CHAPTER 24 Genetic Factors and Periodontal Disease
It is now widely accepted that differences among individuals at risk for developing most diseases have a substantial inherited component. Factors in the environment, such as diet, smoking, preventive care, or exposure to pathogens, interact with each person’s genetic predisposition to determine his or her health outcomes. This complex combination of variables determines if and when a disease affects the person, how fast and how severely symptoms of the disease progress, and how the person responds to different treatments in terms of both side effects and success of alternative therapies. Sometimes, as in diseases such as cystic fibrosis or muscular dystrophy, the genetic component of risk predominates, and differences in environment play only a minor role. In other diseases, factors in the environment are most important, and variation inherited in the person’s deoxyribonucleic acid (DNA) has only an infrequent or minor influence on disease susceptibility or progression. Examples of the latter include infectious diseases, such as human immunodeficiency virus/acquired immunodeficiency syndrome (HIV/AIDS) or cancers, such as mesothelioma (strongly associated with asbestos exposure). The majority of human diseases fall half-way between these two extremes; genes and environment both play very important roles.
Most cases of periodontitis appear to fit this complex genes and environment model. With the exception of a handful of very rare syndromes caused by mutations in single genes, evidence indicates that inherited variation in DNA has a role roughly equal to that of the environment in determining who remains periodontally healthy versus who is affected by this disease. Beyond this broad generalization, however, and despite hundreds of studies of genetic polymorphisms reported to date, our understanding as to which specific genes are important remains extremely limited. We know even less about the likely (though unproven) role that individual genetic differences may have in determining how patients respond to alternative periodontal treatments.
In this chapter, the challenges and barriers that have thus far limited progress in advancing our knowledge of the complex genetics of periodontitis will be reviewed. This requires a clear understanding of not only the architecture of the human genome and the complexity of genetic susceptibility but also the critical roles that statistical power and sample size play in the process of discovery. These latter issues are important for all areas of research, but especially for situations in which a large number of variables need to be evaluated. In genetic studies, over 23,000 genes need to be considered as potential hypotheses or “candidates” for being disease risk factors. Depending on the gene’s size and the frequency of genetic recombination in the gene’s chromosomal region, scientists need to evaluate between a handful or up to several hundred inherited DNA variants in each gene as potential “biomarkers” of disease risk. Each one of these variants essentially amounts to a test of the hypothesis as to whether the variant is associated with disease risk. Furthermore, many complex diseases have been found to be very strongly associated with DNA variation in parts of the human genome where no genes are known to exist, but where the genetic material may have important functional effects nonetheless. Therefore, to fully evaluate the entire human genome, the total number of hypotheses that need to be tested is truly enormous—roughly involving the equivalent of one million independent statistical tests. Only in the past few years have laboratory and computational tools been developed that make this scale of work technically possible, and thus far only one recent study has reported results of a whole genome analysis for periodontitis.
Opportunities for advanced study designs and next-generation genomic technologies to improve understanding of the inherited basis of periodontitis make it likely (although by no means certain) that genetic variation will become an important variable to be routinely considered by practicing dentists within the next 5 to 10 years. If and when this occurs, dentists will need to learn how to access and interpret the massive amounts of information coded in the >3 billion bases of DNA in the human genome and to incorporate this into improved evidence-based practices for periodontal disease prevention, diagnosis, and treatment. Alternatively, new interdisciplinary teams will need to be established with dentists as key members working closely with experts in bioinformatics, genomics, and genetic counseling so that patients will be able to reap the potentially significant benefits of these scientific advances. However the process evolves, it will surely lead to revolutionary changes in both dental education and dental practice in the not-so-distant future.8,23
Genetic and Genomic Methods of the 21st Century
Most health care professionals are aware of many major advances in genetics accomplished during the past 2 decades since the Human Genome Project officially began in 1990.41 Headlines and announcements of “breakthroughs” have appeared regularly in print and television media. Too often, many of these stories seem to promise rapid and significant improvements in health care that are totally unrealistic.11 More cautious assessments are usually not deemed as newsworthy by the media nor is cautious understatement always in the interest of private companies and granting agencies involved in basic research and clinical trials. Consequently, the public has often received an overly optimistic picture of what the future of medical care based on genomic medicine really holds for them. Despite this all-too-common overstatement about rapid translation to clinical practice, advances in technical capabilities for genomic data acquisition and the accumulation of knowledge in the field of genetics have been truly enormous. The eventual impact of this explosion of biologic knowledge on all areas of human health, including those concerning the field of dentistry, over the longer term is certain to be very substantial.
Unfortunately, for reasons explored further in the next section, genomic advances have thus far contributed very little to advancing our understanding of the molecular-pathologic causes of periodontitis or pointed to ways for improving treatment through “individualized” approaches based on patients’ inherited genetic variation. Only recently has the possibility of making major progress been realized based on newly available genomic tools and approaches of Genome Wide Association Studies (GWAS, pronounced “gee-wahs”) and “next generation” DNA sequencing techniques. For these research strategies to be successful, however, they must be combined with further improvement in research definitions of periodontal disease and much larger sample sizes than have been used in most previous genetic studies of this condition. To help improve understanding, some of the commonly used terms in genetics are explained in Table 24-1.
TABLE 24-1 Glossary of Terms Relevant to the Genetics of Periodontal Disease
| Allele | One of several possible alternative forms of a gene caused by small or large differences in the DNA sequence within or near the gene. These differences arise by mutation, and some may affect the function of the gene product (i.e., a protein) or its abundance in different kinds of cells. |
| Autosome | A chromosome that is not a sex chromosome. |
| Autosomal dominant | DNA variation in a gene located on an autosome that has a dominant effect over other forms of variation at this location within the gene. When the dominant DNA sequence is present in combination with some other sequence, the gene’s function is entirely or nearly entirely determined by the dominant sequence while the alternative sequence that occurs on the person’s other chromosome is essentially silent. |
| Autosomal recessive | DNA variation in a gene located on an autosome that has an effect on the gene’s function only when the person has inherited two copies, one from the mother and the other from the father. For example, if an individual has two copies of an abnormal gene that is autosomal recessive, they will be subject to the effects of that gene. |
| Chromosome | A nuclear structure that contains genetic information. Humans have 46 chromosomes, arranged in 23 pairs. There are 22 pairs of autosomes and a pair of sex chromosomes (either XX or XY). |
| Concordance | The probability that a pair of individuals (e.g., twins) both have a certain characteristic (e.g., periodontal disease), given that one of the pair has the characteristic. Presented as a number from 0 to 1, or as a percentage. |
| Dizygotic twins | Twins that have resulted from the fertilization of two separate eggs. They are no more similar to each other (from a genetic perspective) than are nontwin siblings. Nonidentical twins. |
| Epigenetics | Term used to describe the changes in phenotype or gene expression that result from mechanisms other than changes in the underlying DNA sequences (i.e., changes in which the gene is expressed rather than a change in the DNA sequence itself). Nongenetic factors cause the organism’s genes to be expressed differently. |
| Exon | Protein coding regions of DNA. |
| Frameshift mutation | A mutation that results from insertion or deletion of one or more nucleotides into a gene, causing the coding regions to be read in the wrong frame and usually causing the protein produced to be defective in function. |
| Gene | The basic unit of heredity that occupies a specific position (locus) on a chromosome and has specific effect(s) on the phenotype of the organism. A piece of DNA that is transcribed into a molecule of RNA and then translated into a protein. |
| Gene expression | The process by which the information in a gene is utilized via transcription and translation, leading to production of protein. Differences in gene expression can affect the phenotype of the organism including risk of disease. |
| Genetic code | In RNA and DNA, the consecutive nucleotide triplets (codons) that specify the sequence of amino acids for protein synthesis (translation). |
| Genome | The entire hereditary information of an organism. Refers to all the genes and other nongene portions of DNA carried by an individual cell. |
| Genotype | The genetic makeup of an organism or cell, distinct from its expressed features or phenotype. |
| Haplotype | A contraction of the term haploid genotype. Refers to a combination of alleles at multiple loci, which are usually transmitted together on the same region of a chromosome. |
| Heredity | The passing of traits to offspring from parents/ancestors. In biology, the study of heredity is referred to as genetics. As a result of heredity, variation among individuals allows species to evolve by natural selection in response to changes in their environment or by random change over long periods of time. |
| Homozygous | The presence of identical alleles at a specific position in a gene. |
| Heterozygous | The presence of two different alleles at a specific position in a gene. |
| Intron | A DNA region within a gene that is not translated into protein. These intervening (noncoding) portions of DNA or RNA are removed during RNA processing. |
| Isoform | Any of several different forms of the same protein. May be produced from related genes, or may arise from the same gene by alternative splicing. Many isoforms are caused by SNPs. |
| Ligand | A molecule that binds to another molecule, usually binding to a cellular receptor molecule. |
| Linkage | A term to describe the tendency for certain genes to be transmitted from parent to child together because they are located close to each other on the same chromosome. |
| Linkage disequilibrium | The occurrence of specific alleles at different locations in the DNA that are relatively close to each other (linked) more often than would be expected by chance alone (disequilibrium). |
| Locus | The physical location a gene occupies within a chromosome. (Plural: loci). |
| Monozygotic twins | Twins with identical genetic makeup (i.e., identical twins) as a result of fertilization of a single egg that then splits into two embryos. |
| Mutation | Changes in the DNA sequence of the genome can result from errors that occur during DNA replication or meiosis and can be caused by radiation, viruses, mutagenic chemicals. Most mutations have very little or no measurable effect on the gene’s function; some are harmful and a rare few may be advantageous. |
| Nucleotide | Molecules that when linked together make up the structural units of RNA and DNA. They are composed of a phosphate group, the bases adenine, cytosine, guanine, and thymine, and a pentose sugar. In RNA the thymine base is replaced by uracil. |
| Penetrance | The proportion of individuals who have a particular allele/genotype who express an associated trait (phenotype). Genotypes with a high penetrance result in a larger number of individuals in the population with the associated phenotype compared to genotypes with a low penetrance. |
| Phenotype | The observable characteristics displayed by an organism (e.g., morphology, development, gender, eye color, physiologic properties, or behavior). Phenotype results from expression of the organism’s genes, as well as the influence of environmental factors, and interactions between the two. |
| Polymorphism | Polymorphism exists when two or more different phenotypes exist within different individuals of the same population. In the context of genetics, it refers to a region of the genome that varies between individual members of the population in such proportions that the rarest of them cannot be maintained just by recurrent mutation. Polymorphism may be actively maintained in populations by natural selection and also by random drift. |
| Sequencing | Determining in the laboratory the linear arrangement of nucleotides (in RNA or DNA) or amino acids (in proteins). |
| Single nucleotide polymorphism (SNP) | A polymorphism in a gene caused by a change in a single nucleotide in the DNA sequence. A large number of protein isoforms result from SNPs. SNPs occur frequently, approximately every 100-1000 base pairs as a result of deletions, insertions, and substitutions. There are estimated to be >10 million SNPs in the human genome. Many SNPs that occur in genes have no effect on the encoded protein, but some SNPs do influence the function of the protein that the gene produces. An SNP initially arises as a very rare mutation but is considered to be an SNP if it occurs in at least 1% of the population. |
| Signal transduction | A cascade of intracellular events after binding of an extracellular signal (e.g., a hormone or a cytokine) to a receptor on the cell surface. The intracellular cascade can result in changes in gene expression in the nucleus and hence an altered phenotype of the cell (e.g., as a result of different protein production). |
| Splicing | The removal of introns from transcribed RNA. The process of removal can vary, and some exons are skipped or excluded from splicing. This causes production of “splice variants” or “alternatively spliced” protein isoforms, resulting in the formation of different proteins from the same initial RNA. |
| Transcription | RNA synthesis. The process of creating an RNA copy of an equivalent section of DNA is the first step of gene expression, and occurs in the nucleus. The RNA copy that is produced is called messenger RNA (mRNA). |
| Translation | The first stage of protein synthesis. mRNA produced in transcription is decoded to produce an amino acid chain that will later fold into an active protein. Translation occurs in the cytoplasm: ribosomes bind to the mRNA and then facilitate decoding by binding of transfer RNAs (tRNAs) that have complementary anticodon sequences to that of the mRNA. The tRNAs carry specific amino acids that are joined together to form a polypeptide as the mRNA passes through the ribosome. |
DNA, Deoxyribonucleic acid; RNA, ribonucleic acid.
Patterns in Populations and Pedigrees
With all the attention focused on silicon arrays, laser scanners, and the other “glamorous” gadgets of advanced genomic technologies, it is important to consider how much needs to be learned about the genetic basis of a disease before stepping into a DNA laboratory. In fact, a strong foundation of knowledge about a disease’s frequency in different populations and occurrence among closely and distantly related family members (pedigrees) is absolutely essential. Without this foundation, endless gigabytes of DNA sequence data will not enable scientist-clinicians to develop a solid understanding of the causes of a disease. The human genome has not evolved in a test tube nor inside a supercomputer but has existed for millennia in natural populations, transmitted from generation to generation—from parents to child. Only by carefully studying the genetics of a disease in populations and pedigrees can we hope to begin to unearth the complex interactions between genes and the environment that underlie individual differences in disease susceptibility. This field of research is known as genetic epidemiology.
Genetic epidemiologists often first look at whether a disease occurs more often in some human populations than others. These comparisons can include both populations in different geographic areas, as well as racial or ethnic groups living in the same region. Does the disease have more severe symptoms, or more rapid rates of progression, or an earlier age of onset in some populations? Such findings suggest (but do not prove) that genetic differences that are important for the disease may exist among the populations.
Before the massive human migrations in recent centuries, most human populations existed in semiisolation from other populations around the globe. As a consequence of natural selection (differential survival and/or reproduction), populations sometimes adapted genetically to their local environment. The most famous example of adaptation is the sickle cell hemoglobin variant that protects against the infectious disease malaria. This variant is common in populations living in areas where this mosquito-borne parasite has long been endemic because it provides strong protection against the severe symptoms of this disease. The variant persists at high frequency in these populations even though persons who inherit two copies of the mutation (one from mother and one from father) are severely affected by the disease sickle cell anemia. A balance between the benefit of malaria resistance in persons who inherit only one copy of the variant versus the disadvantage of sickle cell disease keeps the variant at relatively high frequencies in populations where malaria is present. Another example of population differentiation by natural selection is the ability to digest the milk sugar lactose as an adult that evolved in Europeans in conjunction with the domestication of dairy cattle over 8000 years ago. In addition to differentiation driven by natural selection as in these examples, a random process called genetic drift also causes populations with little or no migration between them to differentiate genetically over time, so one cannot assume that every population difference observed has a functional biological basis.
Regrettably, comparison of periodontitis in different populations across the globe is extremely challenging because of the lack of calibrated examiners and standardized disease definitions.6 One of the most dramatic population differences in which data quality is not an issue is the observation that both localized and generalized forms of early-onset aggressive periodontitis occur about 10 times more frequently in African Americans compared to Caucasians.28 Human racial and ethnic groups often differ dramatically in frequency of mutations at genes that have major effects on disease risk. For example, cystic fibrosis is caused exclusively by recessive mutations in the CFTR gene and varies in frequency from 1 in 3000 Caucasians to 1 in 15,000 African Americans in the United States (US), whereas only 1 in 350,000 Japanese are affected.44 It is possible that the tenfold higher prevalence of early-onset aggressive periodontitis in African Americans is caused by elevated frequency of high-risk gene variants in this population. However, additional evidence is needed before drawing such a conclusion. Although comparative studies of different populations may provide clues as to possible genetic mechanisms underlying a disease, the environments of the populations may also be dissimilar in important ways. It is possible that variation in diet or in exposure to pathogenic oral bacteria or to some unknown and unmeasured environmental factors could entirely explain the observed difference in frequency of aggressive periodontitis between population groups. Until solid data confirm a genetic basis for population differences, we need to wait before drawing firm conclusions.
Comparison of disease occurrence or severity in identical (monozygotic) versus nonidentical (dizygotic) twins is a very powerful method for distinguishing between effects caused by variation in genes versus factors in the environment. This requires us to make what is usually a reasonable assumption that the environments of pairs of identical twins are no more or less similar than the environments shared by pairs of nonidentical twins. If variation among individuals in disease susceptibility or severity is caused entirely by factors in the environment, then we expect pairs of identical twins to be no more similar to each other than pairs of nonidentical twins. All twins (whether identical or nonidentical) are expected to be more similar to their co-twin on average than to unrelated members of their local population because they were raised in the same family environment (with similar diet, microbial exposures, etc). However, if genetic variation plays an important role in determining the trait, then genetically identical twin pairs will be more similar to each other than nonidentical twin pairs. This is because identical twins share 100% of the same genes, whereas nonidentical twin pairs share only 50% of their parents’ genes on average. Genetic epidemiologists calculate a measure based on these correlations called heritability that estimates the portion of all variation in the trait that is attributable to inherited genetic variation. Traits whose variation is determined entirely by differences in environmental exposures have heritabilities of 0.0, whereas traits with variation attributable solely to inherited genetic differences without any environmental influence have heritabilities of 1.0. Heritabilities are sometimes reported as a percentage ranging from 0% to 100%.
Most human diseases and nondisease traits fall in the middle of this range with heritability ranging between 0.25 and 0.75. For example, type II diabetes was estimated to have a heritability of 0.26 and abnormal glucose tolerance a heritability of 0.61 in one study.45 Note that for it to be feasible to use the twin method with adequate statistical power, the disease has to be fairly common so that the researcher can recruit enough twin pairs in which at least one of the twins is affected by the disease. For periodontal diseases, not surprisingly, only chronic periodontitis occurs frequently enough to have been studied using the twin design. Two twin studies of modest size (110 and 117 pairs) have been reported, and these estimate heritability of measures of chronic periodontitis ranging between 40% to 80%, thus clearly implicating genetic variation in disease risk.38,39 Interestingly, a study of bacteria associated with periodontitis found no difference between identical versus nonidentical twins.40 This suggests (at least for these twins, most of whom did not have severe periodontitis) that inherited variation in risk is not mediated by genes that influence the presence of specific bacteria in subgingival plaque.
Another method used by genetic epidemiologists to understand and distinguish different mechanisms of transmission of diseases through families is called segregation analysis. This is relatively straightforward for traits in which mutation in a single gene causes the disease to develop with nearly 100% certainty in carriers, whereas persons who do not inherit the mutation are at little or no risk. For example, carriers of a single copy of the Huntington’s disease gene mutation or carriers of two copies of a cystic fibrosis gene mutation always develop these diseases if they reach the ages at which symptoms of these conditions normally emerge. By tracking the transmission of these diseases in families, it is obvious, for example, that Huntington’s disease is a dominant single gene disorder because it is transmitted with 50% probability to offspring of affected individuals and thus it is often found occurring across many generations of large pedigrees. By contrast, parents of cystic fibrosis cases are very rarely affected themselves and 25% of the siblings are affected by cystic fibrosis when the disease is present in a nuclear family. This pattern of transmission is expected if a disease is recessive (i.e., it requires inheritance of a mutated gene copy from both parents, who themselves have one normal and one mutated copy and so are not affected). For most common “complex” diseases, however, having a high-risk gene does not automatically lead to development of the disease (a phenomenon called reduced penetrance). Furthermore, several genes or even dozens of different genes may influence disease susceptibility (known as the oligogenic inheritance and genetic heterogeneity). Environmental exposures are also important modifiers of disease risk. Such highly complex combinations of multiple genetic and environmental risk factors make the challenge of deciphering genetic mechanisms by merely observing transmission patterns in families using the segregation analysis approach unfeasible. The limitations of this approach were humorously illustrated a number of years ago in an analysis that facetiously presented evidence of a recessive gene controlling the trait of attending medical school.34 “Risk” for this outcome among first-degree relatives of a doctor was elevated 61 times above that of the general population. More recently, a robust quantitative analysis of the family histories of characters in the Harry Potter series suggested a dominant gene controls inheritance of magic abilities.49 Because the etiology of periodontitis is likely to be highly complex, segregation analyses of this disease that have been reported in the literature should be viewed with considerable skepticism. Unfortunately, the simplifying assumptions required for this method make the results unreliable and potentially misleading. For highly complex diseases, such as most cases of periodontitis, assays at the DNA level need to be combined with careful evaluations of clinical measures among related individuals to derive robust conclusions about a disease’s genetic architecture. Some of the key features of the different techniques for studying the genetics of periodontal disease are explained in Table 24-2.
TABLE 24-2 Techniques for Studying the Genetics of Periodontal Disease
| Candidate gene approach | A gene mapping approach that tests whether one allele of a gene occurs more often in patients with the disease than in subjects without the disease. These methods are also referred to as association analyses and aim to identify which genes are associated with the disease. Candidate genes are chosen on the basis of their known or presumed function (i.e., they have some plausible role in the disease process such as producing a protein that is important in the disease pathogenesis). Conceptually, this makes sense but requires some knowledge of the candidate gene to look for it! |
| Case-control studies | Studies in which the genetic makeup is compared in cases (who have the disease in question) and controls (who do not). The populations need to be carefully matched, otherwise apparent observed differences between cases and controls could arise because of ethnic or geographic variation, for example. |
| Twin studies | Comparisons of traits, including diseases in monozygotic, dizygotic, or usually both types of twins aimed at determining whether variation in the trait among members of a population is caused by genetic variation in inherited DNA sequences, environmental exposures in the subjects’ previous lives, or some combination of both of these processes. Twin studies often measure the concordance rates of twins with a particular trait or disease of interest. Monozygotic (identical) twins are very nearly identical in their DNA, whereas dizygotic (nonidentical) twins share on average half of their DNA as identical sequences inherited from their parents. If a disease has high heritability, identical twins will be more likely to either be both affected or both unaffected (concordant). However, this assumption is complicated in many diseases. A genetic mutation may not have complete penetrance, environmental conditions may contribute to the development of the disease (e.g., one twin may smoke and the other may not). Furthermore, many diseases are polygenic (i.e., caused by alterations in multiple genes). |
| Familial aggregation and relative risk | Many diseases run in families, and the degree of clustering within the family can be estimated by comparing the number of disease cases in relatives of patients to the risk of disease in the general population. Difficulties with this approach relate to the fact that in addition to having many genes in common, family members also share many aspects of a common environment (e.g., diet, nutrition, smoking, infectious organisms, and shared socioeconomic factors). |
| Segregation analyses | Statistical analyses of the patterns of transmission of a disease in families in an attempt to determine the relative likelihood that the disease is caused by a single gene with dominant or recessive inheritance, by multiple genes, or entirely by variation in exposure to risk factors. The observed proportions of offspring who have the trait or disease being evaluated (i.e., the phenotype) are compared with the proportions that are expected in the general population. |
| Linkage analysis | A technique used to map a gene responsible for a trait to a specific location on a chromosome. These studies are based on the fact that genes that are located close to each other on the chromosome tend to be inherited together as a unit. As such, these genes are said to be linked. Because linkage analysis requires use of initially very expensive DNA markers, this was originally only considered justified after finding strong evidence of a genetic basis for a trait using segregation analyses and/or family aggregation studies. A difficulty with linkage analyses is that many diseases are not caused by a single gene of “major” effect but rather by multiple genes of “minor” effect. In the latter situation, multiple genes each contribute a small amount to the phenotype/disease/trait, and the linkage study approach has little power for detection while association analysis methods may still be quite powerful. |
| Genome wide analyses | A Genome Wide Association Study (GWAS) investigates genetic variation across the entire genome simultaneously, with the aim of identifying genetic associations related to a trait or disease of interest. The completion of the Human Genome Project in 2003 and the development of microarray technologies capable of assaying >0.5M SNPs have made GWAS possible. This method has the potential to identify the genetic contributions to common diseases. An important advantage of this approach is, because the entire genome is analyzed, the technique permits the genetics of a disease to be investigated in a nonhypothesis-driven way. It is not necessary to correctly guess which candidate genes are most interesting to evaluate. A GWAS requires that well-characterized cases and controls be identified. A disadvantage of GWAS is that large clinical sample sizes are required to reduce the likelihood of differences between cases and controls being observed simply by chance as a result of the hundreds of thousands of multiple statistical tests required to search the entire human genome. |
Searching for Answers in the DNA
In theory, a genetic marker can be any type of biomolecule or assay that allows us to “read” inherited differences among individuals in their DNA sequences. Blood groups, protein isozymes, and human leukocyte antigens (HLAs) were among the first-developed markers, but even simple traits controlled by single genes, such as eye color, can also serve this purpose. Genomic methods have made these methods obsolete, as researchers can now determine a person’s inherited variation directly at the DNA level for much lower cost and with greater speed and accuracy. So-called “next generation” DNA sequencing methods are projected to enable researchers and clinicians to obtain nearly the entire 3 billion DNA base human genetic blueprint for less than $1000 a few years from now.15,60 At present, most genetic studies use a combination of whole genome arrays that can evaluate up to 1 million variable DNA sites in a single assay combined with lower-throughput methods used for fine mapping of chromosome regions of special interest.48 These regions are said to contain candidate genes of high priority for further investigation either because of the genes’ known biological functions or because results of previous genome-wide surveys indicate strong statistical chances that disease susceptibility genes are located in certain regions of one or more chromosomes. Types of variation include single nucleotide polymorphisms (SNPs, pronounced “snips”) in which one DNA base is substituted for another, small insertions and deletions (“in/dels”) of 1 or more DNA bases, and larger structural changes in the DNA such as inversions (in which a piece of DNA of hundreds or thousands of bases in size is cut out and sewn back into the chromosome in the opposite orientation) and copy number changes (in which a given segment of DNA is either missing or occurs in more than the usual two copies inherited as one copy from each parent).
Equipped with these powerful tools for rapidly measuring DNA variation, the next question is what kinds of study designs are most powerful for identifying the dozen or more variants that influence risk of disease from among the millions of DNA differences that exist between any two individuals in a typical population. One method that has been highly successful in finding molecular defects for simple genetic diseases (caused by mutation in a single gene), such as cystic fibrosis and Huntington’s disease, is linkage analysis.1,7 This gene-mapping strategy requires families with one or more members affected by the disease to be recruited and clinically and molecularly evaluated for a relatively small number of genetic markers. Depending on the type of marker used, as few as 500 markers or up to 10,000 markers distributed evenly across the genome are needed. Investigators usually try to recruit families with two or more close relatives, such as sibling pairs, who are affected by the disease, as well as parents and other siblings who may be unaffected. Under the null hypothesis that a region of the chromosome does not contain genetic variation that influences disease risk, siblings are expected to share identical genetic material inherited from their parents 50% of the time on average. However, if the region of the chromosome being evaluated contains a gene that has a substantial effect on disease risk (e.g., increases risk by tenfold or more), then pairs of siblings who are both affected by the disease will share the chromosome region containing the disease gene substantially more often than 50% of the time, and the null hypothesis of 50% sharing will be statistically rejected if the study has an adequately large enough sample of such families. This simple example illustrates how linkage analysis is performed. In practice, both small and large extended families are studied, including simultaneous evaluation of the sharing of genetic material among both affected and unaffected relatives. Sophisticated mathematical algorithms and computer programs are used to carry out the huge number of calculations required for data analyses.
After achieving many successes using linkage analysis for “simple” diseases caused by mutation of a single gene, this method was extended to complex diseases caused by combinations of multiple susceptibility genes and environmental risk factors. Unfortunately, these conditions proved to be beyond the reach of linkage analysis in most instances. Numerous studies conducted during the 1990s either failed to find any genes or initially positive findings failed to replicate. Linkage studies of very large numbers of carefully diagnosed families for complex diseases, such as orofacial clefting in which twin studies had firmly established heritability of 70%, identified at most a tiny fraction of this genetic variation. Mathematical analyses have subsequently shown that the linkage analysis gene-mapping strategy has extremely low statistical power for complex diseases in which each individual susceptibility gene has a relatively small effect on risk (e.g., twofold or less) and in which there is extensive heterogeneity among different families that have different combinations of susceptibility genes and environmental exposures.50 Consequently, it is not surprising that linkage analysis has only been successfully applied to syndromic forms of periodontitis (summarized later).
Disappointment over this setback to human disease mapping caused by the initially unrecognized limitations of linkage analysis was short-lived. An alternative approach called association analysis was also available, although this had been relegated to studies of HLA and a few other markers of special interest during the prime of linkage approaches.1,7 Mathematical analyses indicated that if we make some reasonable assumptions about the nature of genetic factors in human disease, this method could provide adequate statistical power for finding genes of small-to-modest effect on risk while requiring only moderately large sample sizes that would be feasible to recruit.50 There was, however, one major catch—to search the entire genome using the GWAS method, the number of genetic markers needed was several orders of magnitude greater (500,000 to one million assays needed per subject). Fortunately, advances in molecular assay technologies converged at this time period and several methods of array-based genotyping provided such a capability at an acceptable cost.48
How association studies are used to find disease susceptibility genes is illustrated for the case-control design in Figure 24-1. Association analyses are sometimes referred to as case-control studies, although this is only one of several sampling methods that can be used (including studies of families). Genotype frequencies of an inherited DNA variant for a group of periodontitis cases are statistically compared to the frequencies of the variant in a matched group of periodontally healthy control subjects. If the genotype frequencies differ so greatly that the results are very unlikely to occur by chance, then we conclude that the genotype that is more common in the cases compared to the controls is “associated” with increased disease risk. In Figure 24-1, 59% of the cases have the CC genotype (having inherited a C allele from both of their parents), while only 17% of the healthy controls inherited the CC genotype. Therefore this DNA variant could be used to predict periodontitis risk (but not until after the finding was validated in additional, independent studies). Conversely, we can also say that the AA genotype is “protective” against disease because it occurs much more often in the healthy controls (50%) compared to periodontitis cases (8%). Ideally, the cases and healthy controls are matched as closely as possible for race and ethnicity, smoking behavior, age, gender, etc, so that differences in genotype frequency are likely to be caused by real biologic effects on disease development or progression rather than as artifacts of some kind. For example, it is well known that races and ethnic groups sometimes differ dramatically in genotype frequencies because of their historic isolation in different geographic regions. Consider, for example, a study that had mostly Swedish cases and mostly Italian controls. We know that there are thousands of DNA variants that differ substantially among these populations because of their geographic isolation in human history. Few, if any, of these variants have anything to do with differences in disease risk but could falsely appear to be associated because of the failure to carefully match ethnicity in cases and controls. In practice, this is usually not a problem provided that investigators take reasonable precautions in how cases and controls are selected. It is also now routine practice to use several statistical methods to check for mismatching and then adjust for this in data analyses if it occurs.
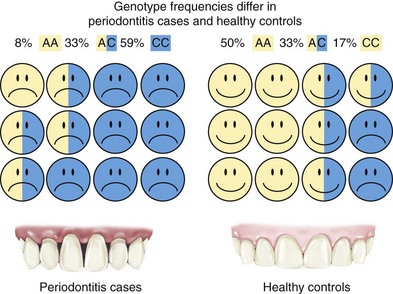
Figure 24-1 Case-control design for a genetic association study. Periodontitis cases shown on the left have frequencies for a hypothetical genetic marker of 8% AA, 33% AC, and 59% CC, which are substantially different from genotype frequencies found in healthy periodontal controls of 50% AA, 33% AC, and 17% CC. In this example, the CC genotype is associated with increased risk because it occurs at much higher frequency in the disease cases, whereas the AA allele is protective because this variant is much more common in the healthy controls.
The good news is now clearly in: association studies have been a boon for discovery of inherited genetic variation important for a wide range of complex diseases, including diabetes, cardiovascular disease, metabolic disorders, obesity, and mental illnesses. A recent review cites 24 genes identified with unquestionable statistical confidence for type 2 diabetes alone and the list continues to grow.57 Most of these genetic polymorphisms with elevated risk are very common in the population (from 5% up to >50%). Although each variant only increases risk slightly (twofold or less), because the risk alleles are so common, they can account for a nontrivial proportion of the occurrence of disease in the population (a measure epidemiologists called attributable risk).
An especially attractive aspect of the GWAS approach is that because the entire human genome is searched, we no longer have to depend on prior hypotheses about the disease’s molecular pathology. In most GWAS studies, about half of the statistically definitive findings point to genes that experts in the field had no suspicion whatsoever were involved in the disease’s etiology. This allows researchers to open up entirely new pathways for investigation that may lead to insights about the disease’s biologic mechanism and suggest novel molecular strategies for pharmaceutical or other therapeutic interventions. In more than a few cases, robust GWAS findings implicate regions of the human genome in which no genes appear to be present, highlighting the limitations of our knowledge even today about basic genome functions.
Although great progress has been made in understanding the etiology of many complex human diseases using GWAS methods, the approach has nevertheless usually failed to account for most of the heritability known to exist for these conditions.12,33 A recent study found that well-established nongenetic diabetes risk factors, such as gender, smoking, family history, body mass index, and blood lipids and glucose, were better predictors of risk than a combination of the top 20 genetic markers for this disease.58 To improve gene-based risk estimates, the missing heritability needs to be found and high hopes are now being placed on the emergence of next generation DNA-sequencing tools. In theory, these may enable researchers to identify the less common (1% to 5%) genetic variants that are predicted to have individual gene effects of greater magnitude on disease risk (>twofold but <tenfold) that cannot readily be found using either GWAS or linkage analysis methods.
Inherited Variation and Risk of Periodontitis
Because of the appropriate focus on the role of bacterial infections in the disease pathogenesis of periodontitis, inherited human genetic variation is often referred to as host defense or (somewhat more broadly) host response. These terms, however, cover only a small portion of the range of gene functions that may be important for periodontitis risk. Many additional biologic processes not directly related to defenses against or responses to infection by microbial pathogens are also likely to play important roles in determining an individual’s susceptibility to this disease.
Periodontitis in Genetic Syndromes and Other Diseases
A number of extremely rare conditions consistently include periodontitis among the array of clinical manifestations that define a syndrome. Many genetic syndromes involve mutations of single genes or larger chromosomal regions. However, a number of syndromes, such as fetal alcohol syndrome, are purely environmental in origin. Some of the syndromes that include periodontitis are caused by mutations in specific genes. For example, mutations in the cathepsin C gene have been shown to cause both Papillon-Lefèvre (Figure 24-2) and Haim-Munk syndromes, some forms of nonsyndromic prepubertal periodontitis, and may also be associated with risk of aggressive periodontitis.43 Periodontitis frequently occurs in some subtypes of Ehlers-Danlos syndrome, Kindler syndrome, Down syndrome (trisomy 21), leukocyte adhesion deficiencies (Figure 24-3), hypophosphatasia, two types of neutropenia, and aplasia of lacrimal and salivary glands. A large tri-racial extended family showed evidence of a single gene causing both early-onset aggressive periodontitis and dentinogenesis imperfecta. The gene has been mapped using linkage to a chromosomal region that contains a dentin matrix protein gene.32 Many of these conditions are so rare that few periodontists see even a single case in a lifetime of practice. However, dentists should be aware that these single gene conditions exist and be prepared to extend clinical evaluations to close relatives and seek the assistance of or referral to appropriately trained genetics counselors or specialists if a patient’s medical history or presentation of multiple symptoms raises the possibility that he or she may be affected. Clinicians can obtain updated information on these conditions by accessing the publicly available Online Mendelian Inheritance in Man database and typing in “periodontitis OR periodontal disease” as query terms.2 Further research is necessary to determine whether inherited variation in the genes that cause these rare syndromes might also influence risk of nonsyndromic forms of aggressive or chronic periodontitis.
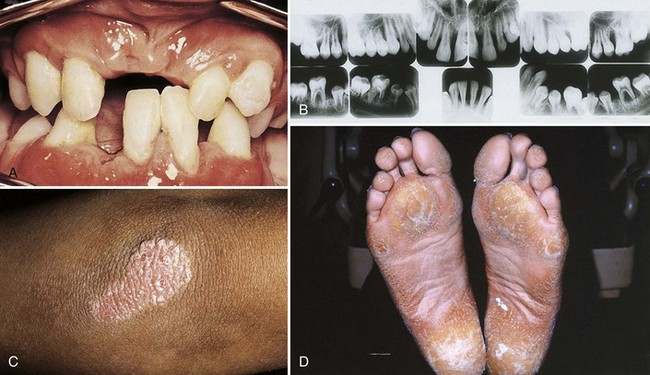
Figure 24-2 Oral (A), radiographic (B), and dermatologic (C and D) findings in Papillon-Lefèvre syndrome. Advanced periodontal disease usually affects the primary and secondary dentition shortly after teeth erupt, and many patients become edentulous while in their teens. Palmar-plantar keratodermas affect the palms of the hands and the plantar surfaces of the feet (D) but can also affect other sites, in this case, the elbow (C).
(Courtesy Dr. Robert J. Gorlin, Minneapolis.)
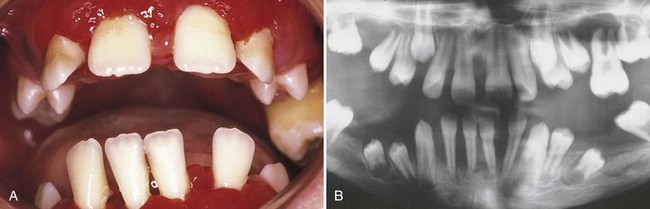
Figure 24-3 Oral (A) and radiographic (B) appearance of a patient with leukocyte adhesion deficiency (LAD). The child was deficient in CD18 (i.e., LAD type I), which results in absent or severely reduced levels of the β-2 integrin molecule. The patient suffered from recurrent infections of the middle ear, tongue, and perirectal areas, as well as the periodontium.
(From Majorana A, Notarangelo LD, Savoldi E et al: Oral Surg Oral Med Oral Pathol Oral Radiol Endod 87:691, 1999.)
Nonsyndromic Aggressive and Chronic Periodontitis
In this section, evidence for association of inherited genetic variation with aggressive and chronic periodontitis will be considered for cases that present without the co-occurrence of anomalies or disorders of other parts of the body or of the subject’s behavior. Such cases are appropriately classified as nonsyndromic periodontitis. This terminology is similar to the way that other human diseases, such as orofacial clefting, have long been recognized as occurring in syndromic and nonsyndromic forms. Elevated risk of periodontitis associated with metabolic conditions, such as diabetes (addressed elsewhere in this book), is more appropriately considered a “co-morbidity” rather than being designated a syndrome.
“A finding of no statistical significance in a well designed, conceptually sound, adequately powered study testing an important hypothesis is likely to provide more useful information than significance in a study that does not meet these criteria.”5
As reviewed above, twin studies have shown that chronic periodontitis has very substantial heritability and we know that aggressive periodontitis aggregates strongly in families. Since aggressive periodontitis occurs so rarely, it is not feasible to perform a twin study to confirm the heritability of this condition. Neither segregation analyses nor gene-mapping linkage studies are capable of providing reliable information about the genetic etiology of a highly complex disease such as periodontitis. However, large numbers of susceptibility genes have been identified for complex disorders, such as diabetes and cardiovascular disease, using association analysis. It seems reasonable to expect that similar successes could be achieved for periodontitis using this approach. In fact, during the past decade, several hundred papers have reported associations of nonsyndromic aggressive and/or chronic periodontitis with polymorphisms in a number of candidate genes. Certain classes of genes, such as cytokines, that have long been a focus of attention by immunologists and cell biologists studying pathogenic mechanisms associated with periodontitis have received the most attention. Early reports of relatively weak associations with variation in interleukin-1 (IL-1) genes led to a large number of attempts to replicate and extend these findings. Unfortunately, with very few exceptions, association studies of periodontitis have been very inadequately powered to detect genetic variation having modest effects on disease risk or progression (i.e., much too small sample sizes). In addition, inconsistency in the methods used to classify subjects as periodontal cases versus controls or to quantitatively measure disease severity and extent greatly limit our ability to draw sound conclusions by comparing results reported in different studies.
The reason why association studies of periodontitis have largely failed will be challenging to fully address. The first issue is straightforward, simply a matter of numbers. It is noteworthy that the successes achieved for many complex diseases, such as diabetes, using the GWAS mapping approach were based on sample sizes involving thousands of cases and controls, with multiple replications by independent teams of investigators. Statistical theory shows that to detect genes of modest effect these large sample sizes are absolutely essential. Using a statistical power calculator developed for case-control studies,47 sample sizes required for 80% power are shown in Figure 24-4 for a study that involves only a single genetic marker, as well as for studies that evaluate 5, 50, or 500 independent genetic markers in which the effects of multiple comparisons need to be accommodated. In this example, we assume that the risk gene acts in a dominant manner, with the high risk allele occurring at frequency of 25% in the population, and that this allele causes risk among carriers to increase by twofold (a greater effect on risk than observed for many susceptibility alleles found in GWAS studies of other complex diseases). Many periodontitis association studies reported in the literature involved multiple markers in each publication, and often the same research team reports positive findings for other genes in subsequent papers. Furthermore, because of the difficulties of publishing negative findings (i.e., when no association is found), many research teams working in this area may assay 50 or more genetic markers over the course of their work over several years. Results shown in Figure 24-4 demonstrate that to obtain 80% power a study of 50 markers would require over 200 cases and 200 controls. Even if a research team assays only 5 SNPs, their study would still require 100 cases and 100 controls in order to achieve adequate power.
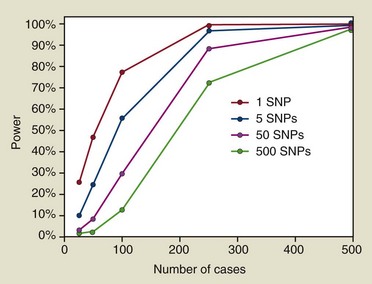
Figure 24-4 Statistical power estimates are shown for a situation in which we hypothesize that a dominantly transmitted disease susceptibility gene that increases risk by twofold and has an allele frequency in the population of 25% is being mapped by a case-control association study. The lines illustrate the loss of statistical power caused by the requirement to adjust for multiple comparisons when a research study involves the evaluation of not just a single genetic marker but as many as 5, 50, or 500 independent genetic polymorphisms. While only 100 cases (and 100 controls) may provide sufficient power if only a single marker is being tested, 250 cases and 250 controls are needed if 5 or 50 SNPs are assayed; a minimum of 500 cases and 500 controls are necessary if a study investigates 500 independent genetic markers.
Our review of the literature identified 307 periodontitis gene association tests with a P-value less than or equal to 0.05 for at least one statistical test performed on the data. These results are summarized in Figure 24-5 in which the x-axis indicates the number of cases included in the study and the P-value for the strongest finding is plotted on the y-axis. When more than one statistical test was reported, only the one with the smallest P-value is shown in this figure, thus in some cases inflating the actual statistical significance, as investigators rarely adjust their findings for these multiple tests when these are reported in a publication. This analysis shows clearly that most association findings have been drastically underpowered if periodontitis is assumed to be a complex disease. The majority (66%) of these association reports for chronic and aggressive periodontitis are based on samples of 100 cases or less, and 41% are based on less than 60 cases. As shown in Figure 24-4, studies of such small sample size have little power to detect a susceptibility gene that increases risk by twofold. Given the added concern about publication bias (i.e., that positive findings are more likely to be accepted for publication), we can have little confidence that even the more statistically significant findings are valid and likely to be independently replicated if based on such very inadequate samples sizes.
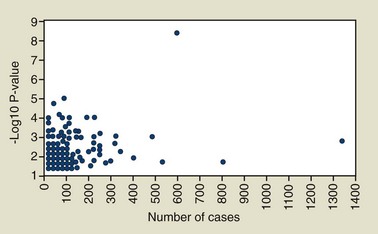
Figure 24-5 Number of cases and the P-values reported for 307 gene association tests for aggressive or chronic periodontitis reported in the literature. Only findings with P-values ≤0.05 are included, and when multiple tests were performed, the strongest association (i.e., the smallest P-value) is the one used for this presentation.
Aside from important lessons about how not to carry out association studies of a complex disease, there are some tentative conclusions that can be drawn from results reported to date. Results of our review and manual curation of 298 publications reporting association findings for periodontitis are presented in Table 24-3 online.

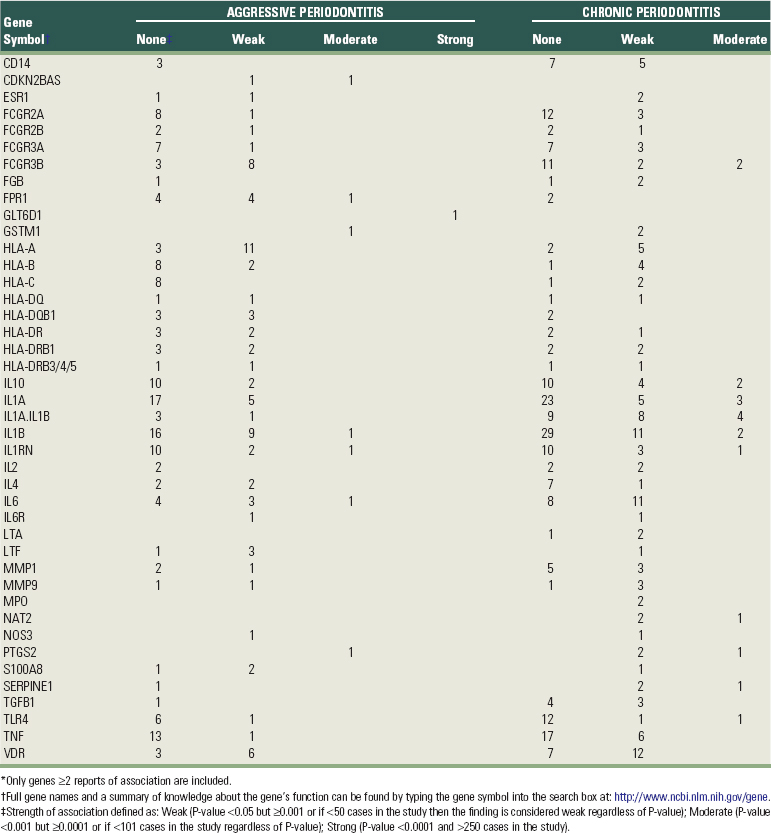
Key findings are condensed in Table 24-4. This summary includes a total of 41 genes plus a combination of the IL-1A and IL-1B genes analyzed jointly. Only genes that had at least two findings classified as “weak” or one finding of association with a “moderate” or “strong” level of support are included in this summary. The technical criteria for these designations are based on a combination of each study’s sample size (assuming larger studies are less likely to produce false positives) and the study’s P-value (statistical significance or type I error) as described in the Table 24-4 footnotes and in online supplementary documentation.
TABLE 24-4 Number of Independent Gene Association Reports for Aggressive Periodontitis and/or Chronic Periodontitis*
Genes in Table 24-4 have usually been evaluated in multiple, independent studies for association with either aggressive or chronic periodontitis or both. Occasionally, these subtypes of the disease have been pooled together, so it is not possible to determine from the publication whether the association involves primarily one subtype or both. With the exception of only the one GWAS study that has been published for this disease, all association tests reported to date involve evaluation of candidate genes that were selected based on known or postulated mechanisms of disease pathogenesis.
For example, when bacteria challenge gingival tissue, CD14 binds lipopolysaccharide and TLR4 plays a key role in pathogen recognition and activation of innate immunity.26 Lactotransferrin (LTF) plays an antimicrobial role as the first line of host defense and can also neutralize endotoxin and inhibit the induction of nuclear factor-κB (NF-κB) in monocytes in response to LPS.24,59 Myeloperoxidase (MPO) is an oxidative enzyme expressed in polymorphonuclear leukocytes. It is involved in the defense against periodontal bacteria and is also able to mediate inflammatory tissue destruction in periodontal disease.35 Glutathione S-transferase mu 1 (GSTM1)10 and N-acetyltransferase 2 (NAT2)25 genes are responsible for detoxification of a wide range of chemicals, including tobacco carcinogens. HLA complex genes56 play a central role in the immune system by presenting extracellular peptides that are important for either self-recognition or initiating immune responses to foreign pathogens. The Fcγ receptor genes (FcγRs) encode receptors for the Fc portion of immunoglobulin G and are involved in the removal of antigen-antibody complexes from the circulation, as well as other antibody-dependent responses.30 Formyl peptide receptor (FPR1) is a G-protein coupled receptor of phagocytic cells interacting with bacterial peptides and mediates chemotaxis, degranulation, and superoxide production involved in inflammation.17 Cytokines, such as interleukins (IL-1, IL-2, IL-4, IL-6, IL-10), tumor necrosis factor (TNF), and lymphotoxin alpha (LTA) play a number of important roles in the immunopathology of periodontal disease.42 Prostaglandin-endoperoxide synthase (PTGS2), also known as cyclooxygenase 2, is the key enzyme in prostaglandin biosynthesis. It is regulated by specific stimulatory events, suggesting that it is responsible for the prostanoid biosynthesis involved in inflammation.20 S100 calcium binding protein A8 (S100A8), the light subunit of calprotectin, is also associated with inflammatory diseases, including periodontitis.27 Fibrinogen (FGB) is an acute-phase protein whose levels are elevated during inflammation and has been associated with cardiovascular disease risk.51 Vitamin D receptor (VDR)19 and estrogen receptor (ESR1)61 are hormone receptors involved in skeletal muscle metabolism, including calcium absorption and bone loss. The matrix metalloproteinases (MMPs) are a group of endogenous proteinases that contribute to degradation of extracellular and basement membrane components.18 CDKN2B antisense RNA (CDKN2BAS) is a nonprotein coding gene of unknown function that has also been reported to be associated with coronary heart disease.52 The one GWAS reported for periodontitis (described in detail later) revealed strong association of a glycosyltransferase gene (GLT6D1) with aggressive periodontitis,54 but there is no clear functional relationship of this gene with periodontal disease pathogenesis.
With few exceptions, there are just as many or more reports in Table 24-4 in which no significant association was found for the gene as there are positive findings. For example, HLA-B has nine negative reports and six weak positive findings. IL-1B has 45 negative reports, 20 weak associations, and 3 moderately supportive findings. The inconsistency among these findings requires a much deeper analysis to understand what may be going on. One possibility is that genetic variation at the candidate gene is not associated with periodontitis. When a large number of studies are performed using a multitude of alternative ways of classifying small numbers of cases versus controls, when multiple alternative statistical analyses are run for the same small data sets, and when there may be a bias against publication of negative results, then a substantial portion of studies should report positive findings, even if no real association exists. Then again, heterogeneity among studies may be real. The negative studies may differ in terms of the racial or ethnic composition of the subjects, and the findings may be valid for some human populations but not others. Different genetic polymorphisms in the candidate gene may be evaluated in the different studies and only some of these may actually be associated with disease risk. Different clinical definitions or sources of information (e.g., clinical attachment loss versus bone loss measured from radiographs) may be used to define cases, or various quantitative measures might be employed, and these sources of variation might also influence the outcome of association tests. With data limited to mostly small studies, it is not currently possible to definitively determine which of these potential explanations applies to most of the findings shown in Table 24-4.
Figure 24-5 shows that a handful of periodontitis association studies have been reported that approach the sample size needed for complex diseases with over 400 cases.14,29,36-37, 52-53 This includes the GWAS in which over 322,825 SNP genetic markers were evaluated for association with risk of aggressive periodontitis.54 In the GWAS, statistical testing was performed in sequence for 3 independent sets of samples with a total of 438 cases and 1320 controls. Interestingly, this relatively powerful analysis failed to yield significant support for “the usual suspect” candidate genes shown in Table 24-4, after adjusting for the large number of hypotheses tested in the analysis. Instead, several novel chromosomal regions and candidate genes were implicated, among the strongest being a glycosyltransferase gene (GLT6D1), one of several glycosyltransferases in the human genome. This had an unadjusted P-value of 0.000000006. However, the P-value was reduced by 3 orders of magnitude to 0.000006 after accounting for the effects of gender, smoking, and diabetes. The adjusted P-value indicates that we should expect to see a difference in genotype frequency of this magnitude by chance alone of about 1 out of 166,666 times under the null hypothesis of no association with disease risk. This may seem to be very strong evidence for rejection of the null hypothesis, but this adjusted finding is actually only a marginal statistical significance in the context of a GWAS. In fact, this sometimes occurs by chance alone in the absence of a valid association, since this study involved testing over 300,000 genetic markers. Because there is no any obvious functional connection of this gene with what we currently know about pathogenesis of periodontitis (the protein is thought to play a role in signalling in development), it might be tempting to further downplay the finding’s significance. However, experience with GWAS studies of other complex diseases has shown that quite often valid gene associations are discovered in pathways that experts in the field did not previously realize were even related to the disease’s biology. This makes such discoveries all the more valuable, assuming that they are definitively and independently replicated, as they offer the potential of revealing completely new avenues for further exploration at the cellular and molecular levels and potentially may also provide novel targets for therapeutic interventions. It will be very interesting and important to see whether the association with GLT6D1 and the other statistically interesting findings reported in this first periodontitis GWAS can be replicated.
Challenges and Opportunities for the Future
The classification of disease used for research studies is an especially difficult challenge to be faced if we are to benefit fully from the opportunities offered by the genomics revolution.46 If we cannot agree which subjects in a study are affected by disease, by a particular subtype of disease, how severely they are affected, or how quantitative measures related to disease should be obtained and analyzed, then the chances of making progress will be very low, regardless of how advanced our molecular or bioinformatics technologies become. This problem is important for all kinds of research, not only genetics, and the issue of diagnosis is addressed elsewhere in this volume. However, genetics may have a unique role to contribute toward solving this dilemma. This may best be illustrated by an example from the early 1980s, in which medical geneticists and oncologists could not agree on classification of the disease neurofibromatosis (NF). “Splitters” argued for up to a dozen different etiologic subtypes, whereas “lumpers” suggested there might be only a single disease with a lot of variation among individuals as a result of differences in environmental exposures and variation in “genetic background” (a term that encompasses the cumulative influence of all other genes distributed throughout the genome in addition to the “major gene”). It had been discovered in research conducted in earlier decades that NF was transmitted in families as a single dominant gene, but the question remained as to how many different genes were involved (locus heterogeneity). Furthermore, even if only one gene was involved, it was possible that different mutations caused unique patterns of signs and symptoms (allelic heterogeneity). The controversy was largely resolved by the discovery of the NF131 and NF255 genes. After these genes were identified, individual patients could be distinguished by DNA testing, and the diagnostic classification controversies were resolved. In hindsight, it became clear that clustering of signs and symptoms fell into two major categories among different types of families and depended on which NF1 or NF2 mutation was involved and on what kind of gene mutation was involved. Although such gene-based clarity is unlikely for a complex multigenic condition such as periodontitis, the potential exists that subtypes of patients may eventually be classified more effectively by examining their DNA to see what kinds of susceptibility genes they inherited.
To move toward this desired outcome, however, we need to use the best strategies available today to classify research subjects into categories such as cases versus controls. Alternatively, we can attempt to use quantitative measures of bone loss or attachment loss to classify research subjects along a more continuous gradient ranging from persons with extremely healthy periodontium with little or no signs of disease to those with tooth loss and very high measures of pocket depth and attachment loss in remaining teeth. Family studies have numerous advantages in genetics, and for these designs, using a quantitative measures approach is especially attractive. When searching for gene associations in unrelated cases and controls, we can decide to select only subjects who are clearly affected as cases and compare these to subjects who clearly are very periodontally healthy assigned as controls. When studying families, however, one has to assign a disease status to all of the family members to fully utilize all of the information in the biologic unit. We may initially recruit the family based on an unambiguous case known as the proband (the subject who makes the family eligible for inclusion in the study), but the handling of parents, siblings, and other close relatives who may be neither clearly periodontally healthy nor clearly diseased is not easy to decide. For example, if we establish a threshold and require that two or more teeth have a minimum of 4 mm attachment loss for subjects to be classified as affected, then how we handle family members who are borderline becomes the challenge—siblings of the proband might have several teeth at 3 mm attachment loss in addition to one tooth with 6 mm loss. Such subjects are close to but not quite over the threshold. They are neither clearly healthy nor clearly diseased, and this makes them very problematic for categorical data analyses that require them to be classified as cases versus controls.
One aspect of the diagnosis challenge that is especially poorly understood is how and why different teeth are affected by periodontitis. Dentists have long recognized that incisors and first molars are more likely to be affected in early-onset disease, but it is not straightforward to incorporate this information into threshold disease definitions. One way to begin to address the problem of variation among teeth is to apply multivariate methods, such as principal components analysis, as was done in a study of aggressive periodontitis.13 This method maximizes the proportion of the total variation in attachment loss among all 28 teeth explained by a limited number of master variables called principal components. In this study, three principal components explained 77.8% of the total variance. The results are displayed in Figure 24-6 using color intensities to help visualize the patterns of correlation among different tooth types. Mixtures of red, green, and blue are “painted” onto the teeth, with each color’s intensity adjusted according to the tooth’s relative “weight” calculated for each of the principal components. This quantitative analysis shows how attachment loss is correlated among different kinds of teeth. First molar teeth are consistently painted yellow color by their principal components scores and these teeth are very different from all of the other teeth in their patterns of attachment loss. There is a gradual change in the patterns of attachment loss for the other teeth extending out from the central incisors (magenta) to the lateral incisors (purple), cuspids (blue), premolars (blue green), and second molars (green). In addition to generally exploring patterns of periodontal disease dispersed in the “geography” of the mouth, because these are studies conducted in families we can also use genetic epidemiologic methods to validate and compare alternative measures of disease.
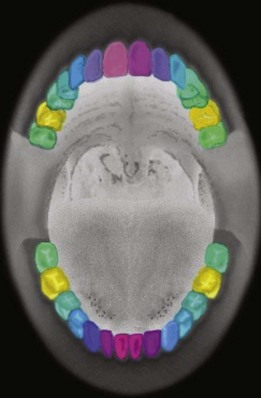
Figure 24-6 Illustration of the similarity and differences among different kinds of teeth in the frequency and severity of attachment loss observed in early onset aggressive periodontitis patients and their unaffected family members using the principal components analysis method.13
Specifically, we can calculate heritability for different quantitative variables such as simple mean attachment loss averaged across all teeth, or averaged for specific groups of teeth, as well as for more complex variables like these principal components. In the study of aggressive periodontitis illustrated in Figure 24-6, the principal component most heavily weighted on first molars had heritability of 30%, which was actually slightly higher than the 26% heritability estimated for a simple mean of attachment loss in first molars. By using heritability and other genetic measures such as association with specific inherited polymorphisms, it may be possible to refine diagnostic and disease classification systems by aligning these according to subgroups of subjects who share homogeneous etiologies.
Learning from the experience of other complex diseases, Sir Isaac Newton famously remarked in 1676: “What Descartes did was a good step. You have added much several ways … If I have seen a little further it is by standing on the shoulders of Giants.”
Although there remains great enthusiasm for GWAS studies, as was noted, even the most successful GWAS results, such as those for type 2 diabetes, have failed to identify most of the genetic variation responsible for the disease. Discovery of over 30 gene associations for type 2 diabetes accounts for a small fraction of the disease’s heritability.12 To track down the missing genetic variation, a great deal of attention is now turning to whole genome DNA-sequencing methods that will be available at very low cost within 3 to 5 years. Sequencing methods also expand capabilities for measuring gene expression in various tissues, and these data, combined with the ever-increasing availability of proteomic assays, will challenge investigators with huge quantities of information. Aside from generating the many gigabytes of data involved in this technology, the greatest challenges of “systems biology” may lie in developing the bioinformatics tools to sift through and identify the key bits of information important for advancing our knowledge from among the vast rising sea of biologic data pouring from our laboratories.4
Strategies for improving our understanding of the genetics of periodontitis, as well as possibilities for translation of this knowledge to benefit patients in the clinic, are being mapped-out today by teams of geneticists, clinicians, and information scientists who are currently focused on major medical conditions. By continuing to learn from the experiences of these early genomic explorers, it is reasonable to hope that dental researchers will be able to avoid some of their mistakes and follow the quickest path to advances in knowledge about our diseases of interest.
Inherited Variation and Treatment of Periodontitis
Pharmacogenomics and Individualized Dentistry
If individuals differ in their susceptibility to disease and especially if underlying the superficial signs and symptoms of a disease there really are etiologically distinct subtypes of disease pathogenesis, then the traditional medical model of “one size fits all” will not be optimal. As shown in Figure 24-7, cases of disease (illustrated by circles with unhappy faces) may differ in their genotype for a gene that determines which treatment works best for each individual. Some cases inherited a G allele from both their mother and their father and have GG genotype, whereas others have GT or TT genotype. The gene might distinguish between different subtypes of disease etiology or pathogenesis, or it might code for an enzyme, transporter, or receptor important for the metabolism of a therapeutic drug and have nothing directly to do with disease susceptibility. In the example illustrated in this figure, treatment method 1 (Tx method 1) has very high success when provided to subjects with the GG or GT genotypes but usually fails for cases with the TT genotype. TT genotypes experience much better outcomes when provided with treatment method 2 (Tx method 2), but this method often fails for subjects with the GT or GT genotypes. Sometimes, concern about severe side effects that limit use of a therapy rather than differences in efficacy and risk of side effects can also be genetically determined. If clinicians are not aware of the relationships between these genotypes and treatment success or side effects, it would appear that neither treatment is consistently effective. Clinicians would be forced to use a trial and error approach—first trying one method and then switching to the other if the first method fails. At best, this causes unnecessary expense and does not provide optimal quality of care. Furthermore, especially for deadly diseases such as cancer, precious time may be lost in halting the advance of the disease and by the time the clinician discovers an individual’s best treatment, it may be too late. This strategy of individualized medicine is already being practiced for an increasing number of diseases.9,16
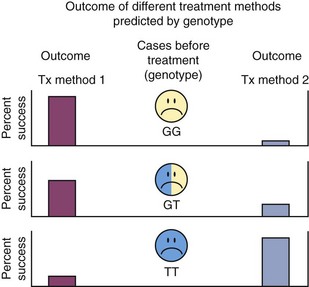
Figure 24-7 Inherited genetic variation determines treatment success. Individuals who inherited the GG or GT genotypes at a gene involved in a biologic pathway related to either the underlying disease susceptibility mechanisms or to the body’s response to the therapy have very good chances for success from treatment method 1 (Tx method 1), but a poor prognosis with treatment method 2 (Tx method 2), while individuals with the TT genotype respond positively to treatment method 2 only.
In dentistry, a test based on genetic variation at IL-1α and IL-1β cytokine and receptor antagonist genes to predict progression and severity of periodontitis has been commercialized and is still offered today, although it does not appear to be widely used. A number of reviews of the large number of studies on the IL-1 gene polymorphisms (see Table 24-3 [online] and Table 24-4) indicate that genetic variation at these loci may be associated with a modest effect on disease risk.21,42 Huynh-Ba et al concluded in 2007 that “there is insufficient evidence to establish if a positive IL-1 genotype status contributes to progression of periodontitis and/or treatment outcomes.”21 Similar lack of supporting evidence for use of IL-1 genetic testing to predict implant success has also been reported.3,22
As genetic testing becomes widespread, especially through “direct to consumer” commercial models, increasing responsibility will fall on dentists to fully understand and counsel their patients on the implications of test results for their treatment options and their risk of developing various dental diseases in the future. Tests for diseases of the oral cavity may likely combine inherited polymorphisms with oral microbial profiles and might also include assays of gene expression or proteomic data measured in saliva or other oral tissues. Few dentists practicing today have been educated to prepare themselves for these future challenges. This major shortfall of knowledge urgently needs to be addressed either by continuing education of today’s dental professionals about advances in knowledge of human genetics and the uses and potential misuses of genetic testing, by establishment of multidisciplinary teams of health practitioners in which dentists work closely with genetics counselors and medical geneticists, or by combinations of these approaches.
Acknowledgments
We thank Olga Korczeniewska for assistance in preparation of the figures and acknowledge support provided by NIH NIDCR grants 5R01DE016057 and 5R01DE018635 and the Foundation of the University of Medicine and Dentistry of New Jersey.
 Science Transfer
Science Transfer
Some rare diseases are associated with a mutation of the gene that controls cathepsin C (a molecule expressed in control of various components of the immune response) that have aggressive periodontitis as part of their manifestation (e.g., Papillon- LeFevre syndrome). Also, there are various groups of genetic abnormalities that result in significant periodontal breakdown (e.g., neutropenia, Down syndrome, Ehlers-Danlos syndrome, and hypophosphatasia).
The human genome has an enormous number of genes that could partially influence the susceptibility of patients to periodontitis, but at present, there is no strong evidence to show any specific genetic pattern that can be absolutely tied to disease activity. The genes controlling interleukin-1 (IL-1) production have a weak connection with periodontal susceptibility in some populations. Genetic investigations using a new genetic identification tool named Genome Wide Association Studies (GWAS) have shown a statistical relationship between periodontitis and the gene GLT6D, which controls glycosyltransferase. The clinical mechanism for this association has not been elucidated.
The most important payoff for clinicians would be to identify those patients whose genetic encoding could influence response to periodontal therapy. Once again, at present, no connections have been confirmed, and the genes controlling IL-1 polymorphism do not apparently play a significant role in prognosis.
References for Table 24-3
1 Agrawal AA, Kapley A, Yeltiwar RK, Purohit HJ. Assessment of single nucleotide polymorphism at IL-1A+4845 and IL-1B+3954 as genetic susceptibility test for chronic periodontitis in Maharashtrian ethnicity. J Periodontol. Sep 2006;77(9):1515-1521.
2 Alley CS, Reinhardt RA, Maze CA, et al. HLA-D and T lymphocyte reactivity to specific periodontal pathogens in type 1 diabetic periodontitis. J Periodontol. Oct 1993;64(10):974-979.
3 Amer A, Singh G, Darke C, Dolby AE. Association between HLA antigens and periodontal disease. Tissue Antigens. Feb 1988;31(2):53-58.
4 An N, Ou-Yang XY, Cao CF, Ye J, Hui RT. [Association of Fc gamma receptors IIIA gene polymorphisms with the susceptibility to periodontitis in Chinese patients]. Beijing Da Xue Xue Bao. Feb 18 2009;41(1):40-43.
5 Astolfi CM, Shinohara AL, da Silva RA, Santos MC, Line SR, de Souza AP. Genetic polymorphisms in the MMP-1 and MMP-3 gene may contribute to chronic periodontitis in a Brazilian population. J Clin Periodontol. Oct 2006;33(10):699-703.
6 Atilla G, Emingil G, Kose T, Berdeli A. TGF-beta1 gene polymorphisms in periodontal diseases. Clin Biochem. Sep 2006;39(9):929-934.
7 Babel N, Cherepnev G, Babel D, et al. Analysis of tumor necrosis factor-alpha, transforming growth factor-beta, interleukin-10, IL-6, and interferon-gamma gene polymorphisms in patients with chronic periodontitis. J Periodontol. Dec 2006;77(12):1978-1983.
8 Baioni CS, de Souza CM, Ribeiro Braosi AP, et al. Analysis of the association of polymorphism in the osteoprotegerin gene with susceptibility to chronic kidney disease and periodontitis. J Periodontal Res. Oct 2008;43(5):578-584.
9 Balndin-Texier A, Gueguin M, Fauchet R, Yardin M, Cathelineau G. [The HLA-A9 antigen and chronic periodontitis]. J Parodontol. Sep 1986;5(3):221-227.
10 Berdeli A, Emingil G, Gurkan A, Atilla G, Kose T. Association of the IL-1RN2 allele with periodontal diseases. Clin Biochem. Apr 2006;39(4):357-362.
11 Berdeli A, Emingil G, Han Saygan B, et al. TLR2 Arg753Gly, TLR4 Asp299Gly and Thr399Ile gene polymorphisms are not associated with chronic periodontitis in a Turkish population. J Clin Periodontol. Jul 2007;34(7):551-557.
12 Berdeli A, Gurkan A, Emingil G, Atilla G, Kose T. Endothelial nitric oxide synthase Glu298Asp gene polymorphism in periodontal diseases. J Periodontol. Aug 2006;77(8):1348-1354.
13 Berglundh T, Donati M, Hahn-Zoric M, Hanson LA, Padyukov L. Association of the -1087 IL 10 gene polymorphism with severe chronic periodontitis in Swedish Caucasians. J Clin Periodontol. Mar 2003;30(3):249-254.
14 Bonfil JJ, Dillier FL, Mercier P, et al. A “case control” study on the role of HLA DR4 in severe periodontitis and rapidly progressive periodontitis. Identification of types and subtypes using molecular biology (PCR.SSO). J Clin Periodontol. Feb 1999;26(2):77-84.
15 Boniotto M, Hazbon MH, Jordan WJ, et al. Novel hairpin-shaped primer assay to study the association of the -44 single-nucleotide polymorphism of the DEFB1 gene with early-onset periodontal disease. Clin Diagn Lab Immunol. Jul 2004;11(4):766-769.
16 Borges MA, de Figueiredo LC, de Brito RBJr, Faveri M, Feres M. Microbiological composition associated with vitamin D receptor gene polymorphism in chronic periodontitis. Braz Oral Res. Apr-Jun 2009;23(2):203-208.
17 Brett PM, Zygogianni P, Griffiths GS, et al. Functional gene polymorphisms in aggressive and chronic periodontitis. J Dent Res. Dec 2005;84(12):1149-1153.
18 Cao Z, Li C, Jin L, Corbet EF. Association of matrix metalloproteinase-1 promoter polymorphism with generalized aggressive periodontitis in a Chinese population. J Periodontal Res. Dec 2005;40(6):427-431.
19 Cao Z, Li C, Zhu G. MMP-1 promoter gene polymorphism and susceptibility to chronic periodontitis in a Chinese population. Tissue Antigens. Jul 2006;68(1):38-43.
20 Chai L, Song YQ, Zee KY, Leung WK. Single nucleotide polymorphisms of complement component 5 and periodontitis. J Periodontal Res. Nov 9 2009.
21 Chen D, Wang Q, Ma ZW, et al. MMP-2, MMP-9 and TIMP-2 gene polymorphisms in Chinese patients with generalized aggressive periodontitis. J Clin Periodontol. May 2007;34(5):384-389.
22 Chung HY, Lu HC, Chen WL, Lu CT, Yang YH, Tsai CC. Gm (23) allotypes and Fcgamma receptor genotypes as risk factors for various forms of periodontitis. J Clin Periodontol. Nov 2003;30(11):954-960.
23 Claudino M, Trombone AP, Cardoso CR, et al. The broad effects of the functional IL-10 promoter-592 polymorphism: modulation of IL-10, TIMP-3, and OPG expression and their association with periodontal disease outcome. J Leukoc Biol. Dec 2008;84(6):1565-1573.
24 Cogen RB, Roseman JM, Al-Joburi W, et al. Host factors in juvenile periodontitis. J Dent Res. Mar 1986;65(3):394-399.
25 Colombo AP, Eftimiadi C, Haffajee AD, Cugini MA, Socransky SS. Serum IgG2 level, Gm(23) allotype and FcgammaRIIa and FcgammaRIIIb receptors in refractory periodontal disease. J Clin Periodontol. Jun 1998;25(6):465-474.
26 Concolino P, Cecchetti F, D’Autilia C, et al. Association of periodontitis with GSTM1/GSTT1-null variants–a pilot study. Clin Biochem. Sep 2007;40(13-14):939-945.
27 Costa AM, Guimaraes MC, de Souza ER, Nobrega OT, Bezerra AC. Interleukin-6 (G-174C) and tumour necrosis factor-alpha (G-308A) gene polymorphisms in geriatric patients with chronic periodontitis. Gerodontology. Jun 25 2009.
28 Costa JE, Gomes CC, Cota LO, et al. Polymorphism in the promoter region of the gene for 5-HTT in individuals with aggressive periodontitis. J Oral Sci. Jun 2008;50(2):193-198.
29 Craandijk J, van Krugten MV, Verweij CL, van der Velden U, Loos BG. Tumor necrosis factor-alpha gene polymorphisms in relation to periodontitis. J Clin Periodontol. Jan 2002;29(1):28-34.
30 Cullinan MP, Sachs J, Wolf E, Seymour GJ. The distribution of HLA-A and -B antigens in patients and their families with periodontosis. J Periodontal Res. Mar 1980;15(2):177-184.
31 Cullinan MP, Westerman B, Hamlet SM, et al. A longitudinal study of interleukin-1 gene polymorphisms and periodontal disease in a general adult population. J Clin Periodontol. Dec 2001;28(12):1137-1144.
32 Cullinan MP, Westerman B, Hamlet SM, et al. Progression of periodontal disease and interleukin-10 gene polymorphism. J Periodontal Res. Jun 2008;43(3):328-333.
33 Dashash M, Nugent J, Baker P, Tansinda D, Blinkhorn F. Interleukin-6 -174 genotype, periodontal disease and adverse pregnancy outcomes: a pilot study. J Clin Immunol. May 2008;28(3):237-243.
34 de Brito Junior RB, Scarel-Caminaga RM, Trevilatto PC, de Souza AP, Barros SP. Polymorphisms in the vitamin D receptor gene are associated with periodontal disease. J Periodontol. Aug 2004;75(8):1090-1095.
35 de Souza AP, Trevilatto PC, Scarel-Caminaga RM, Brito RB, Line SR. MMP-1 promoter polymorphism: association with chronic periodontitis severity in a Brazilian population. J Clin Periodontol. Feb 2003;30(2):154-158.
36 de Souza AP, Trevilatto PC, Scarel-Caminaga RM, de Brito RBJr, Barros SP, Line SR. Analysis of the MMP-9 (C-1562 T) and TIMP-2 (G-418C) gene promoter polymorphisms in patients with chronic periodontitis. J Clin Periodontol. Feb 2005;32(2):207-211.
37 de Souza AP, Trevilatto PC, Scarel-Caminaga RM, de Brito RB, Line SR. Analysis of the TGF-beta1 promoter polymorphism (C-509T) in patients with chronic periodontitis. J Clin Periodontol. Jun 2003;30(6):519-523.
38 de Souza CM, Braosi AP, Luczyszyn SM, et al. Association between vitamin D receptor gene polymorphisms and susceptibility to chronic kidney disease and periodontitis. Blood Purif. 2007;25(5-6):411-419.
39 de Souza RC, Colombo AP. Distribution of FcgammaRIIa and FcgammaRIIIb genotypes in patients with generalized aggressive periodontitis. J Periodontol. Jul 2006;77(7):1120-1128.
40 DeCarlo AA, Grenett H, Park J, Balton W, Cohen J, Hardigan P. Association of gene polymorphisms for plasminogen activators with alveolar bone loss. J Periodontal Res. Aug 2007;42(4):305-310.
41 Diehl SR, Wang Y, Brooks CN, et al. Linkage disequilibrium of interleukin-1 genetic polymorphisms with early-onset periodontitis. J Periodontol. Apr 1999;70(4):418-430.
42 Donati M, Berglundh T, Hytonen AM, Hahn-Zoric M, Hanson LA, Padyukov L. Association of the -159 CD14 gene polymorphism and lack of association of the -308 TNFA and Q551R IL-4RA polymorphisms with severe chronic periodontitis in Swedish Caucasians. J Clin Periodontol. May 2005;32(5):474-479.
43 Drozdzik A. [The effect of environmental and genetic factors in the pathogenesis of periodontitis]. Ann Acad Med Stetin. 2008;54(1):118-126.
44 Drozdzik A, Kurzawski M, Safronow K, Banach J. Polymorphism in interleukin-1beta gene and the risk of periodontitis in a Polish population. Adv Med Sci. 2006;51(Suppl 1):13-17.
45 Duan H, Zhang J, Zhang Y. [The association between IL-1 gene polymorphisms and susceptibility to severe periodontitis]. Hua Xi Kou Qiang Yi Xue Za Zhi. Feb 2002;20(1):48-51.
46 Ehmke B, Kress W, Karch H, Grimm T, Klaiber B, Flemmig TF. Interleukin-1 haplotype and periodontal disease progression following therapy. J Clin Periodontol. Dec 1999;26(12):810-813.
47 Emingil G, Berdeli A, Baylas H, et al. Toll-like receptor 2 and 4 gene polymorphisms in generalized aggressive periodontitis. J Periodontol. Oct 2007;78(10):1968-1977.
48 Emingil G, Berdeli A, Gurkan A, Han Saygan B, Kose T, Atilla G. Gene polymorphisms of tissue plasminogen activator and plasminogen activator inhibitor-1 in Turkish patients with generalized aggressive periodontitis. J Clin Periodontol. Apr 2007;34(4):278-284.
49 Endo M, Tai H, Tabeta K, Kobayashi T, Yamazaki K, Yoshie H. Analysis of single nucleotide polymorphisms in the 5′-flanking region of tumor necrosis factor-alpha gene in Japanese patients with early-onset periodontitis. J Periodontol. Nov 2001;72(11):1554-1559.
50 Fassmann A, Holla LI, Buckova D, Vasku A, Znojil V, Vanek J. Polymorphisms in the +252(A/G) lymphotoxin-alpha and the −308(A/G) tumor necrosis factor-alpha genes and susceptibility to chronic periodontitis in a Czech population. J Periodontal Res. Aug 2003;38(4):394-399.
51 Ferreira SBJr, Trombone AP, Repeke CE, et al. An interleukin-1beta (IL-1beta) single-nucleotide polymorphism at position 3954 and red complex periodontopathogens independently and additively modulate the levels of IL-1beta in diseased periodontal tissues. Infect Immun. Aug 2008;76(8):3725-3734.
52 Fiebig A, Jepsen S, Loos BG, et al. Polymorphisms in the interleukin-1 (IL1) gene cluster are not associated with aggressive periodontitis in a large Caucasian population. Genomics. Nov 2008;92(5):309-315.
53 Firatli E, Kantarci A, Cebeci I, et al. Association between HLA antigens and early onset periodontitis. J Clin Periodontol. Jun 1996;23(6):563-566.
54 Folwaczny M, Glas J, Torok HP, Fricke K, Folwaczny C. Prevalence of the chemokine receptor CCR5-Delta32 gene mutation in periodontal disease. Clin Immunol. Dec 2003;109(3):325-329.
55 Folwaczny M, Glas J, Torok HP, Fricke K, Folwaczny C. The CD14 -159C-to-T promoter polymorphism in periodontal disease. J Clin Periodontol. Nov 2004;31(11):991-995.
56 Folwaczny M, Glas J, Torok HP, Limbersky O, Folwaczny C. Toll-like receptor (TLR) 2 and 4 mutations in periodontal disease. Clin Exp Immunol. Feb 2004;135(2):330-335.
57 Folwaczny M, Glas J, Torok HP, Mauermann D, Folwaczny C. The 3020insC mutation of the NOD2/CARD15 gene in patients with periodontal disease. Eur J Oral Sci. Aug 2004;112(4):316-319.
58 Folwaczny M, Glas J, Torok HP, Mende M, Folwaczny C. Lack of association between the TNF alpha G -308 A promoter polymorphism and periodontal disease. J Clin Periodontol. Jun 2004;31(6):449-453.
59 Folwaczny M, Glas J, Torok HP, et al. Polymorphisms of the interleukin-18 gene in periodontitis patients. J Clin Periodontol. May 2005;32(5):530-534.
60 Folwaczny M, Glas J, Torok HP, et al. Prevalence of the -295 T-to-C promoter polymorphism of the interleukin (IL)-16 gene in periodontitis. Clin Exp Immunol. Oct 2005;142(1):188-192.
61 Fraser DA, Loos BG, Boman U, et al. Polymorphisms in an interferon-gamma receptor-1 gene marker and susceptibility to periodontitis. Acta Odontol Scand. Oct 2003;61(5):297-302.
62 Fu Y, Cao C, Wang S. [Relevance of Fc gamma R polymorphism to the susceptibility of early-onset periodontitis]. Zhonghua Kou Qiang Yi Xue Za Zhi. Nov 1999;34(6):364-366.
63 Fu Y, Korostoff JM, Fine DH, Wilson ME. Fc gamma receptor genes as risk markers for localized aggressive periodontitis in African-Americans. J Periodontol. May 2002;73(5):517-523.
64 Fujigaki Y, Imamura Y, Oomori Y, Ouryouji K, Miyazawa H, Wang PL. Polymorphism of salivary histatin gene and periodontal disease in the Japanese population. J Int Acad Periodontol. Jul 2009;11(3):220-225.
65 Fukusaki T, Ohara N, Hara Y, Yoshimura A, Yoshiura K. Evidence for association between a Toll-like receptor 4 gene polymorphism and moderate/severe periodontitis in the Japanese population. J Periodontal Res. Dec 2007;42(6):541-545.
66 Galbraith GM, Hendley TM, Sanders JJ, Palesch Y, Pandey JP. Polymorphic cytokine genotypes as markers of disease severity in adult periodontitis. J Clin Periodontol. Nov 1999;26(11):705-709.
67 Galbraith GM, Steed RB, Sanders JJ, Pandey JP. Tumor necrosis factor alpha production by oral leukocytes: influence of tumor necrosis factor genotype. J Periodontol. Apr 1998;69(4):428-433.
68 Galicia JC, Tai H, Komatsu Y, Shimada Y, Ikezawa I, Yoshie H. Interleukin-6 receptor gene polymorphisms and periodontitis in a non-smoking Japanese population. J Clin Periodontol. Oct 2006;33(10):704-709.
69 Ge S, Wu YF, Liu TJ, He QM, Zhao L, Meng S. [Correlation between levels of fibrinogen, beta455 g/A fibrinogen gene polymorphism and chronic periodontitis]. Zhonghua Kou Qiang Yi Xue Za Zhi. Feb 2008;43(2):87-91.
70 Geismar K, Enevold C, Sorensen LK, et al. Involvement of interleukin-1 genotypes in the association of coronary heart disease with periodontitis. J Periodontol. Dec 2008;79(12):2322-2330.
71 Glas J, Beynon V, Bachstein B, et al. Increased plasma concentration of surfactant protein D in chronic periodontitis independent of SFTPD genotype: potential role as a biomarker. Tissue Antigens. Jul 2008;72(1):21-28.
72 Glas J, Torok HP, Tonenchi L, et al. A645G (Lys216Glu) polymorphism of the bactericidal/permeability-increasing protein gene in periodontal disease. Int J Immunogenet. Aug 2006;33(4):255-260.
73 Goncalves Lde S, Ferreira SM, Souza CO, Colombo AP. IL-1 gene polymorphism and periodontal status of HIV Brazilians on highly active antiretroviral therapy. Aids. Aug 22 2006;20(13):1779-1781.
74 Goncalves Lde S, Ferreira SM, Souza CO, Colombo AP. Influence of IL-1 gene polymorphism on the periodontal microbiota of HIV-infected Brazilian individuals. Braz Oral Res. Oct-Dec 2009;23(4):452-459.
75 Gonzales JR, Kobayashi T, Michel J, Mann M, Yoshie H, Meyle J. Interleukin-4 gene polymorphisms in Japanese and Caucasian patients with aggressive periodontitis. J Clin Periodontol. May 2004;31(5):384-389.
76 Gonzales JR, Mann M, Stelzig J, Bodeker RH, Meyle J. Single-nucleotide polymorphisms in the IL-4 and IL-13 promoter region in aggressive periodontitis. J Clin Periodontol. Jun 2007;34(6):473-479.
77 Gonzales JR, Michel J, Diete A, Herrmann JM, Bodeker RH, Meyle J. Analysis of genetic polymorphisms at the interleukin-10 loci in aggressive and chronic periodontitis. J Clin Periodontol. Sep 2002;29(9):816-822.
78 Gonzales JR, Michel J, Rodriguez EL, Herrmann JM, Bodeker RH, Meyle J. Comparison of interleukin-1 genotypes in two populations with aggressive periodontitis. Eur J Oral Sci. Oct 2003;111(5):395-399.
79 Gore EA, Sanders JJ, Pandey JP, Palesch Y, Galbraith GM. Interleukin-1beta+3953 allele 2: association with disease status in adult periodontitis. J Clin Periodontol. Oct 1998;25(10):781-785.
80 Goteiner D, Goldman MJ. Human lymphocyte antigen haplotype and resistance to periodontitis. J Periodontol. Mar 1984;55(3):155-158.
81 Guan ZM, Liu JJ, Ma X, Wu DH, Yu J, Huang GQ. [Relationship between interleukin-6 gene-572C/G polymorphism and chronic periodontitis]. Zhonghua Kou Qiang Yi Xue Za Zhi. Jul 2008;43(7):410-413.
82 Gunes S, Sumer AP, Keles GC, et al. Analysis of vitamin D receptor gene polymorphisms in patients with chronic periodontitis. Indian J Med Res. Jan 2008;127(1):58-64.
83 Gunji T, Onouchi Y, Nagasawa T, et al. Functional polymorphisms of the FPR1 gene and aggressive periodontitis in Japanese. Biochem Biophys Res Commun. Dec 7 2007;364(1):7-13.
84 Gurkan A, Emingil G, Saygan BH, et al. Matrix metalloproteinase-2, -9, and -12 gene polymorphisms in generalized aggressive periodontitis. J Periodontol. Dec 2007;78(12):2338-2347.
85 Gurkan A, Emingil G, Saygan BH, et al. Gene polymorphisms of matrix metalloproteinase-2, -9 and -12 in periodontal health and severe chronic periodontitis. Arch Oral Biol. Apr 2008;53(4):337-345.
86 Gurkan A, Emingil G, Saygan BH, et al. Angiotensin-converting enzyme (ACE), angiotensinogen (AGT), and angiotensin II type 1 receptor (AT1R) gene polymorphisms in generalized aggressive periodontitis. Arch Oral Biol. Apr 2009;54(4):337-344.
87 Gurkan A, Emingil G, Saygan BH, et al. Renin-angiotensin gene polymorphisms in relation to severe chronic periodontitis. J Clin Periodontol. Mar 2009;36(3):204-211.
88 Gurkan A, Emingil G, Saygan BH, et al. Tissue plasminogen activator and plasminogen activator inhibitor-1 gene polymorphisms in patients with chronic periodontitis. J Periodontol. Jul 2007;78(7):1256-1263.
89 Gustafsson A, Ito H, Asman B, Bergstrom K. Hyper-reactive mononuclear cells and neutrophils in chronic periodontitis. J Clin Periodontol. Feb 2006;33(2):126-129.
90 Guzeldemir E, Gunhan M, Ozcelik O, Tastan H. Interleukin-1 and tumor necrosis factor-alpha gene polymorphisms in Turkish patients with localized aggressive periodontitis. J Oral Sci. Jun 2008;50(2):151-159.
91 Guzman S, Karima M, Wang HY, Van Dyke TE. Association between interleukin-1 genotype and periodontal disease in a diabetic population. J Periodontol. Aug 2003;74(8):1183-1190.
92 Gwinn MR, Sharma A, De Nardin E. Single nucleotide polymorphisms of the N-formyl peptide receptor in localized juvenile periodontitis. J Periodontol. Oct 1999;70(10):1194-1201.
93 Hart TC, Hart PS, Michalec MD, et al. Localisation of a gene for prepubertal periodontitis to chromosome 11q14 and identification of a cathepsin C gene mutation. J Med Genet. Feb 2000;37(2):95-101.
94 Hart TC, Marazita ML, McCanna KM, Schenkein HA, Diehl SR. Reevaluation of the chromosome 4q candidate region for early onset periodontitis. Hum Genet. Jun 1993;91(5):416-422.
95 Havemose-Poulsen A, Sorensen LK, Bendtzen K, Holmstrup P. Polymorphisms within the IL-1 gene cluster: effects on cytokine profiles in peripheral blood and whole blood cell cultures of patients with aggressive periodontitis, juvenile idiopathic arthritis, and rheumatoid arthritis. J Periodontol. Mar 2007;78(3):475-492.
96 Hennig BJ, Parkhill JM, Chapple IL, Heasman PA, Taylor JJ. Association of a vitamin D receptor gene polymorphism with localized early-onset periodontal diseases. J Periodontol. Sep 1999;70(9):1032-1038.
97 Hennig BJ, Parkhill JM, Chapple IL, Heasman PA, Taylor JJ. Dinucleotide repeat polymorphism in the interleukin-10 gene promoter (IL-10.G) and genetic susceptibility to early-onset periodontal disease. Genes Immun. Aug 2000;1(6):402-404.
98 Hewitt C, McCormick D, Linden G, et al. The role of cathepsin C in Papillon-Lefevre syndrome, prepubertal periodontitis, and aggressive periodontitis. Hum Mutat. Mar 2004;23(3):222-228.
99 Ho YP, Lin YC, Yang YH, Ho KY, Wu YM, Tsai CC. Cyclooxygenase-2 Gene-765 single nucleotide polymorphism as a protective factor against periodontitis in Taiwanese. J Clin Periodontol. Jan 2008;35(1):1-8.
100 Hodge PJ, Riggio MP, Kinane DF. No association with HLA-DQB1 in European Caucasians with early-onset periodontitis. Tissue Antigens. Aug 1999;54(2):205-207.
101 Hodge PJ, Riggio MP, Kinane DF. Failure to detect an association with IL1 genotypes in European Caucasians with generalised early onset periodontitis. J Clin Periodontol. May 2001;28(5):430-436.
102 Holla LI, Buckova D, Fassmann A, Halabala T, Vasku A, Vacha J. Promoter polymorphisms in the CD14 receptor gene and their potential association with the severity of chronic periodontitis. J Med Genet. Nov 2002;39(11):844-848.
103 Holla LI, Fassmann A, Augustin P, Halabala T, Znojil V, Vanek J. The association of interleukin-4 haplotypes with chronic periodontitis in a Czech population. J Periodontol. Oct 2008;79(10):1927-1933.
104 Holla LI, Fassmann A, Benes P, Halabala T, Znojil V. 5 polymorphisms in the transforming growth factor-beta 1 gene (TGF-beta 1) in adult periodontitis. J Clin Periodontol. Apr 2002;29(4):336-341.
105 Holla LI, Fassmann A, Muzik J, Vanek J, Vasku A. Functional polymorphisms in the matrix metalloproteinase-9 gene in relation to severity of chronic periodontitis. J Periodontol. Nov 2006;77(11):1850-1855.
106 Holla LI, Fassmann A, Stejskalova A, Znojil V, Vanek J, Vacha J. Analysis of the interleukin-6 gene promoter polymorphisms in Czech patients with chronic periodontitis. J Periodontol. Jan 2004;75(1):30-36.
107 Holla LI, Fassmann A, Vasku A, et al. Genetic variations in the human gelatinase A (matrix metalloproteinase-2) promoter are not associated with susceptibility to, and severity of, chronic periodontitis. J Periodontol. Jul 2005;76(7):1056-1060.
108 Holla LI, Fassmann A, Vasku A, Znojil V, Vanek J, Vacha J. Interactions of lymphotoxin alpha (TNF-beta), angiotensin-converting enzyme (ACE), and endothelin-1 (ET-1) gene polymorphisms in adult periodontitis. J Periodontol. Jan 2001;72(1):85-89.
109 Holla LI, Jurajda M, Fassmann A, Dvorakova N, Znojil V, Vacha J. Genetic variations in the matrix metalloproteinase-1 promoter and risk of susceptibility and/or severity of chronic periodontitis in the Czech population. J Clin Periodontol. Aug 2004;31(8):685-690.
110 Holla LI, Kankova K, Fassmann A, et al. Distribution of the receptor for advanced glycation end products gene polymorphisms in patients with chronic periodontitis: a preliminary study. J Periodontol. Dec 2001;72(12):1742-1746.
111 Hooshmand B, Hajilooi M, Rafiei A, Mani-Kashani KH, Ghasemi R. Interleukin-4 (C-590T) and interferon-gamma (G5644A) gene polymorphisms in patients with periodontitis. J Periodontal Res. Feb 2008;43(1):111-115.
112 Houshmand B, Rafiei A, Hajilooi M, Mani-Kashani K, Gholami L. E-selectin and L-selectin polymorphisms in patients with periodontitis. J Periodontal Res. Feb 2009;44(1):88-93.
113 Hu KF, Huang KC, Ho YP, et al. Interleukin-10 (-592 C/A) and interleukin-12B (+16974 A/C) gene polymorphisms and the interleukin-10 ATA haplotype are associated with periodontitis in a Taiwanese population. J Periodontal Res. Jun 2009;44(3):378-385.
114 Huang HY, Zhang JC. [Investigation on the association of interleukin-1 genotype polymorphism with chronic periodontitis]. Hua Xi Kou Qiang Yi Xue Za Zhi. Oct 2004;22(5):415-419.
115 Imamura Y, Fujigaki Y, Oomori Y, Kuno T, Ota N, Wang PL. Polymorphism of genes encoding toll-like receptors and inflammatory cytokines in periodontal disease in the Japanese population. J Int Acad Periodontol. Jul 2008;10(3):95-102.
116 Inagaki K, Krall EA, Fleet JC, Garcia RI. Vitamin D receptor alleles, periodontal disease progression, and tooth loss in the VA dental longitudinal study. J Periodontol. Feb 2003;74(2):161-167.
117 Itagaki M, Kubota T, Tai H, Shimada Y, Morozumi T, Yamazaki K. Matrix metalloproteinase-1 and -3 gene promoter polymorphisms in Japanese patients with periodontitis. J Clin Periodontol. Sep 2004;31(9):764-769.
118 Izakovicova Holla L, Buckova D, Fassmann A, Benes P, Znojil V. Plasminogen-activator-inhibitor-1 promoter polymorphism as a risk factor for adult periodontitis in non-smokers. Genes Immun. Aug 2002;3(5):292-294.
119 Izakovicova Holla L, Buckova D, Fassmann A, Roubalikova L, Vanek J. Lack of association between chronic periodontitis and the Toll-like receptor 4 gene polymorphisms in a Czech population. J Periodontal Res. Aug 2007;42(4):340-344.
120 James JA, Poulton KV, Haworth SE, et al. Polymorphisms of TLR4 but not CD14 are associated with a decreased risk of aggressive periodontitis. J Clin Periodontol. Feb 2007;34(2):111-117.
121 Jansson H, Studies on periodontitis and analyses of individuals at risk for periodontal diseases, Swed Dent J Suppl, 180, 2006, 5-49
122 Jansson H, Lyssenko V, Gustavsson A, et al. Analysis of the interleukin-1 and interleukin-6 polymorphisms in patients with chronic periodontitis. A pilot study. Swed Dent J. 2006;30(1):17-23.
123 Jordan WJ, Eskdale J, Lennon GP, et al. A non-conservative, coding single-nucleotide polymorphism in the N-terminal region of lactoferrin is associated with aggressive periodontitis in an African-American, but not a Caucasian population. Genes Immun. Oct 2005;6(7):632-635.
124 Kaarthikeyan G, Jayakumar ND, Padmalatha O, Sheeja V, Sankari M, Anandan B. Analysis of the association between interleukin-1beta (+3954) gene polymorphism and chronic periodontitis in a sample of the south Indian population. Indian J Dent Res. Jan-Mar 2009;20(1):37-40.
125 Kaneko S, Kobayashi T, Yamamoto K, Jansen MD, van de Winkel JG, Yoshie H. A novel polymorphism of FcalphaRI (CD89) associated with aggressive periodontitis. Tissue Antigens. Jun 2004;63(6):572-577.
126 Kang BY, Choi YK, Choi WH, et al. Two polymorphisms of interleukin-4 gene in Korean adult periodontitis. Arch Pharm Res. Jun 2003;26(6):482-486.
127 Kara N, Keles GC, Sumer P, et al. Association of the polymorphisms in promoter and intron regions of the interleukin-4 gene with chronic periodontitis in a Turkish population. Acta Odontol Scand. Oct 2007;65(5):292-297.
128 Kaslick RS, West TL, Chasens AI, Terasaki PI, Lazzara R, Weinberg S. Association between HL-A2 antigen and various periodontal diseases in young adults. J Dent Res. Mar-Apr 1975;54(2):424.
129 Katz J, Goultschin J, Benoliel R, Brautbar C. Human leukocyte antigen (HLA) DR4. Positive association with rapidly progressing periodontitis. J Periodontol. Sep 1987;58(9):607-610.
130 Keles GC, Gunes S, Sumer AP, et al. Association of matrix metalloproteinase-9 promoter gene polymorphism with chronic periodontitis. J Periodontol. Sep 2006;77(9):1510-1514.
131 Kiani Z, Tavakkol-Afshari J, Hojjat H, et al. Polymorphism of IL-1alpha(-889) Gene and Its Association with Aggressive Periodontitis. Iran J Allergy Asthma Immunol. Jun 2009;8(2):95-98.
132 Kim JS, Park JY, Chung WY, Choi MA, Cho KS, Park KK. Polymorphisms in genes coding for enzymes metabolizing smoking-derived substances and the risk of periodontitis. J Clin Periodontol. Nov 2004;31(11):959-964.
133 Kim YJ, Viana AC, Curtis KM, Orrico SR, Cirelli JA, Scarel-Caminaga RM. Lack of association of a functional polymorphism in the interleukin 8 gene with susceptibility to periodontitis. DNA Cell Biol. Apr 2009;28(4):185-190.
134 Kinane DF, Hodge P, Eskdale J, Ellis R, Gallagher G. Analysis of genetic polymorphisms at the interleukin-10 and tumour necrosis factor loci in early-onset periodontitis. J Periodontal Res. Oct 1999;34(7):379-386.
135 Klouda PT, Porter SR, Scully C, et al. Association between HLA-A9 and rapidly progressive periodontitis. Tissue Antigens. Sep 1986;28(3):146-149.
136 Kobayashi T, Ito S, Kuroda T, et al. The interleukin-1 and Fcgamma receptor gene polymorphisms in Japanese patients with rheumatoid arthritis and periodontitis. J Periodontol. Dec 2007;78(12):2311-2318.
137 Kobayashi T, Ito S, Yamamoto K, et al. Risk of periodontitis in systemic lupus erythematosus is associated with Fcgamma receptor polymorphisms. J Periodontol. Mar 2003;74(3):378-384.
138 Kobayashi T, Murasawa A, Ito S, et al. Cytokine gene polymorphisms associated with rheumatoid arthritis and periodontitis in Japanese adults. J Periodontol. May 2009;80(5):792-799.
139 Kobayashi T, Nagata T, Murakami S, et al. Genetic risk factors for periodontitis in a Japanese population. J Dent Res. Dec 2009;88(12):1137-1141.
140 Kobayashi T, Sugita N, van der Pol WL, et al. The Fcgamma receptor genotype as a risk factor for generalized early-onset periodontitis in Japanese patients. J Periodontol. Sep 2000;71(9):1425-1432.
141 Kobayashi T, Westerdaal NA, Miyazaki A, et al. Relevance of immunoglobulin G Fc receptor polymorphism to recurrence of adult periodontitis in Japanese patients. Infect Immun. Sep 1997;65(9):3556-3560.
142 Kobayashi T, Yamamoto K, Sugita N, et al. The Fc gamma receptor genotype as a severity factor for chronic periodontitis in Japanese patients. J Periodontol. Oct 2001;72(10):1324-1331.
143 Kocher T, Sawaf H, Fanghanel J, Timm R, Meisel P. Association between bone loss in periodontal disease and polymorphism of N-acetyltransferase (NAT2). J Clin Periodontol. Jan 2002;29(1):21-27.
144 Komatsu Y, Galicia JC, Kobayashi T, Yamazaki K, Yoshie H. Association of interleukin-1 receptor antagonist +2018 gene polymorphism with Japanese chronic periodontitis patients using a novel genotyping method. Int J Immunogenet. Apr 2008;35(2):165-170.
145 Komatsu Y, Tai H, Galicia JC, et al. Interleukin-6 (IL-6)–373 A9T11 allele is associated with reduced susceptibility to chronic periodontitis in Japanese subjects and decreased serum IL-6 level. Tissue Antigens. Jan 2005;65(1):110-114.
146 Konig J, Ruhling A, Plagmann HC, Meisel P, Kocher T. Influence of interleukin (IL)-1 composite genotype on clinical variables in non-smoking, well-maintained compliant patients with chronic periodontitis. Swed Dent J. 2005;29(1):11-16.
147 Kornman KS, Crane A, Wang HY, et al. The interleukin-1 genotype as a severity factor in adult periodontal disease. J Clin Periodontol. Jan 1997;24(1):72-77.
148 Kratka Z, Bartova J, Krejsa O, Otcenaskova M, Janatova T, Duskova J. Interleukin- 1 gene polymorphisms as assessed in a 10-year study of patients with early-onset periodontitis. Folia Microbiol (Praha). 2007;52(2):183-188.
149 Laine ML, Farre MA, Garcia-Gonzalez MA, et al. [Risk factors in adult periodontitis: polymorphism in the interleukin-1 gene family]. Ned Tijdschr Tandheelkd. Aug 2002;109(8):303-306.
150 Laine ML, Farre MA, Gonzalez G, et al. Polymorphisms of the interleukin-1 gene family, oral microbial pathogens, and smoking in adult periodontitis. J Dent Res. Aug 2001;80(8):1695-1699.
151 Laine ML, Morre SA, Murillo LS, van Winkelhoff AJ, Pena AS. CD14 and TLR4 gene polymorphisms in adult periodontitis. J Dent Res. Nov 2005;84(11):1042-1046.
152 Laine ML, Murillo LS, Morre SA, Winkel EG, Pena AS, van Winkelhoff AJ. CARD15 gene mutations in periodontitis. J Clin Periodontol. Oct 2004;31(10):890-893.
153 Lang NP, Tonetti MS, Suter J, Sorrell J, Duff GW, Kornman KS. Effect of interleukin-1 gene polymorphisms on gingival inflammation assessed by bleeding on probing in a periodontal maintenance population. J Periodontal Res. Apr 2000;35(2):102-107.
154 Li Q, Meng H, Zhang L, et al. Correlation between single nucleotide polymorphisms in a calprotectin subunit gene and risk of periodontitis in a Chinese population. Ann Hum Genet. May 2007;71(Pt 3):312-324.
155 Li QY, Zhao HS, Meng HX, Zhang L, Xu L, Chen ZB. [The relationship between a nucleotide substitution of S100A8 gene and aggressive periodontitis]. Zhonghua Kou Qiang Yi Xue Za Zhi. May 2004;39(3):181-184.
156 Li QY, Zhao HS, Meng HX, Zhang L, Xu L, Chen ZB. [Interleukin-1 polymorphisms in patients with aggressive periodontitis]. Shanghai Kou Qiang Yi Xue. Aug 2005;14(4):333-337.
157 Li QY, Zhao HS, Meng HX, et al. Association analysis between interleukin-1 family polymorphisms and generalized aggressive periodontitis in a Chinese population. J Periodontol. Dec 2004;75(12):1627-1635.
158 Li S, Yang MH, Zeng CA, et al. Association of vitamin D receptor gene polymorphisms in Chinese patients with generalized aggressive periodontitis. J Periodontal Res. Jun 2008;43(3):360-363.
159 Lin L, Pan YP, Yin LY. Study on the correlation of cytokine gene polymorphism with chronic periodontitis. Shanghai Kou Qiang Yi Xue. Dec 2003;12(6):456-459.
160 Loos BG, Fiebig A, Nothnagel M, et al. NOD1 gene polymorphisms in relation to aggressive periodontitis. Innate Immun. Aug 2009;15(4):225-232.
161 Loos BG, Leppers-Van de Straat FG, Van de Winkel JG, Van der Velden U. Fcgamma receptor polymorphisms in relation to periodontitis. J Clin Periodontol. Jul 2003;30(7):595-602.
162 Lopez NJ, Jara L, Valenzuela CY. Association of interleukin-1 polymorphisms with periodontal disease. J Periodontol. Feb 2005;76(2):234-243.
163 Lopez NJ, Valenzuela CY, Jara L. Interleukin-1 gene cluster polymorphisms associated with periodontal disease in type 2 diabetes. J Periodontol. Oct 2009;80(10):1590-1598.
164 Louropoulou A, van der Velden U, Schoenmaker T, Catsburg A, Savelkoul PH, Loos BG. Mannose-binding lectin gene polymorphisms in relation to periodontitis. J Clin Periodontol. Nov 2008;35(11):923-930.
165 Machulla HK, Stein J, Gautsch A, Langner J, Schaller HG, Reichert S. HLA-A, B, Cw, DRB1, DRB3/4/5, DQB1 in German patients suffering from rapidly progressive periodontitis (RPP) and adult periodontitis (AP). J Clin Periodontol. Jun 2002;29(6):573-579.
166 Maney P, Emecen P, Mills JS, Walters JD. Neutrophil formylpeptide receptor single nucleotide polymorphism 348T>C in aggressive periodontitis. J Periodontol. Mar 2009;80(3):492-498.
167 Maney P, Walters JD. Formylpeptide Receptor Single Nucleotide Polymorphism 348T>C and Its Relationship to Polymorphonuclear Leukocyte Chemotaxis in Aggressive Periodontitis. J Periodontol. Sep 2009;80(9):1498-1505.
168 Maria de Freitas N, Imbronito AV, Neves AC, Nunes FD, Pustiglioni FE, Lotufo RF. Analysis of IL-1A(-889) and TNFA(-308) gene polymorphism in Brazilian patients with generalized aggressive periodontitis. Eur Cytokine Netw. Sep 2007;18(3):142-147.
169 McDevitt MJ, Wang HY, Knobelman C, et al. Interleukin-1 genetic association with periodontitis in clinical practice. J Periodontol. Feb 2000;71(2):156-163.
170 Meisel P, Carlsson LE, Sawaf H, Fanghaenel J, Greinacher A, Kocher T. Polymorphisms of Fc gamma-receptors RIIa, RIIIa, and RIIIb in patients with adult periodontal diseases. Genes Immun. Aug 2001;2(5):258-262.
171 Meisel P, Heins G, Carlsson L, et al. Impact of Genetic Polymorphisms on the Smoking-related Risk of Periodontal Disease: the Population-based Study SHIP. Tob Induc Dis. 2003;1(3):197.
172 Meisel P, Krause T, Cascorbi I, et al. Gender and smoking-related risk reduction of periodontal disease with variant myeloperoxidase alleles. Genes Immun. Apr 2002;3(2):102-106.
173 Meisel P, Schwahn C, Gesch D, Bernhardt O, John U, Kocher T. Dose-effect relation of smoking and the interleukin-1 gene polymorphism in periodontal disease. J Periodontol. Feb 2004;75(2):236-242.
174 Meisel P, Siegemund A, Dombrowa S, Sawaf H, Fanghaenel J, Kocher T. Smoking and polymorphisms of the interleukin-1 gene cluster (IL-1alpha, IL-1beta, and IL-1RN) in patients with periodontal disease. J Periodontol. Jan 2002;73(1):27-32.
175 Meisel P, Siegemund A, Grimm R, et al. The interleukin-1 polymorphism, smoking, and the risk of periodontal disease in the population-based SHIP study. J Dent Res. Mar 2003;82(3):189-193.
176 Meisel P, Timm R, Sawaf H, Fanghanel J, Siegmund W, Kocher T. Polymorphism of the N-acetyltransferase (NAT2), smoking and the potential risk of periodontal disease. Arch Toxicol. Aug 2000;74(6):343-348.
177 Mellati E, Arab HR, Tavakkol-Afshari J, Ebadian AR, Radvar M. Analysis of -1082 IL-10 gene polymorphism in Iranian patients with generalized aggressive periodontitis. Med Sci Monit. Nov 2007;13(11):CR510-514.
178 Menezes NG, Colombo AP. Lack of association between the TNF-alpha -308 (G/A) genetic polymorphism and periodontal disease in Brazilians. Braz Oral Res. Oct-Dec 2008;22(4):322-327.
179 Michel J, Gonzales JR, Wunderlich D, Diete A, Herrmann JM, Meyle J. Interleukin-4 polymorphisms in early onset periodontitis. J Clin Periodontol. May 2001;28(5):483-488.
180 Moore S, Ide M, Randhawa M, Walker JJ, Reid JG, Simpson NA. An investigation into the association among preterm birth, cytokine gene polymorphisms and periodontal disease. Bjog. Feb 2004;111(2):125-132.
181 Moreira PR, Costa JE, Gomez RS, Gollob KJ, Dutra WO. The IL1A (-889) gene polymorphism is associated with chronic periodontal disease in a sample of Brazilian individuals. J Periodontal Res. Feb 2007;42(1):23-30.
182 Moreira PR, Costa JE, Gomez RS, Gollob KJ, Dutra WO. TNFA and IL10 Gene Polymorphisms are not Associated with Periodontitis in Brazilians. Open Dent J. 2009;3:184-190.
183 Moreira PR, de Sa AR, Xavier GM, et al. A functional interleukin-1 beta gene polymorphism is associated with chronic periodontitis in a sample of Brazilian individuals. J Periodontal Res. Aug 2005;40(4):306-311.
184 Moreira PR, Lima PM, Sathler KO, et al. Interleukin-6 expression and gene polymorphism are associated with severity of periodontal disease in a sample of Brazilian individuals. Clin Exp Immunol. Apr 2007;148(1):119-126.
185 Naito M, Miyaki K, Naito T, et al. Association between vitamin D receptor gene haplotypes and chronic periodontitis among Japanese men. Int J Med Sci. 2007;4(4):216-222.
186 Nastri L, Caruso F. [Association between interleukin-1 composite genotype and severe periodontitis: case-control study]. Minerva Stomatol. Jun 2003;52(6):253-259.
187 Nibali L, D’Aiuto F, Donos N, et al. Association between periodontitis and common variants in the promoter of the interleukin-6 gene. Cytokine. Jan 2009;45(1):50-54.
188 Nibali L, Donos N, Brett PM, et al. A familial analysis of aggressive periodontitis - clinical and genetic findings. J Periodontal Res. Dec 2008;43(6):627-634.
189 Nibali L, Griffiths GS, Donos N, et al. Association between interleukin-6 promoter haplotypes and aggressive periodontitis. J Clin Periodontol. Mar 2008;35(3):193-198.
190 Nibali L, Parkar M, Brett P, Knight J, Tonetti MS, Griffiths GS. NADPH oxidase (CYBA) and FcgammaR polymorphisms as risk factors for aggressive periodontitis: a case-control association study. J Clin Periodontol. Aug 2006;33(8):529-539.
191 Nibali L, Parkar M, D’Aiuto F, et al. Vitamin D receptor polymorphism (-1056 Taq-I) interacts with smoking for the presence and progression of periodontitis. J Clin Periodontol. Jul 2008;35(7):561-567.
192 Nicu EA, Laine ML, Morre SA, Van der Velden U, Loos BG. Soluble CD14 in periodontitis. Innate Immun. Apr 2009;15(2):121-128.
193 Noack B, Gorgens H, Hempel U, et al. Cathepsin C gene variants in aggressive periodontitis. J Dent Res. Oct 2008;87(10):958-963.
194 Noack B, Gorgens H, Hoffmann T, et al. Novel mutations in the cathepsin C gene in patients with pre-pubertal aggressive periodontitis and Papillon-Lefevre syndrome. J Dent Res. May 2004;83(5):368-370.
195 Noack B, Gorgens H, Lorenz K, Schackert HK, Hoffmann T. TLR4 and IL-18 gene variants in chronic periodontitis: impact on disease susceptibility and severity. Immunol Invest. 2009;38(3-4):297-310.
196 Noack B, Gorgens H, Lorenz K, Ziegler A, Hoffmann T, Schackert HK. TLR4 and IL-18 gene variants in aggressive periodontitis. J Clin Periodontol. Dec 2008;35(12):1020-1026.
197 Ohyama H, Takashiba S, Oyaizu K, et al. HLA Class II genotypes associated with early-onset periodontitis: DQB1 molecule primarily confers susceptibility to the disease. J Periodontol. Sep 1996;67(9):888-894.
198 Pang RY, Chen KY, Zhang JC, Xu CR, Zhang X. [Association of TNFA-308 gene polymorphisms with susceptibility to chronic periodontitis in Chinese patients]. Shanghai Kou Qiang Yi Xue. Dec 2005;14(6):586-589.
199 Papapanou PN, Neiderud AM, Sandros J, Dahlen G. Interleukin-1 gene polymorphism and periodontal status. A case-control study. J Clin Periodontol. May 2001;28(5):389-396.
200 Park KS, Nam JH, Choi J. The short vitamin D receptor is associated with increased risk for generalized aggressive periodontitis. J Clin Periodontol. Aug 2006;33(8):524-528.
201 Park OJ, Shin SY, Choi Y, et al. The association of osteoprotegerin gene polymorphisms with periodontitis. Oral Dis. Jul 2008;14(5):440-444.
202 Parkhill JM, Hennig BJ, Chapple IL, Heasman PA, Taylor JJ. Association of interleukin-1 gene polymorphisms with early-onset periodontitis. J Clin Periodontol. Sep 2000;27(9):682-689.
203 Perez C, Gonzalez FE, Pavez V, et al. The -308 polymorphism in the promoter region of the tumor necrosis factor-alpha (TNF-alpha) gene and ex vivo lipopolysaccharide-induced TNF-alpha expression in patients with aggressive periodontitis and/or type 1 diabetes mellitus. Eur Cytokine Netw. Oct-Dec 2004;15(4):364-370.
204 Peterson RJ, Marsh CL. The relationship of alpha1-antitrypsin to inflammatory periodontal disease. J Periodontol. Jan 1979;50(1):31-35.
205 Pirhan D, Atilla G, Emingil G, Sorsa T, Tervahartiala T, Berdeli A. Effect of MMP-1 promoter polymorphisms on GCF MMP-1 levels and outcome of periodontal therapy in patients with severe chronic periodontitis. J Clin Periodontol. Oct 2008;35(10):862-870.
206 Pirhan D, Atilla G, Emingil G, Tervahartiala T, Sorsa T, Berdeli A. MMP-13 promoter polymorphisms in patients with chronic periodontitis: effects on GCF MMP-13 levels and outcome of periodontal therapy. J Clin Periodontol. Jun 2009;36(6):474-481.
207 Pontes CC, Gonzales JR, Novaes ABJr, et al. Interleukin-4 gene polymorphism and its relation to periodontal disease in a Brazilian population of African heritage. J Dent. Mar 2004;32(3):241-246.
208 Price P, Calder DM, Witt CS, et al. Periodontal attachment loss in HIV-infected patients is associated with the major histocompatibility complex 8.1 haplotype (HLA-A1,B8,DR3). Tissue Antigens. Oct 1999;54(4):391-399.
209 Qian W, Zhang J, Zhang Y. [The relationship between tumor necrosis factor A-308 gene polymorphism and susceptibility of severe periodontitis in adults]. Zhonghua Kou Qiang Yi Xue Za Zhi. Mar 2002;37(2):126-128.
210 Quappe L, Jara L, Lopez NJ. Association of interleukin-1 polymorphisms with aggressive periodontitis. J Periodontol. Nov 2004;75(11):1509-1515.
211 Rabello D, Soedarsono N, Kamei H, et al. CSF1 gene associated with aggressive periodontitis in the Japanese population. Biochem Biophys Res Commun. Sep 1 2006;347(3):791-796.
212 Raunio T, Knuuttila M, Hiltunen L, Karttunen R, Vainio O, Tervonen T. IL-6(-174) genotype associated with the extent of periodontal disease in type 1 diabetic subjects. J Clin Periodontol. Jan 2009;36(1):11-17.
213 Raunio T, Knuuttila M, Karttunen R, Vainio O, Tervonen T. Serum sCD14, polymorphism of CD14(-260) and periodontal infection. Oral Dis. Jun 4 2009.
214 Reichert S, Machulla HK, Klapproth J, et al. Interferon-gamma and interleukin-12 gene polymorphisms and their relation to aggressive and chronic periodontitis and key periodontal pathogens. J Periodontol. Aug 2008;79(8):1434-1443.
215 Reichert S, Machulla HK, Klapproth J, et al. Interleukin-2 -330 and 166 gene polymorphisms in relation to aggressive or chronic periodontitis and the presence of periodontopathic bacteria. J Periodontal Res. Oct 2009;44(5):628-635.
216 Reichert S, Machulla HK, Klapproth J, et al. The interleukin-10 promoter haplotype ATA is a putative risk factor for aggressive periodontitis. J Periodontal Res. Feb 2008;43(1):40-47.
217 Reichert S, Stein J, Gautsch A, Schaller HG, Machulla HK. Gender differences in HLA phenotype frequencies found in German patients with generalized aggressive periodontitis and chronic periodontitis. Oral Microbiol Immunol. Dec 2002;17(6):360-368.
218 Reinholdt J, Bay I, Svejgaard A. Association between HLA-antigens and periodontal disease. J Dent Res. Oct 1977;56(10):1261-1263.
219 Ren XY, Xu L, Meng HX. [Interleukin-1 family polymorphisms in aggressive periodontitis patients and their relatives]. Beijing Da Xue Xue Bao. Feb 18 2008;40(1):28-33.
220 Ren XY, Xu L, Meng HX, et al. Family-based association analysis of S100A8 genetic polymorphisms with aggressive periodontitis. J Periodontal Res. Apr 2009;44(2):184-192.
221 Roshna T, Thomas R, Nandakumar K, Banerjee M. A case-control study on the association of human leukocyte antigen-A*9 and -B*15 alleles with generalized aggressive periodontitis in an Indian population. J Periodontol. Dec 2006;77(12):1954-1963.
222 Sahingur SE, Sharma A, Genco RJ, De Nardin E. Association of increased levels of fibrinogen and the -455G/A fibrinogen gene polymorphism with chronic periodontitis. J Periodontol. Mar 2003;74(3):329-337.
223 Sakellari D, Katsares V, Georgiadou M, Kouvatsi A, Arsenakis M, Konstantinidis A. No correlation of five gene polymorphisms with periodontal conditions in a Greek population. J Clin Periodontol. Nov 2006;33(11):765-770.
224 Sakellari D, Koukoudetsos S, Arsenakis M, Konstantinidis A. Prevalence of IL-1A and IL-1B polymorphisms in a Greek population. J Clin Periodontol. Jan 2003;30(1):35-41.
225 Saleh TA, Stephen L, Kotze M, Pretorius A. The composite interleukin-1 genotype in South Africa. Sadj. May 2009;64(4):170-173.
226 Savarrio L, Donati M, Carr C, Kinane DF, Berglundh T. Interleukin-24, RANTES and CCR5 gene polymorphisms are not associated with chronic adult periodontitis. J Periodontal Res. Apr 2007;42(2):152-158.
227 Scapoli C, Trombelli L, Mamolini E, Collins A. Linkage disequilibrium analysis of case-control data: an application to generalized aggressive periodontitis. Genes Immun. Feb 2005;6(1):44-52.
228 Scarel-Caminaga RM, Trevilatto PC, Souza AP, Brito RB, Camargo LE, Line SR. Interleukin 10 gene promoter polymorphisms are associated with chronic periodontitis. J Clin Periodontol. Jun 2004;31(6):443-448.
229 Scarel-Caminaga RM, Trevilatto PC, Souza AP, Brito RB, Line SR. Investigation of an IL-2 polymorphism in patients with different levels of chronic periodontitis. J Clin Periodontol. Jul 2002;29(7):587-591.
230 Scarel-Caminaga RM, Trevilatto PC, Souza AP, Brito RBJr, Line SR. Investigation of IL4 gene polymorphism in individuals with different levels of chronic periodontitis in a Brazilian population. J Clin Periodontol. Apr 2003;30(4):341-345.
231 Schaefer AS, Richter GM, Groessner-Schreiber B, et al. Identification of a shared genetic susceptibility locus for coronary heart disease and periodontitis. PLoS Genet. Feb 2009;5(2):e1000378.
232 Schaefer AS, Richter GM, Nothnagel M, et al. A 3′ UTR transition within DEFB1 is associated with chronic and aggressive periodontitis. Genes Immun. Oct 15 2009.
233 Schaefer AS, Richter GM, Nothnagel M, et al: A genome-wide association study identifies GLT6D1 as a susceptibility locus for periodontitis, Hum Mol Genet 19(3):553-562, Feb 1
234 Schroder NW, Meister D, Wolff V, et al. Chronic periodontal disease is associated with single-nucleotide polymorphisms of the human TLR-4 gene. Genes Immun. Aug 2005;6(5):448-451.
235 Schulz S, Machulla HK, Altermann W, et al. Genetic markers of tumour necrosis factor alpha in aggressive and chronic periodontitis. J Clin Periodontol. Jun 2008;35(6):493-500.
236 Schulz S, Zissler N, Altermann W, et al. Impact of genetic variants of CD14 and TLR4 on subgingival periodontopathogens. Int J Immunogenet. Dec 2008;35(6):457-464.
237 Scott DA, von Ahsen N, Palmer RM, Wilson RF. Analysis of two common alpha 1-antitrypsin deficiency alleles (PI*Z and PI*S) in subjects with periodontitis. J Clin Periodontol. Dec 2002;29(12):1118-1121.
238 Shapira L, Stabholz A, Rieckmann P, Kruse N. Genetic polymorphism of the tumor necrosis factor (TNF)-alpha promoter region in families with localized early-onset periodontitis. J Periodontal Res. Jun 2001;36(3):183-186.
239 Shimada Y, Tai H, Endo M, Kobayashi T, Akazawa K, Yamazaki K. Association of tumor necrosis factor receptor type 2 +587 gene polymorphism with severe chronic periodontitis. J Clin Periodontol. Jun 2004;31(6):463-469.
240 Shimomura-Kuroki J, Yamashita K, Shimooka S. Tannerella forsythia and the HLA-DQB1 allele are associated with susceptibility to periodontal disease in Japanese adolescents. Odontology. Jan 2009;97(1):32-37.
241 Shirodaria S, Smith J, McKay IJ, Kennett CN, Hughes FJ. Polymorphisms in the IL-1A gene are correlated with levels of interleukin-1alpha protein in gingival crevicular fluid of teeth with severe periodontal disease. J Dent Res. Nov 2000;79(11):1864-1869.
242 Soedarsono N, Rabello D, Kamei H, et al. Evaluation of RANK/RANKL/OPG gene polymorphisms in aggressive periodontitis. J Periodontal Res. Oct 2006;41(5):397-404.
243 Soga Y, Nishimura F, Ohyama H, Maeda H, Takashiba S, Murayama Y. Tumor necrosis factor-alpha gene (TNF-alpha) -1031/-863, -857 single-nucleotide polymorphisms (SNPs) are associated with severe adult periodontitis in Japanese. J Clin Periodontol. Jun 2003;30(6):524-531.
244 Stein J, Reichert S, Gautsch A, Machulla HK. Are there HLA combinations typical supporting for or making resistant against aggressive and/or chronic periodontitis? J Periodontal Res. Oct 2003;38(5):508-517.
245 Stein JM, Smeets R, Reichert S, et al. The role of the composite interleukin-1 genotype in the association between periodontitis and acute myocardial infarction. J Periodontol. Jul 2009;80(7):1095-1102.
246 Struch F, Dau M, Schwahn C, Biffar R, Kocher T, Meisel P. Interleukin-1 gene polymorphism, diabetes, and periodontitis: results from the Study of Health in Pomerania (SHIP). J Periodontol. Mar 2008;79(3):501-507.
247 Sugita N, Kobayashi T, Ando Y, et al. Increased frequency of FcgammaRIIIb-NA1 allele in periodontitis-resistant subjects in an elderly Japanese population. J Dent Res. Mar 2001;80(3):914-918.
248 Sugita N, Yamamoto K, Kobayashi T, et al. Relevance of Fc gamma RIIIa-158V-F polymorphism to recurrence of adult periodontitis in Japanese patients. Clin Exp Immunol. Aug 1999;117(2):350-354.
249 Sumer AP, Kara N, Keles GC, Gunes S, Koprulu H, Bagci H. Association of interleukin-10 gene polymorphisms with severe generalized chronic periodontitis. J Periodontol. Mar 2007;78(3):493-497.
250 Sun JL, Meng HX, Cao CF, et al. Relationship between vitamin D receptor gene polymorphism and periodontitis. J Periodontal Res. Aug 2002;37(4):263-267.
251 Suzuki A, Ji G, Numabe Y, Ishii K, Muramatsu M, Kamoi K. Large-scale investigation of genomic markers for severe periodontitis. Odontology. Sep 2004;92(1):43-47.
252 Suzuki A, Ji G, Numabe Y, et al. Single nucleotide polymorphisms associated with aggressive periodontitis and severe chronic periodontitis in Japanese. Biochem Biophys Res Commun. May 7 2004;317(3):887-892.
253 Tabeta K, Shimada Y, Tai H, et al. Assessment of chromosome 19 for genetic association in severe chronic periodontitis. J Periodontol. Apr 2009;80(4):663-671.
254 Tachi Y, Shimpuku H, Nosaka Y, et al. Vitamin D receptor gene polymorphism is associated with chronic periodontitis. Life Sci. Nov 14 2003;73(26):3313-3321.
255 Tachi Y, Shimpuku H, Nosaka Y, et al. Association of vitamin D receptor gene polymorphism with periodontal diseases in Japanese and Chinese. Nucleic Acids Res Suppl. 2001;1:111-112.
256 Tai H, Endo M, Shimada Y, et al. Association of interleukin-1 receptor antagonist gene polymorphisms with early onset periodontitis in Japanese. J Clin Periodontol. Oct 2002;29(10):882-888.
257 Takahashi K, Ohyama H, Kitanaka M, et al. Heterogeneity of host immunological risk factors in patients with aggressive periodontitis. J Periodontol. Apr 2001;72(4):425-437.
258 Takashiba S, Noji S, Nishimura F, et al. Unique intronic variations of HLA-DQ beta gene in early-onset periodontitis. J Periodontol. May 1994;65(5):379-386.
259 Takeuchi-Hatanaka K, Ohyama H, Nishimura F, et al. Polymorphisms in the 5’ flanking region of IL12RB2 are associated with susceptibility to periodontal diseases in the Japanese population. J Clin Periodontol. Apr 2008;35(4):317-323.
260 Tang Y, Zhang JC, Zhang WH, Pang RY. [The association between Fc gamma receptor IIA gene polymorphism and susceptibility to chronic periodontitis in Chinese Han nationality]. Hua Xi Kou Qiang Yi Xue Za Zhi. Apr 2004;22(2):158-161.
261 Terasaki PI, Kaslick RS, West TL, Chasens AI. Low HL-A2 frequency and periodontitis. Tissue Antigens. May 1975;5(4):286-288.
262 Tervonen T, Raunio T, Knuuttila M, Karttunen R. Polymorphisms in the CD14 and IL-6 genes associated with periodontal disease. J Clin Periodontol. May 2007;34(5):377-383.
263 Thomson WM, Edwards SJ, Dobson-Le DP, et al. IL-1 genotype and adult periodontitis among young New Zealanders. J Dent Res. Aug 2001;80(8):1700-1703.
264 Tian YG, Pan YP, Lin L, Zhang DM, Zhao J. [The relationship of IL-1beta expressed in buccal cells and the polymorphisms of IL-1beta (+3953) with chronic periodontitis]. Shanghai Kou Qiang Yi Xue. Oct 2006;15(5):456-460.
265 Trevilatto PC, Scarel-Caminaga RM, de Brito RBJr, de Souza AP, Line SR. Polymorphism at position -174 of IL-6 gene is associated with susceptibility to chronic periodontitis in a Caucasian Brazilian population. J Clin Periodontol. May 2003;30(5):438-442.
266 Trevilatto PC, Tramontina VA, Machado MA, Goncalves RB, Sallum AW, Line SR. Clinical, genetic and microbiological findings in a Brazilian family with aggressive periodontitis. J Clin Periodontol. Mar 2002;29(3):233-239.
267 Trombone AP, Cardoso CR, Repeke CE, et al. Tumor necrosis factor-alpha -308G/A single nucleotide polymorphism and red-complex periodontopathogens are independently associated with increased levels of tumor necrosis factor-alpha in diseased periodontal tissues. J Periodontal Res. Oct 2009;44(5):598-608.
268 Tsutsumi A, Kobayashi T, Ito S, et al. Mannose binding lectin gene polymorphism and the severity of chronic periodontitis. Nihon Rinsho Meneki Gakkai Kaishi. Feb 2009;32(1):48-52.
269 Unur M, Demirez E, Agachan B, et al. The relationship of oral disturbances of diabetes mellitus patients with paraoxonase gene polymorphisms. Cell Biochem Funct. Dec 2008;26(8):870-873.
270 Ustun K, Alptekin NO, Hakki SS, Hakki EE. Investigation of matrix metalloproteinase-1–1607 1G/2G polymorphism in a Turkish population with periodontitis. J Clin Periodontol. Dec 2008;35(12):1013-1019.
271 Velliyagounder K, Kaplan JB, Furgang D, et al. One of two human lactoferrin variants exhibits increased antibacterial and transcriptional activation activities and is associated with localized juvenile periodontitis. Infect Immun. Nov 2003;71(11):6141-6147.
272 Wagner J, Kaminski WE, Aslanidis C, et al. Prevalence of OPG and IL-1 gene polymorphisms in chronic periodontitis. J Clin Periodontol. Oct 2007;34(10):823-827.
273 Walker SJ, Van Dyke TE, Rich S, Kornman KS, di Giovine FS, Hart TC. Genetic polymorphisms of the IL-1alpha and IL-1beta genes in African-American LJP patients and an African-American control population. J Periodontol. May 2000;71(5):723-728.
274 Wang C, Zhao H, Xiao L, et al. Association between vitamin D receptor gene polymorphisms and severe chronic periodontitis in a Chinese population. J Periodontol. Apr 2009;80(4):603-608.
275 Wang HY, Pan YP. [Screening and analysis of multi-alleles in generalized aggressive periodontitis]. Zhonghua Kou Qiang Yi Xue Za Zhi. Jul 2008;43(7):406-409.
276 Wang HY, Pan YP, Teng D, Zhao J, Lin L. [The relativity between chronic periodontitis and the genetic polymorphisms of vitamin D receptor and estrogen receptor]. Zhonghua Kou Qiang Yi Xue Za Zhi. Apr 2008;43(4):236-239.
277 Wohlfahrt JC, Wu T, Hodges JS, Hinrichs JE, Michalowicz BS. No association between selected candidate gene polymorphisms and severe chronic periodontitis. J Periodontol. Mar 2006;77(3):426-436.
278 Wolf DL, Neiderud AM, Hinckley K, Dahlen G, van de Winkel JG, Papapanou PN. Fcgamma receptor polymorphisms and periodontal status: a prospective follow-up study. J Clin Periodontol. Oct 2006;33(10):691-698.
279 Wu XL, Huang P, Zhou XD. [Association of the progress of chronic periodontitis with interleukin-1B-511 genetic polymorphisms]. Hua Xi Kou Qiang Yi Xue Za Zhi. Dec 2006;24(6):523-526.
280 Wu YM, Juo SH, Ho YP, Ho KY, Yang YH, Tsai CC. Association between lactoferrin gene polymorphisms and aggressive periodontitis among Taiwanese patients. J Periodontal Res. Jun 2009;44(3):418-424.
281 Xiao LM, Yan YX, Xie CJ, et al. Association among interleukin-6 gene polymorphism, diabetes and periodontitis in a Chinese population. Oral Dis. Jun 22 2009.
282 Xie CJ, Xiao LM, Fan WH, Xuan DY, Zhang JC. Common single nucleotide polymorphisms in cyclooxygenase-2 and risk of severe chronic periodontitis in a Chinese population. J Clin Periodontol. Mar 2009;36(3):198-203.
283 Yamamoto K, Kobayashi T, Grossi S, et al. Association of Fcgamma receptor IIa genotype with chronic periodontitis in Caucasians. J Periodontol. Apr 2004;75(4):517-522.
284 Yamazaki K, Tabeta K, Nakajima T, et al. Interleukin-10 gene promoter polymorphism in Japanese patients with adult and early-onset periodontitis. J Clin Periodontol. Sep 2001;28(9):828-832.
285 Yamazaki K, Ueki-Maruyama K, Oda T, et al. Single-nucleotide polymorphism in the CD14 promoter and periodontal disease expression in a Japanese population. J Dent Res. Aug 2003;82(8):612-616.
286 Yasuda K, Sugita N, Kobayashi T, Yamamoto K, Yoshie H. FcgammaRIIB gene polymorphisms in Japanese periodontitis patients. Genes Immun. Dec 2003;4(8):541-546.
287 Yoshie H, Tai H, Kobayashi T, et al. Salivary enzyme levels after scaling and interleukin-1 genotypes in Japanese patients with chronic periodontitis. J Periodontol. Mar 2007;78(3):498-503.
288 Yoshihara A, Sugita N, Yamamoto K, et al. FcgammaRIIIb genotypes and smoking in periodontal disease progression among community-dwelling older adults in Japan. J Periodontol. Feb 2005;76(2):250-255.
289 Yoshihara A, Sugita N, Yamamoto K, Kobayashi T, Miyazaki H, Yoshi H. Analysis of vitamin D and Fcgamma receptor polymorphisms in Japanese patients with generalized early-onset periodontitis. J Dent Res. Dec 2001;80(12):2051-2054.
290 Zhang HX, Xie H, Ren TG. [Relevance of FcgammaRIIIb genotype, IgG G2m(23) factor to the susceptibility of aggressive periodontitis]. Zhonghua Kou Qiang Yi Xue Za Zhi. Mar 2003;38(2):129-131.
291 Zhang JC, Geng HO, Ma WB, Huang P, Pang RY, Zhang YH. [Association of vitamin D receptor gene polymorphisms with the susceptibility to chronic periodontitis of Han nationality]. Zhonghua Kou Qiang Yi Xue Za Zhi. Jan 2005;40(1):50-53.
292 Zhang L, Meng H, Zhao H, et al. Estrogen receptor-alpha gene polymorphisms in patients with periodontitis. J Periodontal Res. Oct 2004;39(5):362-366.
293 Zhang SJ, Yang AL, Zhang JC, Zhang YH, Tang Y. [Association of HLA-DRB1*1501 polymorphism with the susceptibility to severe chronic periodontitis in Chinese Han nationality]. Shanghai Kou Qiang Yi Xue. Jun 2004;13(3):182-185.
294 Zhang Y, Syed R, Uygar C, et al. Evaluation of human leukocyte N-formylpeptide receptor (FPR1) SNPs in aggressive periodontitis patients. Genes Immun. Jan 2003;4(1):22-29.
295 Zhong L, Zhang Y, Zhang J, Yang A, Huang H. [The association of interleukin-1 gene polymorphisms with the susceptibility to chronic periodontitis in Uighur]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. Oct 2002;19(5):405-408.
296 Zhong LJ, Zhang YH, Zhang JC, Feng JH, Yang AL. [The association between interleukin-1 receptor antagonist genotype and chronic periodontitis of Uighur patients]. Zhonghua Kou Qiang Yi Xue Za Zhi. Sep 2003;38(5):370-373.
297 Zhu G, Li C, Cao Z, Corbet EF, Jin L. Toll-like receptors 2 and 4 gene polymorphisms in a Chinese population with periodontitis. Quintessence Int. Mar 2008;39(3):217-226.
298 Zhu XL, Meng HX, Xu L, Zhang L, Chen ZB, Shi D. [Relationship between tumor necrosis factor A-308 gene polymorphism and aggressive periodontitis]. Zhonghua Kou Qiang Yi Xue Za Zhi. May 2007;42(5):268-271.
1 Altshuler D, Daly MJ, Lander ES. Genetic mapping in human disease. Science. 2008;322:881.
2 Amberger J, Bocchini CA, Scott AF, et al. McKusick’s Online Mendelian Inheritance in Man (OMIM). Nucleic Acids Res. 2009;37:D793.
3 Andreiotelli M, Koutayas SO, Madianos PN, et al. Relationship between interleukin-1 genotype and peri-implantitis: a literature review. Quintessence Int. 2008;39:289.
4 Auffray C, Chen Z, Hood L. Systems medicine: the future of medical genomics and healthcare. Genome Med. 2009;1:2.
5 Bernstein BA. An introduction to sample size and power. J Dev Behav Pediatr. 2008;29:516.
6 Borrell LN, Papapanou PN. Analytical epidemiology of periodontitis. J Clin Periodontol. 2005;32(Suppl 6):132.
7 Collins A. Approaches to the identification of susceptibility genes. Parasite Immunol. 2009;31:225.
8 Collins F, Tabak L. A call for increased education in genetics for dental health professionals. J Dent Educ. 2004;68:807.
9 Collins FS. The language of life: dna and the revolution in personalized medicine, ed 1. New York: Harper; 2010.
10 Concolino P, Cecchetti F, D’Autilia C, et al. Association of periodontitis with GSTM1/GSTT1-null variants–a pilot study. Clin Biochem. 2007;40:939.
11 Condit CM. How geneticists can help reporters to get their story right. Nat Rev Genet. 2007;8:815.
12 Dickson SP, Wang K, Krantz L, et al. Rare variants create synthetic genome-wide associations. PLoS Biology. 2010;8:12.
13 Diehl SR, Wu T, Michalowicz BS, et al. Quantitative measures of aggressive periodontitis show substantial heritability and consistency with traditional diagnoses. J Periodontol. 2005;76:279.
14 Fiebig A, Jepsen S, Loos BG, et al. Polymorphisms in the interleukin-1 (IL1) gene cluster are not associated with aggressive periodontitis in a large Caucasian population. Genomics. 2008;92:309.
15 Gilad Y, Pritchard JK, Thornton K. Characterizing natural variation using next-generation sequencing technologies. Trends Genet. 2009;25:463.
16 Ginsburg GS, Willard HF. Genomic and personalized medicine: foundations and applications. Transl Res. 2009;154:277.
17 Gunji T, Onouchi Y, Nagasawa T, et al. Functional polymorphisms of the FPR1 gene and aggressive periodontitis in Japanese. Biochem Biophys Res Commun. 2007;364:7.
18 Gurkan A, Emingil G, Saygan BH, et al. Gene polymorphisms of matrix metalloproteinase-2, -9 and -12 in periodontal health and severe chronic periodontitis. Arch Oral Biol. 2008;53:337.
19 Hennig BJ, Parkhill JM, Chapple IL, et al. Association of a vitamin D receptor gene polymorphism with localized early-onset periodontal diseases. J Periodontol. 1999;70:1032.
20 Ho YP, Lin YC, Yang YH, et al. Cyclooxygenase-2 gene-765 single nucleotide polymorphism as a protective factor against periodontitis in Taiwanese. J Clin Periodontol. 2008;35:1.
21 Huynh-Ba G, Lang NP, Tonetti MS, et al. The association of the composite IL-1 genotype with periodontitis progression and/or treatment outcomes: a systematic review. J Clin Periodontol. 2007;34:305.
22 Huynh-Ba G, Lang NP, Tonetti MS, et al. Association of the composite IL-1 genotype with peri-implantitis: a systematic review. Clin Oral Implants Res. 2008;19:1154.
23 Johnson L, Genco RJ, Damsky C, et al. Genetics and its implications for clinical dental practice and education: report of panel 3 of the Macy study. J Dent Educ. 2008;72:86.
24 Jordan WJ, Eskdale J, Lennon GP, et al. A non-conservative, coding single-nucleotide polymorphism in the N-terminal region of lactoferrin is associated with aggressive periodontitis in an African-American, but not a Caucasian population. Genes Immun. 2005;6:632.
25 Kocher T, Sawaf H, Fanghanel J, et al. Association between bone loss in periodontal disease and polymorphism of N-acetyltransferase (NAT2). J Clin Periodontol. 2002;29:21.
26 Laine ML, Morre SA, Murillo LS, et al. CD14 and TLR4 gene polymorphisms in adult periodontitis. J Dent Res. 2005;84:1042.
27 Li Q, Meng H, Zhang L, et al. Correlation between single nucleotide polymorphisms in a calprotectin subunit gene and risk of periodontitis in a Chinese population. Ann Hum Genet. 2007;71:312.
28 Loe H, Brown LJ. Early onset periodontitis in the United States of America. J Periodontol. 1991;62:608.
29 Loos BG, Fiebig A, Nothnagel M, et al. NOD1 gene polymorphisms in relation to aggressive periodontitis. Innate Immun. 2009;15:225.
30 Loos BG, Leppers-Van de Straat FG, Van de Winkel JG, et al. Fcgamma receptor polymorphisms in relation to periodontitis. J Clin Periodontol. 2003;30:595.
31 Lu-Emerson C, Plotkin SR. The neurofibromatoses. Part 1: NF1. Rev Neurol Dis. 2009;6:E47.
32 MacDougall M, Jeffords LG, Gu TT, et al. Genetic linkage of the dentinogenesis imperfecta type III locus to chromosome 4q. J Dent Res. 1999;78:1277.
33 Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747.
34 McGuffin P, Huckle P. Simulation of mendelism revisited: the recessive gene for attending medical school. Am J Hum Genet. 1990;46:994.
35 Meisel P, Krause T, Cascorbi I, et al. Gender and smoking-related risk reduction of periodontal disease with variant myeloperoxidase alleles. Genes Immun. 2002;3:102.
36 Meisel P, Schwahn C, Gesch D, et al. Dose-effect relation of smoking and the interleukin-1 gene polymorphism in periodontal disease. J Periodontol. 2004;75:236.
37 Meisel P, Siegemund A, Grimm R, et al. The interleukin-1 polymorphism, smoking, and the risk of periodontal disease in the population-based SHIP study. J Dent Res. 2003;82:189.
38 Michalowicz BS, Aeppli D, Virag JG, et al. Periodontal findings in adult twins. J Periodontol. 1991;62:293.
39 Michalowicz BS, Diehl SR, Gunsolley JC, et al. Evidence of a substantial genetic basis for risk of adult periodontitis. J Periodontol. 2000;71:1699.
40 Michalowicz BS, Wolff LF, Klump D, et al. Periodontal bacteria in adult twins. J Periodontol. 1999;70:263.
41 Nebert DW, Zhang G, Vesell ES. From human genetics and genomics to pharmacogenetics and pharmacogenomics: past lessons, future directions. Drug Metab Rev. 2008;40:187.
42 Nikolopoulos GK, Dimou NL, Hamodrakas SJ, et al. Cytokine gene polymorphisms in periodontal disease: a meta-analysis of 53 studies including 4178 cases and 4590 controls. J Clin Periodontol. 2008;35:754.
43 Noack B, Gorgens H, Hempel U, et al. Cathepsin C gene variants in aggressive periodontitis. J Dent Res. 2008;87:958.
44 O’Sullivan BP, Freedman SD. Cystic fibrosis. Lancet. 2009;373:1891.
45 Poulsen P, Kyvik KO, Vaag A, et al. Heritability of type II (non-insulin-dependent) diabetes mellitus and abnormal glucose tolerance–a population-based twin study. Diabetologia. 1999;42:139.
46 Preshaw PM. Definitions of periodontal disease in research. J Clin Periodontol. 2009;36:1.
47 Purcell S, Cherny SS, Sham PC. Genetic Power Calculator: design of linkage and association genetic mapping studies of complex traits. Bioinformatics. 2003;19:149.
48 Ragoussis J. Genotyping technologies for genetic research. Annu Rev Genomics Hum Genet. 2009;10:117.
49 Ramagopalan SV, Knight M, Ebers GC, et al. Origins of magic: review of genetic and epigenetic effects. BMJ. 2007;335:1299.
50 Risch N, Merikangas K. The future of genetic studies of complex human diseases. Science. 1996;273:1516.
51 Sahingur SE, Sharma A, Genco RJ, et al. Association of increased levels of fibrinogen and the -455G/A fibrinogen gene polymorphism with chronic periodontitis. J Periodontol. 2003;74:329.
52 Schaefer AS, Richter GM, Groessner-Schreiber B, et al. Identification of a shared genetic susceptibility locus for coronary heart disease and periodontitis. PLoS Genet. 2009;5:e1000378.
53 Schaefer AS, Richter GM, Nothnagel M, et al. A 3’ UTR transition within DEFB1 is associated with chronic and aggressive periodontitis. Genes Immun. 2010;11:45.
54 Schaefer AS, Richter GM, Nothnagel M, et al. A genome-wide association study identifies GLT6D1 as a susceptibility locus for periodontitis. Hum Mol Genet. 2010;19:553.
55 Selvanathan SK, Shenton A, Ferner R, et al. Further genotype–phenotype correlations in neurofibromatosis 2. Clin Genet. 2009;77:163.
56 Stein JM, Machulla HK, Smeets R, et al. Human leukocyte antigen polymorphism in chronic and aggressive periodontitis among Caucasians: a meta-analysis. J Clin Periodontol. 2008;35:183.
57 Stolerman ES, Florez JC. Genomics of type 2 diabetes mellitus: implications for the clinician. Nat Rev Endocrinol. 2009;5:429.
58 Talmud PJ, Hingorani AD, Cooper JA, et al. Utility of genetic and non-genetic risk factors in prediction of type 2 diabetes: Whitehall II prospective cohort study. BMJ. 2010;340:b4838.
59 Velliyagounder K, Kaplan JB, Furgang D, et al. One of two human lactoferrin variants exhibits increased antibacterial and transcriptional activation activities and is associated with localized juvenile periodontitis. Infect Immun. 2003;71:6141.
60 Voelkerding KV, Dames SA, Durtschi JD. Next-generation sequencing: from basic research to diagnostics. Clin Chem. 2009;55:641.
61 Zhang L, Meng H, Zhao H, et al. Estrogen receptor-alpha gene polymorphisms in patients with periodontitis. J Periodontal Res. 2004;39:362.
Suggested Reading
Collins A. Approaches to the identification of susceptibility genes. Parasite Immunol. May 2009;31(5):225-233.
Collins FS. The language of life: dna and the revolution in personalized medicine. New York: Harper; 2010.
Frazer KA, Murray SS, Schork NJ, Topol EJ. Human genetic variation and its contribution to complex traits. Nat Rev Genet. Apr 2009;10(4):241-251.
Ginsburg GS, Willard HF. Genomic and personalized medicine: foundations and applications. Transl Res. Dec 2009;154(6):277-287.
Kinane DF, Shiba H, Hart TC. The genetic basis of periodontitis. Periodontol 2000. 2005;39:91-117.
Nares S. The genetic relationship to periodontal disease. Periodontol 2000. 2003;32:36-49.
Takashiba S, Naruishi K. Gene polymorphisms in periodontal health and disease. Periodontol 2000. 2006;40:94-106.
Ragoussis J. Genotyping technologies for genetic research. Annu Rev Genomics Hum Genet. 2009;10:117-133.
Rodriguez-Murillo L, Greenberg DA. Genetic association analysis: a primer on how it works, its strengths and its weaknesses. Int J Androl. Dec 2008;31(6):546-556.
Yoshie H, Kobayashi T, Tai H, Galicia JC. The role of genetic polymorphisms in periodontitis. Periodontol 2000. 2007;43:102-132.