in the large intestine. The disease begins in the rectum (proctitis) and may extend proximally to the entire colon (pancolitis). The lesions are limited to mucosal epithelium, are not transmural, and do not involve skip lesions. There is decreased secretion of mucin, which is antimicrobial and provides a protective layer against pathogens. Loss of this protection leads to increased permeability of the mucosa, increased passage of pathogens and other antigens, and stimulation of the gut immune system with an inflammatory response. There is activation of T cells and dendritic cells, triggering the production of proinflammatory cytokines and chemokines, including tumor necrosis factor (TNF)-α, interleukin (IL)-12 and IL-23, toxic oxygen free radicals, and interferon-gamma (IFN-γ), producing damage to the intestinal epithelium. In addition, there is activation of vascular adhesion molecules (integrins, e.g., mucosal addressin cellular adhesion molecule-1 [MadCAM-1]) which promote the trafficking of lymphocytes into the gut, furthering potentiation of the inflammatory response. Some of these molecules have become important targets for treatment.62
The mucosa is inflamed and is involved in a continuous fashion. With milder inflammation, the mucosa is hyperemic and edematous and may appear dark red. In more severe inflammation, the mucosa becomes hemorrhagic, and small erosions form and coalesce into ulcers. Abscess formation, necrosis, and ragged ulceration of the mucosa ensue. Edema and thickening of the muscularis mucosae may narrow the lumen of the involved colon. Mucosal destruction and inflammation cause bleeding, cramping pain, and an urge to defecate. Frequent diarrhea, with passage of small amounts of blood and purulent mucus, is common. Loss of the absorptive mucosal surface and rapid colonic transit time cause large volumes of watery diarrhea.
Clinical Manifestations
The course of UC consists of intermittent periods of remission and exacerbation. Mild UC involves less mucosa, so the frequency of bowel movements, bleeding, and pain is minimal. Severe forms may involve the entire colon and are characterized by abdominal pain, fever, an elevated pulse rate, frequent diarrhea (10 to 20 stools/day), urgency, bloody stools, and continuous, crampy pain; dehydration, weight loss, anemia, and fever result from fluid loss, bleeding, and inflammation. Complications include anal fissures, hemorrhoids, and perirectal abscess. Severe hemorrhage is rare. Edema, strictures, or fibrosis can obstruct the colon. Perforation is an unusual but possible complication. Extraintestinal manifestations include cutaneous lesions (erythema nodosum), polyarthritis, episcleritis, uveitis, disorders of the liver, and alterations in coagulation.63
Evaluation and Treatment
The diagnosis of UC is based on the medical history, clinical manifestations, and laboratory, serologic, imaging, endoscopic, and histology findings. Infectious causes are ruled out by stool culture. Endoscopic evaluation shows an inflamed and hemorrhagic mucosa. Radiologic assessment may show ulceration and irregular mucosa. The laboratory data include low hemoglobin levels, hypoalbuminemia, and low serum potassium levels. The gold standard in making the diagnosis remains biopsy and histology.63 The symptoms of UC are often similar to those of CD, making differential diagnosis challenging.
Treatment is individualized and depends on the severity of symptoms and the extent of mucosal involvement. A goal is to promote mucosal healing and avoid surgery. Mild to moderate disease is treated with 5-aminosalicylate therapy followed by steroids. Immunomodulatory agents are used for failure of first-line treatments or serious recurrent disease including TNF-α–blocking agents (e.g., tacrolimus) or antiadhesion agents (i.e., vedolizumab). New small molecule drugs are being investigated.64 New oral agents have recently been approved (see Emerging Science Box: Oral Treatments for Ulcerative Colitis). Severe, unremitting disease can require hospital admission for administration of intravenous fluids and steroids. Extreme malnutrition may require total parenteral nutrition (TPN). Surgical resection of the colon may be performed if other forms of therapy are unsuccessful or if there are acute serious complications (sepsis, hemorrhage, perforation, or obstruction). Surgical approaches for severe UC include total proctocolectomy with end ileostomy or ileorectal anastomosis, or ileal pouch–anal anastomosis (IPAA). Pouchitis is a common complication of restorative proctocolectomy with IPAA performed as surgical treatment for both UC and CD. There are more frequent bowel movements, urgency to defecate, blood in the stool, incontinence, and abdominal pain. Antibiotic treatment is usually successful, and chronic symptoms may be managed with steroids or immune modulators.65
Crohn Disease
Crohn disease (CD) (granulomatous colitis, ileocolitis, or regional enteritis) is an inflammatory disorder that affects any part of the GI tract from the mouth to the anus. In a small percentage of cases, CD is difficult to differentiate from UC (see Table 41.7). Risk factors associated with CD include smoking, low fiber–high carbohydrate diet, medications such as NSAIDs, and an altered intestinal microbiome.66 CD appears to have a multifactorial etiology in which both genetics and environmental factors manifest the disease. Smoking increases the risk of developing severe disease and may cause a poorer response to treatment. The inflammation is driven by a sustained immune response against luminal bacterial antigens with activation of leukocyte adhesion molecules (integrins), hyperactivity of T cells with excess production of inflammatory cytokines (e.g., IL-12, IL-23, and IL-34), and activation of cytotoxic enzymes resulting in tissue damage.67
Pathophysiology
Inflammation begins in the intestinal submucosa and spreads with discontinuous transmural involvement or “skip lesions” that can involve any part of the GI tract from the mouth to the perianal area. Skip lesions are distinguished by inflamed areas mixed with uninflamed areas, noncaseating granulomas, fistulas, and deep penetrating ulcers. The distal small intestine and proximal large colon are most involved. The ulcerations of CD can produce fissures that extend inflammation into lymphoid tissue. The typical lesion associated with CD is a granuloma or a mass of inflammatory tissue with a cobblestone appearance of inflamed tissue (Fig. 41.12) surrounded by ulceration. Fistula may form in the perianal area between loops of intestine and may extend into the bladder, rectum, or vagina and form intra-abdominal abscesses. Strictures may develop, promoting obstruction.67
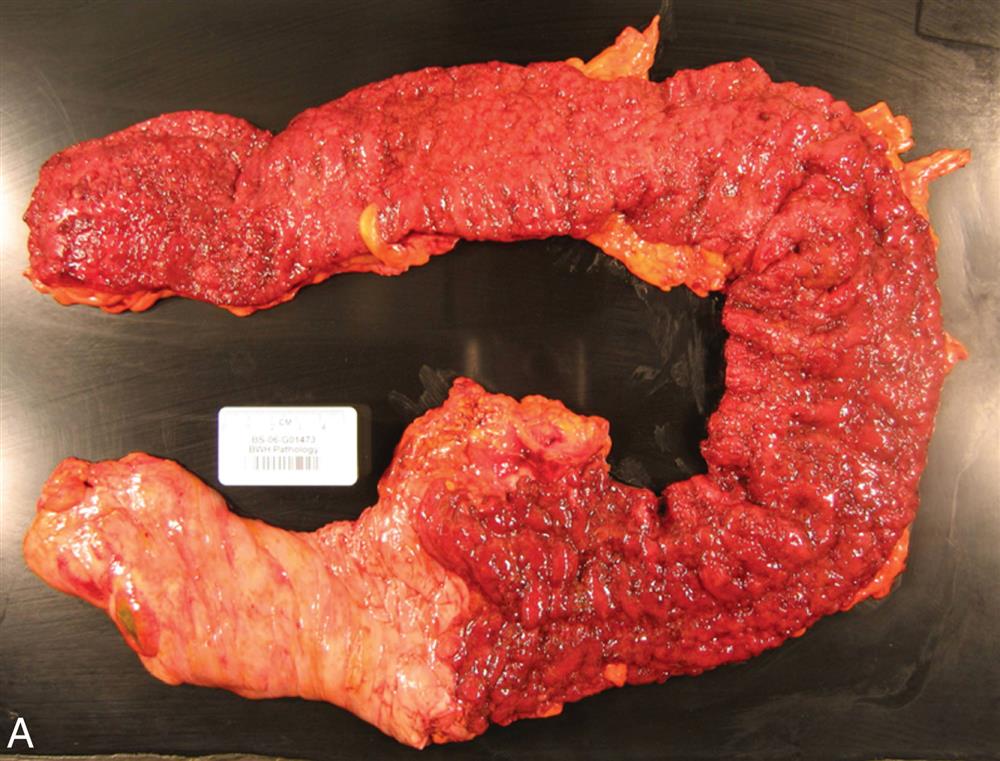
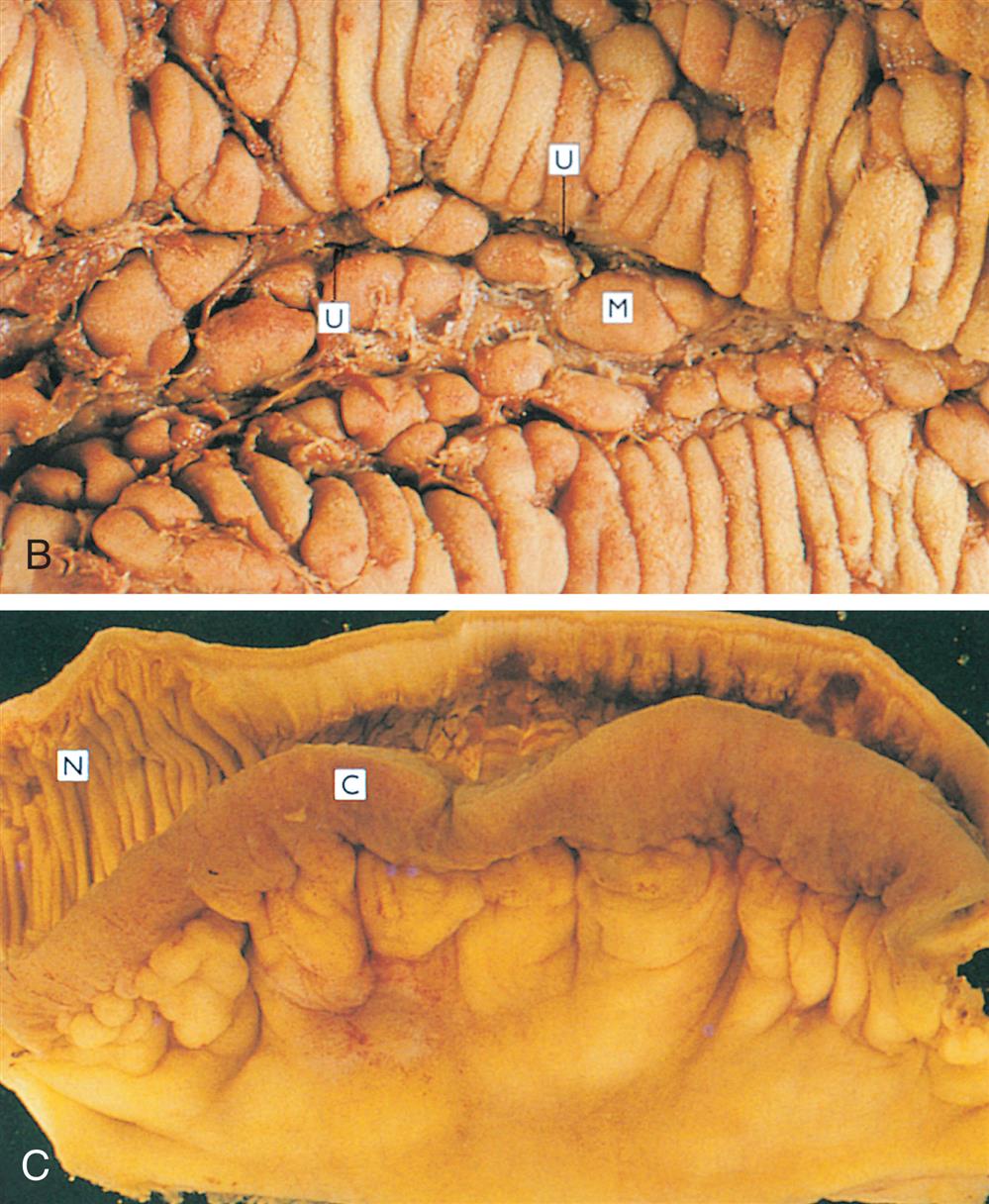
A. Close shot of gross specimen of the colon showing ulcerative colitis. B. Close shot of gross specimen representing longitudinal serpiginous ulcers separated by irregular islands of edematous mucosa. C. Close shot of gross specimen representing ileal segments, the mesenteric fat creeps from the mesentery to surround the bowel wall.
Clinical Manifestations
Individuals with CD may have no specific symptoms for several years. Symptoms vary according to the location of the disease but are similar to those for UC. Diarrhea is one of the most common symptoms, and occasionally rectal bleeding is noted if the colon is involved. Weight loss and abdominal pain accompany CD. Abdominal tenderness may also be noted over the lesions. If the ileum is involved, the individual may be anemic as a result of malabsorption of vitamin B12. There also may be deficiencies in folic acid and vitamin D absorption. In addition, proteins may be lost, leading to hypoalbuminemia. Extraintestinal complications are similar to those occurring in UC. Additional complications include anal fissure, perianal abscess, and fistula. Individuals with CD of long duration also are at risk for intestinal adenocarcinoma. Extraintestinal manifestations include arthropathies, skin, oral, and ocular lesions.68
Evaluation and Treatment
The diagnosis and treatment of CD are similar to the diagnosis and treatment of UC. Imaging of the small intestine is used in the diagnosis of CD, including either a small bowel series or a capsule endoscopy (camera pill). There are no specific biomarkers or definitive treatments for the disease. Smoking cessation is a component of therapy. Steroids are used to induce remission, and immunosuppressants (e.g., thiopurines and methotrexate) are used to sustain remission. Anti–TNF-α, antiintegrins (target the adhesion molecular inhibiting leukocyte migration), and IL inhibitors (target IL-12 and IL-23) are used for the most severe forms of the disease. Surgery may be performed to manage complications, such as fistula, abscess, or obstruction. When treatment involves surgical resection of small intestinal segments, complications related to short bowel syndrome may occur. Complications of short bowel syndrome include malabsorption, diarrhea, and nutritional deficiencies. Since malnutrition affects a significant number of individuals with CD, diet management is a significant component of the care plan.69 Routine colonoscopy for cancer screening should be performed for long-standing colonic disease.66
Microscopic Colitis
Microscopic colitis is a relatively common cause of nonbloody diarrhea. Although the mucosa appears normal, there are two histologic forms: lymphocytic and collagenous. Lymphocytic colitis shows an increase in the number of intraepithelial lymphocytes in the wall of the colon. Collagenous colitis is characterized by a thickened subepithelial collagen layer, with mucosal nodularity and an alteration of the vascular mucosal pattern. Risk factors include age 50 years or older, female sex, weight loss, smoking, use of PPIs, use of NSAIDs, and use of selective serotonin reuptake inhibitors. The cause of microscopic colitis is unknown, and proposed mechanisms include autoimmunity, genetic predisposition, an immune or inflammatory response to luminal antigens, certain medications, and abnormal collagen metabolism.70
The symptoms of frequent daily watery diarrhea are the same for both types and may be accompanied by abdominal pain and weight loss. Diagnosis is made from biopsy. Antidiarrheal agents and budesonide (an oral antiinflammatory steroid) are the best-documented treatments. The disease is negatively associated with CRC.
Irritable Bowel Syndrome
IBS currently is considered a disorder of brain-gut interaction (previously termed a functional GI disorder) characterized by recurrent abdominal pain with altered bowel habits.71 It is estimated that in the United States 10% to 12% of adults suffer with IBS symptoms.72 It is more common in women, with a higher prevalence during youth and middle age. Individuals with symptoms of IBS also are more likely to have anxiety, depression, and a reduced quality of life.73
Pathophysiology.The pathophysiology of IBS is unknown, and there are no specific biomarkers for the disease. There is increasing evidence to explain a multisystem interaction with variables, including infection, gut microbiota, immune activation, serotonin dysregulation, psychological stress, abnormal gut function and diet, as contributing factors to the varying symptom presentations. Mechanisms of pathophysiology are summarized as follows:
- • Visceral hypersensitivity or hyperalgesia may originate in either the peripheral or the central nervous system. The mechanism may be related to a dysregulation of the bidirectional “brain-gut axis” (alterations in gut or central nervous system processing of gut-pain information). Factors include genetic-related changes in the function of serotonin-secreting cells of gut-brain pain modulation, alterations in gut microbiota metabolite production with activation of the gut immune system, increased visceral sensitivity and permeability, and altered motility and secretion.
- • Abnormal GI permeability, motility, secretion, and sensitivity are associated with IBS. Individuals with diarrhea-type IBS have more rapid colonic transit times and increased intestinal permeability. Those with bloating and constipation have delayed transit times and decreased intestinal permeability. The mechanism may be related to dysregulation of the brain-gut axis, alterations in the function of gut neuroendocrine cells or dorsal root ganglion neurons, or changes in the activity of mast cells (produce histamine). Sex hormones may be a contributing factor.
- • Postinflammatory (infectious or noninfectious) IBS is associated with intestinal infection (bacterial enteritis) and low-grade inflammation. Alterations in gut microbiota, immune activation in gut tissues, and changes in intestinal permeability have been proposed.
- • Alteration in gut microbiota (dysbiosis) influences the sensory, motor, and immune systems of the gut and interacts with higher brain centers and may contribute to symptoms of IBS. Small intestine overgrowth of normal gut bacteria may be associated with IBS symptoms in some cases. Nonabsorbable antibiotics and prebiotics and probiotics may be helpful in some individuals.
- • Psychosocial factors (epigenetic factors)—including early life trauma or abuse or emotional stress interacting with neuroendocrine, neuroimmune, autonomic nervous system, and pain modulatory responses—contribute to the symptoms of IBS.
Clinical Manifestations
IBS symptoms may be mild or debilitating. IBS is characterized by lower abdominal pain or discomfort and bloating. IBS can be grouped as diarrhea-predominant, constipation-predominant, or mixed diarrhea/constipation. Symptoms including gas, bloating, fecal urgency, incomplete evacuation, and nausea are usually relieved with defecation and do not interfere with sleep.
Evaluation and Treatment
The diagnosis of IBS is based on signs, symptoms, personal history, and includes the exclusion of structural or biochemical causes of disease such as IBD or intestinal infection. Diagnostic procedures to rule out other causes of symptoms may include endoscopic evaluations, CT scans, abdominal ultrasound, blood tests, and lactose intolerance test. Fecal calprotectin is evaluated in those with suspected IBS and diarrhea symptoms to rule out IBD.
The individual may be evaluated for food allergies, parasites, or bacterial growth. The Rome IV criteria for diagnosing IBS are presented in Box 41.2.
There is no cure for IBS, and treatment is individualized.74 Treatment of symptoms may include laxatives, fiber, antidiarrheals, antispasmodics, prosecretory drugs, low-dose antidepressants, visceral analgesics, and serotonin agonists or antagonists and supportive care is symptom related. Alternative therapies include prebiotics and probiotics to manipulate the microflora. Hypnosis, acupuncture, yoga, cognitive behavioral therapy, and dietary interventions have been used with varying results.75 Research continues to advance the management and understanding of the pathophysiology of this complex syndrome.
Diverticular Disease of the Colon
Diverticula are herniations or sac-like outpouchings of the mucosa and submucosa through the muscle layers, usually located in the wall of the sigmoid colon and they are more common in older adults (Fig. 41.13). They rarely occur in the small intestine. Diverticulosis is asymptomatic diverticular disease. Diverticulitis represents inflammation of the diverticula and occurs in approximately 10% to 15% of cases of diverticular disease of the colon.76 The cause of diverticular disease is unknown, but it is associated with increased intracolonic pressure, abnormal neuromuscular function, and alterations in intestinal motility. Predisposing factors include older age, genetic predisposition, obesity, smoking, diet, lack of physical activity, and medication use (e.g., aspirin and NSAIDs).77 Lack of dietary fiber may or may not contribute to diverticular disease. Altered intestinal microbiota, visceral hypersensitivity, and abnormal colonic motility also may be contributing factors.78
In diverticular disease, the outpouches of mucosa seen in the sigmoid colon appear as slit-like openings from the mucosal surface of the opened bowel. (From Townsend CM, et al. Sabiston textbook of surgery: The biologic basis of modern surgical practice, 21st edition. St. Louis: Elsevier; 2022.)
Pathophysiology
Diverticula can occur anywhere in the GI tract, particularly at weak points in the colon wall, usually where arteries penetrate the tunica muscularis. The most common sites are the left sigmoid colon (more prevalent in Western countries) and the right colon (more prevalent in Asian countries). A common associated finding of the disease is thickening of the circular muscles and shortening of the longitudinal (teniae coli) muscles surrounding the diverticula. Although not characterized as muscle hypertrophy, diverticular disease may cause increased collagen and elastin deposition and is associated with muscle thickening. This contributes to increased intraluminal pressure and herniation. According to the law of Laplace (see Chapter 34), wall pressure increases as the diameter of a cylindrical structure decreases. Therefore pressure within the narrow lumen can increase enough to rupture the diverticula, causing inflammation and diverticulitis. Bacteria and local ischemia also may be contributing factors. Complicated diverticulitis includes abscess, fistula, obstruction, bleeding, or perforation.
Clinical Manifestations
Symptoms of uncomplicated diverticular disease may be vague or absent. Cramping pain of the lower abdomen can accompany constriction of the thickened colonic muscles. Diarrhea, constipation, distention, or flatulence may occur. If the diverticula become inflamed or abscesses form, the individual develops fever, leukocytosis, and tenderness in the lower left quadrant.
Evaluation and Treatment
Diverticula are often discovered during diagnostic procedures performed for other problems. Ultrasound, sigmoidoscopy, or colonoscopy permits direct observation of the lesions. Abdominal CT is used for diagnosis of complicated cases.
An increase in dietary fiber intake often relieves symptoms by increasing bulk and lowering colonic pressure. Uncomplicated diverticulitis is usually treated with bowel rest and a clear, liquid diet, analgesia, and selective use of antibiotics. Complicated cases may require intravenous antibiotics and abscess drainage if needed. In severe cases, bowel resection surgery may be needed with or without a colostomy.79
Appendicitis
Appendicitis is an inflammation of the vermiform appendix, which is a projection from the apex of the cecum. Appendicitis is a medical emergency. It is the most common surgical emergency of the abdomen, usually occurring between 10 and 19 years of age (although it may develop at any age). The incidence in the United States is 10 cases per 10,000 persons.80
Pathophysiology
The exact mechanism of the cause of appendicitis is not well understood. Obstruction of the lumen with stool, tumors, or foreign bodies, with consequent bacterial infection, is the most common theory. The obstructed lumen does not allow drainage of the appendix, and as mucosal secretion continues, intraluminal pressure increases. The increased pressure decreases mucosal blood flow, and the appendix becomes hypoxic. The mucosa ulcerates, promoting bacterial or other microbial invasion, with further inflammation and edema. Inflammation may involve the distal or entire appendix. Gangrene develops from thrombosis of the luminal blood vessels, followed by a periappendicular abscess and perforation resulting in peritonitis in complex cases.80,81
Clinical Manifestations
Epigastric or periumbilical pain is the typical symptom of an inflamed appendix. The pain may be vague at first but will increase in intensity over 3 to 4 hours. It may subside and then migrate to the right lower quadrant, indicating extension of the inflammation to the surrounding tissues. Nausea, vomiting, and anorexia follow the onset of pain, and a low-grade fever is common. Diarrhea occurs in some individuals, particularly children; others have constipation. Perforation, peritonitis, and abscess formation are the most serious complications of appendicitis.
Evaluation and Treatment
In addition to clinical manifestations, there is pain with abdominal palpation and rebound tenderness, usually referred to the right lower quadrant. The white blood cell count is greater than 10,000 cells/mm3, with increased neutrophils and C-reactive protein. Abdominal ultrasound, CT scans, and magnetic resonance imaging (MRI) (particularly for pregnant women and children) assist with diagnostic accuracy and help to rule out nonappendiceal disease. Antibiotics and appendectomy are the treatment for simple appendicitis.82 Treatment for complicated appendicitis (perforation, abscess formation, peritonitis) also includes antibiotics and appendectomy; however, recovery may be more complicated.80,83 There is an increased risk of colon cancer post appendectomy among individuals aged 50 to 54 years, and follow-up colonoscopy is recommended.84
Mesenteric Vascular Insufficiency
Mesenteric vascular insufficiency is rare, with an incidence of approximately 2 to 3 cases per 100,000 persons.85 Three branches of the abdominal aorta supply the stomach and intestines: the celiac artery and the superior and inferior mesenteric arteries. The inferior mesenteric vein drains into the splenic vein, and the splenic vein and superior mesenteric vein join the portal vein. Mesenteric venous thrombosis is the least common of the causes of mesenteric vascular insufficiency. Malignancies, right-sided heart failure, and deep vein thrombosis are risk factors. Mesenteric venous thrombosis presents with abdominal pain and is treated with anticoagulants.
Acute mesenteric arterial insufficiency results in a significant reduction in mucosal blood flow to the large and small intestines.86 Preexisting morbidities include dissecting aortic aneurysms, arterial thrombi, or emboli. Embolic obstruction is associated with atrial fibrillation, mitral valve disease, heart valve prostheses, and myocardial infarction. The superior mesenteric artery has a more direct line of flow from the aorta; therefore emboli enter it more readily than the inferior branch, causing ischemia and necrosis of the small intestine. Ischemia, infarction, and necrosis all alter membrane permeability. Initially, there is increased motility, nausea and vomiting, urgent bowel evacuation, and severe abdominal pain. Ischemia leads to decreased motility and distention. The damaged intestinal mucosa cannot produce enough mucus to protect itself from digestive enzymes. Mucosal alteration causes fluid to move from the blood vessels into the bowel wall and peritoneum. Fluid loss causes hypovolemia and further decreases intestinal blood flow. As intestinal infarction progresses, shock, fever, bloody diarrhea, and leukocytosis develop. Bacteria invade the necrotic intestinal wall, causing gangrene and peritonitis.
Chronic mesenteric ischemia (CMI) is rare. The most common etiology is atherosclerotic stenosis or occlusion of the mesenteric arteries. CMI can be associated with congestive heart failure, acute myocardial infarction, hemorrhage, thrombus formation, or any condition that decreases arterial blood flow. Chronic occlusion is often accompanied by the formation of collateral circulation. The collateral vessels may be able to nourish the resting intestine, but after eating, when the intestine requires more blood, the arterial supply may be insufficient. Ischemia develops, causing cramping abdominal pain or abdominal angina, which is a cardinal symptom reoccurring over a period of 3 months. Some individuals suffer significant weight loss because they stop eating to control the pain. Progressive vascular obstruction eventually causes continuous abdominal pain and necrosis of the intestinal tissue.
The diagnosis of acute and CMI is based on clinical manifestations, laboratory findings, and imaging studies. The diagnosis may be difficult because of the vagueness of symptoms. A bruit can often be heard over a partially occluded artery. Treatment includes aggressive rehydration and the use of antibiotics, anticoagulants, vasodilators, and inhibitors of reperfusion injury. Surgery, including percutaneous stenting or open techniques, is required to remove necrotic tissue, repair sclerosed vessels, and revascularize affected tissue. Acute occlusion is a surgical emergency, and the mortality rate is high (50% to 90%). Early diagnosis and aggressive treatment result in the best survival rates.87,88
Disorders of the accessory Organs of Digestion
The accessory organs of digestion, including the liver, gallbladder, and pancreas, secrete substances necessary for digestion and in the case of the liver carry out metabolic functions needed to maintain life. Disorders of these organs include inflammatory disease, obstruction of ducts, and tumors. (Cancers of the digestive system are described at the end of this chapter.)
Common Complications of Liver Disorders
Of all the accessory organ disorders, acute or chronic liver disease leads to the most significant systemic, life-threatening complications. These complications are common to all liver disorders and include portal hypertension, ascites, hepatic encephalopathy, jaundice, and hepatorenal syndrome (HRS).
Portal Hypertension
Portal hypertension is abnormally high blood pressure in the portal venous system caused by resistance to blood flow. Pressure in this system is normally 3 mm Hg; portal hypertension is an increase to at least 10 mm Hg.
Pathophysiology
Portal hypertension is caused by disorders that obstruct or impede blood flow through any component of the portal venous system or vena cava. Intrahepatic causes result from vascular remodeling with shunts, thrombosis, inflammation, or fibrosis of the sinusoids, as occurs in cirrhosis of the liver, biliary cirrhosis, viral hepatitis, or schistosomiasis (a parasitic infection). Posthepatic causes occur from hepatic vein thrombosis or cardiac disorders that impair the pumping ability of the right side of the heart. This causes blood to collect and increases pressure in the veins of the portal system. The most common cause of portal hypertension is fibrosis and obstruction caused by cirrhosis of the liver. Long-term portal hypertension causes several pathophysiologic problems that are difficult to treat and can be fatal. These problems include varices, splenomegaly, ascites, hepatic encephalopathy, and hepatopulmonary syndrome (HPS) (see Emerging Science Box: Portal Hypertension).
Varices are distended, tortuous collateral veins. Prolonged elevation of pressure in the portal vein causes collateral veins to open between the portal vein and systemic veins. The prolonged pressure is distributed throughout the GI tract and results in transformation into varices, particularly in the lower esophagus and stomach, but also over the abdominal wall (known as the caput medusae [Medusa head]) and rectum (hemorrhoidal varices) (Fig. 41.14). The hyperdynamic circulation in the stomach and esophagus impairs mucosal defenses, promotes inflammation, and disrupts healing with increased risk of mucosal erosion, ulceration, and bleeding.89 Rupture of varices can cause life-threatening hemorrhage.90
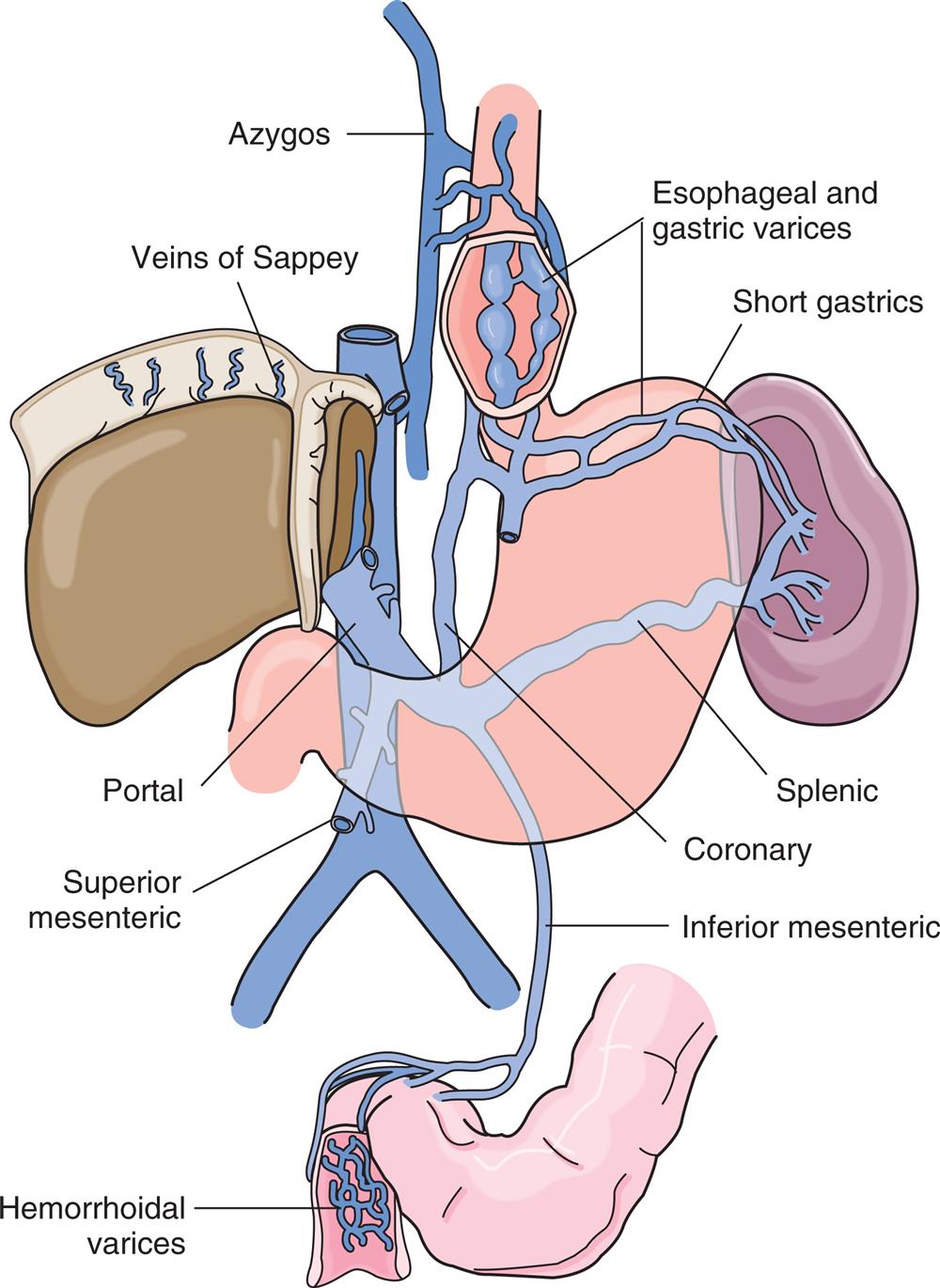
The portal vein, its major tributaries, and the most important shunts (collateral veins) between the portal and caval systems. The shunted blood returns to the systemic venous system, bypassing the liver. (From Monahan FD, et al. Phipps’ medical-surgical nursing: Concepts and clinical practice, 8th edition. St. Louis: Mosby; 2007.)
An illustration depicts the varices encountered in portal hypertension showing the digestive system with labels indicating azygos, esophageal and gastric varices, short gastric, veins of Sappey, portal, superior mesenteric, hemorrhoidal varices, inferior mesenteric, coronary, splenic.
Splenomegaly is enlargement of the spleen caused by increased pressure in the splenic vein, which branches from the portal vein. Thrombocytopenia is the most common symptom of congestive splenomegaly. The enlarged spleen can often times be palpated. HPS and portopulmonary hypertension (PPH) are respiratory complications of liver disease and portal hypertension. The pathophysiology of both is complex and involves different effects of vasoactive substances. In HPS there is pulmonary vasodilation, probably because of the increased nitric oxide synthesis, increased pulmonary venous congestion, and right-to-left shunting that induces hypoxemias. PPH is associated with vasoconstriction and arterial vascular remodeling with thickening and fibrosis of the arterial wall that increases pulmonary artery resistance. Individuals may be asymptomatic, or fatigue, dyspnea, cyanosis, and clubbing may occur with or without signs of right heart failure (jugular venous distention, ascites, and peripheral edema).
Diagnosis includes pulmonary function tests, arterial blood gas analysis, contrast echocardiography, transthoracic echocardiography, and right heart catherization. In PPH, mean pulmonary artery pressure is greater than 25 mm Hg at rest. There is no specific treatment for HPS. Treatment of PPH includes targeting the pulmonary arterial vasculature to reduce pulmonary hypertension with various medication options that include endothelin receptor antagonists, phosphodiesterase type 5 inhibitors, and prostanoids. Liver transplant may be indicated.91
Clinical Manifestations
Hematemesis from bleeding esophageal varices is the most common clinical manifestation of portal hypertension. Bleeding is usually from varices that have developed slowly over a period of years. Slow, chronic bleeding from varices causes anemia or melena. Acute rupture of esophageal varices causes hemorrhage and voluminous vomiting of dark-colored blood. The ruptured varices are usually painless. Rupture is caused by a combination of erosion by gastric acid and elevated venous pressure. Mortality from ruptured esophageal varices ranges from 30% to 60%. Recurrent bleeding of esophageal varices indicates a poor prognosis. Hemorrhoidal varices present as hematochezia with copious rectal bleeding.
Evaluation and Treatment
Portal hypertension is often diagnosed at the time of variceal bleeding and confirmed by upper GI endoscopy and evaluation of portal venous pressure. The individual usually has a history of jaundice, hepatitis, alcoholism, or cirrhosis. Liver elastography is an imaging test that provides a noninvasive measure of liver stiffness and can diagnose the extent of fibrosis.92
Emergency management of bleeding varices includes use of vasopressors and compression of the varices with an inflatable tube or balloon, sclerotherapy, variceal ligation, or portacaval shunt. Surgical construction of transjugular intrahepatic portosystemic shunts (TIPSs) and anastomosis of the portal vein to the inferior vena cava may decompress the varices. This treatment can precipitate encephalopathy. Emergency management will also include stabilization of the individual with fluid resuscitation, red blood cell replacement, and antibiotics. Liver transplantation is the most successful option for liver failure. Nonemergent or long-term management may include nonselective β-blocking drugs to assist reducing the pressure in the portal venous system and to prevent variceal bleeding.93
Ascites
Ascites is the accumulation of fluid in the peritoneal cavity. Ascites traps body fluid in the peritoneal space, from which it cannot escape. Ascites reduces the amount of body fluid available for normal physiologic functions. Cirrhosis is the most common cause of ascites, but other causes include heart failure, constrictive pericarditis, abdominal malignancies, nephrotic syndrome, and malnutrition. Of individuals who develop ascites caused by cirrhosis, 25% die within 1 year. Continued heavy drinking of alcohol is associated with this mortality and is related to decompensated cirrhosis.
Pathophysiology
Several factors contribute to the development of ascites, including portal hypertension, splanchnic vasodilation, decreased synthesis of albumin by the liver, splanchnic arterial vasodilation, and renal sodium and water retention. Portal hypertension causes capillary hydrostatic pressure to exceed capillary osmotic pressure (see Chapter 3), pushing water into the peritoneal cavity. Portal hypertension also increases the production of hepatic lymph, which “weeps” into the peritoneal cavity. Reduced serum albumin levels reduce capillary oncotic pressure adding to the fluid shift. Splanchnic arterial vasodilation is associated with increased nitric oxide produced by the diseased liver and can decrease effective circulating blood volume, activating the renin-angiotensin-aldosterone system and antidiuretic hormone, which in turn promotes renal sodium and water retention. The sodium and water retention expands plasma volume, thereby accelerating portal hypertension and ascites formation. In addition, translocation of bacteria and release of endotoxin cause peritonitis with an inflammatory response that increases splanchnic vasodilation and mesenteric capillary permeability and fluid movement into the peritoneal cavity, promoting ascites. Fig. 41.15 summarizes the mechanisms by which cirrhosis of the liver cause ascites.
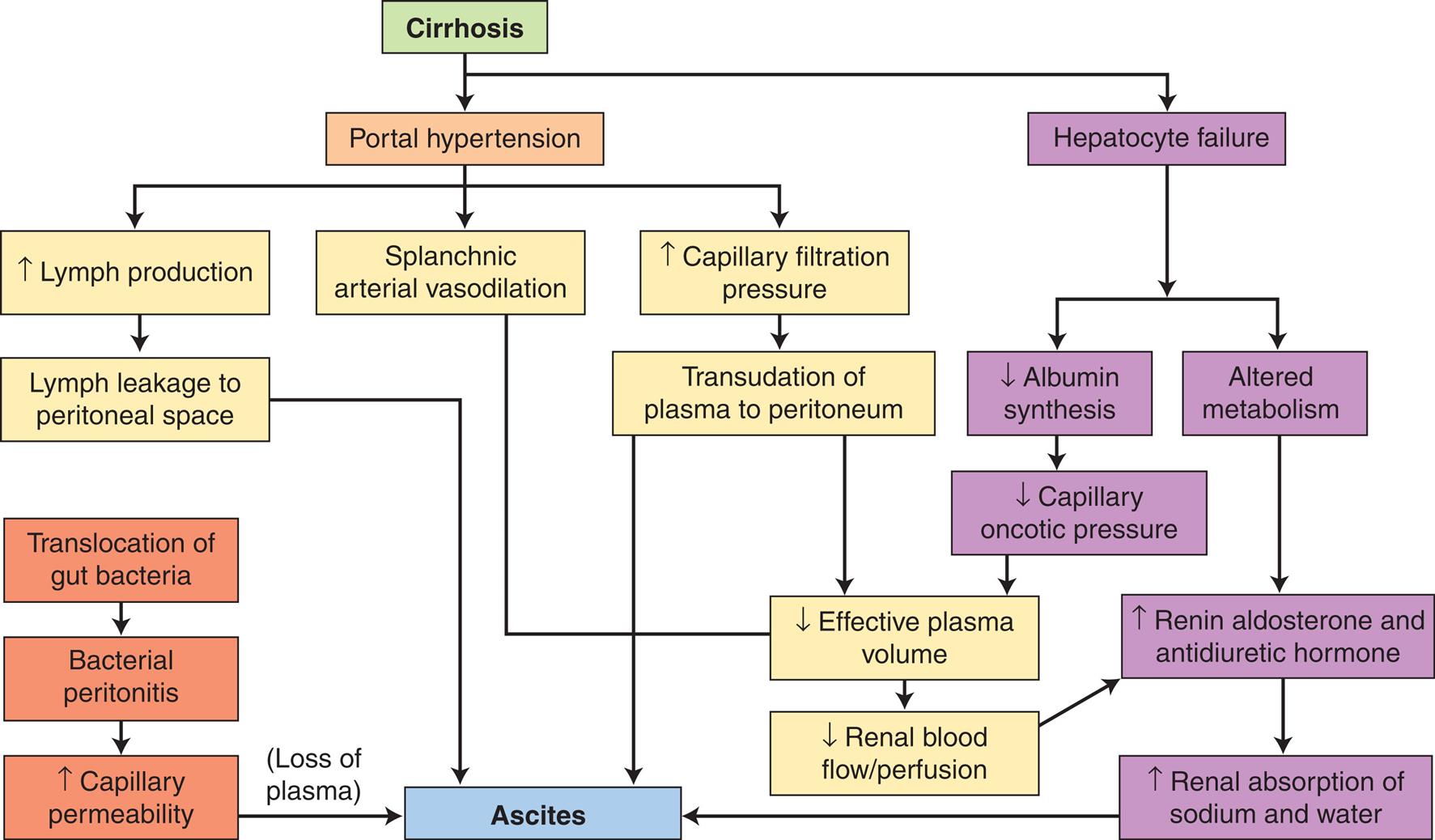
A flow chart summarizes the mechanisms of ascites caused by cirrhosis. The diagram begins as follows. • Cirrhosis leads to portal hypertension and hepatocyte failure. • Portal hypertension leads to increased lymph production, splanchnic arterial vasodilation, and increased capillary filtration pressure. • Increased capillary filtration pressure leads to transudation of plasma to the peritoneum, decreasing effective plasm volume, and renal blood flow, or perfusion, causing ascites. • An increase in lymph production leads to lymph leakage to the peritoneal space causing ascites. • The translocation of gut bacteria leads to bacterial peritonitis, which increases capillary permeability, causing ascites. • Hepatocyte failure leads to a decrease in albumin synthesis and altered metabolism. • Albumin synthesis leads to decreased oncotic capillary pressure causing decreased effective plasma volume and renal blood flow perfusion. •Altered metabolism leads to an increase in renin aldosterone and antidiuretic hormone and an increase in renal absorption of sodium and water, causing ascites.
Clinical Manifestations
The accumulation of ascitic fluid causes abdominal distention, increased abdominal girth, and weight gain (Fig. 41.16). Large volumes of fluid (10 to 20 L) displace the diaphragm and cause dyspnea by decreasing lung capacity. The respiratory rate increases, and the individual may need to assume a sitting position to relieve the dyspnea. Some peripheral edema is usually present. Dilutional hyponatremia may be noted because of excess fluid volume. Approximately 10% of individuals with ascites develop bacterial peritonitis, either spontaneously or because of paracentesis, which causes fever, chills, abdominal pain, decreased bowel sounds, and cloudy ascitic fluid.
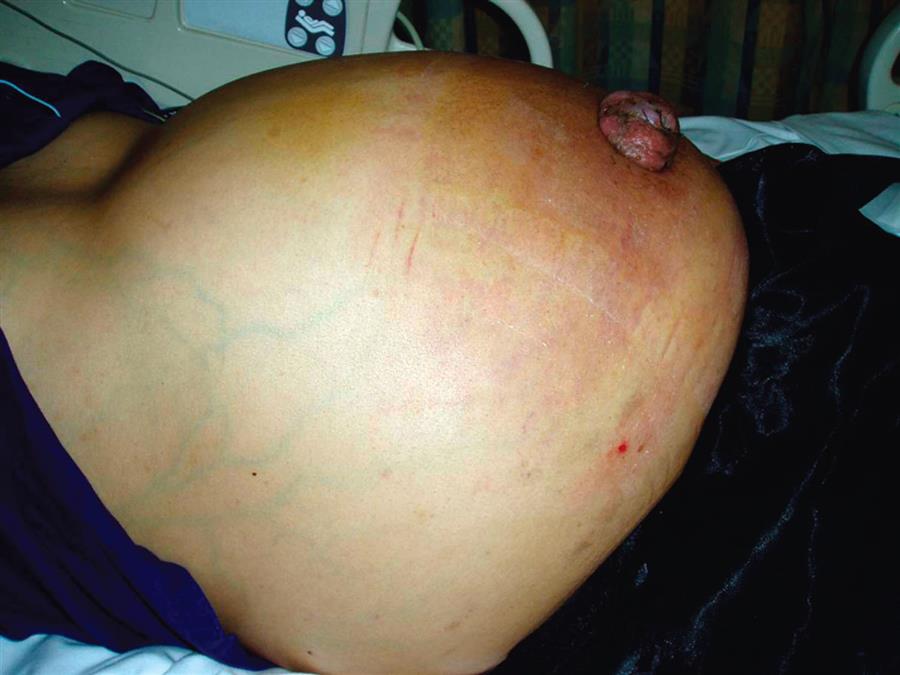
Distended abdomen, dilated upper abdominal veins, and inverted umbilicus are classic manifestations. (From Goldman L, Schafer AI. Goldman’s Cecil medicine, 24th edition. Philadelphia: Saunders; 2012.)
Evaluation and Treatment
The diagnosis is usually based on clinical manifestations and identification of liver disease. Marked abdominal distention is associated with higher grades of ascites. The serum ascites albumin gradient (SAAG) from ascetic fluid analysis is the most specific diagnostic indicator for portal hypertension–related ascites. Ultrasonography, chest and abdominal x-rays, or CT scans are used to evaluate the cause and extent of the ascites and other complications of the disease. Serum electrolyte levels must be carefully monitored as the disease process puts an individual at risk for hyponatremia and hypokalemia. Spontaneous bacterial peritonitis can occur with cirrhotic ascites, the neutrophil count will be elevated in ascetic fluid, and bacterial culture guides antibiotic therapy.
The goal of treatment is to relieve discomfort. If the restoration of liver function is possible (e.g., in ascites caused by viral hepatitis), the ascites diminishes spontaneously. Dietary salt restriction and potassium-sparing diuretics can reduce ascites. Vasopressin receptor-2 antagonists are effective for dilutional hyponatremia. Albumin may be given. Paracentesis is used to aspirate ascitic fluid for bacterial culture, biochemical analysis, and microscopic examination. Maintenance of caloric and protein intake is necessary to prevent malnutrition.
Palliative paracentesis in amounts of 1 or 2 L of ascitic fluid is completed to relieve respiratory distress. However, the removal of too much fluid too quickly relieves pressure on blood vessels and carries the risk of hypotension, shock, or death. Despite repeated paracentesis, ascitic fluid reaccumulates because of the persistent portal hypertension and reduced plasma albumin levels associated with irreversible disease. Peritonitis is treated with antibiotics. Other procedures include placement of a peritoneovenous shunt and TIPS. Individuals with ascites and portal hypertension have a poor prognosis, and liver transplantation is the best treatment option.94
Hepatic Encephalopathy
Hepatic encephalopathy (portal system encephalopathy) is a complex neurologic syndrome characterized by impaired behavioral, cognitive, and motor function. The syndrome may develop rapidly during acute fulminant hepatitis or slowly during the course of cirrhosis and the development of portal hypertension or after portosystemic bypass or shunting.
Pathophysiology
Hepatic encephalopathy results from a combination of biochemical alterations that affect neurotransmission and brain function. Liver dysfunction and the development of collateral vessels that shunt blood around the liver to the systemic circulation permit toxins absorbed from the GI tract and normally removed by the liver, to accumulate and circulate freely to the brain. The accumulated toxins alter cerebral energy metabolism, interfere with neurotransmission, and cause edema. The most hazardous substances are end products of intestinal protein digestion, particularly ammonia, which cannot be converted to urea by the diseased liver. The digestion of blood from leaking or ruptured varices adds to the amount of ammonia present in systemic blood, as does the action of ammonia-forming bacteria in the colon. Ammonia that reaches the brain is metabolized to glutamine, with osmotic disturbances and alterations in cerebral blood flow that interfere with neurotransmitters and cause astrocyte edema or cytotoxic edema and oxidation. Disruption of the blood-brain barrier causes vasogenic edema and contributes to astrocytes swelling, brain edema, and intracranial hypertension. Excessive amounts of gamma-aminobutyric acid (GABA), an inhibitory neurotransmitter from intestinal flora, may contribute to reduced levels of consciousness. Infection, systemic inflammation, hemorrhage, and electrolyte imbalance (including zinc deficiency), constipation, and the use of sedatives and analgesics can precipitate hepatic encephalopathy in the presence of liver disease.95
Clinical Manifestations
Subtle changes in personality, memory loss, irritability, disinhibition, lethargy, and sleep disturbances are common initial manifestations of hepatic encephalopathy. Symptoms then can progress to confusion, disorientation to time and space, asterixis, slow speech, bradykinesia, stupor, convulsions, and coma, which are less frequent. Coma is usually a sign of liver failure and ultimately results in death. Variceal bleeding and ascites may develop concurrently. Symptoms may be episodic, recurrent, or persistent. Hepatic encephalopathy is often associated with bleeding varices and ascites.
Evaluation and Treatment
The diagnosis of hepatic encephalopathy is based on a history of liver disease, clinical manifestations, psychometric tests, and exclusion of other causes of brain dysfunction. Electroencephalography and blood chemistry tests provide supportive data. Tracking levels of serum ammonia can assist to assess treatment effectiveness and liver function, but the test is difficult to perform and may be influenced by hemolysis and severe jaundice.96 Correction of fluid and electrolyte imbalances and withdrawal of depressant drugs metabolized by the liver are the first steps in the treatment of hepatic encephalopathy. Dietary protein is maintained to prevent malnutrition but at levels that reduce blood ammonia levels. Lactulose prevents ammonia absorption in the colon, and polyethylene glycol has also been used with success in reducing ammonia levels.97 Neomycin eliminates ammonia-producing intestinal bacteria but can be nephrotoxic. Glutamase inhibitors reduce gut ammonia. Rifaximin (a nonabsorbable antibiotic) decreases intestinal production of ammonia and may be combined with lactulose or used alone for lactulose nonresponders. Extracorporeal liver support systems remove toxins from the blood and are an option for managing overt hepatic encephalopathy or as a bridge to liver transplantation.96
Jaundice
Jaundice, or icterus, is a yellow or greenish pigmentation of the skin caused by hyperbilirubinemia (plasma bilirubin concentrations greater than 2.5 to 3 mg/dL). Hyperbilirubinemia and jaundice can result from (1) extrahepatic (posthepatic) obstruction to bile flow, (2) intrahepatic obstruction, or (3) prehepatic excessive production of unconjugated bilirubin (i.e., excessive hemolysis of red blood cells) (Fig. 41.17). Jaundice in newborns is caused by impaired bilirubin uptake and conjugation (see Chapter 42).
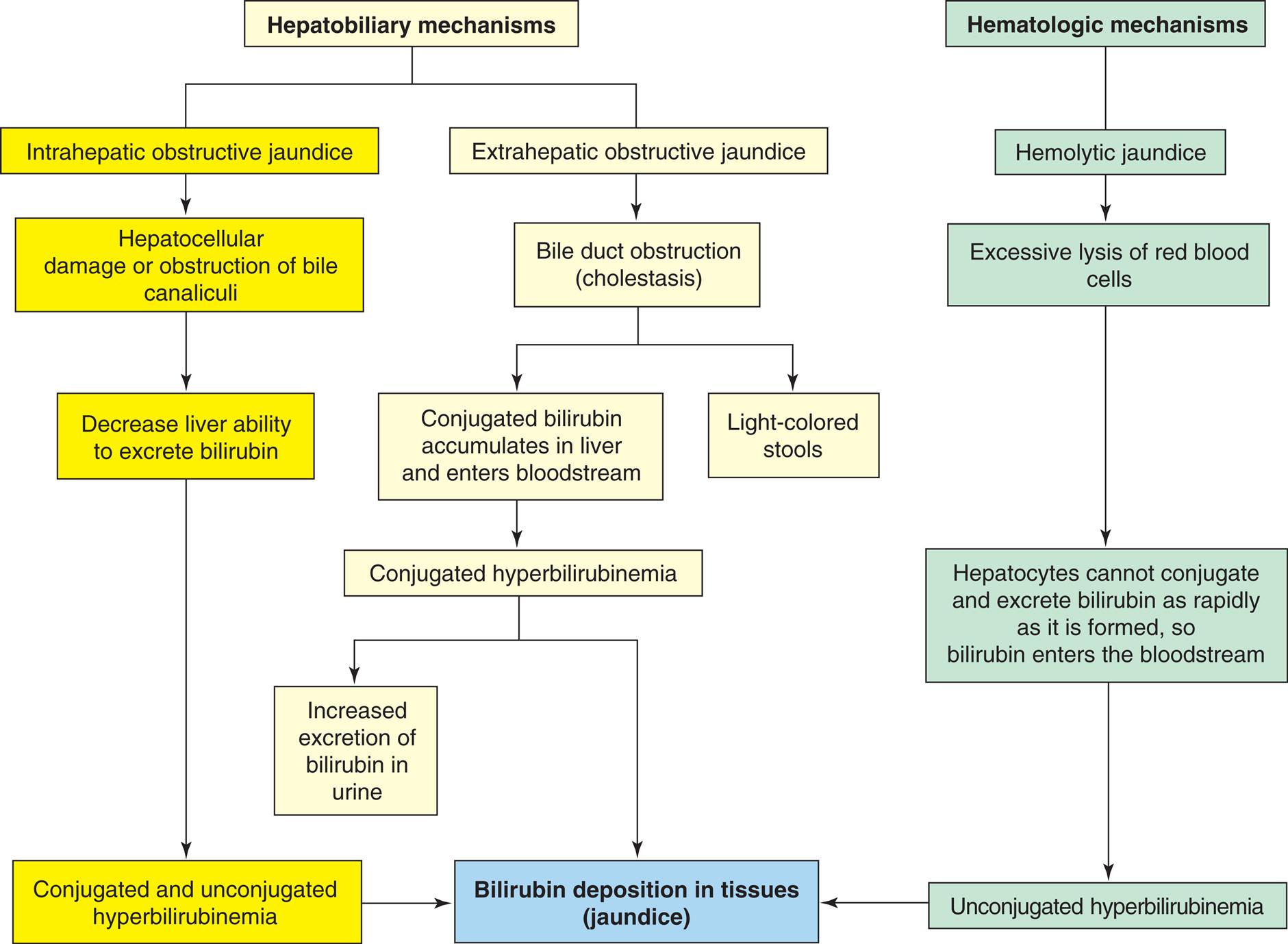
A flow diagram summarizes the mechanism involved in the development of jaundice. The diagram begins as follows. • Hepatobiliary mechanisms involved intrahepatic obstructive jaundice and extrahepatic obstructive jaundice. • Intrahepatic obstructive jaundice leads to hepatocellular damage or obstruction of bile canaliculi. • Hepatocellular damage or obstruction of bile canaliculi leads to decrease in the liver's ability to excrete bilirubin. • Decrease liver ability leads to conjugated unconjugated hyperbilirubinemia causing bilirubin deposition in tissues or jaundice. • Extrahepatic obstructive jaundice leads to bile duct obstruction causes conjugated bilirubin accumulation in the liver and enters the bloodstream and light-colored stools. • Conjugated bilirubin causes conjugated hyperbilirubinemia leading to an increase in excretion of bilirubin in urine. • Hematologic mechanisms cause hemolytic jaundice, leading to excessive lysis of red blood cells. • Excessive lysis of red blood cells leads to hepatocytes to not conjugate and excrete bilirubin as rapidly as it is formed, so bilirubin enters the bloodstream leading to unconjugated hyperbilirubinemia causing bilirubin deposition in tissues.
Pathophysiology
Obstructive jaundice can result from extrahepatic or intrahepatic obstruction.98Extrahepatic obstructive jaundice develops if the common bile duct is occluded. Occlusion may be from a gallstone, tumor, or inflammation. If occluded, bilirubin conjugated by the hepatocytes cannot flow through the obstructed common bile duct into the duodenum. Therefore bilirubin accumulates in the liver and enters the bloodstream, causing hyperbilirubinemia and jaundice. Intrahepatic (hepatocellular) obstructive jaundice involves disturbances in hepatocyte function and obstruction of bile canaliculi. The uptake, conjugation, or excretion of bilirubin can be affected with elevated levels of both conjugated and unconjugated bilirubin. Obstruction of bile canaliculi diminishes flow of conjugated bilirubin into the common bile duct. In mild cases, some of the bile canaliculi open. Consequently, the amount of bilirubin in the intestinal tract may be only slightly decreased.
Excessive hemolysis (destruction) of red blood cells can cause hemolytic jaundice(prehepatic or nonobstructive jaundice). Increased unconjugated bilirubin is formed through metabolism of the heme component of destroyed red blood cells and exceeds the conjugation ability of the liver, causing blood levels of unconjugated bilirubin to rise. Decreased bilirubin uptake or conjugation also causes unconjugated hyperbilirubinemia, as occurs with reaction to some drugs (e.g., rifampin), and in genetic disorders, such as Gilbert syndrome. Unconjugated bilirubin is not water soluble, so it will not be excreted in the urine. The causes of jaundice are summarized in Table 41.8.
Table 41.8
| Type | Mechanism | Causes |
|---|---|---|
| Hemolytic jaundice (predominantly unconjugated bilirubin) | Excessive destruction of erythrocytes | |
| Immune reaction | ||
| Severe infection | ||
| Toxic substances in the circulation (e.g., snake venom) | ||
| Transfusion of incompatible blood | ||
| Obstructive (cholestatic) jaundice (predominantly conjugated bilirubin) | Obstruction to passage of conjugated bilirubin from liver to intestine | Obstruction of bile duct by gallstones or tumor (extrahepatic obstructive jaundice) |
| Obstruction of bile flow through the liver (intrahepatic obstructive jaundice) | ||
| Drugs | ||
| Hepatocellular jaundice (both conjugated and unconjugated bilirubin) | Failure of liver cells (hepatocytes) to conjugate bilirubin and of bilirubin to pass from liver to intestine | Genetic defect of hepatocyte (decreased enzymes), such as occurs in premature infants (see Chapter 42) |
| Hepatitis or biliary cirrhosis |
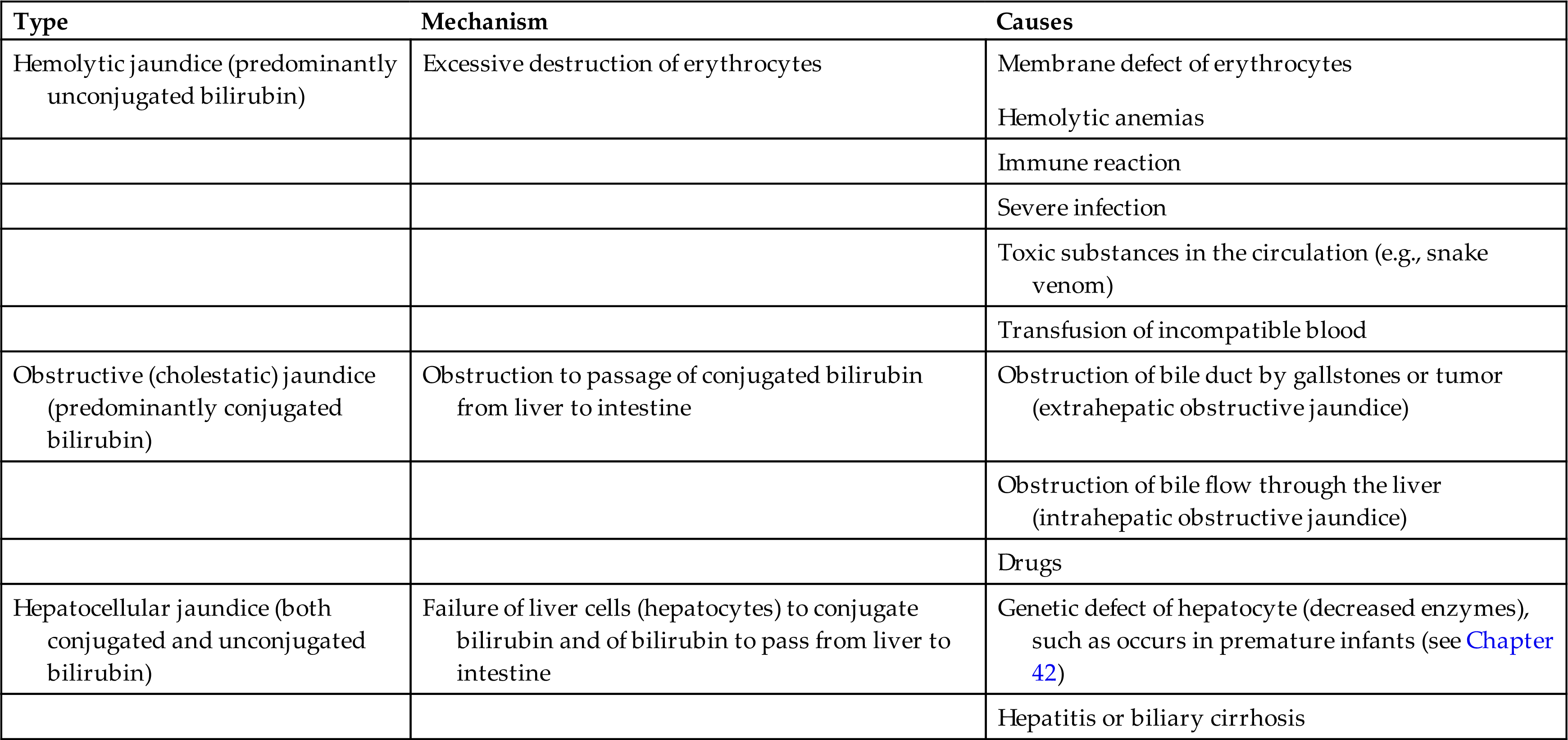
Clinical Manifestations
Conjugated bilirubin is water soluble and appears in the urine. The urine may darken several days before the onset of jaundice. The complete obstruction of bile flow from the liver to the duodenum causes light-colored stools. With partial obstruction, the stools are normal in color and bilirubin is present in the urine.
Fever, chills, and pain often accompany jaundice resulting from viral or bacterial inflammation of the liver. Yellow discoloration may first occur in the sclera of the eye and then progress to the skin as bilirubin attaches to elastic fibers. Pruritus (itching) often accompanies jaundice because bilirubin accumulates in the skin, irritating itch receptors.
Evaluation and Treatment
Laboratory evaluation of serum establishes whether elevated plasma bilirubin is conjugated or unconjugated, or both. The history and physical examination identify underlying disorders, such as cirrhosis, exposure to hepatitis virus, gallbladder or pancreatic disease, or hematologic disorders. The treatment for jaundice consists of correcting the cause.
Hepatorenal Syndrome
HRS is functional renal failure that develops as a complication of advanced liver disease. The renal failure is not caused by primary renal disease or other extrinsic factors but rather by portal hypertension, cardiac impairment, and other circulatory alterations associated with advanced liver disease, such as cirrhosis or fulminant hepatitis with portal hypertension and decreased systemic vascular resistance. HRS is characterized by reduced renal blood flow and glomerular filtration rate.
Pathophysiology
Type 1 HRS–acute kidney injury occurs in less than 2 weeks and accompanies a sudden decrease in blood volume secondary to massive GI or variceal bleeding and hypotension caused by bleeding and peripheral vasodilation associated with failing liver function. Proinflammatory cytokines related to translocation of bacterial to ascetic fluid can promote systemic hypotension and reduced renal blood flow. Hypotension also can be caused by the excessive use of diuretics to treat ascites or decreased cardiac output. The decrease in blood volume and hypotension result in decreased renal perfusion, decreased glomerular filtration, and oliguria (see Chapter 38). The serum creatinine increases to a concentration of greater than 0.3 mg/dL within 48 hours or a urine output less than 0.5 mL/kg body weight for greater than 6 hours.
Type 2 HRS–nonacute kidney injury (HRS-NAKI) develops slowly and is related to ascites resistant to diuretics. Ineffective circulating blood volume causes decreased glomerular filtration and oliguria. Intrarenal vasoconstriction may result from the selective effects of vasoactive substances that accumulate in the blood because of liver failure or as a compensatory response to portal hypertension and the pooling of blood in the splanchnic circulation. Vasoconstriction also may be a compensatory response to portal hypotension and vasodilation in the splanchnic circulation. There are two categories of NAKI. HRS–acute kidney disease (HRS-AKD) is diagnosed when the estimated glomerular filtration rate is less than 60 mL/min/1.73 m2 for less than 3 months and the percent increase in serum creatinine is less than 50% compared with a baseline value obtained within the past 3 months. The kidney usually maintains a normal structure with this condition; HRS–chronic kidney disease (HRS-CKD) is diagnosed when the estimated glomerular filtration rate is less than 60 mL/min/1.73 m2 for more than 3 months. The kidney usually maintains a normal structure, and there is absence of other structural causes of kidney disease.99
Clinical Manifestations
The onset of hepatorenal manifestations may be acute or gradual. Oliguria and complications of advanced liver disease, including jaundice, ascites, peripheral edema, hypotension, and GI bleeding, are usually present. Systolic blood pressure is usually less than 100 mm Hg. Nonspecific symptoms of HRS include anorexia, weakness, and fatigue.
Evaluation and Treatment
Diagnosis of HRS is made by excluding other causes of renal failure. Despite decreased glomerular filtration, serum potassium levels do not become dangerously elevated until the terminal stages of the HRS. Serial changes in serum creatinine provide an index for estimation of GFR, and values increase to 2.5 mg/dL or higher. Guidelines are available for determining AKI or CKD.100 The BUN level increases, and metabolic acidosis develops. Urine osmolality increases, but urine sodium concentrations are less than normal. Urine specific gravity is greater than 1.015.
The prognosis is usually poor and is related to a failing liver, requiring liver transplantation. Secondary problems, including fluid and electrolyte disorders, bleeding, infections, and encephalopathy, are treated. Vasoconstrictors and albumin are often used as first line treatment of HRS. Liver transplant reverses HRS symptoms in most individuals and may be combined with kidney transplant.101
Disorders of the Liver
Acute Liver Failure
Acute liver failure is a rare clinical syndrome resulting in severe impairment or necrosis of liver cells without preexisting liver disease or cirrhosis. Acetaminophen overdose is a leading cause of acute liver failure in the United States.102 N-acetyl cysteine is an available treatment for detoxification and should be given as soon as possible, preferably within 16 hours after the acetaminophen was taken. Acute liver failure also can occur with concurrent liver disease (acute on chronic liver failure), including complication of viral hepatitis, particularly hepatitis B virus (HBV) infection; compounded by infection with the delta virus; as well as metabolic liver disorders (Wilson disease and α1-antitrypsin deficiency; see Chapter 42). Edematous hepatocytes and patchy areas of necrosis and inflammatory cell infiltrates disrupt liver tissue. The death of hepatocytes may also be caused by viral or toxic injury or immunologic and inflammatory damage.
Acute liver failure usually develops 6 to 8 weeks after the initial symptoms of viral hepatitis or a metabolic liver disorder, or within 5 days to 8 weeks of acetaminophen overdose. Anorexia, vomiting, abdominal pain, and progressive jaundice are initial signs, followed by ascites, GI bleeding and hepatic encephalopathy as described in previous sections. Liver function tests reflect liver injury and show elevations in the levels of both direct and indirect serum bilirubin, serum transaminases, and blood ammonia. The prothrombin time is prolonged. Renal failure and pulmonary dysfunction can occur. Treatment of acute liver failure requires rapid evaluation and critical care. The hepatic necrosis is irreversible, and there can be significant mortality. Liver transplantation may be lifesaving. Artificial liver support systems can provide a bridge to transplantation or to allow the liver to recover.103
Autoimmune Hepatitis
Autoimmune hepatitis is a rare chronic, progressive, autoimmune inflammatory liver disease that affects genetically susceptible individuals, usually female adults, and children. The cause is unknown, but certain infections and drugs are thought to trigger the autoimmune response. Autoreactive T cells trigger secretion of proinflammatory cytotoxic cytokines. Serologically, there are two types: type 1 with positivity for antinuclear and/or anti-smooth muscle antibody, and type 2 with anti-liver kidney microsomal type 1 antibody or anti-liver cytosol type 1 antibody. There is hypergammaglobulinemia and an elevation in aspartate and alanine aminotransferase. Biopsy confirms the diagnosis and shows lymphocytic infiltration with interface (parenchymal–connective tissue interface) hepatitis. The individual may be asymptomatic or present with jaundice, fatigue, loss of appetite, amenorrhea, or acute liver failure. Most individuals respond to immunosuppressive drug therapy (e.g., corticosteroids or in combination with azathioprine) with remission within 24 months. Relapses are common with treatment withdrawal. Approximately 10% of cases require liver transplant.104
Cirrhosis
Cirrhosis is an irreversible inflammatory, fibrotic liver disease. The prevalence of liver disease in the United States is approximately 4.5 million, with a mortality rate of 44,358 deaths yearly.105 Hepatitis C, alcohol-related liver disease, nonalcoholic fatty liver disease (NAFLD), and hepatitis B are the most common causes of cirrhosis106; however, many disorders can cause cirrhosis (Box 41.3). Cirrhosis involves the replacement of normal healthy liver tissue with scar tissue.106 The process of cellular injury depends on the cause of cirrhosis; however, not all pathologic mechanisms are clearly understood. Structural changes result from injury (e.g., viruses or toxicity from alcohol) and fibrosis, which is a consequence of infiltration of leukocytes, and release of inflammatory mediators with activation of stellate cells which transdifferentiate into fibrogenic myofibroblasts and promote fibrotic processes. Chaotic fibrosis alters or obstructs biliary channels and blood flow, producing jaundice and portal hypertension. New vascular channels form shunts, and blood from the portal vein bypasses the liver, contributing to portal hypertension, metabolic alterations, and toxin accumulation. The process of regeneration is disrupted by hypoxia, necrosis, atrophy, and, ultimately, liver failure. The formation of fibrous bands and regenerating nodules distorts the architecture of the liver parenchyma and gives the liver a cobbly appearance (Fig. 41.18). The liver may be larger or smaller than normal and is usually firm or hard when palpated.107