CHAPTER 25 Molecular Biology of the Host-Microbe Interaction in Periodontal Diseases
This chapter provides an overview of the molecular biology of the host-parasite relationship from a cellular signalling perspective specific to pathogen-associated lipopolysaccharide (LPS). The implications of periodontal microbiota-associated LPS and other microbiota-associated molecular patterns (MAMPs) on the induction of the innate immune response, toll-like receptor (TLR) signalling, and periodontal pathogenesis are reviewed. The second half of the chapter highlights the pathobiology of periodontal disease and the induction of disease by proinflammatory cytokines that generate host-tissue destruction and alveolar bone loss. The chapter concludes with ramifications for the development of therapeutic strategies that perturb innate immune signal transduction for the treatment of periodontitis.
For the past two decades, the host response to the bacterial challenge originating from the dental biofilm has been considered to play a major role in initiation and tissue destruction of periodontal diseases.176 The significance of host-microbial interactions is reinforced by epidemiologic data indicating different susceptibilities to periodontal disease among individuals, in spite of the long-term presence of oral biofilm.20,21,194 Other studies demonstrating increased susceptibility and greater severity of periodontal disease in individuals with impaired immune response caused by systemic conditions also indicate the significance of the host response to the bacterial challenge.88,209 In the current paradigm of periodontal disease, periodontal pathogens are essential for disease initiation; however, the extent and severity of tissue destruction are largely dependent on the nature of the host-microbial interactions. These interactions are dynamic, since the microbial composition of the dental biofilm and host immune competency can vary widely between patients resulting in differences in host responses and subsequent alveolar bone loss. This concept has evolved in parallel with a more advanced appreciation of the immune response resulting in an increased emphasis focused on mechanisms of host-microbial interactions in periodontal disease pathobiology, as well as on the development of novel therapeutic strategies.
Periodontal diseases provide a unique situation to study microbial-host interactions. Over 500 different microbial species can be found in the oral biofilm;237 however, only a few of those are associated with periodontal disease.293,294 This implicates that the recognition of both nonpathogenic/commensal bacteria and pathogenic bacteria by the host requires vigilance and tolerance mechanisms to mount an appropriate response that can prevent dissemination of infection without inducing an exacerbated reaction that could result in damage to the host tissues. Direct recognition of microbes by the host is mediated by the recognition of MAMPs by pattern-recognition receptors (PRRs).35,37
Host response to periodontal infection requires expression of a number of bioactive agents, including proinflammatory and antiinflammatory cytokines, growth factors, and enzymes that are the result of the activation of multiple signalling pathways. This activation of intracellular signalling may initiate exclusively as an innate immune response associated with PRR-mediated sensing of MAMPs. However, since the oral cavity is one of the most readily colonized postnatal mucosal sites, the long-standing interactions between microbes (commensal and pathogenic) and host results in a “priming” of the immune system with the common presence of adaptive immune cells in the periodontal tissues.
The biologic mediators expressed as a result of PRR-signalling activation include costimulatory molecules involved in the induction of adaptive immunity.37 This results in a cascade of events that will establish very complex cytokine and signalling networks. There is accumulating evidence for a direct role of “classic” innate immunity signalling through PRRs in adaptive immune cells that can modulate the function of these cells. Abundant evidence indicates a key role for the adaptive immune response—humoral and cellular aspects—in mediating the host response to microorganisms existing within the oral biofilm and also in the majority of tissue destruction associated with periodontal diseases.* Even though cells participating in the adaptive immune response are considered to be the primary source of cytokines leading to bone resorption,168 additional data exist showing that periodontal bone loss occurs in the absence of B and T cells, suggesting a role for the innate immune response in periodontal disease initiation or progression.22,25,26
Innate immunity and inflammation are not synonymous; however, inflammation arises primarily in response to infection. More importantly, innate and adaptive immune responses are not mutually exclusive, and the bridge between these arbitrarily and didactically distinguished arms of the immune response has been shortened in recent years. However, considering that this chapter focuses on host-microbe interaction, we will emphasize the direct recognition of MAMPs by cells participating in the immune response and the molecular mechanisms activated downstream of this recognition (Figure 25-1).
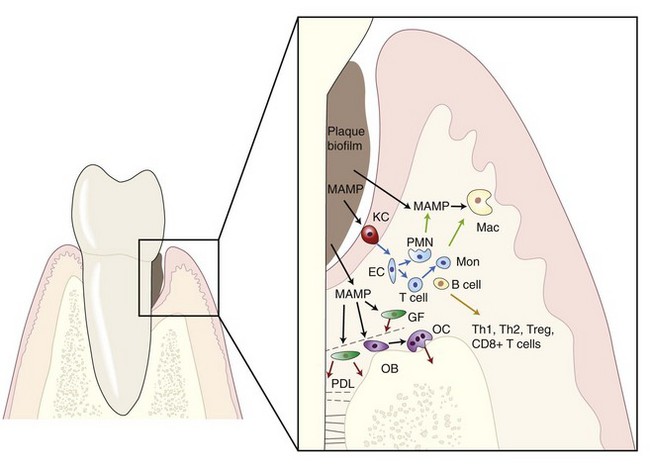
Figure 25-1 Microbial-associated molecular pattern (MAMP) from microorganisms in the dental biofilm activate inflammatory responses in periodontal tissues. Biologic mediators will either affect neighboring cells (blue arrows) by inducing the expression of other mediators (e.g., RANKL) or by triggering chemotaxis (green arrows). Direct damage to periodontal tissues may also result after MAMPs stimulation (red arrows), such as metalloproteinase secretion by gingival and periodontal ligament fibroblasts. MAMP, Microbial-associated molecular patterns; RANKL, receptor activator of nuclear factor (NF) -κB ligand; KC, keratinocyte; Mac, macrophage; GF, gingival fibroblast; PDL, periodontal ligament fibroblast; OB, osteoblast; OC, osteoclast; EC, endothelial cell; PMN, polymorphonuclear neutrophil; Mon, monocyte; T cell, T lymphocyte, B cell, B lymphocyte.
Innate Immunity in Periodontal Diseases
Vigilance and Tolerance
Even though adaptive and innate immune responses are traditionally distinguished, in vivo they are integral parts of the host defenses against infections (i.e., innate immunity is not inactivated once the adaptive response is initiated). Innate immunity is required for the activation of a more specific adaptive immune response, but it also plays an important role in managing host-microbial interactions since the innate immune system is rapidly activated (within minutes) and is responsible for the defense during the initial hours and days of the infection, whereas adaptive immunity requires at least 7 to 10 days before an adequate cellular or humoral response occurs. This role of innate immunity is especially important considering that our host cells are outnumbered 10 : 1 by the microbial cells living on skin and mucosal surfaces. This coexistence, however, is usually pacific and even beneficial for one or both cohabitants but requires both vigilance and tolerance mechanisms that ultimately determine if the host response is to clear the infection, or suppression of the host response is required to maintain the homeostatic equilibrium between host and microbes.
With regard to periodontal diseases, current paradigm indicates that some groups or complexes of bacteria are more strongly associated with the presence of periodontal diseases, but this association is not universally found, as there are individuals harboring the disease-associated complexes that are free of periodontal disease. Moreover, the microbial flora of the oral cavity can include more than 500 different species and there is no uniformity on the level of infection that is sufficient to produce disease. The complexity of the oral microbial flora is particularly intriguing given the limited number of receptors that are able to recognize microbial antigens. Tolerance mechanisms probably play a role in modulating the host responses to commensal/nonpathogenic bacteria. One of the primary challenges of the innate immune system is to discriminate among a large number of periodontal pathogens from the host with a limited number of cell surface receptors. This challenge is compounded because microbial pathogens have the ability to mutate as a mechanism to escape host recognition. The innate immune system has met this challenge through recognition of evolutionarily conserved structures on pathogens that are not present in higher eukaryotes (PRRs). These molecular motifs (MAMPs) have essential roles in the pathogen’s ability to evade host defense and thus are not subject to high mutation rates. MAMPs are shared among various species of microbes but are not expressed by the host. Although many PRRs have been known for years, it was not clear how the innate immune system functioned until the discovery of TLRs, which have proved to be critical for recognition of microbes by the innate immune system and for bridging the innate and acquired immune responses. Interestingly, within the periodontal tissues, the expression of various TLRs appears to be increased in severe disease states.29 Table 25-1 illustrates PRRs and the host cells that express them and their ligands (MAMPs) present in microorganisms.
TABLE 25-1 Pattern-Recognition Receptors (PRRs), Host Cells (Mouse or Human) that Express Them, and Their Ligands (MAMPs) Present in Microorganisms Relevant to Periodontal Diseases
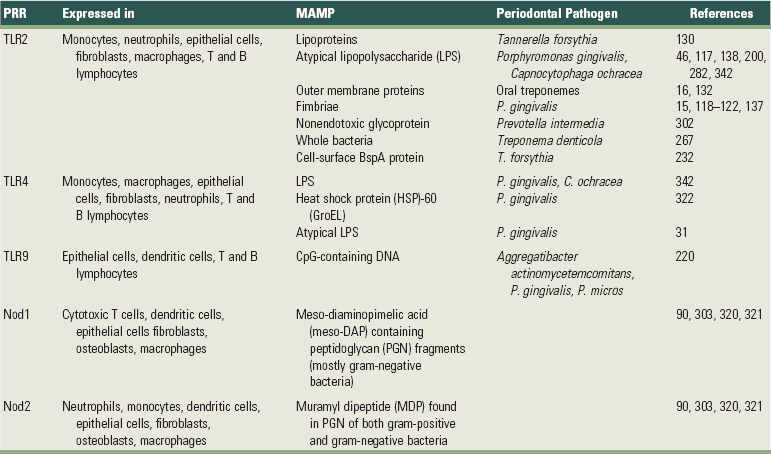
However, there are other PRRs that can be secreted into the plasma as humoral proteins and others that are localized in the cytoplasm as intracellular sensors. Soluble PRRs include various proteins, including collectins, ficolins, and acute-phase pentraxins (e.g., C-reactive protein) that represent functional ancestors of antibodies. These soluble mannose-binding receptors can interact with structures from microbes and activate the complement system via mannan-binding lectin-associated serine kinase (MASP) pathway.269 In fact, direct interaction between complement precursors and microbes can initiate activation through an unclear mechanism that culminates with opsonization and lysis of the microbes. Another example of soluble PRRs is the lectin-complement system, which is involved in recognizing the microbe by the carbohydrate-binding lectin, leading to its subsequent opsonization and phagocytosis.331
Nucleotide-oligomerization domain (NOD) protein-like receptors represent cytoplasmic PRRs and are characterized by C-terminal leucine-rich repeats (LRR domain, similar to the TLRs), an N-terminal caspase-activating recruitment domain (CARD), and a nucleotide-binding domain (NBD). These were initially described as cytosolic TLRs, analogous to the R proteins present in plants.70 NOD proteins are capable of recognizing different peptidoglycan molecules: Nod1 recognizes peptidoglycan containing meso-diaminopimelic acid (meso-DAP) fragments present in most gram-negative and some gram-positive bacteria,106 whereas Nod2 recognizes muramyl dipeptide (MDP), which is found in peptidoglycan from both gram-negative and gram-positive bacteria.207 More recent evidence indicates that NOD proteins, specifically Nod1 and Nod2, are involved in activation of inflammatory gene expression90 and even LPS recognition independently of TLR.152 The relevance of NOD proteins for the immune response is demonstrated by the association between genetic mutations on Nod1 and Nod2 and development of allergic conditions and Crohn’s disease, respectively.148,153 Another type of intracellular PRR is represented by a cytoplasmic protein family, the retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs), which play a critical role in recognizing viral ribonucleic acid (RNA) and induction of type I interferon (IFN) expression. However, since the role of retroviruses in periodontal disease is not clear, RLRs will not be discussed here (for a review of RLRs, see Yoneyama and Fujita340).
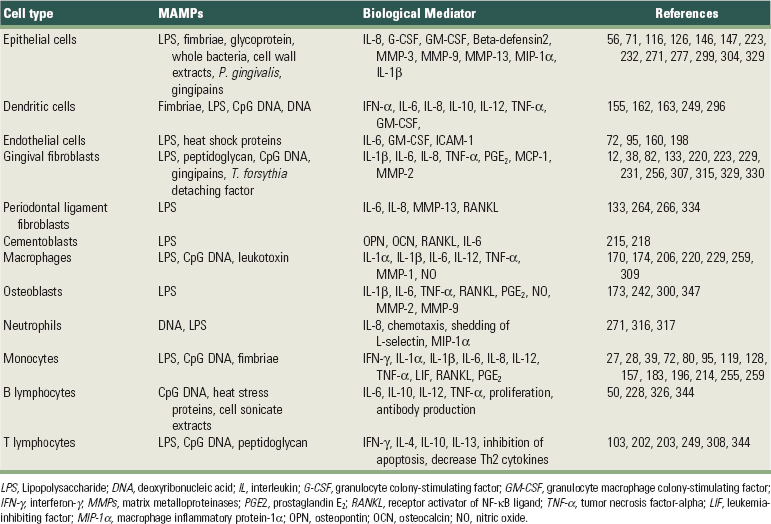
All of these different PRRs represent the necessary armamentaria for recognition of MAMPs by the host and are expressed by a variety of cells that play a role in innate immunity. Importantly, our understanding of the roles of PRRs in inflammation and immune responses has been expanded, so it is now appreciated that these receptors not only recognize various MAMPs to activate innate immune response, but they can also bind to endogenous molecules derived from damaged tissue and have a role in inflammation and adaptive immune responses. The cells involved in innate immunity include macrophages and polymorphonuclear cells as professional phagocytes with the primary function of engulfing and destroying microbes; dendritic cells as professional antigen-presenting cells and activators of adaptive immunity; and natural killer (NK) cells, the innate cytotoxic lymphocytes that recognize and kill host cells that are altered (e.g., tumor cells) or infected with viruses. However, other cell types can also play important roles in innate immunity, since they are able to recognize MAMPs through their PRRs and respond by expressing biologically-active molecules, such as cytokines and matrix metalloproteases (MMPs), that will have an effect on homeostasis of the host tissues in the periodontal microenvironment. Resident, “nonprofessional” cells such as fibroblasts and osteoblasts are also capable of producing a variety of cytokines, such as interleukin-6 (IL-6), prostaglandin E2 (PGE2), MMPs, and receptor activator of NF-κB ligand (RANKL). Table 25-2 presents these biologic mediators elicited by different MAMPs in resident and nonresident cells involved in the pathogenesis of destructive periodontal disease. Because of the sheer proportion of fibroblasts in the periodontal tissues and also to the proximity and relevance of both fibroblasts and osteoblasts to nonmineralized and mineralized tissue turnover, respectively, these cells can play important roles in innate immunity during periodontal diseases. Epithelial cells represent the initial point of contact with MAMPs in the periodontium and play an important role in innate immunity not only because of their function as a physical barrier, but also because these cells are equipped with PRRs and respond to MAMPs by secreting various cytokines and chemokines, including IL-8 and antimicrobial peptides (e.g., beta defensins, cathelicidins).69,156
TABLE 25-2 Biological Mediators Elicited by Different MAMPs in Resident and Nonresident Cells Involved in the Pathogenesis of Destructive Periodontal Disease
Despite the alleged nonspecificity of the innate immune response, it has long been known that there are important quantitative and qualitative differences in cytokine release on stimulation with gram-positive or gram-negative bacteria.
According to the current paradigm of the microbial etiology of periodontal disease, disease initiation depends on shifts in the microbial population of the dental biofilm toward a more complex flora that includes gram-negative and anaerobic species. This implies that some bacterial species in the dental biofilm may not play a role in periodontal disease and in fact, clinical periodontal health is frequently observed in spite of the presence of a dental biofilm. However, the fact that different species of bacteria share common MAMPs (e.g., CpG deoxyribonucleic acid [DNA], LPS, and peptidoglycans) that can potentially trigger an innate immune response suggests that tolerance mechanisms may counterbalance the vigilance aspect of innate immune responses to allow the presence of these commensal, nonpathogenic bacteria. Tolerance mechanisms have been extensively studied in the gut epithelium, since the intestinal mucosa is continuously exposed to innocuous environmental antigens and commensal microorganisms that live in symbiosis with the host (for review, see Artis14). The critical importance of these tolerance mechanisms is exemplified by the fact that its dysregulation is associated with the pathogenesis of various inflammatory conditions, including inflammatory bowel disease and intestinal cancer.40,165
There are some important differences between the oral epithelium and the intestinal mucosa, especially the presence of specialized immune cells subjacent to the mucosal epithelium in the intestines (gut-associated lymphoid tissues [GALT] and Peyer’s patches); however, the concept of a tolerance mechanism that renders the immune system hyporesponsive to commensal bacteria, while retaining the capacity to respond to pathogenic organisms has also been investigated in oral epithelial cells. A comparison of the responses of oral epithelial cells to nonperiodontopathic and periodontopathogenic bacteria demonstrated that, in general, commensal bacteria (such as Streptococcus gordonii or Streptococcus sanguinis) induce expression of antimicrobial peptides without inducing the chemoattractant cytokine IL-8, whereas periodontopathogenic bacteria from the “orange complex” (Fusobacterium nucleatum and Prevotella intermedia) induce strong expression of both antimicrobial peptides and IL-8. Interestingly, the bacteria that are more strongly associated with periodontitis (“red complex” organisms Treponema denticola, Tannerella forsythia, and Porphyromonas gingivalis) tended to suppress the innate immune response by inhibiting the expression of antimicrobial peptides, IL-8, or both simultaneously.156,179,324 Tolerance mechanisms may involve the activation of different signalling pathways, since the activation of NF-κB (which is associated with the expression of many inflammatory genes) was not required for the expression of antimicrobial peptides by oral epithelial cells after stimulation with commensal bacteria, as opposed to the periodontopathogenic microorganisms.56 There is still much to be explored in these tolerance mechanisms, but it is conceivable that commensal bacteria may be beneficial to the host as they induce expression of antimicrobial peptides, which will clear some of the nonpathogenic and pathogenic bacteria while allowing nonpathogenic periodontal bacteria that are resistant to antimicrobial peptides, such as S. gordonii and S. mutans,156,251 to establish themselves in the oral biofilm. The weak induction of IL-8 may function to maintain immune vigilance without causing too much collateral damage to the host tissues. On the other hand, pathogenic bacteria (such as the orange complex microorganisms, F. nucleatum and P. intermedia) that induce both antimicrobial peptides and IL-8 expression may be limiting their survival in the biofilm. Interestingly, the inhibition of expression of antimicrobial peptides and IL-8 in epithelial cells by red complex bacteria (such as P. gingivalis and T. denticola) can represent host-evading strategies that allow them to survive and establish themselves in the oral biofilm.44
Activation of the innate immune system is critical for lymphocyte activation and other immune cells to help clear the infectious microorganisms. However, exuberant production of proinflammatory cytokines leads to severe pathology, including alveolar bone loss.17,76,77,113,114 To help prevent deleterious effects of TLR activation, a number of signalling mechanisms have evolved. These mechanisms include downregulation of surface TLR receptor expression, transcriptional induction of negative regulators such as IL-1 receptor-associated kinase (IRAK-M), suppressor of cytokine signalling-1 (SOCS1), and SH2-containing inositol phosphatase (SHIP), and production of antiinflammatory cytokines, including IL-10 and transforming growth factor-beta (TGF-β).191 In contrast to the release of proinflammatory cytokines and mediators, production of these negative immune regulators occurs over a much longer time frame, providing a vital role in controlling the extent of proinflammatory and antiinflammatory mediators in a proper temporal sequence.191
Cell Signalling Pathways and the Expression of Biologically-Active Mediators in the Innate Immune Response
Once MAMPs are recognized by the appropriate PRRs, a signal is initiated within the cell. This signal is transduced through the cytoplasm and nucleus more commonly by sequential posttranslational modification (e.g., phosphorylation) of a cascade of kinases that are constitutively expressed and ultimately will determine the cell response to the MAMPs detected (Figure 25-2).
TLRs are single-pass transmembrane proteins with N-terminal presenting LRRs that are responsible for the recognition of their ligands and with a C-terminal cytoplasmic domain that is very similar to the cytoplasmic region of the IL-1 receptor,3 which is called the Toll/IL-1 receptor domain (TIR). Thus, subsequent to the recognition of a ligand by TLRs, the signal generated uses pathways similar to those utilized by the IL-1 receptor. TLR signalling was originally described in the context of the activation of the IFN regulatory factor IRF family of transcription factors and NF-κB, leading to the expression of IFN-γ and early-response inflammatory genes, respectively.
Interestingly, the participation of at least four adaptor proteins (MyD88, TRIF, Mal/TIRAP, and TRAM) containing TIR domains that can be recruited by activated TLRs results in important branching of the signal transduction and yields a significant flexibility to TLR signalling by allowing cross-talk with other pathways, notably the MAP kinase pathway. These adaptor proteins are recruited to TLRs by homophilic interactions between their TIR domains and are utilized differently by the TLRs. TLR5, TLR7, and TLR9 were shown to depend on recruitment of MyD88 to signal,136,142 whereas TLR3 is the only TLR that does not use MyD88.337 TLR4, on the other hand, can use all four adaptor proteins: MyD88, TRIF, Mal/TIRAP, and TRAM.336-338 Even though activation of the canonical NF-κB pathway is usually affected by all TLRs, the timing of NF-κB activation,166,167 as well as the additional signalling pathways that are activated by the branching of the signal, varies among TLR receptors and with the participation of different adaptor proteins (Figure 25-2). These variations will ultimately affect the biologic result in terms of gene expression as demonstrated by the finding that even though NF-κB activation is observed after TLR4 stimulation by LPS, this may or may not result in inflammatory gene expression, depending on the adaptor protein used. In wild-type cells, LPS stimulation results in inflammatory cytokine expression, whereas in MyD88-deficient cells, LPS fails to induce cytokine expression. In the absence of MyD88, activation of NF-κB occurs with delayed kinetics in comparison to wild-type cells.166
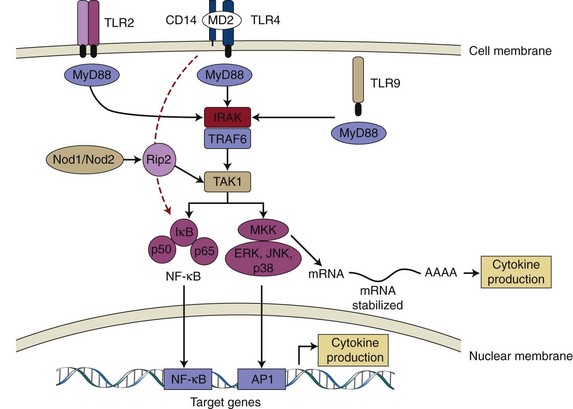
Figure 25-2 Pattern recognition receptors (PRRs) and innate immune signalling. TLR2, TLR4, and TLR9 are depicted as examples of TLR receptors expressed in cells of the periodontal tissues. On ligand binding, all TLRs (except TLR3) recruit adaptor protein MyD88 and activate common upstream activator (IRAK/TRAF6 and TAK1) of NF-κB and MAPKs. TLR4 may also activate NF-κB independently of MyD88 and with delayed kinetics (red dotted line). Nod1/Nod2 are cytosolic PRRs that recognize peptidoglycan fragments of bacterial cell walls and may amplify the TLR-induced activation of signalling pathways. Activated NF-κB and MAPKs translocate to the nucleus and bind to their motifs (NF-κB, AP-1, respectively) in the promoter region of target genes (including early-response and inflammatory genes) and induce their transcription into mRNA, which will ultimately lead to increased cytokine production. p38 MAPK is also involved with post-transcriptional regulation of proinflammatory genes (e.g., IL-6, Cox-2) by modulation of mRNA stability in the cytoplasm. TLR, Toll-like receptor; CD14, cluster of differentiation 14 molecule; MD2, myeloid differentiation protein 2; MyD88, myeloid differentiation primary response gene 88; IRAK, interleukin-1 receptor-associated kinase; TRAF6, TNF receptor-associated factor 6; TAK1, TGF-beta activated kinase 1; MKK, mitogen-activated protein kinase kinase; ERK, extracellular signal-regulated kinase; JNK, c-Jun N-terminal kinase; AP-1, activator protein-1).
The shift in the microbial population present in the oral biofilm from predominantly gram-positive to gram-negative bacteria that is associated with the onset of periodontal disease may lead to different patterns of immune response as a result of the type of TLR predominantly activated. Gram-positive bacteria were shown to activate TLR2, which induced increased expression of IL-8, whereas gram-negative bacteria activated predominantly TLR4, resulting in increased expression of tumor necrosis factor alpha (TNF-α).312
However, some gram-negative microorganisms present in the oral biofilm and associated with periodontal disease are rather unique in their capacity to activate NF-κB via preferential utilization of TLR2.45 Recently, it was reported that most gram-negative bacteria associated with periodontal disease, including P. gingivalis, Tannerella forsythia, Prevotella intermedia, Prevotella nigrescences, Fusobacterium nucleatum, Aggregatibacter actinomycetemcomitans, and Veillonella parvula are all capable of activating TLR2, whereas the latter two microorganisms also activate TLR4.172 Even though all these disease-associated microorganisms activate TLR2 signalling, this pathway can also be activated in vitro by microorganisms present in an oral biofilm composed primarily of gram-positive bacteria and which are common colonizers of the oral biofilm and not associated with clinical signs of periodontal disease.343 The fact that TLR2 is activated by both pathogenic and nonpathogenic microorganisms is an interesting finding and suggests differences in the utilization of adaptor proteins and/or concomitant activation of other TLRs by different MAMPs expressed by the various bacterial species that are present in an oral biofilm associated with disease. These differences can lead to the activation of different signalling pathways and subsequent modulation of the host response.
NOD-like receptors include 23 genes in humans, and even though some of these genes are primarily expressed in macrophages and polymorphonuclear leukocytes (PMNs), the best studied and characterized members Nod1 and Nod2 are expressed by a wide variety of cells, including epithelial cells,318,319 monocytes/macrophages,230 and dendritic cells306 (see Table 25-1). The exact mechanism by which these proteins recognize their ligands is still unknown, and there is no evidence for direct interaction between the ligands and NOD proteins. According to the proposed mechanism of activation of NOD proteins, they are kept in an inactive state by intramolecular interactions between the C-terminal LRR domain and N-terminal fragment CARD and NOD domains. Ligand recognition results in conformational changes that relieve the autoinhibitory intramolecular interactions and allow NOD–domain-dependent nucleotide binding and oligomerization.145 Stimulation of Nod1 and Nod2 will result in activation of NF-κB and mitogen-activated protein kinases (MAPKs), similarly to the activation of TLRs (see Figure 25-2); however, the signal transduction pathways require different signalling intermediates. Activation of Nod2 leads to recruitment of a kinase called receptor-interacting protein 2 (Rip2, also known as RICK), which will bind directly to inhibitor of NF-κB kinase gamma (IKKκ, also known as NF-κB essential modulator [NEMO]) and promote its ubiquitination and activation of the catalytic regulatory subunits of the inhibitor of NF-κB kinase (IKK) complex. Once activated, IKK phosphorylates the inhibitor IkB leading to its degradation by the proteasome, releasing NF-κB and allowing its translocation to the nucleus where it will affect expression of various inflammatory genes.134,234 In this pathway, Rip2 has been shown to be required for Nod1/Nod2 activation of NF-κB,234 but interestingly its kinase activity is not essential.131 The same signalling intermediate Rip2 has also been shown to be involved in Nod1/Nod2-induced MAPK activation by mediating the recruitment of upstream activator of MAPK, TGF-β-activated kinase 1 (TAK1).131 However, even though Rip2 and TAK1 are required for Nod1/Nod2-mediated activation of MAPK, the intermediate steps in the activation of this pathway are not well known.234,281
Since TLRs and NOD proteins are PRRs involved in recognition of bacteria and considering that the signals generated by their activation converge to the same signalling pathways, there may be a synergistic or “amplifying” effect for simultaneous NOD and TLR activation by MAMPs. This cooperation between different PRRs in the activation of NF-κB and MAPK-signalling pathways may increase the sensitivity and potency of the host response to the presence of bacteria.90,91,323 Indeed, there is evidence demonstrating that activation of Nod1/Nod2 has synergistic effects with TLR signalling on the production of proinflammatory cytokines (including IL-1, IL-4, IL-6, IL-10, IL-12, granulocyte macrophage colony-stimulating factor [GM-CSF], and TNF).91,234,306,323 Moreover, it has been suggested that Nod1/Nod2 signalling augments t-helper 1(Th1) polarization induced by TLR signalling, except for TLR2 signals, which inhibit Th1-type cytokines because of IL-10 production.306
Besides their role as PRRs and synergism with TLR signalling, other members of the NOD family of proteins can also affect innate immune response by their role in the formation of the inflammasome, which is an intracellular multiprotein complex that activates caspase-1. Activated caspase-1 will process the inactive pro–IL-1β and pro–IL-18 forms produced and convert them into the biologically active forms secreted.184
Adaptive Immunity in Periodontal Diseases
Activation of Adaptive Immunity in Periodontal Diseases
Besides its role in the immediate detection of infectious agents and toxic/degradation (“danger”) products to elicit an inflammatory response and activate effector processes aimed at the elimination or confinement of microbial invaders, innate immunity also has the pivotal role of initiating and modulating adaptive immune responses. Thus adaptive immunity requires the innate “branch” of immunity to be activated; however, in vivo, this didactic distinction does not exist and the immune response should be understood as a continuum, a constantly adjusting host reaction to the presence of microbes and their MAMPs. In the context of this chapter, the signalling and molecular aspects of the host-microbe interactions related to the adaptive immune response will be emphasized.
Innate immune mechanisms and signalling are not “turned off” once the adaptive immune response is activated but rather are “supplemented” by the latter in the defense against microbes. On the other hand, cells from adaptive immune response also express PRRs and respond to MAMPs, which further emphasizes the dynamic interaction between the two “arms” of the immune response.
In T lymphocytes, TLR agonists have been shown to modulate the expression of costimulatory receptors (such as CD28, CD69, and CD152) and increase their proliferation and IFN-γ production, suggesting that microbes and their MAMPs may also have a direct functional role in the regulation of adaptive immunity.47,292 This is especially important to realize in the context of periodontal disease, which is a chronic inflammatory condition and as such has a dense lymphocytic infiltrate. Ever since the classic studies of the host response in periodontal disease, including the recognition of immunoglobulin-producing plasma cells in the gingival tissues of patients with periodontal disease,43 the classic descriptions of initial, established, and advanced lesions233 and the characterization of the inflammatory infiltrate in the dog model,192 the nature of the inflammatory infiltrate and its association with the inflammatory response and ultimately the quiescence or progression of disease, has been debated.
The adaptive immune response is characterized by the activities of pathogen-specific B and T lymphocytes; however, the cell type primarily responsible for translating innate signals into adaptive immunity is the dendritic cell (DC). In fact, adaptive immunity initiates with DCs recognizing MAMPs in the sites of infection and subsequently migrating into the regional draining lymph nodes to present the processed antigen peptides in the context of major histocompatibility complex (MHC) molecules to naive T lymphocytes. Once inside the lymph nodes, DCs migrate to the T cell areas, seek out antigen-specific T cells, and induce their activation and differentiation into effector cells (helper or cytotoxic cells). The maturation process induced by MAMPs that leads to the migration of DCs to the lymph nodes involves modulation of the expression of various chemokine receptors that render DCs less responsive to inflammatory signals and more responsive to lymphocyte-derived signals. Notably, LPS-induced maturation of DCs has been shown to result in the downregulation of “inflammatory” CCR1 and CCR5 (which responds to macrophage inflammatory protein-1α [MIP-1α], for example) and upregulation of CXCR4 and CCR7 (which respond to CXCL12 and CCL19, respectively).274 These changes ultimately control DC trafficking and migration to the lymph nodes and represent one mechanism, whereby modulation of cell signalling pathways (MAMPs activating PRR signalling versus inflammatory cytokine signalling) can influence the host response. The profound phenotypic changes that DC cells undergo in the process of activation by MAMPs and conversion into antigen-presenting cells is a multistep process that includes not only trafficking and migration into lymph nodes, but also the expression of high levels of MHC bearing the processed microbial-derived peptides that will engage T cell receptors in naive T cells, the expression of costimulatory membrane-bound molecules (such as CD80 and CD86), which are important for T-cell survival and proliferation, and finally the production of mediators (such as IL-12) that act on T cells and induce their terminal differentiation into an effector cell type and their profile of cytokine expression. Ultimately, the integration of these signals (MHC-antigen; costimulatory molecules, cytokines) will determine the fate and nature of the adaptive immune response.161
Interestingly, many of the features of DC activation can be induced by inflammatory cytokines in the absence of MAMPs and PRR signalling. This fact is interpreted by some as supportive of the “danger model” of immunity, in which endogenous danger signals (e.g., toxic products from necrotic, apoptotic, or infected cells, inflammatory cytokines, etc) can also trigger an immune response, similarly to exogenous MAMPs.30,204 There are two important arguments against this concept, as follows:
The signalling aspects involved in DC maturation and activation are especially interesting since most PRR and inflammatory receptor signals converge to similar pathways (NF-κB and MAPK). In spite of similar signalling pathways activated by MAMPs and inflammatory cytokines, MyD88-defficient dendritic cells stimulated with inflammatory cytokines fail to produce IL-12 and other inflammatory cytokines (IL-1β, IL-6, TNF-α, and IFN-β),143 which will have important consequences for the differentiation and activation of naive CD4+ and CD8+ T cells67 and also indirectly affect activation of B cells and the humoral response.297 Among the possible reasons for this differential role of MAMPs and inflammatory cytokines as external signals in spite of the activation of similar signalling pathways are the kinetics of the activation of the signalling pathways and also the other signalling pathways that are simultaneously activated by each external signal. Integration of multiple signal pathways may be required for a given profile of cytokine expression by DCs. Specifically in periodontal diseases, both MAMPs and inflammatory cytokines are usually present to fully activate the DCs, which suggests that there is no impairment to a competent activation of adaptive immunity.
Host-Microbe Interactions in Adaptive Immunity
Since humans are exposed to bacteria in various mucosal sites, including oral mucosa, shortly after birth and this exposure continues throughout our lives, it is reasonable to expect a chronic presence of adaptive immune cells as part of a vigilance mechanism in these sites chronically exposed to microbes and their MAMPs. The crucial role of adaptive immunity to periodontal disease progression is currently undisputed; however, the mechanisms are not fully understood. According to the current paradigm, DCs activated by MAMPs and inflammatory cytokines (also induced by MAMPs in innate immune/resident cells) will initiate an adaptive immune response by driving naive T lymphocytes into a CD8+ (for cytotoxic response) or CD4+ with Th1 or Th2 phenotypes. Recently, more pieces have been added to the puzzle, including the regulatory T lymphocytes (Tregs), which appear to have their inhibitory functions suppressed by activated DCs. Activated T cells and their “specific” cytokine profiles will modulate the inflammatory response and also the activation of B lymphocytes.
The helper subtype of T cells appears to be particularly relevant for periodontal diseases, as shown by studies in which mice lacking CD4+ cells had decreased severity of bone loss induced by P. gingivalis challenge, whereas depletion of CD8+ cells did not have the same effect.24 There has been a long debate on the cytokine profile associated with periodontal disease, and it has been suggested that the outcome (progression of disease or quiescent state) is determined by the predominant T lymphocyte cytokine profile (Th1 or Th2) in the inflamed tissues.144 However, the cytokine profile associated with periodontal disease in vivo varies and includes both Th1- and Th2-type responses. Some studies using in vivo models have associated a Th1-type cytokine profile with progressive periodontal diseases.276,288,311 Compelling evidence for the role of Th1-type response in periodontal disease progression comes from studies in which in periodontal bone loss in rats challenged with A. actinomycetemcomitans was associated with the introduction of T lymphocytes of the Th1 subtype but not with the Th2 subtype T lymphocytes.125 In fact, introduction of T lymphocytes of the Th2 subtype in T cell-depleted rats challenged with P. gingivalis resulted in decreased bone loss, suggesting a protective role for Th2-type responses in periodontal disease.84 Others reported that even if the Th2 response is not protective of bone loss, it does not aggravate the bone loss induced by P. gingivalis challenge, as observed with the Th1-type response.187 In contrast, a Th2 cytokine profile has also been reported as associated with progressive periodontal disease by both in vitro11,290 and in vivo314,339 studies.
Recently, both B and T lymphocytes were shown to express PRRs, more specifically certain types of TLRs. There is still some controversy over the type of TLR and the level of expression, which can be attributed to various experimental methods used, including the procedure to purify lymphocytes, the model (murine or human), and the analysis method (flow cytometry, real time polymerase chain reaction [PCR]). Nevertheless, messenger RNA (mRNA) for TLR1, TLR2, TLR3, TLR4, TLR5, TLR7, and TLR9 was detected in human peripheral T lymphocytes, although the expression levels are highly variable among different studies.47,81,141,345 In spite of the controversy, there are some studies demonstrating a functional role for these PRRs expressed in lymphocytes. In naive T cells, TLR2 and TLR4 may not be functional, since their expression was demonstrated intracellularly,19,177,197 whereas TLR5 was detected in the cell surface.64 However, expression of cell-surface (and functional) TLR2 and TLR4 is regulated in naive human T CD4+ lymphocytes by T-cell receptor (TCR) activation, and the same is true for TLR2 in murine CD4+ and CD8+ T cells.177,193 In contrast, TLR5, constitutively expressed on the cell surface of human CD4+ T lymphocytes, is downregulated by TCR stimulation.64 The mechanism for the TCR-mediated modulation of TLR expression is not known; it may involve induced translocation of intracellular TLRs to the cell surface, or de novo synthesis of membrane-bound TLRs.164 Current evidence suggests that T cells can respond directly to MAMPs through TLR and that TLR signalling in T cells has a costimulatory effect with TCR activation. TLR2 signalling costimulates proliferation and cytokine production by CD4+177,193 and murine CD8+ T cells.62 TLR3 signalling increased survival of activated murine CD4+ T cells,103 whereas TLR9 signalling increased not only survival but also proliferation of CD4+ T cells.104 TLR4 stimulation increased perforin expression in CD4+ T cells, suggesting a role for LPS in the induction of a cytotoxic response.250
Expression of TLRs may also vary in different T-cell subsets. Tregs (CD25+/CD4+) express higher levels of TLR564 and as opposed to CD4+/CD25− T lymphocytes, also express TLR8.243 Treg cells have an important role as negative regulators of immune response and consequently in immune tolerance mechanisms. Their function requires precise control to maintain adequate and effective T cell-mediated immunity.272 Even though the mechanisms for the suppressive effect of Tregs are not well known, they require expression of transcriptional repressor FoxP3.268 During the activation of adaptive immunity, activated DCs not only polarize naive CD4+ T lymphocytes to a Th1 or Th2 subtype, but they also inhibit Tregs. There is evidence that this inhibition of Treg suppressive function involves production of IL-6 by activated DCs and other innate immune cells.225,236 Interestingly, direct stimulation of TLR2 or TLR8, together with TCR activation in Tregs results in increased proliferation and inhibition of FoxP3 expression and consequently of their immune suppressive activity.193,243,305 There is also evidence of modulation of Treg activity according to dose-dependent stimulation of TLR signalling. Low concentrations of flagellin (TLR5 ligand) enhanced the suppressive activity of Tregs (as in an immune tolerance mechanism), whereas higher concentrations of flagellin increased proliferation and cytokine production in CD4+ T cells.64 TLR4 signalling also appears to enhance Treg suppressive functions through IL-10 and TGF-β dependent mechanisms.189
The proportion of B cells is reported to be larger than that of all subsets of T lymphocytes.32 In fact, B–cell-derived plasmocytes and B lymphocytes represent the absolute majority of infiltrating cells in diseased periodontal tissues, and their relative proportion appears to be higher in sites with severe periodontal disease in comparison to sites with moderate-to-mild periodontitis. Besides their classic role as producers of immunoglobulins that identify and bind to MAMPs, B cells also participate as antigen-presenting cells and modulators of host response through the production of cytokines such as TNF-α, IL-6, IL-10, TGF-β, and RANKL.33 In fact, the relevance of B lymphocytes for the pathogenesis of periodontal disease and in particular, for alveolar bone loss has been demonstrated in murine models.124,127 The role of the humoral response in periodontal disease has not been clear until now, and the importance of B cells for periodontal disease pathogenesis is probably largely the result of cytokine production.55,168 Antibodies are considered as primarily defense mechanisms, aimed at neutralizing directly (through inactivation of antigens with or without complement activation) or indirectly (through opsonization of antigens and facilitating phagocytosis) the microbes. However, there is no definite explanation as to why the elevated levels of immunoglobulins against periodontopathogenic bacteria are not sufficient to ameliorate periodontal disease or to prevent its recurrence. Elevated serum titers of antibodies against P. gingivalis have been detected before but did not prevent alveolar bone loss in a murine model of periodontal disease.23 Indeed, elevated levels of immunoglobulins may actually aggravate local destruction of periodontal tissues through complement activation or through self-reactive antibodies.32 It has also been suggested that the high immunoglobulin G (IgG) serum titers against periodontal bacteria may be implicated as one of the possible biologic links between periodontal diseases and systemic diseases such as cardiovascular disease227 or rheumatoid arthritis.262 B cells have been shown to express various TLRs, including TLR1, TLR2, TLR4, TLR6, TLR7, TLR9, and TLR10.141 Recent evidence indicates that a significantly higher proportion of B cells from periodontal tissues and blood from periodontal disease patients expressed TLR4 in comparison to those of periodontally healthy individuals.289 This suggests that expression of some TLRs may be induced, likely by the inflammatory environment resulting from the initial activation of innate immunity by MAMPs. Thus cytokines produced by innate immune response and also the presence of MAMPs would induce TLR expression in B cells, which in turn renders these cells more responsive to the ubiquitously present MAMPs in the periodontally diseased tissue. This concept is supported by the fact that resting B cells express primarily TLR1 and TLR9, with lower proportion of resting B cells expressing TLR2, TLR3, and TLR4.73 Activation of B cells in vitro by CD40 stimulation also increased expression of TLR7, TLR9, and TLR10, and interestingly, costimulation with CD40 ligand and CpG bacterial DNA had a synergistic effect on this induction.41
Importantly, TLR expression by B lymphocytes indicates that similarly to the T lymphocytes, these cells can respond to MAMPs and play a role in the modulation of both innate and adaptive arms of immune response, further stressing the continuity and cooperation between these two aspects of the host response. This cooperation is demonstrated by the reduced antibody production in humans and mice with reduced expression of TLR1 or TLR27; increased proliferation; activation of NF-κB, p38, and c-Jun N-terminal-activated kinases JNK MAPK; and also increased expression of costimulatory molecules (CD86, CD40, or MHC-II) and IL6 by B cells stimulated with TLR9 agonist.129
Pathobiology of Periodontal Disease Progression
Host response to periodontal infection requires expression of a number of bioactive agents, including proinflammatory and antiinflammatory cytokines, growth factors, and enzymes that are the result of the activation of multiple signalling pathways. This activation of intracellular signalling may initiate exclusively as an innate immune response associated with PRR-mediated sensing of MAMPs. However, the biologic mediators expressed as a result of PRR-signalling include costimulatory molecules involved in the induction of adaptive immunity.36 This results in a cascade of events that will establish very complex cytokine and signalling networks.
Innate immunity and inflammation are not synonymous; however, inflammation arises primarily in response to infection. A better understanding of direct host-microbe interactions is especially important to understand how inflammation is initiated in response to microorganisms in the dental biofilm. In this sense, PRR signalling is considered the most important interface between the host and the microbes,36 and as discussed previously in this chapter, it can modulate both innate and adaptive aspects of the host response.
Cytokines and Mediators of Inflammation
Inflammation involves a number of biochemical and cellular alterations. An inappropriate inflammatory response is the cause of many common diseases, including periodontitis. The local inflammatory reaction, in response to bacteria in the dental biofilm, is characterized by an initial increase in blood flow, enhanced vascular permeability, and the influx of cells from the peripheral blood to the gingival crevice. These initial events are triggered by bioactive molecules (such as histamine, bradykinin, PGE2, and nitric oxide) and produced by innate immune cells and resident, nonimmune cells present in the periodontal tissues. PMNs or neutrophils attracted to the area by other bioactive molecules (e.g., IL-8) migrate through the epithelial lining of the gingival sulcus to be the initial defense against invading plaque bacteria and their by-products.195 These cells are nonspecific phagocytes responsible for an acute and rapid defense. Subsequently, there is an increase in the number of monocytes/macrophages, as well as an influx of T and B cells to the area.97
Once activated by cytokines, bioactive molecules, and MAMPs present in the area, infiltrating cells produce other inflammatory mediators that modulate the activity of other cells and affect homeostasis of nonmineralized and mineralized tissues in the periodontium59,111 (Table 25-2 and Figure 25-3). Cytokines responsible for early responses to microbial aggression include IL-1α, IL-1β, IL-6, and TNF-α.185 Other proinflammatory mediators include leukemia-inhibiting factor (LIF); IFN-γ; oncostatin M (OSM); ciliary neurotrophic factor (CNTF); TGF-β; GM-CSF; IL-11, IL-12, IL-17, IL-18, IL-8, and a variety of other chemokines that attract inflammatory cells.49,226,275,276 The net effect of an inflammatory response is determined by the balance between proinflammatory and antiinflammatory cytokines such as IL-4, IL-10, IL-13, IL-16, IFN-α, TGF-β, IL-1Ra, GM-CSF, and soluble receptors for TNF or IL-6.112,186
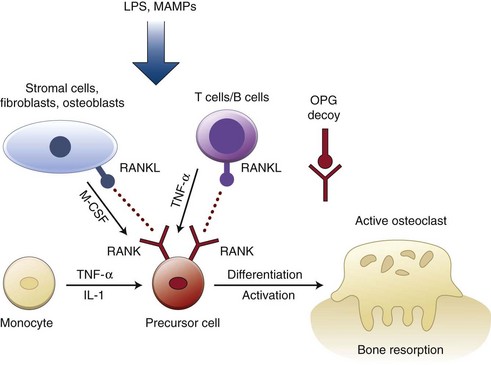
Figure 25-3 Stimuli and factors regulating osteoclastogenesis. Cytokines (interleukin-1 [IL-1], tumor necrosis factor alpha [TNF-α], macrophage-colony stimulating factor [M-CSF]) produced by resident cells (bone stromal cells, fibroblasts, osteoblasts) and immune cells (monocytes, T- and B-lymphocytes) are key regulators of this process. Osteoprotegerin (OPG) acts as a decoy receptor that prevents receptor activator of nuclear factor (NF)-κB ligand (RANKL) from binding to its receptor activator of NF-κB (RANK) on precursor cells and downregulate osteoclastogenesis. Bacterial lipopolysaccharide (LPS) and other microbial-associated molecular patterns (MAMPs) may directly or indirectly initiate expression of RANKL and osteoclastogenesis.
Patients with periodontal inflammation have high concentrations of TNF-α, IL-1β, RANKL, and MMP-13* in the gingival crevicular fluid (GCF). Increased levels of IL-1β, IL-2, IL-6, IL-17, TNF-α, and IFN-γ in gingival tissues are also associated with destructive periodontal disease.110,188
However, in spite of an abundance of publications investigating cytokine profiles associated with periodontal disease, the definitive description of the cytokine response still remains elusive. Among the reasons for the variability of the reports on the cytokine profile are the difficulties in defining periodontal disease and especially in the identification of actively progressing disease sites (i.e., those with ongoing attachment loss and alveolar bone resorption). A gene-expression profiling (or transcriptome analysis) of changes in mRNA levels of over 38,000 genes with 54,000 DNA probes was performed in healthy and diseased periodontal tissues, and demonstrated that over 12,000 probes were differentially regulated in periodontal diseased tissues, including genes involved in inflammation, apoptosis and angiogenesis.78 These data agree with the evidence demonstrating that the cytokine profile associated with periodontal disease in vivo varies and includes both Th1- and Th2-type cytokines. IL-1α, IL-1β, IL-8, and TNF-α mRNA were detected in macrophages present in inflamed gingival tissues,201 whereas Th2 cytokine IL-4 and pleiotropic IL-6 protein were also observed in diseased periodontal tissues.339 A characteristic cytokine profile has been associated with each type of periodontal disease (i.e., inflammation of marginal soft tissues without active bone resorption [gingivitis] or with active bone resorption [periodontitis]). Thus expression of Th1-type cytokines has been associated with gingivitis, whereas Th2 cytokines were found in higher levels in periodontitis-affected tissues,186,314 even though this distinction was not clear because both Th1 and Th2 cytokines were produced in gingivitis- and periodontitis-affected tissues; and the predominant profile may actually represent the current activity of tissue destruction.101,286,335 The relevance of cytokines as biologic mediators of periodontal disease progression is demonstrated by studies showing that conventional periodontal therapy aimed at the reduction of the bacteria and associated metabolic by-products result in their decrease, as shown for IL-1β4,61,313 and TNF-α.154
Once the immune and inflammatory processes are initiated and the complex cytokine network is established, various inflammatory molecules that play a direct role in the degradation of both nonmineralized and mineralized tissues of the periodontium are produced in response to MAMPs and/or to the host-derived cytokines.59,111 One such family of inflammatory molecules is the MMPs, which are released from different cell types present in the lesion, including macrophages, leukocytes, and fibroblasts or other resident cells.178 MMPs are a family of neutral proteases that are zinc/calcium-dependent with an essential role in extracellular matrix (ECM) turnover and degradation. Their activity is regulated at multiple levels (transcriptional, posttranscriptional, and posttranslational) and also by endogenous inducible inhibitors, the tissue inhibitors of MMPs (TIMPs). Collectively, MMPs are capable of degrading virtually all components of ECM, and increased levels have been associated with periodontal disease in both humans61,87,89,180 and animal models.74,102 The same difficulties associated with the determination of disease activity apply to the determination of MMPs in periodontal disease, even though the trend seems to have a positive correlation (i.e., MMPs increase with inflammation and disease activity108) and their detection in saliva has been recently suggested as one possible host-response biomarker of periodontal disease.254 Interestingly, the role of individual MMPs in periodontal disease progression may differ, as MMP-8 was recently shown to have a protective effect on alveolar bone loss induced by P. gingivalis infection in mice182; whereas MMP-13 expression was positively correlated with the severity of inflammation in ligature- and LPS-induced experimental periodontitis in rats74 and also with severity of periodontal disease in humans.291
The relevance of MMPs to the pathogenesis of periodontal diseases is supported by the decrease in bone loss associated with their nonselective inhibition in animal models of periodontal disease252,253 and especially by the improved clinical results observed after periodontal treatment associated with systemic inhibition of MMPs by a sub-antimicrobial dose of doxycycline.99,107,247,248
As periodontal disease progresses, the collagen fibers and connective tissue attachment to the tooth are destroyed, the junctional epithelial cells proliferate apically along the root surface, and these structural changes are reflected clinically as attachment loss and/or increased probing pocket depth. These changes represent not only an increased severity of inflammation but also an apical displacement of the inflammatory infiltrate, which thus is in close proximity to the alveolar bone. This affects homeostasis of bone tissue by triggering the resorptive process that represents the main characteristic of destructive periodontal disease. RANKL plays a pivotal role in bone resorption, since it is involved in osteoclast differentiation, activation, and survival.42,171,310 Osteoprotegerin (OPG) is the endogenous inhibitor of RANKL and functions as its decoy receptor, preventing binding of RANKL to the specific membrane-bound receptor expressed in osteoclast precursor cells (RANK).42,139,301 RANKL is produced as either a membrane-bound or secreted protein by fibroblasts, osteoblasts, chondrocytes, mesenchymal cells, and T- and B-lymphocytes, whereas OPG is secreted primarily by osteoblastic cells, bone marrow stromal cells, and fibroblasts (see Figure 25-3). The ratio between RANKL and OPG is the current paradigm for modulation of coupled bone turnover, and specifically in periodontal disease, this concept is supported by observations demonstrating that patients with advanced periodontitis present with higher levels of RANKL and lower levels of OPG than periodontally healthy patients.65,213 Further support to the relevance of RANKL/OPG cytokine system for bone homeostasis comes from studies in which local delivery of OPG decreases alveolar bone resorption in experimental periodontal disease models.54,159 However, other biologically-active mediators present in the complex network of cytokines established in the periodontal disease microenvironment can play important roles in periodontal disease pathogenesis and alveolar bone resorption, as indicated by studies targeting inhibition of TNF-α.57,79
It is important to consider that there is significant heterogeneity of clinical presentation of periodontal disease in terms of its extent, severity, progression, and response to treatment. This heterogeneity is thought to reflect the high variability of the levels of inflammatory cytokines and bioactive molecules produced by different individuals, which cannot be completely explained by differences in the microbial population of the dental biofilm. Based on susceptibility analysis, individual differences in the host response to MAMPs and to host-derived cytokines that are the result of genetic variations may play an important role in modulating the pathogenesis of periodontal diseases.341 This is supported by epidemiologic studies of the association between various genetic syndromes and periodontal diseases222 and the familial trend for occurrence of some types of aggressive periodontal disease.9,216 There are numerous studies correlating multiple variations in genetic information encoding genes and receptors involved in the immune response and inflammation with periodontal disease,93,151 and these genetic variations are also heterogeneic according to race and ethnic aspects of human populations.13,199,217,273,328 It is interesting to note that there is also evidence suggesting that genetic polymorphisms for the “effector” genes of MMPs18,52,246 and RANKL/OPG235,295 are associated with the severity and extent of periodontal disease. However, the strength of this association, especially for MMPs, is still controversial and may depend on specific types of MMPs, as other studies failed to demonstrate an association between polymorphisms for some MMPs and periodontal disease status.75,115,140
Another important aspect to consider is how the external environment affects the production of inflammatory mediators by the host. Thus, even though there is a “personalized genetic program” controlling how each individual responds to the microbial challenge, this is further modulated by transitory aspects of each individual’s life that can affect production of cytokines and inflammatory mediators, including hormonal imbalances, stress, acquired diseases, medications, and smoking. These are all factors that come into play in determining the individual’s response to the microbial challenge and ultimately, periodontal tissue destruction. Moreover, it has been recently suggested that genetic variations modulating host responses may represent a common trait for susceptibility to periodontitis and systemic diseases such as cardiovascular disease.283
Cell Signalling Events Modulating Inflammatory Mediator Expression
In the complex scenario of host-microbe interactions associated with periodontal disease, the signalling pathways originally shown to be relevant for recognition of stress, inflammatory, and infectious extracellular stimuli are of special interest. Production of cytokines and inflammatory mediators is usually a tightly-controlled process that is initiated by external stimuli or “signals” that are rapidly transduced through the cytoplasm and into the nucleus where gene expression starts with the transcription of DNA into pre-mRNA. From this start to the final assembly of the biologically-active protein, there are a great number of regulatory mechanisms that can affect gene expression and various signalling pathways can participate in many of these mechanisms, both at transcriptional and posttranscriptional levels. Thus cytokine production is a fast and transient process, initiated and controlled by even faster mechanisms represented by the signalling pathways. The fact that a given signalling pathway regulates the expression of various inflammatory mediators is especially important for therapeutic applications if one considers that targeting expression of a single cytokine may not be particularly effective as the result of compensation of its biologic role by other proinflammatory cytokines.
The MAPKs are a group of conserved cytoplasmic kinases that are organized in modules (MAPKKK → MAPKK → MAPK) sequentially activated by dual phosphorylation at tyrosine/threonine residues. Of the four distinct classes of MAPKs described to date in mammals, p38 (α and β isoforms), c-Jun N-terminal-activated kinases (JNK1-3), and extracellular-activated kinases (ERK1,2) are the most studied. Downstream substrates of MAPKs include a variety of transcription factors, RNA-binding proteins, and other kinases (mitogen-activated protein kinase-activated protein kinases [MAPKAPK]) that are involved in regulation of gene expression by transcriptional, posttranscriptional, translational, and posttranslational mechanisms (Figure 25-2).
The signal transducers and activators of transcription (STAT) family of latent transcription factors is involved in many cytokine signalling pathways,150 especially those that use the gp130 receptor. Many cytokines and growth factors (IFNs, interleukins, epidermal growth factor, growth hormone, erythropoietin, and others) exert their biologic functions through the JAK-STAT signal transduction pathway.224,285 Classically, IFNs and interleukins, cytokines with key roles in regulating the immune response, activate enzymes called Janus kinases (JAK1, JAK2, JAK3, and Tyk2), which are associated with the cytoplasmic portion of the transmembrane receptors.149 Activated JAKs phosphorylate the cytoplasmic domain of the receptor leading the activation of its substrates, especially the proteins known as STATs (STAT1-4, 5a, 5b, and 6). On phosphorylation, STATs may form homodimers or heterodimers, which enables them to enter to the nucleus in which they can regulate gene transcription.224 Although individual STAT proteins may be activated by multiple ligands, certain cytokines preferentially activate particular STATs. IFN-γ preferentially activates STAT1 through JAK1/JAK2, and IL-6 activates STAT3 through JAK1. The JAK-STAT pathway is the signalling target of many cytokines that play important roles in inflammation and thus could also be important in periodontal disease, including IFN-γ, IL-1, IL-2, IL-4, IL-6, IL-10, IL-12, and TNF-α.32,259,327 STAT3 can be phosphorylated at tyrosine and serine residues. This dual phosphorylation is needed for full activation, even though it is not known if serine phosphorylation of STAT3 has a role in DNA-binding of STAT3.332 Interestingly, deficient STAT3 signalling in innate immune cells has been recently shown to exacerbate P. gingivalis-induced experimental periodontitis in mice by impairing IL-10 signalling and increasing a Th-1 type response characterized by elevated IL-12 and IL-1α.279
NF-κB consists of five related transcription factors that can form multiple homodimers and heterodimers and regulate inducible gene expression in various physiologic contexts.105 Studies on animal and human diseases have indicated that activation of NF-κB is crucial for the expression of various inflammatory genes.158,278 This role of NF-κB for the inflammatory/immune response has been validated by other studies using genetic approaches and chemical inhibitors.6,211,287 The potential therapeutic applications of NF-κB inhibition have been investigated in clinical trials in cancer2 and in animal models of rheumatoid arthritis.208 Collectively, this information suggests that NF-κB signalling should play an important role in periodontal diseases; this concept is supported not only by gene-profiling analysis demonstrating increased expression of both NF-κB and NF-κB–regulated inflammatory genes in diseased periodontal tissues78 but also by studies in vitro.53
However, in spite of a great deal of information available on the regulation and expression of inflammatory cytokines, there are only a few reports on the signalling pathways activated specifically in periodontal disease in vivo. NF-κB activation has been shown to be associated with increased periodontal disease severity.8 Differences in the activation of signalling pathways in two frequently used murine models of experimentally-induced periodontal disease were reported recently: in both the bacterial LPS injection model and the ligature model, p38 and ERK MAPKs, as well as NF-κB, were activated but with different kinetics. On the other hand, activation of JAK-STAT signalling was only observed with the ligature model.100 However, to date, no studies with NF-κB or STAT inhibitors are available in periodontal disease models, despite the important role of these proteins in the regulation of inflammatory genes.
Interestingly, the proteins comprising many of the signalling pathways are highly conserved among different species of organisms, indicating their fundamental role in many essential physiologic processes. Some of these signalling pathways also have a relevant role in diverse pathologic conditions, demonstrating their multivalency. For instance, the p38 MAPK pathway was originally described as critically important to signal stress, inflammatory, and infectious stimuli, but it is also involved in the control of fundamental processes, including cell proliferation,123 differentiation,190 and migration.135 Nevertheless, many reports indicate its relevance and/or potential therapeutic application in disease processes that involve inflammation and immunity, including rheumatoid arthritis,205,210,325 ischemic heart disease,58 allergies,83,280 chronic obstructive pulmonary diseases,212 Alzheimer’s disease,66 and cancer.257
Surprisingly, in spite of evidence indicating a role of p38 MAPK in all of these diseases, there is a relative paucity of information regarding its role in oral inflammation–related conditions, including temporomandibular joint disorders, chronic oral pain, and inflammatory changes of the oral mucosa.240 p38 MAPK is involved in the regulation of expression of proinflammatory cytokines and enzymes induced by inflammatory and infectious signals in vitro, including IL-6,238,239 MMP-13,264,265 and RANKL263 in resident cells of the periodontium such as fibroblasts and osteoblasts. Based on its involvement in the regulation of these cytokines, it is likely that p38 MAPK plays a role in disease progression, since this signalling pathway is not only one of the main downstream effectors of TLR signalling34,37 but also particularly relevant for the activation and development of adaptive immune responses, as demonstrated by its role in T-cell proliferation and cytokine production258,346 and differentiation of immature T cells into Th1 or Th2 effector cells.60 p38 MAPK is also involved in B-cell activation63 and production of cytokines, including IL-1068 and even modulates IL-4–mediated responses in B cells by crosstalk with STAT6.245 This illustrates the multiple roles of this signalling pathway and how modulation of its activity may have multiple effects both on innate and adaptive immunity. Other signalling pathways that have been shown to be activated and involved in regulation of gene expression during inflammation and immune response, such as Notch,10,94 Wnt,109,244 and PI3-kinase261,333 pathways, participate in host-microbe interactions but have not been studied in the context of periodontal disease.
Therapeutic Strategies for Disrupting Host-Cell Signalling in the Treatment of Periodontal Diseases
A variety of treatment strategies have been developed to target the host response to LPS-mediated tissue destruction. MMP inhibitors, such as low dose formulations of doxycycline, have been used in combination with scaling and root planing48 or surgical therapy.98 In addition, high-risk patient populations, such as those with diabetes, have benefited from systemic administration of MMP inhibitors.51,221,270 Encouraging results have also been shown using soluble antagonists of TNF and IL-1 delivered locally to the periodontal tissues in nonhuman primates.17 Other therapeutic strategies that are being explored are aimed at inhibiting signal transduction pathways involved in inflammation. Pharmacological inhibitors of NF-κB and p38 MAP kinase pathways are actively being developed to manage rheumatoid arthritis and inflammatory bone diseases1,158,181 and have been applied in periodontal disease models with some benefits observed.175,260 Using this novel strategy, inflammatory mediators, including proinflammatory cytokines (IL-1, TNF, IL-6), MMPs and others, would be inhibited at the level of cell signalling pathways required for transcription factor activation necessary for inflammatory gene expression or mRNA stability (Figure 25-2). Indeed, targeting RNA-binding proteins that mediate effects on inflammatory cytokines does have therapeutic value in small animal models of periodontal disease progression.241 These therapies may provide the next wave of adjuvant chemotherapeutics that may be used to manage chronic periodontitis.
 Science Transfer
Science Transfer
A small number of the 500 plus bacterial species found in dental biofilms are related to periodontal destruction. These are gram-negative anaerobic bacteria that reside in the subgingival plaque.
The extent and severity of periodontitis is closely related to the host response to these putative pathologic bacteria. Initially, the host oral bacterial biofilm stimulates the establishment of an innate immune response based on the classical inflammatory cells. With initiation of periodontal disease, dendritic cells signal a more widespread immune reaction (adaptive immune response) that involves not only the B and T lymphocytes, polymorphonuclear leukocytes (PMNs), macrophages, and osteoclasts but also has contributions of signalling molecules and tissue destructive cytokines from resident epithelial cells, fibroblasts, and osteoblasts. The resulting molecular response is complex and interrelated and involves molecular signalling with genetic control of many of the proteins involved. Recent research has unraveled only part of these bioactive networks, and the more that is discovered, the more complicated and diversified are the mechanisms of periodontal destruction.
Most clinicians will have difficulty appreciating all of the signaling pathways and feedback mechanisms that are elucidated, but they should appreciate the importance of these adaptive immune responses in disease progression. New therapies based on regulating tissue destruction by inhibiting such molecules as matrix metalloproteinases (MMPs) with long-term low-dose doxycycline are already in widespread use as supportive therapy for conventional periodontal treatment. Newer approaches soon to be available include antagonists to cytokines and agents to inhibit signalling pathways associated with tissue destructive proteins. Because of the multitudinous molecular mechanisms of tissue destruction, it is unlikely that any one of these approaches will have a singular dramatic therapeutic affect, rather they will be only effectively used in combination with conventional antibacterial treatments such as initial therapy, antimicrobial agents, and periodontal surgery.
1 Adams JL, Badger AM, Kumar S, Lee JC. p38 MAP kinase: molecular target for the inhibition of pro-inflammatory cytokines. Prog Med Chem. 2001;38:1-60.
2 Aghajanian C, Dizon DS, Sabbatini P, et al. Phase I trial of bortezomib and carboplatin in recurrent ovarian or primary peritoneal cancer. J Clin Oncol. 2005;23:5943-5949.
3 Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783-801.
4 Al-Shammari KF, Giannobile WV, Aldredge WA, et al. Effect of non-surgical periodontal therapy on C-telopeptide pyridinoline cross-links (ICTP) and interleukin-1 levels. J Periodontol. 2001;72:1045-1051.
5 Albert ML, Jegathesan M, Darnell RB. Dendritic cell maturation is required for the cross-tolerization of CD8+ T cells. Nat Immunol. 2001;2:1010-1017.
6 Alcamo E, Mizgerd JP, Horwitz BH, et al. Targeted mutation of TNF receptor I rescues the RelA-deficient mouse and reveals a critical role for NF-kappa B in leukocyte recruitment. J Immunol. 2001;167:1592-1600.
7 Alexopoulou L, Thomas V, Schnare M, et al. Hyporesponsiveness to vaccination with Borrelia burgdorferi OspA in humans and in TLR1- and TLR2-deficient mice. Nat Med. 2002;8:878-884.
8 Ambili R, Santhi WS, Janam P, et al. Expression of activated transcription factor nuclear factor-kappaB in periodontally diseased tissues. J Periodontol. 2005;76:1148-1153.
9 American Academy of Periodontology-Research SatC. Periodontal diseases of children and adolescents. Pediatr Dent. 2008;30:240-247.
10 Ando K, Kanazawa S, Tetsuka T, et al. Induction of Notch signaling by tumor necrosis factor in rheumatoid synovial fibroblasts. Oncogene. 2003;22:7796-7803.
11 Aoyagi T, Sugawara-Aoyagi M, Yamazaki K, Hara K. Interleukin 4 (IL-4) and IL-6-producing memory T-cells in peripheral blood and gingival tissue in periodontitis patients with high serum antibody titers to Porphyromonas gingivalis. Oral Microbiol Immunol. 1995;10:304-310.
12 Ara T, Kurata K, Hirai K, et al. Human gingival fibroblasts are critical in sustaining inflammation in periodontal disease. J Periodontal Res. 2009;44:21-27.
13 Armitage GC, Wu Y, Wang HY, et al. Low prevalence of a periodontitis-associated interleukin-1 composite genotype in individuals of Chinese heritage. J Periodontol. 2000;71:164-171.
14 Artis D. Epithelial-cell recognition of commensal bacteria and maintenance of immune homeostasis in the gut. Nat Rev Immunol. 2008;8:411-420.
15 Asai Y, Ohyama Y, Gen K, Ogawa T. Bacterial fimbriae and their peptides activate human gingival epithelial cells through Toll-like receptor 2. Infect Immun. 2001;69:7387-7395.
16 Asai Y, Jinno T, Ogawa T. Oral treponemes and their outer membrane extracts activate human gingival epithelial cells through toll-like receptor 2. Infect Immun. 2003;71:717-725.
17 Assuma R, Oates T, Cochran D, et al. IL-1 and TNF antagonists inhibit the inflammatory response and bone loss in experimental periodontitis. J Immunol. 1998;160:403-409.
18 Astolfi CM, Shinohara AL, da Silva RA, et al. Genetic polymorphisms in the MMP-1 and MMP-3 gene may contribute to chronic periodontitis in a Brazilian population. J Clin Periodontol. 2006;33:699-703.
19 Babu S, Blauvelt CP, Kumaraswami V, Nutman TB. Cutting edge: diminished T cell TLR expression and function modulates the immune response in human filarial infection. J Immunol. 2006;176:3885-3889.
20 Baelum V, Fejerskov O, Karring T. Oral hygiene, gingivitis and periodontal breakdown in adult Tanzanians. J Periodontal Res. 1986;21:221-232.
21 Baelum V, Fejerskov O, Manji F. Periodontal diseases in adult Kenyans. J Clin Periodontol. 1988;15:445-452.
22 Baker PJ, Evans RT, Roopenian DC. Oral infection with Porphyromonas gingivalis and induced alveolar bone loss in immunocompetent and severe combined immunodeficient mice. Arch Oral Biol. 1994;39:1035-1040.
23 Baker PJ, Carter S, Dixon M, et al. Serum antibody response to oral infection precedes but does not prevent Porphyromonas gingivalis-induced alveolar bone loss in mice. Oral Microbiol Immunol. 1999;14:194-196.
24 Baker PJ, Dixon M, Evans RT, et al. CD4(+) T cells and the proinflammatory cytokines gamma interferon and interleukin-6 contribute to alveolar bone loss in mice. Infect Immun. 1999;67:2804-2809.
25 Baker PJ, Garneau J, Howe L, Roopenian DC. T-cell contributions to alveolar bone loss in response to oral infection with Porphyromonas gingivalis. Acta Odontol Scand. 2001;59:222-225.
26 Baker PJ, Howe L, Garneau J, Roopenian DC. T cell knockout mice have diminished alveolar bone loss after oral infection with Porphyromonas gingivalis. FEMS Immunol Med Microbiol. 2002;34:45-50.
27 Baqui AA, Meiller TF, Chon JJ, et al. Granulocyte-macrophage colony-stimulating factor amplification of interleukin-1beta and tumor necrosis factor alpha production in THP-1 human monocytic cells stimulated with lipopolysaccharide of oral microorganisms. Clin Diagn Lab Immunol. 1998;5:341-347.
28 Barksby HE, Nile CJ, Jaedicke KM, et al. Differential expression of immunoregulatory genes in monocytes in response to Porphyromonas gingivalis and Escherichia coli lipopolysaccharide. Clin Exp Immunol. 2009;156:479-487.
29 Beklen A, Hukkanen M, Richardson R, Konttinen YT. Immunohistochemical localization of Toll-like receptors 1-10 in periodontitis. Oral Microbiol Immunol. 2008;23:425-431.
30 Belardelli F, Ferrantini M. Cytokines as a link between innate and adaptive antitumor immunity. Trends Immunol. 2002;23:201-208.
31 Berezow AB, Ernst RK, Coats SR, et al. The structurally similar, penta-acylated lipopolysaccharides of Porphyromonas gingivalis and Bacteroides elicit strikingly different innate immune responses. Microb Pathog. 2009.
32 Berglundh T, Donati M. Aspects of adaptive host response in periodontitis. J Clin Periodontol. 2005;32(Suppl 6):87-107.
33 Berglundh T, Donati M, Zitzmann N. B cells in periodontitis: friends or enemies? Periodontol 2000. 2007;45:51-66.
34 Beutler B. Innate immune sensing of microbial infection: the mechanism and the therapeutic challenge. Wien Med Wochenschr. 2002;152:547-551.
35 Beutler B. Toll-like receptors: how they work and what they do. Curr Opin Hematol. 2002;9:2-10.
36 Beutler B. Innate immune responses to microbial poisons: discovery and function of the Toll-like receptors. Annu Rev Pharmacol Toxicol. 2003;43:609-628.
37 Beutler B, Hoebe K, Du X, Ulevitch RJ. How we detect microbes and respond to them: the Toll-like receptors and their transducers. J Leukoc Biol. 2003;74:479-485.
38 Bodet C, Andrian E, Tanabe S, Grenier D. Actinobacillus actinomycetemcomitans lipopolysaccharide regulates matrix metalloproteinase, tissue inhibitors of matrix metalloproteinase, and plasminogen activator production by human gingival fibroblasts: a potential role in connective tissue destruction. J Cell Physiol. 2007;212:189-194.
39 Bostanci N, Allaker R, Johansson U, et al. Interleukin-1alpha stimulation in monocytes by periodontal bacteria: antagonistic effects of Porphyromonas gingivalis. Oral Microbiol Immunol. 2007;22:52-60.
40 Bouma G, Strober W. The immunological and genetic basis of inflammatory bowel disease. Nat Rev Immunol. 2003;3:521-533.
41 Bourke E, Bosisio D, Golay J, et al. The toll-like receptor repertoire of human B lymphocytes: inducible and selective expression of TLR9 and TLR10 in normal and transformed cells. Blood. 2003;102:956-963.
42 Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423:337-342.
43 Brandtzaeg P, Kraus FW. Autoimmunity and periodontal disease. Odontol Tidskr. 1965;73:281-393.
44 Brissette CA, Pham TT, Coats SR, et al. Treponema denticola does not induce production of common innate immune mediators from primary gingival epithelial cells. Oral Microbiol Immunol. 2008;23:474-481.
45 Brozovic S, Sahoo R, Barve S, et al. Porphyromonas gingivalis enhances FasL expression via up-regulation of NFkappaB-mediated gene transcription and induces apoptotic cell death in human gingival epithelial cells. Microbiology. 2006;152:797-806.
46 Burns E, Bachrach G, Shapira L, Nussbaum G. Cutting Edge: TLR2 is required for the innate response to Porphyromonas gingivalis: activation leads to bacterial persistence and TLR2 deficiency attenuates induced alveolar bone resorption. J Immunol. 2006;177:8296-8300.
47 Caron G, Duluc D, Fremaux I, et al. Direct stimulation of human T cells via TLR5 and TLR7/8: flagellin and R-848 up-regulate proliferation and IFN-gamma production by memory CD4+ T cells. J Immunol. 2005;175:1551-1557.
48 Caton JG, Ciancio SG, Blieden TM, et al. Subantimicrobial dose doxycycline as an adjunct to scaling and root planing: post-treatment effects. J Clin Periodontol. 2001;28:782-789.
49 Cavaillon JM. Cytokines and macrophages. Biomed Pharmacother. 1994;48:445-453.
50 Champaiboon C, Yongvanitchit K, Pichyangkul S, Mahanonda R. The immune modulation of B-cell responses by Porphyromonas ginginvalis and interleukin-10. J Periodontol. 2000;71:468-475.
51 Chang K, Rani AS, Kumar S. Plasminogen activator activity is decreased in rat gingiva during diabetes. J Periodontol. 1996;67:743-747.
52 Chen D, Wang Q, Ma ZW, et al. MMP-2, MMP-9 and TIMP-2 gene polymorphisms in Chinese patients with generalized aggressive periodontitis. J Clin Periodontol. 2007;34:384-389.
53 Chen D, Nie M, Fan MW, Bian Z. Anti-inflammatory activity of curcumin in macrophages stimulated by lipopolysaccharides from Porphyromonas gingivalis. Pharmacology. 2008;82:264-269.
54 Chen R, Kanzaki H, Chiba M, et al. Local osteoprotegerin gene transfer to periodontal tissue inhibits lipopolysaccharide-induced alveolar bone resorption. J Periodontal Res. 2008;43:237-245.
55 Choi Y, Woo KM, Ko SH, et al. Osteoclastogenesis is enhanced by activated B cells but suppressed by activated CD8(+) T cells. Eur J Immunol. 2001;31:2179-2188.
56 Chung WO, Dale BA. Innate immune response of oral and foreskin keratinocytes: utilization of different signaling pathways by various bacterial species. Infect Immun. 2004;72:352-358.
57 Cirelli JA, Park CH, MacKool K, et al. AAV2/1-TNFR:Fc gene delivery prevents periodontal disease progression. Gene Ther. 2009;16:426-436.
58 Clark JE, Sarafraz N, Marber MS. Potential of p38-MAPK inhibitors in the treatment of ischaemic heart disease. Pharmacol Ther. 2007;116:192-206.
59 Cochran DL. Inflammation and bone loss in periodontal disease. J Periodontol. 2008;79:1569-1576.
60 Cook R, Wu CC, Kang YJ, Han J. The role of the p38 pathway in adaptive immunity. Cell Mol Immunol. 2007;4:253-259.
61 Correa FO, Goncalves D, Figueredo CM, et al. The short-term effectiveness of non-surgical treatment in reducing levels of interleukin-1beta and proteases in gingival crevicular fluid from patients with type 2 diabetes mellitus and chronic periodontitis. J Periodontol. 2008;79:2143-2150.
62 Cottalorda A, Verschelde C, Marcais A, et al. TLR2 engagement on CD8 T cells lowers the threshold for optimal antigen-induced T cell activation. Eur J Immunol. 2006;36:1684-1693.
63 Craxton A, Shu G, Graves J, Saklatvala J, et al. p38 MAPK is required for CD40-induced gene expression and proliferation in B lymphocytes. J Immunol. 1998;161:3225-3226.
64 Crellin NK, Garcia RV, Hadisfar O, et al. Human CD4+ T cells express TLR5 and its ligand flagellin enhances the suppressive capacity and expression of FOXP3 in CD4+CD25+ T regulatory cells. J Immunol. 2005;175:8051-8059.
65 Crotti T, Smith MD, Hirsch R, et al. Receptor activator NF kappaB ligand (RANKL) and osteoprotegerin (OPG) protein expression in periodontitis. J Periodontal Res. 2003;38:380-387.
66 Culbert AA, Skaper SD, Howlett DR, et al. MAPK-activated protein kinase 2 deficiency in microglia inhibits pro-inflammatory mediator release and resultant neurotoxicity. Relevance to neuroinflammation in a transgenic mouse model of Alzheimer disease. J Biol Chem. 2006;281:23658-23667.
67 Curtsinger JM, Lins DC, Mescher MF. Signal 3 determines tolerance versus full activation of naive CD8 T cells: dissociating proliferation and development of effector function. J Exp Med. 2003;197:1141-1151.
68 Dadgostar H, Zarnegar B, Hoffmann A, et al. Cooperation of multiple signaling pathways in CD40-regulated gene expression in B lymphocytes. Proc Natl Acad Sci U S A. 2002;99:1497-1502.
69 Dale BA. Periodontal epithelium: a newly recognized role in health and disease. Periodontol 2000. 2002;30:70-78.
70 Dangl JL, Jones JD. Plant pathogens and integrated defence responses to infection. Nature. 2001;411:826-833.
71 Darveau RP, Belton CM, Reife RA, Lamont RJ. Local chemokine paralysis, a novel pathogenic mechanism for Porphyromonas gingivalis. Infect Immun. 1998;66:1660-1665.
72 Darveau RP, Arbabi S, Garcia I, Bainbridge B, Maier RV. Porphyromonas gingivalis lipopolysaccharide is both agonist and antagonist for p38 mitogen-activated protein kinase activation. Infect Immun. 2002;70:1867-1873.
73 Dasari P, Nicholson IC, Hodge G, et al. Expression of toll-like receptors on B lymphocytes. Cell Immunol. 2005;236:140-145.
74 de Aquino SG, Guimaraes MR, Stach-Machado DR, et al. Differential regulation of MMP-13 expression in two models of experimentally induced periodontal disease in rats. Arch Oral Biol. 2009;54:609-617.
75 de Souza AP, Trevilatto PC, Scarel-Caminaga RM, et al. Analysis of the MMP-9 (C-1562 T) and TIMP-2 (G-418C) gene promoter polymorphisms in patients with chronic periodontitis. J Clin Periodontol. 2005;32:207-211.
76 Delima AJ, Oates T, Assuma R, et al. Soluble antagonists to interleukin-1 (IL-1) and tumor necrosis factor (TNF) inhibits loss of tissue attachment in experimental periodontitis. J Clin Periodontol. 2001;28:233-240.
77 Delima AJ, Karatzas S, Amar S, Graves DT. Inflammation and tissue loss caused by periodontal pathogens is reduced by interleukin-1 antagonists. J Infect Dis. 2002;186:511-516.
78 Demmer RT, Behle JH, Wolf DL, et al. Transcriptomes in healthy and diseased gingival tissues. J Periodontol. 2008;79:2112-2124.
79 Di Paola R, Mazzon E, Muia C, et al. Effects of etanercept, a tumour necrosis factor-alpha antagonist, in an experimental model of periodontitis in rats. Br J Pharmacol. 2007;150:286-297.
80 Doherty DE, Zagarella L, Henson PM, Worthen GS. Lipopolysaccharide stimulates monocyte adherence by effects on both the monocyte and the endothelial cell. J Immunol. 1989;143:3673-3679.
81 Dolganiuc A, Garcia C, Kodys K, Szabo G. Distinct Toll-like receptor expression in monocytes and T cells in chronic HCV infection. World J Gastroenterol. 2006;12:1198-1204.
82 Dongari-Bagtzoglou AI, Ebersole JL. Production of inflammatory mediators and cytokines by human gingival fibroblasts following bacterial challenge. J Periodontal Res. 1996;31:90-98.
83 Duan W, Wong WS. Targeting mitogen-activated protein kinases for asthma. Curr Drug Targets. 2006;7:691-698.
84 Eastcott JW, Yamashita K, Taubman MA, et al. Adoptive transfer of cloned T helper cells ameliorates periodontal disease in nude rats. Oral Microbiol Immunol. 1994;9:284-289.
85 Ebersole JL, Taubman MA, Smith DJ, et al. Human serum antibody responses to oral microorganisms. IV. Correlation with homologous infection. Oral Microbiol Immunol. 1987;2:53-59.
86 Ejeil AL, Gaultier F, Igondjo-Tchen S, et al. Are cytokines linked to collagen breakdown during periodontal disease progression? J Periodontol. 2003;74:196-201.
87 Ejeil AL, Igondjo-Tchen S, Ghomrasseni S, et al. Expression of matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs) in healthy and diseased human gingiva. J Periodontol. 2003;74:188-195.
88 Feller L, Lemmer J. Necrotizing periodontal diseases in HIV-seropositive subjects: pathogenic mechanisms. J Int Acad Periodontol. 2008;10:10-15.
89 Figueredo CM, Areas A, Miranda LA, et al. The short-term effectiveness of non-surgical treatment in reducing protease activity in gingival crevicular fluid from chronic periodontitis patients. J Clin Periodontol. 2004;31:615-619.
90 Franchi L, Warner N, Viani K, Nunez G. Function of Nod-like receptors in microbial recognition and host defense. Immunol Rev. 2009;227:106-128.
91 Fritz JH, Girardin SE, Fitting C, et al. Synergistic stimulation of human monocytes and dendritic cells by Toll-like receptor 4 and NOD1- and NOD2-activating agonists. Eur J Immunol. 2005;35:2459-2470.
92 Fujii S, Liu K, Smith C, et al. The linkage of innate to adaptive immunity via maturing dendritic cells in vivo requires CD40 ligation in addition to antigen presentation and CD80/86 costimulation. J Exp Med. 2004;199:1607-1618.
93 Fukusaki T, Ohara N, Hara Y, et al. Evidence for association between a Toll-like receptor 4 gene polymorphism and moderate/severe periodontitis in the Japanese population. J Periodontal Res. 2007;42:541-545.
94 Fung E, Tang SM, Canner JP, et al. Delta-like 4 induces notch signaling in macrophages: implications for inflammation. Circulation. 2007;115:2948-2956.
95 Galdiero M, de l’Ero GC, Marcatili A. Cytokine and adhesion molecule expression in human monocytes and endothelial cells stimulated with bacterial heat shock proteins. Infect Immun. 1997;65:699-707.
96 Gamonal J, Acevedo A, Bascones A, et al. Levels of interleukin-1 beta, -8, and -10 and RANTES in gingival crevicular fluid and cell populations in adult periodontitis patients and the effect of periodontal treatment. J Periodontol. 2000;71:1535-1545.
97 Gamonal J, Acevedo A, Bascones A, et al. Characterization of cellular infiltrate, detection of chemokine receptor CCR5 and interleukin-8 and RANTES chemokines in adult periodontitis. J Periodontal Res. 2001;36:194-203.
98 Gapski R, Barr JL, Sarment DP, et al. Effect of systemic matrix metalloproteinase inhibition on periodontal wound repair: a proof of concept trial. J Periodontol. 2004;75:441-452.
99 Gapski R, Hasturk H, Van Dyke TE, et al. Systemic MMP inhibition for periodontal wound repair: results of a multi-centre randomized-controlled clinical trial. J Clin Periodontol. 2009;36:149-156.
100 Garcia de Aquino S, Manzolli Leite FR, Stach-Machado DR, et al. Signaling pathways associated with the expression of inflammatory mediators activated during the course of two models of experimental periodontitis. Life Sci. 2009;84:745-754.
101 Garlet GP, Martins WJr, Ferreira BR, et al. Patterns of chemokines and chemokine receptors expression in different forms of human periodontal disease. J Periodontal Res. 2003;38:210-217.
102 Garlet GP, Cardoso CR, Silva TA, et al. Cytokine pattern determines the progression of experimental periodontal disease induced by Actinobacillus actinomycetemcomitans through the modulation of MMPs, RANKL, and their physiological inhibitors. Oral Microbiol Immunol. 2006;21:12-20.
103 Gelman AE, Zhang J, Choi Y, Turka LA. Toll-like receptor ligands directly promote activated CD4+ T cell survival. J Immunol. 2004;172:6065-6073.
104 Gelman AE, LaRosa DF, Zhang J, et al. The adaptor molecule MyD88 activates PI-3 kinase signaling in CD4+ T cells and enables CpG oligodeoxynucleotide-mediated costimulation. Immunity. 2006;25:783-793.
105 Ghosh S, Hayden MS. New regulators of NF-kappaB in inflammation. Nat Rev Immunol. 2008;8:837-848.
106 Girardin SE, Boneca IG, Carneiro LA, et al. Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science. 2003;300:1584-1587.
107 Golub LM, McNamara TF, Ryan ME, et al. Adjunctive treatment with subantimicrobial doses of doxycycline: effects on gingival fluid collagenase activity and attachment loss in adult periodontitis. J Clin Periodontol. 2001;28:146-156.
108 Goncalves LD, Oliveira G, Hurtado PA, et al. Expression of metalloproteinases and their tissue inhibitors in inflamed gingival biopsies. J Periodontal Res. 2008;43:570-577.
109 Gordon MD, Nusse R. Wnt signaling: multiple pathways, multiple receptors, and multiple transcription factors. J Biol Chem. 2006;281:22429-22433.
110 Gorska R, Gregorek H, Kowalski J, et al. Relationship between clinical parameters and cytokine profiles in inflamed gingival tissue and serum samples from patients with chronic periodontitis. J Clin Periodontol. 2003;30:1046-1052.
111 Graves D. Cytokines that promote periodontal tissue destruction. J Periodontol. 2008;79:1585-1591.
112 Graves DT, Delima AJ, Assuma R, et al. Interleukin-1 and tumor necrosis factor antagonists inhibit the progression of inflammatory cell infiltration toward alveolar bone in experimental periodontitis. J Periodontol. 1998;69:1419-1425.
113 Graves DT. The potential role of chemokines and inflammatory cytokines in periodontal disease progression. Clin Infect Dis. 1999;28:482-490.
114 Graves DT, Cochran D. The contribution of interleukin-1 and tumor necrosis factor to periodontal tissue destruction. J Periodontol. 2003;74:391-401.
115 Gurkan A, Emingil G, Saygan BH, et al. Gene polymorphisms of matrix metalloproteinase-2, -9 and -12 in periodontal health and severe chronic periodontitis. Arch Oral Biol. 2008;53:337-345.
116 Gursoy UK, Kononen E, Uitto VJ. Stimulation of epithelial cell matrix metalloproteinase (MMP-2, -9, -13) and interleukin-8 secretion by fusobacteria. Oral Microbiol Immunol. 2008;23:432-434.
117 Hajishengallis G, Martin M, Schifferle RE, Genco RJ. Counteracting interactions between lipopolysaccharide molecules with differential activation of toll-like receptors. Infect Immun. 2002;70:6658-6664.
118 Hajishengallis G, Martin M, Sojar HT, et al. Dependence of bacterial protein adhesins on toll-like receptors for proinflammatory cytokine induction. Clin Diagn Lab Immunol. 2002;9:403-411.
119 Hajishengallis G, Genco RJ. Downregulation of the DNA-binding activity of nuclear factor-kappaB p65 subunit in Porphyromonas gingivalis fimbria-induced tolerance. Infect Immun. 2004;72:1188-1191.
120 Hajishengallis G, Sojar H, Genco RJ, DeNardin E. Intracellular signaling and cytokine induction upon interactions of Porphyromonas gingivalis fimbriae with pattern-recognition receptors. Immunol Invest. 2004;33:157-172.
121 Hajishengallis G, Tapping RI, Harokopakis E, et al. Differential interactions of fimbriae and lipopolysaccharide from Porphyromonas gingivalis with the Toll-like receptor 2-centred pattern recognition apparatus. Cell Microbiol. 2006;8:1557-1570.
122 Hajishengallis G, Wang M, Liang S. Induction of distinct TLR2-mediated proinflammatory and proadhesive signaling pathways in response to Porphyromonas gingivalis fimbriae. J Immunol. 2009;182:6690-6696.
123 Han J, Lee JD, Bibbs L, Ulevitch RJ. A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science. 1994;265:808-811.
124 Han X, Kawai T, Eastcott JW, Taubman MA. Bacterial-responsive B lymphocytes induce periodontal bone resorption. J Immunol. 2006;176:625-631.
125 Han X, Kawai T, Taubman MA. Interference with immune-cell-mediated bone resorption in periodontal disease. Periodontol 2000. 2007;45:76-94.
126 Han YW, Shi W, Huang GT, et al. Interactions between periodontal bacteria and human oral epithelial cells: Fusobacterium nucleatum adheres to and invades epithelial cells. Infect Immun. 2000;68:3140-3146.
127 Harada Y, Han X, Yamashita K, et al. Effect of adoptive transfer of antigen-specific B cells on periodontal bone resorption. J Periodontal Res. 2006;41:101-107.
128 Hartmann G, Krieg AM. CpG DNA and LPS induce distinct patterns of activation in human monocytes. Gene Ther. 1999;6:893-903.
129 Hartmann G, Krieg AM. Mechanism and function of a newly identified CpG DNA motif in human primary B cells. J Immunol. 2000;164:944-953.
130 Hasebe A, Yoshimura A, Into T, et al. Biological activities of Bacteroides forsythus lipoproteins and their possible pathological roles in periodontal disease. Infect Immun. 2004;72:1318-1325.
131 Hasegawa M, Fujimoto Y, Lucas PC, et al. A critical role of RICK/RIP2 polyubiquitination in Nod-induced NF-kappaB activation. EMBO J. 2008;27:373-383.
132 Hashimoto M, Asai Y, Ogawa T. Treponemal phospholipids inhibit innate immune responses induced by pathogen-associated molecular patterns. J Biol Chem. 2003;278:44205-44213.
133 Hatakeyama J, Tamai R, Sugiyama A, et al. Contrasting responses of human gingival and periodontal ligament fibroblasts to bacterial cell-surface components through the CD14/Toll-like receptor system. Oral Microbiol Immunol. 2003;18:14-23.
134 Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195-2224.
135 Hedges JC, Dechert MA, Yamboliev IA, et al. A role for p38(MAPK)/HSP27 pathway in smooth muscle cell migration. J Biol Chem. 1999;274:24211-24219.
136 Hemmi H, Kaisho T, Takeuchi O, et al. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat Immunol. 2002;3:196-200.
137 Hiramine H, Watanabe K, Hamada N, Umemoto T. Porphyromonas gingivalis 67-kDa fimbriae induced cytokine production and osteoclast differentiation utilizing TLR2. FEMS Microbiol Lett. 2003;229:49-55.
138 Hirschfeld M, Weis JJ, Toshchakov V, et al. Signaling by toll-like receptor 2 and 4 agonists results in differential gene expression in murine macrophages. Infect Immun. 2001;69:1477-1482.
139 Hofbauer LC, Heufelder AE. Role of receptor activator of nuclear factor-kappaB ligand and osteoprotegerin in bone cell biology. J Mol Med. 2001;79:243-253.
140 Holla LI, Fassmann A, Vasku A, et al. Genetic variations in the human gelatinase A (matrix metalloproteinase-2) promoter are not associated with susceptibility to, and severity of, chronic periodontitis. J Periodontol. 2005;76:1056-1060.
141 Hornung V, Rothenfusser S, Britsch S, et al. Quantitative expression of toll-like receptor 1-10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J Immunol. 2002;168:4531-4537.
142 Hoshino K, Kaisho T, Iwabe T, et al. Differential involvement of IFN-beta in Toll-like receptor-stimulated dendritic cell activation. Int Immunol. 2002;14:1225-1231.
143 Hou B, Reizis B, DeFranco AL. Toll-like receptors activate innate and adaptive immunity by using dendritic cell-intrinsic and -extrinsic mechanisms. Immunity. 2008;29:272-282.
144 Houri-Haddad Y, Wilensky A, Shapira L. T-cell phenotype as a risk factor for periodontal disease. Periodontol 2000. 2007;45:67-75.
145 Hsu LC, Ali SR, McGillivray S, et al. A NOD2-NALP1 complex mediates caspase-1-dependent IL-1beta secretion in response to Bacillus anthracis infection and muramyl dipeptide. Proc Natl Acad Sci U S A. 2008;105:7803-7808.
146 Huang GT, Haake SK, Kim JW, Park NH. Differential expression of interleukin-8 and intercellular adhesion molecule-1 by human gingival epithelial cells in response to Actinobacillus actinomycetemcomitans or Porphyromonas gingivalis infection. Oral Microbiol Immunol. 1998;13:301-309.
147 Huang GT, Kim D, Lee JK, et al. Interleukin-8 and intercellular adhesion molecule 1 regulation in oral epithelial cells by selected periodontal bacteria: multiple effects of Porphyromonas gingivalis via antagonistic mechanisms. Infect Immun. 2001;69:1364-1372.
148 Hysi P, Kabesch M, Moffatt MF, et al. NOD1 variation, immunoglobulin E and asthma. Hum Mol Genet. 2005;14:935-941.
149 Ihle JN, Kerr IM. Jaks and Stats in signaling by the cytokine receptor superfamily. Trends Genet. 1995;11:69-74.
150 Ihle JN. The Stat family in cytokine signaling. Curr Opin Cell Biol. 2001;13:211-217.
151 Imamura Y, Fujigaki Y, Oomori Y, et al. Polymorphism of genes encoding toll-like receptors and inflammatory cytokines in periodontal disease in the Japanese population. J Int Acad Periodontol. 2008;10:95-102.
152 Inohara N, Ogura Y, Chen FF, et al. Human Nod1 confers responsiveness to bacterial lipopolysaccharides. J Biol Chem. 2001;276:2551-2554.
153 Inohara N, Ogura Y, Fontalba A, et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn’s disease. J Biol Chem. 2003;278:5509-5512.
154 Iwamoto Y, Nishimura F, Soga Y, et al. Antimicrobial periodontal treatment decreases serum C-reactive protein, tumor necrosis factor-alpha, but not adiponectin levels in patients with chronic periodontitis. J Periodontol. 2003;74:1231-1236.
155 Jakob T, Walker PS, Krieg AM, et al. Activation of cutaneous dendritic cells by CpG-containing oligodeoxynucleotides: a role for dendritic cells in the augmentation of Th1 responses by immunostimulatory DNA. J Immunol. 1998;161:3042-3049.
156 Ji S, Kim Y, Min BM, Han SH, Choi Y. Innate immune responses of gingival epithelial cells to nonperiodontopathic and periodontopathic bacteria. J Periodontal Res. 2007;42:503-510.
157 Jiang Y, Mehta CK, Hsu TY, Alsulaimani FF. Bacteria induce osteoclastogenesis via an osteoblast-independent pathway. Infect Immun. 2002;70:3143-3148.
158 Jimi E, Aoki K, Saito H, et al. Selective inhibition of NF-kappa B blocks osteoclastogenesis and prevents inflammatory bone destruction in vivo. Nat Med. 2004;10:617-624.
159 Jin Q, Cirelli JA, Park CH, et al. RANKL inhibition through osteoprotegerin blocks bone loss in experimental periodontitis. J Periodontol. 2007;78:1300-1308.
160 Jirik FR, Podor TJ, Hirano T, et al. Bacterial lipopolysaccharide and inflammatory mediators augment IL-6 secretion by human endothelial cells. J Immunol. 1989;142:144-147.
161 Joffre O, Nolte MA, Sporri R, Reis e Sousa C. Inflammatory signals in dendritic cell activation and the induction of adaptive immunity. Immunol Rev. 2009;227:234-247.
162 Jotwani R, Pulendran B, Agrawal S, Cutler CW. Human dendritic cells respond to Porphyromonas gingivalis LPS by promoting a Th2 effector response in vitro. Eur J Immunol. 2003;33:2980-2986.
163 Jotwani R, Cutler CW. Fimbriated Porphyromonas gingivalis is more efficient than fimbria-deficient P. gingivalis in entering human dendritic cells in vitro and induces an inflammatory Th1 effector response. Infect Immun. 2004;72:1725-1732.
164 Kabelitz D. Expression and function of Toll-like receptors in T lymphocytes. Curr Opin Immunol. 2007;19:39-45.
165 Karin M, Lawrence T, Nizet V. Innate immunity gone awry: linking microbial infections to chronic inflammation and cancer. Cell. 2006;124:823-835.
166 Kawai T, Adachi O, Ogawa T, et al. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity. 1999;11:115-122.
167 Kawai T, Takeuchi O, Fujita T, et al. Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IFN-regulatory factor 3 and the expression of a subset of lipopolysaccharide-inducible genes. J Immunol. 2001;167:5887-5894.
168 Kawai T, Matsuyama T, Hosokawa Y, et al. B and T lymphocytes are the primary sources of RANKL in the bone resorptive lesion of periodontal disease. Am J Pathol. 2006;169:987-998.
169 Kawai T, Paster BJ, Komatsuzawa H, et al. Cross-reactive adaptive immune response to oral commensal bacteria results in an induction of receptor activator of nuclear factor-kappaB ligand (RANKL)-dependent periodontal bone resorption in a mouse model. Oral Microbiol Immunol. 2007;22:208-215.
170 Kelk P, Claesson R, Chen C, et al. IL-1beta secretion induced by Aggregatibacter (Actinobacillus) actinomycetemcomitans is mainly caused by the leukotoxin. Int J Med Microbiol. 2008;298:529-541.
171 Khosla S. Minireview: the OPG/RANKL/RANK system. Endocrinology. 2001;142:5050-5055.
172 Kikkert R, Laine ML, Aarden LA, van Winkelhoff AJ. Activation of toll-like receptors 2 and 4 by gram-negative periodontal bacteria. Oral Microbiol Immunol. 2007;22:145-151.
173 Kikuchi T, Matsuguchi T, Tsuboi N, et al. Gene expression of osteoclast differentiation factor is induced by lipopolysaccharide in mouse osteoblasts via Toll-like receptors. J Immunol. 2001;166:3574-3579.
174 Kim SJ, Choi EY, Kim EG, et al. Prevotella intermedia lipopolysaccharide stimulates release of tumor necrosis factor-alpha through mitogen-activated protein kinase signaling pathways in monocyte-derived macrophages. FEMS Immunol Med Microbiol. 2007;51:407-413.
175 Kirkwood K, Li F, Rogers J, et al. A p38 alpha selective mitogen-activated protein kinase inhibitor prevents periodontal bone loss. J Pharmacol Exp Ther. 2007;320:56-63.
176 Kirkwood KL, Cirelli JA, Rogers JE, Giannobile WV. Novel host response therapeutic approaches to treat periodontal diseases. Periodontol 2000. 2007;43:294-315.
177 Komai-Koma M, Jones L, Ogg GS, et al. TLR2 is expressed on activated T cells as a costimulatory receptor. Proc Natl Acad Sci U S A. 2004;101:3029-3034.
178 Kornman KS, Page RC, Tonetti MS. The host response to the microbial challenge in periodontitis: assembling the players. Periodontol 2000. 1997;14:33-53.
179 Krisanaprakornkit S, Kimball JR, Weinberg A, et al. Inducible expression of human beta-defensin 2 by Fusobacterium nucleatum in oral epithelial cells: multiple signaling pathways and role of commensal bacteria in innate immunity and the epithelial barrier. Infect Immun. 2000;68:2907-2915.
180 Kubota T, Itagaki M, Hoshino C, et al. Altered gene expression levels of matrix metalloproteinases and their inhibitors in periodontitis-affected gingival tissue. J Periodontol. 2008;79:166-173.
181 Kumar S, Votta BJ, Rieman DJ, et al. IL-1- and TNF-induced bone resorption is mediated by p38 mitogen activated protein kinase. J Cell Physiol. 2001;187:294-303.
182 Kuula H, Salo T, Pirila E, et al. Local and systemic responses in matrix metalloproteinase 8-deficient mice during Porphyromonas gingivalis-induced periodontitis. Infect Immun. 2009;77:850-859.
183 Lai WC, Zhou M, Shankavaram U, Peng G, Wahl LM. Differential regulation of lipopolysaccharide-induced monocyte matrix metalloproteinase (MMP)-1 and MMP-9 by p38 and extracellular signal-regulated kinase 1/2 mitogen-activated protein kinases. J Immunol. 2003;170:6244-6249.
184 Lamkanfi M, Kanneganti TD, Franchi L, Nunez G. Caspase-1 inflammasomes in infection and inflammation. J Leukoc Biol. 2007;82:220-225.
185 Lamster IB, Novak MJ. Host mediators in gingival crevicular fluid: implications for the pathogenesis of periodontal disease. Crit Rev Oral Biol Med. 1992;3:31-60.
186 Lappin DF, MacLeod CP, Kerr A, et al. Anti-inflammatory cytokine IL-10 and T cell cytokine profile in periodontitis granulation tissue. Clin Exp Immunol. 2001;123:294-300.
187 Leshem O, Kashino SS, Goncalves RB, et al. Th1 biased response to a novel Porphyromonas gingivalis protein aggravates bone resorption caused by this oral pathogen. Microbes Infect. 2008;10:664-672.
188 Lester SR, Bain JL, Johnson RB, Serio FG. Gingival concentrations of interleukin-23 and -17 at healthy sites and at sites of clinical attachment loss. J Periodontol. 2007;78:1545-1550.
189 Lewkowicz P, Lewkowicz N, Sasiak A, Tchorzewski H. Lipopolysaccharide-activated CD4+CD25+ T regulatory cells inhibit neutrophil function and promote their apoptosis and death. J Immunol. 2006;177:7155-7163.
190 Li Y, Jiang B, Ensign WY, Vogt PK, Han J. Myogenic differentiation requires signalling through both phosphatidylinositol 3-kinase and p38 MAP kinase. Cell Signal. 2000;12:751-757.
191 Liew FY, Xu D, Brint EK, O’Neill LA. Negative regulation of toll-like receptor-mediated immune responses. Nat Rev Immunol. 2005;5:446-458.
192 Lindhe J, Hamp SE, Loe H. Plaque induced periodontal disease in beagle dogs. A 4-year clinical, roentgenographical and histometrical study. J Periodontal Res. 1975;10:243-255.
193 Liu H, Komai-Koma M, Xu D, Liew FY. Toll-like receptor 2 signaling modulates the functions of CD4+ CD25+ regulatory T cells. Proc Natl Acad Sci U S A. 2006;103:7048-7053.
194 Loe H, Anerud A, Boysen H, Morrison E. Natural history of periodontal disease in man. Rapid, moderate and no loss of attachment in Sri Lankan laborers 14 to 46 years of age. J Clin Periodontol. 1986;13:431-445.
195 Madianos PN, Papapanou PN, Sandros J. Porphyromonas gingivalis infection of oral epithelium inhibits neutrophil transepithelial migration. Infect Immun. 1997;65:3983-3990.
196 Mahanonda R, Sa-Ard-Iam N, Charatkulangkun O, et al. Monocyte activation by Porphyromonas gingivalis LPS in aggressive periodontitis with the use of whole-blood cultures. J Dent Res. 2004;83:540-545.
197 Mansson A, Adner M, Cardell LO. Toll-like receptors in cellular subsets of human tonsil T cells: altered expression during recurrent tonsillitis. Respir Res. 2006;7:36.
198 Mao S, Maeno N, Matayoshi S, et al. The induction of intercellular adhesion molecule-1 on human umbilical vein endothelial cells by a heat-stable component of Porphyromonas gingivalis. Curr Microbiol. 2004;48:108-112.
199 Maria de Freitas N, Imbronito AV, Neves AC, et al. Analysis of IL-1A(-889) and TNFA(-308) gene polymorphism in Brazilian patients with generalized aggressive periodontitis. Eur Cytokine Netw. 2007;18:142-147.
200 Martin M, Katz J, Vogel SN, Michalek SM. Differential induction of endotoxin tolerance by lipopolysaccharides derived from Porphyromonas gingivalis and Escherichia coli. J Immunol. 2001;167:5278-5285.
201 Matsuki Y, Yamamoto T, Hara K. Detection of inflammatory cytokine messenger RNA (mRNA)-expressing cells in human inflamed gingiva by combined in situ hybridization and immunohistochemistry. Immunology. 1992;76:42-47.
202 Mattern T, Thanhauser A, Reiling N, et al. Endotoxin and lipid A stimulate proliferation of human T cells in the presence of autologous monocytes. J Immunol. 1994;153:2996-3004.
203 Mattern T, Flad HD, Brade L, et al. Stimulation of human T lymphocytes by LPS is MHC unrestricted, but strongly dependent on B7 interactions. J Immunol. 1998;160:3412-3418.
204 Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991-1045.
205 Mbalaviele G, Anderson G, Jones A, De Ciechi P. Inhibition of p38 mitogen-activated protein kinase prevents inflammatory bone destruction. J Pharmacol Exp Ther. 2006;317:1044-1053.
206 McCoy SL, Kurtz SE, Hausman FA, et al. Activation of RAW264.7 macrophages by bacterial DNA and lipopolysaccharide increases cell surface DNA binding and internalization. J Biol Chem. 2004;279:17217-17223.
207 McDonald C, Inohara N, Nunez G. Peptidoglycan signaling in innate immunity and inflammatory disease. J Biol Chem. 2005;280:20177-20180.
208 McIntyre KW, Shuster DJ, Gillooly KM, et al. A highly selective inhibitor of I kappa B kinase, BMS-345541, blocks both joint inflammation and destruction in collagen-induced arthritis in mice. Arthritis Rheum. 2003;48:2652-2659.
209 Mealey BL, Rose LF. Diabetes mellitus and inflammatory periodontal diseases. Curr Opin Endocrinol Diabetes Obes. 2008;15:135-141.
210 Medicherla S, Ma JY, Mangadu R, et al. A selective p38 alpha mitogen-activated protein kinase inhibitor reverses cartilage and bone destruction in mice with collagen-induced arthritis. J Pharmacol Exp Ther. 2006;318:132-141.
211 Mendes Sdos S, Candi A, Vansteenbrugge M, et al. Microarray analyses of the effects of NF-kappaB or PI3K pathway inhibitors on the LPS-induced gene expression profile in RAW264.7 cells: synergistic effects of rapamycin on LPS-induced MMP9-overexpression. Cell Signal. 2009;21:1109-1122.
212 Mercer BA, D’Armiento JM. Emerging role of MAP kinase pathways as therapeutic targets in COPD. Int J Chron Obstruct Pulmon Dis. 2006;1:137-150.
213 Mogi M, Otogoto J, Ota N, Togari A. Differential expression of RANKL and osteoprotegerin in gingival crevicular fluid of patients with periodontitis. J Dent Res. 2004;83:166-169.
214 Nagasawa T, Kobayashi H, Kiji M, et al. LPS-stimulated human gingival fibroblasts inhibit the differentiation of monocytes into osteoclasts through the production of osteoprotegerin. Clin Exp Immunol. 2002;130:338-344.
215 Nemoto E, Darveau RP, Foster BL, et al. Regulation of cementoblast function by P. gingivalis lipopolysaccharide via TLR2. J Dent Res. 2006;85:733-738.
216 Nibali L, Donos N, Brett PM, et al. A familial analysis of aggressive periodontitis – clinical and genetic findings. J Periodontal Res. 2008;43:627-634.
217 Nikolopoulos GK, Dimou NL, Hamodrakas SJ, Bagos PG. Cytokine gene polymorphisms in periodontal disease: a meta-analysis of 53 studies including 4178 cases and 4590 controls. J Clin Periodontol. 2008;35:754-767.
218 Nociti FHJr, Foster BL, Barros SP, et al. Cementoblast gene expression is regulated by porphyromonas gingivalis lipopolysaccharide partially via toll-like receptor-4/MD-2. J Dent Res. 2004;83:602-607.
219 Nolte MA, Leibundgut-Landmann S, Joffre O, Reis e Sousa C. Dendritic cell quiescence during systemic inflammation driven by LPS stimulation of radioresistant cells in vivo. J Exp Med. 2007;204:1487-1501.
220 Nonnenmacher C, Dalpke A, Zimmermann S, et al. DNA from periodontopathogenic bacteria is immunostimulatory for mouse and human immune cells. Infect Immun. 2003;71:850-856.
221 Novak MJ, Johns LP, Miller RC, Bradshaw MH. Adjunctive benefits of subantimicrobial dose doxycycline in the management of severe, generalized, chronic periodontitis. J Periodontol. 2002;73:762-769.
222 Nualart Grollmus ZC, Morales Chavez MC, Silvestre Donat FJ. Periodontal disease associated to systemic genetic disorders. Med Oral Patol Oral Cir Bucal. 2007;12:E211-215.
223 O’Brien-Simpson NM, Pathirana RD, Walker GD, Reynolds EC. Porphyromonas gingivalis RgpA-Kgp proteinase-adhesin complexes penetrate gingival tissue and induce proinflammatory cytokines or apoptosis in a concentration-dependent manner. Infect Immun. 2009;77:1246-1261.
224 O’Shea JJ, Gadina M, Schreiber RD. Cytokine signaling in 2002: new surprises in the Jak/Stat pathway. Cell. 2002;109(Suppl):S121-S131.
225 Oberg HH, Wesch D, Grussel S, et al. Differential expression of CD126 and CD130 mediates different STAT-3 phosphorylation in CD4+CD25- and CD25high regulatory T cells. Int Immunol. 2006;18:555-563.
226 Offenbacher S, Salvi GE. Induction of prostaglandin release from macrophages by bacterial endotoxin. Clin Infect Dis. 1999;28:505-513.
227 Offenbacher S, Beck JD. A perspective on the potential cardioprotective benefits of periodontal therapy. Am Heart J. 2005;149:950-954.
228 Ogawa T. Immunobiological properties of chemically defined lipid A from lipopolysaccharide of Porphyromonas (Bacteroides) gingivalis. Eur J Biochem. 1994;219:737-742.
229 Ogawa T, Asai Y, Yamamoto H, et al. Immunobiological activities of a chemically synthesized lipid A of Porphyromonas gingivalis. FEMS Immunol Med Microbiol. 2000;28:273-281.
230 Ogura Y, Inohara N, Benito A, et al. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-kappaB. J Biol Chem. 2001;276:4812-4818.
231 Ohguchi Y, Ishihara Y, Ohguchi M, et al. Capsular polysaccharide from Actinobacillus actinomycetemcomitans inhibits IL-6 and IL-8 production in human gingival fibroblast. J Periodontal Res. 2003;38:191-197.
232 Onishi S, Honma K, Liang S, et al. Toll-like receptor 2-mediated interleukin-8 expression in gingival epithelial cells by the Tannerella forsythia leucine-rich repeat protein BspA. Infect Immun. 2008;76:198-205.
233 Page RC, Schroeder HE. Pathogenesis of inflammatory periodontal disease. A summary of current work. Lab Invest. 1976;34:235-249.
234 Park JH, Kim YG, McDonald C, et al. RICK/RIP2 mediates innate immune responses induced through Nod1 and Nod2 but not TLRs. J Immunol. 2007;178:2380-2386.
235 Park OJ, Shin SY, Choi Y, et al. The association of osteoprotegerin gene polymorphisms with periodontitis. Oral Dis. 2008;14:440-444.
236 Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299:1033-1036.
237 Paster BJ, Boches SK, Galvin JL, et al. Bacterial diversity in human subgingival plaque. J Bacteriol. 2001;183:3770-3783.
238 Patil C, Zhu X, Rossa CJr, Kim YJ, Kirkwood KL. p38 MAPK regulates IL-1beta induced IL-6 expression through mRNA stability in osteoblasts. Immunol Invest. 2004;33:213-233.
239 Patil C, Rossa CJr, Kirkwood KL. Actinobacillus actinomycetemcomitans lipopolysaccharide induces interleukin-6 expression through multiple mitogen-activated protein kinase pathways in periodontal ligament fibroblasts. Oral Microbiol Immunol. 2006;21:392-398.
240 Patil CS, Kirkwood KL. p38 MAPK signaling in oral-related diseases. J Dent Res. 2007;86:812-825.
241 Patil CS, Liu M, Zhao W, et al. Targeting mRNA stability arrests inflammatory bone loss. Mol Ther. 2008;16:1657-1664.
242 Pelt P, Zimmermann B, Ulbrich N, Bernimoulin JP. Effects of lipopolysaccharide extracted from Prevotella intermedia on bone formation and on the release of osteolytic mediators by fetal mouse osteoblasts in vitro. Arch Oral Biol. 2002;47:859-866.
243 Peng G, Guo Z, Kiniwa Y, et al. Toll-like receptor 8-mediated reversal of CD4+ regulatory T cell function. Science. 2005;309:1380-1384.
244 Pereira C, Schaer DJ, Bachli EB, et al. Wnt5A/CaMKII signaling contributes to the inflammatory response of macrophages and is a target for the antiinflammatory action of activated protein C and interleukin-10. Arterioscler Thromb Vasc Biol. 2008;28:504-510.
245 Pesu M, Aittomaki S, Takaluoma K, et al. p38 Mitogen-activated protein kinase regulates interleukin-4-induced gene expression by stimulating STAT6-mediated transcription. J Biol Chem. 2002;277:38254-38261.
246 Pirhan D, Atilla G, Emingil G, et al. Effect of MMP-1 promoter polymorphisms on GCF MMP-1 levels and outcome of periodontal therapy in patients with severe chronic periodontitis. J Clin Periodontol. 2008;35:862-870.
247 Preshaw PM, Hefti AF, Novak MJ, et al. Subantimicrobial dose doxycycline enhances the efficacy of scaling and root planing in chronic periodontitis: a multicenter trial. J Periodontol. 2004;75:1068-1076.
248 Preshaw PM, Novak MJ, Mellonig J, et al. Modified-release subantimicrobial dose doxycycline enhances scaling and root planing in subjects with periodontal disease. J Periodontol. 2008;79:440-452.
249 Pulendran B, Kumar P, Cutler CW, et al. Lipopolysaccharides from distinct pathogens induce different classes of immune responses in vivo. J Immunol. 2001;167:5067-5076.
250 Raffeiner B, Dejaco C, Duftner C, et al. Between adaptive and innate immunity: TLR4-mediated perforin production by CD28null T-helper cells in ankylosing spondylitis. Arthritis Res Ther. 2005;7:R1412-1420.
251 Raj PA, Antonyraj KJ, Karunakaran T. Large-scale synthesis and functional elements for the antimicrobial activity of defensins. Biochem J. 2000;347(Pt 3):633-641.
252 Ramamurthy NS, Rifkin BR, Greenwald RA, et al. Inhibition of matrix metalloproteinase-mediated periodontal bone loss in rats: a comparison of 6 chemically modified tetracyclines. J Periodontol. 2002;73:726-734.
253 Ramamurthy NS, Xu JW, Bird J, et al. Inhibition of alveolar bone loss by matrix metalloproteinase inhibitors in experimental periodontal disease. J Periodontal Res. 2002;37:1-7.
254 Ramseier CA, Kinney JS, Herr AE, et al. Identification of pathogen and host-response markers correlated with periodontal disease. J Periodontol. 2009;80:436-446.
255 Reddi K, Poole S, Nair S, et al. Lipid A-associated proteins from periodontopathogenic bacteria induce interleukin-6 production by human gingival fibroblasts and monocytes. FEMS Immunol Med Microbiol. 1995;11:137-144.
256 Reddi K, Wilson M, Poole S, et al. Relative cytokine-stimulating activities of surface components of the oral periodontopathogenic bacterium Actinobacillus actinomycetemcomitans. Cytokine. 1995;7:534-541.
257 Rennefahrt U, Janakiraman M, Ollinger R, Troppmair J. Stress kinase signaling in cancer: fact or fiction? Cancer Lett. 2005;217:1-9.
258 Rincon M, Enslen H, Raingeaud J, et al. Interferon-g expression by Th1 effector T cells mediated by the p38 MAP kinase signaling pathway. EMBO J. 1998;17:2817-2829.
259 Roberts FA, Richardson GJ, Michalek SM. Effects of Porphyromonas gingivalis and Escherichia coli lipopolysaccharides on mononuclear phagocytes. Infect Immun. 1997;65:3248-3254.
260 Rogers JE, Li F, Coatney DD, et al. A p38 mitogen-activated protein kinase inhibitor arrests active alveolar bone loss in a rat periodontitis model. J Periodontol. 2007;78:1992-1998.
261 Rommel C, Camps M, Ji H. PI3K delta and PI3K gamma: partners in crime in inflammation in rheumatoid arthritis and beyond? Nat Rev Immunol. 2007;7:191-201.
262 Rosenstein ED, Greenwald RA, Kushner LJ, Weissmann G. Hypothesis: the humoral immune response to oral bacteria provides a stimulus for the development of rheumatoid arthritis. Inflammation. 2004;28:311-318.
263 Rossa C, Ehmann K, Liu M, Patil C, Kirkwood KL. MKK3/6-p38 MAPK signaling is required for IL-1beta and TNF-alpha-induced RANKL expression in bone marrow stromal cells. J Interferon Cytokine Res. 2006;26:719-729.
264 Rossa CJr, Liu M, Patil C, Kirkwood KL. MKK3/6-p38 MAPK negatively regulates murine MMP-13 gene expression induced by IL-1beta and TNF-alpha in immortalized periodontal ligament fibroblasts. Matrix Biol. 2005;24:478-488.
265 Rossa CJr, Liu M, Bronson P, Kirkwood KL. Transcriptional activation of MMP-13 by periodontal pathogenic LPS requires p38 MAP kinase. J Endotoxin Res. 2007;13:85-93.
266 Rossa CJr, Liu M, Kirkwood KL. A dominant function of p38 mitogen-activated protein kinase signaling in receptor activator of nuclear factor-kappaB ligand expression and osteoclastogenesis induction by Aggregatibacter actinomycetemcomitans and Escherichia coli lipopolysaccharide. J Periodontal Res. 2008;43:201-211.
267 Ruby J, Rehani K, Martin M. Treponema denticola activates mitogen-activated protein kinase signal pathways through Toll-like receptor 2. Infect Immun. 2007;75:5763-5768.
268 Rudensky AY, Gavin M, Zheng Y. FOXP3 and NFAT: partners in tolerance. Cell. 2006;126:253-256.
269 Runza VL, Schwaeble W, Mannel DN. Ficolins: novel pattern recognition molecules of the innate immune response. Immunobiology. 2008;213:297-306.
270 Ryan ME, Usman A, Ramamurthy NS, et al. Excessive matrix metalloproteinase activity in diabetes: inhibition by tetracycline analogues with zinc reactivity. Curr Med Chem. 2001;8:305-316.
271 Ryu OH, Choi SJ, Linares AM, et al. Gingival epithelial cell expression of macrophage inflammatory protein-1alpha induced by interleukin-1beta and lipopolysaccharide. J Periodontol. 2007;78:1627-1634.
272 Sakaguchi S. Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat Immunol. 2005;6:345-352.
273 Sakellari D, Katsares V, Georgiadou M, et al. No correlation of five gene polymorphisms with periodontal conditions in a Greek population. J Clin Periodontol. 2006;33:765-770.
274 Sallusto F, Schaerli P, Loetscher P, et al. Rapid and coordinated switch in chemokine receptor expression during dendritic cell maturation. Eur J Immunol. 1998;28:2760-2769.
275 Salvi GE, Beck JD, Offenbacher S. PGE2, IL-1 beta, and TNF-alpha responses in diabetics as modifiers of periodontal disease expression. Ann Periodontol. 1998;3:40-50.
276 Salvi GE, Brown CE, Fujihashi K, et al. Inflammatory mediators of the terminal dentition in adult and early onset periodontitis. J Periodontal Res. 1998;33:212-225.
277 Sandros J, Karlsson C, Lappin DF, et al. Cytokine responses of oral epithelial cells to Porphyromonas gingivalis infection. J Dent Res. 2000;79:1808-1814.
278 Sarkar FH, Li Y, Wang Z, Kong D. NF-kappaB signaling pathway and its therapeutic implications in human diseases. Int Rev Immunol. 2008;27:293-319.
279 Sasaki H, Suzuki N, Kent RJr, et al. T cell response mediated by myeloid cell-derived IL-12 is responsible for Porphyromonas gingivalis-induced periodontitis in IL-10-deficient mice. J Immunol. 2008;180:6193-6198.
280 Sasaki Y, Aiba S. Dendritic cells and contact dermatitis. Clin Rev Allergy Immunol. 2007;33:27-34.
281 Sato S, Sanjo H, Takeda K, et al. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat Immunol. 2005;6:1087-1095.
282 Sawada N, Ogawa T, Asai Y, et al. Toll-like receptor 4-dependent recognition of structurally different forms of chemically synthesized lipid As of Porphyromonas gingivalis. Clin Exp Immunol. 2007;148:529-536.
283 Schaefer AS, Richter GM, Groessner-Schreiber B, et al. Identification of a shared genetic susceptibility locus for coronary heart disease and periodontitis. PLoS Genet. 2009;5:e1000378.
284 Schierano G, Pejrone G, Brusco P, et al. TNF-alpha TGF-beta2 and IL-1beta levels in gingival and peri-implant crevicular fluid before and after de novo plaque accumulation. J Clin Periodontol. 2008;35:532-538.
285 Schindler CW. Series introduction. JAK-STAT signaling in human disease. J Clin Invest. 2002;109:1133-1137.
286 Seymour GJ, Gemmell E. Cytokines in periodontal disease: where to from here? Acta Odontol Scand. 2001;59:167-173.
287 Sha WC, Liou HC, Tuomanen EI, Baltimore D. Targeted disruption of the p50 subunit of NF-kappa B leads to multifocal defects in immune responses. Cell. 1995;80:321-330.
288 Shapira L, van Dyke TE, Hart TC. A localized absence of interleukin-4 triggers periodontal disease activity: a novel hypothesis. Med Hypotheses. 1992;39:319-322.
289 Shin H, Zhang Y, Jagannathan M, et al. B cells from periodontal disease patients express surface Toll-like receptor 4. J Leukoc Biol. 2009;85:648-655.
290 Sigusch B, Klinger G, Glockmann E, Simon HU. Early-onset and adult periodontitis associated with abnormal cytokine production by activated T lymphocytes. J Periodontol. 1998;69:1098-1104.
291 Silva N, Dutzan N, Hernandez M, et al. Characterization of progressive periodontal lesions in chronic periodontitis patients: levels of chemokines, cytokines, matrix metalloproteinase-13, periodontal pathogens and inflammatory cells. J Clin Periodontol. 2008;35:206-214.
292 Simone R, Floriani A, Saverino D. Stimulation of human CD4 T lymphocytes via TLR3, TLR5 and TLR7/8 up-regulates expression of costimulatory and modulates proliferation. Open Microbiol J. 2009;3:1-8.
293 Socransky SS, Haffajee AD, Cugini MA, et al. Microbial complexes in subgingival plaque. J Clin Periodontol. 1998;25:134-144.
294 Socransky SS, Haffajee AD, Ximenez-Fyvie LA, Feres M, Mager D. Ecological considerations in the treatment of Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis periodontal infections. Periodontol 2000. 1999;20:341-362.
295 Soedarsono N, Rabello D, Kamei H, et al. Evaluation of RANK/RANKL/OPG gene polymorphisms in aggressive periodontitis. J Periodontal Res. 2006;41:397-404.
296 Sparwasser T, Koch ES, Vabulas RM, et al. Bacterial DNA and immunostimulatory CpG oligonucleotides trigger maturation and activation of murine dendritic cells. Eur J Immunol. 1998;28:2045-2054.
297 Sporri R, Reis e Sousa C. Inflammatory mediators are insufficient for full dendritic cell activation and promote expansion of CD4+ T cell populations lacking helper function. Nat Immunol. 2005;6:163-170.
298 Stashenko P, Jandinski JJ, Fujiyoshi P, et al. Tissue levels of bone resorptive cytokines in periodontal disease. J Periodontol. 1991;62:504-509.
299 Stathopoulou PG, Benakanakere MR, Galicia JC, Kinane DF. The host cytokine response to Porphyromonas gingivalis is modified by gingipains. Oral Microbiol Immunol. 2009;24:11-17.
300 Suda K, Udagawa N, Sato N, et al. Suppression of osteoprotegerin expression by prostaglandin E2 is crucially involved in lipopolysaccharide-induced osteoclast formation. J Immunol. 2004;172:2504-2510.
301 Suda T, Takahashi N, Udagawa N, et al. Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocr Rev. 1999;20:345-357.
302 Sugawara S, Yang S, Iki K, et al. Monocytic cell activation by Nonendotoxic glycoprotein from Prevotella intermedia ATCC 25611 is mediated by toll-like receptor 2. Infect Immun. 2001;69:4951-4957.
303 Sugawara Y, Uehara A, Fujimoto Y, et al. Toll-like receptors, NOD1, and NOD2 in oral epithelial cells. J Dent Res. 2006;85:524-529.
304 Sugiyama A, Uehara A, Iki K, et al. Activation of human gingival epithelial cells by cell-surface components of black-pigmented bacteria: augmentation of production of interleukin-8, granulocyte colony-stimulating factor and granulocyte-macrophage colony-stimulating factor and expression of intercellular adhesion molecule 1. J Med Microbiol. 2002;51:27-33.
305 Sutmuller RP, den Brok MH, Kramer M, et al. Toll-like receptor 2 controls expansion and function of regulatory T cells. J Clin Invest. 2006;116:485-494.
306 Tada H, Aiba S, Shibata K, et al. Synergistic effect of Nod1 and Nod2 agonists with toll-like receptor agonists on human dendritic cells to generate interleukin-12 and T helper type 1 cells. Infect Immun. 2005;73:7967-7976.
307 Takeshita A, Imai K, Hanazawa S. CpG motifs in Porphyromonas gingivalis DNA stimulate interleukin-6 expression in human gingival fibroblasts. Infect Immun. 1999;67:4340-4345.
308 Tam V, O’Brien-Simpson NM, Chen YY, et al. The RgpA-Kgp proteinase-adhesin complexes of Porphyromonas gingivalis Inactivate the Th2 cytokines interleukin-4 and interleukin-5. Infect Immun. 2009;77:1451-1458.
309 Tani Y, Tani M, Kato I. Extracellular 37-kDa antigenic protein from Actinobacillus actinomycetemcomitans induces TNF-alpha, IL-1 beta, and IL-6 in murine macrophages. J Dent Res. 1997;76:1538-1547.
310 Teitelbaum SL. Osteoclasts: what do they do and how do they do it? Am J Pathol. 2007;170:427-435.
311 Teng YT, Mahamed D, Singh B. Gamma interferon positively modulates Actinobacillus actinomycetemcomitans-specific RANKL+ CD4+ Th-cell-mediated alveolar bone destruction in vivo. Infect Immun. 2005;73:3453-3461.
312 Tietze K, Dalpke A, Morath S, et al. Differences in innate immune responses upon stimulation with gram-positive and gram-negative bacteria. J Periodontal Res. 2006;41:447-454.
313 Toker H, Poyraz O, Eren K. Effect of periodontal treatment on IL-1beta, IL-1ra, and IL-10 levels in gingival crevicular fluid in patients with aggressive periodontitis. J Clin Periodontol. 2008;35:507-513.
314 Tokoro Y, Matsuki Y, Yamamoto T, et al. Relevance of local Th2-type cytokine mRNA expression in immunocompetent infiltrates in inflamed gingival tissue to periodontal diseases. Clin Exp Immunol. 1997;107:166-174.
315 Tomi N, Fukuyo Y, Arakawa S, Nakajima T. Pro-inflammatory cytokine production from normal human fibroblasts is induced by Tannerella forsythia detaching factor. J Periodontal Res. 2008;43:136-142.
316 Trevani AS, Chorny A, Salamone G, et al. Bacterial DNA activates human neutrophils by a CpG-independent pathway. Eur J Immunol. 2003;33:3164-3174.
317 Tsukahara Y, Lian Z, Zhang X, et al. Gene expression in human neutrophils during activation and priming by bacterial lipopolysaccharide. J Cell Biochem. 2003;89:848-861.
318 Uehara A, Sugawara Y, Kurata S, et al. Chemically synthesized pathogen-associated molecular patterns increase the expression of peptidoglycan recognition proteins via toll-like receptors, NOD1 and NOD2 in human oral epithelial cells. Cell Microbiol. 2005;7:675-686.
319 Uehara A, Fujimoto Y, Fukase K, Takada H. Various human epithelial cells express functional Toll-like receptors, NOD1 and NOD2 to produce anti-microbial peptides, but not proinflammatory cytokines. Mol Immunol. 2007;44:3100-3111.
320 Uehara A, Takada H. Functional TLRs and NODs in human gingival fibroblasts. J Dent Res. 2007;86:249-254.
321 Uehara A, Imamura T, Potempa J, et al. Gingipains from Porphyromonas gingivalis synergistically induce the production of proinflammatory cytokines through protease-activated receptors with Toll-like receptor and NOD1/2 ligands in human monocytic cells. Cell Microbiol. 2008;10:1181-1189.
322 Ueki K, Tabeta K, Yoshie H, Yamazaki K. Self-heat shock protein 60 induces tumour necrosis factor-alpha in monocyte-derived macrophage: possible role in chronic inflammatory periodontal disease. Clin Exp Immunol. 2002;127:72-77.
323 van Heel DA, Ghosh S, Butler M, et al. Synergistic enhancement of Toll-like receptor responses by NOD1 activation. Eur J Immunol. 2005;35:2471-2476.
324 Vankeerberghen A, Nuytten H, Dierickx K, Quirynen M, et al. Differential induction of human beta-defensin expression by periodontal commensals and pathogens in periodontal pocket epithelial cells. J Periodontol. 2005;76:1293-1303.
325 Wada Y, Nakajima-Yamada T, Yamada K, et al. R-130823, a novel inhibitor of p38 MAPK, ameliorates hyperalgesia and swelling in arthritis models. Eur J Pharmacol. 2005;506:285-295.
326 Wagner M, Poeck H, Jahrsdoerfer B, et al. IL-12p70-dependent Th1 induction by human B cells requires combined activation with CD40 ligand and CpG DNA. J Immunol. 2004;172:954-963.
327 Walker JG, Ahern MJ, Coleman M, et al. Expression of Jak3, STAT1, STAT4, and STAT6 in inflammatory arthritis: unique Jak3 and STAT4 expression in dendritic cells in seropositive rheumatoid arthritis. Ann Rheum Dis. 2006;65:149-156.
328 Walker SJ, Van Dyke TE, Rich S, Kornman KS, di Giovine FS, Hart TC. Genetic polymorphisms of the IL-1alpha and IL-1beta genes in African-American LJP patients and an African-American control population. J Periodontol. 2000;71:723-728.
329 Warner RL, Bhagavathula N, Nerusu KC, et al. Matrix metalloproteinases in acute inflammation: induction of MMP-3 and MMP-9 in fibroblasts and epithelial cells following exposure to pro-inflammatory mediators in vitro. Exp Mol Pathol. 2004;76:189-195.
330 Watanabe A, Takeshita A, Kitano S, Hanazawa S. CD14-mediated signal pathway of Porphyromonas gingivalis lipopolysaccharide in human gingival fibroblasts. Infect Immun. 1996;64:4488-4494.
331 Weis WI, Taylor ME, Drickamer K. The C-type lectin superfamily in the immune system. Immunol Rev. 1998;163:19-34.
332 Wen Z, Darnell JEJr. Mapping of Stat3 serine phosphorylation to a single residue (727) and evidence that serine phosphorylation has no influence on DNA binding of Stat1 and Stat3. Nucleic Acids Res. 1997;25:2062-2067.
333 Williams DL, Ozment-Skelton T, Li C. Modulation of the phosphoinositide 3-kinase signaling pathway alters host response to sepsis, inflammation, and ischemia/reperfusion injury. Shock. 2006;25:432-439.
334 Yamaji Y, Kubota T, Sasaguri K, et al. Inflammatory cytokine gene expression in human periodontal ligament fibroblasts stimulated with bacterial lipopolysaccharides. Infect Immun. 1995;63:3576-3581.
335 Yamamoto M, Fujihashi K, Hiroi T, et al. Molecular and cellular mechanisms for periodontal diseases: role of Th1 and Th2 type cytokines in induction of mucosal inflammation. J Periodontal Res. 1997;32:115-119.
336 Yamamoto M, Sato S, Hemmi H, et al. Essential role for TIRAP in activation of the signalling cascade shared by TLR2 and TLR4. Nature. 2002;420:324-329.
337 Yamamoto M, Sato S, Hemmi H, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301:640-643.
338 Yamamoto M, Sato S, Hemmi H, et al. TRAM is specifically involved in the Toll-like receptor 4-mediated MyD88-independent signaling pathway. Nat Immunol. 2003;4:1144-1150.
339 Yamazaki K, Nakajima T, Gemmell E, et al. IL-4- and IL-6-producing cells in human periodontal disease tissue. J Oral Pathol Med. 1994;23:347-353.
340 Yoneyama M, Fujita T. RNA recognition and signal transduction by RIG-I-like receptors. Immunol Rev. 2009;227:54-65.
341 Yoshie H, Kobayashi T, Tai H, Galicia JC. The role of genetic polymorphisms in periodontitis. Periodontol 2000. 2007;43:102-132.
342 Yoshimura A, Kaneko T, Kato Y, et al. Lipopolysaccharides from periodontopathic bacteria Porphyromonas gingivalis and Capnocytophaga ochracea are antagonists for human toll-like receptor 4. Infect Immun. 2002;70:218-225.
343 Yoshioka H, Yoshimura A, Kaneko T, et al. Analysis of the activity to induce toll-like receptor (TLR)2- and TLR4-mediated stimulation of supragingival plaque. J Periodontol. 2008;79:920-928.
344 Yun PL, DeCarlo AA, Collyer C, Hunter N. Modulation of an interleukin-12 and gamma interferon synergistic feedback regulatory cycle of T-cell and monocyte cocultures by Porphyromonas gingivalis lipopolysaccharide in the absence or presence of cysteine proteinases. Infect Immun. 2002;70:5695-5705.
345 Zarember KA, Godowski PJ. Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J Immunol. 2002;168:554-561.
346 Zhang FX, Kirschning CJ, Mancinelli R, et al. Bacterial lipopolysaccharide activates nuclear factor-kappaB through interleukin-1 signaling mediators in cultured human dermal endothelial cells and mononuclear phagocytes. J Biol Chem. 1999;274:7611-7614.
347 Zou W, Bar-Shavit Z. Dual modulation of osteoclast differentiation by lipopolysaccharide. J Bone Miner Res. 2002;17:1211-1218.
Suggested Reading
Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783-801.
Artis D. Epithelial-cell recognition of commensal bacteria and maintenance of immune homeostasis in the gut. Nat Rev Immunol. 2008;8:411-420.
Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423:337-342.
Graves D. Cytokines that promote periodontal tissue destruction. J Periodontol. 2008;79(8 Suppl):1585-1591.
Kabelitz D. Expression and function of Toll-like receptors in T lymphocytes. Curr Opin Immunol. 2007;19:39-45.
Karin M, Lawrence T, Nizet V. Innate immunity gone awry: linking microbial infections to chronic inflammation and cancer. Cell. 2006;124:823-835.
Kirkwood KL, Cirelli JA, Rogers JE, Giannobile WV. Novel host response therapeutic approaches to treat periodontal diseases. Periodontol 2000. 2007;43:294-315.
Patil CS, Kirkwood KL. p38 MAPK signaling in oral-related diseases. J Dent Res. 2007;86:812-825.
Sarkar FH, Li Y, Wang Z, Kong D. NF-kappaB signaling pathway and its therapeutic implications in human diseases. Int Rev Immunol. 2008;27:293-319.