chapter 14 Urologic Aspects of AIDS and HIV Infection
The acquired immunodeficiency syndrome (AIDS) is the most severe manifestation of infection with human immunodeficiency virus (HIV). AIDS is defined by development of serious opportunistic infections, neoplasms, or other life-threatening conditions resulting from progressive immunosuppression caused by HIV infection.
After AIDS was described in 1981 the number of cases increased rapidly (Steinbrook, 2004; World Health Organization, 2004). An estimated 30.6 million to 36.1 million people worldwide are living with HIV infection. More than 20 million have died of AIDS (Joint United Nations Program on HIV/AIDS, 2004; UNAIDS/WHO, 2008). In 2007 alone, 2.5 million people were infected and 2.1 million died of AIDS. Of all people 15 to 49 years of age worldwide, 1.1% are now infected with HIV (Steinbrook, 2004).
HIV/AIDS Epidemiology
In less than 15 years HIV infection reached pandemic proportions, with AIDS reported in over 190 countries (UNAIDS/WHO, 2008). The impact of HIV infection is much different in the developing world than in the industrialized world. At one time AIDS was the leading cause of death among 25- to 44-year old men in Western European and North American cities and was the third most common cause of death among young women (Carael et al, 2004; Steinbrook, 2004). With effective antiretroviral therapy, deaths attributed to AIDS are declining rapidly. Worldwide, AIDS-related deaths declined from 2.9 million to 2.1 million from 2004 to 2007 (UNAIDS/WHO, 2008).
In contrast, in many African cities AIDS represents the leading cause of death and of years of potential lost in men and the second most important cause of death in women. Life expectancy in the most affected sub-Saharan countries was reduced as much as 15 years by the year 2000, compared with projections of life expectancy in those not infected with HIV.
Worldwide Perspective
The three main modes of HIV transmission have changed little: unprotected intercourse, contact with blood, and transmission from mother to child. Direct blood contact, such as sharing drug-injection equipment, results in the most efficient transmission. Globally, “unprotected” sexual intercourse between men and women is the predominant mode of HIV transmission (World Health Organization, 2004).
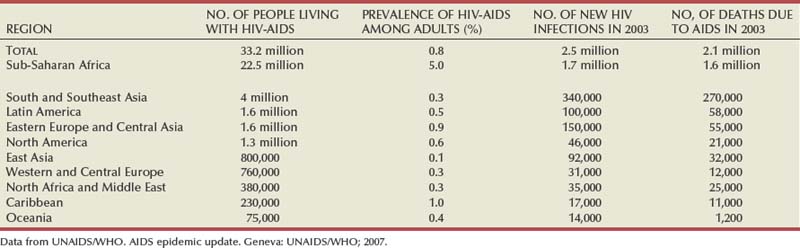
The burden of HIV infection is greatest in the developing world (Steinbrook, 2004). Two thirds of HIV-infected persons are in Africa, where the epidemic exploded during the 1990s, and one fifth are in Asia, where the epidemic has been growing steadily (Steinbrook, 2004). Eight of nine countries with the most HIV-infected people are in sub-Saharan Africa. Estimates for India range from 2.2 to 7.6 million, and for China they are from 430,000 to 1.5 million. In comparison, an estimated 1.2 million people are living with HIV in the United States, 860,000 in the Russian Federation, and 680,000 in Brazil. Statistics highlight global disparities in availability of therapy. Overall, 1.6 million people died of AIDS in sub-Saharan Africa in 2007. By comparison, in Western and Central Europe, only 12,000 people died of AIDS in 2007 (Table 14–1) (Joint United Nations Program on HIV/AIDS, 2004; UNAIDS/WHO, 2007; Quinn, 2008).
Developed World Perspective
The prevalence of HIV has continued to rise in the United States with estimates as high as 1.2 million Americans living with HIV, nearly 75% of whom are adult men (Carael et al, 2004; Centers for Disease Control and Prevention, 2004; Steinbrook, 2004; Heyns et al, 2009). Early in the epidemic most infections occurred among men who have sex with men, but the incidence in this group leveled off by 1985-1987. However, HIV prevalence levels of 7% to 9% are still found among young homosexual and bisexual men in cities such as San Francisco and New York. The largest decline in the proportion of AIDS cases in the United States has occurred among homosexual and bisexual men, whereas cases acquired by heterosexual transmission have increased. Despite antiretroviral therapy, blood screening, and treatment of sexually transmitted infections the number of infections has remained at a plateau of 40,000 new HIV infections per year in the United States over the past decade (Mayer and Safren, 2004).
The HIV prevalence among injecting drug users has been increasing steadily, but with large regional differences (Carael et al, 2004). Since the late 1980s on the U.S. west coast about 90% of people with AIDS are men who have sex with men, whereas on the northeastern coast most new HIV infections occurred among injecting drug users. Young adults belonging to ethnic minorities (including men who have sex with men) are at considerably greater risk of infection than they were 5 years ago. For example, African-Americans make up only 12% of the U.S. population but were affected in 47% of AIDS cases reported in 2000.
Dynamics of the HIV Epidemic: Importance of Urologic Risk Factors
HIV epidemics may occur suddenly, reflecting circumstances that are not fully understood (Carael et al, 2004). For example, HIV seroprevalence among injecting drug users in Bangkok increased from zero in 1985-1986 to 16% in 1988 and 40% to 60% in 1992.
A number of the major risk factors are urologic (Table 14–2) (Carael et al, 2004). Early epidemiologic studies identified major risk factors, especially unprotected sexual intercourse with multiple partners or an infected partner, presence of sexually transmitted infections (STIs), or a history of STIs (Van de Perre et al, 1985; Kreiss et al, 1986; Cameron et al, 1989; Laga et al, 1994). More recent studies have highlighted sex differences in HIV transmission. For many monogamous women the main risk factor may be the sexual behavior of their steady partner (UNAIDS/WHO, 2008).
Table 14–2 HIV Infection Risk Associated with Sexual Behaviors Compared with Blood Exposure
| ROUTE/TYPE OF EXPOSURE | RISK OF INFECTION MEAN/RANGE (%) |
|---|---|
| Transfusion of contaminated blood | 84-100 |
| Intravenous drug use (needle sharing) | 0.8 |
| Receptive anal intercourse | 0.3-0.8 |
| Insertive anal intercourse | 0.04-0.1 |
| Occupational needlestick exposure | 0.28-0.33 |
| Insertive vaginal intercourse | 0.03-0.09 |
| Receptive vaginal intercourse | 0.005-0.02 |
| Insertive oral intercourse | 0.003-0.008 |
| Receptive oral intercourse | 0.006-0.02 |
Data from Henderson et al, 1986; Kaplan and Heimer, 1994; Royce et al, 1997; Varghese et al, 2002; and Henderson, 2004.
Variable Rates of Sexual Transmission
Multiple cofactors affect HIV transmission, which is why estimates vary on the relative risk for specific exposures (see Table 14–2) (Royce et al, 1997; Vernazza and Eron, 1997). The probability of HIV transmission associated with unprotected vaginal sexual intercourse is not constant from one contact to another: estimates range from 0.0005 to 0.002 per episode, in the absence of cofactors (Carael et al, 2004).
A recent meta-analysis of observational studies provided risk estimates for per sexual act transmission of HIV-1 among heterosexuals (Boily et al, 2009). The study included 43 publications, based on work in 25 different study populations. The pooled risk estimates suggest that female-to-male (0.04% per act [95% confidence interval [CI] 0.01-0.14]) and male-to-female (0.08% per act [95% CI 0.06-0.11]) HIV transmission rates are low in high-income countries. In contrast, much higher HIV transmission rates occur in low-income countries with female-to-male (0.38% per act [95% CI 0.13-1.10]) and male-to-female (0.30% per act [95% CI 0.14-0.63]) HIV transmission rates, even in the absence of commercial sex exposure. In multivariate analyses, transmission rates were influenced by gender, setting (high- vs. low-income countries), and antenatal HIV prevalence. As expected, the pooled risk estimate was higher for receptive anal intercourse (1.7% per act [95% CI 0.3-8.9]) and for the presence or history of genital ulcers in either couple member (5.3% per act [95% CI 1.4-19.5]).
Sexually Transmitted Infections
The presence of STIs suggests a marked risk of concurrent HIV. First, the modes of transmission of HIV and other STIs are similar. Second, genital ulcers as well as nonulcerative STIs facilitate HIV transmission (Cameron et al, 1989). Ulcerative STIs, including herpes, syphilis, and chancroid, enhance susceptibility to HIV per sexual act. Nonulcerative STIs, including gonorrhea, chlamydial infections, and trichomoniasis, are independent risk factors for HIV, with relative risks of 2.7 to 3.5 (Laga et al, 1994). Because syphilis facilitates acquisition and transmission of HIV, recent outbreaks of syphilis among men who have sex with men in major U.S. cities and reported increases in sexual behavior have raised concerns about potential increases in HIV transmission (Centers for Disease Control and Prevention, 2004).
Several studies report conflicting results for aggressive diagnosis and treatment of STIs to limit HIV infection in high-risk populations. One study from Uganda employed syndromic management of STIs in an area where the epidemic was in its early stages with less than 1% of the adult population being infected at study inception. HIV incidence decreased in communities where the intervention was undertaken compared with control communities (Grosskurth et al, 1995). Different findings were reported in a study from Tanzania that employed periodic mass STI treatment of at-risk adults. This study found no decrease in HIV incidence (Wawer et al, 1999). In the later study the epidemic was more advanced with more than 16% of the adult population being infected at study inception. In addition there was a high rate of concomitant genital herpes that was not treated. Taken together, these findings suggest that efforts to decrease HIV spread through treating STIs should focus on specific treatment for individual patients, with aggressive diagnosis and treatment of STIs that are common in the community. The potential benefit of STI control programs may also prove greatest in areas of lower HIV prevalence. In communities where the epidemic is widespread, the likelihood of encountering a new partner who is HIV infected may be substantial, so the benefit of modifying a cofactor may be more limited (Mayer and Safren, 2004).
Antiretroviral Therapy and Genital Secretions
Only a small proportion of sexual exposures result in HIV transmission (Anderson and May, 1988). Transmission may occur by exposure to cell-free or cell-associated virus, and different factors may affect expression of virus concentrations in different body fluids (i.e., blood, semen, or cervicovaginal secretions) (Krieger et al, 1998; Chakraborty et al, 2001; Mayer and Safren, 2004). Although lower blood HIV levels are associated with lower transmission rates (Quinn et al, 2000), antiretroviral therapy may not make HIV-infected people noninfectious. In fact, sexual transmission of multidrug-resistant HIV has been well documented (Boden et al, 1999; Little et al, 1999). These observations underscore the need for HIV-infected patients to practice safer sex even if they are on therapy.
Circumcision Status
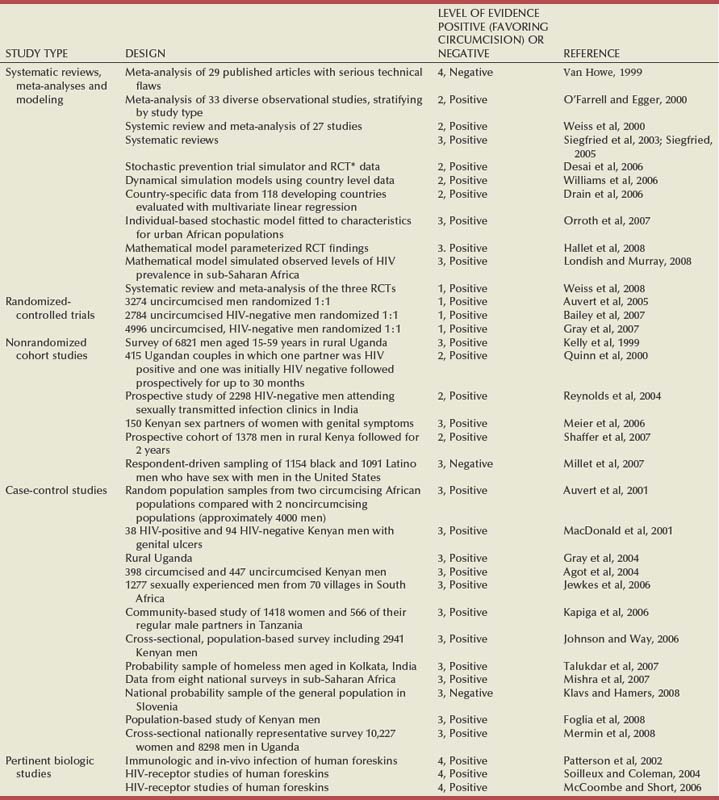
Three lines of evidence support the conclusion that circumcised men are at lower risk for HIV infection: biologic evidence, data from observational studies supported by meta-analyses, and the results of three randomized clinical trials supported by meta-analyses (Table 14–3) (Weiss et al, 2000).
Table 14–3 Evidence Table for Studies of Male Circumcision and HIV Infection Risk That Include Original Data, Systematic Reviews or Meta-Analysis, Expert Opinion, or Other Human Data (1999-2008)
The biologic mechanisms that could explain the increased risk of HIV among uncircumcised men include an increased rate of inflammatory conditions, susceptibility of the mucosal surface of the prepuce to trauma, and the longer survival of pathogens in the warm, moist subpreputial space. The inner foreskin is particularly susceptible to HIV infection owing to lack of keratinization and the high density of HIV target cells that are readily accessible (Patterson et al, 2002; Soilleux and Coleman, 2004; McCoombe and Short, 2006).
The hypothesis that male circumcision might protect against HIV infection was first suggested in 1986 (Fink, 1986). Subsequent support was provided by ecologic studies in areas with low prevalence of male circumcision and high HIV prevalence in sub-Saharan Africa in the late 1980s (Moses et al, 1999) and later across 118 developing countries (Drain et al, 2006). Further evidence comes from two systematic reviews of the observational study data comparing HIV infection rates in circumcised and uncircumcised men from the same populations (Weiss et al, 2000; Siegfried, 2005). One review was restricted to 27 studies from sub-Saharan Africa (Weiss et al, 2000), and the other was a global review including 37 studies (Siegfried, 2005). Circumcised men had substantially lower risk of HIV infection. Meta-analysis of the 15 studies that adjusted for potential confounders showed this reduction to be large and highly significant (adjusted risk ratio of 0.42, 95% CI 0.34-0.54) (Weiss et al, 2000). Subsequent studies have found similar large risk reductions among circumcised men (Weiss et al, 2008).
Although provocative, observational studies cannot prove causality. Three randomized clinical trials of adult male circumcision among consenting, healthy men in South Africa (Auvert et al, 2005), Kenya (Bailey et al, 2007), and Uganda (Gray et al, 2007) were started in 2002-2003. All three trials were stopped early after recommendations by independent data and safety monitoring boards when interim analyses found highly significant decreases in HIV infection risk among participants randomly assigned to circumcision.
The three trials enrolled a total of 10,908 uncircumcised, HIV-negative adult men. Participants were randomly assigned to circumcision or control arms and then observed for up to 2 years. Retention rates were high (86% to 92%). Subsequent meta-analysis using a random-effects model of the trial results, following the QUORUM statement recommendations, found no evidence of heterogeneity among the trials (Weiss et al, 2008). The overall risk ratio was 0.42 (95% CI 0.31-0.57), corresponding to a protective effect of 58% (95% CI 43%-69%), which was identical to that found in the observational studies (58%, 95% CI 46%-66%).
The true protection provided by male circumcision may be better estimated by an “as-treated” analysis, assigning outcomes according to the actual circumcision status of participants. All participants did not adhere to the study arm to which they were randomly assigned. Meta-analysis of the “as-treated” results of the three trials shows even stronger protection against HIV infection in the circumcision group (summary risk ratio of 0.35; 95% CI 0.24-0.54) (Weiss et al, 2008). Additional data have suggested that circumcision is both highly effective at reducing costs of HIV and AIDS and has a very low complication rate (Krieger et al, 2005, 2007; Gray et al, 2007). In summary, the positive findings in the male circumcision trials are in contrast to recent negative results with other HIV prevention interventions, including microbicides, the female diaphragm and gel, treatment to suppress genital herpes infections, and, most recently, an adenovirus-based HIV vaccine. There is a need to provide safe male circumcision services for high-risk populations because this is one of few proven HIV prevention strategies. In addition to other health benefits, male circumcision provides a much-needed addition to the limited HIV prevention armamentarium. Male circumcision is possibly the oldest and the most common surgical procedure. The evidence from biologic studies, observational studies, randomized controlled clinical trials, meta-analyses, and cost-effectiveness studies is conclusive; the challenges of implementation in high-risk populations must now be faced.
Gynecologic Factors
Female genital tract tissues vary in susceptibility to HIV infection. The stratified vaginal epithelium contains fewer cells with coreceptors that can bind HIV (Patterson et al, 1998). Thus, vaginal mucosa is less susceptible to HIV infection than the endocervix, which has a thinner, highly vascular layer, containing a much higher concentration of HIV-binding cells. Physiologic events that result in ectropion (i.e., increased exposure of the endocervix), such as the use of hormonal contraceptives or occult Chlamydia trachomatis infection, increase susceptibility to HIV (Mostad et al, 2000; Moscicki et al, 2001).
Specific Sexual Behaviors
Calculation of the precise risk for infection for each type of exposure is imprecise because many cofactors alter the amount of HIV in the genital tract (Mayer and Safren, 2004). Factors such as a source with a high plasma viral load (Quinn et al, 2000) or concomitant STI can greatly increase the average per-contact risk (Cohen et al, 1997; Wawer et al, 1999). Data used to generate per-contact risks are often based on cohort studies in which individuals recollect their risk during previous time intervals (often every 3 to 6 months). Recollections are often obtained about behaviors under the influence of drugs or alcohol.
Data from the developed world suggest that men are more likely to transmit HIV to their female partners (see Table 14–2) (Mayer and Safren, 2004). However, studies from Africa suggest that rates of male-to-female and female-to male transmission were similar (Quinn et al, 2000). Reasons for this apparent difference in the efficiency of female-to-male transmission may include different rates of male circumcision or the prevalence of other STIs. For anal intercourse the insertive partner is less likely to acquire HIV from an infected receptive partner than vice versa but there appear to be sufficient susceptible cells in the distal male urethra and foreskin such that an insertive partner is still at substantial risk. One study suggested that, on average, receptive anal intercourse was more than seven times as efficient at transmitting HIV as insertive anal intercourse (DeGruttola et al, 1989).
Oral intercourse, either fellatio or cunnilingus, is a much less efficient way to acquire HIV. However, case reports show that oral exposure to ejaculate may transmit HIV (Mayer and Safren, 2004). The efficiency of oral transmission is less than that of unprotected vaginal intercourse, in the realm of less than 1 per 1000 contacts. However, animal studies demonstrate that HIV can be transmitted orally via lymphoid tissue in the oropharynx. Although HIV has been identified in pre-ejaculatory secretions there are no reliable case reports of HIV transmission through exposure to pre-ejaculate without exposure to semen.
HIV Virology and Targets for Antiviral Therapy
The Virion
Although there are two HIV viruses, in this chapter the focus is on HIV-1 because there have been very few cases of HIV-2 in the developed world. HIV-2 is transmitted less readily than HIV-1 and the virus is less virulent. In this review, the term HIV refers to the clinical manifestations of infection caused by HIV-1.
HIV has a spherical shape with an outer envelope, variable surface projections, and an icosahedral capsid containing ribonucleoprotein complexed with a core shell (Fig. 14–1) (Liang and Wainberg, 2004). The HIV projections consist of envelope glycoproteins loosely associated with a lipid bilayer. Beneath the lipid layer the matrix protein covers the internal surface of the viral coat. The capsid protein constitutes the shell of the cone-shaped core, and the nucleocapsid protein forms part of a nucleoid structure that also consists of reverse transcriptase, integrase, and two copies of the single-stranded viral genomic RNA (Liang and Wainberg, 2004).
HIV Replication Cycle
The HIV replication cycle is diagrammed in Figure 14–2 (Liang and Wainberg, 2004).
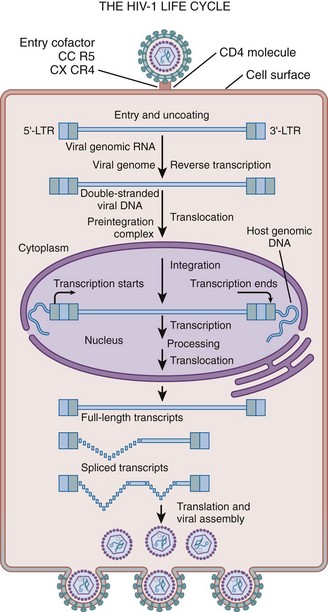
Figure 14–2 HIV replication cycle. The diagram illustrates the stages of HIV replication, including viral entry, uncoating, reverse transcription, integration, expression of proviral genome, viral assembly, and particle release.
(From Liang C, Wainberg MA. Virology of HIV. In: Cohen J, Powderly WG, Berkley SF, et al, editors. Infectious diseases. vol. 2, 2nd ed. Edinburgh: Mosby; 2004. p. 1251–5.)
Viral Attachment, Fusion, and Uncoating
HIV initially attaches to the CD4 receptor on susceptible host cells. High-affinity interactions occur between the viral envelope glycoprotein and a specific region of the CD4 molecule (Pollard and Malim, 1998) on the surface of immature T cells and mature CD4+ T-helper cells. In addition to CD4, a variety of coreceptors and host immune cell membrane proteins may promote HIV entry into target cells after the initial binding (Cocchi et al, 1996; Liang and Wainberg, 2004).
Efforts are underway to develop compounds that antagonize HIV entry into susceptible cells by interfering with either the CD4 receptor or the coreceptors. Cell entry probably occurs by fusion of viral and cell membranes mediated by the viral transmembrane protein. Following fusion the virion is uncoated by a proteolytic event mediated by the virion-encoded protease. A number of protease inhibitors that inhibit this step have proven very effective antiretroviral agents.
Reverse Transcription
After entry, viral RNA is converted into DNA, which is then integrated into host cell chromosomal DNA. This process of reverse transcription occurs within 4 to 6 hours of infection and takes place mainly in the cytoplasm catalyzed by virion-encoded reverse transcriptase. The final products of reverse transcription are double-stranded viral DNA molecules. This distinctive viral reverse transcription step has been the target of a number of effective antiretroviral agents.
Integration
The newly transcribed double-stranded DNAs are then transported into the nucleus where viral DNA is integrated into host cell chromosomal DNA, a process catalyzed by viral integrase. After integration, HIV proviral DNA remains permanently associated with the host genetic material for as long as the cell lives. The viral integrase is currently under active investigation as a potential target for antiretroviral therapy.
Viral Gene Expression, Packaging, and Assembly
HIV exploits the host cell transcription and translation machineries to generate its own gene products. The primary RNA transcripts of the provirus are made by host cell RNA polymerase II. Cellular activation and proliferation signals result in the binding of transcription factors to the long terminal repeat and lead to increased rates of initiation of transcription. Tat and Rev are two key virion-encoded proteins that upregulate viral gene expression and replication, whereas HIV accessory proteins, including Nef, Vif, Vpu, and Vpr, are crucial determinants of HIV virulence (Emerman and Malim, 1998). Host cellular ribosomes translate proviral mRNA into viral proteins (Liang and Wainberg, 2004). The HIV glycosaminoglycan proteins are the driving force for virus assembly.
Therapeutic Considerations
Viral reverse transcriptase and protease enzymes have been the main targets of anti-HIV therapy. Viral reverse transcriptase has an exceedingly high error rate (i.e., about one mutation/virus replication event) (Liang and Wainberg, 2004); thus mutants are constantly generated that have the potential for drug resistance. Drug resistance to HIV occurs when these mutations result in altered forms of HIV reverse transcriptase and protease that function yet are not inhibited by antiviral agents.
Pathogenesis of HIV Infection
The prognosis and outcome of HIV infection changed dramatically with availability of highly active antiretroviral combination therapy (HAART). HAART has delayed the natural history and rate of progression, prolonging survival. However, initial optimism for immune reconstitution has been thwarted by information about HIV’s pathogenic mechanisms and the limitations of HAART. It is now clear that eradication of the virus is not possible with available antiviral drugs.
Natural History of HIV Infection
The course of HIV infection is highly variable. In the absence of treatment the typical course occurs over 8 to 12 years. Three distinct phases have been defined: primary infection, chronic asymptomatic infection, and overt AIDS (Pierre-Alexandre and Pantaleo, 2004).
Primary HIV Infection
Primary HIV infection is a transient symptomatic illness of variable severity. This acute syndrome occurs in 40% to 90% of patients. Clinical signs and symptoms generally appear 2 to 4 weeks after infection. The clinical presentation may mimic acute mononucleosis or other acute febrile illnesses, emphasizing the nonspecific nature of these symptoms and the difficulty of early diagnosis. The self-limited syndrome generally lasts less than 14 days.
Diagnosis of primary HIV infection depends on diagnostic testing. There are three laboratory characteristics: high plasma viremia often greater than 1 million HIV RNA copies/mL, a decrease in the blood CD4+ T-cell count, and large increase in the blood CD8+ T-cell count. Subsequently, a marked decline in plasma viremia coincides with resolution of the clinical syndrome (Kahn and Walker, 1998). This decrease in viral load also correlates with the appearance of the virus-specific immune responses, particularly HIV-specific cytotoxic T cells, indicating that immune responses downregulate virus replication (Pantaleo et al, 1994; Musey et al, 1997). Standard laboratory assays used to diagnose HIV infection are usually negative during acute infection.
Chronic Asymptomatic HIV Infection
Primary HIV infection is followed by a long phase of clinical latency, usually lasting around 10 years. During this phase patients have neither signs nor symptoms. Relatively stable HIV replication levels and CD4+ T-cell counts characterize this chronic phase.
This apparent clinical “stability” is deceptive. Although viral levels and the blood CD4 T-cell count are stable, this stability is only apparent in the blood. Virus replication and accumulation of extracellular virus trapped in the follicular dendritic cell network continue actively in the lymphoid tissue, where progressive anatomic and functional deterioration occurs, leading to impaired specific immune responses (Pantaleo et al, 1993, 1998). Eventually, immunologic deterioration is reflected by a rapid increase in viremia levels and a drop in CD4+ T-cell counts, resulting in transition to overt AIDS.
Overt AIDS
AIDS represents the end stage of HIV infection. This advanced stage of HIV disease is marked by low CD4+ T-cell counts (<2 to 300 cells/µL) and appearance of constitutional symptoms. This phase may be complicated by the development of AIDS-defining opportunistic infections or malignancies. Without antiretroviral therapy, AIDS usually leads to death in 2 to 3 years. The risk for death and opportunistic infections increases significantly with CD4+ T-cell counts less than 50 cells/µL.
Highly Variable Clinical Course
The clinical course of HIV infection is variable (Pierre-Alexandre and Pantaleo, 2004). Conceptually, patients may be classified in four categories: typical progressors, rapid progressors, slow progressors, or nonprogressors. In 60% to 70% of HIV-infected patients the median time between infection and the development of AIDS is 10 to 11 years, in the absence of therapy. These infected persons are “typical progressors.” Their clinical course is described earlier.
Ten to 20 percent of HIV infections progress rapidly. These “rapid progressors” develop AIDS less than 5 years after infection. After primary infection these patients’ plasma virus levels are often more than 105 HIV-1 RNA copies/mL and their CD4+ T-cell counts start to decrease much earlier and more rapidly. HIV-specific immune responses are either never detected or are lost rapidly after the transition from the acute to the chronic phase of infection.
At the other extreme, 5% to 15% of HIV-infected people are “slow progressors,” remaining free of AIDS for more than 15 years in the absence of therapy. Their CD4+ T-cell counts remain stable, frequently greater than 500 cells/µL, and plasma HIV RNA levels are usually less than 10,000 copies/mL.
The slow progressors group includes a subgroup of long-term nonprogressors. About 1% of HIV-infected people appear to fall into this category. Long-term nonprogressors have documented HIV infections for at least 8 to 10 years without antiretroviral therapy and have no signs of disease progression (e.g., constant high CD4+ T-cell counts and either low [500 to 1000 HIV RNA copies/mL] or very low [<50 copies/mL] plasma virus levels).
Early Pathogenic Events Determine the Natural History
The most common route of HIV infection is sexual transmission at the genital mucosa (Royce et al, 1997). Animal models provide important insights into the early pathogenic events. After intravaginal infection of rhesus monkeys, tissue dendritic cells (i.e., Langerhans cells that reside in the lamina propria adjacent to the vaginal epithelium) are the first potential target cells of HIV (Spira et al, 1996). These dendritic cells constitute a complex and highly developed system of antigen-presenting cells that are able to prime naive T cells. The ability of dendritic cells to attract and prime naive T cells can be explained by expression of a type II membrane protein with an external mannose binding, C-type lectin domain, named DC-Sign (Steinman, 2000). It has been suggested that interaction between DC-Sign and the intracellular adhesion molecule-3 represents the initial contact between dendritic cells and resting T cells, a critical step for initiation of T-cell immune responses. Dendritic cells express high levels of specific chemokines that target naive rather than memory T cells (Cameron et al, 1996).
After encountering HIV below the vaginal epithelium, Langerhans cells either can be infected or can pick up HIV virions (Steinman, 2000). Thus DC-Sign–positive dendritic cells play the major role in the delivery of HIV to T cells, greatly amplifying the viral infection (Zhu et al, 1996). Subsequently dendritic cells migrate to the internal iliac lymph nodes where they target the T-cell areas and then present the viral antigens to activated virus-specific T cells. Therefore, dendritic cells play a key role, both in priming the initial HIV-specific immune response and in transporting HIV to the draining lymphoid tissue. Rapid recruitment and spreading of target cells (i.e., activated CD4+ T cells) confers a major advantage to HIV because these events occur before the appearance of virus-specific immune responses.
Within 2 days after infection, HIV can be detected in the draining lymphoid tissue. The virus then disseminates rapidly throughout the lymphoid system. Afterward, HIV enters the bloodstream, where viral replication can be detected in plasma 5 days after infection in the animal model. In humans the time from mucosal infection and initial plasma viremia varies, ranging from 4 to 11 days (Pierre-Alexandre and Pantaleo, 2004). The clinical importance of these observations is that the risk of infection is increased by conditions that decrease genital mucosal barriers, especially lesions caused by inflammatory or infectious diseases, such as cervicitis, urethritis, and genital ulcers.
Critical Role of Lymphoid Tissue in Primary HIV Infection
Events in the lymphoid tissue play a central role in HIV infection. During primary infection the peak number of virus-expressing cells in the lymph node occurs at the same time as the peak of plasma viremia or shortly precedes it (Brodie et al, 1999; Musey et al, 1999; Pierre-Alexandre and Pantaleo, 2004). With the transition from primary to chronic infection there is a switch from individual virus-expressing cells to virus trapped by the follicular dendritic cell network of lymph node germinal centers. This trapped virus becomes the dominant form of virus in lymph nodes, and this event is associated with a dramatic decrease in the number of individual cells expressing viral RNA, reflecting the role of the host immune system in partially containing HIV infection (Pantaleo et al, 1998).
Virus Escape and Establishment of Chronic HIV Infection
Early in primary HIV infection, vigorous virus-specific immune responses may contribute to both control of the initial peak of virus replication and reduction in plasma viremia (Musey et al, 1997; Kahn and Walker, 1998; Pantaleo et al, 1993, 1998). However, HIV-specific immune responses cannot control HIV or block the eventual progression to symptomatic disease. HIV differs from other viruses by targeting a broad spectrum of effector components of the antiviral immune response very early after infection (during the primary phase) and by rendering these mechanisms ineffective or reshaping them (Soudeyns and Pantaleo, 1999).
HIV escapes the host immune response by both virologic and immunologic mechanisms. Virologic mechanisms include formation of a stable pool of latently infected CD4+ T cells containing HIV that is capable of replicating (Finzi et al, 1997; Wong et al, 1997), the genetic variability of HIV (Coffin, 1995), and trapping of infectious virions on the surface of follicular dendritic cells (Pantaleo et al, 1994; Chun et al, 1997; Wong et al, 1997). Initiation of HAART very early during primary HIV infection does have a significant impact on the size of this pool of CD4+ T cells. Decay of this pool of infected CD4+ T cells is very slow and not much influenced by the effective suppression of virus replication. This reservoir of replication-competent virus represents a major obstacle to HIV eradication and long-term control of virus replication (Chun et al, 1997; Finzi et al, 1997; Wong et al, 1997).
Genetic variability is another efficient mechanism by which HIV escapes the host immune response. During both primary and established chronic infection, rapid mutations ensure that the virus is able to escape both the humoral and the cell-mediated virus-specific immune responses.
HIV also reshapes antiviral mechanisms by trapping of the virus on the surface of follicular dendritic cells in lymphoid germinal centers. For most infections, formation of immune complexes and their attachment to the follicular dendritic cell network represent mechanisms leading to clearance of the pathogen and to maintenance of effective immune responses. However, with HIV infection these mechanisms lead to formation of a stable reservoir of infectious virions, representing a continuous source for infection of CD4+ T cells that ultimately results in the destruction of the lymphoid tissue (Musey et al, 1997).
HIV escapes from the immune response through multiple immunologic mechanisms that include deletion of HIV-specific CD4+ T-cell clones (Rosenberg et al, 1997), deletion of HIV-specific cytotoxic CD8+ T-cell clones, generation of virus escape mutants (Soudeyns and Pantaleo, 1999), egress of cytotoxic T cells from lymph nodes, impaired function of antigen-presenting cells (Soudeyns and Pantaleo, 1999), and interference with the humoral neutralizing response.
In summary, virus escape and establishment of chronic HIV infection reflect the ability of HIV to target the host’s antiviral immune response and reshape it into a self-defense mechanism. Primary infection is critical in the pathogenesis of HIV infection because events that occur during this stage determine both the pattern and the rate of disease progression.
Virologic Set Point
During the transition from primary to chronic infection, HIV plasma RNA levels reach a virologic set point that predicts the rate of disease progression (Mellors et al, 1996). The virologic set point varies among HIV-infected individuals and tends to remain stable in the same person during the chronic phase. The virologic set point that a person attains is determined by both the mechanisms involved in the establishment of chronic infection and by host factors that can modulate the course of HIV disease. The plasma RNA load is the most accurate predictor of disease progression (Mellors et al, 1996). The higher the plasma viral RNA load, the greater is the risk of rapid progression to AIDS and death.
Prospects for HIV Eradication
Free HIV virions are eliminated with a half-life of 6 hours. In contrast, productively infected cells have a half-life of 1.6 days. It was hoped that HIV eradication was achievable within 2 to 3 years (Perelson et al, 1997). This goal could only be realized with complete and sustained suppression of HIV replication and if there were no pool of long-lived, latently infected cells to serve as a virus reservoir (Chun et al, 1997; Finzi et al, 1997; Wong et al, 1997).
Data from HIV-infected patients on long-term HAART significantly challenged these theories. There appears to be a pool of latent HIV–infected resting CD4+ T cells that persists in HIV-infected patients who adhered to HAART for up to 3 years (Chun et al, 1997; Finzi et al, 1997; Wong et al, 1997). This pool is composed of quiescent memory CD4+ T cells with a longer half-life than the original estimate of 1 to 4 weeks. More importantly, these cells contain replication-competent HIV proviral DNA. After activation these cells are able to support efficient viral replication.
The extent of this HIV reservoir is quantitatively limited, estimated between 50,000 and 5 million resting memory CD4+ T cells for the whole body. However, this pool of cells has a half-life of 3 to 6 months, dramatically increasing estimates of the potential time required for HIV eradication.
The reservoir of HIV-infected cells originates from productively infected cells during primary infection (Pierre-Alexandre and Pantaleo, 2004). Initiation of HAART as early as 10 days after the onset of symptoms of primary HIV infection rapidly controls plasma viremia but does not preclude generation of this viral reservoir. These observations emphasize the rapidity with which HIV viral reservoirs are established and the limited ability of HAART to interfere with this pathogenic process. This pool of HIV-infected cells represents a major obstacle to HIV eradication.
In addition to their role as a viral reservoir, HIV-infected, long-lived, resting memory CD4+ T cells possess the ability to support virus production, albeit at low levels. Patients on HAART with viral loads below detectable levels with usual clinical assays may still have viral replication detected by more sensitive tests (Pierre-Alexandre and Pantaleo, 2004). In these studies, 40% to 60% of patients still had low levels of viremia after 36 to 48 weeks of HAART.
Viral sanctuaries represent another potential source of residual HIV replication. Sanctuaries are defined as cells and organs where the HIV can be sheltered or where HAART does not achieve therapeutic concentrations. Tissue sanctuaries include the lymphoid tissue, mucosa-associated lymphoid tissue, the genital organs, and the central nervous system, where the achievable concentrations of antiviral drugs (especially protease inhibitors) may be suboptimal. These sites may serve both as potential sources of low-level virus replication and as reservoirs of latently infected cells (Pierre-Alexandre and Pantaleo, 2004). Presence of viral sanctuaries, including the reproductive tract, with limited bioavailability of antiviral drugs further complicates the issue of HIV eradication. Current estimates are that it may take 5 to 10 years or more to eliminate HIV, considering a half-life of 4 months for the long-lived infected CD4+ cells and provided that effective and durable suppression of viral replication is achieved by HAART.
Tests to Diagnose and Monitor HIV Infection: What the Urologist Should Know
HIV was first isolated in 1983, and the first diagnostic tests were marketed in 1985. Tests for diagnosis and monitoring of HIV infection have improved constantly (Kuritzkes, 2004; Reiss et al, 2004). Few areas in medicine have witnessed such rapid and widespread adaptation of molecular tools to everyday practice. Fortunately the many tests for diagnosis and monitoring HIV infection can be considered in three categories: diagnostic tests, viral load, and resistance assays.
Diagnostic Tests and Testing Algorithms
Assays have been developed to detect HIV antibodies in serum, whole blood, saliva, and urine (Kuritzkes, 2004). Most laboratories screen for anti-HIV-1 and anti-HIV-2 antibodies using an enzyme-linked immunosorbent assay (ELISA) based on antigens from viral lysates, recombinant or synthetic. Current third-generation HIV ELISAs have sensitivity and specificity approaching 100%. In contrast to earlier tests that only detected IgG antibodies these tests detect all classes of anti-HIV antibodies, substantially shortening the time to diagnosis after acute infection. A variety of rapid tests and “home tests” have also been developed.
Despite sensitivity of more than 99%, a reactive HIV ELISA has a relatively low positive predictive value in low-risk populations. Thus the current testing algorithm includes a confirmatory test to exclude false-positive results. The Western blot is most widely used for confirmation. Some laboratories prefer immunoblotting for confirmation because immunoblots are simpler to standardize and are more sensitive in cases of recent seroconversion (Kuritzkes, 2004).
Clinical Application
After acute infection, HIV RNA can be detected from day 12. HIV RNA assays (see later) have a sensitivity of 100% in diagnosing acute infection but at the cost of lowered specificity (97%) (Kuritzkes, 2004). The first antibodies can generally be detected on day 21. However, development of a positive HIV antibody test can vary according to the patient and infecting strain. Beyond week 6 after infection antibodies are detectable in almost all patients (Kuritzkes, 2004). Because reactivity by the Western blot test lags seroconversion by ELISA, a positive ELISA in a patient with a negative or evolving Western blot assay can provide evidence of recent infection.
Testing is a two-stage process. Samples positive by an initial screening assay are retested to exclude clerical or laboratory error, and then a confirmatory assay is performed on repeatedly reactive sera to verify that the antibodies are directed against HIV. Confirmation of a reactive ELISA by a positive Western blot establishes the diagnosis of HIV infection. A negative Western blot test suggests a false-positive ELISA or an acute infection.
Viral Load Monitoring
Assays to quantify HIV RNA, termed the viral load, led to our current understanding of HIV pathogenesis and helped establish complete suppression of HIV replication as the ultimate goal for antiretroviral therapy (Kuritzkes, 2004). These assays are now standard. Performance characteristics of the commercially available assays are similar. Current commercial assays have a lower limit of quantitation of 50 to 80 HIV RNA copies/mL.
Clinical Application
Plasma HIV RNA levels correlate with clinical stage (Kuritzkes, 2004). Patients with symptomatic HIV infection or AIDS have significantly higher virus loads than patients with asymptomatic infection. Plasma HIV RNA load is also a powerful predictor of the risk of disease progression and death at all stages of disease. Current guidelines recommend obtaining two plasma HIV RNA measurements to determine the baseline or “set point” virus load before initiating antiretroviral therapy.
After antiretroviral therapy the change in plasma HIV RNA provides important clinical information. Several large clinical trials showed significant correlations between the plasma HIV RNA reduction from baseline and the clinical benefit (Marschner et al, 1998). The nadir in plasma HIV RNA levels is a marker for the duration of virus suppression (Raboud et al, 1999). The early response to treatment predicts long-term outcome. Conversely, increasing plasma HIV RNA level suggests treatment failure.
Increasing plasma HIV RNA level is the harbinger of potential development of retroviral drug resistance (Reiss et al, 2004). This is not necessarily accompanied by an immediate decline in blood CD4+ lymphocyte numbers or by the development of clinical symptoms. Regular monitoring with plasma HIV RNA levels facilitates timely detection of drug-resistant HIV in patients on HAART.
HIV Drug-Resistance Assays
Various assays are available to assess drug resistance (Kuritzkes, 2004; Reiss et al, 2004). These assays can be considered as either genotypic or phenotypic assays. Genotypic assays evaluate HIV nucleotide sequences to detect critical drug-resistance mutations. Phenotypic assays estimate the concentration of antiviral drugs necessary to inhibit virus replication in vitro. Each approach has potential advantages and disadvantages. An important limitation is that both approaches measure characteristics of the predominant viral species but do not indicate the presence of minor species that may emerge as resistant variants during subsequent treatment.
Clinical Application
Clinical trials demonstrate the value of drug-resistance testing (Kuritzkes, 2004; Reiss et al, 2004). The risk of virologic failure during salvage therapy was reduced by 30% to 50% for each drug in the new regimen that was predicted to have activity (De Gruttola et al, 2001; Kuritzkes, 2004). Drug-resistance remained an independent predictor of the likelihood of treatment failure even after controlling for treatment history.
Most experts recommend drug-resistance testing to guide the selection of salvage therapy for patients whose therapy is failing and in pregnant women (Hirsch et al, 2000). Resistance testing is also recommended for patients with acute HIV infection. Some experts also recommend that evidence for increasing transmission of drug-resistant HIV (Little et al, 2002) provides a rationale for testing of all patients before initiation of antiretroviral therapy where affordable (Kuritzkes, 2004).
Urologic Manifestations of HIV Infection
Nonmalignant Conditions
The genitalia may be involved in either early or late stages of HIV infection (Kho et al, 2004).
Primary HIV Infection
Primary HIV infection may be characterized by an acute exanthem beginning 1 to 5 weeks after infection. Characteristic symptoms include fatigue, fever, and night sweats, accompanied by lymphadenopathy. The skin eruption consists of erythematous, round-to-oval macules and papules. The syndrome resolves with complete recovery.
Sexually Transmitted Infections
In many populations the pattern of HIV acquisition parallels that of STIs (Quinn et al, 1988). Thus, testing for HIV is recommended in anyone with a diagnosed STI or at risk for STI (Centers for Disease Control and Prevention, 2002). Accurate diagnosis of STIs is of exceptional importance in individuals at high risk for HIV infection because presence of an STI increases the risk of both transmitting and acquiring HIV infection. These infections may have more atypical and prolonged clinical manifestations in people with HIV infection.
Genital Herpesvirus
Herpes simplex virus (HSV) infection is common in HIV-infected persons (Centers for Disease Control and Prevention, 2002). In these populations HSV types 1 and 2 cause recurrent, severe, painful vesicles with an erythematous base. Untreated lesions may enlarge becoming confluent ulcerations that persist with secondary bacterial infection. Sometimes ulceration occurs without well-defined vesicles ever being noted (Kho et al, 2004). Specific diagnosis is established by viral culture, other diagnostic tests, or biopsy.
In patients with intact immune function the course of HSV is similar to that of other HSV-infected patients. Immunocompromised patients may have more prolonged and severe infections (Centers for Disease Control and Prevention, 2002). Episodic or suppressive therapy with oral antiviral agents is often beneficial (Table 14–4) (Centers for Disease Control and Prevention, 2002). Severe cases merit parenteral therapy with intravenously administered acyclovir (5 to 10 mg/kg every 8 hours) (Centers for Disease Control and Prevention, 2002).
Table 14–4 Recommended Regimens for Treating Genital Herpes Virus Infections in HIV-Infected Patients
| Episodic Treatment Regimens | ||
|---|---|---|
| DRUG | DOSAGE | DURATION |
| Acyclovir | 400 mg orally three times a day | 5-10 days |
| Acyclovir | 200 mg orally 5 times a day | 5-10 days |
| Famciclovir | 500 mg orally twice a day | 5-10 days |
| Valacyclovir | 1 g orally twice a day | 5-10 days |
| Drug Suppressive Therapy | |
|---|---|
| DRUG | DOSAGE |
| Acyclovir | 400-800 mg orally twice or three times a day |
| Famciclovir | 500 mg orally twice a day |
| Valacyclovir | 500 mg orally twice a day |
From Centers for Disease Control and Prevention. Sexually transmitted diseases treatment guidelines 2002. MMWR Morb Mortal Wkly Rep 2002;5:1.
HSV resistance should be suspected if lesions persist or recur despite antiviral therapy. Viral culture and resistance testing should be obtained if possible (Centers for Disease Control and Prevention, 2002). Patients with acyclovir-resistant HSV strains require alternative agents, such as foscarnet (40 mg/kg intravenously every 8 hours until clinical resolution) or topical cidofovir gel (1% applied to lesions daily for 5 days), which is a preparation that is not commercially available but must be compounded by a pharmacy.
HSV infection increases HIV replication in persons infected with both viruses (Van de Perre et al, 2008). Studies have also shown that anti-HSV treatment with acyclovir reduced HIV in genital secretions among HIV- and HSV-coinfected persons (Dunne et al, 2008). Thus, it was disappointing that randomized clinical trials found that acyclovir as HSV suppressive therapy did not decrease the incidence of HIV infection in HSV-positive, high-risk HIV-negative women (Watson-Jones et al, 2008).
Human Papillomavirus
Warts can develop in unusual locations, such as on the lips, tongue, and oral mucosa in addition to the genitalia. Such lesions are treated using standard regimens (cryotherapy, podofilox, imiquimod, podophyllin resin, laser, or excisional surgery) (Centers for Disease Control and Prevention, 2002). However, in immunosuppressed patients genital lesions are resistant to treatment and these patients may be at higher risk for recurrence (Centers for Disease Control and Prevention, 2002).
In men, human papillomavirus (HPV) affects the penis, urethra, scrotum, perineal, and rectal mucosa as condylomata acuminata, which are usually recognized as soft sessile lesions with finger-like projections (Fig. 14–3). In women the spectrum of clinical disease is broad with vulvar, vaginal, and cervical condylomata. Other clinical presentations of HPV infection include bowenoid papulosis and epidermodysplasia verruciformis. Bowenoid papulosis presents as small, brown, flat papules affecting the perianal and genital areas. Epidermodysplasia verruciformis consists of a widespread eruption of pink-red, flat, wartlike lesions distributed mostly on sun-exposed skin (Berger et al, 1991).
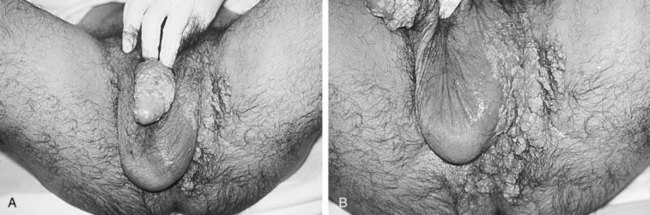
Figure 14–3 A and B, AIDS patient with five active urogenital viral infections. Extensive genital condylomata associated with areas of dysplasia and squamous intraepithelial neoplasia related to coinfection with human papillomavirus. Shedding of HIV, cytomegalovirus, and hepatitis B virus was documented in the patient’s semen. Herpes simplex virus type 2 was cultured from active ulcers in the patient’s inguinal crease.
HPV infection increases the risk for carcinoma, especially in HIV-infected hosts. These lesions include cervical intraepithelial neoplasia in women and squamous cell carcinoma in men. Whenever extensive warts develop the patient should be screened for HIV infection. Because there is an increased risk of anal cancer in HIV-infected homosexual men, screening for anal squamous intraepithelial neoplasia by cytology is recommended by some authorities. Until more data on the natural history of anal squamous intraepithelial neoplasia and the efficacy of treatment are available, routine screening is not recommended (Centers for Disease Control and Prevention, 2002). Differential diagnosis of atypical and/or extensive genital warts includes squamous cell carcinoma in-situ and squamous cell carcinoma. These cases are best diagnosed by biopsy. Treatment by various excisional procedures is often effective, but recurrence occurs frequently.
Syphilis
A high prevalence of syphilis is found in certain HIV-infected populations, particularly men who have sex with men (Quinn et al, 1988; Centers for Disease Control and Prevention, 2002). In HIV-infected patients, primary infection with Treponema pallidum presents as the typical chancre. Secondary syphilis may present in classic papulosquamous form with involvement of palms, soles, and mucous membranes, but unusual presentations are common, such as verrucous plaques, extensive oral ulcerations, keratoderma, deep cutaneous nodules, and widespread gummata (Kho et al, 2004). The disease may progress faster from secondary to tertiary syphilis in HIV-infected patients. Early central nervous system relapse also can be more common.
Unusual serologic responses have been observed among HIV-infected persons with syphilis, usually among persons with higher than expected titers. False-negative tests have also been reported (Centers for Disease Control and Prevention, 2002). Patients with clinical findings suggesting possible syphilis but with nonreactive or atypical serologic tests should have alternative diagnostic tests, such as biopsy of lesions, darkfield examination, or direct fluorescent antibody staining for diagnosis.
HIV-infected persons with early syphilis appear to be at increased risk for neurologic complications and higher risk for treatment failure with standard regimens. For primary and secondary syphilis in HIV-infected patients the current recommendation is intramuscular injection of benzathine penicillin G (2.4 million units). At least one dose is recommended and some authorities recommend three doses at weekly doses, as recommended for late syphilis. Careful follow-up is recommended with repeated serologic studies for at least 24 months after treatment.
Chancroid
In the United States, chancroid usually occurs in discrete outbreaks. Chancroid is a cofactor for HIV transmission. Definitive diagnosis requires culture of Haemophilus ducreyi on special culture. Although polymerase chain reaction–based tests are described, none is approved by the U.S. Food and Drug Administration. Probable diagnosis can be made if the patient has painful genital ulcers, no evidence of Treponema pallidum infection, and a negative test for HSV. The combination of a painful ulcer and tender inguinal adenopathy, symptoms occurring in one third of patients, suggests a diagnosis of chancroid. When accompanied by suppurative inguinal adenopathy these signs are almost pathognomonic.
Recommended regimens for chancroid in HIV-infected patients are either ciprofloxacin, 500 mg orally twice daily for 3 days, or erythromycin base, 500 mg orally three times daily for 7 days (Centers for Disease Control and Prevention, 2002). Alternative drugs are either azithromycin, 1 g orally as a single dose, or ceftriaxone, 250 mg as a single intramuscular dose. HIV-infected patients with chancroid should be monitored closely because, as a group, these patients are more likely to experience treatment failure and to have ulcers that heal more slowly. HIV-infected patients may require longer courses of therapy, and treatment failures can occur with any regimen.
Urethritis
As the first rheumatic disease reported in HIV-infected patients, Reiter syndrome, consisting of uveitis, urethritis, and arthritis often presents in incomplete form (Solomon et al, 1991). The link between Reiter syndrome and AIDS is poorly understood. The syndrome, particularly the urethral discharge, is usually refractory to antibiotic therapy.
Molluscum Contagiosum
Molluscum contagiosum is caused by a sexually transmitted poxvirus. This condition reportedly develops in 10% to 20% of AIDS patients (Kho et al, 2004). Characteristic lesions are umbilicated, dome-shaped, translucent 2- to 4-mm papules. Lesions can develop in any part of the body, especially the face and genital areas. In AIDS patients these lesions are widespread and may attain immense size (Izu et al, 1994). Most HIV-infected patients with extensive molluscum contagiosum have CD4+ counts less than 250 cells/mL. In such immunosuppressed patients the diagnosis of molluscum contagiosum should be confirmed by histologic examination because the clinical appearance may simulate more serious infections such as cutaneous pneumocystosis, histoplasmosis, Penicillium marneffei infection, cryptococcosis, or cutaneous mycobacterial infection. Molluscum contagiosum is treated with cryotherapy, electrodissection, curettage, or topical application of keratolytic preparations (Kho et al, 2004).
HIV-Related Genitourinary Tract Infections
The genitourinary tract may be involved as the site or sequelae of HIV-related opportunistic infections (Randazzo et al, 1986; Stamm et al, 1988; Miles et al, 1989; Shevchuk et al, 1989, 1999; Pudney and Anderson, 1991; Wilson et al, 1992; Buzelin et al, 1994; Leibovitch and Goldwasser, 1994; Swartz et al, 1994; Trauzzi et al, 1994; Cespedes et al, 1995; Timmerman et al, 1995; Appel et al, 2004; Kuritzkes, 2004).
Renal Infections
There has been a rise in drug-resistant tuberculosis in the United States. Persons with or at risk for HIV infection appear to be at increased risk for active infection with drug-resistant strains of Mycobacterium tuberculosis. Persons coinfected with HIV and M. tuberculosis have a greater likelihood of developing clinical tuberculosis, including renal and other extrapulmonary disease, that may be more difficult to diagnose and to treat (Weiss et al, 1998).
Other renal infectious manifestations of AIDS include cytomegalovirus, Aspergillus, and Toxoplasma infections. Cytomegalovirus is a common opportunistic infection in immunocompromised patients. Renal infection is most commonly noted in widespread disease in association with acute tubular necrosis (Kwan and Lowe, 1995). Although it has been suggested that renal cytomegalovirus infection might facilitate the development of HIV-associated nephropathy (HIVAN), an autopsy study of 75 kidneys suggested that the two findings are unrelated (Nadasdy et al, 1992). Aspergillosis and toxoplasmosis are common opportunistic infections of the kidney. Both organisms are treated with systemic therapy. Serious infections such as abscesses require percutaneous drainage, open drainage, or nephrectomy.
Prostatitis
The prostate is the site for varied opportunistic infections in HIV-infected patients. In one study bacterial prostatitis was diagnosed in 17 (8%) of 209 men hospitalized for HIV (Leport et al, 1989). The clinical presentation varies based on the severity of the infection but includes fever, obstructive and irritative voiding symptoms, and a tender prostate on digital rectal examination with possible fluctuance. Superimposed urinary tract infections occurred in 22% of AIDS patients in one study (Kaplan et al, 1987). Diagnosis requires broad cultures including aerobes, anaerobes, fungi, and mycobacteria. Prostatic abscesses require open, transrectal or endoscopic drainage. Bacterial prostatitis requires prolonged courses of antibiotics with high levels of prostatic penetration. Fungal prostatitis is initially treated with two-agent therapy (amphotericin and flucytosine) or one of the newer antifungal drugs, with long-term suppression for persistent or recurrent infections.
Epididymitis and Orchitis
The most common intrascrotal pathology in AIDS is testicular atrophy secondary to endocrine imbalances, febrile episodes, malnutrition, toxic effects of therapeutic agents, and testicular infections (Leibovitch and Goldwasser, 1994). Autopsy studies have demonstrated opportunistic testicular infection in up to 39% of AIDS patients (De Paepe et al, 1990). One study (Shevchuk et al, 1989) of 80 autopsies in AIDS patients demonstrated that 2 of 11 cases with systemic toxoplasmosis involved the testes, 4 of 48 cases of systemic cytomegalovirus infection involved the prostate whereas 1 involved the testes, and 1 of 27 cases of systemic candidiasis involved the prostate. Most patients’ testes exhibited marked spermatogenic arrest, germ cell degeneration, peritubular fibrosis, and Leydig cell depletion, nonspecific findings that likely reflect severe systemic disease (Shevchuk et al, 1999). Other immunocompromised patients develop symptomatic genitourinary tract infections with both common and unusual organisms, for example, epididymitis caused by Candida (Swartz et al, 1994) or cytomegalovirus (Randazzo et al, 1986). Patients may be less likely to respond to medical therapy and to require surgery (Coburn, 1998).
Clinically, scrotal infections usually present as epididymo-orchitis. Relapse is common and may result in persistent symptoms or fulminant infection with abscess formation. Treatment requires initial antibiotic treatment followed by long-term maintenance suppression. The differential diagnosis of epididymitis or orchitis that does not respond to conventional therapy includes fungal, mycobacterial, and other opportunistic infections, in addition to testicular tumors (see later).
Impetigo, Abscesses, Cellulitis, Lymphadenitis, and Necrotizing Fasciitis
Impetigo, usually caused by Staphylococcus aureus, occurs in the inguinal area as in other common sites (the face, shoulders, and axilla). Typically the infection begins with painful red macules, progressing to pustules that rupture producing a characteristic honey-colored crust (Kho et al, 2004). Soft tissue and deep-seated infections, including cellulitis, abscesses, and necrotizing fasciitis may develop in HIV-infected patients.
Necrotizing fasciitis of the genitalia, also known as Fournier gangrene, has a propensity to occur in immunocompromised hosts (Corman et al, 1999). This aggressive, progressive, necrotizing fasciitis has been documented as the presenting finding in previously undiagnosed AIDS patients (McKay and Waters, 1994). Treatment depends on rapid diagnosis, wide surgical debridement, hemodynamic support, and prolonged antibiotic therapy. Diverting colostomy and partial or complete scrotectomy may prove necessary during operative debridement.
Voiding Dysfunction
Because HIV infection affects both the central and peripheral nervous systems, voiding dysfunction is common in patients with advanced infection (Hermieu et al, 1996). Urinary retention is the most common voiding dysfunction (54%) seen by urologists, although detrusor hyperreflexia (27%) and outflow obstruction (18%) have been documented during urodynamic evaluation of AIDS patients with voiding symptoms (Travers and Huybers, 1987; Kane et al, 1996). Standard pharmacologic measures are preferred for patients with mild dysfunction, and routine urodynamic investigation has proven to be of low yield (Gyrtrup et al, 1995). In our experience, voiding complaints often reflect the degree of systemic impairment; such symptoms often improve substantially after effective antiretroviral therapy. For patients in urinary retention, intermittent catheterization is the treatment of choice. When the patient lacks sufficient manual dexterity, indwelling urethral or suprapubic catheterization is employed.
Urolithiasis
Urinary calculi have been associated with several treatments of HIV infection. The strongest association is with protease inhibitors, especially indinavir (Daudon and Jungers, 2004). Kidney and ureteral stones occur in 2% to 3% of patients taking indinavir (Lerner et al, 1998; Katner et al, 2002), but rates as high as 12% to 24% have been reported (Hermieu et al, 1999; Reiter et al, 1999; Meraviglia et al, 2002). Renal secretion appears to increase urinary supersaturation with formation of indinavir urinary crystals, especially with indinavir dosages of more than 600 mg (Vella and Floridia, 2004). The stones are nonopaque and may be associated with minimal findings on noncontrast CT examination (Gentle et al, 1997; Blake et al, 1998; Schwartz et al, 1999).
The authors’ experience is consistent with recommendations for conservative treatment in most cases, consisting of hydration, analgesics, and temporary cessation of indinavir (Hermieu et al, 1999; Kohan et al, 1999). Indications for intervention include persistent fever, intractable pain, inability to tolerate oral hydration, or obstruction of a solitary kidney. Indinavir crystals develop at pH 7.0 and can be dissolved when the pH is lowered to 4.0. This suggests that short-term urinary acidification may be valuable for stone dissolution (Kalaitzis et al, 2002). After resolution of the acute symptoms and passage of the stone, indinavir therapy can be restarted with aggressive oral hydration (Clayman, 1998). Urinary calculi have also been associated with use of other protease inhibitors, such as nelfinavir (Engeler et al, 2002) and saquinavir (Green et al, 1998).
HIV-infected patients on protease inhibitors also form stones that do not contain protease inhibitors. One study of 24 HIV-infected patients with urinary calculi found that only 3 (29%) of 14 on indinavir had indinavir-containing stones (Nadler et al, 2003). Of 10 patients evaluated, 8 had metabolic abnormalities. Thus metabolic evaluation may help prevent future stone episodes while avoiding the need to alter antiretroviral regimens in most patients.
Sulfadiazine therapy for toxoplasmosis has also been associated with obstructive nephropathy (Farina et al, 1995; Colebunders et al, 1999; Crespo et al, 2000). The obstructing calculi often appear radiolucent. Initial therapy should include hydration, discontinuation of the sulfadiazine, and urinary alkalinization (Crespo et al, 2000). Invasive interventions should be reserved for patients who fail such medical management (Colebunders et al, 1999).
HIV-Associated Nephropathy
AIDS-related renal failure was first reported in the 1980s and has now become known as HIV-associated nephropathy (HIVAN) (Rao et al, 1984). This glomerular disease has emerged as a significant medical renal disease in HIV-infected patients. The high prevalence and severity of HIVAN among certain populations has made this diagnosis almost synonymous with HIV-associated chronic kidney disease (Appel et al, 2004; Winston et al, 2008). More recently the clinical course of HIVAN has been substantially improved by the widespread use of HAART (Lucas et al, 2004). Nonetheless, HIVAN remains a significant cause of morbidity and mortality among infected individuals.
Epidemiology
HIVAN has been reported more often on the U.S. east coast than the west coast. In urban east coast populations the prevalence of HIVAN approached 90% in nephrotic HIV-infected patients compared with 2% in San Francisco, where most patients were white homosexuals (Appel et al, 2004; Ross and Klotman, 2004).
HIVAN occurs more frequently among HIV-infected black patients, with a black-to-white ratio of 12 : 1, and represents the third leading cause of end-stage renal disease among 20- to 64- year-old blacks (following only diabetes and hypertension). Blacks also appear to have more severe renal disease with heavier proteinuria, higher rate of the nephrotic syndrome, and greater renal insufficiency. Intravenous drug use has been the most common risk factor associated with HIVAN, but the disease has been seen in all HIV risk groups (Appel et al, 2004). There is no relationship between HIVAN and patient age, duration of HIV infection, or types of opportunistic infections or malignancies (Appel et al, 2004). The prevalence of HIVAN is reported to be 3.5% in HIV-infected clinic patients and 6.9% in autopsy series of HIV-infected patients (Shahinian et al, 2000). As expected, hypertension and diabetes mellitus remain the most common risk factors for the development of chronic kidney disease, leading to a 10-fold increased risk of chronic kidney disease and accounting for more than 70% of cases of end-stage renal disease. Data from the Multicenter AIDS Cohort Study suggest that HIV-infected individuals are four times more likely to have diabetes compared with control subjects. Thus the association between HIV, HIVAN, and chronic kidney disease is complex (Brown et al, 2005; Winston et al, 2008).
Clinical Presentation
HIVAN usually occurs in patients with low CD4 counts, but overt AIDS is not a prerequisite (Appel et al, 2004). HIVAN typically presents as nephrotic range proteinuria and renal insufficiency (Appel et al, 2004). Other characteristics typical of the nephrotic syndrome are common in some series, including edema, hypoalbuminemia, hypercholesterolemia, and hypertension. Patients may also present with sub–nephrotic-range proteinuria and urinary sediment findings of microhematuria and sterile pyuria. Renal ultrasound evaluation shows echogenic kidneys with preserved or enlarged size despite severe renal insufficiency (Appel et al, 2004).
Pathology and Pathogenesis
Diagnosis of HIVAN is based on a renal biopsy specimen showing characteristic focal segmental glomerulosclerosis with collapsing features and related mesangiopathies (Appel et al, 2004). Patients with HIVAN have a higher percentage of glomerular collapse, less hyalinosis, and greater visceral cell swelling than patients with classic, idiopathic focal segmental glomerulosclerosis or heroin nephropathy.
There are conflicting data on the potential for direct HIV infection of renal glomerular cells in vivo, but HIV appears to be able to infect glomerular endothelial cells and to a lesser degree mesangial cells in vitro (Marras et al, 2002; Appel et al, 2004). HIV replication may not be necessary for development of HIVAN, and it is likely that a viral gene product or indirect effects on host cytokine production are involved in the pathogenesis.
Course and Treatment
During the early years of the epidemic HIVAN was characterized by rapid progression to end-stage renal disease (Appel et al, 2004). There was an almost universal requirement for dialysis within 1 year of diagnosis (Appel et al, 2004).
With the introduction of HAART the rise in new cases of end-stage renal disease due to HIVAN slowed markedly (Kirchner, 2002; Appel et al, 2004). There are reports of patients with biopsy-documented or presumed HIVAN who experienced dramatic resolution of nephrotic syndrome and improved glomerular filtration rate on therapy (Appel et al, 2004). Antiretroviral therapy also appears to delay progression of HIVAN to end-stage renal disease. Thus antiretroviral therapy is recommended for patients with early histologic lesions and milder proteinuria without renal deterioration. Antiretroviral drugs need to be selected with caution given their propensity to cause or potentate nephrotic syndrome and must be dose adjusted in cases of renal insufficiency (Winston et al, 2008). Other therapies that have been recommended include angiotensin-converting enzyme inhibitors, angiotensin II receptor blockers, and immunosuppressive therapy in selected patients (Appel et al, 2004).
Abnormal Urinalysis
Abnormalities on urinalysis are relatively common in HIV-infected persons. These findings include hematuria, pyuria, bacteriuria, and proteinuria. One study suggests that hematuria occurs in 25% of HIV-infected persons (Cespedes et al, 1995). Although hematuria may be related to many causes, genitourinary tumors appear to be uncommon, particularly in young men. Thus, complete urologic evaluation may safely be omitted in young men with asymptomatic microscopic hematuria (Cespedes et al, 1995). In contrast, the authors have seen a number of HIV-infected patients with urologic malignancies, including bladder, renal, and other tumors, presenting with hematuria. These observations suggest that complete investigation is indicated in older patients and those with significant hematuria. Proteinuria is also common in certain populations of HIV-infected patients (see earlier).
Neoplasms
Many neoplasms occur in HIV-infected patients (Kho et al, 2004; Tirelli and Vaccher, 2004). Vascular and lymphoreticular neoplasms have been associated most strongly with HIV infection, particularly Kaposi sarcoma (KS) and non-Hodgkin lymphoma (NHL) (Cadogan and Dalgleish, 2008). The risks of KS and NHL are increased 1000- and 100-fold, respectively, among HIV-infected patients compared with the general population (Tirelli and Vaccher, 2004). KS, primary central nervous system lymphoma, systemic intermediate/high-grade B-cell NHL, and invasive cervical cancer have been designated as AIDS-defining illnesses. Other malignancies reported to be associated with HIV infection include Hodgkin disease, high-grade anal epithelial lesions, nonmelanomatous skin cancer, myeloma, and lung and testicular carcinoma (International Collaboration on HIV and Cancer, 2000; Tirelli and Vaccher, 2004). HAART radically changed the clinical spectrum of HIV infection in industrialized countries. During the mid 1990s, rates of opportunistic infections, KS, primary central nervous system lymphoma, and NHL decreased significantly (International Collaboration on HIV and Cancer, 2000; Grulich et al, 2001).
Kaposi Sarcoma
KS is the most common vascular neoplasm and a significant cause of morbidity and mortality in certain HIV-infected populations (Cockerell, 1996).
Classification and Clinical Presentation
KS is classified into classic and epidemic forms. The classic form was first described in 1827 (Kho et al, 2004) and occurs mainly in elderly Mediterranean men, black equatorial Africans, and patients with lymphoma or immune deficiencies. In contrast, epidemic KS is associated with AIDS.
KS ranges from an indolent to an aggressive disease with substantial morbidity and mortality. Typical patients present with disseminated skin lesions, often with lymph node and visceral involvement (Tirelli and Vaccher, 2004). Any skin area may be involved, including the genitalia. Three types of skin lesions are described: macule or patch, plaque, and nodule (Kho et al, 2004). Typical macules are oriented along skin cleavage lines and may be mistaken for blushes, purpura, or nevi. With time the lesions darken and develop into raised, firm, indurated, purplish plaques reflecting the presence of abundant blood vessels, extravasated erythrocytes, and siderophages. Some lesions may ulcerate. Nodular lesions are dome-shaped elevated lesions that are usually purple. On palpation they are firm and may be ulcerated.
Epidemiology
KS incidence rates started to decline in the late 1980s and then dramatically lessened with the introduction of HAART in the mid 1990s (International Collaboration on HIV and Cancer, 2000; Grulich et al, 2001; Tirelli and Vaccher, 2004). Meta-analysis of 47,936 HIV-infected individuals from North America, Europe, and Australia found that the KS rate ratio for 1997-1999 versus 1992-1996 was 0.3 (International Collaboration on HIV and Cancer, 2000). The risk of KS among male homosexuals is more than 100,000-fold higher than other HIV-infected populations (Tirelli and Vaccher, 2004).
A new herpesvirus, called KS-associated herpesvirus or human herpesvirus 8 (Chang et al, 1994), is essential for all forms of KS. KS-associated herpesvirus transmission correlates with a history of STIs and number of male sexual partners. Coinfection with HIV and KS-associated herpesvirus increases the risk of developing KS 10,000-fold compared with KS-associated herpesvirus infection alone. The probability of developing KS after coinfection with both KS-associated herpesvirus and HIV approaches 50% over 10 years (Chang et al, 1994).
Pathology and Pathogenesis
KS is an proliferative disease characterized by angiogenesis, endothelial spindle cell growth (KS cells), inflammatory cell infiltration, and edema (Tirelli and Vaccher, 2004). These lesions reflect immune dysregulation characterized by CD8+ T-cell activation, production of T-helper-1 cytokines, and angiogenic factors. This process induces generalized activation of endothelial cells.
Diagnosis and Management
An experienced observer can often make a presumptive diagnosis but skin biopsy is necessary to confirm the diagnosis of KS.
No specific therapy is curative (Tirelli and Vaccher, 2004). Treatment needs to be individualized, based on the patient’s clinical and immunologic status. HAART results in clinical improvement of KS lesions and prolongation of time to treatment failure (Lebbe et al, 1998; Levine and Tulpule, 2001). Numerous anecdotal reports document KS regression on HAART alone. Anti-KS activity of HAART appears to reflect immune system reconstitution and, to a lesser extent, suppression of HIV replication. HIV protease inhibitors are also potent antiangiogenic molecules that may affect KS pathogenesis (Sgadari et al, 2002).
Localized, cutaneous KS lesions can be treated using radiation, laser, cryotherapy, or intralesional injections of antineoplastic drugs (Levine and Tulpule, 2001). Cytotoxic chemotherapy is indicated for patients who do not respond to HAART and patients with life-threatening or visceral disease. A wide variety of drugs, including Vinca alkaloids (vincristine, vinblastine), bleomycin, and doxorubicin individually and in combination have produced tumor regression in 21% to 59% of patients, with lower rates for monotherapy. Paclitaxel is the newest systemic agent approved for use in the patient in whom KS has relapsed (Levine and Tulpule, 2001). Corticosteroid therapy should be avoided because they induce development of KS and may worsen preexisting KS lesions.
Prognosis for HIV-infected patients with KS depends primarily on their immune status and, secondarily, on the clinical stage of the KS lesions. Median survival of patients with CD4+ T-cell counts greater than or equal to 150/µL and early-stage disease is 35 months. Patients with CD4+ T-cell counts less than 150/µL have poor median survival (13 months for early-stage disease and 12 months for advanced-stage disease, respectively) (Levine and Tulpule, 2001).
Non-Hodgkin Lymphoma
A number of lymphoreticular malignancies of both B and T cells develop in HIV-infected patients. Most are based in the lymph nodes and reticuloendothelial system. The genital skin and genitourinary organs may be involved primarily or secondarily (Mearini et al, 2002; Kho et al, 2004; Tirelli and Vaccher, 2004). Patients usually present with advanced disease associated with a short median survival time. NHL is the most common lymphoreticular neoplasm.
Epidemiology and Clinical Presentation
NHL incidence decreased significantly after the introduction of HAART (Tirelli and Vaccher, 2004). Systemic NHL incidence decreased less than KS and later than the other AIDS-defining illnesses. Consequently, lymphoma has become the most common AIDS-associated cancer among patients receiving HAART (International Collaboration on HIV and Cancer, 2000; Grulich et al, 2001; Tirelli and Vaccher, 2004).
HIV-infected patients characteristically have widespread disease at presentation (Tirelli and Vaccher, 2004). They frequently have systemic symptoms, including fever, night sweats, and weight loss of more than 10%. At diagnosis of NHL, approximately 75% of HIV-infected patients have advanced disease with frequent involvement of extranodal sites. Any body site may be affected, including the genitourinary tract.
Diagnosis and Treatment
NHL should be diagnosed by histologic examination of an incisional or excisional biopsy specimen. Treatment consists of the usual therapy for systemic lymphoma. Optimal therapy has not been defined, and whether intensive or conservative chemotherapy regimens are indicated remains controversial. Prognosis for HIV-infected patients with lymphoma is poor, with a median survival of 5 to 10 months after diagnosis (Tirelli and Vaccher, 2004).
Squamous Genital Cancers: Role of Human Papillomavirus
HIV infection is associated with HPV-related anogenital neoplasms (Tirelli and Vaccher, 2004). These lesions include cancers and precancerous lesions of the cervix, vulva, anus, and penis. The risk of all HPV-associated cancers and their in-situ precursor lesions in both women and men is elevated across all HIV exposure categories (Tirelli and Vaccher, 2004). Risk factors include multiple sexual partners, cigarette smoking, and STIs, particularly HPV infection.
HPV sequences are found in more than 99% of cervical squamous cell carcinomas and in most anal cancers, with HPV type 16 present in 50% and types 18, 31, and 45 present in another 30%. High-grade intraepithelial precursor lesions have comparable rates of similar HPV types. HPV-associated oncogenesis results from upregulated expression of viral-encoded transforming proteins, particularly E6 and E7. These proteins interact with and inactivate the host cell tumor suppressor gene products, including retinoblastoma and TP53. This results in upregulated cell cycle progression, deficient DNA repair, and, eventually, malignant transformation (Tirelli and Vaccher, 2004). The degree of HIV-related immunosuppression corresponds with the occurrence and severity of these anogenital tumors.
Testicular Cancer
There may be an increased incidence of germ cell and non–germ cell testicular tumors associated with HIV infection (Wilkinson and Carroll, 1990; Wilson et al, 1992; Kwan and Lowe, 1995). Retrospective review of 3000 patients enrolled in an HIV clinic found a 50 times greater rate of testicular neoplasms than in the general population (Wilson et al, 1992). Compared with HIV-uninfected patients there is a greater risk of tumor bilaterality and a greater risk of high-grade testicular lymphoma (Armenakas et al, 1992). More recent data suggest that the increased risk of testicular germ cell tumors is more modest, whereby HIV-infected men are approximately two times more likely to develop seminoma (Standardized Incidence Ratio 1.9, 95% CI 1.6-2.2) but not more likely to develop nonseminomatous tumors (Goedert et al, 2007).
The therapeutic dilemma is that all accepted treatments for testicular neoplasms, except surveillance, cause additional immune suppression. Furthermore, patients with advanced HIV/AIDS often tolerate radiation and chemotherapy poorly. This may require major modifications of standard treatment protocols, resulting in decreased effectiveness. Despite these limitations, most experts recommend that HIV-infected patients with testicular neoplasms should receive standard treatment, as indicated by the tumor histology and stage (Wilkinson and Carroll, 1990).
Occupational Risks for HIV Infection in Urology
For reasons that are not completely understood, occupational HIV infection remains relatively uncommon (Henderson, 2004b).
Epidemiology
The Centers for Disease Control and Prevention has recorded only 57 instances of documented occupational HIV infection. For each case the worker sustained an occupational exposure, had a negative baseline serum sample, and then developed serologic evidence of HIV infection. In addition there have been 138 instances of probable or possible occupational HIV infection among U.S. health care workers (Centers for Disease Control and Prevention, 2004). Of adults with AIDS, 5.1% have occurred in health care workers, including 122 surgeons. Approximately 60 cases of “possible or probable” occupational infection have been reported from outside the United States (Henderson, 2004b). There were no documented cases of occupationally acquired AIDS/HIV infection among surgeons and 6 possible cases of occupational transmission among surgeons.
Risks Associated with Specific Exposures
Of the 57 health care workers who seroconverted following occupational exposures 48 (84%) had percutaneous (puncture/cut injury) exposures; 5 (9%) had mucocutaneous (mucous membrane and/or skin) exposures; 2 (4%) had both percutaneous and mucocutaneous exposure; and 2 (4%) had an unknown route of exposure. Forty-nine health care workers were exposed to HIV-infected blood, 3 to concentrated virus in a laboratory, 1 to visibly bloody fluid, and 4 to an unspecified fluid (Centers for Disease Control and Prevention, 2004).
Many occupational cases of HIV transmission share common features. Most are percutaneous exposures, especially injection needlestick injuries. All clinical cases occurred after exposure to blood or grossly blood-stained fluids from HIV-infected patients. As yet, no case has been documented following a needlestick injury with a solid surgical needle (Henderson, 2004b).
A retrospective, case-control study from the United States, France, and the United Kingdom identified five risk factors for occupational infection: deep as compared with superficial exposure (P < .0001), blood visible on the device causing the exposure (P = .0014), placement of the device in a source-patient’s vein or artery (P = .0028), death of the source-patient within 60 days of the exposure (P = .0011), and no postexposure chemoprophylaxis with zidovudine (P = .0026) (Cardo et al, 1997). The first four factors are likely related to an increased inoculum.
The importance of an inoculum effect is supported by other data. Transfusion of a unit of blood from an HIV-infected donor is associated with virtually 100% risk for infection (Ward et al, 1987). The depth of a percutaneous exposure was an independent risk factor for occupational HIV infection (Cardo et al, 1997). In addition, all needlestick exposures associated with occupational transmission have been caused by hollow-bore needles (i.e., injection as compared with suturing needles), which contain more blood than solid needles.
Combining available data, health care workers have sustained more than 6800 percutaneous exposures to sharp devices contaminated with blood or other blood-stained body fluids from HIV-infected patients (Ippolito et al, 1999; Henderson, 2004b). Twenty-one occupational HIV infections were documented, resulting in a risk of transmission per injury of 0.31% (Table 14–5). Thus, 1 in 324 parenteral exposures resulted in occupational HIV infection.
Table 14–5 Risk for HIV Infection Associated with Occupational Exposure
| TYPE OF EXPOSURE | PERCUTANEOUS | MUCOUS MEMBRANE |
|---|---|---|
| No. of longitudinal studies | 27 | 21 |
| No. of exposures | 6807 | 2761 |
| No. of documented infections | 21 | 0/1 |
| Infection rate per exposure | 0.031% | 0-0.11% |
From Henderson DK: Preventing occupational infection with HIV in the health care environment. In Cohen J, Powderly WG, Berkley SF, et al, editors. Infectious diseases. 2nd ed, vol. 22. Edinburgh: Mosby; 2004. p. 1217–22.
Other exposures represent a lower risk for infection. In rare circumstances, mucous membrane or cutaneous exposures may produce occupational infection (Chamberland et al, 1995). To date, more than 2700 exposures have been followed prospectively, with only one seroconversion (Ippolito et al, 1993). Thus, a maximum risk estimate for HIV infection is 0.11% per mucous membrane exposure. Occupational exposures other than percutaneous and mucous membrane exposures are even less likely to result in infection. Prospective studies (Friedland et al, 1990; Gershon et al, 1990) that include hundreds of person-years of follow-up have not identified a single instance of HIV transmission. Although exposures to other fluids are likely to be associated with some occupational risk, this risk is below measurable limits.
Interventions to Decrease Occupational Risk
Adherence to current guidelines might prevent one third of parenteral occupational exposures (Marcus, 1988). Universal Precautions are based on the principle that health care workers should treat blood and blood-stained body fluids from all patients as potentially infectious (Centers for Disease Control and Prevention, 1988). The CDC also recommends Standard Precautions, focusing on the bidirectional spread of organisms to and from patients and health care providers. The recommendations emphasize that blood and blood-containing body fluids represent risks to health care workers and that health care workers should use barriers and take precautions to prevent occupational exposures. These recommendations are effective. For example, the act of piercing a latex glove with a needle covered with blood may reduce the blood inoculum by as much as 50% (Henderson, 2004b).
Once exposure to HIV has occurred, the exposed site should be allowed to bleed freely and first aid should be administered. The wound should be cleansed and decontaminated as soon as patient safety permits. Wounds should be washed with soap and water and then irrigated with sterile saline, a disinfectant, or other suitable solution.
Postexposure Prophylaxis
Data support postexposure chemoprophylaxis to reduce the risk of occupational HIV transmission. Postexposure prophylaxis has proven effective in animal models (Henderson and Gerberding, 1989; Henderson, 2004a). The case-control study described earlier found that zidovudine chemoprophylaxis was associated with an 80% reduction in the risk for occupational infection (Cardo et al, 1997).
Current recommendations include a three-drug regimen for the most severe occupational exposures (i.e., zidovudine, lamivudine, and one of four additional agents) and a two-drug regimen for lesser exposures (i.e., zidovudine plus lamivudine or one of two other alternative two-drug regimens) (Centers for Disease Control and Prevention, 2001). If the source patient is (or recently has been) receiving antiretroviral therapy, some authorities recommended use of alternative regimens (Henderson, 2004a).
There are two major problems with antiretroviral prophylaxis following occupational exposure to HIV. First, these drugs have substantial toxicity. Data on healthy individuals, especially for the newer agents, are extremely limited (Henderson, 2004a). Studies of antiretroviral prophylaxis after occupational HIV exposures in health care workers have demonstrated substantial side effects, often limiting completion of the regimen. Recently, use of nevirapine has been contraindicated because of the risk of severe liver toxicity, including hepatic failure. Virtually all marketed antiretroviral agents have potential for carcinogenicity, teratogenicity, and mutagenicity. Second, there have been documented failures of antiretroviral prophylaxis. Cases of zidovudine chemoprophylaxis failure (eight of which have clinical relevance) have appeared in the literature (Henderson, 2004a). Thus, the potential risks and benefits of post-exposure prophylaxis must be considered on a case-by-case basis.
Antiretroviral Therapy
A diagram of antiretroviral therapy in HIV infection is presented in Figure 14–4 (Vella and Floridia, 2004).
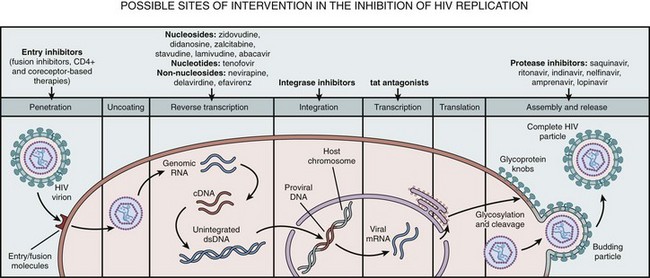
Figure 14–4 Antiretroviral therapy for HIV infection. Diagram illustrating potential sites of action for drug therapy.
(From Vella S, Florida M. HIV therapy: antiviral therapy. In: Cohen J, Powderly SF, Berkley SF, et al, editors. Infectious diseases. 2nd ed, vol. 2. Edinburgh: Mosby; 2004. p. 1387–98).
Suppression of HIV replication is associated with a significant delay in the progression to AIDS and increased survival. Thus, the goal of antiretroviral treatment should be to obtain maximal and sustained suppression of HIV replication (Vella and Floridia, 2004).
The main tools for monitoring antiretroviral treatment are the plasma HIV RNA level and CD4+ T-cell count (O’Brien et al, 1996). Previously untreated patients should reach undetectable plasma HIV RNA levels within 4 to 6 months accompanied by a concomitant rise in CD4+ counts. In this context “undetectable plasma HIV RNA level” does not mean eradication of the virus at other sites (lymphoid tissues, central nervous system, or genital tract) (Perelson et al, 1997). Eradication of HIV from these viral reservoirs and sanctuaries is not yet a realistic goal.
From Monotherapy to Highly Active Antiretroviral Therapy (HAART)
In 1987 zidovudine, the first effective antiretroviral drug and prototype of the nucleoside reverse transcriptase inhibitors (NRTIs), was introduced. Zidovudine monotherapy remained the mainstay of treatment until it became evident that benefit was limited (Concorde, 1994). Clinical trials demonstrated that two-drug regimens induced a more profound and prolonged effect on viral load, CD4+ T-cell counts, delayed clinical progression, and increased patient survival (Hammer et al, 1996; Deeks et al, 1997). In 1995 protease inhibitors became available. Combined with NRTIs, protease inhibitors produced a further reduction of HIV replication, often to undetectable levels (Deeks et al, 1997). Non-nucleoside reverse transcriptase inhibitors (NNRTIs), introduced after 1996, also proved highly effective. Availability of a rapidly increasing number of drugs and the evolving information has made HIV treatment extremely complex. The objective of antiretroviral therapy is to prevent disease progression and prolong survival while maintaining quality of life. Long-term nonprogression will be achieved by reducing plasma viral load below 50 copies/mL on a long-term basis. Combinations of antiretroviral agents with minimal or no overlapping toxicity and demonstrated antiviral additive to synergistic effect is recommended to maximize the antiviral response (Montaner et al, 2004). Current recommendations from the International AIDS Society-USA Panel note that the initial treatment regimen must be individualized but should generally include a protease inhibitor (efavirenz) plus two NRTIs (tenofovir/emtricitabine or abacavir/lamivudine) (Hammer et al, 2008).
Available Drugs
The optimal treatment of HIV infection is a potent combination of drugs, termed highly active antiretroviral therapy (HAART). Most HAART regimens are based on a dual nucleoside “backbone” plus an NNRTI or a protease inhibitor (with the option of adding low-dose ritonavir to another protease inhibitor for pharmacoenhancement.) Three-nucleoside regimens including abacavir are increasingly used because these regimens are easy to administer and are dual-class sparing (i.e., allow deferred use of protease inhibitors and NNRTIs).
Currently antiretroviral drugs include 7 NRTIs (zidovudine, didanosine, zalcitabine, stavudine, lamivudine, emtricitabine, and abacavir), 1 NRTI (tenofovir disoproxil fumarate), 3 NNRTIs (nevirapine, delavirdine, and efavirenz), and 10 HIV protease inhibitors (saquinavir, ritonavir, indinavir, nelfinavir, amprenavir, fosamprenavir, tipranavir, darunavir, atazanavir, and lopinavir). Other drugs have recently been approved by the U.S. Food and Drug Administration for treating HIV. These drugs may prove particularly useful in cases of viral resistance to standard regimens and include a fusion inhibitor (enfuviritide), an entry inhibitor (maraviroc), and an HIV integrase strand transfer inhibitor (raltegravir) (Table 14–6) (He et al, 2008; Hicks and Gulick, 2009; Hunt and Romanelli, 2009).
Table 14–6 Antiviral Drugs Available for HAART Combination Regimens
| CLASS | DRUG | TRADE NAME |
|---|---|---|
| Nucleoside reverse transcriptase inhibitors (NRTIs) | Zidovudine (AZT) | Retrovir*† |
| Didanosine (ddI) | Videx | |
| Zalcitabine (ddC) | Hivid | |
| Lamivudine (3TC) | Epivir*† | |
| Stavudine (d4T) | Zerit | |
| Abacavir (ABC) | Ziagen† | |
| Emtricitabine (FTC) | Emtriva‡ | |
| Nucleotide reverse transcriptase inhibitors | Tenofovir | Viread |
| Non-nucleoside reverse transcriptase inhibitors (NNRTIs) | Delaviridine (DLV) | Rescriptor |
| Nevirapine (NVP) | Viramune | |
| Efavirenz (EFV) | Sustiva | |
| Protease inhibitors | Saquinavir (SAQ) | Invirase |
| Ritonavir (RTV) | Norvir | |
| Nelfinavir (NFV) | Viracept | |
| Indinavir (IDV) | Crixivan | |
| Amprenavir (APV) | Agenerase | |
| Fosamprenavir (FOS-APV) | Lexiva | |
| Lopinavir (LPV) | Kaletra§ | |
| Tipranavir (TPV) | Aptivus | |
| Darunavir | Prezista | |
| Atazanavir (ATV) | Reyataz | |
| Fusion inhibitors | Enfuviritide (T-20) | Fuzeon |
| Entry inhibitors | Maraviroc | Selzentry |
| HIV integrase strand transfer inhibitors | Raltegravir | Isentress |
* Also available as Combivir (300 mg AZT + 150 mg 3TC).
† Also available as Trizivir (300 mg AZT + 150 mg 3TC + 300 mg ABV).
‡ Also available as Truvada (300 mg FTC + 200 mg tenofovir).
§ Only available as Kaletra (133.3 mg lopinavir +33.3 mg ritonavir).
From Vella S, Floridia M. HIV therapy: antiviral therapy. In: Cohen J, Powderly WG, Berkley SF, et al, editors. Infectious diseases. 2nd ed, vol. 2. Edinburgh: Mosby; 2004. p. 1387–98.
Nucleoside Reverse Transcriptase Inhibitors and Non-Nucleoside Reverse Transcriptase Inhibitors
NRTIs act through at least two mechanisms. First, as “chain terminators,” they block viral DNA elongation, stopping addition of further nucleosides. This mechanism depends on intracellular phosphorylation of NRTIs to the corresponding triphosphates. Second, they act by competition/binding of the reverse transcriptase; NNRTIs act only through this mechanism and not as “chain terminators.”
Protease Inhibitors
Protease inhibitors act by binding to the catalytic site of HIV aspartic protease. This enzyme is critical for post-translational processing of the polyprotein products into the functional core proteins and viral enzymes. By inhibiting this step, protease inhibitors release immature, noninfectious viral particles. However, unlike the NRTIs, which provide no protection in established HIV infection, protease inhibitors are also active in chronically infected cells. Protease inhibitors are combined with NRTIs or with other protease inhibitors, taking advantage of a boosting effect on drug concentrations that occurs when protease inhibitors are administered together.
Significant Urologic Side Effects
Of the NRTIs, zalcitabine is associated with peripheral neuropathy and painful penile ulcers. Of the protease inhibitors, ritonavir should be used with caution among patients at high risk of bleeding. Indinavir is a potent protease inhibitor that has been associated with urolithiasis (see earlier). In addition to drug-specific adverse events, long-term use HAART has highlighted a number of previously unrecognized adverse events. Some appear to be associated with a specific class of drugs, such as mitochondrial toxicity and lactic acidosis with NRTIs or hypoglycemia with protease inhibitors. Fat distribution changes (commonly described as lipodystrophy) represent a major clinical event. This syndrome presents as atrophic changes (peripheral fat loss involving face and limbs resembling Cushing disease), hypertrophic changes (visceral or dorsocervical fat accumulation, breast enlargement), or both.
New Investigational Agents
Even with potent, multidrug regimens, virologic failure is frequent. This may depend on various factors, including low adherence, drug-resistant HIV strains, low drug levels in the plasma or tissues, and other undetermined factors. Targeted strategies are being developed to improve treatment efficacy, including development of new drugs with improved formulation and administration schedules, increased potency, and reduced toxicity. A number of agents in each of the available drug classes as well as drugs with novel mechanisms of action are being developed. Desirable characteristics of this new generation of drugs include easier administration schedule, perfected pharmacokinetics, potency, tolerability, and the ability to inhibit HIV viruses that are resistant to the existing agents.
Evolving HIV Treatment Strategies
Growing appreciation of the difficulties of taking antiretroviral therapy on a long-term basis, the adverse effects of many regimens, and their negative impact on quality of life have led to reevaluation of this early intervention strategy. Because “eradication” of HIV is not on the horizon, the goal of therapy must now be redirected toward the long-term management of a chronic infection (Vella and Floridia, 2004).
The decision to start treatment is now based first on the patient’s disease stage, because it is clear that a symptomatic patient should be treated. For an asymptomatic patient, the decision is often driven by the CD4+ lymphocyte count and the plasma HIV RNA level (Montaner et al, 2004). The CD4+ T lymphocyte count can be regarded as an immediate predictor of prognosis, whereas the plasma HIV RNA level indicates the level of actual HIV replication. Other elements include the patient’s commitment to therapy and knowledge of the limitations of current regimens.
Despite the effectiveness of HAART the optimal time to initiate therapy remains unclear. Although current antiviral therapies have proven ability to suppress viral load and improve immune system function, several questions remain. Specific concerns remain about early initiation of HAART and patient compliance, drug toxicities, drug resistance, and the marked costs associated with prolonged therapy (Wilkin and Gulick, 2008). Despite the absence of randomized clinical trials, several large cohort studies have demonstrated decreased AIDS-related morbidity and mortality when HAART is started early in the course of infection, when CD4 cell counts are higher. Recent data from the ART Cohort Collaboration suggest that individuals in whom therapy is initiated with a CD4 count of 200 to 350 cells/µL have a 5-year mortality rate that is 40% higher than those in whom therapy is started with counts greater than 350 cells/µL (May et al, 2007). The rationale for early therapy is augmented by studies suggesting decreased rates of non–AIDS-defining conditions, such as cancers, neuropathy, and anemia when antiviral medications are initiated at these higher CD4 levels. Fear that early therapy may increase costs of HIV treatment prompted multiple studies of the cost-effectiveness of early antiviral treatment, with sensitivity analyses using varying CD4 count levels. These studies suggest that early treatment is cost effective and offers significant improvements in quality-adjusted life years (Wilkin and Gulick, 2008).
Strategies on the Horizon
In addition to a plethora of new antiretroviral agents targeting the HIV replication cycle there are a number of strategies under active investigation to prevent HIV infection or to improve therapy. Two promising approaches are development of immune-based strategies and vaccines.
Immune-Based Strategies That May Be Combined with HAART
Because eradication of HIV with HAART probably cannot be achieved in less than 50 to 60 years (Montaner et al, 2004) the possibility of adding additional treatment to ensure immune restoration holds considerable appeal (Pollard and Onorato, 2004). Various strategies are under investigation to both enhance HIV-specific immune responses and to stimulate the general immune responses, such as therapeutic vaccination, structured treatment interruption, transfer of HIV-specific cell populations to enhance cell-mediated immunity, and transfer of pooled immune sera from donors who have HIV infection or of monoclonal antibodies directed against HIV. Such strategies may be of particular value for treating patients with later-stage infection, whose high levels of viremia and previous exposure to therapy place them at greatest risk of developing resistance to therapeutic agents (Pollard and Onorato, 2004).
Vaccines to Prevent HIV Infection
Considerable efforts are being expended to develop an effective vaccine to prevent HIV infection (Rosenberg et al, 1997; Amara et al, 2001; Shiver et al, 2002; Fast et al, 2004). HIV represents a formidable challenge. Its immune evasion mechanisms include molecular tricks to avoid neutralization by antibody, downregulation of immune functions, direct destruction of the CD4 T cells that support both antibody and effector T-cell responses, and a very rapid mutation rate that gives rise to virus strains lacking the antigenic sites to which effective immune responses are directed.
Nevertheless, several lines of evidence suggest that vaccines may prove effective in preventing HIV infection or in preventing progression to AIDS (Fast et al, 2004). First, HIV infection occurs after only a small fraction of all exposures, suggesting that a modest immune defense might be effective. Second, the natural history of HIV infection is that after an initial phase of rapid viral replication immune responses may control HIV replication and slow its pathogenic effects for many years. Third, in primate models some vaccines can prevent infection or slow viral replication and disease progression. Immunization that induces mucosal responses is particularly effective against mucosal challenges, as would occur with sexual exposure (Amara et al, 2001; Shiver et al, 2002). A number of vaccines have been tested and, lacking credible immunogenicity data, abandoned. Several more promising vaccines have recently entered trials or are expected to do so soon.
Bailey RC, Plummer FA, Moses S. Male circumcision and HIV prevention: current knowledge and future research directions. Lancet Infect Dis. 2001;1:223.
Boily MC, Baggaley R, Wang L, et al. Heterosexual risk of HIV-1 infection per sexual act: systematic review and meta-analysis of observational studies. Lancet Infect Dis. 2009;9:118.
Centers for Disease Control and Prevention. Updated U.S. Public Health Service guidelines for the management of occupational exposures to HBV, HCV, and HIV and recommendations for postexposure prophylaxis. MMWR Morb Mortal Wkly Rep. 2001;50:1.
Daudon M, Jungers P. Drug-induced renal calculi: epidemiology, prevention and management. Drugs. 2004;64:245.
Krieger JN, Nirapathpongporn A, Chaiyaporn M, et al. Vasectomy and human immunodeficiency virus type 1 in semen. J Urol. 1998;159:820.
Patterson BK, Landay A, Siegel JN, et al. Susceptibility to human immunodeficiency virus-1 infection of human foreskin and cervical tissue grown in explant culture. Am J Pathol. 2002;161:867.
Pierre-Alexandre B, Pantaleo G. Pathogenesis: immunopathogenesis of HIV-1 infection. In: Cohen J, Powderly WG, Berkley SF, et al, editors. Infectious diseases, vol. 2. Edinburgh: Mosby; 2004:1235-1249.
Quinn TC, Wawer MJ, Sewankambo N, et al. Viral load and heterosexual transmission of human immunodeficiency virus type 1. Rakai Project Study Group. N Engl J Med. 2000;342:921.
Ross MJ, Klotman PE. HIV-associated nephropathy. AIDS. 2004;18:1089.
Vella S, Floridia M. HIV therapy: antiviral therapy. In: Cohen J, Powderly WG, Berkley SF, et al, editors. Infectious diseases, vol. 2. Edinburgh: Mosby; 2004:1387-1398.
Weiss HA, Halpernr D, Bailey RC, et al. Male circumcision for HIV prevention: from evidence to action? AIDS. 2008;22:567.
World Health Organization. The world health report 2004—changing history. http://www.who.int/whr, 2004. [accessed May 2004]
Agot KE, Ndinya-Achola JO, Kreiss JK, Weiss NS. Risk of HIV-1 in rural Kenya: a comparison of circumcised and uncircumcised men. Epidemiology. 2004;15(2):157-163.
Amara RR, Villinger F, Altman JD, et al. Control of a mucosal challenge and prevention of AIDS by a multiprotein DNA/MVA vaccine. Science. 2001;292:69-74.
Anderson RM, May RM. Epidemiological parameters of HIV transmission. Nature. 1988;333(6173):514-519.
Appel GB, Radhakishnan J, D’Agati D. Secondary glomerular disease. In: Brenner BM, editor. Brenner and Rector’s The Kidney. 7th ed. Philadelphia: WB Saunders; 2004:1381-4182.
Armenakas NA, Schevchuk MM, Brodherson M, Fracchia JA. AIDS presenting as primary testicular lymphoma. Urology. 1992;40(2):162-164.
Auvert B, Buve A, Ferry B, et al. Ecological and individual level analysis of risk factors for HIV infection in four urban populations in sub-Saharan Africa with different levels of HIV infection. AIDS. 2001;15(Suppl 4):S15-S30.
Auvert B, Buvé A, Lagarde E, et al. Male circumcision and HIV infection in four cities in sub-Saharan Africa. AIDS. 2001;15(Suppl 4):S31-S40.
Auvert B, Taljaard D, Lagarde E, et al. Randomized, controlled intervention trial of male circumcision for reduction of HIV infection risk: the ANRS 1265 Trial. PLoS Med. 2005;2(11):e298.
Bailey RC, Moses S, Parker CB, et al. Male circumcision for HIV prevention in young men in Kisumu, Kenya: a randomised controlled trial. Lancet. 2007;369(9562):643-656.
Berger TG, Sawchuk WS, Leonardi C, et al. Epidermodysplasia verruciformis–associated papillomavirus infection complicating human immunodeficiency virus disease. Br J Dermatol. 1991 Jan;124(1):79-83.
Blake SP, McNicholas MM, Raptopoulos V. Nonopaque crystal deposition causing ureteric obstruction in patients with HIV undergoing indinavir therapy [see comments]. AJR Am J Roentgenol. 1998;171(3):717-720.
Boden D, Hurley A, Zhang L, et al. HIV-1 drug resistance in newly infected individuals. JAMA. 1999;282(12):1135-1141.
Boily MC, Baggaley RF, Wang L, et al. Heterosexual risk of HIV-1 infection per sexual act: systematic review and meta-analysis of observational studies. Lancet Infect Dis. 2009;9(2):118-129.
Brodie SJ, Lewinsohn DA, Patterson BK, et al. In vivo migration and function of transferred HIV-1-specific cytotoxic T cells. Nat Med. 1999;5(1):34-41.
Brown TT, Cole SR, Li X, et al. Antiretroviral therapy and the prevalence and incidence of diabetes mellitus in the multicenter AIDS cohort study. Arch Intern Med. 2005;165(10):1179-1184.
Buzelin F, Karam G, Moreau A, et al. Testicular tumor and the acquired immunodeficiency syndrome. Eur Urol. 1994;26(1):71-76.
Cadogan M, Dalgleish AG. HIV induced AIDS and related cancers: chronic immune activation and future therapeutic strategies. Adv Cancer Res. 2008;101:349-395.
Cameron DW, Simonsen JN, D’Costa LJ, et al. Female to male transmission of human immunodeficiency virus type 1: risk factors for seroconversion in men. Lancet. 1989;2(8660):403-407.
Cameron P, Pope M, Granelli-Piperno A, Steinman RM. Dendritic cells and the replication of HIV-1. J Leukoc Biol. 1996;59(2):158-171.
Carael M, Schwartlander B, Piot P. Epidemiology of HIV infection. In: Cohen J, Powderly WG, Berkley SF, Calandra T, editors. Infectious diseases. 2nd ed. Edinburgh: Mosby; 2004:1197-1207.
Cardo DM, Culver DH, Ciesielski CA, et al. A case-control study of HIV seroconversion in health care workers after percutaneous exposure. Centers for Disease Control and Prevention Needlestick Surveillance Group. N Engl J Med. 1997;337(21):1485-1490.
Centers for Disease Control and Prevention. Update: universal precautions for prevention of transmission of human immunodeficiency virus, hepatitis B virus, and other bloodborne pathogens in health-care settings. MMWR Morb Mortal Wkly Rep. 1988;37:377-388.
Centers for Disease Control and Prevention. Updated US Public Health Service guidelines for the management of occupational exposures to HBV, HCV, and HIV and recommendations for postexposure prophylaxis. MMWR Morb Mortal Wkly Rep. 2001;50:1-52.
Centers for Disease Control and Prevention. Sexually transmitted diseases treatment guidelines 2002. MMWR Morb Mortal Wkly Rep. 2002;51(RR-6):1-80.
Centers for Disease Control and Prevention. Surveillance of healthcare personnel with HIV/AIDS, as of December 2002. http://www.cdc.gov/ncidod/hip/BLOOD/hivpersonnel.htm, 2004. [accessed 14.08.04]
Centers for Disease Control and Prevention. Trends in primary and secondary syphilis and HIV infections in men who have sex with men—San Francisco and Los Angeles, California, 1998–2002. MMWR Morb Mortal Wkly Rep. 2004;53:575-578.
Cespedes RD, Peretsman SJ, Blatt SP. The significance of hematuria in patients infected with the human immunodeficiency virus. J Urol. 1995;154:1455-1456.
Chakraborty H, Sen PK, Helms RW, et al. Viral burden in genital secretions determines male-to-female sexual transmission of HIV-1: a probabilistic empiric model. AIDS. 2001;15(5):621-627.
Chamberland ME, Ciesielski CA, Howard RJ, et al. Occupational risk of infection with human immunodeficiency virus. Surg Clin North Am. 1995;75(6):1057-1070.
Chang Y, Cesarman E, Pessin MS, et al. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science. 1994;266(5192):1865-1869.
Chun TW, Carruth L, Finzi D, et al. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature. 1997;387(6629):183-188.
Clayman RV. Crystalluria and urinary tract abnormalities associated with indinavir. J Urol. 1998;160(2):633.
Coburn M. Urological manifestations of HIV infection. AIDS Res Hum Retroviruses. 1998 Apr;14(Suppl 1):S23-S25.
Cocchi F, DeVico AL, Garzino-Demo A, et al. The V3 domain of the HIV-1 gp120 envelope glycoprotein is critical for chemokine-mediated blockade of infection. Nat Med. 1996;2(11):1244-1247.
Cockerell CJ. Mucocutaneous neoplasms in patients with human immunodeficiency virus infection. Semin Diagn Pathol. 1996;13(1):19-39.
Coffin JM. HIV population dynamics in vivo: implications for genetic variation, pathogenesis, and therapy. Science. 1995;267(5197):483-489.
Cohen MS, Hoffman IF, Royce RA, et al. Reduction of concentration of HIV-1 in semen after treatment of urethritis: implications for prevention of sexual transmission of HIV-1. AIDSCAP Malawi Research Group. Lancet. 1997;349(9069):1868-1873.
Colebunders R, Depraetere K, De Droogh E, et al. Obstructive nephropathy due to sulfa crystals in two HIV seropositive patients treated with sulfadiazine. JBR-BTR. 1999;82(4):153-154.
Concorde. MRC/ANRS randomised double-blind controlled trial of immediate and deferred zidovudine in symptom-free HIV infection. Concorde Coordinating Committee. Lancet. 1994;343(8902):871-881.
Corman JM, Moody JA, Aronson WJ. Fournier’s gangrene in a modern surgical setting: improved survival with aggressive management. BJU Int. 1999;84(1):85-88.
Crespo M, Quereda C, Pascual J, et al. Patterns of sulfadiazine acute nephrotoxicity. Clin Nephrol. 2000;54(1):68-72.
Daudon M, Jungers P. Drug-induced renal calculi: epidemiology, prevention and management. Drugs. 2004;64(3):245-275.
De Gruttola VG, Clax P, DeMets DL, et al. Considerations in the evaluation of surrogate endpoints in clinical trials: summary of a National Institutes of Health workshop. Control Clin Trials. 2001;22(5):485-502.
De Paepe ME, Guerrieri C, Waxman M. Opportunistic infections of the testis in the acquired immunodeficiency syndrome. Mt Sinai J Med. 1990;57(1):25-29.
Deeks SG, Smith M, Holodniy M, Kahn JO. HIV-1 protease inhibitors: a review for clinicians. JAMA. 1997;277(2):145-153.
DeGruttola V, Seage GR3rd, Mayer KH, Horsburgh CRJr. Infectiousness of HIV between male homosexual partners. J Clin Epidemiol. 1989;42(9):849-856.
Desai K, Boily MC, Garnett GP, et al. The role of sexually transmitted infections in male circumcision effectiveness against HIV—insights from clinical trial simulation. Emerg Themes Epidemiol. 2006;3:19.
Drain PK, Halperin DT, Hughes JP, et al. Male circumcision, religion, and infectious diseases: an ecologic analysis of 118 developing countries. BMC Infect Dis. 2006;6:172.
Dunne EF, Whitehead S, Sternberg M, et al. Suppressive acyclovir therapy reduces HIV cervicovaginal shedding in HIV- and HSV-2-infected women, Chiang Rai, Thailand. J Acquir Immune Defic Syndr. 2008;49(1):77-83.
Emerman M, Malim MH. HIV-1 regulatory/accessory genes: keys to unraveling viral and host cell biology. Science. 1998;280(5371):1880-1884.
Engeler DS, John H, Rentsch KM, et al. Nelfinavir urinary stones. J Urol. 2002;167(3):1384-1385.
Farina LA, Palou Redorta J, Chechile Toniolo G. Reversible acute renal failure due to sulfonamide-induced lithiasis in an AIDS patient. Arch Esp Urol. 1995;48(4):418-419.
Fast PE, Gilmour J, Gupta K. HIV vaccines: research and development. In: Cohen J, Powderly WG, Berkley SF, et al, editors. Infectious diseases. 2nd ed. Edinburgh: Mosby; 2004:1223-1230.
Fink AJ. A possible explanation for heterosexual male infection with AIDS. N Engl J Med. 1986;315:1167.
Finzi D, Hermankova M, Pierson T, et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science. 1997;278(5341):1295-1300.
Foglia G, Sateren WB, Renzullo PO, et al. High prevalence of HIV infection among rural tea plantation residents in Kericho, Kenya. Epidemiol Infect. 2008;136(5):694-702.
Friedland G, Kahl P, Saltzman B, et al. Additional evidence for lack of transmission of HIV infection by close interpersonal (casual) contact. AIDS. 1990;4(7):639-644.
Gentle DL, Stoller ML, Jarrett TW, et al. Protease inhibitor-induced urolithiasis. Urology. 1997;50(4):508-511.
Gershon RR, Vlahov D, Nelson KE. The risk of transmission of HIV-1 through non-percutaneous, non-sexual modes—a review. AIDS. 1990;4(7):645-650.
Goedert JJ, Purdue MP, McNeel TS, et al. Risk of germ cell tumors among men with HIV/acquired immunodeficiency syndrome. Cancer Epidemiol Biomarkers Prev. 2007;16(6):1266-1269.
Gray R, Azire J, Serwadda D, et al. Male circumcision and the risk of sexually transmitted infections and HIV in Rakai, Uganda. AIDS. 2004;18(18):2428-2430.
Gray RH, Kigozi G, Serwadda D, et al. Male circumcision for HIV prevention in men in Rakai, Uganda: a randomised trial. Lancet. 2007;369(9562):657-666.
Gray RH, Li X, Kigozi G, et al. The impact of male circumcision on HIV incidence and cost per infection prevented: a stochastic simulation model from Rakai, Uganda. AIDS. 2007;21(7):845-850.
Green ST, McKendrick MW, Schmid ML, et al. Renal calculi developing de novo in a patient taking saquinavir. Int J STD AIDS. 1998;9(9):555.
Grosskurth H, Mosha F, Todd J, et al. Impact of improved treatment of sexually transmitted diseases on HIV infection in rural Tanzania: randomised controlled trial. Lancet. 1995;346(8974):530-536.
Grulich AE, Li Y, McDonald AM, et al. Decreasing rates of Kaposi’s sarcoma and non-Hodgkin’s lymphoma in the era of potent combination anti-retroviral therapy. AIDS. 2001;15(5):629-633.
Gyrtrup HJ, Kristiansen VB, Zachariae CO, et al. Voiding problems in patients with HIV infection and AIDS. Scand J Urol Nephrol. 1995;29(3):295-298.
Hallett TB, Singh K, Smith JA, et al. Understanding the impact of male circumcision interventions on the spread of HIV in southern Africa. PLoS One. 2008;3(5):e2212.
Hammer SM, Eron JJJr, Reiss P, et al. Antiretroviral treatment of adult HIV infection: 2008 recommendations of the International AIDS Society—USA panel. JAMA. 2008;300(5):555-570.
Hammer SM, Katzenstein DA, Hughes MD, et al. A trial comparing nucleoside monotherapy with combination therapy in HIV-infected adults with CD4 cell counts from 200 to 500 per cubic millimeter. AIDS Clinical Trials Group Study 175 Study Team. N Engl J Med. 1996;335(15):1081-1090.
He Y, Cheng J, Lu H, et al. Potent HIV fusion inhibitors against Enfuvirtide-resistant HIV-1 strains. Proc Natl Acad Sci U S A. 2008;105(42):16332-16337.
Henderson DK. Postexposure prophylaxis for nonoccupational HIV exposure. In: Cohen J, Powderly WG, Berkley SF, et al, editors. Infectious diseases. 2nd ed. Edinburgh: Mosby; 2004:1231-1234.
Henderson DK. Preventing occupational infection with HIV in the health care environment. In: Cohen J, Powderly WG, Berkley SF, et al, editors. Infectious diseases. 2nd ed. Edinburgh: Mosby; 2004:1217-1222.
Henderson DK, Gerberding JL. Prophylactic zidovudine after occupational exposure to the human immunodeficiency virus: an interim analysis. J Infect Dis. 1989;160(2):321-327.
Henderson DK, Saah AJ, Zak BJ, et al. Risk of nosocomial infection with human T-cell lymphotropic virus type III/lymphadenopathy-associated virus in a large cohort of intensively exposed health care workers. Ann Intern Med. 1986;104(5):644-647.
Hermieu J, Prevot M, Ravery V, et al. Urolithiasis and the protease inhibitor indinavir. Eur Urol. 1999;35(3):239-241.
Hermieu JF, Delmas V, Boccon-Gibod L. Micturition disturbances and human immunodeficiency virus infection. J Urol. 1996;156(1):157-159.
Heyns CF, Groeneveld AE, Sigarroa NB. Urologic complications of HIV and AIDS. Nat Clin Pract Urol. 2009;6(1):32-43.
Hicks C, Gulick RM. Raltegravir: the first HIV type 1 integrase inhibitor. Clin Infect Dis. 2009;48(7):931-939.
Hirsch MS, Brun-Vezinet F, D’Aquila RT, et al. Antiretroviral drug resistance testing in adult HIV-1 infection: recommendations of an International AIDS Society—USA Panel. JAMA. 2000;283(18):2417-2426.
Hunt JS, Romanelli F. Maraviroc, a CCR5 coreceptor antagonist that blocks entry of human immunodeficiency virus type 1. Pharmacotherapy. 2009;29(3):295-304.
International Collaboration on HIV and Cancer. Highly active antiretroviral therapy and incidence of cancer in human immunodeficiency virus-infected adults. J Natl Cancer Inst. 2000;92:1188-1201.
Ippolito G, Puro V, De Carli G. The risk of occupational human immunodeficiency virus infection in health care workers. Italian Multicenter Study. The Italian Study Group on Occupational Risk of HIV infection. Arch Intern Med. 1993;153(12):1451-1458.
Ippolito G, Puro V, Heptonstall J, et al. Occupational human immunodeficiency virus infection in health care workers: worldwide cases through September 1997. Clin Infect Dis. 1999;28(2):365-383.
Izu R, Manzano D, Gardeazabal J, Diaz-Perez JL. Giant molluscum contagiosum presenting as a tumor in an HIV-infected patient. Int J Dermatol. 1994;33(4):266-267.
Jewkes R, Dunkle K, Nduna M, et al. Factors associated with HIV seropositivity in young, rural South African men. Int J Epidemiol. 2006;35(6):1455-1460.
Johnson K, Way A. Risk factors for HIV infection in a national adult population: evidence from the 2003 Kenya Demographic and Health Survey. J Acquir Immune Defic Syndr. 2006;42(5):627-636.
Joint United Nations Program on HIV/AIDS. 2004 Report on the global AIDS epidemic. Geneva: United Nations; July 2004.
Kahn JO, Walker BD. Acute human immunodeficiency virus type 1 infection. N Engl J Med. 1998;339(1):33-39.
Kalaitzis C, Dimitriadis G, Tsatidis T, et al. Treatment of indinavir sulfate induced urolithiasis in HIV-positive patients. Int Urol Nephrol. 2002;34(1):13-15.
Kane CJ, Bolton DM, Connolly JA, Tanagho EA. Voiding dysfunction in human immunodeficiency virus infections. J Urol. 1996;155(2):523-526.
Kapiga SH, Sam NE, Mlay J, et al. The epidemiology of HIV-1 infection in northern Tanzania: results from a community-based study. AIDS Care. 2006;18(4):379-387.
Kaplan EH, Heimer R. HIV incidence among needle exchange participants: estimates from syringe tracking and testing data. J Acquir Immune Defic Syndr. 1994;7(2):182-189.
Kaplan MS, Wechsler M, Benson MC. Urologic manifestations of AIDS. Urology. 1987;30:441-443.
Katner HP, Paar DP, Nadler JP, et al. Open-label study of a twice-daily indinavir 800-mg/ritonavir 200-mg regimen in HIV-infected adults failing a protease inhibitor regimen. J Acquir Immune Defic Syndr. 2002;31(5):483-487.
Kelly R, Kiwanuka N, Wawer MJ, et al. Age of male circumcision and risk of prevalent HIV infection in rural Uganda. AIDS. 1999;13(3):399-405.
Kho TK, Bandel C, Cockrell CJ. Dermatologic manifestations of HIV infection. In: Cohen J, Powderly WG, Berkley SF, et al, editors. Infectious diseases. 2nd ed. Edinburgh: Mosby; 2004:1322-1332.
Kirchner JT. Resolution of renal failure after initiation of HAART: 3 cases and a discussion of the literature. AIDS Read. 2002;12(3):103-105. 110–12
Klavs I, Hamers FF. Male circumcision in Slovenia: results from a national probability sample survey. Sex Transm Infect. 2008;84(1):49-50.
Kohan AD, Armenakas NA, Fracchia JA. Indinavir urolithiasis: an emerging cause of renal colic in patients with human immunodeficiency virus. J Urol. 1999;161(6):1765-1768.
Kreiss JK, Koech D, Plummer FA, et al. AIDS virus infection in Nairobi prostitutes. Spread of the epidemic to East Africa. N Engl J Med. 1986;314(7):414-418.
Krieger JN, Bailey RC, Opeya J, et al. Adult male circumcision: results of a standardized procedure in Kisumu District, Kenya. BJU Int. 2005;96(7):1109-1113.
Krieger JN, Bailey RC, Opeya JC, et al. Adult male circumcision outcomes: experience in a developing country setting. Urol Int. 2007;78(3):235-240.
Krieger JN, Nirapathpongporn A, Chaiyaporn M, et al. Vasectomy and human immunodeficiency virus type 1 in semen. J Urol. 1998;159(3):820-825. discussion 825–6
Kuritzkes DR. Diagnostic tests for HIV infection and resistance assays. In: Cohen J, Powderly WG, Berkley SF, et al, editors. Infectious diseases. 2nd ed. Edinburgh: Mosby; 2004:1369-1378.
Kwan DJ, Lowe FC. Genitourinary manifestations of the acquired immunodeficiency syndrome. Urology. 1995;45(1):13-27.
Laga M, Alary M, Nzila N, et al. Condom promotion, sexually transmitted diseases treatment, and declining incidence of HIV-1 infection in female Zairian sex workers. Lancet. 1994;344(8917):246-248.
Lebbe C, Blum L, Pellet C, et al. Clinical and biological impact of antiretroviral therapy with protease inhibitors on HIV-related Kaposi’s sarcoma. AIDS. 1998;12(7):F45-F49.
Leibovitch I, Goldwasser B. The spectrum of acquired immune deficiency syndrome–associated testicular disorders. Urology. 1994;44(6):818-824.
Leport C, Rousseau F, Perronne C, et al. Bacterial prostatitis in patients infected with the human immunodeficiency virus. J Urol. 1989;141:334-336.
Lerner LB, Cendron M, Rous SN. Nephrolithiasis from indinavir, a new human immunodeficiency virus drug. J Urol. 1998;159(6):2074. discussion 2075
Levine AM, Tulpule A. Clinical aspects and management of AIDS-related Kaposi’s sarcoma. Eur J Cancer. 2001;37(10):1288-1295.
Liang C, Wainberg MA. Virology of HIV. In: Cohen J, Powderly WG, Berkley SF, et al, editors. Infectious diseases. 2nd ed. Edinburgh: Mosby; 2004:1251-1255.
Little SJ, Daar ES, D’Aquila RT, et al. Reduced antiretroviral drug susceptibility among patients with primary HIV infection. JAMA. 1999;282(12):1142-1149.
Little SJ, Holte S, Routy JP, et al. Antiretroviral-drug resistance among patients recently infected with HIV. N Engl J Med. 2002;347(6):385-394.
Londish GJ, Murray JM. Significant reduction in HIV prevalence according to male circumcision intervention in sub-Saharan Africa. Int J Epidemiol. 2008;37(6):1246-1253.
Lucas GM, Eustace JA, Sozio S, et al. Highly active antiretroviral therapy and the incidence of HIV-1-associated nephropathy: a 12-year cohort study. AIDS. 2004;18(3):541-546.
MacDonald KS, Malonza I, Chen DK, et al. Vitamin A and risk of HIV-1 seroconversion among Kenyan men with genital ulcers. AIDS. 2001;15(5):635-639.
Marcus R. Surveillance of health care workers exposed to blood from patients infected with the human immunodeficiency virus. N Engl J Med. 1988;319(17):1118-1123.
Marras D, Bruggeman LA, Gao F, et al. Replication and compartmentalization of HIV-1 in kidney epithelium of patients with HIV-associated nephropathy. Nat Med. 2002;8(5):522-526.
Marschner IC, Collier AC, Coombs RW, et al. Use of changes in plasma levels of human immunodeficiency virus type 1 RNA to assess the clinical benefit of antiretroviral therapy. J Infect Dis. 1998;177(1):40-47.
May M, Sterne JA, Sabin C, et al. Prognosis of HIV-1-infected patients up to 5 years after initiation of HAART: collaborative analysis of prospective studies. AIDS. 2007;21(9):1185-1197.
Mayer KH, Safren SA. Prevention of HIV transmission through behavioral and biological interventions. In: Cohen J, Powderly WG, Berkley SF, et al, editors. Infectious diseases. 2nd ed. Edinburgh: Mosby; 2004:1209-1215.
McCoombe SG, Short RV. Potential HIV-1 target cells in the human penis. AIDS. 2006;20(11):1491-1495. 13
McKay TC, Waters WB. Fournier’s gangrene as the presenting sign of an undiagnosed human immunodeficiency virus infection [see comments]. J Urol. 1994;152(5 pt 1):1552-1554.
Mearini E, Zucchi A, Costantini E, et al. Primary Burkitt’s lymphoma of bladder in patient with AIDS. J Urol. 2002;167(3):1397-1398.
Meier AS, Bukusi EA, Cohen CR, Holmes KK. Independent association of hygiene, socioeconomic status, and circumcision with reduced risk of HIV infection among Kenyan men. J Acquir Immune Defic Syndr. 2006;43(1):117-118.
Mellors JW, Rinaldo CRJr, Gupta P, et al. Prognosis in HIV-1 infection predicted by the quantity of virus in plasma. Science. 1996;272(5265):1167-1170.
Meraviglia P, Angeli E, Del Sorbo F, et al. Risk factors for indinavir-related renal colic in HIV patients: predictive value of indinavir dose/body mass index. AIDS. 2002;16(15):2089-2093.
Mermin J, Musinguzi J, Opio A, et al. Risk factors for recent HIV infection in Uganda. JAMA. 2008;300(5):540-549.
Miles BJ, Melser M, Farah R, et al. The urological manifestations of the acquired immunodeficiency syndrome. J Urol. 1989;142:771-773.
Millett GA, Ding H, Lauby J, et al. Circumcision status and HIV infection among Black and Latino men who have sex with men in 3 US cities. J Acquir Immune Defic Syndr. 2007;46(5):643-650.
Mishra V, Assche SB, Greener R, et al. HIV infection does not disproportionately affect the poorer in sub-Saharan Africa. AIDS. 2007;21(Suppl 7):S17-S28.
Montaner JSG, Phillips P, Montressori V. Principles of management in the developed world. In: Cohen J, Powderly WG, Berkley SF, et al, editors. Infectious diseases. 2nd ed. Edinburgh: Mosby; 2004:1379-1386.
Moscicki AB, Ma Y, Holland C, Vermund SH. Cervical ectopy in adolescent girls with and without human immunodeficiency virus infection. J Infect Dis. 2001;183(6):865-870.
Moses S, Nagelkerke NJ, Blanchard J. Analysis of the scientific literature on male circumcision and risk for HIV infection. Int J STD AIDS. 1999;10(9):626-628.
Mostad SB, Kreiss JK, Ryncarz AJ, et al. Cervical shedding of herpes simplex virus in human immunodeficiency virus-infected women: effects of hormonal contraception, pregnancy, and vitamin A deficiency. J Infect Dis. 2000;181(1):58-63.
Musey L, Hughes J, Schacker T, et al. Cytotoxic-T-cell responses, viral load, and disease progression in early human immunodeficiency virus type 1 infection. N Engl J Med. 1997;337(18):1267-1274.
Musey LK, Krieger JN, Hughes JP, et al. Early and persistent human immunodeficiency virus type 1 (HIV-1)-specific T helper dysfunction in blood and lymph nodes following acute HIV-1 infection. J Infect Dis. 1999;180(2):278-284.
Nadasdy T, Miller KW, Johnson LD, et al. Is cytomegalovirus associated with renal disease in AIDS patients? Mod Pathol. 1992;5(3):277-282.
Nadler RB, Rubenstein JN, Eggener SE, et al. The etiology of urolithiasis in HIV infected patients. J Urol. 2003;169(2):475-477.
O’Brien WA, Hartigan PM, Martin D, et al. Changes in plasma HIV-1 RNA and CD4+ lymphocyte counts and the risk of progression to AIDS. Veterans Affairs Cooperative Study Group on AIDS. N Engl J Med. 1996;334(7):426-431.
O’Farrell N, Egger M. Circumcision in men and the prevention of HIV infection: a ‘meta-analysis’ revisited. Int J STD AIDS. 2000;11(3):137-142.
Orroth KK, Freeman EE, Bakker R, et al. Understanding the differences between contrasting HIV epidemics in east and west Africa: results from a simulation model of the Four Cities Study. Sex Transm Infect. 2007;83(Suppl 1):15-16.
Pantaleo G, Cohen OJ, Schacker T, et al. Evolutionary pattern of human immunodeficiency virus (HIV) replication and distribution in lymph nodes following primary infection: implications for antiviral therapy. Nat Med. 1998;4(3):341-345.
Pantaleo G, Demarest JF, Soudeyns H, et al. Major expansion of CD8+ T cells with a predominant V beta usage during the primary immune response to HIV. Nature. 1994;370(6489):463-467.
Pantaleo G, Graziosi C, Demarest JF, et al. HIV infection is active and progressive in lymphoid tissue during the clinically latent stage of disease. Nature. 1993;362(6418):355-358.
Pantaleo G, Graziosi C, Demarest JF, et al. Role of lymphoid organs in the pathogenesis of human immunodeficiency virus (HIV) infection. Immunol Rev. 1994;140:105-130.
Patterson BK, Landay A, Andersson J, et al. Repertoire of chemokine receptor expression in the female genital tract: implications for human immunodeficiency virus transmission. Am J Pathol. 1998;153(2):481-490.
Patterson BK, Landay A, Siegel JN, et al. Susceptibility to human immunodeficiency virus-1 infection of human foreskin and cervical tissue grown in explant culture. Am J Pathol. 2002;161(3):867-873.
Perelson AS, Essunger P, Cao Y, et al. Decay characteristics of HIV-1–infected compartments during combination therapy. Nature. 1997;387(6629):188-191.
Pierre-Alexandre B, Pantaleo G. Pathogenesis: immunopathogenesis of HIV-1 infection. In: Cohen J, Powderly WG, Berkley SF, et al, editors. Infectious diseases. 2nd ed. Edinburgh: Mosby; 2004:1235-1249.
Pollard RB, Onorato M. Immunobased therapies. In: Cohen J, Powderly WG, Berkley SF, et al, editors. Infectious diseases. 2nd ed. Edinburgh: Mosby; 2004:1399-1403.
Pollard VW, Malim MH. The HIV-1 Rev protein. Annu Rev Microbiol. 1998;52:491-532.
Pudney J, Anderson D. Orchitis and human immunodeficiency virus type 1 infected cells in reproductive tissues from men with the acquired immune deficiency syndrome. Am J Pathol. 1991;139(1):149-160.
Quinn TC. HIV epidemiology and the effects of antiviral therapy on long-term consequences. AIDS. 2008;22(Suppl 3):S7-12.
Quinn TC, Glasser D, Cannon RO, et al. Human immunodeficiency virus infection among patients attending clinics for sexually transmitted diseases. N Engl J Med. 1988;318(4):197-203.
Quinn TC, Wawer MJ, Sewankambo N, et al. Viral load and heterosexual transmission of human immunodeficiency virus type 1. Rakai Project Study Group. N Engl J Med. 2000;342(13):921-929.
Raboud JM, Rae S, Hogg RS, et al. Suppression of plasma virus load below the detection limit of a human immunodeficiency virus kit is associated with longer virologic response than suppression below the limit of quantitation. J Infect Dis. 1999;180(4):1347-1350.
Randazzo RF, Hulette CM, Gottlieb MS, Rajfer J. Cytomegaloviral epididymitis in a patient with the acquired immune deficiency syndrome. J Urol. 1986;136:1095-1097.
Rao TK, Filippone EJ, Nicastri AD, et al. Associated focal and segmental glomerulosclerosis in the acquired immunodeficiency syndrome. N Engl J Med. 1984;310(11):669-673.
Reiss P, Boucher C, Vella S. The role of resistance testing. In: Cohen J, Powderly WG, Berkley SF, et al, editors. Infectious diseases. 2nd ed. Edinburgh: Mosby; 2004:1404-1405.
Reiter WJ, Schon-Pernerstorfer H, Dorfinger K, et al. Frequency of urolithiasis in individuals seropositive for human immunodeficiency virus treated with indinavir is higher than previously assumed. J Urol. 1999;161(4):1082-1084.
Reynolds SJ, Shepherd ME, Risbud AR, et al. Male circumcision and risk of HIV-1 and other sexually transmitted infections in India. Lancet. 2004;363(9414):1039-1040.
Rosenberg ES, Billingsley JM, Caliendo AM, et al. Vigorous HIV-1-specific CD4+ T cell responses associated with control of viremia. Science. 1997;278(5342):1447-1450.
Ross MJ, Klotman PE. HIV-associated nephropathy. AIDS. 2004;18(8):1089-1099.
Royce RA, Sena A, Cates WJr, Cohen MS. Sexual transmission of HIV. N Engl J Med. 1997;336(15):1072-1078.
Schwartz BF, Schenkman N, Armenakas NA, Stoller ML. Imaging characteristics of indinavir calculi. J Urol. 1999;161(4):1085-1087.
Sgadari C, Barillari G, Toschi E, et al. HIV protease inhibitors are potent anti-angiogenic molecules and promote regression of Kaposi sarcoma. Nat Med. 2002;8(3):225-232.
Shaffer DN, Bautista CT, Sateren WB, et al. The protective effect of circumcision on HIV incidence in rural low-risk men circumcised predominantly by traditional circumcisers in Kenya: two-year follow-up of the Kericho HIV Cohort Study. J Acquir Immune Defic Syndr. 2007;45(4):371-379.
Shahinian V, Rajaraman S, Borucki M, et al. Prevalence of HIV-associated nephropathy in autopsies of HIV-infected patients. Am J Kidney Dis. 2000;35(5):884-888.
Shevchuk M, de Silza M, Armenakas N, et al. The male genital tract in AIDS. J Urol. 1989;141:354A. [abstract 738, Eighty-fourth annual meeting of the American Urological Association]
Shevchuk MM, Pigato JB, Khalife G, et al. Changing testicular histology in AIDS: its implication for sexual transmission of HIV. Urology. 1999;53(1):203-208.
Shiver JW, Fu TM, Chen L, et al. Replication-incompetent adenoviral vaccine vector elicits effective anti-immunodeficiency-virus immunity. Nature. 2002;415(6869):331-335.
Siegfried N. Does male circumcision prevent HIV infection? PLoS Med. 2005;2(11):e393.
Siegfried N, Muller M, Volmink J, et al. Male circumcision for prevention of heterosexual acquisition of HIV in men. Cochrane Database Syst Rev 2003(3):CD003362.
Soilleux EJ, Coleman N. Expression of DC-SIGN in human foreskin may facilitate sexual transmission of HIV. J Clin Pathol. 2004;57(1):77-78.
Solomon G, Brancato L, Winchester R. An approach to the human immunodeficiency virus–positive patient with a spondyloarthropathic disease. Rheum Dis Clin North Am. 1991;17(1):43-58.
Soudeyns H, Pantaleo G. The moving target: mechanisms of HIV persistence during primary infection. Immunol Today. 1999;20(10):446-450.
Spira AI, Marx PA, Patterson BK, et al. Cellular targets of infection and route of viral dissemination after an intravaginal inoculation of simian immunodeficiency virus into rhesus macaques. J Exp Med. 1996;183(1):215-225.
Stamm W, Handsfield H, Rompalo A, et al. The association between genital ulcer disease and acquisition of HIV infection in homosexual men. JAMA. 1988;260:1429-1433.
Steinbrook R. The AIDS epidemic in 2004. N Engl J Med. 2004;351:115-117.
Steinman RM. DC-SIGN: a guide to some mysteries of dendritic cells. Cell. 2000;100(5):491-494.
Swartz DA, Harrington P, Wilcox R. Candidal epididymitis treated with ketoconazole. N Engl J Med. 1994;319:1485.
Talukdar A, Khandokar MR, Bandopadhyay SK, Detels R. Risk of HIV infection but not other sexually transmitted diseases is lower among homeless Muslim men in Kolkata. AIDS. 2007;21(16):2231-2235. 18
Timmerman JM, Northfelt DW, Small EJ. Malignant germ cell tumors in men infected with the human immunodeficiency virus: natural history and results of therapy. J Clin Oncol. 1995;13(6):1391-1397.
Tirelli U, Vaccher E. Neoplastic disease. In: Cohen J, Powderly WG, Berkley SF, et al, editors. Infectious diseases. 2nd ed. Edinburgh: Mosby; 2004:1313-1317.
Trauzzi SJ, Kay CJ, Kaufman DG, Lowe FC. Management of prostatic abscess in patients with human immunodeficiency syndrome. Urology. 1994;43(5):629-633.
Travers GR, Huybers M. Neurological complications in acquired immune deficiency syndrome. Axone. 1987;8(4):107-111.
UNAIDS/WHO. AIDS epidemic update. Geneva: UNAIDS/WHO; 2007.
UNAIDS/WHO. Report on the global HIV/AIDS epidemic. Geneva: UNAIDS/WHO; 2008.
Van de Perre P, Clumeck N, Carael M, et al. Female prostitutes: a risk group for infection with human T-cell lymphotropic virus type III. Lancet. 1985;2(8454):524-527.
Van de Perre P, Segondy M, Foulongne V, et al. Herpes simplex virus and HIV-1: deciphering viral synergy. Lancet Infect Dis. 2008;8(8):490-497.
Van Howe RS. Circumcision and HIV infection: review of the literature and meta-analysis. Int J STD AIDS. 1999;10(1):8-16.
Varghese B, Maher JE, Peterman TA, et al. Reducing the risk of sexual HIV transmission: quantifying the per-act risk for HIV on the basis of choice of partner, sex act, and condom use. Sex Transm Dis. 2002;29(1):38-43.
Vella S, Floridia M. HIV therapy: antiviral therapy. In: Cohen J, Powderly WG, Berkley SF, et al, editors. Infectious diseases. 2nd ed. Edinburgh: Mosby; 2004:1387-1398.
Vernazza PL, Eron JJJr. Probability of heterosexual transmission of HIV. J Acquir Immune Defic Syndr Hum Retrovirol. 1997;14(1):85-86.
Ward JW, Deppe DA, Samson S, et al. Risk of human immunodeficiency virus infection from blood donors who later developed the acquired immunodeficiency syndrome. Ann Intern Med. 1987;106(1):61-62.
Watson-Jones D, Weiss HA, Rusizoka M, et al. Effect of herpes simplex suppression on incidence of HIV among women in Tanzania. N Engl J Med. 2008;358(15):1560-1571.
Wawer MJ, Sewankambo NK, Serwadda D, et al. Control of sexually transmitted diseases for AIDS prevention in Uganda: a randomised community trial. Rakai Project Study Group. Lancet. 1999;353(9152):525-535.
Weiss HA, Halpernr D, Bailey RC, et al. Male circumcision for HIV prevention: from evidence to action? AIDS. 2008;22(5):567-574.
Weiss HA, Quigley MA, Hayes RJ. Male circumcision and risk of HIV infection in sub-Saharan Africa: a systematic review and meta-analysis. AIDS. 2000;14(15):2361-2370.
Weiss SG2nd, Kryger JV, Nakada SY, Uehling DT. Genitourinary tuberculosis. Urology. 1998;51(6):1033-1034.
Wilkin TJ, Gulick RM. When to start antiretroviral therapy? Clin Infect Dis. 2008;47(12):1580-1586.
Wilkinson M, Carroll PR. Testicular carcinoma in patients positive and at risk for human immunodeficiency virus. J Urol. 1990;144:1157-1159.
Williams BG, Lloyd-Smith JO, Gouws E, et al. The potential impact of male circumcision on HIV in sub-Saharan Africa. PLoS Med. 2006;3(7):e262.
Wilson WT, Frenkel E, Vuitch F, Sagalowsky AI. Testicular tumors in men with human immunodeficiency virus. J Urol. 1992;147(4):1038-1040.
Winston J, Deray G, Hawkins T, et al. Kidney disease in patients with HIV infection and AIDS. Clin Infect Dis. 2008;47(11):1449-1457.
Wong JK, Hezareh M, Gunthard HF, et al. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science. 1997;278(5341):1291-1295.
World Health Organization. The world health report 2004—changing history. http://wwwwhoint/whr, 2004. [accessed May 2004]
Zhu T, Wang N, Carr A, et al. Genetic characterization of human immunodeficiency virus type 1 in blood and genital secretions: evidence for viral compartmentalization and selection during sexual transmission. J Virol. 1996;70(5):3098-3107.