chapter 95 Epidemiology, Etiology, and Prevention of Prostate Cancer
Epidemiology
Incidence and Mortality Trends
Incidence
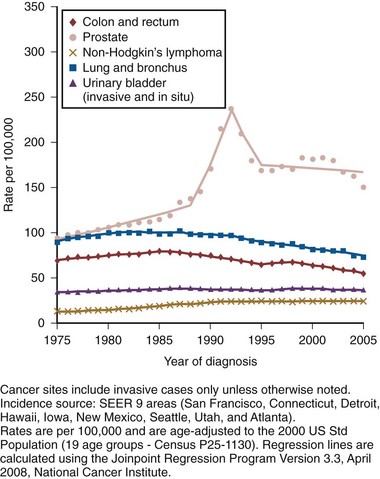
Prostate cancer has been the most common noncutaneous malignancy in U.S. men since 1984, now accounting for one quarter of all such cancers (American Cancer Society, 2008). The estimated lifetime risk of disease is 16.72%, with a lifetime risk of death at 2.57%. Prostate cancer incidence varies by race/ethnicity, with African-Americans at highest risk (Table 95–1). The incidence of prostate cancer peaked in 1992 (approximately 5 years after introduction of the prostate-specific antigen [PSA] screening test), fell precipitously until 1995, increased slowly until 1995 at a slope similar to that observed prior to the PSA era, and has decreased again in recent years (Fig. 95–1). The years in which peak rates were observed can be differentiated by race: 1992 for whites (237.8 per 100,000 men) and 1993 for African-Americans in (343.1 per 100,000). The decrease in incidence between 1992 and 1995 has been attributed to the “cull effect” of identifying previously unknown cancers in the population by the use of the PSA test, followed by a return to baseline where fewer cases were detected in previously screened individuals (Stephenson et al, 1996). For 2008, the American Cancer Society estimated 186,320 new cases of prostate cancer in the United States (American Cancer Society, 2008).
Table 95–1 Prostate Cancer Incidence and Mortality by Race/Ethnicity, United States, 2000–2004
| INCIDENCE* | MORTALITY* | |
|---|---|---|
| White | 161.4 | 25.6 |
| African-American | 255.5 | 62.3 |
| Hispanic/Latino | 140.8 | 21.2 |
| Asian-American and Pacific Islander | 96.5 | 11.3 |
| American Indian and Alaska Native | 68.2 | 21.5 |
* Per 100,000, age adjusted to the 2000 U.S. standard population.
Data from American Cancer Society. Cancer facts and figures 2008. <http://www.cancer.org/acs/groups/content/@nho/documents/document/2008cafffinalsecuredpdf.pdf>; 2008 [accessed 06.04.11].
Mortality
Prostate cancer mortality rates in the United States rose slowly between 1973 and 1990 (Fig. 95–2). This may have resulted from a gradual increase in the number of biologically lethal cancers or a decreasing use or effectiveness of therapy during this interval. In the early 1990s, an abrupt rise in mortality was observed. This increase may have been caused by an increase in attribution bias occurring when the National Center for Health Statistics made a change from manual to automated methods for assignment of cause of death (Feuer et al, 1999). Subsequent to 1991, the peak mortality year, steady declines in prostate cancer mortality were reported. The magnitude of this decline is nearly 2.5 times larger than the increase in mortality seen as a result of attribution bias; thus it seems likely that the declines in prostate cancer mortality in the United States since 1991 are real and clinically significant (Stephenson, 2005). In 2008, the American Cancer Society estimated 28,660 prostate cancer–related deaths in the United States, for an approximate annual rate of 23.3 per 100,000 population, representing a 41% decrease from the peak in 1991 (American Cancer Society, 2008). Furthermore, the mortality rate for prostate cancer in white men in the United States has declined to a level lower than that observed prior to the introduction of PSA-based screening in 1987 (Tarone et al, 2000).
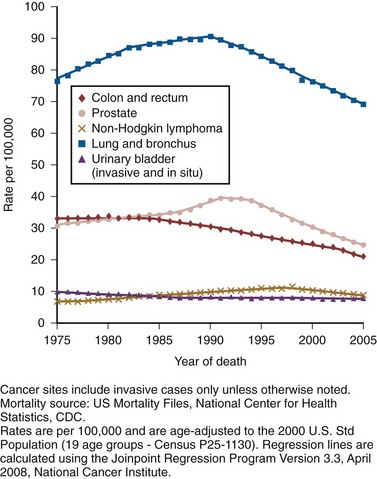
Figure 95–2 Age-adjusted cancer death rates for men, United States, 1975-2005.
(From Surveillance, Epidemiology, and End Results Program, 1975-2001, Division of Cancer Control and Population Sciences, National Cancer Institute, 2008.)
The observed decline in mortality since 1991 is unlikely to be explained by PSA screening alone. Based on our current beliefs concerning lead time, the decline would have occurred too soon after the initiation of screening (Etzioni et al, 1999). One hypothesis is that it is the result of the more aggressive treatment of prostate cancer that began in the 1980s (Walsh, 2000). Rates of both radical prostatectomy (RP) and radiation therapy rose steadily through the 1980s (pre-PSA era), whereas hormone therapy and no-treatment rates remained stable (Stephenson, 2005). Outcomes for patients treated in the 1980s should be reflected in the mortality data of the 1990s, while outcomes for patients treated in the PSA era (the 1990s) have had less time to affect recent mortality data. Given the long natural history of low-stage cancers detected in the PSA era, their treatment would not be expected to have a substantial effect on mortality statistics for another 5 to 10 years. Additional observation time is necessary to determine if screening, PSA-induced stage migration, and more aggressive use of therapy have contributed to declining mortality.
Racial Differences
Although anthropologists accept that there are subtle biologic differences among populations, commonly used categories, such as African-American, white, and Hispanic, are social and cultural descriptors that have no defined biologic basis. Observed disease-related differences between groups defined in this fashion may therefore be more reflective of common environmental exposure, diet, lifestyle, and attitudes toward health care than of differences in genetic structure or function. Recognizing these caveats, it is noteworthy that African-American men have the highest reported incidence of prostate cancer in the world, with a relative incidence of 1.6 compared with white men in the United States (American Cancer Society, 2008). Although African-Americans have experienced a greater decline in mortality than white men since the early 1990s, their death rates remain more than 2.4 times higher than whites. A recent meta-analysis quantifies this difference, demonstrating a higher risk of biochemical recurrence (risk ratio = 1.34, 95% confidence interval [CI] = 1.23 to 1.46), prostate cancer–specific death (risk ratio = 1.29, 95% CI = 1.13 to 1.47), and all-cause mortality (risk ratio = 1.35, 95% CI = 1.23 to 1.48) (Evans et al, 2008). These differences were not fully explained by differences in comorbidity, PSA screening, or access to health care. One study that analyzed Medicare data found shorter survival intervals for African-Americans: 1.8 years shorter with localized disease treated by RP, 0.7 years shorter after radiation, and 1 year shorter in those choosing watchful waiting, findings that persist after adjusting for other covariates, including education and income levels (Godley et al, 2003).
Many biologic, environmental, and social hypotheses have been advanced to explain these differences, ranging from postulated differences in genetic predisposition; differences in mechanisms of tumor initiation, promotion, and/or progression; higher fat diets, higher serum testosterone levels, or higher body mass index; structural, financial, and cultural barriers to screening, early detection, and aggressive therapy; and physician bias. Differences in screening rates between whites and African-Americans may play a role in explaining the differences in mortality, although the 2005 National Health Interview Survey demonstrated that among men ages 40 to 49 years, African-American men were more likely to have had a PSA test than white men, and there were no significant differences in PSA screening rates by race/ethnicity in men ages 50 to 79 years (Ross et al, 2008). Studies have consistently shown that African-American men are more likely to receive androgen-deprivation therapy, expectant management or external beam radiation therapy, and are less likely to undergo radical prostatectomy compared with white men (Underwood et al, 2005). Even among those treated with “watchful waiting,” African- American men may receive less intensive follow-up (Shavers et al, 2004). There are currently no data that clearly indicate any of these hypotheses are the determinants of the observed differences in incidence or mortality, and it seems likely that the source of the disparity is multifactorial. Recent observations suggest that the incidence of organ-confined disease at diagnosis among African-Americans is increasing, that the disparity in mortality is lessening in the PSA era, and that those with organ-confined disease can be cured at a high rate regardless of race (Powell et al, 2004; American Cancer Society, 2008).
The incidence of prostate cancer in other ethnic groups is lower than that of whites and African-Americans. Comparative data for prostate cancer–related incidence and mortality are now available for these groups (see Table 95–1).
Worldwide Incidence and Mortality
Incidence rates show that prostate cancer is the fifth most common malignancy worldwide and the second most common in men (Parkin et al, 2005). Prostate cancer makes up 11.7% of new cancer cases overall, 19% in developed countries, and 5.3% in developing countries. Its incidence varies widely between countries and ethnic populations, with disease rates differing by more than 100-fold. The lowest yearly incidence rates occur in Asia (1.9 cases per 100,000 in China) and the highest in North America and Scandinavia, especially in African-Americans (249 cases per 100,000) (Parkin et al, 2005; American Cancer Society, 2008). As in the United States, prostate cancer incidence has increased in many countries since the early 1990s, with the largest increases in high-risk countries (Hsing et al, 2000). Although much of the increase can be correlated with the introduction of PSA, some of the increase predates screening and is perhaps partly due to the increasing diagnosis of latent cancers following transurethral resection of the prostate (Potosky et al, 1990; Oliver et al, 2001). Large increases have also been noted in low-risk countries between 1973 and 1992 (e.g., 104% among Chinese in Singapore and 84% in Miyagi, Japan) (Hsing et al, 2000). Mortality also varies widely among countries, being highest in the Caribbean (28 per 100,000 per year) and lowest in Southeast Asia, China, and North Africa (<5 per 100,000 per year) (Parkin et al, 2005). Mortality rates increased slowly for most countries between 1985 and 1995 (Quinn and Babb, 2002). The CONCORD study, a worldwide population-based analysis of cancer survival in five continents, recently analyzed international differences in survival for breast, colorectal, and prostate cancer (Coleman et al, 2008). Using data from cancer registries, age-standardized 5-year survival rates were found to vary greatly, ranging from 80% or higher in the United States (92%), Australia, and Canada, to less than 40% in Denmark, Poland, and Algeria.
There are multiple potential explanations for the worldwide and ethnic variations in prostate cancer incidence and mortality. Access to and quality of health care, the accuracy of cancer registries, and the penetrance of PSA screening all affect how rates of disease are reported. Before reliable data were available from African countries, rates of prostate cancer in Africa were thought to be much the same as those in Asia. However, in Uganda and Nigeria, prostate cancer is very common, and in Nigeria it is the most common cancer in men (Gronberg, 2003). Interestingly, there has been a more marked decline in prostate cancer mortality in countries with a higher uptake of PSA screening when compared with countries where routine screening is not widely performed (Collin et al, 2008). Possible explanations for the observed mortality differences include international variability in treatment approaches and bias related to the misattribution of cause of death.
Environment also plays an important role in modulating prostate cancer risk around the world. Japanese and Chinese men in the United States have a higher risk of developing and dying from prostate cancer than do their relatives in Japan and China (Muir et al, 1991; Shimizu et al, 1991). Likewise, prostate cancer incidence and mortality have increased in Japan as the country has become more Westernized (Hsing et al, 2000). It is important to note, however, that Asian-Americans have a lower prostate cancer incidence than white or African-American men, indicating that genetics still plays a role in determining prostate cancer predisposition.
Age at Diagnosis
Prostate cancer is rarely diagnosed in men younger than 50 years old, accounting for only 2% of all cases (Jani et al, 2008). The median age at diagnosis is 68 years, with 63% diagnosed after age 65 (Ries et al, 2011). At 85 years of age, the cumulative risk of clinically diagnosed prostate cancer ranges from 0.5% to 20% worldwide, despite autopsy evidence of microscopic lesions in approximately 30% of men in the fourth decade, 50% of men in the sixth decade, and more than 75% of men older than 85 years (Sakr et al, 1993; Gronberg, 2003). PSA-based screening has induced an important age migration effect; the incidence of prostate cancer in men 50 to 59 years of age has increased by 50% between 1989 and 1992 (Hankey et al, 1999), with important implications for deciding on the need for, type of, and complications after therapy.
Stage at Diagnosis
In addition to changes in prostate cancer incidence and mortality over the last several decades, there has been a substantial shift to more favorable stage at presentation in men with newly diagnosed disease. This clinical stage migration is largely if not exclusively accounted for by PSA screening (Catalona et al, 1993; Mettlin et al, 1993). Since the introduction of PSA testing, the incidence of local-regional disease has increased, whereas the incidence of metastatic disease has decreased (Newcomer et al, 1997). Nonpalpable cancers (American Joint Committee on Cancer [AJCC] clinical stage T1c) now account for 60% to 75% of newly diagnosed disease (Derweesh et al, 2004; Gallina et al, 2008). Clinical stage migration has also been associated with improvements in 5- and 10-year disease-specific survival, which for all stages combined now are 99% and 91%, respectively (American Cancer Society, 2008).
The use of PSA has also resulted in a substantial downward pathologic stage migration as evidenced by an increase in the proportion of patients with organ-confined disease (Catalona et al, 1993; Jhaveri et al, 1999; Derweesh et al, 2004;) and a decrease in the proportion with seminal vesicle involvement (Gallina et al, 2008). These observations have been consistent across the United States and Europe (Noldus et al, 2000; Gallina et al, 2008). Since 1995, however, a slowing in this trend has been observed, suggesting a diminishing effect of PSA screening on pathologic stage migration (Fig. 95–3) (Dong et al, 2007b). The improvement in pathologic stage has been seen for clinical stages T1-T3 tumors and all tumor grades (Fig. 95–4) and has resulted in improved cancer-specific survival after external radiation or surgery for patients treated late in the PSA era (Jhaveri et al, 1999; Derweesh et al, 2004; Kupelian et al; 2005).
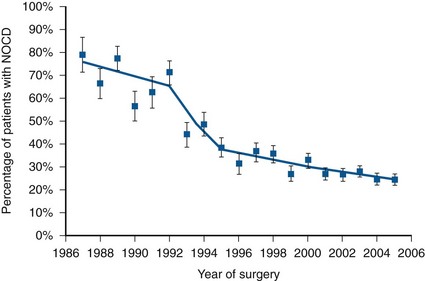
Figure 95–3 Declining rate of extracapsular extension (resulting in increased rate of organ-confined disease) on radical prostatectomy specimens at the Cleveland Clinic, 1987-2005. Trends in pathologic stage migration with joinpoint regression analysis. Annual change: 1987 to 1992: −2.9%; 1992 to 1995: −16.9%; 1995 to 2005: −4.2%. NOCD, non–organ-confined disease.
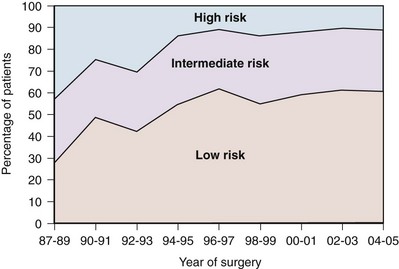
Figure 95–4 Risk stratification of surgical population over time. Low risk: prostate-specific antigen (PSA) less than 10, and stage T1 or T2a, and biopsy Gleason score = 6 or lower. Intermediate risk: PSA 10 to 20, or stage T2b or T2c, or biopsy Gleason score = 7. High risk: PSA more than 20, or stage T3, or biopsy Gleason score = 8 or higher, or any two or more intermediate risk factors.
Effect of Screening on Mortality
Recently, the results of two large randomized trials assessing the effect of PSA screening on prostate cancer mortality were published. The first examined results from the Prostate, Lung, Colorectal, and Ovarian (PLCO) cancer screening trial and reported on 76,693 men aged 55 to 74 years at 10 U.S. centers receiving either annual screening or usual care (Andriole et al, 2009). After 7 years of follow-up, no difference in prostate cancer mortality was detected between the groups, with an incidence of 2.0 deaths per 10,000 person-years (50 deaths) in the screening group and 1.7 deaths per 10,000 person-years (44 deaths) in the control group (rate ratio = 1.13, 95% CI = 1.16 to 1.29). However, the trial has been criticized for its relatively short follow-up, given the long natural history for prostate cancer and because more than half the men in the control arm actually had a PSA test done outside of the trial. The second trial, the European Randomized Study of Screening for Prostate Cancer (ERSPC), included 162,243 men between the ages of 55 and 69 years randomized to either PSA screening every 4 years or no screening (Schroder et al, 2009). After a median follow-up of 9 years, screening reduced the rate of death from prostate cancer by 20% (rate ratio = 0.80, 95% CI = 0.65 to 0.98). However, they estimated that to prevent one prostate cancer death, one would need to screen 1410 men (95% CI = 1142 to 1721), and treat 48 additional cases of prostate cancer.
The results of these trials highlight two important points. First, overall, prostate cancer is an indolent disease with a very low cause-specific death rate and will only impact life expectancy in a minority of men. Second, the morbidity of treatment needs to be strongly considered when evaluating population screening. Prior to the release of the above-mentioned studies, an evidence update for the U.S. Preventative Task Force on the benefits and harms of PSA screening concluded that false-positive PSA test results cause psychologic adverse effects for up to 1 year and that the benefits of screening remain uncertain (Lin et al, 2008). On the other hand, in 2009 the American Urological Association (AUA) released a Best Practice Statement suggesting that early detection of and risk assessment for prostate cancer should continue to be offered to asymptomatic men 40 years of age or older who have a life expectancy of at least 10 years (American Urological Association, 2009).
Key Points
Epidemiology

Risk Factors
Although the specific causes of prostate cancer initiation and progression are not yet known, considerable evidence suggests that both genetics and environment play a role in the origin and evolution of this disease. Traditional and molecular epidemiology have identified a number of potential risk factors associated with the development of prostate cancer.
Familial and Genetic Influences
Ample epidemiologic evidence suggests that prostate cancer has both a familial and genetic component. The first reports of a familial clustering were published in the mid-20th century and suggested that the risk of developing prostate cancer was higher in those with an affected first-degree relative (Woolf, 1960). Subsequent case control and cohort studies have confirmed this observation (Eeles et al, 1997). Twin studies have also suggested a genetic component, with higher rates of concordance for monozygotic than dizygotic brothers (Gronberg et al, 1994; Ahlbom et al, 1997; Page et al, 1997). Results of a meta-analysis demonstrate that relative risk increases according to the number of affected family members, their degree of relatedness, and the age at which they were affected (Table 95–2) (Zeegers et al, 2003). A consistent finding is the increased risk when a brother is affected compared with when a father is affected. This is consistent with either X-linked or recessive genetic components (Monroe et al, 1995). However, the bias associated with the fact that brothers of affected men are more likely to seek prostate cancer screening may also explain the differential association.
Table 95–2 Family History and Risk of Prostate Cancer
| FAMILY HISTORY | RELATIVE RISK | 95% CONFIDENCE INTERVAL |
|---|---|---|
| None | 1 | |
| Father affected | 2.17 | 1.90-2.49 |
| Brother affected | 3.37 | 2.97-3.83 |
| First-degree family member affected, age less than 65 years at diagnosis | 3.34 | 2.64-4.23 |
| Greater than two first-degree relatives affected | 5.08 | 3.31-7.79 |
| Second-degree relative affected | 1.68 | 1.07-2.64 |
Data derived from meta-analysis assessing risk of prostate cancer for relatives of patients with prostate carcinoma (Zeegers, Jellema A, Ostrer H. Empiric risk of prostate carcinoma for relatives of patients with prostate carcinoma: a meta-analysis. Cancer 2003;97:1894–03).
For investigative purposes, prostate cancer may be conveniently divided into three phenotypes: sporadic, familial, and hereditary. Sporadic cancers occur in individuals with a negative family history. Familial prostate cancer is defined as cancer in a man with more than one affected relative. Hereditary prostate cancer is a subset of the familial form and has been operationally defined as nuclear families with more than three affected members, prostate cancer in three successive generations, or two affected individuals diagnosed with cancer before age 55 (Carter et al, 1993). Although most prostate cancer is likely to be polygenic in origin, the existence of true hereditary prostate cancer is suggested by three epidemiologic observations: (1) relatives of patients < 55 years old are at higher risk of getting prostate cancer than those with older affected relatives; (2) there is stronger familial clustering in families with early onset prostate cancer; and (3) the number of affected family members and their age at onset are the most important determinants of risk among relatives. Sporadic cancers account for about 85% of all prostate cancers and about 15% are familial and/or hereditary. Hereditary prostate cancer accounts for 43% of early onset disease (55 years of age or younger) but only 9% of all cancers that occur by 85 years of age (Carter et al, 1992).
A number of studies have suggested a familial coaggregation of prostate cancer with breast cancer (Thiessen, 1974; Tulinius et al, 1992; Goldgar et al, 1994). The two major susceptibility genes in breast cancer, BRCA1 (17q21) and BRCA2 (13q12), have also been studied in prostate cancer. Two large epidemiologic studies have estimated the risk of prostate cancer in men who are carriers of specific BRCA1 founder mutations to be double that of noncarriers, with a cumulative risk of 30% by 80 years of age (Struewing et al, 1997; Warner et al, 1999). Moreover, the BRCA2 gene has provided consistent association with an increased risk of prostate cancer, estimated at 5- to 7-fold, and may play a more important role in early onset disease (Breast Cancer Linkage Consortium, 1999; Edwards et al, 2003). However, due to their rarity, these mutations only contribute to a small fraction of hereditary prostate cancer cases.
Linkage studies have identified a number of candidate prostate cancer susceptibility genes, including RNaseL (hereditary prostate cancer-1 [HPC1] region, 1q23-25) (Carpten et al, 2002), ELAC2 (HPC2 region, 17p) (Tavtigian et al, 2001), and MSR1 (8p22-23) (Xu et al, 2002). Segregation studies have also suggested the existence of prostate cancer susceptibility loci on chromosomes 1q42.2-43 (PCAP) (Berthon et al, 1998), 1p36 (CAPB, also linked to brain tumors) (Gibbs et al, 1999), and Xq27-28 (Xu et al, 1998), but the gene or genes linked to these regions have not been identified. Many other genetic loci and prostate cancer susceptibility genes have been more recently identified, and the list is continually growing (Online Mendelian Inheritance in Man, 2009).
More recently, genome-wide association studies (GWAS) have emerged as a new approach to indentify alleles associated with prostate cancer without prior knowledge of position or function (Wellcome Trust Case Control Consortium, 2007). Genotype frequencies are compared among cases and controls at DNA sequence variations called single nucleotide polymorphisms (SNPs). Using this technique, studies have demonstrated SNPs on 8q24 and 17q, as well as on chromosomes 3, 6, 7, 10, 11, 19, and X, to be associated with prostate cancer risk (Amundadottir et al, 2006; Gudmundsson et al, 2007; Eeles et al, 2008). A worldwide consortium of 13 groups was recently formed to compile the large series necessary to provide accurate estimates of the risks associated with genetic variants and to evaluate the combined association of these variants (Kote-Jarai et al, 2008). The combined risks associated with each pair of SNPs were found to be consistent with a multiplicative risk model. In total, they determined that the 15 susceptibility variants studied only explained 16% of the familial risk of prostate cancer. This indicates that, to date, only a small piece of the prostate cancer genetic puzzle has been discovered, and with the continued improvement in genetic technology, it is likely that the number of known susceptibility genes will increase. A commercially available genetic test, which assesses nine genetic variants associated with prostate cancer risk, is currently offered to the general public (deCODE Genetics, Reykjavík, Iceland). Although the test is being marketed as a preventative tool, its clinical utility in the general population requires further study.
Of the known susceptibility genes, HPC1 is the best characterized. The existence of HPC1 was suggested by a genome-wide scan of families with hereditary prostate cancer (Smith et al, 1996) and later confirmed by linkage studies (Cooney et al, 1997; Eeles et al, 1998). A recent genome-wide association screen also identified an association between this locus and sporadic prostate cancer (Nam et al, 2008). HPC1 was mapped to the human gene that encodes for the antiviral and pro-apoptotic enzyme RNaseL (Carpten et al, 2002). RNaseL is part of the innate immune system that responds to a pathogen-associated molecular pattern (PAMP) to induce degradation of viral and cellular RNAs, thereby blocking viral infections (Klein and Silverman, 2008). It is the terminal enzyme of the 2-5A synthetase family, an RNA degradation pathway that plays an important role in mediating the biologic effects of interferons, especially in response to viral infection. 2-5A binds with high affinity to RNaseL, converting it from its inactive form as a monomer to a potent dimer that degrades single-stranded RNA, thus preventing viral replication, interfering with protein synthesis, and causing caspase-mediated apoptosis (Fig. 95–5) (Silverman, 2003). RNaseL knockout mice are more prone to viral infections (Silverman, 2003).
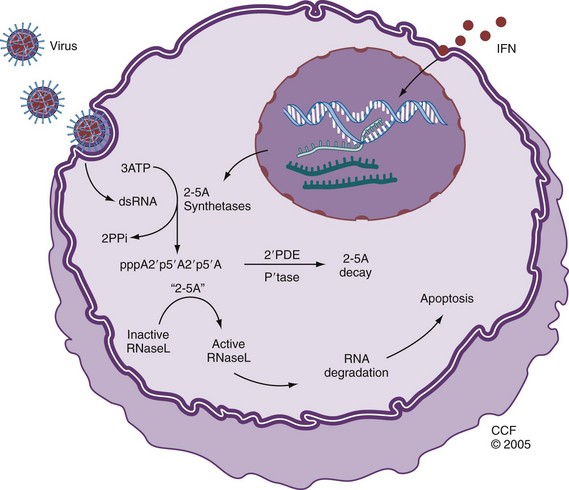
Figure 95–5 Cellular effects of RNaseL. In response to interferon (IFN) and viral infection, a family of oligoadenylate synthetases are activated, resulting in the conversion of ATP to a series of short 2′- to 5′- linked oligoadenylates (2-5A). 2-5A binds with high affinity to RNaseL, converting it from its inactive form as a monomer to a potent dimer that degrades ssRNA and causes caspase-mediated apoptosis of the host cell. Prostate cancer cells deficient in RNaseL are resistant to apoptosis.
(Reprinted with permission, Cleveland Clinic Center for Medical Art & Photography © 2005-2011. All Rights Reserved.)
A variety of inactivating and missense mutations of RNaseL have been identified in families with hereditary prostate cancer (Xiang et al, 2003). Of these, SNP R462Q, resulting from an arginine to glutamine substitution, has been shown to be associated with an increased risk of prostate cancer (Casey et al, 2002); men and cell lines with this allelic variant have been shown to have reduced RNaseL activity leading to deficient apoptosis (Carpten et al, 2002; Xiang et al, 2003; Malathi et al, 2004), presumed to lead to an accumulation of genetic defects ultimately resulting in cancer formation.
The epidemiologic data suggest that HPC1 is a rare autosomal dominant gene that has high penetrance, meaning that while it does not account for many prostate cancers, an individual carrier is highly likely to be affected by prostate cancer. Although cancers with specific mutations of the HPC1/RNaseL gene have been reported to present with higher-grade and more advanced stages (Gronberg et al, 1997; Goode et al, 2001; Rennert et al, 2005), other mutations have not shown such an association (Larson et al, 2008).
The biologic functions of the other known susceptibility genes fall into several classes. As with other malignancies, these genes include mediators of inflammation (e.g., MIC1, MSR1), antioxidant enzymes (e.g., MSR1, PON1), DNA repair (e.g., OGG, CHEK2, and BRCA2), susceptibility to infection (e.g., MSR1, RNaseL, and TLR4), and apoptosis (e.g., RNaseL).
Key Points
Risk Factors
Inflammation, Infection, and Genetic Susceptibility
It is estimated that infections cause almost 20% of all cancers worldwide (American Cancer Society, 2008). Chronic inflammation leading to cellular hyperproliferation to replace damaged tissue contributes to the development of infection-associated cancers of the colon, esophagus, stomach, bladder, and liver (Coussens and Werb, 2002; De Marzo et al, 2007). Accumulating epidemiologic, histologic, and genetic evidence suggests that a similar process may underlie the development of prostate cancer.
Some evidence suggests that prostate cancer may have an infectious etiology. Two meta-analyses examining 34 case-control studies reported statistically significant associations of prostate cancer with a history of sexually transmitted infection (RR = 1.4) or prostatitis (OR = 1.57) (Dennis et al, 2002b; Dennis and Dawson, 2002). Supportive evidence is provided by studies demonstrating positive associations of antibodies against syphilis, human papillomavirus (HPV), and human herpesvirus-8 (HHV-8) with prostate cancer (De Marzo et al, 2007). Case-only or case-control studies have also reported higher plasma concentrations of acute-phase reactants and proinflammatory cytokines in men with prostate cancer (De Marzo et al, 2007). Two studies have demonstrated evidence of viral pathogens in human prostate tissue, including HPV, human herpes simplex virus type 2 (HSV2), cytomegalovirus (CMV), and HHV-8 (Strickler and Goedert, 2001; Zambrano et al, 2002; Samanta et al, 2003).
However, recent studies assessing the association between infection and prostate cancer have shown mixed results. In the Health Professionals Follow-up Study (Sutcliffe et al, 2006), a prospective study of 51,529 American male health professionals aged 40 to 75 years, no association was found between a self-reported history of gonorrhea or syphilis and prostate cancer, although the incidence of sexually transmitted infections was very low in this population. In addition, no overall correlation between prostate cancer and clinical prostatitis was found. Similarly in this cohort, no associations were observed between Chlamydia trachomatis, HPV-16, HPV-18, and HPV-33 antibody seropositivity and prostate cancer (Sutcliffe et al, 2007a). Conversely, in a small case-control study in African-American men (Sarma et al, 2006), a history of gonorrhea or prostatitis increased the odds of prostate cancer by 1.78-fold (95% CI = 1.13 to 2.79) and 4.93-fold (95% CI = 2.79 to 8.74), respectively, even after adjusting for potential confounders. Furthermore, men reporting 25 or more sexual partners had 2.80 times the odds of being diagnosed with cancer (95% CI = 1.29 to 6.09) compared with men with five or fewer partners.
Inflammatory infiltrates and the histologic lesion called proliferative inflammatory atrophy (PIA) are frequent in clinical prostate specimens (De Marzo et al, 1999). PIA is a spectrum of lesions characterized by epithelial atrophy, low apoptotic index, and an increased proliferative index usually associated with inflammatory infiltrates (Putzi and De Marzo, 2000). Inflammation in PIA may include mononuclear infiltrates in the stroma and macrophages and/or neutrophils in the glandular lumen or epithelium. Macrophages activated by interferon-γ secrete proinflammatory cytokines and reactive nitrogen species (e.g., nitric oxide). Inducible nitric oxide synthase, which catalyzes the generation of nitric oxide, is overexpressed in macrophages in PIA but not in normal epithelium (Nelson et al, 2003). PIA appears to be a regenerative lesion appearing as a consequence of infection or cell trauma resulting from oxidant damage, hypoxia, infection, or autoimmunity, and its hyperproliferative state may lead to cancer. PIA is often found adjacent to high-grade prostatic intraepithelial neoplasia (HGPIN) or early cancer (Putzi and De Marzo, 2000), and there is an identifiable genetic pathway between PIA, HGPIN, and cancer (Shah et al, 2001; Nakayama et al, 2003; Nelson et al, 2003).
The previously described genetic and histologic observations in prostate cancer strongly suggest that compromised cellular defenses against inflammatory oxidants may initiate and/or perpetuate prostatic carcinogenesis. Oxidative stress is mediated by reactive oxygen and nitrogen species that bind DNA and cause mutations, and oxidant stresses from exogenous and endogenous sources are implicated in the accumulation of DNA damage that occurs with aging and subsequently leads to malignant change (Coussens and Werb, 2002). Cellular defense mechanisms against this process include front-line antioxidant enzymes, which scavenge reactive oxygen and nitrogen species and prevent mutations; DNA repair enzymes; and the ability to undergo apoptosis if the DNA damage is too severe to repair. A recent model proposes that inherited and acquired defects in cellular defense mechanisms against infection and oxidative stress allow prostate cancer to develop. This model suggests that, as in other inflammatory-mediated cancers, persistent infection leads to chronic inflammation, and that defects in antioxidant enzymes, DNA repair mechanisms, and apoptosis lead to cancer. Subsequent expression by tumor cells of α-methylacyl-CoA racemase (Kumar-Sinha et al, 2004), an enzyme that oxidizes branched-chain fatty acids from dietary sources, results in generation of hydrogen peroxide, which may contribute to continued oxidative stress, and tumor growth. This model is illustrated in Figure 95–6. In addition to providing a framework for further experimental study, this model gives a theoretical rationale for the use of antioxidants as chemopreventative agents (see later).
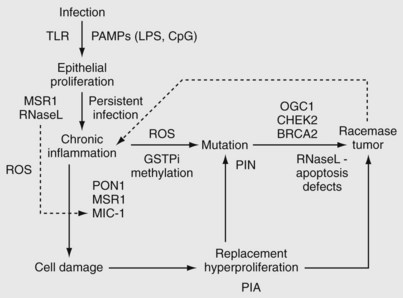
Figure 95–6 Model of prostate cancer pathogenesis related to infection, chronic inflammation, and defects in cellular defense against oxidative stress. Defects in Toll-like receptor (TLR) genes lead to immortalization of benign prostatic epithelium, and allelic variants of RNaseL and MSR1 allow persistent infection, leading to a proinflammatory cellular microenvironment. Defects in genes that control the intensity of the inflammatory response (MSR1 and MIC1, the latter of which is regulated by RNaseL) permit a robust inflammatory response. Defects in cellular defenses against the resultant oxidative stress, including deficiencies in antioxidant enzymes (allelic variants in PON1, methylation of GSTP1), DNA repair (OGG1, CHEK2, BRCA2), and apoptotic defects (RNaseL), lead to cancer through the histologic intermediates proliferative inflammatory atrophy (PIA) and prostatic intraepithelial neoplasia (PIN). Established tumors make racemase, an enzyme that converts fatty acids to free radicals, creating an autocrine growth loop based on perpetuation of oxidative stress. CpG, DNA CpG island repeats (representing viral genomes in this context); LPS, bacterial lipopolysaccarhide; PAMP, pathogen-associated molecular profile; ROS, reactive oxygen species.
Like cervical cancer, which is characterized by viral infection coincident with the onset of sexual activity, histologic evidence of preinvasive lesions, and a latency period of 15 to 20 years before the development of invasive cancer, the accumulating histologic, epidemiologic, and genetic evidence suggests the possibility that at least some prostate cancer cases have a viral pathogenesis. The epidemiologic data linking prostate cancer risk to prior infections and the association of a functionally important mutation in the antiviral HPC1/RNaseL gene support this hypothesis. Recently reported studies have identified a previously undescribed γ-retrovirus, xenotropic murine leukemia–related virus (XMRV), from prostate cancer tissue in men with the R462Q allelic variant of HPC1/RNaseL. The initial experiment demonstrated the presence of viral genomes for XMRV in prostate tumors in 8 of 20 patients with the R462Q SNP in RNaseL, but in only 1 of 62 wild-type or heterozygous patients (Urisman et al, 2006). XMRV infections were visualized by immunohistochemistry and fluorescence in-situ hybridizations in prostatic stromal cells adjacent to the tumor. A full-length, replication-competent, and infectious viral molecular clone of XMRV was subsequently cloned from prostate tissue cDNA and has been used for in-vitro studies (Dong et al, 2007a). In addition, XMRV integration sites have been mapped in host chromosomal DNA (Kim et al, 2008), suggesting that modulation of cancer-related gene function by XMRV could be important. The presence of XMRV in prostate tissue has been recently confirmed by two other groups, including evidence of expression of XMRV proteins in malignant prostatic epithelium using a rabbit polyclonal antibody (Fischer et al, 2008; Singh Laboratory, 2010). Together these observations suggest that XMRV infects and persists in the prostate when there is a deficiency in RNaseL activity, providing a proposed mechanism by which XMRV could be oncogenic in genetically susceptible individuals.
Other infectious agents have also been implicated in prostate cancer. BK virus (BKV), a member of the polyomavirus family, infects almost 90% of the human population by early childhood and resides in a subclinical persistent state in the urinary tracts of healthy individuals. BKV transforms rodent cells in culture, causes kidney tumors in transgenic mice, and immortalizes primary human cells alone or in the presence of other oncogenes such as c-RAS and adenovirus E1A (Das et al, 2008). The occurrence of mutations in TP53 and RB1 in the early stages of prostate cancer is relatively low, suggesting the possibility that a virus, such as BKV (encoding tumor antigens that inactivate these tumor suppressors), may play a role in the etiology of prostate cancer. BKV DNA has been demonstrated in epithelial cells of benign and PIA ducts of cancerous prostate specimens, and BKV large tumor antigen (Tag) expression has been observed specifically in atrophic epithelial cells, PIA, and prostatic intraepithelial neoplasia (PIN) but not in cancer nor in the epithelium of normal prostates (Das et al, 2004, 2008). These observations suggest a role for BKV in PIA and the early development of prostate cancer. Furthermore, in a study examining the bacterial flora of prostate cancer, Sfanos and Isaacs (2008) demonstrated the presence of 83 distinct microorganisms by sequencing bacterial 16S rDNA in core samples of 30 prostate cancers, most of which were not found by routine culture methods. If XMRV, BKV, or specific bacteria are proven to be oncogenic, their effects may be preventable by development of a protective vaccine.
Key Points
Inflammation, Infection, and Genetic Susceptibility
Molecular Epidemiology
Molecular epidemiologic approaches have identified many biomarkers of exposure measured in blood or other tissues and evaluated them in relation to incidence or mortality. These biomarkers capture aspects of diet, environmental contaminants, and factors for which concentrations are partly genetically determined. A brief survey of major molecular epidemiologic studies relating to prostate cancer is presented.
Gene Fusions
Gene fusions resulting from chromosomal translocations are the most common genetic alteration in human cancers (Futreal et al, 2004). These were previously thought to be the oncogenic mechanism exclusively limited to hematologic malignancies and sarcomas, as exemplified by the BCR-ABL1 fusion protein in chronic myeloid leukemia. In 2005, recurrent genomic rearrangements in prostate cancer were identified, resulting in the fusion of the 5′ untranslated end of TMPRSS2 (21q22.2, an androgen-responsive, prostate-specific, transmembrane serine protease gene) to members of the ETS family of oncogenic transcription factors (Tomlins et al, 2005). In over two dozen published reports using PSA-screened patient cohorts, the most common fusion identified in localized prostate cancer involves TMPRSS2 fused to ERG (ETS-related gene, 21q22.3) in approximately 50% of patients (Kumar-Sinha et al, 2008). However, this gene fusion may be less common (15%) in population-based cohorts (Demichelis et al, 2007). Gene fusions involving other members of the ETS family, most commonly ETV1 (ETS variant gene 1, 7q21.2), but also ETV4 and ETV5, are likely present in less than 10% of prostate cancer samples collectively (Kumar-Sinha et al, 2008).
The TMPRSS2 gene is prostate specific, and is expressed in both benign and malignant prostatic epithelium. TMPRSS2 expression has been shown to be induced by androgens in hormone-responsive prostate cancer cell lines (Lin et al, 1999). Therefore it is hypothesized that TMPRSS2 drives ETS gene overexpression in prostate cancers positive for TMPRSS2-ETS gene fusions (Fig. 95–7) (Kumar-Sinha et al, 2008). Although some studies have demonstrated an association between TMPRSS2-ERG with high pathologic stage (Mehra et al, 2007), higher recurrence rates (Nam et al, 2007), and prostate cancer–specific death (Demichelis et al, 2007), others have failed to report such clinical correlations. It is notable, however, that the presence of TRMPSS-ETS gene fusion was associated with a markedly higher risk of death in men managed by watchful waiting in one Swedish cohort (Demichelis et al, 2007).
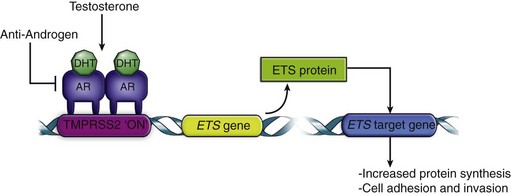
Figure 95–7 Role of gene fusion in prostate cancer. Androgen-mediated upregulation of ETS genes (for example, ERG or ETV1) leads to the activation of ETS target genes involved in increased protein synthesis, and cell adhesion and invasion. DHT, dihydrotestosterone.
Currently, the clinical application of this discovery is uncertain. Given that ETS-positive and ETS-negative tumors have distinct morphologic features, expression signatures, and potentially different clinical outcomes, some have proposed classifying prostate cancer as two categories: ETS gene fusion–positive and ETS gene fusion–negative disease (Kumar-Sinha et al, 2008). Because these gene fusions do not occur in benign prostate tissue, a sensitive and specific screening test based on their presence could be developed for the detection of prostate cancer. Eventually, gene fusion status may be used to decide the need for initial or repeat biopsy, for enrollment in therapeutic trials, or for surveillance after therapy. It also follows that targeting ERG activity in fusion-positive prostate cancers may provide an effective therapeutic approach for this subset of patients. Presently, attempts at identifying small molecular inhibitors of the ERG pathway that could serve as therapeutic agents are underway. At this time, the emerging consensus suggests that TRMPSS-related gene fusions are highly specific for the presence of prostate cancer, but their utility as prognostic markers requires further study.
Androgens
Androgens influence the development, maturation, and maintenance of the prostate, affecting both proliferation and differentiation of the luminal epithelium. There is little doubt that exposure of the prostate at key developmental times to androgens plays an important role in prostate carcinogenesis. Androgens are also important in the maintenance of established cancers, as supported by the historical observation that the majority of prostate cancers initially respond to androgen-deprivation therapy, and more recently by results of the Prostate Cancer Prevention Trial, which indicated that inhibition of the conversion of testosterone to the more potent dihydrotestosterone by finasteride reduces the incidence of prostate cancer by approximately 25% (Thompson et al, 2003). In addition, genetic polymorphisms of the androgen receptor (AR) (Balic et al, 2002; Taplin et al, 2003), the 5α-reductase type 2 isoenzyme (Makridakis and Reichardt, 2004), and the genes involved in biosynthesis of testosterone have also been implicated in prostate carcinogenesis (Chang et al, 2002).
High serum androgen levels have long been hypothesized to be a risk factor for prostate cancer. However, studies examining this association have been inconsistent, with some studies finding an association between specific hormones and prostate cancer risk. Recently, a collaborative group performed a pooled analysis of 18 prospective studies assessing this association using data from 3886 men with prostate cancer and 6438 control subjects (Endogenous Hormones, Prostate Cancer Collaborative Group, 2008). The collaborative analysis failed to detect an association between the risk of prostate cancer and serum concentrations of testosterone, calculated free testosterone, dihydrotestosterone, dehydroepiandrostenedione sulfate, androstenedione, androstanediol glucuronide, estradiol, or calculated free estradiol. The only positive finding in this study was a modest inverse association between prostate cancer risk and serum concentration of sex-hormone binding globulin. This study suggests that one-time measurements of serum sex hormones in adulthood are not a good measure of prostate cancer risk.
Estrogens
Estrogens have both direct and indirect effects on prostatic growth and development and have been postulated to play a role in prostate cancer initiation and progression. Traditionally, estrogens have been considered protective against prostate cancer and have been used as a treatment for advanced disease. This treatment effect is primarily through a negative feedback on the hypothalamo-pituitary-gonadal axis, and also through a direct inhibitory effect of estrogens on prostate epithelial cell growth. However, there is increasing evidence from animal studies that estrogens may act as procarcinogens in the prostate (Leav et al, 1988). Aromatase-knockout mice cannot produce 17β-estradiol locally in the prostate, and despite elevated testosterone and dihydrotestosterone, they do not develop prostate cancer (McPherson et al, 2001). There is evidence across multiple species that estrogens acting through stromal estrogen receptor (ER) α may contribute to prostate carcinogenesis and cancer progression (Prins and Korach, 2008). ERα expression is silenced in early prostate cancers, and re-emerges with disease progression. Prostate epithelial ERβ may play an important role in initiation of prostate cancer, with loss of ERβ potentially contributing to disease progression in organ-confined disease (Prins and Korach, 2008). In addition, the re-emergence of ERβ expression in metastatic prostate cancer also suggests a potential role in progression to castrate-resistant disease. Age-related prostatic disease parallels increases in serum estrogen levels, and there is a low incidence of prostate cancer in cultures with diets rich in phytoestrogens (Denis et al, 1999). However, the data on serum estrogen levels and prostate cancer risk are mixed (Barrett-Connor et al, 1990; Gann et al, 1996; Chen et al, 2003). Complicating interpretation of serum measurements is the fact that estradiol can be produced from testosterone by intraprostatic aromatase (Risbridger et al, 2003).
Insulin-Like Growth Factor Axis
Insulin-like growth factor (IGF)-1 is a peptide hormone that promotes growth in adolescence and childhood and is correlated with adult lean body mass (Severson et al, 1988). IGF-1 promotes proliferation and inhibits apoptosis in normal prostate and tumor cells in vitro (Cohen et al, 1991; Cohen et al, 1994). IGF-1 circulates bound to binding proteins, the most prevalent of which is insulin-like growth factor binding protein (IGFBP)-3. In the prostate IGFBP-3 promotes apoptosis and may mediate growth inhibition by 1,25-dihydroxyvitamin D (Boyle et al, 2001). IGFBP-3 can be cleaved by PSA, reducing its pro-apoptotic activity (Koistinen et al, 2002).
Results of epidemiologic studies suggest an association between IGFs and their associated binding proteins (IGFBPs) with prostate cancer risk. A recent combined analysis of individual patient data from 12 studies attempted to quantify the magnitude of the association (Roddam et al, 2008). The study included data on 3700 men with prostate cancer and 5200 control subjects. They found a positive correlation between serum concentration of IGF-1 and subsequent prostate cancer risk. The OR in the highest versus lowest quintile was 1.38 (95% CI = 1.19 to 1.60). Serum IGFBP-3 concentration was not found to be independently associated with prostate cancer risk; the observed association was secondary to its association with IGF-1 levels. Neither IGF-2 nor IGFBP-2 concentrations were associated with prostate cancer risk.
Leptin
Leptin, a peptide hormone produced by adipocytes, contributes to the control of body weight by appetite suppression and modulating energy use (Friedman, 2002). Obese men become leptin resistant and exhibit elevated plasma leptin (Chu et al, 2001). Epidemiologic studies assessing the association between circulating leptin concentrations with prostate cancer have yielded mixed results (Chung and Leibel, 2006). A genetic study found that prostate cancer patients were almost 5 times more likely to carry a polymorphism that results in increased leptin expression compared with controls (Ribeiro et al, 2004). There is significant evidence, however, suggesting that leptin plays a role in the development of advanced prostate cancer (Ribeiro et al, 2006). Leptin has been shown to stimulate proliferation of the androgen-independent prostate cancer cell lines DU145 and PC-3 (Somasundar et al, 2004; Deo et al, 2008). In addition, leptin appears to induce expression of vascular endothelial growth factor (VEGF) and basal fibroblast growth factor (BFGF), and stimulate cell migration (Frankenberry et al, 2004). Energy imbalance, perhaps mediated by leptin, is emerging as a possible contributor to the progression of prostate cancer to metastasis and death (Calle et al, 2003; Platz et al, 2003).
Vitamin D, Vitamin D Receptor, and Calcium
Vitamin D (1,25 dihydroxyvitamin D3) is an essential vitamin that is a part of the steroid hormone superfamily. Human sources include both dietary intake and conversion from an inactive to active vitamin D in the skin through sunlight exposure. Interest in vitamin D as a determinant of prostate cancer risk comes from several epidemiologic observations (Peehl et al, 2003):
In addition, prostate cancer cells express the vitamin D receptor, and several studies have demonstrated an antiproliferative effect of vitamin D on prostate cancer cell lines by inducing cell cycle arrest (Krishnan et al, 2003).
Many studies show no or a weak association between vitamin D levels and prostate cancer risk (Chan and Giovannucci, 2001; Freedman et al, 2002; Jacobs et al, 2004; Ahn J et al, 2008). The Cancer Prevention Study II Nutrition Cohort, a prospective cohort of 65,321 men demonstrated a modestly increased relative risk of 1.2 for total calcium intake (dietary and with supplements) and 1.6 for high dietary calcium intake alone (≥2000 vs. <700 mg/day), but not for dairy intake (Rodriguez et al, 2003). The results suggest that very high calcium intake, above daily recommendation, may modestly increase risk. These conflicting results regarding vitamin D, calcium, and prostate cancer risk may be explained by variants in the vitamin D receptor (VDR). Polymorphisms resulting in a VDR with lower activity have been associated with increased risk for prostate cancer, as well as with increased risk of biochemical recurrence following radical prostatectomy (Oakley-Girvan et al, 2004; Williams et al, 2004; John et al, 2005).
Other Influences
Sexual Activity/Sexually Transmitted Diseases
Sexual activity has been hypothesized to expose the prostate to infectious agents, which may increase the risk of prostate cancer, akin to the causal relationship between HPV and cervical cancer in women. Some studies have found a link between sexually transmissible infections (STIs) and prostate cancer risk (Fernandez et al, 2005; Sarma et al, 2006), although not consistently (Patel et al, 2005; Huang et al, 2008). A meta-analysis of 6022 cases of prostate cancer and 7,320 controls found significant associations between prostate cancer and any STI (odds ratio [OR] = 1.48, 95% CI = 1.26 to 1.73), gonorrhea (OR = 1.35, 95% CI = 1.05 to 1.83), and human papillomavirus (OR = 1.39, 95% CI = 1.12 to 2.06) but not for syphilis (Taylor et al, 2005). Studies have also suggested a protective association between prostate cancer and frequency of ejaculation, with RR ranging from 0.66 to 0.89 (Giles et al, 2003; Leitzmann et al, 2004). In the Giles study, the protective effect was seen in men in their 20s, who reported greater than 5 ejaculations per week; the large prospective cohort study by Leitzmann demonstrated a protective effect for men in their 20s and 40s, reporting greater than or equal to 21 ejaculations per month in the previous year and as a lifetime average. The biologic basis for this effect is not known.
Vasectomy
A relationship between vasectomy and prostate cancer risk was initially suggested in 1993 with a relative risk of 1.6 based on two large cohort studies (Giovannucci et al, 1993a, 1993b). Risk increased with time so that men who underwent vasectomy at an early age had a higher risk. A recent meta-analysis reported a pooled relative risk of 1.37, with a linear trend suggesting a 10% increase for each additional 10 years since vasectomy (Dennis et al, 2002a). On the other hand a large, population-based, case-control study found no association between prostate cancer and vasectomy, nor with time since vasectomy (Cox et al, 2002). The biologic mechanism by which vasectomy might predispose to cancer is unknown, although presence of antisperm antibodies, and decreased seminal androgen concentrations or secretory activity have been proposed. The magnitude of the relative risk is small enough to potentially be explained by inadequate selection of controls and/or selection bias, because men who have undergone vasectomy may be more likely to seek follow-up with a urologist. After excluding studies that were likely biased, one analysis found that the association became nonsignificant (Millard, 1999).
Smoking
Cigarette smoke may be a risk factor for prostate cancer, because it is a source of cadmium exposure, increases circulating androgen levels, and causes significant cellular oxidative stress. Both case-control and cohort studies have produced conflicting results and none have demonstrated a clear dose-response relationship, although some recent studies have suggested an association with more advanced stage at diagnosis and increased prostate cancer–related mortality (Bostwick et al, 2004).
Diet
Descriptive epidemiologic studies of migrants, geographic variations, and temporal studies suggest that dietary factors may contribute to prostate cancer development (Bostwick et al, 2004). The incidence of latent prostate cancers is similar around the world, but the incidence of clinically manifest cancers differs, with Asians having the lowest rates of clinical prostate cancer. Thus the most convincing evidence for the role of the diet and other environmental factors in modulating prostate cancer risk comes from migration studies showing an increased incidence of prostate cancer in first-generation immigrants to the United States from Japan and China (Muir et al, 1991; Shimizu et al, 1991). These observations suggest that diet may play a role in converting latent tumors into clinically manifest ones. A strong positive correlation exists between prostate cancer incidence and the corresponding rates of several other diet-related cancers, including breast and colon cancers (Bostwick et al, 2004).
Dietary Fat
Prostate cancer incidence and mortality rates around the world correlate highly with the average level of fat consumption, especially for polyunsaturated fats (Bostwick et al, 2004). Potential mechanisms of action include fat-induced changes in the hormonal milieu, induction of oxidative stress, and/or insulin-like growth factor-1 (IGF-1). High levels of dietary fat stimulate proliferation of prostate cancer cells both in vitro and in vivo, and animal models have shown that a fat-free diet can reduce the growth of androgen-dependent tumors in the Dunning model (Clinton et al, 1988; Wang et al, 1995; Aronson et al, 1999).
Epidemiologic studies have suggested a moderate to strong association between total and specific fats and the risk of developing prostate cancer (Chan et al, 2005). However, results from large prospective studies showed no association between dietary fat intake and prostate cancer risk (Park et al, 2007; Wallstrom et al, 2007; Crowe et al, 2008). A meta-analysis of observational studies demonstrated only a weak association between higher intake of total fat and risk of prostate cancer (RR = 1.2, 95% CI = 1.1 to 1.3), and noted a large degree of heterogeneity between studies (Dennis et al, 2004). Observations on the association of dietary fat and risk may have alternative explanations. Diets high in meats, which are sources of fat, are also usually low in vegetables containing nutrients that may protect against prostate cancer. Furthermore, meats and dairy products contain other constituents, such as zinc (Zn) and calcium (Ca), that may affect prostate cancer risk.
Obesity
Obesity as measured by body mass index (BMI) has been suggested as a risk factor for prostate cancer, because of their common occurrence in middle-aged men and clear links to colon and breast cancer risk (Giovannucci, 1995; Madigan et al, 1998). White fat in mammals serves not only as an important energy reservoir but also as an endocrine organ, with secretion of cytokines and agents with cytokine-like activity (tumor necrosis factor [TNF]-α, interleukin [IL]-1β, IL-6, IL-8, IL-10, transforming growth factor [TGF]-β), as well as their soluble receptors (Trayhurn and Wood, 2004). Several studies have shown that BMI and waist circumference show significant positive correlation with markers of oxidative stress (Keaney et al, 2003; Furukawa et al, 2004). Treatment of obesity through reduction in fat intake and increased exercise has been shown to reduce oxidative stress, suggesting that lifestyle modification could be important in reducing the risk of prostate cancer (Roberts et al, 2002).
Studies examining the association between adult body mass index and the risk of prostate cancer have yielded conflicting results. Two recent meta-analyses of observational studies have estimated the RR of prostate cancer to be 1.05 (95% CI = 1.01 to 1.08) and 1.03 (95% CI = 1.00 to 1.07) per 5 kg/m2 increment in BMI (MacInnis and English, 2006; Renehan et al, 2008). Recently, three large prospective studies, examining the association between obesity and prostate cancer risk by stage and/or grade at diagnosis, suggested that obesity was associated with a lower risk of low-grade disease but a greater risk of high-grade disease (Gong et al, 2006; Rodriguez et al, 2007; Wright et al, 2007). Potential explanations for the latter observation include the association of obesity with higher serum estradiol, insulin, free IGF-1, and leptin levels, and lower free testosterone and adiponectin levels, which have also been associated with more aggressive prostate cancer (Buschemeyer and Freedland, 2007). Another possible explanation is detection bias. Higher BMI has been shown to be associated with lower serum PSA (Baillargeon et al, 2005), which in obese men could lead to fewer prostate biopsies. However, given that the association between obesity and high-grade disease was also observed in the Prostate Cancer Prevention Trial, a study in which all men underwent biopsy, it is unlikely that detection bias is solely responsible for this finding (Gong et al, 2006).
Alcohol Consumption
Alcohol consumption and risk of prostate cancer is of interest because of its association with other cancers, its effect on estrogen and testosterone, and the high content of polyphenolic compounds with antioxidant activity in red wine. Although initial studies suggested an association between alcohol intake and prostate cancer risk (Sesso et al, 2001; Velicer et al, 2006), and a possible protective effect of red wine (Schoonen et al, 2005), recent large studies have failed to detect an association between total alcohol intake and the incidence of prostate cancer (Schoonen et al, 2005; Baglietto et al, 2006; Sutcliffe et al, 2007b; Rohrmann et al, 2008), suggesting that alcohol does not contribute appreciably to the etiology of this disease.
Key Points
Molecular Epidemiology
Etiology and Molecular Genetics
Prostate cancer is unique among solid tumors in that it exists in two forms: a histologic or clinically occult form, which can be identified in approximately 30% of men greater than 50 years old and 60% to 70% of men greater than 80 years old, and a clinically evident form, which affects approximately one of six U.S. men. Latent prostate cancer is believed to have a similar prevalence worldwide and among all ethnicities, whereas the incidence of clinical prostate cancer varies dramatically between and within different countries. For this reason, an understanding of prostate cancer etiology must encompass the steps leading to both the initiation of histologic cancer and progression to clinically evident disease. The exact molecular relationship between latent and clinical cancers is not known, and it is likely that the progression from latent to clinically evident cancer is a biologic continuum with overlap in the associated molecular events. Mutations, downregulation by promoter methylation and other mechanisms, and protein modification have all been implicated in progression of prostate cancer (Fig. 95–8).
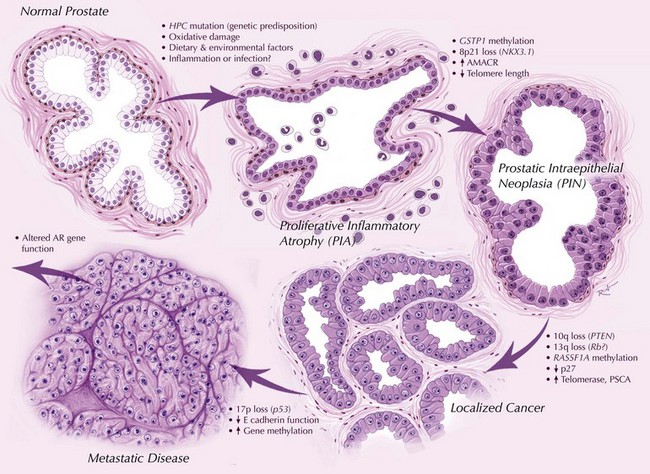
Figure 95–8 Contemporary model of prostate cancer progression. Genetic predisposition, oxidative damage, and inflammatory changes are associated with earliest steps of prostate cancer development. Downregulation of caretaker genes, such as GSTP1, by aberrant promoter methylation may increase potential for neoplastic transformation. Chromosomal loss and telomere shortening may also contribute to genetic instability and progression to invasive disease. Further methylation changes, loss of tumor suppressor gene function and additional mutational events are associated with metastases and androgen independence.
(From Gonzalgo ML, Isaacs WB. Molecular pathways to prostate cancer. J Urol 2003;170:2444–52.)
The Influence of Androgens
As previously discussed, androgens play an important role in prostate carcinogenesis. The primary androgen of the prostate is dihydrotestosterone (DHT), irreversibly catalyzed from testosterone by 5α-reductase. DHT binds to intracytoplasmic AR with much greater affinity than testosterone, and binding of DHT to the AR enhances translocation of the steroid-receptor complex into the nucleus and activation of androgen response elements (Steers, 2001). There are two isoenzymes of 5α-reductase, the products of two separate genes. Type 1 5α-reductase is expressed primarily in the skin and liver and, to a lesser extent, in the prostate, while the type 2 enzyme is expressed predominantly in prostate epithelium and other genital tissues (Andriole et al, 2004b).
Functional type 2 5α-reductase is a prerequisite for normal development of the prostate and external genitalia in males, and insufficient exposure of the prostate to DHT appears to protect against the development of prostate cancer. Transrectal ultrasonography (TRUS) in males with inherited 5α-reductase deficiency demonstrates miniscule prostatic tissue, and biopsies demonstrate stroma but no epithelium (Imperato-McGinley and Zhu, 2002). In addition to the lack of enzyme activity, a lack of testosterone may also protect against the development of prostate cancer, as evidenced by the atrophic prostates seen in men after surgical castration (Wilson and Roehrborn, 1999).
Although exposure of the prostate to androgens seems to be prerequisite for later development of prostate cancer, the duration and magnitude of androgen exposure needed to set the stage for carcinogenesis is unknown. As mentioned above, studies assessing the association between serum testosterone or sex hormone–binding globulin (SHBG) and prostate cancer have been inconsistent. Single measures of serum testosterone levels may not accurately reflect the amount of bioavailable testosterone; furthermore, they do not accurately quantify the testosterone exposure in the individual over the years it takes to develop prostate cancer. Future research will likely improve our understanding of the factors that may affect tissue concentrations of androgen at the biochemical and molecular level.
Stem Cells
Stem cells are required for the maintenance of high-cell turnover–tissues where cells continually need to be replaced, and like most epithelial organs, the prostate is believed to contain stem cells capable of multilineage differentiation. The existence of prostatic stem cells is suggested by studies demonstrating the ability of prostate epithelium to regenerate when androgen is administered to castrated rodents (Isaacs and Coffey, 1989; Bui and Reiter, 1998; Tsujimura et al, 2002). Supportive evidence includes observations that (1) basal and luminal cells of prostate glands have distinct phenotypes as measured by immunohistochemical marker expression, especially for various forms of keratin; (2) cell culture experiments demonstrate that some prostate cancer cells are self-replicating while others are not; and (3) stem cell–enriched populations of cells can produce three-dimensional structures with basal cells and fully differentiated luminal cells in nude mice (Taylor and Risbridger, 2008). These observations led to the development of a stem cell theory for prostate cancer that postulated that an androgen-independent stem cell produces androgen-independent transit-amplifying (TA) cells that, in turn, give rise to androgen-dependent, fully differentiated luminal secretory cells (Isaacs and Coffey, 1989). Although it was originally believed that prostatic stem cells reside within the basal epithelium, recent models have challenged this notion (Tsujimura et al, 2002). A recent study has suggested that TROP2 may be a marker for a subpopulation of basal cells with stem cell characteristics (Goldstein et al, 2008). Once their biology is fully understood, stem cells are likely to be an attractive target for both prevention and therapy.
Epigenetic Changes
Epigenetic events affect gene expression without altering the actual sequence of DNA. Known mechanisms include DNA hyper- and hypomethylation, chromatin remodeling, histone modification, and RNA interference (Dobosy et al, 2007). A variety of genes implicated in prostate cancer initiation and progression are affected by these processes, including hypermethylation of hormonal response genes (ERαA, ERβ, and RARβ), genes controlling the cell cycle (CYCLIND2, 14-3-3σ), tumor cell invasion/tumor architecture genes (CD44), DNA repair genes (GSTpi, GPX3, and GSTM1), tumor suppressor genes (APC, RASSF1α, DKK3, TP16INK4α, E-cadherin, and TP57WAF1), signal transduction genes (EDNRB and SFRP1), and inflammatory response genes (PTGS/COX2); hypomethylation of CAGE, HPSE, and PLAU; histone hypoacetylation of CAR, CPA3, RARB, and VDR; and histone methylation of GSTP1 and PSA (Li et al, 2005). Clinical studies have shown that quantitative methylation analysis of the GSTpi, APC, PTGS2, RASSF1α, MDR1, TP16, and MGMT genes can improve sensitivity and specificity for the diagnosis of cancer, and efforts to develop global gene methylation profiles in serum, urine, and tissue as an adjunct to predicting risk of cancer, determining the need for rebiopsy, and tumor aggressiveness are underway (Dobosy et al, 2007). Furthermore, agents that can inhibit or reverse the effects of DNA methyl transferases (DNMTs) and histone deacetylases (HDACs) and restore normal gene expression are under study for both prevention and therapy.
Cyclooxygenase
Inflammatory cells in the prostate produce a variety of compounds designed to eradicate infectious microorganisms, many of which have potential to cause oxidative DNA damage. These compounds include superoxide, hydrogen peroxide, oxygen free radicals, and peroxynitrite (Potts and Pasqualotto, 2003). The inflammatory response stimulates the production of prostaglandins that are part of the eicosanoid family, which includes long-chain polyunsaturated fatty acids. Cyclooxygenase (COX) enzymes catalyze the rate-limiting step of prostaglandin synthesis, converting arachidonic acid to the intermediate prostaglandin G2 (Zha et al, 2004). Two isoforms of COX (COX-1 and COX-2) have been identified (DeWitt and Smith; 1988, Hla and Neilson, 1992). COX-1 is constitutively expressed in many tissues and cell types and helps mediate normal cellular physiologic functions. COX-2 is an inducible enzyme that mediates acute and chronic inflammation, pain, and cellular repair mechanisms. COX-2 expression is rapidly induced in response to inflammatory or mitogenic stimuli, cytokines, tumor promoters, and growth factors (Zha et al, 2004; Patel et al, 2008). Prostaglandins resulting from COX-2 expression mediate a variety of responses to tissue injury and hypoxia that promote tumor formation, including inhibition of apoptosis, promotion of cellular proliferation, and angiogenesis (Patel et al, 2008). Most studies suggest increased COX-2 expression in prostate cancer and HGPIN compared with benign prostatic epithelium (Patel et al, 2008). Epidemiologic studies and clinical observations suggest that nonsteroidal anti-inflammatory drugs (NSAIDs) and certain selective COX-2 inhibitors may reduce the risk of prostate cancer (Aparicio Gallego et al, 2007). However, the recent observation of increased cardiovascular morbidity associated with long-term COX-2 inhibitor use led to the early closure of a drug sponsored prostate cancer prevention trial in 2004. Further clinical trials of COX-2 inhibitors may be difficult to complete, and the expected benefit must be weighed against the potential risks of the drug in the target population.
Somatic Mutations Associated with Tumor Initiation and Progression
A number of somatic mutations in tumor DNA are acquired during clonal expansion of nascent tumors as a result of mitotic errors, defects in DNA repair mechanisms, loss of apoptotic ability, and other mechanisms. A variety of genes are so affected in prostate cancer.
Androgen Receptor
As discussed earlier, polymorphisms of the AR are linked epidemiologically to prostate cancer risk. There is also growing evidence that the AR itself may in fact be oncogenic under certain circumstances (Scher and Sawyers, 2005). Alterations in AR signaling can occur at various levels, all of which could potentially contribute to the development and/or progression of prostate cancer. These include (1) changes in ligand concentration in tumor cells (e.g., intracrine mechanism) (Labrie et al, 1993), (2) increased AR protein expression (e.g., this has been detected in castrate-resistant disease, leading to increased sensitivity to low ligand levels) (Bubendorf et al, 1999), (3) mutations leading to altered structure and function (shown to be present in 50% of prostate cancers) (Bostwick et al, 2004), (4) changes in coregulatory molecules (e.g., coactivator proteins such as ARA54 and ARA70 can selectively enhance the activity of AR to alternative or low concentrations of ligand, or lead to independent activation) (Scher and Sawyers, 2005), and (5) factors leading to activation of the receptor independent of the ligand (e.g., many of the growth factors and tyrosine kinases implicated in prostate cancer initiation and progression may exert their effects by cross talk with the AR) (Culig et al, 1994; Craft et al, 1999). A recent study shows that knockout of AR causes regression of even castrate-resistant prostate cancer, suggesting that AR is biologically and clinically important even after androgen deprivation (Snoek et al, 2009).
NKX3-1
NKX3-1 is an androgen-regulated and prostate-specific gene located on chromosome 8p21 and belongs to the homeobox gene family (He et al, 1997; Prescott et al, 1998). Mice deficient in NKX3.1 show defects in prostate ductal morphogenesis and secretory protein production and with age develop dysplasia, PIN, and cancer (Bhatia-Gaur et al, 1999; Tanaka et al, 2000; Abdulkadir et al, 2002). Loss of either one or both NKX3-1 alleles becomes increasingly common in prostate lesions as they progress from PIN to local, metastatic, and androgen-independent tumors (Bowen et al, 2000).
PTEN
PTEN on chromosome 10q23 is among the best studied tumor suppressor genes in prostate cancer. Genetic, functional, and molecular studies suggest a role of PTEN in both initiation and progression of prostate cancer (Dong, 2006). PTEN deletion has been shown to induce prostate cancer in mice (Wang et al, 2003). Loss of PTEN function is seen in PIN and is associated with a high Gleason score and advanced stage (Rubin et al, 2000).
Classical Oncogenes
Genetic alterations in the tumor suppressor genes RB1 and TP53 are commonly present in metastatic or hormone-refractory disease, suggesting a role in tumor progression. Alterations in c-MYC, c-ERBB2, and BCL-2 have also been observed in advanced and hormone-refractory prostate cancer but not commonly in lower-stage and -grade cancers, suggesting that mutations of proto-oncogenes usually occur as secondary events associated with tumor progression (Gurumurthy et al, 2001; Fossa et al, 2002; Qian et al, 2002).
Telomerase
Eukaryotic chromosomes have telomeres on their ends that consist of repeating units of 6 base pairs (TTAGGG) that protect the chromosomal ends from inappropriate recombination. Normal cellular aging is characterized by the progressive loss of these repeated elements, and consequently telomere length decreases successively with every cell replication. Critical telomere shortening leads to chromosomal instability, further leading to an increased incidence of cancers due to chromosomal fusions, breakage, and rearrangements (Blasco et al, 1997). There is increasing evidence that telomere shortening during cell division and oxidative cell damage, possibly as a result of chronic inflammation, may play a role in prostate carcinogenesis (Meeker, 2006). Prostate cancer and high-grade PIN appear to result in an activation of the telomerase enzyme, a reverse transcriptase that maintains or increases telomere length (Meeker et al, 2002; Meeker, 2006). In addition to prostate cancer causation, telomeres may also play a role in disease progression, and studies suggest that telomerase suppression may contribute to the emergence of androgen-independent disease (Meeker, 2006).
Glutathione-S-Transferase
Reactive oxygen species are inactivated by a host of protective enzymes, including glutathione-S-transferase (GST), glutathione peroxidase, and superoxide dismutase. The expression of one of these enzymes, GSTP1, is absent in approximately 70% of PIN and virtually all cases of prostate cancer, making it the most common genetic alteration in this malignancy (Nelson et al, 2003). This loss of expression results from hypermethylation of CpG islands in the GSTP1 promoter and causes a defect in cellular defense against oxidant stress, leading to a higher mutation rate. Additionally, GSTP1 hypermethylation has been identified in prostate biopsies with histologic findings of PIN (Nakayama et al, 2004). DNA methylation assays that may soon be used in diagnostic testing of serum and tissue for prostate cancer are under development.
TP27
TP27 belongs to the CIP/KIP family of cyclin-dependent kinase (CDK) inhibitors and regulates progression of the cell cycle from G1 to S phase. Loss of TP27 may accelerate tumorigenesis by enabling cells to progress unregulated through the cell cycle. Mice deficient in TP27 develop prostate hyperplasia, and mice deficient in both TP27 and PTEN have a high incidence of prostate cancer (Cordon-Cardo et al, 1998; Di Cristofano et al, 2001). Studies in humans have found loss of heterozygosity for TP27 in 50% of patients with metastatic prostate cancer (Kibel et al, 2000). Studies of patients undergoing RP have found an association between loss of TP27 expression and increased risk of biochemical recurrence (Guo et al, 1997; Cordon-Cardo et al, 1998).
Vascular Endothelial Growth Factor
Vascular endothelial growth factor (VEGF), which is an important mediator of tumor angiogenesis, is expressed in a majority of prostate cancers, and increased expression has been associated with disease progression and survival (Green et al, 2007; Peyromaure et al, 2007). VEGF is regulated by androgen, suggesting a potential pathway by which androgen regulates prostate cancer growth (Joseph and Isaacs, 1997). Several SNPs in the VEGF gene have been associated with protein expression; however to date, studies examining the influence of these polymorphisms on prostate cancer risk have been inconsistent (Langsenlehner et al, 2008). Therapeutic inhibition of the VEGF receptor signaling pathway is currently being investigated in the treatment of prostate cancer.
E-Cadherin
The E-cadherin gene, which is located on chromosome 16q22.1, encodes a transmembrane glycoprotein that mediates intercellular adhesion as well as cell signaling (Takeichi, 1991). E-cadherin expression is reduced in a significant percentage of prostate cancers, particularly in poorly differentiated tumors, and E-cadherin expression in prostate cancer correlates inversely with grade, stage, metastasis, recurrence, and survival (Umbas et al, 1992; Bostwick et al, 2004). However, studies of the association between SNPs of E-cadherin and prostate cancer risk have yielded conflicting results.
α-Methylacyl-CoA Racemase (AMACR)
AMACR (racemase) is an enzyme that plays a critical role in the metabolism of branched-chain fatty acids. AMACR expression has been shown to decrease in metastatic prostate cancer compared with localized disease, and is associated with cancer-specific survival (Rubin et al, 2005). In addition, studies suggest that polymorphisms within the AMACR gene may be associated with prostate cancer risk (Levin et al, 2007), although further experimental studies are needed to better characterize this association.
Prostate-Specific Membrane Antigen
Prostate-specific membrane antigen (PSMA) is a transmembrane glycoprotein that is expressed by prostate cancer epithelium and on the neovasculature of other solid tumors (Ghosh et al, 2005). Overexpression of PSMA is observed after androgen withdrawal and in hormone-refractory disease, and is the basis of clinical imaging studies using anti-PSMA monoclonal antibodies. Because of its restricted tissue expression, PSMA is being actively investigated as an immunotherapeutic target (Schulke et al, 2003).
Epidermal Growth Factor and Epidermal Growth Factor Receptor
There is substantial evidence that epidermal growth factor (EGF) signaling through the epidermal growth factor receptor (EGFR) is important in prostate cancer. EGF levels are frequently elevated, and EGFR is commonly overexpressed in prostate cancer cells (Vicentini et al, 2003). In addition, EGFR expression increases as the disease progresses to hormone resistance (Di Lorenzo et al, 2002). A genetic polymorphism in the EGF gene has been shown to be associated with progression-free survival in patients on androgen-deprivation therapy, identifying a potential therapeutic target in these patients (Teixeira et al, 2008).
EZH2
EZH2 is a histone-lysine methyltransferase with activity dependent on its association with other components of the polycomb repressive complexes 2 (PRC2). EZH2 levels are increasingly elevated during prostate cancer progression (LaTulippe et al, 2002). EZH2 expression in localized prostate cancer has also been shown to be associated with poor prognosis and disease recurrence (Varambally et al, 2002; van Leenders et al, 2007).
Key Points
Etiology and Molecular Genetics
Chemoprevention
Rationale
As previously discussed, prostate carcinogenesis is a multistep molecular process induced by genetic and epigenetic changes that disrupt pathways controlling the balance between cell proliferation, apoptosis, differentiation, and senescence. The presence of precursor lesions that represent intermediate stages between normal and malignant cells—as long as 20 years before the appearance of cancer, coupled with the age-dependent incidence of most cancers—suggests that carcinogenesis occurs slowly during a protracted interval. In theory, this provides the opportunity to intervene before a malignancy is established, using lifestyle changes (dietary alterations, smoking cessation, exercise) or by chemoprevention, defined as the use of natural or synthetic agents that reverse, inhibit, or prevent the development of cancer. The goal of primary chemoprevention is to decrease the incidence of a given cancer, simultaneously reducing both treatment-related side effects and mortality. Effective chemoprevention requires the use of nontoxic agents that inhibit specific molecular steps in the carcinogenic pathway.
Prostate cancer is an attractive and appropriate target for primary prevention because of its incidence, prevalence, and disease-related mortality (see Table 95–1). The burden of prostate cancer can also be measured in other terms. A recent study of complications after surgical therapy for localized disease in an unselected population-based cohort reported that, at greater than 18 months following RP, 8.4% of men were incontinent, and 41.9% reported that their sexual performance was a moderate-to-large problem (Stanford et al, 2000). In a prospective cohort study comparing outcomes 5 years after radiation or surgery to age-matched controls, the authors concluded that “…declines in urinary and sexual function domains after diagnosis and treatment of localized cancer far exceeded any effects from aging…” (Hoffman et al, 2004). A large study assessing health-related quality of life after primary treatment for prostate cancer found that although each treatment modality was associated with a distinct pattern of change in quality-of-life domains, they all had a significant effect on urinary, sexual, bowel, and/or hormonal function (Sanda et al, 2008). Although many single-institution studies have reported better results in highly selected cohorts of treated patients, it is clear that the majority of men treated for localized disease in the community pay a substantial price to be cured, and fear of cancer recurrence remains substantial for as long as 2 years after treatment (Mehta et al, 2003). It seems self-evident that an effective prevention strategy would spare many men this burden of diagnosis and cure.
The molecular pathogenesis of prostate cancer also lends itself to a primary prevention strategy. Histologic lesions, including atypical small acinar proliferation (ASAP), PIA, and PIN contain both genetic and epigenetic changes intermediate between normal prostatic epithelium and prostate cancer. Clinically evident prostate cancer is rare in men younger than 50 years, whereas prostatic intraepithelial neoplasia is apparent at autopsy in men younger than 30 years. Furthermore, the prevalence of PIN is similar in populations at much different risks of developing clinically evident cancer, suggesting that external environmental influences are important and potentially modifiable.
Clinical Trial Design
Target populations appropriate for primary prevention studies can be subdivided into those with low, intermediate, and high risk of developing prostate cancer based on current epidemiologic evidence (Klein and Meyskens, 2001). Each group has its own clinical characteristics that lend advantages and disadvantages for trial design, end points, and statistical analysis (Table 95–3). Based on the hypothesis that the specific molecular mechanisms underlying the development and/or progression of disease in each risk group and model may be unique and that different agents may be useful for each situation, it seems apparent that multiple potential chemopreventative agents should be tried in all risk groups and clinical models to best define which agents appear promising for large-scale studies. Testing of potentially active agents for secondary prevention in clinical models of patients with active disease is also appropriate (Table 95–4). The presurgical model allows for pre- and post-treatment tissue biopsies and is thus useful for proof-of-principle demonstrations that a given agent is affecting its intended molecular target. The other models may yield useful strategies for the management of patients in specific clinical situations. The molecular mechanisms that underlie disease progression is likely different for each primary and secondary target population, and it may not be possible to generalize the results from a particular trial to other clinical scenarios.
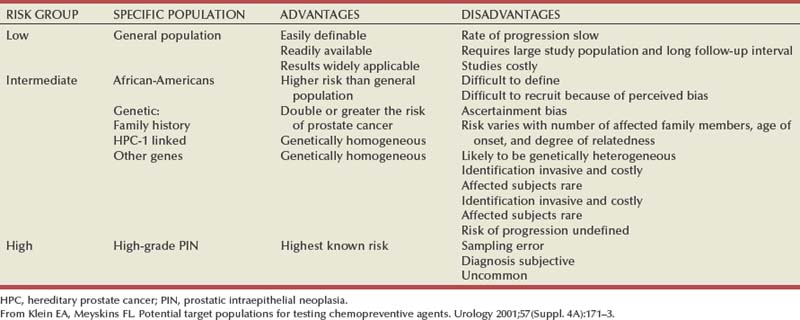
Table 95–4 Models of Secondary Prevention
| MODEL | ADVANTAGES | DISADVANTAGES |
|---|---|---|
| Presurgical | ||
| Elevated PSA/negative biopsy | ||
| Adverse pathology after RP | ||
| Rising PSA after RP or RT |
PSA, prostate-specific antigen; RP, radical prostatectomy; RT, radiation therapy.
From Klein EA, Meyskins FL: Potential target populations for testing chemopreventive agents. Urology 2001;57(Suppl. 4A):171–3.
Numerous observations in the epidemiologic literature suggest associations between various dietary, lifestyle, genetic, and nontraditional factors and the risk for developing prostate cancer. It is generally not practical to quantitate all of these factors in the course of a prevention trial, but if the trial is large enough, the effects of these factors are likely to be equally and randomly distributed across all study arms. Most trials are designed to collect data relevant to these factors for secondary analysis and analysis of potential confounds for unexpected results.
Prostate Cancer Prevention Trial
The most significant event in chemoprevention of prostate cancer occurred with the publication of the results of the Prostate Cancer Prevention Trial (PCPT) (Thompson et al, 2003). This landmark study, opened in 1993, was the first large-scale population-based trial to test a chemopreventative strategy in men at risk for prostate cancer. The PCPT was based on two observations: (1) Androgens are required for the development of prostate cancer and (2) men with congenital deficiency of type 2 5α-reductase are unaffected by benign prostatic hyperplasia (BPH) and prostate cancer. The PCPT tested the hypothesis that treatment with finasteride, which induces an acquired deficiency of type 2 5α-reductase, would lower intraprostatic DHT levels and thereby prevent prostate cancer. In the PCPT, 18,882 men greater than or equal to 55 years of age with a normal digital rectal examination (DRE) and a prostate-specific antigen (PSA) level of less than or equal to 3.0 ng/mL were randomly assigned to treatment with finasteride (5 mg/day) or a placebo for 7 years (Fig. 95–9). Prostate biopsy was recommended if the annual PSA level, adjusted for the effect of finasteride, exceeded 4.0 ng/mL or if the DRE was abnormal. The primary end point was the prevalence of prostate cancer during the 7 years of the study, as diagnosed by either for-cause biopsies (abnormal DRE or PSA) or by end-of-study biopsy. The trial was stopped approximately 15 months early by an independent data safety and monitoring committee, because the primary end point of a 25% risk reduction on the finasteride arm was reached, and sensitivity analyses suggested that additional follow-up would not change that outcome.
Figure 95–9 Design of the Prostate Cancer Prevention Trial. DRE, digital rectal examination; PSA, prostate-specific antigen.
The main findings of the PCPT are as follows:
Additional important observations were as follows: (1) the risk reduction in the finasteride arm was seen in both clinically apparent tumors (those diagnosed “for cause” because of an elevated PSA or abnormal DRE) and end-of-study biopsies; (2) there were an equal number of deaths due to prostate cancer (five) in each study arm; (3) 98% of the tumors were clinically localized; and (4) finasteride-treated glands were 25% smaller than those in the placebo arm.
A number of relevant observations can be made about the results of the trial. Most surprising was the 24.4% prevalence of prostate cancer in the placebo arm, 4 times higher than the 6% assumed for the trial design. This discrepancy can be explained by the fact that the 6% assumption was based on Surveillance, Epidemiology, and End Results (SEER) incidence estimates, which are derived from clinically evident cases and not on the prevalence in men undergoing biopsy for no reason other than being on the trial. The incidence of clinically evident cancers detected “for cause” by elevations in PSA or abnormal PSA was 7.2% at 7 years, roughly what was estimated by use of the SEER data. Another observation was a marked effect of finasteride on the prevalence of Gleason sum 6 tumors, no effect on the prevalence of Gleason sum 7 tumors, and a slight increase in the prevalence of Gleason sum 8 to 10 tumors. The higher incidence of high-grade tumors was restricted to those men undergoing “for-cause” biopsy. Finally, secondary analyses of the PCPT trial have demonstrated an overall improved sensitivity of DRE (Thompson et al, 2007b), as well as a higher sensitivity and area under the curve for PSA for the diagnosis of prostate cancer in the finasteride arm (Thompson et al, 2006). However, given that the latter finding was demonstrated for all grades, the bias would lead to a greater detection of all grades of prostate cancer with finasteride.
There are two areas of continued debate over the results of the PCPT. The first is whether Gleason sum 6 cancers that were “prevented” are biologically significant, which can be defined as destined to metastasize or kill the host. A review of the pathologic characteristics of prostate biopsies from men in the placebo and finasteride groups of the PCPT found that 75% of all cancers and 62% of cancers with a Gleason score less than or equal to 6 met the accepted biopsy criteria for clinically significant tumors (Epstein et al, 1994; Lucia et al, 2008). Although the proportion of insignificant cancers was directly correlated with serum PSA, even at the lowest PSA values (0 to 1.0 ng/mL) almost half of the tumors met the definition of clinical significance. Moreover, an analysis of the Cancer of the Prostate Strategic Urologic Research Endeavor (CaPSURE) found that of men with prostate cancer who met this definition, 91% choose some form of definitive therapy instead of watchful waiting (Barocas et al, 2008). Viewed in the context of “clinical relevance” as defined by current urologic practice, preventing grade 6 tumors by finasteride also prevents the anxiety, cost, and morbidity associated with their treatment. From a public health perspective, preventing the “burden of cure” in newly diagnosed patients should be added as a positive to the 25% reduction in risk of diagnosis and significant reduction in urinary symptoms associated with finasteride use.
The second question still under debate is whether the increased prevalence of higher-grade tumors is real or artifactual. Finasteride is known to change the appearance of prostatic epithelium in a way that could bias interpretation, and the Gleason grading system has never been validated on glands treated with antiandrogenic agents (Civantos et al, 1996). However, when this was examined in the PCPT, degenerative hormonal changes in high-grade biopsies were equivalent between the finasteride and placebo groups (Lucia et al, 2007). Another potential explanation for the observed increase in high-grade tumors in the finasteride group is ascertainment bias. As stated above, finasteride has been shown to increase the sensitivity of PSA and DRE, as well as decrease prostate volume by approximately 25%. Data suggest that having a smaller prostate results in more high-grade cancers diagnosed at biopsy (Kulkarni et al, 2006). This was demonstrated in the PCPT, where biopsy identified a greater proportion of patients with high-grade disease present at prostatectomy in the finasteride group than in the placebo group (Lucia et al, 2007). Therefore when accounting for all biases, the adjusted prostate cancer rates were estimated to be 21.1% for the placebo group and 14.7% in the finasteride group, a 30% risk reduction for all cancers (relative risk [RR] = 0.70, 95% CI = 0.64 to 0.76), and a nonstatistically significant 14% increase in high-grade cancer (Redman et al, 2008). Using known predictive factors to estimate true tumor grade in those men who did not undergo end-of-study biopsy, and incorporating grading information from those who had radical prostatectomies, the rate of high-grade cancer was estimated to be 8.2% and 6.0% in the placebo and finasteride groups, respectively, suggesting that finasteride may in fact reduce the risk of high-grade disease (27% risk reduction [RR = 0.73, 95% CI = 0.56 to 0.96]) (Fig. 95–10). Using a different analysis methodology in an independent analysis, another study concurred that the rate of true high-grade disease may have been lower in the finasteride group compared with the placebo group (Pinsky et al, 2008). In March 2009, the American Urological Society and the American Society for Clinical Oncology jointly published guidelines based on expert review of the available evidence, stating that use of finasteride for the prevention of cancer should be discussed with at-risk men (Kramer et al, 2009).
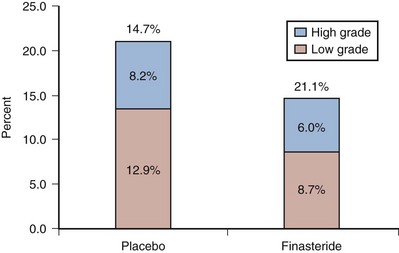
Figure 95–10 Estimated bias-adjusted fractions of total subjects with low-grade and high-grade prostate cancers in the finasteride and placebo arms of the Prostate Cancer Prevention Trial.
Deciding whether or not the advantages of taking finasteride (Table 95–5) outweigh the potential disadvantages is not a simple task. An initial mathematical model suggested a baseline 5 : 1 benefit to risk ratio, as defined by the number of cancers prevented for each excess high-grade cancer in those taking finasteride (Klein et al, 2005). Another study using a person-years–saved model estimated that the use of finasteride as a preventative could reduce the lifetime risk of prostate cancer for a U.S. man from 17% to 13%, while saving more than 250,000 lives every 10 years (Unger et al, 2004). However, more recently published decision-analytic models examining finasteride chemoprevention found that its widespread use is unlikely to be cost effective when considering the usually indolent natural history of treated prostate cancer (Zeliadt et al, 2005; Svatek et al, 2006, 2008). Nonetheless, they suggested that chemoprevention may be cost effective in high-risk populations.
Table 95–5 Benefits and Costs of Finasteride When Used to Prevent Prostate Cancer
| Benefits |
| Costs |
In addition to the prevention of prostate cancer, 5α-reductase inhibitors (5ARIs) have other benefits that need to be considered. As mentioned earlier, finasteride improves the sensitivity of PSA (Thompson et al, 2006) and DRE (Thompson et al, 2007b) in prostate cancer screening, decreases the risk of high-grade prostatic intraepithelial neoplasia (HR = 0.79, 95% CI = 0.70 to 0.89) (Thompson et al, 2007a), and may be effective in the treatment and possibly prevention of chronic nonbacterial prostatitis (Nickel et al, 2004). Furthermore, benign prostatic hyperplasia (BPH) treatment trials in patients with moderate to severe lower urinary tract symptoms demonstrated: (1) a reduction in AUA symptom sores by 20% to 39% compared with 13% to 29% for the placebo, (2) a reduction in the risk of acute urinary retention by one half in long-term trials (RR = 0.51, 95% CI = 0.38 to 0.69; absolute risk reduction [ARR] = 2.2%), and (3) a reduction in the risk of surgical intervention due to BPH progression by one half (RR = 0.50, 95% CI = 0.43 to 0.58, ARR = 1.7%) (Wilt et al, 2008).
On the other hand, adverse effects that are more common with finasteride than with the placebo include impaired sexual or erectile function and endocrine effects. Pooled data from randomized trials indicate absolute differences of 2% (95% CI = 1 to 2) for gynecomastia, 3% (95% CI = 1 to 6) for decreased libido, 4% (95% CI = 1 to 8) for erectile dysfunction, and 4% (95% CI = 8 to 17) for reduced volume of ejaculate (Wilt et al, 2008). Sexual dysfunction was also assessed in the PCPT participants during the 7-year trial period. As measured by the Sexual Activity Scale in a companion quality-of-life (QOL) study to the PCPT, comparing baseline scores over the course of 7 years, finasteride increased sexual dysfunction relative to the placebo by a mere 3.21 points (95% CI = 2.83 to 3.59 on a scale of 0 to 100), a clinically meaningless effect.
Other 5α-Reductase Inhibitors
A second large-scale trial of another 5ARI, dutasteride, completed accrual in the spring of 2005. This agent inhibits both type 1 and type 2 forms of 5α-reductase, is antiandrogenic, and promotes death of prostate cancer cell lines (Lazier et al, 2004), and has been shown to reduce the risk of prostate cancer in men treated for lower urinary tract symptoms related to benign prostatic enlargement when compared with the placebo (Andriole et al, 2004c). The Eligibility for the Reduction by Dutasteride of Prostate Events (REDUCE) trial included men aged greater than or equal to 50 and less than or equal to 75 years who had PSA scores from 2.5 to 10 ng/mL, prostate volumes less than or equal to 80 cc, and one prior negative prostate biopsy within 6 months of enrollment, thus representing a group at high risk for cancer on subsequent biopsy (Andriole et al, 2004a). The primary end point of REDUCE is the prevalence of cancer on study-mandated prostate biopsies performed at 2 and 4 years after entry. The trial accrued 8231 men, of whom 6726 (82.6%) underwent at least one biopsy and 1516 (22.5%) were diagnosed with prostate cancer. Dutasteride reduced the risk of prostate cancer over 4 years by 23% (857 in the placebo arm vs. 659 in the dutasteride arm, P < .0001). Interestingly, no significant increase in Gleason sum 8 to 10 tumors was observed in the study (19 in the placebo arm vs. 29 in dutasteride arm, P = .15). Preliminary analyses also suggest that dutasteride enhanced the utility of PSA as a diagnostic test for prostate cancer, demonstrated beneficial effects on BPH outcomes (relative risk reductions of 77% for acute urinary retention and 73% for BPH-related surgery), and was generally well tolerated (15% drug-related adverse events in the placebo arm vs. 22% in dutasteride arm). The fact that the results of REDUCE were congruent with those of the PCPT with respect to the magnitude of risk reduction, benefits for BPH end points, minimal toxicity, and no issues related to tumor grade suggests a class effect for 5ARIs and that these agents should be used more liberally for prevention of prostate cancer.
Antioxidants and the Selenium and Vitamin E Cancer Prevention Trial (SELECT)
Selenium
Selenium (Se) is an essential trace element occurring in both organic and inorganic forms. The organic form is found predominantly in grains, fish, meat, poultry, eggs, and dairy products and enters the food chain through plant consumption. There is marked geographic variability of Se in food related to local soil content. The strongest evidence for a protective effect of Se came from the Nutritional Prevention of Cancer Trial, a randomized study of oral selenized yeast in patients with nonmelanoma skin cancer, which showed a 65% reduction in the prostate cancer incidence when compared with the placebo (Clark et al, 1996). In that trial, 1312 participants took the equivalent of 200 µg yeast per day versus the placebo, and with a mean follow-up of 4.5 years, the incidence of prostate cancer was reduced in the Se arm by 65% compared with the placebo. Of note, the effect was strongest for those with a PSA value less than 4 ng/mL and those with the lowest serum Se levels at study entry (Duffield-Lillico et al, 2003).
Vitamin E
Vitamin E is a family of naturally occurring, essential, fat-soluble vitamin compounds that functions as the major lipid-soluble antioxidant in cell membranes. The most active form of vitamin E is α-tocopherol; it is also among the most abundant, is widely distributed in nature, and is the predominant form in human tissues. Alpha-tocopherol may influence the development of cancer through several mechanisms, including induction of cell cycle arrest and through direct antiandrogen activity (Ni et al, 2003; Thompson and Wilding, 2003). The Alpha-Tocopherol, Beta-Carotene Cancer Prevention Trial (ATBC), a randomized, placebo-controlled trial of α-tocopherol (50 mg/day) and beta-carotene (20 mg/day) alone or in combination in male smokers, with a primary end point of lung cancer incidence and mortality (Albanes et al, 1996), on secondary analysis found a statistically significant 32% reduction in prostate cancer incidence and a 41% lower mortality in those receiving α-tocopherol (Albanes et al, 1995).
Selenium and Vitamin E Cancer Prevention Trial (SELECT)
The accumulated epidemiologic and biologic evidence that Se and vitamin E may prevent prostate cancer led to the design and launch of SELECT, the Selenium and Vitamin E Cancer Prevention Trial (Klein et al, 2001; Lippman et al, 2005). SELECT was a phase III, randomized, double-blind, placebo-controlled, population-based trial designed to test the efficacy of selenium and vitamin E alone and in combination in the prevention of prostate cancer. It was the largest cancer prevention trial ever performed. Eligibility criteria include age greater than or equal to 50 years for African-Americans, greater than or equal to 55 years for whites, a DRE not suspicious for cancer, serum PSA less than or equal to 4 ng/mL, and normal blood pressure. Randomization was equally distributed among four study arms (selenium + placebo, vitamin E + placebo, selenium + vitamin E, and placebo + placebo). Although the study duration was planned for 12 years, as a result of a planned interim analysis in August 2008, an independent data and safety monitoring committee recommended discontinuation of the study because the data convincingly demonstrated no effect on the risk of prostate cancer by either agent alone or in combination, and no chance of a beneficial effect of the hypothesized magnitude with continued supplementation (Fig. 95–11) (Lippman et al, 2009). There were statistically nonsignificant increased risks of prostate cancer in the vitamin E group (HR = 1.13, 99% CI = 0.95 to 1.35, P = .06) and type 2 diabetes mellitus in the selenium group (RR = 1.07, 95% CI 0.94 to 1.22, P = .16). However, neither of these findings was observed in the combination group. Secondary analyses also showed no effect on the risks of lung, colorectal, or overall cancer incidence, no effect on cardiovascular events, and no effect on overall survival. Null results for the effect of vitamin E on prostate cancer risk were also reported in the Physicians’ Health Study II (PHS), a randomized trial of vitamins E (400 IU every other day) and C (500 mg daily) versus placebo in the prevention of prostate and other cancers (Gaziano et al, 2009). Together, these results suggest that neither selenium nor vitamin E should be used in the hope of preventing prostate or other cancers.
Figure 95–11 Cumulative incidence of prostate cancer detection over time by intervention group: results from the Selection and Vitamin E Cancer Prevention Trial.
(From Lippman SM, Klein EA, Goodman PJ, et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial [SELECT]. JAMA 2009;301:39–51.)
Other Agents
Selective Estrogen Receptor Modulators (SERMs)
SERMs possess both agonistic and antagonistic estrogen-like activity and have been shown to repress prostate cancer growth in several transgenic mouse models. In the transgenic mouse model of prostate cancer (TRAMP), toremifene reduces the incidence of high-grade PIN and cancer in an estrogen-dependent, androgen-independent mechanism (Raghow et al, 2002). A randomized, double-blind, placebo-controlled trial is currently open to assess the activity of toremifene against high-grade PIN and prostate cancer incidence.
Soy
Legumes play an important role in the traditional diets of Asian countries, where prostate cancer incidence is low, but only a minor role in the West, where the incidence is the highest worldwide. Soybeans are unique among legumes because they are a concentrated source of isoflavones, which have weak estrogenic activity. The major isoflavone components of soy, including genistein, daidzein, and their metabolites, inhibit benign and malignant prostatic epithelial cell growth, downregulate androgen-regulated genes, and reduce tumor growth in animal models (Cohen et al, 2003; Hedlund et al, 2003; Yu et al, 2003). Migration studies and lower prostate cancer rates in Asian men with higher soy intake also support the role of soy as an anticancer agent (Severson et al, 1989). However, results from small clinical trials have not shown promise thus far. Dietary trials conducted in men prior to prostatectomy (Dalais et al, 2004), as well as in men on active surveillance (Kumar et al, 2004; Ornish et al, 2005), showed modest or no decrease in PSA values (the primary end point of these studies) with soy intake.
Lycopene
Lycopene is a red-orange carotenoid found primarily in tomatoes, tomato-derived products, and other red fruits and vegetables. Lycopene is a highly unsaturated acyclic isomer of β-carotene, is the predominant carotenoid in human plasma, and possesses potent antioxidant activity. Lycopene has been shown to inhibit the growth of benign and malignant prostatic epithelial cells in vitro (Obermuller-Jevic et al, 2003). There is mixed epidemiologic evidence that lycopene consumption is associated with a lower risk of prostate cancer (Giovannucci and Clinton, 1999). A meta-analysis of observational studies reported a 23% reduction in prostate cancer risk with high tomato and lycopene intake (Etminan et al, 2004). However, a nested case-control study in the Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial prospectively examined the intake of more than 25 tomato-containing foods in 29,361 men and found no correlation with the incidence of prostate cancer (Peters et al, 2007). To date, results of initial clinical trials have demonstrated potential beneficial activity of lycopene in prostate cancer; however, treatment length has been relatively short, participants have included men at various stages of disease, and surrogate markers have been used rather than clinical meaningful end points (Bunker et al, 2007; Dahan et al, 2008). Currently, no phase III trials examining the role of lycopene in prostate cancer prevention are being conducted.
Green Tea (Camellia sinensis)
Green tea has been suggested as a prostate cancer preventative based on epidemiologic observations of a low incidence of prostate cancer among Asians with a high dietary intake. Previous work has focused on the effects of polyphenols (i.e., flavanols; also know as catechins), which accounts for 30% to 40% of extractable solids from dried green tea leaves. Prostate cancer cell culture experiments have demonstrated that the major polyphenolic constituent of green tea, (−)-epigallocatechin-3-gallate (EGCG), induces apoptosis, cell-growth inhibition, and cell-cycle dysregulation (Adhami et al, 2003). In the TRAMP model, oral infusion of a polyphenolic fraction isolated from green tea significantly inhibited tumor development and metastasis (Brusselmans et al, 2003; Hastak et al, 2003). A small randomized, double-blind, placebo controlled trial of green tea catechin tablets in 60 men with HGPIN demonstrated cancer at 1-year biopsy in 9 patients in the placebo group, compared with only 1 patient in the treatment group (Bettuzzi et al, 2006). Another small trial in men with hormone-refractory prostate cancer found only minimal clinical activity (Choan et al, 2005). Confirmatory trials are needed to better assess the role of green tea consumption in prostate cancer prevention.
Statins
Statins are widely used cholesterol-lowering drugs given for the treatment and prevention of atherosclerotic cardiovascular disease. They inhibit 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA), the rate-limiting enzyme in cholesterol biosynthesis. Statins are hypothesized to play a role in the prevention of cancer by inhibiting inflammation, angiogenesis, cell proliferation, migration/adhesion, invasion, and/or by preferentially promoting apoptosis in tumor cells (Hamilton and Freedland, 2008). Observational studies of statin use demonstrate high heterogeneity with mixed results for prostate cancer risk. Statin users have been shown to have lower serum PSA levels than nonusers (Cyrus-David et al, 2005; Hamilton et al, 2008), suggesting an anticancer effect, but potentially leading to fewer prostate biopsies, and thus biasing results of epidemiologic and clinical studies. A meta-analysis of six randomized clinical trials, six cohort, and seven case-control studies found no association between statin use and overall prostate cancer incidence but did find a protective association with advanced prostate cancer (RR = 0.77, 95% CI = 0.64 to 0.93) (Bonovas et al, 2008). Statin users tend to be healthier and more medically compliant than nonusers, making them more likely to undergo PSA screening and possibly resulting in earlier cancer detection. Further research is needed to help determine the role, if any, of statins in the prevention of prostate cancer.
Key Points
Chemoprevention
Conclusion
It is clear that the process of prostate carcinogenesis is complex. The interplay of constitutional, behavioral, molecular, and environmental factors—against the backdrop of various processes occurring during aging—interact to set in motion a series of events that ultimately are manifest through the diagnosis of prostate cancer. The understanding of this process is complicated by the various confounds associated with the diagnosis of the disease in clinical trials, general populations, and epidemiologic studies. Additionally, the current inability to distinguish biologically significant from biologically insignificant disease complicates full understanding of the interaction of these factors.
Prostate cancer is an attractive target for chemoprevention, because of its ubiquity, treatment-related morbidity, long latency between premalignant lesions and clinically evident cancer, and defined molecular pathogenesis. The PCPT provides the first firm evidence that this cancer can be prevented by a relatively nontoxic oral agent. The current body of evidence argues against a routine recommendation of any dietary or nutritional supplement for the prevention of prostate cancer.
Andriole GL, Grubb RL3rd, Buys SS, et al. Mortality results from a randomized prostate-cancer screening trial. N Engl J Med. 2009;360:1310-1319.
Gaziano JM, Glynn RJ, Christen WG, et al. Vitamins E and C in the prevention of prostate and total cancer in men: the Physicians’ Health Study II randomized controlled trial. JAMA. 2009;301:52-62.
Kramer BS, Hagerty KL, Justman S, et al. Use of 5-alpha-reductase inhibitors for prostate cancer chemoprevention: American Society of Clinical Oncology/American Urological Association 2008 Clinical Practice Guideline. J Clin Oncol. 2009;27:1502-1516.
Lin K, Lipsitz R, Miller T, et al. Benefits and harms of prostate-specific antigen screening for prostate cancer: an evidence update for the U.S. Preventive Services Task Force. Ann Intern Med. 2008;149:192-199.
Lippman SM, Klein EA, Goodman PJ, et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA. 2009;301:39-51.
Redman MR, Tangen C, Goodman PJ, et al. Finasteride does not increase the risk of high-grade prostate cancer: a bias-adjusted modeling approach. Cancer Prev Res. 2008;1:174. 174–81
Schroder FH, Hugosson J, Roobol MJ, et al. Screening and prostate-cancer mortality in a randomized European study. N Engl J Med. 2009;360(13):1320-1328.
Thompson IM, Goodman PJ, Tangen CM, et al. The influence of finasteride on the development of prostate cancer. N Engl J Med. 2003;349:215-224.
Tomlins SA, Rhodes DR, Perner S, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644-648.
Witte JS. Prostate cancer genomics: towards a new understanding. Nat Rev Genet. 2009;10:77-82.
Abdulkadir SA, Magee JA, Peters TJ, et al. Conditional loss of Nkx3.1 in adult mice induces prostatic intraepithelial neoplasia. Mol Cell Biol. 2002;22:1495-1503.
Adhami VM, Ahmad N, Mukhtar H. Molecular targets for green tea in prostate cancer prevention. J Nutr. 2003;133:2417S-2424S.
Ahlbom A, Lichtenstein P, Malmstrom H, et al. Cancer in twins: genetic and nongenetic familial risk factors. J Natl Cancer Inst. 1997;89:287-293.
Ahn J, Peters U, Albanes D, et al. Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial Project Team. Serum vitamin D concentration and prostate cancer risk: a nested case-control study. J Natl Cancer Inst. 2008;100:796-804.
Albanes D, Heinonen OP, Huttunen JK, et al. Effects of alpha-tocopherol and beta-carotene supplements on cancer incidence in the alpha-tocopherol beta-carotene cancer prevention study. Am J Clin Nutr. 1995;62:1427S-1430S.
Albanes D, Heinonen OP, Taylor PR, et al. Alpha-tocopherol and beta-carotene supplements and lung cancer incidence in the alpha-tocopherol, beta-carotene cancer prevention study: effects of base-line characteristics and study compliance. J Natl Cancer Inst. 1996;88:1560-1570.
American Cancer Society. Cancer facts and figures 2008. http://www.cancer.org/acs/groups/content/@nho/documents/document/2008cafffinalsecuredpdf.pdf. 2008 [accessed 06.04.11]
American Urological Association. Prostate-specific antigen best practice statement: 2009 update. www.auanet.org/content/media/psa09.pdf, 2009. [accessed 16.06.11]
Amundadottir LT, Sulem P, Gudmundsson J, et al. A common variant associated with prostate cancer in European and African populations. Nat Genet. 2006;38:652-658.
Andriole G, Bostwick D, Brawley O, et al. Chemoprevention of prostate cancer in men at high risk: rationale and design of the Reduction by Dutasteride of Prostate Cancer Events (REDUCE) trial. J Urol. 2004;172:1314-1317.
Andriole G, Bruchovsky N, Chung LW, et al. Dihydrotestosterone and the prostate: the scientific rationale for 5alpha-reductase inhibitors in the treatment of benign prostatic hyperplasia. J Urol. 2004;172:1399-1403.
Andriole GL, Grubb RL3rd, Buys SS, et al. Mortality results from a randomized prostate-cancer screening trial. N Engl J Med. 2009;360:1310-1319.
Andriole GL, Roehrborn C, Schulman C, et al. Effect of dutasteride on the detection of prostate cancer in men with benign prostatic hyperplasia. Urology. 2004;64:537-541.
Aparicio Gallego G, Diaz Prado S, Jimenez Fonseca P, et al. Cyclooxygenase-2 (COX-2): a molecular target in prostate cancer. Clin Transl Oncol. 2007;9:694-702.
Aronson WJ, Tymchuk CN, Elashoff RM, et al. Decreased growth of human prostate LNCaP tumors in SCID mice fed a low-fat, soy protein diet with isoflavones. Nutr Cancer. 1999;35:130-136.
Baglietto L, Severi G, English DR, et al. Alcohol consumption and prostate cancer risk: results from the Melbourne Collaborative Cohort Study. Int J Cancer. 2006;119:1501-1504.
Baillargeon J, Pollock BH, Kristal AR, et al. The association of body mass index and prostate-specific antigen in a population-based study. Cancer. 2005;103:1092-1095.
Balic I, Graham ST, Troyer DA, et al. Androgen receptor length polymorphism associated with prostate cancer risk in Hispanic men. J Urol. 2002;168:2245-2248.
Barocas DA, Cowan JE, Smith JAJr, et al. What percentage of patients with newly diagnosed carcinoma of the prostate are candidates for surveillance? An analysis of the CaPSURE database. J Urol. 2008;180:1330-1334.
Barrett-Connor E, Garland C, McPhillips JB, et al. A prospective, population-based study of androstenedione, estrogens, and prostatic cancer. Cancer Res. 1990;50:169-173.
Berthon P, Valeri A, Cohen-Akenine A, et al. Predisposing gene for early-onset prostate cancer, located on chromosome 1q42.2-43. Am J Hum Genet. 1998;62:1416-1420.
Bettuzzi S, Brausi M, Rizzi F, et al. Chemoprevention of human prostate cancer by oral administration of green tea catechins in volunteers with high-grade prostate intraepithelial neoplasia: a preliminary report from a one-year proof-of-principle study. Cancer Res. 2006;66:1234-1240.
Bhatia-Gaur R, Donjacour AA, Sciavolino PJ, et al. Roles for Nkx3.1 in prostate development and cancer. Genes Dev. 1999;13:966-977.
Blasco MA, Lee HW, Hande MP, et al. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell. 1997;91:25-34.
Bonovas S, Filioussi K, Sitaras NM. Statin use and the risk of prostate cancer: a metaanalysis of 6 randomized clinical trials and 13 observational studies. Int J Cancer. 2008;123:899-904.
Bostwick DG, Burke HB, Djakiew D, et al. Human prostate cancer risk factors. Cancer. 2004;101:2371-2490.
Bowen C, Bubendorf L, Voeller HJ, et al. Loss of NKX3.1 expression in human prostate cancers correlates with tumor progression. Cancer Res. 2000;60:6111-6115.
Boyle BJ, Zhao XY, Cohen P, Feldman D. Insulin-like growth factor binding protein-3 mediates 1 alpha,25-dihydroxyvitamin d(3) growth inhibition in the LNCaP prostate cancer cell line through p21/WAF1. J Urol. 2001;165:1319-1324.
Breast Cancer Linkage Consortium. Cancer risks in BRCA2 mutation carriers. J Natl Cancer Inst. 1999;91:1310-1316.
Brusselmans K, De Schrijver E, Heyns W, et al. Epigallocatechin-3-gallate is a potent natural inhibitor of fatty acid synthase in intact cells and selectively induces apoptosis in prostate cancer cells. Int J Cancer. 2003;106:856-862.
Bubendorf L, Kononen J, Koivisto P, et al. Survey of gene amplifications during prostate cancer progression by high-throughput fluorescence in situ hybridization on tissue microarrays. Cancer Res. 1999;59:803-806.
Bui M, Reiter RE. Stem cell genes in androgen-independent prostate cancer. Cancer Metastasis Rev. 1998;17:391-399.
Bunker CH, McDonald AC, Evans RW, et al. A randomized trial of lycopene supplementation in Tobago men with high prostate cancer risk. Nutr Cancer. 2007;57:130-137.
Buschemeyer WC3rd, Freedland SJ. Obesity and prostate cancer: epidemiology and clinical implications. Eur Urol. 2007;52:331-343.
Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med. 2003;348:1625-1638.
Carpten J, Nupponen N, Isaacs S, et al. Germline mutations in the ribonuclease L gene in families showing linkage with HPC1. Nat Genet. 2002;30:181-184.
Carter BS, Beaty TH, Steinberg GD, et al. Mendelian inheritance of familial prostate cancer. Proc Natl Acad Sci U S A. 1992;89:3367-3371.
Carter BS, Bova GS, Beaty TH, et al. Hereditary prostate cancer: epidemiologic and clinical features. J Urol. 1993;150:797-802.
Casey G, Neville PJ, Plummer SJ, et al. RNASEL Arg462Gln variant is implicated in up to 13% of prostate cancer cases. Nat Genet. 2002;32:581-583.
Catalona WJ, Smith DS, Ratliff TL, Basler JW. Detection of organ-confined prostate cancer is increased through prostate-specific antigen-based screening. JAMA. 1993;270:948-954.
Chan JM, Gann PH, Giovannucci EL. Role of diet in prostate cancer development and progression. J Clin Oncol. 2005;23:8152-8160.
Chan JM, Giovannucci EL. Dairy products, calcium, and vitamin D and risk of prostate cancer. Epidemiol Rev. 2001;23:87-92.
Chang BL, Zheng SL, Hawkins GA, et al. Joint effect of HSD3B1 and HSD3B2 genes is associated with hereditary and sporadic prostate cancer susceptibility. Cancer Res. 2002;62:1784-1789.
Chen C, Weiss NS, Stanczyk FZ, et al. Endogenous sex hormones and prostate cancer risk: a case-control study nested within the carotene and retinol efficacy trial. Cancer Epidemiol Biomarkers Prev. 2003;12:1410-1416.
Choan E, Segal R, Jonker D, et al. A prospective clinical trial of green tea for hormone refractory prostate cancer: an evaluation of the complementary/alternative therapy approach. Urol Oncol. 2005;23:108-113.
Chu NF, Spiegelman D, Yu J, et al. Plasma leptin concentrations and four-year weight gain among US men. Int J Obes Relat Metab Disord. 2001;25:346-353.
Chung WK, Leibel RL. The links between obesity, leptin, and prostate cancer. Cancer J. 2006;12:178-181.
Civantos F, Soloway MS, Pinto JE. Histopathological effects of androgen deprivation in prostatic cancer. Semin Urol Oncol. 1996;14:22-31.
Clark LC, Combs GFJr, Turnbull BW, et al. Effects of selenium supplementation for cancer prevention in patients with carcinoma of the skin. A randomized controlled trial. Nutritional Prevention of Cancer Study Group. JAMA. 1996;276:1957-1963.
Clinton SK, Palmer SS, Spriggs CE, Visek WJ. Growth of dunning transplantable prostate adenocarcinomas in rats fed diets with various fat contents. J Nutr. 1988;118:908-914.
Cohen LA, Zhao Z, Pittman B, Scimeca J. Effect of soy protein isolate and conjugated linoleic acid on the growth of dunning R-3327-AT-1 rat prostate tumors. Prostate. 2003;54:169-180.
Cohen P, Peehl DM, Lamson G, Rosenfeld RG. Insulin-like growth factors (IGFs), IGF receptors, and IGF-binding proteins in primary cultures of prostate epithelial cells. J Clin Endocrinol Metab. 1991;73:401-407.
Cohen P, Peehl DM, Rosenfeld RG. The IGF axis in the prostate. Horm Metab Res. 1994;26:81-84.
Coleman MP, Quaresma M, Berrino F, et al. Cancer survival in five continents: a worldwide population-based study (CONCORD). Lancet Oncol. 2008;9:730-756.
Collin SM, Martin RM, Metcalfe C, et al. Prostate-cancer mortality in the USA and UK in 1975-2004: an ecological study. Lancet Oncol. 2008;9:445-452.
Cooney KA, McCarthy JD, Lange E, et al. Prostate cancer susceptibility locus on chromosome 1q: a confirmatory study. J Natl Cancer Inst. 1997;89:955-959.
Cordon-Cardo C, Koff A, Drobnjak M, et al. Distinct altered patterns of p27KIP1 gene expression in benign prostatic hyperplasia and prostatic carcinoma. J Natl Cancer Inst. 1998;90:1284-1291.
Coussens LM, Werb Z. Inflammation and cancer. [See comment]. Nature. 2002;420:860-867.
Cox B, Sneyd MJ, Paul C, et al. Vasectomy and risk of prostate cancer. JAMA. 2002;287:3110-3115.
Craft N, Shostak Y, Carey M, Sawyers CL. A mechanism for hormone-independent prostate cancer through modulation of androgen receptor signaling by the HER-2/neu tyrosine kinase. Nat Med. 1999;5:280-285.
Crowe FL, Key TJ, Appleby PN, et al. Dietary fat intake and risk of prostate cancer in the European prospective investigation into cancer and nutrition. Am J Clin Nutr. 2008;87:1405-1413.
Culig Z, Hobisch A, Cronauer MV, et al. Androgen receptor activation in prostatic tumor cell lines by insulin-like growth factor-I, keratinocyte growth factor, and epidermal growth factor. Cancer Res. 1994;54:5474-5478.
Cyrus-David MS, Weinberg A, Thompson T, Kadmon D. The effect of statins on serum prostate specific antigen levels in a cohort of airline pilots: a preliminary report. J Urol. 2005;173:1923-1925.
Dahan K, Fennal M, Kumar NB. Lycopene in the prevention of prostate cancer. J Soc Integr Oncol. 2008;6:29-36.
Dalais FS, Meliala A, Wattanapenpaiboon N, et al. Effects of a diet rich in phytoestrogens on prostate-specific antigen and sex hormones in men diagnosed with prostate cancer. Urology. 2004;64:510-515.
Das D, Shah RB, Imperiale MJ. Detection and expression of human BK virus sequences in neoplastic prostate tissues. Oncogene. 2004;23:7031-7046.
Das D, Wojno K, Imperiale MJ. BK virus as a cofactor in the etiology of prostate cancer in its early stages. J Virol. 2008;82:2705-2714.
De Marzo AM, Marchi VL, Epstein JI, Nelson WG. Proliferative inflammatory atrophy of the prostate: implications for prostatic carcinogenesis. Am J Pathol. 1999;155:1985-1992.
De Marzo AM, Platz EA, Sutcliffe S, Xu J, et al. Inflammation in prostate carcinogenesis. Nat Rev Cancer. 2007;7:256-269.
Demichelis F, Fall K, Perner S, et al. TMPRSS2:ERG gene fusion associated with lethal prostate cancer in a watchful waiting cohort. Oncogene. 2007;26:4596-4599.
Denis L, Morton MS, Griffiths K. Diet and its preventive role in prostatic disease. Eur Urol. 1999;35:377-387.
Dennis LK, Dawson DV. Meta-analysis of measures of sexual activity and prostate cancer. Epidemiology. 2002;13:72-79.
Dennis LK, Dawson DV, Resnick MI. Vasectomy and the risk of prostate cancer: a meta-analysis examining vasectomy status, age at vasectomy, and time since vasectomy. Prostate Cancer Prostatic Dis. 2002;5:193-203.
Dennis LK, Lynch CF, Torner JC. Epidemiologic association between prostatitis and prostate cancer. Urology. 2002;60:78-83.
Dennis LK, Snetselaar LG, Smith BJ, et al. Problems with the assessment of dietary fat in prostate cancer studies. Am J Epidemiol. 2004;160:436-444.
Deo DD, Rao AP, Bose SS, et al. Differential effects of leptin on the invasive potential of androgen-dependent and -independent prostate carcinoma cells. J Biomed Biotechnol. 2008;2008:163902.
Derweesh IH, Kupelian PA, Zippe C, et al. Continuing trends in pathological stage migration in radical prostatectomy specimens. Urol Oncol. 2004;22:300-306.
DeWitt DL, Smith WL. Primary structure of prostaglandin G/H synthase from sheep vesicular gland determined from the complementary DNA sequence. Proc Natl Acad Sci U S A. 1988;85:1412-1416.
Di Cristofano A, De Acetis M, Koff A, et al. Pten and p27KIP1 cooperate in prostate cancer tumor suppression in the mouse. Nat Genet. 2001;27:222-224.
Di Lorenzo G, Tortora G, D’Armiento FP, et al. Expression of epidermal growth factor receptor correlates with disease relapse and progression to androgen-independence in human prostate cancer. Clin Cancer Res. 2002;8:3438-3444.
Dobosy JR, Roberts JL, Fu VX, Jarrard DF. The expanding role of epigenetics in the development, diagnosis and treatment of prostate cancer and benign prostatic hyperplasia. J Urol. 2007;177:822-831.
Dong JT. Prevalent mutations in prostate cancer. J Cell Biochem. 2006;97:433-447.
Dong B, Kim S, Hong S, et al. An infectious retrovirus susceptible to an IFN antiviral pathway from human prostate tumors. Proc Natl Acad Sci U S A. 2007;104:1655-1660.
Dong F, Reuther AM, Magi-Galluzzi C, et al. Pathologic stage migration has slowed in the late PSA era. Urology. 2007;70:839-842.
Duffield-Lillico AJ, Dalkin BL, Reid ME, et al. Selenium supplementation, baseline plasma selenium status and incidence of prostate cancer: an analysis of the complete treatment period of the nutritional prevention of cancer trial. BJU Int. 2003;91:608-612.
Edwards SM, Kote-Jarai Z, Meitz J, et al. Two percent of men with early-onset prostate cancer harbor germline mutations in the BRCA2 gene. Am J Hum Genet. 2003;72:1-12.
Eeles RA, Dearnaley DP, Ardern-Jones A, et al. Familial prostate cancer: the evidence and the cancer research Campaign/British prostate group (CRC/BPG) UK familial prostate cancer study. Br J Urol. 1997;79:8-14.
Eeles RA, Durocher F, Edwards S, et al. Linkage analysis of chromosome 1q markers in 136 prostate cancer families. The cancer research Campaign/British prostate group U.K. familial prostate cancer study collaborators. Am J Hum Genet. 1998;62:653-658.
Eeles RA, Kote-Jarai Z, Giles GG, et al. Multiple newly identified loci associated with prostate cancer susceptibility. Nat Genet. 2008;40:316-321.
Endogenous Hormones, Prostate Cancer Collaborative GroupRoddam AW, Allen NE, et al. Endogenous sex hormones and prostate cancer: a collaborative analysis of 18 prospective studies. J Natl Cancer Inst. 2008;100:170-183.
Epstein JI, Walsh PC, Carmichael M, Brendler CB. Pathologic and clinical findings to predict tumor extent of nonpalpable (stage T1c) prostate cancer. JAMA. 1994;271:368-374.
Etminan M, Takkouche B, Caamano-Isorna F. The role of tomato products and lycopene in the prevention of prostate cancer: a meta-analysis of observational studies. Cancer Epidemiol Biomarkers Prev. 2004;13:340-345.
Etzioni R, Legler JM, Feuer EJ, et al. Cancer surveillance series: interpreting trends in prostate cancer—part III: quantifying the link between population prostate-specific antigen testing and recent declines in prostate cancer mortality. J Natl Cancer Inst. 1999;91:1033-1039.
Evans S, Metcalfe C, Ibrahim F, et al. Investigating black-white differences in prostate cancer prognosis: a systematic review and meta-analysis. Int J Cancer. 2008;123:430-435.
Fernandez L, Galan Y, Jimenez R, et al. Sexual behaviour, history of sexually transmitted diseases, and the risk of prostate cancer: a case-control study in Cuba. Int J Epidemiol. 2005;34:193-197.
Feuer EJ, Merrill RM, Hankey BF. Cancer surveillance series: interpreting trends in prostate cancer—part II: cause of death misclassification and the recent rise and fall in prostate cancer mortality. J Natl Cancer Inst. 1999;91:1025-1032.
Fischer N, Hellwinkel O, Schulz C, et al. Prevalence of human gammaretrovirus XMRV in sporadic prostate cancer. J Clin Virol. 2008;43:277-283.
Fossa A, Lilleby W, Fossa SD, et al. Independent prognostic significance of HER-2 oncoprotein expression in pN0 prostate cancer undergoing curative radiotherapy. Int J Cancer. 2002;99:100-105.
Frankenberry KA, Somasundar P, McFadden DW, Vona-Davis LC. Leptin induces cell migration and the expression of growth factors in human prostate cancer cells. Am J Surg. 2004;188:560-565.
Freedman DM, Dosemeci M, McGlynn K. Sunlight and mortality from breast, ovarian, colon, prostate, and non-melanoma skin cancer: a composite death certificate based case-control study. Occup Environ Med. 2002;59:257-262.
Friedman JM. The function of leptin in nutrition, weight, and physiology. Nutr Rev. 2002;60:S1-S14.
Furukawa S, Fujita T, Shimabukuro M, et al. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest. 2004;114:1752-1761.
Futreal PA, Coin L, Marshall M, et al. A census of human cancer genes. Nat Rev Cancer. 2004;4:177-183.
Gallina A, Chun FK, Suardi N, et al. Comparison of stage migration patterns between Europe and the USA: an analysis of 11 350 men treated with radical prostatectomy for prostate cancer. BJU Int. 2008;101:1513-1518.
Gann PH, Hennekens CH, Ma J, et al. Prospective study of sex hormone levels and risk of prostate cancer. J Natl Cancer Inst. 1996;88:1118-1126.
Gaziano JM, Glynn RJ, Christen WG, et al. Vitamins E and C in the prevention of prostate and total cancer in men: the Physicians’ Health Study II randomized controlled trial. JAMA. 2009;301:52-62.
Ghosh A, Wang X, Klein E, Heston WD. Novel role of prostate-specific membrane antigen in suppressing prostate cancer invasiveness. Cancer Res. 2005;65:727-731.
Gibbs M, Stanford JL, McIndoe RA, et al. Evidence for a rare prostate cancer-susceptibility locus at chromosome 1p36. Am J Hum Genet. 1999;64:776-787.
Giles GG, Severi G, English DR, et al. Sexual factors and prostate cancer. BJU Int. 2003;92:211-216.
Giovannucci E. Insulin and colon cancer. Cancer Causes Control. 1995;6:164-179.
Giovannucci E, Ascherio A, Rimm EB, et al. A prospective cohort study of vasectomy and prostate cancer in US men. JAMA. 1993;269:873-877.
Giovannucci E, Clinton SK. Tomatoes, lycopene, and cancer: review of the epidemiologic literature. J Natl Cancer Inst. 1999;91:317-331.
Giovannucci E, Tosteson TD, Speizer FE, et al. A retrospective cohort study of vasectomy and prostate cancer in US men. JAMA. 1993;269:878-882.
Godley PA, Schenck AP, Amamoo MA, et al. Racial differences in mortality among Medicare recipients after treatment for localized prostate cancer. J Natl Cancer Inst. 2003;95:1702-1710.
Goldgar DE, Easton DF, Cannon-Albright LA, Skolnick MH. Systematic population-based assessment of cancer risk in first-degree relatives of cancer probands. J Natl Cancer Inst. 1994;86:1600-1608.
Goldstein AS, Lawson DA, Cheng D, et al. Trop2 identifies a subpopulation of murine and human prostate basal cells with stem cell characteristics. Proc Natl Acad Sci U S A. 2008;105:20882-20887.
Gong Z, Neuhouser ML, Goodman PJ, et al. Obesity, diabetes, and risk of prostate cancer: results from the prostate cancer prevention trial. Cancer Epidemiol Biomarkers Prev. 2006;15:1977-1983.
Goode EL, Stanford JL, Peters MA, et al. Clinical characteristics of prostate cancer in an analysis of linkage to four putative susceptibility loci. Clin Cancer Res. 2001;7:2739-2749.
Green MM, Hiley CT, Shanks JH, et al. Expression of vascular endothelial growth factor (VEGF) in locally invasive prostate cancer is prognostic for radiotherapy outcome. Int J Radiat Oncol Biol Phys. 2007;67:84-90.
Gronberg H. Prostate cancer epidemiology. [See comment]. Lancet. 2003;361:859-864.
Gronberg H, Damber L, Damber JE. Studies of genetic factors in prostate cancer in a twin population. J Urol. 1994;152:1484-1487.
Gronberg H, Isaacs SD, Smith JR, Carpten JD, et al. Characteristics of prostate cancer in families potentially linked to the hereditary prostate cancer 1 (HPC1) locus. JAMA. 1997;278:1251-1255.
Gudmundsson J, Sulem P, Steinthorsdottir V, et al. Two variants on chromosome 17 confer prostate cancer risk, and the one in TCF2 protects against type 2 diabetes. Nat Genet. 2007;39:977-983.
Guo Y, Sklar GN, Borkowski A, Kyprianou N. Loss of the cyclin-dependent kinase inhibitor p27(Kip1) protein in human prostate cancer correlates with tumor grade. Clin Cancer Res. 1997;3:2269-2274.
Gurumurthy S, Vasudevan KM, Rangnekar VM. Regulation of apoptosis in prostate cancer. Cancer Metastasis Rev. 2001;20:225-243.
Hamilton RJ, Freedland SJ. Rationale for statins in the chemoprevention of prostate cancer. Curr Urol Rep. 2008;9:189-196.
Hamilton RJ, Goldberg KC, Platz EA, Freedland SJ. The influence of statin medications on prostate-specific antigen levels. J Natl Cancer Inst. 2008;100:1511-1518.
Hankey BF, Feuer EJ, Clegg LX, et al. Cancer surveillance series: interpreting trends in prostate cancer—part I: evidence of the effects of screening in recent prostate cancer incidence, mortality, and survival rates. J Natl Cancer Inst. 1999;91:1017-1024.
Hastak K, Gupta S, Ahmad N, et al. Role of p53 and NF-kappaB in epigallocatechin-3-gallate-induced apoptosis of LNCaP cells. Oncogene. 2003;22:4851-4859.
He WW, Sciavolino PJ, Wing J, et al. A novel human prostate-specific, androgen-regulated homeobox gene (NKX3.1) that maps to 8p21, a region frequently deleted in prostate cancer. Genomics. 1997;43:69-77.
Hedlund TE, Johannes WU, Miller GJ. Soy isoflavonoid equol modulates the growth of benign and malignant prostatic epithelial cells in vitro. Prostate. 2003;54:68-78.
Hla T, Neilson K. Human cyclooxygenase-2 cDNA. Proc Natl Acad Sci U S A. 1992;89:7384-7388.
Hoffman RM, Gilliland FD, Penson DF, et al. Cross-sectional and longitudinal comparisons of health-related quality of life between patients with prostate carcinoma and matched controls. Cancer. 2004;101:2011-2019.
Hsing AW, Tsao L, Devesa SS. International trends and patterns of prostate cancer incidence and mortality. Int J Cancer. 2000;85:60-67.
Huang WY, Hayes R, Pfeiffer R, et al. Sexually transmissible infections and prostate cancer risk. Cancer Epidemiol Biomarkers Prev. 2008;17:2374-2381.
Imperato-McGinley J, Zhu YS. Androgens and male physiology: the syndrome of 5alpha-reductase-2 deficiency. Mol Cell Endocrinol. 2002;198:51-59.
Isaacs JT, Coffey DS. Etiology and disease process of benign prostatic hyperplasia. Prostate Suppl. 1989;2:33-50.
Jacobs ET, Giuliano AR, Martinez ME, et al. Plasma levels of 25-hydroxyvitamin D, 1,25-dihydroxyvitamin D and the risk of prostate cancer. J Steroid Biochem Mol Biol. 2004;89–90:533-537.
Jani AB, Johnstone PA, Liauw SL, et al. Age and grade trends in prostate cancer (1974-2003): a surveillance, epidemiology, and end results registry analysis. Am J Clin Oncol. 2008;31:375-378.
Jhaveri FM, Klein EA, Kupelian PA, et al. Declining rates of extracapsular extension after radical prostatectomy: evidence for continued stage migration. J Clin Oncol. 1999;17:3167-3172.
John EM, Schwartz GG, Koo J, et al. Sun exposure, vitamin D receptor gene polymorphisms, and risk of advanced prostate cancer. Cancer Res. 2005;65:5470-5479.
Joseph IB, Isaacs JT. Potentiation of the antiangiogenic ability of linomide by androgen ablation involves down-regulation of vascular endothelial growth factor in human androgen-responsive prostatic cancers. Cancer Res. 1997;57:1054-1057.
Keaney JFJr, Larson MG, Vasan RS, et al. Obesity and systemic oxidative stress: clinical correlates of oxidative stress in the framingham study. Arterioscler Thromb Vasc Biol. 2003;23:434-439.
Kibel AS, Faith DA, Bova GS, Isaacs WB. Loss of heterozygosity at 12P12-13 in primary and metastatic prostate adenocarcinoma. J Urol. 2000;164:192-196.
Kim S, Kim N, Dong B, et al. Integration site preference of xenotropic murine leukemia virus-related virus, a new human retrovirus associated with prostate cancer. J Virol. 2008;82:9964-9977.
Klein EA, Meyskens FLJr. Potential target populations and clinical models for testing chemopreventive agents. Urology. 2001;57:171-173.
Klein EA, Silverman R. Inflammation, infection, and prostate cancer. Curr Opin Urol. 2008;18:315-319.
Klein EA, Tangen CM, Goodman PJ, et al. Assessing benefit and risk in the prevention of prostate cancer: the prostate cancer prevention trial revisited. J Clin Oncol. 2005;23:7460-7466.
Klein EA, Thompson IM, Lippman SM, et al. SELECT: the next prostate cancer prevention trial. Selenum and Vitamin E Cancer Prevention Trial. J Urol. 2001;166:1311-1315.
Koistinen H, Paju A, Koistinen R, et al. Prostate-specific antigen and other prostate-derived proteases cleave IGFBP-3, but prostate cancer is not associated with proteolytically cleaved circulating IGFBP-3. Prostate. 2002;50:112-118.
Kote-Jarai Z, Easton DF, Stanford JL, et al. Multiple novel prostate cancer predisposition loci confirmed by an international study: the PRACTICAL consortium. Cancer Epidemiol Biomarkers Prev. 2008;17:2052-2061.
Kramer BS, Hagerty KL, Justman S, et al. Use of 5-alpha-reductase inhibitors for prostate cancer chemoprevention: American Society of Clinical Oncology/American Urological Association 2008 Clinical Practice Guideline. J Clin Oncol. 2009;27:1502-1516.
Krishnan AV, Peehl DM, Feldman D. Inhibition of prostate cancer growth by vitamin D: regulation of target gene expression. J Cell Biochem. 2003;88:363-371.
Kulkarni GS, Al-Azab R, Lockwood G, et al. Evidence for a biopsy derived grade artifact among larger prostate glands. J Urol. 2006;175:505-509.
Kumar NB, Cantor A, Allen K, et al. The specific role of isoflavones in reducing prostate cancer risk. Prostate. 2004;59:141-147.
Kumar-Sinha C, Shah RB, Laxman B, et al. Elevated alpha-methylacyl-CoA racemase enzymatic activity in prostate cancer. Am J Pathol. 2004;164:787-793.
Kumar-Sinha C, Tomlins SA, Chinnaiyan AM. Recurrent gene fusions in prostate cancer. Nat Rev Cancer. 2008;8:497-511.
Kupelian P, Kuban D, Thames H, et al. Improved biochemical relapse-free survival with increased external radiation doses in patients with localized prostate cancer: the combined experience of nine institutions in patients treated in 1994 and 1995. Int J Radiat Oncol Biol Phys. 2005;61:415-419.
Labrie F, Dupont A, Simard J, et al. Intracrinology: the basis for the rational design of endocrine therapy at all stages of prostate cancer. Eur Urol. 1993;24:94-105.
Langsenlehner T, Langsenlehner U, Renner W, et al. Single nucleotide polymorphisms and haplotypes in the gene for vascular endothelial growth factor and risk of prostate cancer. Eur J Cancer. 2008;44:1572-1576.
Larson BT, Magi-Galluzzi C, Casey G, et al. Pathological aggressiveness of prostatic carcinomas related to RNASEL R462Q allelic variants. J Urol. 2008;179:1344-1348.
LaTulippe E, Satagopan J, Smith A, et al. Comprehensive gene expression analysis of prostate cancer reveals distinct transcriptional programs associated with metastatic disease. Cancer Res. 2002;62:4499-4506.
Lazier CB, Thomas LN, Douglas RC, et al. Dutasteride, the dual 5alpha-reductase inhibitor, inhibits androgen action and promotes cell death in the LNCaP prostate cancer cell line. Prostate. 2004;58:130-144.
Leav I, Ho SM, Ofner P, et al. Biochemical alterations in sex hormone-induced hyperplasia and dysplasia of the dorsolateral prostates of noble rats. J Natl Cancer Inst. 1988;80:1045-1053.
Leitzmann MF, Platz EA, Stampfer MJ, et al. Ejaculation frequency and subsequent risk of prostate cancer. JAMA. 2004;291:1578-1586.
Levin AM, Zuhlke KA, Ray AM, et al. Sequence variation in alpha-methylacyl-CoA racemase and risk of early-onset and familial prostate cancer. Prostate. 2007;67:1507-1513.
Li LC, Carroll PR, Dahiya R. Epigenetic changes in prostate cancer: implication for diagnosis and treatment. J Natl Cancer Inst. 2005;97:103-115.
Lin B, Ferguson C, White JT, et al. Prostate-localized and androgen-regulated expression of the membrane-bound serine protease TMPRSS2. Cancer Res. 1999;59:4180-4184.
Lin K, Lipsitz R, Miller T, et al. Benefits and harms of prostate-specific antigen screening for prostate cancer: an evidence update for the U.S. preventive services task force. Ann Intern Med. 2008;149:192-199.
Lippman SM, Goodman PJ, Klein EA, et al. Designing the Selenium and Vitamin E Cancer Prevention Trial (SELECT). J Natl Cancer Inst. 2005;97:94-102.
Lippman SM, Klein EA, Goodman PJ, et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA. 2009;301:39-51.
Lucia MS, Darke AK, Goodman PJ, et al. Pathologic characteristics of cancers detected in the prostate cancer prevention trial: implications for prostate cancer detection and chemoprevention. Cancer Prev Res. 2008;1:167. 167–73
Lucia MS, Epstein JI, Goodman PJ, et al. Finasteride and high-grade prostate cancer in the prostate cancer prevention trial. J Natl Cancer Inst. 2007;99:1375-1383.
MacInnis RJ, English DR. Body size and composition and prostate cancer risk: systematic review and meta-regression analysis. Cancer Causes Control. 2006;17:989-1003.
Madigan MP, Troisi R, Potischman N, et al. Serum hormone levels in relation to reproductive and lifestyle factors in postmenopausal women (United States). Cancer Causes Control. 1998;9:199-207.
Makridakis NM, Reichardt JK. Molecular epidemiology of androgen-metabolic loci in prostate cancer: predisposition and progression. J Urol. 2004;171:S25-S28.
Malathi K, Paranjape JM, Ganapathi R, Silverman RH. HPC1/RNASEL mediates apoptosis of prostate cancer cells treated with 2′,5′-oligoadenylates, topoisomerase I inhibitors, and tumor necrosis factor-related apoptosis-inducing ligand. Cancer Res. 2004;64:9144-9151.
McPherson SJ, Wang H, Jones ME, et al. Elevated androgens and prolactin in aromatase-deficient mice cause enlargement, but not malignancy, of the prostate gland. Endocrinology. 2001;142:2458-2467.
Meeker AK. Telomeres and telomerase in prostatic intraepithelial neoplasia and prostate cancer biology. Urol Oncol. 2006;24:122-130.
Meeker AK, Hicks JL, Platz EA, et al. Telomere shortening is an early somatic DNA alteration in human prostate tumorigenesis. Cancer Res. 2002;62:6405-6409.
Mehra R, Tomlins SA, Shen R, et al. Comprehensive assessment of TMPRSS2 and ETS family gene aberrations in clinically localized prostate cancer. Mod Pathol. 2007;20:538-544.
Mehta SS, Lubeck DP, Pasta DJ, Litwin MS. Fear of cancer recurrence in patients undergoing definitive treatment for prostate cancer: results from CaPSURE. J Urol. 2003;170:1931-1933.
Mettlin C, Murphy GP, Lee F, et al. Characteristics of prostate cancers detected in a multimodality early detection program. The investigators of the American Cancer Society—national prostate cancer detection project. Cancer. 1993;72:1701-1708.
Millard PS. Review: bias may contribute to the association between vasectomy and prostate cancer. Evidence-Based Medicine. 1999;4:92.
Monroe KR, Yu MC, Kolonel LN, et al. Evidence of an X-linked or recessive genetic component to prostate cancer risk. Nat Med. 1995;1:827-829.
Muir CS, Nectoux J, Staszewski J. The epidemiology of prostatic cancer. Geographical distribution and time-trends. Acta Oncol. 1991;30:133-140.
Nakayama M, Bennett CJ, Hicks JL, et al. Hypermethylation of the human glutathione-S-transferase-pi gene (GSTP1) CpG island is present in a subset of proliferative inflammatory atrophy lesions but not in normal or hyperplastic epithelium of the prostate: a detailed study using laser-capture microdissection. Am J Pathol. 2003;163:923-933.
Nakayama M, Gonzalgo ML, Yegnasubramanian S, et al. GSTP1 CpG island hypermethylation as a molecular biomarker for prostate cancer. J Cell Biochem. 2004;91:540-552.
Nam RK, Sugar L, Wang Z, et al. Expression of TMPRSS2:ERG gene fusion in prostate cancer cells is an important prognostic factor for cancer progression. Cancer Biol Ther. 2007;6:40-45.
Nam RK, Zhang WW, Loblaw DA, et al. A genome-wide association screen identifies regions on chromosomes 1q25 and 7p21 as risk loci for sporadic prostate cancer. Prostate Cancer Prostatic Dis. 2008;11:241-246.
Nelson WG, De Marzo AM, Isaacs WB. Prostate cancer. N Engl J Med. 2003;349:366-381.
Newcomer LM, Stanford JL, Blumenstein BA, Brawer MK. Temporal trends in rates of prostate cancer: declining incidence of advanced stage disease, 1974 to 1994. J Urol. 1997;158:1427-1430.
Ni J, Chen M, Zhang Y, et al. Vitamin E succinate inhibits human prostate cancer cell growth via modulating cell cycle regulatory machinery. Biochem Biophys Res Commun. 2003;300:357-363.
Nickel JC, Downey J, Pontari MA, et al. A randomized placebo-controlled multicentre study to evaluate the safety and efficacy of finasteride for male chronic pelvic pain syndrome (category IIIA chronic nonbacterial prostatitis). BJU Int. 2004;93:991-995.
Noldus J, Graefen M, Haese A, et al. Stage migration in clinically localized prostate cancer. Eur Urol. 2000;38:74-78.
Oakley-Girvan I, Feldman D, Eccleshall TR, et al. Risk of early-onset prostate cancer in relation to germ line polymorphisms of the vitamin D receptor. Cancer Epidemiol Biomarkers Prev. 2004;13:1325-1330.
Obermuller-Jevic UC, Olano-Martin E, Corbacho AM, et al. Lycopene inhibits the growth of normal human prostate epithelial cells in vitro. J Nutr. 2003;133:3356-3360.
Oliver SE, May MT, Gunnell D. International trends in prostate-cancer mortality in the “PSA ERA”. Int J Cancer. 2001;92:893-898.
Online Mendelian Inheritance in Man. #176807: prostate cancer. www.ncbi.nlm.nih.gov/omim/176807, 2009. [accessed 06.04.11]
Ornish D, Weidner G, Fair WR, et al. Intensive lifestyle changes may affect the progression of prostate cancer. J Urol. 2005;174:1065-1069.
Page WF, Braun MM, Partin AW, et al. Heredity and prostate cancer: a study of World War II veteran twins. Prostate. 1997;33:240-245.
Park SY, Murphy SP, Wilkens LR, et al. Fat and meat intake and prostate cancer risk: the multiethnic cohort study. Int J Cancer. 2007;121:1339-1345.
Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74-108.
Patel DA, Bock CH, Schwartz K, et al. Sexually transmitted diseases and other urogenital conditions as risk factors for prostate cancer: a case-control study in Wayne County, Michigan. Cancer Causes Control. 2005;16:263-273.
Patel MI, Kurek C, Dong Q. The arachidonic acid pathway and its role in prostate cancer development and progression. J Urol. 2008;179:1668-1675.
Peehl DM, Krishnan AV, Feldman D. Pathways mediating the growth-inhibitory actions of vitamin D in prostate cancer. J Nutr. 2003;133:2461S-2469S.
Peters U, Leitzmann MF, Chatterjee N, et al. Serum lycopene, other carotenoids, and prostate cancer risk: A nested case-control study in the prostate, lung, colorectal, and ovarian cancer screening trial. Cancer Epidemiol Biomarkers Prev. 2007;16:962-968.
Peyromaure M, Camparo P, Badoual C, et al. The expression of vascular endothelial growth factor is associated with the risk of cancer progression after radical prostatectomy. BJU Int. 2007;99:1150-1153.
Pinsky P, Parnes H, Ford L. Estimating rates of true high-grade disease in the prostate cancer prevention trial. Cancer Prev Res (Phila Pa). 2008;1:182-186.
Platz EA, Leitzmann MF, Michaud DS, et al. Interrelation of energy intake, body size, and physical activity with prostate cancer in a large prospective cohort study. Cancer Res. 2003;63:8542-8548.
Potosky AL, Kessler L, Gridley G, et al. Rise in prostatic cancer incidence associated with increased use of transurethral resection. J Natl Cancer Inst. 1990;82:1624-1628.
Potts JM, Pasqualotto FF. Seminal oxidative stress in patients with chronic prostatitis. Andrologia. 2003;35:304-308.
Powell IJ, Banerjee M, Bianco FJ, et al. The effect of race/ethnicity on prostate cancer treatment outcome is conditional: a review of Wayne State University data. J Urol. 2004;171:1508-1512.
Prescott JL, Blok L, Tindall DJ. Isolation and androgen regulation of the human homeobox cDNA, NKX3.1. Prostate. 1998;35:71-80.
Prins GS, Korach KS. The role of estrogens and estrogen receptors in normal prostate growth and disease. Steroids. 2008;73:233-244.
Putzi MJ, De Marzo AM. Morphologic transitions between proliferative inflammatory atrophy and high-grade prostatic intraepithelial neoplasia. Urology. 2000;56:828-832.
Qian J, Hirasawa K, Bostwick DG, et al. Loss of p53 and c-myc overrepresentation in stage T(2-3)N(1-3)M(0) prostate cancer are potential markers for cancer progression. Mod Pathol. 2002;15:35-44.
Quinn M, Babb P. Patterns and trends in prostate cancer incidence, survival, prevalence and mortality. Part I: international comparisons. BJU Int. 2002;90:162-173.
Raghow S, Hooshdaran MZ, Katiyar S, Steiner MS. Toremifine prevents prostate cancer in the transgenic adenocarcinoma of mouse prostate model. Cancer Res. 2002;62:1370-1376.
Redman MR, Tangen C, Goodman PJ, et al. Finasteride does not increase the risk of high-grade prostate cancer: a bias-adjusted modeling approach. Cancer Prev Res. 2008;1:174. 174–81
Renehan AG, Tyson M, Egger M, et al. Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. [See comment]. Lancet. 2008;371:569-578.
Rennert H, Zeigler-Johnson CM, Addya K, et al. Association of susceptibility alleles in ELAC2/HPC2, RNASEL/HPC1, and MSR1 with prostate cancer severity in European American and African American men. Cancer Epidemiol Biomarkers Prev. 2005;14:949-957.
Ribeiro R, Lopes C, Medeiros R. The link between obesity and prostate cancer: the leptin pathway and therapeutic perspectives. [See comment]. Prostate Cancer Prostatic Dis. 2006;9:19-24.
Ribeiro R, Vasconcelos A, Costa S, et al. Overexpressing leptin genetic polymorphism (-2548 G/A) is associated with susceptibility to prostate cancer and risk of advanced disease. Prostate. 2004;59:268-274.
Ries LAG, Melbert D, Krapcho M, et al. SEER cancer statistics review, 1975-2007. www.seer.cancer.gov/csr/1975_2007/index.html, 2011. [accessed 06.04.11]
Risbridger GP, Bianco JJ, Ellem SJ, McPherson SJ. Oestrogens and prostate cancer. Endocr Relat Cancer. 2003;10:187-191.
Roberts CK, Vaziri ND, Barnard RJ. Effect of diet and exercise intervention on blood pressure, insulin, oxidative stress, and nitric oxide availability. Circulation. 2002;106:2530-2532.
Roddam AW, Allen NE, Appleby P, et al. Insulin-like growth factors, their binding proteins, and prostate cancer risk: analysis of individual patient data from 12 prospective studies. Ann Intern Med. 2008;149:461-471.
Rodriguez C, Freedland SJ, Deka A, et al. Body mass index, weight change, and risk of prostate cancer in the cancer prevention study II nutrition cohort. Cancer Epidemiol Biomarkers Prev. 2007;16:63-69.
Rodriguez C, McCullough ML, Mondul AM, et al. Calcium, dairy products, and risk of prostate cancer in a prospective cohort of United States men. Cancer Epidemiol Biomarkers Prev. 2003;12:597-603.
Rohrmann S, Linseisen J, Key TJ, et al. Alcohol consumption and the risk for prostate cancer in the European prospective investigation into cancer and nutrition. Cancer Epidemiol Biomarkers Prev. 2008;17:1282-1287.
Ross LE, Berkowitz Z, Ekwueme DU. Use of the prostate-specific antigen test among U.S. men: findings from the 2005 National Health Interview Survey. Cancer Epidemiol Biomarkers Prev. 2008;17:636-644.
Rubin MA, Bismar TA, Andren O, et al. Decreased alpha-methylacyl CoA racemase expression in localized prostate cancer is associated with an increased rate of biochemical recurrence and cancer-specific death. Cancer Epidemiol Biomarkers Prev. 2005;14:1424-1432.
Rubin MA, Gerstein A, Reid K, et al. 10q23.3 loss of heterozygosity is higher in lymph node-positive (pT2-3,N+) versus lymph node-negative (pT2-3,N0) prostate cancer. Hum Pathol. 2000;31:504-508.
Sakr WA, Haas GP, Cassin BF, et al. The frequency of carcinoma and intraepithelial neoplasia of the prostate in young male patients. J Urol. 1993;150:379-385.
Samanta M, Harkins L, Klemm K, et al. High prevalence of human cytomegalovirus in prostatic intraepithelial neoplasia and prostatic carcinoma. J Urol. 2003;170:998-1002.
Sanda MG, Dunn RL, Michalski J, et al. Quality of life and satisfaction with outcome among prostate-cancer survivors. N Engl J Med. 2008;358:1250-1261.
Sarma AV, McLaughlin JC, Wallner LP, et al. Sexual behavior, sexually transmitted diseases and prostatitis: the risk of prostate cancer in black men. J Urol. 2006;176:1108-1113.
Scher HI, Sawyers CL. Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J Clin Oncol. 2005;23:8253-8261.
Schoonen WM, Salinas CA, Kiemeney LA, Stanford JL. Alcohol consumption and risk of prostate cancer in middle-aged men. Int J Cancer. 2005;113:133-140.
Schroder FH, Hugosson J, Roobol MJ, et al. Screening and prostate-cancer mortality in a randomized European study. N Engl J Med. 2009;360:1320-1328.
Schulke N, Varlamova OA, Donovan GP, et al. The homodimer of prostate-specific membrane antigen is a functional target for cancer therapy. Proc Natl Acad Sci U S A. 2003;100:12590-12595.
Sesso HD, Paffenbarger RSJr, Lee IM. Alcohol consumption and risk of prostate cancer: the Harvard Alumni Health Study. Int J Epidemiol. 2001;30:749-755.
Severson RK, Grove JS, Nomura AM, Stemmermann GN. Body mass and prostatic cancer: a prospective study. BMJ. 1988;297:713-715.
Severson RK, Nomura AM, Grove JS, Stemmermann GN. A prospective study of demographics, diet, and prostate cancer among men of Japanese ancestry in Hawaii. Cancer Res. 1989;49:1857-1860.
Sfanos KS, Isaacs WB. An evaluation of PCR primer sets used for detection of propionibacterium acnes in prostate tissue samples. Prostate. 2008;68:1492-1495.
Shah R, Mucci NR, Amin A, et al. Postatrophic hyperplasia of the prostate gland: neoplastic precursor or innocent bystander? Am J Pathol. 2001;158:1767-1773.
Shavers VL, Brown M, Klabunde CN, et al. Race/ethnicity and the intensity of medical monitoring under “watchful waiting” for prostate cancer. Med Care. 2004;42:239-250.
Shimizu H, Ross RK, Bernstein L, et al. Cancers of the prostate and breast among Japanese and white immigrants in Los Angeles County. Br J Cancer. 1991;63:963-966.
Silverman RH. Implications for RNase L in prostate cancer biology. Biochemistry. 2003;42:1805-1812.
Singh Laboratory. Welcome to the Singh laboratory. www.path.utah.edu/labs/singh, 2010. [accessed 04.06.11]
Smith JR, Freije D, Carpten JD, et al. Major susceptibility locus for prostate cancer on chromosome 1 suggested by a genome-wide search. Science. 1996;274:1371-1374.
Snoek R, Cheng H, Margiotti K, et al. In vivo knockdown of the androgen receptor results in growth inhibition and regression of well-established, castration-resistant prostate tumors. Clin Cancer Res. 2009;15:39-47.
Somasundar P, Frankenberry KA, Skinner H, et al. Prostate cancer cell proliferation is influenced by leptin. J Surg Res. 2004;118:71-82.
Stanford JL, Feng Z, Hamilton AS, et al. Urinary and sexual function after radical prostatectomy for clinically localized prostate cancer: the prostate cancer outcomes study. JAMA. 2000;283:354-360.
Steers WD. 5alpha-reductase activity in the prostate. Urology. 2001;58:17-24.
Stephenson RA. Prostate cancer overdiagnosis and overtreatment: analysis of US mortality and SEER incidence. Trends in the PSA and pre-PSA eras. In: Klein EA, editor. Management of prostate cancer. Totowa (NJ): Humana Press; 2005:3-13.
Stephenson RA, Smart CR, Mineau GP, et al. The fall in incidence of prostate carcinoma. On the down side of a prostate specific antigen induced peak in incidence—data from the Utah Cancer Registry. Cancer. 1996;77:1342-1348.
Strickler HD, Goedert JJ. Sexual behavior and evidence for an infectious cause of prostate cancer. Epidemiol Rev. 2001;23:144-151.
Struewing JP, Hartge P, Wacholder S, et al. The risk of cancer associated with specific mutations of BRCA1 and BRCA2 among Ashkenazi Jews. N Engl J Med. 1997;336:1401-1408.
Sutcliffe S, Giovannucci E, De Marzo AM, et al. Gonorrhea, syphilis, clinical prostatitis, and the risk of prostate cancer. Cancer Epidemiol Biomarkers Prev. 2006;15:2160-2166.
Sutcliffe S, Giovannucci E, Gaydos CA, et al. Plasma antibodies against chlamydia trachomatis, human papillomavirus, and human herpesvirus type 8 in relation to prostate cancer: a prospective study. Cancer Epidemiol Biomarkers Prev. 2007;16:1573-1580.
Sutcliffe S, Giovannucci E, Leitzmann MF, et al. A prospective cohort study of red wine consumption and risk of prostate cancer. Int J Cancer. 2007;120:1529-1535.
Svatek RS, Lee JJ, Roehrborn CG, et al. The cost of prostate cancer chemoprevention: a decision analysis model. Cancer Epidemiol Biomarkers Prev. 2006;15:1485-1489.
Svatek RS, Lee JJ, Roehrborn CG, et al. Cost-effectiveness of prostate cancer chemoprevention: a quality of life-years analysis. Cancer. 2008;112:1058-1065.
Takeichi M. Cadherin cell adhesion receptors as a morphogenetic regulator. Science. 1991;251:1451-1455.
Tanaka M, Komuro I, Inagaki H, et al. Nkx3.1, a murine homolog of Ddrosophila bagpipe, regulates epithelial ductal branching and proliferation of the prostate and palatine glands. Dev Dyn. 2000;219:248-260.
Taplin ME, Rajeshkumar B, Halabi S, et al. Androgen receptor mutations in androgen-independent prostate cancer: cancer and leukemia group B study 9663. J Clin Oncol. 2003;21:2673-2678.
Tarone RE, Chu KC, Brawley OW. Implications of stage-specific survival rates in assessing recent declines in prostate cancer mortality rates. Epidemiology. 2000;11:167-170.
Tavtigian SV, Simard J, Teng DH, et al. A candidate prostate cancer susceptibility gene at chromosome 17p. Nat Genet. 2001;27:172-180.
Taylor ML, Mainous AG3rd, Wells BJ. Prostate cancer and sexually transmitted diseases: a meta-analysis. Fam Med. 2005;37:506-512.
Taylor RA, Risbridger GP. The path toward identifying prostatic stem cells. Differentiation. 2008;76:671-681.
Teixeira AL, Ribeiro R, Cardoso D, et al. Genetic polymorphism in EGF is associated with prostate cancer aggressiveness and progression-free interval in androgen blockade-treated patients. Clin Cancer Res. 2008;14:3367-3371.
Thiessen EU. Concerning a familial association between breast cancer and both prostatic and uterine malignancies. Cancer. 1974;34:1102-1107.
Thompson IM, Chi C, Ankerst DP, et al. Effect of finasteride on the sensitivity of PSA for detecting prostate cancer. J Natl Cancer Inst. 2006;98:1128-1133.
Thompson IM, Goodman PJ, Tangen CM, et al. The influence of finasteride on the development of prostate cancer. N Engl J Med. 2003;349:215-224.
Thompson IM, Lucia MS, Redman MW, et al. Finasteride decreases the risk of prostatic intraepithelial neoplasia. J Urol. 2007;178:107-109.
Thompson IM, Tangen CM, Goodman PJ, et al. Finasteride improves the sensitivity of digital rectal examination for prostate cancer detection. J Urol. 2007;177:1749-1752.
Thompson TA, Wilding G. Androgen antagonist activity by the antioxidant moiety of vitamin E, 2,2,5,7,8-pentamethyl-6-chromanol in human prostate carcinoma cells. Mol Cancer Ther. 2003;2:797-803.
Tomlins SA, Rhodes DR, Perner S, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644-648.
Trayhurn P, Wood IS. Adipokines: inflammation and the pleiotropic role of white adipose tissue. Br J Nutr. 2004;92:347-355.
Tsujimura A, Koikawa Y, Salm S, et al. Proximal location of mouse prostate epithelial stem cells: a model of prostatic homeostasis. J Cell Biol. 2002;157:1257-1265.
Tulinius H, Egilsson V, Olafsdottir GH, Sigvaldason H. Risk of prostate, ovarian, and endometrial cancer among relatives of women with breast cancer. BMJ. 1992;305:855-857.
Umbas R, Schalken JA, Aalders TW, et al. Expression of the cellular adhesion molecule E-cadherin is reduced or absent in high-grade prostate cancer. Cancer Res. 1992;52:5104-5109.
Underwood W3rd, Jackson J, Wei JT, et al. Racial treatment trends in localized/regional prostate carcinoma: 1992-1999. Cancer. 2005;103:538-545.
Unger JM, LeBlanc M, Thompson IM, Coltman CAJr. The person-years saved model and other methodologies for assessing the population impact of cancer-prevention strategies. Urol Oncol. 2004;22:362-368.
Urisman A, Molinaro RJ, Fischer N, et al. Identification of a novel gammaretrovirus in prostate tumors of patients homozygous for R462Q RNASEL variant. PLoS Pathog. 2006;2:e25.
van Leenders GJ, Dukers D, Hessels D, et al. Polycomb-group oncogenes EZH2, BMI1, and RING1 are overexpressed in prostate cancer with adverse pathologic and clinical features. Eur Urol. 2007;52:455-463.
Varambally S, Dhanasekaran SM, Zhou M, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419:624-629.
Velicer CM, Kristal A, White E. Alcohol use and the risk of prostate cancer: results from the VITAL cohort study. Nutr Cancer. 2006;56:50-56.
Vicentini C, Festuccia C, Gravina GL, et al. Prostate cancer cell proliferation is strongly reduced by the epidermal growth factor receptor tyrosine kinase inhibitor ZD1839 in vitro on human cell lines and primary cultures. J Cancer Res Clin Oncol. 2003;129:165-174.
Wallstrom P, Bjartell A, Gullberg B, et al. A prospective study on dietary fat and incidence of prostate cancer (Malmo, Sweden). Cancer Causes Control. 2007;18:1107-1121.
Walsh PC. Cancer surveillance series: Interpreting trends in prostate cancer—part I: evidence of the effects of screening in recent prostate cancer incidence, mortality, and survival rates. J Urol. 2000;163:364-365.
Wang S, Gao J, Lei Q, et al. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell. 2003;4:209-221.
Wang Y, Corr JG, Thaler HT, et al. Decreased growth of established human prostate LNCaP tumors in nude mice fed a low-fat diet. J Natl Cancer Inst. 1995;87:1456-1462.
Warner E, Foulkes W, Goodwin P, et al. Prevalence and penetrance of BRCA1 and BRCA2 gene mutations in unselected Ashkenazi Jewish women with breast cancer. J Natl Cancer Inst. 1999;91:1241-1247.
Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661-678.
Williams H, Powell IJ, Land SJ, et al. Vitamin D receptor gene polymorphisms and disease free survival after radical prostatectomy. Prostate. 2004;61:267-275.
Wilson JD, Roehrborn C. Long-term consequences of castration in men: lessons from the skoptzy and the eunuchs of the Chinese and Ottoman courts. J Clin Endocrinol Metab. 1999;84:4324-4331.
Wilt TJ, MacDonald R, Hagerty K, et al. Five-alpha-reductase inhibitors for prostate cancer prevention. Cochrane Database Syst Rev 2008;2:CD007091.
Woolf CM. An investigation of the familial aspects of carcinoma of the prostate. Cancer. 1960;13:739-744.
Wright ME, Chang SC, Schatzkin A, et al. Prospective study of adiposity and weight change in relation to prostate cancer incidence and mortality. Cancer. 2007;109:675-684.
Xiang Y, Wang Z, Murakami J, et al. Effects of RNase L mutations associated with prostate cancer on apoptosis induced by 2′,5′-oligoadenylates. Cancer Res. 2003;63:6795-6801.
Xu J, Meyers D, Freije D, et al. Evidence for a prostate cancer susceptibility locus on the X chromosome. Nat Genet. 1998;20:175-179.
Xu J, Zheng SL, Komiya A, et al. Germline mutations and sequence variants of the macrophage scavenger receptor 1 gene are associated with prostate cancer risk. Nat Genet. 2002;32:321-325.
Yu L, Blackburn GL, Zhou JR. Genistein and daidzein downregulate prostate androgen-regulated transcript-1 (PART-1) gene expression induced by dihydrotestosterone in human prostate LNCaP cancer cells. J Nutr. 2003;133:389-392.
Zambrano A, Kalantari M, Simoneau A, et al. Detection of human polyomaviruses and papillomaviruses in prostatic tissue reveals the prostate as a habitat for multiple viral infections. Prostate. 2002;53:263-276.
Zeegers MP, Jellema A, Ostrer H. Empiric risk of prostate carcinoma for relatives of patients with prostate carcinoma: a meta-analysis. Cancer. 2003;97:1894-1903.
Zeliadt SB, Etzioni RD, Penson DF, et al. Lifetime implications and cost-effectiveness of using finasteride to prevent prostate cancer. Am J Med. 2005;118:850-857.
Zha S, Yegnasubramanian V, Nelson Wg, et al. Cyclooxygenases in cancer: progress and perspective. Cancer Lett. 2004;215:1-20.